SEARCH RESULTS FOR: Inflammation
systemic-lupus-erythematosus-gastrointestinal-manifestations

esophageal-gastric-varices

Legend:
Pathophysiology Mechanism
Sign/Symptom/Lab Finding
Complications
• Venous drainage of spleen backed up into gastric anastomoses
Tachycardia and hypotension
Anemia Death Melena Coffee ground emesis Hematemesis Bright red blood per rectum")
central-retinal-artery-occlusion-pathogenesis-and-clinical-findings
 (i.e. Valvular, arrhythmias, congenital defects) (i.e. OCP, Protein C&S deficiency, ATIII deficiency) (i.e. leukemia/lymphoma, sickle cell, polycythemia) Endothelial cell damage Abnormal blood flow 1` coagulation and/or 1 blood viscosity and creates hypercoagulable state causing localized stasis 4, anti-coagulation inflammation
Abbreviations: • GCA — Giant Cell Arteritis • SLE — Systemic Lupus Erythematosus • GPA — granulomatosis with polyangitis • OCP — Oral contraceptive pill • ATIII — Anti-thrombin Ill
Thrombus formation
Blockage of central retinal artery
Central Retinal Artery Occlusion (CRAO)
Authors: Graeme Prosperi-Porta Reviewers: Stephanie Cote Usama Malik Johnathan Wong* * MD at time of publication Carotid Artery Atherosclerosis
Atherosclerotic plaque dislodges from carotid artery
The retina becomes pale 4, perfusion of retinal Slow retinal artery blood Acute retinal edema Ganglion cells and axons from NI, perfusion arterioles due to upstream flow allows for caused by ischemia results death due to ischemia CRAO segmentation of the blood column in a blurred appearance of the retina results in disc pallor seen months after CRAO The choroidal vessels supplying the macula via the posterior ciliary artery become more prominent within a background of retinal pallor")
infectious-esophagitis-pathogenesis-and-clinical-findings

Herpes Simplex Infection of squamous cells and macrophages
Colonization facilitated by use of antacid therapy
Nuclear Large Superficial Squamous Macrophage inclusion bodies esophageal ulceration ulcers cell inclusion bodies aggregation Legend: Pathophysiology Mechanism Sign/Symptom/Lab Finding Complications
Spores and pseudohyphae seen on biopsy
• Invas.on of underlying blood vessels
• White plaques over erythematous base
'1' neutrophils due to inflammatory response")
Primary Spontaneous Pneumothorax: Pathogenesis and clinical findings

Malnutrition Smoking
Structurally compromised lung parenchyma
Notes: • PSPs usually occur at rest • Respiratory symptoms vary in severity • Suspect thoracic endometriosis in young women with recurrent PSPs that coincide with menstruation • *Pathophysiology of tension pneumothorax is described in a separate slide
Air leaks into the subcutaneous tissue
Subcutaneous emphysema
Authors: Lauren Hampton Reviewers: Kening (Midas) Kang Natalie Morgunov Sadie Kutz Usama Malik Leila Barss* * MD at time of publication
Thoracic Ischemia endometriosis
Mechanical forces of respiration create blebs Inflammation disrupts mesotheial and/or bullae ~ cell layer of the visceral pleura
47
Spontaneous rupture of blebs or bullae
47
Sudden onset pleuritic chest pain
71r
Primary spontaneous pneumothorax: Presence or introduction of air in the pleural space in a patient WITHOUT diagnosed or clinically apparent lung disease Tachycardia Abbreviations: Communication occurs between the alveoli and pleural space • FLCN- Folliculin gene • HCY- Homocystinuria • MFS- Marfan syndrome Alveolar pressure > pleural pressure • CTD- Connective tissue disease • PSP- Primary spontaneous Air from the lungs enters the pleural space pneumothorax • V/Q- ventilation/perfusion Air separates the chest from /1` intrapleural pressure • Sp02- oxygen saturation the lung parenchyma Small areas of lung collapse Blood flow to areas of On affected side: under un-opposed intrinsic atelectasis is maintained -• Si, chest wall expansion elastic recoil while ventilation 4, t resonance to percussion Si, or absent tactile fremitus Si, or absent breath sounds 4, lung compliance Shunting and V/Q mismatch Pleural line on chest x-ray 1` work of breathing Tension pneumothorax* Accessory muscle use, 4• Sp02, Sudden onset dyspnea, Tachycardia Hypotension, Juglar venous Tachypnea distension, Pulsus paradoxus")
Asthma Acute Exacerbation: Pathogenesis and Treatment
: An episode of increased symptoms due to decreases in airflow
Abbreviations • PCO2: Partial pressure of CO, in arterial blood • PEF: Peak expiratory flow • SABA: Short-acting beta-2 agonists • Sp02 : Blood oxygen saturation level
Mild to moderate exacerbation: PEF 50% of predicted
Titrate O2 toSpO2, 92%, give SABA & steroids ■
Good response: symptoms resolved, PEF > 80%
[Treat at home with SABA as needed and steroids
Dyspnea
Bronchoconstriction
1` Residual volume and 1` PCO2
Respiratory failure
1` Air trapping causes '1' intra-alveolar pressure
Severe exacerbation: PEF 50% of predicted Educate patient regarding medications, Loss of Pulsus inhaler technique & [consciousness paradoxus follow up with primary care provider I
Legend: Pathophysiology Mechanism
Titrate O2 to402 93%, give SABA, steroids & magnesium sulfate
Sign/Symptom/Lab Finding
{Worsening symptoms and/or respiratory failure: Do not delay intubation, send to ICU, give SABA, steroids & magnesium sulfate
Authors: Luke Gagnon Reviewers: Midas (Kening) Kang Usama Malik Lian Szabo* * MD at time of publication
4, Delivery of oxygen rich air to alveoli 4, Oxygenation of blood
Drowsy and confused
Central cyanosis
• Tachycardia
Pneumothorax
[Depending on 1 severity: Observation or place chest tube")
Bronchiectasis Pathogenesis and clinical findings
, MAC complex infection, COPD, allergic bronchopulmonary aspergillosis, chronic infections
Irreversibly dilated bronchi
Chronic bronchial infection and inflammation
1
Easily collapsible airways
I Bronchiectasis (persistent and progressive damage to lungs)
Chronic cough (mucopurulent)
Defect in immunity and/or mucus clearance
Persistent bacteria in airway (commonly Pseudomonas/Staph aureus)
Inflammatory response
Rhinosinusitis
Abbreviations: • A1AT — Alpha-1-antitrypsin • COPD — Chronic Obstructive Pulmonary Disease • HIV — Human Immunodeficiency Virus • MAC — Membrane Attack Complex • VQ— Ventilation/Perfusion ratio
Legend:
Pathophysiology Mechanism
Fever
Sign/Symptom/Lab Finding
Failure to thrive (children)
Authors: Rebecca (Becky) Phillips Reviewers: Midas (Kening) Kang Usama Malik Eric Leung* * MD at time of publication
Notes: • Can be focal (single lobe/segment) or diffuse (both lungs) • Mainly in elderly • 1% prevalence in children
Tissue damage
Epithelial destruction of airways
Further impairment of bacterial clearance
Persistent inspiratory adventitious sounds (crackles > wheezing)
Complications
Structural damage to bronchial walls
Obstructive pulmonary function tests
Hemoptysis
Chest pain
VQ mismatch and 4, gas exchange
4, oxygenation
Digital clubbing (rare)
Fatigue Dyspnea
Cyanosis (uncommon)")
Benign Prostatic Hyperplasia: Pathogenesis and medications

Note: MoA not fully established
PDE-5 inhibitors (e.g. tadalafil)
Bladder and prostate smooth-muscle a-1 receptor antagonism
—110.
Relaxation of bladder outlet and prostate smooth-muscle
Authors: Michael Korostensky Reviewers: Alex Tang Usama Malik Dr. Jay Lee* * MD at time of publication
Acronyms: 5-ARI = 5-a reductase inhibitors COX = cyclooxygenase DHT = dihydrotestosterone GnRH = gonadotropin-releasing hormone LUTS = lower urinary tract symptoms PDE-5-mediated cGMP degradation in prostate smooth-Improved urinary outflow muscle and associated vascular supply Relaxation of prostate smooth-muscle MoA = mechanism of action NSAID = nonsteroidal anti-inflammatory drugs PDE-5 = phosphodiesterase-5 -NO
5-ARIs (e.g. dutasteride)
LHRH receptor antagonists (e.g. cetrorel ix)
P3-adrenergic agonists (e.g. mirabegron)
anticholinergics (e.g. oxybutynin)
NSAIDs
1` Bladder pressures
Pathophysiology Mechanism
5-a-reductase activity
1, Conversion of testosterone into DHT
4, Progression of LUTS
1, Testosterone secretion from testicular Leydig cells 1, LH secretion from pituitary GnRH antagonism DHT production Relaxation of detrusor Bladder muscle 1` capacity Improved LUTS
Acetylcholine antagonism at muscarinic receptors Relaxation of bladder outlet smooth-muscle 1` volume to first detrusor contraction 4, Prostaglandin release Analgesia and 4, Prostatic ,f, COX activity ► inflammation —110. Bladder smooth-muscle hyperplasia (detrusor thickening) /1` Sensitivity (i.e. overactive detrusor) -1110. 1, Volume to first detrusor contraction LUTS")
Celiac Disease: Pathogenesis and clinical findings

Genetic predisposition -■ (Northern European, Down's syndrome, Associated with HLA DQ 2,8)
Note: *The anti-TTG antibody is an IgA anti-body, therefore if the patient is IgA-deficient, absence of anti-TTG does not rule out celiac
Anti-TTG in serum*
Anti-TTG reacts with TTG in skin
Deposition of anti-TTG in renal glomeruli
Dermatitis herpetiformis
Chronic Kidney Disease
Small Bowel Biopsy: Crypts of bowel become enlarged (hyperplasia) with architectural change, villous shortening
Legend: Pathophysiology
Mechanism
Exposure to prolamins (proteins found in wheat, rye, oats, barley)
TTG alters prolamin Altered protein fits more easily into HLA
HLA activates adaptive immunity
IgA generated against prolamin-TTG
Wheat prolamin (gliandin) interacts with and activates zonulin signalling
Gut epithelium becomes more porous
Large dietary proteins in epithelium disrupt tight junctions
Author: Matthew Harding Yan Yu Peter Bishay Reviewers: Dean Percy Jason Baserman Usama Malik Kerri Novak* * MD at time of publication
Inflammation of Intestinal epithelium Inflammation disrupts structure of bowel mucosa Mechanism Unknown Lymphocytes migrate to site of inflammation Extraintestinal Complications: Arthropathy Ataxia (gluten associated) Infertility Mild Hepatitis Villi of intestine atrophy Risk of Microscopic Colitis (50x) Malabsorption Extraintestinal manifestations: Chronic watery Fatigue Failure to thrive, weight loss Anemia (Fe, B12, folate) Peripheral neuropathy (B12, Ca) Ataxia (Ca) Dysrhythmia (Ca, K) Osteoporosis (Vit D, Ca) diarrhea + Intestinal manifestations steatorrhea: Pale, foul-smelling Abdominal bloating Steatorrhea (fat in stool) Diarrhea")
Orbital Cellulitis: Pathogenesis and clinical findings
 Periorbital cellulitis1,2
Hematogenous spread
Contiguous spread of infection
Pathogens reach orbital tissue (posterior to the orbital septum)
Spreads to periorbital tissue (anterior to the orbital septum)
Localized inflammation
Conjunctival chemosisa
Eyelid and periorbital edema
Pain on palpation
Induration
Warmth
Orbital Cellulitis Inflammation of orbital tissue Proptosis
Spreads to surrounding structures
Subperiosteal abscess Brain abscess Cavernous sinus thrombosis Meningitis Subdural empyema Orbital abscess
Notes:
Impinges on ocular muscles
Impaired extra- ocular movements
Pain with eye
movement or opthalmoplegia
Definitions:
Impinges on nerves
Afferent pupillary defect
Decreased visual acuity
Exposes cornea
Corneal drying and scarring
a. Chemosis: Edema of the bulbar conjunctiva
b. Panopthalmitis: inflammation of all coats of the eye including intraocular structures.
c. Endopthalmitis: inflammation of the interior of the eye.
1. See slide on Periorbital Cellulitis for how sinusitis can lead to the development of periorbital cellulitis
2. The micro-organism responsible for periorbital cellulitis varies depending on how the pathogen was introduced to the system.
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published November 5, 2018 on www.thecalgaryguide.com")
Periorbital Cellulitis: Pathogenesis and Clinical Findings

Note: Also referred to as preseptal cellulitis
Dacryoadenitisa Conjunctivitisb
Acute chalazionc
Dacryocystitisd Hordeolume
Streptococcus pneumoniae, Moraxella catarrhalis, non-typable Haemophilus influenza (most common organisms)
Abrasion Insect bite
Burns Trauma
Local infection
Contiguous spread of infection
Sinusitis
Otitis media Hematogenous spread
Local break in skin Micro-organisms enter
Definitions:
Note:
Eye exam should reveal normal:
- extra-ocular
movements and globe
position
- pupillary reflex and
visual acuity
If any are abnormal, the presentation is no longer considered periorbital cellulitis, as the infection has likely spread beyond the preseptal compartment/orbital septum.
If the eye cannot be assessed, the patient NEEDS a CT scan.
Pathogens reach dermis and subcutaneous periorbital tissue
Periorbital Cellulitis
a. Dacryoadenitis: infection of the lacrimal glands
b. Conjunctivitis: inflammation of the conjunctiva
c. Chalazion: a benign, painless bump or nodule inside the upper or lower eyelid which results from healed internal hordeolums that are no longer infectious.
d. Dacryocystitis: an infection of the lacrimal sac, secondary to obstruction of the nasolacrimal duct at the junction of lacrimal sac.
e. Hordeolum: localized infection or inflammation of the eyelid margin involving hair follicles of the eyelashes or meibomian glands.
Spreads beyond preseptal compartment/orbital septum
Involves the orbit Orbital cellulitis
See slide on Orbital Cellulitis: Pathogenesis and clinical findings
Localized inflammation
Pain on palpation
Induration
Warmth
Eyelid and periorbital edema
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published November 5, 2018 on www.thecalgaryguide.com")
Mastoiditis: Pathogenesis and clinical findings

Distal middle ear is physically connected to mastoid air spaces
Pathogens spread from middle ear to the mastoid air spaces
Mucosa lining the mastoid becomes inflamed
Mastoiditis 1
Infection persists
Accumulation of pus in mastoid cavities
↑ pressure Formation of abscess cavities
Dissection of pus into adjacent areas
Infection spreads
Into intracranial compartment
See slide on Acute Otitis Media (AOM): Pathogenesis and Clinical Findings in Children
Post- operation
Trauma Infection
Notes:
1. Most common suppurative complication of AOM
Tenderness, erythema, swelling and fluctuance over the mastoid process
Inflammation spreads to external auditory canal
Cranial Nerve VII anatomically near mastoid
Cranial Nerve VIII anatomically near mastoid air space
Destroys bony septae b/t air cells (visible on CT)
Mastoid abscess
Swelling of external auditory canal
Mastoid inflammation disrupts nerve
Mastoid inflammation disrupts nerve
Petrositis
Facial nerve palsy
Sensorineural hearing loss Labyrinthitis
Osteomyelitis of the calvaria
Into adjacent bones
Underneath the periosteum Subperiosteal abscess Pinna is pushed out and
of the temporal bone
Into the neck beneath the attachment of the sternocleidomastoid and digastric muscles
forward
Dural venous thrombosis Temporal lobe abscess Meningitis
Epidural abscess Subdural abscess Cerebellar abscess
Bezold abscess
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published November 5, 2018 on www.thecalgaryguide.com")
Sinusitis: Pathogenesis and clinical findings

Venous insufficiency- Signs and symptoms

Reactive Neutrophilia- Pathogenesis and Clinical Findings

Primary Myelofibrosis pathogenesis and clinical findings

within hematopoietic
stem cells
Non-driver mutations in
other myeloid genes
(e.g., LNK, CBL, TET2,
ASXL1, IDH)
Constitutive activation of
cellular proliferation
pathways
↑ cell signaling
↑ gene transcription and
expression
Cellular proliferation and
resistance to apoptosis
Proliferation of abnormal
megakaryocytes
↑ neutrophil engulfment
by megakaryocytes
↑ growth factor release
by megakaryocytes
Stimulation of
fibroblasts
Stimulation of
endothelial cells
New blood vessel formation
↑ osteoprotegerin Unbalanced osteoblast
proliferation Osteosclerosis
Fibrosis of the
bone marrow
Anemia
Bleeding and
bruising
Infections
Fatigue and pallor
Bone pain
Increased cell
turnover
Tumor lysis
syndrome
Cachexia, night sweats,
fever/chills, malaise
Expanding
marrow pushing
against bone
Extramedullary
hematopoiesis Hepatomegaly
Portal
hypertension
Splenomegaly
↑ LDH
Thrombocytosis
Leukocytosis
Secretion of
coagulation
inducing cytokines
Arterial and
venous
thromboembolism
↓ blood cell
production
&
leukoerythroblastosis
Thrombocytopenia
Leukopenia
↑ K+, PO4
2-, uric acid
↓ Ca2+
Bone pain
Periostitis
Immature granulocyte and
erythroid precursors with blasts
↑ cytokine
production
(+)
↑ sequestration of blood cells
Disseminated
intervascular
coagulation
(see MAHA slide)
Definitions:
Periostitis – Inflammation of the
membrane surrounding bone
Osteosclerosis – Abnormal hardening
and increased in density of bone")
Hemorrhoids - Pathogenesis and Clinical Findings

Increased Intra-Abdominal Pressure
I.e. pregnancy, constipation, chronic straining,
lifting, cirrhosis
Hemorrhoids: Pathogenesis and clinical findings
Dilations originate from inferior
hemorrhoidal venous plexus
Vascular cushions engorge
along anal canal
Legend: Published March 30, 2019 on www.Pathophysiology Mechanism Sign/Symptom/Lab Finding Complications thecalgaryguide.com
Authors:
Aleeza Manucot
Reviewers:
Yoyo Chan
Sean Doherty
Dr. Sylvain Coderre*
* MD at time of publication
Supporting tissues of anal cushions weaken,
disintegrate, or deteriorate
Inflammatory reaction
occurs, involving vascular
wall and connective tissue
Thrombosis
Pain
↑ mucus secretions or fecal
soiling of prolapsing
hemorrhoids
Cushion epithelium erodes via
damage from compression
Painless
rectal
bleeding
Bleeding without prolapse
Prolapse with spontaneous
reduction
Prolapse requiring manual
reduction
Irreducible
1st degree
2nd degree
3rd degree
4th degree
Infarction and thrombosis
Acute severe pain
Anal cushions prolapse (downwardly slide)
into rectum or open space
Dentate line: divides
the upper two thirds
and lower third
of the anal canal
EXTERNAL Hemorrhoids
- Found distal to the dentate line
- Somatic innervation
Somatic nerve
receptors activated
Sebaceous glands
↑ secretions around
area of hemorrhoid
Itching Perianal
irritation
Swelling
Inflammation creates
prothrombotic state
Hemorrhoids")
gastroesophageal-reflux-disease-gerd-complications
: Complications
Esophageal stricture
disease
Esophagitis
Esophageal
adenocarcinoma
Barrett’s esophagus
GERD
Reflux of gastric content into distal esophagus
Damage to squamous
esophageal epithelium
Legend: Published March 30, 2019 on www.Pathophysiology Mechanism Sign/Symptom/Lab Finding Complications thecalgaryguide.com
Authors:
Wendy Wang
Reviewers:
Yoyo Chan
Sean Doherty
Dr. Sylvain Coderre*
* MD at time of publication
Squamous esophageal
epithelium undergoes
metaplasia to become
columnar epithelium
This predisposes cells to
premalignant changes
(dysplasia)
Collagen is deposited
where ulcers heal
Asthma/Chronic Cough
Chronic Laryngitis
Laryngeal and
Tracheal Stenosis
Extra-esophageal Complications Esophageal Complications
Airway becomes
irritated
Fibroblasts proliferate
and deposit granulation
tissue in airway
Tissue deposition
leads to narrowing of
laryngeal and
tracheal space
Damage to pharyngeal
lining and airway
Esophageal tissue repeatedly
exposed to stomach acid
Pro-inflammatory cells and cytokines
are recruited to the area
Definitions:
• Metaplasia: abnormal change in the
nature of a tissue
• Pro-inflammatory cells and cytokines:
Mediators of inflammation. Examples
of cells include macrophages and T
cells, cytokines include IL-17, IL-2, IL-4
Over time, collagen fibers
contract
Bronchoconstriction
↑ vagal
tone
↑ bronchial
reactivity
Cough sensory
nerve endings are
stimulated
Vagal reflex
is activated
Activation of
cough center in
brainstem
↑ inflammation of
squamous epithelium
Ulcers form in esophagus")
Polyarteritis Nodosa (PAN): Pathogenesis and Clinical Findings
: Pathogenesis and clinical findings
Environmental triggers
Infectious/viral agents (commonly Hepatitis B)
Medical Comorbidities Malignancies (most commonly hairy-cell leukemia)
Immunogenetic Predisposition: patient is genetically predisposed to a dysregulated immune response
Fever
↑ ESR and CRP
Postulate 1
Viral antigen-antibody complexes deposit in vasculature, causing lesions and activating cellular inflammatory response
Authors: Nela Cosic, Yan Yu* Reviewers: Sean Doherty Martin Atkinson*
* MD at time of publication
Palpable or necrotic purpura
Malignant Hypertension
Renal Insufficiency
Myocardial ischemia
Heart failure Diffuse myalgias
Postulate 2
Viral replication causes direct injury to vascular endothelial cells
↑ Anti- endothelial cell autoantibodies (AECA)
Altered cytokine profile (↑TNF-α, IL-1β, IFN-α, IL- 2)à↑ T-cell mediated immune response
Weight metabolism Loss
Autoimmune attack on various areas of the body
Malaise and/or Arthralgias (knees, ankles, elbows, wrists)
Orchitis: Testicular pain, erythema and/or swelling
Small intestine perforation GI Manifestations
Non-specific abdo pain
GI hemorrhage
Peripheral sensory changes: Distal mononeuropathy
multiplex
Polyarteritis Nodosa (PAN)
Focal segmental necrotizing leukocytoclastic vasculitis of medium or small-sized arteries
Inflammation of arteries damages the vascular endothelium of those arteries
Inflammation predisposes formation of arterial thromboses
Blockage of arteriesà tissue ischemia and possible necrosis (tissue cell death)
↑ basal
Arterial aneurysms
Inflamed subcutaneous arteries
Inflamed renal artery
àluminal narrowing and reduced blood flow to kidneys
Inflamed coronary artery à luminal narrowing, occlusion, thromboses
Segmental inflammation of muscular arteries, stimulating surrounding nociceptors. Muscle ischemia develops long-term.
Ischemia/necrosis of the testicles
Ischemia/necrosis of the small intestine
Ischemic vasculitic nerve damage: Immune complex deposition within vessel walls of arteries traveling with nerves leads to persistent vascular inflammation and ischemia of associated nerve
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published April 18, 2019 on www.thecalgaryguide.com")
Crohn's Disease

Inflammation of the GI tract lining
- Inflammation is “transmural”, spanning the entire thickness of the intestinal wall from luminal mucosa to the serosa.
- The inflammation occurs anywhere in the GI tract from the oral mucosa to the anal mucosa (from ‘gums to bum’) in skip lesion pattern.
Atrophy, scarring of the intestinal villi
Inflammatory cytokines destroy the mucosa epithelial cells of the GI tract wall, causing cell apoptosis and ulceration
↑ permeability of the blood vessels supplying the GI tract wall
Chronic inflammation impairs healing responses
Dysregulated wound healingàexcess
extracellular matrix deposition
Fibrosis leads to scar tissue and thickening of all layers of the GI tract
Strictures
Inflammation is systemic, affecting:
Joints Arthropathy Erythema
Impaired absorption of nutrients
Weight loss
Prolonged GI bleeding
Anemia
Transporter proteins responsible for Na+ reabsorption gradually disappear from the epithelium
More sodium (and thus water) is
retained in the GI tract lumen
Microperforations can penetrate through the intestinal wall
Anal fistulae (“holes” connecting the anus to the skin, bladder, peritoneum, small bowel, etc.)
Continued inflammation and/or infection can lead to:
Leakage of fluid out of capillaries into the GI tract
Luminal edema and swelling
Narrowing of GI lumenàbowel obstruction
Skin
Mouth Eyes
Liver
nodosum, pyoderma gangreno- sum
>5 canker sores
Uveitis
Iritis, scleritis
Sclerosing cholangitis
↓ fat absorption
Fatty acids (negatively charged) bind Ca2+, freeing oxalate from Ca2+
↑ oxalate absorbed into blood & filtered by kidney
Calcium oxalate kidney stones
Diarrhea
Abdominal cramping and pain
(see Bowel Obstruction page for full mechanism
Anal abscesses Inflammatory masses
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Re-Published June 15, 2019 on www.thecalgaryguide.com")
Ulcerative Colitis

Immune response against the GI tract. (Unclear mechanism, but thought to be mediated by cytokine release and neutrophil infiltration)
Inflammation of the GI tract epithelial lining
- Starting at the rectum and moves up the colon and is continuous (does not invade the small intestine)
- Inflammation affects the mucosal and submucosal only
Diarrhea, abdo pain and cramping causing avoidance of food
Weight loss
Apoptosis of GI tract mucosa
Transporter proteins responsible for Na+ reabsorption gradually disappear from the epithelium
Ulceration, into the anus, and more severe
Prolonged Bleeding - GI and anus
Anemia, often iron deficiency
Inflammation ↑ permeability of the blood vessels supplying the GI tract wall
Fluid leak out of capillaries into GI tract wall, causes edema and swelling
Swelling narrows the GI tract lumen, causing bowel obstruction
Inflammation, ulceration, or infection at the anus (all involve the RECTUM!)
Anal irritation stimulates autonomic and somatic nerves leading up to the brain, causing the pt to want to defecate
Tenesmus, urgency, frequency (feeling or urgency to defecate, but little stool is produced)
Joints Skin
Arthroplasty/ joint pain
Erythema nodosum, pyoderma gangrenosum
Mouth >5 canker sores
More sodium (and thus water) is retained in the GI tract lumen
Bloody Diarrhea, usually bloody due to anal bleeding and ulceration bleeding
Abdominal Cramping and pain (see Bowel Obstruction page for full mechanism)
Eyes (uvea, iris, sclera)
Liver Blood
Uveitis
Iritis, scleritis
Sclerosing Cholangitis
Autoimmune hemolytic anemia
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Re-Published May 5, 2019 on www.thecalgaryguide.com")
perforated-viscous
:
Author:
Yan Yu
Reviewers:
Michael Blomfield, Tony Gu, Dean Percy, Danny Guo Maitreyi Ramran* * MD at initial time of publication
Chest X-Ray (CXR)
Pathogenesis and Clinical Findings
Diverticulitis
Crohn’s disease Peptic ulcer (H. pylori
infection, NSAID use, ICU stress, etc)
Appendicitis
Malignant neoplasm
Irritates visceral peritoneum, stimulates autonomic nerves
Severe inflammation causes destruction of GI tract mucosa
Over time, Perforation of the GI tract wall
Bowel contents (air, fluids) released into peritoneal cavity
Massive peritoneal inflammation
Diagnostic investigations if a GI perforation is suspected
Dull diffuse abdominal pain
Severe, Sharp abdominal pain with peritoneal signs
Abdominal X-ray
Irritation of parietal peritoneum, stimulates somatic nerves
• Abdominal X-ray
• Intra-peritoneal air will coat the GI tract surfaces, giving them a faint white outline
under X-ray
• Chest X-ray of upright patient (Diagnostic)
• Intra-peritoneal air will rise above the peritoneal fluid when pt is upright, accumulating under the right hemi-diaphragm.
• Note: air under left hemi-diaphragm = normal gastric bubble
• CT? Most patients with suspected GI perforation will get a CT scan, but this is not the diagnostic gold standard (and access to CT can be limited, especially in rural settings)
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Re-Published June 30, 2019 on www.thecalgaryguide.com")
acanthosis-nigricans-pathogenesis-and-clinical-findings

Appendicitis

The appendix is anatomically located in the RLQ; appendicitis may be confused with disorders of surrounding structures: Gynecological Diseases
• RuleoutpregnancywithHCG pregnancy test
• Rupturedovariancyst
• Ectopicpregnancy
• Mittelschmerz(mid-cycle
pain)
Gastro-intestinal Diseases
• Meckel’sdiverticulum (presents identically to appendicitis; surgically located 2 feet from ileocecal valve; mostly seen in children)
• Diverticulitis(presentsasleft sided appendicitis)
Non-GI Abdominal Issues
• Mesentericadenitisinkids <15: swollen mesenteric lymph nodes
• Renalcolic
Obstruction of appendiceal lumen (by fecalith, fibrosis, neoplasia, foreign bodies or lymph nodes in kids)
Appendix distension and spasms
↑ lumen pressure, ↓ blood flow to appendix
Ischemia, tissue necrosis, loss of appendix structural integrity
Bacterial invasion of the appendix wall, causing transmural inflammationandnecrosis
Stretching of visceral peritoneum, stimulation of autonomic nerves T9-T10
Progression of inflammation over several days (variable length of time)
Irritation of parietal peritoneum, stimulation of somaticnerves
If appendix not surgically removed
Perforation of colon wall, causing peritonitis, abscesses or death
Note: Symptoms hugely variable. Only 30% present with classic history. Diagnosis is mostly clinical. Further investigations:
CBC: Leukocytosis (due to inflammatory response) CT: Gold standard test. Thickened visceral membrane with enhancing (white) rim due to ↑ blood flow
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Re-Published July 27, 2019 on www.thecalgaryguide.com")
Acute GI Related Abdominal Pain

Bowel stretching, pulling, contracting
Abdominal pain type:
Diffuse, non-localized Dull, crampy, periodic Not associated with movement
Patient may writhe around, trying to get rid of the pain
Mesentery Intestinal lumen
Parietal peritoneum
(innervated by somatic nerves)
Cross-section of the GI tract
Cuts, structural damage, and inflammation in the bowel
Important Notes
• Acute abdominal pain can also result from non-
gastrointestinal causes, such as kidney stones, female reproductive tract issues, and urinary tract issues. For simplicity’s sake, only the GI-related acute abdominal pain disorders are listed here.
• The DDx of visceral abdominal pain is broad. Please consult relevant sections of the Calgary Black Book for the DDx.
• Keep in mind that visceral abdominal pain can also be caused by the “acute abdomen” diseases (if the diseases are presenting in their initial phases).
• • •
• • •
Abdominal pain type:
Sharp, well-localized
Excruciatingly painful, persistent Associated with movement of bowels
Patient often lies still to avoid abdominal vibration
Peritoneal signs
Abdominal guarding, pain with abdominal vibration (coughing, shaking, percussion, palpation)
Transition from diffuse to localized pain can indicate disease progression (e.g. from visceral to parietal peritoneal inflammation)
Note: bowel obstruction may or may not present as acute abdominal pain
Bowel Infarction
Appendicitis Diverticulitis
Acute Cholecystitis
Acute Pancreatitis
Perforated Ulcer
DDx of an “acute abdomen”:
A sudden, non-traumatic disorder of the abdomen that needs urgent diagnosis and treatment. Each topic will be further explored in their respective slides.
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Re-Published July 27, 2019 on www.thecalgaryguide.com")
virchows-triad-and-deep-vein-thrombosis-dvt
:
Authors: Dean Percy Yan Yu Reviewers: Tristan Jones Ryan Brenneis Man-Chiu Poon* Maitreyi Raman* * MD at time of publication
Pregnancy, Oral Contraceptives (OCP)
Pathogenesis and Complications
Platelet Activation
Increased clot formation
Hypercoagulable State
↑ ability for the blood to coagulate upon stimulation
Inherited Disorders
Congenital defect in coagulation (ie. Factor V Leiden, Factor II
mutation, Protein S/C deficiency) ↑ blood clotting ability
Estrogen promotes
hypercoagulability, especially in presence of other risk factors
Notes:
• Venous thrombus causes pulmonary embolism, arterial thrombus causes stroke
• Previous DVT is risk factor for current DVT
Trauma/Surgery
Malignancy
Abnormal release of coagulation-promoting cytokines
Systemic injuryà activation of coagulation cascade
Hypertension
Bacteria Artificial Valve
Physically damages blood vessel walls
Adhere/invade vessel wall
Abnormal surface
Vessel Injury
Exposes tissue factor on damaged cells and subendothelium for vWF binding
Virchow’s Triad
Venous Stasis
Low blood flow rate over site of vessel injury, concentrating blood clotting factors at that site
Fat contains more aromatase, converts more androgens to estrogen
Sedentary lifestyle, poor venous return
Obesity
Clot formation typically occurs in leg veins
Deep, large veins allow for blood pooling (stasis, hypercoagulability) Venous return from legs often against gravity (stasis)
Valves in leg veins prone to backflow (stasis)
↓ muscle motion = ↓ venous blood flow
Fracture, immobilization, bedrest, long vehicle/airplane ride
Destruction of vein valve by clot
Venous Insufficiency
Clot prevents blood from returning to heart. Blood accumulating in the leg results in unilateral leg edema and venous inflammation (redness, warmth, tenderness)
1. 2. 3.
Clot embolizes to the lungs
Thromboembolus
-*Pulmonary embolism (acute life threatening complication)
-Chronic thromboembolic pulmonary hypertension
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Re-Published September 1, 2019 on thecalgaryguide.com")
Acute-Pancreatitis
![Acute Pancreatitis: Pathogenesis and Clinical Findings
Authors: Yan Yu Reviewers: Laura Craig Noriyah AlAwadhi Ryan Brenneis Maitreyi Raman* * MD at time of publication
Associated signs due to intra- abdominal hemorrhage from an unknown mechanism (classically associated with pancreatitis, but happens in <1% of cases):
Note:
It is not enough to just diagnose “acute pancreatitis”. Full management requires determining underlying etiology with further work-up.
Alcohol
↑ Toxic metabolites within pancreas and Spincter of Oddi Spasms
Gallstones
Migration to common bile duct blocks Sphincter of Oddi
Hypertriglyceridemia
Unknown
mechanism (rare)
Idiopathic
Further investigations:
CBC: Cell counts elevated, due to sever hypovolemia
Serum [Lipase]: Gold Standard Diagnostic Test; rupture of pancreatic cells releases lipase into circulation
Pancreatic secretions back up, ↑ pressure within pancreas
Hypercalcemia (Rare; Ca2+ depositions in bile ducts block outflow of pancreatic secretions)
Since pancreas is retroperitoneal, somatic
nerves in the parietal peritoneum are directly stimulated
Inflammation triggers cytokine release
Inflamed pancreas irritates adjacent intestines, causing ileus
Inflamed, more permeable blood vessels leak fluid into pancreas
• •
Cullen’s sign (bruising in peri-umbilical region) Grey-Turner’s sign (bruises along both flanks)
Sudden, severe epigastric pain (with peritoneal signs), radiates to the center of the back
Fever, nausea/vomiting
(general signs of inflammation)
Diminished bowel sounds Profound dehydration
(flat JVP, hypotension, tachycardia, oliguria) – may happen, not always
1. Pressure compresses pancreatic blood vessels, causing tissue ischemia.
2. Activation of inactive proteases (zymogens) digesting pancreatic tissue
Necrosis (death) of pancreatic cells
Inflammation self- perpetuates
Massive systemic inflammatory response
2 main complications, usually detected on CT;
may happen, but not always
1. Pancreatic pseudocyst (enlargement of the
pancreas due to fluid accumulation)
2. Pancreatic necrosis/abscesses (death of a part of the pancreas)
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Re-published September 1, 2019 on thecalgaryguide.com](http://calgaryguide.ucalgary.ca/wp-content/uploads/2019/09/Acute-Pancreatitis.jpg "Acute Pancreatitis: Pathogenesis and Clinical Findings
Authors: Yan Yu Reviewers: Laura Craig Noriyah AlAwadhi Ryan Brenneis Maitreyi Raman* * MD at time of publication
Associated signs due to intra- abdominal hemorrhage from an unknown mechanism (classically associated with pancreatitis, but happens in <1% of cases):
Note:
It is not enough to just diagnose “acute pancreatitis”. Full management requires determining underlying etiology with further work-up.
Alcohol
↑ Toxic metabolites within pancreas and Spincter of Oddi Spasms
Gallstones
Migration to common bile duct blocks Sphincter of Oddi
Hypertriglyceridemia
Unknown
mechanism (rare)
Idiopathic
Further investigations:
CBC: Cell counts elevated, due to sever hypovolemia
Serum [Lipase]: Gold Standard Diagnostic Test; rupture of pancreatic cells releases lipase into circulation
Pancreatic secretions back up, ↑ pressure within pancreas
Hypercalcemia (Rare; Ca2+ depositions in bile ducts block outflow of pancreatic secretions)
Since pancreas is retroperitoneal, somatic
nerves in the parietal peritoneum are directly stimulated
Inflammation triggers cytokine release
Inflamed pancreas irritates adjacent intestines, causing ileus
Inflamed, more permeable blood vessels leak fluid into pancreas
• •
Cullen’s sign (bruising in peri-umbilical region) Grey-Turner’s sign (bruises along both flanks)
Sudden, severe epigastric pain (with peritoneal signs), radiates to the center of the back
Fever, nausea/vomiting
(general signs of inflammation)
Diminished bowel sounds Profound dehydration
(flat JVP, hypotension, tachycardia, oliguria) – may happen, not always
1. Pressure compresses pancreatic blood vessels, causing tissue ischemia.
2. Activation of inactive proteases (zymogens) digesting pancreatic tissue
Necrosis (death) of pancreatic cells
Inflammation self- perpetuates
Massive systemic inflammatory response
2 main complications, usually detected on CT;
may happen, but not always
1. Pancreatic pseudocyst (enlargement of the
pancreas due to fluid accumulation)
2. Pancreatic necrosis/abscesses (death of a part of the pancreas)
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Re-published September 1, 2019 on thecalgaryguide.com")
Endometritis

Uterine tenderness
Pelvic and/or abdominal pain
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published November 26, 2019 on www.thecalgaryguide.com")
Viral-Hepatitis
 of hepatocytes
Infection with chronic viruses (HBV and HCV) persist over time and additional symptoms may develop
RUQ pain/tenderness
If infection is prolonged or severe, inflammation becomes systemic
Release of hepatocyte’s cellular contents into the bloodstream
Infection with acute viruses (HAV and HEV) resolve over time, and the symptoms above normalize
Notes:
• HDV can only infect people with concomitant HBV infection
• HAV and HBV vaccines are the only ones that currently exist
• Not all patients with viral hepatitis will develop each of these symptoms. The presentations vary.
Fever, nausea, vomiting ↑ serum ALT, AST
↓ Hepatic metabolic activity (e.g. reduction of gluconeogenesis) ↓Serum Glucose
↓ Synthesis of plasma proteins (albumin, clotting factors, etc) ↓ Albumin, ↑ INR
Abbreviations:
• HAV - Hepatitis A Virus
• HBV - Hepatitis B Virus
• HCV - Hepatitis C Virus
• HEV - Hepatitis E Virus
• RUQ - Right Upper Quadrant
• ALT - Alanine Aminotransferase
• AST - Aspartate Aminotransferase
• INR - International Normalized Ratio
↓ Bilirubin clearance from blood, bilirubin ends up under the skin Jaundice Portal Hypertension
Encephalopathy, Splenomegaly, Esophageal Varices, Ascites, Caput Medusae, Edema
Encephalopathy, Muscle Wasting, Metabolic Bone Disease, Terry’s Nails, Ascites, Bruising, Clubbing, Edema
Spider Nevi, Altered Hair Patterns, Testicular Atrophy, Gynecomastia, Palmar Erythema
Progressive deterioration in liver function, possibly ending up in cirrhosis. (See slide on “Cirrhosis: pathogenesis and complications” for more details on mechanisms and full explanations.)
Hepatic Insufficiency Hyperestrogenism
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Re-Published January 12, 2020 on www.thecalgaryguide.com")
C5-C9-deficiency
 causes Secondary (acquired) causes
All are autosomal recessive Biologic therapy ex. eculizumab Absence or suboptimal functioning of
Abbreviations:
• MAC: Membrane Attack
Complex
• CH50: Classic Hemolytic
Complement Test
• AH50: Alternative Hemolytic
Complement Test
• CNS: Central Nervous System • CSF: Cerebrospinal Fluid
≥1 terminal complement proteins
C5-C9 deficiency
Inability to form MAC
↑ susceptibility to systemic Neisseria infection
Commonly N. meningitidis Rarely N. monorrhoeae
Nasopharyngeal colonization of N. meningitidis, ↑ susceptibility to bacteremia
CH50 ± AH50 assay No lysis
Note:
Total complement activity assay <10% activity C5, C6, C7, C9 <50% activity C8
Varied bactericidal action via other complement proteins
• Risk of invasive meningococcal disease is 1000-10000X higher in C5-C9 deficiency than in the general population
• Reason is unknown
• C5-C9 deficient patients are not at greater
risk for contracting other gram (-) infections • Clinical meningitis in C5-C9 deficiency is less
severe and fatality is rare
Bacterial lysis
Especially gram (-)ve bacteria like Neisseria
Bacteria cross the blood-brain barrier, causing swelling and damaging brain tissue
Fatigue, fever, headache, altered mental status, etc.
Inflammation of CSF and meninges
Activation of dura and pia mater fibres
Headache, neck stiffness
Bacteria release toxins
Damage to surface blood vessels
Maculopapular rash
Exact mechanism unknown
Recurrent meningitis
CNS damage due Sepsis to recurrent
meningitis
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published February 16, 2019 on www.thecalgaryguide.com")
Infarctus du myocarde: Antécédents médicaux

Auteur: Yan Yu Traductrice: Olivia Genereux Réviseurs: Sean Spence Tristan Jones Nanette Alvarez* Marie Giroux* *MD au moment de publication
Sang du ventricule gauche reflue à l’oreille[e gauche et finalement s'accumule dans le système vasculaire pulmonaire
Haute pression du système vasculaire pulmonaire force fluide hors des capillaires et dans l'interstitium pulmonaire et les alvéoles
Inflammation myocardique locale
Médiateurs inflammatoires irritent les nerfs cardiaques (plexus cardiaque)
̄ Fonc8on systolique (myocarde nécro6que ne peut pas se contracter suffisamment)
Invasion généralisée des cytokines inflammatoires
Cytokines agissent les régulateurs de température hypothalamique
Fièvre légère
Irritation des nerfs sympathiques afférents (T1-T4)
Signal entre moelle épinière, a/n dermatomes T1-T4
Cerveau perçoit irritation des nerfs comme douleur des dermatomes T1-T4
Douleur écrasante, pression, oppression de la poitrine: Souvent rétrosternale, avec radiation à épaule, cou et l'intérieur des deux bras (D>G) (Apparition: au repos, crescendo)
IrritaNon des branches cardiaques du nerf vague
AcNvaNon réponse vasovagale
Fatigue, étourdissements, nausée, vomissement
↓ volume systolique (VS) ↓ debit cardiaque (DC)
Activité sympathique (pour essayer de maintenir DC)
Sueurs (diaphorèse)
Peau moite
VasoconstricNon généralisé
Vasoconstriction des artérioles de peaux
Peau froide
Interstitium
pulmonaire
mou ↓
compliance
pulmonaire
d
Fluide compresse les voies respiratoires, ↑ résistance au flux d’air
Abrévia8ons:
• a/n – au niveau
À Noter: Douleur myocardique ischémique peut présenter différemment entre les patients, mais les symptômes récurrents habituellement présentent les mêmes pour un patient donné.
Muscles respiratoires travaillent plus fort pour venNler les poumons
Essoufflement
(difficulté à respirer)
Légende:
Physiopathologie
Mécanisme
Signe/Symptôme/Résultats de Laboratoire
Complications
Publié Janvier 20, 2013 sur www.thecalgaryguide.com")
acute-pancreatitis-complications
:
Interstitial edematous pancreatitis
Local accumulation of fluid in the pancreas
<2 weeks after onset
Acute peripancreatic fluid collection (not encapsulated)
Walled off by fibrous & granulation tissue
>2 weeks after onset
Pancreatic pseudocyst
(completely encapsulated)
Peritoneal irritation à pain
Large cyst can (very rarely) compress surrounding bowel
Acute Pancreatitis
Inflammatory cytokines are released from damaged pancreas
If recurrentàchronic pancreatitis (see relevant slide)
Inflammation damages pancreatic exocrine
cellsàInappropriate release of pancreatic enzymes into surrounding tissue & vasculature àdigesting pancreatic parenchyma
Authors: Nissi Wei, *Yan Yu Reviewers: Dean Percy, Miles Mannas, Varun Suresh, Brandon Hisey, *Kerri Novak, *Sylvain Coderre * MD at time of publication
complete resolution (most cases)
Necrotic tissue is vulnerable to
infection (esp. Gram neg GI bacteria)
inflammation & necrosis activate cytokine cascade
Severe, necrosis (15%): Necrotizing pancreatitis
Local infection
Severe pancreatic inflammation shifts body fluid into retroperitoneal spaceàintravascular volume depletion
Systemic Inflammatory Response Syndrome (SIRS) (see relevant slide)
Organ failure (may be sole feature on presentation)
Stagnant fluid can more easily become infected
Infection spreads to bloodstream
Cardiac failure Hypovolemic shock Renal failure
Local accumulation of fluid & necrosis in the pancreas
< 4 weeks after onset:
Acute necrotic collection (not encapsulated)
Walled off by fibrous & granulation tissue
> 4 weeks after onset
walled-off necrosis
(completely encapsulated)
When treated with excess fluid resuscitation:
Intra- abdominal hypertension
Respiratory failure (ARDS)
Disseminated intravascular coagulation (DIC)
Bowel obstruction Gastric outlet (see relevant slide) obstruction
Infected pancreatic necrosis
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published September 20, 2016, updated September 7, 2020 on www.thecalgaryguide.com")
Multiple-Sclerosis-on-Brain-MRI
 on Brain MRI
Authors: Evan Allarie Davis Maclean Viesha Ciura* Reviewers: Yan Yu* * MD at time of publication
infiltrates in the infratentorial region
Note: variation in findings exist. The findings shown here are not exhaustive but are some of the most common areas implicated on brain MRI in MS. Most common sites that lesions are observed are: juxtacortical regions, periventricular, infratentorial, spinal cord, and the optic nerve. However lesions can occur anywhere there is myelin in the CNS.
Active inflammation and destruction allows for gadolinium contrast to cross the blood- brain barrier, which can be visualized as a marker of active inflammation in MS
Classic optic nerve (CN II) lesion seen in optic neuritis. Gadolinium enhancement (↑ signal
intensity of lesion after gadolinium injection) in this T1 image demonstrates active inflammation
Image Credits: Dr. Viesha Ciura
For further pathogenesis, see the Calgary Guide slide: Multiple Sclerosis (MS): Pathogenesis and Clinical Findings
Inflammation within the central nervous system (CNS) T-cell, B-cell, and macrophages infiltrate CNS
Infiltration occurs through veinsàlocal inflammation Perivenular infiltrates around the medullary veins
(perpendicular to ventricles)
Inflammation and blood-brain barrier destruction ↑ extravascular inflammatory fluid around lesions, which appears hyperintense/bright
Loss of neuronal/axonal density in affected region, replaced by gliosis over time
Lesions extend out around veins, creating characteristic ovoid lesions
Classic hyperintense T2/FLAIR perpendicular periventricular plaques following the medullary veins – ‘Dawson’s Fingers’
Middle cerebellar peduncle T2/FLAIR hyperintense lesion in classic location for MS
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published October 25, 2020 on www.thecalgaryguide.com")
Bronchiolitis-updated
 - but can be others such as rhinovirus,
adenovirus, parainfluenza, influenza, and coronaviruses - initially colonizes the nasopharyngeal mucosa Virus travels via the epithelium to the lower airways to the terminal bronchioles (small airways)
Authors: Nick Baldwin Rebecca Lindsay Reviewers: Kayla Nelson, Yan Yu Timothy Fu, Danielle Nelson* *MD at time of publication
Upper airway mucosal inflammation
Bronchiolitis
(bronchiole inflammation)
Apnea
(cessation of breathing; via unknown mechanism, potentially apnea reflex)
RSV-fusion protein facilitates fusion of the virus to the host cell and directs viral penetration as well as facilitates fusion of the infected cell with its healthy neighbors
Forms syncytia (multinucleated cells)
Cytokines are released into circulation
↑ thermo- regulatory set- point at the hypothalamus
Mild Fever
Copious coryza
(nasal discharge)
Protein and fluid leak into nasopharyngeal interstitium
↑ Capillary permeability
Protein and fluid leak into the bronchiole interstitium, accumulating around airway walls
Airway wall becomes thickened, more readily apparent on x-ray
Peribronchial cuffing
(X-ray finding: bronchi appear like thickened ‘cuffs’ when viewed head-on)
Inflammation stimulates the upregulation of mucous secreting goblet cells
↑ mucous production
Mucous within alveoli ↑ intra- alveolar surface tensionà collapsing alveolar walls
During inspiration, air enters the collapsed alveoli if airway is not yet occluded
↑ intra-alveolar pressure causes the alveoli to suddenly pop open
Inspiratory crackles on auscultation
Syncytia slough off the bronchial epithelium into airways
Airways become narrower and occlude
Disruption of the ciliated epithelial cells (which transport mucous out of the airways and into the pharynx, to be swallowed or evacuated)
↓ mucous clearance from airways
Excess airway mucous triggers cough reflex
Cough
Interstitial edema
Nasal Congestion
Air is absorbed distal to occlusion (gas trapping)
When all air is absorbed, alveoli collapse (resorptive atelectasis)
↓ gas exchange between blood and air in remaining alveoli
↓ O2 saturation & ↑ CO2 content of blood
Narrower airways, especially during expiration, causes audible turbulent airflow
Wheeze on auscultation
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Re-Published November 29, 2020 on www.thecalgaryguide.com")
Potassium-Sparing-Diuretics-Mechanism-of-Action-and-Side-Effects
 Inhibit Na+ channels (involved in Na+ reabsorption) in the luminal membrane of the principal cells in the cortical collecting duct
Aldosterone Antagonists (Spironolactone and Eplerenone) Competitively blocks mineralocorticoid receptors and ↓ aldosterone effect in the renal tubules and throughout the body
↓ aldosterone effectà↓ expression of basolateral Na+/K+ pumps & luminal epithelium Na+ channels on the principal cells of cortical collecting duct
Spironolactone’s molecular structure is similar to that of steroid
hormonesàspironolactone can also block androgen receptors
Anti-androgenic effects
(due to ↓ androgen function in reproductive organs, skin, and on brain centers)
Triamterene can form triamterene crystals
and granular casts (unclear mechanism)
↓ Na+ reabsorption by principal cells
↓ in serum Na+ concentration: Hyponatremia
↓ K+ pumped out into the tubule by principal cells
Crystals & casts obstruct tubular lumen → inflammation (unclear mechanism)
Triamterene crystals are
directly cytotoxic to tubular cells
Only ~2-5% of Na+ filtered by the glomerulus is normally reabsorbed in the
cortical collecting ductàthe ↑ in Na+ retained in cortical collecting duct is mild
Water follows Na+ to maintain a balanced osmotic pressureà↑ in water in cortical collecting duct available for excretion
↑ positive charges in lumen relative to surroundings generates an electropositive tubular lumen
Unfavorable electrical gradient ↓ secretion of positive charged ions into electropositive lumen
↓ libido
Menstrual abnormalities (in women)
↓ acne Gynecomastia
(in men)
↓ secretion of K+
↑ in serum K+ concentration:
Hyperkalemia
Cardiac Arrythmias (See relevant slide on Hyperkalemia: Clinical Findings)
Interstitial inflammation and tubular injury
Drug-induced Nephrotoxicity
Mild Diuresis
↓ blood volume
↓ Blood pressure/ Hypotension
↓ secretion of H+
↑ in serum H+ concentration:
Metabolic Acidosis
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published February 15, 2021 on www.thecalgaryguide.com")
AAA-Pathogenesis
: Pathogenesis
Different parts of the aorta have different embryologic origins
Atherosclerosis
Hypertension
Age > 65
Progressive deterioration of aorta structural integrity over life span
Connective Tissue Disease
Structurally abnormal protein or protein organization in aorta
Autoimmunity
Infection (e.g Chlamydia, Mycoplasma pneumoniae, Helicobacter pylori, human cytomegalovirus, herpes simplex virus)
Antigens (substance that causes immune response) on virus or bacteria resemble local proteins in abdominal aorta
Antibodies produced in response to infection inappropriately target host cells in the aorta
Antibodies tag cells in the abdominal aorta for destruction by T-lymphocytes
Immune-mediated destruction of aorta
Smoking
Genetics
Unclear mechanisms
Subacute (not clinically detectable) inflammation of aortic tissue
Inflammatory cytokines are released and immune cells are recruited
↑ pressure on aorta and other vessel walls
Infiltration of vessel wall by lymphocytes and macrophages
Production of enzymes that break down elastin & collagen proteins (which provide most tensile strength to aorta)
Aorta susceptible to damage
Degradation of aortic connective tissue
Biomechanical stress on vessels
Authors: Olivia Genereux Davis Maclean Reviewers: Jason Waechter* Amy Bromley* Yan Yu* *MD at time of publication
The exact mechanisms are complex, debatable, and an area of intensive research – the 3 mechanisms and associated pathophysiology presented here are generally thought to be the main causes of abdominal aortic aneurysms
Infrarenal aorta has poorly developed vaso vasorum (dedicated blood supply to vessel wall)
Infrarenal aorta relies solely on nutrient diffusion from aortic blood that crosses abdominal aorta
Infrarenal aortic wall has fewer “lamellar” units (fibromuscular units) than other regions of the aorta
Infrarenal aorta is less elastic & less able to distribute stress
Loss of smooth muscle cells & thinning of tunica media
Destruction of elastin in tunica media
Normal layers of the aortic wall
↓ aortic tensile strength (ability to withstand stretching) Aorta expands and dilates due to internal pressure
Tunica Intima (inner-most tissue layer of aorta)
Tunica Media (layers of elastic
tissue (elastin) and muscle fibers)
Adventitia (thin outermost collagenous layer)
(longitudinal section)
Aortic aneurysms are usually infrarenal (85%)
Abdominal Aortic Aneurysm
Infrarenal aorta more prone to ischemia and has impaired repair potential
Abnormal, irreversible dilation of a focal area of abdominal aorta (area of aorta between diaphragm & aortic bifurcation) to twice the diameter of adjacent normal artery segment
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Re-Published February 27, 2021 on www.thecalgaryguide.com")
Cubital-Tunnel-Syndrome-Ulnar-Neuropathy
: Pathogenesis and clinical findings
Osteoarthritis
Rheumatoid arthritis
Diabetes mellitus (see Calgary Guide slide on Diabetic Neuropathy)
Degenerative bone & joint changes
Autoimmune damage to joints
Abnormal bone structure/lesions
Idiopathic
Hyperglycemia causes nerve damage (complex mechanisms)
Elbow trauma
Repetitive elbow flexion
Authors: Chris Oleynick Alexandros Mouratidis Yan Yu* Reviewers: Annalise Abbott Sean Crooks Davis Maclean Hannah Koury Jeremy LaMothe* Ian Auld* * MD at time of publication
Inflammation or edema in cubital tunnel (space between medial epicondyle of humerus and olecranon of ulna) where ulnar nerve is found
↓ size of the cubital tunnelà↑ pressure on internal contents (e.g. ulnar nerve)
Axonal conduction is interrupted
Myelin sheath is damaged
↓ blood supply to nerve
Weakness of adductor pollicus longus (works to adduct thumb)
↑ compensatory activity of flexor
pollicis longus with pinching
↓ activity of hypothenar muscles (which move the 5th digit)
↓ activity of 5th digit palmar interosseus muscles (which adduct the 5th digit)
Cubital Tunnel syndrome: compression neuropathy
Paresthesia
Abnormal sensations of skin (“pins & needles”, tingling, burning, and/or numbness) in ulnar nerve sensory distribution
ulnar nerve
↓ activity of muscles innervated by ulnar nerve (medial forearm flexors, hypothenar muscles, interossei muscles, adductor pollicis longus)
+ Tinel sign
over cubital tunnel (tapping at medial elbow causes discomfort and/or paresthesia)
Weak pinch & ↓ grip strength
+ Froment’s sign
(thumb hyperflexion compensates for weak pinch)
Hypothenar atrophy
+ Wartenburg’s sign
(involuntary, excess abduction of 5th digit)
of the
Altered sensation in areas innervated by ulnar nerve (medial forearm and wrist, 5th digit, and medial half of 4th digit)
+ elbow flexion test
(flexing elbow & extending wrist further ↓ volume of the cubital tunnelà paresthesia in ulnar nerve sensory distribution
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
First published January 12, 2017, re-published February 28, 2021 on www.thecalgaryguide.com")
Achilles-Tendon-Rupture

Impaired ability to heal from microtrauma
Mechanical factors
Abnormal biomechanics:
bowleg, flatfoot, excessive supination, obesity, change in terrain
Participation in vigorous exercise
Excessive heat generation
(hyperthermia) in Achilles tendon induces changes in the extracellular matrix
Intra-tendinous collagen degeneration, disorientation, and thinningàcompromised Achilles tendon integrity
Sudden shear stress and high tension in an already weakened Achilles tendon, stretching it beyond its capacity
Haglund’s deformity: enlargement of the calcaneal postero- superior tuberosity
Bony enlargement rubs on soft tissue and causes inflammation of Achilles tendon
↓ tendon strength from
changes in extracellular matrix
Episodic participation or sudden ↑ in exercise
Irregular use renders Achilles tendon weaker
Microtrauma to the
and less flexible Achilles tendon on Achilles Tendon
Abnormal loading
Sudden, forced dorsiflexion of foot
Eccentric contraction of the gastrocnemius-soleus complex
The gastrocnemius muscle normally attaches to Achilles tendon to control plantar flexion of the foot
With the Achilles tendon ruptured, contraction of the gastrocnemius muscle cannot induce plantar flexion
Achilles tendon rupture
Achilles tendon innervated by the sural nerve and tibial nerve
Pain over rupture site (e.g. “like being kicked in the back of the leg”)
Antalgic gate: abnormal gait developed to avoid pain while walking
High tension at the origin and insertion points of the tendon causes the ends of the tear to separate
The Achilles tendon contains blood vessels. Rupturing the tendon also ruptures the blood vessels withinàblood leaks out into tendon rupture site
Bruising and swelling over rupture site
↓ resting ankle plantar flexion (Observed in prone position with knees flexed at 90 degrees)
Passive contraction (e.g. squeezing) of the gastrocnemius does not cause plantar flexion
+ Thompson (calf squeeze) test
Weak ankle plantar flexion
Lack of push-off in gait
Audible “pop” at time of injury
Palpable gap between rupture ends
Difficulty ambulating
↓ muscle use over time
Calf atrophy
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Re-Published March 21, 2021 on www.thecalgaryguide.com")
Thyroïdite

Symptômes généraux de l’hyperthyroïdie
Post-natal
Médicaments/ Radiation
Thyroïdite bactérienne
Subaigüe (post-virale)
Abbréviations:
• TSH- Hormone stimulant de la
thyroïde
Inflammation de la glande thyroïde
Plusieurs méchanismes
Brianna Ghali Sylvain Coderre* Phillippe Couillard*
Translator:
Infiltration de la glande thyroïde avec des globules blancs
Destruction immunitaire de la glande thyroïde
↑ libération de T4 et T3 stockés par glande thyroïde endommagée
durant 2-6 semaines (phase hyperthyroïde)
↓ sécrétion de T4 et T3 de la glande thyroïde endommagée
durant des semaines-mois (phase hypothyroïde)
Souvent une résolution à un état euthyroïde (normal), mais peut aussi rester dans un état hypothyroïde
↑ T4/T3 inhibe la
libération de la TSH par la ↓ sécrétion TSH
Réaction inflammatoire altère la fonction folliculaire normale (production T4/T3 et importation d’iode)
glande pituitaire
↓ Absorption de l'iode par la glande thyroïde
↓ Capture de l'iode radioactif à la scintigraphie de médecine nucléaire
Plusieurs méchanismes Symptômes généraux de l’hypothyroïdie
↓ T4/T3 stimule la
liberation de la TSH par la ↑ sécrétion TSH
Dommage altère la fonction folliculaire normale, (production T4/T3 et importation d’iode)
glande pituitaire
↓ Absorption de l'iode par la glande thyroïde
↓ Capture de l'iode radioactif à la scintigraphie de médecine nucléaire
Légende:
Pathophysiologie
Méchanisme
Signes/Symptômes/Résultats labo
Complications
Publié June 19, 2013 on www.thecalgaryguide.com")
Fat-Embolism-Syndrome

Non-trauma related (rare)
Long bone fracture
Pelvic fracture
Orthopedic Trauma
Intraosseous access
Soft tissue injuries
Chest compressions
Bone marrow transplant
Pancreatitis
Diabetes mellitus
Fat from bone marrow is disrupted and leaks into bloodstream via damaged blood vessels
Fat globules obstruct dermal capillaries
Capillaries rupture
Blood leaks into the skin
Petechial rash
Non-Orthopedic Trauma (less common)
Fat from injured adipose tissue is released from adipocytes into bloodstream
Metabolic disturbance mobilizes stored fat and moves it into circulation
Fat Embolism Syndrome
the presence of fat globules in circulation
Fat globules damage blood vessel walls
Platelets stick to damaged areas Platelet aggregation
↑ circulating free fatty acids
↑ inflammatory cytokines (TNF, IL1, IL6)
↑ serum C Reactive Protein (an acute phase reactant)
C reactive protein binds to lipid vesicles in circulation
↑ formation of lipid complexes in the blood
Obstruction of cerebral vasculature
↓ blood flow and oxygen delivery to brain tissue
Neurological findings: ranging from ↓ level of consciousness to seizures
Notes:
Large quantities of fat globules can obstruct pulmonary vasculature
Blood clots form throughout the body
Disseminated intravascular coagulopathy
Back up of blood into right heart àRight ventricular dysfunction
↓ pulmonary arterial blood flow à↓ gas exchange in the lungs
Higher CO2 & lower O2 levels in blood àdetected by chemoreceptors
Chemoreceptors stimulate respiratory centre in the brain to ↑ rate of respiration
Dyspnea / Tachypnea
Authors: Tabitha Hawes Reviewers: Hannah Koury, Alyssa Federico, Davis MacLean, Mehul Gupta, Yan Yu*, Jeremy Lamothe* * MD at time of publication
• Underlined findings indicate classic triad of symptoms (petechial rash, neurologic findings, dyspnea/tachypnea)
• Clinical presentation of fat embolism syndrome is variable and may present with any or all of these findings
↓ pumping of blood into systemic circulation
Hypotension Obstructive shock
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published July 19, 2021 on www.thecalgaryguide.com")
Dry-Eye-Syndrome-Clinical-Findings
:
Clinical Findings
If dry eye is chronic or severe (e.g Sjögren’s syndrome) corneal ulcerations or scarring may occur
Impaired visual acuity/ Blindness (does not improve with lubrication)
See Calgary Guide: “Dry Eye Syndrome (Keratoconjunctivitis sicca): Pathogenesis” for explanation of etiology and pathogenesis
Dry Eye Syndrome (Keratoconjunctivitis sicca): Disease of the ocular surface and tears characterized by loss of tear film homeostasis, tear film hyperosmolality and inflammation
Tear film instability & hyperosmolarity
Ocular surface inflammation: Immune cell (specifically T-Cell) destruction of corneal and conjunctival epithelium
↓ quantity and/or quality of tear film
Inflammatory factors vasodilate conjunctival vessels, making the eye look redder in appearance
Conjunctival injection
Red eye
Corneal surface irregularities
Light rays pass through
disrupted tear film and ocular surface structures as they enter the eye
Degraded image quality as light passes through the eye
Impaired visual acuity
(improves with lubrication)
↓ lubrication over the eye surface
↑ friction on the eye during blinking or when opening eyes after sleep
May cause corneal abrasion (See Calgary Guide: Corneal Abrasion)
↑ stimulation of corneal and conjunctival nerve endings
Irritation and damage to corneal and conjunctival cells
↑ stimulation of nerve endings
Chronic inflammation surrounding corneal nerves in conjunctiva and epithelium can lead to decreased neuronal firing thresholds (Neurosensory dysfunction / hyperalgesia)
↑ activity of the afferent portion of corneal reflex arc (responsible for reflex tearing: tearing in response to irritation of the eye)
Foreign body sensation
(scratching / feeling as if a foreign body, like a grain of sand, is in the eye)
Minor (typically non-painful stimuli) may cause pain due to hypersensitive corneal nerves
Corneal neuropathic pain (pain not fully explained by ocular surface changes, often resistant to standard treatments, sometimes still present following ocular surface anaesthesia if central pathways affected)
Pain with temperature change and wind exposure
Exact mechanism unknown
Photophobia
Burning sensation Paradoxical reflex tearing (↑ tearing, which does
not improve dry eye due to altered tear composition, poor surface coverage or overflow of temporary tearing out of the eye
Authors: Michael Penny, O.D. Davis Maclean Yan Yu*
Reviewers: Natalie Arnold Saleel Jivraj, O.D. Adam Muzychuk* Victor Penner* *MD at time of publication
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published August 6. 2021 on www.thecalgaryguide.com")
COPD Acute Exacerbations
, Zesheng Ye (叶泽生), Yonglin Mai (麦泳琳)*, Kerri Johannson* * MD at time of publication
Notes:
• The triggers for acute exacerbations of COPD are unknown in approximately 1/3 of cases
• Changes in pulmonary function (e.g. FEV1) are poorly sensitive in the individual diagnosis of acute exacerbations of COPD due to individual variability
Acute Bacterial Infection
Activation of proinflammatory cytokines and recruitment of neutrophils
Acute Viral Infection
Epithelial cell secretion of cytokines and ↑ airway lymphocythemia
Acute ↑ in airway inflammation
(accumulation of inflammatory cells and release of harmful mediators, such as reactive oxygen species and proteases, into the lung tissue/airways)
Air pollution
(including cigarette smoke)
↑ reactive oxygen species in lungs
Airway inflammation ↑ secretions that accumulate in the airway lumen
Airway wall edema
Limits outflow of air from lungs on expiration
Airway bronchoconstriction in response to inflammation
Constricted airways create audible turbulent airflow on expiration
Systemic spread of inflammatory markers via the bloodstream cause inflammation throughout the body
↑ Cardiac Morbidity
↑ CRP
Worsening of respiratory symptoms
Irritation of cough reflexes in airways
Cough
↑ Sputum production and sputum purulence
↑ Wheeze Unexpired air becomes trapped in
the lungsàDynamic Hyperinflation
Bloodflow to lungs continue to perfuse under- ventilated regions of lungs àV/Q Mismatch
Lungs are larger àmore blood
remains in lungs à↓ preload
Acute Respiratory Failure
Tachypnea
↑ Dyspnea
Accessory Muscle Use
Attempts to restore normal arterial CO2 and O2 levels
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published April 21, 2019, updated October 6, 2021 on www.thecalgaryguide.com")
Hypercortisolemia
: Clinical Findings
Cortisol is a net catabolic hormone affecting many body systems, serving to release energy into the blood in response to stress. Excess cortisol also impacts circulation and impairs immune function. Excess Serum Cortisol affects:
Authors: Samin Dolatabadi, Yan Yu* Reviewers: Meena Assad, Amanda Henderson, Brooke Fallis, David Campbell* * MD at time of publication
Bone and Calcium Metabolism
Kidney and vasculature
Excess cortisol in the renal tubule saturates the enzyme 11β- HSD2, which converts cortisol to cortisone
Capacity of body to convert cortisol to cortisone is exceeded
Excess cortisol can mimic aldosterone
and bind to mineralocorticoid receptors (cortisone can’t bind to these receptors)
↑ Aldosterone effect → ↑ Na+ reabsorption from the cortical collecting duct into blood vessels
Liver and Peripheral Tissue
Cortisol ↑ gluco- neogenesis in liver, and ↑ insulin resistance by body tissue (unclear mechanisms)
Hyper- glycemia
Reproductive System
Cortisol exerts negative feedback on hypothalamus
à↓ gonadotropin releasing hormone (GnRH) secretion
↓ GnRH → ↓ LH/FSH → ↓ estrogen and testosterone production (especially important in females)
Infertility, ↓ Libido, Irregular Menses
Adipose Tissue
Cortisol ↑ fat breakdown (lipolysis)
Selective expression of cortisol receptor on different adipose tissuesàcentral, facial, dorsal fat is less broken down than in other areas (mechanism unclear)
Combined with cortisol ↑ appetite:
Skin & Connective Tissue
↑ Serum cortisol à↓ Fibroblast proliferation → ↓ Collagen synthesis
Skin atrophy with loss of connective tissue
Muscle
↑ Proteolysis & ↓ Protein synthesisà↓ muscle growth and function
Immune System
Normal serum cortisol protects against damaging effects of uncontrolled inflammatory and immune responses
↑ Serum cortisolà over-suppression of inflammation and impaired cell- mediated immunity
↑ Serum cortisol leads to ↓ Intestinal Ca2+ absorption and ↓ renal Ca2+ reabsorption
↓ Serum Ca2+ ↑ PTH secretion
↑ Serum cortisolà↑ RANKL:OPG ratio
↓ Osteoblast activity & ↑ Osteoclast activity
Cardiac muscle
Cardio- myopathy, Heart Failure
Skeletal
muscle, especially upper arms & thighs (for unclear reasons)
Proximal Muscle Weakness
Easy Bruising
↑ abdominal size stretches the fragile skin to become thinneràvenous blood of the underlying dermis becomes visible
Purple Striae
If hypertension is chronic
Ca2+ resorption from bone into the blood
Osteoporosis
Supraclavicular & Dorsal Fat Pads
Central Obesity
Poor Wound Healing
Susceptibility to infection
Round Face (Moon Face)
Abbreviations:
• RANKL – Receptor activator of nuclear factor kappa-Β ligand • OPG – Osteoprotegerin
• 11β-HSD2 – 11β-hydroxysteroid dehydrogenase type 2
Water follows Na+ into blood vessels to balance the osmotic pressure between the blood and renal tubules
Water reabsorption → Expansion of blood volume
Hypertension
Both primary Cushing’s (e.g. adenomas that extend into zona reticularis of the adrenal cortex) and central/secondary Cushing’s (e.g. ↑ ACTH stimulation of zona reticularis) are associated with ↑ adrenal androgen secretion
Removal of positively charged Na+ from tubular lumen creates a negative luminal environment
K+ follows the electrical gradient and is secreted into tubular lumen
↓ Serum K+ concentration
Hypokalemia
Arrhythmia, Paralysis, Cramps
(see Hypokalemia: Clinical Findings slide)
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published October 17, 2021 on www.thecalgaryguide.com
Hirsutism, Acne")
Acne Vulgaris Complications
, leukotrienes (LTC4, LTD4), and thromboxane A2 are released in response to inflammation
Atrophic Acne Scars
Collagen degradation and disordered deposition during healing
Formation of round-
rectangular depressions with defined edges
Box Car Scar
True scars with textural change, very slow autoresolution over time
The pigmentary changes and true scar formation resulting from acne may be more psychologically distressing than the original acne lesions
Healing processes favoring deposition of Type 1 & Type 3 collagen
Proliferation beyond the borders of original acne lesion
Keloid Formation
Healing processes favoring deposition of Type 4 collagen
Growth remains within the margins of acne lesion
Hypertrophic Scar Formation
Dermal inflammation: Due to disruption of basal cell layer, melanin released and trapped by macrophages in the dermis.
Epidermal inflammation: ↑ melanin production and transfer of melanin to keratinocytes
Microvascular dilatation and ↑presence of erythrocytes
Erythematous macules appear where acne was present
Post- inflammatory Erythema (PIE). More common in Fitzpatrick Skin Types I-III
Formation of V- shaped tracts with sharp margins
Ice Pick Scar
Formation of a
shallow edged depression layer anchored to dermal layer and subcutis
Rolling Scar
Pigmented
macules or patches appear where acne was present
Post-inflammatory Hyperpigmentation (PIH). More common in
Fitzpatrick Skin Types III-VI
No textural change, slow
autoresolution over time
True scars with textural change, do not resolve over time
Psychosocial Concerns (Depression, Anxiety, Social Isolation)
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published October 19, 2021 on www.thecalgaryguide.com")
Vestibular Neuritis
 ganglion
Inflammatory cell infiltration leads to degeneration and atrophy of the vestibular nerve
Superior Vestibular Neuritis
(40-48%) Inflammatory cells traverse through only one long bony canal, easier for inflammatory cell infiltration
Loss of afferent innervation from the superior and horizontal semicircular canal (SCC), utricle, and parts of the saccule
Combined Superior Vestibular Neuritis and Inferior Vestibular Neuritis (34-56%)
Inferior Vestibular Neuritis (1.3-18%) Inflammatory cells must pass through two separate bony canals, making inflammatory cell infiltration more difficult
Loss of afferent innervation from the posterior semicircular canal (SCC) and saccule
Utricular degeneration
Displacement of otoliths/ otoconia (commonly into the posterior SCC)
BPPV
(can occur several months after onset of neuritis)
Loss of horizontal SCC afferent neuron signaling to the brain
Unilateral Loss or ↓ of normal nystagmus response to Caloric Testing (insertion of cold and warm water/air into the ear canal while supine)
Loss of utricular afferent neuron signaling to the brain
↓ or absent Ocular Vestibular- Evoked Myogenic Potentials (VEMPs)
and Normal Cervical VEMPs
↓ unilateral vestibular input to the brain leads to acute phase symptoms (over time, brain can compensate which allows for some symptom improvement)
Loss of posterior SCC and saccular afferent neuron signaling to the brain
↓ or absent
Cervical
VEMPs, but Ocular VEMPs are normal
↓ or absent input to the ipsilateral vestibular nuclei elicits a vestibular nucleus response similar to contralateral SCC excitation
Acute-Phase Spontaneous Nystagmus Fast phase beats away from the affected side (3- 10 days)
And
Loss of ocular fixation on Head Impulse Test
↓ or absent saccular input to the lateral vestibular nuclei (e.g., lateral vestibulospinal tract) results in the loss of lower limb postural adjustability
Postural Instability
(e.g., abnormal Romberg/sharpened Romberg, Fukuda step test)
Bilateral mismatch of vestibular
information to the brain
Peripheral Vertigo (lasts several hours to days; rapid onset, severe, constant)
Nausea and Vomiting
Only the vestibular
part of the vestibulo- cochlear nerve is affected, not the cochlear nerve
No Hearing
Loss or Tinnitus
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published October 19, 2021 on www.thecalgaryguide.com")
COPD-发病机制
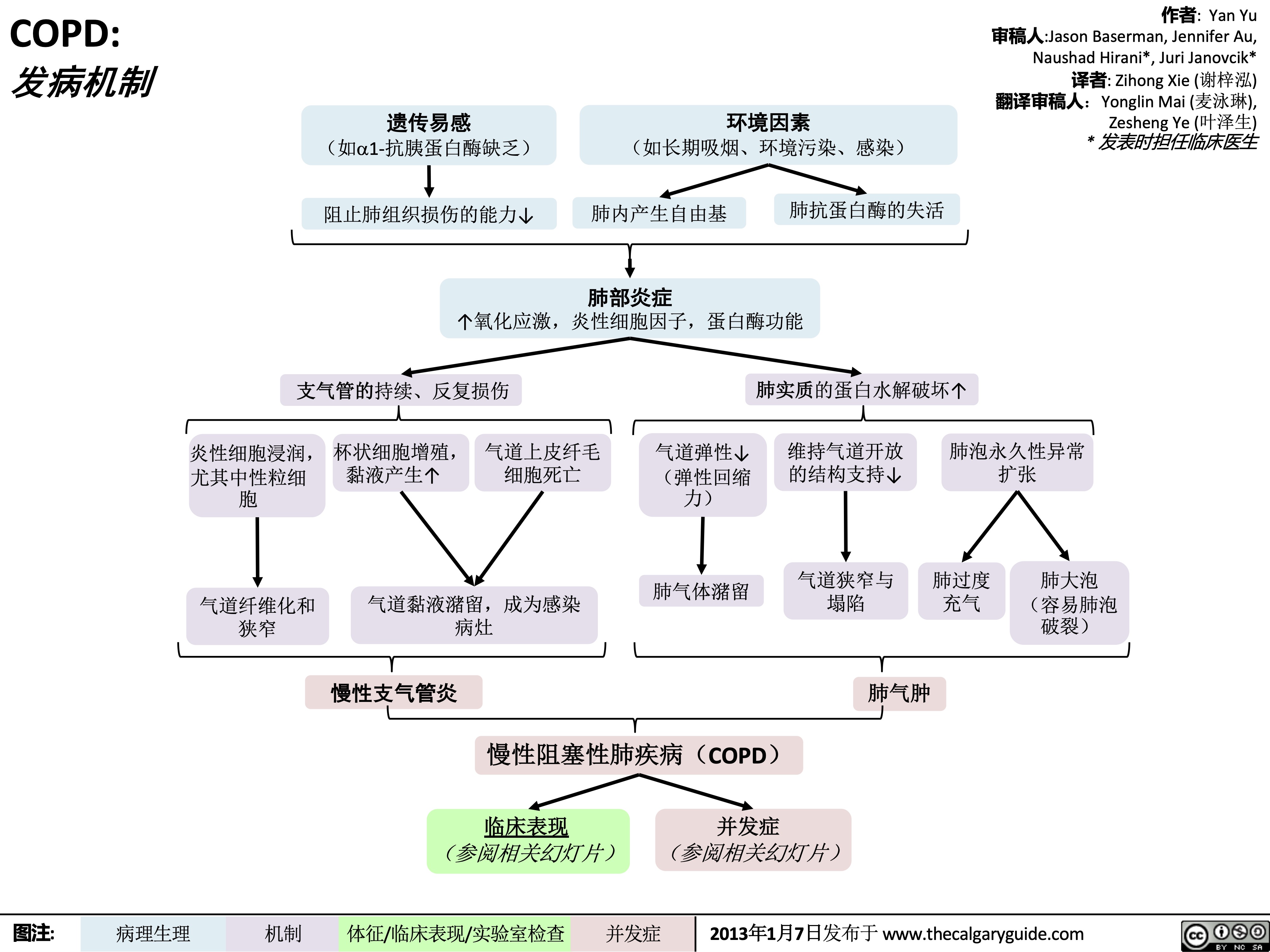 45
(on ABGs)
Ventilation- perfusion mismatch
High A-a gradient
(calculated from ABGs)
Low, flat diaphragm, >10 posterior ribs
(on frontal CXR)
High TLC and VC
(on spirometry)
• •
PaO2: partial pressure of O2 in arterial blood PaCO2: partial pressure of CO2 in arterial blood
• In the setting of fever and productive cough, especially if lung field opacifications are seen on CXR: consider sputum gram stain and culture to rule out pneumonia.
Air does not block X-ray beams, will appear black on X-ray film
Chronic hypercapnia makes breathing centers less sensitive to the high PaCO2 stimulus for breathing, & more reliant on the low PaO2 stimulus
(“CO2 retention”)
Give O2 carefully to these patients (high PaO2 may suppress patients’ hypoxic respiratory drive, ↓ their breathing, & ↑↑↑ PaCO2)
↑ retrosternal air space
(on lateral CXR)
Hyper-lucent
(darker) lung fields, ↓ lung markings (on frontal CXR)
• Arterial Blood Gasses (ABGs)
• Chest X-Ray (CXR): frontal and
lateral
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published January 7, 2013 on www.thecalgaryguide.com
COPD: !"#$ 气流阻塞
肺泡通气↓ 呼气时,胸膜腔正压挤压气 道à 阻塞↑
作者: Yan Yu 审稿人: Jason Baserman, Jennifer Au, Naushad Hirani*, Juri Janovcik* 译者:Zihong Xie (谢梓泓) 翻译审稿人: Yonglin Mai (麦泳琳), Zesheng Ye (叶泽生) * 发表时担任临床医生
慢性阻塞性肺疾病 (COPD)
肺组织损伤
没有弹性回缩力将
气体排出肺
肺实质与血管分布减少导 致气体交换面积↓
弥散功能↓ (肺功能检查)
更多的CO2残留 并扩散到血液中
高碳酸血症: PaCO2 > 45
(动脉血气)
血流灌注通气不良的肺泡
时无法获得足够的氧气
总呼气时长较正常长
FEV1/FEV < 0.7
(肺功能检查)
肺无法完全排空
更多空气潴留在肺部
(肺过度充气)
低氧血症: PaO2 < 70mmHg
(动脉血气)
通气-灌注不匹配
肺泡-动脉氧分压差↑ (可通过动脉血气分析计算得出)
横膈低平, 下移至第10肋后端 及以下部位 (胸部正位片)
TLC与VC增大 (肺功能检查)
缩写: • • FEV1: 1秒用 •
VC:肺活量
PaO2: 动脉血 力呼气量 氧分压
空气不会阻挡X射线, 在X光片上呈现为黑色
慢性高碳酸血症使呼吸中枢对PaCO2 刺激呼吸的敏感性下降 & 更依赖于低PaO2的刺激 (“二氧化碳潴留”)
给患者吸氧时需注意(高PaO2
可能会抑制患者低氧时对呼吸的 刺激,使呼吸驱动↓ & PaCO2↑↑↑ )
• FVC: 用力肺 • 活量
• TLC:肺总量 慢阻肺相关检查 :
PaCO2: 动脉 血二氧化碳 分压
胸骨后间隙↑
(胸部侧位片) 肺纹理↓
• 肺功能检查
• 动脉血气分析(Arterial Blood Gasses, ABGs)
• 胸部正侧位片
• 当患者发热和湿咳,特别是胸片上见肺野不清晰时:
肺透亮度↑, (胸部正位片)
考虑进行痰革兰氏染色及痰培养以排除肺炎可能
图注:
病理生理
机制
体征/临床表现/实验室检查
并发症
2013年1月7日发布于 www.thecalgaryguide.com
COPD: Complications Lung inflammation
Chronic Obstructive Pulmonary Disease (COPD)
Airway obstruction ↓ inhaled air in alveoli and terminal bronchioles
Rupture of emphasematous bullae on surface of lung
Inhaled air leaks into pleural cavity and is trapped there
Pneumothorax
Feeling a loss of control over one’s life, and hopelessness for the future
Goblet cell proliferation, ↑ mucus production
Death of airway
epithelium ciliated cells
↓ oxygenation of the blood passing through the lungs
Chronic hypoxemia
Kidneys compensate by ↑ erythropoietin (EPO) production
↑ Hemoglobin and red blood cell synthesis
Polycythemia (secondary)
Hypoxic alveoli cause the pulmonary arterioles perfusing them to reflexively vasoconstrict
Since most alveoli in the lungs are hypoxic, hypoxic vasoconstriction occurs across entire lung
Vasoconstriction ↑ blood pressure within lung vasculature
Pulmonary hypertension
↑ workload of the right ventricle (to pump against higher pressures)
To compensate, the right ventricle progressively hypertrophies and dilates, but over time its output ↓
Cor pulmonale
(Right heart failure in isolation, not due to Left heart failure)
Mucus trapped in airways, serve as nidus for infection
Acute exacerbation of COPD (AECOPD)
Pneumonia
The chronic, systemic inflammation in COPD is a hyper-metabolic state that consumes calories
Macro-nutrient deficiency
Trouble with respiration lead to inactivity and deconditioning
Wasting, muscle atrophy
More inactivity and deconditioning perpetuates the cycle
Depression
Author: Yan Yu Reviewers: Jason Baserman Naushad Hirani* Juri Janovcik* * MD at time of publication
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published January 7, 2013 on www.thecalgaryguide.com
COPD: !"# 肺部炎症
杯状细胞增殖, 气道上皮纤毛 粘液产生↑ 细胞死亡
黏液潴留呼吸道,成为感
染的病灶
慢性阻塞性肺疾病 (COPD) 气道阻塞à 吸入肺泡和终末细
肺大疱破裂
吸入的空气渗入
并潴留于胸腔
气胸
感觉生活失控,对未
来感到绝望
抑郁
作者: Yan Yu 审稿人: Jason Baserman, Naushad Hirani*, Juri Janovcik* 译者: Zihong Xie (谢梓泓) 翻译审稿人: Yonglin Mai (麦泳琳), Zesheng Ye (叶泽生) * 发表时担任临床医生
支气管的空气 ↓
流经肺的血液进行气 缺氧的肺泡à灌注肺泡的肺小动
慢性阻塞性肺疾 病急性加重期 (AECOPD)
肺炎
体交换↓ 慢性低氧血症
肾脏合成促红细胞 生成素进行代偿↑
血红蛋白与红 细胞合成↑
红细胞增多症 (继发性)
脉发生反射性血管收缩
肺大部分肺泡缺氧à整个肺 都出现缺氧性血管收缩
肺血管收缩 à 肺血管压力↑ 肺动脉高压
↑ 右心室负荷(泵血时对抗高压) 为了代偿,右心室逐渐肥大和扩张,
但随着病程进展,右心室输出量 ↓
肺心病 (单独出现右心衰竭,非左心衰)
COPD所致的慢性全身 呼吸困难导致活 性炎症会使机体处于高 动量减少和活动
代谢状态,消耗能量 耐量降低
宏量营养 素缺乏症
消瘦,肌肉萎缩
运动量下降和活动耐量
的降低造成恶性循环
图注:
病理生理
机制
体征/临床表现/实验室检查
并发症
2013年1月7日发布于 www.thecalgaryguide.com
" title="COPD: 发病机制
作者: Yan Yu 审稿人:Jason Baserman, Jennifer Au, Naushad Hirani*, Juri Janovcik* 译者: Zihong Xie (谢梓泓) 翻译审稿人:Yonglin Mai (麦泳琳), Zesheng Ye (叶泽生) * 发表时担任临床医生
/012
(如a1-抗胰蛋白酶缺乏) 阻止肺组织损伤的能力↓
+,-.
(如长期吸烟、环境污染、感染)
肺内产生自由基
34*5
肺抗蛋白酶的失活
↑氧化应激,炎性细胞因子,蛋白酶功能
支气管的持续、反复损伤
炎性细胞浸润, 杯状细胞增殖, 气道上皮纤毛 尤其中性粒细 黏液产生↑ 细胞死亡
气道弹性↓ (弹性回缩
肺实质的蛋白水解破坏↑ 维持气道开放 肺泡永久性异常
的结构支持↓ 扩张
胞 力)
肺气体潴留 气道狭窄与 肺过度 肺大泡
气道黏液潴留,成为感染 狭窄 病灶
塌陷 充气
肺气肿
(容易肺泡 破裂)
气道纤维化和
%&'()*
慢性阻塞性肺疾病(COPD)
临床表现 并发症 (参阅相关幻灯片) (参阅相关幻灯片)
图注:
病理生理
机制
体征/临床表现/实验室检查
并发症
2013年1月7日发布于 www.thecalgaryguide.com
COPD: Clinical Findings Lung tissue
Chronic Obstructive Pulmonary Disease (COPD)
damage
↓ elastic recoil to push air out of lungs on expiration
Lungs don’t fully empty, air is trapped in alveoli (lung hyperinflation)
↑ lung volume means diaphragm is tonically contracted (flatter)
If occurring around airways
Airflow obstruction
↑ mucus production
↓ number of epithelial ciliated cells to clear away the mucus (the cells have been killed by airway inflammation)
Chronic cough with sputum
Author: Yan Yu Reviewers: Jason Baserman Jennifer Au Naushad Hirani* Juri Janovcik* * MD at time of publication
During expiration, positive pleural pressure squeezes on airwaysà↑ obstruction
↓ ventilation of alveoli
↓ oxygenation of blood (hypoxemia)
↓ perfusion of body tissues (i.e. brain, muscle)
Fatigue; ↓ exercise tolerance
Total expiration time takes longer than normal
Prolonged expiration
More effort needed to ventilate larger lungs
Respiratory muscles must work harder to breathe
Turbulent airflow in narrower airways is heard on auscultation
Expiratory Wheeze
Diaphragm can’t flatten much further to generate deep breaths
To breathe, chest wall must expand out more
Dyspnea
Shortness of breath, especially on exertion
Breathes are rapid & shallow
If end-stage:
Chronic fatigue causes deconditioning
Muscle weakness & wasting
Barrel chest
If end-stage: diaphragm will be “flat”. Continued
Patient tries to expire against higher mouth air pressure, forcing airways to open wider
Pursed-lip breathing
Patient breathes with accessory muscles as well as diaphragm to try to improve airflow
inspiratory effort further contracts diaphragmà pull the lower chest wall inwards
Hoover’s sign
(paradoxical shrinking of lower chest during inspiration)
Tripod sitting position (activates pectoral muscles)
Neck (SCM, scalene) muscles contracted
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published January 7, 2013 on www.thecalgaryguide.com
COPD: !"#$
慢性阻塞性肺疾病 (COPD) 如果出现在气道周围 气流阻塞
肺不能完全排空
气体,气体潴留
于肺泡(肺过度
充气)
总呼气时长大于 正常时长
呼气相延长
肺组织损伤
呼气时,将空气排出肺外 的弹性回缩力↓
肺不能完全排空气体,
气体潴留于肺泡内
(肺过度充气)
肺容积↑,膈肌紧张 性收缩(膈肌平坦)
呼气时,胸膜腔正压挤压气道 à 气道阻塞↑
肺泡通气↓ 血液氧合↓ (低
氧血症)
身体组织灌注 量↓ (比如脑、 肌肉)
疲劳; 运动耐量↓
黏液生成↑ 清除黏液的上皮纤
毛细胞数量↓ (受 气道炎症损伤)
慢性咳嗽伴咳 痰
作者: Yan Yu
审稿人: Jason Baserman, Jennifer Au, Naushad Hirani*, Juri Janovcik* 译者: Zihong Xie (谢梓泓) 翻译审稿人: Yonglin Mai (麦泳琳),
Zesheng Ye (叶泽生)
* 发表时担任临床医生
容积较大 的肺需要
更加努力 才能通气
呼吸肌必须
更用力才能 呼吸
听诊闻及狭窄气
道中的湍流气流
呼气喘鸣音
呼吸困难 气促,尤其是劳累
膈肌无法进一步收缩以
产生深呼吸
呼吸浅快
为了呼吸,
胸壁必须延
展得更大
桶状胸
晚期病人:
患者试图在较高的口 慢性疲劳导致 患者动用辅助呼吸肌和膈肌呼吸,
腔内气压下进行呼气, 活动耐量下降 从而使气道更开放
以改善气流
晚期病人:膈肌 “平坦” ,持续吸气进一步压 缩膈肌à 向内拉季肋部胸壁
胡佛征 (吸气时,胸廓下侧季肋部内收)
缩唇呼吸
肌肉无力 & 消瘦
端坐呼吸 (调动胸肌)
颈部肌肉收
缩(胸锁乳
突肌、斜角
肌)
图注:
病理生理
机制
体征/临床表现/实验室检查
并发症
2013年1月7日发布于 www.thecalgaryguide.com
COPD: Findings on Investigations
Chronic Obstructive Pulmonary Disease (COPD)
Author: Yan Yu Reviewers: Jason Baserman Jennifer Au Naushad Hirani* Juri Janovcik* * MD at time of publication
Airflow obstruction
Lung tissue damage
↓ ventilation of alveoli
Blood perfusing ill- ventilated alveoli does not receive normal amounts of oxygen
During expiration, positive pleural pressure squeezes on airwaysà↑ obstruction)
No elastic recoil to push air out of lungs
Loss of lung parenchyma and vasculature ↓ surface area for gas exchange
↓ diffusion capacity
(on spirometry)
Hypoxemia: PaO2 < 70mmHg (on ABGs)
Abbreviations:
• FEV1: Forced expiratory volume in 1 second
• FVC: Forced vital capacity
• TLC: Total lung capacity
• VC: Vital Capacity
Investigations for COPD :
• Spirometry (Pulmonary function test)
Total expiration time takes longer than normal
FEV1/FEV < 0.7
(on spirometry)
Lungs don’t fully empty
More air trapped within lungs (hyperinflation)
More CO2 remains and diffuses into the blood
Hypercapnia: PaCO2 > 45
(on ABGs)
Ventilation- perfusion mismatch
High A-a gradient
(calculated from ABGs)
Low, flat diaphragm, >10 posterior ribs
(on frontal CXR)
High TLC and VC
(on spirometry)
• •
PaO2: partial pressure of O2 in arterial blood PaCO2: partial pressure of CO2 in arterial blood
• In the setting of fever and productive cough, especially if lung field opacifications are seen on CXR: consider sputum gram stain and culture to rule out pneumonia.
Air does not block X-ray beams, will appear black on X-ray film
Chronic hypercapnia makes breathing centers less sensitive to the high PaCO2 stimulus for breathing, & more reliant on the low PaO2 stimulus
(“CO2 retention”)
Give O2 carefully to these patients (high PaO2 may suppress patients’ hypoxic respiratory drive, ↓ their breathing, & ↑↑↑ PaCO2)
↑ retrosternal air space
(on lateral CXR)
Hyper-lucent
(darker) lung fields, ↓ lung markings (on frontal CXR)
• Arterial Blood Gasses (ABGs)
• Chest X-Ray (CXR): frontal and
lateral
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published January 7, 2013 on www.thecalgaryguide.com
COPD: !"#$ 气流阻塞
肺泡通气↓ 呼气时,胸膜腔正压挤压气 道à 阻塞↑
作者: Yan Yu 审稿人: Jason Baserman, Jennifer Au, Naushad Hirani*, Juri Janovcik* 译者:Zihong Xie (谢梓泓) 翻译审稿人: Yonglin Mai (麦泳琳), Zesheng Ye (叶泽生) * 发表时担任临床医生
慢性阻塞性肺疾病 (COPD)
肺组织损伤
没有弹性回缩力将
气体排出肺
肺实质与血管分布减少导 致气体交换面积↓
弥散功能↓ (肺功能检查)
更多的CO2残留 并扩散到血液中
高碳酸血症: PaCO2 > 45
(动脉血气)
血流灌注通气不良的肺泡
时无法获得足够的氧气
总呼气时长较正常长
FEV1/FEV < 0.7
(肺功能检查)
肺无法完全排空
更多空气潴留在肺部
(肺过度充气)
低氧血症: PaO2 < 70mmHg
(动脉血气)
通气-灌注不匹配
肺泡-动脉氧分压差↑ (可通过动脉血气分析计算得出)
横膈低平, 下移至第10肋后端 及以下部位 (胸部正位片)
TLC与VC增大 (肺功能检查)
缩写: • • FEV1: 1秒用 •
VC:肺活量
PaO2: 动脉血 力呼气量 氧分压
空气不会阻挡X射线, 在X光片上呈现为黑色
慢性高碳酸血症使呼吸中枢对PaCO2 刺激呼吸的敏感性下降 & 更依赖于低PaO2的刺激 (“二氧化碳潴留”)
给患者吸氧时需注意(高PaO2
可能会抑制患者低氧时对呼吸的 刺激,使呼吸驱动↓ & PaCO2↑↑↑ )
• FVC: 用力肺 • 活量
• TLC:肺总量 慢阻肺相关检查 :
PaCO2: 动脉 血二氧化碳 分压
胸骨后间隙↑
(胸部侧位片) 肺纹理↓
• 肺功能检查
• 动脉血气分析(Arterial Blood Gasses, ABGs)
• 胸部正侧位片
• 当患者发热和湿咳,特别是胸片上见肺野不清晰时:
肺透亮度↑, (胸部正位片)
考虑进行痰革兰氏染色及痰培养以排除肺炎可能
图注:
病理生理
机制
体征/临床表现/实验室检查
并发症
2013年1月7日发布于 www.thecalgaryguide.com
COPD: Complications Lung inflammation
Chronic Obstructive Pulmonary Disease (COPD)
Airway obstruction ↓ inhaled air in alveoli and terminal bronchioles
Rupture of emphasematous bullae on surface of lung
Inhaled air leaks into pleural cavity and is trapped there
Pneumothorax
Feeling a loss of control over one’s life, and hopelessness for the future
Goblet cell proliferation, ↑ mucus production
Death of airway
epithelium ciliated cells
↓ oxygenation of the blood passing through the lungs
Chronic hypoxemia
Kidneys compensate by ↑ erythropoietin (EPO) production
↑ Hemoglobin and red blood cell synthesis
Polycythemia (secondary)
Hypoxic alveoli cause the pulmonary arterioles perfusing them to reflexively vasoconstrict
Since most alveoli in the lungs are hypoxic, hypoxic vasoconstriction occurs across entire lung
Vasoconstriction ↑ blood pressure within lung vasculature
Pulmonary hypertension
↑ workload of the right ventricle (to pump against higher pressures)
To compensate, the right ventricle progressively hypertrophies and dilates, but over time its output ↓
Cor pulmonale
(Right heart failure in isolation, not due to Left heart failure)
Mucus trapped in airways, serve as nidus for infection
Acute exacerbation of COPD (AECOPD)
Pneumonia
The chronic, systemic inflammation in COPD is a hyper-metabolic state that consumes calories
Macro-nutrient deficiency
Trouble with respiration lead to inactivity and deconditioning
Wasting, muscle atrophy
More inactivity and deconditioning perpetuates the cycle
Depression
Author: Yan Yu Reviewers: Jason Baserman Naushad Hirani* Juri Janovcik* * MD at time of publication
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published January 7, 2013 on www.thecalgaryguide.com
COPD: !"# 肺部炎症
杯状细胞增殖, 气道上皮纤毛 粘液产生↑ 细胞死亡
黏液潴留呼吸道,成为感
染的病灶
慢性阻塞性肺疾病 (COPD) 气道阻塞à 吸入肺泡和终末细
肺大疱破裂
吸入的空气渗入
并潴留于胸腔
气胸
感觉生活失控,对未
来感到绝望
抑郁
作者: Yan Yu 审稿人: Jason Baserman, Naushad Hirani*, Juri Janovcik* 译者: Zihong Xie (谢梓泓) 翻译审稿人: Yonglin Mai (麦泳琳), Zesheng Ye (叶泽生) * 发表时担任临床医生
支气管的空气 ↓
流经肺的血液进行气 缺氧的肺泡à灌注肺泡的肺小动
慢性阻塞性肺疾 病急性加重期 (AECOPD)
肺炎
体交换↓ 慢性低氧血症
肾脏合成促红细胞 生成素进行代偿↑
血红蛋白与红 细胞合成↑
红细胞增多症 (继发性)
脉发生反射性血管收缩
肺大部分肺泡缺氧à整个肺 都出现缺氧性血管收缩
肺血管收缩 à 肺血管压力↑ 肺动脉高压
↑ 右心室负荷(泵血时对抗高压) 为了代偿,右心室逐渐肥大和扩张,
但随着病程进展,右心室输出量 ↓
肺心病 (单独出现右心衰竭,非左心衰)
COPD所致的慢性全身 呼吸困难导致活 性炎症会使机体处于高 动量减少和活动
代谢状态,消耗能量 耐量降低
宏量营养 素缺乏症
消瘦,肌肉萎缩
运动量下降和活动耐量
的降低造成恶性循环
图注:
病理生理
机制
体征/临床表现/实验室检查
并发症
2013年1月7日发布于 www.thecalgaryguide.com
" />
45
(on ABGs)
Ventilation- perfusion mismatch
High A-a gradient
(calculated from ABGs)
Low, flat diaphragm, >10 posterior ribs
(on frontal CXR)
High TLC and VC
(on spirometry)
• •
PaO2: partial pressure of O2 in arterial blood PaCO2: partial pressure of CO2 in arterial blood
• In the setting of fever and productive cough, especially if lung field opacifications are seen on CXR: consider sputum gram stain and culture to rule out pneumonia.
Air does not block X-ray beams, will appear black on X-ray film
Chronic hypercapnia makes breathing centers less sensitive to the high PaCO2 stimulus for breathing, & more reliant on the low PaO2 stimulus
(“CO2 retention”)
Give O2 carefully to these patients (high PaO2 may suppress patients’ hypoxic respiratory drive, ↓ their breathing, & ↑↑↑ PaCO2)
↑ retrosternal air space
(on lateral CXR)
Hyper-lucent
(darker) lung fields, ↓ lung markings (on frontal CXR)
• Arterial Blood Gasses (ABGs)
• Chest X-Ray (CXR): frontal and
lateral
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published January 7, 2013 on www.thecalgaryguide.com
COPD: !"#$ 气流阻塞
肺泡通气↓ 呼气时,胸膜腔正压挤压气 道à 阻塞↑
作者: Yan Yu 审稿人: Jason Baserman, Jennifer Au, Naushad Hirani*, Juri Janovcik* 译者:Zihong Xie (谢梓泓) 翻译审稿人: Yonglin Mai (麦泳琳), Zesheng Ye (叶泽生) * 发表时担任临床医生
慢性阻塞性肺疾病 (COPD)
肺组织损伤
没有弹性回缩力将
气体排出肺
肺实质与血管分布减少导 致气体交换面积↓
弥散功能↓ (肺功能检查)
更多的CO2残留 并扩散到血液中
高碳酸血症: PaCO2 > 45
(动脉血气)
血流灌注通气不良的肺泡
时无法获得足够的氧气
总呼气时长较正常长
FEV1/FEV < 0.7
(肺功能检查)
肺无法完全排空
更多空气潴留在肺部
(肺过度充气)
低氧血症: PaO2 < 70mmHg
(动脉血气)
通气-灌注不匹配
肺泡-动脉氧分压差↑ (可通过动脉血气分析计算得出)
横膈低平, 下移至第10肋后端 及以下部位 (胸部正位片)
TLC与VC增大 (肺功能检查)
缩写: • • FEV1: 1秒用 •
VC:肺活量
PaO2: 动脉血 力呼气量 氧分压
空气不会阻挡X射线, 在X光片上呈现为黑色
慢性高碳酸血症使呼吸中枢对PaCO2 刺激呼吸的敏感性下降 & 更依赖于低PaO2的刺激 (“二氧化碳潴留”)
给患者吸氧时需注意(高PaO2
可能会抑制患者低氧时对呼吸的 刺激,使呼吸驱动↓ & PaCO2↑↑↑ )
• FVC: 用力肺 • 活量
• TLC:肺总量 慢阻肺相关检查 :
PaCO2: 动脉 血二氧化碳 分压
胸骨后间隙↑
(胸部侧位片) 肺纹理↓
• 肺功能检查
• 动脉血气分析(Arterial Blood Gasses, ABGs)
• 胸部正侧位片
• 当患者发热和湿咳,特别是胸片上见肺野不清晰时:
肺透亮度↑, (胸部正位片)
考虑进行痰革兰氏染色及痰培养以排除肺炎可能
图注:
病理生理
机制
体征/临床表现/实验室检查
并发症
2013年1月7日发布于 www.thecalgaryguide.com
COPD: Complications Lung inflammation
Chronic Obstructive Pulmonary Disease (COPD)
Airway obstruction ↓ inhaled air in alveoli and terminal bronchioles
Rupture of emphasematous bullae on surface of lung
Inhaled air leaks into pleural cavity and is trapped there
Pneumothorax
Feeling a loss of control over one’s life, and hopelessness for the future
Goblet cell proliferation, ↑ mucus production
Death of airway
epithelium ciliated cells
↓ oxygenation of the blood passing through the lungs
Chronic hypoxemia
Kidneys compensate by ↑ erythropoietin (EPO) production
↑ Hemoglobin and red blood cell synthesis
Polycythemia (secondary)
Hypoxic alveoli cause the pulmonary arterioles perfusing them to reflexively vasoconstrict
Since most alveoli in the lungs are hypoxic, hypoxic vasoconstriction occurs across entire lung
Vasoconstriction ↑ blood pressure within lung vasculature
Pulmonary hypertension
↑ workload of the right ventricle (to pump against higher pressures)
To compensate, the right ventricle progressively hypertrophies and dilates, but over time its output ↓
Cor pulmonale
(Right heart failure in isolation, not due to Left heart failure)
Mucus trapped in airways, serve as nidus for infection
Acute exacerbation of COPD (AECOPD)
Pneumonia
The chronic, systemic inflammation in COPD is a hyper-metabolic state that consumes calories
Macro-nutrient deficiency
Trouble with respiration lead to inactivity and deconditioning
Wasting, muscle atrophy
More inactivity and deconditioning perpetuates the cycle
Depression
Author: Yan Yu Reviewers: Jason Baserman Naushad Hirani* Juri Janovcik* * MD at time of publication
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published January 7, 2013 on www.thecalgaryguide.com
COPD: !"# 肺部炎症
杯状细胞增殖, 气道上皮纤毛 粘液产生↑ 细胞死亡
黏液潴留呼吸道,成为感
染的病灶
慢性阻塞性肺疾病 (COPD) 气道阻塞à 吸入肺泡和终末细
肺大疱破裂
吸入的空气渗入
并潴留于胸腔
气胸
感觉生活失控,对未
来感到绝望
抑郁
作者: Yan Yu 审稿人: Jason Baserman, Naushad Hirani*, Juri Janovcik* 译者: Zihong Xie (谢梓泓) 翻译审稿人: Yonglin Mai (麦泳琳), Zesheng Ye (叶泽生) * 发表时担任临床医生
支气管的空气 ↓
流经肺的血液进行气 缺氧的肺泡à灌注肺泡的肺小动
慢性阻塞性肺疾 病急性加重期 (AECOPD)
肺炎
体交换↓ 慢性低氧血症
肾脏合成促红细胞 生成素进行代偿↑
血红蛋白与红 细胞合成↑
红细胞增多症 (继发性)
脉发生反射性血管收缩
肺大部分肺泡缺氧à整个肺 都出现缺氧性血管收缩
肺血管收缩 à 肺血管压力↑ 肺动脉高压
↑ 右心室负荷(泵血时对抗高压) 为了代偿,右心室逐渐肥大和扩张,
但随着病程进展,右心室输出量 ↓
肺心病 (单独出现右心衰竭,非左心衰)
COPD所致的慢性全身 呼吸困难导致活 性炎症会使机体处于高 动量减少和活动
代谢状态,消耗能量 耐量降低
宏量营养 素缺乏症
消瘦,肌肉萎缩
运动量下降和活动耐量
的降低造成恶性循环
图注:
病理生理
机制
体征/临床表现/实验室检查
并发症
2013年1月7日发布于 www.thecalgaryguide.com
" title="COPD: 发病机制
作者: Yan Yu 审稿人:Jason Baserman, Jennifer Au, Naushad Hirani*, Juri Janovcik* 译者: Zihong Xie (谢梓泓) 翻译审稿人:Yonglin Mai (麦泳琳), Zesheng Ye (叶泽生) * 发表时担任临床医生
/012
(如a1-抗胰蛋白酶缺乏) 阻止肺组织损伤的能力↓
+,-.
(如长期吸烟、环境污染、感染)
肺内产生自由基
34*5
肺抗蛋白酶的失活
↑氧化应激,炎性细胞因子,蛋白酶功能
支气管的持续、反复损伤
炎性细胞浸润, 杯状细胞增殖, 气道上皮纤毛 尤其中性粒细 黏液产生↑ 细胞死亡
气道弹性↓ (弹性回缩
肺实质的蛋白水解破坏↑ 维持气道开放 肺泡永久性异常
的结构支持↓ 扩张
胞 力)
肺气体潴留 气道狭窄与 肺过度 肺大泡
气道黏液潴留,成为感染 狭窄 病灶
塌陷 充气
肺气肿
(容易肺泡 破裂)
气道纤维化和
%&'()*
慢性阻塞性肺疾病(COPD)
临床表现 并发症 (参阅相关幻灯片) (参阅相关幻灯片)
图注:
病理生理
机制
体征/临床表现/实验室检查
并发症
2013年1月7日发布于 www.thecalgaryguide.com
COPD: Clinical Findings Lung tissue
Chronic Obstructive Pulmonary Disease (COPD)
damage
↓ elastic recoil to push air out of lungs on expiration
Lungs don’t fully empty, air is trapped in alveoli (lung hyperinflation)
↑ lung volume means diaphragm is tonically contracted (flatter)
If occurring around airways
Airflow obstruction
↑ mucus production
↓ number of epithelial ciliated cells to clear away the mucus (the cells have been killed by airway inflammation)
Chronic cough with sputum
Author: Yan Yu Reviewers: Jason Baserman Jennifer Au Naushad Hirani* Juri Janovcik* * MD at time of publication
During expiration, positive pleural pressure squeezes on airwaysà↑ obstruction
↓ ventilation of alveoli
↓ oxygenation of blood (hypoxemia)
↓ perfusion of body tissues (i.e. brain, muscle)
Fatigue; ↓ exercise tolerance
Total expiration time takes longer than normal
Prolonged expiration
More effort needed to ventilate larger lungs
Respiratory muscles must work harder to breathe
Turbulent airflow in narrower airways is heard on auscultation
Expiratory Wheeze
Diaphragm can’t flatten much further to generate deep breaths
To breathe, chest wall must expand out more
Dyspnea
Shortness of breath, especially on exertion
Breathes are rapid & shallow
If end-stage:
Chronic fatigue causes deconditioning
Muscle weakness & wasting
Barrel chest
If end-stage: diaphragm will be “flat”. Continued
Patient tries to expire against higher mouth air pressure, forcing airways to open wider
Pursed-lip breathing
Patient breathes with accessory muscles as well as diaphragm to try to improve airflow
inspiratory effort further contracts diaphragmà pull the lower chest wall inwards
Hoover’s sign
(paradoxical shrinking of lower chest during inspiration)
Tripod sitting position (activates pectoral muscles)
Neck (SCM, scalene) muscles contracted
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published January 7, 2013 on www.thecalgaryguide.com
COPD: !"#$
慢性阻塞性肺疾病 (COPD) 如果出现在气道周围 气流阻塞
肺不能完全排空
气体,气体潴留
于肺泡(肺过度
充气)
总呼气时长大于 正常时长
呼气相延长
肺组织损伤
呼气时,将空气排出肺外 的弹性回缩力↓
肺不能完全排空气体,
气体潴留于肺泡内
(肺过度充气)
肺容积↑,膈肌紧张 性收缩(膈肌平坦)
呼气时,胸膜腔正压挤压气道 à 气道阻塞↑
肺泡通气↓ 血液氧合↓ (低
氧血症)
身体组织灌注 量↓ (比如脑、 肌肉)
疲劳; 运动耐量↓
黏液生成↑ 清除黏液的上皮纤
毛细胞数量↓ (受 气道炎症损伤)
慢性咳嗽伴咳 痰
作者: Yan Yu
审稿人: Jason Baserman, Jennifer Au, Naushad Hirani*, Juri Janovcik* 译者: Zihong Xie (谢梓泓) 翻译审稿人: Yonglin Mai (麦泳琳),
Zesheng Ye (叶泽生)
* 发表时担任临床医生
容积较大 的肺需要
更加努力 才能通气
呼吸肌必须
更用力才能 呼吸
听诊闻及狭窄气
道中的湍流气流
呼气喘鸣音
呼吸困难 气促,尤其是劳累
膈肌无法进一步收缩以
产生深呼吸
呼吸浅快
为了呼吸,
胸壁必须延
展得更大
桶状胸
晚期病人:
患者试图在较高的口 慢性疲劳导致 患者动用辅助呼吸肌和膈肌呼吸,
腔内气压下进行呼气, 活动耐量下降 从而使气道更开放
以改善气流
晚期病人:膈肌 “平坦” ,持续吸气进一步压 缩膈肌à 向内拉季肋部胸壁
胡佛征 (吸气时,胸廓下侧季肋部内收)
缩唇呼吸
肌肉无力 & 消瘦
端坐呼吸 (调动胸肌)
颈部肌肉收
缩(胸锁乳
突肌、斜角
肌)
图注:
病理生理
机制
体征/临床表现/实验室检查
并发症
2013年1月7日发布于 www.thecalgaryguide.com
COPD: Findings on Investigations
Chronic Obstructive Pulmonary Disease (COPD)
Author: Yan Yu Reviewers: Jason Baserman Jennifer Au Naushad Hirani* Juri Janovcik* * MD at time of publication
Airflow obstruction
Lung tissue damage
↓ ventilation of alveoli
Blood perfusing ill- ventilated alveoli does not receive normal amounts of oxygen
During expiration, positive pleural pressure squeezes on airwaysà↑ obstruction)
No elastic recoil to push air out of lungs
Loss of lung parenchyma and vasculature ↓ surface area for gas exchange
↓ diffusion capacity
(on spirometry)
Hypoxemia: PaO2 < 70mmHg (on ABGs)
Abbreviations:
• FEV1: Forced expiratory volume in 1 second
• FVC: Forced vital capacity
• TLC: Total lung capacity
• VC: Vital Capacity
Investigations for COPD :
• Spirometry (Pulmonary function test)
Total expiration time takes longer than normal
FEV1/FEV < 0.7
(on spirometry)
Lungs don’t fully empty
More air trapped within lungs (hyperinflation)
More CO2 remains and diffuses into the blood
Hypercapnia: PaCO2 > 45
(on ABGs)
Ventilation- perfusion mismatch
High A-a gradient
(calculated from ABGs)
Low, flat diaphragm, >10 posterior ribs
(on frontal CXR)
High TLC and VC
(on spirometry)
• •
PaO2: partial pressure of O2 in arterial blood PaCO2: partial pressure of CO2 in arterial blood
• In the setting of fever and productive cough, especially if lung field opacifications are seen on CXR: consider sputum gram stain and culture to rule out pneumonia.
Air does not block X-ray beams, will appear black on X-ray film
Chronic hypercapnia makes breathing centers less sensitive to the high PaCO2 stimulus for breathing, & more reliant on the low PaO2 stimulus
(“CO2 retention”)
Give O2 carefully to these patients (high PaO2 may suppress patients’ hypoxic respiratory drive, ↓ their breathing, & ↑↑↑ PaCO2)
↑ retrosternal air space
(on lateral CXR)
Hyper-lucent
(darker) lung fields, ↓ lung markings (on frontal CXR)
• Arterial Blood Gasses (ABGs)
• Chest X-Ray (CXR): frontal and
lateral
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published January 7, 2013 on www.thecalgaryguide.com
COPD: !"#$ 气流阻塞
肺泡通气↓ 呼气时,胸膜腔正压挤压气 道à 阻塞↑
作者: Yan Yu 审稿人: Jason Baserman, Jennifer Au, Naushad Hirani*, Juri Janovcik* 译者:Zihong Xie (谢梓泓) 翻译审稿人: Yonglin Mai (麦泳琳), Zesheng Ye (叶泽生) * 发表时担任临床医生
慢性阻塞性肺疾病 (COPD)
肺组织损伤
没有弹性回缩力将
气体排出肺
肺实质与血管分布减少导 致气体交换面积↓
弥散功能↓ (肺功能检查)
更多的CO2残留 并扩散到血液中
高碳酸血症: PaCO2 > 45
(动脉血气)
血流灌注通气不良的肺泡
时无法获得足够的氧气
总呼气时长较正常长
FEV1/FEV < 0.7
(肺功能检查)
肺无法完全排空
更多空气潴留在肺部
(肺过度充气)
低氧血症: PaO2 < 70mmHg
(动脉血气)
通气-灌注不匹配
肺泡-动脉氧分压差↑ (可通过动脉血气分析计算得出)
横膈低平, 下移至第10肋后端 及以下部位 (胸部正位片)
TLC与VC增大 (肺功能检查)
缩写: • • FEV1: 1秒用 •
VC:肺活量
PaO2: 动脉血 力呼气量 氧分压
空气不会阻挡X射线, 在X光片上呈现为黑色
慢性高碳酸血症使呼吸中枢对PaCO2 刺激呼吸的敏感性下降 & 更依赖于低PaO2的刺激 (“二氧化碳潴留”)
给患者吸氧时需注意(高PaO2
可能会抑制患者低氧时对呼吸的 刺激,使呼吸驱动↓ & PaCO2↑↑↑ )
• FVC: 用力肺 • 活量
• TLC:肺总量 慢阻肺相关检查 :
PaCO2: 动脉 血二氧化碳 分压
胸骨后间隙↑
(胸部侧位片) 肺纹理↓
• 肺功能检查
• 动脉血气分析(Arterial Blood Gasses, ABGs)
• 胸部正侧位片
• 当患者发热和湿咳,特别是胸片上见肺野不清晰时:
肺透亮度↑, (胸部正位片)
考虑进行痰革兰氏染色及痰培养以排除肺炎可能
图注:
病理生理
机制
体征/临床表现/实验室检查
并发症
2013年1月7日发布于 www.thecalgaryguide.com
COPD: Complications Lung inflammation
Chronic Obstructive Pulmonary Disease (COPD)
Airway obstruction ↓ inhaled air in alveoli and terminal bronchioles
Rupture of emphasematous bullae on surface of lung
Inhaled air leaks into pleural cavity and is trapped there
Pneumothorax
Feeling a loss of control over one’s life, and hopelessness for the future
Goblet cell proliferation, ↑ mucus production
Death of airway
epithelium ciliated cells
↓ oxygenation of the blood passing through the lungs
Chronic hypoxemia
Kidneys compensate by ↑ erythropoietin (EPO) production
↑ Hemoglobin and red blood cell synthesis
Polycythemia (secondary)
Hypoxic alveoli cause the pulmonary arterioles perfusing them to reflexively vasoconstrict
Since most alveoli in the lungs are hypoxic, hypoxic vasoconstriction occurs across entire lung
Vasoconstriction ↑ blood pressure within lung vasculature
Pulmonary hypertension
↑ workload of the right ventricle (to pump against higher pressures)
To compensate, the right ventricle progressively hypertrophies and dilates, but over time its output ↓
Cor pulmonale
(Right heart failure in isolation, not due to Left heart failure)
Mucus trapped in airways, serve as nidus for infection
Acute exacerbation of COPD (AECOPD)
Pneumonia
The chronic, systemic inflammation in COPD is a hyper-metabolic state that consumes calories
Macro-nutrient deficiency
Trouble with respiration lead to inactivity and deconditioning
Wasting, muscle atrophy
More inactivity and deconditioning perpetuates the cycle
Depression
Author: Yan Yu Reviewers: Jason Baserman Naushad Hirani* Juri Janovcik* * MD at time of publication
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published January 7, 2013 on www.thecalgaryguide.com
COPD: !"# 肺部炎症
杯状细胞增殖, 气道上皮纤毛 粘液产生↑ 细胞死亡
黏液潴留呼吸道,成为感
染的病灶
慢性阻塞性肺疾病 (COPD) 气道阻塞à 吸入肺泡和终末细
肺大疱破裂
吸入的空气渗入
并潴留于胸腔
气胸
感觉生活失控,对未
来感到绝望
抑郁
作者: Yan Yu 审稿人: Jason Baserman, Naushad Hirani*, Juri Janovcik* 译者: Zihong Xie (谢梓泓) 翻译审稿人: Yonglin Mai (麦泳琳), Zesheng Ye (叶泽生) * 发表时担任临床医生
支气管的空气 ↓
流经肺的血液进行气 缺氧的肺泡à灌注肺泡的肺小动
慢性阻塞性肺疾 病急性加重期 (AECOPD)
肺炎
体交换↓ 慢性低氧血症
肾脏合成促红细胞 生成素进行代偿↑
血红蛋白与红 细胞合成↑
红细胞增多症 (继发性)
脉发生反射性血管收缩
肺大部分肺泡缺氧à整个肺 都出现缺氧性血管收缩
肺血管收缩 à 肺血管压力↑ 肺动脉高压
↑ 右心室负荷(泵血时对抗高压) 为了代偿,右心室逐渐肥大和扩张,
但随着病程进展,右心室输出量 ↓
肺心病 (单独出现右心衰竭,非左心衰)
COPD所致的慢性全身 呼吸困难导致活 性炎症会使机体处于高 动量减少和活动
代谢状态,消耗能量 耐量降低
宏量营养 素缺乏症
消瘦,肌肉萎缩
运动量下降和活动耐量
的降低造成恶性循环
图注:
病理生理
机制
体征/临床表现/实验室检查
并发症
2013年1月7日发布于 www.thecalgaryguide.com
" />
COPD-临床表现
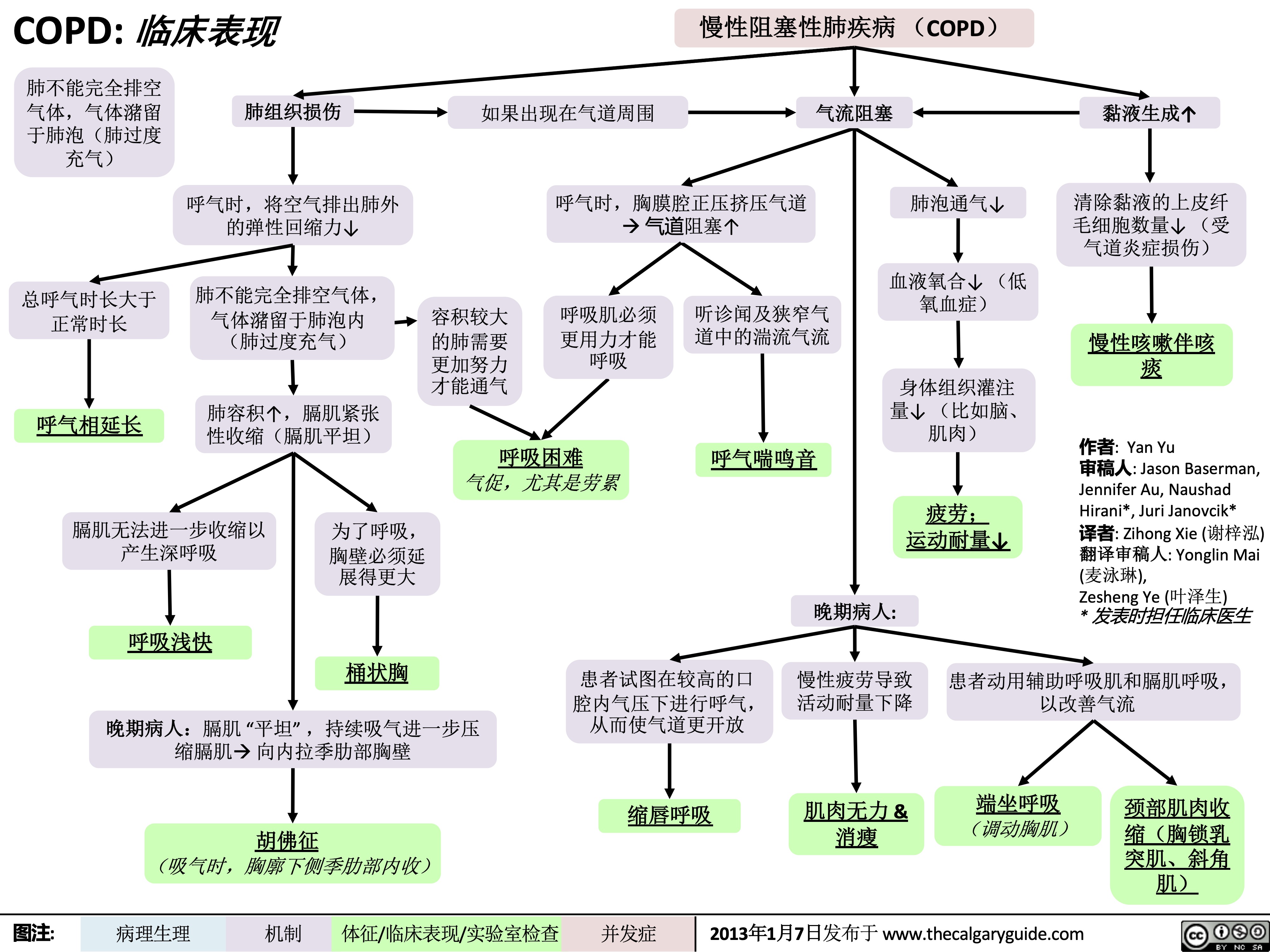 45
(on ABGs)
Ventilation- perfusion mismatch
High A-a gradient
(calculated from ABGs)
Low, flat diaphragm, >10 posterior ribs
(on frontal CXR)
High TLC and VC
(on spirometry)
• •
PaO2: partial pressure of O2 in arterial blood PaCO2: partial pressure of CO2 in arterial blood
• In the setting of fever and productive cough, especially if lung field opacifications are seen on CXR: consider sputum gram stain and culture to rule out pneumonia.
Air does not block X-ray beams, will appear black on X-ray film
Chronic hypercapnia makes breathing centers less sensitive to the high PaCO2 stimulus for breathing, & more reliant on the low PaO2 stimulus
(“CO2 retention”)
Give O2 carefully to these patients (high PaO2 may suppress patients’ hypoxic respiratory drive, ↓ their breathing, & ↑↑↑ PaCO2)
↑ retrosternal air space
(on lateral CXR)
Hyper-lucent
(darker) lung fields, ↓ lung markings (on frontal CXR)
• Arterial Blood Gasses (ABGs)
• Chest X-Ray (CXR): frontal and
lateral
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published January 7, 2013 on www.thecalgaryguide.com
COPD: !"#$ 气流阻塞
肺泡通气↓ 呼气时,胸膜腔正压挤压气 道à 阻塞↑
作者: Yan Yu 审稿人: Jason Baserman, Jennifer Au, Naushad Hirani*, Juri Janovcik* 译者:Zihong Xie (谢梓泓) 翻译审稿人: Yonglin Mai (麦泳琳), Zesheng Ye (叶泽生) * 发表时担任临床医生
慢性阻塞性肺疾病 (COPD)
肺组织损伤
没有弹性回缩力将
气体排出肺
肺实质与血管分布减少导 致气体交换面积↓
弥散功能↓ (肺功能检查)
更多的CO2残留 并扩散到血液中
高碳酸血症: PaCO2 > 45
(动脉血气)
血流灌注通气不良的肺泡
时无法获得足够的氧气
总呼气时长较正常长
FEV1/FEV < 0.7
(肺功能检查)
肺无法完全排空
更多空气潴留在肺部
(肺过度充气)
低氧血症: PaO2 < 70mmHg
(动脉血气)
通气-灌注不匹配
肺泡-动脉氧分压差↑ (可通过动脉血气分析计算得出)
横膈低平, 下移至第10肋后端 及以下部位 (胸部正位片)
TLC与VC增大 (肺功能检查)
缩写: • • FEV1: 1秒用 •
VC:肺活量
PaO2: 动脉血 力呼气量 氧分压
空气不会阻挡X射线, 在X光片上呈现为黑色
慢性高碳酸血症使呼吸中枢对PaCO2 刺激呼吸的敏感性下降 & 更依赖于低PaO2的刺激 (“二氧化碳潴留”)
给患者吸氧时需注意(高PaO2
可能会抑制患者低氧时对呼吸的 刺激,使呼吸驱动↓ & PaCO2↑↑↑ )
• FVC: 用力肺 • 活量
• TLC:肺总量 慢阻肺相关检查 :
PaCO2: 动脉 血二氧化碳 分压
胸骨后间隙↑
(胸部侧位片) 肺纹理↓
• 肺功能检查
• 动脉血气分析(Arterial Blood Gasses, ABGs)
• 胸部正侧位片
• 当患者发热和湿咳,特别是胸片上见肺野不清晰时:
肺透亮度↑, (胸部正位片)
考虑进行痰革兰氏染色及痰培养以排除肺炎可能
图注:
病理生理
机制
体征/临床表现/实验室检查
并发症
2013年1月7日发布于 www.thecalgaryguide.com
COPD: Complications Lung inflammation
Chronic Obstructive Pulmonary Disease (COPD)
Airway obstruction ↓ inhaled air in alveoli and terminal bronchioles
Rupture of emphasematous bullae on surface of lung
Inhaled air leaks into pleural cavity and is trapped there
Pneumothorax
Feeling a loss of control over one’s life, and hopelessness for the future
Goblet cell proliferation, ↑ mucus production
Death of airway
epithelium ciliated cells
↓ oxygenation of the blood passing through the lungs
Chronic hypoxemia
Kidneys compensate by ↑ erythropoietin (EPO) production
↑ Hemoglobin and red blood cell synthesis
Polycythemia (secondary)
Hypoxic alveoli cause the pulmonary arterioles perfusing them to reflexively vasoconstrict
Since most alveoli in the lungs are hypoxic, hypoxic vasoconstriction occurs across entire lung
Vasoconstriction ↑ blood pressure within lung vasculature
Pulmonary hypertension
↑ workload of the right ventricle (to pump against higher pressures)
To compensate, the right ventricle progressively hypertrophies and dilates, but over time its output ↓
Cor pulmonale
(Right heart failure in isolation, not due to Left heart failure)
Mucus trapped in airways, serve as nidus for infection
Acute exacerbation of COPD (AECOPD)
Pneumonia
The chronic, systemic inflammation in COPD is a hyper-metabolic state that consumes calories
Macro-nutrient deficiency
Trouble with respiration lead to inactivity and deconditioning
Wasting, muscle atrophy
More inactivity and deconditioning perpetuates the cycle
Depression
Author: Yan Yu Reviewers: Jason Baserman Naushad Hirani* Juri Janovcik* * MD at time of publication
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published January 7, 2013 on www.thecalgaryguide.com
COPD: !"# 肺部炎症
杯状细胞增殖, 气道上皮纤毛 粘液产生↑ 细胞死亡
黏液潴留呼吸道,成为感
染的病灶
慢性阻塞性肺疾病 (COPD) 气道阻塞à 吸入肺泡和终末细
肺大疱破裂
吸入的空气渗入
并潴留于胸腔
气胸
感觉生活失控,对未
来感到绝望
抑郁
作者: Yan Yu 审稿人: Jason Baserman, Naushad Hirani*, Juri Janovcik* 译者: Zihong Xie (谢梓泓) 翻译审稿人: Yonglin Mai (麦泳琳), Zesheng Ye (叶泽生) * 发表时担任临床医生
支气管的空气 ↓
流经肺的血液进行气 缺氧的肺泡à灌注肺泡的肺小动
慢性阻塞性肺疾 病急性加重期 (AECOPD)
肺炎
体交换↓ 慢性低氧血症
肾脏合成促红细胞 生成素进行代偿↑
血红蛋白与红 细胞合成↑
红细胞增多症 (继发性)
脉发生反射性血管收缩
肺大部分肺泡缺氧à整个肺 都出现缺氧性血管收缩
肺血管收缩 à 肺血管压力↑ 肺动脉高压
↑ 右心室负荷(泵血时对抗高压) 为了代偿,右心室逐渐肥大和扩张,
但随着病程进展,右心室输出量 ↓
肺心病 (单独出现右心衰竭,非左心衰)
COPD所致的慢性全身 呼吸困难导致活 性炎症会使机体处于高 动量减少和活动
代谢状态,消耗能量 耐量降低
宏量营养 素缺乏症
消瘦,肌肉萎缩
运动量下降和活动耐量
的降低造成恶性循环
图注:
病理生理
机制
体征/临床表现/实验室检查
并发症
2013年1月7日发布于 www.thecalgaryguide.com
" title="COPD: 临床表现
作者: Yan Yu 审稿人:Jason Baserman, Jennifer Au, Naushad Hirani*, Juri Janovcik* 译者: Zihong Xie (谢梓泓) 翻译审稿人:Yonglin Mai (麦泳琳), Zesheng Ye (叶泽生) * 发表时担任临床医生
/012
(如a1-抗胰蛋白酶缺乏) 阻止肺组织损伤的能力↓
+,-.
(如长期吸烟、环境污染、感染)
肺内产生自由基
34*5
肺抗蛋白酶的失活
↑氧化应激,炎性细胞因子,蛋白酶功能
支气管的持续、反复损伤
炎性细胞浸润, 杯状细胞增殖, 气道上皮纤毛 尤其中性粒细 黏液产生↑ 细胞死亡
气道弹性↓ (弹性回缩
肺实质的蛋白水解破坏↑ 维持气道开放 肺泡永久性异常
的结构支持↓ 扩张
胞 力)
肺气体潴留 气道狭窄与 肺过度 肺大泡
气道黏液潴留,成为感染 狭窄 病灶
塌陷 充气
肺气肿
(容易肺泡 破裂)
气道纤维化和
%&'()*
慢性阻塞性肺疾病(COPD)
临床表现 并发症 (参阅相关幻灯片) (参阅相关幻灯片)
图注:
病理生理
机制
体征/临床表现/实验室检查
并发症
2013年1月7日发布于 www.thecalgaryguide.com
COPD: Clinical Findings Lung tissue
Chronic Obstructive Pulmonary Disease (COPD)
damage
↓ elastic recoil to push air out of lungs on expiration
Lungs don’t fully empty, air is trapped in alveoli (lung hyperinflation)
↑ lung volume means diaphragm is tonically contracted (flatter)
If occurring around airways
Airflow obstruction
↑ mucus production
↓ number of epithelial ciliated cells to clear away the mucus (the cells have been killed by airway inflammation)
Chronic cough with sputum
Author: Yan Yu Reviewers: Jason Baserman Jennifer Au Naushad Hirani* Juri Janovcik* * MD at time of publication
During expiration, positive pleural pressure squeezes on airwaysà↑ obstruction
↓ ventilation of alveoli
↓ oxygenation of blood (hypoxemia)
↓ perfusion of body tissues (i.e. brain, muscle)
Fatigue; ↓ exercise tolerance
Total expiration time takes longer than normal
Prolonged expiration
More effort needed to ventilate larger lungs
Respiratory muscles must work harder to breathe
Turbulent airflow in narrower airways is heard on auscultation
Expiratory Wheeze
Diaphragm can’t flatten much further to generate deep breaths
To breathe, chest wall must expand out more
Dyspnea
Shortness of breath, especially on exertion
Breathes are rapid & shallow
If end-stage:
Chronic fatigue causes deconditioning
Muscle weakness & wasting
Barrel chest
If end-stage: diaphragm will be “flat”. Continued
Patient tries to expire against higher mouth air pressure, forcing airways to open wider
Pursed-lip breathing
Patient breathes with accessory muscles as well as diaphragm to try to improve airflow
inspiratory effort further contracts diaphragmà pull the lower chest wall inwards
Hoover’s sign
(paradoxical shrinking of lower chest during inspiration)
Tripod sitting position (activates pectoral muscles)
Neck (SCM, scalene) muscles contracted
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published January 7, 2013 on www.thecalgaryguide.com
COPD: !"#$
慢性阻塞性肺疾病 (COPD) 如果出现在气道周围 气流阻塞
肺不能完全排空
气体,气体潴留
于肺泡(肺过度
充气)
总呼气时长大于 正常时长
呼气相延长
肺组织损伤
呼气时,将空气排出肺外 的弹性回缩力↓
肺不能完全排空气体,
气体潴留于肺泡内
(肺过度充气)
肺容积↑,膈肌紧张 性收缩(膈肌平坦)
呼气时,胸膜腔正压挤压气道 à 气道阻塞↑
肺泡通气↓ 血液氧合↓ (低
氧血症)
身体组织灌注 量↓ (比如脑、 肌肉)
疲劳; 运动耐量↓
黏液生成↑ 清除黏液的上皮纤
毛细胞数量↓ (受 气道炎症损伤)
慢性咳嗽伴咳 痰
作者: Yan Yu
审稿人: Jason Baserman, Jennifer Au, Naushad Hirani*, Juri Janovcik* 译者: Zihong Xie (谢梓泓) 翻译审稿人: Yonglin Mai (麦泳琳),
Zesheng Ye (叶泽生)
* 发表时担任临床医生
容积较大 的肺需要
更加努力 才能通气
呼吸肌必须
更用力才能 呼吸
听诊闻及狭窄气
道中的湍流气流
呼气喘鸣音
呼吸困难 气促,尤其是劳累
膈肌无法进一步收缩以
产生深呼吸
呼吸浅快
为了呼吸,
胸壁必须延
展得更大
桶状胸
晚期病人:
患者试图在较高的口 慢性疲劳导致 患者动用辅助呼吸肌和膈肌呼吸,
腔内气压下进行呼气, 活动耐量下降 从而使气道更开放
以改善气流
晚期病人:膈肌 “平坦” ,持续吸气进一步压 缩膈肌à 向内拉季肋部胸壁
胡佛征 (吸气时,胸廓下侧季肋部内收)
缩唇呼吸
肌肉无力 & 消瘦
端坐呼吸 (调动胸肌)
颈部肌肉收
缩(胸锁乳
突肌、斜角
肌)
图注:
病理生理
机制
体征/临床表现/实验室检查
并发症
2013年1月7日发布于 www.thecalgaryguide.com
COPD: Findings on Investigations
Chronic Obstructive Pulmonary Disease (COPD)
Author: Yan Yu Reviewers: Jason Baserman Jennifer Au Naushad Hirani* Juri Janovcik* * MD at time of publication
Airflow obstruction
Lung tissue damage
↓ ventilation of alveoli
Blood perfusing ill- ventilated alveoli does not receive normal amounts of oxygen
During expiration, positive pleural pressure squeezes on airwaysà↑ obstruction)
No elastic recoil to push air out of lungs
Loss of lung parenchyma and vasculature ↓ surface area for gas exchange
↓ diffusion capacity
(on spirometry)
Hypoxemia: PaO2 < 70mmHg (on ABGs)
Abbreviations:
• FEV1: Forced expiratory volume in 1 second
• FVC: Forced vital capacity
• TLC: Total lung capacity
• VC: Vital Capacity
Investigations for COPD :
• Spirometry (Pulmonary function test)
Total expiration time takes longer than normal
FEV1/FEV < 0.7
(on spirometry)
Lungs don’t fully empty
More air trapped within lungs (hyperinflation)
More CO2 remains and diffuses into the blood
Hypercapnia: PaCO2 > 45
(on ABGs)
Ventilation- perfusion mismatch
High A-a gradient
(calculated from ABGs)
Low, flat diaphragm, >10 posterior ribs
(on frontal CXR)
High TLC and VC
(on spirometry)
• •
PaO2: partial pressure of O2 in arterial blood PaCO2: partial pressure of CO2 in arterial blood
• In the setting of fever and productive cough, especially if lung field opacifications are seen on CXR: consider sputum gram stain and culture to rule out pneumonia.
Air does not block X-ray beams, will appear black on X-ray film
Chronic hypercapnia makes breathing centers less sensitive to the high PaCO2 stimulus for breathing, & more reliant on the low PaO2 stimulus
(“CO2 retention”)
Give O2 carefully to these patients (high PaO2 may suppress patients’ hypoxic respiratory drive, ↓ their breathing, & ↑↑↑ PaCO2)
↑ retrosternal air space
(on lateral CXR)
Hyper-lucent
(darker) lung fields, ↓ lung markings (on frontal CXR)
• Arterial Blood Gasses (ABGs)
• Chest X-Ray (CXR): frontal and
lateral
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published January 7, 2013 on www.thecalgaryguide.com
COPD: !"#$ 气流阻塞
肺泡通气↓ 呼气时,胸膜腔正压挤压气 道à 阻塞↑
作者: Yan Yu 审稿人: Jason Baserman, Jennifer Au, Naushad Hirani*, Juri Janovcik* 译者:Zihong Xie (谢梓泓) 翻译审稿人: Yonglin Mai (麦泳琳), Zesheng Ye (叶泽生) * 发表时担任临床医生
慢性阻塞性肺疾病 (COPD)
肺组织损伤
没有弹性回缩力将
气体排出肺
肺实质与血管分布减少导 致气体交换面积↓
弥散功能↓ (肺功能检查)
更多的CO2残留 并扩散到血液中
高碳酸血症: PaCO2 > 45
(动脉血气)
血流灌注通气不良的肺泡
时无法获得足够的氧气
总呼气时长较正常长
FEV1/FEV < 0.7
(肺功能检查)
肺无法完全排空
更多空气潴留在肺部
(肺过度充气)
低氧血症: PaO2 < 70mmHg
(动脉血气)
通气-灌注不匹配
肺泡-动脉氧分压差↑ (可通过动脉血气分析计算得出)
横膈低平, 下移至第10肋后端 及以下部位 (胸部正位片)
TLC与VC增大 (肺功能检查)
缩写: • • FEV1: 1秒用 •
VC:肺活量
PaO2: 动脉血 力呼气量 氧分压
空气不会阻挡X射线, 在X光片上呈现为黑色
慢性高碳酸血症使呼吸中枢对PaCO2 刺激呼吸的敏感性下降 & 更依赖于低PaO2的刺激 (“二氧化碳潴留”)
给患者吸氧时需注意(高PaO2
可能会抑制患者低氧时对呼吸的 刺激,使呼吸驱动↓ & PaCO2↑↑↑ )
• FVC: 用力肺 • 活量
• TLC:肺总量 慢阻肺相关检查 :
PaCO2: 动脉 血二氧化碳 分压
胸骨后间隙↑
(胸部侧位片) 肺纹理↓
• 肺功能检查
• 动脉血气分析(Arterial Blood Gasses, ABGs)
• 胸部正侧位片
• 当患者发热和湿咳,特别是胸片上见肺野不清晰时:
肺透亮度↑, (胸部正位片)
考虑进行痰革兰氏染色及痰培养以排除肺炎可能
图注:
病理生理
机制
体征/临床表现/实验室检查
并发症
2013年1月7日发布于 www.thecalgaryguide.com
COPD: Complications Lung inflammation
Chronic Obstructive Pulmonary Disease (COPD)
Airway obstruction ↓ inhaled air in alveoli and terminal bronchioles
Rupture of emphasematous bullae on surface of lung
Inhaled air leaks into pleural cavity and is trapped there
Pneumothorax
Feeling a loss of control over one’s life, and hopelessness for the future
Goblet cell proliferation, ↑ mucus production
Death of airway
epithelium ciliated cells
↓ oxygenation of the blood passing through the lungs
Chronic hypoxemia
Kidneys compensate by ↑ erythropoietin (EPO) production
↑ Hemoglobin and red blood cell synthesis
Polycythemia (secondary)
Hypoxic alveoli cause the pulmonary arterioles perfusing them to reflexively vasoconstrict
Since most alveoli in the lungs are hypoxic, hypoxic vasoconstriction occurs across entire lung
Vasoconstriction ↑ blood pressure within lung vasculature
Pulmonary hypertension
↑ workload of the right ventricle (to pump against higher pressures)
To compensate, the right ventricle progressively hypertrophies and dilates, but over time its output ↓
Cor pulmonale
(Right heart failure in isolation, not due to Left heart failure)
Mucus trapped in airways, serve as nidus for infection
Acute exacerbation of COPD (AECOPD)
Pneumonia
The chronic, systemic inflammation in COPD is a hyper-metabolic state that consumes calories
Macro-nutrient deficiency
Trouble with respiration lead to inactivity and deconditioning
Wasting, muscle atrophy
More inactivity and deconditioning perpetuates the cycle
Depression
Author: Yan Yu Reviewers: Jason Baserman Naushad Hirani* Juri Janovcik* * MD at time of publication
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published January 7, 2013 on www.thecalgaryguide.com
COPD: !"# 肺部炎症
杯状细胞增殖, 气道上皮纤毛 粘液产生↑ 细胞死亡
黏液潴留呼吸道,成为感
染的病灶
慢性阻塞性肺疾病 (COPD) 气道阻塞à 吸入肺泡和终末细
肺大疱破裂
吸入的空气渗入
并潴留于胸腔
气胸
感觉生活失控,对未
来感到绝望
抑郁
作者: Yan Yu 审稿人: Jason Baserman, Naushad Hirani*, Juri Janovcik* 译者: Zihong Xie (谢梓泓) 翻译审稿人: Yonglin Mai (麦泳琳), Zesheng Ye (叶泽生) * 发表时担任临床医生
支气管的空气 ↓
流经肺的血液进行气 缺氧的肺泡à灌注肺泡的肺小动
慢性阻塞性肺疾 病急性加重期 (AECOPD)
肺炎
体交换↓ 慢性低氧血症
肾脏合成促红细胞 生成素进行代偿↑
血红蛋白与红 细胞合成↑
红细胞增多症 (继发性)
脉发生反射性血管收缩
肺大部分肺泡缺氧à整个肺 都出现缺氧性血管收缩
肺血管收缩 à 肺血管压力↑ 肺动脉高压
↑ 右心室负荷(泵血时对抗高压) 为了代偿,右心室逐渐肥大和扩张,
但随着病程进展,右心室输出量 ↓
肺心病 (单独出现右心衰竭,非左心衰)
COPD所致的慢性全身 呼吸困难导致活 性炎症会使机体处于高 动量减少和活动
代谢状态,消耗能量 耐量降低
宏量营养 素缺乏症
消瘦,肌肉萎缩
运动量下降和活动耐量
的降低造成恶性循环
图注:
病理生理
机制
体征/临床表现/实验室检查
并发症
2013年1月7日发布于 www.thecalgaryguide.com
" />
45
(on ABGs)
Ventilation- perfusion mismatch
High A-a gradient
(calculated from ABGs)
Low, flat diaphragm, >10 posterior ribs
(on frontal CXR)
High TLC and VC
(on spirometry)
• •
PaO2: partial pressure of O2 in arterial blood PaCO2: partial pressure of CO2 in arterial blood
• In the setting of fever and productive cough, especially if lung field opacifications are seen on CXR: consider sputum gram stain and culture to rule out pneumonia.
Air does not block X-ray beams, will appear black on X-ray film
Chronic hypercapnia makes breathing centers less sensitive to the high PaCO2 stimulus for breathing, & more reliant on the low PaO2 stimulus
(“CO2 retention”)
Give O2 carefully to these patients (high PaO2 may suppress patients’ hypoxic respiratory drive, ↓ their breathing, & ↑↑↑ PaCO2)
↑ retrosternal air space
(on lateral CXR)
Hyper-lucent
(darker) lung fields, ↓ lung markings (on frontal CXR)
• Arterial Blood Gasses (ABGs)
• Chest X-Ray (CXR): frontal and
lateral
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published January 7, 2013 on www.thecalgaryguide.com
COPD: !"#$ 气流阻塞
肺泡通气↓ 呼气时,胸膜腔正压挤压气 道à 阻塞↑
作者: Yan Yu 审稿人: Jason Baserman, Jennifer Au, Naushad Hirani*, Juri Janovcik* 译者:Zihong Xie (谢梓泓) 翻译审稿人: Yonglin Mai (麦泳琳), Zesheng Ye (叶泽生) * 发表时担任临床医生
慢性阻塞性肺疾病 (COPD)
肺组织损伤
没有弹性回缩力将
气体排出肺
肺实质与血管分布减少导 致气体交换面积↓
弥散功能↓ (肺功能检查)
更多的CO2残留 并扩散到血液中
高碳酸血症: PaCO2 > 45
(动脉血气)
血流灌注通气不良的肺泡
时无法获得足够的氧气
总呼气时长较正常长
FEV1/FEV < 0.7
(肺功能检查)
肺无法完全排空
更多空气潴留在肺部
(肺过度充气)
低氧血症: PaO2 < 70mmHg
(动脉血气)
通气-灌注不匹配
肺泡-动脉氧分压差↑ (可通过动脉血气分析计算得出)
横膈低平, 下移至第10肋后端 及以下部位 (胸部正位片)
TLC与VC增大 (肺功能检查)
缩写: • • FEV1: 1秒用 •
VC:肺活量
PaO2: 动脉血 力呼气量 氧分压
空气不会阻挡X射线, 在X光片上呈现为黑色
慢性高碳酸血症使呼吸中枢对PaCO2 刺激呼吸的敏感性下降 & 更依赖于低PaO2的刺激 (“二氧化碳潴留”)
给患者吸氧时需注意(高PaO2
可能会抑制患者低氧时对呼吸的 刺激,使呼吸驱动↓ & PaCO2↑↑↑ )
• FVC: 用力肺 • 活量
• TLC:肺总量 慢阻肺相关检查 :
PaCO2: 动脉 血二氧化碳 分压
胸骨后间隙↑
(胸部侧位片) 肺纹理↓
• 肺功能检查
• 动脉血气分析(Arterial Blood Gasses, ABGs)
• 胸部正侧位片
• 当患者发热和湿咳,特别是胸片上见肺野不清晰时:
肺透亮度↑, (胸部正位片)
考虑进行痰革兰氏染色及痰培养以排除肺炎可能
图注:
病理生理
机制
体征/临床表现/实验室检查
并发症
2013年1月7日发布于 www.thecalgaryguide.com
COPD: Complications Lung inflammation
Chronic Obstructive Pulmonary Disease (COPD)
Airway obstruction ↓ inhaled air in alveoli and terminal bronchioles
Rupture of emphasematous bullae on surface of lung
Inhaled air leaks into pleural cavity and is trapped there
Pneumothorax
Feeling a loss of control over one’s life, and hopelessness for the future
Goblet cell proliferation, ↑ mucus production
Death of airway
epithelium ciliated cells
↓ oxygenation of the blood passing through the lungs
Chronic hypoxemia
Kidneys compensate by ↑ erythropoietin (EPO) production
↑ Hemoglobin and red blood cell synthesis
Polycythemia (secondary)
Hypoxic alveoli cause the pulmonary arterioles perfusing them to reflexively vasoconstrict
Since most alveoli in the lungs are hypoxic, hypoxic vasoconstriction occurs across entire lung
Vasoconstriction ↑ blood pressure within lung vasculature
Pulmonary hypertension
↑ workload of the right ventricle (to pump against higher pressures)
To compensate, the right ventricle progressively hypertrophies and dilates, but over time its output ↓
Cor pulmonale
(Right heart failure in isolation, not due to Left heart failure)
Mucus trapped in airways, serve as nidus for infection
Acute exacerbation of COPD (AECOPD)
Pneumonia
The chronic, systemic inflammation in COPD is a hyper-metabolic state that consumes calories
Macro-nutrient deficiency
Trouble with respiration lead to inactivity and deconditioning
Wasting, muscle atrophy
More inactivity and deconditioning perpetuates the cycle
Depression
Author: Yan Yu Reviewers: Jason Baserman Naushad Hirani* Juri Janovcik* * MD at time of publication
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published January 7, 2013 on www.thecalgaryguide.com
COPD: !"# 肺部炎症
杯状细胞增殖, 气道上皮纤毛 粘液产生↑ 细胞死亡
黏液潴留呼吸道,成为感
染的病灶
慢性阻塞性肺疾病 (COPD) 气道阻塞à 吸入肺泡和终末细
肺大疱破裂
吸入的空气渗入
并潴留于胸腔
气胸
感觉生活失控,对未
来感到绝望
抑郁
作者: Yan Yu 审稿人: Jason Baserman, Naushad Hirani*, Juri Janovcik* 译者: Zihong Xie (谢梓泓) 翻译审稿人: Yonglin Mai (麦泳琳), Zesheng Ye (叶泽生) * 发表时担任临床医生
支气管的空气 ↓
流经肺的血液进行气 缺氧的肺泡à灌注肺泡的肺小动
慢性阻塞性肺疾 病急性加重期 (AECOPD)
肺炎
体交换↓ 慢性低氧血症
肾脏合成促红细胞 生成素进行代偿↑
血红蛋白与红 细胞合成↑
红细胞增多症 (继发性)
脉发生反射性血管收缩
肺大部分肺泡缺氧à整个肺 都出现缺氧性血管收缩
肺血管收缩 à 肺血管压力↑ 肺动脉高压
↑ 右心室负荷(泵血时对抗高压) 为了代偿,右心室逐渐肥大和扩张,
但随着病程进展,右心室输出量 ↓
肺心病 (单独出现右心衰竭,非左心衰)
COPD所致的慢性全身 呼吸困难导致活 性炎症会使机体处于高 动量减少和活动
代谢状态,消耗能量 耐量降低
宏量营养 素缺乏症
消瘦,肌肉萎缩
运动量下降和活动耐量
的降低造成恶性循环
图注:
病理生理
机制
体征/临床表现/实验室检查
并发症
2013年1月7日发布于 www.thecalgaryguide.com
" title="COPD: 临床表现
作者: Yan Yu 审稿人:Jason Baserman, Jennifer Au, Naushad Hirani*, Juri Janovcik* 译者: Zihong Xie (谢梓泓) 翻译审稿人:Yonglin Mai (麦泳琳), Zesheng Ye (叶泽生) * 发表时担任临床医生
/012
(如a1-抗胰蛋白酶缺乏) 阻止肺组织损伤的能力↓
+,-.
(如长期吸烟、环境污染、感染)
肺内产生自由基
34*5
肺抗蛋白酶的失活
↑氧化应激,炎性细胞因子,蛋白酶功能
支气管的持续、反复损伤
炎性细胞浸润, 杯状细胞增殖, 气道上皮纤毛 尤其中性粒细 黏液产生↑ 细胞死亡
气道弹性↓ (弹性回缩
肺实质的蛋白水解破坏↑ 维持气道开放 肺泡永久性异常
的结构支持↓ 扩张
胞 力)
肺气体潴留 气道狭窄与 肺过度 肺大泡
气道黏液潴留,成为感染 狭窄 病灶
塌陷 充气
肺气肿
(容易肺泡 破裂)
气道纤维化和
%&'()*
慢性阻塞性肺疾病(COPD)
临床表现 并发症 (参阅相关幻灯片) (参阅相关幻灯片)
图注:
病理生理
机制
体征/临床表现/实验室检查
并发症
2013年1月7日发布于 www.thecalgaryguide.com
COPD: Clinical Findings Lung tissue
Chronic Obstructive Pulmonary Disease (COPD)
damage
↓ elastic recoil to push air out of lungs on expiration
Lungs don’t fully empty, air is trapped in alveoli (lung hyperinflation)
↑ lung volume means diaphragm is tonically contracted (flatter)
If occurring around airways
Airflow obstruction
↑ mucus production
↓ number of epithelial ciliated cells to clear away the mucus (the cells have been killed by airway inflammation)
Chronic cough with sputum
Author: Yan Yu Reviewers: Jason Baserman Jennifer Au Naushad Hirani* Juri Janovcik* * MD at time of publication
During expiration, positive pleural pressure squeezes on airwaysà↑ obstruction
↓ ventilation of alveoli
↓ oxygenation of blood (hypoxemia)
↓ perfusion of body tissues (i.e. brain, muscle)
Fatigue; ↓ exercise tolerance
Total expiration time takes longer than normal
Prolonged expiration
More effort needed to ventilate larger lungs
Respiratory muscles must work harder to breathe
Turbulent airflow in narrower airways is heard on auscultation
Expiratory Wheeze
Diaphragm can’t flatten much further to generate deep breaths
To breathe, chest wall must expand out more
Dyspnea
Shortness of breath, especially on exertion
Breathes are rapid & shallow
If end-stage:
Chronic fatigue causes deconditioning
Muscle weakness & wasting
Barrel chest
If end-stage: diaphragm will be “flat”. Continued
Patient tries to expire against higher mouth air pressure, forcing airways to open wider
Pursed-lip breathing
Patient breathes with accessory muscles as well as diaphragm to try to improve airflow
inspiratory effort further contracts diaphragmà pull the lower chest wall inwards
Hoover’s sign
(paradoxical shrinking of lower chest during inspiration)
Tripod sitting position (activates pectoral muscles)
Neck (SCM, scalene) muscles contracted
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published January 7, 2013 on www.thecalgaryguide.com
COPD: !"#$
慢性阻塞性肺疾病 (COPD) 如果出现在气道周围 气流阻塞
肺不能完全排空
气体,气体潴留
于肺泡(肺过度
充气)
总呼气时长大于 正常时长
呼气相延长
肺组织损伤
呼气时,将空气排出肺外 的弹性回缩力↓
肺不能完全排空气体,
气体潴留于肺泡内
(肺过度充气)
肺容积↑,膈肌紧张 性收缩(膈肌平坦)
呼气时,胸膜腔正压挤压气道 à 气道阻塞↑
肺泡通气↓ 血液氧合↓ (低
氧血症)
身体组织灌注 量↓ (比如脑、 肌肉)
疲劳; 运动耐量↓
黏液生成↑ 清除黏液的上皮纤
毛细胞数量↓ (受 气道炎症损伤)
慢性咳嗽伴咳 痰
作者: Yan Yu
审稿人: Jason Baserman, Jennifer Au, Naushad Hirani*, Juri Janovcik* 译者: Zihong Xie (谢梓泓) 翻译审稿人: Yonglin Mai (麦泳琳),
Zesheng Ye (叶泽生)
* 发表时担任临床医生
容积较大 的肺需要
更加努力 才能通气
呼吸肌必须
更用力才能 呼吸
听诊闻及狭窄气
道中的湍流气流
呼气喘鸣音
呼吸困难 气促,尤其是劳累
膈肌无法进一步收缩以
产生深呼吸
呼吸浅快
为了呼吸,
胸壁必须延
展得更大
桶状胸
晚期病人:
患者试图在较高的口 慢性疲劳导致 患者动用辅助呼吸肌和膈肌呼吸,
腔内气压下进行呼气, 活动耐量下降 从而使气道更开放
以改善气流
晚期病人:膈肌 “平坦” ,持续吸气进一步压 缩膈肌à 向内拉季肋部胸壁
胡佛征 (吸气时,胸廓下侧季肋部内收)
缩唇呼吸
肌肉无力 & 消瘦
端坐呼吸 (调动胸肌)
颈部肌肉收
缩(胸锁乳
突肌、斜角
肌)
图注:
病理生理
机制
体征/临床表现/实验室检查
并发症
2013年1月7日发布于 www.thecalgaryguide.com
COPD: Findings on Investigations
Chronic Obstructive Pulmonary Disease (COPD)
Author: Yan Yu Reviewers: Jason Baserman Jennifer Au Naushad Hirani* Juri Janovcik* * MD at time of publication
Airflow obstruction
Lung tissue damage
↓ ventilation of alveoli
Blood perfusing ill- ventilated alveoli does not receive normal amounts of oxygen
During expiration, positive pleural pressure squeezes on airwaysà↑ obstruction)
No elastic recoil to push air out of lungs
Loss of lung parenchyma and vasculature ↓ surface area for gas exchange
↓ diffusion capacity
(on spirometry)
Hypoxemia: PaO2 < 70mmHg (on ABGs)
Abbreviations:
• FEV1: Forced expiratory volume in 1 second
• FVC: Forced vital capacity
• TLC: Total lung capacity
• VC: Vital Capacity
Investigations for COPD :
• Spirometry (Pulmonary function test)
Total expiration time takes longer than normal
FEV1/FEV < 0.7
(on spirometry)
Lungs don’t fully empty
More air trapped within lungs (hyperinflation)
More CO2 remains and diffuses into the blood
Hypercapnia: PaCO2 > 45
(on ABGs)
Ventilation- perfusion mismatch
High A-a gradient
(calculated from ABGs)
Low, flat diaphragm, >10 posterior ribs
(on frontal CXR)
High TLC and VC
(on spirometry)
• •
PaO2: partial pressure of O2 in arterial blood PaCO2: partial pressure of CO2 in arterial blood
• In the setting of fever and productive cough, especially if lung field opacifications are seen on CXR: consider sputum gram stain and culture to rule out pneumonia.
Air does not block X-ray beams, will appear black on X-ray film
Chronic hypercapnia makes breathing centers less sensitive to the high PaCO2 stimulus for breathing, & more reliant on the low PaO2 stimulus
(“CO2 retention”)
Give O2 carefully to these patients (high PaO2 may suppress patients’ hypoxic respiratory drive, ↓ their breathing, & ↑↑↑ PaCO2)
↑ retrosternal air space
(on lateral CXR)
Hyper-lucent
(darker) lung fields, ↓ lung markings (on frontal CXR)
• Arterial Blood Gasses (ABGs)
• Chest X-Ray (CXR): frontal and
lateral
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published January 7, 2013 on www.thecalgaryguide.com
COPD: !"#$ 气流阻塞
肺泡通气↓ 呼气时,胸膜腔正压挤压气 道à 阻塞↑
作者: Yan Yu 审稿人: Jason Baserman, Jennifer Au, Naushad Hirani*, Juri Janovcik* 译者:Zihong Xie (谢梓泓) 翻译审稿人: Yonglin Mai (麦泳琳), Zesheng Ye (叶泽生) * 发表时担任临床医生
慢性阻塞性肺疾病 (COPD)
肺组织损伤
没有弹性回缩力将
气体排出肺
肺实质与血管分布减少导 致气体交换面积↓
弥散功能↓ (肺功能检查)
更多的CO2残留 并扩散到血液中
高碳酸血症: PaCO2 > 45
(动脉血气)
血流灌注通气不良的肺泡
时无法获得足够的氧气
总呼气时长较正常长
FEV1/FEV < 0.7
(肺功能检查)
肺无法完全排空
更多空气潴留在肺部
(肺过度充气)
低氧血症: PaO2 < 70mmHg
(动脉血气)
通气-灌注不匹配
肺泡-动脉氧分压差↑ (可通过动脉血气分析计算得出)
横膈低平, 下移至第10肋后端 及以下部位 (胸部正位片)
TLC与VC增大 (肺功能检查)
缩写: • • FEV1: 1秒用 •
VC:肺活量
PaO2: 动脉血 力呼气量 氧分压
空气不会阻挡X射线, 在X光片上呈现为黑色
慢性高碳酸血症使呼吸中枢对PaCO2 刺激呼吸的敏感性下降 & 更依赖于低PaO2的刺激 (“二氧化碳潴留”)
给患者吸氧时需注意(高PaO2
可能会抑制患者低氧时对呼吸的 刺激,使呼吸驱动↓ & PaCO2↑↑↑ )
• FVC: 用力肺 • 活量
• TLC:肺总量 慢阻肺相关检查 :
PaCO2: 动脉 血二氧化碳 分压
胸骨后间隙↑
(胸部侧位片) 肺纹理↓
• 肺功能检查
• 动脉血气分析(Arterial Blood Gasses, ABGs)
• 胸部正侧位片
• 当患者发热和湿咳,特别是胸片上见肺野不清晰时:
肺透亮度↑, (胸部正位片)
考虑进行痰革兰氏染色及痰培养以排除肺炎可能
图注:
病理生理
机制
体征/临床表现/实验室检查
并发症
2013年1月7日发布于 www.thecalgaryguide.com
COPD: Complications Lung inflammation
Chronic Obstructive Pulmonary Disease (COPD)
Airway obstruction ↓ inhaled air in alveoli and terminal bronchioles
Rupture of emphasematous bullae on surface of lung
Inhaled air leaks into pleural cavity and is trapped there
Pneumothorax
Feeling a loss of control over one’s life, and hopelessness for the future
Goblet cell proliferation, ↑ mucus production
Death of airway
epithelium ciliated cells
↓ oxygenation of the blood passing through the lungs
Chronic hypoxemia
Kidneys compensate by ↑ erythropoietin (EPO) production
↑ Hemoglobin and red blood cell synthesis
Polycythemia (secondary)
Hypoxic alveoli cause the pulmonary arterioles perfusing them to reflexively vasoconstrict
Since most alveoli in the lungs are hypoxic, hypoxic vasoconstriction occurs across entire lung
Vasoconstriction ↑ blood pressure within lung vasculature
Pulmonary hypertension
↑ workload of the right ventricle (to pump against higher pressures)
To compensate, the right ventricle progressively hypertrophies and dilates, but over time its output ↓
Cor pulmonale
(Right heart failure in isolation, not due to Left heart failure)
Mucus trapped in airways, serve as nidus for infection
Acute exacerbation of COPD (AECOPD)
Pneumonia
The chronic, systemic inflammation in COPD is a hyper-metabolic state that consumes calories
Macro-nutrient deficiency
Trouble with respiration lead to inactivity and deconditioning
Wasting, muscle atrophy
More inactivity and deconditioning perpetuates the cycle
Depression
Author: Yan Yu Reviewers: Jason Baserman Naushad Hirani* Juri Janovcik* * MD at time of publication
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published January 7, 2013 on www.thecalgaryguide.com
COPD: !"# 肺部炎症
杯状细胞增殖, 气道上皮纤毛 粘液产生↑ 细胞死亡
黏液潴留呼吸道,成为感
染的病灶
慢性阻塞性肺疾病 (COPD) 气道阻塞à 吸入肺泡和终末细
肺大疱破裂
吸入的空气渗入
并潴留于胸腔
气胸
感觉生活失控,对未
来感到绝望
抑郁
作者: Yan Yu 审稿人: Jason Baserman, Naushad Hirani*, Juri Janovcik* 译者: Zihong Xie (谢梓泓) 翻译审稿人: Yonglin Mai (麦泳琳), Zesheng Ye (叶泽生) * 发表时担任临床医生
支气管的空气 ↓
流经肺的血液进行气 缺氧的肺泡à灌注肺泡的肺小动
慢性阻塞性肺疾 病急性加重期 (AECOPD)
肺炎
体交换↓ 慢性低氧血症
肾脏合成促红细胞 生成素进行代偿↑
血红蛋白与红 细胞合成↑
红细胞增多症 (继发性)
脉发生反射性血管收缩
肺大部分肺泡缺氧à整个肺 都出现缺氧性血管收缩
肺血管收缩 à 肺血管压力↑ 肺动脉高压
↑ 右心室负荷(泵血时对抗高压) 为了代偿,右心室逐渐肥大和扩张,
但随着病程进展,右心室输出量 ↓
肺心病 (单独出现右心衰竭,非左心衰)
COPD所致的慢性全身 呼吸困难导致活 性炎症会使机体处于高 动量减少和活动
代谢状态,消耗能量 耐量降低
宏量营养 素缺乏症
消瘦,肌肉萎缩
运动量下降和活动耐量
的降低造成恶性循环
图注:
病理生理
机制
体征/临床表现/实验室检查
并发症
2013年1月7日发布于 www.thecalgaryguide.com
" />
Chronic Cough Pathogenesis_2021

Infection
IP; likely chronic ↑ cough receptors commonly pertussis- related or post-viral
Asthma
↑ EDN
↑ MBP levels in RT
Neoplasm
Bronchiectasis
COPD
GERD
Allergic Rhinitis
↑ sputum production
and accumulation
Damage from chronic inflammation causes poor mucus clearance
Inhalants trigger ↑ cytokines ↑ mucus
Aspiration of refluxed microdroplets
Irritation by post- nasal drip
↑ inflammatory mediators in RT
Mechanical obstructions (i.e. mediastinal masses, neoplasms)
Foreign body Presence irritation of irritants
Stimulation of sensory nerve receptors expressed on nerve endings penetrating the epithelia of the upper RT
Abbreviations:
• RT: respiratory tract
• IP: indefinite
pathophysiology
• GERD: Gastro-esophageal
reflux disease
• EDN: Eosinophil-derived
neurotoxin
• MBP: Major basic protein
• COPD: Chronic
Obstructive Pulmonary Disease
Complications: Syncope, insomnia, hemoptysis, rib fractures
Slowly adapting receptors
Respond to mechanical forces during breathing
Stimulation of the vagus nerve
Activation of nucleus tractus solitarius in central respiratory generator (brainstem)
Generation of efferent cough signal
Chronic cough: Cough of over 8 weeks duration with no dyspnea or fever
C-fibre receptors
Respond to chemical stimuli (bradykinin, capsaicin, acid/alkaline solutions, mannitol, hypertonic saline, and other inhaled irritants)
Authors: Arsalan Ahmad Lance Bartel Reviewers: Midas (Kening) Kang Usama Malik Ciara Hanly Yonglin Mai (麦泳琳) Yan Yu*, Eric Leung* * MD at time of publication
Rapidly adapting receptors
Responds to mechanical stimuli, such as physical obstruction of the airways
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published Feb 06, 2018, updated Dec 4, 2021 on www.thecalgaryguide.com")
hip-osteoarthritis-pathogenesis-and-clinical-findings

Trauma
Infection (e.g. septic arthritis)
Anatomical abnormality (e.g. developmental hip dysplasia)
↑ Cartilage stiffness and degeneration ↑ Friction in the hip joint with movement
Changes in normal hip architecture Abnormal load on the hip joint
Radiographic changes
See Osteoarthritis (OA): X-Ray Features slide
Repeated attempts to repair cartilage damage and bone
Bone spur (bony growth) formation in joint
Joint fluid accumulation from mild inflammation of the joint
Stiffness
Limited joint movement
Hip Osteoarthritis
Multifactorial condition that manifests as degeneration of cartilage, bone, and synovium in the hip joint
↓ Cartilage between femoral head and acetabulum ↑ Bone on bone contact
Mechanical symptoms (crepitus, locking, clicking, catching)
Bones rubbing together activates nociceptors within the hip joint
Nociceptors further aggravated by actions that ↑ bone on bone contact (e.g. prolonged loading, movements that ↓ joint space)
Worsening pain in the groin region
Favour the affected leg to avoid pain while walking
↓ Space for the femoral head to move
↓ Range of motion of affected hip joint
Limited range of motion when walking
Antalgic gait
↓ Use of muscles around the affected hip
Muscular atrophy around the hip
C Sign: patient identifies location of pain by cupping lateral hip with one hand, which creates a “C” shape with that hand
Pain in groin region
↑ Sensitivity of surrounding nerves
Radiating pain to buttocks and knee
Inactivity
Hip instability when walking
Muscle contractures
↑ Falls
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published March 6, 2022 on www.thecalgaryguide.com")
cough-physiology
* Yan Yu* Stephen Field* * MD at time of publication
Systemic immune reaction
Inflammation of alveoli Cytokine release
Cytokines bind to sensory neurons
Neurons ↑ receptor sensitivity and number
↑ sensation of stimuli Ionic flux across neuronal
membrane
Vagus neurons depolarize
Thinly myelinated axons (Aδ fibers)
General chemicals, heat, cold, and hydrogen ions
Transient receptor potential vanilloid (TRPV)/Transient receptor potential ankyrin (TRPA) receptors activate
TRPV/TRPA activation opens ion channels on vagus neurons
Unmyelinated axons (C fibers)
carry depolarization
Vagus nerves conduct depolarization to
carry depolarization
SUMMARY
Afferent pathways activated
Cough center (medulla)
Efferent pathways Respiratory muscles
nucleus of the solitary tract
Medullary nuclei activated, stimulates:
Phrenic nerves
Diaphragm contraction
Spinal nerves
External intercostal contraction
Vagal nerves
Glottis closure
Spinal nerves
Thoracic, abdominal, and pelvic muscle contraction
Vagal nerves
Glottis opens, allowing forceful expiration
Intrathoracic pressure and volume decrease
Volume in lungs increases
Intrathoracic pressure increases
Mechanical Cough
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published Sept 1, 2019, updated Dec 5, 2021 on www.thecalgaryguide.com")
proximal-biceps-tendon-rupture
 place more stress on the proximal biceps tendon
Repetitive microtrauma and inflammation of biceps tendon Compromised biceps tendon integrity from intra-tendinous collagen degeneration, disorientation, and thinning
Increased involvement in physical labour and hobbies
Impaired cell proliferation and healing after injury
Exact mechanisms remain unclear
Bicep tendon involved in shoulder stabilization, elbow flexion, and forearm supination
Biceps muscle cannot contract with as much force when one head is detached
Heavy lifting or fall on outstretched handà sudden tension in the proximal biceps tendon
Proximal biceps tendon rupture
Long head of proximal biceps tendon detaches from bony attachment on supraglenoid tubercle
Audible “pop” at time of injury
No tension to hold proximal bicep tendon head in place
Proximal bicep tendon head retracts distally
Biceps muscle fatigue and cramping
Weakness in shoulder flexion, elbow flexion & supination
Nerve damage (musculocutaneous nerve) during injury
Local inflammatory response at site of injury
Surrounding blood vessels damaged during injury
Blood collection under skin surface
Immediate bruising over rupture site
Sudden and sharp pain at shoulder with initial injury
Inflammatory response aggravates musculocutaneous nerve
Swelling over rupture site
+ Speed test: affected elbow extended and forearm supinated. Shoulder flexed against resistanceàpain at bicipital groove
+ Neer test: on affected side, scapula stabilized while arm passively flexed and internally rotatedàpain at bicipital groove
+ Yergeson test: affected elbow flexed at 90° and forearm pronated. Forearm supinated against resistance àpain at bicipital groove
“Popeye” deformity:
mass in distal upper arm due to excessive
retraction of biceps muscle distally, away from its origin
Palpable gap
between proximal biceps tendon and bicipital groove
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published April 20, 2022 on www.thecalgaryguide.com")
distal-biceps-tendon-rupture
 during forearm pronation
Repetitive compressionà tendon degeneration
↑ Involvement in physical labour and hobbies
Exact mechanisms remain unclear
Impaired cell proliferation
Biceps tendon inserts at the radial tuberosity to control forearm supination and flexion
Compromised biceps tendon integrity from intra-tendinous collagen degeneration, disorientation, and thinning
Sudden eccentric force to flexed elbow (forced lengthening of a contracted bicep muscle)àtension in the distal biceps tendon
Biceps muscle cannot contract when distal tendon is detached from insertion site
Distal biceps tendon rupture
Complete detachment of distal biceps tendon at the attachment site on the radial tuberosity
Audible “pop” at time of injury
Weak forearm supination and flexion
+ Ruland biceps squeeze test:
Affected elbow held in 60-80° of flexion, with forearm slightly pronated. Distal biceps muscle squeezed by practitioner à forearm does not supinate
Local inflammatory response at site of injury
Inflammatory response aggravates musculo- cutaneous nerve
Pain over rupture site after initial injury
Swelling over rupture site
Nerve damage (musculocut aneous nerve) during injury
Sudden and sharp pain at
elbow at initial injury
Surrounding blood vessels damaged during injury
Blood collects under skin surface
Immediate bruising over rupture site
Biceps tendon retracts proximally
High tension during tendon retraction
Fragment of bone torn off from tendon insertion site on radial tuberosity
Avulsion fracture of
radial tuberosity
Connective tissue attachment (aponeurosis) torn during injury
Palpable gap between distal end of biceps tendon & radial tuberosity
+ Hook test: Elbow actively flexed to 90° and forearm supinated. Index finger of examiner cannot be placed beneath distal aspect of biceps tendon (>1 cm deep)
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published April 20, 2022 on www.thecalgaryguide.com")
abnormal-uterine-bleeding-aub-pathogenesis-and-clinical-findings
 release from hypothalamus
↑Prolactin (see Feedback Loop: Prolactin)
Exogenous estrogen (e.g. estrogen-only birth control)
↑ Peripheral adipose tissue
↑ Aromatase (enzyme present in adipose tissue)
↑ Conversion of androgens to estrogens (aromatization)
Excessive stress, exercise, low body mass (mechanism unclear)
↓ Hypothalamic gonadotropin releasing hormone secretion
↓Luteinizing hormone and follicle stimulating hormone release from pituitary
↑ Estrogen
Heavier bleeding
Angiogenesis in tumour tissue
Polycystic ovarian syndrome (see slide)
Pelvic inflammatory disease
Immature hypothalamic- pituitary-ovarian axis
(an immature axis is transient in most females)
↓ Positive feedback of estrogen in late follicular stage
No LH surge
Adenomyosis (endometrial tissue grows into uterus muscular wall)
Endometrial polyps
Retained products of contraception
Infection
Inflammation of endometrium
Endometrium is more fragile
Anovulatory Bleeding
Leiomyoma (benign tumour in myometrium)
Tara Shannon, Hannah Yaphe, Dr. Sarah Glaze* * MD at time of publication
Ovarian Scarring
Foreign bodies
Intrauterine device perforation
Physical trauma
Impaired follicle maturation
Anovulation
Corpus luteum does not form
No ovarian progesterone production
↑ Estrogen to progesterone ratio
Proliferative effect of estrogen unopposed
Endometrial proliferation
No progesterone to organize growing endometrium
Disorganized endometrium overgrows and sloughs off
Irregular bleeding
Menopause
Premature ovarian insufficiency
Intermittent congestion of polyp blood supply
Transient ischemia and slight polyp necrosis
Altered growth factor production
Vascular dysregulation and leakier vessels
Coagulopathies (e.g. von Willebrand disease)
Impaired hemostasis
Invasion of normal tissue
↓ Estrogen (hypoestrogenism)
Atrophy of endometrium and vulvovaginal tissue
Dry endometrial surfaces (↓ fluid to prevent friction)
Endometrial carcinoma
Micro- erosions of epithelium
Inflammation
Evidence unclear
Heavier and more irregular bleeding
Spotting
Heavier and more irregular bleeding
Light bleeding and spotting
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published May 2, 2022 on www.thecalgaryguide.com")
rhumatisme-psoriasique-pathogenese-et-resultats-cliniques
![rhumatisme-psoriasique-pathogenese-et-resultats-cliniques
Psoriatic Arthritis: Pathogenesis and clinical findings
Auteur:
Payam Pournazari
Rédacteurs:
Yan Yu Scott Rapske Liam Martin* Traducteurs: Dianne Ganeswaran Stephen Williams Sylvain Coderre* * MD au moment de publication
Note:
L’arthrite rhumatoïde peut survenir avant ou après le psoriasis (la pathogenèse est la même).
Augmentation de l’activité ostéoclastique
Érosion de l’os sous- chondral mais formation de nouvelle osseuse ailleurs dans l’articulation
Radiographie: la périostite; les syndesmophytes, érosions, déformation du crayon en godets (les articulations IP), ankylose des articulations IP
HLA B27, Cw6 et d’autres sous- types
Antécédents familiaux positifs
Activation des cellules T (mécanisme inconnu)
Infiltration du tissu synovial de l’articulation par les cellules T et B, cellules tueuses naturelles et les macrophages
Production augmentée des molécules inflammatoires (les facteurs de nécrose tumorale [TNFs], les interleukines, etc.) qui agissent de façon systémique (dans tout le corps)
Dans les tendons et le tissu conjonctif
Inflammation des enthèses et du tissu conjonctif provoque le gonflement
Dans la peau
Réaction immunitaire détruisant les structures de la peau et des ongles (Voir diapositive Psoriasis)
Le psoriasis en plaques sur la peau, dépressions punctiformes, onychorrhexie, taches d’huile, onycholyse, décoloration et hyperkératose
Angiogenèse et vascularite dans les vaisseaux sanguins de la membrane synoviale
Dans les articulations
Les cytokines inflammatoires stimulent les nocicepteurs locaux
1. Oligoarthrites - asymétriques
2. Arthrite des articulations IPD
3. Polyarthrite rhumatoïde -
symétrique
4. Atteinte axiale (raison
inconnue, probablement en raison de « biomécanique », « innervation », et
« vascularisation ».
5. Arthrite mutilante (grave et destructrice)
L’enthésite
(Douleur/sensibilité à l’insertion du ligament dans l’os)
La dactylite (Inflammation de tout le doigt – les tissus mous et les articulations sont enflammés
Légende:
Physiopathologie
Mécanisme
Signe/Symptôme/Résultats de Laboratoire
Complications
Publié 10 November 2012 sur www.thecalgaryguide.com](https://calgaryguide.ucalgary.ca/wp-content/uploads/2022/05/Psoriatic-Arthritis-PsA-Pathclinical.jpg "rhumatisme-psoriasique-pathogenese-et-resultats-cliniques
Psoriatic Arthritis: Pathogenesis and clinical findings
Auteur:
Payam Pournazari
Rédacteurs:
Yan Yu Scott Rapske Liam Martin* Traducteurs: Dianne Ganeswaran Stephen Williams Sylvain Coderre* * MD au moment de publication
Note:
L’arthrite rhumatoïde peut survenir avant ou après le psoriasis (la pathogenèse est la même).
Augmentation de l’activité ostéoclastique
Érosion de l’os sous- chondral mais formation de nouvelle osseuse ailleurs dans l’articulation
Radiographie: la périostite; les syndesmophytes, érosions, déformation du crayon en godets (les articulations IP), ankylose des articulations IP
HLA B27, Cw6 et d’autres sous- types
Antécédents familiaux positifs
Activation des cellules T (mécanisme inconnu)
Infiltration du tissu synovial de l’articulation par les cellules T et B, cellules tueuses naturelles et les macrophages
Production augmentée des molécules inflammatoires (les facteurs de nécrose tumorale [TNFs], les interleukines, etc.) qui agissent de façon systémique (dans tout le corps)
Dans les tendons et le tissu conjonctif
Inflammation des enthèses et du tissu conjonctif provoque le gonflement
Dans la peau
Réaction immunitaire détruisant les structures de la peau et des ongles (Voir diapositive Psoriasis)
Le psoriasis en plaques sur la peau, dépressions punctiformes, onychorrhexie, taches d’huile, onycholyse, décoloration et hyperkératose
Angiogenèse et vascularite dans les vaisseaux sanguins de la membrane synoviale
Dans les articulations
Les cytokines inflammatoires stimulent les nocicepteurs locaux
1. Oligoarthrites - asymétriques
2. Arthrite des articulations IPD
3. Polyarthrite rhumatoïde -
symétrique
4. Atteinte axiale (raison
inconnue, probablement en raison de « biomécanique », « innervation », et
« vascularisation ».
5. Arthrite mutilante (grave et destructrice)
L’enthésite
(Douleur/sensibilité à l’insertion du ligament dans l’os)
La dactylite (Inflammation de tout le doigt – les tissus mous et les articulations sont enflammés
Légende:
Physiopathologie
Mécanisme
Signe/Symptôme/Résultats de Laboratoire
Complications
Publié 10 November 2012 sur www.thecalgaryguide.com")
acute-cholecystitis
![Acute Cholecystitis: Pathogenesis and clinical findings
Gallstone blocks the cystic duct, backing up bile into the gallbladder
Gallstones causing physical trauma to gallbladder wall
Irritation of adjacent diaphragm, stimulates phrenic nerve (C3-C5)
Activates stretch receptors of visceral peritoneum, stimulates foregut autonomic nerves (T5-T8)
Inflammatory mediator (i.e. prostaglandin) release by gallbladder and systemic inflammatory response
Thickened gallbladder wall on ultrasound (gold standard test)
On inspiration, the diaphragm pushes the gallbladder downward
Irritation of parietal peritoneum, stimulates somatic nerves
↑ Permeability of vessels with systemic inflammation, which leak
fluid from the blood into the interstitial space
Radiating pain to the back and right shoulder
Dull, diffuse abdominal pain referred to the epigastric region
Fever, nausea/vomiting, tachycardia
Positive Murphy’s sign (pain upon palpation of right upper quadrant [RUQ] on inspiration)
Persistent RUQ pain, abdominal guarding and peritoneal signs
Dehydration
Authors: Yan Yu, Vina Fan Reviewers: Dean Percy, Mirna Matta, Crystal Liu, Ben Campbell Maitreyi Raman* * MD at time of publication
Inflammation self-perpetuates
Irritation of inner gallbladder wall/mucosa
↑ Gallbladder lumen pressure
Intraluminal pressure exceeds arterial pressure
↓ Blood flow to gallbladder
Gallbladder ischemia
Local inflammation, loss of gallbladder mucosal integrity
Bacterial invasionàtransmural inflammation of gallbladder
Without treatment, prolonged ischemia and inflammation of the gallbladder
Gallbladder gangrene (20%)
Gallbladder perforation (20%)
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published August 4 2019, updated May 16 2022 on www.thecalgaryguide.com](https://calgaryguide.ucalgary.ca/wp-content/uploads/2015/05/Acute-Cholecystitis-2022.jpg "Acute Cholecystitis: Pathogenesis and clinical findings
Gallstone blocks the cystic duct, backing up bile into the gallbladder
Gallstones causing physical trauma to gallbladder wall
Irritation of adjacent diaphragm, stimulates phrenic nerve (C3-C5)
Activates stretch receptors of visceral peritoneum, stimulates foregut autonomic nerves (T5-T8)
Inflammatory mediator (i.e. prostaglandin) release by gallbladder and systemic inflammatory response
Thickened gallbladder wall on ultrasound (gold standard test)
On inspiration, the diaphragm pushes the gallbladder downward
Irritation of parietal peritoneum, stimulates somatic nerves
↑ Permeability of vessels with systemic inflammation, which leak
fluid from the blood into the interstitial space
Radiating pain to the back and right shoulder
Dull, diffuse abdominal pain referred to the epigastric region
Fever, nausea/vomiting, tachycardia
Positive Murphy’s sign (pain upon palpation of right upper quadrant [RUQ] on inspiration)
Persistent RUQ pain, abdominal guarding and peritoneal signs
Dehydration
Authors: Yan Yu, Vina Fan Reviewers: Dean Percy, Mirna Matta, Crystal Liu, Ben Campbell Maitreyi Raman* * MD at time of publication
Inflammation self-perpetuates
Irritation of inner gallbladder wall/mucosa
↑ Gallbladder lumen pressure
Intraluminal pressure exceeds arterial pressure
↓ Blood flow to gallbladder
Gallbladder ischemia
Local inflammation, loss of gallbladder mucosal integrity
Bacterial invasionàtransmural inflammation of gallbladder
Without treatment, prolonged ischemia and inflammation of the gallbladder
Gallbladder gangrene (20%)
Gallbladder perforation (20%)
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published August 4 2019, updated May 16 2022 on www.thecalgaryguide.com")
clostridium-difficile-infection-pathogenesis-and-clinical-findings
 Infection
Authors: Ryan Brenneis, Sravya Kakumanu Reviewers: Yoyo Chan, Sean Doherty, Vina Fan, Ben Campbell, Dr. Steve Vaughan*, Dr. Sylvain Coderre* * MD at time of publication
Community exposure
Infected close contacts
Nosocomial exposure (most common)
Poor hand hygiene and sanitization of surfaces and medical equipment
Nosocomial risk factors
Any antibiotic use
(Especially clindamycin, fluoroquinolones, penicillins, cephalosporins)
↑ Antibiotic resistant strains
Presence of pre-disposing risk factors
(Note: do not need to be present for infection)
Recent GI surgery
Chemotherapy that has antimicrobial and immunosuppressive effects
Usage of medications that reduce stomach acid (↑ pH)
↑ C. diff spores on surfaces and personnel
Contact exposure
Environmental exposure
to C. diff carriers
Inoculation of GI tract
Disruption of normal gut microbiome allowing C. diff overgrowth
Comorbidities
(>65 years old, cirrhosis, inflammatory bowel disease, enteral feeding, obesity)
via fecal-oral route
Clostridium difficile Infection of GI Tract
Spores unaffected by antibiotics germinate post-antibiotic treatment
Infection recurrence
Pseudomembranous colitis on endoscopy
(colonic ulcerations potentially seen with severe infection)
Hypotension Acute kidney injury
Release of C. diff toxin A and B inactivates Rho and Ras GTPases in colonic epithelial cells (colonocytes)
(Rho and Ras GTPases control cytoskeletal dynamics and gene expression)
Cytoskeletal disorganization and arrest of RNA synthesis causes necrosis of colonocytes and triggers host immune response
Neutrophil chemotaxis and activation
↑ Inflammation of colon
Disruption of tight junctions between colonocytes
Release of fluid into intestinal lumen and inability of colon to reabsorb it
Toxic megacolon
Bowel perforation
Bloody stool
(<10% of patients)
Abdominal cramps
Large bowel dilation from muscle paralysis
Inflammation and destruction of underlying smooth muscle
Breakdown of colonocyte cell membranes
Inflammation of visceral peritoneum
Volume depletion
Watery diarrhea: ↑ frequency, small volume
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published March 30, 2019, updated May 16, 2022 on www.thecalgaryguide.com")
constatations-classiques-de-sclerose-en-plaques-sep-sur-irm-cerebrale
 sur IRM cérébrale
Auteurs: Evan Allarie Davis Maclean Viesha Ciura* Rédacteurs: Yan Yu* Traducteurs: Liana Martel Eddy Lang* * MD au moment de la publication
Infiltrations dans la région infratentorielle
Perte de la densité neuronale/axonale dans la région affectée, remplacée par une gliose au fil du temps
Remarque : les constatations peuvent varier.
Les constatations présentées ici ne sont pas exhaustives, mais constituent certaines des zones les plus fréquemment mises en évidence par l'IRM du cerveau dans la SEP. Les sites les plus communément observés sont : les régions juxtacorticales, périventriculaires, infratentorielles, la moelle épinière et le nerf optique. Cependant, les lésions peuvent apparaître partout où il y a de la myéline dans le SNC.
L'inflammation et la destruction actives permettent au contraste de gadolinium de traverser la barrière hémato-encéphalique, ce qui peut être visualisé comme un marqueur de l'inflammation active dans la SEP
Lésion classique du nerf optique (CN II) en cas de névrite optique. Rehaussement par Gadolinium (↑ intensité du signal de la lésion après l’injection de gadolinium) sur cette image en T1 démontre une inflammation active
Image Credits: Dr. Viesha Ciura
Pour plus de détails sur la pathogenèse, voir la diapositive du Guide de Calgary: Multiple Sclerosis (MS): Pathogenesis and Clinical Findings
Inflammation dans le système nerveux central (SNC)
Les cellules T, les cellules B et les macrophages infiltrent le SNC
Infiltration se produit autour des veinsàinflammation locale Infiltrats périveineux autour des veines médullaires
(perpendiculaires aux ventricules)
Inflammation et destruction de la barrière hémato-encéphalique ↑ liquide inflammatoire extravasculaire autour des lésions, qui apparaît hyperintense/brillant
Lésions s’étendent autour des veines, créant les lésions ovoïdes charactéristiques
Plaques hyperintenses perpendiculaires périventriculaires classiques en T2/FLAIR suivant les veins médullaires – ‘Doigts de Dawson’
Lésion hyperintense du pédoncle cérebelleux moyen en T2/FLAIR dans une localisation classique de la SEP
Légende:
Physiopathologie
Mécanisme
Signe/Symptôme/Résultats de Laboratoire
Complications
Publié 25 octobre 2020 sur www.thecalgaryguide.com")
pleural-effusions-pathogenesis-and-anterior-posterior-chest-x-ray-findings

Excess fluid accumulation within pleural space that is gravity- dependent/free- flowing
Large accumulation of fluid pressing against lung tissue and mediastinum
Fluid layering (i.e. opacification) between lung and chest wall
Author: Sravya Kakumanu Reviewers: Reshma Sirajee, Tara Shannon *Stephanie Nguyen, *Shelley Spaner * MD at time of publication
Transudative
pleural effusion
*See Pathogenesis and Clinical Findings of
Transudative Pleural Effusions slide
Exudative pleural
effusion
*See Pathogenesis and Clinical Findings of
Exudative Pleural Effusions slide
Fluid accumulation at bottom of pleural space in anterior- posterior chest x- ray
Lung atelectasis (lung collapse)
Fluid is denser than air and appears white on x-ray
↓ Pressure and occupied space in thoracic cavity
Meniscus sign (concave line above opacification)
Opacification of affected pleural space
Blunting of costophrenic angles
Diaphragmatic silhouette sign (sharp diaphragm edge obscured by fluid)
Mediastinal shift towards atelectic lung
Contralateral mediastinal shift (when no lung atelectasis)
Contralateral tracheal deviation
Pleural infections
↑ Inflammation at infection site à↑ permeability of parietal pleura endothelium
Bacterial invasion into pleural space from pulmonary parenchyma
↑ Migration of neutrophils and release of inflammatory cytokines in pleural space
Inflammation ↑ activation of coagulation cascade and ↓ fibrinolytic activity
↑ Deposition of fibrin clots/membranes within pleural space creating septations
Loculated pleural effusion/empyema (fluid stuck within septations within the pleural space)
Opacification does not follow gravity
Effusion does not move in decubitus view
Pleural thickening (whitening of lung perimeter from fibrin deposition - more visible on CT scan)
More rounded margins with chest wall/fissures
Note: ~175-500mL of fluid may need to accumulate before being visible on an Anterior-Posterior Chest X-Ray
Image credit: https://radiopaedia.org/cases/pleural-effusion-7
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published May 22, 2022 on www.thecalgaryguide.com")
gout-pathogenesis-of-x-ray-findings
 crystals
Uric acid crystallizes due to lower temperature, change in pH, mechanical stress and other synovial factors
Crystals found more commonly in 1st MTP joint > ankle > wrist
MSU deposits within the bone producing
intra-osseus tophi (stone-like deposits of crystals)
MSU deposits in soft tissue and bone
↑ Inflammatory marker recruitment causes granulomatous inflammation
Osteoclasts (↑ bone resorption) are activated, and osteoblasts (↑ bone formation) are inhibited at
site of inflammation Marked localized bone loss
‘Rat Bite’ erosions
Intra-osseus bone lesion
(rare and non-specific for gout)
Joint effusions may be seen on x-ray as an early finding in the acute phase of gout
Soft Tissue Tophi
(appear cloudy and can hide other x-ray findings)
Normal bone density and preserved joint space
(until late in the disease)
Overhanging
sclerotic margins
Bone remodelled in an outward fashion to create the edge
Punched out lytic bone lesions Appear as circular ‘hole punches’ in bone
Note: Repeated episodes of gout must occur for 5 to 10 years before most joint changes may be seen on x-ray
Image credit: Radiopaedia
Legend:
Pathophysiology
Mechanism
Sign/Radiographic Findings
Complications
Published June 7, 2022 on www.thecalgaryguide.com")
Epilepsy Pathogenesis

Neoplasm
Metabolic
Inborn errors of metabolism
Inherited enzyme deficiencies
Neurodegenerative
Alzheimer’s Disease
Protein-rich plaque build-up and brain atrophy
Inflammation and formation of scar tissue irritates neural tissue
Infiltration of mass, grey matter irritation
Damage to the brain and subsequent alteration of neuronal circuitry
Neurotransmitter imbalances (i.e. ↑ glutamate, ↓ GABA, ↓ serotonin, ↓ dopamine, ↓ noradrenaline)
Abbreviations:
• HSV1 – Herpes Simplex Virus 1 • VZV – Varicella Zoster Virus
↑ Inflammatory cytokines (e.g. interleukin-6, tumor necrosis factor-α)
Altered neurogenesis and gliosis
Ion channel and receptor dysfunction causes imbalance of ion channel charges
Authors: Keerthana Pasumarthi Christopher Li Reviewers: Negar Tehrani, Ephrem Zewdie, Ran (Marissa) Zhang, Carlos R. Camara-Lemarroy* * MD at time of publication
Neurons fire in burst activity (referred to as paroxysmal depolarization shift) and often in groups (hypersynchrony)
Abnormal neural activities eventually create self-reinforcing circuits and transform neural network over time
Injuries from
falls, bumps, operating heavy machinery
Involuntary contractions Loss of consciousness
Post-ictal confusion
Recurrent seizures
Sudden Unexplained Death of Epilepsy Patient (SUDEP)
Loss of airway reflexes
Aspiration of saliva or food contents
Aspiration Pneumonia
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published July 5, 2022 on www.thecalgaryguide.com")
exudative-pleural-effusions-pathogenesis-and-lab-findings

Lung infection signals inflammatory response
Systemic Lupus Rheumatoid Erythematosus (SLE) arthritis (RA)
Autoimmune antibodies localize to pleura
Pleural tumors (primary or secondary from metastatic cancer)
Inflammatory cells migrate to affected site and release cytokines
↑ Permeability of pleural capillaries ↑ Fluid leakage across capillaries
Exudative Pleural Effusion
Cancer invades lymphatic drainage of pleural space (PS)
↓ Drainage of pleural fluid (PF) from pleural space
If infectious etiology
Tumor invasion = inflammatory response
See Pleural Effusions: X-ray Findings and Physical Exam Findings of Lung Diseases slides
↑ Permeable pleural capillaries allow ↑ protein and cell leakage into pleural space
If pleural tumour: Release of cancer cells into pleural space
Cancer cells on PF cytology 60-75% sensitive for malignancy
If rheumatoid arthritis:
Release of auto-antibodies into pleural space
Auto-antibodies initiate inflammatory response in pleural space
↑ Inflammatory cells have ↑ glucose metabolism in pleural space
Sterile PF with mildly elevated white blood cells, normal pH, normal glucose ↑ Inflammation at infection site damages endothelium of pleura
Pleural Infection Stage I: Simple Parapneumonic Effusion
↑ Inflammatory cells and bacterial cells in pleural space have ↑ glucose metabolism in pleural space
Bacterial invasion from infected parenchymaà pleural space
< 40mg/dL glucose in PF
↑ Activation of coagulation cascade and ↓ fibrinolytic activity
↑ Deposition of fibrin
clots/membranes within pleural
space creates loculated effusion
(compartmentalized effusion due to septations in pleural space)
↑ Production of
lactate
dehydrogenase
(LDH)
(LDH maintains NAD+ supply during ↑ glucose metabolism)
↑ CO2 production pH of PF < 7.20
↑ CO2 production pH of PF < 7.20
< 3.3mmol/L glucose in PF
Pleural Infection Stage II: Complicated Parapneumonic Effusion
Loculated effusion OR bacteria present OR ↓ pH + ↓ glucose
↑ Fibroblast proliferation creates thickened pleura ↑ Pus in pleural space Pleural Infection Stage III/Empyema: Loculated effusion and pus in pleural space
PF/serum protein ratio ≥ 0.5
PF/serum LDH ratio ≥ 0.6
Light’s Criteria: Any criteria can be met to be an exudative pleural effusion
PF LDH ≥ 2/3 upper limit of normal
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published August 9, 2022 on www.thecalgaryguide.com")
thrombose-veineuse-profonde-soupconne-tvp-pathogenese-et-complications

Auteurs: Dean Percy Yan Yu Rédacteur: Tristan Jones Julia Heighton Man-Chiu Poon* Lynn Savoie* Traducteurs: Sophia Shah Sylvain Coderre* *MD à la publication
La grossesse, contraceptifs oraux
Pathogénèse et complications
Activation plaquettaire
↑ formation de caillots
Maladies héréditaires
Anomalie congénitale de la coagulation (p.e. facteur V Leiden, facteur II muté, déficit en protéine S/C) ↑ capacité de coagulation
Notes:
• TVP provoque une embolie pulmonaire, le thrombus artériel provoque un accident vasculaire cérébral
• TVP précédente est un facteur de risque de TVP courante
Malignité
Libération anormale de cytokines favorisant la coagulation
Traumatisme/opération
Blessure systémiqueà activation de la cascade de coagulation
État d'hypercoagulabilité↑ capacité du sang à coaguler lors de la stimulation
Oestrogène favorise l'hypercoagulabilité, surtout en présence d'autres risques
Hypertension Bactéries
Valve artificielle
Endommage les parois des vaisseaux
Adhèrent/ envahissent / vaisseaux
Surface anormale
Blessure de vaisseau
Expose le facteur tissulaire sur les cellules endommagées /sous-endothélium pour la liaison au FvW
Triade de Virchow
Stase veineuse
↓ débit sanguins au site de la lésion
vasculaireà concentration des facteurs de coagulation sanguine sur ce site
La graisse contient plus d'aromatase: ↑ d'androgènesà œstrogènes
mode de vie sédentaire, mauvais retour veineux
Obésité
Les caillots se produisent généralement dans les veines des jambes
1. Les veines larges et profondes permettent l'accumulation de sang
2. Retour veineux des jambes est contre la gravité
3. Valves dans les veines des jambes enclins au refoulement
↓ mouvement musculaire = ↓ débit sanguin
Fracture, immobilisation, alitement, long tour d’avion/ de véhicule
Destruction de la valve veineuse par un caillot
Insuffisance veineuse
Les caillots empêchent le sang à retourner au cœur. L'accumulation de sang dans une jambe entraîne l’œdème unilatéral et l’inflammation veineuse (rougeur, chaleur, sensibilité)
Le caillot s'embolie dans les poumons
Thromboembole
-*Embolie pulmonaire (complication aiguë mortelle)
- Hypertension pulmonaire thromboembolique chronique
Légende:
Physiopathologie
Mécanisme
Signe/Symptôme/Résultats de Laboratoire
Complications
Publié 15 juin 2019 sur www.thecalgaryguide.com")
patellar-tendon-rupture-pathogenesis-and-clinical-findings

Repetitive activities involving rapid knee flexion and extension
Tendon inflammation and micro-tearing
Fluoroquinolone (antibiotic class) use
Smoking
Immobilization of knee
↓ Muscle and tendon flexibility and strength
Steroid use (including intraarticular injections)
Changes in collagen fibrils composing tendon (exact mechanism unknown)
Disrupted blood supply to tendon and poor healing
Compromised integrity and strength of the tendon
Sudden heavy load on flexed knee while foot is planted
Quadriceps muscles contract while lengthening (eccentric contraction)àforce exceeds tendon strength
Patellar tendon innervated by common peroneal (fibular) nerve
Nociceptors (pain receptors) within tendon activated by tendon rupture
Pain and tenderness below the patella
Collagen fibres in tendon tear
Tearing or popping sensation at time of injury
Patellar Tendon Rupture
Tendon/Ligament detaches from bony attachment on patella or tibial tubercle No connection of quadriceps to tibia via patellar tendon
↑ Force on tibial tubercle during tendon detachment
Avulsion fracture of tibial tubercle
Trauma to tendon ruptures surrounding blood vessels
Coagulation cascade and inflammatory mediators released at rupture site
Quadriceps muscles unable to stabilize patellar tracking
Instability of knee joint
Quadriceps muscle tension pulls patella upwards with no resistance from patellar tendon
Patellar tendon cannot use distal attachment site for leverage
Knee extensor muscles unable to effectively contract
Knee buckling
Abnormal gait
Patella alta
(high sitting patella) on X-ray
Palpable gap below the patella
Bruising below the patella
Swelling below the patella
↑ Risk of falls
Loss of patellar reflex
Inability to perform a straight leg raise (hip flexes and knee cannot remain straight)
Hematoma (blood collection under the skin)
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published August 28, 2022 on www.thecalgaryguide.com")
ascending-cholangitis-pathogenesis-clinical-findings

Bacteria ascends into biliary tract from duodenum via Sphincter of Oddi
Gallstone in the common bile duct
Stricture of biliary/hepatic ducts
Biliary / pancreatic duct malignancy
Biliary obstruction (partial bile duct obstruction)
↓ Excretion of bilirubin into
bileà ↑ bilirubin in blood
↑ Serum bilirubin
Deposition of bilirubin in the skin and mucous membranes
Jaundice*†
Parasites in bile duct
(E.g. Clonorchis)
Complication of endoscopic retrograde cholangiopancreatography (ERCP)
Bile accumulates in biliary tract
Bile duct dilation on ultrasound
Sludge (bile precipitants) impact bile ducts worsening obstruction
Obstruction & detergents in bile inflame ductal mucosa. Inflammation then spreads to adjacent structures
Stimulates phrenic (C3-C5) and foregut autonomic nerves (T5-T8)
Dull right upper quadrant pain*† radiating to back and right shoulder
↑ Intra-biliary pressure
Impaired forward flowà
↑ backflow of bile
Impaired bile secretion damages ductal epithelium of the biliary tract
Damaged ductal epithelium leaks ALP and GGT (enzymes) into blood
↑ Serum ALP and GGT
↑ Permeability of bile ductules
Reduced flushing of bile out to duodenum
Inflammatory response triggered
Fever*† ↑ WBCs
Tachycardia Hypotension† Confusion† Oliguria
Bacterial translocation from biliary tract into blood
Bacteremia
Massive systemic inflammatory response
Septic Shock
(see Distributive Shock slide)
* Charcot’s triad ~20% of cases | † Reynolds’ pentad ~7% of cases
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
First published November 18, 2015; Updated August 31, 2022 on www.thecalgaryguide.com")
acute-lower-gi-bleeds-pathogenesis-and-clinical-findings

Intestinal vessels are stretched over the domes of the diverticula
Angiodysplasia
Formation of
dilated, thin- walled vessels in GI tract mucosa/ submucosa
Colorectal Cancer
Infectious Colitis
Invasion of bacteria and/or bacterial toxins into intestinal wall
Cell damage and cell death
Sloughing off of intestinal epithelial cells
Ischemic Colitis
↓ Blood flow to a portion of the colon
Lack of oxygen delivery to colon wall
Necrosis (cell death) in the colon wall
Inflammatory Bowel Diseases (Note: these are chronic diseases and rarely present with acute lower GI bleed)
New, extremely friable blood vessels develop within the tumor
Malignant tissue invades the colon wall and disrupts colonic blood vessels
Crohn Disease
Immune- mediated full thickness inflammation of bowel wall
Ulcer formation and disruption of intestinal vessels
Ulcerative Colitis
Recurrent immune- mediated inflammation of colon mucosa
Authors:
Miranda Schmidt Illustrator:
Mizuki Lopez Reviewers:
Vina Fan,
Ben Campbell, Kerri Novak*
* MD at initial time of publication
Acute Lower GI Bleed
↑ Risk for vessel damage and rupture
Blood travels rapidly through GI tract
Hematochezia
(bright red blood per rectum)
May result in significant blood loss
Blood from the small intestine or right colon travels a longer distance through the GI tract
Bacteria in the GI tract has time to oxidize hemoglobin in the blood
Oxidization makes blood a darker color
Melena (rare in a lower GI bleed) (tarry black stool)
Inferior Vena Cava
Diaphragm
Esophagus
Loss of red
blood cells results in a loss of hemoglobin (a component of red blood cells)
Fluid from the extravascular space moves into the blood vessels to maintain vascular volume
Fluid is administered in a healthcare setting to compensate for blood loss
Hypovolemic Shock (rare in a lower GI bleed) (↓ Oxygen delivery to tissues due to low blood volume)
See “Hypovolemic Shock” slide for signs and symptoms
Lower GI Bleeds
are intra-luminal GI tract bleeds that occur anywhere distal to the ligament of Treitz (transition between duodenum and jejunum)
After 24 hours, the addition of fluid to the intravascular space dilutes hemoglobin in the blood
Normocytic Anemia
Duodenum
Ligament of Treitz
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published August 31, 2022 on www.thecalgaryguide.com")
quadriceps-tendon-rupture-pathogenesis-and-clinical-findings

Quadricep muscles atrophy
↓ Quadricep muscle strength
Smoking
Chronic disease (e.g. renal failure, gout, rheumatoid arthritis, and diabetes)
Fluoroquinolone (antibiotic class) use
Changes in collagen fibrils composing tendonàimpaired tendon strength (exact mechanism unknown)
Quadriceps tendon shortens
↓ Quadricep tendon flexibility
Disrupted blood supply to quadriceps tendon and poor healing
Compromised integrity of the quadriceps tendon and ↓ strength
Sudden heavy load on flexed knee while foot is plantedàquadriceps muscles contract while lengthening (eccentric contraction)
Force on quadriceps tendon exceeds its strength
Quadriceps Tendon Rupture
Tendon detaches from bony attachment on patella
↑ Force on patella during quadriceps tendon detachment
Collagen fibres in quadriceps tendon tear
No connection of quadriceps to tibia via quadriceps tendon
Patellar tendon tension pulls patella downwards with no resistance from quadriceps muscles
Quadriceps tendon innervated by common peroneal (fibular) nerve
Nociceptors (pain receptors) within tendon activated by tendon rupture
Pain and tenderness above the patella
Trauma to tendon ruptures blood vessels
Coagulation cascade and inflammatory mediators released at rupture site
Quadriceps muscles unable to stabilize patellar tracking
Knee joint instability
Patella baja (low sitting patella) on X-ray
Palpable gap above the patella
Loss of patellar reflex
Patellar tendon cannot use proximal attachment site for leverage
Knee extensor muscles unable to effectively contract
Inability to perform a straight leg raise (hip flexes while knee cannot remain straight)
Bruising above the patella
Swelling above the patella
Avulsion fracture of patella
Tearing or popping
sensation at time of injury
Knee buckling
Abnormal gait
↑ Risk of falls
Hematoma (blood collection under the skin)
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published October 2, 2022 on www.thecalgaryguide.com")
microscopic-colitis-pathogenesis-and-clinical-findings
)
Downregulation of intestinal tight junction proteins
↓ Epithelial barrier function
↑ Transmucosal permeability
Infiltration of bacteria or antigens
↑ Expression of nuclear factor kappa B (NF-kB) in colonic epithelial cells
NF-kB increases transcriptional activity in specific genes
↑ Inflammatory molecules
(histamine, prostaglandins, and nitric oxide) in epithelium
predisposition Unknown trigger
↑ Transforming growth factor beta 1 (TGF- β1)
and vascular endothelial growth factor (VEGF) expression in colonic epithelial cells
Equilibrium shift to more production of collagen
(fibrinogenesis) than breakdown (fibrinolysis)
Drug exposure (e.g., non-
steroidal anti- (+)
inflammatory drugs (NSAIDs), proton pump inhibitors (PPIs))
Inflammation and accumulation of immature subepithelial collagen matrix
Subtype: Collagenous Colitis
Intestinal mucosal inflammation
Subtype: Lymphocytic Colitis
characterized by a colonic subepithelial characterized by intraepithelial
collagen band on histology
lymphocytic infiltrates on histology
Microscopic Colitis
Intestinal epithelial damage
Malabsorption of nutrients (including bile acids)
Weight loss
↑ Intestinal transmucosal permeability
Water drawn into lumen through osmosis
Ions and water back leak into intestinal lumen
Non-bloody watery diarrhea
Dehydration and electrolyte disturbances
Continuous release of inflammatory mediators
Peripheral afferent nerve sensitization
Visceral hypersensitivity
Abdominal pain
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published December 8, 2018, Updated October 18, 2022 on www.thecalgaryguide.com")
copd-findings-on-posterior-anterior-and-lateral-chest-x-ray-findings
 See Pathogenesis of COPD Slides
Findings of COPD may not be apparent on chest X-ray early within the disease
Lung inflammation causes proteolytic destruction of lung parenchyma (part of lungs involved in gas exchange)
Alveolar walls are damagedàformation of fragile enlarged sacs of air called bullae
Rupture of bullae due to weak alveolar walls
Air leaves alveola and enters pleural space
Pneumothorax
See Primary Spontaneous Pneumothorax slide
Vasoconstriction of pulmonary arteries to shunt blood to better ventilated areas
Pulmonary hypertension
See Pathogenesis of Pulmonary Hypertension slide
↑ Pressure within pulmonary arteries
Enlargement of pulmonary arteries
Hilar enlargement Lung hyperinflation
(defined as 10 posterior ribs above the diaphragm level on the midclavicular line)
↑ Lucency of the lung
field, ↓ lung markings
(unless COPD is of chronic bronchitis phenotype)
↑ Size of retrosternal airspace
↑ Anterior – posterior (AP) diameter (“Barrel Chest”)
Flattening of hemidiaphragms
12 3 4
5 6
7
8
9 10
Poorly ventilated areas of lung
Hyperinflation as air becomes trapped in damaged lungs
↑ Total volume of air within the chest
Airway fibrosisà bronchial wall thickening (yellow arrow heads)
Air is less dense than soft tissue and vessels so it appears black
↑ Pressure anteriorly on sternum
↑ Pressure posteriorly on thoracic wall
↑ Pressure posteriorly on diaphragms
PA
Bullae sometimes visible on X-ray (focal areas of lucency with spread out vascular markings)
Image credit: Bronchial wall thickening image courtesy of Dr. Ashley Davidoff
Image credit: Radiopaedia
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published October 26, 2022 on www.thecalgaryguide.com")
Granulomatosis with Polyangiitis Pathogenesis

Environmental exposures
Silica dust, cigarette smoke, infections (Staphylococcus aureus)
Genetic factors
Alpha-1 antitrypsin deficiency, proteinase 3 gene mutation
↑ Production of cytokines and antineutrophil cytoplasmic antibodies (ANCAs) (mechanism unknown) Cytokines bind to endothelial cells (that line blood vessels) and neutrophils, priming them
Proteinase 3 (PR3), an enzyme that degrades extracellular matrix proteins, migrates from neutrophil granules to neutrophil cell surface
Circulating ANCAs bind to PR3 on neutrophils
PR3 stimulates maturation of dendritic cells in lungs
Dendritic cells present antigen (PR3) to naïve CD4+ T cells in peripheral lymph nodes
T cells differentiate into type 1 and type 17 helper T cells (Th1 and Th17 cells)
Th1 and Th17 cells secrete cytokines (interferon γ (INF-γ) and tumor necrosis factor α (TNF-α)) in lungs
Secreted cytokines trigger macrophage maturation
Formation of granulomas (giant cells with central necrosis surrounded by plasma cells, lymphocytes, and dendritic cells) primarily in lungs and upper airways
ANCA-activated neutrophils release proinflammatory cytokines, attracting more neutrophils to endothelium (blood vessel wall)
ANCA-activated neutrophils undergo firm adhesion to endothelium
ANCAs stimulate ↑ secretion of proteolytic enzymes and reactive oxygen species from neutrophils
Endothelial damage and tissue injury
Granulomatosis with polyangiitis (GPA)
ANCA-associated vasculitis affecting medium and small-sized arteries, associated with necrotizing granulomas
Constitutional symptoms with involvement of multiple organ systems
(see slide on clinical findings)
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published December 4, 2022 on www.thecalgaryguide.com
Granulomatosis with polyangiitis: Clinical findings Inflammation-mediated endothelial damage and granuloma formation
(see slide on pathogenesis)
Granulomatosis with polyangiitis (GPA)
Author:
Oswald Chen
Reviewers:
Ben Campbell
*Liam Martin
* MD at time of publication
ANCA-associated vasculitis affecting medium and small-sized arteries, associated with necrotizing granulomas
Constitutional symptoms
Fever, unintentional weight loss, night sweats, arthralgias
Skin involvement
Inflammation of cutaneous vessels
Systemic inflammation obstructing blood flow, with granulomatous lesions primarily in upper airways and lungs
Ear, nose, and throat involvement
↑ C-reactive protein (CRP) and erythrocyte sedimentation rate (ESR) (markers of inflammation)
Necrotizing granulomas on biopsy of affected tissue
Positive PR3-ANCA/c-ANCA blood test (antibodies present in ~90% of patients, described in GPA Pathogenesis slide)
Renal involvement
Inflammation of renal vessels
Rupture of basement membrane (layer that filters blood from glomerular capillaries into Bowman’s capsule)
Pauci-immune glomerulonephritis (see Nephritic Syndrome slide)
Rapidly progressive glomerulonephritis
Eye involvement
Inflammation of ocular tissue
Conjunctivitis
Scleritis/ episcleritis (painful red eye)
Lower respiratory tract involvement
Inflammation of pulmonary vessels
Vessel occlusion and ischemia
Skin necrosis
Vessels burst and blood pools under skin
Round and retiform (net- like) palpable purpura of lower extremities
Granulomatous destruction of nasal cartilage
Collapse of nasal bridge
Saddle nose deformity
Inflammation of paranasal sinus and nasal cavity vessels
↓ Perfusion of lungs
Dyspnea
Damage to interstitial capillaries
Hemoptysis
Diffuse alveolar hemorrhage
Rhinitis/ sinusitis
Granulomatous obstruction of eustachian tube
Otitis media (see Otitis Media slide)
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published December 4, 2022 on www.thecalgaryguide.com")
Granulomatosis with polyangiitis: Clinical findings

Granulomatosis with polyangiitis (GPA)
Author:
Oswald Chen
Reviewers:
Ben Campbell
*Liam Martin
* MD at time of publication
ANCA-associated vasculitis affecting medium and small-sized arteries, associated with necrotizing granulomas
Constitutional symptoms
Fever, unintentional weight loss, night sweats, arthralgias
Skin involvement
Inflammation of cutaneous vessels
Systemic inflammation obstructing blood flow, with granulomatous lesions primarily in upper airways and lungs
Ear, nose, and throat involvement
↑ C-reactive protein (CRP) and erythrocyte sedimentation rate (ESR) (markers of inflammation)
Necrotizing granulomas on biopsy of affected tissue
Positive PR3-ANCA/c-ANCA blood test (antibodies present in ~90% of patients, described in GPA Pathogenesis slide)
Renal involvement
Inflammation of renal vessels
Rupture of basement membrane (layer that filters blood from glomerular capillaries into Bowman’s capsule)
Pauci-immune glomerulonephritis (see Nephritic Syndrome slide)
Rapidly progressive glomerulonephritis
Eye involvement
Inflammation of ocular tissue
Conjunctivitis
Scleritis/ episcleritis (painful red eye)
Lower respiratory tract involvement
Inflammation of pulmonary vessels
Vessel occlusion and ischemia
Skin necrosis
Vessels burst and blood pools under skin
Round and retiform (net- like) palpable purpura of lower extremities
Granulomatous destruction of nasal cartilage
Collapse of nasal bridge
Saddle nose deformity
Inflammation of paranasal sinus and nasal cavity vessels
↓ Perfusion of lungs
Dyspnea
Damage to interstitial capillaries
Hemoptysis
Diffuse alveolar hemorrhage
Rhinitis/ sinusitis
Granulomatous obstruction of eustachian tube
Otitis media (see Otitis Media slide)
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published December 4, 2022 on www.thecalgaryguide.com")
Erythema multiforme (EM): Pathophysiology and clinical findings
: Pathophysiology and clinical findings Infectious agents (cause >90% of EM)
Herpes simplex virus types 1 and 2
Mononuclear cells transport herpes simplex virus DNA fragments to keratinocytes in epidermis
Herpes simplex virus-specific CD4 T-cells of the immune system are recruited to the epidermis
CD4 T-cells release interferon-gamma (a pro-inflammatory cytokine) in response to viral antigens in keratinocytes in the epidermis
Lymphocytes infiltrate along the dermal-epidermal junction of the skin Lymphocytes create an inflammatory and edematous environment Keratinocytes irreversibly degenerate and undergo apoptosis and necrosis Formation of inflammatory epidermal lesions
Authors: Ayaa Alkhaleefa Reviewers: Ben Campbell Damilola Omotajo Jori Hardin* *MD at time of publication
Drugs (a rare cause of EM)
Non-steroidal anti- inflammatory drugs i.e. diclofenac sodium
Antibiotics i.e. TMP- SMX
Topical 5% Imiquimod, used to treat actinic keratoses
Activate tumour-necrosis-factor- alpha, an inflammatory cytokine
Skin
Epidermal layer
Dermal-Epidermal
Junction Dermal layer
Triggering agent (infection, drugs)
Immune-mediated inflammation and epidermal tissue damage
Erythema multiforme
Inflammation ↑ capillary blood flow in dermis
Red colouring of skin in the area
Centre of lesions
contain a necrotic core
Inflammatory edema causes margins around necrosis to form a raised and pale ring
Inflammation stimulates cutaneous itch receptors
Erythematous, papular, target-shaped, pruritic lesions on mucosal and acral (palms of hands, soles of feet) sites and face
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published November 29, 2022 on www.thecalgaryguide.com")
rheumatoid-pneumoconiosis-caplans-syndrome-pathogenesis-and-clinical-findings
: Pathogenesis and clinical findings
Authors: Christopher Li Keerthana Pasumarthi Reviewers: Daniela Urrego, Ben Campbell, Liam Martin* * MD at time of publication
Dust-exposed occupation (e.g. coal, asbestos, silica)
↑ Accumulation of dust particles in lungs
Dust (e.g. silica) mobilizes from the lungs to other organs such as the kidneys, lymph nodes, and spleen
Accumulation of silica in other organs which activate the immune system to secrete cytokines, chemokines, and lysosomal enzymes
Activation of antigen- presenting and antibody- producing cells
Increased risk to produce autoantibodies
Increased risk for autoimmune conditions such as rheumatoid arthritis
See slide: Rheumatoid Arthritis (RA): Extra- articular manifestations for mechanism of systemic inflammation
See slide Pneumoconioses: Pathogenesis and clinical findings for mechanism
Note: Rheumatoid arthritis may precede lung nodules or develop later in the course
Rheumatoid arthritis
Systemic autoimmune inflammatory disease that mainly involves synovial joints
(+)
Alveolar macrophages and neutrophils ingest dust particles
↑ Inflammation and cytokine production in pulmonary parenchyma (e.g. interleukin-1, tumor necrosis factor-α)
Rheumatoid pneumoconiosis
Interstitial lung disease, with characteristic nodules, resulting from the interaction between dust inhalation and rheumatoid arthritis inflammation
Cytokines recruit histiocytes, neutrophils, lymphocytes, and fibroblasts to the area to produce a zone of inflammation around the dust-containing cell
Necrosis and apoptosis of dust-containing cells, macrophages, and surrounding collagen
Necrotic cells are digested by new macrophages to restart the process
Production of nodules that contain a central necrotic area surrounded by alternate layers of dust and necrotic tissue (Caplan nodules)
Hyperactive immune response to foreign materials in lungs
Multiple concentric rings of dust seen histologically on light microscopy
0.5-5 cm rounded opacities on chest X-ray in periphery of lungs
See slide on
Pneumoconioses
for symptoms and pulmonary function test results
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published November 17, 2022 on www.thecalgaryguide.com")
Acute and Chronic Gastritis: Pathogenesis and clinical findings

Noninfectious
NSAIDs, alcohol, gastric reflux
Stress
Critical illness, trauma
Gastric ulceration (see Peptic Ulcer Disease slide)
↓ Gastric mucosal defense
(↓ secretion of prostaglandins and mucus)
Acid erodes gastric mucosa
Inflammatory cells (primarily neutrophils) infiltrate site of injury
Acute Gastritis
Acute inflammation of gastric mucosa
Inflammatory cells (primarily lymphocytes and plasma cells) accumulate in gastric mucosa
Autoimmune Metaplastic Environmental Metaplastic Atrophic Gastritis (AMAG) Atrophic Gastritis (EMAG)
Atrophic Gastritis
Chronic inflammation of gastric mucosa
Inflammatory cells destroy gastric glandular epithelial cells
Gastric mucosal atrophy and metaplasia (replacement of gastric mucosal cells with intestinal epithelial cells, commonly goblet cells)
Hypochlorhydria
(parietal cell loss → ↓ hydrochloric acid secretion)
↓ Iron absorption
Iron deficiency anemia
(see Iron Deficiency Anemia slide)
Activation of chemoreceptors and mechanoreceptors
Activation
of visceral nociceptors
Visceral afferents stimulate chemoreceptor trigger zone of medulla
Nausea/ vomiting
Autoimmune
Associated with human leukocyte antigens HLA-B8 and HLA-DR3, which are proteins expressed on immune cell surface
Antibodies destroy parietal cells and intrinsic factor in gastric body and fundus
Epigastric pain (typically burning or gnawing) Dyspepsia (abdominal discomfort after
eating, often with early satiety and bloating)
Parietal cell loss →
↓ intrinsic factor →
↓ vitamin B12 absorption
Vitamin B12 deficiency
(see Vitamin B12 Deficiency slide)
Macrocytic Peripheral anemia neuropathy
↓ Gastric acidity leads to ↓ inhibition of G cells in antrum Hypergastrinemia (↑ gastrin release from G cells)
Gastrin binds to cholecystokinin-B (CCK-B) receptors on parietal and enterochromaffin-like (ECL) cells of gastric body
↑ CCK-B signaling leads to ↑ cell proliferation and ↓ cell death (↓ apoptotic activity)
Hyperplasia (↑ number of cells)
Low-grade dysplasia (disordered growth of epithelium) High-grade dysplasia
Carcinoid tumor (0.7% of cases) Malignancy arising from ECL cells
Gastric adenocarcinoma (0.3% of cases) Malignancy arising from gastric epithelial cells
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published November 15, 2022 on www.thecalgaryguide.com")
Achalasia: Findings on Fluoroscopy with Barium Swallow
 tract during fluoroscopy)
Primary Achalasia
(idiopathic)
Secondary (Pseudo) Achalasia
(due to another disease process such as tumor, Chaga’s disease, diabetes mellitus)
Achalasia
Secondary disease processes can cause denervation or nerve dysfunction of esophageal myenteric plexus (EMP). Tumors can obstruct the lumen and infiltrate the EMP.
Visual differences from primary achalasia
Look for fixed abnormalities and/or mucosal irregularities and shouldering in the setting of a tumor
Nerves controlling the lower esophagus are damaged, making it difficult for food and liquids to pass the esophagus
See “Achalasia: Pathogenesis and Clinical Findings” for full pathogenesis of primary and secondary achalasia
Inflammation and degeneration of nerves in the wall of esophagus
Dysfunction of the esophageal myenteric plexus (network of nerves in the GI tract responsible for peristalsis and sphincter relaxation)
Incomplete relaxation of the Loss of peristalsis in distal lower esophageal sphincter (LES) esophagus
Barium has difficulty passing through the LES to the stomach
Barium outlines the GI tract, following esophageal abnormalities
Normal findings
Esophageal Dilatation
Occurs upstream of narrow LES
Bird Beak Sign / Rat Tail Sign
Smooth narrowing of the distal esophagus
Incomplete LES relaxation
Barium Swallow may be normal in some patients with achalasia. Esophageal manometry (gold standard for diagnosis) or upper endoscopy can be considered instead.
Image Source: Radiopaedia
Legend:
Pathophysiology
Mechanism
Radiographic Findings
Complications
Published November 9, 2022 on www.thecalgaryguide.com")
popliteal-bakers-cyst-pathogenesis-and-clinical-findings
 Cyst: Pathogenesis and clinical findings
Authors: Liam Thompson, Megan Ure Reviewers: M. Patrick Pankow, Tara Shannon, Reza Ojaghi, Usama Malik, Dr. Gerhard Kiefer* * MD at time of publication
Repeated contraction of muscles surrounding the bursaàmicro trauma to bursa
Inflammatory response
Idiopathic
(common: kids 4-7 years old)
Bursa located between medial head of gastrocnemius and semimembranosus tendon
Accumulation of inflammatory fluid into the bursa forming a cyst
Meniscal Tear
Tear creates a channel between joint capsule and bursa posterior to the knee joint
Rheumatoid Arthritis
Auto-immune mediated inflammation of knee joint
Osteoarthritis
Destruction of articular cartilageà inflammation within knee joint See Osteoarthritis: Pathogenesis slide
Damage to joint capsule and accumulation of inflammatory fluid
Proteolytic enzyme dysregulation Local muscle action influences fluid flow and pressure changes
Extra- articular Popliteal Cyst
Fluid filled sac behind knee
Usually asymptomatic
(resolves spontaneously)
Synovial joint herniates into popliteal fossa (most commonly medially)àaccumulation of fluid (synovial + inflammatory) into synovial herniation, forming a cyst
Knee flexion
Intra-articular Popliteal Cyst
Known as “Baker’s cyst”
Knee extension
↑ Muscle tension around cyst
Channel between knee joint and cyst gradually closes with ↑ tensionàat full extension, no fluid flow between the joint and cyst (trapping the fluid in the cyst)
↓ Muscle tension around cyst
Channel between knee joint and cyst opens
Knee joint pressure becomes negative
↑ Fluid flow from cyst into knee joint
↓ Fluid in popliteal cyst
Knee joint pressure becomes positiveà ↑ Fluid flow from knee joint into cyst
(Posterior tibial nerve located lateral to the popliteal cyst and enervates posterior knee, calf, and bottom of foot)
Posterior tibial nerve becomes entrapped by mass (cyst)
Enlarging mass in posterior knee
Cyst reaches maximum volume
Pressure within cyst exceeds tensile strength of surrounding sac
Fluid drains distally into calf
Popliteal vein located lateral to cyst
↑ Venous pressure distal to cyst
Popliteal vein occluded by mass
Venous pooling distal to site of occlusion
↓ Space in posterior calf compartmentàcompression of calf muscles and posterior tibial nerve
Ankle dorsiflexion ↑ pressure in compartment
Positive Homan’s sign
(discomfort with passive ankle dorsiflexion)
Posterior plantar numbness
Pain receptors (nociceptors) activated
Posterior knee pain/pressure
Cyst Rupture Abrupt and intense pain
Stimulates inflammatory response
Fluid pushed from veins into interstitial space
Swelling and erythema (redness) in distal calf and foot
Pseudo-Thrombophlebitis
worse with extension or physical activity
(shares symptoms with deep vein thrombosis but no associated clot)
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published Feb 10, 2018, updated Nov 19, 2022 on www.thecalgaryguide.com")
hypovolemic-shock

Inflammation (pancreatitis, cirrhosis, post-operative, etc.)
Inflammatory mediators vessel permeability and fluid leaks out
Trauma
Ruptured vessels leak fluid into potential spaces
Hemorrhagic losses
(GI bleed, postpartum hemorrhage, etc.)
↓ Intravascular volume
↓ Venous return to the heart
↓ Cardiac output (blood pumped from the heart)
Hypovolemic Shock
↓ Oxygen delivery to tissues due to low blood volume
Insufficient organ perfusion
Non-Hemorrhagic losses
(dehydration, GI losses, skin losses / burns, renal losses, etc.)
‘Third Spacing’ of fluid
(fluid located outside the intravascular or intracellular space; large collections can occur in the pelvis, thorax, GI tract, long bones of children, intra-abdominally, retroperitoneally)
P = Q x R; less ‘flow’ in the vessels (Q), with vessels not constricting enough to maintain resistance (R)à pressure (P) will drop
↓ Blood Pressure
Caution: young, healthy individuals can maintain blood pressure during circulatory collapse with cardiac output and vasoconstriction; do not use blood pressure as an indicator of shock severity in children
Carotid sinus baroreceptors sense low blood pressure ↓ Carotid sinus inhibition of sympathetic nervous system Release of sympathetic catecholamines (epinephrine and
↓ Pressure in venous circulation
Brain
Heart
Kidneys
↓ Blood in the right internal jugular vein
↓ Oxygen delivery to the brain
↓ Myocardial contractility (from lactic acidosis)
↓ Blood flow to kidneys
↓ Jugular Venous Pressure
Catecholamines bind to beta-1 receptors in the sinoatrial node of the heart
Beta-1 receptor activation causes ↑ heart rate
Tachycardia
norepinephrine)
Catecholamines bind to and stimulate alpha-1 receptors in peripheral vessels
Vasoconstriction of peripheral vessels
↓ Blood flow to peripheral tissue
Catecholamines bind to and stimulate beta receptors in sweat glands
Diaphoresis
(sweating)
In all body tissues
Inadequate oxygen delivery
↓ ATP production
↑ Anaerobic metabolism
↓ Body temperature
Impaired neurological functioning
Renal ischemia
Activation of the renin-angiotensin aldosterone system
↓ Glomerular filtration rate
↓ Clearance of lactic acid by the kidney
↑ Lactic acid production
↓ Rate of activity of clotting enzymes
Lactic Acidosis
Unknown mechanism
Coagulopathy Hypothermia
Trauma Triad of Death
↑ Capillary Cold, mottled refill time extremities
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published January 24, 2013, updated December 4, 2022 on www.thecalgaryguide.com")
Ischemic Stroke: Pathogenesis
 of basal or brainstem penetrating arteries
Large artery atherosclerosis
Cholesterol plaque ↓ diameter of intra- or extracranial vessel
Cardiac embolism
Blood clot in heart breaks free, travels to brain
Other
E.g. volume loss, severe infection
Unknown
E.g. 2 or more mechanisms
Modest ↓ in O2 at penumbra (see figure)
Authors: Mizuki Lopez Andrea Kuczynski Illustrator: Mizuki Lopez Reviewers: Sina Marzoughi Usama Malik Hannah Mathew Ran (Marissa) Zhang Andrew M Demchuk* Gary M. Klein* * MD at time of publication
Significant ↓ in O2 at ischemic core (see figure)
↑ Anaerobic metabolism ↓ ATP
Production
Dysfunction of Na+/K+ ATPase pump (for 1 ATP molecule, 3 Na+ moved out of cell, 2 K+ moved into cell)
H2O influx following Na+ Cerebral edema
Compression of vessels and surrounding tissue damages blood-brain barrier
↑ Permeability of damaged blood-brain barrier
Infiltration by peripheral immune cells
Immune cells release inflammatory cytokines
↓ Cerebral Blood Flow
Penumbra Ischemic core
Metabolic demands are greater than supply of ATP
Cell death
Microglia (resident neural immune cells) activate to clean dead cell debris
Microglia release inflammatory cytokines (TNFα, IFγ, IL-1β)
Cytokines lead to astrocyte activation (support cells for neurons)
Astrocytes release more inflammatory cytokines
Inflammation of brain tissue
↑ Na+, Ca2+ influx, K+ outflux
↓ Glutamate (excitatory neurotransmitter) reuptake by astrocytes (support cells for neurons)
↑ Glutamate in extracellular fluid
Spreading depolarization from core (unclear mechanism)
Activate biochemical pathways including glutamate receptor activation
↑ Glutamate activity
Activate glutamate receptors that conduct Ca2+
↑ Ca2+ influx into neuron
Activation of catabolic proteases, lipases, nucleases in neuron
Dysfunction of neuronal protein synthesis and activity
Neuronal cell death
↑ Volume of dead (infarcted) brain tissue
Neurons depolarize and release glutamate
Reversal of Na+ Dependent Glutamate Reuptake Transporters on astrocytes (normally 3 Na++ 1 H+ + 1 glutamate into cell, for 2 K+ out)
↑ Glutamate in extracellular fluid
Stroke symptoms (e.g. weakness, slurred speech, visual field losses, autonomic dysfunction)
(see Ischemic Stroke: Impairment by Localization stroke slide)
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published November 14, 2017; updated November 6, 2022 on www.thecalgaryguide.com")
Cirrhosis
 Hepatitis D occurs with hepatitis B
Alcohol related liver disease
Non-alcoholic steatohepatitis
Autoimmune: autoimmune hepatitis, primary biliary & sclerosing cholangitis
Genetic: hemochromatosis, Wilson’s disease, α1- antitrypsin deficiency
Toxic: drugs may rarely cause chronic liver disease or cirrhosis
Severe or chronic hepatocyte injury overrides regenerative capacity → Fibrosis
Cirrhosis
Mild fibrosis may reverse with treatment of underlying cause
Nodular/shrunken liver on ultrasound
↑ Liver stiffness on transient elastography Diffuse fibrosis with nodular regeneration on biopsy
Fibrotic liver provides ↑ resistance to blood flow
Portal hypertension
Irreversible formation of fibrosis, within which hepatic cell regeneration is restricted to form nodules of poorly-functioning cells
Inflammation → epigenetic changes, oncogene mutations
Hepatocellular carcinoma
↑ Serum α-fetoprotein (sensitivity 50%, specificity 99%)
Fibrosis disrupts normal function of hepatic lobules
Hepatic insufficiency
↓ Liver synthetic function
↓ Thrombopoietin synthesis
Thrombocytopenia
↓ Conjugation of bilirubin,
↓ secretion of bilirubin into bile ducts, and
↓ drainage of bilirubin into hepatic duct
Accumulation of serum bilirubin >30 μmol/L
Jaundice
Scleral icterus, jaundiced frenulum
↑ Pressure in portal venous circulation
Portosystemic shunts
Increased flow to esophageal, rectal, and splenic veins
Vascular stretch → endothelial vasodilator (e.g. NO) release
Vasodilators enter systemic circulation
Pulmonary vasodilation
Blood cells in pulmonary vasculature have ↓ time for gas exchange
Hepatopulmonary syndrome (rare)
Dyspnea or hypoxemia, worsened when upright
↓ Synthesis of clotting factors (V, VII, IX, X, XI, XII, fibrinogen, prothrombin) and anticoagulant proteins (antithrombin, proteins C/S)
Unpredictable imbalance of hemostatic and anticoagulant factors
Elevated INR
± coagulopathy
Impaired metabolism of waste products and toxins
Toxins (mainly ammonia) accumulate and cross the blood- brain barrier
Hepatic encephalopathy
Day-night reversal, asterixis, delirium
Varices
(dilated veins in the esophagus, stomach, or rectum)
Variceal bleed
GI bleed and hypovolemia
Backflow of blood into spleen
Splenomegaly
Enlarged spleen sequesters (traps) blood cells
Cytopenias
(eg. thrombocytopenia or anemia), petechiae, easy bruising
↓ Blood flow to kidneys, which sense ↓ effective blood volume
Kidneys retain water and Na+
↑ Hydrostatic pressure in splanchnic vessels
↓ Albumin synthesis
↓ Capillary oncotic pressure
Net fluid flux out of vasculature into interstitial space
Ascites
(fluid in peritoneal cavity)
Peripheral edema
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published August 7, 2022, updated November 6, 2022 on www.thecalgaryguide.com")
Hidradenitis Suppurativa

Defective notch signaling pathway (a regulator of many cell processes)
Hormones (excess androgen activity)
Smoking (nicotine exposure)
Skin to skin friction
Systemic inflammation
Hidradenitis Suppurativa:
Pathogenesis and Clinical Findings
Other unknown genetic factors
Follicular occlusion
HS nodule
Epidermal layer
Dermal- Epidermal Junction
Dermal layer
Inflammatory cytokine- mediated activation of nociceptors
Inflammatory cytokines
Sebaceous gland
Apocrine sweat gland
Hair follicle
Pilosebaceous- apocrine unit
Changes in gene expression of hair follicle and apocrine gland anti-microbial peptides
Accumulation of corneocytes (dead keratinocytes) and sebum àfollicular occlusion and rupture
↑ Interleukin-36 (IL-36, a cytokine) Altered keratinocyte differentiation
Hair follicle hyperkeratinization
Abnormal structure and function of the pilosebaceous- apocrine unit
Infiltration of normal skin flora microbes, macrophages, dendritic cells, and Th17 cells
Increased density of nicotinic acetylcholine receptors in pilosebaceous-apocrine unit (see figure)
Mechanism not fully understood
Hyperhidrosis
Pain
Innate immune cells produce inflammatory cytokines: tumour necrosis factor alpha (TNF-α) and various interleukins (IL-1, IL-6, IL-12, IL-17, IL-22 and IL-23)
IL-17 promotes neutrophil migration into the skin
Formation of neutrophil extracellular traps (NETs)
B-cell activation and IgG autoantibody formation against skin tissue antigens
Scarring
Ongoing inflammation impairs wound healing
Granulocyte infiltration and activation, release of granulocyte colony-stimulating factor
Accumulation of keratin, purulent and/or serosanguinous fluid in dermis
Inflammatory papules, nodules, and abscesses
Pro-inflammatory cytokine-mediated response, leading to epithelial hyperplasia with increased fibrosis and collagen remodelling in the dermis
Formation of epithelial tissue tunnels in the dermis with two cavities on either end that open to the skin surface and fill with fluid or keratin
Hidradenitis Suppurativa
Chronic Inflammatory skin disease that occurs in areas with a high density of apocrine sweat glands, including the axilla, underneath the breasts, groin, and buttocks
Sinus tracts (dermal connections between lesions)
Double-ended comedones
Authors: Leah Johnston Reviewers: Mehul Gupta Lauren Lee Stephen Williams Ben Campbell Laurie Parsons* * MD at time of publication
Wound drainage and odour
Psychological distress
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published January 30, 2023 on www.thecalgaryguide.com")
Inhalational Injury

Acute hypoxemia
Loss of consciousness
Death
Upper airway injury (above vocal cords)
Cell lysis & necrosis
Local release of inflammatory substances
↑ Vascular permeability à edema of tissues in upper airway
Damage to lower respiratory tract cilia
↓ Mucus clearance from alveoli
Parenchymal injury
(delayed reaction dependent on severity of burn)
Alveolar epithelial and endothelial barrier irritation/damage
Inflammatory response
Carbon monoxide poisoning
Cyanide poisoning
Cyanide binds to mitochondrial cytochrome oxidase a3
CO binds more strongly to hemoglobin than O2
↑ Carboxyhemoglobin in blood, ↓ free hemoglobin available to bind O2 in the lungs
↑ Affinity for O2 on remaining binding sites in hemoglobin (i.e. hemoglobin binds O2 more strongly and is slow to release it)
↓ O2 delivered to tissues
Inhibition of cytochrome c oxidase
Mucous obstructs airways
Air retained
distal to the obstruction is resorbed from nonventilated alveoli
Regions without gas collapse i.e atelectasis
↑ Risk of infection
↓ Mitochondrial respiratory chain function
Impaired oxidative phosphorylation & cellular energy production
Cellular dysfunction in high metabolic tissues
Nitric oxide acts as a vasodilator in alveolar arterioles
Loss of hypoxic vasoconstriction (constriction of arterioles in alveoli due to ↓ O2)
Reactive inflammation & bronchoconstriction
Stridor
Complete airway obstruction
↑ Vascular permeability leading to fluid leakage into interstitium and alveoli
Blood flow to poorly ventilated alveoli is maintained
Ventilation/perfusion (V/Q) mismatch (regions of lung not effectively ventilated despite being well perfused by blood)
Acute Respiratory Distress Syndrome See ARDS Pathogenesis slide
Hypoxemia
Lower airway edema
Wheezing Coughing
Impaired brain function
Muscle weakness
Impaired heart function
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
First published November 10, 2019; Updated on February 12, 2023 on www.thecalgaryguide.com")
Central Retinal Vein Occlusion

↓ Anticoagulation (e.g., Protein C or S def) and/or ↑ coagulation (e.g., malignancy, Polycythemia Vera)
Non-ischemic (perfused) CRVO
Glaucoma
(a disease characterized by ↑ intraocular pressure)
Optic disc drusen
(calcified nodules located within the optic disk – the most anterior part of the optic nerve)
Diabetes
Dyslipidemia
Hypertension
Vasculitis
(inflammation of blood vessels; e.g., sarcoid, Systemic Lupus Erythematosus)
Endothelial damage
Pupillary responses do not vary between both eyes when a light is shone in one eye at a time
Relative afferent pupillary defect (RAPD) mild or absent
Lost vascular wall integrity
Normal retinal electrical response to a light stimuli
Normal
electro- retinography (ERG) with b- wave >60%
Few scattered Intra-retinal hemorrhages
↓ In retinal electrical response to a light stimuli
Pupils respond differently to a light stimulus shone in one eye at a time
Severe ↓ in visual acuity (Snellen acuity of <20/400)
Visual field deficits
↓ b-wave amplitude (<60%) on ERG
RAPD present
↑ Pressure compromises retinal vein outflow
Central retinal atherosclerosis (build-up of fat & cholesterol plaque in arteries) & hardening
Atherosclerotic changes in the central retinal artery compress the central retinal vein (since they are both held together in region of the optic disc)
Venous stasis (stagnation of blood flow) ↑ Likelihood of thrombus formation
Central Retinal Vein Occlusion (CRVO)
A common retinal vascular disease characterized by blockage of the main vein that drains blood from the retina
CRVO classified based on the degree of perfusion (as seen through retinal angiography)
Ischemic (nonperfused) CRVO
↑ Intraluminal venous pressure
Dilated tortuous retinal veins
Normal visual fields
Vascular wall integrity lost
Four-quadrant hemorrhages described as “blood and thunder” on fundus exam
Obstruction due to thrombus
↓ Retinal capillary perfusion causes ischemia
Hypoxia in the retinal tissues
↑ Release of vascular endothelial growth factor to revascularize diseased tissue & overcome hypoxic conditions
Angiogenesis (formation of new blood vessels)
Intraretinal infarcts
“Cotton wool spots” on fundus exam
Venous capillary fluid/protein leakage
Mild retina, macula and disc edema
Moderate ↓in visual acuity often (Snellen acuity >20/400)
New vessel proliferation can occur in the anterior chamber of the eye
This change can block the outflow path of the aqueous humor in the eye
Neovascular glaucoma
Neovascular vessels are structurally different in comparison to regular retinal vasculature
Neovascular vessels are more fragile
↑ Likelihood of rupture Vitreous hemorrhage
Neovascular vessels lack tight junctions
↑ Fluid leakage
Retina, macula and disc edema
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
First Published June 28, 2017, updated March 4, 2023 on www.thecalgaryguide.com")
Pyogenic Brain Abscess on MRI
 transcellularly, paracellularly or by a Trojan Horse mechanism
Authors: Omer Mansoor, Aly Valji, Nameerah Wajahat Reviewers: Mao Ding, Reshma Sirajee, James Scott* *MD at time of publication
R
Transcellular
Pathogen invades BBB cell directly to enter
Paracellular
Pathogen goes between BBB cells by disrupting tight junctions
Trojan Horse
Pathogen bypasses BBB by hiding inside a macrophage cell
Ring Enhancing Lesion Sensitive, but not specific for a Brain Abscess
Pathogen causes parenchymal inflammation and progresses through four stages of infection
Axial T1 + Gadolinium MRI Head. Pyogenic Brain Abscess showing infection at Stage 3 or 4. T1 highlights fat, whereas T2 highlights fat and water*.
R
Stage 1: Early Cerebritis Focal infection (2-3 days) with no pus formation
Stage 2: Late Cerebritis Progressive (1 week) infection of abscess
Stage 3: Early Encapsulation
In 1-2 weeks, pus is formed from the pathogen
Stage 4: Late Encapsulation After 2 weeks, abscess shrinks in size as it necroses
Pathogen causes acute inflammatory changes of swelling (edema) and vascular congestion
No fibrous capsule is formed yet and infection is not well localized on a T1 MRI
Increased edema could be seen on T2 MRI
Poor Demarcation and Vasogenic Edema
Abscess is not well-defined with minimal enhancement on T1 but can have increased signal on T2
Fibrous capsule is formed by surrounding healthy brain tissue that walls off abscess
Capsule is made of granulation tissue and fat, which lights up non-specifically on a T1 MRI
Use Diffusion Weighted Imaging (DWI) to distinguish abscess from other brain lesions
DWI measures water diffusion in different directions, specifically diseased areas where water movement is restricted
Restricted Diffusion Edema and inflammation allow water to move freely (hyperintense signal)
*Imaging Source: Feraco et al., 2020: https://jptcp.com/index.php/jptcp/article/download/688/685?inline=1
DWI Sequence Axial MRI Head. Same Pyogenic Brain Abscess as above. DWI is the mainstay imaging sequence for diagnosing a pyogenic brain abscess*.
Legend:
Pathophysiology
Mechanism
Radiographic Findings
Complications
Published Mar 25, 2023 on www.thecalgaryguide.com")
Acute Pulmonary Embolism on CTPA

Virchow’s Triad:
Hypercoagulability, venous stasis, vascular endothelial injury
Image Source: European Society of Radiology
Image: Polo mint sign on axial CTPA.
Image Source: Journal of The Indian Academy of Echocardiography
Image: Railway sign on axial CTPA. Image Source: Moore et al. 2018
Image: Pleural effusion and pulmonary infarction on axial CTPA.
Authors: Aly Valji, Nameerah Wajahat, Omer Mansoor Reviewers: Reshma Sirajee, Sravya Kakumanu, Victória Silva, Mao Ding Vincent Dinculescu* *MD at time of publication
Deep Vein Thrombosis (DVT): Majority of pulmonary embolism (PE) arise from DVT: Clot travels via inferior vena cava → right atrium → right ventricle → pulmonary arteries/arterioles
See “Virchow’s Triad and Deep Vein Thrombosis” for full pathogenesis
Polo Mint Sign
Clot visualized in short axis, Filling defect entirely surrounded by IV contrast creating a circle (polo mint)
Railway Sign
Clot visualized in long axis, Filling defect surrounded by IV contrast on two sides creating the appearance of a railway track
Type I or II Ventilation/Perfusion respiratory failure (V/Q) mismatch
Reverse Halo Sign
Pulmonary infarction leads to wedge shaped opacity with a rim of consolidation (black arrows) surrounding “ground glass” (red arrows)
Pulmonary Embolism: Clot in pulmonary arteries
See “Signs and Symptoms of Pulmonary Embolism” for full presentation
Saddle Embolus: Large clot over the pulmonary trunk bifurcation Lobar/Segmental/Subsegmental Embolus: Clot within the pulmonary arteries of the lungs
Blockage of pulmonary arteries = ↓ Blood flow, ↑ Right heart pressure
↓ Gas exchange b/w lungs and blood
Ischemia of lung tissue → infarction → inflammation of dead tissue
↑ Right heart filling and expansion
Left heart filling impaired
↓ Cardiac output due to ↓ Left heart filling
“Massive PE” = sustained systemic hypotension or bradycardia (SBP < 90 mmhg, HR < 40 bpm)
Saddle Embolus
Filling defect due to blockage of bifurcation. IV contrast appears white, and embolus appears grey
Pleural Effusion
Exudative Pleural Effusion
Tissue inflammation → ↑ blood vessel permeability = Leakage of fluid into pleural space
Transudative Pleural Effusion
↑ Hydrostatic pressure from right heart congestion = Pushes fluid into pleural space
Image Source: Samra et al. 2017
Image: Saddle embolus on axial CTPA.
Legend:
Pathophysiology
Mechanism
Radiographic Findings
Complications
Published March 28, 2023 on www.thecalgaryguide.com")
Coronary Artery Bypass Graft CABG Indications
: Indications
Author: Breanne Gordulic Reviewers: Miranda Schmidt Ben Campbell Sunawer Aujla Angela Kealey* * MD at time of publication
Symptomatic multivessel (≥ 3 vessels) coronary artery disease (MVCAD) or complex MVCAD
Acute coronary syndrome (ACS)
Left main coronary artery disease
Multivessel (≥ 3 vessels) CAD and diabetes
Cardiac surgery required for other pathology
Multivessel CAD, LV dysfunction and congestive heart failure (CHF)
Complex CAD includes stenosed vein grafts, bifurcation lesions, calcified lesions, total occlusions, ostial lesions
STEMI initial treatment is PCI/thrombolysis
CABG outcomes compared to percutaneous coronary intervention (PCI) in MVCAD include ↑ survival in diabetes, ↑ survival with LV dysfunction, ↓ repeat revascularization, ↓ myocardial infarction, ↓ stroke
↑ Risk of failure in complex CAD with PCI
Rapid reperfusion to myocardium most important in STEMI to decrease myocardial damage
CABG can be considered for residual stenoses 6-8 weeks later
NSTEMI or unstable angina (UA) with MVCAD involving at least three vessels including the proximal left anterior descending (LAD)
Left main coronary artery divides into left anterior descending (LAD) and left circumflex (LCx) which supplies 2/3 of myocardium
↑ Survival Myocardial infarction from left main artery occlusion Death
Left main stenosis
Ventricular dysrhythmias
Ongoing ischemia
LV dysfunction Hemodynamic instability
↑ risk of PCI
CABG has mortality benefit
↓ Number of operations
↑ Risk of cardiovascular disease in diabetes
Revascularization indicated along with other cardiac surgery
Multivessel CAD with >90% stenosis and CHF
LV ejection fraction <35%
↑ Risk of atherosclerosis from hyperglycemia and dyslipidemia
CABG bypasses several atherosclerotic plaques in coronary arteries
↑ Durability
↑ Complete perfusion
Valve stenosis or regurgitation Septal defect
Aortic root or arch pathology
Combination procedure
Evidence of ischemia at rest
Evidence of
impaired LV function at rest
↓ All-cause mortality in CABG vs medical management
Chronic obstructive pulmonary disease
Abbreviations:
• ACS- acute coronary syndrome. Acute reduction in
blood flow to heart muscle resulting in cell death. • CAD- coronary artery disease. Narrowing or blockage of the coronary arteries by plaque
• NSTEMI- myocardial infarction (heart attack) with no ST elevation on electrocardiogram
• PCI- percutaneous coronary intervention. A balloon tipped catheter is used to open blocked coronary arteries; a stent may be placed.
• STEMI- myocardial infarction with ST segment elevation on electrocardiogram
Consider revascularization (restore blood flow to blocked or narrowed blood vessels) of coronary arteries to increase perfusion to myocardium (heart muscle)
Coronary artery bypass graft recommended
Assess surgical risk and comorbidities with evaluation by heart team that includes both a cardiac surgeon and interventional cardiologist (SYNTAX Trial)
Individual management plan for patients with comorbidities that increase mortality
Frailty
Chronic Renal Failure
↑ Inflammation and deregulated angiogenesis affects all organ systems
↓ Physiologic reserve and ↓ ability to recover from acute stress
↑ Pneumonia
↑ Respiratory and Renal Failure
↑ Stroke
↑ In hospital mortality
↓ Survival two years after surgery
Use Society of Thoracic Surgeons Score, EuroSCORE, or SYNTAX II Score to predict patient outcome with anatomy, disease severity, and preoperative characteristics
Coronary Artery Bypass Graft
Surgery to take healthy blood vessels from the body and connect them proximally and distally to blocked coronary arteries
Cardiopulmonary bypass, fluid overload, ↑ renal vasoconstriction and ↓ renal oxygenation from rewarming
Kidney injury
↑ End stage kidney disease
Blood flow restored to
ischemic myocardium
↓ Angina
↑ Quality of life ↑ LV function ↑ Survival
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published April 12, 2023 on www.thecalgaryguide.com")
OA Clinical findings

Damage of normal cartilage under abnormal loading
Authors: Sean Spence Modhawi Alqanaie Reviewers: Yan Yu Jennifer Au Mao Ding Gary Morris* * MD at time of publication
Single large traumatic event or repeated microtrauma
Damage to normal articular cartilage under normal loading (force put on a joint)
Genetic anomalies in cartilage production and inborn errors of cartilage metabolism
Damage of abnormal cartilage under normal joint loading
Destruction/attrition of articular cartilage
Osteoarthritis
A degenerative joint disease that can affect both load bearing joints (knee, hip) as well as in smaller joints (proximal inter-phalangeal, carpometacarpal joints in hand)
Repeated physical joint trauma
Aberrant bone deposition secondary to wear on subchondral bone
Formation of osteophytes (bony projections) and subchondral sclerosis (bone thickening)
Lack of cartilage
Direct contact between bony processes with movement of the joint
Inflammation alters the chemical milieu of joint tissue
Synovial fluid forced into bone
Damage to subchondral (under cartilage) blood vessels
Subchondral fractures
cytokines regulate hyaluronic acid synthase
Synovium makes lower molecular weight hyaluronic acids (a potent proinflammatory molecule)
↓ synovial fluid viscosity
↑ risk of infection
Increased secretion of proteolytic enzymes
↑ joint fluid production
Joint effusion
Disruption of normal joint architecture
Palpable bony hypertrophy
(ex. Bouchard’s nodes (bony bumps on the middle joints of the finger))
Impaction of osteophyte with normal joint structures during movement
Physical disability
Crepitus
(grating sound in a joint)
Joint movement with reduced lubrication stimulates joint nociceptors
Stimulation of joint nociceptors (sensory receptors that detect damaging stimuli) in subchondral bone
Pain with use of joint
Pain with motion
↓ Range of motion
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published November 1, 2012, updated May 18, 2023 on www.thecalgaryguide.com")
diverticulosis-vs-diverticulitis-distinguishing-features
 contributing to reduced gut motility
↑Intraluminal pressure in the colon
Herniation of colonic mucosa and submucosa through circular muscle at points of weakness to form outpouchings
Diverticulosis
Presence of outpouchings in the colon (diverticuli)
Note: In Western populations, most diverticulosis is left-sided, whereas in Asian populations, these outpouchings are more often right-sided.
Mucosal abrasion or micro-perforation by ↑ intraluminal pressure or dense food particles
Bacterial overgrowth,
dysbiosis and passage into the lamina propria
Feces collects in diverticuli
Gut bacteria metabolize undigested material and produce gas
Stretching of colon Bloating wall irritates and
visceral afferent flatulence nerves
Episodic abdominal discomfort and cramping
Blood vessels in
the mucosa and submucosa (vasa recta) are stretched over the diverticuli, and may rupture from ↑ pressure
Diverticular bleed
Painless hematochezia (passage of fresh blood from the rectum)
Inflammatory cytokines activate clotting factors
Clotting of blood in vessels supplying diverticula
Inflammatory cytokine release (IL-6, TNF-α)
Cytokines enter systemic circulation
Edema in the bowel wall
Irritation of adjacent parietal peritoneum and somatic nerves
Left lower quadrant (LLQ) pain, guarding
Small abrasions are walled off by pericolic fat and mesentery
↑WBC
Fever
Inflammation may spread to nearby organs, leading to ulceration and abnormal connections between organs
Pericolic abscess
No hematochezia
Local ischemia and focal necrosis resulting in loss of integrity of the bowel wall
Perforation
Generalized peritonitis
Colonic obstruction (rare)
Fistulae (rare): i.e. colovesical, coloenteric
Stricture/fibros is formation with healing
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published October 12, 2016; Updated July 30, 2023 on www.thecalgaryguide.com")
Knee Osteoarthritis

Articular trauma
Inflammatory disease or infection (e.g. Rheumatoid or septic arthritis)
Obesity and ↑leptin ↑ Chondrocytes,
inflammatory mediators, and metalloproteinases
Extracellular matrix degradation
↑ Knee joint loading forces
Metabolic syndromes (e.g. diabetes mellitus)
↑ Oxidative stress and insulin resistance
Low-grade systemic inflammation
↓ Elasticity and ↑ degradation of cartilage
↑ Friction in knee joint with movement
↓ Cartilage along femoral groove and posterior surface of patella
Pain, catching, and crepitus (crackling/clicking sound) in the patellofemoral joint
Inability/difficulty with kneeling or climbing stairs
Abnormal distribution of forces accumulate and stress articular surface
↑ Damage/laxity to soft tissue structures stabilizing knee joint
Knee Osteoarthritis
(Multifactorial entity characterized by cartilage breakdown, deterioration of connective tissue, and bone deformities)
↓ Cartilage between distal femur and proximal tibia ↓ Joint spaceàto articular dysfunction
Radiographic changes
See Osteoarthritis (OA): X-Ray Features slide
Repeated attempts to repair cartilage and joint disruption
Subchondral bone thickening (sclerosis) under joint cartilage and bone spur (osteophyte) formation around joint line
Rotational/antero-posterior instability and ↑ external adduction moments during walking
Alterations in proteoglycans, fiber arrangement, and collagen composition in soft tissue structures within/around knee joint
↑ Shear forces and medial compartment narrowing erode and pinch soft tissue structures within the knee joint
Cruciate ligament degeneration
Weakened passive stabilizers of the knee joint
Knee giving way and instability (falls)
Meniscal tears, if large àprevents knee extension/flexion
Locking of the knee
Joint line tenderness:
Patient points to area of tenderness/pain reproducible upon palpation
Anatomical axis of hip, knee, and ankle joints ↑ loading medially
Medial > lateral joint line tenderness
↑ Joint friction activating nociceptors in the surrounding anatomical tissues
Injury and inflammation ↑ nociceptive responses in soft tissue structures and subchondral bone within knee joint
Nociceptive feedback to brain inhibits activity of motor cortex neurons controlling muscles around the knee
↓ Motor output and muscle activation over time
↓ Muscle strength/endurance, lower limb muscle use, functional ability (walking, stairs, etc.)
Joint inflammationà accumulation of fluid within joint
Stiffness, swelling, redness, and pain
Limited joint space reduces range of motion for femur to roll/slide on tibia
↓ Knee flexion and extension
Flexion contracture and antalgic gait
Reduced weight acceptance of the joint and surrounding muscles/tendons
↓ Mobility and physical dysfunction
Muscular atrophy
Reduced function of active stabilizers of the knee joint (quadriceps, adductors, hamstrings)
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published July 30, 2023 on www.thecalgaryguide.com")
Rotator Cuff Disease
 of glenohumeral joint
Tensile forces
Repeated eccentric tension from overhead activities
Trauma, sports, and occupation
↑ Torque, compression, and translational stresses
Metabolic syndromes
Reactive oxygen species
interact with ↑ glucose forming advanced glycation end-products (AGEs) which accumulate in soft tissues
Smoking
Impingement syndromes
Vessel damage, ischemia, tenocyte apoptosis
Macro-trauma causing an acute, complete tear in the rotator cuff muscle(s)
↓Dynamic stability (from rotator cuff and periscapular muscles) and range of motion of the shoulder at the glenohumeral joint
↑ Bone on bone contact of proximal humeral head and boney structures of the scapula
Subacromial bursa degeneration
↓ Protection of underlying supraspinatus muscle from attrition between humeral head and acromion
Rotator Cuff Syndrome
(Inflammation, impingement, or tearing of one or more of the four muscles/tendons of the rotator cuff: supraspinatus, subscapularis, infraspinatus, teres minor)
Repetitive loading and micro-tearing of tendon/muscle fibers
↑ Oxidative stressors and inflammatory cascades
↓ Vascularity of rotator cuff structures
Radiographic changes: See Rotator Cuff Disease: X-ray and ultrasound features slide, in addition to: calcific tendonitis, calcification of in the coracohumeral ligament, and hooked acromion (calcification from tendon pulling)
In some cases, soft tissues enclose/surround shoulder joint capsule thicken (fibrose) and tighten
Degenerative joint disease and rotator cuff arthropathy
Proximal humeral head migration and ↓ subacromial space
Inflammation and insufficient healing of rotator cuff structures, which may lead to:
Supraspinatus (shoulder abduction) degeneration
Pain, shoulder stiffness, ↓ active AND passive range of motion
Adhesive Capsulitis (frozen shoulder)
Infraspinatus and teres minor (external rotation) degeneration
Rotator cuff tendons become inflamed and irritated as they rub against acromion
Subacromial impingement
Subscapularis (internal rotation) degeneration
+Lift-off test: Inability to hold dorsum of the hand off lumbar spine while internally rotating shoulder
↓ Shoulder strength and muscular atrophy
Pain with passive shoulder flexion beyond 90°
Winging of the scapula during arm adduction
+Empty-can test: Weakness and/or arm depression with resisted abduction with arm internally rotated in 90°
+Drop-arm test: Inability to maintain shoulder in abducted position at 90° and/or adduct the arm in a controlled manner (resulting in ”dropping”)
Weakness to resisted external rotation with elbow in 90° flexion, inability to keep arm externally rotated (infraspinatus)
+Hornblower’s sign: decreased external rotation strength in arm abduction (suggests additional teres minor tear)
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published July 30, 2023 on www.thecalgaryguide.com")
Mitral Regurgitation Pathogenesis and clinical findings

Myocarditis
Left ventricular dilation displaces papillary muscles
Dilation of the Tethering of mitral valve annulus chordae tendineae
↑ Volume and pressure in left atrium
↑ Volume pushed back into left ventricle
Dilated left ventricle
Apical impulse on palpation and auscultation
↓ Forward flow of blood out of heart
Blood backs up into pulmonary circulation
↑ Intravascular hydrostatic pressure in pulmonary vessels
Fluid extravasates out of vessels and into the lungs
Papillary muscle rupture
Mitral valve leaflets flail
Mitral valve prolapse
Structurally abnormal valve
Connective tissue disorders
Weak valve leaflets
Rheumatic heart disease
Dilatation of the mitral valve annulus, inflammation of leaflets
Infective endocarditis
Vegetations form on valve leaflets
Mitral Regurgitation
Blood consistently flows backward throughout systole
Holosystolic murmur, radiates to axilla, ↑ with afterload (e.g. making a fist)
Backflow of blood from left ventricle to left atrium due to impaired mitral valve closure
S3 heart sound
Myocardial remodeling
↓ Muscle efficiency
↓ Left ventricle systolic function
↓ O2 saturation, tachypnea, wheeze, ↑ work of breathing, crackles, frothy sputum (if severe)
Congestive heart failure
↓ Stroke volume ejected into aorta
↓ Cardiac output
↓ Organ perfusion ↓ O2 to kidney
Activation of renin-angiotensin- aldosterone system
↑ Reabsorption of water by kidneys
↑ Intravascular hydrostatic pressure systemically
Peripheral edema
Injury to kidney parenchyma
↓ ability for kidney to clear creatinine
↑ Serum creatinine
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published Feb 3, 2018, updated Oct 15, 2023 on www.thecalgaryguide.com")
Death Cardiovascular Respiratory and Neurologic Mechanisms

Hypoxemia (Type I Respiratory Failure): low dissolved oxygen in blood (PaO2)
Lungs can’t oxygenate blood fast enough
Lungs can’t rid blood of CO2 fast enough
Hypercapnia / hypercarbia (Type II Respiratory Failure): elevated dissolved CO2 in blood (PaCO2)
Cerebral vasodilation
Toxins: e.g. cyanide, pesticides, arsenic Severe anemia
Distributive problems:
Systemic inflammation (sepsis, anaphylaxis, pancreatitis), adrenal insufficiency, vasodilatory drugs
Obstructive problems: Cardiac tamponade*, tension pneumothorax* or massive pulmonary embolism*
Hypovolemic* problems (low blood volume): Hemorrhage, dehydration, widespread skin disruption or burns
Cardiac valve dysfunction
Myocardial infarction* or cardiomyopathy
Cardiac arrhythmia or heart block
Disturbed electrical activity in cardiomyocytes
Peripheral metabolic disturbances
Hypokalemia*, Hyperkalemia* Acidosis* (including renal failure) Hypothermia*
Toxins* (e.g. cocaine, beta blockers, tricyclics) Severe thyroid derangement
Inappropriate systemic vasodilation
Adjacent forces impair heart filling
Low cardiac preload
Low stroke volume (SV; depends on valves, contractility, preload)
Decreased systemic vascular resistance (SVR)
Low blood pressure (BP = CO x SVR)
Decreased cardiac output (CO = SV x HR)
Disseminated intravascular coagulationàwidespread thrombi that occlude blood flow (also causes hemorrhage, see relevant box at left)
Methemoglobinemia: some hemoglobin gets stuck in a state that can’t carry O2
Hemoglobin has reduced capacity to carry or release O2
Drugs: e.g. dapsone, nitrates
Carbon monoxide poisoning
Circulatory collapse / shock: inadequate perfusion of tissue with blood
Respiratory collapse: blood has insufficient useable O2 content
Ventricular fibrillation (VF) or pulseless ventricular tachycardia (VT)
Hypoxia*: inadequate O2 delivery or utilization in tissues
Hypoxia creates metabolic disturbances that impair cardiac cells. Alternatively, any of the preceding conditions marked with (*) can directly trigger cardiac arrest first
Pulseless Electrical Activity (PEA): organized activity on ECG with no cardiac output (can be preceded or mimicked by pseudo-PEA, in which there is still some output on ultrasound)
Low atmospheric pressure or oxygen content Severe lung disease
Asthma, COPD, interstitial lung disease, congestive heart failure, pulmonary hypertension, pulmonary embolism, lung collapse / atelectasis
Acute respiratory distress syndrome
Pneumonia, aspiration pneumonitis, inhalational injury, systemic inflammation, drowning
Severe hypoventilation
Respiratory fatigue, advanced COPD, chest wall disorders, neuromuscular disorders, upper airway obstruction, toxins (e.g. opioids, botulism)
Can degenerate at any time
Asystole: no cardiac electrical activity or output
Death
Respiratory arrest: cessation of breathing
Inability to protect airway
Decreased level of consciousness
Note
This is a broad overview of the many scenarios that can result in death. For detailed explanations of the various disease mechanisms, refer to the corresponding slides.
* = reversible causes of cardiac arrest (Hs and Ts)
Author:
Ben Campbell
Reviewers:
Yan Yu*
Huma Ali*
* MD at time of publication
Bradycardia
(low heart rate, HR)
Unopposed parasympathetic stimulation of heart (can also cause vasodilation, see Distributive problems)
Disruption of spinal cord sympathetic control
Injury to cervical or upper thoracic spinal cord
Irreversible cessation of cardiac, respiratory, and brain function
Prolonged seizure initially causes increased cardiovascular activity, until the system fatigues
Disruption of respiratory control center in medulla
Expanding skull contents squeeze brainstem (herniation)
Increased intracranial pressure
Edema from intracranial hemorrhage, trauma, brain mass
Edema, inflammation, hypoxia and/or metabolic derangements cause diffuse neuron dysfunction
Central nervous system infection
Dementia, particularly with delirium
Massive ischemic stroke
Seizure
activity prevents or alters breathing
Metabolic disturbances that affect the central nervous system Hypoglycemia Hypocalcemia, hypercalcemia Hyponatremia, hypernatremia Uremia
Acute liver failure (hyperNH4) Many drugs / toxins Withdrawal (e.g. EtOH)
Status epilepticus
Brainstem lesion (e.g. stroke, neoplasm, inflammatory)
Nervous System Insult
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published November 11, 2023 on www.thecalgaryguide.com
Respiratory System Insult
Cardiovascular System Insult Cardiogenic problems")
Non-Alcoholic Fatty Liver Disease
)
Steatohepatitis: chronic inflammatory and apoptotic climate in the hepatocytes (in the absence of alcohol consumption, termed Non-Alcoholic Steatohepatitis (NASH))
Fibrosis of the Liver: excessive scarring of liver tissue resulting from chronic inflammation, although liver architecture is largely intact
Fat droplets form and grow in the hepatocytes
Hepatic mitochondria increase their workload in attempt to break down the excess free fatty acids through beta-oxidation
↑ in cellular workload creates more reactive oxygen speciesà Inflammation and apoptosis of hepatocytes
On-going inflammation damages hepatic stellate cells (the primary extracellular matrix–producing cells of the liver) causing the release of fibrinogenic cytokines
Cirrhosis of the liver: normal lobular structure distorts and is replaced by regenerating nodules and bridging septa, disrupting normal liver blood flow
Deposition of fibrotic
material and collagen within the perisinusoidal spaces of the liver
Decompensated Cirrhosis Hepatocellular carcinoma
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published November 25, 2023 on www.thecalgaryguide.com")
RICE mechanism of action
: Mechanism of action
Healing of an injury requires pain and inflammatory control to encourage activity. The goal of Rest, Ice, Compression, Elevation (RICE) is to
decrease inflammation. Though activity itself will contribute to pain and inflammation, it is integral to the rehabilitation process.
Authors: Matthew Roberts Emma Windfeld Reviewers: Alexander Arnold Amanda Eslinger Shyla Bharadia Mao Ding Bradley Jacobs* * MD at time of publication
Rest: (weight bearing or stressful motion discontinued)
Further damage to affected tissues from mechanical stress is prevented
Ice: (applied to injury)
Compression:
(wrap applied to injured area)
Mechanical force applied to tissue
Excess fluid is pushed back into capillaries and lymph network
Elevation:
(limb raised above heart)
Blood flow to tissue is constricted
Mechanism not well understood
↓ delivery of inflammatory mediators such as polymorphonuclear neutrophils and macrophages to injured site
↓ production of inflammatory cytokines (pro-inflammatory substances) such as Tumor Necrosis Factor-α, Platelet Derived Growth Factor, Epidermal Growth Factor, and Transforming Growth Factor-β
↓ Inflammation
Gravity ↑ venous blood return to systemic circulation
↓ edema (accumulation of fluid in interstitium)
Early initiation of injury-specific rehabilitative exercises improves range of motion, strength, and proprioception
Stress to targeted area induces inflammation that, when tightly regulated, is integral to repair
Injured muscle, tendon, bone, or ligament is strengthened
↓ Pain
↑Range of motion and therefore function
Early recovery from injury
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published Dec 9, 2013, updated Oct 15, 2023 on www.thecalgaryguide.com
Rest, Ice, Compression, Elevation (RICE): Mechanism of action
Healing of an injury requires pain and inflammatory control to encourage activity. The goal of Rest, Ice, Compression, Elevation (RICE) is to decrease
inflammation. Though activity itself will contribute to pain and inflammation, it is integral to the rehabilitation process.
Authors: Matthew Roberts Emma Windfeld Reviewers: Alexander Arnold Amanda Eslinger Shyla Bharadia Bradley Jacobs* * MD at time of publication
Rest: (weight bearing or stressful motion discontinued)
Further damage to affected tissues from mechanical stress is prevented
Ice: (applied to injury)
Compression: (wrap applied to injured area)
Mechanical force applied to tissue
Excess fluid is pushed back into capillaries and lymph network
Elevation: (limb raised above heart)
Gravity ↑ venous blood return to systemic circulation
Blood flow to tissue is constricted
↓ delivery of inflammatory mediators such as polymorphonuclear neutrophils and macrophages to injured site
Mechanism not well understood
↓ production of inflammatory cytokines (pro- inflammatory substances) such as Tumor Necorsis Factor-α, Platelet Derived Growth Factor, Epidermal Growth Factor, and Transforming Growth Factor-β
↓ Inflammation
↓ Pain
↓ edema (accumulation of fluid in interstitium)
↑Range of motion and therefore function
Early initiation of injury- specific rehabilitative exercises improves range of motion, strength, and proprioception
Stress to targeted area induces inflammation that, when tightly regulated, is integral to repair
Injured muscle, tendon, bone, or ligament is strengthened
Early recovery from injury
Legend: Published MONTH, DAY, YEAR on www.thecalgaryguide.com
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
RICE: Mechanism of action
Authors: Matthew Roberts Emma Windfeld Reviewers: Alexander Arnold Amanda Eslinger Shyla Bharadia Bradley Jacobs* * MD at time of publication
Rest
(weight bearing or stressful motion discontinued)
Ice
(applied to injury)
Compression
(wrap applied to injured area)
Elevation
(limb raised above heart)
Constricts blood flow to tissue
Mechanism not well understood
Mechanical force applied to tissue
↓ Delivery of polymorphonuclear neutrophils and macrophages to injured site
Excess fluid pushed back into capillaries and lymph network
Gravity ↑ venous blood return to systemic circulation
Prevents further damage to affected tissues from mechanical stress
↓ Production of inflammatory cytokines
↓ Edema (accumulation of fluid in interstitium)
↓ Inflammation
↓ Pain
↑Range of motion and therefore function
Early initiation of injury-specific rehabilitative exercises to improve range of motion, strength, and proprioception
Stress to targeted area induces inflammation that, when tightly regulated, is integral to repair
Injured muscle, tendon, bone, or ligament is strengthened
Early recovery from injury
Legend: Published MONTH, DAY, YEAR on www.thecalgaryguide.com
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications")
Aspiration Pneumonia
 (e.g. stroke)
Altered level of consciousness and impaired cough/clearance
Tube Poor Alcohol and Substance Medications Neurologic diseases
Esophageal and gastric motility disorders
Impaired swallowing
Chronic obstructive pulmonary disorder
feeding oral health
Bacteria adhere to epithelial surfaces and ↑ risk of airway and lung bacterial colonization
Aspirated oropharyngeal and gastric contents can also contain bacteria
↓ Elimination and clearance of foreign bacteria from airway and lung
Macroaspiration (large volume aspiration) of oropharyngeal bacteria, during eating and drinking
Bacteria and fluid fill bronchi and alveolar space
Aspiration Pneumonia
Alterations to lung microbial flora
An infectious lung process caused by inhalation of foreign bacterial and oropharyngeal and gastric contents
Aspiration of acidic fluid and pneumonia causative pathogen (typically anaerobes or bacteria in normal oral flora) with resultant inflammation
Infiltrate develops in a gravity-dependent pattern in patches around bronchi segments.
Produces proinflammatory cytokines, (e.g. tumor necrosis factor-alpha, and interleukin-1)
Hypothalamic production of prostaglandin E2 results in thermogenesis
Fever
Authors: Luiza Radu
Reviewers: Mao Ding, *Yan Yu, *Jonathan Liu *MD at time of publication
Aspiration to the right lung more common due to large diameter and more vertical orientation of the right main bronchus
Crackles and ↑ lung vibrations (fremitus) on auscultation
Productive Cough
Impaired alveolar gas exchange
Chemoreceptor detection of ↓ pO2 triggers
↑ ventilation
Hypoxemia
Dyspnea
Consolidation in lower lobes (particularly superior segments) and posterior segments of upper lobes
If untreated, a
pus-filled lung cavity develops (e.g. abscess)
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published Jan 11, 2024 on www.thecalgaryguide.com")
Shoulder Impingement Syndrome
 on the anterolateral acromion and coracoacromial ligament
X-Ray: Normal Ultrasound: +/- Tendinopathy, muscle atrophy
Pain with
overhead movement
Pain at night
(e.g., sleep position, gravity)
Pain with lifting
(e.g., weight- training, groceries)
Pain (between 60°-120°) with passive shoulder abduction
+ Painful arc Test
Pain with passive shoulder flexion
+ Neer’s Test
Pain with passive shoulder flexion (to 90°) + internal rotation
+ Hawkins-Kennedy test
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
First published May 27, 2018; updated Jan 11, 2024 on www.thecalgaryguide.com")
Arachnoid Cysts MRI Findings

Cerebrospinal Fluid (CSF) collection within arachnoid membrane adhesions following trauma, infection, inflammation or surgery (secondary)
Arachnoid cyst (fluid-filled sac) formation within the layers of the arachnoid membrane, outside of brain parenchyma (for full pathogenesis, see Calgary Guide slide Arachnoid Cysts: Pathogenesis and clinical findings)
Use Diffusion Weighted Imaging (DWI) and Fluid-Attenuated Inversion Recovery (FLAIR) to distinguish from other cysts
Isointense to CSF on T1 and T2
Arachnoid cyst contents should appear isointense to CSF on each MR sequence, including diffusion-weighted imaging
Axial T2 MRI Head. Clearly demarcated hyperintense (bright) arachnoid cyst is seen (red arrows)
Axial T1 MRI Head. Clearly demarcated hypointense (dark) arachnoid cyst is seen (red arrows)
FLAIR allows for suppression of free water signal to enhance fluid with ↑ protein concentration
Arachnoid cysts contain CSF-like fluid with very little to no protein
Complete suppression on FLAIR
Cysts are mostly fluid and contains no protein, so it is suppressed and appears darker in FLAIR images.
Axial FLAIR MRI Head. Hypointense cyst is seen on FLAIR (red arrows), showing low protein content in CSF-like fluid inside arachnoid cyst
DWI measures water diffusion in different directions and whether there is restriction on the direction of flow
Fluid within the cyst can flow freely, with no restriction on the direction of movement
Non-restricted diffusion
There is no directional restriction on flow within the cyst (unrestricted diffusion), so it appears dark on DWI.
Axial DWI MRI Head. Hypointense cyst is seen on DWI (red arrows), showing unrestricted diffusion within arachnoid cyst
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published Jan 20, 2024 on www.thecalgaryguide.com")
Arachnoid Cysts Pathogenesis and clinical findings
-like fluid is trapped within the erroneous membrane
Formation of a primary, congenital arachnoid cyst (most common cause)
Head trauma, intracranial hemorrhage, or infection
Inflammation and deposition of cellular matrix Adhesion of the arachnoid membrane
CSF accumulates in the subarachnoid space (space between the arachnoid mater and pia mater)
Formation of a secondary arachnoid cyst (less common)
Cyst remains stable in size and does not expand (most common)
Patients are asymptomatic
Arachnoid cyst is diagnosed incidentally on unrelated neuroimaging (see Calgary Guide slide Arachnoid Cysts: Findings on MRI)
Arachnoid Cyst
Cyst grows in size and expands (rare but more common in children under four years of age)
Cyst exerts pressure on other structures (mass effect)
Suprasellar region
Cyst ruptures into the subdural space (rare)
CSF-like fluid accumulates in the subdural space
Subdural hygroma (collection of non-bloody CSF)
Intracranial hypertension (↑ intracranial pressure)
Generalized symptoms
Middle fossa
Compression and irritation of the temporal cortex
Seizures
Focal symptoms depending on cyst location
Cerebellopontine angle
Compression of vestibulocochlear nerve (Cranial Nerve VIII)
Compression and interruption of cochlear blood supply
Cyst presses on the third ventricle and aqueduct
buildup of CSF in the ventricles
Obstructive hydrocephalus
Cyst presses on the optic chiasm, hypothalamus, and pituitary
Visual impairments and endocrinopathies
Headache (most common)
Vomiting
Nausea
Progressive hearing loss
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published Jan 25, 2024 on www.thecalgaryguide.com")
Avascular Necrosis AVN of the Femoral Head Findings on MRI
 of the Femoral Head: Findings on MRI
Traumatic or atraumatic disruption of blood supply to the proximal femur (for full pathogenesis, see Calgary Guide slide Avascular Necrosis: Pathogenesis and clinical findings)
Ischemia of the femoral head (usually unilateral for traumatic and bilateral for non-traumatic)
Prolonged anoxia (total oxygen deprivation) within the femoral head
Cell death (necrosis) of osteocytes and marrow cells in the femoral head, forming a focal lesion (sequestrum)
Histiocytes and giant cells (immune cells) aggregate around the sequestrum, forming a “reactive zone” around the periphery of the sequestrum
Apoptotic osteocytes in the anoxic reactive zone cannot be phagocytosed leading to dysregulated bone remodeling and osteosclerosis (hardening of bone and ↑ bone mineralization and density due to ↓ resorption and ↑ bone formation)
Femoral head becomes progressively weaker while the mechanical load on it remains the same
Progressive femoral head/subchondral bone collapse Osteoarthritis
Areas with ↑ fluid content appear darker on T1w images
Areas with ↑ fluid content appear brighter on T2w images
Areas with ↑ bone density and ↓ fat content appear darker on T1w images
Areas with ↑ bone density and ↓ fat content appear darker on T2w images
Basic MRI Physiology
Edema and inflammation increases fluid content
Sclerotic areas have ↑ bone density and thus ↓ fat content
Location of Signs
Inflamed reactive zones are darker on T1w images
Inflamed reactive zones are brighter on T2w images
Sclerotic areas are darker on both T1w and T2w images
Authors: George S. Tadros Reviewers: Matthew Hobart, Mao Ding Shahab Marzoughi David Cornell* * MD at time of publication
T1w
Signs on both T1-weighted and T2-weighted images are most commonly seen on the superior anterolateral aspect of the femoral head
Single dark band on T1-weighted MRI
A single band-like crescentic lesion of low signal intensity is seen on T1-weighted MRI images (white arrows). This band represents the edematous reactive zone between the necrotic and normal tissue, and typically extends to the subchondral plate
Double Line Sign on T2-weighted fat-saturated MRI
An outer distal low signal intensity line is seen (white arrows) representing reactive bone sclerosis
Image source: radiologymasterclass
T2w
An inner proximal high signal intensity line is also seen (red arrows) representing vascular and repair tissue at the periphery of the sequestrum
Image source: radiologymasterclass
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published Jan 27, 2024 on www.thecalgaryguide.com")
Bacterial Infections from Transfusion
 recognize bacteria
Immune cells release pyrogens (either bacterial components or signalling molecules) upon recognition
Pyrogens bind to receptors on the hypothalamus
Hypothalamus ↑ body temperature set point
Body generates and conserves heat to reach set point
Immune cells release pro- inflammatory cytokines (messenger protein)
Cytokines activate sympathetic nervous system via hypothalamus
Adrenal glands release stress hormones (epinephrine and norepinephrine) into bloodstream
Stress hormones bind to receptors on cardiomyocytes (heart muscle cells)
↑ Heart contractility
↑ Heart rate
Cytokines circulate throughout the body
Cytokines interact with endothelial cells of the blood vessels
↑ Nitric oxide production
Relaxes smooth muscle cells of blood vessel walls
Vasodilation (↑ blood vessel diameter)
Hypotension
Systemic inflammation
Stimulate nerve endings in muscles
Stimulate nerve endings in gastrointestinal tract’s lining
Blood brain barrier’s integrity is compromised
Nitric oxide reacts with oxygen to form reactive nitrogen species
Tissue damage
Fatigue
Weakness Muscle aches
Nausea and vomiting
Immune and inflammatory cells enter the brain
Authors:
Arzina Jaffer
Kayleigh Yang
Reviewers:
Nimaya De Silva
Raafi Ali
Michelle J. Chen
Yan Yu*
Kareem Jamani*
* MD at time of publication
Inflammation in the brain
Activates pain processing centers in the brain
Headache
Damages neurons and disrupts cell communication
Confusion and altered mental state
Shivering
Fever
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published January 30, 2024 on www.thecalgaryguide.com")
Acute Otitis Externa Complications
: Complications
Acute Otitis Externa (AOE)
Authors: Charmaine Szalay-Anderson Vaneeza Moosa Reviewers: Shayan Hemmati Shahab Marzoughi Ben Campbell Justin Lui* * MD at time of publication
Spread to subcutaneous tissue
Chronic otitis externa (>6 weeks)
Chronic inflammation of the outer ear
Fibroblast activation for collagen and extracellular matrix components production for tissue repair
Excess accumulation of tissue
Ear canal fibrosis (thickening)
Ear canal stenosis (narrowing)
Damage/obstruction to ear canal structures with impaired fluid drainage & pressure buildup
Inflammation of the outer ear
Recurrent or non-resolving acute otitis externa Dissemination of infection
Spread to connective tissue and cartilage
Perichondritis (inflammation of ear cartilage)
Spread of Pseudomonas aeruginosa
in an immunocompromised host or due to antibiotic resistance
Rapid infectious spread through soft tissue to mastoid and/or temporal bone
Malignant (necrotizing) otitis externa *can be life threatening
Inflammation of connective tissue and bony structures
Spread to
tympanic membrane
Myringitis (inflammation of tympanic membrane)
Swelling and thinning of tissue
Tympanic membrane perforation (tear)
Immune reaction with inflammation
Dead white blood cell, bacteria & tissue debris accumulation in the ear canal
Pus formation with purulent otorrhea (discharge from ear)
Localized pus accumulation
Abscess
Ear canal blockage
Periauricular/ pinna (outer ear) cellulitis
Facial cellulitis
Erosion of temporal bone decreasing bony sound conduction
Permanent conductive hearing loss
Direct toxicity of pathogens to surrounding nerves
Cranial nerve (CN) VII (facial) palsy (+/- CN X, XI, XII)
Out-of- proportion primary otalgia (ear pain)
Sensation of fullness in the ear
Temporary hearing loss
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published Dec 4, 2022; updated Feb 7, 2024 on www.thecalgaryguide.com")
Infective endocarditis
 drug use (mostly causes right sided endocarditis)
Bacteria enter the bloodstream (bacteremia)
In subacute cases, valvular abnormality usually present beforehand
In all cases, vegetation forms on affected valve
Immune complex deposit in kidney
Immune complexes cause vasculitis in retinal vessels
Immune complexes deposit subcutaneously
↓ Blood flow to organs perfused by the obstructed arteries
Valve endothelium is damaged
Bacteria adhere to thrombi on the cardiac valve endothelium
Infective Endocarditis
Infection of the thrombus typically produces a vegetation on the flow surface of a valve
Immune complexes (complexes of antibody bound to antigen) form secondary to infection
Generalized immune response
Malaise Chills Fever (> 38°C)
Author:
Sean Spence
George S. Tadros Reviewers:
Yan Yu
Jason Baserman
Danny Guo
Steve Vaughan*
*MD at time of publication
Parts of vegetation embolize systemically, obstructing arteries
Infection destroys infected valve
Smaller emboli block smaller vessels on hands/feet
Microinfarctions
Mitral regurgitation, Aortic stenosis, Aortic insufficiency
(Valve involvement: Mitral > Aortic > Tricuspid)
Vegetation seen on ultrasound/echocardiogram Damage to Glomerulonephritis
glomeruli (Inflammation of glomeruli) Roth’s spots (retinal hemorrhages with pale centers
due to coagulated fibrin)
Osler nodes (tender, raised, red lesions found on the hands and feet)
Organ infarction (tissue death)
Splinter hemorrhages (small red streaks under nails)
Janeway lesions (non-tender, red macules/nodules on palms/soles – only a few millimeters wide)
Regurgitation (blood leaks back through the insufficient valve despite it being closed)
Valve unable to fulfill normal functions (valve insufficiency)
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published Aug 20, 2013, updated Feb 24, 2024 on www.thecalgaryguide.com")
Onychomycosis

Proximal Subungual
White Superficial
Tinea infection (e.g. Tinea Pedis, Corporis, Capitis)
Infection spreads to distal hyponychial space
Dermatophytes colonize local tissue in nail plate and nail bed
Dermatophytes feed on keratinized tissue
General Symptoms (All Subtypes)
Spongiosis (Intercellular edema)
Acanthosis (Thickening of stratum spinosum layer of epidermis)
Hyperkeratosis (Thickening of stratum corneum In effort to rid infection)
Papillomatosis (Projections of dermal papillae)
Secondary damage to nail matrix
Loss of nail
Keratinocytes produce an acute, low-grade inflammatory cytokine response
Onychomycosis
Dermatophytic infection of the nail bed
Inflammation promotes ↑ fluid to tissues for ↑ immune cell delivery
Widespread inflammation thickens parts of the epidermis in efforts to shed the infection
Inflammation and epidermal hyperplasia (↑ growth of cells) influence local dermal papillae (group of cells just beneath the hair follicle) to proliferate and project above the skin
Distal Subungual
Superinfecting bacteria or other fungi proliferate beneath the compromised nail imparting a yellowish appearance
Distal Subungual Subtype
(Thick yellow nails, keratin and debris accumulate distally underneath nail plate)
Dermatophytes invade the proximal end of the nail plate
Dermatophytes penetrate through the cuticle to the newly forming nail plate moving distally
Proximal Subungual Subtype (Whitish discolouration of nail plate that begins proximally and moves distally, indicative of immunosuppression)
Fungi predominantly invade various areas of the superficial nail plate layers eventually joining together
White Superficial Subtype (Chalky white scale that spreads slowly beneath nail plate, well-defined “white islands” that coalesce as disease progresses)
The entirety of the nail plate is infected by the dermatophytes
Widespread inflammation thickens the nail plate as well as beneath the nail (subungual hyperkeratosis) in efforts to shed the infection
Total Dystrophic Subtype (End-stage nail disease, entire nail becomes thick and dystrophic)
Local spread of infection Dermatophytes spread causing cracks in the skin deeper into toe
Abnormal keratinization in hyponychium
Keratin accumulates between nail plate and hyponychium
Fissure (splits in the skin)
Bacteria enters lymphatics and bloodstream
Cellulitis Sepsis
Onycholysis (nail plate separates from nail bed)
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published Mar 13, 2024 on www.thecalgaryguide.com
Dermatophyte Onychomycosis: Pathogenesis and clinical findings
Authors: Holly Zahary Loreman Reviewers: Mina Youakim Elise Hansen Shahab Marzoughi Jodi Hardin* * MD at time of publication
Host Risk Factors
Environmental Risk Factors
Immuno- compromised
↓ Immune response to infection
Older age
Peripheral vascular disease
Reduced blood circulation
Diabetes
Pre-existing nail dystrophy
Previous nail trauma
Integrity of nail unit is compromised
Micro- traumatic pressure on nail
Dark, warm shoe environment
Optimal conditions for fugal growth
Exposure to tinea pedis or onychomycosis
Direct spread of infection to nail unit
High blood sugar favoring infection
Dermatophytes invade corneocytes on stratum corneum, the uppermost non-living layer of keratinized skin
Compromise/breaking of hyponychial seal or cuticle (connection between hyponychium and nail plate)
Proximal Subungual
White Superficial
Distal Subungual
Superinfecting bacteria or other fungi proliferate beneath the compromised nail imparting a yellowish appearance
Distal Subungual Subtype
(Thick yellow nails, keratin and debris accumulate distally underneath nail plate)
Infection spreads to distal hyponychial space
Dermatophytes colonize local tissue in nail plate and nail bed
Dermatophytes feed on keratinized tissue
Keratinocytes produce an acute, low-grade inflammatory cytokine response
Onychomycosis
Dermatophytic infection of the nail bed
Inflammation promotes ↑ fluid to tissues for ↑ immune cell delivery
Widespread inflammation thickens parts of the epidermis in efforts to shed the infection
Inflammation and epidermal hyperplasia (↑ growth of cells) influence local dermal papillae (group of cells just beneath the hair follicle) to proliferate and project above the skin
General Symptoms (All Subtypes)
Spongiosis (Intercellular edema)
Acanthosis (Thickening of stratum spinosum layer of epidermis)
Hyperkeratosis (Thickening of stratum corneum In effort to rid infection)
Papillomatosis (Projections of dermal papillae)
Secondary damage to nail matrix
Loss of nail
Tinea infection (e.g. Tinea Pedis, Corporis, Capitis)
Dermatophytes invade the proximal end of the nail plate
Dermatophytes penetrate through the cuticle to the newly forming nail plate moving distally
Proximal Subungual Subtype (Whitish discolouration of nail plate that begins proximally and moves distally, indicative of immunosuppression)
Fungi predominantly invade various areas of the superficial nail plate layers eventually joining together
White Superficial Subtype (Chalky white scale that spreads slowly beneath nail plate, well-defined “white islands” that coalesce as disease progresses)
The entirety of the nail plate is infected by the dermatophytes
Widespread inflammation thickens the nail plate as well as beneath the nail (subungual hyperkeratosis) in efforts to shed the infection
Total Dystrophic Subtype (End-stage nail disease, entire nail becomes thick and dystrophic)
Dermatophytes spread deeper into toe
Bacteria enters lymphatics and bloodstream
Abnormal keratinization in hyponychium
Keratin accumulates between nail plate and hyponychium
Local spread of infection causing cracks in the skin
Fissure (splits in the skin)
Cellulitis Sepsis
Onycholysis (nail plate separates from nail bed)
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published MONTH, DAY, YEAR on www.thecalgaryguide.com
Dermatophyte Onychomycosis: Pathogenesis and clinical findings
Authors: Holly Zahary Loreman Reviewers: Mina Youakim Elise Hansen Shahab Marzoughi Jodi Hardin* * MD at time of publication
Host Risk Factors
Environmental Risk Factors
Immuno- Older compromised age
↓ Immune response to infection
Peripheral vascular disease
Reduced blood circulation
Diabetes
Pre-existing nail dystrophy
Previous nail trauma
Integrity of nail unit is compromised
Micro- traumatic pressure on nail
Dark, warm shoe environment
Optimal conditions for fugal growth
Exposure to tinea pedis or onychomycosis
Direct spread of infection to nail unit
High blood sugar favoring infection
Dermatophytes invade corneocytes on stratum corneum, the uppermost non-living layer of keratinized skin
Compromise/breaking of hyponychial seal or cuticle (connection between hyponychium and nail plate)
Tinea infection (e.g. Tinea Pedis, Corporis, Capitis)
Infection spreads to distal hyponychial space
Dermatophytes colonize local tissue in nail plate and nail bed
Dermatophytes feed on keratinized tissue
Keratinocytes produce an acute, low-grade inflammatory cytokine response
Onychomycosis
Dermatophytic infection of the nail bed
General Symptoms (All Subtypes)
Spongiosis (Intercellular edema)
Acanthosis (Thickening of stratum spinosum layer of epidermis)
Papillomatosis
(Projections of dermal papillae)
Hyperkeratosis (Thickening of stratum corneum In effort to rid infection)
Secondary damage to nail matrix
Loss of nail
Proximal Subungual
White Superficial
Distal Subungual
Distal Subungual Subtype
(Thick yellow nails, keratin and debris accumulate distally underneath nail plate)
Proximal Subungual Subtype (Whitish discolouration of nail plate that begins proximally and moves distally, indicative of immunosuppression)
White Superficial Subtype (Chalky white scale that spreads slowly beneath nail plate, well-defined “white islands” that coalesce as disease progresses)
Total Dystrophic Subtype (End-stage nail disease, entire nail becomes thick and dystrophic)
Local spread of infection causing cracks in the skin
Dermatophytes spread deeper into toe
Abnormal keratinization in hyponychium
Keratin accumulates between nail plate and hyponychium
Onycholysis (nail plate separates from nail bed)
Fissure (splits in the skin)
Bacteria enters lymphatics and bloodstream
Cellulitis Sepsis
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Legend:
Published MONTH, DAY, YEAR on www.thecalgaryguide.com
Dermatophyte Onychomycosis: Pathogenesis and clinical findings
Authors: Holly Zahary Loreman Reviewers: Mina Youakim Elise Hansen Shahab Marzoughi Jodi Hardin* * MD at time of publication
Host Risk Factors
Environmental Risk Factors
Immuno- Older compromised age
↓ Immune response to infection
Peripheral vascular disease
Reduced blood circulation
Diabetes
Pre-existing nail dystrophy
Previous nail trauma
Integrity of nail unit is compromised
Micro- traumatic pressure on nail
Dark, warm shoe environment
Optimal conditions for fugal growth
Exposure to tinea pedis or onychomycosis
Direct spread of infection to nail unit
High blood sugar favoring infection
Dermatophytes invade corneocytes on stratum corneum, the uppermost non-living layer of keratinized skin
Compromise/breaking of hyponychial seal or cuticle (connection between hyponychium and nail plate)
Tinea infection (e.g. Tinea Pedis, Corporis, Capitis)
Infection spreads to distal hyponychial space
Dermatophytes colonize local tissue in nail plate and nail bed
Dermatophytes feed on keratinized tissue
Keratinocytes produce an acute, low-grade inflammatory cytokine response
Onychomycosis
Dermatophytic infection of the nail bed
Proximal Subungual
White Superficial
General Symptoms (All Subtypes)
Spongiosis (Intercellular edema)
Acanthosis (Thickening of stratum spinosum layer of epidermis)
Papillomatosis
(Projections of dermal papillae)
Hyperkeratosis (Thickening of stratum corneum In effort to rid infection)
Secondary damage to nail matrix
Loss of nail
Distal Subungual
Distal Subungual Subtype
(Thick yellow nails, keratin and debris accumulate distally underneath nail plate)
Proximal Subungual Subtype (Whitish discolouration of nail plate that begins proximally and moves distally, indicative of immunosuppression)
White Superficial Subtype (Chalky white scale that spreads slowly beneath nail plate, well- defined “white islands” that coalesce as disease progresses)
Total Dystrophic Subtype (End-stage nail disease, entire nail becomes thick and dystrophic)
Local spread of infection causing cracks in the skin
Dermatophytes spread deeper into toe
Abnormal keratinization in hyponychium
Keratin accumulates between nail plate and hyponychium
Onycholysis (nail plate separates from nail bed)
Bacteria enters lymphatics and bloodstream
Fissure (splits in the skin)
Cellulitis
Sepsis
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Legend:
Published MONTH, DAY, YEAR on www.thecalgaryguide.com
Dermatophyte Onychomycosis (Tinea Unguium): Pathogenesis, clinical findings,
Authors: Holly Zahary Loreman Reviewers: Elise Hansen Name Name* * MD at time of publication
and complications
Host Risk Factors
Environmental Risk Factors
Immuno- compromised
↓ immune response to infection
Older age
Peripheral vascular disease
Diabetes
Pre-existing nail dystrophy
Previous Nail Trauma
Integrity of nail unit is compromised
Micro-traumatic pressure on nail
Dark, warm shoe environment
Optimal conditions for fugal growth
Exposure to tinea pedis or onychomycosis
Direct spread of infection to nail unit
Reduced blood circulation
High blood sugar, favoring infection
Tinea pedis infection (see ‘Tinea Capitis, Tinea Corporis, and Tinea Pedis’)
Infection spreads to distal hyponychial space Dermatophytes colonize local tissue in nail plate and nail bed Dermatophytes feed on keratinized tissue
Proximal Subungual
White Superficial
Dermatophytes invade corneocytes on stratum corneum, the uppermost non-living layer of keratinized skin
Compromise/breaking of hyponychial seal or cuticle (connection between hyponychium and nail plate)
Keratinocytes produce an acute, low-grade inflammatory cytokine response
Onychomycosis (Tinea Unguium)
(dermatophytic infection of the nail bed)
Distal Subungual
General Symptoms (All Subtypes)
Spongiosis
Intercellular edema
Acanthosis
Thickening of stratum spinosum layer of epidermis
Papillomatosis
Projections of dermal papillae
Hyperkeratosis
Thickening of stratum corneum In effort to rid infection
Secondary damage to nail matrix
Loss of nail
Distal Subungual Subtype
Thick yellow nails, keratin and debris accumulate distally underneath nail plate
Proximal Subungual Subtype
Whitish discolouration of nail plate that begins proximally and moves distally, indicative of immunosuppression
White Superficial Subtype
Chalky white scale that spreads slowly beneath nail plate, well-defined “white islands” that coalesce as disease progresses
Total Dystrophic Subtype End-stage nail disease, entire nail becomes thick and dystrophic
Local spread of infection causing cracks in the skin
Dermatophytes spread deeper into toe
Abnormal keratinization in hyponychium
Keratin accumulates between nail plate and hyponychium
Onycholysis (nail plate separates from nail bed)
Tissue Damage
Cellulitis
Sepsis
Bacteria enters lymphatics and bloodstream
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Legend:
Published MONTH, DAY, YEAR on www.thecalgaryguide.com")
Neonatal Necrotising Enterocolitis in Premature Neonates
 in Premature Neonates:
Pathogenesis and clinical findings
Prematurity risk factors ↓ Intestinal motility
↑ Intestinal stasis allows bacteria more time to proliferate
Bacterial overgrowth in gut
↓ Goblet cells in intestinal epithelium
↓ Intestinal mucus layer production leads to impaired mechanical defense against pathogenic bacteria
Immature tight junctions in intestinal epithelium
↑ Permeability of intestinal epithelial barrier
↑ Toll-like receptor 4 (TLR4) expression on intestinal epithelial cells
Aberrant bacterial colonization of gut
Impaired gut barrier allows for ↑ bacterial translocation across intestinal epithelium
TLR4 on intestinal mesentery endothelial cells bind lipopolysaccharides (LPS) on Gram-negative gut bacteria
Immune cells release proinflammatory mediators (TNF, IL-12, IL-18)
Cytokines mediate ↑ enterocyte apoptosis (including enteric stem cells) and ↓ enterocyte proliferation
Intestinal mucosa healing is impaired, leading to local inflammation & injury
TLR4 on intestinal epithelial cells binds LPS from Gram-negative gut bacteria
Authors: Rachel Bethune Naima Riaz Reviewers: Nicola Adderley Michelle J. Chen Kamran Yusuf* Jean Mah* * MD at time of publication
Endothelial nitric oxide synthase expression is reduced
Vasoconstriction from ↓ NO reduces blood flow to intestines
Prolonged ↓ in O2 perfusion results in irreversible intestinal mucosal cell death (necrosis)
Gas escapes into abdominal cavity
Leakage of intestinal contents irritates parietal peritoneum
Bacteria enter bloodstream
Pneumo- peritoneum
Abdominal distention
Peritonitis
Sepsis
Blood from tissue damage mixes with intestinal contents
Bloody stool
Intestinal sensory neurons detect damage and send signals to medullary vomiting centre
Bilious vomiting
Damaged intestinal cells are unable to absorb nutrients
Short gut syndrome
Persistent intestinal mucosal injury creates penetrating lesions through intestinal wall
Intestinal perforation
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published May 6, 2019; updated Mar 21, 2024 on www.thecalgaryguide.com")
Lichen Sclerosus
: Pathogenesis and clinical findings
Authors: Mina Youakim Reviewers: Elise Hansen Sunawer Aujla Shahab Marzoughi Jori Hardin* * MD at time of publication
Histamine receptor binding stimulates sensory nerve endings
Pruritus (itching)
Unknown triggers
Genetic predisposition (HLA-DQ7 and HLA-DR12)
Chronic inflammation (i.e. chronic infection, chronic toxin exposure) and trauma
Medications (e.g. carbamazepine, pembrolizumab, nivolumab, ipilimumab)
↑ Activation of CD4+ and CD8+ T cells released from macrophages (white blood cell) in the perineum and genital skin tissue infiltrate into the dermal-epidermal junction
T-cells proliferate in a horizontal linear formation Pro-inflammatory response activation
Fibroblasts (contributes to the formation of connective tissue) proliferate and persist producing altered collagen under the epidermis
Collagen deposits and hyalinizes (transforms into acellular translucent material) beneath the epidermal layer
Sclerotic plaques (localized areas of thickened skin)
T cells release of pro-inflammatory cytokines (interleukins and transforming growth factor β)
↑ Oxidative stress and cell damage
Progressive basal layer degeneration thins the overall skin thickness
Mast cells respond to increased need for immune cell flow to area
Nitric oxide is released when histamine binds to vascular receptors
Localized area appears red
Erythema (reddening of the skin)
Erosion/ulceration
Skin fissures (linear cleavage of skin)
Localized histamine release
Nitric oxide induces localized vasodilation (↑ blood flow)
Normal Skin
Lichen Sclerosus
Epidermal layer
Basal layer
Dermal-Epidermal Junction Dermal layer
Hypopigmented patches (localized, pale areas of skin)
Epidermal atrophy (crinkling paper-type skin appearance)
Thinned skin is weakened and prone to physical stress or trauma
↑ fluid in the extracellular space due to capillary leakage from ↑ blood flow
Superficial dermal edema (swelling)
Fibrotic tissue deposition following healing of damaged tissue
Atrophic Epidermal layer
Hyalinized collagen deposits
Basal layer degeneration Band of T-cell infiltrate
Dermal layer
Scarring
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published Mar 25, 2024 on www.thecalgaryguide.com
Lichen Sclerosus (genital manifestation): Pathogenesis and clinical findings
Authors: Mina Youakim Reviewers: Elise Hansen Sunawer Aujla Shahab Marzoughi Jori Hardin* * MD at time of publication
Histamine receptor binding stimulates sensory nerve endings
Pruritus (itching)
Unknown triggers
Genetic predisposition (HLA-DQ7 and HLA-DR12)
Chronic inflammation (i.e. chronic infection, chronic toxin exposure) and trauma
Medications (e.g. carbamazepine, pembrolizumab, nivolumab, ipilimumab)
↑ Activation of CD4+ and CD8+ T cells released from macrophages (white blood cell) in the perineum and genital skin tissue infiltrate into the dermal-epidermal junction
T-cells proliferate in a horizontal linear formation Pro-inflammatory response activation
Fibroblasts (contributes to the formation of connective tissue) proliferate and persist producing altered collagen under the epidermis
Collagen deposits and hyalinizes (transforms into acellular translucent material) beneath the epidermal layer
Sclerotic plaques (localized areas of thickened skin)
T cells release of pro-inflammatory cytokines (interleukins and transforming growth factor β)
↑ Oxidative stress and cell damage
Mast cells respond to increased need for immune cell flow to area
Nitric oxide is released when histamine binds to vascular receptors
↑ fluid in the extracellular space due to capillary leakage from ↑ blood flow
Superficial dermal edema (swelling)
Epidermal atrophy (crinkling paper- type skin appearance)
Thinned skin is weakened and prone to physical stress or trauma
Localized histamine release
Nitric oxide induces localized vasodilation (↑ blood flow)
Localized area appears red
Erythema (reddening of the skin)
Erosion/ulceration
Skin fissures (linear cleavage of skin)
Normal Skin
Lichen Sclerosus
Epidermal layer
Basal layer
Dermal-Epidermal Junction Dermal layer
Atrophic Epidermal layer
Hyalinized collagen deposits
Basal layer degeneration Band of T-cell infiltrate
Dermal layer
Progressive basal layer degeneration thins the overall skin thickness
Hypopigmented patches (localized, pale areas of skin)
Fibrotic tissue deposition following healing of damaged tissue
Scarring
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published MONTH, DAY, YEAR on www.thecalgaryguide.com
Chronic inflammation (i.e. chronic
Reviewers:
Dermal layer
Unknown triggers
Genetic predisposition (HLA-DQ7 and HLA-DR12)
infection, chronic toxin exposure) and trauma
Medications (e.g. carbamazepine, pembrolizumab, nivolumab, ipilimumab)
Elise Hansen Sunawer Aujla Shahab Marzoughi Jori Hardin* * MD at time of publication
↑ Activation of CD4+ and CD8+ T cells released from macrophages (white blood cell) in the perineum and genital skin tissue infiltrate into the dermal-epidermal junction
T-cells proliferate in a horizontal linear formation
Pro-inflammatory response activation
↑ Expression of MicroRNA-155 (enhances pro-inflammatory response and ↓ expression of tumor suppression genes)
Fibroblasts (contributes to the formation of connective tissue) proliferate and persist producing altered collagen
Collagen deposits and hyalinizes (transforms into acellular translucent material) beneath the epidermal layer
Sclerotic plaques (localized areas of thickened skin)
T cells release of pro-inflammatory cytokines (interleukins and transforming growth factor β)
↑ Oxidative stress and cell damage
Mast cells respond to increased need for immune cell flow to area
Nitric oxide is released when histamine binds to vascular receptors
↑ fluid in the extracellular space due to capillary leakage from ↑ blood flow
Superficial dermal edema (swelling)
Epidermal atrophy (crinkling paper- type skin appearance)
Thinned skin is weakened and prone to physical stress or trauma
Lichen Sclerosus
Localized histamine release
Nitric oxide induces localized vasodilation (↑ blood flow)
Localized area appears red
Erythema (reddening of the skin)
Erosion/ulceration
Skin fissures (linear cleavage of skin)
Atrophic Epidermal layer
Hyalinized collagen deposits
Basal layer degeneration Band of T-cell infiltrate
Histamine receptor binding stimulates sensory nerve endings
Pruritus (itching)
Normal Skin
Progressive basal layer degeneration thins the overall skin thickness
Hypopigmented patches (localized, pale areas of skin)
Epidermal layer
Basal layer
Dermal-Epidermal Junction
Fibrotic tissue deposition following healing of damaged tissue
Scarring
Chronic inflammation (i.e. chronic
Reviewers:
Dermal layer
Unknown triggers
Genetic predisposition (HLA-DQ7 and HLA-DR12)
infection, chronic toxin exposure) and trauma
Medications (e.g. carbamazepine, pembrolizumab, nivolumab, ipilimumab)
Elise Hansen Sunawer Aujla Shahab Marzoughi Jori Hardin* * MD at time of publication
↑ Activation of CD4+ and CD8+ T cells released from macrophages in the perineum and genital skin tissue infiltrate into the dermal-epidermal junction
T-cells proliferate in a horizontal linear formation Pro-inflammatory response activation
↑ Expression of MicroRNA-155 (short segment of RNA which enhances pro- inflammatory response and ↓ expression of tumor suppression genes)
Fibroblasts proliferate and persist producing altered collagen
Collagen deposits and hyalinizes (transforms into acellular translucent material) beneath the epidermal layer
Sclerotic plaques (localized areas of thickened skin)
T cells release of pro-inflammatory cytokines (interleukins and transforming growth factor β)
↑ Oxidative stress and cell damage
Progressive basal layer degeneration thins the overall skin thickness
Hypopigmented patches (localized, pale areas of skin)
Epidermal layer
Basal layer
Dermal-Epidermal Junction
Mast cells respond to increased need for immune cell flow to area
Nitric oxide is released when histamine binds to vascular receptors
Superficial dermal edema (swelling)
Epidermal atrophy (crinkling paper- type skin appearance)
Localized histamine release
Nitric oxide induces localized vasodilation (↑ blood flow)
Localized area appears red
Erythema (reddening of the skin)
Histamine receptor binding stimulates sensory nerve endings
Pruritus (itching)
Normal Skin
Lichen Sclerosus
Skin fissures (linear cleavage of skin)
Erosion/ulceration
Fibrotic tissue deposition following healing of damaged tissue
Scarring
Atrophic Epidermal layer
Hyalinized collagen deposits
Basal layer degeneration Band of T-cell infiltrate
Reviewers:
Dermal layer
Unknown triggers
Genetic predisposition (HLA-DQ7 and HLA-DR12)
Chronic inflammation (i.e. chronic infection, chronic toxin exposure) and trauma
Medications (e.g. carbamazepine, pembrolizumab, nivolumab, ipilimumab)
Elise Hansen Sunawer Aujla Shahab Marzoughi Jori Hardin* * MD at time of publication
↑ Activation and infiltration of CD4+ and CD8+ T cells into the dermal-epidermal junction
T-cells proliferate in a band (horizontal linear) formation Pro-inflammatory response activation
↑ Expression of MicroRNA-155 (short segment of RNA which enhances pro-inflammatory response and ↓ expression of tumor suppression genes)
Fibroblasts proliferate and persist producing altered collagen
Collagen deposits and hyalinizes (transforms into acellular translucent material) beneath the epidermal layer
Sclerotic plaques (localized areas of thickened skin)
T cells release of pro-inflammatory cytokines (interleukins and transforming growth factor β)
↑ Oxidative stress and cell damage
Progressive basal layer degeneration thins the epidermis
Hypopigmented patches (localized, pale areas of skin)
Epidermal layer
Basal layer
Dermal-Epidermal Junction
Mast cells respond to increased need for immune cell flow to area
Nitric oxide is released when histamine binds to vascular receptors
Superficial dermal edema (swelling)
Epidermal atrophy (crinkling paper- type skin appearance)
Localized histamine release
Histamine receptor binding stimulates sensory nerve endings
Pruritus (itching)
Skin fissures (linear cleavage of skin)
Nitric oxide induces localized vasodilation (↑ blood flow)
Localized area appears red
Erythema (reddening of the skin)
Scarring
Atrophic Epidermal layer
Hyalinized collagen deposits
Basal layer degeneration Band of T-cell infiltrate
Erosion/ulceration
Fibrotic tissue deposition following healing of damaged tissue
Normal Skin
Lichen Sclerosus
Chronic inflammation (i.e. chronic
Elise Hansen
Unknown triggers
Genetic predisposition infection, chronic toxin exposure) Medications (e.g. carbamazepine, (HLA-DQ7 and HLA-DR12) and trauma pembrolizumab, nivolumab, ipilimumab)
↑ Activation and infiltration of CD4+ and CD8+ T cells into the dermal-epidermal junction
Sunawer Aujla Shahab Marzoughi Name Name* * MD at time of publication
T-cells proliferate in a band (horizontal linear) formation
Pro-inflammatory response activation (increase in pro-inflammatory cytokines such as Interleukins 1-alpha and 1-beta)
↑ Expression of MicroRNA-155 (short segment of RNA which enhances pro-inflammatory response and ↓ expression of tumor suppression genes)
Fibroblasts proliferate and persist producing altered collagen
T cells release of pro-inflammatory cytokines (interleukins and transforming growth factor β)
↑ Oxidative stress and cell damage
Progressive basal layer degeneration thins the epidermis
Hypopigmented patches (localized, pale areas of skin)
Histamine receptor binding stimulates sensory nerve endings
Localized area appears red
Localized histamine release
Localized vasodilation (↑ blood flow)
Superficial dermal edema (swelling)
Pruritus
Erythema
Collagen deposits and hyalinizes (transforms into acellular translucent material) beneath the epidermal layer
Normal Skin
Sclerotic plaques (localized areas of thickened skin)
Epidermal layer
Basal layer
Dermal-Epidermal
Junction Dermal layer
Skin fissures (linear cleavage of skin)
Bleeding
Erosion/Ulceration
Epidermal atrophy (crinkling paper- type skin appearance)
Lichen Sclerosus
Atrophic Epidermal layer
Hyalinized collagen deposits
Basal layer degeneration
Band of T-cell infiltrate
Dermal layer
Fibrotic tissue deposition following healing of damaged tissue
Scarring
Lichen Sclerosus (genital manifestation): Pathogenesis and clinical findings
Authors: Mina Youakim Reviewers: Elise Hansen Sunawer Aujla Shahab Marzoughi Name Name* * MD at time of publication
Pruritus
Histamine receptor binding stimulates sensory nerve endings
Localized area appears red
Erythema
Unknown triggers
Genetic predisposition (HLA-DQ7 and HLA-DR12)
Chronic inflammation and trauma
Medications (e.g. carbamazepine, pembrolizumab, nivolumab, ipilimumab)
↑ Activation and infiltration of CD4+ and CD8+ T cells into the dermal-epidermal junction
T-cells proliferate in a band formation Pro-inflammatory response activation
↑ Expression of MicroRNA-155 (short segment of RNA which enhances pro-inflammatory response and ↓ expression of tumor suppression genes)
Fibroblasts proliferate and persist producing altered collagen
T cells release of pro-inflammatory cytokines (interleukins and transforming growth factor β)
Localized histamine release
Localized vasodilation (↑ blood flow)
Superficial dermal edema (swelling)
Collagen deposits and hyalinizes beneath the atrophic epidermal layer
Hypopigmented patches (localized, pale areas of skin)
↑ Oxidative stress and cell damage
Progressive basal layer degeneration thins the epidermis
Skin fissures
Lichen Sclerosus
Epidermal atrophy (crinkling paper- type skin appearance)
Sclerotic plaques (localized areas of thickened skin)
Bleeding Scarring
Erosion/Ulceration
Normal Skin
Epidermal layer
Basal layer
Dermal-Epidermal
Junction Dermal layer
Atrophic Epidermal layer
Hyalinized collagen deposits
Basal layer degeneration
Band of T-cell infiltrate
Dermal layer
Legend:
Published MONTH, DAY, YEAR on www.thecalgaryguide.com
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications")
Post-Renal Acute Kidney Injury AKI
: Pathogenesis and clinical findings
Blood clot or cellular debris
Foreign body
Neurogenic bladder
Obstruction intrinsic to urine excretion system
Nephrolithiasis (kidney stones)
Anatomical defect(s)
Intra- abdominal adhesions
Retroperitoneal fibrosis (scar-like tissue)
Benign or malignant masses
Prostate cancer
Benign prostatic hyperplasia
↓ Urine flow across point of obstruction Obstruction extrinsic to urine excretion system
Urine buildup distends urine collecting system (hydronephrosis)
Compression of renal vasculature due to mass effect
↑ Volume/pressure proximal to obstruction
↑ Intratubular pressure
↓ Pressure gradient between glomerular afferent arteriole and Bowman’s space
Casts occlude tubules
↓ Filtration of plasma into nephrons
↓ Glomerular Filtration Rate (GFR)
Obstruction is relieved ↓ Intratubular pressure Rapid GFR ↑
Rapid diuresis of fluid and electrolytes
Dilated pelvicalyceal system on ultrasound
↓ Urine output
↓ Venous drainage
and arterial supply
Local ischemia and inflammation of kidney
Impaired resorption, excretion, and fine tuning by tubules
Acute Tubular Necrosis (ATN) with granular casts
↓ Urine output
↓ Clearance of free water and solutes
↑ Intravascular volume
↑ Serum creatinine
↓ Medullary solute concentration, ischemia, ↓ response of collecting ducts to antidiuretic hormone
Lasts > 24hrs
Post-obstructive diuresis causes hypovolemia and electrolyte derangements
Authors: David Campbell, Matthew Hobart Reviewers: Raafi Ali, Luiza Radu Huneza Nadeem, Marissa Zhang, Julian Midgley* * MD at time of publication
Resolves < 24hrs in euvolemia
Physiologic post- obstructive diuresis
↑ Na+ and Cl- delivery to distal convoluted tubule is sensed by macula densa
Secretion of adenosine by macula densa
Adenosine constricts afferent arterioles
↓ GFR
↓ Renal clearance of drugs and waste products
↑ Venous hydrostatic pressure
↑ Volume in arterial system overwhelms pressure regulation mechanisms
Hypertension
Fluid extravasation from veins and capillaries
Generalized Edema
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published Mar 25, 2024 on www.thecalgaryguide.com")
Irritant Contact Dermatitis Pathogenesis and Clinical Findings

Hyperkeratotic skin is less amenable to skin stretching and pressure
Skin fissures (cracks in the skin)
Irritant agents
(abrasives, cleaning solution, oxidative & reducing agents, dust, soils, water)
Acute exposure triggers inflammatory response
Keratinocytes undergo cytotoxic damage with ↑ neutrophil & cytokine release
Common occupational exposures (housekeeping, cleaning, catering, medical/dental, construction)
Risk factors
(atopy, fair skin, low temperature, low humidity)
Stimulation of local nociceptors (free nerve endings extend into the mid epidermis)
Perivascular (around the blood vessel) inflammation causes histamine release from mast cells
Damaged keratinocytes are destroyed via
apoptosis (programmed cell death)
Epidermal Necrosis (death of epidermal tissue)
Shedding of necrotic tissue
Ulceration (deep open wound on skin)
Burning
pain (uncomfortable stinging sensation)
Pruritus (itching)
Histamine causes local blood vessel dilation and ↑ blood flow to the area of skin affected
Erythema (area appears red from ↑ blood flow)
Burning & Itching Spongiosis
Neutrophils Neutrophils
Histamine causes local blood vessel walls to have
↑ permeability, thereby ↑ leakage of fluid
Spongiosis
(↑ fluid between keratinocytes in the epidermis)
Fluid continues to build up from ongoing inflammation
Vesiculation (fluid collections in the epidermis)
Long-term skin scratching causes chronic irritation which eventually hardens the skin
Lichenification
(thick, hardened patches of skin)
↑ Overall keratin production
Hyperkeratosis (thickening of the outermost skin layer)
Further fluid buildup bursts vesicles leaving behind erosions and dried crust on the epidermis
Crust (scaling over the skin)
Lichenification
Erosions (open sore on skin)
Ulcer
Epidermis
Perivascular Inflammation
Hyperkeratosis
Dermis
Dermal-epidermal junction
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published Oct 19, 2016; updated Mar 30, 2024 on www.thecalgaryguide.com")
Deep Partial Thickness Burns Pathogenesis and Clinical Findings

Ionizing radiation gets into contact with DNA
Damage to keratinocytes
Fire (Flash fire or direct contact with flame)
Contact (Hot solid objects)
Scalding (Hot liquid via immersion, spill or splash)
Chemical (Strong acid, alkali or irritant gas)
Electrical (Contact with exposed electrical wiring/appliances)
Author and Illustrator: Amanda Eslinger Maharshi Gandhi Reviewers:
Alexander Arnold
Shahab Marzoughi
Duncan Nickerson*
* MD at time of publication
Direct transfer of heat energy
Coagulation necrosis (cell death due to ischemia) is induced
Deep Partial Thickness Burn
Injury to the epidermal layer and both the papillary and a portion of the reticular layer of the dermis
Transfer of heat energy & direct injury to cellular membranes
Epidermis
Papillary dermis Reticular dermis
Sub- cutaneous Tissue
↑ vascular permeability due to damage
Fluid leak results in edema between dermal & epidermal layer
Blisters (a bubble on the skin containing fluid)
Thin epidermal layer forming fluid-filled vesicle breaks open
Cutaneous capillary bed is destroyed
Blood flow to injured area is compromised
Pressing on the skin doesn’t easily force blood cells out of the area
Non-blanchable skin
Vasodilation in fascia (a thin casing of connective tissue) underlying subcutaneous tissue
Inflammatory mediators such as cytokines and prostaglandins activate fibroblasts
Collagen deposition and subsequently induration (thickening of skin)
Some healthy dermal appendages surrounded by islands of undamaged epithelial cells
Possible spontaneous re-epithelialization in 2-9 weeks
During re-epithelization of burns there can be excessive deposition of collagen and other extracellular matrix components
Thick raised scars in dermis due to excess collagen deposition
Somatosensory structures (nociceptors, thermoreceptors, and mechanoreceptors) are completely injured within the dermis
Analgesia of impacted area (the inability to feel pain)
Ongoing Inflammation and dilated blood vessels
Red Induration (Thickened skin appearing red)
Compromised blood flow and more extensive tissue damage
White Induration (Thickened skin appearing white)
Moist Wound
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published Dec 2, 2013, updated Apr 20, 2024 on www.thecalgaryguide.com")
Costochondritis

Infection spreads through blood to ribcage and chest wall
Age-related degeneration of cartilage within ribcage joints
Immune cells present in joints secrete proinflammatory cytokines
Cytokines sensitize nociceptors (pain receptors) in joints
Repetitive trauma or strain to chest wall muscles (e.g. chronic cough, chest wall injury, overexertion of chest wall muscles)
Excessive stretch and tearing of ribcage cartilage and ligaments
Autoimmune disease (e.g. rheumatoid arthritis)
Antigen presenting cells display self-antigens to CD4+ T cells
Mechanical stimuli (e.g. Damaged cells release cytokines Pathogens activate immune cells at site of stretch) activate nociceptors that recruit immune cells inflammation
Costochondritis
Benign inflammation of the ribcage (costal) cartilage, particularly at the costosternal junctions (connection between sternum and cartilage) and the costochondral junctions (connection between the cartilage and rib)
Authors:
Michelle J. Chen Reviewers:
Raafi Ali
Yan Yu*
Gerhard Kiefer*
* MD at time of publication
Cartilage, ligament, or muscle injury
Presence of foreign pathogen in costal cartilage
Injury or pathogen activates inflammatory cascade in the ribcage cartilage
Immune cells proliferate so that they exceed the available space in the cartilage matrix of the ribcage
Cartilage swelling compresses intercostal nerves
Compression acts as a mechanical stimulus to activate nociceptors
Sharp, localized pain reproducible upon palpation over ribcage
Immune cells secrete cytokines
Cytokines activate nociceptors so that they become more sensitive to mechanical stimuli
Muscle or ligament stretch from normal use activates sensitized nociceptors
Pain increases with inspiration or cough
Natural resolution of inflammation over time
Macrophages phagocytose apoptotic cells (immune and cartilage cells) and cellular debris
Immune cells release factors that degrade proinflammatory cytokines
Fewer cytokines reduce immune cell recruitment into costal cartilage
Pain usually self-resolves
Immune cells release lipid molecules that bind to the same cells (autocrine signaling) to inhibit further production of inflammatory cytokines and chemokines
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published Apr 29, 2024 on www.thecalgaryguide.com")
Acute Otitis Media Complications
 accumulates in mastoid air spaces
Untreated middle ear infection allows further bacterial proliferation
Persistent effusion
causes ion channel changes in inner ear
Composition of endolymph and perilymph in inner ear changes
Vestibular/Labyrinth dysfunction
Feelings of Vertigo imbalance
Purulent discharge from middle ear through perforation
Otorrhea (ear discharge)
↑ Pressure in middle ear
Pressure stretches tympanic membrane
Cytokines reach hypothalamus through the bloodstream
Hypothalamus responds to stimulation and ↑ thermoregulatory set-point
High-grade fever
Infection spreads into bloodstream
Sepsis
Cytokines alter metabolism pathways of neurotransmitters in the brain
Cerebral cortex dysfunction
Infection spreads to cranium
Intracranial complications (e.g., meningitis, brain abscess, thrombus)
Author: Jody Platt Stephanie de Waal Reviewers: Yan Yu Elizabeth De Klerk William Kim Annie Pham Michelle J. Chen Danielle Nelson* * MD at time of publication
Helper T cells and macrophages release inflammatory cytokines into the bloodstream
Perforation of tympanic membrane
↓ Conduction of sound waves
Conductive hearing loss
Pressure in middle ear diffuses out of perforation
↓ Tympanic membrane stretching
Otalgia (ear pain) fades
Accumulated debris compresses cranial nerve VII
Facial nerve palsy
Mastoid abscess
Febrile seizures
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published Feb 28, 2013, updated Apr 29, 2024 on www.thecalgaryguide.com")
Sickle Cell Disease Pathogenesis Clinical Findings and Complications
 Liu Priyanka Grewal Reviewers: Alexander Arnold Luiza Radu JoyAnne Krupa Yan Yu* Lynn Savoie* * MD at time of publication
Hemoglobin S (HbS) variant formed instead of normal Hemoglobin A (HbA)
Hb electrophoresis shows approximately 45% HbS, 52% HbA (ααββ), 2% HbA
Hb electrophoresis shows approximately 90% HbS, 8% HbF, & 2% HbA .
No HbA present
(ααδδ), & 1% HbF (ααγγ;2 fetal hemoglobin)
Heterozygous: point mutation in one of the two chromosomes (Hb AS)
Sickle cell trait
(asymptomatic unless severely hypoxic)
Homozygous: point mutation in both chromosomes (Hb SS)
Sickle cell disease
An inherited blood disorder characterized by defective hemoglobin that leads to red blood cells sickling
2
Dehydration Hypoxemia
Acidosis
↓ Volume of RBC cytoplasm
↓ O2 Saturation of Hb
O morereadily 2
released from Hb in low pH environment
↑ Concentration of deoxygenated HbSinred blood cells (RBCs)
Hydrophilic glutamate→ hydrophobic valine substitution makes HbS less soluble in the cytoplasm & more prone to polymerization & precipitation in its deoxygenated state
↑ Concentration of deoxygenated Hb in RBC leads to ↑ polymerization rate Polymerized & precipitated HbS forms long needle-like fibers
RBC shape becomes sickled
Sickle cells on peripheral blood smear
Vaso-occlusion
(sickled RBCs lodge in small vessels, blocking bloodflow to organs & tissues)
Blockage of venous outflow
Occlusion of vessels in lungs ↑ pulmonary blood pressure
Fluid extravasates into interstitial tissue leading to pulmonary edema
Acute chest syndrome (chest pain, hypoxemia (↓blood oxygen), etc.)**
to the penis:
to the spleen:
Priapism (persistent, painful erection)
Splenic Sequestration (blood pools in spleenà splenomegaly & hypotension)
Extravascular hemolysis (macrophages in the spleen phagocytose sickled RBCs)
Normocytic anemia
↑Marrow erythropoiesis (RBC production) to compensate for hemolysis
Infarction of bone
Pain crises
If occurring in hands
Dactylitis (inflammation of digits)
Blockage of arteries ↓ oxygenation of organs & tissues
Vaso-occlusion of the splenic artery
Splenic infarction (↓ blood supply leads to tissue death)
RBC inclusions (structures found in RBCs) not removed by spleen
Howell-Jolly bodies (RBC DNA remnants) on blood smear
Vaso-occlusion of other arteries (cranial, renal, etc)
Stroke, renal failure
↑ RBC breakdown
↑ Unconjugated bilirubin released from RBC breakdown
RBC precursors (reticulocytes) are released into the blood stream
Reticulocytosis (increased number of immature red blood cells) on peripheral blood smear
Patient’s RBC level becomes dependent on increased marrow activity
Bone marrow infarction or viral infections (i.e. Parvovirus B19) suppresses bone marrow activity
Aplastic crises (profound anemia)
** See corresponding Calgary Guide slide(s)
↑ Serum level of unconjugated bilirubin
Some of the circulating unconjugated bilirubin deposits in the skin
Jaundice (yellowing of skin)
↑ Conjugation of bilirubin in liver
↑Amounts of conjugated bilirubin released into bile
Gallstone formation
Cholelithiasis (presence of gallstones in the gallbladder)
Spleen releases invasive encapsulated bacteria (eg. Haemophilus influenzae, Streptococcus pneumoniae, Neisseria meningitidis) into circulation, which causes infections
Meningitis
Sepsis
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published Sept 16, 2013, updated Apr 29, 2024 on www.thecalgaryguide.com")
Wiskott-Aldrich Syndrome
: Pathogenesis and clinical findings X-linked recessive inheritance (mostly males impacted) Spontaneous DNA mutation
Genetic mutations in Wiskott-Aldrich Syndrome Protein (WASP) impair actin cytoskeleton remodeling in hematopoietic cells (immature blood cells)
Authors: Mina Mina Reviewers: Hana Osman Mao Ding Maharshi Gandhi Shahab Marzoughi Louis-Philippe Girard* * MD at time of publication
Megakaryocytes (platelet precursor cells) depend on the cytoskeleton changing shape to form pseudopodia (arm-like projections) which bud off forming cellular fragments (platelets)
Abnormal cytoskeleton reorganization leads to ineffective thrombocytopoiesis (production of platelets)
Microthrombocytopenia (↓ platelet size and quantity)
Issues with formation of a platelet plug due to abnormal platelets (dysfunction of primary hemostasis)
Reduced ability for platelet adhesion, activation, and/or aggregation
Natural killer cells depend on the cytoskeleton reorganization to form immunological synapses (communication) with body cells in order to have effective surveillance
T cells depend on the cytoskeleton remodeling to form pseudopods which allow them to synapse with other cells when a pathogen is encountered
Defective immune synapse formation
↓ Cancer immunosurveillance
↑ Risk of malignancy
Helper T cells cannot activate B cells which generate antibodies (immunoglobulins) to destroy pathogens
Regulatory T cells can not sufficiently downregulate effector cells which typically limit the immune response and prevent autoimmune conditions
↑ Risk of autoimmune diseases
Recurrent opportunistic infections
Immune dysregulation
Impaired immune response
↓ Number and function of T cells
Immune responses contribute to Type 2 immune responses
Atopic dermatitis (AD; chronic inflammation that causes itchy skin)
↑ Immunoglobulin (Ig) A
Epistaxis (nosebleed)
Red blood cells leak from capillaries
Menorrhagia (heavy menstrual bleeding)
Petechiae (small, flat, red spots that appear on the skin)
Inflammation in AD triggers release of interleukins (modulatory proteins during inflammatory and immune responses) leading to Immunoglobulin (Ig) class switching
↑ IgE levels
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published Apr 20, 2024 on www.thecalgaryguide.com")
Scabies pathogenesis and clinical findings

Authors: Heena Singh Amanda Eslinger Nirav Saini Reviewers: Shahab Marzoughi Danny Guo Yan Yu Richard Haber* * MD at time of publication
Mechanical irritation and immunological reaction
Mast cell activation and histamine release
Itch
(> 10 Mites)
Stratum corneum
Stratum lucidum Stratum granulosum
Stratum spinosum Stratum basale
Females lay 2-3 ova/day which hatch in the stratum corneum in pockets after 3 days
The larvae molt and mature for 2 weeks and mate within the pockets of the stratum corneum
Following mating, female mites begin burrowing
Cycle is propagated as new female mites create more burrows in stratum corneum
Superficial, Linear, Tortuous (i.e., Twisted) +/- Scaly Burrows
↑ Exposure to mite antigens such as Sar s 14.3 and Sar s 14.2
Type 2 helper T-cells produce proteins IL-4, IL-5, IL-13
↑ Serum Immunoglobulin G & E ↑ Eosinophil Count in Peripheral Blood Inappropriate T helper-type immune response in skin (mechanism unknown)
Antigens forming from mite feces, ova, or decomposing bodies
Type IV hypersensitivity reaction (delayed immune response 2-4 weeks following initial contact)
Skin barrier dysfunction from inflammation and histamine release
Pre-existing immunosuppression Chronic inflammatory response Allergic reaction to ↑ mite activity at night
Uncontrollable Itch (Worse At Night) + Excoriations (Scratch Marks)
Urticarial (Hive-Like) Crusted Papules Eczematous Plaques
Skin breakdown Opening for bacteria (commonly S. aureus) 2° Bacterial infection Pustules
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published Oct 17, 2013; updated Jun 9, 2024 on www.thecalgaryguide.com")
Posterior Cruciate Ligament PCL Injury Pathogenesis and Clinical Findings
 Injury: Pathogenesis and clinical findings
Authors: Luc Wittig Stephanie de Waal Michelle J. Chen Reviewers: Reza Ojaghi Usama Malik Sunawer Aujla Nojan Mannani Mankirat Bhogal Dr. R. Buckley* Dr. Gerhard Kiefer* * MD at time of publication
Sport-related trauma
Dashboard knee injury from a motor vehicle collision
Tibia contacts dashboard at high velocity with knee in flexed position
Tibia is forced posteriorly, relative to knee joint The PCL undergoes sudden & forceful strain
Hyperextension injury (knee joint moves past its normal extension limit)
Extreme tibia movement anteriorly past knee joint overstretches PCL
Hard fall on flexed knee
Posterior Cruciate Ligament Injury
Strain & overstretching of the PCL, which connects the anterior distal femur to the posterior proximal tibia to prevent posterior translocation of the tibia relative to the femur, can result in a partial or complete tear. The tear can be isolated (rare) or be one of multiple ligaments that are torn (multi-ligamentous tear).
Mild injurious force causes a partial tear
Intact portion of PCL prevents extreme tibial translocation posterior to the femur. Torn portion still allows 1-5 mm of posterior tibial translocation (Grade 1)
Positive posterior drawer test
Knee injury tests
• Posterior drawer test – Push against leg below
the knee to test for posterior tibial translocation
relative to femur
• Lachman test – Pull leg below the knee to test
for anterior tibial translocation relative to femur • McMurray test – Tests for medial and lateral
meniscus tears
• Varus & valgus stress test – Tests for medial and
Strong injurious force causes a complete, isolated tear
PCL is unable to prevent posterior translocation of the tibia relative to the femur, allowing 6-10 mm of posterior tibial translocation (Grade 2)
Positive posterior Negative Lachman, McMurray, drawer test and varus & valgus stress test
Very strong injurious force causes a multi- ligamentous tear and damage to the knee joint capsule (capsuloligamentous injury)
Absence of stability from PCL combined with absence of stability from other knee ligaments allows for > 10 mm of posterior tibial translocation (Grade 3)
Positive posterior Positive Lachman OR McMurray drawer test OR varus & valgus stress test
PCL unable to stabilize knee by preventing posterior tibial translocation (chronic PCL insufficiency)
Body uses quadriceps tendon for knee stabilization to compensate for PCL insufficiency
Inflammation from injury contributes to progressive degenerative articular changes
lateral collateral ligament tears
Post-traumatic patellofemoral pain
Osteoarthritis
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published Sept 20, 2018; updated Jun 9, 2024 on www.thecalgaryguide.com")
Migraines and auras pathogenesis and clinical findings

Personal triggers (e.g., lack of food, emotional stress)
“Cortical Spreading Depolarization” followed by “Cortical Spreading Depression” across one cortical hemisphere
Cortical spreading depression
creates “auras” via unknown mechanism(s)
Expanding scotoma (area of visual blurriness)
Spreading paraesthesias (numbness that travels across body)
Dysphasic language (trouble finding words)
Brainstem symptoms (e.g., vertigo, tinnitus)
Motor symptoms (e.g., hemiplegia)
Triggering of a depolarization wave in a unilateral region of the cortex with associated ↑ blood flow to that region
Neurons and glia release K+ into extracellular space that spreads depolarization wave to nearby cortical areas
Ion gradient imbalances cause Prolonged vasoconstriction neurons in original cortical area to and ↓ blood flow
swell and become inhibited
Activation of the hypothalamus (responsible for maintaining homeostasis)
Prodromal symptoms (↑ thirst, hunger, yawning, ↓ cognitive function)
Author:
Yan Yu
Braxton Phillips
Reviewers:
Shahab Marzoughi
Owen Stechishin
Dustin Anderson
Scott Jarvis*
Sina Marzoughi*
* MD at time of publication
Release of hypothalamic neurotransmitters (e.g., orexins, neuropeptide Y) at the trigeminalcervical complex (TCC) of the brainstem and cervical spinal cord
Depolarizing wave passes over pseudounipolar neurons (neurons with two axons) of the trigeminal ganglion which synapse in brainstem & dura matter
These neurotransmitters reduce the activation threshold of spinal trigeminal nucleus neurons in the TCC
Neuropeptides (e.g., calcitonin gene-related peptide, pituitary adenylate cyclase-activating polypeptide)
are released at the TTC, triggering local inflammation (termed neurogenic inflammation)
Ongoing neurogenic inflammation activates secondary nociceptive neurons in the TTC
Central sensitization (↓ response threshold of secondary nociceptive neurons in the TTC)
Brain perceives referred pain from the face as the TTC also receives convergent nociceptive input from the face
Facial allodynia (pain with normally non-painful stimuli)
Serotonin & histamine are released on dural blood vessels triggering neurogenic inflammation in the dura matter
Ongoing neurogenic inflammation activates primary nociceptive neurons in the dura matter
Peripheral sensitization (↓ response threshold of primary nociceptive neurons around the dural blood vessels_
Unknown mechanisms no longer believed to be related to dural blood vessel dilation
Unilateral throbbing headache
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published Nov 22, 2012, updated Jul 1, 2024 on www.thecalgaryguide.com")
Legg Calve Perthes Disease

Early interruption of blood supply to femoral head
↓ Venous outflow from intraosseous vasculature ↑ pressure within bone
Fragmentation stage (8 months)
Body replaces resorbed necrotic bone with structurally weaker bone
Backup of blood within bone vasculature impedes arterial inflow
Temporary interruption of blood supply to proximal femoral epiphysis primarily during childhood
Residual stage
Differential growth of proximal femoral epiphysis
Overgrowth of greater trochanter
Healing stage (4 years)
Body gradually replaces necrotic bone with stronger, more dense bone
Lack of O2 & nutrients disrupts normal bone growth
Bone tissue begins dying from loss of O2 & nutrients (osteonecrosis)
Osteonecrosis is associated with inflammation of synovial tissue (synovitis) at the femoral head
Metaphyseal cyst (area of bone resorption) grows on proximal femoral neck metaphysis
Growth plate abnormalities
Femoral bone head deformity
Limited hip internal rotation & abduction
Antalgic gait
Femoral epiphysis collapses laterally and extrudes from acetabulum
Femoral head now composed of dense and less dense bone
Scattered radiolucency & radiodensity on plain radiographs
Femoral head deformity becomes set in place and limits hip range of motion
Femoral bone head deformity
Limb length discrepancy
Normal bone density on plain radiographs
Abnormal articulo- trochanteric distance (vertical distance between highest point of greater trochanter & highest point of femoral head)
Remodeling of femoral head & acetabulum seen on plain radiographs
Growth plate irregularity
↓ Range of motion
Widened medial joint space
Antalgic gait
Femoral head is displaced laterally
Fragmented epiphysis on plain radiographs
Groin pain & referred pain to anteromedial thigh or knee region
Authors: Janelle Wai Michelle J. Chen Reviewers: Patrick Pankow Nojan Mannani Kirat Bhogal Dr. Gerhard Kiefer* * MD at time of publication
Findings on magnetic resonance imaging (MRI) & plain radiographs
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published July 19, 2024 on www.thecalgaryguide.com")
Acute Otitis Media Pathogenesis and Clinical Findings in Children

Exposure to tobacco smoke
Impaired macrophage function in nasopharynx
Lack of immunizations
Lack of breastfeeding
Infant does not receive antibodies from breast milk
Overcrowding
Age 6 – 16 months
Immune system deficiencies
Close proximity of kids & ↓ sanitation (e.g. daycare)
↑ Infection risk Upper Respiratory Tract Infection (i.e. bacterial (Streptococcus pneumoniae, Haemophilus influenzae, Moraxella catarrhalis), viral)
Immature Eustachian tube anatomy which facilitates pathogen transmission to middle ear
Author: Jody Platt Stephanie de Waal Reviewers: Yan Yu Elizabeth De Klerk William Kim Annie Pham Michelle J. Chen Danielle Nelson* * MD at time of publication
Abnormal anatomical structure (e.g. cleft palate)
↑ Nasopharyngeal streptococcus pneumoniae
Lack of immunity to pathogens
↑ Colonization of nasopharynx with bacterial pathogens
Inflammation & edema of respiratory mucosa including the nose, nasopharynx, and Eustachian tube
Obstruction of the Eustachian tube
Air from middle ear resorbs into circulation which creates a low-pressure environment
Negative pressure gradient pulls viral/bacterial pathogens into middle ear
Degenerating white blood cells, tissue debris, and microorganisms accumulate & develops into purulent effusion
Inflammation & infection of middle ear
Complications of acute otitis media**
↑ Pressure in middle ear
Stretching of tympanic membrane
Helper T cells & macrophages release cytokines into the bloodstream
Cytokines trigger hypothalamus to ↑ thermoregulatory set-point
Effusion behind tympanic membrane
Effusion obstructs visualization of ossicles
Neutrophils infiltrate middle ear & phagocytose pathogens
Pus accumulates behind the tympanic membrane
Blood vessels of the tympanic membrane vasodilate
Bulging tympanic membrane
Otalgia (ear pain)
Discomfort disrupts daily activities in young children
Fever
Irritable Poor feeding
Tympanic membrane erythema
Painful blisters on tympanic membrane (e.g. Bullous myringitis)
Can persist for up to 3 months after infection resolves
Loss of tympanic membrane landmarks (i.e. handle of malleus, light reflex)
** See corresponding Calgary Guide slide
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published Feb 28, 2013; updated Aug 25, 2024 on www.thecalgaryguide.com")
Low Ankle Sprain

Excessive stress to medial ankle deltoid ligament
Ankle plantar flexion and inversion beyond normal range (most common)
Excessive stress to lateral ankle ligament(s)
Associated fracture of malleolus or subtalar joint
Bone pain, inability to weight bear
Recruitment of inflammatory cells to damaged area
↑ Local pro- inflammatory cytokines
↑ Capillary permeability & vasodilation to damaged area
Collagen fibers in ligament(s) rupture
Low Ankle Sprain
(medial or lateral)
Local blood vessels tear
Blood leaks into surrounding tissue
Grade I
Mild injury
Grade II
Moderate injury
Grade III
Severe injury
Minimal ligament disruption
Incomplete ligament tear
Complete ligament tear
No joint laxity
Joint laxity
Gross joint laxity
Mechanical and functional instability
Decreased ankle joint space
Ankle Impingement
(compression of soft and/or bony structures in joint)
Chronic ankle instability
Talus subluxation with anterior force on heel
+ Anterior drawer test
Recurrent sprains
Synovial inflammation & hypertrophy
Remodeling of collagen and bone
Degenerative changes
(e.g., osteophytes, subchondral sclerosis)
Damage to mechanoreceptors in muscles surrounding ankle joint
Bruising
Swelling
Focal tenderness over torn ligament(s)
Pain on injury and with weight bearing
Impaired proprioception & neuromuscular control
Nociceptors activated by trauma and inflammatory factors
Falls Antalgic gait
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published Nov 9, 2014; updated Aug 15, 2023 on www.thecalgaryguide.com")
Hypomagnesemia
![Hypomagnesemia: Physiology
Hyperglycemia
An increased amount of glucose enters renal tubules as glomerulus performs blood filtration
↑ [Solute] in renal tubules from ↑ glucose content exerts osmotic force that pulls water & electrolytes, including Mg2+, into renal tubules
↑ Urinary Mg2+ excretion
Lack of insulin
Lack of insulin receptor signaling in distal convoluted tubule (DCT) ↓ glucose uptake from renal tubules
Hypercalcemia
Ca2+ binds to Ca2+ sensing receptors on thick ascending limb (TAL) of loop of Henle, where resorption of Ca2+ & Mg2+ occurs
Receptor activation ↓ Na-K- 2Cl (NKCC) transporter activity which maintains electro- chemical gradient in TAL
Passive paracellular resorption of Ca2+ and Mg2+, dependent on electrochemical gradient, ↓
↑ Extra- cellular fluid
↓ Resorption of Na+ & H2O from renal tubules
Genetic disorders (e.g. Bartter syndrome, familial hypomagnesemia)
Medications (e.g. loop & thiazide diuretics, certain antibiotics, calcineurin inhibitors)
Some metabolic byproducts of these drugs are nephrotoxic
Inability to absorb free fatty acids (FFAs)
Mg2+, which associates with FFAs, is not absorbed through the gut
Steatorrhea (fat in the stool)
Mal- absorption (often due to inflammation or infection) & diarrhea
Acute pancreatitis
↓ Lipase secretion from pancreas ↑ levels of undigested fats in small intestine
↓ Passive Mg resorption from
tubules
Mg saponification in necrotic fat
2+
Varying mechanisms causing defective Mg2+ re-absorption (e.g. impacts to PCT, TAL, DCT disrupting transporters and ion shifting; ↓ gut resorption of Mg2+)
Nutrients & 2+
electrolytes are lost in stool
undergoes
Renal loss of magnesium
Gastrointestinal loss of magnesium
Hypomagnesemia
Serum [Mg2+] < 0.7 mmol/L
Impairs production and release of parathyroid hormone responsible for ↑ blood Ca2+ Hypocalcemia
Muscle cells are unable to activate Mg2+ dependent ATP hydrolysis
Impairs muscle relaxation and reduces the ability to stop muscular contraction
Lack of Ca2+ disrupts neurotransmitter release and neuronal signaling
Impairs rapid depolarization and repolarization during muscle contraction
Neuromuscular excitability (large, rapid change in membrane voltage due to small stimulus)
Delirium
Apathy
QRS widening and peaking of T waves on ECG
Torsade de Pointes
Constant muscle contraction compresses blood vessels
Reduced blood supply to hands, wrists, feet, and ankles
Trousseau sign (carpopedal spasm with inflation of BP cuff)
Authors: Caroline Kokorudz Reviewers: Shyla Bharadia Allesha Eman Michelle J. Chen Dr. Adam Bass* * MD at time of publication
Chvostek sign (facial muscle twitch with cheek touch)
Seizures
Tetany
Weakness
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published Oct 4, 2024 on www.thecalgaryguide.com](https://calgaryguide.ucalgary.ca/wp-content/uploads/2024/10/Hypomagnesia.jpg "Hypomagnesemia: Physiology
Hyperglycemia
An increased amount of glucose enters renal tubules as glomerulus performs blood filtration
↑ [Solute] in renal tubules from ↑ glucose content exerts osmotic force that pulls water & electrolytes, including Mg2+, into renal tubules
↑ Urinary Mg2+ excretion
Lack of insulin
Lack of insulin receptor signaling in distal convoluted tubule (DCT) ↓ glucose uptake from renal tubules
Hypercalcemia
Ca2+ binds to Ca2+ sensing receptors on thick ascending limb (TAL) of loop of Henle, where resorption of Ca2+ & Mg2+ occurs
Receptor activation ↓ Na-K- 2Cl (NKCC) transporter activity which maintains electro- chemical gradient in TAL
Passive paracellular resorption of Ca2+ and Mg2+, dependent on electrochemical gradient, ↓
↑ Extra- cellular fluid
↓ Resorption of Na+ & H2O from renal tubules
Genetic disorders (e.g. Bartter syndrome, familial hypomagnesemia)
Medications (e.g. loop & thiazide diuretics, certain antibiotics, calcineurin inhibitors)
Some metabolic byproducts of these drugs are nephrotoxic
Inability to absorb free fatty acids (FFAs)
Mg2+, which associates with FFAs, is not absorbed through the gut
Steatorrhea (fat in the stool)
Mal- absorption (often due to inflammation or infection) & diarrhea
Acute pancreatitis
↓ Lipase secretion from pancreas ↑ levels of undigested fats in small intestine
↓ Passive Mg resorption from
tubules
Mg saponification in necrotic fat
2+
Varying mechanisms causing defective Mg2+ re-absorption (e.g. impacts to PCT, TAL, DCT disrupting transporters and ion shifting; ↓ gut resorption of Mg2+)
Nutrients & 2+
electrolytes are lost in stool
undergoes
Renal loss of magnesium
Gastrointestinal loss of magnesium
Hypomagnesemia
Serum [Mg2+] < 0.7 mmol/L
Impairs production and release of parathyroid hormone responsible for ↑ blood Ca2+ Hypocalcemia
Muscle cells are unable to activate Mg2+ dependent ATP hydrolysis
Impairs muscle relaxation and reduces the ability to stop muscular contraction
Lack of Ca2+ disrupts neurotransmitter release and neuronal signaling
Impairs rapid depolarization and repolarization during muscle contraction
Neuromuscular excitability (large, rapid change in membrane voltage due to small stimulus)
Delirium
Apathy
QRS widening and peaking of T waves on ECG
Torsade de Pointes
Constant muscle contraction compresses blood vessels
Reduced blood supply to hands, wrists, feet, and ankles
Trousseau sign (carpopedal spasm with inflation of BP cuff)
Authors: Caroline Kokorudz Reviewers: Shyla Bharadia Allesha Eman Michelle J. Chen Dr. Adam Bass* * MD at time of publication
Chvostek sign (facial muscle twitch with cheek touch)
Seizures
Tetany
Weakness
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published Oct 4, 2024 on www.thecalgaryguide.com")
Acute Diverticulitis

Obstipation (inability to pass any feces)
Inherent weakness in the muscle layers of the colonic wall associated with immature collagen fibers and diminished wall elasticity
Mucosal and submucosal layers of the colon wall push through a weak spot of the circular muscle layer
Formation of diverticulum (sac-like protrusion of the colonic wall)
Continued stress on diverticula causes micro- perforations of the diverticulum
Inflammation of diverticula
Mesentery and pericolic fat (fat surrounding the colon) attempt to wall off inflammation or perforations
Stool bacteria escapes the colon
Formation of abscess (accumulation of pus in response to the bacteria )
Pro-inflammatory cytokine release from nearby adipose tissue
Hypothalamic thermoregulatory center increases core temperature set point
Fever
Irritation of parietal peritoneum
Inflamed vessels are more permeable and fluid leaks from colonic vessels into the abdominal cavity
Chronic low-grade inflammation triggers activation of pro-fibrotic factors and fibroblasts
Excess production of extracellular matrix proteins
Stimulation of somatic nerves sends pain signals to the brain
Lower left quadrant abdominal pain
Peritoneal signs (abdominal guarding, rigidity, rebound tenderness)
↓ Total circulating blood volume
↑ heart rate
↓ Jugular venous pressure
Orthostatic hypotension (low blood pressure when standing after sitting/lying down)
Gastrointestinal strictures and/or colonic obstruction
Accumulation of micro- perforations further weakens intestinal wall
Complete bowel perforation (medical emergency)
Development of an abnormal connection (fistula) through the bladder, vagina, skin, or gut
Abscesses
Accumulation of fibrotic tissue narrows the intestinal lumen
Fibrosis of colon
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Re-Published Oct 4, 2024 on thecalgaryguide.com")
Epilepsy in Older Adults
, primarily ischemic stroke, intracerebral hemorrhage, subarachnoid hemorrhage
Ischemic and hemorrhagic injuries cause inflammation and nerve cell degeneration
Glial cells (astrocytes and oligodendrocytes) proliferate around the lesion area to repair the damaged tissue
Glial scar formation impedes neuronal reconnection and growth
Alzheimer’s or Vascular Dementia
Central nervous system disease (e.g. traumatic brain injury, prior meningitis, mass)
Medications associated with hyponatremia (e.g. diuretics, antidepressants, antipsychotics, etc.)
Cerebral edema
Increased intracranial pressure
Compression on structures and blood vessels
Sleep deprivation
Tau or amyloid deposition (abnormal protein aggregates in brain)
Small vessel disease
Increased delta wave activity
Heightened neural excitability
Decreased seizure threshold
Elevated stress hormones (e.g. cortisol)
Increased neuronal excitability and decreased inhibition
Areas of tissue death, white matter changes & cortical irritability
Structural and electrical brain changes
Epilepsy: Neurological disorder characterized by increased susceptibility to recurrent unprovoked seizures
Excessive, hypersynchronous & oscillatory network function Imbalance between excitatory and inhibitory activity
Resultant seizure activity
Atypical Seizure pattern; e.g. seem confused, stare into space, wander, make unusual movements, inability to answer questions
Often atypical location in brain (limbic or neocortical)
Focal seizures are more common than generalized
Postictal paresis can last for days & disorientation, hyperactivity, wandering and incontinence may persist for 1 week
Neurotransmitter dysregulation, neural network disruption, genetic factors, psychosocial factors
Psychiatric comorbidities
Authors: Anna Crone
Reviewers: Anika Zaman,
Rachel Carson, Raafi Ali, Luiza Radu, Gary Michael K Klein*
* MD at time of publication
Widespread structural changes and hippocampal atrophy
Dementia
Sub-optimal treatment results in ongoing and more frequent epileptic seizures
Status epilepticus (higher mortality among older adults)
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published Oct 4, 2024 on www.thecalgaryguide.com")
Cholesteatoma of middle ear
: Pathogenesis and clinical findings
Authors: Emma Holmes Angela Mak Reviewers: Stephanie Cote Vaneeza Moosa Shahab Marzoughi William Kim Sunawer Aujla Kristine Anne Smith* * MD at time of publication
Tympanic membrane perforation (e.g., acute otitis media, trauma)
Squamous epithelium invasion & migration into middle ear
Eustachian tube dysfunction (e.g., craniofacial abnormalities)
Negative pressure
Invagination of tympanic membrane
Retraction pocket with keratin trapping & ingrowth
Inflammation (e.g., upper respiratory tract infection, rhinitis)
Mucosal lining of middle ear become hyperproliferative
Squamous metaplasia
Basal cell hyperplasia
Invagination & epithelial ingrowth in basement membrane of the middle ear
Theory of implantation due to trauma (e.g., during surgery)
Implantation of skin into the
middle ear through a defect in the eardrum
Traps moisture
Bacterial infection
Erosion of external auditory canal
Conductive hearing loss
Labyrinthine fistula (abnormal communication between the inner ear and the surrounding structures)
Leakage of perilymph
Cholesteatoma
Destructive growth of keratinizing squamous epithelium in the middle ear
Bone erosion allows for secondary infection from outside the middle ear
Chronic otitis media
Migration of bacteria from middle to inner ear
Intracranial infection (e.g., meningitis, parenchymal abscess)
Acute otitis media
Accumulation of keratinous debris
Release of inflammatory molecules
Release of osteoclasts
Secretion of acid and proteinases
Erosion of temporal bone
Pressure change in the middle ear
Aural polyp (growth in external or middle ear canal)
Vertigo
Coalescent mastoiditis (bone is remodeled and resorbed from pressure necrosis, inflammation, and increased osteoclastic activity)
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published Oct 4, 2024 on www.thecalgaryguide.com")
Complex Regional Pain Syndrome
: Pathogenesis and clinical findings
Type 1 origin: CRPS arises spontaneously or from trauma without confirmed damage to nerves (e.g. surgery, nerve compression, fracture, tissue trauma, ischemia, sprain)
Type 2 origin: CRPS arises from trauma with confirmed evidence of nerve damage
Intense psychological stress is associated with ↑ severity of CRPS
Peripheral nerve endings release ↑ levels of neuropeptides (e.g. substance P, bradykinin, calcitonin gene-related peptide)
Vasodilation & protein extravasation in tissues
↑ Perfusion to motor cortex
↓ Grey matter volume in cortical pain regions
↓ Cortical grey matter volume & ↓ perfusion of cortex in limbic & sensorimotor areas
Dysregulation of sympathetic nerve fibres Autonomic dysfunction
Central nerve fibres ↑ release of proinflammatory cytokines while ↓ release of anti- inflammatory cytokines
Central sensitization (threshold at which a central nerve transmits a signal is lowered so that it more readily transmits signals)
Mechanical hyperalgesia (hallmark of central sensitization)
Authors:
Jessica Hammal
Calvin Howard
Reviewers:
Sina Marzoughi
Michelle J. Chen
Scott Jarvis*
* MD at time of publication
Nociceptors (nerve endings detecting pain) on skin become activated
↑ Cytokine & nerve growth factor release
Primary afferent (sensory) neurons release ↑ levels of inflammatory neuropeptides
Peripheral sensitization (threshold at which a nerve transmits a signal is lowered so that it more readily transmits signals)
Widespread neurogenic inflammation (inflammation due to activation of peripheral nerve fibres)
Sustained and dysregulated pro-inflammatory state
Hyperalgesia (↑ sensitivity to feeling pain)
Sweat gland related changes: Edema, sweating changes, asymmetric sweating
Allodynia (pain from a stimulus that doesn’t normally cause pain)
Trophic changes (wasting of skin, muscle, tissues; thinning of bones; thickening/thinning of nails)
Motor dysfunction
↓ Range of motion
Immobile due to severity of pain
Impaired vasodilation & vasoconstriction
Temperature asymmetry
Altered skin color (red, blue, pale)
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published Oct 28, 2024 on www.thecalgaryguide.com")
Renal manifestations of SLE
: Renal Manifestations
Authors: Madison Turk Reviewers: Modhawi Alqanaie Mao Ding Luiza Radu Glen Hazlewood* * MD at time of publication
Genetic susceptibility and potential environmental triggers (smoking, silica, Epstein-Barr Virus, hormones, ect)
↓Clearance of dead cell debris in the body
(exact mechanisms unknown; See slide on pathogenesis of SLE)
Extracellular exposure of nuclear proteins (which bind DNA and regulate gene expression)
Immune cells respond to nuclear proteins as if they are non-self, as they usually don’t ‘see’ them
Systemic Lupus Erythematosus
An autoimmune disease characterized by anti-nuclear-antibody production resulting in widespread inflammation and tissue damage in varying affected organs, including one, or a combination of, joints, skin, brain, lungs, kidneys, and blood vessels
Production of auto-antibodies against self nuclear proteins
IC deposition in renal vessels
Activation of inflammatory cells against IC’s causing vascular injury/inflammation
↑Clot formation in vessels leading to ↓vessel lumen diameter and ↓blood flow
↑Reninàcleavage of angiotensinogen to angiotensin Ià cleavage by angiotensin converting enzyme into angiotensin II
Vessel constriction to ↑blood pressure (to ↑ renal blood flow)
IC deposition in tubule basement membrane of the kidney
Formation of immune complexes (IC) (collections of antigen(s), antibodies, and/or complement proteins bound together)
Tubulointerstitial nephritis: (Inflammation of the renal tubules and interstitium, sparing the glomeruli)
Renal ischemia
IC deposition in the glomerulus
Activation of complement, initiating an inflammatory response
Recruitment of myeloid cells (monocytes/ neutrophils)
Production of reactive oxygen species, cytokines and release of cytotoxic granules
Tubular inflammation and damage resulting in ↓sodium reabsorption
Renal release of reninà ↑ angiotensin I/II and resulting ↑ aldosterone
↑Sodium reabsorption, and potassium excretion in the collecting duct
↓Potassium secretion from the Distal Convoluted Tubule
Hyperkalemia Hypokalemia
⍺-intercalated cell damage
↓Acid excretion
Metabolic acidosis
End stage kidney disease
(all manifestations can result in this)
Hypertension
Tissue damage from the inflammatory response
Glomerular nephritis (inflammation/damage of the filtering part of the kidneys)
Fibrosis (thickening or scarring of tissue)
Mesangial and parietal cell proliferation
Podocyte injury
↑blood and protein excretion due to damage to the glomerular filtration membrane
↓protein in blood Edema
Proteinuria Hematuria
↓Functional renal tissue to filter creatinine from blood
↓ Glomerular filtration rate ↑ Plasma creatinine
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published Nov 11, 2024 on www.thecalgaryguide.com")
Diabetic Polyneuropathy

↓ Insulin induces expression of neurotrophic factors (i.e. nerve growth factor, brain-derived neurotrophic factor, neurotrophin-3, insulin-like growth factor (IGF), & vascular endothelial growth factor (VEGF))
↓ Stimulus for peripheral nerve maintenance & repair
Impaired peripheral nerve repair
Hyperglycemia
(↑ Intracellular glucose levels)
↑ Glycolysis (breakdown of glucose)
↑ Intermediates & products of glycolysis (i.e. Protein Kinase C, AGE, polyols (sorbitol & fructose), NADH)
Protein Kinase C Pathway activated & ↑ inflammation in endothelium, vascular smooth muscle, fibroblasts of blood vessels
Prothrombotic state with ↑ vascular fibrosis, ↑ smooth muscle proliferation, ↑ chance of blood clots, ↑ impaired endothelial- mediated vasodilation
Arterial stiffening & ischemia
Advanced Glycation End Products (AGEs): end products of glycolysis that have carbohydrates attached
AGEs bind AGE receptors on endothelium & white blood cells
↑ Inflammation, vascular permeability, procoagulant activity & monocyte influx
Immune cells generate toxic reactive oxygen species as part of their defense system
Polyol pathway activated in kidney, retina, nerves
Excess glucose converted to sorbitol & fructose
Metabolism of sorbitol & fructose produces intermediates that undergo auto-oxidation
↑ Reactive oxygen species
↑ NADH (reducing agent) overloads the electron transport chain
↑ Leakage of electrons in the electron transport chain
↑ Free radical formation (type of reactive oxygen species that is toxic to cells & tissues when in excess)
↑ Oxidative stress
Diabetic Polyneuropathy (damage to & loss of peripheral nerve function due to nerve hypoxia & impaired repair mechanisms)
Sensory axonal loss
Damage to small myelinated fibers
Impaired pain, light touch & temperature sensation
Pain, paresthesia (pins & needles feeling), dysthesia (burning, tingling, itching)
X-ray: destruction of weight-bearing foot joints
Damage to large myelinated fibers
Loss of vibratory sensation in glove & stocking pattern distribution; altered proprioception
Claw toe deformity
Charcot Foot: weakening of bones, joints, soft tissues causes pain insensitivity
Distal motor axonal loss
↓ Ability of axons to transmit signals to central nervous system to foot muscles
↓ Foot muscle strength,
↓ Coordination & imbalance between toe extensors & flexors
Weight shift creates pressure points
Fissures in skin of weight-bearing areas
Infection & chronic ulceration
Damage to nerves that control contractions of stomach muscles
Gastroparesis: stomach muscles weakened, ↓ motility & delayed transit of contents
Autonomic neuropathy
Damage to nerves that control heart blood flow, contractions & contractility
Damage to nerves that control blood flow and arousal responses for sexual function
Erectile dysfunction
Orthostatic or postural hypotension
Exercise intolerance
Resting tachycardia
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published Apr 28, 2014; updated Nov 16, 2024 on www.thecalgaryguide.com
Diabetic Polyneuropathy: Pathogenesis and clinical findings
Authors: Gurreet Bhandal Amanda Eslinger Reviewers: Raafi Ali Mark Elliott Jaye Platnich Alexander Arnold Haotian Wang Hanan Bassyouni* * MD at time of publication
Hypoinsulinemia
(↓ Intracellular insulin levels)
↓ Insulin induces expression of neurotrophic factors (i.e. nerve growth factor, brain-derived neurotrophic factor, neurotrophin-3, insulin-like growth factor (IGF), & vascular endothelial growth factor (VEGF))
↓ Stimulus for peripheral nerve maintenance & repair
Impaired peripheral nerve repair
Hyperglycemia
(↑ Intracellular glucose levels)
Protein Kinase C Pathway in endothelium, vascular smooth muscle, fibroblasts of blood vessels
↑ Inflammation
Prothrombotic state with ↑ chance of blood clots, ↑ vasoconstriction & ↑ arterial stiffening
Ischemia
↑ Glycolysis (breakdown of glucose)
Advanced Glycation End Products (AGEs): end products of glycolysis that have carbohydrates attached
AGEs bind cellular receptors
Inflammation, vascular permeability, procoagulant activity & monocyte influx
Immune cells generate toxic reactive oxygen species as part of their defense system
↑ Intermediates & products of glycolysis (i.e. Protein Kinase C, AGE, sorbitol & fructose, NADH)
Polyol Pathway in kidney, retina, nerves
Excess glucose converted to sorbitol & fructose
Metabolism of sorbitol & fructose produces intermediates that undergo auto-oxidation
↑ Reactive oxygen species
↑ Leakage of electrons when NADH (reducing agent) overloads the electron transport chain
↑ Free radical formation (type of reactive oxygen species that is toxic to cells & tissues when in excess)
↑ Oxidative stress
Diabetic Polyneuropathy (damage to & loss of peripheral nerve function due to nerve hypoxia & impaired repair mechanisms)
Sensory axonal loss
Damage to small myelinated fibers
Impaired pain, light touch & temperature sensation
Pain, paresthesia (pins & needles feeling), dysthesia (burning, tingling, itching)
X-ray: destruction of weight-bearing foot joints
Damage to large myelinated fibers
Loss of vibratory sensation in glove & stocking pattern distribution; altered proprioception
Claw toe deformity
Charcot Foot: weakening of bones, joints, soft tissues causes pain insensitivity
Distal motor axonal loss
↓ Ability of axons to transmit signals to central nervous system to foot muscles
↓ Foot muscle strength,
↓ Coordination & imbalance between toe extensors & flexors
Weight shift creates pressure points
Fissures in skin of weight-bearing areas
Infection & chronic ulceration
Damage to nerves that control contractions of stomach muscles
Gastroparesis: stomach muscles weakened, ↓ motility & delayed transit of contents
Autonomic neuropathy
Damage to nerves that control heart blood flow, contractions & contractility
Damage to nerves that control blood flow and arousal responses for sexual function
Erectile dysfunction
Orthostatic or postural hypotension
Exercise intolerance
Resting tachycardia
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published April 28th, 2014 on www.thecalgaryguide.com
Diabetic Polyneuropathy: Pathogenesis and clinical findings
Authors: Gurreet Bhandal Amanda Eslinger Reviewers: Mark Elliott Jaye Platnich Alexander Arnold Haotian Wang Hanan Bassyouni* * MD at time of publication
Hypoinsulinemia
↓ intracellular insulin levels
↓ Neurotrophic factors (i.e. nerve growth factor, brain-derived neurotrophic factor, neurotrophin-3, IGF, & VEGF)
↓ stimulus for peripheral nerve maintenance and repair
Impaired peripheral nerve repair
Sensory axonal loss Small myelinated fibers
Impaired pain, light touch & temperature sensation
Pain, paresthesias or tingling and numbness, dysthesias where sense of touch is distorted
Hyperglycemia
↑ Intracellular glucose levels
This produces an excess of glycolysis intermediates & products of glycolysis (i.e. sorbitol & fructose; AGE; Protein Kinase C)
PKC Pathway in endothelium, vascular
smooth muscle, fibroblasts of blood vessels
↑ inflammation
Prothrombotic state with ↑ chance of blood clots; vasoconstriction & arterial stiffening
Ischemia
Advanced Glycation End Products: The body “glycates” end products of glycolysis, which means that some end products have a carbohydrate added to them
Polyol Pathway
in kidney, retina, nerves
Excess glucose is converted to sorbitol and fructose, which accumulates in cells
↑ Oxidative stress
↑ Free Radical Formation
AGE’s bind cellular receptors inducing inflammation, vascular
permeability, procoagulant activity & monocyte influx
Abbreviations:
AGE – Advanced Glycation End Products
IGF - Insulin-like Growth Factor
VEGF - Vascular Endothelial Growth Factor PKC – Protein Kinase C
Autonomic neuropathy
Nerve hypoxia & impaired repair mechanisms leading to dysfunction & loss of peripheral nerves Distal motor axonal loss
Large myelinated fibers
Atrophy of intrinsic foot muscles
Fissures in skin of weight-bearing areas
Imbalance between toe extensors & flexors
Weight shift results in pressure points
Infection & chronic ulceration
Gastroparesis: stomach muscles weakened with ↓ motility
Claw toe deformity
Erectile dysfunction
Altered proprioception
X-ray: destruction of weight-bearing foot joints
Loss of vibratory sensation in glove & stocking pattern distribution
Charcot Foot: weakening of bones, joints, soft tissues causes pain insensitivity
Cardiac: Resting tachycardia, exercise intolerance, orthostatic or postural hypotension
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published April 28th, 2014 on www.thecalgaryguide.com
Diabetic Polyneuropathy: Pathogenesis and clinical findings
Authors: Amanda Eslinger Reviewers: Mark Elliott Jaye Platnich Alexander Arnold Haotian Wang Hanan Bassyouni* * MD at time of publication
Hypoinsulinemia
↓ Neurotrophic factors (i.e. nerve growth factor, brain-derived neurotrophic factor, neurotrophin-3, IGF, & VEGF)
↓ stimulus for peripheral nerve maintenance and repair
Impaired peripheral nerve repair
Sensory axonal loss
Small myelinated fibers
Impaired pain, light touch & temperature sensation
Pain, paresthesias, dysthesias
Hyperglycemia
↑ Intracellular glucose levels
This produces of glycolysis
an excess of glycolysis intermediates & products (i.e. sorbitol & fructose; AGE; Protein Kinase C)
PKC Pathway
PKC pathway activation results in ↑ inflammation
Prothrombotic state; vasoconstriction & arterial stiffening
Ischemia
Advanced Glycation End Products: The body “glycates” end products of glycolysis, which means that some end products have a carbohydrate added to them
AGE’s bind cellular receptors inducing inflammation, vascular
permeability, procoagulant activity & monocyte influx
Polyol Pathway
(i.e. sorbitol & fructose)
Excess glucose is converted to sorbitol, which accumulates in cells
↑ Oxidative stress
↑ Free Radical Formation
Nerve hypoxia & impaired repair mechanisms leading to dysfunction & loss of peripheral nerves Distal motor axonal loss
Abbreviations:
AGE – Advanced Glycation End Products
IGF - Insulin-like Growth Factor
VEGF - Vascular Endothelial Growth Factor PKC – Protein Kinase C
Autonomic neuropathy
Large myelinated fibers Altered
proprioception
Atrophy of intrinsic foot muscles
Fissures in skin of weight-bearing areas
Infection & chronic ulceration
Imbalance between toe extensors & flexors
Weight shift results in pressure points
Gastroparesis
Claw toe deformity
Erectile dysfunction
X-ray: destruction of weight-bearing foot joints
Loss of vibratory sensation in glove & stocking pattern distribution
Charcot Foot
Cardiac: Resting tachycardia, exercise intolerance, orthostatic hypotension
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published April 28th, 2014 on www.thecalgaryguide.com")
Open Fractures
 or low-energy force on diseased bone
Force applied to bone exceeds strength of bone resulting in periosteal stripping and subsequent soft tissue and neurovascular destruction
Open Fractures
Also known as “compound fractures” and classified with the Gustilo-Anderson classification system (Types I, II, III), these are fractures in which the skin is penetrated and bone is exposed to the external environment. Comminuted fractures have ≥ 2 breaks in the bone.
Inside-out (bone) or outside-in (external) penetration of skin to create a wound
Skin tearing creates vacuum-like effect pulling debris into wound
Minimal comminution
Bone penetrates skin to create a wound < 1 cm in diameter (Type I)
Smaller wound creates minimal opportunities for pathogen entry and contamination
Moderate comminution
Bone penetrates skin to create a wound 1-10 cm in diameter (Type II)
Moderate wound creates some opportunity for pathogen entry and contamination
Extensive comminution
Bone penetrates skin to create a wound > 10 cm in diameter (Type III)
Large wound creates ample opportunity for pathogen entry and extensive contamination
Type IIIA (adequate soft tissue for bone coverage)
Type IIIB
(soft tissue damage with periosteal stripping)
Type IIIC (vascular injuries, potential amputation)
Displacement/shortening/ angulation/rotation of fracture fragment
Improper bone healing
Bone deformity
Authors: Meaghan MacKenzie Holly Zahary Loreman Nojan Mannani Reviewers: Annalise Abbott Usama Malik Michelle J. Chen Dr. Prism Schneider* Dr. Jared Topham* * MD at time of publication
Pain & lack of mechanical load bearing axis
Inability to weight bear
Decreased mobility promotes stasis of venous blood flow & intravascular vessel wall damage
Deep vein thrombosis
Potential progression to pulmonary embolism
Bleeding or inflammation within fascia
Muscle atrophy
Compartment syndrome (↑ pressure in muscle) **
Open wound exposes bone
Infiltration of debris & contaminants
Infection of soft tissues or bone (osteomyelitis)
Initial injury damages blood vessels
Initial injury damages nerves
↓ Sensation distal to injury
↓ Limb function & proprioception
↓ Pulses distal to injury
Amputation
↓ Blood flow to bone
Avascular necrosis (bone tissue death)
Limb ischemia
↓ Blood flow & oxygen delivery to tissues
Compartment syndrome**
Delayed union (bone healing) on serial radiographs
Non-union (bone fails to heal) on serial radiographs
**See corresponding Calgary Guide slide on Acute Compartment Syndrome
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published Nov 16, 2017; updated Nov 21, 2024 on www.thecalgaryguide.com")
Statins Mechanisms and Side Effects

↓ Mevalonate acid (cholesterol precursor) production in the mevalonate pathway (metabolic processes that produce cholesterol)
↓ Cholesterol ubiquinone (CoQ10) production
↓ Ubiquinone (protective of cellular oxidative effects)
Impaired mitochondrial function
↓ Cyclooxygenase-2 (COX2) expression (enzyme involved in the production of prostaglandins)
↓ Availability of
arachidonic acid (derivative of LDL cholesterol breakdown)
↓ Thromboxane A2 (lipid with prothrombotic properties)
↓ Platelet adhesion
↓ Thrombogenicity (tendency to generate blood clots)
Improved endothelial function
Improved myocardial blood flow
↓ Intrinsic cholesterol biosynthesis in the liver
Compensatory mechanisms
↓ Isoprenoid production (electron & proton carriers)
↓ Isoprenoid intermediates
↓Inflammatory signaling proteins
↓Proliferation of macrophages
↓ Inflammation
Upregulation of hepatic LDL receptors
↑ Hepatic LDL uptake
↓ Serum low-density lipoprotein (LDL)
↑ Apolipoprotein A1 (component of HDL) production
↑ High-density lipoprotein (HDL) Carries serum
cholesterol back to liver
↓ Serum apolipoprotein B levels (acts as a tag for LDL, facilitating uptake by LDL receptors)
↑ Oxidative Stress Hepatocyte damage & inflammation ↑ Serum liver enzymes Hepatotoxicity
↓ ATP
↓ Serum cholesterol
↓ Muscle membrane stability
Rhabdomyolysis (muscle breakdown)
Author: Rupali Manek, Andrew Wu, Rafael Sanguinetti, Luiza Radu Reviewers: Joshua Dian, Laura Byford-Richardson, Alexander Ah-Chi Leung* * MD at time of publication
Stabilization of plaques
↓ Atherosclerosis (plaque build-up in arteries)
↓ Coronary events & mortality
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Physiological outcome
Published Jan 9, 2017; updated Nov 22, 2024 on www.thecalgaryguide.com")
Hypocalcemia Pathogenesis

Autoimmune (e.g., autoimmune polyglandular syndrome)
Infiltrative (e.g., hemochro matosis, Wilson’s disease)
Congenital (e.g., DiGeorge)
Hypomagnesemia
Magnesium is needed to produce PTH
Malabsorption
↓ Small bowel surface area
↓ Absorption of 25-OH Vitamin D (calcidiol)
↓ Conversion of calcidiol to 1-25 (OH)2 Vitamin D (calcitriol) by 1-α- hydroxylase
↓ Calcitriol
(impacts GI absorption of calcium)
Chronic Renal Failure
↓ Renal parenchymal mass (site of 1-α- hydroxylase production)
Vitamin D Dependent Type 1 Rickets
Mutation in the gene that encodes 1-α- hydroxylase
↓ Sun exposure
↓ 1-α-hydroxylase Pancreatitis
Destruction or atrophy of parathyroid gland
↓ Parathyroid hormone (PTH)
Calcitriol resistance (e.g., Vitamin D Type 2 rickets)
Inflammation of pancreas
↑ Release of lipase enzyme into serum
↑ Breakdown of fat by lipase
↑ Free fatty acids in serum
↑ Ca2+ binding to negatively charged tail of free fatty acids
↑ Parathyroid hormone (PTH)
PTH resistance
Genetic mutation causes body to not respond to PTH
↓ Renal calcium reabsorption
↑ Calcium excretion
↓ Osteoclast activity
↓ Calcium release from bone
↓ Action of PTH on intestinal cells
↓ Absorption of calcium in the small intestine
↑ Serum phosphate (PO4) (e.g. tumor lysis, rhabdomyolysis)
↑ Ca2+
binding to
negatively charged PO4
↑ Serum pH
↓ H+ in serum binding to proteins
↑ Ca2+ binding to proteins
Calcium sequestration
↓ Absorption of calcium in the small intestine
Hypocalcemia
Author: Breanna Fang Reviewers: Gurreet Bhandal, Raafi Ali, Luiza Radu Samuel Fineblit* * MD at time of publication
Serum calcium levels below 2.10mmol/L
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published Nov 22, 2024 on www.thecalgaryguide.com")
Acquired Inguinal Hernias

Collagen deficiency
↑ Protease activity Smoking Aging
Malnutrition Collagen breakdown
Long-term glucocorticoid use
Fibroblast (cells which produce collagen) suppression causing ↓ collagen production
Thinning of skin & soft tissues
Incomplete obliteration of the processus vaginalis (peritoneal tunnel through which the testes migrate toward the scrotum during embryological development)
Failed closure leaves an opening for organs to herniate through
Intestinal loops entrapped within herniation
Weakening of connective tissues
Indirect Inguinal Hernia
Protrusion of abdominal and/or pelvic contents through deep inguinal ring
Protrusion of abdominal and/or pelvic contents through the Hesselbach triangle (anatomic area defined by the rectus abdominis medially, inferior epigastric vessels laterally, and the inguinal ligament inferiorly)
Herniated contents become entrapped
Inguinal mass +/- pain that is worse with straining
Abdominal distension & tenderness
Rhythmic contractions of the intestine attempting to push contents past the blockage site
Colicky pain
Incarceration (Surgical Emergency)
Reduced vascular supply to entrapped contents
Strangulation (Surgical Emergency)
Constriction & ischemia of hernial contents
Gangrenous skin changes (swelling, blue/purple colour, ↑ temperature, ulceration, ↓ sensation)
Inflammation of the herniated contents to larger than the hernial opening
Irreducible hernia (a hernia which cannot be manually reduced)
Bowel Obstruction (Surgical Emergency)
Obstruction of intestinal lumen impairs passage of intestinal contents
Stimulation of stretch receptors in the bowel wall
Nausea +/- vomiting
Herniated tissue undergoes necrosis (irreversible cell death), perforating the gastrointestinal wall
Obstipation (complete inability to pass stool or gas)
Bowel perforation (surgical emergency)
Leakage of gastrointestinal contents allows bacteria to infect the abdominal cavity
Sudden ↑ pain
Abscess (localized collection of pus)
Peritonitis (inflammation of the peritoneum)
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published Jun 3, 2018; updated dec 29, 2024 on www.thecalgaryguide.com")
Barretts Esophagus

Authors: Sophia Khan Reviewers: Claire Song Shahab Marzoughi Sylvain Coderre* * MD at time of publication
Reflux of stomach acid, bile salts + digestive enzymes
through lower esophageal sphincter (located at junction between esophagus and stomach)
Chronic reflux exposure to squamous epithelium (cells lining distal esophagus)
Squamous epithelial cells release inflammatory cytokines: interleukin-8 and interleukin-1beta
Inflammation of squamous epithelium
Adaptive changes of squamous epithelium to prevent damage by acidic reflux
Migration of stem cells in gastric cardia (proximal region of the stomach) into distal esophagus
Replacement of esophageal squamous epithelium by gastric columnar epithelium
Conversion (metaplasia) of normal esophageal squamous epithelium into abnormal gastric columnar epithelium with interspersed goblet cells
Z line (the squamocolumnar junction between the esophagus and stomach) is irregular and displaced proximally on esophagus on endoscopy
Heartburn (retrosternal burning sensation from stomach reflux)
Regurgitation (involuntary expulsion
of stomach content back into esophagus)
Development of goblet cells (intestinal mucus-producing cells) within esophageal epithelium
Abnormal presence of goblet cells on histology of distal esophagus
Abnormal presence of columnar epithelial cells on histology of distal esophagus
↑ Atypical proliferation &
↓ apoptosis of Barrett’s epithelial cells
Dysplasia (disordered growth of cells with the
potential to develop into cancer) of Barrett’s epithelium
Esophageal adenocarcinoma
Improper division of nuclear chromatin (packaged DNA)
Cellular nuclei increase in size due to retained chromatin from improper division
Excess chromatin absorbs more stain during histology
Double- stranded DNA breaks
Nuclear enlargement on histology
Continued acidic reflux causes oxidative DNA damage
Hyperchromatic (darkly stained) nuclei on histology
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published Dec 15, 2024 on www.thecalgaryguide.com")
Malignant Esophagitis

Chronic gastric reflux (acidic partially- digested stomach contents)
Conversion of normal esophageal squamous epithelium to metaplastic columnar epithelium
Gastroesophageal Reflux Disease
Recurrent regurgitation of gastric content
Chronic Irritation & damage to the esophageal mucosa
Lesions and plaques develop in distal esophagus
Malignancy develops in the mucus-producing glandular cells in the lower esophagus
Smoking
Carcinogenic tobacco condensate is swallowed
Irritation of the the esophageal lining
Heavy alcohol consumption
Mutation in alcohol- dehydrogenase, which breaks down ethanol
↓ Alcohol breakdown
Irritation of the esophageal mucosa & lining
↑ Inflammation of the squamous epithelium located in the upper & middle esophagus
Achalasia (Esophageal smooth muscle motility disorder)
Food stasis in the esophagus ↑ Bacteria production
Lactic acid production & fermentation damages esophageal mucosa
Chronic inflammation in the esophageal epithelium
Malignant cells invade the aorta & tracheobronchial region
↑ Spread & growth of cancer in other organ systems
Metastatic invasion into the aorta, lungs, liver, bones, & kidneys through the lymph nodes
↑ Spread & growth of cancer in other organ systems
↑ Metabolic demand
Unintentional weight loss
Generalized chest pain
Hoarseness Cough
Esophageal adenocarcinoma (malignancy of the glandular cells of the esophagus)
Esophageal squamous cell carcinoma (malignancy of the squamous epithelium of the esophagus)
↑ Metabolic demand
↑ Weight loss
Chronic GI bleeding
Iron-deficiency anemia
↑ Ulceration
Exposure to stomach acid
↑ Tumor- induced inflammation
↑ Fibrosis
Irregular stricture of the upper & middle esophagus
Dysphagia
Esophageal wall narrowing
Abnormal reflexes of GI tract
Dysphagia
Indigestion
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published Dec 15, 2024 on www.thecalgaryguide.com")
Pituitary Tumour Classification and Clinical Outcomes
 (lack of hormone-producing cells or due to gene mutations)
Authors: Chris Oleynick Caroline Kokorudz Reviewers: Amyna Fidai Laura Byford- Richardson Joseph Tropiano Julia Gospodinov Luiza Radu Hanan Bassyouni* * MD at time of publication
hormones (70%)
Relative abundance & predisposition for lactotroph (prolactin-producing), somatotroph (growth hormone – producing), thyrotroph (TSH producing) & corticotroph (adrenocorticotropic hormone – producing) cells to form adenomas (epithelial cell tumours)
↑ Lactotroph cells
↑ Prolactin (PRL) (most common)
Hyper- prolactinemia (high prolactin blood levels)
↑ Somatotroph cells
↑ Growth hormone (GH)
Acromegaly* (excess tissue growth & metabolic dysfunction)
↑ Corticotroph cells
↑ Adrenocortico- tropic hormone (ACTH)
Cushing disease* (prolonged exposure to excess cortisol)
↑ Thyrotroph cells
↑ Thyroid stimulating hormone (TSH)
Central hyper- thyroidism (↑ T4 & T3)
Large size (macro) (>10mm on MRI) may cause mass effect (tissue compression from mass)
Small size (micro) (<10mm on MRI) unlikely to cause mass effect (compression to surrounding structures)
Asymptomatic
Headache
Nausea & vomiting
Compresses pituitary gland & impairs blood supply & function of pituitary cells
Impinges pituitary stalk & disrupts hormone transport from hypothalamus to the anterior and posterior pituitary
Compresses surrounding structures
↑ Pressure & stretching of dural mater
Stretching of the meninges activates mechanoreceptors (stretch receptors) in GI tract
Pituitary hormone deficiency (typically in left-right order with mass effect):
Dopamine release obstruction
↓ Inhibition of prolactin secretion
Antidiuretic hormone (ADH) obstruction
Inferior tumour growth
Growth into sphenoid sinus
Lateral growth of the tumor compresses surrounding cranial nerves
Superior tumour growth
↓ GH
↓ Protein production & muscle cell proliferation
↓ Luteinizing hormone (LH) (triggers ovulation & sex hormone synthesis) & follicle stimulating hormone (FSH) (stimulates ovarian follicles & sperm growth)
↓ (TSH)
↓ ACTH (stimulates cortisol & androgen release)
Cranial nerve (CN) II (optic)
↓ Visual acuity
Diplopia (double vision)
CN III, (oculomotor) IV (trochlear), or VI (abducens)
Ptosis* (drooping upper eyelid)
CN V (trigeminal) branches V1 & V2
Facial numbness
Ophthalmoplegia (eye muscle paralysis)
Compresses optic chiasma
Bitemporal hemianopsia (decreased lateral peripheral vision)
Tumour extends into hypothalamus
Hypothalamic dysfunction due to damage to hypothalamic cells
Disruption of multiple regulatory systems (i.e. sleep-wake cycles, appetite, temperature)
Tumour occludes ventricles
Tumour obstructs CSF flow
↑ Intracranial pressure
Hydrocephalus* (abnormal buildup of CSF in ventricles of the brain) and papilledema
Bacteria migrates from sinus flora into sphenoid sinus
Cerebrospinal fluid (CSF) leaks into throat
Post-nasal drip and nasopharyngeal mass
Stunted growth and short stature in children
↓ Muscle mass & fatigue
Diabetes insipidus* (excessive urination & extreme thirst)
Central hypothyroidism* (↓ T4 & T3)
Hyperprolactinemia
Meningitis* (inflammation of the meningeal layers of central nervous system)
*See relevant Calgary Guide slide
Hypogonadotropic hypogonadism (↓ Sex hormones)
Adrenal insufficiency* (↓ 8 AM cortisol)
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published Oct 1, 2027; updated Jan 5, 2025 on www.thecalgaryguide.com")
Central Precocious Puberty
, trauma, infection, ischemia
Damage to hypothalamic & pituitary structures (i.e. infarct or traumatic sheering forces often causing inflammation and ultimately cell death)
Septo-optic dysplasia (early brain development disorder)
Abnormal pituitary & hypothalamus development (unknown pathogenesis)
Psychosocial stress
International adoption from developing countries
Rapid increase in nutrition
Rapid catch-up growth
Hormonal and metabolic changes
Endocrine disrupting chemicals (ex. Phthalates, bisphenol A)
Hypothalamic inflammation
Suppression of inhibitory and activation of stimulatory elements of the GnRH production pathway
Idiopathic familial central precocious puberty from different genetic mutations
primarily impacting tissues derived from ectoderm)
Makorin RING- finger protein 3( MKRN3) mutation (repressor of GnRH secretion)
Kisspeptin protein and/or receptor mutation (regulates puberty & reproductive function)
Extended Kisspeptin protein availability and/or ↑ production
Kisspeptin receptors signal directly to gonadotropin releasing hormone (GnRH) neurons to release GnRH into portal system
Early ↑ or inappropriate GnRH pulses
Neurofibromatos is T1 (NF1) (causes benign tumors to grow in the brain along nerves)*
Tumor development involving the optic chiasm (>15% of individuals with NF1) may impact hypothalamic function
Damage to and/or disruption of normal hypothalamic & pituitary function
Tuberous sclerosis autosomal dominant disorder (rare genetic disease that causes growth of benign tumors in brain & body)
Dysregulated cell division & growth
Hamartomas form as benign mass composed of cells similar to those surrounding it, occurs in many areas including hypothalamic hamartoma (HH)
HH acts as an accessory, unregulated hypothalamus
Autonomous (unregulated) pulsatile GnRH release
Sturge-Weber Syndrome (vascular disorder causing capillary-venus malformations impacting the face, brain, eyes)
Vascular malformation throughout the brain including the hypothalamus
Impaired blood flow to the hypothalamus
Hypothalamic ischemia
Hypothalamus dysregulation and premature activation of GnRH release
Disruption of excitation and inhibition balance in the CNS
Hypothalamic- pituitary dysregulation
Reduced inhibition of GnRH secretion
Increased corticotropin- releasing hormone
Adrenal gland production & release of androgens by unknown mechanism that play a role in maturation of the HPG axis
Adrenarche (start of androgen production). Including: acne, body odor, & pubarche (pubic hair growth)
Dysregulated GnRH pulses
Dysregulated luteinizing hormone (LH) & follicle simulating hormone (FSH) release
Early GnRH release
Early FSH secretion from pituitary gland
↑ Uterine volume & growth of ovarian follicles
Cyclical ovulation
Premature menarche (first menses < 7 years)
Early LH secretion from pituitary
↑ Gonadal androgen production
↑ Estrogen release from ovaries
gland
**See Calgary Guide slide - “Brain Neoplasms”
↑ Skeletal growth
↑ Bone mineral density
Thelarche (breast development)
Authors: Joanna Keough Reviewers: Run Xuan (Karen) Zeng Luiza Radu Samuel Fineblit* *MD at time of publication
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published Jan 28, 2025 on www.thecalgaryguide.com")
Osgood Schlatter Disease

Patellar tendon inserts more proximally or broadly on tibia
↓ Patellar tendon moment arm length causes ↑ biomechanical forces on tibial tubercle
Patella alta (superior displacement of the patella within the trochlear groove) *temporal relationship uncertain
Quadriceps require ↑ forces to achieve full extension
Patellar tendon overuse
↑ Patellar tendon tension
↑ Traction stress on the patellar tendon insertion at the cartilaginous tibial tubercle apophysis in one or both knees
Osgood-Schlatter Disease
Self-limiting osteochondrosis or traction apophysitis (inflammation of the growth plate) of the proximal tibial tubercle at the insertion of the patellar tendon
Tibial tubercle apophysis hypertrophies & develops chronic micro-avulsions (tibial apophyseal ossification centers & cartilage is pulled off the tibial metaphysis)
↑ Inflammation at injury site Proximal patellar tendon micro- Repetitive strain/forces on the proximal tibial apophysis
Tibial apophysis undergoes partial arrest
Asymmetric proximal tibial epiphyseal growth abnormality (posterior > anterior)
Genu recurvatum (knee hyperextension) (rare)
due to traction apophysitis avulses from the tibial apophysis
overcomes bone-tendon attachment strength
Activated immune cells release cytokines at injury site
Sensitization of nociceptors in periosteum and surrounding tissues
Tenderness and Antalgic swelling of gait
patellar tendon
Proximal tibial apophyseal fragmentation (visible on X-ray)
Avulsion fracture of tibial tubercle
Calcium is abnormally deposited in the tendon
Calcific tendinopathy in patellar tendon (visible on X-ray)
Anterior knee pain that worsens with exercise
Inflammatory mediators recruit osteoblast precursors to stabilize injury site
↑ Osteoblast activity drives bone remodeling and formation of ossicles (small pieces of bone)
Bone remodeling causes united ossicles (ossicle fusion with tibial tuberosity)
Ununited ossicle imbedded within patellar tendon (visible on X-ray)
Bony prominence of tibial tubercle (visible on X-ray)
Residual ununited ossicle remains after apophysis fuses with proximal tibial metaphysis
Persistent visible prominence after proximal tibial apophyseal closure
Authors: Emily J. Doucette Reviewers: Michelle J. Chan Yan Yu* Gerhard Kiefer* * MD at time of publication
Persistent pain & discomfort with ↓ strength & function
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published Feb 16, 2025 on www.thecalgaryguide.com")
Tonsillitis Pathogenesis and clinical findings

Group A Streptococci (GAS) (most common bacteria)
Group B, C & G Strep,
Fusobacterium necrophorum
Age 5-15 (tonsils have ↑ role in immune function at this age)
Tonsillitis
Inflammation of the tonsils
Infectious agent exposure
Susceptible host Pathogen colonizes the oropharynx
Acute suppurative disease
Immune cells release proinflammatory cytokines & antibodies
Inflammatory mediators ↑ vascular permeability of tonsils
Leakage of protein & fluid into surrounding tissue
Regional nodes receive ↑ lymph
Enlarged anterior cervical nodes
Sinusitis**
Pharyngitis**
Local spread of pathogen
Acute otitis media**
Pneumonia**
Cervical lymphadenitis
Bacteria spread from
tonsils into lymphatic system & bloodstream
Bacteremia
F. necrophorum
invades lateral pharyngeal space & soft tissue in neck
Thrombosis forms in peritonsillar vein
Thrombosis extends into internal jugular vein
Lemierre’s syndrome
Systemic inflammatory cytokines disrupt hypothalamic regulation
Fever
Additional immune cells are recruited to facilitate immune response
Macrophages phagocytize pathogen
Bacteria invade distant tissue & elicit local inflammatory response
Hepatitis Osteomyelitis
Infective endocarditis
Bacteria illicit systemic response
Sepsis
Tonsillar tissue become swollen & irritated
Tonsillar hypertrophy
Localized collection of pus forms
Immune cells cause inadvertent cellular injury & hemolysis
Palatal petechiae
Meningitis
Products of immune response & cellular debris are deposited into tonsillar tissue
Peritonsillar or Tonsillar retropharyngeal abscess** exudate
** See corresponding Calgary Guide slide
Toxin-mediated disease
Bacteria release exotoxins into bloodstream
Inflammatory mediators & cytokines are overactivated (cytokine storm)
Skin has local inflammatory response
Toxic shock syndrome**
Scarlet fever**
Post-infectious disease
Antibodies to GAS cross react with host tissue
Acute rheumatic fever** Post-strep glomerulonephritis
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published Nov 5, 2018; updated Mar 14, 2025 on www.thecalgaryguide.com")
Acute Infectious Mononucleosis
, Cytomegalovirus
(CMV), human immunodeficiency
virus (HIV), etc) through saliva
(most commonly in adolescents
& young adults aged 15-24)
Virus infects epithelial
cells of the oropharynx
Virus infects & immortalizes
circulating B lymphocytes
Virus replicates in infected B-cells
Immune cell proliferation
in lymphatic system
(primarily lymph nodes & spleen)
Infected B-cells enter
systemic circulation
Systemic inflammatory
response activated
Inflammation &
edema of sinuses
Bilateral periorbital &/or palpebral edema
Pharyngitis**
(often with grey/white
exudative secretions)
Palatal petechiae
Blood vessel damage
in the soft palate
Edema of soft palate & tonsils
Airway obstruction
↑ Immunoglobulin M (IgM) antibodies
to EBV viral capsid antigen (VCA) Positive IgM-VCA
Immortalized B-cells
produce ↑ antibodies
Production of heterophile antibodies
(weakly reactive & non-specific)
Positive monospot (heterophile antibody)
test (↓ sensitivity in children <4 years)
Virus may remain dormant in B-cells
Latent infection with periodic reactivation
Generalized lymphadenopathy (enlarged lymph nodes)
Massive cervical, mediastinal or hilar lymphadenopathy
Posterior cervical
lymphadenopathy
Platelets sequestered (trapped) within spleen &
overactive spleen (hypersplenism) discards ↑ platelets
Thrombocytopenia
(↓ platelets)
Weak reticular tissue in the spleen stretches
Splenomegaly Splenic rupture
& becomes more susceptible to injury
Inflammatory response ↑ energy demand
Fatigue
Longstanding impacts from
unknown mechanism Chronic Fatigue Syndrome
Inflammatory cytokines
released into circulation
↑ Thermo-regulatory
set-point at hypothalamus Fever
Lymphocytosis (↑ lymphocytes)
(markedly CD8+ T-cells & NK cells) Immune cells infiltrate the liver Hepatomegaly
↑ Aminotransferases
Leukocytosis (↑ leukocytes)
CD8+ T-cells & NK cells
indirectly damage hepatocytes
↑ Bilirubin & jaundice**
Systemic immune response &/or
antibiotic hypersensitivity reaction
CD8+ T-cells
respond to virus
Atypical lymphocytes
on blood smear
Generalized maculopapular rash
Authors:
Ayden Hansen, Griselle Leon
Reviewers:
Charissa Chen, Emily J. Doucette
Danielle Nelson*
* MD at time of publication
Published Sept 3, 2015; updated Apr 22, 2025 on www.thecalgaryguide.com
Pathophysiology Mechanism
Sign/Symptom/Lab Finding Complications")
Trachoma

Mechanical vectors: Eye-seeking
flies (e.g. Musca sorbens)
Chlamydia trachomatis infection (serotype A, B & C)
Trachoma
Chronic keratoconjunctivitis (inflammation of the conjunctiva & cornea) caused by recurrent infection with C. trachomatis
Active Phase
Bacteria infects the conjunctival epithelial cells
Redness, irritation & mucopurulent discharge
Immune response triggers inflammation of the upper tarsal conjunctiva
Herbert’s pits (small, sunken scars at the limbus (border
between cornea & sclera) from healed follicles
Corneal lesions
Pannus (invasion by superficial vessels) at the limbus
Lymphoid follicles consisting mainly of B & T cells form in the upper tarsal conjunctiva
Trachomatous inflammation - follicular: Presence of five or
more white or yellow follicles, each 0.5 mm or bigger in size
Inflammation leads to papillae formation with dilated blood vessels, infiltration
of lymphocytes, plasma cells & neutrophils & proliferation of epithelial cells
Small, raised bumps with a central blood vessel in the upper conjunctiva
Papillary hypertrophy causes conjunctival thickening & opaqueness Trachomatous inflammation - intense: Papillae obscure deep tarsal blood vessels
Repeated re-infection or untreated infections
Cicatricial (scarring) Phase
Proliferation of fibroblasts & deposition of collagen leads to scarring (fibrosis)
Trachomatous scarring: White, horizontal lines or bands on upper tarsal conjunctiva
Scarring blocks
meibomian gland ducts
Conjunctival epithelium atrophy
& goblet cell destruction Subconjunctival
fibrosis
Entropion (eyelid turns inward)
Cornea ulcers
& scarring
↑ Tear evaporation
↓ Tear production
Scar contraction distorts
upper tarsal plate
Dry eye
Trachomatous trichiasis
(eyelashes grow inward
toward the eye)
Corneal
abrasion
Corneal opacity
& blindness
Published Sept, 2 2015; updated May 7, 2025 on www.thecalgaryguide.com
Legend: Pathophysiology Mechanism
Sign/Symptom/Lab Finding Complications")
Meckels Diverticulum
 forms & is trapped in diverticulum
Foreign body enters diverticulum & becomes trapped inside
Incomplete vitelline duct obliteration (~5-7 weeks gestation)
Diverticulum sags into
Intussusception (section
bowel lumen & acts as lead
of intestine slides into
Meckel’s Diverticulum
point for intussusception
adjacent region)
Persistent remnant of vitelline duct containing all 3 layers of
ileal mucosa (asymptomatic in 96-98% of cases)
Diverticulum remains
attached to umbilicus by
persistent fibrous band
Volvulus (loop of ileum twists
around diverticulum)
Ectopic tissue present
within diverticulum
Diverticulum goes
through abdominal
wall defect
Littre’s Hernia
(diverticulum
in hernial sac)
Diverticulum
becomes
incarcerated
Ectopic gastric mucosa
produces acid secretions
Ectopic pancreatic tissue
secretes digestive enzymes
Ileal mucosa adjacent to
diverticulum become ulcerated
Inflammatory mediators
disrupt hypothalamic
regulation
Small bowel
becomes
obstructed
Perforation of
diverticulum
Brisk painless
bleeding from
small bowel
Chronic
inflammation
& ulceration
Fever
Nausea/
vomiting
Small bowel
necrosis
Hematochezia
(bright red blood
per rectum)
↓ Effective
arterial blood
volume
Mucosa undergoes
dysplastic & neoplastic
transformation
Complete
erosion through
ileal mucosa
Small bowel
perforation
Hypotension
Neuroendocrine
tumour or other
cancer
Diverticulum becomes
blocked & triggers
local inflammatory
response
Meckel’s Diverticulitis
Inflammation of Meckel’s
Diverticulum
Diverticulum is
distended due to
inflammation
Visceral afferent
nerves are stimulated
by intestinal stretch
Abdominal pain
Chronic painless
bleeding from
small bowel
Melena (partially
digested blood in
stool)
Iron-deficiency
anemia
Authors:
Taylor Krawec, Merry Faye Graff
Reviewers:
Emily J. Doucette, Sylvain Coderre*
* Indicates MD at time of publication
Legend: Pathophysiology Mechanism
Sign/Symptom/Lab Finding Complications
Intestinal contents
leak into peritoneum
Peritonitis
Systemic inflammatory
response to peritonitis
Published May 31, 2025 on www.thecalgaryguide.com
Sepsis")
Heparin-Induced Thrombocytopenia

Platelets aggregate
within the bloodstream
↓ Free unbound
platelets in bloodstream
Mild Thrombocytopenia:
platelet count between
100 x 109/L - 150 x 109/L
Fewer platelets
available for thrombosis
(formation of a
pathological blood clot)
Self limiting with
no thrombotic risk
Legend: Heparin-Induced Thrombocytopenia (HIT): Pathogenesis and clinical findings
Obesity Hypertension Macrovascular disease Hyperlipidemia
Heparin (often unfractionated)
complexes with platelet factor 4 (PF4)
on the platelet surface, due to
heparin’s negative charge & PF4’s
affinity for long-chain polysaccharides
Immune-Mediated:
Type 2 (within 5-15 days
of heparin administration)
Anti-heparin/PF4
Immunoglobulin G
antibodies binds to
the heparin-PF4
complex in the blood
Splenic macrophages
remove platelet-
immunoglobulin G
complexes in the spleen
Thrombocytopenia:
Platelet count < 100 x
109/L (though < 20 x
109/L should prompt
consideration of
other diagnoses)
↑ Vascular inflammation & endothelial
dysfunction activate platelets
Authors:
Kelle Edgar, Hadi Hassan
Sergio F. Sharif
Reviewers:
Haotian Wang, Jessica Asgarpour
Yan Yu*, Lynn Savoie*
Kareem Jamani*
* MD at time of publication
Activated platelets
release PF4
Anti-heparin/PF4
antibodies activate
adjacent platelets
Activated
platelets release
inflammatory
mediators
Circulating anti-
heparin/PF4 antibodies
bind adjacent cellular
targets
Anti-heparin/PF4
antibodies bind
endogenous heparan
sulfate on endothelium
Activated
endothelium
releases
procoagulants
thrombin &
tissue factor
Activated
platelets release
procoagulants
(factors
promoting blood
clotting)
Free platelets are
consumed from
formation of thrombi
Hypercoagulable state
(↑ risk of thrombosis)
↓ Platelets available
to clot at different
area of the body
Thrombosis in
coronary, peripheral
or cerebral arteries
Bleeding (rare)
Myocardial
infarction
Acute
ischemia
Stroke
Inflammatory
mediators disrupt
hypothalamic body
thermoregulation
Fever/
chills
Inflammatory mediators
promote vasodilation of
arterioles under the skin
↑ Blood to
surface of skin
Flushing of skin
Venous thrombosis
in lower leg or lung
Deep vein
thrombosis
Pulmonary
embolism
Microthrombi occlude
vessels surrounding
injection site, limiting
nutrient supply
Necrosis (tissue death)
at heparin injection site
Published Aug 29, 2014; updated Jun 16, 2025 on www.thecalgaryguide.com
Pathophysiology Mechanism
Sign/Symptom/Lab Finding Complications")
Varicella Zoster Virus
 - Chicken Pox: Pathogenesis and clinical findings Exposure to
individual with
VZV infection
VZV enters
respiratory tract
& conjunctiva
VZV enters
dendritic
cells (DC)
DCs travel to
regional
lymph nodes
Primary viremia
Authors:
Morgan Garland,
Sean Spence
VZV replicates
VZV proteins ↑ major
↓ MHCI
Reviewers:
in nasopharynx
histocompatibility
expression
Yan Yu, Danny Guo,
& CD4+ T cells
complex class I
on CD4+ T
Emily J. Doucette,
(MHCI) degradation
cell surface
Laurie Parsons*,
James D. Kellner*
*MD at time of publication
Acute phase
VZV evades the immune system
VZV travels through the blood in CD4+ T cells
Immune cells release
cytokines to fight the
virus (stronger in adults)
Fluid & cellular debris
accumulate between
epidermal layers
Generalized pruritic (itchy) vesicular
rash at different stages of development
(macule, papule, vesicle)
Immune system activates
& releases inflammatory
compounds
VZV replicates in liver,
spleen & lymph nodes
Erythema
(redness) & pain
Scratching lesions
introduces bacteria
Fibroblasts lay down
collagen to heal skin lesions
Resets thermostat
in hypothalamus
Systemic
response
Tissue damage
in oropharynx
Direct
inflammation in
neural tissues
Cerebellar ataxia
(more common
in children)
Bacterial superinfection from S.
aureus & Streptococcus pyogenes
(more common in children)
Scarring
Fever
(prodromal)
Malaise
(prodromal)
Sore throat
(prodromal)
Encephalitis (more
common in adults)
Abscess
Cellulitis
Toxic shock syndrome (TSS)
Secondary viremia
Immune cells release cytokines to
fight infection (stronger in adults)
Inflammation & fluid
build up in lungs
VZV travels through the blood in CD4+ T
cells expressing skin homing receptors
Fluid & cellular
debris accumulate
in damaged alveoli Lungs more susceptible to
secondary bacterial infection
Pneumonia** (more
common in adults)
VZV colonizes
keratinocytes
over several days
VZV colonizes
neurons & glial
cells in the brain
VZV colonizes
alveolar
epithelial cells
Resolution & latency
VZV tracks along
neurons from sites of
infection to dorsal root
ganglia & cranial nerves
↓ MHCI expression
in sensory ganglia
Adaptive
immune system
controls viremia
↓ Viral gene expression
Resolution of VZV
infection &
development of
lifelong immunity
Direct viral toxicity (DNA damage, mitochondrial
dysfunction, & protein misfolds in infected cells)
Apoptosis & dysfunction of infected cells
Latent
infection
Stress or immune suppression may
precipitate reactivation of VZV later in life Shingles**
**See corresponding Calgary Guide slide
Pathophysiology Mechanism
Published Nov 12, 2012; updated Jun 22, 2025 on www.thecalgaryguide.com
Legend: Sign/Symptom/Lab Finding Complications")
Unconjugated Hyperbilirubinemia
 factor
incompatibility
Maternal sensitization
against Rh-positive fetal cells
Maternal antibodies against Rh
factor attack fetal RBCs
ABO incompatibility
Native maternal antibodies against non-
native blood types attack fetal RBCs
Sepsis** Widespread systemic inflammation Cytokines & complement factors damage RBC membranes
Disseminated intravascular
coagulation** (clotting
proteins become overactive)
Clots form in
systemic circulation
Small clots shear & damage
RBCs in circulation
Red blood cell
(RBC) enzyme
defects
Glucose-6 phosphatase
dehydrogenase (G6PD)
deficiency**
G6PD protects RBCs against
oxidative damage
↑ RBC sensitivity to oxidative
stress (during acute stressors)
Pyruvate kinase
deficiency (PKD)
Abnormal glycolysis & cellular
energy production
↓ RBCs life spans
RBC membrane
defects
Hereditary spherocytosis
RBCs are abnormally round (spherocytes)
RBC
membranes
easily damaged
in circulation
Bilverdin reductase
Hereditary elliptocytosis
RBCs are abnormally elongated or oval
converts bilverdin to
Sickle cell
Abnormal hemoglobin (HbS)
RBCs become abnormally
unconjugated bilirubin
disease**
polymerizes under ↓ O2
sickle shaped
Hemoglobinopathies
Thalassemia's**
Defective hemoglobin
chains in RBCs
Irregularly
shaped RBCs
trapped in
spleen
RBC
sequestration
↑ Production of RBCs (Polycythemia**)
Accumulation of blood (i.e.
Cephalohematoma, hemorrhage)
↑ RBC load &
turnover
Causes of ↓
hepatocellular
bilirubin clearance
Genetic defects in
uridine diphosphate
glucuronosyltransfe
rase (UGT) enzyme
(conjugates
bilirubin in liver)
Gilbert syndrome
Unconjugated bilirubin
↓ UGT production
remains insoluble & cannot
Crigler–Najjar
be excreted in bile
syndrome Type II
Crigler–Najjar
syndrome Type I
No UGT production
Breast milk jaundice Breast milk contains β-glucuronidase
Causes of ↑
entero-hepatic
bilirubin circulation
Intestinal obstruction
Obstruction blocks bile flow from liver to intestines Breastfeeding
jaundice
Inadequate milk intake (volume
depletion/dehydration)
↑ Reabsorption of bilirubin
in the intestines
**See corresponding Calgary Guide slide
Legend: Macrophages engulf
old or damaged RBCs
in spleen & liver
↑ Hemolysis (RBC
breakdown)
RBCs release cellular
contents including heme
(from hemoglobin)
Heme oxygenase
converts heme to
bilverdin
↑ Unconjugated
(indirect) bilirubin
accumulation in blood
↑ Total serum
bilirubin levels
Excess bilirubin
deposition into elastin-
rich tissues (eg. skin,
sclera) due to ↑
affinity for elastin
Pathologic neonatal jaundice
(yellow discoloration of skin
within first 24 hours of life)
Pathophysiology Mechanism
Sign/Symptom/Lab Finding Complications
Published July 13, 2025 on www.thecalgaryguide.com
Authors:
Merry Faye Graff
Khushi Arora
Reviewers:
Annie Pham
Emily J. Doucette
Danielle Nelson*
* MD at time of publication
Hemolytic
anemia**
↓ Bilirubin
conjugation
β-glucuronidase deconjugates bilirubin
Bilirubin builds up
in hepatic system
Unconjugated
bilirubin crosses
blood-brain barrier
Bilirubin-induced
neurologic
dysfunction (BIND)
Kernicterus (bilirubin-
induced neurological
damage)
Scleral icterus
(yellowing of
sclera in eyes)")
Lupus Muco-cutaneous Manifestations

Environmental factors
(ultraviolet radiation,
chemicals, Epstein Barr virus
infection)
Estrogen and prolactin
hormones at birth
Cell damage reduces clearance of
intracellular contents and disrupts
tolerance of immune cells towards
self-tissue
Genetic predisposition (HLA-DR2
& HLA-DR3): Increased
autoimmune disorders
Authors:
Heena Singh
Kevin Zhan
Reviewers:
Bill Ressl
Matthew Harding
Luiza Radu
Liam Martin*
Glen Hazlewood*
*MD at time of publication
Other autoimmune diseases
such as thyroid disorders and
Sjorgen’s disease
Scaring alopecia (hair loss with
scarring)
Antibodies bind self-antigens & form soluble
immune complexes (ICx) that deposit and
promote inflammation in joints, skin & kidney
Circulating ICx cause
vascular/muco- membranous
changes
ICx activate and use up complement
to induce tissue damage
↓ Complement levels (C3/C4) in
blood (proteins involved in
immune response)
**See corresponding Calgary Guide slide
Legend: Mechanism
B cells are activated to
produce autoantibodies
towards intracellular contents
↑ Anti-DNA antibodies
(anti-Ro, anti-La, anti-
Sm, anti-RNP)
ICx stimulate Langerhan cells and
keratinocytes to release cytokines which
induce localized inflammatory response
Subacute cutaneous
lupus: scaly annular
lesions and plaques form
in sun exposed areas
Red plaques with keratotic scaling that
extend inflammation onto hair follicles
resulting in follicular plugging
Discoid rash (chronic,
scaring)
Ultraviolet light ↑ cell death of keratinocytes within the skin
Abnormal removal by phagocytes ↑ local inflammation on skin
ICx & immunoglobulin deposit at the
dermal-epidermal junction (not
restricted to mucous membranes)
Mouth/nasal
ulcers
Inflammation destroys hair follicles
through complex mechanism
Alopecia**
Vasospasm of blood vessels in skin dermal layer (↓ release
and activity of nitric oxide on endothelium as a vasodilator ) &
↓ venous circulation
Malar (butterfly) rash
Photosensitivity
Livedo reticularis (net-like blue
skin discoloration)
Vasculitis
Raynaud’s phenomenon:
white/blue/red discoloration of
toes and fingers induced by
stress and cold
Pathophysiology Sign/Symptom/Lab Finding Complications
Published May 23, 2013; updated Aug 12, 2025 on www.thecalgaryguide.com")
Chronic Thromboembolic Pulmonary Hypertension CTEPH Pathogenesis
: Pathogenesis
History of ↑
hypercoagulability from ↑
plasma clotting factor VIII
Impaired clot breakdown
from fibrin being resistant to
plasmin-mediated lysis
Impaired angiogenesis from
improper vascular endothelial
growth factor function
Inflammation from chronic
conditions or infected
medical implants
Legend: ↑ Incidence &
persistence of clots
Splenectomy
(etiology unknown)
Pro-thrombotic &
impaired healing state
Fibrin clots embed into blood vessel walls of pulmonary vasculature
Pulmonary vasculature undergoes cellular remodeling in response to remnant clots
Hypoxia & growth
factors drives
smooth muscle
proliferation
Chronic growth factors
trigger faulty
angiogenesis in
pulmonary vasculature
Clots activate platelets to
release platelet derived
& vascular endothelial
growth factors
Clots activate T cells to
release pro-inflammatory
cytokines Interleukin-6 &
tissue necrosis factor-α
Cytokines activate
macrophages to
release transforming
growth factor-β (TGF-β)
TGF-β triggers
fibroblasts to
deposit extra cellular
matrix (ECM)
Abnormal cell proliferation narrows lumen & impairs vasodilation Chronic inflammation & ECM deposits cause vessel thickening & fibrosis
Vascular obstruction & fibrosis ↓ pulmonary
arterial cross-sectional area & ↑ vascular resistance
CT pulmonary angiography shows
pulmonary artery occlusions with
clots, webs, & stenotic lesions
↑ Blood pressure in unoccluded vessels of the pulmonary
vasculature causes pulmonary hypertension**
↑ Pulmonary vascular resistance reduces
cardiac output, especially during exertion
CTEPH (Chronic Thromboembolic pulmonary hypertension):
Prolonged occlusion of the pulmonary arteries due to intraluminal fibrosis of
thromboembolic material from unresolved PE clots
Exertional dyspnea**
(difficulty breathing with
physical activity)
Progressive ↑ right ventricular pressure causes right
atrial dilation & right ventricle hypertrophy
Echocardiogram shows enlarged
right atrium & right ventricle
Right heart failure**
**See corresponding Calgary Guide Slide(s)
Authors:
Dinusha T. Senaratne
Devansh Bhatt
Reviewers:
Midas (Kening) Kang
Usama Malik
Sergio Sharif
Natalie Morgunov*
Lian Szabo*
Jonathan Liu*
* MD at time of publication
Published Feb 7, 2018; updated Aug 13, 2025 on www.thecalgaryguide.com
Pathophysiology Mechanism
Sign/Symptom/Lab Finding Complications")
Cystic Fibrosis

Cystic Fibrosis (CF): Pathogenesis, clinical findings, and complications
Cystic Fibrosis Transmembrane Regulator (CFTR) autosomal recessive gene mutation on chromosome 7
CFTR protein (transmembrane chloride ion
channel found in exocrine tissue) dysfunction
Mutated CFTR
proteins prevent
Cl- reabsorption
in sweat glands
↑ Secretion of
Cl- into sweat
↑ Sweat Cl-
concentration
Mutated CFTR proteins in duct epithelial
tissue of other parts of the body prevent
diffusion of Cl- into secretions
↓ Cl- diffusion into peri-ciliary fluid
↓ Water composition of peri-ciliary fluid
↓ Clearance of mucociliary secretions
Secretions accumulate in secretory
passages throughout the body
Inhibition of sperm transport
(obstructive azoospermia)
Male
infertility
Upper Respiratory Tract Manifestations
Retained secretions
in sinuses
Failure to clear
bacteria in sinuses
Persistent neutrophilic inflammation triggers
tissue remodeling & mucosal overgrowth
Bacterial
proliferation
Nasal
polyps
Chronic
sinusitis
Pancreatic Manifestations
Trapped digestive
enzymes degrade
pancreatic tissue
Pancreatic tissue
damage triggers
inflammation,
scarring & fatty
tissue replacement
Islet cell damage
& destruction
Cystic-fibrosis related
diabetes (CFRD)
Lower Respiratory Tract Manifestations
Retained secretions
in airways
Bacterial proliferation
in lower airway
Airway infection
& inflammation
Chronic
productive cough
Signs of obstructive lung disease (lung hyperinflation
on x-ray & abnormal pulmonary function tests)
Bronchitis ±
bronchiectasis**
↓ Production & secretion of
pancreatic enzymes into GI
tract (pancreatic insufficiency)
Fat & protein malabsorption
Failure to
thrive
↓ Absorption of
fat-soluble vitamins
Steatorrhea
(↑ fat in stool)
Vitamin D
deficiency
Vitamin K
deficiency**
Rickets**
Osteoporosis**
Coagulopathies
Hepatic Manifestations
Delayed passage of bile
through biliary tree
↑ Loss of bile acids in stool
Inflammatory hepatic
response
↑ Production of lithogenic bile (bile
supersaturated with cholesterol)
Biliary cirrhosis with
portal hypertension
Cholelithiasis**
Gastrointestinal (GI) Manifestations
↓ Movement of
intestinal contents
In newborns:
Meconium ileus
In children/adults: Distal ileal
obstruction syndrome (DIOS)
↑ Retention
of meconium
↑ Reabsorption
of bilirubin
Prolonged jaundice
in neonates
**See corresponding Calgary Guide slide
Legend: Sign/Symptom/Lab Finding Complications
Pathophysiology Mechanism
Published Jan 21, 2013; updated Aug 20, 2025 on www.thecalgaryguide.com
Reproductive Manifestations
Degeneration of Wolffian duct derivatives
(vas deferens, epididymis, & seminal vesicles)
Inhibition of sperm transport
(obstructive azoospermia)
Male
infertility
Cystic Fibrosis (CF): Pathogenesis, clinical findings, and complications
Cystic Fibrosis Transmembrane Regulator (CFTR) autosomal recessive gene mutation on chromosome 7
CFTR protein (a transmembrane chloride ion
channel that is found in exocrine tissue) dysfunction
Authors:
Navdeep Goraya, Spencer Montgomery
Reviewers:
Yan Yu, Kayla Nelson, Emily J. Doucette,
Mark Montgomery*, Danielle Nelson*
*MD at time of publication
Mutated CFTR
proteins prevent
Cl- reabsorption
in sweat glands
↑ Secretion of
Cl- into sweat
↑ Sweat Cl-
concentration
Mutated CFTR proteins in duct epithelial
tissue of other parts of the body prevent
diffusion of Cl- into secretions
↓ Cl- diffusion into peri-ciliary fluid
↓ Water composition of peri-ciliary fluid
↓ Clearance of
mucociliary secretions
Secretions accumulate in secretory
passages throughout the body
Upper Respiratory Tract Manifestations
Retained secretions
in sinuses
Failure to clear
bacteria in sinuses
Persistent neutrophilic inflammation triggers
tissue remodeling & mucosal overgrowth
Bacterial
proliferation
Nasal
polyps
Chronic
sinusitis
Lower Respiratory Tract Manifestations
Retained secretions
in airways
Bacterial proliferation
in lower airway
Airway infection
& inflammation
Chronic
productive cough
Signs of obstructive lung disease (lung hyperinflation
on x-ray & abnormal pulmonary function tests)
Bronchitis ±
bronchiectasis**
Pancreatic Manifestations
Pancreas unable to
secrete digestive enzymes
into GI tract (pancreatic
insufficiency)
Fat & protein
malabsorption
↓ Absorption of
fat-soluble vitamins
Failure to
thrive
↓ Serum Vitamin D
Osteoporosis**
Trapped digestive
enzymes degrade
pancreatic tissue
Tissue damage
triggers inflammation,
scarring & fatty tissue
replacement
Islet cell
destruction
Cystic-fibrosis related
diabetes (CFRD)
Hepatic Manifestations
Delayed passage of bile
through biliary tree
Inflammatory hepatic
response
Cirrhosis** & portal
hypertension
Gastrointestinal Manifestations
↓ Movement of
intestinal contents
In newborns:
Meconium ileus
In children/adults: Distal ileal
obstruction syndrome (DIOS)
↑ Retention
of meconium
↑ Reabsorption
of bilirubin
Prolonged jaundice
in neonates
Legend: Pathophysiology Mechanism
Sign/Symptom/Lab Finding Complications
**See corresponding Calgary Guide slide
Published January 21, 2013 on www.thecalgaryguide.com
Please only review slide 1 – slides 3-7 are previous draft
versions.
Thank you!
Authors:
Spencer Montgomery, Navdeep Goraya
Reviewers:
Yan Yu, Kayla Nelson, Emily J. Doucette,
Mark Montgomery*, Name Name*
*MD at time of publication
In the vas deferens
in utero
Cystic Fibrosis: Pathogenesis, clinical findings, and complications
Cystic Fibrosis Transmembrane Regulator (CFTR) autosomal recessive gene mutation on chromosome 7
CFTR protein (a transmembrane chloride ion
channel that is found in exocrine tissue) dysfunction
Mutated CFTR
proteins prevent
Cl- reabsorption
in sweat glands
↑ Secretion of
Cl- into sweat
↑ Sweat Cl-
concentration
Mutated CFTR proteins in duct epithelial
tissue of other parts of the body prevent
diffusion of Cl- into secretions
↓ Cl- diffusion into peri-ciliary fluid
↓ Water composition of peri-ciliary fluid
↓ Clearance of
mucociliary secretions
Secretions accumulate in secretory
passages throughout the body
Degeneration of vas deferens, Wolffian
ducts & associated structures
Infertility in
affected males
In upper
respiratory
tract
Retained
secretions
in sinuses
Failure to clear
bacteria in
airways
Persistent neutrophilic inflammation triggers
tissue remodeling & mucosal overgrowth
Bacterial
proliferation
Chronic
sinusitis
Nasal polyps
In lower
respiratory
tract
Chronic
productive cough
Retained
secretions in
airways
Bacterial
proliferation
Airway
infection &
inflammation
Signs of obstructive lung disease (lung hyperinflation
on x-ray & abnormal pulmonary function tests)
Bronchitis ±
bronchiectasis**
In pancreas
Pancreas unable to secrete
digestive enzymes into GI tract
(pancreatic insufficiency)
Fat & protein
malabsorption
↓ Absorption of fat-
soluble vitamins
Failure to
thrive
↓ Serum Vitamin D
Osteoporosis**
Trapped digestive
enzymes degrade
pancreatic tissue
Tissue damage triggers
inflammation, scarring
& fatty tissue
replacement
Islet cell
destruction
Cystic-fibrosis related
diabetes (CFRD)
In biliary tree
Delayed
passage of bile
Inflammatory hepatic
response
Cirrhosis** &
portal
hypertension
In GI tract
↓ Movement
of intestinal
contents
In children/adults: Distal ileal
obstruction syndrome (DIOS)
In newborns:
Meconium
ileus
↑ Retention
of meconium
↑ Reabsorption
of bilirubin
Prolonged
jaundice in
neonates
Legend: Pathophysiology Mechanism
Sign/Symptom/Lab Finding Complications
Published January 21, 2013 on www.thecalgaryguide.com
Authors:
Spencer Montgomery, Navdeep Goraya
Reviewers:
Yan Yu, Kayla Nelson, Emily J. Doucette,
Mark Montgomery*, Name Name*
*MD at time of publication
In the vas deferens
in utero
Cystic Fibrosis: Pathogenesis, clinical findings, and complications
Cystic Fibrosis Transmembrane Regulator (CFTR) autosomal recessive gene mutation on chromosome 7
CFTR protein (a transmembrane chloride ion
channel that is found in exocrine tissue) dysfunction
Mutated CFTR
proteins prevent
Cl- reabsorption
in sweat glands
↑ Secretion of
Cl- into sweat
↑ Sweat Cl-
concentration
Mutated CFTR proteins in duct epithelial
tissue of other parts of the body prevent
diffusion of Cl- into secretions
↓ Cl- diffusion into peri-ciliary fluid
↓ Water composition of peri-ciliary fluid
↓ Clearance of
mucociliary secretions
Secretions accumulate in secretory
passages throughout the body
Degeneration of vas deferens, Wolffian
ducts & associated structures
Infertility in
affected males
In upper
respiratory
tract
Retained
secretions
in sinuses
Failure to clear
bacteria in
airways
Persistent neutrophilic inflammation triggers
tissue remodeling & mucosal overgrowth
Bacterial
proliferation
Chronic
sinusitis
Nasal polyps
In lower
respiratory
tract
Chronic
productive cough
Retained
secretions in
airways
Bacterial
proliferation
Airway
infection &
inflammation
Signs of obstructive lung disease (lung hyperinflation
on x-ray & abnormal pulmonary function tests)
Bronchitis ±
bronchiectasis**
Trapped digestive
enzymes degrade
pancreatic tissue
Inflammation
Scarring & fatty
tissue infiltration
Islet cell
destruction
Type II Diabetes
Mellitus**
In pancreas
Pancreas unable to secrete
digestive enzymes into GI tract
(pancreatic insufficiency)
Fat & protein
malabsorption
↓ Absorption of fat-
soluble vitamins
Failure to
thrive
↓ Serum Vitamin D
Osteoporosis**
In biliary tree
Delayed
passage of bile
Inflammatory hepatic
response
Cirrhosis** &
portal
hypertension
In GI tract
↓ Movement
of intestinal
contents
In children/adults: Distal ileal
obstruction syndrome (DIOS)
In newborns:
Meconium
ileus
↑ Retention
of meconium
↑ Reabsorption
of bilirubin
Prolonged
jaundice in
neonates
Legend: Pathophysiology Mechanism
Sign/Symptom/Lab Finding Complications
Published January 21, 2013 on www.thecalgaryguide.com
In the vas deferens
in utero
Retained secretions
in sinuses
Cystic Fibrosis: Pathogenesis, clinical findings, and complications
Cystic Fibrosis Transmembrane Regulator (CFTR) autosomal recessive gene mutation on chromosome 7
CFTR protein (a transmembrane chloride ion
channel that is found in exocrine tissue) dysfunction
Mutated CFTR
proteins prevent
Cl- reabsorption
in sweat glands
↑ Secretion of
Cl- into sweat
↑ Sweat Cl-
concentration
Mutated CFTR proteins in duct epithelial
tissue of other parts of the body prevent
diffusion of Cl- into secretions
↓ Cl- diffusion into peri-ciliary fluid
↓ Water composition of peri-ciliary fluid
↓ Clearance of
mucociliary secretions
Accumulation of secretions in
secretory passages throughout the
body obstructing these passages
Authors:
Spencer Montgomery, Navdeep Goraya
Reviewers:
Yan Yu, Kayla Nelson,
Emily J. Doucette, Mark Montgomery*
*MD at time of publication
Degeneration of vas deferens, Wolffian
ducts & associated structures
Infertility in
affected males
In upper
respiratory
tract
Failure to clear
bacteria in
airways
Bacterial
proliferation
Chronic
sinusitis
Nasal polyps
In lower
respiratory
tract
Chronic
productive
cough
Retained
secretions in
airways
Bacterial
proliferation
Airway
infection &
inflammation
Signs of obstructive lung disease i.e. lung
hyperinflation on x-ray & abnormal pulmonary
function tests
Bronchitis ±
bronchiectasis
Trapped digestive
enzymes degrade
pancreatic tissue
Inflammation
Scarring & fatty
tissue infiltration
Islet cell
destruction
Type II Diabetes
Mellitus
In pancreas
Pancreas unable to secrete
digestive enzymes into GI tract
(pancreatic insufficiency)
Fat and protein
malabsorption
↓ Absorption of fat-
soluble vitamins
Failure to
thrive
↓Serum Vitamin D
Osteoporosis
In biliary tree
Delayed
passage
of bile
Inflammatory hepatic
response
Cirrhosis & portal
hypertension
In GI tract
↓ Movement
of intestinal
contents
In children/adults: Distal ileal
obstruction syndrome (DIOS)
In newborns:
Meconium
ileus
↑ Retention
of meconium
↑ Reabsorption
of bilirubin
Prolonged
jaundice in
neonates
Legend: Pathophysiology Mechanism
Sign/Symptom/Lab Finding Complications
Published January 21, 2013 on www.thecalgaryguide.com
In the vas
deferens
in utero
Cystic Fibrosis: Pathogenesis, clinical findings, and complications
Cystic Fibrosis Transmembrane Regulator (CFTR) autosomal recessive gene mutation on chromosome 7
CFTR protein (a transmembrane chloride ion channel that is found in
exocrine tissue) dysfunction
Chloride channel no longer allows Cl- transport
CFTR proteins in
sweat glands reabsorb
Cl-
CFTR proteins in duct epithelial tissue of
other parts of the body facilitate diffusion
of Cl- into secretions
↓Reabsorption
↓Cl- diffusion into peri-ciliary fluid
↓Water composition of peri-ciliary fluid
↑Secretion of Cl-
into sweat
↓Clearance of mucociliary secretions
↑Sweat Cl-
concentration
Accumulation of secretions in secretory
passages throughout the body obstructing
these passages
Degeneration of vas deferens, Wolffian
ducts & associated structures
Authors:
Spencer Montgomery, Navdeep Goraya
Reviewers:
Yan Yu, Kayla Nelson,
Emily J. Doucette, Mark Montgomery*
*MD at time of publication
Infertility in
affected males
In upper
respiratory
tract
Retained secretions
in sinuses
Nasal polyps
Bacterial
proliferation
Chronic
sinusitis
In lower
respiratory
tract
Chronic
productive
cough
Retained
secretions in
airways
Bacterial
proliferation
Signs of obstructive lung disease
i.e. lung hyperinflation on x-ray &
abnormal pulmonary function
tests
Airway
infection &
inflammation
Bronchitis ±
bronchiectasis
Trapped digestive
enzymes degrade
pancreatic tissue
Inflammation
Scarring & fatty
tissue infiltration
Islet cell
destruction
Type II Diabetes
Mellitus
In pancreas
Pancreas unable to secrete
digestive enzymes into GI tract
(pancreatic insufficiency)
Fat and protein
malabsorption
↓Absorption of fat-
soluble vitamins
Failure to
thrive
↓Serum Vitamin D
Osteoporosis
In biliary tree
Delayed
passage
of bile
Inflammatory hepatic
response
Cirrhosis & portal
hypertension
In GI tract
↓Movement
of intestinal
contents
In children/adults: Distal ileal obstruction
syndrome (DIOS)
In
newborns:
Meconium
ileus
↑Retention
of meconium
↑Reabsorption
of bilirubin
Prolonged
jaundice
in
neonates
Legend: Pathophysiology Mechanism
Sign/Symptom/Lab Finding Complications
Published January 21, 2013 on www.thecalgaryguide.com
In the vas
deferens
in utero
Degeneration of
vas deferens,
Wolffian ducts
and associated
structures
Infertility in
affected males
Legend: Cystic Fibrosis: Pathogenesis, clinical findings, and complications
Mutation of Cystic Fibrosis Transmembrane Regulator (CFTR) gene on chromosome 7 à
Dysfunction of the CFTR protein (a transmembrane chloride ion channel that is found in exocrine tissue)
Author: Spencer Montgomery
Reviewers: Yan Yu, Kayla
Nelson, Mark Montgomery*
* MD at time of publication
Chloride channel no longer allows Cl- transport
In sweat glands, CFTR
proteins are
responsible for the
reabsorption of Cl-
In duct epithelial tissue of other parts of
the body, CFTR proteins facilitate diffusion
of Cl- into secretions
Notes:
• The CFTR mutation exhibits an autosomal recessive inheritance pattern
• > 1700 different CFTR gene mutations are identified, ∆F508 mutation accounts for
~67% of cases in Caucasians.
• Cystic fibrosis is diagnosed based presence of ↑ sweat chloride concentration,
disease causing CFTR mutations, & symptoms of ≥ 1 associated organ system
↓ Cl- diffusion into peri-ciliary fluid →
↓water composition of peri-ciliary fluid
In children/adults: Distal ileal
obstruction syndrome (DIOS)
↓ reabsorption =
↑secretion of Cl-
into sweat
In GI
tract
↓movement
of intestinal
contents
↓ clearance of mucociliary secretions
In
newborns:
Meconium
ileus
↑ retention of
meconium → ↑
reabsorption of
bilirubin
Prolonged
jaundice in
neonates
↑ Sweat chloride
concentration
Accumulation of secretions in secretory
passages throughout the body,
obstructing these passages
In biliary
tree
Delayed passage of bile →
inflammatory hepatic response
Cirrhosis & portal
hypertension
In upper respiratory tract
In pancreas
Trapped digestive
enzymes degrade
pancreatic tissue
Nasal
polyps
Retained
secretions in
sinuses →
bacterial
proliferation
Pancreas unable to secrete
digestive enzymes into GI tract
(pancreatic insufficiency)
Fat and
protein mal-
absorption
↓ absorption of fat
soluble vitamins
Inflammation →
scarring & fatty
tissue infiltration
→ islet cell
destruction
Chronic
sinusitis
↓ serum Vit. D
Type II Diabetes
Mellitus
Failure to
thrive
Osteoporosis
Published January 21, 2013 on www.thecalgaryguide.com
In lower respiratory tract
Chronic
productive
cough
Retained secretions in airways → bacterial proliferation
à Airway infection & inflammation
Persistent respiratory tract infections
Can progress to chronic bronchitis ± bronchiectasis
(This is the biggest cause of death in CF)
Pathophysiology Signs of obstructive lung dx: i.e.
Lung hyperinflation (on x-ray),
Abnormal pulmonary function
tests
Mechanism
Sign/Symptom/Lab Finding Complications")
Hypocalcemia Physiology
![Hypocalcemia: Physiology
Hypomagnesemia**
Pseudohypoparathyroidism
(genetic resistance to PTH)
Sepsis** or severe illness
Authors:
Serra Thai,
Ryan Dion
Reviewers:
Jessica Hammal,
Michelle J. Chen,
Emily J. Doucette,
Hanan Bassyouni*
* MD at time of
publication
Parathyroid gland hypofunction
from surgical removal,
autoimmune disease, or congenital
disease (e.g. DiGeorge Syndrome)
Impaired Mg-dependent
generation of cyclic
adenosine monophosphate
↑ Systemic
inflammation
↑ Calcium
sequestration into
cells (mechanism of
action unknown)
↓ Liver function &
albumin synthesis
↓ Or inappropriately normal
parathyroid hormone (PTH)
in circulation
↓ PTH receptor (PTHR) sensitivity
↓ Albumin-bound
calcium in blood
↓ PTHR signaling in kidneys
↓ PTHR signaling in
osteoblastic lineage
Vitamin D deficiency** (e.g.
cells within bones
↓ intake, malabsorption)
False hypocalcemia (↓
total serum calcium with
normal Ca2+ levels)
Acute pancreatitis**
↓ Sodium-hydrogen
exchanger 3 (NHE3)
activity & expression
in proximal tubule
Chronic
kidney
disease
(CKD)**
↓ 1-⍺ hydroxylase
enzyme (converts
inactive vitamin D to
active form) activity
in kidneys
↑ Claudin14 (tight
junction membrane
proteins) expression in
thick ascending limb (TAL)
of the loop of Henle
↓ Transcription of
calcium
transporter genes
(TRPV5, calbindin
D28K, NCX) in
distal convoluted
tubule
↓ Nuclear factor
kappa B ligand
(RANKL) expression
& binding to
receptors on
osteoclast
precursor cells
Pancreatic enzymes are
prematurely activated
↓ Sodium
reabsorption
triggers
electrochemical
gradient
changes
↓ Glomerular
filtration due
to ↓ kidney
function
↓ Activated
vitamin D
(calcitriol)
synthesis
Claudin14 binds cation
channels composed of
Claudin16 & 19 in TAL
tight junctions
Lipase released from
pancreatic
autodigestion breaks
down peripancreatic fat
↓ Renal phosphate
filtration from
blood
↓ Binding of vitamin
D regulated calcium
transporters in
duodenum & jejunum
Binding blocks
paracellular Ca & Mg
transport from tubule
into vasculature
through tight junctions
↓ Probability
of calcium
transport
channels
opening
↓ Osteoclast
differentiation
(responsible
for breaking
down bone)
Free fatty acids bind
ionized Ca2+ to form
insoluble calcium
soaps (saponification)
↓ Circulating ionized
calcium in blood
↑ Phosphate-calcium
crystal formation
↓ Gastrointestinal
↓ Renal reabsorption
↓ Bone resorption
of calcium
of calcium
reabsorption of calcium Hypocalcemia**
(serum [Ca2+]
<2.1mmol/L+)
**See corresponding Calgary Guide slide
Legend: Pathophysiology Mechanism
Sign/Symptom/Lab Finding Complications
Published Aug 23, 2025 on www.thecalgaryguide.com
Hypocalcemia: Physiology
Authors:
Serra Thai
Ryan Dion
Reviewers:
Jessica Hammal
Michelle J. Chen
Emily J. Doucette
Hanan Bassyouni*
* MD at time of
publication
Parathyroid gland dysfunction
from surgical removal,
autoimmune disease, or
congenital disease (e.g. DiGeorge)
↓ Parathyroid hormone (PTH) in circulation
↓ PTHR signaling in kidneys
Chronic kidney
disease (CKD)
↓ Renal
blood flow
↓ Glomerular
filtration
↓ Sodium-hydrogen
exchanger 3 (NHE3)
activity & expression
in proximal tubule
↓ Sodium
reabsorption
Electrochemical
gradient changes
↓ Phosphate
excretion
↑ Phosphate-calcium
complex formation
**See corresponding Calgary Guide slide
Legend: Pathophysiology Mechanism
Hypomagnesemia**
Pseudohypoparathyroidism
(genetic resistance to PTH)
Sepsis** or severe illness
Impaired Mg-dependent
generation of cyclic
adenosine monophosphate
↑ Inflammation
↓ PTH receptor (PTHR) sensitivity
↑ Calcium
sequestration
into cells
(unknown
mechanism of
action)
↓ Liver
function
Vitamin D deficiency (e.g.
↓ intake, malabsorption)
↓ 1-⍺ hydroxylase
enzyme (converts
inactive vitamin D to
active form) activity in
kidneys
↑ Claudin14 (tight
junction membrane
protein) proteins
are expressed in
the thick ascending
loop of Henle
↓ Transcription of
calcium transporter
genes (TRPV5,
calbindin D28K,
NCX) in the distal
convoluted tubule
↓ Synthesis of activated
vitamin D (calcitriol)
Claudin14 binds to
cation channels
composed of
Claudin16 & 19 found
↓ Binding of vitamin D
in TAL tight junctions
regulated calcium
transporters (TRPV6,
calbindin9k, PMCa2B,
NCX2) in the duodenum
& jejunum
Binding blocks paracellular
Ca & Mg transport from
tubule into vasculature
through tight junctions
between cells
↓ Probability
of calcium
transport
channels
opening
↓ Gastrointestinal
reabsorption of Ca
↓ Renal reabsorption of calcium
↓ Synthesis
of albumin
↓ PTHR signaling in
osteoblastic lineage
cells in bones
↓ Nuclear factor kappa
B ligand (RANKL)
expression & binding to
receptors on osteoclast
precursor cells
↓ Osteoclast
differentiation
(responsible for
breaking down bone)
↓ Bone resorption
↓ Albumin-
bound calcium
with normal free
(biologically
active) Ca levels
False
hypocalcemia
Acute pancreatitis
↓ Lipase secretion
from pancreas
↑ Undigested fats in
small intestine
Free fatty acids
percipitate calcium
Hypocalcemia**
(serum [Ca2+]
<2.1mmol/L+)
Published MONTH, DAY, YEAR on www.thecalgaryguide.com
Sign/Symptom/Lab Finding Complications
Hypomagnesemia*
Impaired magnesium-
dependent generation
of cyclic adenosine
monophosphate
↓ PTH receptor (PTHR) sensitivity
Hypocalcemia: Physiology
Authors:
Serra Thai
Ryan Dion
Reviewers:
Jessica Hammal
Michelle J. Chen
* MD at time of publication
Parathyroid gland dysfunction
from surgical removal,
autoimmune disease, or
congenital disease (e.g. DiGeorge)
Pseudohypoparathyroidism
(genetic resistance to PTH)
Sepsis or severe illness
↓ Parathyroid hormone (PTH) in circulation
↓ PTHR signaling in kidneys
Vitamin D deficiency (e.g.
↓ intake, malabsorption)
↓ Sodium-hydrogen
exchanger 3 (NHE3)
activity & expression in
proximal tubule
↓ 1-⍺ hydroxylase
enzyme (converts
inactive vitamin D to its
active form) activity in
the kidneys
↑ Claudin14 (tight
junction membrane
protein) proteins
are expressed in
the thick ascending
loop of Henle
↓ Transcription of
calcium transporter
genes (TRPV5,
calbindin D28K,
NCX) in the distal
convoluted tubule
Chronic kidney
disease
↓ Sodium
reabsorption
↓ Synthesis of activated
vitamin D (calcitriol)
↓ Renal
blood flow
Electrochemical
gradient changes
Claudin14 binds to
cation channels
composed of
Claudin16 & 19 found
in TAL tight junctions
↓ Glomerular
filtration
↓ Phosphate
excretion
↓ Binding of vitamin D
regulated calcium
transporters (TRPV6,
calbindin9k, PMCa2B,
NCX2) in the duodenum
& jejunum
Binding blocks paracellular
Ca (and Mg) transport
from the tubule into
vasculature through tight
junctions between cells
↓ Probability
of calcium
transport
channels
opening
↑ Phosphate-calcium
complex formation
↓ Gastrointestinal
reabsorption of Ca
↓ Renal reabsorption of calcium
*See corresponding Calgary Guide slide: “Hypomagnesemia: Physiology”
** See corresponding Calgary Guide slide: “Hypoca;cemia: Clinical Findings”
Legend: ↑ Inflammation
↑ Ca
sequestration
into cells
(unknown
mechanism of
action)
↓ Liver
function
↓ Synthesis
of albumin
↓ PTHR signaling in
osteoblastic lineage
cells in bones
↓ Nuclear factor kappa
B ligand (RANKL)
expression and binding
to its receptors on
osteoclast precursor
cells
↓ Differentiation of
osteoclasts which are
responsible for
breaking down
(resorbing) bone
↓ Bone resorption
↓ Albumin-
bound calcium
with normal
free
(biologically
active) Ca
levels
False
hypocalcemia
Acute pancreatitis
↓ Lipase secretion
from pancreas
↑ Undigested fats in
small intestine
Free fatty acids
percipitate calcium
Hypocalcemia**
(serum [Ca2+]
<2.1mmol/L+)
Complications
Published MONTH, DAY, YEAR on www.thecalgaryguide.com
Pathophysiology Mechanism
Sign/Symptom/Lab Finding
Hypocalcemia: Physiology
Authors:
Serra Thai
Reviewers:
Jessica Hammal
Michelle J. Chen
* MD at time of publication
Parathyroid gland dysfunction
from surgical removal,
autoimmune disease, or
congenital disease (e.g. DiGeorge)
Hypomagnesemia**
Pseudohypoparathyroidism
(genetic resistance to PTH)
Sepsis or severe illness
Impaired magnesium-
dependent generation
of cyclic adenosine
monophosphate
↑ Inflammation
↓ Parathyroid hormone (PTH) in circulation
↓ PTH receptor (PTHR) sensitivity
↑ Ca
sequestration
into cells
(unknown
mechanism of
action)
↓ Liver
function
↓ Synthesis
of albumin
↓ PTHR signaling in kidneys
Vitamin D deficiency (e.g.
↓ intake, malabsorption)
↓ Sodium-hydrogen
exchanger 3 (NHE3)
activity & expression in
proximal tubule
↓ 1-⍺ hydroxylase
enzyme (converts
inactive vitamin D to its
active form) activity in
the kidneys
↑ Claudin14 (tight
junction membrane
protein) proteins
are expressed in
the thick ascending
loop of Henle
↓ Transcription of
calcium transporter
genes (TRPV5,
calbindin D28K,
NCX) in the distal
convoluted tubule
Chronic kidney
disease
↓ Sodium
reabsorption
↓ Synthesis of activated
vitamin D (calcitriol)
↓ Renal
blood flow
Electrochemical
gradient changes
Claudin14 binds to
cation channels
composed of
Claudin16 & 19 found
in TAL tight junctions
↓ Glomerular
filtration
↓ Phosphate
excretion
↓ Binding of vitamin D
regulated calcium
transporters (TRPV6,
calbindin9k, PMCa2B,
NCX2) in the duodenum
& jejunum
Binding blocks paracellular
Ca (and Mg) transport
from the tubule into
vasculature through tight
junctions between cells
↓ Probability
of calcium
transport
channels
opening
↑ Phosphate-calcium
complex formation
↓ Gastrointestinal
reabsorption of Ca
↓ Renal reabsorption of calcium
↓ PTHR signaling in
osteoblastic lineage
cells in bones
↓ Nuclear factor kappa
B ligand (RANKL)
expression and binding
to its receptors on
osteoclast precursor
cells
↓ Differentiation of
osteoclasts which are
responsible for
breaking down
(resorbing) bone
↓ Bone resorption
↓ Albumin-
bound calcium
with normal
free Ca levels
due to
homeostatic
mechanisms
False
hypocalcemia
Acute pancreatitis
↓ Lipase secretion
from pancreas
↑ Undigested fats in
small intestine
Free fatty acids
percipitate calcium
Hypocalcemia**
(serum [Ca2+]
<2.1mmol/L+)
Published MONTH, DAY, YEAR on www.thecalgaryguide.com
**See corresponding Calgary Guide slide: “Hypomagnesemia: Physiology”
Legend: Pathophysiology Mechanism
Sign/Symptom/Lab Finding Complications
Hypocalcemia: Physiology
Hypomagnesemia**
Authors:
Serra Thai
Reviewers:
Jessica Hammal
* MD at time of publication
Parathyroid gland
dysfunction: removal,
autoimmune,
congenital (DiGeorge)
Vitamin D
deficiency:
↓intake,
malabsorption
Pseudohypoparathyroidism
Sepsis/severe illness
↓PTHR
activation in
kidneys
CKD
↓ Renal
blood flow
↓ Glomerular
filtration
↓ Sodium-
hydrogen
exchanger 3
(NHE3) activity &
expression in
proximal tubule
↓ Sodium
reabsorption
Electrochemical
gradient changes
↓ Phosphate
excretion
↑ Phosphate calcium
complex formation
**See corresponding Calgary Guide slide(s)
Legend: ↓ 1-alpha
hydroxylase
enzyme activity
↓ Synthesis of
activated vitamin
D (calcitriol)
↓ Binding of vitamin
D regulated calcium
transporters (TRPV6,
calbindin9k, PMCa2B,
NCX2) in the
duodenum & jejunum
↓ Gastrointestinal
reabsorption of
calcium
Pathophysiology Mechanism
Sign/Symptom/Lab Finding Impaired magnesium
dependent generation
of cyclic adenosine
monophosphate ↑ Inflammation
↓ Parathyroid
hormone (PTH)
↑ Claudin14 (tight
junction membrane
protein) activity in
the thick ascending
loop of Henle
↑ Binding of
claudin14 to
claudin16
Inhibition of claudin
16 & 19 from
forming cation
permeable pores in
the tight junctions
↑ Calcium
sequestration
↓ Liver
function
into cells
(mechanism
of action
↓ Synthesis
of albumin
↓ PTH receptor
remains
(PTHR) sensitivity ↓ Albumin-
unknown)
bound calcium
with normal
↓ PTHR
activation in
bones
free Ca levels
due to
homeostatic
mechanisms
↓ Transcription
of calcium
transporter
genes (TRPV5,
calbindin D28K,
NCX) in the distal
convoluted
tubule
↓ Probability
of calcium
transport
channels
opening
↓ Renal reabsorption
of calcium
↓ Osteoblast
stimulation
↓ Nuclear factor
kappa b ligand
(RANKL) secretion
↓ Nuclear factor
kappa b receptor
(RANK) binding on
osteoclast
precursors cells
↓Osteoclast
production
↓ Bone
resorption
False
Hypocalcemia
Acute pancreatitis
↓ Lipase secretion
from pancreas
↑ Undigested fats in
small intestine
Free fatty acids
percipitate calcium
Hypocalcemia**
(serum [Ca2
<2.1mmol/L+])
Published MONTH, DAY, YEAR on www.thecalgaryguide.com
Complications
Hypocalcemia: Physiology
Hypomagnesemia**
Authors:
Serra Thai
Pseudohypoparathyroidism
Reviewers:
Jessica Hammal
* MD at time of publication
Vitamin D
deficiency: ↓
intake,
malabsorption
↓ 1-alpha
hydroxylase
enzyme activity
↓ Synthesis of
activated vitamin
D (calcitriol)
↓ Binding of
vitamin D regulated
calcium transporters
(TRPV6, calbindin9k,
PMCa2B, NCX2) in
the duodenum &
jejunum
↓ Gastrointestinal
reabsorption of
calcium
Parathyroid gland
dysfunction: removal,
autoimmune,
congenital (DiGeorge)
CKD
↓ Renal
blood flow
↓ Glomerular
filtration
**See corresponding Calgary Guide slide(s)
Legend: Sepsis/severe illness
Impaired magnesium
dependent generation of cyclic
adenosine monophosphate ↑ Inflammation
↓ Signaling proteins
↓ Parathyroid
hormone (PTH)
↓ PTH receptor
(PTHR) sensitivity
↑ Calcium
sequestration
into cells
(mechanism
of action
remains
unknown)
Pathophysiology Mechanism
↓PTHR1 activation
in kidneys
↓ Activation of G-
coupled protein
signaling pathways
↑ Expression of
sodium-phosphate
co-transporters
(NaPiIIa & NaPIIc)
in proximal tubule
↓ Expression &
phosphorylation of
transient receptor
potential vanilloid
(TRPV5, calcium
transporter channel) in
distal convoluted tubule
↓ PTHR1 activation
in bones
↓ Osteoblast
↑ Claudin14 (tight
stimulation
junction membrane
protein) activity in
the thick ascending
loop of Henle
↓ Nuclear
factor kappa b
ligand (RANKL)
secretion
↑ Binding of
claudin14 to
claudin16
↓ Nuclear
factor kappa b
receptor
Inhibition of
(RANK) binding
on osteoclast
↓ Phosphate
excretion
Electrochemical
↓ Amount of
claudin 16 & 19
gradient
TRPV5 & ↓
from forming
precursors cells
changes
probability of
cation-permeable
channels
pores in the tight
↑ Phosphate
opening
junctions
calcium
↓Osteoclast
production
complex
formation
↓ Paracellular
transport of
calcium ↓ Renal reabsorption
of calcium
↓ Bone
resorption
Published MONTH, DAY, YEAR on www.thecalgaryguide.com
↓ Liver
function
↓ Synthesis
of albumin
↓ Albumin-
bound calcium
with normal
free calcium
levels due to
homeostatic
mechanisms
False
Hypocalcemia
Acute
pancreatitis
↓ Lipase
secretion
from pancreas
↑ Undigested
fats in small
intestine
Free fatty acids
percipitate
calcium
Hypocalcemia**
(serum [Ca2
<2.1mmol/L+])
Sign/Symptom/Lab Finding Complications
Hypocalcemia: Physiology
Parathyroid gland
dysfunction: removal,
autoimmune,
congenital (DiGeorge)
Hypomagnesemia**
Impaired magnesium
dependent generation
of cyclic adenosine
monophosphate
↓ Parathyroid
hormone (PTH)
↓ PTH
sensitivity
Vitamin D
deficiency:
↓intake,
malabsorption
↓ Enzyme 1-alpha
hydroxylase activity
↓ Synthesis of
activated vitamin
D (calcitriol)
↓ Binding of
vitamin D
regulated calcium
transporters in
the duodenum &
jejunum
↓ Gastrointestinal
reabsorption of
calcium
Legend: ↓PTH receptor
activation in
kidneys
↑ Claudin14
activity in the
thick ascending
loop of Henle
Inhibition of
claudin 16 & 19
from forming
cation permeable
pores in the tight
junctions
↓ Transcription
of calcium
transporter
genes in the
distal
convoluted
tubule
↓ Probability
of calcium
transport
channels
opening
↓ Renal reabsorption
of calcium
↓ Sodium-hydrogen
exchanger 3 (NHE3)
activity & expression in
proximal tubule
↓ Sodium
reabsorption
Electrochemical
gradient changes
↓ Phosphate excretion
↑ Phosphate calcium
complex formation
Pathophysiology Mechanism
Sign/Symptom/Lab Finding Complications
↑ Inflammation
Authors:
Serra Thai
Reviewers:
Jessica Hammal
* MD at time of publication
Sepsis/severe illness
↑ Calcium
sequestration
into cells
↓ Liver
synthesis of
albumin
↓ Albumin-
calcium
binding
False
Hypocalcemia
Acute pancreatitis
↓ Lipase
secretion from
pancreas
↑ Levels of
undigested
fats in small
intestine
Free fatty
acids
percipitate
calcium
↓ PTH receptor
activation in
bones
↓ Osteoblast
stimulation
↓ RANKL
ligand
secretion
↓ RAANK
receptor
binding
↓Osteoclast
production
↓ Bone
resorption
Hypocalcemia
(serum [Ca2+]
<2.1mmol/L)
Published MONTH, DAY, YEAR on www.thecalgaryguide.com
Hypocalcemia: Physiology
Vitamin D
deficiency:
↓intake,
malabsorption
Parathyroid gland
dysfunction: removal,
autoimmune,
congenital (DiGeorge)
Hypomagnesemia**
Impaired magnesium
dependent generation
of cyclic adenosine
monophosphate
↓ Parathyroid
hormone (PTH)
↓ PTH
sensitivity
↓ Enzyme 1-alpha
hydroxylase activity
↓ Synthesis of
activated vitamin
D (calcitriol)
↓ Binding of
vitamin D
regulated calcium
transporters in
the duodenum &
jejunum
↓ Gastrointestinal
reabsorption of calcium
Legend: ↓PTH receptor
activation in
kidneys
↑ Claudin14
activity in the
thick ascending
loop of Henle
Inhibition of
claudin 16 & 19
from forming
cation permeable
pores in the tight
junctions
↓ Transcription
of calcium
transporter
genes in the
distal
convoluted
tubule
↓ Probability
of calcium
transport
channels
opening
↓ Renal reabsorption
of calcium
↓ Sodium-hydrogen
exchanger 3 (NHE3)
activity & expression in
proximal tubule
↓ Sodium
reabsorption
Electrochemical
gradient changes
↓ Phosphate excretion
↑ Phosphate calcium
complex formation
Pathophysiology Mechanism
Sign/Symptom/Lab Finding Complications
↑ Inflammation
Sepsis/severe illness
↑ Calcium
sequestration
into cells
↓ Liver
synthesis of
albumin
↓ Albumin-
calcium
binding
False
Hypocalcemia
Acute pancreatitis
↓ Lipase
secretion from
pancreas
↑ Levels of
undigested
fats in small
intestine
Free fatty
acids
percipitate
calcium
↓ PTH receptor
activation in
bones
↓ Osteoblast
stimulation
↓ RANKL
ligand
secretion
↓ RAANK
receptor
binding
↓Osteoclast
production
↓ Bone
resorption
Hypocalcemia
(serum [Ca2+] <2.1mmol/L)
Authors:
Name Name
Name Name*
Reviewers:
Name Name
Name Name*
* MD at time of publication
Published MONTH, DAY, YEAR on www.thecalgaryguide.com
Hypocalcemia: Physiology
Chronic kidney
disease
↓ Renal
blood flow
↓ Glomerular
filtration
Parathyroid gland
dysfunction: removal,
autoimmune,
congenital (DiGeorge)
Vitamin D deficiency:
↓intake, malabsorption
Sepsis/severe illness
↓ NHE3 activity and
expression in
proximal tubule
↓PTH receptor
activation in
kidneys
↑ Claudin14
activity in the
thick ascending
loop of Henle
↓ Parathyroid
hormone (PTH)
↓ Transcription of
calcium transporter
genes (TRPV5, calbindin
D28K, NCX) in the distal
convoluted tubule
↓ Enzyme 1-alpha
hydroxylase activity
↓ PTH receptor
activation in
bones
↓ osteoblast
stimulation
↓ RANKL ligand
secretion
↑ Inflammation ↓ PTH sensitivity
↓ Glomerular
filtration
↓ Liver
synthesis of
serum albumin
False
Hypocalcemia
↑ Calcium
sequestration
into cells**
↓ Lipase secretion
from pancreas
Acute pancreatitis
Legend: Pathophysiology Mechanism
Sign/Symptom/Lab Finding ↓ Albumin-
bound calcium
Current mechanism
is unknown
↑ levels of undigested
fats in small intestine
Complications
Hypomagnesemia Multiple blood
transfusions
↓ Sodium
reabsorption
Electrochemical
gradient changes
↓ Phosphate
excretion
↑ Phosphate
calcium complex
formation
Inhibits claudin16 and 19
from forming cation
permeable pores in the
tight junctions
↓Probability of
calcium transport
channels opening
↓ Synthesis of activated
vitamin D (calcitriol)
↓ RAANK
receptor
binding
↓ Calcium
reabsorption
↓ Binding of vitamin D
regulated calcium
transporters (TRPV6,
calbindin9K, PMCa2B,
NCX2) in the duodenum
and jejunum
↓osteoclast
production
↓ Bone
resorption
↓ Serum calcium
Hypocalcemia
<2.1 mmol/L
↑ Precipitation of calcium
by free fatty acids
Authors:
Name Name
Name Name*
Reviewers:
Name Name
Name Name*
* MD at time of publication
Published MONTH, DAY, YEAR on www.thecalgaryguide.com
https://journals.physiology.org/doi/full/10.1152/physrev.00003.2004?rfr_dat=cr_pub++0pubmed&url_ver=Z39.88-
2003&rfr_id=ori%3Arid%3Acrossref.org – Calcium Absoprtion across Epithelia
https://academic.oup.com/endo/article-abstract/137/1/13/2498579 - PTH and Calcium Signaling Pathways
https://www.pnas.org/doi/10.1073/pnas.1616733114 - Information on Claudin 14
https://onlinelibrary-wiley-com.ezproxy.lib.ucalgary.ca/doi/full/10.1111/apha.13959 - effects of PTH on renal calcium and
phosphate handling
https://www.ncbi.nlm.nih.gov/books/NBK430912/ - Hypocalcemia overview + causes + pathophys
https://www.orthobullets.com/basic-science/9010/bone-signaling-and-rankl - information about bone signaling
https://www.sciencedirect.com/science/article/abs/pii/S0889852921000682?via%3Dihub – Calcium homeostasis article
https://pubmed.ncbi.nlm.nih.gov/3012979/#:~:text=Abstract,intestine%2C%20require%20the%20parathyroid%20hormone.
vitamin D metabolism and function
–
https://pmc.ncbi.nlm.nih.gov/articles/PMC2669834/#:~:text=Calcium%20is%20actively%20absorbed%20from,for%20proper
%20mineralization%20of%20bone.
– more vitamin D metabolism and specific receptors
https://jidc.org/index.php/journal/article/view/32903236/2331 - sepsis + hypocalcemia](https://calgaryguide.ucalgary.ca/wp-content/uploads/2025/08/Hypocalcemia-Pathogenesis.jpg "Hypocalcemia: Physiology
Hypomagnesemia**
Pseudohypoparathyroidism
(genetic resistance to PTH)
Sepsis** or severe illness
Authors:
Serra Thai,
Ryan Dion
Reviewers:
Jessica Hammal,
Michelle J. Chen,
Emily J. Doucette,
Hanan Bassyouni*
* MD at time of
publication
Parathyroid gland hypofunction
from surgical removal,
autoimmune disease, or congenital
disease (e.g. DiGeorge Syndrome)
Impaired Mg-dependent
generation of cyclic
adenosine monophosphate
↑ Systemic
inflammation
↑ Calcium
sequestration into
cells (mechanism of
action unknown)
↓ Liver function &
albumin synthesis
↓ Or inappropriately normal
parathyroid hormone (PTH)
in circulation
↓ PTH receptor (PTHR) sensitivity
↓ Albumin-bound
calcium in blood
↓ PTHR signaling in kidneys
↓ PTHR signaling in
osteoblastic lineage
Vitamin D deficiency** (e.g.
cells within bones
↓ intake, malabsorption)
False hypocalcemia (↓
total serum calcium with
normal Ca2+ levels)
Acute pancreatitis**
↓ Sodium-hydrogen
exchanger 3 (NHE3)
activity & expression
in proximal tubule
Chronic
kidney
disease
(CKD)**
↓ 1-⍺ hydroxylase
enzyme (converts
inactive vitamin D to
active form) activity
in kidneys
↑ Claudin14 (tight
junction membrane
proteins) expression in
thick ascending limb (TAL)
of the loop of Henle
↓ Transcription of
calcium
transporter genes
(TRPV5, calbindin
D28K, NCX) in
distal convoluted
tubule
↓ Nuclear factor
kappa B ligand
(RANKL) expression
& binding to
receptors on
osteoclast
precursor cells
Pancreatic enzymes are
prematurely activated
↓ Sodium
reabsorption
triggers
electrochemical
gradient
changes
↓ Glomerular
filtration due
to ↓ kidney
function
↓ Activated
vitamin D
(calcitriol)
synthesis
Claudin14 binds cation
channels composed of
Claudin16 & 19 in TAL
tight junctions
Lipase released from
pancreatic
autodigestion breaks
down peripancreatic fat
↓ Renal phosphate
filtration from
blood
↓ Binding of vitamin
D regulated calcium
transporters in
duodenum & jejunum
Binding blocks
paracellular Ca & Mg
transport from tubule
into vasculature
through tight junctions
↓ Probability
of calcium
transport
channels
opening
↓ Osteoclast
differentiation
(responsible
for breaking
down bone)
Free fatty acids bind
ionized Ca2+ to form
insoluble calcium
soaps (saponification)
↓ Circulating ionized
calcium in blood
↑ Phosphate-calcium
crystal formation
↓ Gastrointestinal
↓ Renal reabsorption
↓ Bone resorption
of calcium
of calcium
reabsorption of calcium Hypocalcemia**
(serum [Ca2+]
<2.1mmol/L+)
**See corresponding Calgary Guide slide
Legend: Pathophysiology Mechanism
Sign/Symptom/Lab Finding Complications
Published Aug 23, 2025 on www.thecalgaryguide.com
Hypocalcemia: Physiology
Authors:
Serra Thai
Ryan Dion
Reviewers:
Jessica Hammal
Michelle J. Chen
Emily J. Doucette
Hanan Bassyouni*
* MD at time of
publication
Parathyroid gland dysfunction
from surgical removal,
autoimmune disease, or
congenital disease (e.g. DiGeorge)
↓ Parathyroid hormone (PTH) in circulation
↓ PTHR signaling in kidneys
Chronic kidney
disease (CKD)
↓ Renal
blood flow
↓ Glomerular
filtration
↓ Sodium-hydrogen
exchanger 3 (NHE3)
activity & expression
in proximal tubule
↓ Sodium
reabsorption
Electrochemical
gradient changes
↓ Phosphate
excretion
↑ Phosphate-calcium
complex formation
**See corresponding Calgary Guide slide
Legend: Pathophysiology Mechanism
Hypomagnesemia**
Pseudohypoparathyroidism
(genetic resistance to PTH)
Sepsis** or severe illness
Impaired Mg-dependent
generation of cyclic
adenosine monophosphate
↑ Inflammation
↓ PTH receptor (PTHR) sensitivity
↑ Calcium
sequestration
into cells
(unknown
mechanism of
action)
↓ Liver
function
Vitamin D deficiency (e.g.
↓ intake, malabsorption)
↓ 1-⍺ hydroxylase
enzyme (converts
inactive vitamin D to
active form) activity in
kidneys
↑ Claudin14 (tight
junction membrane
protein) proteins
are expressed in
the thick ascending
loop of Henle
↓ Transcription of
calcium transporter
genes (TRPV5,
calbindin D28K,
NCX) in the distal
convoluted tubule
↓ Synthesis of activated
vitamin D (calcitriol)
Claudin14 binds to
cation channels
composed of
Claudin16 & 19 found
↓ Binding of vitamin D
in TAL tight junctions
regulated calcium
transporters (TRPV6,
calbindin9k, PMCa2B,
NCX2) in the duodenum
& jejunum
Binding blocks paracellular
Ca & Mg transport from
tubule into vasculature
through tight junctions
between cells
↓ Probability
of calcium
transport
channels
opening
↓ Gastrointestinal
reabsorption of Ca
↓ Renal reabsorption of calcium
↓ Synthesis
of albumin
↓ PTHR signaling in
osteoblastic lineage
cells in bones
↓ Nuclear factor kappa
B ligand (RANKL)
expression & binding to
receptors on osteoclast
precursor cells
↓ Osteoclast
differentiation
(responsible for
breaking down bone)
↓ Bone resorption
↓ Albumin-
bound calcium
with normal free
(biologically
active) Ca levels
False
hypocalcemia
Acute pancreatitis
↓ Lipase secretion
from pancreas
↑ Undigested fats in
small intestine
Free fatty acids
percipitate calcium
Hypocalcemia**
(serum [Ca2+]
<2.1mmol/L+)
Published MONTH, DAY, YEAR on www.thecalgaryguide.com
Sign/Symptom/Lab Finding Complications
Hypomagnesemia*
Impaired magnesium-
dependent generation
of cyclic adenosine
monophosphate
↓ PTH receptor (PTHR) sensitivity
Hypocalcemia: Physiology
Authors:
Serra Thai
Ryan Dion
Reviewers:
Jessica Hammal
Michelle J. Chen
* MD at time of publication
Parathyroid gland dysfunction
from surgical removal,
autoimmune disease, or
congenital disease (e.g. DiGeorge)
Pseudohypoparathyroidism
(genetic resistance to PTH)
Sepsis or severe illness
↓ Parathyroid hormone (PTH) in circulation
↓ PTHR signaling in kidneys
Vitamin D deficiency (e.g.
↓ intake, malabsorption)
↓ Sodium-hydrogen
exchanger 3 (NHE3)
activity & expression in
proximal tubule
↓ 1-⍺ hydroxylase
enzyme (converts
inactive vitamin D to its
active form) activity in
the kidneys
↑ Claudin14 (tight
junction membrane
protein) proteins
are expressed in
the thick ascending
loop of Henle
↓ Transcription of
calcium transporter
genes (TRPV5,
calbindin D28K,
NCX) in the distal
convoluted tubule
Chronic kidney
disease
↓ Sodium
reabsorption
↓ Synthesis of activated
vitamin D (calcitriol)
↓ Renal
blood flow
Electrochemical
gradient changes
Claudin14 binds to
cation channels
composed of
Claudin16 & 19 found
in TAL tight junctions
↓ Glomerular
filtration
↓ Phosphate
excretion
↓ Binding of vitamin D
regulated calcium
transporters (TRPV6,
calbindin9k, PMCa2B,
NCX2) in the duodenum
& jejunum
Binding blocks paracellular
Ca (and Mg) transport
from the tubule into
vasculature through tight
junctions between cells
↓ Probability
of calcium
transport
channels
opening
↑ Phosphate-calcium
complex formation
↓ Gastrointestinal
reabsorption of Ca
↓ Renal reabsorption of calcium
*See corresponding Calgary Guide slide: “Hypomagnesemia: Physiology”
** See corresponding Calgary Guide slide: “Hypoca;cemia: Clinical Findings”
Legend: ↑ Inflammation
↑ Ca
sequestration
into cells
(unknown
mechanism of
action)
↓ Liver
function
↓ Synthesis
of albumin
↓ PTHR signaling in
osteoblastic lineage
cells in bones
↓ Nuclear factor kappa
B ligand (RANKL)
expression and binding
to its receptors on
osteoclast precursor
cells
↓ Differentiation of
osteoclasts which are
responsible for
breaking down
(resorbing) bone
↓ Bone resorption
↓ Albumin-
bound calcium
with normal
free
(biologically
active) Ca
levels
False
hypocalcemia
Acute pancreatitis
↓ Lipase secretion
from pancreas
↑ Undigested fats in
small intestine
Free fatty acids
percipitate calcium
Hypocalcemia**
(serum [Ca2+]
<2.1mmol/L+)
Complications
Published MONTH, DAY, YEAR on www.thecalgaryguide.com
Pathophysiology Mechanism
Sign/Symptom/Lab Finding
Hypocalcemia: Physiology
Authors:
Serra Thai
Reviewers:
Jessica Hammal
Michelle J. Chen
* MD at time of publication
Parathyroid gland dysfunction
from surgical removal,
autoimmune disease, or
congenital disease (e.g. DiGeorge)
Hypomagnesemia**
Pseudohypoparathyroidism
(genetic resistance to PTH)
Sepsis or severe illness
Impaired magnesium-
dependent generation
of cyclic adenosine
monophosphate
↑ Inflammation
↓ Parathyroid hormone (PTH) in circulation
↓ PTH receptor (PTHR) sensitivity
↑ Ca
sequestration
into cells
(unknown
mechanism of
action)
↓ Liver
function
↓ Synthesis
of albumin
↓ PTHR signaling in kidneys
Vitamin D deficiency (e.g.
↓ intake, malabsorption)
↓ Sodium-hydrogen
exchanger 3 (NHE3)
activity & expression in
proximal tubule
↓ 1-⍺ hydroxylase
enzyme (converts
inactive vitamin D to its
active form) activity in
the kidneys
↑ Claudin14 (tight
junction membrane
protein) proteins
are expressed in
the thick ascending
loop of Henle
↓ Transcription of
calcium transporter
genes (TRPV5,
calbindin D28K,
NCX) in the distal
convoluted tubule
Chronic kidney
disease
↓ Sodium
reabsorption
↓ Synthesis of activated
vitamin D (calcitriol)
↓ Renal
blood flow
Electrochemical
gradient changes
Claudin14 binds to
cation channels
composed of
Claudin16 & 19 found
in TAL tight junctions
↓ Glomerular
filtration
↓ Phosphate
excretion
↓ Binding of vitamin D
regulated calcium
transporters (TRPV6,
calbindin9k, PMCa2B,
NCX2) in the duodenum
& jejunum
Binding blocks paracellular
Ca (and Mg) transport
from the tubule into
vasculature through tight
junctions between cells
↓ Probability
of calcium
transport
channels
opening
↑ Phosphate-calcium
complex formation
↓ Gastrointestinal
reabsorption of Ca
↓ Renal reabsorption of calcium
↓ PTHR signaling in
osteoblastic lineage
cells in bones
↓ Nuclear factor kappa
B ligand (RANKL)
expression and binding
to its receptors on
osteoclast precursor
cells
↓ Differentiation of
osteoclasts which are
responsible for
breaking down
(resorbing) bone
↓ Bone resorption
↓ Albumin-
bound calcium
with normal
free Ca levels
due to
homeostatic
mechanisms
False
hypocalcemia
Acute pancreatitis
↓ Lipase secretion
from pancreas
↑ Undigested fats in
small intestine
Free fatty acids
percipitate calcium
Hypocalcemia**
(serum [Ca2+]
<2.1mmol/L+)
Published MONTH, DAY, YEAR on www.thecalgaryguide.com
**See corresponding Calgary Guide slide: “Hypomagnesemia: Physiology”
Legend: Pathophysiology Mechanism
Sign/Symptom/Lab Finding Complications
Hypocalcemia: Physiology
Hypomagnesemia**
Authors:
Serra Thai
Reviewers:
Jessica Hammal
* MD at time of publication
Parathyroid gland
dysfunction: removal,
autoimmune,
congenital (DiGeorge)
Vitamin D
deficiency:
↓intake,
malabsorption
Pseudohypoparathyroidism
Sepsis/severe illness
↓PTHR
activation in
kidneys
CKD
↓ Renal
blood flow
↓ Glomerular
filtration
↓ Sodium-
hydrogen
exchanger 3
(NHE3) activity &
expression in
proximal tubule
↓ Sodium
reabsorption
Electrochemical
gradient changes
↓ Phosphate
excretion
↑ Phosphate calcium
complex formation
**See corresponding Calgary Guide slide(s)
Legend: ↓ 1-alpha
hydroxylase
enzyme activity
↓ Synthesis of
activated vitamin
D (calcitriol)
↓ Binding of vitamin
D regulated calcium
transporters (TRPV6,
calbindin9k, PMCa2B,
NCX2) in the
duodenum & jejunum
↓ Gastrointestinal
reabsorption of
calcium
Pathophysiology Mechanism
Sign/Symptom/Lab Finding Impaired magnesium
dependent generation
of cyclic adenosine
monophosphate ↑ Inflammation
↓ Parathyroid
hormone (PTH)
↑ Claudin14 (tight
junction membrane
protein) activity in
the thick ascending
loop of Henle
↑ Binding of
claudin14 to
claudin16
Inhibition of claudin
16 & 19 from
forming cation
permeable pores in
the tight junctions
↑ Calcium
sequestration
↓ Liver
function
into cells
(mechanism
of action
↓ Synthesis
of albumin
↓ PTH receptor
remains
(PTHR) sensitivity ↓ Albumin-
unknown)
bound calcium
with normal
↓ PTHR
activation in
bones
free Ca levels
due to
homeostatic
mechanisms
↓ Transcription
of calcium
transporter
genes (TRPV5,
calbindin D28K,
NCX) in the distal
convoluted
tubule
↓ Probability
of calcium
transport
channels
opening
↓ Renal reabsorption
of calcium
↓ Osteoblast
stimulation
↓ Nuclear factor
kappa b ligand
(RANKL) secretion
↓ Nuclear factor
kappa b receptor
(RANK) binding on
osteoclast
precursors cells
↓Osteoclast
production
↓ Bone
resorption
False
Hypocalcemia
Acute pancreatitis
↓ Lipase secretion
from pancreas
↑ Undigested fats in
small intestine
Free fatty acids
percipitate calcium
Hypocalcemia**
(serum [Ca2
<2.1mmol/L+])
Published MONTH, DAY, YEAR on www.thecalgaryguide.com
Complications
Hypocalcemia: Physiology
Hypomagnesemia**
Authors:
Serra Thai
Pseudohypoparathyroidism
Reviewers:
Jessica Hammal
* MD at time of publication
Vitamin D
deficiency: ↓
intake,
malabsorption
↓ 1-alpha
hydroxylase
enzyme activity
↓ Synthesis of
activated vitamin
D (calcitriol)
↓ Binding of
vitamin D regulated
calcium transporters
(TRPV6, calbindin9k,
PMCa2B, NCX2) in
the duodenum &
jejunum
↓ Gastrointestinal
reabsorption of
calcium
Parathyroid gland
dysfunction: removal,
autoimmune,
congenital (DiGeorge)
CKD
↓ Renal
blood flow
↓ Glomerular
filtration
**See corresponding Calgary Guide slide(s)
Legend: Sepsis/severe illness
Impaired magnesium
dependent generation of cyclic
adenosine monophosphate ↑ Inflammation
↓ Signaling proteins
↓ Parathyroid
hormone (PTH)
↓ PTH receptor
(PTHR) sensitivity
↑ Calcium
sequestration
into cells
(mechanism
of action
remains
unknown)
Pathophysiology Mechanism
↓PTHR1 activation
in kidneys
↓ Activation of G-
coupled protein
signaling pathways
↑ Expression of
sodium-phosphate
co-transporters
(NaPiIIa & NaPIIc)
in proximal tubule
↓ Expression &
phosphorylation of
transient receptor
potential vanilloid
(TRPV5, calcium
transporter channel) in
distal convoluted tubule
↓ PTHR1 activation
in bones
↓ Osteoblast
↑ Claudin14 (tight
stimulation
junction membrane
protein) activity in
the thick ascending
loop of Henle
↓ Nuclear
factor kappa b
ligand (RANKL)
secretion
↑ Binding of
claudin14 to
claudin16
↓ Nuclear
factor kappa b
receptor
Inhibition of
(RANK) binding
on osteoclast
↓ Phosphate
excretion
Electrochemical
↓ Amount of
claudin 16 & 19
gradient
TRPV5 & ↓
from forming
precursors cells
changes
probability of
cation-permeable
channels
pores in the tight
↑ Phosphate
opening
junctions
calcium
↓Osteoclast
production
complex
formation
↓ Paracellular
transport of
calcium ↓ Renal reabsorption
of calcium
↓ Bone
resorption
Published MONTH, DAY, YEAR on www.thecalgaryguide.com
↓ Liver
function
↓ Synthesis
of albumin
↓ Albumin-
bound calcium
with normal
free calcium
levels due to
homeostatic
mechanisms
False
Hypocalcemia
Acute
pancreatitis
↓ Lipase
secretion
from pancreas
↑ Undigested
fats in small
intestine
Free fatty acids
percipitate
calcium
Hypocalcemia**
(serum [Ca2
<2.1mmol/L+])
Sign/Symptom/Lab Finding Complications
Hypocalcemia: Physiology
Parathyroid gland
dysfunction: removal,
autoimmune,
congenital (DiGeorge)
Hypomagnesemia**
Impaired magnesium
dependent generation
of cyclic adenosine
monophosphate
↓ Parathyroid
hormone (PTH)
↓ PTH
sensitivity
Vitamin D
deficiency:
↓intake,
malabsorption
↓ Enzyme 1-alpha
hydroxylase activity
↓ Synthesis of
activated vitamin
D (calcitriol)
↓ Binding of
vitamin D
regulated calcium
transporters in
the duodenum &
jejunum
↓ Gastrointestinal
reabsorption of
calcium
Legend: ↓PTH receptor
activation in
kidneys
↑ Claudin14
activity in the
thick ascending
loop of Henle
Inhibition of
claudin 16 & 19
from forming
cation permeable
pores in the tight
junctions
↓ Transcription
of calcium
transporter
genes in the
distal
convoluted
tubule
↓ Probability
of calcium
transport
channels
opening
↓ Renal reabsorption
of calcium
↓ Sodium-hydrogen
exchanger 3 (NHE3)
activity & expression in
proximal tubule
↓ Sodium
reabsorption
Electrochemical
gradient changes
↓ Phosphate excretion
↑ Phosphate calcium
complex formation
Pathophysiology Mechanism
Sign/Symptom/Lab Finding Complications
↑ Inflammation
Authors:
Serra Thai
Reviewers:
Jessica Hammal
* MD at time of publication
Sepsis/severe illness
↑ Calcium
sequestration
into cells
↓ Liver
synthesis of
albumin
↓ Albumin-
calcium
binding
False
Hypocalcemia
Acute pancreatitis
↓ Lipase
secretion from
pancreas
↑ Levels of
undigested
fats in small
intestine
Free fatty
acids
percipitate
calcium
↓ PTH receptor
activation in
bones
↓ Osteoblast
stimulation
↓ RANKL
ligand
secretion
↓ RAANK
receptor
binding
↓Osteoclast
production
↓ Bone
resorption
Hypocalcemia
(serum [Ca2+]
<2.1mmol/L)
Published MONTH, DAY, YEAR on www.thecalgaryguide.com
Hypocalcemia: Physiology
Vitamin D
deficiency:
↓intake,
malabsorption
Parathyroid gland
dysfunction: removal,
autoimmune,
congenital (DiGeorge)
Hypomagnesemia**
Impaired magnesium
dependent generation
of cyclic adenosine
monophosphate
↓ Parathyroid
hormone (PTH)
↓ PTH
sensitivity
↓ Enzyme 1-alpha
hydroxylase activity
↓ Synthesis of
activated vitamin
D (calcitriol)
↓ Binding of
vitamin D
regulated calcium
transporters in
the duodenum &
jejunum
↓ Gastrointestinal
reabsorption of calcium
Legend: ↓PTH receptor
activation in
kidneys
↑ Claudin14
activity in the
thick ascending
loop of Henle
Inhibition of
claudin 16 & 19
from forming
cation permeable
pores in the tight
junctions
↓ Transcription
of calcium
transporter
genes in the
distal
convoluted
tubule
↓ Probability
of calcium
transport
channels
opening
↓ Renal reabsorption
of calcium
↓ Sodium-hydrogen
exchanger 3 (NHE3)
activity & expression in
proximal tubule
↓ Sodium
reabsorption
Electrochemical
gradient changes
↓ Phosphate excretion
↑ Phosphate calcium
complex formation
Pathophysiology Mechanism
Sign/Symptom/Lab Finding Complications
↑ Inflammation
Sepsis/severe illness
↑ Calcium
sequestration
into cells
↓ Liver
synthesis of
albumin
↓ Albumin-
calcium
binding
False
Hypocalcemia
Acute pancreatitis
↓ Lipase
secretion from
pancreas
↑ Levels of
undigested
fats in small
intestine
Free fatty
acids
percipitate
calcium
↓ PTH receptor
activation in
bones
↓ Osteoblast
stimulation
↓ RANKL
ligand
secretion
↓ RAANK
receptor
binding
↓Osteoclast
production
↓ Bone
resorption
Hypocalcemia
(serum [Ca2+] <2.1mmol/L)
Authors:
Name Name
Name Name*
Reviewers:
Name Name
Name Name*
* MD at time of publication
Published MONTH, DAY, YEAR on www.thecalgaryguide.com
Hypocalcemia: Physiology
Chronic kidney
disease
↓ Renal
blood flow
↓ Glomerular
filtration
Parathyroid gland
dysfunction: removal,
autoimmune,
congenital (DiGeorge)
Vitamin D deficiency:
↓intake, malabsorption
Sepsis/severe illness
↓ NHE3 activity and
expression in
proximal tubule
↓PTH receptor
activation in
kidneys
↑ Claudin14
activity in the
thick ascending
loop of Henle
↓ Parathyroid
hormone (PTH)
↓ Transcription of
calcium transporter
genes (TRPV5, calbindin
D28K, NCX) in the distal
convoluted tubule
↓ Enzyme 1-alpha
hydroxylase activity
↓ PTH receptor
activation in
bones
↓ osteoblast
stimulation
↓ RANKL ligand
secretion
↑ Inflammation ↓ PTH sensitivity
↓ Glomerular
filtration
↓ Liver
synthesis of
serum albumin
False
Hypocalcemia
↑ Calcium
sequestration
into cells**
↓ Lipase secretion
from pancreas
Acute pancreatitis
Legend: Pathophysiology Mechanism
Sign/Symptom/Lab Finding ↓ Albumin-
bound calcium
Current mechanism
is unknown
↑ levels of undigested
fats in small intestine
Complications
Hypomagnesemia Multiple blood
transfusions
↓ Sodium
reabsorption
Electrochemical
gradient changes
↓ Phosphate
excretion
↑ Phosphate
calcium complex
formation
Inhibits claudin16 and 19
from forming cation
permeable pores in the
tight junctions
↓Probability of
calcium transport
channels opening
↓ Synthesis of activated
vitamin D (calcitriol)
↓ RAANK
receptor
binding
↓ Calcium
reabsorption
↓ Binding of vitamin D
regulated calcium
transporters (TRPV6,
calbindin9K, PMCa2B,
NCX2) in the duodenum
and jejunum
↓osteoclast
production
↓ Bone
resorption
↓ Serum calcium
Hypocalcemia
<2.1 mmol/L
↑ Precipitation of calcium
by free fatty acids
Authors:
Name Name
Name Name*
Reviewers:
Name Name
Name Name*
* MD at time of publication
Published MONTH, DAY, YEAR on www.thecalgaryguide.com
https://journals.physiology.org/doi/full/10.1152/physrev.00003.2004?rfr_dat=cr_pub++0pubmed&url_ver=Z39.88-
2003&rfr_id=ori%3Arid%3Acrossref.org – Calcium Absoprtion across Epithelia
https://academic.oup.com/endo/article-abstract/137/1/13/2498579 - PTH and Calcium Signaling Pathways
https://www.pnas.org/doi/10.1073/pnas.1616733114 - Information on Claudin 14
https://onlinelibrary-wiley-com.ezproxy.lib.ucalgary.ca/doi/full/10.1111/apha.13959 - effects of PTH on renal calcium and
phosphate handling
https://www.ncbi.nlm.nih.gov/books/NBK430912/ - Hypocalcemia overview + causes + pathophys
https://www.orthobullets.com/basic-science/9010/bone-signaling-and-rankl - information about bone signaling
https://www.sciencedirect.com/science/article/abs/pii/S0889852921000682?via%3Dihub – Calcium homeostasis article
https://pubmed.ncbi.nlm.nih.gov/3012979/#:~:text=Abstract,intestine%2C%20require%20the%20parathyroid%20hormone.
vitamin D metabolism and function
–
https://pmc.ncbi.nlm.nih.gov/articles/PMC2669834/#:~:text=Calcium%20is%20actively%20absorbed%20from,for%20proper
%20mineralization%20of%20bone.
– more vitamin D metabolism and specific receptors
https://jidc.org/index.php/journal/article/view/32903236/2331 - sepsis + hypocalcemia")
Endometriosis

Transfer of endometrial tissue away
from the uterus during pelvic surgery,
vaginal delivery, or cesarean section
Sampson’s theory: Retrograde
flow of endometrial tissue
through fallopian tubes
Long menstrual flows
(↑ retrograde menstruation)
Coelomic metaplasia theory:
Undifferentiated mesothelial cells
from the peritoneal cavity
differentiate into endometrial cells
during fetal development. May also
occur in patients with testes
Implantation theory: Menstrual endometrium
implants onto pelvic structures
↓ Cellular antioxidant
capacity (↑ cell damage)
Alcohol use
(mechanism unclear)
Early menarche (↑
estrogen exposure)
Stem cell theory: Endometrial stem/
Embryonic rest theory: Residual
progenitor cells from menstrual
embryonic cells from Müllerian
Dissemination theory: Endometrial
blood or neonatal uterine bleeding
or Wolffian ducts are misplaced
tissue transported through bloodstream
implant in the pelvic cavity
during organogenesis
or lymphatics to extrauterine structures
(e.g., brain, pericardium)
Cells differentiate into endometrial-like tissue
Ovary releases estrogen
& progesterone
Endometriosis
Endometrial glands & stroma (structural support tissue) found outside the uterus
Ectopic endometrial-like
tissue on bladder
Ectopic endometrial-like tissue in posterior cul-de-sac
Monthly proliferation
(by estrogen) and
stabilization (by
progesterone) of
endometrial tissue
Chronic inflammation of surrounding tissues & fibrosis/adhesion formation
Tissue inflammation & fibrosis/adhesions push on & displace pelvic organs
Ectopic tissue
abnormally
activates
nerve signaling
pathways
responsible for
bladder
contraction
Bladder wall
contractions
activate
nociceptors
(pain
sensors)
Penetrative intercourse involving the
posterior vagina applies ↑ pressure to
tethered & immobile pelvic structures
Feces moves down colon into
rectum & applies pressure to
the tethered rectum
Monthly
progesterone
withdrawal
Dyspareunia (pain with
deep intercourse)
Dyschezia (pain with
bowel movements)
↑ Urinary
frequency &
urgency
Pain with
micturition
(urination)
Endometrial tissue sloughs
off from a basal tissue layer
(menstruation**)
Extra-uterine endometrial-
like tissue cannot evacuate
the implantation site
Retrograde menstruation in ovary ↑ the
presence of ectopic endometrial-like tissue
& blood (mechanism poorly understood)
Old blood fills
endometrial-like
tissue on ovary
Endometrioma
(chocolate cyst)
Chronic cyclical local
inflammation (release of
pro-inflammatory chemicals)
Nociceptors near ectopic
endometrial-like tissue activate
Pain signals & release of inflammatory
mediators subsides over time
Cyclic dysmenorrhea
(menstrual pain)
Repair of inflammation
Ectopic endometrial-like tissue
becomes fibrotic (scarred)
Significant ↑ scar tissue
Nodule formation
↓ Hormone levels
post-menopause
Accumulation of
inflammatory fluid
within scar tissue
↑ Risk of scar tissue obstructing
or kinking fallopian tubes
Endometrial tissue
stops proliferating
Hormone-dependent symptoms
(e.g., dysmenorrhea) cease
Cyst formation
Embryos unable to pass through
fallopian tube to uterus
Author:
Joshua Seto
Kayla Nelson
Reviewers:
Yan Yu
Sean Spence
Jessica Revington
Infertility
Colin Birch*, Rachel Wang*
* MD at time of publication
**See corresponding Calgary Guide slide
Legend: Published December 20, 2013, revised September 24,
2025 on www.thecalgaryguide.com
Pathophysiology Mechanism
Sign/Symptom/Lab Finding Complications")
Hydrocephalus in pediatrics

Narrowing
blocks CSF
flow from 3rd
to 4th ventricle
Congenital anomalies blocking CSF flow
Atresia of
foramen of
Monro (channels
connecting
paired lateral
ventricles & 3rd
ventricle
narrow)
Narrowing
blocks CSF flow
from lateral
ventricles to 3rd
ventricle
Posterior fossa
malformations (4th
ventricle closure
failure) (e.g.
Dandy-Walker
Malformations)
Persistence of
Blake's pouch (cyst
in 4th ventricle)
obstructs CSF
outflow from 4th
ventricle
Brain mass/tumor
(e.g. medulloblastoma, pinealoma)
Obstructs the passage of CSF from
ventricles to subarachnoid space
Hydrocephalus in Pediatrics: Pathogenesis and complications
Intracranial or subarachnoid
hemorrhage (brain bleed)
Blood accumulates in ventricles
or subarachnoid space
Accumulated red blood cells
break down over time & release
hemoglobin, bilirubin, etc.
Blood clots form in
subarachnoid spaces &
around ventricular outlets
Developmental cysts
(CSF filled sacs
located between the
brain & arachnoid
Infection in brain
membrane – e.g.
tissue or meninges
Arachnoid Cysts)
(e.g., meningitis)
Breakdown products trigger
inflammation of ependymal
lining & arachnoid membranes
Immune cells respond to
infection & accumulate
Inflammation stimulates
transforming growth factor-
beta (TGF-β) & neutrophil
extracellular traps (NETs)
Exudate & pus form
in subarachnoid
spaces & around
ventricular outlets
TGF-β promotes
fibrosis in
subarachnoid space
NETs obstruct
meningeal
lymphatic vessels
Scarring & obliteration
of arachnoid villi
Obstruction impairs
cerebrospinal fluid
(CSF) drainage
through lymphatics
Choroid plexus tumors
Choroid plexus epithelial
cells are overactive
Sacs compress or
obstruct
interventricular
foramina, 3rd or 4th
ventricle, aqueduct
of Sylvius, etc.
(location dependent)
Congenital absence
of arachnoid villi
Impaired CSF
resorption
Normal flow of CSF is
physically blocked
Overactivity produces excessive CSF
Communicating hydrocephalus (CSF accumulation due to impaired
absorption or increased production in the subarachnoid spaces)
Obstructive hydrocephalus (CSF accumulation due to
structural blockage of CSF flow within the ventricular system)
Hydrocephalus
Active distension of the ventricles in the brain due to excessive accumulation of CSF
Pathophysiology Mechanism
Sign/Symptom/Lab Finding Complications
Published Sept 29, 2025 on www.thecalgaryguide.com
Chiari II
malformation
(downward
herniation of
cerebellum,
brainstem, & 4th
ventricle through
foramen magnum)
Herniated
structures block
CSF outflow to
spinal cord
Authors:
Abisola Adegbulugbe,
Lexyn Iliscupidez
Reviewers:
Annie Pham,
Merry Faye Graff,
Emily J. Doucette,
Jean Mah*
*MD at time of publication
Legend:")
Reactive infectious mucocutaneous eruption
: Pathogenesis & clinical findings
Bacterial or viral respiratory infection
(most common infectious agent is
Mycoplasma pneumoniae)
↑ Proliferation of polyclonal B cells
(antibodies secreted by different B cell
lines) & ↑ antibody production
Antibodies cross-react (react to multiple
antigens) with antigens in host tissue
causing inflammation & tissue damage
Systemic & local
inflammatory response
targets infectious agent
RIME prodrome (early symptoms
indicating a forthcoming illness)
Fever
Malaise
Cough Dyspnea
Inflammation & tissue damage activate
inappropriate immune responses at distal sites
Authors:
Sonia Czyz
Reviewers:
Shahab Marzoughi
Jessica Revington
Michele Ramien*
* MD at time of publication
Normal Mucosa
Complement system activates as part
of the immune response & produces
various complement proteins
Epithelium
Lamina
propria
Complement proteins
C3a & C5a attract other
immune cells to the site
of infection & initiate ↑
inflammatory responses
Complement protein
C3b tags pathogens
& infectious agents
to help immune cells
identify them
↑ Leukocyte infiltrate (accumulation) in tissue & ↑
vascular permeability of surrounding tissues
Skin tissue experiences immune-mediated damage
from immune cells & active complement proteins
Cutaneous lesions (abnormal surface
skin growths or skin changes)
Mucositis in RIME
Legend: Pathophysiology Mechanism
Antibodies bind to antigens &
form active immune complexes
to attack infectious agents
Immune complexes circulate in
the body to target infectious
agents. Antigen excess forms
additional immune complexes
Immunoglobulin G
antibodies specific to the
infectious agent cross-
react with antigens on
keratinocytes (keratin-
producing skin cells) &
mucosal cells in the
epidermis (outermost
skin layer)
Additional immune complexes
continue circulating in the
absence of an infectious agent
& subsequently deposit in skin
tissue & mucous membranes
Antibodies
& immune
complexes
Leukocyte
infiltrate
Destructive inflammatory response induced at mucosal
& skin sites with no target pathogen or infectious agent
Complement
activation
Mucosal sites experience localized intense inflammation & ongoing
immune-mediated damage from active complement proteins
Severe mucositis (inflammation &
ulceration of the mucous membranes)
Acute hemorrhagic (excessively bleeding) crusts & erosions, ulcers, or
vesiculobullous lesions (fluid-filled blisters) in multiple mucosal sites
Published Nov 8, 2025 on www.thecalgaryguide.com
Full-thickness
epithelium
necrosis
(tissue death)
Lamina
propria
Sign/Symptom/Lab Finding Complications
Mucositis in RIME
Reactive Infectious Mucocutaneous Eruption (RIME): Pathogenesis and clinical findings
Normal Mucosa
Epithelium
Bacterial or viral respiratory infection; most
common infection is Mycoplasma
pneumoniae
Systemic and local
inflammatory response
towards invading microbe
Lamina
propria
Polyclonal B cell proliferation
and antibody production
Full-thickness
epithelium necrosis
Prodrome: cough, dyspnea, malaise, fever ~1
week prior to mucocutaneous eruption
The antibodies cross-react with host tissue
causing inflammation and tissue damage
Lamina
propria
Authors:
Sonia Czyz
Reviewers:
Shahab Marzoughi
* MD at time of publication
Inappropriate immune
responses at distal sites
Molecular
mimicry
Immune complex
activation
Complement
activation
I would remove all of this
Immunoglobulin G
antibodies specific for
invading microbe cross-
react with antigens on
keratinocytes and
mucosal cells in the
epidermis
Circulating immune
complexes form in
antigen excess
C3a, C5a, and C3b
produced
Deposition in
skin and mucous
membrane
↑ Leukocyte
infiltrate and
vascular
permeability
Severe mucositis
(inflammation and
ulceration of the
mucous
membranes
Destructive
inflammatory
response at mucosal
and skin sites
Antibodies
& immune
complexes
Leukocyte
infiltrate
Complement
activation
Acute occurrence of hemorrhagic
crusts and erosions, ulcers, or
vesiculobullous lesions in 2 or
more mucosal sites
Localized inflammation and
immune response activate
complement system which
damage mucosal tissues
Immune
response leads
to painful, red
nodules
typically on
the shins
Triggering localized, intense
inflammation and immune-
mediated damage in these
specific mucosal sites.
Legend: Widespread
systemic
immune
response
Erythema
nodosum
Urticaria
Immune response
results in hives or
itchy welts on the
skin.
Pathophysiology Mechanism
Sign/Symptom/Lab Finding Complications
Cutaneous
lesions
Published MONTH, DAY, YEAR on www.thecalgaryguide.com")
Acute Tubular Necrosis

Septic shock** Cardiogenic shock**
Cholesterol embolization
syndrome (rare)
Severe hypotension
↓ Mean arterial pressure
reduces renal perfusion pressure
which ↓ renal blood flow
Renal vascular autoregulation
fails to maintain renal perfusion
Tubular cell hypoxia & depletion
of adenosine triphosphate (ATP)
(primary energy carrier in cells)
Cell ion pump failure, cell swelling,
& cell membrane disruption
Ischemic tubular necrosis (renal tubular
cell injury secondary to ↓ blood flow)
Myoglobinuria
Hemoglobinuria
Aminoglycosides
Ethylene glycol
Amphotericin
Cisplatin
↓ Efficiency of kidney reabsorption & excretion
Kidney removes ↓
potassium (K+) from
the blood volume
Hyperkalemia** (high K+
concentration in the blood)
↑ Serum K+ levels
induce abnormal
cardiac electrical
conduction patterns &
excessive cardiac
excitability
Fatal cardiac
dysrhythmia (irregular
heartbeat pattern)
Kidney removes
↓ urea from the
blood volume
Kidney removes ↓
creatinine from the
blood volume
Azotemia (↑ serum
urea & creatinine)
Uremic toxins
systemically
impair
neutrophil
function
Uremic toxins (indoxyl
sulfate or paracresyl
sulfate) induce oxidative
stress in the brain through
accumulation of reactive
oxygen species (ROS)
↑ Risk of infection
ROS damage neuronal membranes & ion channels &
induce dysfunctional mitochondria in the brain
Authors:
Adam Bubelenyi
Reviewers:
Britney Wong
Luiza Radu
Jessica Revington
Veronica Hammer*
Braden Manns*
* MD at time of publication
**See corresponding Calgary Guide slide
Dysfunctional brain mitochondria cannot
properly metabolize purines & urea
Brain cannot use any metabolic or cellular pathways requiring ATP
↓ Cerebral neuronal signaling
Legend: Pathophysiology Mechanism
Sign/Symptom/Lab Finding Intravenous (IV)
administration of
iodinated
contrast medium
Tumor lysis syndrome
Cancer cell apoptosis (cell death)
releases ↑ uric acid & phosphate
Heme-containing
pigments
Medications
Uric acid & calcium phosphate
crystals precipitate in renal tubules
Nephrotoxic substances damage renal tubular cells through unique mechanisms
Lipid peroxidation
(oxidative degradation) of
lipid bilayer weakens renal
tubular cell membranes
Damage to mitochondria in tubular
cells causes depletion of ATP
Formation of oxygen free
radicals in renal tubular cells
Impairment of ATP-
dependent processes
Activation of pro-inflammatory
cytokines & enzymes in the kidney ↑ Renal tubular cell
permeability
Disruption of protein synthesis &
enzyme function in renal tubular cells
Degradation of tubular cell
membrane lipids & proteins
Free radicals
oxidize tubular
cell proteins &
impair structure
& function
Toxic tubular necrosis (renal tubular injury due to nephrotoxic substances)
Acute Tubular Necrosis
Acute kidney injury via renal tubular cell damage & cell death
Denudation (removal of surface tissue layers) &
erosion of the tubular basement membrane
Kidney removes ↓
sodium (Na+) & water
from the blood volume
↓ Bicarbonate
reabsorption from
the proximal tubule
of the kidney
Necrotic tubular cells fall into tubular lumen
Ongoing tubule damage
↓ ability to concentrate
urine solutes
↓ Bicarbonate
neutralization of
acids in the blood
Necrotic
tubular cells
release
intracellular
contents
Obstruction of
tubular lumen
impedes
glomerular
filtration
↓ Urine osmolality
(low urine solute
concentration)
↑ Water in blood
volume dilutes
Na+ serum
concentration
Metabolic acidosis
(blood pH <7.35)
Muddy brown
granular casts
↓ Glomerular
filtration rate (GFR)
Hyponatremia** (low Na+
concentration in the blood)
↑ Na+ retention in
the circulatory blood
volume & ↓ Na+
excretion in the urine
↓ Urine output
Accumulation of ROS activates microglia
& releases pro-inflammatory cytokines
↑ Fluid retention in
the circulatory system
Aggressive IV fluid
rehydration in septic
shock or cardiogenic
shock patients
Cerebral inflammation ↑ oxidative
damage to neurons & glial cells
Fluid overload
Uremic encephalopathy
Pulmonary edema (excess
fluid accumulation in lung
alveoli & interstitium)
Published November 15, 2025 on www.thecalgaryguide.com
Complications
Disruption of
DNA & RNA
synthesis
within renal
tubular cells
Tubular epithelial
cells in urine
Epithelial cell
casts in urine
(cast composed
of epithelial
cells embedded
in a matrix)
↓ Ability to clear
bacteria in urine
↑ Risk of urinary
tract infection")
Infection-Related Glomerulonephritis
 trigger the inflammatory cascade
Common infection sites include pharyngitis (infection of the mucous membranes in the oropharynx) &
endocarditis (inflammation of the endocardium) but bacterial infections can originate anywhere in the body
In streptococcal infections, bacteria release nephritis (inflammation of the kidney) associated plasmin receptor antigens (NAPlr) & streptococcal
pyrogenic exotoxin B antigens (SPeB) into the systemic circulation from the infection site. Other bacterial infections may have different pathophysiology
Immune response generated against circulating NAPlr & SPeB antigens includes
↑ production of IgG antibodies (critical for immune response & memory)
IgG antibodies bind with self-antigens (molecules
recognized by the immune system as belonging to the
host) including plasmin & glomerular proteins (proteins
found in the kidney’s filtering unit, the glomerulus)
IgG antibodies activate plasmin
(enzyme responsible for breaking
down protein in the kidneys)
Plasmin degrades
protein in the kidneys
IgG antibodies bind to
glomerular proteins in
the kidney & form
immune complexes in
glomeruli basement
membrane & sub-
epithelial podocytes
(highly specialized
cells in the glomerular
filtration barrier)
Degradation of key
glomerulus components,
including the glomerular
basement membrane
(GBM) & mesangial matrix
Damage to key
glomerulus
components leads to
abnormal regulation
of blood filtration
↓ Selective
permeability of the
glomerular membrane
IgG antibodies bind with NAPIr & SPeB antigens &
form immune complexes (antibody & antigen
combinations involved in immune system signalling)
Immune complexes circulate & deposit in glomeruli
basement membrane & sub-epithelial podocytes
RBCs escape through
the glomerulus into
renal tubules &
progress into the urine
Dysmorphic
hematuria (blood
in the urine
characterized by
misshapen RBCs)
Author:
Joanna Keough
Reviewers:
Britney Wong
Luiza Radu
Jessica Revington
Veronica Hammer*
Louis Girard*
*MD at time of publication
**See corresponding Calgary Guide slide
Legend: Pathophysiology Mechanism
Immune complexes activate
complement (C3, oxidizing
agents, proteases)
Complement activation helps ↑
proliferation of glomerular mesangial
cells (cells with specialized proteins
capable of producing motile forces)
Mesangial cells produce
extracellular matrix in
the glomerulus
Excess
extracellular
matrix
blocks
glomerular
capillaries
Mesangial cells release chemoattractants
(signalling molecules) into the glomerulus
& attract various immune cells
Lymphocytes follow
chemoattractants to the
glomerulus & activate
Neutrophils follow chemoattractants to the
glomerulus & release lysosomal enzymes
Lymphocytes accumulate in the glomerulus
& obstruct glomerular capillaries
Impaired blood flow through glomerular capillaries
Capillary injury Podocyte injury or death
Infection-Related Glomerulonephritis
Glomerular kidney damage sustained after a bacterial or viral infection
↓ Glomerular
filtration rate (GFR)
Creatinine builds
up in the blood
↑ Glomerular membrane permeability allows
large molecules to pass through the membrane
(e.g., red blood cells (RBCs), protein)
↓ GFR impairs the kidney’s
ability to filter & excrete fluid
↓ Estimated GFR
Prolonged
(eGFR)
renal damage
Protein escapes into renal tubules
& progresses into the urine
Chronic kidney disease
Proteinuria (loss of
protein through the urine)
↑ Fluid retention
in systemic
circulation & ↓
renal blood flow
Impaired
potassium
excretion
↓ Urine
output
Impaired
acid waste
product
excretion
Urine contains ↑ large
proteins (e.g., albumin)
Activation of the
intrarenal renin-
angiotensin-aldosterone
system (RAAS)**
Hyperkalemia
(↑ serum
potassium)
Oliguria
(↓ urine
volume)
Metabolic
acidosis**
Albuminuria (loss of
albumin through the urine)
Intrarenal RAAS activation
leads to ↑ angiotensin II
(Ang II) production
↑ Sodium & water retention in systemic
circulation & ↓ excretion in urine (↓ GFR)
Hypertension**
Edema (swelling due to ↑ fluid in body tissues)
Circulating blood volume exerts ↑ pressure on blood vessels & forces the heart to pump harder
Left ventricle thickens & enlarges over time & demonstrates
progressive cardiac injury (e.g., fibrosis, loss of cardiac muscle cells)
Impaired cardiac output
Heart failure
Sign/Symptom/Lab Finding Complications
Published November 15, 2025 on www.thecalgaryguide.com")
Pyelonephritis

Obstruction of bladder outlet
(congenital/acquired)
High blood glucose damages
nerves controlling the bladder,
as well as blood vessels
supplying bladder nerves
Glycosuria (elevated levels
of glucose in urine) create
nutritive environment for
bacterial growth
Excessive urine buildup
increases pressure exerted
against the bladder, pushing
urine from bladder up to ureters
Authors:
Sergio F. Sharif
Reviewers:
Michelle J. Chen
Jessica Revington
Yan Yu*
Juliya Hemmett*
Brandon Christensen*
* MD at time of publication
Shorter urethra
allows for fecal
microbes to more
readily enter the
urinary tract
Catheter inoculates deep
urinary tract with bacteria
& facilitates organisms to
enter bladder
Neurogenic bladder (decreased
bladder control due to nerve damage)
is unable to appropriately release
urine, leading to urinary retention &
failure to clear residing bacteria
Bacteremia
(bacterial infection
in bloodstream)
Bacteria initially colonize the lower urinary tract, then migrate up the tract towards
one or both kidneys (most commonly Gram-negative rods, e.g., Escherichia coli)
Bacteria, such as
Staphylococcus species,
spread from another site
of infection to kidneys
via bloodstream
Acute Pyelonephritis
Inflammation of the kidney interstitium due to upper urinary tract infection, usually bacterial
Collective contribution of
mechanisms stated below, if severe
Sepsis**
Concurrent cystitis
irritates urinary
tract (see lower
UTI slide)**
Bacteria expressing virulence factors (e.g., P fimbriae in Escherichia coli) exploit
host susceptibility & infect upper urinary tract (ureters & kidneys)
Positive urine culture
Dysuria (pain & burning
while urinating)
Bacteria trigger IL-8 release by local
immune cells in the upper urinary tract,
triggering recruitment of neutrophils to
renal tubules & interstitium
Neutrophils degranulate in the infected
tissue, phagocytose bacteria & form
sacrificial neutrophil extracellular traps
(resulting in neutrophil death) in
attempt to clear infection
Neutrophil-mediated
immune response
damages surrounding
kidney tissue (tubular
injury)
Acute
kidney
injury**
Immune cells at the site of infection release cytokines
(e.g., IL-1, TNF), which disseminate systemically
Cytokines act in the brain in
concert with unclear
mechanisms relating to tissue
injury & inflammation
triggered by infection (see
Neurotransmitters &
Pharmacology behind Nausea
and Vomiting slide**)
Cytokines bind
receptors in the
anterior hypothalamus,
triggering pathways
ultimately leading to
heat-generating
responses (e.g.,
shivering)
Cytokines promote
the growth &
release of white
blood cells from
bone marrow into
bloodstream
Dead neutrophils collect
in renal tubules near
infection & attach to
assemblies of
mucoprotein (a renal
tubular epithelium protein
secreted into tubules)
Pyuria (WBCs in
urine)
Nausea/vomiting
Fever
Leukocytosis
(↑ WBCs)
Urine white blood cell
(WBC) casts (cylindrical
particles with WBCs
seen on microscopy)
Costovertebral
Flank pain CT KUB -
angle tenderness
findings may
include focal
or diffuse
involvement,
fat stranding,
gas formation,
&/or other
complications
including
renal scarring
**See corresponding
and abscesses
Calgary Guide slide(s)
Complications
Published November 23, 2025 on www.thecalgaryguide.com
Unclear mechanism
involving systemic
inflammation
worsens mental
status in susceptible
individuals
Altered level of
consciousness
(especially in elderly)
Legend: Pathophysiology Mechanism
Sign/Symptom/Lab Finding")
Hypothyroidism

Iodine
deficiency
Infiltrative disease affecting
thyroid gland cells
Iatrogenic or medication-
related thyroid dysfunction
Hypothyroid phase of thyroiditis
(inflammation of the thyroid)
Pituitary gland dysfunction
(central hypothyroidism)
Progressive autoimmune
destruction of thyroid gland cells
↓ T3/T4 levels
activate the
hypothalamus-
pituitary-thyroid
axis & ↑ TSH
release by the
pituitary gland
Primary
hypothyroidism
(↑ TSH and ↓
T3/T4)
↓ T3/T4 levels
cause ↓ expression
of TH-dependent
myocardial enzymes
essential for cardiac
contractility
Cardiac myocytes
(cardiac muscle
cells) experience ↓
contractility
↓ Heart rate
↑ Bleeding
Impaired function of the thyroid gland & ↓ thyroid hormone (TH) secretion
Pituitary gland secretes ↓ levels of
thyroid stimulating hormone (TSH)
Hypothyroidism
Low or significantly ↓ secretion of triiodothyronine (T3) & thyroxine (T4) from the thyroid gland
↓ T3/T4 levels ↓
production of nitric oxide
(vasodilator) & cause
systemic vasoconstriction
of blood vessels
Systemic vaso-
constriction ↑
systemic
vascular
resistance
↑ Systemic vascular
resistance à ↑
intrarenal vascular
resistance & contributes
to ↓ renal perfusion
↓ T3/T4 levels reduce the rate of metabolic reactions in the body
including ↓ rates of carbohydrate, fat, & protein metabolism
↓ T3 levels allow fibroblasts
(connective tissue cells) to synthesize ↑
glycosaminoglycans (GAGs – structural
cell molecules capable of attracting
large amounts of water)
↓ Basal metabolic rate (base energy
required to sustain bodily functions)
↑ Diastolic blood pressure
(diastolic hypertension)
↓ Renal blood
flow à ↓
glomerular
filtration rate
(GFR)
↓ GFR
impairs renal
clearance of
excess GAGs
↑ GAG deposits
& water retention
in body tissues
↓ Energy to support physiological function
Narrowed pulse
pressure (↓
difference
between systolic
& diastolic blood
pressure)
Compensatory
mechanisms to
manage blood
volume &
circulation during
↓ T3/T4 levels
are overwhelmed
in the presence
of an infection or
systemic trauma
(e.g., heart
attack, stroke)
↓ GFR impairs
removal of excess free
water from the blood
volume by the kidneys
↑ GAG
deposits &
swelling in
skin tissue
↑ GAG deposits
in the larynx
swell & stiffen
vocal cords
↓ Skin gland
activity & ↓
sebum (oil) &
sweat
production
↑
Fatigue
↓ Motility of the
gastrointestinal
(GI) tract causes
↓ movement of
digested food &
↓ rate of waste
excretion
↓ TH levels & ↓
metabolic rate
impair neurologic
function (e.g.,
neurotransmitter
signaling & neuron
functioning)
Thickened skin
Hoarse speech
Dry skin
↑ Sodium (Na+) & water
retention in blood volume
↓ Perspiration
(sweating)
Constipation
Neurologic
symptoms (e.g.
depression,
somnolence)
Abnormally heavy menses
Excessive water retention
dilutes serum Na+ levels in
severe hypothyroidism
↓ Caloric expenditure at rest & with activity
↑ Risk of infertility
Myxedema coma**
(extreme manifestation
of hypothyroidism)
Hyponatremia
(↓ Na+)
↑ Fluid
retention in
body tissues
Weight gain
Musculoskeletal
symptoms (e.g.,
muscle cramps,
joint pain & ↓
muscle tone)
Published June 19, 2013, revised November 23, 2025 on www.thecalgaryguide.com
Normal or ↓
TSH levels
when T3/T4
levels are low
Authors:
Sophia Khan, Ayden Hansen, Jessica
Revington, Rupali Manek, Jaye
Platnich
Reviewers:
Shahab Marzoughi, Gurreet
Bhandal, Raafi Ali, Matthew
Harding, Mark Elliott, Hanan
Bassyouni*, Breanna McSweeney*
* MD at time of publication
**See corresponding Calgary Guide
slides
↓ T3 levels lead to
↓ lipoprotein lipase
(lipid metabolism
enzyme) activity
↑ Triglyceride
levels in blood
↓ TH levels & ↓
metabolic rate
impair cold
tolerance &
thermogenesis
(heat production)
mechanisms
Cold
extremities
(e.g. hands,
feet)
↓ Cold
tolerance
↓ Activation
of the T3-
mediated low-
density
lipoprotein
(LDL) receptor
gene
↓ LDL
receptor
expression
for LDL
clearance
↑ LDL
cholesterol
levels in
the blood
↓ T3/T4 levels ↓
coagulation factor
synthesis by the liver
↓ T3/T4 levels disrupt
the menstrual cycle &
cause anovulation
(absence of ovulation)
Legend: Hyperlipidemia
Pathophysiology Mechanism
Sign/Symptom/Lab Finding Complications")
Infectious Large Bowel Diarrhea

Contaminated
food, water,
poor
sanitation
Eggs,
contaminated
Undercooked
poultry
Undercooked
pork
Contaminated
food, water
produce,
undercooked
poultry
Unpasteurized
milk
Undercooked
beef,
contaminated
vegetables
Antibiotic use,
healthcare
associated
infection
Cytomegalovirus
S. enteritidis, S.
Enteroinvasive
Enterohemorrhagic
typhimurium
E. histolytica Y. enterocolitica
E. coli (EIEC)
Shigella
E. coli (EHEC)
C. jejuni C. difficile
Individual ingests an adequate
amount of organism
Bacteria employ protective
mechanisms to survive gastric acid
Organism adheres to colonic wall &
colonizes the large bowel
Organism releases a toxin that
disrupts the colon (enterotoxin)
Invading organisms activate local immune surveillance
Toxins A & B (from C. diff) disrupt
tight junctions between colonocytes
Toxin enters bloodstream
Release of pro-inflammatory cytokines triggers colonic inflammation
Cytokines
Inflammation
&/or toxins
stimulates
act on
nerve endings
hypothalamus
in rectal wall
to trigger
temperature
change
(pyrogens)
Inflammation
dysregulates
enteric nervous
system
Inflammation
spreads to
visceral
peritoneum
Inflammation &
invading bacteria
breaks down
colonic mucosa
Triggers
apoptosis of
colonocytes
Massive
neutrophilic
infiltration
Toxin reaches the
chemoreceptor
trigger zone in
medulla
Shiga toxins
(from Shigella or
EHEC) bind to
Gb3 receptors
on endothelial
cells
Pseudomembranous colitis on endoscopy
Abnormal
peristalsis &
spasms
Diffuse abdominal
pain & cramping Nausea & vomiting
Toxin enters cells
via endocytosis
Fever
Colonic epithelium loses
Na⁺ resorptive channels
Colonic epithelial cells
secrete ↑ Cl⁻ into lumen
↑ Sense of urgency & incomplete
evacuation (Tenesmus)
Inflammation &
organisms disrupts
blood vessels & mucosa
develops ulcers
Intestine absorbs ↓
fluid from lumen
Intestine secretes ↑
fluid into lumen
Shiga toxin
activates
platelets
(mechanism
unclear)
Toxin inhibits
protein
synthesis &
induces
apoptosis
Authors: Merry Faye Graff, Noriyah Al
Awadhi, Yan Yu
Reviewers: Paul Ratti, Jason Baserman,
Sergio F. Sharif Kerri Novak*, Sylvain
Coderre*
*Indicates MD at time of publication
**See corresponding Calgary Guide slide
Small volume diarrhea
Ulcers bleed
into colonic
lumen
Volume depletion
Platelet activation & endothelial cell
damage promote microthrombi
formation within systemic vasculature
Dehydration
Hypotension
Bloody stool (can be
nocturnal)
Hemolytic uremic syndrome**
Legend: Pathophysiology Mechanism
Sign/Symptom/Lab Finding Complications
Published Aug 7, 2012; updated Nov 23, 2025 on www.thecalgaryguide.com")
Kawasaki Disease

External trigger such as infection or environmental factor
(particulate matter, seasonal airborne antigenic triggers possible)
Kawasaki Disease (KD)
Acute systemic, inflammatory illness affecting medium-sized arteries (especially coronary arteries), diagnosed clinically with
presence of fever & 4/5 of: conjunctivitis, polymorphous rash, cervical lymphadenopathy, mucosal involvement,
edema/erythema on hands/feet. KD is the most common cause of acquired heart disease in children in developed countries.
Authors:
Taylor Krawec, Sunni Ho,
Zarrukh Baig, Nissi Wei
Reviewers:
Merry Faye Graff, Emily J. Doucette,
Zaini Sarwar, Mandy Ang,
Charissa Chen, Taj Jadavji*,
Susanne Benseler*,
Danielle Nelson*
* MD at time of publication
Acute Phase
Cytokines disrupt hypothalamic
thermoregulation
Fever
Innate immune
cells around
medium-sized
vessels (especially
coronary arteries)
are inappropriately
activated
Neutrophils
infiltrate
tunica
adventitia
(outer
vessel wall)
Coronary
arteritis
Cytokines enter
circulation &
mediate systemic
inflammation
Cervical lymphadenopathy
Immune cells infiltrate additional tissue
(e.g., lymph nodes, myocardium, pericardium)
Myocarditis Pericarditis
Local
cytokine
release
Inflammation of
tunica media &
intima (middle &
inner vessel walls)
Endothelial activation
in small vessels ↑
vasodilation & vessel
permeability
↑ Blood flow,
plasma leakage
& perivascular
infiltration of
skin & mucosa
Strawberry tongue
Cracked lips
Edema & erythema of hands/feet
Bilateral non-exudative conjunctivitis
Non-vesicular rash
Uveitis
Patchy destruction all 3 vessel layers (necrotizing
arteritis) but with vessel wall remaining intact
Early coronary changes on echocardiogram
Subacute Phase / Chronic Vasculitis
Vessel walls lose
structural integrity
Coronary artery aneurysm
(rarely other aneurysms)
Extensive adaptive immune-mediated destruction
of intima, media, elastic lamina & smooth muscle
Adaptive immune cells
(e.g., eosinophils, plasma
cells, lymphocytes) drive
ongoing inflammation
Damaged vessels disrupt blood flow
Thrombus
formation
Acute
myocardial
infarction**
Systemic inflammation stimulates
megakaryocyte proliferation in bone marrow ↑ Platelet production Thrombocytosis
Inflammation damages dermis & epidermis
Gradual resolution of edema from acute
phase & initiation of dermal/epidermal repair
Periungal desquamation
(peeling of skin around nails)
Luminal Myofibroblast Infiltration
↓ Baseline
coronary perfusion
Coronary flow cannot ↑ to match myocardial
O2 demand when needed (demand ischemia)
Exertional chest
pain or dyspnea
Myofibroblasts replace
inflammatory infiltrates
in vessel walls during
chronic healing phase
Myofibroblast
proliferation
thickens intima
Affected arteries
become narrowed
(coronary arteries
most common)
Exercise intolerance
Ischemic myocardium
Coronary artery stenosis Arrhythmias
is electrically unstable
Legend: Pathophysiology Mechanism
Sign/Symptom/Lab Finding Complications
Published Oct 10, 2014; updated Dec 12 2025 on www.thecalgaryguide.com")
Inflammatory Versus Degenerative Joint Disease
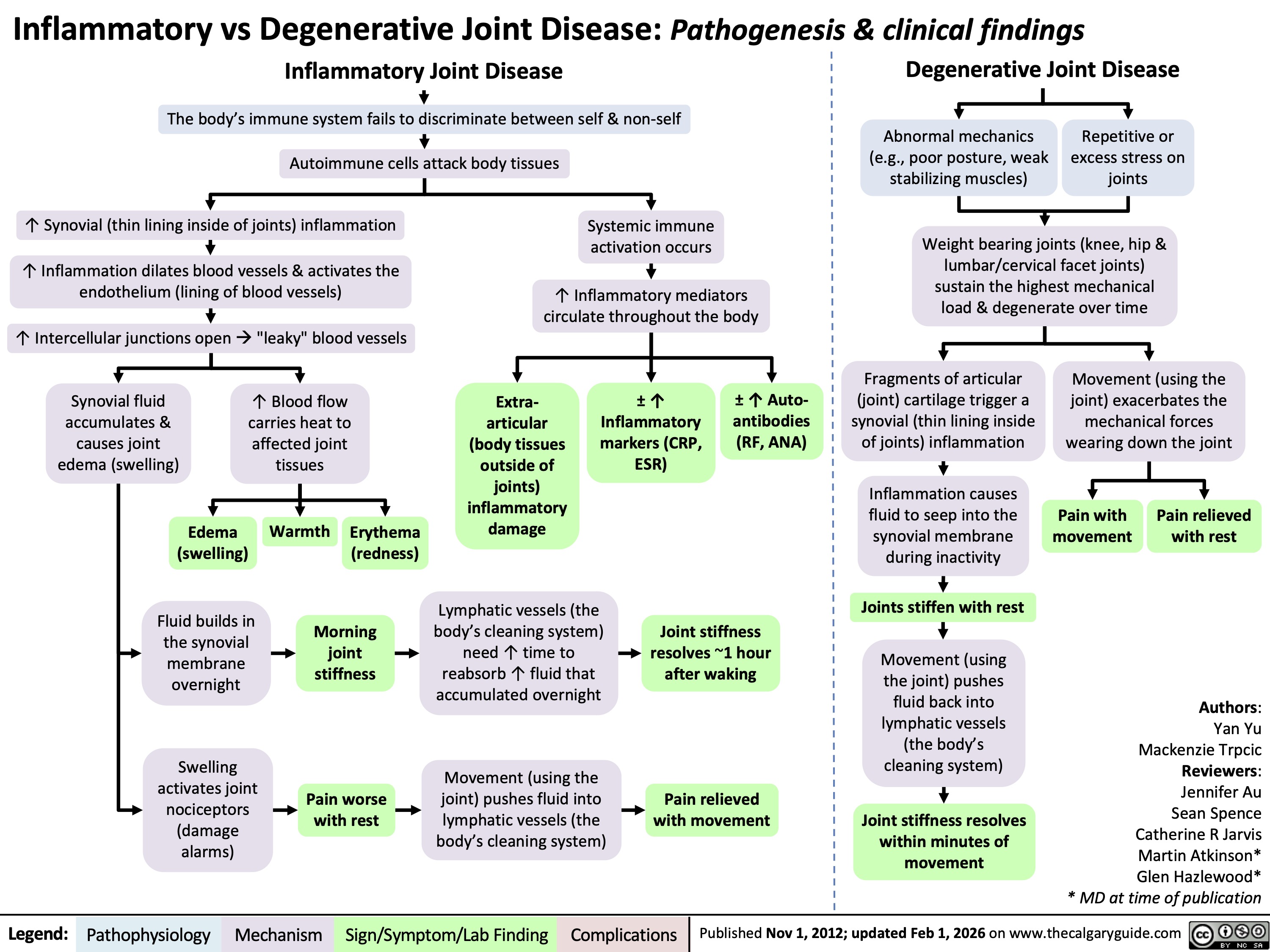 1hr
after waking
Legend: Mechanism
Sign/Symptom/Lab Finding Systemic
immune
activation occurs
↑
Inflammatory
mediators
circulate
throughout the
body
Extra-
articular
involvement
(e.g., skin,
eyes, lung,
heart,
kidneys) ±
abnormal
inflammatory
markers (CRP,
ESR) &
autoantibodi
es (RF, ANA)
Pathophysiology Complications
Degenerative Joint Disease
Abnormal mechanics or repetitive/excessive
loading stresses the joints
Joint structures degrade over time
Movement
(using the
joint)
exacerbates
the
mechanical
forces that are
wearing down
the joint
Pain with
movement
Pain relieved
with rest
Fragments of broken
articular cartilage
triggers a mild
synovial (thin lining
inside of the joint)
inflammation that
seeps fluid into the
synovial membrane
during inactivity
Joints stiffen with rest
Moving the joint
physically pushes
inflammatory fluid
back into lymphatic
vessels (body's clean-
up system) within
minutes
Joint stiffness resolves
within minutes of
movement
Published on www.thecalgaryguide.com
Authors:
Yan Yu
Mackenzie Trpcic
Reviewers:
Jennifer Au
Sean Spence
Martin Atkinson*
Catherine R Jarvis
* MD at time of
publication
The weight-
bearing joints
(i.e., knee, hip, &
lumbar/cervical
facet joints)
sustain the
highest
mechanical load
& degeneration
Distinguishing between Inflammatory and Degenerative joint disease
Inflammatory Joint
Disease
Auto-immune attack of body tissues, due to
failure of the body’s immune system to
discriminate between self and non-self.
Degenerative Joint
Disease
Repetitive, excess, or abnormal mechanical
forces on joints over time, leading to physical
breakdown of the joint.
Authors:
Yan Yu
Reviewers:
Jennifer Au
Sean Spence
Martin Atkinson*
* MD at time of
publication
Within joints, severe synovial inflammation
dilates local blood vessels, and causes the
vessel endothelium to be more leaky.
Accumulation of abundant
inflammatory synovial fluid
at rest or overnight
Swelling stimulates
joint nociceptors
Pain with rest
Moving the joint
physically pushes fluid
back into lymphatics,
relieving the painful
swelling.
Pain relieved with
motion
Legend: Overnight fluid
accumulation in the
synovial membrane
Morning stiffness
Lots of fluid, takes
more time (hours)
to be reabsorbed
by lymphatics
Stiffness resolves
after >1hr
Blood carries
heat and
looks red
Warm, Red,
Swollen
Joints
Pathophysiology Mechanism
Sign/Symptom/Lab Finding Inflammation is
oftentimes
systemic, not
localized
exclusively within
the joint (i.e. in
Rheumatoid
arthritis, Lupus,
etc)
Extra-articular
manifestations;
positive
inflammatory
markers (CRP,
ESR, RF, ANA)
Motion (using
the joint)
exacerbates the
mechanical
forces that are
wearing down
the joint
Fragments of
broken articular
cartilage triggers a
mild synovial
inflammation that
seeps fluid into the
synovial membrane
during inactivity
Pain with motion
(relieved by rest)
Joint stiffness
after inactivity
Moving the joint physically
pushes inflammatory fluid
back into lymphatics within
minutes.
Note: one big difference btw
the two is the degree of joint
inflammation (synovitis)
present in the joint capsule
Stiffness is short-lived,
quickly relieved by
movement
Published November1,2012 on www.thecalgaryguide.com
Larger, weight-
bearing joints tend to
bear the brunt of the
mechanical forces
Most commonly
affects the knee,
hip, and L/C-spine
facet joints
Complications" title="Legend: Inflammatory vs Degenerative Joint Disease: Pathogenesis & clinical findings
Inflammatory Joint Disease
The body’s immune system fails to discriminate between self & non-self
Autoimmune cells attack body tissues
↑ Synovial (thin lining inside of joints) inflammation
↑ Inflammation dilates blood vessels & activates the
endothelium (lining of blood vessels)
↑ Intercellular junctions open à "leaky" blood vessels
Synovial fluid
accumulates &
causes joint
edema (swelling)
↑ Blood flow
carries heat to
affected joint
tissues
Edema
(swelling)
Warmth
Erythema
(redness)
Fluid builds in
the synovial
membrane
overnight
Morning
joint
stiffness
Swelling
activates joint
nociceptors
(damage
alarms)
Pain worse
with rest
Systemic immune
activation occurs ↑ Inflammatory mediators
circulate throughout the body
Extra-
articular
(body tissues
outside of
joints)
inflammatory
damage
± ↑
Inflammatory
markers (CRP,
ESR)
± ↑ Auto-
antibodies
(RF, ANA)
Lymphatic vessels (the
body’s cleaning system)
need ↑ time to
reabsorb ↑ fluid that
accumulated overnight
Joint stiffness
resolves ~1 hour
after waking
Movement (using the
joint) pushes fluid into
lymphatic vessels (the
body’s cleaning system)
Pain relieved
with movement
Degenerative Joint Disease
Abnormal mechanics
(e.g., poor posture, weak
stabilizing muscles)
Repetitive or
excess stress on
joints
Weight bearing joints (knee, hip &
lumbar/cervical facet joints)
sustain the highest mechanical
load & degenerate over time
Fragments of articular
(joint) cartilage trigger a
synovial (thin lining inside
of joints) inflammation
Movement (using the
joint) exacerbates the
mechanical forces
wearing down the joint
Inflammation causes
fluid to seep into the
synovial membrane
during inactivity
Pain with
movement
Pain relieved
with rest
Joints stiffen with rest
Movement (using
the joint) pushes
fluid back into
lymphatic vessels
(the body’s
cleaning system)
Joint stiffness resolves
within minutes of
movement
Authors:
Yan Yu
Mackenzie Trpcic
Reviewers:
Jennifer Au
Sean Spence
Catherine R Jarvis
Martin Atkinson*
Glen Hazlewood*
* MD at time of publication
Published Nov 1, 2012; updated Feb 1, 2026 on www.thecalgaryguide.com
Pathophysiology Mechanism
Sign/Symptom/Lab Finding Complications
Final slide is the 1st slide
Legend: Inflammatory vs Degenerative Joint Disease: Pathogenesis & clinical findings
Inflammatory Joint Disease
Degenerative Joint Disease
The body’s immune system fails to discriminate between self & non-self
Autoimmune cells attack body tissues
Abnormal mechanics
(e.g., poor posture, weak
stabilizing muscles)
Repetitive or
excess stress on
joints
↑ Synovial (thin lining inside of joints)
inflammation dilates blood vessels
Systemic immune
activation occurs ↑ Endothelium (lining of blood vessels) activation
opens intercellular junctions (“leaky” blood vessels”)
↑ Inflammatory mediators
circulate throughout the body
Weight bearing joints (knee, hip &
lumbar/cervical facet joints)
sustain the highest mechanical
load & degenerate over time
Synovial fluid
accumulates &
causes joint
edema (swelling)
Edema
(swelling)
↑ Blood flow
carries heat to
affected joint
tissues
Extra-
articular
(body tissues
outside of
± ↑
Inflammatory
markers (CRP,
ESR)
± ↑ Auto-
antibodies
(RF, ANA)
Fragments of articular
(joint) cartilage trigger a
synovial (thin lining inside
of joints) inflammation
Movement (using the
joint) exacerbates the
mechanical forces
wearing down the joint
joints)
inflammatory
Inflammation causes
Warmth Pain relieved
Erythema
damage
fluid to seep into the
Pain with
(redness)
synovial membrane
movement
with rest
during inactivity
Fluid builds in
the synovial
membrane
overnight
Morning
joint
stiffness
Lymphatic
vessels (cleaners)
need ↑ time to
reabsorb ↑ fluid
that accumulated
overnight
Joints stiffen
with rest
Joint stiffness resolves
~1 hour after waking
Movement (using
the joint) pushes
fluid back into
lymphatic vessels
(cleaners)
Swelling
activates joint
nociceptors
(damage
alarms)
Pain worse
with rest
Movement (using
the joint) pushes
fluid into
lymphatic vessels
(cleaners)
Pain relieved with
movement
Joint stiffness
resolves within
minutes of
movement
Authors:
Yan Yu
Mackenzie Trpcic
Reviewers:
Jennifer Au
Sean Spence
Martin Atkinson*
Catherine R Jarvis
* MD at time of
publication
Published on www.thecalgaryguide.com
Pathophysiology Mechanism
Sign/Symptom/Lab Finding Complications
Inflammatory vs Degenerative Joint Disease: Pathogenesis & clinical findings
Inflammatory Joint Disease
Degenerative Joint Disease
The body’s immune system fails to discriminate between self & non-self
Auto-immune cells attack body tissues
Abnormal mechanics
(e.g., poor posture, weak
stabilizing muscles)
Repetitive or
excess stress on
joints
↑ Synovial (thin lining inside of joints)
inflammation dilates blood vessels
Systemic immune
activation occurs ↑ Endothelium (lining of blood vessels) activation
opens intercellular junctions (“leaky” blood vessels”)
↑ Inflammatory mediators
circulate throughout the body
Weight bearing joints (knee, hip &
lumbar/cervical facet joints)
sustain the highest mechanical
load & degenerate over time
Synovial fluid accumulates &
leads to joint edema (swelling)
↑ Blood flow carries heat
to affected joint tissues
Swelling
activates
joint
nociceptors
(damage
alarms)
Fluid
builds in
the
synovial
membrane
overnight
Edema
(swelling)
Warmth
Erythema
(redness)
Extra-articular
(body tissues
outside of
joints)
inflammatory
damage
± ↑
Inflammatory
markers (CRP,
ESR) & auto-
antibodies
(RF, ANA)
Movement (using the
joint) exacerbates the
mechanical forces
wearing down the joint
Pain with
movement
Pain relieved
with rest
Pain worse
with rest
Morning
joint stiffness
Movement
(using the
joint) pushes
fluid into
lymphatic
vessels
(cleaners)
Lymphatic
vessels
(cleaners)
need ↑ time
to reabsorb
↑ fluid
overnight
Pain relieved
with
movement
Joint stiffness
resolves ~1 hour
after waking
Authors:
Yan Yu
Mackenzie Trpcic
Reviewers:
Jennifer Au
Sean Spence
Martin Atkinson*
Catherine R Jarvis
* MD at time of
publication
Legend: Pathophysiology Mechanism
Sign/Symptom/Lab Finding Complications
Fragments of articular
(joint) cartilage trigger a
synovial (thin lining inside
of joints) inflammation
Inflammation causes
fluid to seep into the
synovial membrane
during inactivity
Joints stiffen
with rest
Movement (using
the joint) pushes
inflammatory fluid
back into lymphatic
vessels (cleaners)
Joint stiffness
resolves within
minutes of
movement
Published on www.thecalgaryguide.com
Inflammatory vs Degenerative Joint Disease: Pathogenesis & clinical findings
Inflammatory Joint Disease
The body’s immune system fails to discriminate between self and non-self
Auto-immune cells attack body tissues
↑ Synovial (thin lining inside of joints)
inflammation dilates blood vessels
↑ Endothelium (lining inside of
blood vessels) permeability
Synovial fluid accumulates &
leads to joint edema
(swelling)
Systemic immune
activation occurs
↑ Inflammatory mediators
circulate throughout the
body
Extra-articular
involvement
(body tissues
outside of
joints)
Elevated
inflammatory
markers (CRP,
ESR) & auto-
antibodies
(RF, ANA)
Degenerative Joint Disease
Abnormal
mechanics (e.g.,
poor posture,
weak stabilizing
muscles)
Repetitive/excess
stress on joints
Weight bearing joints (knee, hip &
lumbar/cervical facet joints)
sustain the highest mechanical
load & degenerate over time
Movement (using the joint)
exacerbates the mechanical
forces wearing down the
joint
Pain with
movement
Pain
relieved
with
rest
Authors:
Yan Yu
Mackenzie Trpcic
Reviewers:
Jennifer Au
Sean Spence
Martin Atkinson*
Catherine R Jarvis
* MD at time of
publication
Swelling activates
pain-sensitive joint
nociceptors
(damage alarms)
Pain worse with rest
Joint movement
pushes fluid into
lymphatic vessels
(body's clean-up
system)
Pain relieved
with movement
Legend: ↑ Blood flow carries heat to
affected joint tissues
Edema
(swelling)
Warmth
Erythema
(redness)
Fluid gradually builds in the
synovial membrane overnight
Morning joint stiffness
Lymphatic vessels
(body's clean-up system)
need ↑ time to reabsorb
↑ fluid that accumulated
overnight
Joint stiffness resolves ~1
hour after waking
Fragments of articular
(joint) cartilage trigger a
synovial (thin lining inside
of joints) inflammation
Inflammation causes fluid
to seep into the synovial
membrane during inactivity
Joints stiffen with rest
Moving the joint
physically pushes
inflammatory fluid back
into lymphatic vessels
(body's clean-up
system)
Joint stiffness resolves
within minutes of
movement
Pathophysiology Mechanism
Sign/Symptom/Lab Finding Complications
Published on www.thecalgaryguide.com
JOINT DISEASE: Inflammatory vs. Degenerative
Inflammatory Joint Disease
The body’s immune system fails to discriminate between self and non-self
Auto-immune cells attack body tissues
↑ Synovial (thin lining inside of the joint)
inflammation dilates blood vessels
↑ Endothelium (inner lining of blood
vessels) permeability
Synovial fluid accumulates &
leads to joint edema (swelling)
↑ Blood flow carries
heat to affected joint
tissues
Swelling activates
pain-sensitive joint
nociceptors
(damage alarms)
Fluid accumulates
progressively in the
synovial membrane
overnight
Edema
(swelling)
Warmth
Inflammatory pain
worse with rest
Morning joint
stiffness
Erythema
(redness)
Joint movement physically
pushes fluid back into
lymphatic vessels (body's
clean-up system)
Lymphatic vessels (body's
clean-up system) need ↑
time to reabsorb the ↑ fluid
that accumulated overnight
Inflammatory pain relieved
with activity
Joint stiffness resolves > 1hr
after waking
Legend: Mechanism
Sign/Symptom/Lab Finding Systemic
immune
activation occurs
↑
Inflammatory
mediators
circulate
throughout the
body
Extra-
articular
involvement
(e.g., skin,
eyes, lung,
heart,
kidneys) ±
abnormal
inflammatory
markers (CRP,
ESR) &
autoantibodi
es (RF, ANA)
Pathophysiology Complications
Degenerative Joint Disease
Abnormal mechanics or repetitive/excessive
loading stresses the joints
Joint structures degrade over time
Movement
(using the
joint)
exacerbates
the
mechanical
forces that are
wearing down
the joint
Pain with
movement
Pain relieved
with rest
Fragments of broken
articular cartilage
triggers a mild
synovial (thin lining
inside of the joint)
inflammation that
seeps fluid into the
synovial membrane
during inactivity
Joints stiffen with rest
Moving the joint
physically pushes
inflammatory fluid
back into lymphatic
vessels (body's clean-
up system) within
minutes
Joint stiffness resolves
within minutes of
movement
Published on www.thecalgaryguide.com
Authors:
Yan Yu
Mackenzie Trpcic
Reviewers:
Jennifer Au
Sean Spence
Martin Atkinson*
Catherine R Jarvis
* MD at time of
publication
The weight-
bearing joints
(i.e., knee, hip, &
lumbar/cervical
facet joints)
sustain the
highest
mechanical load
& degeneration
Distinguishing between Inflammatory and Degenerative joint disease
Inflammatory Joint
Disease
Auto-immune attack of body tissues, due to
failure of the body’s immune system to
discriminate between self and non-self.
Degenerative Joint
Disease
Repetitive, excess, or abnormal mechanical
forces on joints over time, leading to physical
breakdown of the joint.
Authors:
Yan Yu
Reviewers:
Jennifer Au
Sean Spence
Martin Atkinson*
* MD at time of
publication
Within joints, severe synovial inflammation
dilates local blood vessels, and causes the
vessel endothelium to be more leaky.
Accumulation of abundant
inflammatory synovial fluid
at rest or overnight
Swelling stimulates
joint nociceptors
Pain with rest
Moving the joint
physically pushes fluid
back into lymphatics,
relieving the painful
swelling.
Pain relieved with
motion
Legend: Overnight fluid
accumulation in the
synovial membrane
Morning stiffness
Lots of fluid, takes
more time (hours)
to be reabsorbed
by lymphatics
Stiffness resolves
after >1hr
Blood carries
heat and
looks red
Warm, Red,
Swollen
Joints
Pathophysiology Mechanism
Sign/Symptom/Lab Finding Inflammation is
oftentimes
systemic, not
localized
exclusively within
the joint (i.e. in
Rheumatoid
arthritis, Lupus,
etc)
Extra-articular
manifestations;
positive
inflammatory
markers (CRP,
ESR, RF, ANA)
Motion (using
the joint)
exacerbates the
mechanical
forces that are
wearing down
the joint
Fragments of
broken articular
cartilage triggers a
mild synovial
inflammation that
seeps fluid into the
synovial membrane
during inactivity
Pain with motion
(relieved by rest)
Joint stiffness
after inactivity
Moving the joint physically
pushes inflammatory fluid
back into lymphatics within
minutes.
Note: one big difference btw
the two is the degree of joint
inflammation (synovitis)
present in the joint capsule
Stiffness is short-lived,
quickly relieved by
movement
Published November1,2012 on www.thecalgaryguide.com
Larger, weight-
bearing joints tend to
bear the brunt of the
mechanical forces
Most commonly
affects the knee,
hip, and L/C-spine
facet joints
Complications" />
1hr
after waking
Legend: Mechanism
Sign/Symptom/Lab Finding Systemic
immune
activation occurs
↑
Inflammatory
mediators
circulate
throughout the
body
Extra-
articular
involvement
(e.g., skin,
eyes, lung,
heart,
kidneys) ±
abnormal
inflammatory
markers (CRP,
ESR) &
autoantibodi
es (RF, ANA)
Pathophysiology Complications
Degenerative Joint Disease
Abnormal mechanics or repetitive/excessive
loading stresses the joints
Joint structures degrade over time
Movement
(using the
joint)
exacerbates
the
mechanical
forces that are
wearing down
the joint
Pain with
movement
Pain relieved
with rest
Fragments of broken
articular cartilage
triggers a mild
synovial (thin lining
inside of the joint)
inflammation that
seeps fluid into the
synovial membrane
during inactivity
Joints stiffen with rest
Moving the joint
physically pushes
inflammatory fluid
back into lymphatic
vessels (body's clean-
up system) within
minutes
Joint stiffness resolves
within minutes of
movement
Published on www.thecalgaryguide.com
Authors:
Yan Yu
Mackenzie Trpcic
Reviewers:
Jennifer Au
Sean Spence
Martin Atkinson*
Catherine R Jarvis
* MD at time of
publication
The weight-
bearing joints
(i.e., knee, hip, &
lumbar/cervical
facet joints)
sustain the
highest
mechanical load
& degeneration
Distinguishing between Inflammatory and Degenerative joint disease
Inflammatory Joint
Disease
Auto-immune attack of body tissues, due to
failure of the body’s immune system to
discriminate between self and non-self.
Degenerative Joint
Disease
Repetitive, excess, or abnormal mechanical
forces on joints over time, leading to physical
breakdown of the joint.
Authors:
Yan Yu
Reviewers:
Jennifer Au
Sean Spence
Martin Atkinson*
* MD at time of
publication
Within joints, severe synovial inflammation
dilates local blood vessels, and causes the
vessel endothelium to be more leaky.
Accumulation of abundant
inflammatory synovial fluid
at rest or overnight
Swelling stimulates
joint nociceptors
Pain with rest
Moving the joint
physically pushes fluid
back into lymphatics,
relieving the painful
swelling.
Pain relieved with
motion
Legend: Overnight fluid
accumulation in the
synovial membrane
Morning stiffness
Lots of fluid, takes
more time (hours)
to be reabsorbed
by lymphatics
Stiffness resolves
after >1hr
Blood carries
heat and
looks red
Warm, Red,
Swollen
Joints
Pathophysiology Mechanism
Sign/Symptom/Lab Finding Inflammation is
oftentimes
systemic, not
localized
exclusively within
the joint (i.e. in
Rheumatoid
arthritis, Lupus,
etc)
Extra-articular
manifestations;
positive
inflammatory
markers (CRP,
ESR, RF, ANA)
Motion (using
the joint)
exacerbates the
mechanical
forces that are
wearing down
the joint
Fragments of
broken articular
cartilage triggers a
mild synovial
inflammation that
seeps fluid into the
synovial membrane
during inactivity
Pain with motion
(relieved by rest)
Joint stiffness
after inactivity
Moving the joint physically
pushes inflammatory fluid
back into lymphatics within
minutes.
Note: one big difference btw
the two is the degree of joint
inflammation (synovitis)
present in the joint capsule
Stiffness is short-lived,
quickly relieved by
movement
Published November1,2012 on www.thecalgaryguide.com
Larger, weight-
bearing joints tend to
bear the brunt of the
mechanical forces
Most commonly
affects the knee,
hip, and L/C-spine
facet joints
Complications" title="Legend: Inflammatory vs Degenerative Joint Disease: Pathogenesis & clinical findings
Inflammatory Joint Disease
The body’s immune system fails to discriminate between self & non-self
Autoimmune cells attack body tissues
↑ Synovial (thin lining inside of joints) inflammation
↑ Inflammation dilates blood vessels & activates the
endothelium (lining of blood vessels)
↑ Intercellular junctions open à "leaky" blood vessels
Synovial fluid
accumulates &
causes joint
edema (swelling)
↑ Blood flow
carries heat to
affected joint
tissues
Edema
(swelling)
Warmth
Erythema
(redness)
Fluid builds in
the synovial
membrane
overnight
Morning
joint
stiffness
Swelling
activates joint
nociceptors
(damage
alarms)
Pain worse
with rest
Systemic immune
activation occurs ↑ Inflammatory mediators
circulate throughout the body
Extra-
articular
(body tissues
outside of
joints)
inflammatory
damage
± ↑
Inflammatory
markers (CRP,
ESR)
± ↑ Auto-
antibodies
(RF, ANA)
Lymphatic vessels (the
body’s cleaning system)
need ↑ time to
reabsorb ↑ fluid that
accumulated overnight
Joint stiffness
resolves ~1 hour
after waking
Movement (using the
joint) pushes fluid into
lymphatic vessels (the
body’s cleaning system)
Pain relieved
with movement
Degenerative Joint Disease
Abnormal mechanics
(e.g., poor posture, weak
stabilizing muscles)
Repetitive or
excess stress on
joints
Weight bearing joints (knee, hip &
lumbar/cervical facet joints)
sustain the highest mechanical
load & degenerate over time
Fragments of articular
(joint) cartilage trigger a
synovial (thin lining inside
of joints) inflammation
Movement (using the
joint) exacerbates the
mechanical forces
wearing down the joint
Inflammation causes
fluid to seep into the
synovial membrane
during inactivity
Pain with
movement
Pain relieved
with rest
Joints stiffen with rest
Movement (using
the joint) pushes
fluid back into
lymphatic vessels
(the body’s
cleaning system)
Joint stiffness resolves
within minutes of
movement
Authors:
Yan Yu
Mackenzie Trpcic
Reviewers:
Jennifer Au
Sean Spence
Catherine R Jarvis
Martin Atkinson*
Glen Hazlewood*
* MD at time of publication
Published Nov 1, 2012; updated Feb 1, 2026 on www.thecalgaryguide.com
Pathophysiology Mechanism
Sign/Symptom/Lab Finding Complications
Final slide is the 1st slide
Legend: Inflammatory vs Degenerative Joint Disease: Pathogenesis & clinical findings
Inflammatory Joint Disease
Degenerative Joint Disease
The body’s immune system fails to discriminate between self & non-self
Autoimmune cells attack body tissues
Abnormal mechanics
(e.g., poor posture, weak
stabilizing muscles)
Repetitive or
excess stress on
joints
↑ Synovial (thin lining inside of joints)
inflammation dilates blood vessels
Systemic immune
activation occurs ↑ Endothelium (lining of blood vessels) activation
opens intercellular junctions (“leaky” blood vessels”)
↑ Inflammatory mediators
circulate throughout the body
Weight bearing joints (knee, hip &
lumbar/cervical facet joints)
sustain the highest mechanical
load & degenerate over time
Synovial fluid
accumulates &
causes joint
edema (swelling)
Edema
(swelling)
↑ Blood flow
carries heat to
affected joint
tissues
Extra-
articular
(body tissues
outside of
± ↑
Inflammatory
markers (CRP,
ESR)
± ↑ Auto-
antibodies
(RF, ANA)
Fragments of articular
(joint) cartilage trigger a
synovial (thin lining inside
of joints) inflammation
Movement (using the
joint) exacerbates the
mechanical forces
wearing down the joint
joints)
inflammatory
Inflammation causes
Warmth Pain relieved
Erythema
damage
fluid to seep into the
Pain with
(redness)
synovial membrane
movement
with rest
during inactivity
Fluid builds in
the synovial
membrane
overnight
Morning
joint
stiffness
Lymphatic
vessels (cleaners)
need ↑ time to
reabsorb ↑ fluid
that accumulated
overnight
Joints stiffen
with rest
Joint stiffness resolves
~1 hour after waking
Movement (using
the joint) pushes
fluid back into
lymphatic vessels
(cleaners)
Swelling
activates joint
nociceptors
(damage
alarms)
Pain worse
with rest
Movement (using
the joint) pushes
fluid into
lymphatic vessels
(cleaners)
Pain relieved with
movement
Joint stiffness
resolves within
minutes of
movement
Authors:
Yan Yu
Mackenzie Trpcic
Reviewers:
Jennifer Au
Sean Spence
Martin Atkinson*
Catherine R Jarvis
* MD at time of
publication
Published on www.thecalgaryguide.com
Pathophysiology Mechanism
Sign/Symptom/Lab Finding Complications
Inflammatory vs Degenerative Joint Disease: Pathogenesis & clinical findings
Inflammatory Joint Disease
Degenerative Joint Disease
The body’s immune system fails to discriminate between self & non-self
Auto-immune cells attack body tissues
Abnormal mechanics
(e.g., poor posture, weak
stabilizing muscles)
Repetitive or
excess stress on
joints
↑ Synovial (thin lining inside of joints)
inflammation dilates blood vessels
Systemic immune
activation occurs ↑ Endothelium (lining of blood vessels) activation
opens intercellular junctions (“leaky” blood vessels”)
↑ Inflammatory mediators
circulate throughout the body
Weight bearing joints (knee, hip &
lumbar/cervical facet joints)
sustain the highest mechanical
load & degenerate over time
Synovial fluid accumulates &
leads to joint edema (swelling)
↑ Blood flow carries heat
to affected joint tissues
Swelling
activates
joint
nociceptors
(damage
alarms)
Fluid
builds in
the
synovial
membrane
overnight
Edema
(swelling)
Warmth
Erythema
(redness)
Extra-articular
(body tissues
outside of
joints)
inflammatory
damage
± ↑
Inflammatory
markers (CRP,
ESR) & auto-
antibodies
(RF, ANA)
Movement (using the
joint) exacerbates the
mechanical forces
wearing down the joint
Pain with
movement
Pain relieved
with rest
Pain worse
with rest
Morning
joint stiffness
Movement
(using the
joint) pushes
fluid into
lymphatic
vessels
(cleaners)
Lymphatic
vessels
(cleaners)
need ↑ time
to reabsorb
↑ fluid
overnight
Pain relieved
with
movement
Joint stiffness
resolves ~1 hour
after waking
Authors:
Yan Yu
Mackenzie Trpcic
Reviewers:
Jennifer Au
Sean Spence
Martin Atkinson*
Catherine R Jarvis
* MD at time of
publication
Legend: Pathophysiology Mechanism
Sign/Symptom/Lab Finding Complications
Fragments of articular
(joint) cartilage trigger a
synovial (thin lining inside
of joints) inflammation
Inflammation causes
fluid to seep into the
synovial membrane
during inactivity
Joints stiffen
with rest
Movement (using
the joint) pushes
inflammatory fluid
back into lymphatic
vessels (cleaners)
Joint stiffness
resolves within
minutes of
movement
Published on www.thecalgaryguide.com
Inflammatory vs Degenerative Joint Disease: Pathogenesis & clinical findings
Inflammatory Joint Disease
The body’s immune system fails to discriminate between self and non-self
Auto-immune cells attack body tissues
↑ Synovial (thin lining inside of joints)
inflammation dilates blood vessels
↑ Endothelium (lining inside of
blood vessels) permeability
Synovial fluid accumulates &
leads to joint edema
(swelling)
Systemic immune
activation occurs
↑ Inflammatory mediators
circulate throughout the
body
Extra-articular
involvement
(body tissues
outside of
joints)
Elevated
inflammatory
markers (CRP,
ESR) & auto-
antibodies
(RF, ANA)
Degenerative Joint Disease
Abnormal
mechanics (e.g.,
poor posture,
weak stabilizing
muscles)
Repetitive/excess
stress on joints
Weight bearing joints (knee, hip &
lumbar/cervical facet joints)
sustain the highest mechanical
load & degenerate over time
Movement (using the joint)
exacerbates the mechanical
forces wearing down the
joint
Pain with
movement
Pain
relieved
with
rest
Authors:
Yan Yu
Mackenzie Trpcic
Reviewers:
Jennifer Au
Sean Spence
Martin Atkinson*
Catherine R Jarvis
* MD at time of
publication
Swelling activates
pain-sensitive joint
nociceptors
(damage alarms)
Pain worse with rest
Joint movement
pushes fluid into
lymphatic vessels
(body's clean-up
system)
Pain relieved
with movement
Legend: ↑ Blood flow carries heat to
affected joint tissues
Edema
(swelling)
Warmth
Erythema
(redness)
Fluid gradually builds in the
synovial membrane overnight
Morning joint stiffness
Lymphatic vessels
(body's clean-up system)
need ↑ time to reabsorb
↑ fluid that accumulated
overnight
Joint stiffness resolves ~1
hour after waking
Fragments of articular
(joint) cartilage trigger a
synovial (thin lining inside
of joints) inflammation
Inflammation causes fluid
to seep into the synovial
membrane during inactivity
Joints stiffen with rest
Moving the joint
physically pushes
inflammatory fluid back
into lymphatic vessels
(body's clean-up
system)
Joint stiffness resolves
within minutes of
movement
Pathophysiology Mechanism
Sign/Symptom/Lab Finding Complications
Published on www.thecalgaryguide.com
JOINT DISEASE: Inflammatory vs. Degenerative
Inflammatory Joint Disease
The body’s immune system fails to discriminate between self and non-self
Auto-immune cells attack body tissues
↑ Synovial (thin lining inside of the joint)
inflammation dilates blood vessels
↑ Endothelium (inner lining of blood
vessels) permeability
Synovial fluid accumulates &
leads to joint edema (swelling)
↑ Blood flow carries
heat to affected joint
tissues
Swelling activates
pain-sensitive joint
nociceptors
(damage alarms)
Fluid accumulates
progressively in the
synovial membrane
overnight
Edema
(swelling)
Warmth
Inflammatory pain
worse with rest
Morning joint
stiffness
Erythema
(redness)
Joint movement physically
pushes fluid back into
lymphatic vessels (body's
clean-up system)
Lymphatic vessels (body's
clean-up system) need ↑
time to reabsorb the ↑ fluid
that accumulated overnight
Inflammatory pain relieved
with activity
Joint stiffness resolves > 1hr
after waking
Legend: Mechanism
Sign/Symptom/Lab Finding Systemic
immune
activation occurs
↑
Inflammatory
mediators
circulate
throughout the
body
Extra-
articular
involvement
(e.g., skin,
eyes, lung,
heart,
kidneys) ±
abnormal
inflammatory
markers (CRP,
ESR) &
autoantibodi
es (RF, ANA)
Pathophysiology Complications
Degenerative Joint Disease
Abnormal mechanics or repetitive/excessive
loading stresses the joints
Joint structures degrade over time
Movement
(using the
joint)
exacerbates
the
mechanical
forces that are
wearing down
the joint
Pain with
movement
Pain relieved
with rest
Fragments of broken
articular cartilage
triggers a mild
synovial (thin lining
inside of the joint)
inflammation that
seeps fluid into the
synovial membrane
during inactivity
Joints stiffen with rest
Moving the joint
physically pushes
inflammatory fluid
back into lymphatic
vessels (body's clean-
up system) within
minutes
Joint stiffness resolves
within minutes of
movement
Published on www.thecalgaryguide.com
Authors:
Yan Yu
Mackenzie Trpcic
Reviewers:
Jennifer Au
Sean Spence
Martin Atkinson*
Catherine R Jarvis
* MD at time of
publication
The weight-
bearing joints
(i.e., knee, hip, &
lumbar/cervical
facet joints)
sustain the
highest
mechanical load
& degeneration
Distinguishing between Inflammatory and Degenerative joint disease
Inflammatory Joint
Disease
Auto-immune attack of body tissues, due to
failure of the body’s immune system to
discriminate between self and non-self.
Degenerative Joint
Disease
Repetitive, excess, or abnormal mechanical
forces on joints over time, leading to physical
breakdown of the joint.
Authors:
Yan Yu
Reviewers:
Jennifer Au
Sean Spence
Martin Atkinson*
* MD at time of
publication
Within joints, severe synovial inflammation
dilates local blood vessels, and causes the
vessel endothelium to be more leaky.
Accumulation of abundant
inflammatory synovial fluid
at rest or overnight
Swelling stimulates
joint nociceptors
Pain with rest
Moving the joint
physically pushes fluid
back into lymphatics,
relieving the painful
swelling.
Pain relieved with
motion
Legend: Overnight fluid
accumulation in the
synovial membrane
Morning stiffness
Lots of fluid, takes
more time (hours)
to be reabsorbed
by lymphatics
Stiffness resolves
after >1hr
Blood carries
heat and
looks red
Warm, Red,
Swollen
Joints
Pathophysiology Mechanism
Sign/Symptom/Lab Finding Inflammation is
oftentimes
systemic, not
localized
exclusively within
the joint (i.e. in
Rheumatoid
arthritis, Lupus,
etc)
Extra-articular
manifestations;
positive
inflammatory
markers (CRP,
ESR, RF, ANA)
Motion (using
the joint)
exacerbates the
mechanical
forces that are
wearing down
the joint
Fragments of
broken articular
cartilage triggers a
mild synovial
inflammation that
seeps fluid into the
synovial membrane
during inactivity
Pain with motion
(relieved by rest)
Joint stiffness
after inactivity
Moving the joint physically
pushes inflammatory fluid
back into lymphatics within
minutes.
Note: one big difference btw
the two is the degree of joint
inflammation (synovitis)
present in the joint capsule
Stiffness is short-lived,
quickly relieved by
movement
Published November1,2012 on www.thecalgaryguide.com
Larger, weight-
bearing joints tend to
bear the brunt of the
mechanical forces
Most commonly
affects the knee,
hip, and L/C-spine
facet joints
Complications" />
Necrosis versus Apoptosis

Hypoxia
(↓ Oxygen
delivered to tissues)
Physical agents
(extreme temperatures,
trauma, radiation)
Bacterial, viral, or
fungal infections
&/or microbe toxins
Necrosis
Uncontrolled & pathologic cell death
Harmful stimuli trigger ↓ ATP (cellular energy)
↓ ATP leads to ion pump failure & ↑ cell permeability (“leakiness”)
↑ Cellular ion influx causes cell to swell & lyse (rupture its plasma membrane)
Intracellular contents spill into the extracellular space
Intracellular contents activate inflammasomes (cytoplasmic multiprotein complexes)
Pro-inflammatory cytokine interleukin-1β is secreted & activates the immune system
Excessive cytokine release provokes a large inflammatory response
Inflammation damages surrounding healthy tissues & organs
Ischemia (↓ Blood
flow) to organs
except brain
↓ Oxygen denatures
enzymes à dead cell
skeletons cannot breakdown
Cells remain
“coagulated”
(firm) for days
Coagulative
necrosis
↓ Blood flow to limb(s)
+/- bacterial invasion
Gangrenous
necrosis
↑ Pain or ↓ feeling in affected limb(s)
Skin color changes (red, purple, black)
Damaged adipocytes (fat cells) release
triglyceride (fat) contents that group into oil cysts
Scars form &
calcify (harden)
Fat necrosis
Damaged endothelium (blood
vessel wall) becomes leaky
Fibrin (blood protein) deposits in
endothelium with immune complexes
Fibrinoid
necrosis
Brain ischemia &/or
infection kills cells
Digestive enzymes transform dead
tissue into creamy liquid pus
Liquefactive necrosis
Coagulative + liquefactive necrosis mechanisms à granular debris
Caseous necrosis
Legend: Pathophysiology Mechanism
Sign/Symptom/Lab Finding Complications
-----------------------------------------------------------------------------------------------------
Authors:
Tyra Fernandes
Catherine R Jarvis
Reviewers:
Mitchell Chorney
Sarah Smith*
* MD at time of publication
Apoptosis
Programmed cell death via an energy-dependent process
Extrinsic pathway: cellular stress or
damage signals in the extracellular
environment trigger apoptosis
Intrinsic pathway: internal
cellular stressors (damage from
chemotherapy, UV radiation,
drugs) trigger apoptosis
Ligands such as TNF-α (tumor
necrosis factor-alpha, produced by
macrophages) & FasL (Fas ligand,
produced by T cells) bind to apoptotic
receptors on the cell’s surface
Pro-apoptotic factors recognize
cellular stressors
Cytochrome C releases from the
intermembrane space of the
mitochondria to the cytoplasm
Ligand binding activates initiator
caspases (protease (protein
breakdown)-like enzymes) 2, 8, 9, 10
Cytochrome C activates Caspase-9
Initiator caspases activate
effector caspases 3, 6, 7
Caspase-9 activates
effector caspases 3 & 7
Effector caspases begin execution pathway
Caspases degrade the cell’s nuclear envelope & DNA
à DNA repair is inhibited & cell shrinks
Cellular shrinkage forms apoptotic bodies
Phagocytes engulf apoptotic bodies to prevent inflammation
Ineffective phagocytosis à inflammation
Published Feb 8, 2026 on www.thecalgaryguide.com
Final Slide is 1st
Necrosis vs Apoptosis: Pathogenesis and clinical findings
Toxic chemicals
(poisons, drug
toxicities)
Hypoxia
(↓ Oxygen
delivered to tissues)
Physical agents
(extreme temperatures,
trauma, radiation)
Toxin release
from bacteria,
viruses, or fungi
Necrosis
Uncontrolled & pathologic cell death
Harmful stimuli trigger ↓ ATP (cellular energy)
↓ ATP leads to ion pump failure & ↑ cell permeability
↑ Cellular ion influx causes cell to swell & lyse (rupture its plasma membrane)
Intracellular contents spill into the extracellular space
Intracellular contents activate inflammasomes (cytoplasmic multiprotein complexes)
Pro-inflammatory cytokine interleukin-1β is secreted and activates the immune system
Excessive cytokine release provokes a large inflammatory response
Inflammation damages surrounding healthy tissues & organs
Ischemia (↓ Blood
flow) to organs
except brain
↓ Oxygen denatures
enzymes à cell skeleton
cannot be broken down
Cell is preserved
in “coagulated”
state for days
Coagulative
necrosis
↓ Blood flow to limbs
+/- bacterial invasion
Gangrenous
necrosis
↑ Pain or ↓ feeling in limb
Skin color changes (red, purple, black)
Damaged adipocytes (fat cells) release
triglycerides (fat) contents that group into oil cysts
Scars form &
calcify (harden)
Fat necrosis
Damaged endothelium (blood
vessel wall) becomes leaky
Fibrin (blood protein) deposits in
endothelium with immune complexes
Fibrinoid
necrosis
Brain ischemia &/or
infection kills cells
Digestive enzymes transform dead
tissue into creamy liquid pus
Liquefactive necrosis
Coagulative & liquefactive necrosis mixture à granular debris
Caseous necrosis
Legend: Pathophysiology Mechanism
Sign/Symptom/Lab Finding -----------------------------------------------------------------------------------------------------
Complications
Authors:
Tyra Fernandes
Catherine R Jarvis
Reviewers:
Mitchell Chorney
Sarah Smith*
* MD at time of publication
Apoptosis
Programmed cell death via an energy-dependent process
Extrinsic pathway: cellular stress or
damage signals in the extracellular
environment trigger apoptosis
Intrinsic pathway: internal
cellular stressors (chemotherapy,
UV radiation, drugs, misfolded
proteins) trigger apoptosis
Ligands such as TNF-α (tumor
necrosis factor-alpha, produced by
macrophages) & FasL (Fas ligand,
produced by T cells) bind to apoptotic
receptors on the cell’s surface
Pro-apoptotic factors recognize
cellular stressors
Cytochrome C releases from the
intermembrane space of the
mitochondria to the cytoplasm
Ligand binding activates initiator
caspases (protease (protein
breakdown)-like enzymes) 2, 8, 9, 10
Cytochrome C activates Caspase-9
Initiator caspases activate
effector caspases 3, 6, 7
Caspase-9 activates
effector caspases 3 & 7
Effector caspases begin execution pathway
Caspases degrade the cell’s nuclear envelope & DNA
à DNA repair is inhibited & cell shrinks
Cellular shrinkage forms apoptotic bodies
Phagocytes engulf apoptotic bodies to prevent inflammation
Ineffective phagocytosis à inflammation
Published MONTH, DAY, YEAR on www.thecalgaryguide.com
Necrosis vs Apoptosis
Toxic chemicals
(poisons, drug
toxicities)
Hypoxia
(↓ Oxygen
delivered to tissues)
Physical agents
(extreme temperatures,
trauma, radiation)
Biological agents
(bacteria,
viruses, fungi)
Necrosis
Non-physiologic & uncontrolled cell death
Noxious (harmful) stimuli trigger increased cell permeability and ion pump failure
Cell swells & ruptures its plasma membrane à cell lysis (breakdown)
Intracellular contents spill into the extracellular space
Intracellular contents activate inflammasomes (cytoplasmic multiprotein complexes)
Pro-inflammatory cytokine interleukin-1β is secreted and activates the immune system
Excessive cytokine release provokes a large inflammatory response
Inflammation damages surrounding healthy tissues & organs
Acute tubular
necrosis
(damaged kidney
tubule cells)
Gangrene
(body tissue death)
Myocardial infarction
(heart attack)
Steatohepatitis
(progressive
liver disease)
↓ Blood flow to a
Myocardium (heart
portion of the body
muscle) is deprived
Necrotic renal
(usually limbs)
of oxygen &
(kidney) tubular
becomes damaged
epithelial cells
deteriorate &
slough off
↑ Extracellular
matrix proteins
(especially
collagen) are
produced and
deposit in liver
Granular casts
on urine
microscopy
↑ Pain
or
↓ Feeling
Skin
color
change
(red,
purple,
black)
↑ Blood
troponin
levels
↓ Heart
function
Liver fibrosis
(scarring)
Legend: Authors:
Tyra Fernandes
Catherine R Jarvis
Reviewers:
-----------------------------------------------------------------------------------------------------
Pathophysiology Mechanism
Sign/Symptom/Lab Finding Complications
Mitchell Chorney
Name Name*
* MD at time of publication
Apoptosis
Programmed cell death via an energy-dependent process
Extrinsic Pathway: Apoptotic signals
from other cells cause initiation
Intrinsic Pathway: Internal cellular
stressors (chemotherapy, UV
radiation, drugs, misfolded
proteins) cause initiation
Ligands such as TNF-α (tumor necrosis
factor-alpha, produced by
macrophages) & FasL (Fas ligand,
produced by T cells) bind to apoptotic
receptors on the cell’s surface
Pro-apoptotic factors recognize
cellular stressors
Cytochrome P releases from the
intermembrane space of the
Ligand binding activates initiator
mitochondria to the cytoplasm
caspases (protease (protein
breakdown)-like enzymes) 2, 8, 9, 10
Cytochrome P activates Caspase-9
Initiator caspases activate
effector caspases 3, 6, 7
Caspase-9 activates caspases
3 & 7
Execution Pathway
Caspases degrade the cell’s nuclear envelope & DNA
à DNA repair is inhibited & cell shrinks
Cellular shrinkage forms apoptotic bodies
Phagocytes engulf apoptotic bodies to prevent inflammation
Ineffective phagocytosis à inflammation
Published MONTH, DAY, YEAR on www.thecalgaryguide.com
Necrosis vs Apoptosis
Hypoxia
Chemical agents
Physical agents Biological agents
Necrosis
Non-physiologic & uncontrolled
cell death
Oncolysis (ion pump failure and
increased cell permeability
cause cell swelling) occurs due
to noxious stimuli
Plasma membrane ruptures and
cell is lysed (broken down)
Intracellular contents spill into
the extracellular space
Authors:
Tyra Fernandes
Reviewers:
Mitchell Chorney
Catherine R Jarvis
Name Name*
* MD at time of publication
Renal tubule is damaged due to
reduced oxygen supply and toxicity
Granular casts on urine microscopy
Acute tubular necrosis (kidney
disorder of the tubule cells
Extracellular matrix proteins accumulate
in liver causing collagen deposition
Liver fibrosis
Steatohepatitis
(progressive liver disease)
Intracellular contents activate
inflammasomes (cytoplasmic
multiprotein complexes)
Myocardium (heart muscle) is
deprived of oxygen
↑ Blood troponin levels
Myocardial infarction
-----------------------------------------------------------------------------------------------------
Apoptosis
Programmed cell death via an
energy-dependent process
Extrinsic Pathway: Apoptotic signals
from other cells cause initiation
Ligands such as TNF-α (tumor necrosis
factor-alpha, produced by
macrophages) and FasL (Fas ligand,
produced by T cells) bind to apoptotic
receptors on the cell’s surface
Ligand binding activates initiator
caspases (protease-like
enzymes) 2, 8, 9, 10
Initiator caspases activate
effector caspases 3, 6, 7
Intrinsic Pathway: Internal cellular
stressors (chemotherapy, UV
radiation, drugs, & misfolded
proteins) cause initiation
Pro-apoptotic factors recognize
cellular stressors
Cytochrome P releases from the
intermembrane space of the
mitochondria to the cytoplasm
Cytochrome P activates Caspase-9
Caspase-9 activates caspases
3 & 7
Pro-inflammatory cytokine
interleukin-1β is secreted and
activates the immune system
Excessive cytokine release
provokes a large inflammatory
response
Legend: Inflammatory
damage to the
surrounding
healthy tissues
Microscopic
Effects:
Pathophysiology Mechanism
Sign/Symptom/Lab Finding Complications
Execution Pathway
Caspases degrade the cell’s nuclear envelope. DNA
fragments inhibit DNA repair, which shrinks the cell
Cellular shrinkage forms apoptotic bodies
Phagocytes engulf apoptotic bodies to prevent inflammation
Ineffective phagocytosis à inflammation
Published MONTH, DAY, YEAR on www.thecalgaryguide.com
Necrosis vs Apoptosis
Authors:
Tyra Fernandes
Reviewers:
Mitchell Chorney
Catherine R Jarvis
Hypoxia, physical agents,
chemical agents, biologic agents
Necrosis (Non-physiologic
& uncontrolled cell death)
Oncolysis (cell swelling due to
the failure of ion pumps and
increased cell permeability)
occurs due to noxious stimuli
Plasma membranes rupture
leading to cell lysis
Intracellular contents spill into
the extracellular space
Inflammasomes (cytoplasmic
multiprotein complexes) are
activated
Pro-inflammatory cytokine
interleukin-1β is secreted and
activates the immune system
Excessive cytokine release
provokes a large inflammatory
response
Legend: Microscopic Effects:
Steatohepatitis
Collagen deposition due
to the accumulation of
extracellular matrix
proteins
Liver fibrosis
Myocardial
Infarction
Ischemic or toxic
injury to the renal
tubule
Granular casts on
urine microscopy
Acute tubular
necrosis
Oxygen supply
deprivation to the
myocardium
Elevated blood
troponin levels
-----------------------------------------------------------------------------------------------------
Extrinsic Pathway: Apoptotic Signals
from other cells cause initiation
Ligands such as TNF-α (tumor necrosis
factor-alpha, produced by
macrophages) and FasL (Fas ligand,
produced by T cells) bind to apoptotic
receptors on the cell’s surface
Ligand binding activates initiator
caspases (protease-like
enzymes) 2, 8, 9, 10
Initiator caspases activate
effector caspases 3, 6, 7
Apoptosis (Programmed
cell death via an energy-
dependent process)
Name Name*
* MD at time of publication
Intrinsic Pathway: Internal cellular
stressors chemotherapy, UV
radiation, drugs, & misfolded
proteins) cause initiation
Pro-apoptotic factors recognize
cellular stressors
Cytochrome P releases from the
intermembrane space of the
mitochondria to the cytoplasm
Cytochrome P activates Caspase-9
Caspase-9 activates caspases 3 and
7
Execution Pathway
Cellular shrinkage
forms apoptotic
bodies
Inflammatory damage to
surrounding healthy tissues à
potential organ failure
Pathophysiology Mechanism
Sign/Symptom/Lab Finding Complications
The nuclear envelope
degrades and DNA
fragments which
inhibits DNA repair
which shrinks the cell
Phagocytes engulf
apoptotic bodies to
prevent inflammation
Ineffective phagocytosis
à inflammation
Published MONTH, DAY, YEAR on www.thecalgaryguide.com
Authors:
Necrosis vs Apoptosis
Tyra Fernandes
Reviewers:
Mitchell Chorney
Necrosis Apoptosis
Catherine R Jarvis
Hypoxia: ischemia, respiratory
insufficiency
Physical agents: trauma, radiation,
Non-physiologic &
temperature, electrical shock
uncontrolled cell death
Biological agents: bacteria, viruses, fungi
Chemical agents: poisons, drugs,
Cell swelling (oncosis) due to
occupational exposures
the failure of ion pumps and
increased cell permeability
Collagen deposition leading
to liver fibrosis
Cell lysis due to the rupture of
the plasma membrane
Steatohepatitis
Microscopic
Spillage of intracellular contents
Effects:
into the extracellular space
Acute tubular
Myocardial
Activation of cytoplasmic
necrosis
Infarction
multiprotein complexes, known
as inflammasomes
Ischemic or toxic
Oxygen supply
injury to the renal
deprivation to the
tubule
myocardium
Secretion of pro-inflammatory
cytokine interleukin-1β, which
-----------------------------------------------------------------------------------------------------
Name Name*
* MD at time of publication
Programmed cell death via
an energy-dependent
process
Extrinsic pathway Intrinsic pathway
The cell receives apoptotic signals
Initiated by internal cellular
from other cells
stressors such as chemotherapy
drugs, UV radiation, and misfolded
Apoptosis is activated via ligands
proteins
binding to apoptotic receptors on
the cell surface
Pro-apoptotic factors recognize
cellular stressors
Ligand binding activates initiator
caspases (2, 8, 9, 10), which are
Cytochrome P releases from the
protease-like enzymes
intermembrane space of the
mitochondria to the cytoplasm
Examples of ligands include: TNF-α
(produced by macrophages) and
Caspase-9 is activated
FasL (produced by T cells)
Activation of downstream effector
Effector caspases (3, 6, 7) are
caspases 3 and 7
activated
activates the immune system
Excessive cytokine release
provokes a large inflammatory
response
Legend: Granular casts on
urine microscopy
Elevated blood
troponin levels
Damage to surrounding
healthy tissues may lead to
organ failure
Prevention of
inflammation
Execution pathway
The nuclear envelope
degrades and DNA fragments
which inhibits DNA repair
Cellular shrinkage
forms apoptotic
bodies
Phagocy
engulfment of
apoptotic bodies
Published MONTH, DAY, YEAR on www.thecalgaryguide.com
Pathophysiology Mechanism
Sign/Symptom/Lab Finding Complications
Necrosis versus apoptosis
Necrosis
Non-physiologic &
uncontrolled cell death due
to noxious stimuli
Cell swelling (oncosis) à cell
lysisà spillage of
intracellular contents à
tissue damage
Inflammatory response,
leading to inflammasome
activation and the secretion of
pro-inflammatory cytokine
interleukin-1β
Macroscopic
Effects:
Hypoxia: ischemia, respiratory insufficency
Physical agents: trauma, radiation,
temperature, electrical shock
Biological agents: bacteria, viruses, fungi
Chemical agents: poisons, drugs,
occupational exposures
Acute tubular
necrosis due to
chemical agents
Myocardial
infarction due to
hypoxia
Microscopic
Effects:
Stroke due to
hypoxia in the brain
Gangrene due to
hypoxia of the limbs
Steatohepatitis due
to chemical agents
-----------------------------------------------------------------------------------------------------
Apoptosis
Programmed cell death via
an energy-dependent
process
Execution phase: pro-& anti-
apoptotic proteins mediate
cell breakdown by caspases
Caspase activity blocks DNA
repair à leading to
fragmented DNA and cell
shrinkage
Apoptosis is
inhibited in cancer
the overexpression
of anti-apoptotic
proteins
Authors:
Tyra Fernandes
Reviewers:
Mitchell Chorney
Name Name*
* MD at time of publication
Extrinsic pathway: immune cell activation
activates executioner caspases; ligands such
as TNF-α & FasL bind to receptors on the cell
membrane
Intrinsic pathway: internal cellular factors
cause pro-apoptotic factors to increase
mitochondrial membrane permeability.
Cytochrome c is released from the inner
mitochondrial membrane, which ultimately
activates caspases
Cellular fragments are
engulfed by immune cells
which prevents an
inflammatory response
Organism death via
necrosis
Menstruation Embryogenesis
Endometrial
shedding of the
inner lining of
the uterus is an
apoptotic
mechanism
The tissue
between the
fingers and toes
degrades via
apoptosis in
utero à
separation of
the digits
Legend: Pathophysiology Mechanism
Sign/Symptom/Lab Finding Abbreviations:
• IL-1β– interleukin-1β
• TNF-α– tumour necrosis factor - alpha
• FasL– CD95 ligand; mediates apoptosis
Published MONTH, DAY, YEAR on www.thecalgaryguide.com
Complications
References
• https://next.amboss.com/us/article/VP0GdT?q=apoptosis#Y0d79977cce8fc2c548ac720f8060d667
• https://www.ncbi.nlm.nih.gov/books/NBK557627/
• https://www.ncbi.nlm.nih.gov/books/NBK499821/
• https://pmc.ncbi.nlm.nih.gov/articles/PMC5855670/
• https://www.sciencedirect.com/topics/pharmacology-toxicology-and-pharmaceutical-science/oncosis
• https://www.nature.com/articles/s41421-020-0167-x
• https://pmc.ncbi.nlm.nih.gov/articles/PMC3714593/
• https://www.ncbi.nlm.nih.gov/books/NBK537076/
Hypoxia
Physical agents
Biological agents
Chemical agents")
Coxsackie, Echo, and Entero-viruses; including Hand, Foot, & Mouth Disease

Virus replicates in intestinal &
pharyngeal lymphoid tissue &
spreads to regional lymph
nodes
Virus spreads to various target
tissues via blood (viremia)
Systemic inflammatory
cytokines trigger hypothalamus
to ↑ temperature
Fever
Sore, swollen
throat
Viral replication
creates vesicles in oral
mucosa, which rapidly
rupture to create
ulcers
Enanthem in
mucous
membranes
of mouth
Sores &
inflammation
cause painful
swallowing,
preventing
fluid intake
Clusters of macules & papules
(immunopathologic response) and vesicles
(pathogenic response) develops on palms of
hands, & soles of feet
Dehydration
EV-A71 targets
pulmonary epithelial
cells and thus proliferate
in lung tissue
Brainstem encephalitis or systemic
inflammation possibly ↑ vascular
permeability in lungs (exact
mechanism unknown)
Pulmonary hemorrhage
Pulmonary edema
Legend: Virus replicates in
squamous epithelial cells
via SCARB2 proteins, which
are abundant in the hands,
feet & mouth
Virus causes keratinocytes to swell
and degenerate and burst, resulting
in edema of the papillary dermis
EV-A71 targets specific neuronal &
neuromuscular proteins, thus
proliferating in central nervous
system and muscle
Virus causes infection &
swelling of the meninges
& cerebrospinal fluid
Viral meningitis
EV-A71 attaches to white blood
cells, passes through the blood
brain barrier, & triggers brain
inflammation
Brainstem encephalitis
Possible injury to nail
matrix from damage to
keratinocytes in nail bed
(exact mechanism
unknown)
Onychomadesis
(nail shedding)
Pathophysiology Mechanism
Sign/Symptom/Lab Finding Complications
Authors:
Alua Kulenova
Reviewers:
Emily J. Doucette
Brandon Christensen*
*MD at time of publication
**See corresponding Calgary Guide slide
Published Feb 17, 2026 on www.thecalgaryguide.com
hand foot mouth hfm disease")
Cryoglobulinemia

(↑ Lymphocyte
(blood) cancers
infection (HCV)
attacking) conditions
cerebral
Cerebral
threshold
production) conditions
(brain)
vasculitis Ischemic stroke (clot blocks
Inflamed vasculature
arteries
↓ cerebral blood flow
cerebral blood flow)
Acute ↑ B cell activity ↑
Chronic B cell activation ↑
Deposits in
Retinal
Retinal artery hemorrhage (severe bleeding)
immunoglobulin (Ig) production
immunoglobulin (Ig) production
retinal arteries
vasculitis Retinal artery infarct (↓ oxygenation à tissue death)
of eyes
Tingling
Type II:
Type III:
Deposits damage & narrow walls of
↓ Blood flow
monoclonal (recognize 1
polyclonal
tiny blood vessels supplying
to nerve cells Numbness
Type I:
site on antigen) IgM, IgG or
(recognize >1
nutrients to peripheral nerves
Weakness
monoclonal (recognize 1
IgA & polyclonal (recognize
site on antigen)
site on antigen) IgM or IgG
>1 site on antigen) Igs
IgG & IgM
Deposits in
Alveolar
alveolar (lung air
capillaries are
sacs) capillaries
destroyed
Exposure to temperatures <37°C
Immune complexes deposit & form cryoglobulin
(abnormal protein) in small/medium-sized vessels
Cryoglobulins activate complement (factors that
enhance innate (first-line) immune system responses)
Deposits in
cardiac (heart)
tissue
Deposits obstruct
mesenteric (abdominal)
vasculature
Myocardial (heart muscle)
inflammation, necrosis &
fibrosis (scarring)
Mesenteric
vasculitis
Alveolar
Dyspnea (short of breath)
hemorrhage
(severe bleeding) Hemoptysis (coughing blood)
Hypoxia (↓ Tissue oxygenation)
Myocarditis (inflamed myocardium)**
Inflamed vasculature
Gastrointestinal
à ↑ rupture risk
bleed
Mesenteric ischemia (↓
↓ Mesenteric
blood flow
oxygen to intestines) à
scars & narrows intestines
Complement factors are
consumed (activated &
used)
Immune complexes
(primarily type I)
occlude vessels
Immune complexes (primarily
types II & III) deposit in vessels à
endothelial (vessel wall) damage &
vasculitis (inflammation)
Abdominal pain
Abdominal cramps
Bowel obstruction
Deposits damage glomeruli
(kidney capillary clusters) à
triggers inflammation
↑ Permeability
(“leakiness”) in
damaged capillaries
Hematuria (blood in urine)
Proteinuria (protein in urine)
↓ Complement
factors C3 & C4
Blood in obstructed
vessel areas becomes
hyper-viscous (thick)
Hyperviscosity
syndrome
Inflammation ↑ proliferation
of glomerular basement
Blood stasis &
vascular congestion
↓ Oxygen
delivered to skin
Local hypoxia (↓
oxygen delivery)
Visual disturbances
Confusion
Headache
Membranoproliferative glomerulonephritis
(immune-mediated kidney inflammation)
Joint swelling
Joint stiffness
↑ Hydrostatic
pressure ruptures
capillaries
Livedo reticularis
(reddish-blue patchy
skin discolouration)
Reactive
vasoconstriction
(narrowed vessels)
Deposits in synovial (joint
connective tissue) membrane Deposits
damage post-
capillary
venules in
dermis (skin)
Red blood
cells leak
into
surrounding
tissue
Mucosal (moist lining of
Raynaud phenomena (extremities
exposed tissues) bleeding
become white & numb
Vasoconstriction
obstructs blood vessels
Ischemia (restricted
blood supply to tissues)
Synovial edema (swelling) & thickening
Non-blanchable
(non-color-
fading) purpura
(palpable purple
skin spots)
Necrosis (cell &
tissue death)**
Authors:
Julia Fox, Catherine R Jarvis
Reviewers:
Navdeep Goraya, Glen Hazlewood*
* MD at time of publication
**See corresponding Calgary Guide slide(s)
Legend: Pathophysiology Mechanism
Complications
Published Mar 1, 2026 on www.thecalgaryguide.com
↓ Blood
flow
Sign/Symptom/Lab Finding")