SEARCH RESULTS FOR: renal
Nephrotic Syndrome: Pathogenesis and Clinical Findings
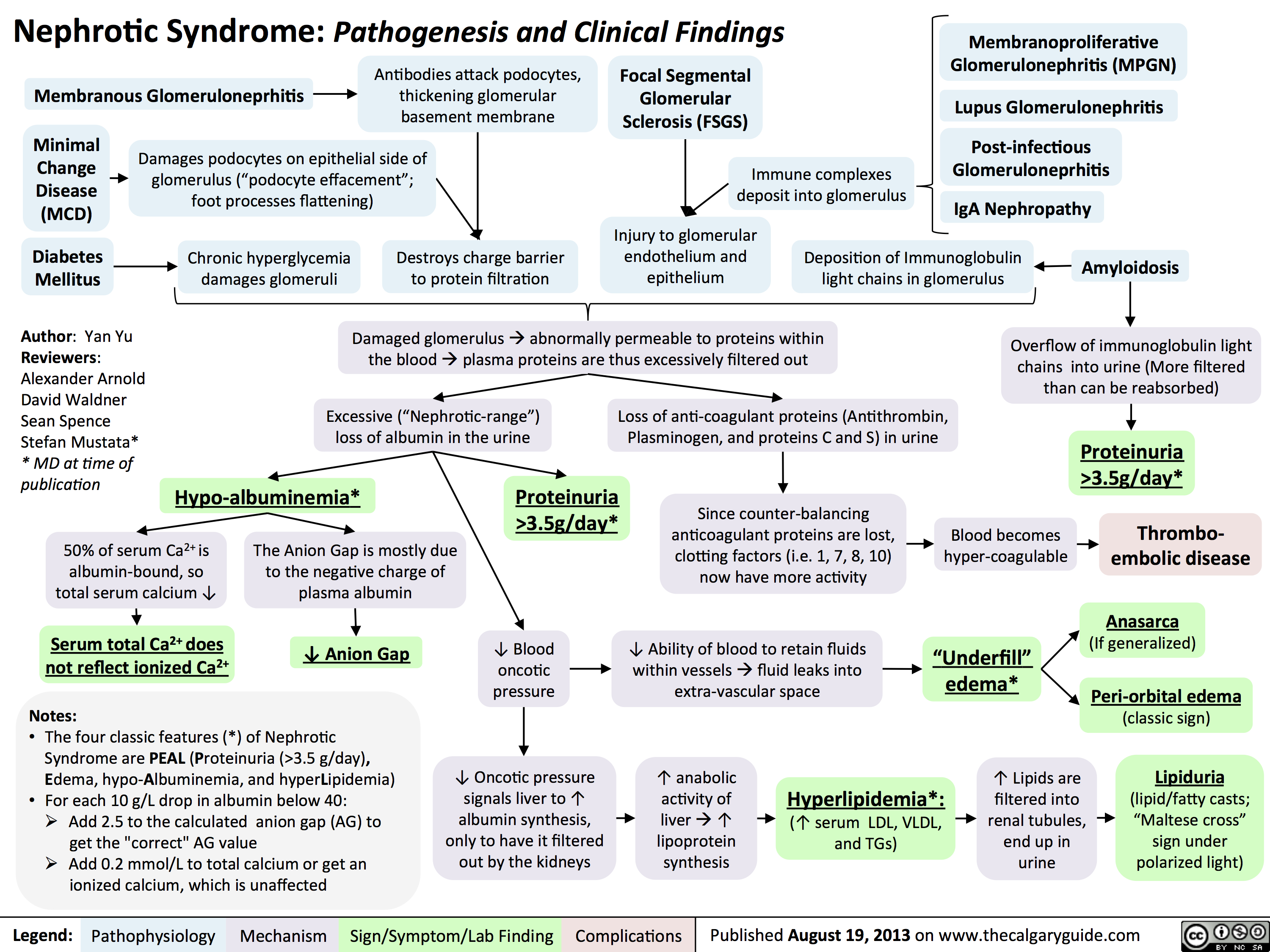 3.5g/day*? Ability of blood to retain fluids within vessels ? fluid leaks into extra-vascular spaceInjury to glomerular endothelium and epitheliumImmune complexes deposit into glomerulusDamaged glomerulus ? abnormally permeable to proteins within the blood ? plasma proteins are thus excessively filtered out? Oncotic pressure signals liver to ? albumin synthesis, only to have it filtered out by the kidneys? anabolic activity of liver ? ? lipoprotein synthesisHyperlipidemia*:(? serum LDL, VLDL, and TGs)Lipiduria(lipid/fatty casts; "Maltese cross" sign under polarized light)Since counter-balancing anticoagulant proteins are lost, clotting factors (i.e. 1, 7, 8, 10) now have more activityThrombo-embolic diseaseBlood becomes hyper-coagulable? Lipids are filtered into renal tubules, end up in urineMembranoproliferative Glomerulonephritis (MPGN)Lupus Glomerulonephritis Post-infectious GlomeruloneprhitisIgA NephropathyDamages podocytes on epithelial side of glomerulus ("podocyte effacement"; foot processes flattening)Diabetes MellitusChronic hyperglycemia damages glomeruliDeposition of Immunoglobulin light chains in glomerulusAmyloidosisAnasarca(If generalized)Peri-orbital edema (classic sign)Focal Segmental Glomerular Sclerosis (FSGS)Membranous GlomeruloneprhitisAntibodies attack podocytes, thickening glomerular basement membraneOverflow of immunoglobulin light chains into urine (More filtered than can be reabsorbed)Proteinuria >3.5g/day*The Anion Gap is mostly due to the negative charge of plasma albumin? Anion GapNotes: The four classic features (*) of Nephrotic Syndrome are PEAL (Proteinuria (>3.5 g/day), Edema, hypo-Albuminemia, and hyperLipidemia)For each 10 g/L drop in albumin below 40:Add 2.5 to the calculated anion gap (AG) to get the "correct" AG valueAdd 0.2 mmol/L to total calcium or get an ionized calcium, which is unaffected50% of serum Ca2+ is albumin-bound, so total serum calcium ? Serum total Ca2+ does not reflect ionized Ca2+ ? Blood oncotic pressure" title="Destroys charge barrier to protein filtrationNephrotic Syndrome: Pathogenesis and Clinical FindingsAuthor: Yan YuReviewers:Alexander ArnoldDavid WaldnerSean SpenceStefan Mustata** MD at time of publicationLegend:Published August 19, 2013 on www.thecalgaryguide.comMechanismPathophysiologySign/Symptom/Lab FindingComplicationsExcessive ("Nephrotic-range") loss of albumin in the urineHypo-albuminemia*Loss of anti-coagulant proteins (Antithrombin, Plasminogen, and proteins C and S) in urineMinimal Change Disease (MCD)"Underfill" edema*Proteinuria >3.5g/day*? Ability of blood to retain fluids within vessels ? fluid leaks into extra-vascular spaceInjury to glomerular endothelium and epitheliumImmune complexes deposit into glomerulusDamaged glomerulus ? abnormally permeable to proteins within the blood ? plasma proteins are thus excessively filtered out? Oncotic pressure signals liver to ? albumin synthesis, only to have it filtered out by the kidneys? anabolic activity of liver ? ? lipoprotein synthesisHyperlipidemia*:(? serum LDL, VLDL, and TGs)Lipiduria(lipid/fatty casts; "Maltese cross" sign under polarized light)Since counter-balancing anticoagulant proteins are lost, clotting factors (i.e. 1, 7, 8, 10) now have more activityThrombo-embolic diseaseBlood becomes hyper-coagulable? Lipids are filtered into renal tubules, end up in urineMembranoproliferative Glomerulonephritis (MPGN)Lupus Glomerulonephritis Post-infectious GlomeruloneprhitisIgA NephropathyDamages podocytes on epithelial side of glomerulus ("podocyte effacement"; foot processes flattening)Diabetes MellitusChronic hyperglycemia damages glomeruliDeposition of Immunoglobulin light chains in glomerulusAmyloidosisAnasarca(If generalized)Peri-orbital edema (classic sign)Focal Segmental Glomerular Sclerosis (FSGS)Membranous GlomeruloneprhitisAntibodies attack podocytes, thickening glomerular basement membraneOverflow of immunoglobulin light chains into urine (More filtered than can be reabsorbed)Proteinuria >3.5g/day*The Anion Gap is mostly due to the negative charge of plasma albumin? Anion GapNotes: The four classic features (*) of Nephrotic Syndrome are PEAL (Proteinuria (>3.5 g/day), Edema, hypo-Albuminemia, and hyperLipidemia)For each 10 g/L drop in albumin below 40:Add 2.5 to the calculated anion gap (AG) to get the "correct" AG valueAdd 0.2 mmol/L to total calcium or get an ionized calcium, which is unaffected50% of serum Ca2+ is albumin-bound, so total serum calcium ? Serum total Ca2+ does not reflect ionized Ca2+ ? Blood oncotic pressure" />
3.5g/day*? Ability of blood to retain fluids within vessels ? fluid leaks into extra-vascular spaceInjury to glomerular endothelium and epitheliumImmune complexes deposit into glomerulusDamaged glomerulus ? abnormally permeable to proteins within the blood ? plasma proteins are thus excessively filtered out? Oncotic pressure signals liver to ? albumin synthesis, only to have it filtered out by the kidneys? anabolic activity of liver ? ? lipoprotein synthesisHyperlipidemia*:(? serum LDL, VLDL, and TGs)Lipiduria(lipid/fatty casts; "Maltese cross" sign under polarized light)Since counter-balancing anticoagulant proteins are lost, clotting factors (i.e. 1, 7, 8, 10) now have more activityThrombo-embolic diseaseBlood becomes hyper-coagulable? Lipids are filtered into renal tubules, end up in urineMembranoproliferative Glomerulonephritis (MPGN)Lupus Glomerulonephritis Post-infectious GlomeruloneprhitisIgA NephropathyDamages podocytes on epithelial side of glomerulus ("podocyte effacement"; foot processes flattening)Diabetes MellitusChronic hyperglycemia damages glomeruliDeposition of Immunoglobulin light chains in glomerulusAmyloidosisAnasarca(If generalized)Peri-orbital edema (classic sign)Focal Segmental Glomerular Sclerosis (FSGS)Membranous GlomeruloneprhitisAntibodies attack podocytes, thickening glomerular basement membraneOverflow of immunoglobulin light chains into urine (More filtered than can be reabsorbed)Proteinuria >3.5g/day*The Anion Gap is mostly due to the negative charge of plasma albumin? Anion GapNotes: The four classic features (*) of Nephrotic Syndrome are PEAL (Proteinuria (>3.5 g/day), Edema, hypo-Albuminemia, and hyperLipidemia)For each 10 g/L drop in albumin below 40:Add 2.5 to the calculated anion gap (AG) to get the "correct" AG valueAdd 0.2 mmol/L to total calcium or get an ionized calcium, which is unaffected50% of serum Ca2+ is albumin-bound, so total serum calcium ? Serum total Ca2+ does not reflect ionized Ca2+ ? Blood oncotic pressure" title="Destroys charge barrier to protein filtrationNephrotic Syndrome: Pathogenesis and Clinical FindingsAuthor: Yan YuReviewers:Alexander ArnoldDavid WaldnerSean SpenceStefan Mustata** MD at time of publicationLegend:Published August 19, 2013 on www.thecalgaryguide.comMechanismPathophysiologySign/Symptom/Lab FindingComplicationsExcessive ("Nephrotic-range") loss of albumin in the urineHypo-albuminemia*Loss of anti-coagulant proteins (Antithrombin, Plasminogen, and proteins C and S) in urineMinimal Change Disease (MCD)"Underfill" edema*Proteinuria >3.5g/day*? Ability of blood to retain fluids within vessels ? fluid leaks into extra-vascular spaceInjury to glomerular endothelium and epitheliumImmune complexes deposit into glomerulusDamaged glomerulus ? abnormally permeable to proteins within the blood ? plasma proteins are thus excessively filtered out? Oncotic pressure signals liver to ? albumin synthesis, only to have it filtered out by the kidneys? anabolic activity of liver ? ? lipoprotein synthesisHyperlipidemia*:(? serum LDL, VLDL, and TGs)Lipiduria(lipid/fatty casts; "Maltese cross" sign under polarized light)Since counter-balancing anticoagulant proteins are lost, clotting factors (i.e. 1, 7, 8, 10) now have more activityThrombo-embolic diseaseBlood becomes hyper-coagulable? Lipids are filtered into renal tubules, end up in urineMembranoproliferative Glomerulonephritis (MPGN)Lupus Glomerulonephritis Post-infectious GlomeruloneprhitisIgA NephropathyDamages podocytes on epithelial side of glomerulus ("podocyte effacement"; foot processes flattening)Diabetes MellitusChronic hyperglycemia damages glomeruliDeposition of Immunoglobulin light chains in glomerulusAmyloidosisAnasarca(If generalized)Peri-orbital edema (classic sign)Focal Segmental Glomerular Sclerosis (FSGS)Membranous GlomeruloneprhitisAntibodies attack podocytes, thickening glomerular basement membraneOverflow of immunoglobulin light chains into urine (More filtered than can be reabsorbed)Proteinuria >3.5g/day*The Anion Gap is mostly due to the negative charge of plasma albumin? Anion GapNotes: The four classic features (*) of Nephrotic Syndrome are PEAL (Proteinuria (>3.5 g/day), Edema, hypo-Albuminemia, and hyperLipidemia)For each 10 g/L drop in albumin below 40:Add 2.5 to the calculated anion gap (AG) to get the "correct" AG valueAdd 0.2 mmol/L to total calcium or get an ionized calcium, which is unaffected50% of serum Ca2+ is albumin-bound, so total serum calcium ? Serum total Ca2+ does not reflect ionized Ca2+ ? Blood oncotic pressure" />
Upper Urinary Tract infection (UUTI): Pathogenesis and Clinical Findings

Hypercalcemia - Clinical Findings
![Yu, Yan - Hypercalcemia - Clinical Findings - FINAL.pptx
Hypercalcemia: Clinical FindingsAuthor: Yan YuReviewers:David WaldnerSean SpenceGreg Kline** MD at time of publicationLegend:Published May 7, 2013 on www.thecalgaryguide.comMechanismPathophysiologySign/Symptom/Lab FindingComplicationsHypercalcemia(serum [Ca2+] > 2.5mmol/L)Na+ channels on neuronal membranes become more resistant to opening (resists Na+ influx)Cognitive dysfunctionIf precipitation occurs in the urinary tract...Fatigue? contractility of GI tract smooth muscle? K+ movement out of TAL epithelial cells into the tubule lumen Alters charge balance across the cell membraneCa2+ precipitates with PO43- throughout the bodyDetected by the Ca-Sensing-Receptor (CaSR) on Thick Ascending Limb (TAL) epithelial cells? neuronal action potential generationSluggish neuronal activity...? appetiteConstipationFlank painInhibit insertion of Renal Outer Medullary K+ (ROMK) channels on TAL's luminal membrane? K+ in TAL lumen to drive Na+/Cl- reabsorption through the Na-K-Cl Cotransporter (NKCC)? Na/Cl in tubule lumen ? osmotically draws water into lumen? drinking (polydipsia)? Urine volume (polyuria)Rationale for the CaSR-pathway: ECF has enough Ca2+, no need for more K+ to be excreted into the tubule lumen to create a more + charge there that drives Ca2+ reabsorptionBehavior compensates to prevent dehydrationKidney stones (nephrolithiasis)Constantly feeling full because of reduced GI motilityCa2+ directly inhibits the insertion of aquaporin channels in the collecting duct membraneLess water reabsorbed into the renal vasculatureMore water remains in the tubule filtrateMuscle Weakness...in central nervous system:...at neuromuscular junction:A rhyme to help you recall the manifestations of one specific cause of hypercalcemia, primary hyperparathyroidism:Bones (Calcium levels are high often due to ? resorption from bones)Stones (? Calcium-containing kidney stones)Groans (GI and skeletal muscle issues) Psychic Moans (Cognitive dysfunction from neuronal disturbances)Note: sick/ICU patients have ? serum albumin, due to ? synthesis from a sick liver. Their lab Ca2+ values can be](http://calgaryguide.ucalgary.ca/wp-content/uploads/2015/05/Hypercalcemia-Clinical-Findings.jpg "Yu, Yan - Hypercalcemia - Clinical Findings - FINAL.pptx
Hypercalcemia: Clinical FindingsAuthor: Yan YuReviewers:David WaldnerSean SpenceGreg Kline** MD at time of publicationLegend:Published May 7, 2013 on www.thecalgaryguide.comMechanismPathophysiologySign/Symptom/Lab FindingComplicationsHypercalcemia(serum [Ca2+] > 2.5mmol/L)Na+ channels on neuronal membranes become more resistant to opening (resists Na+ influx)Cognitive dysfunctionIf precipitation occurs in the urinary tract...Fatigue? contractility of GI tract smooth muscle? K+ movement out of TAL epithelial cells into the tubule lumen Alters charge balance across the cell membraneCa2+ precipitates with PO43- throughout the bodyDetected by the Ca-Sensing-Receptor (CaSR) on Thick Ascending Limb (TAL) epithelial cells? neuronal action potential generationSluggish neuronal activity...? appetiteConstipationFlank painInhibit insertion of Renal Outer Medullary K+ (ROMK) channels on TAL's luminal membrane? K+ in TAL lumen to drive Na+/Cl- reabsorption through the Na-K-Cl Cotransporter (NKCC)? Na/Cl in tubule lumen ? osmotically draws water into lumen? drinking (polydipsia)? Urine volume (polyuria)Rationale for the CaSR-pathway: ECF has enough Ca2+, no need for more K+ to be excreted into the tubule lumen to create a more + charge there that drives Ca2+ reabsorptionBehavior compensates to prevent dehydrationKidney stones (nephrolithiasis)Constantly feeling full because of reduced GI motilityCa2+ directly inhibits the insertion of aquaporin channels in the collecting duct membraneLess water reabsorbed into the renal vasculatureMore water remains in the tubule filtrateMuscle Weakness...in central nervous system:...at neuromuscular junction:A rhyme to help you recall the manifestations of one specific cause of hypercalcemia, primary hyperparathyroidism:Bones (Calcium levels are high often due to ? resorption from bones)Stones (? Calcium-containing kidney stones)Groans (GI and skeletal muscle issues) Psychic Moans (Cognitive dysfunction from neuronal disturbances)Note: sick/ICU patients have ? serum albumin, due to ? synthesis from a sick liver. Their lab Ca2+ values can be")
Pathogenesis of Anxiety Disorders
Sense of foreboding or apprehensionActivation of hypothalamus-pituitary-adrenal cortex axisPredisposition to anxiety: Imbalance and/or abnormal functioning of norepinephrine, serotonin, dopamine, and gamma-aminobutyric acid (GABA) Female gender (may be related to hormonal factors, less internal locus of control, greater reporting rates)Other biological theories (under investigation)Prefrontal Cortex modulation of amygdala impairedUnpleasant tensionAmygdala maladaptively activates fear response? Cortisol releaseChronic activation of stress hormones over time causes death of neurons in the hippocampusAnxiety Disorders:A maladaptive emotional state causing fear, worry, and excessive stress, characterized by:Perceived environmental threatHippocampus and Cingulate Gyrus abnormally process threatActivation of autonomic nervous system and adrenal medulla? Epinephrine releaseHippocampus shrinks in sizeAbility of hippocampus to normally integrate environmental stimuli is further compromisedMood dysregulationMemory impairmentStrong association between anxiety disorders and depressionMeasurable ? in Brain-Derived Neurotrophic factor (BDNF)BDNF value correlates with the degree of neuronal loss in the hippocampus
97 kB / 173 words")
Pre-Renal Acute Kidney Injury Pathogenesis

Hyperosmolar Hyperglycemic State
![Hyperosmolar Hyperglycemic State (HHS)
Note: HHS is only seen in Type II DM patients!
Note: In patients with either DKA or HHS, always look for an underlying cause (i.e. an infection)
Author: Yan Yu Reviewers:
Peter Vetere
Gill Goobie
Hanan Bassyouni* * MD at time of publication
Alters total body water & ion osmosis
Inadequate insulin production, insulin resistance, non- adherence to insulin Tx
Relative Insulin deficit
Stresses that ↑ Insulin demand: infections, pneumonia, MI, pancreatitis, etc)
Hyperglycemia
(Very high blood [glucose], higher than in DKA)
When blood [glucose] > 12mmol/L, glucose filtration > reabsorption, ↑ urine [glucose]
Glucosuria
Glucose in filtrate promotes osmotic diuresis: large- volume urine output
Polyuria
Dehydration
(↓ JVP, orthostasis: postural hypotension/ postural tachycardia, ↑ resting HR)
Some insulin still present, but not enoughsome glucose is utilized by muscle/fat cells, some remain in the blood
Cells not “starved”, but still need more energy
↑ release of Catabolic hormones: Glucagon, Epinephrine, Cortisol, GH
Body tries to ↑ blood [glucose], to hopefully ↑ cell glucose absorption
Hypothalamic cells sense low intra-cellular glucose, triggering feelings of hunger
Polyphagia
Note: the presence of some insulin directly inhibits lipolysis; thus, in HHS there is no ketone body production, and no subsequent metabolic acidosis and ketouria (unlike in DKA). If ketones are detected in an HHS patient it’s likely secondary to starvation or other mechanisms.
↓ ECF volume, ↑ ECF osmolarity (i.e. hypernatremia)
↑ Gluconeogenesis ↑ Glycogenolysis (in liver)
↓ Protein synthesis, ↑ proteolysis
(in muscle)
↑ Gluconeogenic substrates for liver If the patient doesn’t drink enough
water to replenish lost blood volume If pt is alert and
Electrolyte imbalance
water is accessible
Water osmotically leaves neurons, shrinking them
Neural damage: delirium, lethargy, seizure, stupor, coma
↓ renal perfusion, ↓ GFR
Renal Failure
(pre-renal cause; see relevant slides)
Polydipsia Note: in HHS, body K+ is lost via osmotic diuresis. But diffusion of K+ out of cells
may cause serum [K+] to be falsely normal/elevated. To prevent hypokalemia, give IV KCl along with IV insulin as soon as serum K+ <5.0mmol/L. But ensure patient has good renal function/urine output first, to avoid iatrogenic hyperkalemia!
Note: Electrolyte imbalances (i.e. hyperkalemia, hypernatremia) are worsened by the acute renal failure commonly coexisting with DKA/HHS
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published November 3, 2016 on www.thecalgaryguide.com](http://calgaryguide.ucalgary.ca/wp-content/uploads/2015/05/Hyperosmolar-Hyperglycemic-State-HHS.jpg "Hyperosmolar Hyperglycemic State (HHS)
Note: HHS is only seen in Type II DM patients!
Note: In patients with either DKA or HHS, always look for an underlying cause (i.e. an infection)
Author: Yan Yu Reviewers:
Peter Vetere
Gill Goobie
Hanan Bassyouni* * MD at time of publication
Alters total body water & ion osmosis
Inadequate insulin production, insulin resistance, non- adherence to insulin Tx
Relative Insulin deficit
Stresses that ↑ Insulin demand: infections, pneumonia, MI, pancreatitis, etc)
Hyperglycemia
(Very high blood [glucose], higher than in DKA)
When blood [glucose] > 12mmol/L, glucose filtration > reabsorption, ↑ urine [glucose]
Glucosuria
Glucose in filtrate promotes osmotic diuresis: large- volume urine output
Polyuria
Dehydration
(↓ JVP, orthostasis: postural hypotension/ postural tachycardia, ↑ resting HR)
Some insulin still present, but not enoughsome glucose is utilized by muscle/fat cells, some remain in the blood
Cells not “starved”, but still need more energy
↑ release of Catabolic hormones: Glucagon, Epinephrine, Cortisol, GH
Body tries to ↑ blood [glucose], to hopefully ↑ cell glucose absorption
Hypothalamic cells sense low intra-cellular glucose, triggering feelings of hunger
Polyphagia
Note: the presence of some insulin directly inhibits lipolysis; thus, in HHS there is no ketone body production, and no subsequent metabolic acidosis and ketouria (unlike in DKA). If ketones are detected in an HHS patient it’s likely secondary to starvation or other mechanisms.
↓ ECF volume, ↑ ECF osmolarity (i.e. hypernatremia)
↑ Gluconeogenesis ↑ Glycogenolysis (in liver)
↓ Protein synthesis, ↑ proteolysis
(in muscle)
↑ Gluconeogenic substrates for liver If the patient doesn’t drink enough
water to replenish lost blood volume If pt is alert and
Electrolyte imbalance
water is accessible
Water osmotically leaves neurons, shrinking them
Neural damage: delirium, lethargy, seizure, stupor, coma
↓ renal perfusion, ↓ GFR
Renal Failure
(pre-renal cause; see relevant slides)
Polydipsia Note: in HHS, body K+ is lost via osmotic diuresis. But diffusion of K+ out of cells
may cause serum [K+] to be falsely normal/elevated. To prevent hypokalemia, give IV KCl along with IV insulin as soon as serum K+ <5.0mmol/L. But ensure patient has good renal function/urine output first, to avoid iatrogenic hyperkalemia!
Note: Electrolyte imbalances (i.e. hyperkalemia, hypernatremia) are worsened by the acute renal failure commonly coexisting with DKA/HHS
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published November 3, 2016 on www.thecalgaryguide.com")
chronic-hypertensive-retinopathy-pathogenesis-and-clinical-findings
 • 1° HTN
Retinal Detachment
Vitreous Hemorrhage
Central/Branch Retinal Artery/Vein Occlusions
Risk Factors for 2° HTN (ex. Hyperaldosterone, Cushing's, Acromegaly, Chronic Kidney Disease, Obstructive Sleep Apnea, Diabetes Mellitus, Hypo/Hyper-thyroid, Adrenal Hyperplasia, Renal Artery Stenosis)
2° HTN
Ophthalmic Artery Hypertension ,17
Stage 1: Mild/vasoconstrictive
Stage 2: Moderate/sclerotic
Stage 3: Severe/exudative
Stage 4: Malignant
Abbreviations: • HTN — Hypertension • BRB — Blood-retinal barrier • RPE — Retinal pigment epithelium
Legend:
Pathophysiology
Mechanism
Acute and chronic vasospasm
Authors: Graeme Prosperi-Porta Reviewers: Stephanie Cote Usama Malik Johnathan Wong* * MD at time of publication
Diffuse and focal arterial narrowing and vascular tortuosity
Atherosclerosis and hyalinization causes arteriolar wall thickening resulting in a diffuse light reflex appearing red-brown coloured
Thickening of the arteriolar wall and/or sclerotic thickening at the arteriole/venule crossing compresses the underlying venule
BRB breakdown causes dot/blot hemorrhages in the inner retina and flame hemorrhages in the nerve fiber layer
Serum proteins and lipids leakage from damaged BRB appears as white or yellow areas with sharp margins
Occlusion of the terminal retinal arterioles causes fluffy white ischemic lesions in the inner retinal nerve fiber layer
Hyper-pigmented patches surrounded by a hypo-pigmented ring due to RPE clumping around atrophic areas in the choroid
Sign/Symptom/Lab Finding
lschemia of optic disc arterioles causes optic nerve swelling and blurred disc margins. Leakage of optic disc arterioles causes hemorrhage and disc edema.
Complications
Copper Wiring
AV nicking
Retinal Hemorrhages
Yellow Hard Exudates
Cotton-wool Spots
Elschnig's Spots
Papilledema")
Serotonin Syndrome Pathogenesis and Clinical Findings
, opioids, antiemetic agents, triptans, weight loss agents, drugs of abuse (e.g. cocaine, amphetamines)
Therapeutic drug use
• Drug interactions (esp. combo of serotonergic agents) Serotonin Syndrome Variable combination of mental status changes, autonomic instability, and neuromuscular hyperactivity ranging from mild to life-threatening with an abrupt onset (within minutes to hours) after medication ingestion and most cases resolving within 24 hours of cessation of offending medication
Intentional self-poisoning
Excessive serotonergic activity at 5-HT receptors centrally (brainstem) and peripherally
serotonin synthesis and release
serotonin reuptake and metabolism
IN receptor agonism and sensitivity
4,
Drug-induced changes in the relative ratio of non-serotonergic neurotransmitters (e.g. increase in noradrenaline)
Altered Mental Status
Anxiety, confusion, agitation, hypervigilance, pressured speech, delirium, coma
Autonomic Instability
Shivering, diaphoresis, fever, diarrhea, tachycardia, mydriasis, hypertension
Authors: Preeti Kar Reviewers: Erika Russell Usama Malik Aaron Mackie* * MD at time of publication
Notes: The Hunter Serotonin Toxicity Criteria is used to make a clinical diagnosis • History of serotonergic agent taken within past 5 weeks + any of the following clinical features: • Spontaneous clonus • Inducible clonus and either agitation or diaphoresis • Ocular clonus and either agitation or diaphoresis • Tremor and hyperreflexia • Hypertonia, temperature > 38 °C, and either ocular clonus or inducible clonus
Neuromuscular Hyperactivity
Hyperreflexia, muscle rigidity (esp. lower extremities), myoclonus, tremor, incoordination, trismus*, opisthotonus*, ocular clonus*, seizures
*Notes: • Trismus or lockjaw, is the reduced opening of the jaw • Opisthotonus is an abnormal body position where the person is usually rigid and arches their back, with their head thrown backwards • Ocular Clonus is rhythmic or equal movements of both eyes; should be distinguished from nystagmus which has a fast and slow component
Legend: Pathophysiology Mechanism
Sign/Symptom/Lab Finding
Abbreviations: • 5-HT = serotonin • SSRI = selective serotonin reuptake inhibitors • SNRI = selective noradrenaline reuptake inhibitors • MOAI = monoamine oxidase inhibitors • TCA = tricyclic antidepressants")
2nd gen antipsychotics (Slovenian translation) - FINAL VERSION
: primeri: klozapin, olanzapin, kvetiapin, risperidon, paliperidon itd.
antagonisti dopaminskih D2 receptorjev zavirajo delovanje DA na D2 receptorjih po celotnih mo2ganih
antagonisti serotoninskih 2A receptorjev zavirajo delovanje 5-HT na 5-HT2A receptorjih po celotnih mo2ganih
antagonisti serotoninskih 2C receptorjev klozapin, olanzapin, kvetiapin
antagonisti ACh M1 receptorjev zavirajo delovanje ACh po telesu (v ustih, prebavilih, o6eh, mo2ganih)
antagonisti aradrenoceptorjev v oiilju dilatacija gladkih migic v stenah arteriol
antagonisti histaminskih H1 receptorjev zavirajo delovanje histamina po telesu
sano-presnovni kink' mehanizem neznan
Legenda:
patofiziologija mehanizem
Pomni: posledica velikih razlik v afiniteti za receptorska vezavna mesta so svojevrstni terapevtski in varnostni profili atipicnih a nti psi hoti kov. Kloza pi n med vsemi velja za najud nkovitejSega, a i ma tudi najve6 neZelenih uC'inkov, vkljUuja agranulocitozo (0,5-2 %). Tako predstavlja terapijo drugega izbora, potrebno je red no spremljanje bolnikove krvne slike.
mezolimbiEna pot DA blokada > 5-HT blokado
nigrostriatna pot 5-HT blokada > DA blokado
4, pozitivnih simptomov terapevtski ueinek
blokada 5-HT vodi v sprokanje DA v striatumu
tubero- blokada 5-HT zavre sprokanje infundibularna pot prolaktina v adenohipofizi
mezokortikalna pot
blokada 5-HT2c receptorjev stimulira sprokanje DA in NA v prefrontalni skorji
zamegljen vid
kognitivna upolasnjenost
sposobnost vzdrievanja krvnega tlaka
omotica
apetit
T tel. tee
ortostatska hipotenzija
tveganje za debelost
trigliceridi na tee
inzulinska odpornost —■ diabetes tipa 2
znak/simptom/laboratorijska najdba
4, halucinacii
blodeni
avtorica: Sara Meunier pregledala: Yan Yu, Aaron Mackie*
* dr. med. ob objavi
prevedel in priredil: Jan KejZar, dr. med., specializant psihiatrije pregledala: doc. dr. Brigita Novak Sarotar, dr. med., spec. psih.
4, ekstrapiramidnih simptomov (EPS)*
4, hiperprolaktinemije*
prokognitivni in antidepresivni ainki
4, kognitivnih simptomov* 4, negativnih simptomov*
* Glede na anti-psihotike prve generacije, glej ustreznogradivo!
okrajgave: 5-HT - serotonin DA - dopamin NA - noradrenalin ACh - acetilholin
visoko presnovno tveganje: klozapin, olanzapin zmerno presnovno tveganje: risperidon, kvetiapin nizko presnovnotveganje:antipsihotikitretjegeneracije
1 tveganje za diabeti6no ketoacidozo pri bolnikih z visokim tveganjem tveganje za srbo-iilne dogodke tveganje za prezgodnjo smrt
zaplet Objavljeno 15. junija 2017 na www.thecalgaryguide.com.")
BMR (Slovenian translation) - FINAL VERSION
 )
Pomni: • Bipolar motnja tipa 1: maniba epizoda in/ali depresivna epizoda: priblrino 10 epizod manije in depresije v 2ivljenju bolnika v primeru nezdravljene bolezni • Bipolar motnja tipa 2: hipomaniba epizoda in depresivna epizoda • hipomaniba epizoda predstavlja bla2jo obliko manije, tj. brez funkcionalne okrnjenosti, brez psihotibih simptomov, brez hospitalizacije in v trajanju vsaj 4 zaporednih dni
znak/simptom/laboratorijska najdba
avtorici: Briana Cassetta, Sara Meunier pregledali: Yan Yu, Alexander Arnold, Philip Stokes*
* dr. med. ob objavi
prevedel in priredil: Jan Kejiar, dr. med., specializant psihiatrije pregledala: doc. dr. Brigita Novak Sarotar, dr. med., spec. psih.
depresivna epizoda najmanj 2 tedna
depresivno razpoloienje
socialnega urriika
obEutkov krivde, tesnobe
brezupa
samomorilnih misli in vedenja
anhedonija
teiave s pozornostjo
A v spanju, utrujenost
A tel. teie, apetita
psihomotorna agitira nost ali upolasnjenost
zaplet Objavljeno 30. junija 2017 na www.thecalgaryguide.com.")
Bupropion (Slovenian translation) - FINAL VERSION
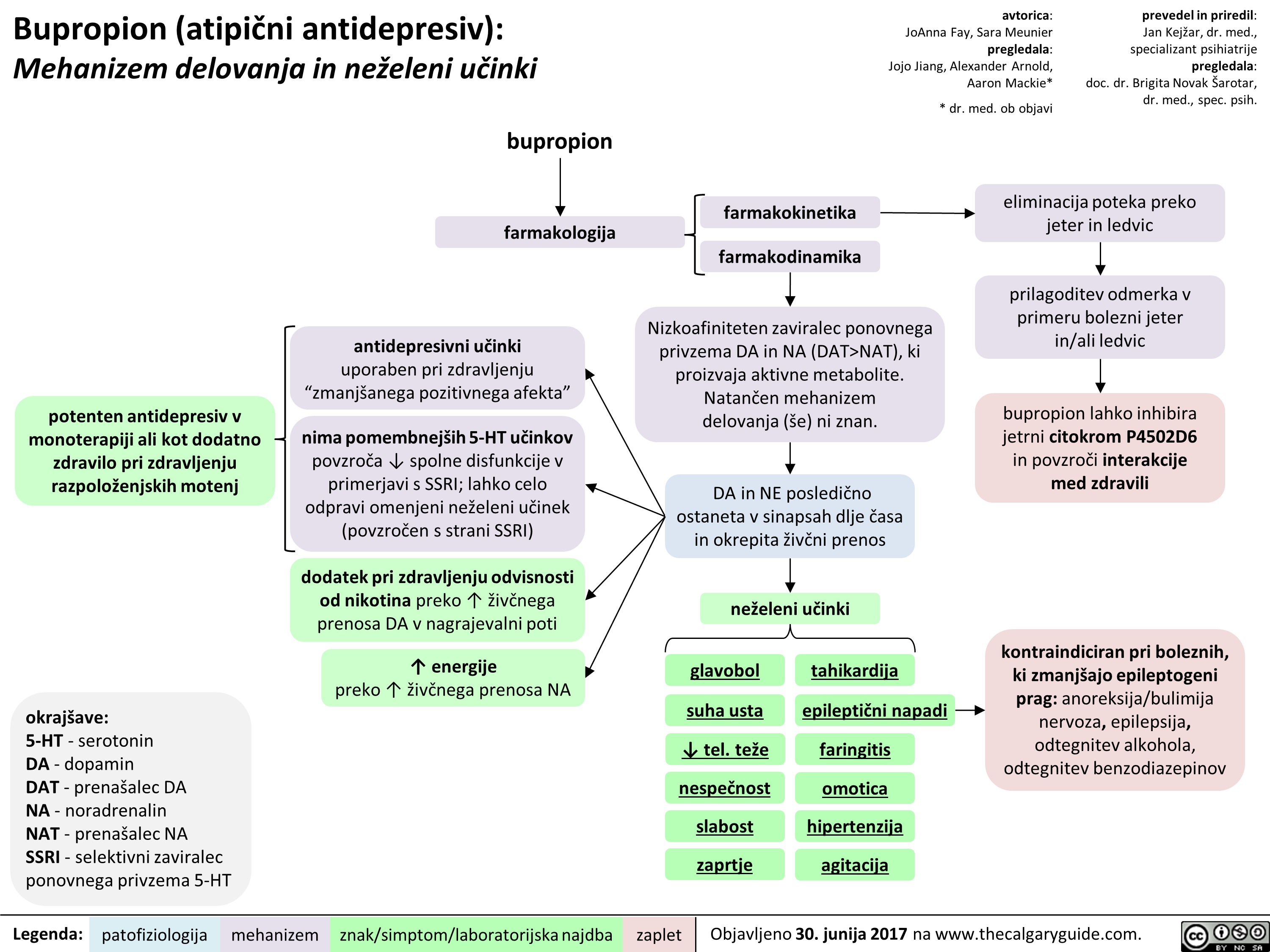 NAT), ki proizvaja aktivne metabolite. Natan'6en mehanizem delovanja (se) ni znan.
znak/simptom/laboratorijska najdba
DA in NE posledi6no ostaneta v sinapsah dlje Casa in okrepita 2iv6ni prenos
neieleni udnki
glavobol
suha usta
4, tel. tee nespeEnost slabost zaprtie
tahikardija
epileptiEni napadi
faringitis
omotica hipertenziia agitacija
prevedel in priredil: Jan Kejiar, dr. med., specializant psihiatrije pregledala: doc. dr. Brigita Novak Sarotar, dr. med., spec. psih.
eliminacija poteka preko jeter in ledvic
prilagoditev odmerka v primeru bolezni jeter in/ali ledvic
bupropion lahko inhibira jetrni citokrom P4502D6 in povzrod interakcije med zdravili
kontraindiciran pri boleznih, ki zmanIgajo epileptogeni prag: anoreksija/bulimija nervoza, epilepsija, odtegnitev alkohola, odtegnitev benzodiazepinov
zaplet Objavljeno 30. junija 2017 na www.thecalgaryguide.com.
" title="Bupropion (atipiEni antidepresiv): Mehanizem delovanja in neieleni utinki
potenten antidepresiv v monoterapiji all kot dodatno zdravilo pri zdravljenju razpoloienjskih motenj
okrajgave: 5-HT - serotonin DA - dopamin DAT - prenagalec DA NA - noradrenalin NAT - prenagalec NA SSRI - selektivni zaviralec ponovnega privzema 5-HT
Legenda:
bupropion
farmakologija
antidepresivni udnki uporaben pri zdravljenju "zmaniganega pozitivnega afekta"
nima pomembnelgih 5-HT udnkov povzraa spolne disfunkcije v primerjavi s SSRI; lahko celo odpravi omenjeni neieleni udnek (povzraen s strani SSRI)
dodatek pri zdravljenju odvisnosti od nikotina preko T iive'nega prenosa DA v nagrajevalni poti
energije preko T 2ivbega prenosa NA
patofiziologija mehanizem
farmakokinetika
farmakodinamika
avtorica: JoAnna Fay, Sara Meunier pregledala: Jojo Jiang, Alexander Arnold, Aaron Mackie*
* dr. med. ob objavi
Nizkoafiniteten zaviralec ponovnega privzema DA in NA (DAT>NAT), ki proizvaja aktivne metabolite. Natan'6en mehanizem delovanja (se) ni znan.
znak/simptom/laboratorijska najdba
DA in NE posledi6no ostaneta v sinapsah dlje Casa in okrepita 2iv6ni prenos
neieleni udnki
glavobol
suha usta
4, tel. tee nespeEnost slabost zaprtie
tahikardija
epileptiEni napadi
faringitis
omotica hipertenziia agitacija
prevedel in priredil: Jan Kejiar, dr. med., specializant psihiatrije pregledala: doc. dr. Brigita Novak Sarotar, dr. med., spec. psih.
eliminacija poteka preko jeter in ledvic
prilagoditev odmerka v primeru bolezni jeter in/ali ledvic
bupropion lahko inhibira jetrni citokrom P4502D6 in povzrod interakcije med zdravili
kontraindiciran pri boleznih, ki zmanIgajo epileptogeni prag: anoreksija/bulimija nervoza, epilepsija, odtegnitev alkohola, odtegnitev benzodiazepinov
zaplet Objavljeno 30. junija 2017 na www.thecalgaryguide.com.
" />
NAT), ki proizvaja aktivne metabolite. Natan'6en mehanizem delovanja (se) ni znan.
znak/simptom/laboratorijska najdba
DA in NE posledi6no ostaneta v sinapsah dlje Casa in okrepita 2iv6ni prenos
neieleni udnki
glavobol
suha usta
4, tel. tee nespeEnost slabost zaprtie
tahikardija
epileptiEni napadi
faringitis
omotica hipertenziia agitacija
prevedel in priredil: Jan Kejiar, dr. med., specializant psihiatrije pregledala: doc. dr. Brigita Novak Sarotar, dr. med., spec. psih.
eliminacija poteka preko jeter in ledvic
prilagoditev odmerka v primeru bolezni jeter in/ali ledvic
bupropion lahko inhibira jetrni citokrom P4502D6 in povzrod interakcije med zdravili
kontraindiciran pri boleznih, ki zmanIgajo epileptogeni prag: anoreksija/bulimija nervoza, epilepsija, odtegnitev alkohola, odtegnitev benzodiazepinov
zaplet Objavljeno 30. junija 2017 na www.thecalgaryguide.com.
" title="Bupropion (atipiEni antidepresiv): Mehanizem delovanja in neieleni utinki
potenten antidepresiv v monoterapiji all kot dodatno zdravilo pri zdravljenju razpoloienjskih motenj
okrajgave: 5-HT - serotonin DA - dopamin DAT - prenagalec DA NA - noradrenalin NAT - prenagalec NA SSRI - selektivni zaviralec ponovnega privzema 5-HT
Legenda:
bupropion
farmakologija
antidepresivni udnki uporaben pri zdravljenju "zmaniganega pozitivnega afekta"
nima pomembnelgih 5-HT udnkov povzraa spolne disfunkcije v primerjavi s SSRI; lahko celo odpravi omenjeni neieleni udnek (povzraen s strani SSRI)
dodatek pri zdravljenju odvisnosti od nikotina preko T iive'nega prenosa DA v nagrajevalni poti
energije preko T 2ivbega prenosa NA
patofiziologija mehanizem
farmakokinetika
farmakodinamika
avtorica: JoAnna Fay, Sara Meunier pregledala: Jojo Jiang, Alexander Arnold, Aaron Mackie*
* dr. med. ob objavi
Nizkoafiniteten zaviralec ponovnega privzema DA in NA (DAT>NAT), ki proizvaja aktivne metabolite. Natan'6en mehanizem delovanja (se) ni znan.
znak/simptom/laboratorijska najdba
DA in NE posledi6no ostaneta v sinapsah dlje Casa in okrepita 2iv6ni prenos
neieleni udnki
glavobol
suha usta
4, tel. tee nespeEnost slabost zaprtie
tahikardija
epileptiEni napadi
faringitis
omotica hipertenziia agitacija
prevedel in priredil: Jan Kejiar, dr. med., specializant psihiatrije pregledala: doc. dr. Brigita Novak Sarotar, dr. med., spec. psih.
eliminacija poteka preko jeter in ledvic
prilagoditev odmerka v primeru bolezni jeter in/ali ledvic
bupropion lahko inhibira jetrni citokrom P4502D6 in povzrod interakcije med zdravili
kontraindiciran pri boleznih, ki zmanIgajo epileptogeni prag: anoreksija/bulimija nervoza, epilepsija, odtegnitev alkohola, odtegnitev benzodiazepinov
zaplet Objavljeno 30. junija 2017 na www.thecalgaryguide.com.
" />
Left Heart Failure: Pathophysiology (Neurohormonal Activation)

Frank Starling Mechanism • The Frank Starling mechanism of the heart represents the relationship between preload (EDV) and SV • As preload (EDV) increases, SV increases, because higher volumes of blood in the ventricles stretch the cardiac fibers and increases cardiac contraction during systole. However, volume overload causes reduced SV.
Myocardial Dysfunction: • Left ventricular Compensatory 4, SV 4A, CO 4 Mechanisms
4, BP
Important equations: • BP = CO x SVR • CO = SV x HR Cardiac hemodynamics: • Stroke volume is affected by three factors 1) Preload (end-diastolic volume (EDV)) 2) Afterload (resistance to LV ejection) 3) Contractility (inherent strength of contraction of LV myocytes) Definition of heart failure: • Myocardial dysfunction (systolic or diastolic) results in decreased CO, such that the heart cannot meet the body's metabolic demands or can only do so at elevated filling pressures
Anti-diuretic hormone (ADH) activation: Arginine 4, BP 4 carotid sinus Vasopressin --• and aortic arch (V2) receptor baroceptors activation activation 4 I` ADH release
RAAS System: 4, BP 4 t release of renin from the juxtaglomerular kidney cells due to renal hypoperfusion
SNS System: In response to CO, 4 SNS t release of (catecholamines) norepinephrine and epinephrine
Adrenal Glands: Aldosterone release
Angiotensin II Type 1 receptor activation
al receptor activation
13, receptor activation
—•
Renal Collecting Ducts: t H2O retention
Renal Distal —• Tubules: t Na+ & H2O retention
Heart: Activation of fibroblasts 4 collagen synthesis and hypertrophy
Blood Vessels: Peripheral vasoconstriction 4 SVR
Heart: Chronic p, receptor activation 4 Ca2+ overload myocyte apoptosis
Heart: Increase HR to maintain normal CO
Maladaptive Response: t preload (EDV), —• volume overload
Abbreviations: • SV — Stroke volume • CO — Cardiac output • SVR — Systemic vascular resistance • BP — Blood pressure • RAAS — Renin-Angiotensin-Aldosterone System • SNS — Sympathetic nervous system
tin systemic and pulmonary congestion via the Frank-Starling Mechanism
Maladaptive 1` resistance Response: t BP, —• against LV afterload ejection 4 4, SV
Maladaptive Response: Adverse LV remodelling
Maladaptive Response: t myocardial oxygen demand and 4, diastolic time
4, contractility —• of the heart
4, coronary blood flow 4 myocardial ischemia
Physical signs and symptoms of congestive heart failure (see relevant slide)
Authors: Sunny Fong Reviewers: —• I Jack Fu Usama Malik Dr. Jason Waechter* *MD at time of publication")
Anksiozne motnje: patogeneza tesnobe

Depresivna epizoda/motnja: patogeneza in klinične najdbe

pomanjkanje tesnih, zaupnih odnosov
druibeni dejavniki
Prevalenca 10-15 % pri 2enskah in 5-12 % pri mogkih. Razmerje med 2enskami in mc&imi 2:1. Pogosteja pri razvezanih, laenih in samskih ter tistih z nizkim druThenoekonomskim stanjem.
depresivna epizoda/motnja simptomi (po DSM 5 merilih) prisotni tekom istega dvotedenskega obdobja in pomenijo odklon od premorbidne ravni funkcioniranja drugi simptomi pomanjkanje libida tesnoba razpolo2enje '6ez dan niha (ang. diurnal variation) huje pozimi (sezonska depresivna motnja) halucinacije (psihotiCna depresija) blodnje (psihotiena depresija) anhedonija (izguba zanimanj/u2itkov) depresivno razpolo2enje nejeknost ali povaan apetit s pridrdienimi spremembami telesne te2e (4, ali 1`) anergija (utrujenost) nespanost ali preva spanja okrajgave: 5-HT - serotonin NA - noradrenalin nastop v mesecu po porodu (poporodna depresija) obC'utki niC'vrednosti ali pretirane krivde psihomotorit'na upaasnjenost ali agitiranost upad spoznavnih sposobnosti (zlasti motnje pozornosti) ponavljajae se misli na smrt/samomor")
Serotoninski sindrom: patogeneza in klinične najdbe
, opioidi, antiemetiki, triptani, zdravila za izgubo tel. te2e, ilegalne psihoaktivne snovi (npr. kokain, amfetamini)
terapevtska raba
1
interakcije (se posebej pri kombinacijah serotoninergikov) namerna samozastrupitev
serotoninski sindrom raznolika kombinacija sprememb v psihiZnem stanju, vegetativne nestabilnosti in iivZnomigiZne hiperaktivnosti, ki sega od blage do iivljenje ogroiajoL'e z nenadnim nastopom (v nekaj minutah do urah) po administraciji zdravil(a), v vecini primerov pa se razregi v <24 urah po prekinitvi jemanja odgovorne snovi
prekomerna serotoninergitha aktivnost 5-HT receptorjev centralno (v moiganskem deblu) in periferno
sinteza in privzem in T receptorski agonizem sprogEanje 5-HT presnova 5-HT in ok'utljivost
z zdravili povzraene spremembe v relativnem razmerju ne-serotoninergithih nevrotransmitorjev (npr. pove6anje konc. noradrenalina)
spremenjeno psihreno stanje
tesnoba, zmedenost, agitacija, hipervigilnost, dolgoveznost, delirij, koma
vegetativna nestabilnost
mrzlica/drgetanje, 6ezmerno potenje, vraina, driska, tahikardija, ra8irjene zenice, hipertenzija
avtorica: Preeti Kar pregledali: Erika Russell, Usama Malik, Aaron Mackie*
* dr. med. ob objavi
prevedel in priredil: Jan Kejiar, dr. med., specializant psihiatrije pregledala: doc. dr. Brigita Novak Sarotar, dr. med., spec. psih.
Pomni: za klinicno postavitev diagnoze uporabljamo Hunterjeva merila serotoninske toksiZnosti: - zgodovina jemanja serotoninergithih snovi v preteklih 5 tednih + kateri koli od naslednjih pogojev: • spontani klonus • inducirani klonus in bodisi agitacija bodisi L'ezmerno potenje • aesni klonus in bodisi agitacija bodisi L'ezmerno potenje • tremor in hiperrefleksija • hipertonija, tel. temperatura >38 °C in bodisi aesni klonus bodisi inducirani klonus
ihgnomigiZna hiperaktivnost
hiperrefleksija, mR")
Zaviralci privzema serotonina in noradrenalina (SNRI): mehanizem delovanja in neželeni učinki
: mehanizem delovanja in neieleni utinki
zaviralci privzema serotonina in noradrenalina predstavnika: venlafaksin, duloksetin
konc. 5-HT in NA v 02S/P2S
farmakologija
farmakokinetika farmakodinamika
nenadna prekinitev jemanja zdravila ali vnos
avtorici: JoAnna Fay, Sara Meunier pregledali: Jojo Jiang, Alexander Arnold, Jessica Asgarpour, Aaron Mackie*
iz krvnega obtoka jih odstranijo jetra
zavirajo prenagalec za privzem NA (NET) in prenaalec za privzem 5-HT (SERT)
5-HT in NA ostaneta v sinapsah dlje in podaVata oz. okrepita iiv6ni prenos
odtegnitev namerno predoziranje nenamerno povzrocene interakcije nepri6akovan odziv na terapevtski odmerek prekometna v aktivnost 02S/P2S sindrom 1` 5-HT omotica 5-HT serotoninski omotica driska nejegZnost nespeEnost nespanost slabost utrujenost glavobol vegetativna * iivEnomiKiEne kognitivne hiperaktivnost nepravilnosti spremembe vrtoglavica potencialno iivljenje ogroiajoE spolne hipertenzija olesni klonus agitacija motn'e porast tahikardija tremor akatizra tel. tee raigir-ene hiperrefleksija zenice migiEni klonus
Legenda: patofiziologija
mehanizem
znak/simptom/laboratorijska najdba
1` NA
slabost
Zezmerno potenie
* dr. med. ob objavi
prevedel in priredil: Jan Kejiar, dr. med., specializant psihiatrije pregledala: doc. dr. Brigita Novak Sarotar, dr. med., spec. psih.
v primeru ietrnih bolezni in/ali saasne rabe zaviralcev/sproncev CYPD26 ie potrebna prilagoditev odmerka
izboljganje depresivne in anksiozne simptomatike 1 blokada ponovnega privzema 5-HT in NA v sinapsah pa u6inka SNRI ne pojasni v celoti
posredno SNRI povzro6ijo sproManja nevrozaMitnih proteinov, denimo BDNF, ki bi naj imel po podatkih §tudij protivnetno delovanje
za dosego kliniEnega uZinka ie potrebnih 3-8 tednov all veZ zdravlienia
okralgave in opombe: 5-HT— serotonin NA— noradrenalin BDNF — mo2ganski nevrotrofiEni faktor 025 — osrednji 2ivEni sistem P2S — periferni 2ivEni sistem *vegetativen — nanagajoE se na avtonomni 2ivEni sistem")
Socialna anksiozna motnja: patogeneza in klinične najdbe

Impetigo Pathogenesis and clinical findings

Fisiologi sistem Renin-Angiotensin-Aldosteron (RAAS)

Hipoperfusi Ginjal
Regangan baroreseptor pada dinding arteriol aferen
Hipotensi/ Hipovolemia
9 Pengiriman NaCI ke Makula Densa
9 Tekanan dirasakan oleh Baroreseptor Jantung & Arteri
Katekolamin di sirkulasi
Aktivitas saraf simpatis pada arteriol aferen
Stimulasi reseptorn-adrenergik pada arteriol aferen
Keseimbangan Glomerulotubuler (Mekanisme intrinsik ginjal diluar RAAS) *Hanya sebagian dari mekanisme tersebut
Angiotensin II 4 konstriksi arteriol eferen
01% Tekanan hidrostatik glomerulus
Jumlah cairan yang disaring
+ Konsentrasi sisa protein darah di glomerulus
Darah kental bergerak dari glomerulus menuju kapiler peritubuler
it pada kapiler peritubuler
9 Tekanan hidrostatik peritubular
Legenda:
Patofisiologi Mekanisme
Pelepasan Renin oleh sel-sel Jukstaglomerular pada arteriol aferen 4 berujung pada T Angiotensin II
Sekresi aldosterone dari korteks adrenal
1
Insersi KNaE pada Sel Prinsipal di DK
■
Aktivitas transporter NHE3 di dalam PCT
Reabsorpsi Na ke dalam darah, menarik H2O ke dalam darah melalui osmosis
Reabsorpsi H2O langsung ke dalam sirkulasi darah (via aliran gradien tekanan onkotik dan hidrostatik H2O)
Tanda/Gejala/Penunjang
Komplikasi
Author: David Waldner Reviewers: Yan Yu Sean Spence Sophia Chou* Penerjemah: M Harmen Reza S* * MD (dokter) pada saat publikasi
Hati secara normal mensintesis angiotensinogen dengan laju basal, melepaskannya ke dalam sirkulasi darah:
Vasokonstri ksi arteri sistemik
Tekanan darah
Daftar singkatan: • ACE: Angiotensin Converting Enzyme (disintesis oleh Ginjal dan Paru) • DK: Duktus Kolektivus • PCT: Tubulus Konturtus Proksimal • NHE3: Sodium Hydrogen Exchanger (Antiport) 3 • KNaE: Kanal Natrium (Sodium) Epitel • n:Tekanan onkotik")
Celiac Disease: Pathogenesis and clinical findings

Genetic predisposition -■ (Northern European, Down's syndrome, Associated with HLA DQ 2,8)
Note: *The anti-TTG antibody is an IgA anti-body, therefore if the patient is IgA-deficient, absence of anti-TTG does not rule out celiac
Anti-TTG in serum*
Anti-TTG reacts with TTG in skin
Deposition of anti-TTG in renal glomeruli
Dermatitis herpetiformis
Chronic Kidney Disease
Small Bowel Biopsy: Crypts of bowel become enlarged (hyperplasia) with architectural change, villous shortening
Legend: Pathophysiology
Mechanism
Exposure to prolamins (proteins found in wheat, rye, oats, barley)
TTG alters prolamin Altered protein fits more easily into HLA
HLA activates adaptive immunity
IgA generated against prolamin-TTG
Wheat prolamin (gliandin) interacts with and activates zonulin signalling
Gut epithelium becomes more porous
Large dietary proteins in epithelium disrupt tight junctions
Author: Matthew Harding Yan Yu Peter Bishay Reviewers: Dean Percy Jason Baserman Usama Malik Kerri Novak* * MD at time of publication
Inflammation of Intestinal epithelium Inflammation disrupts structure of bowel mucosa Mechanism Unknown Lymphocytes migrate to site of inflammation Extraintestinal Complications: Arthropathy Ataxia (gluten associated) Infertility Mild Hepatitis Villi of intestine atrophy Risk of Microscopic Colitis (50x) Malabsorption Extraintestinal manifestations: Chronic watery Fatigue Failure to thrive, weight loss Anemia (Fe, B12, folate) Peripheral neuropathy (B12, Ca) Ataxia (Ca) Dysrhythmia (Ca, K) Osteoporosis (Vit D, Ca) diarrhea + Intestinal manifestations steatorrhea: Pale, foul-smelling Abdominal bloating Steatorrhea (fat in stool) Diarrhea")
Kallmann Syndrome and Normosmotic Idiopathic Hypogonadotropism: Pathogenesis and Clinical Findings
 and Normosmic Idiopathic Hypogonadotropic Hypogonadism (nIHH): Pathogenesis and clinical findings
Authors: Danielle Lynch Reviewers: Nicola Adderley Josephine Ho* * MD at time of publication
Abbreviations:
GnRH – gonadotropin- releasing hormone
LH – luteinizing hormone FSH - follicle stimulating hormone
Notes:
• 4 male : 1 female
• KS, nIHH, and isolated
anosmia exist on a
spectrum
• KS has X-linked recessive,
autosomal dominant, autosomal recessive,
oligogenic, and idiopathic
forms
• KS and nIHH are also
associated with gene- specific features such as
cleft lip/palate, oculomotor synkinesis, hearing loss, and unilateral renal aplasia
• Diagnosis typically occurs around pubertal age, but may be earlier in males if micropenis and cryptorchidism are present at birth
Mutations in KAL1, NELF, and PROKR2
Abnormal olfactory bulb development
Mutations in FGFR1, FGF8, PROK2, PROKR2, HS6ST1, CHD7, WRD11, and SEMA3A or idiopathic
Mutations in
GNRH1, KISS1, KISS1R, TAC3, TACR3
Impaired migration of olfactory neurons from olfactory bulb to hypothalamus
Impaired migration of GnRH neurons from olfactory bulb to hypothalamus
Impaired GnRH neuron activity
Kallmann syndrome
(idiopathic hypogonadotropic hypogonadism with anosmia)
Normosmic idiopathic hypogonadotropic hypogonadism
Anosmia
Olfactory bulb aplasia
Abnormal /absent olfactory bulb on MRI
↓ GnRH ↓LH and ↓FSH
Delayed or absent puberty
Infertility
↓estrogen
In males: ↓testosterone
Cryptorchidism
Delayed epiphyseal plate closure
Micropenis (5-10%)
Uninhibited long bone growth
↑ length hands, arms, legs
Delayed bone age (hand/wrist x-ray)
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published September 29, 2018 on www.thecalgaryguide.com")
Feedback Loop: Adrenocorticotropic Hormone (ACTH)

Authors: Rhiannon Brett Reviewers: Andrea Kuczynski Bernard Corenblum* * MD at time of publication
Posterior Pituitary Gland
ADH
-
-
-
Hypothalamus
CRH
+
Anterior Pituitary Gland
ACTH, MSH, other hormones produced from POMC
ACTH
+
Adrenal Cortex
Activate ACTHR
Activate cAMP Activate PKA Activate Zona Fasciculata
Cortisol
Abbreviations:
CRH: Corticotropin Releasing Hormone
ADH: Anti-diuretic Hormone
ACTH: Adrenocorticotropic Hormone
MSH: Melanocyte Stimulating Hormone
POMC: Pro-Opiomelanocortin
ACTHR: ACTH Receptor
cAMP: Cyclic Adenosine Monophosphate
PKA: Protein Kinase A
DHEA(S): Dehydroepiandrosterone (sulfonated form) F: Female
M: Male
Excess: hirsutism, acne, oily skin, oligo- or amenorrhea, virilization in F
Deficiency: symptoms not typically seen due to gonadal production
Note:
Activate Zona Reticularis Activate Zona Glomerulosa
(minor effect)
Excess: central obesity, hirsutism, violaceous striae, weakness
Deficiency: weight loss, fatigue, weakness, nausea/vomiting, diarrhea
DHEA(S) Aldosterone
Specific to primary adrenal insufficiency: ↑ pigmentation, hyperkalemia
• Virilization is a red flag for an androgen- secreting tumor
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published October 2, 2018 on www.thecalgaryguide.com")
intrauterine-growth-restriction-iugr-pathogenesis
: Pathogenesis
Authors: Ricki Hagen Reviewers: Jaimie Bird Sarah McQuillan* * MD at time of publication
Abbreviations:
• DM: diabetes mellitus
• HTN: hypertension • IUGR: intrauterine growth restriction
• SGA: small for gestational age
• SLE: systemic lupus erythematosus
• TORCH: Toxoplasmosis, Others, Rubella, CMV, HSV
Maternal Factors
Maternal-Fetal Factors Placental malformations
(Ex. previa, accreta, infarction, abnormal implantation, ischemia)
Gestational HTN/ Preeclampsia
Multiple gestation Gestational DM
Fetal Factors Structural anomalies
(often comorbid with cytogenetic disorders)
Congenital infections
(Ex. TORCH)
Inborn errors of metabolism
Chromosomal disorders/ genetic syndromes
Multiple unclear intrinsic fetal mechanisms
Note:
Teratogenic medications (Ex. Warafin, Valproic Acid, Folic Acid Antagonists)
High altitude living
Smoking, ETOH and/or drug use
Malnutrition/ Low pre-gestational weight
Multiple unclear extrinsic fetal mechanisms
Medical conditions
(Ex: chronic HTN, cyanotic heart disease, severe chronic anemia, kidney disease)
Autoimmune conditions (Ex. Type 1 DM, SLE)
Decreased uteroplacental blood flow
Nutrient supply to fetus compromised
Reduction of total body mass, bone
mineral content, and muscle mass
Blood flow redirected away from vital organs to brain, placenta, heart and adrenal glands
Reduction of overall fetal size to increase survival
IUGR
Failure to reach genetically determined growth potential
• IUGR is not synonymous with SGA • Constitutional SGA is due to
paternal and maternal factors such as height, weight, ethnicity, and parity; it is not associated with increased risk for infant mortality or morbidity
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published October, 30, 2018 on www.thecalgaryguide.com")
fetal-alcohol-spectrum-disorder-pathogenesis-and-clinical-findings

Maternal alcohol consumption during pregnancy (greatest risk with binge drinking and daily/chronic intake)
Prenatal alcohol exposure to fetus via placenta-umbilical transport
Notes:
Fetal Alcohol Spectrum Disorder with Sentinel Facial Features (confirmed or unknown PAE)
Fetal Alcohol Spectrum Disorder without Sentinel Facial Features
• There is no known safe amount of prenatal alcohol exposure
(PAE). Although PAE below the threshold of >7 drinks/week or > 2 binge episodes (one binge episode is >4 drinks in one sitting) has not been associated with neurodevelopmental effects, there is insufficient objective evidence to deem this level of alcohol exposure safe.
Sentinel Facial Features
(confirmed PAE) Alcohol exposure between Alcohol acts directly as a teratogen
Vasoconstriction of placental-umbilical unit ̄ blood flow and ̄ oxygen delivery to fetus
Notes (continued):
• A multidisciplinary team is necessary for an accurate and comprehensive diagnosis and subsequent recommendations. Canadian Guidelines for diagnosis, 4-digit code, the APP Toolkit, University of Washington Lip- Philtrum Guide are different methods of assessing and diagnosing patients.
• While not specific to FASD, congenital abnormalities (e.g. heart defects, renal problems, auditory/visual impairments, skeletal defects) and growth deficits (prenatal and/or postnatal height or weight ≤ 10th percentile) are often associated with this disorder.
Smooth philtrum
Thin upper lip
day 15 and 22 of & indirectly via its metabolites pregnancy (most often)
Neuronal cell death & disruption of migration & proliferation Altered signaling, neurotransmitter imbalance, & neural connectivity Evidence of impairment in 3+ neurodevelopmental domains
Small palpebral fissures
(≥2 SDs below the mean for age & sex or < 3rd percentile)
Language Memory Attention Motor skills Cognition Affect regulation
Academic achievement
Executive function (including hyperactivity & impulsivity)
Neuroanatomy/ Neurophysiology (e.g. seizure disorder, abnormal brain structure as seen on brain imaging, microcephaly (head circumference ≤ 10th percentile))
Adaptive behavior, social skills, or social communication
Failure to reach age-appropriate milestones, challenges at school, aggression, delinquency, mental health challenges, substance use disorders
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published October 30, 2018 on www.thecalgaryguide.com")
Vesicoureteric reflux (VUR): Pathogenesis and clinical findings
: Pathogenesis and clinical findings
Authors: Nicola Adderley Reviewers: Emily Ryznar *Lindsay Long * MD at time of publication
Abnormal function
Abnormal anatomy
Neurogenic bladder (e.g. cerebral palsy, constipation, spinal injury, iatrogenic)
Non-neurogenic bladder (neuropsychological)
Lower urinary tract abnormality (posterior urethral valves, meatal stenosis)
Bladder outlet obstruction
↑ pressure distorts UVJ
Upper urinary tract abnormality (ureters)
UVJ abnormality
Incomplete closure of UVJ during bladder contraction
Abbreviations
• UVJ - ureterovesicular junction • UTI – urinary tract infection
Failure of bladder sphincter to relax during bladder contraction
Vesicoureteric reflux (VUR):
Back flow of urine from the bladder into one or both ureters +/- kidneys
Migration of lower urinary tract bacteria to kidneys
Bacterial invasion of renal parenchyma
Upper UTI (pyelonephritis)
Incomplete emptying of bladder during Abnormal
↑ pressure in bladder
Bladder dilates
Dilated bladder on U/S
urination
Bacteria in bladder are not cleared during urination
voiding habits
↑ bladder capacity
Renal scarring
↓ functional renal tissue
*Chronic kidney disease (↓ GFR,
hypertension, proteinuria)
Flank tenderness
Fever, dysuria, urgency, frequency
Lower UTI (cystitis) Urinary stasis
Cloudy, foul- smelling urine
Urethral stricture
Notes
Urgency, dysuria, frequency
• First febrile UTI in an infant should trigger a work-up for VUR
• High likelihood of spontaneous resolution • *Late complication of severe VUR
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published November 19, 2018 on www.thecalgaryguide.com")
Anticonvulsants as Mood Stabilizers: Mechanism and Side-effect
, thrombocytopenia
Abbreviations: • VSSCs: Voltage-Sensitive Sodium Channels")
Microangiopathic Hemolytic Anemia: Pathogenesis and clinical findings

↓ Inhibition of complement
Immune inflammatory
↑ Pressure in
↑ Extrinsic clotting pathway activation
TTP
(See TTP-HUS Slide)
Disruption of
endothelial cell
activation perfusion/ischemia response metabolism vasculature ↑Thrombin Deficient
Placental under-
renal afferent
↑ Complement pathway activation
MAC-mediated cell lysis
Cytokine release
Endothelial injury
Shear stress on endothelium
production
↑ Fibrin clot production and deposition in small vessels
ADAMTS13 protease enzyme
↓ Cleavage and ↑ accumulation of VWF multimers
Abbreviations:
• HELLP - Hemolysis, Elevated Liver Enzymes, Low Platelets • HUS - Hemolytic-Uremic Syndrome
• DIC - Disseminated Intravascular Coagulation
• TTP - Thrombotic Thrombocytopenic Purpura
• MAC - Membrane Attack Complex
• STEC - Shiga Toxin-Producing Escherichia coli
• VWF - Von Willebrand Factor
Pro-thrombotic Environment (see Virchow’s Triad Slide)
Platelet aggregation Mechanical obstruction of the vessel lumen
Hypercoagulable state
Thrombocytopenia
↑ RBC production in bone marrow
↑ Reticulocytes
Hemoglobin released from damaged RBCs Shearing of RBCs Anemia
Binds to haptoglobin Conversion to bilirubin in liver
↓ Free haptoglobin ↑Indirect bilirubin
Shistocytes Jaundice
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published August 13, 2018 on www.thecalgaryguide.com")
Hypernatremia Physiology
![Hypernatremia: Physiology Unreplaced H2O loss
Hypodipsia
H2O shift into cells
Severe exercise, electroshock induced seizures
Transient ↑ cell osmolality
Na+ overload
Inappropriate IV hypertonic solution, salt poisoning
Abbreviations:
H2O: Water
GI: Gastrointestinal
DM: Diabetes Mellitus
DI: Diabetes Insipidus
Na+: Sodium ion
IV: Intravenous
ADH: Antidiuretic Hormone LOC: Level of Consciousness
Skin
Sweat, burns
GI
Vomiting, bleeding, osmotic diarrhea
Fluid [Na+] < serum [Na+]
↑ H2O loss compared to Na+ loss
Renal
DM, Mannitol, Diuretics
Absent thirst mechanism
Hypothalamic lesion impairs normal drive for H2O intake
Nephrogenic
↑ renal resistance to ADH
H2O Deprivation Test + no AVP response
↓ access to H2O
DI
Central
↓ ADH secretion
H2O Deprivation Test + AVP response
↑ [Na+] 10- 15 mEq/L within a few minutes
Weakness, irritability, seizures, coma
↑ thirst, ↓ urinary frequency and volume
Note:
Hypernatremia
Serum [Na+] > 145 mmol/L
Intracranial hemorrhage
Headache, vomiting, ↓ LOC
• Plasma [Na+] is regulated by water intake/excretion, not by changes in [Na+].
• Effects on plasma [Na+] of IV fluids or loss of bodily fluids is determined by the tonicity of the fluid, not the osmolality.
Authors: Mannat Dhillon Reviewers: Andrea Kuczynski Kevin McLaughlin* * MD at time of publication
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published January 11, 2019 on www.thecalgaryguide.com](http://calgaryguide.ucalgary.ca/wp-content/uploads/2019/01/Hypernatremia-Physiology-.jpg "Hypernatremia: Physiology Unreplaced H2O loss
Hypodipsia
H2O shift into cells
Severe exercise, electroshock induced seizures
Transient ↑ cell osmolality
Na+ overload
Inappropriate IV hypertonic solution, salt poisoning
Abbreviations:
H2O: Water
GI: Gastrointestinal
DM: Diabetes Mellitus
DI: Diabetes Insipidus
Na+: Sodium ion
IV: Intravenous
ADH: Antidiuretic Hormone LOC: Level of Consciousness
Skin
Sweat, burns
GI
Vomiting, bleeding, osmotic diarrhea
Fluid [Na+] < serum [Na+]
↑ H2O loss compared to Na+ loss
Renal
DM, Mannitol, Diuretics
Absent thirst mechanism
Hypothalamic lesion impairs normal drive for H2O intake
Nephrogenic
↑ renal resistance to ADH
H2O Deprivation Test + no AVP response
↓ access to H2O
DI
Central
↓ ADH secretion
H2O Deprivation Test + AVP response
↑ [Na+] 10- 15 mEq/L within a few minutes
Weakness, irritability, seizures, coma
↑ thirst, ↓ urinary frequency and volume
Note:
Hypernatremia
Serum [Na+] > 145 mmol/L
Intracranial hemorrhage
Headache, vomiting, ↓ LOC
• Plasma [Na+] is regulated by water intake/excretion, not by changes in [Na+].
• Effects on plasma [Na+] of IV fluids or loss of bodily fluids is determined by the tonicity of the fluid, not the osmolality.
Authors: Mannat Dhillon Reviewers: Andrea Kuczynski Kevin McLaughlin* * MD at time of publication
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published January 11, 2019 on www.thecalgaryguide.com")
Hyponatremia- Physiology
![Hyponatremia: Physiology
Authors: Mannat Dhillon Reviewers: Andrea Kuczynski Kevin McLaughlin* * MD at time of publication
Abnormal Renal H2O Handling (hypo-osmolar serum)
AKI/CKD Heart failure
↓ renal blood flow
↓ glomerular filtration
GFR < 25 mL/min, ↓ urine dilution ↑ H2O retention
Note:
• Plasma [Na+] is regulated by water intake/excretion, not by changes in [Na+].
• Artifactual hyponatremia can be differentiated by a normal or hyperosmolar serum.
Appropriate ADH secretion
↓ EABV
Hypovolemia: losses via GI, renal, skin, 3rd spacing, bleeding
Hypervolemia: heart failure, cirrhosis
↑ Na+/H2O absorption at PCT
↓ EABV, ↑ H2O retention
Urine [Na+] < 20 mmol/L
Hereditary: tubular disorders
(Bartter, Gitlemann syndromes).
Thiazide diuretics
Inappropriate: SIADH, hypothyroidism, AI
Normal EABV
Anti-diuresis
Primary polydipsia, eating disorder
↑ H2O or ↓ solute intake
↓ Osmoles
Impaired desalination
Block NCC
↑ H2O retention ↑ Na+/K+ excretion
Hyponatremia
Serum [Na+] < 135 mmol/L
Urine osmolality > 100 mmol/L
Urine osmolality < 100 mmol/L
Cerebral edema, ↑ intracranial pressure, vasoconstriction
If hypovolemic: ↓ JVP, ↓ blood pressure
Lethargy, altered mental status
Abbreviations:
AKI: Acute Kidney Injury
CKD: Chronic Kidney Disease
GFR: Glomerular Filtration Rate
H2O: Water
PCT: Proximal Convoluted Tubule
EABV: Effective Arterial Blood Volume
NCC: Na+/Cl- Co-Transporter
SIADH: Syndrome of Inappropriate ADH Secretion AI: Adrenal Insufficiency
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published January 11, 2019 on www.thecalgaryguide.com](http://calgaryguide.ucalgary.ca/wp-content/uploads/2019/01/Hyponatremia-Physiology-.jpg "Hyponatremia: Physiology
Authors: Mannat Dhillon Reviewers: Andrea Kuczynski Kevin McLaughlin* * MD at time of publication
Abnormal Renal H2O Handling (hypo-osmolar serum)
AKI/CKD Heart failure
↓ renal blood flow
↓ glomerular filtration
GFR < 25 mL/min, ↓ urine dilution ↑ H2O retention
Note:
• Plasma [Na+] is regulated by water intake/excretion, not by changes in [Na+].
• Artifactual hyponatremia can be differentiated by a normal or hyperosmolar serum.
Appropriate ADH secretion
↓ EABV
Hypovolemia: losses via GI, renal, skin, 3rd spacing, bleeding
Hypervolemia: heart failure, cirrhosis
↑ Na+/H2O absorption at PCT
↓ EABV, ↑ H2O retention
Urine [Na+] < 20 mmol/L
Hereditary: tubular disorders
(Bartter, Gitlemann syndromes).
Thiazide diuretics
Inappropriate: SIADH, hypothyroidism, AI
Normal EABV
Anti-diuresis
Primary polydipsia, eating disorder
↑ H2O or ↓ solute intake
↓ Osmoles
Impaired desalination
Block NCC
↑ H2O retention ↑ Na+/K+ excretion
Hyponatremia
Serum [Na+] < 135 mmol/L
Urine osmolality > 100 mmol/L
Urine osmolality < 100 mmol/L
Cerebral edema, ↑ intracranial pressure, vasoconstriction
If hypovolemic: ↓ JVP, ↓ blood pressure
Lethargy, altered mental status
Abbreviations:
AKI: Acute Kidney Injury
CKD: Chronic Kidney Disease
GFR: Glomerular Filtration Rate
H2O: Water
PCT: Proximal Convoluted Tubule
EABV: Effective Arterial Blood Volume
NCC: Na+/Cl- Co-Transporter
SIADH: Syndrome of Inappropriate ADH Secretion AI: Adrenal Insufficiency
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published January 11, 2019 on www.thecalgaryguide.com")
adrenergic-agonists-for-treating-hypotensionlow-blood-pressure
 β1-receptor
activation on
cardiac
myocytes
↑ contractility
α1-receptor
activation on
the smooth
muscle of blood
vessel walls
↑ intra-cellular Ca2+
in these cells, ↑
their contraction
Ephedrine
Direct
effect
Indirect
effect
↑ release of endogenous norepinephrine
from the adrenal medulla (see above)
Mimics epinephrine
(see above)
Primary
Indications:
anaphylaxis,
cardiac arrest
Primary
Indications:
Hypovolemic states
(e.g. blood loss),
Low systemic
vascular resistance
states (e.g. sepsis,
Anesthesia-induced
hypotension)
Primary
Indications:
mainly used in
Anesthesiainduced
hypotension
↑ intracellular
Ca2+ ↑ rate of
myocyte
contraction
Phenylephrine
↑ Cardiac
Output
↑ heart rate
↑ strength
of myocyte
contraction
↑ arterial
wall tone
↑
stroke
volume
Pushes more
blood to flow
back to heart
(↑ preload)
↑ systemic vascular
resistance (SVR)
More blood in
ventricles stretch
myocytes more
optimally for ↑
contractility
(Frank Starling
Law)
↑ venous
wall tone
↑ Blood
Pressure
Nonspecific activation of α and β receptors on other areas of the body
(e.g. on the autonomic nervous system) as well as on cardiac myocytes
Hypertension
Cardiac Dysrythmias: e.g.
palpitations, ventricular fibrillation
Tremors Cardiac
arrest
All four
drugs")
Hyperkalemia- Physiology
![Hyperkalemia: Physiology ↓ Renal Excretion
↑ Intake
↓ Intracellular Shift
Acute and chronic kidney disease; CHF
Principal Cell Dysfunction (TTKG < 7)
ACEi/ARB; AI; heparin
Hypovolemia (TTKG > 7)
↓ EABV
↓ distal flow of Na+ and H2O
Urine [Na+] < 20 mmol/L
Cell lysis
↑ osmolarity H2O efflux
Solvent drag
β2 inhibition α1 stimulation
Digoxin ↓ A
NAGMA ↓ insulin
↓ NHE1 activity
Diabetic nephropathy; NSAIDs
↓ A: ↓ R
K+ sparing diuretics; voltage- dependent RTA
↓ Na+/K+ ATPase activity
↓ GFR
↓ A: ↑ R ↑ A: ↑ R
↓ CCD K+ secretion
↑ K+ availability
↑ K+ release
↓ intracellular K+ influx
Chronic: Desensitize voltage- gated Na+ channels and ↓ membrane excitability
ECG: Peaked T-waves, ↑ PR interval, flat/absent P-wave, ↑ QRS, QRST “sine wave”
Hyperkalemia
Serum [K+] > 5.1 mmol/L
Acute: ↑ extracellular [K+] makes the RMP less (-)
Abbreviations:
A: Aldosterone
AI: Adrenal Insufficiency
CCD: Cortical Collecting Duct
CHF: Congestive Heart Failure
EABV: Effective Arterial Blood Volume H+: Hydrogen ion
K+: Potassium ion
Na+: Sodium ion
NAGMA: Normal Anion Gap Metabolic Acidosis
NSAIDs: Non-steroidal anti-inflammatory drugs Note:
Muscle weakness or paralysis, ↓ urinary acid excretion
R: Renin
RTA: Renal Tubular Acidosis
RMP: Resting Membrane Potential TTKG: Transtubular Potassium Gradient
• Pseudohyperkalemia should always be excluded; can be caused by thrombocytosis, leukocytosis or improper blood withdrawal technique.
Authors: Mannat Dhillon Joshua Low Reviewers: Andrea Kuczynski Kevin McLaughlin* * MD at time of publication
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published March 6, 2019 on www.thecalgaryguide.com](http://calgaryguide.ucalgary.ca/wp-content/uploads/2019/03/Hyperkalemia-Physiology-.jpg "Hyperkalemia: Physiology ↓ Renal Excretion
↑ Intake
↓ Intracellular Shift
Acute and chronic kidney disease; CHF
Principal Cell Dysfunction (TTKG < 7)
ACEi/ARB; AI; heparin
Hypovolemia (TTKG > 7)
↓ EABV
↓ distal flow of Na+ and H2O
Urine [Na+] < 20 mmol/L
Cell lysis
↑ osmolarity H2O efflux
Solvent drag
β2 inhibition α1 stimulation
Digoxin ↓ A
NAGMA ↓ insulin
↓ NHE1 activity
Diabetic nephropathy; NSAIDs
↓ A: ↓ R
K+ sparing diuretics; voltage- dependent RTA
↓ Na+/K+ ATPase activity
↓ GFR
↓ A: ↑ R ↑ A: ↑ R
↓ CCD K+ secretion
↑ K+ availability
↑ K+ release
↓ intracellular K+ influx
Chronic: Desensitize voltage- gated Na+ channels and ↓ membrane excitability
ECG: Peaked T-waves, ↑ PR interval, flat/absent P-wave, ↑ QRS, QRST “sine wave”
Hyperkalemia
Serum [K+] > 5.1 mmol/L
Acute: ↑ extracellular [K+] makes the RMP less (-)
Abbreviations:
A: Aldosterone
AI: Adrenal Insufficiency
CCD: Cortical Collecting Duct
CHF: Congestive Heart Failure
EABV: Effective Arterial Blood Volume H+: Hydrogen ion
K+: Potassium ion
Na+: Sodium ion
NAGMA: Normal Anion Gap Metabolic Acidosis
NSAIDs: Non-steroidal anti-inflammatory drugs Note:
Muscle weakness or paralysis, ↓ urinary acid excretion
R: Renin
RTA: Renal Tubular Acidosis
RMP: Resting Membrane Potential TTKG: Transtubular Potassium Gradient
• Pseudohyperkalemia should always be excluded; can be caused by thrombocytosis, leukocytosis or improper blood withdrawal technique.
Authors: Mannat Dhillon Joshua Low Reviewers: Andrea Kuczynski Kevin McLaughlin* * MD at time of publication
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published March 6, 2019 on www.thecalgaryguide.com")
autosomal-dominant-polycystic-kidney-disease-adpkd
:
Pathogenesis,
Clinical Findings,
and Complications
Author:
Yan Yu*
Reviewers:
David Waldner*
Sean Spence*
Andrew Wade*
* MD at time of
publication
Legend: Published April 14, 2019 on www.Pathophysiology Mechanism Sign/Symptom/Lab Finding Complications thecalgaryguide.com
One theory (mechanism unclear): these mutations in the polycystin
gene result in dysfunctional Ca2+ channels on epithelial cells
PKD1 mutation
(~78%)
Abnormal Ca2+ entry disrupts intracellular Ca2+ signaling
In the Kidney: all segments of the nephron develop cysts: sacs of flattened epithelium
filled with proteinaceous fluid, replacing normal parenchyma with dysfunctional tissue
PKD2 mutation
(~15%)
Expansive cell
proliferation
Low urine
Osmolality
(< 500
mmol/kg)
Abnormally expandable
basement membranes
PKD3 mutation
(rare)
↑ fluid
secretion
95% inherited, autosomal dominant mutations
5% spontaneous mutations
In adults, the same pathophysiology occurs
in epithelial tissue throughout the body
When pH of urine <5.5, uric acid is in
its protonated form & is less soluble
àprecipitates uric acid stones
Nephrolithiasis
Urine accumulates within cysts Cyst growth
Pyelonephritis
Damaged
tubules
leak
proteins
into filtrate
Proteinuria
Flank pain
Inability to
concentrate
urine
Low urine
specific
gravity
(<1.010)
¯ NH3 production,
↑ acidity of renal
tubule (¯ pH)
Activates
nociceptors Cyst hemorrhage
Urine stasis à
precipitation of
CaOxalate stones
within cysts
Multiple Renal Cysts
(bilaterally, in cortex and
medulla, on Ultrasound)
In the Brain:
expansion &
weakening of
cerebral
arterial walls
“Berry
Aneurysms” 9-
12%
(ask about this
on Family Hx!)
In many organs:
epithelial tissue
expansion &
fluid secretion
In the Heart:
abnormal valve
collagen matrix
Valve prolapse
& regurgitation
Liver (90%),
spleen/pancreas
(5-10%), thyroid
(rare) cysts
In seminal vesicles:
Cyst formation disrupts
sperm motility
Infertility
In GI tract: Cyst
formation
Herniations,
diverticuli
Dysfunctional
collecting ducts
Abdominal mass
(enlarged kidneys)
(may be palpable)
Blood leaks into
renal tubules
Hematuria (isomorphic)
Bacteria
accumulate
in the static
urine
Dysfunctional
proximal tubules
Unknown
mechanisms à
¯ urine citrate
àmore urine
Ca2+ binds to
oxalate than to
citrate à↑ Ca-
Oxalate stones
Note: PKD1 mutations have
more severe prognosis than
PKD2 mutations (earlier
disease onset, larger cysts)
Vessels
tear
more
easily
Stretches
renal
capsule
Cysts compress renal vasculature
¯ Glomerular perfusion
To ↑ perfusion à
kidneys activate RAAS
Hypertension")
Polyarteritis Nodosa (PAN): Pathogenesis and Clinical Findings
: Pathogenesis and clinical findings
Environmental triggers
Infectious/viral agents (commonly Hepatitis B)
Medical Comorbidities Malignancies (most commonly hairy-cell leukemia)
Immunogenetic Predisposition: patient is genetically predisposed to a dysregulated immune response
Fever
↑ ESR and CRP
Postulate 1
Viral antigen-antibody complexes deposit in vasculature, causing lesions and activating cellular inflammatory response
Authors: Nela Cosic, Yan Yu* Reviewers: Sean Doherty Martin Atkinson*
* MD at time of publication
Palpable or necrotic purpura
Malignant Hypertension
Renal Insufficiency
Myocardial ischemia
Heart failure Diffuse myalgias
Postulate 2
Viral replication causes direct injury to vascular endothelial cells
↑ Anti- endothelial cell autoantibodies (AECA)
Altered cytokine profile (↑TNF-α, IL-1β, IFN-α, IL- 2)à↑ T-cell mediated immune response
Weight metabolism Loss
Autoimmune attack on various areas of the body
Malaise and/or Arthralgias (knees, ankles, elbows, wrists)
Orchitis: Testicular pain, erythema and/or swelling
Small intestine perforation GI Manifestations
Non-specific abdo pain
GI hemorrhage
Peripheral sensory changes: Distal mononeuropathy
multiplex
Polyarteritis Nodosa (PAN)
Focal segmental necrotizing leukocytoclastic vasculitis of medium or small-sized arteries
Inflammation of arteries damages the vascular endothelium of those arteries
Inflammation predisposes formation of arterial thromboses
Blockage of arteriesà tissue ischemia and possible necrosis (tissue cell death)
↑ basal
Arterial aneurysms
Inflamed subcutaneous arteries
Inflamed renal artery
àluminal narrowing and reduced blood flow to kidneys
Inflamed coronary artery à luminal narrowing, occlusion, thromboses
Segmental inflammation of muscular arteries, stimulating surrounding nociceptors. Muscle ischemia develops long-term.
Ischemia/necrosis of the testicles
Ischemia/necrosis of the small intestine
Ischemic vasculitic nerve damage: Immune complex deposition within vessel walls of arteries traveling with nerves leads to persistent vascular inflammation and ischemia of associated nerve
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published April 18, 2019 on www.thecalgaryguide.com")
Secondary Polycythemia

Poor lung function
Renal artery stenosis
↓ blood flow to kidney
Kidney senses ↓ O2
High affinity Hb or CO poisoning
Hb does not easily unload O2
Tissues become hypoxic
Tumors (e.g. renal, hepatic, lung)
Secretes EPO in an unregulated way, as a “paraneoplastic syndrome”
High altitude
Obstructive sleep apnea
Episodic airway obstruction during sleep
Intermittent hypoxia
Cyanotic heart disease
Shunting of blood
Venous and arterial blood mixes
Poorly oxygenated blood
↓ O2 partial pressure
↑ EPO production independent of O2 (inappropriate response)
↑ EPO production due to hypoxia (appropriate response)
Abbreviations:
• Hb- Hemoglobin
• EPO- Erythropoietin • ILD- Interstitial Lung
Disease
• COPD- Chronic
Obstructive Pulmonary Disease
“Endogenous causes” of high EPO
Secondary Polycythemia
“Exogenous causes” of high EPO
Testosterone therapy Iatrogenic EPO (results in ↑ EPO synthesis administration
within the body)
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Re-Published May 5, 2019 on www.thecalgaryguide.com")
Appendicitis

The appendix is anatomically located in the RLQ; appendicitis may be confused with disorders of surrounding structures: Gynecological Diseases
• RuleoutpregnancywithHCG pregnancy test
• Rupturedovariancyst
• Ectopicpregnancy
• Mittelschmerz(mid-cycle
pain)
Gastro-intestinal Diseases
• Meckel’sdiverticulum (presents identically to appendicitis; surgically located 2 feet from ileocecal valve; mostly seen in children)
• Diverticulitis(presentsasleft sided appendicitis)
Non-GI Abdominal Issues
• Mesentericadenitisinkids <15: swollen mesenteric lymph nodes
• Renalcolic
Obstruction of appendiceal lumen (by fecalith, fibrosis, neoplasia, foreign bodies or lymph nodes in kids)
Appendix distension and spasms
↑ lumen pressure, ↓ blood flow to appendix
Ischemia, tissue necrosis, loss of appendix structural integrity
Bacterial invasion of the appendix wall, causing transmural inflammationandnecrosis
Stretching of visceral peritoneum, stimulation of autonomic nerves T9-T10
Progression of inflammation over several days (variable length of time)
Irritation of parietal peritoneum, stimulation of somaticnerves
If appendix not surgically removed
Perforation of colon wall, causing peritonitis, abscesses or death
Note: Symptoms hugely variable. Only 30% present with classic history. Diagnosis is mostly clinical. Further investigations:
CBC: Leukocytosis (due to inflammatory response) CT: Gold standard test. Thickened visceral membrane with enhancing (white) rim due to ↑ blood flow
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Re-Published July 27, 2019 on www.thecalgaryguide.com")
iga-vasculitis-henoch-scholein-purpura-pathogenesis-and-clinical-findings
 : Pathogenesis and clinical findings
Authors: Mia Koegler Nela Cosic Reviewers: Crystal Liu Yan Yu* Martin Atkinson* * MD at time of publication
Infectious Agents
50% have preceding upper respiratory tract infections, i.e., influenza virus or Group A Strep
Drugs
I.e., antibiotics (penicillin, erythromycin), NSAIDs and biologics (tumor necrosis factor α inhibitors)
Immunogenetic and cellular predisposition
Various genetic polymorphisms alter cell- mediated immune response, IgA levels elevated in 50% of people
↑ Circulating galactose-deficient IgA1 (GD-IgA1). Deficiency in galactosylation of IgAà↓ IgA serum clearanceàadhesion of IgA complexes, which then deposit into the endothelial lining of blood vesselsàattraction of various inflammatory cells to the area:
Formation of Secretion of Interleukin 8 (IL8) - cytokine that induces Neutrophils infiltrate Activation of complement immune complexes neutrophilic chemotaxis and macrophage phagocytosis the tissue site factors (C3, C4)
Leukocytoclastic vasculitis (histopathologic term for small vessels inflamed by neutrophilic autoimmune response)
Inflamed cutaneous vessels become enlarged in clusters
Symmetrical palpable purpura (red/purple, non- blanchable papules) distributed on lower limbs and buttocks areas
Cutaneous small vessel vasculitis (100%)
Inflamed gastric vessels - hemorrhage and edema within bowel wall
Gastrointestinal (85%)
Colicky abdominal pain (commonly in the periumbilical region), nausea, vomiting
Gastrointestinal
GI bleeding (hematemesis, melena), Intussusception
Glomerular mesangial proliferation and inflammation
↑ mast cell deposition in joints
Joints (60-85%)
Arthralgia's (common), arthritis (especially knees and ankles)
Arthralgia often transient. No permanent sequelae
Sympathetic nervous system activation
Glomerulosclerosis, tubulointerstitial and podocyte damage
Renal tissue ischemia
↑ Na sensitivity in renal tubules (↑ Na and water retention)
Renal (10-50%)
Increased renin secretion
HTN, nephrotic/nephritic syndrome, renal insufficiency
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published September 1, 2019 on www.thecalgaryguide.com")
Varicocele
.
• Small varicoceles can be identified by preforming the Valsalva maneuver (decreases venous return).
• Unilateral right varicoceles are uncommon and should be investigated for underlying pathology causing obstruction.
Primary
Anatomically: the left spermatic vein drains into the left renal vein
Nutcracker Effect: The left renal vein can get pinched by the abdominal aorta and superior mesenteric artery
Backup of blood in left renal vein ↑ pressure in left spermatic vein
Secondary
Renal cell carcinoma or retroperitoneal masses
Inferior vena cava thrombus
External compression of spermatic vein
Obstruction of blood flow
↑ spermatic vein pressure
Vein valve leaflet failure & retrograde bloodflow back towards testicle
Dilation of pampiniform plexus and scrotal vein plexus
Varicocele
↑ scrotal blood volume ↑ volume in a closed
space
↑ pressure and distension of scrotal layers
↑ scrotal vein plexus pressure
Compliant veins distend, becoming visible through scrotum
Blood heats up the structures it flows through
Scrotal hyperthermia
Unsuitable environment for spermatogenesis
Loss of germ cell mass
Bag of Worms Sign
Dull ache/heaviness
Decreased fertility Testicular atrophy
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published November 26, 2019 on www.thecalgaryguide.com")
Multiple-Myeloma
 immunoglobulins
Normal SPEP: no spike in the gamma (γ) region
Notes:
• MGUSismuchmorecommon than MM!
• “Plasmacell”:Blymphocytes that produce antibodies/ immunoglobulins
Stimulation by specific antigens
Genetic changes/mutations accumulate over time in one type of plasma cell
Abbreviations/Definitions: • SPEP - Serum Protein
Electrophoresis
• Ig – Immunoglobulin • Monoclonal – “of one
specific genetic strain or subtype”
One type of plasma cell starts to proliferate abnormally
Monoclonal Gammopathy of Undetermined Significance (MGUS) (Premalignant, mild monoclonal plasma cell proliferation; asymptomatic)
In 1-2% of cases, further cytogenetic changes over time stimulate further proliferation of this plasma cell line
Multiple Myeloma (MM)
(extensive monoclonal B lymphocyte proliferation, causing end organ damage)
Slightly more monoclonal plasma cells will produce slightly more monoclonal Immunoglobulins
Small spike in the gamma (γ) region of the SPEP (less prominent compared to the spike in MM)
More monoclonal plasma cells result in far greater amounts of monoclonal immunoglobulins being secreted
Large spike in gamma (γ) region of the SPEP
Ig light chains accumulate in the tubules of kidney nephrons
Light chain casts obstruct tubules
Renal Insufficiency (↓GFR)
Osteoblasts ↑ expression of RANK-ligand (RANKL, an apoptosis regulator), and ↓ expression of Osteoprotegerin (OPG, a decoy receptor for RANKL)
↑ osteoclast activity vs osteoblast activityàbone loss
Clonal plasma cells overrun normal bone marrow, crowding out production of red blood cells, ↓ red blood cell counts
Anemia
Authors: Tristan Jones, Tyler Anker, Yan Yu Reviewers: Jennifer Au, Crystal Liu, Man- Chiu Poon*, Lynn Savoie* * MD at time of publication
Damaged bones hurt, & become more brittle
Bony pain, pathologic fractures
Osteolytic bone lesions
Osteoclasts release calcium from bone and into blood
Hypercalcemia
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published January 12, 2020 on www.thecalgaryguide.com")
Diabetic-Nephropathy

↑ Intrarenal angiotensin II
↑ Advanced glycation end products (AGE)
Shunting of glucose through non- glycolytic pathways (e.g. polyol)
Activation of Protein Kinase C (PKC) pathway
Excessive production and accumulation of glycolytic intermediates (e.g. sorbitol, hexosamine, succinate)
hyperglycemia
Succinate via GPR91
↑ Free radical production (oxidative stress)
Activation of cellular signalling, transcription factors and cytokines (e.g. TGF-β-Smad-MAPK, IGF-1, NF-κB)
↑NADPH oxidase activity
↑Blood volume ↑ Blood pressure ↑ Renal perfusion
Relative afferent arteriole dilatation, efferent arteriole constriction
Initial ↑ in glomerular filtration rate (GFR)
Podocyte loss/injury
Authors: Steven Chen Shannon Gui Yan Yu* Reviewers: Julia Heighton Ryan Brenneis Sophia Chou* * MD at time of publication
Initial glomerular hyperfiltration at time of diagnosis
↓ Production of matrix metallo- proteinases
Aberrant extracellular matrix (ECM) protein expression and accumulation
Sheer stress to glomeruli à pressure-induced damage
↑ Glomerular basement membrane permeability to proteins like albumin
↓ Extracellular matrix regulation
“Metabolic Pathway”
Mesangial matrix expansion
Kimmelstiel-
Wilson lesions (pink
hyaline nodules due to accumulation of damaged proteins)
Tubular fibrosis
Scarred glomeruli are less able to effectively filter blood
↓ in glomerular filtration rate (GFR)
Albuminuria
Usually occurs after ~5 years from time of diagnosis in T1DM; can occur at time of diagnosis in T2DM
Abbreviations
IGF: Insulin-like growth factor
MAPK: Mitogen-activated protein kinases NADPH: Nicotinamide adenine dinucleotide phosphate
NF-κB: Nuclear factor kappa-light-chain- enhancer of activated B cells
TGF-β: Transforming growth factor-β
Protein endocytosis into tubular cells causing inflammation
“Hemodynamic Pathway”
Diabetic Nephropathy
Overt diabetic nephropathy may take upwards of 15-25 years to develop
Note: The mechanisms presented here have been simplified. The cross- talk and signaling between the metabolic and hemodynamic factors do not manifest in a step-wise fashion, but rather occur in parallel.
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published May 3, 2020 on www.thecalgaryguide.com")
GU-changes-in-pregnancy

Mechanism unclear, possibly 1) ↑ circulating anti- angiogenic factorsà ↑ permeability of glomerular basement membrane, or 2) ↑ plasma volumeà↓ oncotic pressure of plasma at the glomerulus
Proteinuria
> 300 mg/day, abnormal in pregnancy
↑ hCG level
Dilation of renal vasculature
↑ renal vascular & interstitial volume
↑ filtration surface area
Overwhelming load of glucose to proximal tubule
↑ urine output
↑ relaxin secreted by placenta
Mechanism not well understood
↓ osmotic threshold for ADH release & thirst
↑ ADH secretion & ↑ oral hydration
Incomplete reabsorption of glucose
Glucosuria
Encourage bacterial growth in the urine
Urinary tract infection (e.g. cystitis, pyelonephritis)
↑ serum progesterone
Uterine rotation as uterus enlarge due to presence of large bowel
Ureter compression (R > L)
Ureterovesical reflux (back-flow of urine into the ureters/kidneys)
Dilatation of ureters (R > L) (hydroureter) & renal pelvis (hydronephrosis)
Progesterone competes with aldosterone
↓ sodium reabsorption
↓ plasma sodium concentration
Hyponatremia of pregnancy* (pathological if < 130 mEq/L)
*Despite factors favoring sodium excretion, there is a net retention of sodium during pregnancy from adaptation of the renal tubules
↑ urinary stasis
↑ excretion of creatinine and urea in urine
↓ serum creatinine and urea
↓ ureteral toneà ↑potential for ureter dilation
Mechanism not well understood
↓ peristalsis of ureters
↑ urinary frequency (voiding > 7x/day)
↑ nocturia (voiding ≥ 2x/night)
↓ oncotic pressure of plasma intra-vascularly àwater leaves blood, enters interstitial tissue, and stays there in gravity-dependent regions of the body
Pedal +/- ankle edema
Author: Simonne Horwitz Reviewers: Claire Lothian, Crystal Liu, Ronald Cusano*, Candace O’Quinn*, Yan Yu* * MD at time of publication
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published June 28, 2020 on www.thecalgaryguide.com")
acute-pancreatitis-complications
:
Interstitial edematous pancreatitis
Local accumulation of fluid in the pancreas
<2 weeks after onset
Acute peripancreatic fluid collection (not encapsulated)
Walled off by fibrous & granulation tissue
>2 weeks after onset
Pancreatic pseudocyst
(completely encapsulated)
Peritoneal irritation à pain
Large cyst can (very rarely) compress surrounding bowel
Acute Pancreatitis
Inflammatory cytokines are released from damaged pancreas
If recurrentàchronic pancreatitis (see relevant slide)
Inflammation damages pancreatic exocrine
cellsàInappropriate release of pancreatic enzymes into surrounding tissue & vasculature àdigesting pancreatic parenchyma
Authors: Nissi Wei, *Yan Yu Reviewers: Dean Percy, Miles Mannas, Varun Suresh, Brandon Hisey, *Kerri Novak, *Sylvain Coderre * MD at time of publication
complete resolution (most cases)
Necrotic tissue is vulnerable to
infection (esp. Gram neg GI bacteria)
inflammation & necrosis activate cytokine cascade
Severe, necrosis (15%): Necrotizing pancreatitis
Local infection
Severe pancreatic inflammation shifts body fluid into retroperitoneal spaceàintravascular volume depletion
Systemic Inflammatory Response Syndrome (SIRS) (see relevant slide)
Organ failure (may be sole feature on presentation)
Stagnant fluid can more easily become infected
Infection spreads to bloodstream
Cardiac failure Hypovolemic shock Renal failure
Local accumulation of fluid & necrosis in the pancreas
< 4 weeks after onset:
Acute necrotic collection (not encapsulated)
Walled off by fibrous & granulation tissue
> 4 weeks after onset
walled-off necrosis
(completely encapsulated)
When treated with excess fluid resuscitation:
Intra- abdominal hypertension
Respiratory failure (ARDS)
Disseminated intravascular coagulation (DIC)
Bowel obstruction Gastric outlet (see relevant slide) obstruction
Infected pancreatic necrosis
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published September 20, 2016, updated September 7, 2020 on www.thecalgaryguide.com")
insuffisance-cardiaque-gauche-les-resultats-de-lexamen-physique

Dysfonction systolique
̄ Perfusion tissulaire (au niveau cerebral, renal etc.)
Vasoconstriction périphérique àdetournement limité du volume d’ejection systolique vers les organes principaux
Extrémités froides, cyanose périphérique
̄ Du débit urinaire
̄ De l’état de conscience
Hypoxie tissulaire la respiration anaérobique, production d’acide lactique (acidose)
Les centres respiratoires tentent de compenser
Expulsion des fluides des capillaires vers les alvéolesà oedème pulmonaire transudatif
La congestion vasculaire et les alvéoles oedématiées compriment les voies respiratoires, provoquant une turbulence du flux d'air
Un flux d'air turbulent est entendu lors de l'auscultation
Respiration sifflante (wheezing)
Flux turbulent dans le ventricule gauche distendu au début de la diastole
B3
Choc
de la pointe diffuse
̄ Du flux sanguin artériel pendant la systole
̄ Tension artérielle en systole
De la
tension artérielle lors de la diastole
̄ Pouls cardiaque
Fermeture des valves pulmonaires avec une force supérieure à la normale
B2 pulmonaire
̄ De l’oxygenation sanguine
Le transsudat obstrue les petites voies aériennes et alvéolaires
Si le ventricule gauche est en surcharge de pression ou si hypertrophié (i.e. sténose aortique, hypertension sévère)
Choc de la pointe soutenue
De l'activité du système sympathique (pour tenter de rétablir le débit cardiaque)
Pendant l'inspiration, de petites zones de compartiments aériennes s'ouvrent
Crépitements pulmonaires
(ils sont généralement bilatéraux et surtout au niveau de la base pulmonaire)
Auteur: Yan Yu
Editeurs: Sean Spence, Jason Baserman, Nanette Alvarez*
Traductrice/Traducteur:
Emma Hofland-Burry, Jean-François Lemay* *MD au moment de la publication
Activité des glandes sudoripares
Diaphorèse
Activité cardiaque
Tachycardie
Fréquence respiratoire
Tachypnée
Remarque: l'insuffisance cardiaque gauche étant la cause principale de l'insuffisance cardiaque droite, elle peut également se présenter par des signes d'insuffisance cardiaque droite. Les autres caractéristiques de l'insuffisance cardiaque gauche dépendent de la cause sous-jacente.
Si la pression vasculaire artérielle pulmonaire est chroniquement élevéeàporvequera un surmenage prolongé de la function du ventricule droit:
Insuffisance cardiaque droite: Le patient présente des conséquences directes de sa congestion veineuse systémique: œdème
périphérique (i.e.cheville), épanchements pleuraux, congestion hépatique/ascites (s.v.p. voir la diapositive correspondante)
Publié le 10 Janvier 2013 à www.thecalgaryguide.com")
Pathogenesis-of-Female-Infertility
 from hypothalamus
↓ release of Luteinizing hormone (LH) & Follicle stimulating hormone (FSH) by pituitary
↓ release of estrogen by ovaries
Anovulation (oocyte is not released)
Fewer follicles available to ovulate
* Causes of Hyperprolactinemia include: prolactinoma (prolactin-producing tumor), hypothalamic infiltrate or mass, chest wall irritation, hypothyroidism, renal or liver disease (↓ prolactin clearance), dopamine antagonists that ↑ prolactin secretion (antipsychotics, anti- depressants, anti-emetics)
Polycystic ovary syndrome (see PCOS: Pathogenesis and Clinical findings)
↑ androgen production & ↑ estrogen earlier in the menstrual cycle
↓ FSHà↓ follicle growth
↑ rate of follicle depletion
Oocyte not available every month for fertilization
Premature ovarian insufficiency due to unexplained causes, chemotherapy, radiation, autoimmune ovarian destruction, Turner’s & Fragile X Syndromes
Damage in germ cells that accumulates over a woman’s lifetime
Age-related changes in quality of granulosa cells surrounding oocyte
Genetic damage accumulates, such as ↑ rates of meiotic nondisjunction (failure of chromosomes to separate during gamete cell division)
Tubal occlusion or ↓ transport of oocyte tubal cilia dysfunction through fallopian tube
↓ quality of oocytes
Normal transport of oocyte & sperm through fallopian tube is impaired
↓ facilitation of sperm transportation
Inhibits normal zygote implantation
Chlamydial or gonorrhoeal pathogens
Previous tubal surgery or ectopic pregnancy surgery tissue removal ↓ transport of oocyte through fallopian tube
Female Infertility
Previous abdominal infection or surgery Endometriosis
Congenital malformations or trauma / surgery to cervix
Uterine leiomyomata (benign smooth muscle monoclonal tumor) or polyp
Intrauterine procedures
Pelvic adhesions (scar-like tissue that tether together abdominal organs) may distort the shape and normal anatomy of the fallopian tube
Ectopic endometrial cells implant & Local inflammatory response grow along pathway of egg/sperm further ↓ egg/sperm mobility
Inability of cervix to produce normal mucus, and/or sperm physically unable to enter the cervix
Submucosal or intracavitary component disrupts uterine lining
Trauma to basalis layer of endometrium
Intrauterine scarring or synechiae (adhesions)
↓ vascularization & endometrial regrowth
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published October 25, 2020 on www.thecalgaryguide.com")
Tumour-Lysis-Syndrome

Though rare, aggressive tumours can spontaneously lyse without treatment
Intracellular potassium released into bloodstream
Intracellular phosphate released
Hyperphosphatemia
↑ serum phosphate
Intracellular lactate dehydrogenase (LDH) released
↑ serum LDH
Intracellular nucleic acids released
Nucleic acids metabolized to uric acid
Hyperuricemia
↑ serum uric acid
Hyperkalemia
↑ serum potassium
(see Hyperkalemia: clinical findings)
Uric acid (a crystallizing substance) ↑ precipitation of calcium phosphate
↑ filtration of poorly soluble uric acid into acidic environment of renal tubules
Serum phosphate binds serum calcium, forming solid calcium phosphate precipitate crystals
High levels of calcium phosphate ↑ uric acid precipitation
Uric acid precipitates as crystals and deposits in kidney tubules and collecting ducts
Tubular injury and/or intraluminal obstruction
Endothelial dysfunction in renal vasculature
Renal inflammation, vasoconstriction, and
impaired renal vascular autoregulation
Decreased renal filtration
Acute Kidney Injury
(see Acute Kidney Injury Overview)
Crystal deposition in the heart
Depletion of soluble calcium
Hypocalcemia
Calcium phosphate crystals deposit in kidneys
Main mechanism of Acute Kidney Injury
Cardiac Arrythmias
↓ serum calcium
(see Hypocalcemia: clinical findings)
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published October 25, 2020 on www.thecalgaryguide.com")
Primary-Aldosteronism

↑ Serum aldosterone level
↑ Circulating aldosterone activates mineralocorticoid receptors on cardiac myocytes
↑ Transcription of proinflammatory and profibrotic genes in cardiac myocytes
Cardiac fibrosis and hypertrophy
In the rest of the body, K+ moves from intracellular to extracellular environment (down it's concentration gradient) to compensate for decreased serum K+
Loss of positively charged K+ànegative intracellular chargeàcells take up H+ to remain electrostatically neutral
↑ Expression of epithelial sodium channels in
principal cells of cortical collecting duct
↑ Na+ reabsorption from cortical collecting duct into blood vessels
+ Water follows Na
into the blood vessels to balance the osmotic pressure between the blood and the renal tubules
↑ blood volume within the volume-
constrained space of blood vessels
↑ Activation of Na+/K+ ATPase in principle cells of cortical collecting duct
Removal of positively charged Na+ from tubular lumenà lumen becomes electronegative versus the interstitial space & inside tubular epithelial cells
Positively charged K+ follows the electrical gradient and is secreted into tubular lumen
↓ Serum K+ concentration
↑ Expression of H+ ATPase in the apical (luminal)
membrane of intercalated cell of cortical collecting duct
H+ is secreted into cortical collecting duct → renal loss of H+ from the body
Metabolic Alkalosis
(blood becomes more basic; ↑ serum pH)
Hypertension
Muscle weakness, fatigue, polyuria
↓ pH in intercalated cell
↓ pH in cells of proximal convoluted tubule
↑ loss of H+ through kidneys
H+ secretion into cortical collecting duct
↑ H+ ATPase activity in intercalated cell
Activation of glutaminase in proximal convoluted tubule
HCO3- pumped into blood
↑ production of HCO3- (product of glutamine breakdown)
↑ breakdown of glutamine
Hypokalemia
(See Hypokalemia: Clinical Findings slide)
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published January 16, 2020 on www.thecalgaryguide.com")
Beta-Blockers-Mechanism-of-Action-and-Side-Effects
 to these receptors, ↓ their normal adrenergic tone
Beta-2 receptor antagonism Beta-1 receptor antagonism
Lungs Eyes Central nervous system Heart Kidneys ↓ cAMP (intracellular messenger) productionàcomplex, tissue-specific intracellular mechanisms resulting in a variety of effects in different tissues:
Throughout body tissue
Epinephrine (via cAMP) indirectly ↑ the activity of the Na+/K+ pump on cell membranes (a pump that moves 3 Na+ out of cells per 2 K+ moved into cells)
Blocking epinephrine from binding
the beta-2 receptor and producing cAMPà↓ activity of Na+/K+ pump à↓K+ moved into cells
↑ proportion of K+ now resides in extracellular fluid, detectable in serum (total body K+ remains the same)
Hyperkalemia (see Calgary Guide: Hyperkalemia – Clinical findings)
Blocking sympathetic hormonesà↓ relaxation of smooth muscle circumferentially wrapped around airways
↑ resting airway muscle toneà bronchoconstriction
↑ resistance to airflow
Wheezing, dyspnea, chest tightness
Exacerbation of underlying airway disease (e.g. asthma)
↓ ciliary epithelium’s production of aqueous humor (fluid that fills anterior chamber of the eye)
Reduced intraocular pressure
Blocking adrenergic response mediated by epinephrine and norepinephrine (e.g. the physiologic “fight- or-flight” response to stress)
↓ tremor, irritability, anxiety
↓ ability to produce adrenergic symptoms in response to hypoglycemia
Hypoglycemia unawareness
Coronary perfusion pressure = diastolic blood pressure in aorta – LV end diastolic pressure
↓ inotropy (contractility of cardiac muscle)
↓ chronotropy (heart rate and conduction velocity)
↓ renin releaseà↓ creation of angiotensin II & aldosterone
+ ↓ reabsorption of Na
and H2O in nephron
↑ urinary Na+ & H2O loss
↓ total blood volume
Decompensation of acute heart failure
Dizziness and fatigue Hypotension (Blood pressure = cardiac
output x systemic vascular resistance)
↓ O2 demand of myocardial tissue
Bradycardia
Inability to ↑ heart rate in response to stress (e.g. shock, sepsis)
↓ stroke volume
↓ cardiac output
Beta blockers ↓ diastolic blood pressure, & thus may ↓ coronary perfusion pressure
Before giving beta blockers, ensure blood pressure isn’t too low
Otherwise, may worsen acute myocardial ischemia
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published Jan 14, 2021, updated Feb 7, 2021 on www.thecalgaryguide.com")
Potassium-Sparing-Diuretics-Mechanism-of-Action-and-Side-Effects
 Inhibit Na+ channels (involved in Na+ reabsorption) in the luminal membrane of the principal cells in the cortical collecting duct
Aldosterone Antagonists (Spironolactone and Eplerenone) Competitively blocks mineralocorticoid receptors and ↓ aldosterone effect in the renal tubules and throughout the body
↓ aldosterone effectà↓ expression of basolateral Na+/K+ pumps & luminal epithelium Na+ channels on the principal cells of cortical collecting duct
Spironolactone’s molecular structure is similar to that of steroid
hormonesàspironolactone can also block androgen receptors
Anti-androgenic effects
(due to ↓ androgen function in reproductive organs, skin, and on brain centers)
Triamterene can form triamterene crystals
and granular casts (unclear mechanism)
↓ Na+ reabsorption by principal cells
↓ in serum Na+ concentration: Hyponatremia
↓ K+ pumped out into the tubule by principal cells
Crystals & casts obstruct tubular lumen → inflammation (unclear mechanism)
Triamterene crystals are
directly cytotoxic to tubular cells
Only ~2-5% of Na+ filtered by the glomerulus is normally reabsorbed in the
cortical collecting ductàthe ↑ in Na+ retained in cortical collecting duct is mild
Water follows Na+ to maintain a balanced osmotic pressureà↑ in water in cortical collecting duct available for excretion
↑ positive charges in lumen relative to surroundings generates an electropositive tubular lumen
Unfavorable electrical gradient ↓ secretion of positive charged ions into electropositive lumen
↓ libido
Menstrual abnormalities (in women)
↓ acne Gynecomastia
(in men)
↓ secretion of K+
↑ in serum K+ concentration:
Hyperkalemia
Cardiac Arrythmias (See relevant slide on Hyperkalemia: Clinical Findings)
Interstitial inflammation and tubular injury
Drug-induced Nephrotoxicity
Mild Diuresis
↓ blood volume
↓ Blood pressure/ Hypotension
↓ secretion of H+
↑ in serum H+ concentration:
Metabolic Acidosis
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published February 15, 2021 on www.thecalgaryguide.com")
AAA-Pathogenesis
: Pathogenesis
Different parts of the aorta have different embryologic origins
Atherosclerosis
Hypertension
Age > 65
Progressive deterioration of aorta structural integrity over life span
Connective Tissue Disease
Structurally abnormal protein or protein organization in aorta
Autoimmunity
Infection (e.g Chlamydia, Mycoplasma pneumoniae, Helicobacter pylori, human cytomegalovirus, herpes simplex virus)
Antigens (substance that causes immune response) on virus or bacteria resemble local proteins in abdominal aorta
Antibodies produced in response to infection inappropriately target host cells in the aorta
Antibodies tag cells in the abdominal aorta for destruction by T-lymphocytes
Immune-mediated destruction of aorta
Smoking
Genetics
Unclear mechanisms
Subacute (not clinically detectable) inflammation of aortic tissue
Inflammatory cytokines are released and immune cells are recruited
↑ pressure on aorta and other vessel walls
Infiltration of vessel wall by lymphocytes and macrophages
Production of enzymes that break down elastin & collagen proteins (which provide most tensile strength to aorta)
Aorta susceptible to damage
Degradation of aortic connective tissue
Biomechanical stress on vessels
Authors: Olivia Genereux Davis Maclean Reviewers: Jason Waechter* Amy Bromley* Yan Yu* *MD at time of publication
The exact mechanisms are complex, debatable, and an area of intensive research – the 3 mechanisms and associated pathophysiology presented here are generally thought to be the main causes of abdominal aortic aneurysms
Infrarenal aorta has poorly developed vaso vasorum (dedicated blood supply to vessel wall)
Infrarenal aorta relies solely on nutrient diffusion from aortic blood that crosses abdominal aorta
Infrarenal aortic wall has fewer “lamellar” units (fibromuscular units) than other regions of the aorta
Infrarenal aorta is less elastic & less able to distribute stress
Loss of smooth muscle cells & thinning of tunica media
Destruction of elastin in tunica media
Normal layers of the aortic wall
↓ aortic tensile strength (ability to withstand stretching) Aorta expands and dilates due to internal pressure
Tunica Intima (inner-most tissue layer of aorta)
Tunica Media (layers of elastic
tissue (elastin) and muscle fibers)
Adventitia (thin outermost collagenous layer)
(longitudinal section)
Aortic aneurysms are usually infrarenal (85%)
Abdominal Aortic Aneurysm
Infrarenal aorta more prone to ischemia and has impaired repair potential
Abnormal, irreversible dilation of a focal area of abdominal aorta (area of aorta between diaphragm & aortic bifurcation) to twice the diameter of adjacent normal artery segment
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Re-Published February 27, 2021 on www.thecalgaryguide.com")
VITT
: Pathogenesis and Clinical Findings
Current leading theory: COVID-19 viral vector vaccines (Johnson and Johnson and AstraZeneca) contain an anionic molecule (currently unspecified), which binds to the cationic platelet factor 4 (PF4) molecule in the bloodstream, forming vaccine/PF4 complexes
Authors: Brooke Fallis Reviewers: Yan Yu* Katie Lin* * MD at time of publication
Spleen macrophages remove antibody/platelet complexes from circulation
Fewer unbound platelets in circulation detected on complete blood count (CBC)
Thrombocytopenia: Platelets <150x!")
disseminated-intravascular-coagulation
: Pathogenesis and clinical findings
Authors: Emily Wildman Mehul Gupta Sean Spence Yan Yu* Reviewers: Kiera Pajunen Wendy Yao Tristan Jones Man-Chiu Poon* Lynn Savoie* * MD at time of publication
Sepsis
Microorganisms express pathogen- associated molecular patterns (PAMPs)
Severe Trauma
Damaged endothelial cells release damage- associated molecular patterns (DAMPs)
Solid and Hematologic Malignancies
Cancer cells express tissue factor and release procoagulants
Immune cells recognize DAMPs and PAMPs and begin expressing tissue factor and releasing procoagulants
Systemic activation of coagulation cascade
Activation of coagulation cascade results in the factor X mediated conversion of large quantities of prothrombin (factor II) to its active form, thrombin (factor IIa)
Thrombin cleaves fibrinogen (factor I) circulating in blood to an activate form known as fibrin (factor 1a)
↓ Serum fibrinogen
Diffuse coagulation causes imbalances between coagulation and anticoagulation pathways
Consumption of coagulation factors (including platelets, fibrinogen, prothrombin, factor V, factor VIII) exceeds rate of production
Fibrin, in conjunction with platelets, form widespread thrombi within vasculature
Fibrin thrombi accumulate in the microvasculature and shear transiting red blood cells
Microangiopathic hemolytic anemia (MAHA)
↑ thrombi formation ↑ fibrinolysis, a parallel process that naturally degrades fibrin thrombi
Relative deficiency of coagulation factors leads to a bleeding diathesis (tendency to bleed) despite widespread clots
↑ Prothrombin time (PT)
↑ Partial thromboplastin time (PTT)
↑ D-dimer Fibrin thrombi
↑ Fibrin degradation products
Platelets are used up forming thrombià less free platelets in circulation
↓ Platelets on Complete Blood Count
accumulate in microvasculature of major organs
Deposition of thrombi occlude blood flow resulting in ischemia of organ parenchyma
Lack of coagulation factors can predispose spontaneous hemorrhage in various organs
Multiple organ dysfunction syndrome (MODS) - (renal failure, hepatic dysfunction, stroke, pulmonary disease)
Clinical manifestations of bleeding, including petechiae (pinpoint red spots on skin), ecchymoses (bruising), weeping wound sites, bleeding mucous membranes
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
First published Aug 7, 2012, updated July 27, 2019 & Aug 29, 2021 on www.thecalgaryguide.com")
Primary-Adrenal-Insufficiency

Note: “Primary” refers to pathology in the gland that produces the functional hormone (in this case, the adrenal gland), as opposed to “secondary” or “tertiary” which refers to pathology in glands that indirectly control the primary gland.
Bilateral adrenal Autoimmune damage (cancer,
↓ Androgen levels in women (in whom adrenal glands are the primary source)
↓ Libido
↓ Axillary and pubic hair
Muscle/joint pain
Anorexia, weight loss
Abdominal pain, nausea, vomiting
Diseases bleeding, etc)
↓ Androgen production from the zona reticularis
↓ Cortisol production from the zona fasciculata
↓ Serum cortisol
Patients could present with many severe symptoms of
adrenal insufficiency, e.g. low blood pressure refractory to fluid resuscitation
Significant bilateral damage to the adrenal glands
Entire adrenal cortex function impaired
↓ Serum levels of adrenal steroid hormones exert positive feedback effect on pituitary gland, ↑ pituitary gland’s production of adrenocorticotropic hormone (ACTH)
↑ Serum levels of ACTH
The same gene that encodes ACTH also encodes melanocyte stimulating hormone (MSH)
Coincidental ↑ in MSH production by the pituitary
↓ Serum DHEAS, testosterone and androstenedione
Unknown mechanism(s)
Malaise
Adrenal Crisis
↓ Cortisol mediated gluconeogenesis and glycogenolysis
↑ Cortisol Releasing Hormone (CRH) from the hypothalamus
Lack of cortisol-driven vasoconstriction of blood vesselsà↑ Vasodilation
↓ Blood pressure, postural dizziness
Water follows sodiumà ↑ excretion of water
↓ Sodium resorbed from kidney tubulesà↑ Sodium excretion
↓ Potassium pumped into kidney tubules à↑ potassium retained in serum
Hypoglycemia (↓ blood glucose)
↑↑ Antidiuretic hormone (ADH) release
Kidneys reabsorb water à↑ total body water
Hyponatremia (↓ serum sodium)
↑ Salt craving
Hyperkalemia (high serum potassium level)
↓ Aldosterone production from the zona glomerulosa
↓ Serum aldosterone
MSH triggers ↑ production of melanin by the melanocytes in skin
↓ Excretion of hydrogen ions from kidneys (See type IV RTA slide for full mechanism)
Metabolic acidosis (low pH, low bicarbonate)
↓ Sodium channel and Na/K ATPase
expression on principle cells of the kidney
Hyperpigmentation of the skin and mucosal surfaces
Authors: Jaye Platnich, Brooke Fallis, Yan Yu* Reviewers: Mark Elliott, Alexander Arnold, Hanan Bassyouni*, David Campbell* * MD at time of publication
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published November 13, 2013, updated October 1, 2021 on www.thecalgaryguide.com")
Hypercortisolemia
: Clinical Findings
Cortisol is a net catabolic hormone affecting many body systems, serving to release energy into the blood in response to stress. Excess cortisol also impacts circulation and impairs immune function. Excess Serum Cortisol affects:
Authors: Samin Dolatabadi, Yan Yu* Reviewers: Meena Assad, Amanda Henderson, Brooke Fallis, David Campbell* * MD at time of publication
Bone and Calcium Metabolism
Kidney and vasculature
Excess cortisol in the renal tubule saturates the enzyme 11β- HSD2, which converts cortisol to cortisone
Capacity of body to convert cortisol to cortisone is exceeded
Excess cortisol can mimic aldosterone
and bind to mineralocorticoid receptors (cortisone can’t bind to these receptors)
↑ Aldosterone effect → ↑ Na+ reabsorption from the cortical collecting duct into blood vessels
Liver and Peripheral Tissue
Cortisol ↑ gluco- neogenesis in liver, and ↑ insulin resistance by body tissue (unclear mechanisms)
Hyper- glycemia
Reproductive System
Cortisol exerts negative feedback on hypothalamus
à↓ gonadotropin releasing hormone (GnRH) secretion
↓ GnRH → ↓ LH/FSH → ↓ estrogen and testosterone production (especially important in females)
Infertility, ↓ Libido, Irregular Menses
Adipose Tissue
Cortisol ↑ fat breakdown (lipolysis)
Selective expression of cortisol receptor on different adipose tissuesàcentral, facial, dorsal fat is less broken down than in other areas (mechanism unclear)
Combined with cortisol ↑ appetite:
Skin & Connective Tissue
↑ Serum cortisol à↓ Fibroblast proliferation → ↓ Collagen synthesis
Skin atrophy with loss of connective tissue
Muscle
↑ Proteolysis & ↓ Protein synthesisà↓ muscle growth and function
Immune System
Normal serum cortisol protects against damaging effects of uncontrolled inflammatory and immune responses
↑ Serum cortisolà over-suppression of inflammation and impaired cell- mediated immunity
↑ Serum cortisol leads to ↓ Intestinal Ca2+ absorption and ↓ renal Ca2+ reabsorption
↓ Serum Ca2+ ↑ PTH secretion
↑ Serum cortisolà↑ RANKL:OPG ratio
↓ Osteoblast activity & ↑ Osteoclast activity
Cardiac muscle
Cardio- myopathy, Heart Failure
Skeletal
muscle, especially upper arms & thighs (for unclear reasons)
Proximal Muscle Weakness
Easy Bruising
↑ abdominal size stretches the fragile skin to become thinneràvenous blood of the underlying dermis becomes visible
Purple Striae
If hypertension is chronic
Ca2+ resorption from bone into the blood
Osteoporosis
Supraclavicular & Dorsal Fat Pads
Central Obesity
Poor Wound Healing
Susceptibility to infection
Round Face (Moon Face)
Abbreviations:
• RANKL – Receptor activator of nuclear factor kappa-Β ligand • OPG – Osteoprotegerin
• 11β-HSD2 – 11β-hydroxysteroid dehydrogenase type 2
Water follows Na+ into blood vessels to balance the osmotic pressure between the blood and renal tubules
Water reabsorption → Expansion of blood volume
Hypertension
Both primary Cushing’s (e.g. adenomas that extend into zona reticularis of the adrenal cortex) and central/secondary Cushing’s (e.g. ↑ ACTH stimulation of zona reticularis) are associated with ↑ adrenal androgen secretion
Removal of positively charged Na+ from tubular lumen creates a negative luminal environment
K+ follows the electrical gradient and is secreted into tubular lumen
↓ Serum K+ concentration
Hypokalemia
Arrhythmia, Paralysis, Cramps
(see Hypokalemia: Clinical Findings slide)
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published October 17, 2021 on www.thecalgaryguide.com
Hirsutism, Acne")
Diabetische Ketoazidose (DKA)

Betrifft hauptsächlich Patienten mit Diabetes Mellitus Typ 1: Bestimmte Situationen (z.B. Infektionen) erhöhen den Insulinbedarf, welcher durch fehlende Produktion/unzureichende Substitution nicht gedeckt werden kann
Autor: Yan Yu Rezensenten: Peter Vetere, Gill Goobie, Sean Spence, Hanan Bassyouni* Übersetzung: Sarah Schwarz Übersetzungsprüfung: Dr. Gesche Tallen * MD zum Zeitpunkt der Veröffentlichung
Hyperglykämie
(erhöhter Blutzucker)
Bei >12mmol/L, Glukosefiltration > - resorption, Glukose verbleibt im Urin
Glukosurie
Glukose ist osmotisch wirksam und zieht große Mengen Wasser mit sich (Osmotische Diurese)
Polyurie
Schwere Dehydrierung(bis zu 4-5L)
(ZVD↓, Orthostase: posturale Hypotension/ posturale Tachykardie, Ruhepuls ↑ )
Insulinmangel enthemmt die Lipolyse, sodass der Körper Energie aus Triglyceriden produziert
Freisetzung von freien Fettsäuren aus Fettgewebe
Absoluter
Insulinmangel
Hypothalamische Zellen induzieren bei geringem intrazellulären Glukosespiegel ein starkes Hungergefühl
Glukose bleibt im Blut & kann nicht durch Muskel- /Fettgewebe verstoffwechselt werden
“Ausgehungerte” Zellen triggern die Freisetzung kataboler Hormone: Glucagon, Katecholamine, Cortisol, Somatotropin
Damit intrazellulärer Glukosespiegel ↑, erhöht der Körper den Blutzucker
Hydrolyse von freien Fettsäure in
der Leber (Ketogenese) Polyphagie
↓ Proteinsynthese, ↑ Proteolyse (in Muskelgewebe)
↑ Gluconeogenese, ↑ Glykogenolyse (in der Leber)
Acetyl Co-A
Energiequelle für “ausgehungerte” Zellen
Beachte: Neben Glukose sind Ketonkörper die einzigen Energieträger die von Nervenzellen metabolisiert werden können. Deshalb werden sie vom Körper bei geringem Glukoseangebot produziert.
Ketonkörper
( β-Hydroxybutyrat, Acetoacetat, Aceton, akkumulieren im Blut)
Ketonurie
Metabolische Azidose
(↑Anionenlücke: Ketonkörper verbrauchen den HCO3--Puffer)
Substrate der Gluconeogenese↑
Durch ↓ Extrazellulärflüssigkeit, werden Ketonkörper konzentriert → Azidose
Stört das enterische Nervensystem, Magenentleerung ↓, Ileus
Bauchschmerzen, Übelkeit, Erbrechen
(Dehydration↑)
Abatmen von CO2 um Azidose auszugleichen
Kussmaul- Atmung (Tiefe, schnelle Züge)
Abatmen von Keton- körpern
Azeton- geruch der Atemluft
Beeinträchtigung elektrischer Signale zum Gehirn, Rücken- mark und Nervenzellen
Perfusion des Gehirns, Rückenmark, Nervenzellen↓
Verschiebung des Wasser- und Elektrolythaushalts
Falls Trinkwasser vorhanden
Polydipsie Beachte: Während der DKA verliert der Körper K+ durch
Elektrolytstörung
Schwäche, Delirium +/- Koma
osmotische Diurese und Erbrechen. Da gleichzeitig allerdings K+ aus den Zellen diffundiert, kann das Serumkalium unauffällig/erhöht sein. Um Hypokaliämien zu verhindern sollte bei K+ <5.0mmol/L KCl zusammen mit Insulin i.v. verabreicht werden. Cave: gute Nierenfunktion des Patienten
Therapie: 1) +++ Flüssigkeit. 2) Insulin + KCl. 3) Auffüllen der Anionenlücke. 4) Auslösende Faktoren identifizieren. 5) Wenig PO4 (typischerweise wenige Stunden bis einen Tag nach der Ketoazidose durch ↑ ATP-Produktion.)
Legende:
Pathophysiologie
Mechanismen
Symptome/Klinische Befunde
Komplikationen
Publikationsdatum: 17 Juni 2013 auf www.thecalgaryguide.com
Hyperosmolares/ Hyperglykämisches Koma (HHS)
Beachte: betrifft nur Patienten mit Diabetes mellitus Typ II
Beachte: Es sollte stets nach auslösenden Faktoren z.B. einer Infektion gesucht werden
Autor: Yan Yu Rezensenten:
Peter Vetere
Gill Goobie
Hanan Bassyouni* Übersetzung:
Sarah Schwarz Übersetzungsprüfung: Dr. Gesche Tallen
* MD zum Zeitpunkt der Veröffentlichung
Verschiebung des Wasser- und Elektrolythaushalts
Elektrolytstörung
Unzureichende Insulin- produktion, Insulinresistenz, Nichtansprechen auf Insulintherapie
Relativer Insulinmangel
Situationen mit ↑Insulinbedarf: Infekt, Pneumonie, Herzinfarkt, Pancreatitis, etc.)
Hyperglykämie
(Stark erhöhter Blutzucker, höher als bei der DKA)
Bei >12mmol/L, Glukosefiltration > - resorption, Glukose verbleibt im Urin
Glukosurie
Glukose ist osmotisch wirksam und zieht große Mengen Wasser mit sich (Osmotische Diurese)
Polyurie Dehydration
(ZVD↓, Orthostase: posturale Hypotension/posturale Tachykardie, Ruhepuls ↑ )
Durch das wenige noch vorhandene Insulin, wird ein Teil der Glukose von Muskel- /Fettgewebe verstoffwechselt, ein Teil verbleibt im Blut
Körperzellen brauchen eine weitere Energiequelle
Freisetzung kataboler Hormone: Glucagon, Katecholamine, Cortisol, Somatrotropin
Damit intrazellulärer Glukosespiegel ↑, erhöht der Körper den Blutzucker
Hypothalamische Zellen induzieren bei geringem intrazellulären Glukosespiegel ein starkes Hungergefühl
Polyphagie
Beachte: Durch das wenige noch
vorhandene Insulin wird die Lipolyse gehemmt und werden keine Ketonkörper produziert. Es kommt nicht zur Azidose und Ketonurie (Gegensatz zur DKA). Sollte es doch zur Ketourie kommen, ist das meist Folge von Hungerzuständen oder anderen Mechanismen
Extrazellulärflüssigkeit ↓ mit erhöhter Osmolarität (z.B. Hypernatriämie)
↑ Gluconeogenese, ↑ Glykogenolyse (in der Leber)
↓ Proteinsynthese, ↑ Proteolyse (in Muskelgewebe)
Substrate der Gluconeogenese ↑
Sollte der Patient nicht ausreichend trinken um das Volumendefizit auszugleichen
Falls Trinkwasser vorhanden
Polydipsie
Wasser verlässt Zellen entlang des osmotischen Gradienten, Neuronen schrumpfen
Neuronaler Schaden: Delirium, Krampfanfall, Benommenheit, Koma
Renale Perfusion↓, GFR ↓
Nierenversagen
(prä-renal, siehe entsprechende Folie)
Beachte: Während der DKA verliert der Körper K+ durch osmotische Diurese. Da gleichzeitig allerdings K+ aus den Zellen diffundiert, kann das Serumkalium unauffällig/erhöht sein. Um Hypokaliämien zu verhindern sollte bei K+ <5.0mmol/L KCl zusammen mit Insulin i.v. verabreicht werden.
Cave: gute Nierenfunktion des Patienten
Beachte: Elektrolytstörungen (z.B. Hyperkaliämien, Hypernatriämien) verschlechtern sich bei akutem Nierenversagen, welches bei der DKA &HK häufig auftritt
Legende:
Pathophysiologie
Mechanismen
Symptome/Klinische Befunde
Komplikationen
Veröffentlicht: 3. November 2016 auf www.thecalgaryguide.com")
Hyperosmolares/ Hyperglykämisches Koma (HHS)

Betrifft hauptsächlich Patienten mit Diabetes Mellitus Typ 1: Bestimmte Situationen (z.B. Infektionen) erhöhen den Insulinbedarf, welcher durch fehlende Produktion/unzureichende Substitution nicht gedeckt werden kann
Autor: Yan Yu Rezensenten: Peter Vetere, Gill Goobie, Sean Spence, Hanan Bassyouni* Übersetzung: Sarah Schwarz Übersetzungsprüfung: Dr. Gesche Tallen * MD zum Zeitpunkt der Veröffentlichung
Hyperglykämie
(erhöhter Blutzucker)
Bei >12mmol/L, Glukosefiltration > - resorption, Glukose verbleibt im Urin
Glukosurie
Glukose ist osmotisch wirksam und zieht große Mengen Wasser mit sich (Osmotische Diurese)
Polyurie
Schwere Dehydrierung(bis zu 4-5L)
(ZVD↓, Orthostase: posturale Hypotension/ posturale Tachykardie, Ruhepuls ↑ )
Insulinmangel enthemmt die Lipolyse, sodass der Körper Energie aus Triglyceriden produziert
Freisetzung von freien Fettsäuren aus Fettgewebe
Absoluter
Insulinmangel
Hypothalamische Zellen induzieren bei geringem intrazellulären Glukosespiegel ein starkes Hungergefühl
Glukose bleibt im Blut & kann nicht durch Muskel- /Fettgewebe verstoffwechselt werden
“Ausgehungerte” Zellen triggern die Freisetzung kataboler Hormone: Glucagon, Katecholamine, Cortisol, Somatotropin
Damit intrazellulärer Glukosespiegel ↑, erhöht der Körper den Blutzucker
Hydrolyse von freien Fettsäure in
der Leber (Ketogenese) Polyphagie
↓ Proteinsynthese, ↑ Proteolyse (in Muskelgewebe)
↑ Gluconeogenese, ↑ Glykogenolyse (in der Leber)
Acetyl Co-A
Energiequelle für “ausgehungerte” Zellen
Beachte: Neben Glukose sind Ketonkörper die einzigen Energieträger die von Nervenzellen metabolisiert werden können. Deshalb werden sie vom Körper bei geringem Glukoseangebot produziert.
Ketonkörper
( β-Hydroxybutyrat, Acetoacetat, Aceton, akkumulieren im Blut)
Ketonurie
Metabolische Azidose
(↑Anionenlücke: Ketonkörper verbrauchen den HCO3--Puffer)
Substrate der Gluconeogenese↑
Durch ↓ Extrazellulärflüssigkeit, werden Ketonkörper konzentriert → Azidose
Stört das enterische Nervensystem, Magenentleerung ↓, Ileus
Bauchschmerzen, Übelkeit, Erbrechen
(Dehydration↑)
Abatmen von CO2 um Azidose auszugleichen
Kussmaul- Atmung (Tiefe, schnelle Züge)
Abatmen von Keton- körpern
Azeton- geruch der Atemluft
Beeinträchtigung elektrischer Signale zum Gehirn, Rücken- mark und Nervenzellen
Perfusion des Gehirns, Rückenmark, Nervenzellen↓
Verschiebung des Wasser- und Elektrolythaushalts
Falls Trinkwasser vorhanden
Polydipsie Beachte: Während der DKA verliert der Körper K+ durch
Elektrolytstörung
Schwäche, Delirium +/- Koma
osmotische Diurese und Erbrechen. Da gleichzeitig allerdings K+ aus den Zellen diffundiert, kann das Serumkalium unauffällig/erhöht sein. Um Hypokaliämien zu verhindern sollte bei K+ <5.0mmol/L KCl zusammen mit Insulin i.v. verabreicht werden. Cave: gute Nierenfunktion des Patienten
Therapie: 1) +++ Flüssigkeit. 2) Insulin + KCl. 3) Auffüllen der Anionenlücke. 4) Auslösende Faktoren identifizieren. 5) Wenig PO4 (typischerweise wenige Stunden bis einen Tag nach der Ketoazidose durch ↑ ATP-Produktion.)
Legende:
Pathophysiologie
Mechanismen
Symptome/Klinische Befunde
Komplikationen
Publikationsdatum: 17 Juni 2013 auf www.thecalgaryguide.com
Hyperosmolares/ Hyperglykämisches Koma (HHS)
Beachte: betrifft nur Patienten mit Diabetes mellitus Typ II
Beachte: Es sollte stets nach auslösenden Faktoren z.B. einer Infektion gesucht werden
Autor: Yan Yu Rezensenten:
Peter Vetere
Gill Goobie
Hanan Bassyouni* Übersetzung:
Sarah Schwarz Übersetzungsprüfung: Dr. Gesche Tallen
* MD zum Zeitpunkt der Veröffentlichung
Verschiebung des Wasser- und Elektrolythaushalts
Elektrolytstörung
Unzureichende Insulin- produktion, Insulinresistenz, Nichtansprechen auf Insulintherapie
Relativer Insulinmangel
Situationen mit ↑Insulinbedarf: Infekt, Pneumonie, Herzinfarkt, Pancreatitis, etc.)
Hyperglykämie
(Stark erhöhter Blutzucker, höher als bei der DKA)
Bei >12mmol/L, Glukosefiltration > - resorption, Glukose verbleibt im Urin
Glukosurie
Glukose ist osmotisch wirksam und zieht große Mengen Wasser mit sich (Osmotische Diurese)
Polyurie Dehydration
(ZVD↓, Orthostase: posturale Hypotension/posturale Tachykardie, Ruhepuls ↑ )
Durch das wenige noch vorhandene Insulin, wird ein Teil der Glukose von Muskel- /Fettgewebe verstoffwechselt, ein Teil verbleibt im Blut
Körperzellen brauchen eine weitere Energiequelle
Freisetzung kataboler Hormone: Glucagon, Katecholamine, Cortisol, Somatrotropin
Damit intrazellulärer Glukosespiegel ↑, erhöht der Körper den Blutzucker
Hypothalamische Zellen induzieren bei geringem intrazellulären Glukosespiegel ein starkes Hungergefühl
Polyphagie
Beachte: Durch das wenige noch
vorhandene Insulin wird die Lipolyse gehemmt und werden keine Ketonkörper produziert. Es kommt nicht zur Azidose und Ketonurie (Gegensatz zur DKA). Sollte es doch zur Ketourie kommen, ist das meist Folge von Hungerzuständen oder anderen Mechanismen
Extrazellulärflüssigkeit ↓ mit erhöhter Osmolarität (z.B. Hypernatriämie)
↑ Gluconeogenese, ↑ Glykogenolyse (in der Leber)
↓ Proteinsynthese, ↑ Proteolyse (in Muskelgewebe)
Substrate der Gluconeogenese ↑
Sollte der Patient nicht ausreichend trinken um das Volumendefizit auszugleichen
Falls Trinkwasser vorhanden
Polydipsie
Wasser verlässt Zellen entlang des osmotischen Gradienten, Neuronen schrumpfen
Neuronaler Schaden: Delirium, Krampfanfall, Benommenheit, Koma
Renale Perfusion↓, GFR ↓
Nierenversagen
(prä-renal, siehe entsprechende Folie)
Beachte: Während der DKA verliert der Körper K+ durch osmotische Diurese. Da gleichzeitig allerdings K+ aus den Zellen diffundiert, kann das Serumkalium unauffällig/erhöht sein. Um Hypokaliämien zu verhindern sollte bei K+ <5.0mmol/L KCl zusammen mit Insulin i.v. verabreicht werden.
Cave: gute Nierenfunktion des Patienten
Beachte: Elektrolytstörungen (z.B. Hyperkaliämien, Hypernatriämien) verschlechtern sich bei akutem Nierenversagen, welches bei der DKA &HK häufig auftritt
Legende:
Pathophysiologie
Mechanismen
Symptome/Klinische Befunde
Komplikationen
Veröffentlicht: 3. November 2016 auf www.thecalgaryguide.com")
Nephrotisches Syndrom: Pathogenese und klinische Befunde

Lupus-Nephritis
Postinfektiöse Glomeruloneprhitis
IgA Nephropathie
Membranöse Glomerulonephritis
Antikörper greifen Podozyten an, Verdickung der glomerulären Basalmembran
Fokal-segmentale Glomerulosklerose (FSSGN)
Schädigung des glomerulären Endo- und Epithels
Minimal Change Glomerulo- nephritis (MCNG)
Diabetes mellitus
Autor: Yan Yu Rezensenten: Alexander Arnold David Waldner
Sean Spence
Stefan Mustata* Übersetzung:
Sarah Schwarz Übersetzungsprüfung: Gesche Tallen*
* MD zum Zeitpunkt der Veröffentlichung
Podozyten-Schädigung auf der epithelialen Seite des Glomerulums (Abflachung der Podozytenfortsätze)
Glomeruläre Immunkomplex- ablagerungen
Chronische Hyperglykämien schädigen das Glomerulum
Geschädigter Proteinfilter (v.a. für geladene Proteine)
Ablagerungen von Immunglobulin-Leichtketten im Glomerulum
Amyloidose
Geschädigte Glomeruli --> gestörte Filterbarriere v.a. für Proteine --> übermäßige Filtration von Plasmaproteinen
Vermehrte renale Ausscheidung von Immunglobulin-Leichtketten (Filtration>Resorption)
Hypoalbuminämie*
Übermäßiger Verlust an Albumin über den Urin
Proteinurie >3.5g/Tag*
Verlust an Antikoagulationsproteinen (Antithrombin, Plasminogen, Protein C & S) über den Urin
Koagulations- /Gerinnungsfaktoren (z.B. 1,7,8, 10) sind in Überzahl
Proteinurie >3.5g/Tag*
Thrombophilie
Anasarka
(Generalisiertes Ödem)
Lidödem
(klassisches Frühzeichen)
Lipidurie
(zeigt unter
gekreuztem polarisiertem Licht eine Malteserkreuz- form)
50% des Serumkalziums sind an Albumin gebunden, sodass Serumkalzium- spiegel ↓
Serum- Ca2+ repräsentiert nicht mehr das Gesamt-Ca2+
Beachte:
• DerklassischeSymptomkomplex(*)desnephrotischen Syndroms besteht aus: Proteinurie (>3,5g/Tag), Ödemen, Hypoalbuminämie,Hyperlipidämie
• Fürjede10g/LmitAlbumin<40:
➔ Addiere 2.5 zur errechneten Anionenlücke um
dessen “wahren” Wert zu bekommen
➔ Addiere 0,2mmol/L zum Gesamt-Ca um den
Wert des ionisierten Kalziums zu errechnen
Blut neigt zur Bildung von Thromben
Ödeme*
↑ Renale Filtrationder Lipide und Ausscheidung über den Urin
Die Anionenlücke ergibt sich hauptsächlich aus negativ geladenem Serumalbumin
Anionenlücke↓
Kolloid- osmotischer Druck ↓
Flüssigkeit kann nicht mehr in den Blutgefäßen gehalten werden und diffundiert ins Gewebe
Signalisiert der Leber die Albuminproduktion zuerhöhen,Albumin wird aber weiterhin über die Nieren verloren
Synthese- arbeit der Leber↑, auch ↑ Lipoprotein- synthese
Hyperlipidämie*:
(Serum-LDL, -VLDL, - TG ↑ )
Legende:
Pathophysiologie
Mechanismen
Symptome/Klinische Befunde
Komplikationen
Veröffentlicht: 19. August 2013 auf www.thecalgaryguide.com")
impetigo-patogenesis-y-hallazgos-clinicos

Psuedocholinesterase Deficiency

Break down or inhibit the pseudocholinesterase enzymeà↓ pseudo- cholinesterase activity
Note:
Pseudocholinesterase has many synonymous
names including butyrylcholinesterase, BChE, BuChE, plasma esterase, plasma cholinesterase, and serum cholinesterase
Malignancy Abnormal gene
expressionà↓ protein synthesis
Acquired
Liver disease
↓ liver’s ability to synthesize proteins
Malnutrition
↓ molecular precursors for protein production
Renal Disease
Mechanism unclear
Fluid Overload
Hemodilution of circulating proteins
Hereditary
BChE (Butyrylcholinesterase) gene mutation
↓ production or production of non-functional pseudo- cholinesterase by the liver
↓ synthesis of pseudocholinesterase by the liver
Pseudocholinesterase Deficiency: reduced levels of functional pseudocholinesterase in plasma and tissues
Impaired ability to hydrolyze ester linkages of substrates like neuromuscular blocking agents (e.g. succinylcholine, mivacurium, diamorphine, acetylsalicylic acid, methylprednisolone, cocaine, heroin )
Prolonged
binding of neuromuscular blocking agent to nicotinic cholinergic receptors in neuromuscular junctions
↑ patient’s susceptibility to side effects from drugs with ester linkages
Mivacurium administered to produce muscle paralysis
Succinylcholine administered
to produce muscle paralysis
Competitively binds to acetylcholine nicotinic receptor
Irreversibly binds to acetylcholine nicotinic receptor
Active site of post-synaptic acetylcholine receptor is blocked
Continuous depolarization
of skeletal muscle
Acetylcholine cannot act on receptor = skeletal muscle can’t depolarize
Skeletal muscle unable to repolarize
Skeletal muscle paralysis
Acetylcholine released cannot trigger an action potential
↑ duration of muscle fiber paralysis
Since respiratory muscles are affectedà Apnea requiring sedation and respiratory assistance for up to several hours
Extended paralysis and/or anesthesia
Authors: Evan Allarie Yan Yu* Reviewers: Stephen Chrusch Brooke Fallis Melinda Davis* * MD at time of publication
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published December 4, 2021 on www.thecalgaryguide.com")
Primary Aldosteronism Pathogenesis
à↑ aldosterone
Overproduction of aldosterone, independent from renin-angiotensin- aldosterone system (RAAS)
Ectopic aldosterone secreting tumor
adrenocortical tissue outside of the adrenal glands produce aldosterone
Familial hyperaldosteronism (FH) Type I
Rare mutation causing ACTH- sensitive aldosterone production in the zona fasciculata
Aldosterone binds its receptor on the principal cells located in the collecting duct of the kidney
↑ Na+ and K+ channel insertion on luminal surface of the principal cell and ↑ Na/K ATPase activity on basolateral surface
Na+ follows the concentration gradient and moves into the principal cell cytoplasmàK+ moves into the collecting duct to maintain electroneutrality
Aldosterone binds its receptor on the alpha intercalated cells located in the late distal tubule and collecting duct of the kidney
↑ H+ ATPase and Na/H+ ATPase activity on the luminal surface of alpha intercalated cellsà↑ H+ excretion
H+ loss permits HCO3- to move down the electrochemical gradient across the luminal surfaceà↑ HCO3- resorption into peritubular capillaries
Metabolic alkalosis
K+ efflux (or hypokalemia of any etiology) is counterbalanced by influx of H+ into tubular cells to maintain electroneutrality
↓ pH in tubular cells activates glutaminase (a pH dependent enzyme) generating glutamate from glutamine
Glutamate within renal tubules dissociates into NH4+, HCO3- Intracellular acidosis within tubular cells
Disrupted cellular signaling within the collecting duct results in reduced APQ2 translocation to luminal surface
Authors: Kyle Moxham Reviewers: Emily Wildman Austin Laing Yan Yu* Matthew Harding* * MD at time of publication
Hypertension
↑ Na+ reabsorption
H2O follows active reabsorption
of solute into circulationà ↑ effective arterial blood volume (EABV)
Chronic EABV expansion resets hypothalamic osmotic sensitive ADH releaseàdecreased ADH production
↑ K+ excretion
Hypokalemia (see our Hypokalemia: Clinical Findings Slide)
↓ Na+ delivery to macula densa in the distal convoluted tubule
↓Endogenous renin secretion by juxtaglomerular apparatus while aldosterone continues to be independently produced
Polydipsia & Polyuria
Mild hypernatremia
↓ Renin: Aldosterone ratio
Nephrogenic diabetes insipidus: ↓ urinary concentrating ability
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published December 4, 2021 on www.thecalgaryguide.com")
Normal anion gap metabolic acidosis
, the distal nephron secretes one corresponding urine Cl- anion
Urine NH4+ cannot be measured, so measured
urine Cl- exceeds measured urine (Na+ + K+)
Negative urine net charge (Urine (Na+ + K+) - Urine Cl-)
Type II RTA RTA: renal tubular acidosis ↓ activity of proximal
Type I RTA
↓ activity of H+ ATPase on luminal surface of ⍺- intercalated cell in the collecting duct
Impaired ⍺-intercalated cell function
↓ H+ secretion into urine by ⍺- intercalated cell
↑ H+ concentration in plasmaà↓ in blood pH
Activation of blood bicarbonate buffer system
Compensatory ↓ in plasma HCO3- (in response to ↑ in plasma H+)
Type IV RTA
↓ aldosterone production or aldosterone resistant state
↓ Na+ channels on luminal surface of principal cell in the collecting duct
Impaired principal cell function
↓ Na+ reabsorption by principal cell
↑ Na+ remains in collecting duct lumen
↓ negative charge in collecting duct lumen
↓ K+ secretion into urine by principal cell
↑ K+ in plasma
Hyperkalemia
Normal anion gap
tubule transporters, pumps or enzymes
Impaired proximal tubule cell function
reabsorption
Complex and Incompletely understood mechanisms
Hypokalemia
Distal nephron function is impairedàdistal nephron is unable to secrete excess acid into the urine in the form of NH4+
↓ in urinary NH + 4
secretion is accompanied by corresponding ↓ in urinary Cl- secretion
Measured urine (Na+ + K+) exceeds measured urine Cl-
Positive urine net charge (Urine (Na+ + K+) - Urine Cl-)
↓ HCO3
-
in proximal tubule
↑ HCO3- loss through urine
↓ HCO3- concentration in plasma
Relative ↑ in plasma H+ compared to plasma HCO3-
Compensatory ↑ in CO2 exhalation by lungs (since CO2 is acidic once metabolized in the blood)
Since Anion gap = Na+ - Cl- - HCO3-,
1 to 1 replacement of Cl- for HCO - in the 3
plasma results in anion gap unchanged
↓ ability to buffer excess acid
Activation of the membrane Cl-/HCO3- exchanger in the collecting duct
Exchanger works to ↑ Cl- levels in the plasma by
one Cl- for each ↓ of one HCO3- in the plasma
Metabolic Acidosis
↓ Plasma partial pressure of CO2
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published December 21, 2021 on www.thecalgaryguide.com")
Renal Artery Stenosis
 of the renal artery
Renal Artery Stenosis can be unilateral or bilateral
Authors: Samin Dolatabadi, Yan Yu* Reviewers: Meena Assad, Jessica Krahn Timothy Fu, Brooke Fallis, Juliya Hemmett* * MD at time of publication
↓ Pressure perfusing the kidney
↑ RAAS (renin-angiotensin- aldosterone system) activation
↓ pressure gradient in glomerulus
↓ Glomerular filtration rate (GFR)
↑ Secretion of aldosterone
Turbulent blood flow through area of stenosis
Abdominal bruit on side of affected kidney(s)
↑ Secretion of Angiotensin IIà ↑ Systemic vasoconstriction
Hypertension
↑ Expression of epithelial sodium channels in cortical collecting duct
↑ Blood volume within volume- constrained space of blood vessels
↓ Renal blood flowà ischemic renal injury
Atrophy and fibrosis of affected kidney(s)
Unilateral stenosis à Kidney size asymmetry (≥1.5cm difference)
↓ Positively charged Na+ in lumen à Electronegative lumen compared to the interstitial/tubular epithelial cells
K+ follows the electrical gradient and is secreted into the electronegative tubular lumen
↓ Serum K+ concentration
Hypokalemia
*Note: In unilateral renal artery stenosis, the contralateral (normal) kidney can compensate for the increase in renal perfusion pressure caused by hypertension by increasing sodium excretion (pressure natriuresis), preventing flash pulmonary edema.
↑ NHE3 (Sodium Hydrogen Exchanger 3) activity in proximal collecting tubule
↑ Na+ and water reabsorption from renal tubular lumen into blood vessels
Chronic left ventricle pressure overload àLeft ventricle hypertrophy (see Left Heart Failure: Pathogenesis Slide)
Systolic dysfunction → ↓ left ventricle stroke volume
Normally, ↑ in renal perfusion pressureà↑ Na+ excretion and water loss (pressure natriuresis)
In bilateral renal artery stenosis*, ↓ GFRàinability for kidney to ↑ renal Na+ & water excretion à volume overload
Acute increase in afterload or preload → sudden ↑ in left ventricular filling pressures → blood backup into lungs
Pulmonary vasculature hypertension → ↑ fluid filtration across the pulmonary endothelium into interstitium and alveolar spaces
Flash (rapid onset) pulmonary edema
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published December 30, 2021 on www.thecalgaryguide.com")
Membranous Nephropathy
 Membranous Nephropathy
Secondary Membranous Nephropathy
Drugs, infections, auto-immune diseases, and malignancies make antigens
Blood tests for Anti- PLA2R antibodies
are positive in ~70% of cases
Autoantibodies are made to antigens expressed near or on podocyte surfaces or planted in the glomerular capillary wall (ie. PLA2R; THSD7A; NELL-1)
IgG antibodies filter through the glomerulus
Autoimmune: (e.g. Systemic Lupus Erythematous, thyroiditis)
Drugs: (e.g. NSAIDs, gold, penicillamine, captopril)
Malignancies: (ie. Prostate, Colon)
Infections: (e.g. Hepatitis B, Syphilis)
Circulating antigens filter through the glomerulus and between the GBM and podocytes
Antibodies & antigens form immunocomplexes, which lodge Immunofluorescence shows diffuse “granular”
between the glomerular basement membrane & podocytes
deposits of immunocomplexes throughout GBM
Basement membrane is formed between and around immunocomplex deposits
Immunocomplexes activate complement and the assembly of the Membrane attack complex (MAC)
MAC creates holes in podocyte plasma membranes
Stimulates the release/activation of proteases and oxidases from glomerular podocytes and mesangial cells
GBM appears thick on light microscopy
“Spike and dome” pattern appears with silver stain
Podocyte effacement
Damage to negatively charged foot processes damages the charge barrier of the glomerulus that repels negatively charged molecules
Increased filtration of large negatively charged molecules, especially albumin
Induces ↑ hepatic lipoprotein synthesis and ↓ lipoprotein catabolism
Hyperlipidemia
(↑ serum LDL, VLDL, and triglycerides)
↑ lipid filtration through glomerulus
Lipiduria (fatty casts)
*Hypo-albuminemia
↓ oncotic pressure in capillaries
Fluid leaks into interstitial space
↑ Filtration of Proteins C and S and antithrombin
Hypercoagulable state
Thrombosis *Edema (especially
*Proteinuria
↑ Filtration of immunoglobulins
Immunosuppression
Infections
↑ Filtration of Plasminogen
Plasminogen converted to plasmin in the cortical collecting duct via urokinase- type plasminogen activator
Plasmin activates the epithelial sodium channel
↓ intravascular volume
Hypotension
Pre-Renal Acute Kidney Injury
Underfill edema
(see slide)
peri-orbital, scrotal, labial, and extremities)
Overfill
edema
(see slide)
↑ Na+ and water reabsorption
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published MONTH, DAY, YEAR on www.thecalgaryguide.com")
Overview of Calcium Phosphate Vitamin D Physiology
 and ↓ expression of osteoprotegerin (OPG), a decoy receptor for RANKL
RANKL binds to receptor activator of nuclear factor kappa-B (RANK) on osteoclasts
↑ Osteoclast differentiation and activity
↑ Ca2+ resorption from bone into blood
Sensed by Calcium-Sensing Receptor on Chief Cells of the Parathyroid Gland
− −
↑ Serum phosphate (PO4) ↓ Serum calcium (Ca2+)
Parathyroid Gland releases Parathyroid Hormone (PTH) into the blood, which acts on the kidneys and the bones
Kidney
↑ Activation of Transient Receptor Potential Vanilloid subfamily member 5 (Ca2+ channel)
in the distal convoluted tubule
↓ Expression of 24-hydroxylase enzyme (which functions to catabolize calcitriol)
↑ Expression of 1α- hydroxylase enzyme
↑ Conversion of 25-hydroxy vitamin D (Calcidiol) to the
active form, 1,25-dihydroxy vitamin D (Calcitriol)
↑ Calcitriol Small Intestine
↑ Endocytosis of sodium phosphate co-transporters NaPi-2a and NaPi-2c, which reabsorb PO4 from ultrafiltrate in the renal tubules
↓ Reabsorption of PO 4
↓ Serum PO
Calcitriol negatively feeds back on parathyroid gland to inhibit PTH production
↑ Expression of sodium phosphate co- transporters NaPi-2a and NaPi-2b that absorb PO4 from intestinal lumen
4
↑ Expression of apical epithelial Ca2+ channels (Transient Receptor Potential Vanilloid subfamily member 6)
↑ Entry of Ca2+ on apical side of enterocytes
↑ Reabsorption of Ca2+ in the distal convoluted tubule through Ca2+ channels
↑ Reabsorption of PO
↑ Expression of cytoplasmic Calbindin-D
↑ Transport of Ca2+ across enterocytes
↑ Absorption of Ca2+ in small intestine
↑ Serum Ca+2
↑ Expression of basolateral plasma membrane calcium ATPase
↑ Extrusion of Ca2+ from enterocytes into bloodstream
4
Authors: Samin Dolatabadi, Hannah Yaphe Reviewers: Amanda Henderson, Meena Assad, Brooke Fallis, Yan Yu*, Hanan Bassyouni* * MD at time of publication
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
First published Oct 15, 2017, updated Nov 11, 2021 on www.thecalgaryguide.com")
Minimal Change Disease
 present in serum
Immunofluorescence test negative
Idiopathic/Primary Minimal Change Disease
No identifiable extraglomerular disease process causes this condition
Secondary Minimal Change Disease
Infections, NSAIDS, neoplasms via unclear mechanisms
Minimal change to glomerulus seen on light microscopy
Podocyte effacement seen on electron microscopy
Abnormal T Cell activation and release of cytokines (sometimes called permeability factors) that are toxic to podocytes
Podocyte foot processes efface (become flattened) or fuse together
Damage to negatively charged foot processes damages the charge barrier of the glomerulus that repels negatively charged molecules
↑ filtration of larger negatively charged molecules, such as low-molecular weight proteins like albumin, from the blood into the renal tubular filtrate
Induces ↑ hepatic lipoprotein synthesis and ↓ lipoprotein catabolism
Hyperlipidemia
(↑ serum LDL, VLDL, and triglycerides)
↑ lipid filtration through glomerulus
Lipiduria (fatty casts)
Hypo-albuminemia
↓ oncotic pressure in capillaries
Fluid leaks into interstitial space
↑ Filtration of Proteins C and S and antithrombin
Hypercoagulable state
Proteinuria
↑ Filtration of immunoglobulins
Immunosuppression
Infections
↑ Filtration of Plasminogen
Plasminogen converted to plasmin in the cortical collecting duct via urokinase- type plasminogen activator
Plasmin activates the epithelial sodium channel
↓ intravascular volume
Hypotension
Pre-Renal Acute Kidney Injury
Underfill edema
(see slide)
Thrombosis Edema (especially peri-
orbital, scrotal, labial, and extremities)
Overfill
edema
(see slide)
↑ Na+ and water reabsorption
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published December 30, 2021 on www.thecalgaryguide.com")
ACE inhibitors
 inhibitors: Mechanism of Action and Clinical Findings Angiotensin Converting Enzyme (ACE) Inhibitors
Inhibits the conversion of angiotensin I to angiotensin IIà↓ serum angiotensin II levels
Inhibits breakdown of bradykinin
↓ Activation of zona glomerulosa region of the adrenal cortex
↓ Aldosterone secretion from the adrenal cortex
↓ Expression of basolateral Na+/K+ pump
and ↓ insertion of luminal Na+ channels in principal cells of the cortical collecting duct in kidney
↓ K+ secretion into urine
↑ Serum K+ concentration
Hyperkalemia
↓ Activation of Na+/H+ exchanger in proximal convoluted tubule (PCT) of the kidney
↓ Na+ absorption into PCT and ↓ H+ excretion into lumen
↓ Na+ absorption into the blood
↓ Renal H2O reabsorption via osmosis
↓ Activation of paraventricular nuclei located in the hypothalamus
↓ Secretion of anti- diuretic hormone (ADH)
into the capillaries of the posterior pituitary gland
↓ Aquaporin channel insertion at cortical collecting duct in kidney
↓ Renal H2O reabsorption
à↑ serum bradykinin levels Vasodilation of efferent
arteriole in kidneys
↓ Hydrostatic pressure within the glomerular capillaries
↑ Activation of cyclooxygenase-2 pathway
↑ Prostaglandin production
↓ Activation of angiotensin II type 1 receptors on surface of arteriolar smooth muscle
↓ Activity of G protein-coupled receptor intracellular secondary messenger cascade
↓ Systemic arteriolar vasoconstriction
↓ Total peripheral resistance (resistance
to blood flow by systemic vasculature)
↓ Blood Pressure
↓ total blood flow perfusing kidneys
Renal hypoxia
↑ Systemic arteriolar vasodilation
↓ Total peripheral resistance
↓ Blood Pressure
↓ Concentration of protein
travelling through tubules for excretion
↓ Fraction of H2O filtered from glomerular capillaries into bowman’s capsule (start of the renal tubule)
↓ Filtration of blood contents through glomerular membranes
Sensitization of airway vagal afferent receptors
Stimulation of the cough
center located in the medulla oblongata
Cough
↓ Protein in urine
High K+ levels surrounding the heart muscle cellsàchronic depolarization
of the cardiomyocyte cell membrane àalters cardiomyocyte conduction (complex mechanisms)
Cardiac Arrythmias (see relevant slide on Hyperkalemia: Clinical Findings)
↓ Effective arterial blood volume
Authors:
Nicole Brockman Reviewers:
Emily Wildman, Austin Laing Adam Bass*, Yan Yu*
* MD at time of publication
Renal tubules have relatively less water compared with the peritubular capillaries
↓ H2O reabsorption from the renal tubule back into the blood
↑ Serum creatinine, ↓ estimated glomerular filtration rate
Renal nephron injury and dysfunction
Acute Kidney Injury
(rise in creatinine >26.5umol/L in <7 days)
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published January 10, 2022 on www.thecalgaryguide.com")
complications-of-chronic-kidney-disease-ckd

Authors: Samin Dolatabadi, Brooke Fallis Reviewers: Jessica Krahn, Meena Assad, Yan Yu* Juliya Hemmett* * MD at time of publication
Abnormalities of kidney structure or function that is present for 3 or more months
Kidney tubules atrophy & kidney interstitial tissue undergo fibrosis
Kidney damage ↓ erythropoietin production (normally a kidney function)
↓stimulation of bone marrow → ↓ red blood cell production
Build up of toxic substances (e.g. urea, guanidine, and indoxyl sulfate)
↓ Conversion of calcidiol to calcitriol in by kidney
↓ Calcitriol levels in blood
↓ Ca+2 absorption from small intestine
Hypocalcemia
↓ Glomerular Filtration Rate
↑ Vascular calcification and endothelial dysfunction (e.g. changes in permeability, clotting response to inflammation, amongst other mechanisms)
Atherosclerotic disease (see “Complications of Atherosclerosis” slide)
↓ Lipoprotein lipase, apoA-1, and Lecithin-cholesterol acyltransferase
activity acting on serum lipoproteins (complex mechanisms)
↓ Lipoprotein clearance from blood
Dyslipidemia (↑ LDL, ↑ triglycerides, ↓ HDL)
Toxic damage ↓ red blood cell survival
Anemia
Uremic Syndrome
↓ Renal excretion of phosphate
↑ phosphate remaining in bloodstream
Hyperphosphatemia
↑ serum phosphate binds with ionized calcium
↓ Renal excretion of K+
↑ K+ remaining in bloodstream
Hyperkalemia Edema
↓ Renal excretion of ammonium
Reduced filtration capability ↑ organic anions remaining in blood→ ↑ anion gap
↓ Renal excretion of salt and water
↑ in extracellular fluid → systemic volume overload
↓ calcium in blood ↓ inhibition of Parathyroid Hormone (PTH) release
↑ PTH in blood
Secondary hyperparathyroidism
Metabolic Acidosis
Hypertension
Pitting
Chronic kidney disease – mineral and bone disorder (CKD-MBD)
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published Jan 4, 2022 on www.thecalgaryguide.com")
NSAIDs and the Kidney Nephrotoxicity

Authors: Kyle Moxham Mehul Gupta Reviewers: Emily Wildman Yan Yu* Adam Bass* * MD at time of publication
Inhibition of Cyclooxygenase COX-1 (expressed in kidney) and COX-2 (expressed in kidney and sites of inflammation)
NSAID induced nephrotoxicity: associated with chronic NSAID usage independent of dosage
COX inhibition ↑ conversion of arachidonic acid (AA) to leukotrienes, causing systemic T-cell dysfunction (unknown mechanism)
Type IV systemic hypersensitivity (delayed
T helper cell mediated) reaction to drug exposure
T cells release inflammatory cytokines into the bloodstream
(see Calgary Guide slide on NSAIDs and the Kidney: Mechanism of Action and Side Effects)
T cells infiltrate the renal interstitium, sparing the glomeruli and blood vessels
Overproduction of cytokines by T cells causing inflammation,
tissue damage, and cell death, of the renal intersitium
Drug Induced Acute interstitial nephritis (AIN): a type of immune-mediated tubulointerstitial injury
Activated T-cells infiltrate the glomerulus and cause podocyte injury (epithelial cells attached to the glomerular basement membrane)
Membranous nephropathy:
nephrotic syndrome involving autoimmune glomerular basement membrane thickening & complete podocyte effacement (seen on kidney biopsy)
Minimal change disease: nephrotic syndrome caused by autoimmune podocyte effacement (seen on kidney biopsy)
Cytokine mediated activation and
proliferation of immune cells like macrophages and eosinophils
Cytokines travel to hypothalamus,
causing change in the body’s thermal set point
Fever
Podocyte effacement allows for serum proteins to across the glomerulus into the tubular lumen (see Calgary Guide slide on Nephrotic Syndrome for full mechanisms)
Repeated NSAID exposure causing
recurrent unrecognized AIN and damage of the kidney
Chronic interstitial nephritis /analgesic nephropathy
Infiltrating immune cells (predominantly neutrophils) are filtered into/enter the renal tubules, and form clumps (“casts”) within the tubulesà casts are then released into urine
WBC cast on urinalysis
↑ Blood eosinophils
Immune cells infiltrate the
dermis and epidermis of the skin
Rash
Abnormal quantities of protein present in urine
Protein present in the blood is improperly filtered into the filtrate at the glomerulus
Protein- Creatinine
Ratio >3.5mg/mg
Proteinuria >3.5g/d
Hypo- albuminemia
↓ plasma oncotic pressure resulting in fluid extravasation into the interstitium (see Calgary guide slide on Edema for full mechanisms)
Pitting Edema
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published January 13, 2022 on www.thecalgaryguide.com")
Overfill Edema Pathogenesis

Aberrant filtration of proteins including plasminogen
(glycoprotein in the systemic circulation involved in the dissolution of fibrin blood clots)
↑ Plasminogen concentration in tubular fluid
Conversion of plasminogenà plasmin by urokinase-type plasminogen activator in the cortical collecting tubule
Plasmin activates epithelium
sodium channels (involved in Na+ reabsorption) in principal cells of the cortical collecting duct
Nonsteroidal Anti-inflammatory Drugs
Inhibition of cyclooxygenase throughout body (enzyme that converts arachidonic acidàprostaglandins)
↓ Prostaglandins throughout body
Thiazolidines
↓ Prostaglandin- mediated inhibition of Na+ and Cl- transport in ascending loop of Henle and collecting ducts
↑ Na+ reabsorption from kidney tubules back into blood
↓ Prostaglandin-mediated vasodilation in kidneys
↓ Pressure of blood perfusing kidneys → ↓ pressure gradient between glomerulus and bowman’s capsule
↓ Glomerular filtration rate
↓ Renal excretion of salt & water
↑ Salt and water retention
↑ Effective arterial blood volume
↑ blood pressure in veins
Unclear mechanism but possible theories include upregulation of epithelium sodium channels in the cortical collecting tubule and involvement of other transporters in the proximal tubule and cortical collecting tubule
Acute Renal Failure Chronic Renal Failure
↑ Hydrostatic pressure within capillariesàexceeds hydrostatic pressure within interstitial spaceàfluid moves from capillaries into interstitial space
Overfill Edema
Abnormal accumulation of fluid in the interstitial space where urine Na+ >40meq/L
Authors: Samin Dolatabadi Reviewers: Meena Assad, Jessica Krahn, Brooke Fallis, Ran (Marissa) Zhang, Yan Yu*, Juliya Hemmett* * MD at time of publication
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published January 23, 2022 on www.thecalgaryguide.com")
NSAIDs and the Kidney mechanism of action and side effects
 or angiotensin II receptor blockers (ARB)
AECi and ARBs act to ↓ renin- angiotensin-aldosterone system (RAAS) activation
Reduced angiotensin (AT) 2 activity at its receptors on efferent arteriole of the nephron
Net vasodilation of efferent arterioles ↓ in glomerular pressure
↓ blood perfusing kidney tissueà↑ hypoxemia & renal ischemia
Pre-existing ↓effective arterial blood volume (EABV) from
dehydration, GI loss, diuretics, CKD, CHF, cirrhosis, etc.
Decreased EABV triggers endogenous renal autoregulation, resulting in norepinephrine (NE) mediated vasoconstriction of afferent arteriole of the nephron
Net ↓ in renal blood flow and ↓ in glomerular pressure
↓ volume of blood filtered by the glomeruli per unit time
Non-steroidal anti-inflammatory drugs (NSAIDs)
Inhibition of Cyclooxygenase COX-1 (expressed in kidney) and COX-2 (expressed in kidney and sites of inflammation)
↓ Renal prostaglandin (PG) synthesis: local hormones involved in renal homeostasis
NSAID induced nephrotoxicity:
associated with chronic usage independent of dosage
(see Calgary Guide slide on NSAIDs and the Kidney: NSAID induced nephrotoxicity)
↓ intrarenal PG reduces inhibitory effects of PG over ADH in cortical colleting duct (CCD) of the kidneyà↓ antagonism on ADH activity
↑ ADH activity causes insertion of more
aquaporins (water channels) in the collecting duct of renal tubules
Net ↑ in volume of water reabsorbed into the blood
↓ vasodilatory effect of PG at the afferent arteriole of the nephron
↓ Glomerular filtration rate (GFR)
↓ PG signalling results in ↓ renin secretion at juxtaglomerular apparatus
Low renin levelsà↓ conversion of angiotensinogen into its AT1 form and, by extension, ACE mediated conversion of AT1 to AT2
↓ ACE 2 signalling leads to ↓ levels of aldosterone in the serum
↓ Na+ and K+ channel insertion on apical surface and ↓ Na/K ATPase activity on basolateral surface of principal cells
↓ K+ excretion into urine, and ↓ Na+ reabsorption
back into the blood, at the late DCT and collecting duct of the kidney
Pre-renal Acute Kidney Injury (AKI): kidney injury due to renal hypoperfusion
Prolonged and/or severe ischemia causes cell death and aggregation of tubular epithelial cells of the kidney with subsequent inability to reabsorb luminal Na+
Renal dehydration predisposes
precipitation of uromodulin protein
Renal tubules mold uromodulin into cylindrical structures known as “casts”. Casts that contain only uromodulin protein are known as “hyaline casts”
Hyaline cast seen on urinalysis
Hypoperfusion of the kidney activates the renin- angiotensin- aldosterone system (RAAS)
↑ amount of sodium (Na) reabsorbed from the filtrate (less Na excreted)
Fractional excretion of Na <1%
Papillary necrosis: ureteral passage of sloughed ischemic tissue causing ureteral obstruction
Acute tubular necrosis: a type of kidney injury causing damage to the tubules
Hypertension
Post-renal AKI: a type of kidney injury due to
obstruction of the urinary tract
Distal distal obstruction of the urinary tract causes fluid to accumulate within the kidneysàenlarging the kidneys
Renal Ultrasound shows hydronephrosis (enlarged kidneys)
Damaged tubule epithelial cells slough into the tubular lumen
Epithelial cell breakdown in the tubular lumen
releases uromodulin & other proteins, which aggregate into “casts” (cylindrical imprints of the renal tubule). The varied protein content of these casts result in them having a coarse, granular appearance
Damaged tubular epithelial cells are unable to properly reabsorb sodium
↓ amount of sodium (Na) reabsorbed from filtrate into the blood (more Na excreted)
Fractional excretion of Na >2%
True excess of free water relative to Na+ in the blood
Excess free water ↑ venous hydrostatic pressure (see Calgary Guide slide on edema for full mechanisms)
Pitting Edema
Hyperkalemia
Hyponatremia
Coarse granular casts seen on urinalysis
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published January 29, 2022 on www.thecalgaryguide.com")
Hypokalemia Physiology
![Hypokalemia: Physiology
Authors: Samin Dolatabadi, Ran (Marissa) Zhang, Mannat Dhillon Reviewers: Meena Assad, Yan Yu*, Juliya Hemmett*
Beta-2 receptor stimulation
(e.g. Salbutamol)
↑ Red blood cell production
↑ Na+/K+ ATPase activity in skeletal muscle cells (moves K+ into the cell & Na+ out of cell)
↑ K+ entry into skeletal muscle cells
* MD at time of publication
Abbreviations:
• EABV – Effective Arterial
Blood Volume
• ENaC – Epithelial Sodium
Channel
• HCL – Hydrochloric acid • HCO3- –Bicarbonate ion
↑ K+ uptake by new red blood cells
↑ Intracellular shift of K+ into cells
Refeeding Syndrome Exogenous insulin
↓ K+ dietary intake
(rare cause in isolation)
↑ Insulin in response to carbohydrate load
↑ Na+/K+ ATPase activity in skeletal muscle & hepatic cells
↑ K+ entry into skeletal muscle & hepatic cells
↓ K+ availability for gastrointestinal absorption
Hypokalemia (Serum [K+] < 3.5 mmol/L)
↑ Renal K+ secretion
K+ follows the electrical gradient into tubular lumen
↑ Electronegativity of tubular lumen
↑ Na+ reabsorptionin principal cellsà Cl- left behind in tubular lumen of kidneys
Gastric acid depletionà ↓HCl
Loss of H+àShift in bicarbonate buffer system to ↑ plasma HCO3-
Plasma HCO3- above reabsorptive capacity of the proximal tubule
↑ HCO3- in the distal tubular lumen of kidneys
Vomiting
Diarrhea Laxatives
Renin secreting tumour Hyperaldosteronism Renal artery stenosis Loop and Thiazide
diuretics
Bartter’s and Gittelman’s syndrome
Liddle syndrome
Extracellular fluid volume depletion
↓ EABV ↑ Renin secretion
↓ Afferent arteriole pressure perfusing kidneys
Renin-Angiotensin- Aldosterone System (RAAS) activationà ↑ Aldosterone release from the adrenal cortex
↑ Expression of ENaC (Na+ reabsorption) in principal cells of the cortical collectingduct)
+ ↑Na &
water excretion in kidneys
↓ EABV
Genetic condition leading to inability to degrade ENaC channels in principal cells of the cortical collecting duct
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published March 6, 2019, updated Jan 23, 2022 on www.thecalgaryguide.com
Hypokalemia: Physiology
Authors: Samin Dolatabadi Reviewers: Meena Assad Dr. Juliya Hemmett* * MD at time of publication
TTKG > 4 with N/↑ EABV in hypokalemia is inappropriate and a principal cell problem.
β2 Stimulation (e.g., Salbutamol)
↑ Na+/K+ ATPase activity
↑ K+ entry into cell
↑ RBC Production
↑ Cell production
↑ K+ uptake by new cells
↓ Extracellular ↑ Insulin in response to H+
carbohydrate load
↑ Na+/H+ antiporter activity (movement of H+ out of cell and Na+ into cell)
↑ Intracellular Na+
↑ Na+/K+ ATPase activity ↑ K+ entry into cell
↑ Intracellular Shift of K+
Notes:
Refeeding Syndrome
Insulin
Alkalemia
•
Abbreviations:
• CCD – Cortical Collecting Duct
• EABV – Effective Arterial Blood Volume
• RAAS – Renin-Angiotensin-Aldosterone System • TTKG – Trans-tubular Potassium Gradient
• ENaC – Epithelial Sodium Channel
↓ K+ Intake (Rare cause in isolation)
↓ K+ availability
Diarrhea, Vomiting, Laxatives
↑ Gastrointestinal loss of K+
Polyuria
↑ Renal loss of K+ (TTKG < 4 as principal cell is working appropriately but small amount of K+ is lost per urination)
Hypokalemia (Serum [K+] < 3.5 mmol/L)
Liddle Syndrome Hyperaldosteronism Renin Secreting Tumour
Renal artery stenosis
Loop and Thiazide Diuretics
Bartter’s and Gittelman’s Syndrome
Genetic condition leading to inability to degrade ENaC channels ↑ Renin
↑ Renal K+ secretion
K+ follows the electrical gradient
Electronegative lumen
↓ Pressure perfusing the kidney
RAAS activation RAAS activation
↑ Aldosterone
↑ Na+ and water excretion
↓ EABV
↑ Expression of ENaC in principal cells of CCD
↑ Na+ reabsorption
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published March 6, 2019 on www.thecalgaryguide.com](https://calgaryguide.ucalgary.ca/wp-content/uploads/2019/03/Hypokalemia-Physiology.jpg "Hypokalemia: Physiology
Authors: Samin Dolatabadi, Ran (Marissa) Zhang, Mannat Dhillon Reviewers: Meena Assad, Yan Yu*, Juliya Hemmett*
Beta-2 receptor stimulation
(e.g. Salbutamol)
↑ Red blood cell production
↑ Na+/K+ ATPase activity in skeletal muscle cells (moves K+ into the cell & Na+ out of cell)
↑ K+ entry into skeletal muscle cells
* MD at time of publication
Abbreviations:
• EABV – Effective Arterial
Blood Volume
• ENaC – Epithelial Sodium
Channel
• HCL – Hydrochloric acid • HCO3- –Bicarbonate ion
↑ K+ uptake by new red blood cells
↑ Intracellular shift of K+ into cells
Refeeding Syndrome Exogenous insulin
↓ K+ dietary intake
(rare cause in isolation)
↑ Insulin in response to carbohydrate load
↑ Na+/K+ ATPase activity in skeletal muscle & hepatic cells
↑ K+ entry into skeletal muscle & hepatic cells
↓ K+ availability for gastrointestinal absorption
Hypokalemia (Serum [K+] < 3.5 mmol/L)
↑ Renal K+ secretion
K+ follows the electrical gradient into tubular lumen
↑ Electronegativity of tubular lumen
↑ Na+ reabsorptionin principal cellsà Cl- left behind in tubular lumen of kidneys
Gastric acid depletionà ↓HCl
Loss of H+àShift in bicarbonate buffer system to ↑ plasma HCO3-
Plasma HCO3- above reabsorptive capacity of the proximal tubule
↑ HCO3- in the distal tubular lumen of kidneys
Vomiting
Diarrhea Laxatives
Renin secreting tumour Hyperaldosteronism Renal artery stenosis Loop and Thiazide
diuretics
Bartter’s and Gittelman’s syndrome
Liddle syndrome
Extracellular fluid volume depletion
↓ EABV ↑ Renin secretion
↓ Afferent arteriole pressure perfusing kidneys
Renin-Angiotensin- Aldosterone System (RAAS) activationà ↑ Aldosterone release from the adrenal cortex
↑ Expression of ENaC (Na+ reabsorption) in principal cells of the cortical collectingduct)
+ ↑Na &
water excretion in kidneys
↓ EABV
Genetic condition leading to inability to degrade ENaC channels in principal cells of the cortical collecting duct
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published March 6, 2019, updated Jan 23, 2022 on www.thecalgaryguide.com
Hypokalemia: Physiology
Authors: Samin Dolatabadi Reviewers: Meena Assad Dr. Juliya Hemmett* * MD at time of publication
TTKG > 4 with N/↑ EABV in hypokalemia is inappropriate and a principal cell problem.
β2 Stimulation (e.g., Salbutamol)
↑ Na+/K+ ATPase activity
↑ K+ entry into cell
↑ RBC Production
↑ Cell production
↑ K+ uptake by new cells
↓ Extracellular ↑ Insulin in response to H+
carbohydrate load
↑ Na+/H+ antiporter activity (movement of H+ out of cell and Na+ into cell)
↑ Intracellular Na+
↑ Na+/K+ ATPase activity ↑ K+ entry into cell
↑ Intracellular Shift of K+
Notes:
Refeeding Syndrome
Insulin
Alkalemia
•
Abbreviations:
• CCD – Cortical Collecting Duct
• EABV – Effective Arterial Blood Volume
• RAAS – Renin-Angiotensin-Aldosterone System • TTKG – Trans-tubular Potassium Gradient
• ENaC – Epithelial Sodium Channel
↓ K+ Intake (Rare cause in isolation)
↓ K+ availability
Diarrhea, Vomiting, Laxatives
↑ Gastrointestinal loss of K+
Polyuria
↑ Renal loss of K+ (TTKG < 4 as principal cell is working appropriately but small amount of K+ is lost per urination)
Hypokalemia (Serum [K+] < 3.5 mmol/L)
Liddle Syndrome Hyperaldosteronism Renin Secreting Tumour
Renal artery stenosis
Loop and Thiazide Diuretics
Bartter’s and Gittelman’s Syndrome
Genetic condition leading to inability to degrade ENaC channels ↑ Renin
↑ Renal K+ secretion
K+ follows the electrical gradient
Electronegative lumen
↓ Pressure perfusing the kidney
RAAS activation RAAS activation
↑ Aldosterone
↑ Na+ and water excretion
↓ EABV
↑ Expression of ENaC in principal cells of CCD
↑ Na+ reabsorption
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published March 6, 2019 on www.thecalgaryguide.com")
metabolic-alkalosis-pathogenesis
![Metabolic Alkalosis: Pathogenesis
Authors: Wazaira Khan Reviewers: Jessica Krahn, Emily Wildman, Austin Laing, Huneza Nadeem, Ran (Marissa) Zhang, Adam Bass* * MD at time of publication
Primary hyperaldosteronism E.g., aldosterone- secreting mass, adrenal hyperplasia
Secondary hyperaldosteronism E.g., renin-secreting mass, renal artery stenosis
Pseudo- hypoaldosteronism E.g., Liddle’s Syndrome
Unregulated aldosterone production in adrenal cortex
Excess aldosterone suppresses renin production
Unregulated renin production by juxtaglomerular cells
Sustained ↑ in mineralocorticoid (aldosterone) activity
Insertion of epithelial sodium channels on principal cells in collecting duct
↑ Water retention by the kidney
↑ Na+ reabsorption by principal cells
↑ EABV
• ↑ Jugular venous pressure • Hypertension
• Urine Na+ > 40 mEq/L
Low renin, high aldosterone state
Activation of renin- angiotensin-aldosterone system (RAAS)
Release of aldosterone from adrenal cortex
High renin, high aldosterone state
↑ Negative charge in collecting duct lumen
K+ leaks into collecting duct lumen by principal cell to maintain electroneutrality
Hypokalemia
Intracellular K+ leaks out of any cell in the body to compensate for low serum K+ levels
Extracellular H+ enters cell to maintain electroneutrality
Intracellular acidosis
Activation of compensatory acid secreting mechanisms in kidney
Impaired tubular function
E.g., loop/thiazide diuretics, Bartter’s/ Gitelman’s Syndrome
Upper GI Loss
E.g., vomiting, nasogastric suction
Unregulated epithelial sodium channel activity in collecting duct mimicking aldosterone function
↑ in Na+ and water retention à RAAS inhibition
↓ Na+ and Cl- reabsorption in thick ascending limb or distal convoluted tubule
Low renin, low aldosterone state
↑ Na+, Cl- and water secretion through kidney
RAAS activation
See “Physiology of RAAS” slide
↓ EABV • • •
Temporary ↑ in mineralocorticoid (aldosterone) activity
↓ Jugular venous pressure Orthostatic hypotension Dry mucous membranes Urine Na+ < 20 mEq/L
•
Loss of fluid through GI tract
↓ HCl delivery to small intestine
↑ NH + secretion and 4
↑ HCO3- reabsorption in proximal tubule
↑ H+ secretion in cortical collecting duct
Loss of gastric contents, including HCl
↓ HCO3- secretion by pancreas, liver and intestines to neutralize HCl
Loss of intrinsic acid to neutralize HCO3- ↑ Plasma [HCO3-]
Metabolic Alkalosis
Arterial blood gas pH > 7.40 Plasma [HCO3-] > 24 mEq/L
Effective Arterial Blood Volume (EABV): component of arterial blood volume that is effectively perfusing organs
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published May 31, 2022 on www.thecalgaryguide.com](https://calgaryguide.ucalgary.ca/wp-content/uploads/2022/05/Metabolic-Alkalosis-1.jpg "Metabolic Alkalosis: Pathogenesis
Authors: Wazaira Khan Reviewers: Jessica Krahn, Emily Wildman, Austin Laing, Huneza Nadeem, Ran (Marissa) Zhang, Adam Bass* * MD at time of publication
Primary hyperaldosteronism E.g., aldosterone- secreting mass, adrenal hyperplasia
Secondary hyperaldosteronism E.g., renin-secreting mass, renal artery stenosis
Pseudo- hypoaldosteronism E.g., Liddle’s Syndrome
Unregulated aldosterone production in adrenal cortex
Excess aldosterone suppresses renin production
Unregulated renin production by juxtaglomerular cells
Sustained ↑ in mineralocorticoid (aldosterone) activity
Insertion of epithelial sodium channels on principal cells in collecting duct
↑ Water retention by the kidney
↑ Na+ reabsorption by principal cells
↑ EABV
• ↑ Jugular venous pressure • Hypertension
• Urine Na+ > 40 mEq/L
Low renin, high aldosterone state
Activation of renin- angiotensin-aldosterone system (RAAS)
Release of aldosterone from adrenal cortex
High renin, high aldosterone state
↑ Negative charge in collecting duct lumen
K+ leaks into collecting duct lumen by principal cell to maintain electroneutrality
Hypokalemia
Intracellular K+ leaks out of any cell in the body to compensate for low serum K+ levels
Extracellular H+ enters cell to maintain electroneutrality
Intracellular acidosis
Activation of compensatory acid secreting mechanisms in kidney
Impaired tubular function
E.g., loop/thiazide diuretics, Bartter’s/ Gitelman’s Syndrome
Upper GI Loss
E.g., vomiting, nasogastric suction
Unregulated epithelial sodium channel activity in collecting duct mimicking aldosterone function
↑ in Na+ and water retention à RAAS inhibition
↓ Na+ and Cl- reabsorption in thick ascending limb or distal convoluted tubule
Low renin, low aldosterone state
↑ Na+, Cl- and water secretion through kidney
RAAS activation
See “Physiology of RAAS” slide
↓ EABV • • •
Temporary ↑ in mineralocorticoid (aldosterone) activity
↓ Jugular venous pressure Orthostatic hypotension Dry mucous membranes Urine Na+ < 20 mEq/L
•
Loss of fluid through GI tract
↓ HCl delivery to small intestine
↑ NH + secretion and 4
↑ HCO3- reabsorption in proximal tubule
↑ H+ secretion in cortical collecting duct
Loss of gastric contents, including HCl
↓ HCO3- secretion by pancreas, liver and intestines to neutralize HCl
Loss of intrinsic acid to neutralize HCO3- ↑ Plasma [HCO3-]
Metabolic Alkalosis
Arterial blood gas pH > 7.40 Plasma [HCO3-] > 24 mEq/L
Effective Arterial Blood Volume (EABV): component of arterial blood volume that is effectively perfusing organs
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published May 31, 2022 on www.thecalgaryguide.com")
pheochromocytoma-pathogenesis-and-clinical-findings
 Zhang Hanan Bassyouni* * MD at time of publication
↑ Blood pressure results in the activation of neural pain receptors
Sustained or paroxysmal 2o hypertension (↑ blood pressure)
Pallor
Tachycardia (↑ heart rate), palpitations
Diaphoresis (excessive sweating)
Hyperglycemia Weight loss, fatigue
Familial Disorders (10%)
E.g., Multiple Endocrine Neoplasm 2 Syndrome (MEN2) types A and B, Neurofibromatosis 1 (NF-1), Von Hippel Lindau Syndrome (VHL), familial pheochromocytoma
Dysfunction of various tumor suppressor and/or oncogene proteins
Uncontrolled proliferation of the chromaffin cells in the medulla of the adrenal gland(s)
Adenoma formation (10% bilateral)
Over-production of epinephrine and
norepinephrine from the adrenal adenoma(s) or extra- adrenal tumour (10%)
Detectable metanephrines (epinephrine/norepinephrine breakdown products) in both plasma and urine
Sporadic DNA mutations arising from
DNA damage from exposure to mutagens, malignancy (10%), or during DNA replication
Detectable mutations on the Von Hippel Linda (VHL), (Rearranged During Transcription) RET, (Succinate Dehydrogenase) SDH, and/or other tumour suppressor or oncogenes
Visible adrenal mass on CT scan Heart attack, stroke, or death (Note: These
tumors can be fatal therefore screening is essential in patients with adrenal masses or 2o hypertension)
Episodic hyper-activity of the sympathetic nervous system
Hyper-stimulation of G protein-coupled receptors involved in catabolic metabolic processes
Headaches
↑ Vasoconstriction of peripheral blood vessels
Hyper-stimulation of adrenergic receptors on cardiac myocytes
↑ Secretion from the eccrine sweat glands
Panic, tremor, anxiety
↑ Ability to mobilize glucose into the bloodstream through enhanced lipolysis, glycogenolysis and gluconeogenesis
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published February 20, 2014, updated June 27, 2022 on www.thecalgaryguide.com")
Epilepsy Pathogenesis

Neoplasm
Metabolic
Inborn errors of metabolism
Inherited enzyme deficiencies
Neurodegenerative
Alzheimer’s Disease
Protein-rich plaque build-up and brain atrophy
Inflammation and formation of scar tissue irritates neural tissue
Infiltration of mass, grey matter irritation
Damage to the brain and subsequent alteration of neuronal circuitry
Neurotransmitter imbalances (i.e. ↑ glutamate, ↓ GABA, ↓ serotonin, ↓ dopamine, ↓ noradrenaline)
Abbreviations:
• HSV1 – Herpes Simplex Virus 1 • VZV – Varicella Zoster Virus
↑ Inflammatory cytokines (e.g. interleukin-6, tumor necrosis factor-α)
Altered neurogenesis and gliosis
Ion channel and receptor dysfunction causes imbalance of ion channel charges
Authors: Keerthana Pasumarthi Christopher Li Reviewers: Negar Tehrani, Ephrem Zewdie, Ran (Marissa) Zhang, Carlos R. Camara-Lemarroy* * MD at time of publication
Neurons fire in burst activity (referred to as paroxysmal depolarization shift) and often in groups (hypersynchrony)
Abnormal neural activities eventually create self-reinforcing circuits and transform neural network over time
Injuries from
falls, bumps, operating heavy machinery
Involuntary contractions Loss of consciousness
Post-ictal confusion
Recurrent seizures
Sudden Unexplained Death of Epilepsy Patient (SUDEP)
Loss of airway reflexes
Aspiration of saliva or food contents
Aspiration Pneumonia
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published July 5, 2022 on www.thecalgaryguide.com")
etomidate

↓ Adrenal cortisol production
↓ Serum cortisol available for stress response
Transient adrenal steroid insufficiency
↑ Morbidity & mortality in trauma/critically ill patients (Contraindicated in sepsis)
Inhibits nitric oxide signaling
Transient cerebral vasoconstriction
↓ Cerebral blood flow
↓ Oxygen delivery to brain
↓ Cerebral metabolic rate
Suppression of electrical activity in brain
Flat electroencephalogram
Binds to GABAA
(inhibitory CNS neurotransmitter) receptor
Authors:
Cindy Chang
Ran (Marissa) Zhang Reviewers:
Melissa King
Brooke Fallis
Julia Haber*
* MD at time of publication
Abbreviations:
• CNS - Central Nervous
System
• GABAA- Gamma-
aminobutyric acid type A • TRPA1- Transient
receptor potential type A1
Higher selectivity for specific GABAA receptor subtypes compared to other induction agents
Positively modulates GABAA receptor (GABAA receptor activated at lower concentrations of GABA )
↓ Cerebral blood volume
Activation of fewer GABAA receptor subtypes in neurons and cardiovascular structures compared to other induction agents
↓ Intracranial pressure ↑ Hemodynamic stability
(minimal changes in blood pressure or heart rate)
Myoclonus (brief, involuntary, irregular muscle twitch)
↑ Spontaneous CNS neuron firing to skeletal muscle
Prolonged opening of GABAA receptors (acts as ion channel for chloride)
Inhibits activity of nerve cells in the reticular activating system
Activation of peripheral nociceptor receptors
↑ Involuntary muscle contractions
Influx of chlorideàHyperpolarization of nerve membranes in reticular activating system
(brainstem neuronal network that regulates arousal and sleep- wake transitions)
Depression of arousal & loss of consciousness
Induction of general anesthesia
(no analgesic effect)
Direct activation of TRPA1 cation channels (key receptor in pain pathway)
Stinging pain with injection
(Treat with co-administration of local anesthetic)
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published July 9, 2022 on www.thecalgaryguide.com")
complicaciones-de-la-enfermedad-renal-cronica

hyperkalemia-↓-renal-excretion-pathophysiology
![Hyperkalemia (↓ Renal Excretion): Pathophysiology
Non-steroidal anti- inflammatory drugs (NSAIDs)
Inhibition of prostaglandins which promote renin secretion (see NSAIDs and the Kidney: Mechanism of Action and Side Effects slide)
Diabetic Nephropathy
Acute (AIN)and chronic (CIN) interstitial nephritis
Immune-mediated damage of the kidney tubule and interstitium
Damage to distal tubule leads to aldosterone resistance at principal cell
Aldosterone cannot ↑ EnaC insertion on principal cell of CCD
Epithelial sodium channel (ENaC) blockers
Acute kidney injury and chronic kidney disease
↓ Effective arterial blood volume (volume of blood effectively perfusing tissue)
↓ Oxygen perfusion to renal tissue causing renal ischemia
Autonomic neuropathy ↓ sympathetic drive to produce renin
Chronic juxtaglomerular cell damage ↓ synthesis of renin
Blockage of ENaC on the principal cells of the CCD
Angiotensin converting enzyme inhibitors (ACEi) and angiotensin receptor blockers (ARBs)
Angiotensin II is either not formed (ACEi), or blocked at its receptor (ARBs)
Angiotensin II cannot stimulate the release of aldosterone from the adrenal cortex
Adrenal Insufficiency
Adrenal gland cannot produce sufficient amounts of aldosterone
↓ Renin secretion by the afferent arteriole prevents RAAS activation
↓ Aldosterone release from adrenal cortex
↓ ENaC (Na+ reabsorption channel) expression on principal cells of the cortical collecting duct (CCD)
Damage to kidney causes renal impairment and ↓ glomerular filtration rate
↓ Glomerular filtrate production means ↓ tubular flow rate
↓ Na+ delivery to the distal tubule
↓ Na+ reabsorption by ENaCs at the principal cell in CCD
↓ Na+ and water reabsorption by ENaCs in CCD ↓ Effective arterial blood volume (EABV)
Activates renin-angiotensin-aldosterone system (RAAS) leads to ↑ renin secretion in the afferent arteriole (see Physiology of the renin-angiotensin-aldosterone system (RAAS) slide)
↑ Na+ and water loss in tubular lumen
↑ Positive charge in tubular lumen
↓ Electronegativity gradient in tubular lumen
of CCD
↓ K+ excretion by the principal cell in the CCD as there is less of a electronegative gradient
↑ Accumulation of K+ in blood Hyperkalemia
Serum [K+] > 5.1 mmol/L
See Hyperkalemia: Clinical Findings slide
In the case of diabetic nephropathy and NSAIDS ↓ EABV does not stimulate RAAS and therefore aldosterone production
↓ Renin
↓ Aldosterone
↑ Renin secretion activates an ↑ in aldosterone production but aldosterone action at the principal cell is blocked because of ENaCs or resistance in AIN and CIN
↑ Renin
↑ Aldosterone
In the case of ACEi, ARBs, or adrenal insufficiency ↑ renin secretion does not lead to ↑ aldosterone
↑ Renin
↓ Aldosterone
Note: as described in the above flow chart, measuring serum renin and aldosterone levels can be used to help diagnose the cause of hyperkalemia.
Authors: Mannat Dhillon, Joshua Low, Emily Wildman, Huneza Nadeem Reviewers: Andrea Kuczynski, Marissa (Ran) Zhang, Adam Bass*, Kevin McLaughlin* * MD at time of publication
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published Aug 2, 2022 on www.thecalgaryguide.com](https://calgaryguide.ucalgary.ca/wp-content/uploads/2022/08/Hyperkalemia-renal-excretion.jpg "Hyperkalemia (↓ Renal Excretion): Pathophysiology
Non-steroidal anti- inflammatory drugs (NSAIDs)
Inhibition of prostaglandins which promote renin secretion (see NSAIDs and the Kidney: Mechanism of Action and Side Effects slide)
Diabetic Nephropathy
Acute (AIN)and chronic (CIN) interstitial nephritis
Immune-mediated damage of the kidney tubule and interstitium
Damage to distal tubule leads to aldosterone resistance at principal cell
Aldosterone cannot ↑ EnaC insertion on principal cell of CCD
Epithelial sodium channel (ENaC) blockers
Acute kidney injury and chronic kidney disease
↓ Effective arterial blood volume (volume of blood effectively perfusing tissue)
↓ Oxygen perfusion to renal tissue causing renal ischemia
Autonomic neuropathy ↓ sympathetic drive to produce renin
Chronic juxtaglomerular cell damage ↓ synthesis of renin
Blockage of ENaC on the principal cells of the CCD
Angiotensin converting enzyme inhibitors (ACEi) and angiotensin receptor blockers (ARBs)
Angiotensin II is either not formed (ACEi), or blocked at its receptor (ARBs)
Angiotensin II cannot stimulate the release of aldosterone from the adrenal cortex
Adrenal Insufficiency
Adrenal gland cannot produce sufficient amounts of aldosterone
↓ Renin secretion by the afferent arteriole prevents RAAS activation
↓ Aldosterone release from adrenal cortex
↓ ENaC (Na+ reabsorption channel) expression on principal cells of the cortical collecting duct (CCD)
Damage to kidney causes renal impairment and ↓ glomerular filtration rate
↓ Glomerular filtrate production means ↓ tubular flow rate
↓ Na+ delivery to the distal tubule
↓ Na+ reabsorption by ENaCs at the principal cell in CCD
↓ Na+ and water reabsorption by ENaCs in CCD ↓ Effective arterial blood volume (EABV)
Activates renin-angiotensin-aldosterone system (RAAS) leads to ↑ renin secretion in the afferent arteriole (see Physiology of the renin-angiotensin-aldosterone system (RAAS) slide)
↑ Na+ and water loss in tubular lumen
↑ Positive charge in tubular lumen
↓ Electronegativity gradient in tubular lumen
of CCD
↓ K+ excretion by the principal cell in the CCD as there is less of a electronegative gradient
↑ Accumulation of K+ in blood Hyperkalemia
Serum [K+] > 5.1 mmol/L
See Hyperkalemia: Clinical Findings slide
In the case of diabetic nephropathy and NSAIDS ↓ EABV does not stimulate RAAS and therefore aldosterone production
↓ Renin
↓ Aldosterone
↑ Renin secretion activates an ↑ in aldosterone production but aldosterone action at the principal cell is blocked because of ENaCs or resistance in AIN and CIN
↑ Renin
↑ Aldosterone
In the case of ACEi, ARBs, or adrenal insufficiency ↑ renin secretion does not lead to ↑ aldosterone
↑ Renin
↓ Aldosterone
Note: as described in the above flow chart, measuring serum renin and aldosterone levels can be used to help diagnose the cause of hyperkalemia.
Authors: Mannat Dhillon, Joshua Low, Emily Wildman, Huneza Nadeem Reviewers: Andrea Kuczynski, Marissa (Ran) Zhang, Adam Bass*, Kevin McLaughlin* * MD at time of publication
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published Aug 2, 2022 on www.thecalgaryguide.com")
presentation-of-sah
![Subarachnoid Hemorrhage: Clinical Findings
Sudden bleeding into space surrounding the brain (for pathogenesis, see Subarachnoid Hemorrhage: Pathogenesis)
Authors: Jason An, M. Patrick Pankow Reviewers: Owen Stechishin, Dave Nicholl, Haotian Wang, Hannah Mathew, Ran (Marissa) Zhang, Yan Yu*, Cory Toth* * MD at time of publication
Bleed into subarachnoid space
Subarachnoid Hemorrhage (SAH)
Posterior hypothalamus ischemia (↓ Blood flow and oxygen)
Red blood cell lysis from energy depletion or complement activation
Release of spasmogens (spasm inducing agents)
Cerebral vasospasm (narrowing of arteries from persistent contraction) ↓ blood flow
Cerebral ischemia
Release catecholamines (hormones from the adrenal gland; e.g., epinephrine, norepinephrine)
↑ Intracellular calcium
Release of antidiuretic hormone
Antidiuretic hormone acts on the distal convoluted tubule and collecting duct in kidney to reabsorb water
Dilution of serum sodium
Hyponatremia (low blood sodium levels)
Release of epileptogenic (potential seizure causing agents) into cerebral circulation
Seizure
Products from blood breakdown in cerebral spinal fluid
Irritation of meninges (membranes surrounding the brain)
Aseptic meningitis (non-infectious inflammation)
Meningismus
(neck pain + rigidity)
Cerebral infarction (death of tissue)
Obstructs cerebral spinal fluid flow and absorption at subarachnoid granulations
Hydrocephalus (fluid build up in ventricles)
↓ Level of consciousness
Reduced cerebral blood flow
Dilation of cranial vessels to ↑ blood flow
Rapid ↑ internal carotid artery intracranial pressure
Refer to Increased Intracranial Pressure: Clinical Findings slide
Internal carotid artery
Pituitary ischemia
Hypopituitarism
[underactive pituitary gland, failing to produce 1+ pituitary hormone(s)]
Refer to hypopituitarism slides
Myocardial disruption
Left ventricle dysfunction
↑ Pressure in left heart
Blood forced backwards into pulmonary veins
↑ Pulmonary blood pressure
Fluid from blood vessels leaks into lungs
Dysrhythmias (disturbance in rate/rhythm of heart) causing ↓ cardiac output
Syncope
(loss of consciousness due to ↓ blood flow to the brain)
Pulmonary edema
(excess accumulation of fluid in lung)
Cerebral hypoperfusion
Sudden ↑in blood volume
Vessels and meninges suddenly stretch
Thunderclap Headache (worst headache of patient's life)
Shortness of breath
Reactive cerebral hyperemia (excess blood in vessels supplying the brain)
Artery specific findings:
Rapid ↑ internal carotid artery intracranial pressure
Middle cerebral artery
Posterior communicating artery
Compression of outer CN3 Compression of inner CN3
Anterior communicating artery
Nonreactive pupil
Gaze palsy
(eye deviates down and out)
Diplopia
(double vision)
Ptosis
(drooping of upper eyelid)
Frontal lobe ischemia
Avolition
(complete lack of motivation)
Ischemia of motor strip pertaining to the legs
Bilateral leg weakness
Motor strip ischemia
Hemiparesis
(weakness/ inability to move one side of the body)
Ischemia of parietal association areas (brain regions integral for motor control of the eyes, the extremities and spatial cognition)
Aphasia
(impaired ability to speak and/or understand language)/ neglect
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published July 1, 2014, updated August 10, 2022 on www.thecalgaryguide.com](https://calgaryguide.ucalgary.ca/wp-content/uploads/2015/05/SAH-Clinical-Findings-2022.jpg "Subarachnoid Hemorrhage: Clinical Findings
Sudden bleeding into space surrounding the brain (for pathogenesis, see Subarachnoid Hemorrhage: Pathogenesis)
Authors: Jason An, M. Patrick Pankow Reviewers: Owen Stechishin, Dave Nicholl, Haotian Wang, Hannah Mathew, Ran (Marissa) Zhang, Yan Yu*, Cory Toth* * MD at time of publication
Bleed into subarachnoid space
Subarachnoid Hemorrhage (SAH)
Posterior hypothalamus ischemia (↓ Blood flow and oxygen)
Red blood cell lysis from energy depletion or complement activation
Release of spasmogens (spasm inducing agents)
Cerebral vasospasm (narrowing of arteries from persistent contraction) ↓ blood flow
Cerebral ischemia
Release catecholamines (hormones from the adrenal gland; e.g., epinephrine, norepinephrine)
↑ Intracellular calcium
Release of antidiuretic hormone
Antidiuretic hormone acts on the distal convoluted tubule and collecting duct in kidney to reabsorb water
Dilution of serum sodium
Hyponatremia (low blood sodium levels)
Release of epileptogenic (potential seizure causing agents) into cerebral circulation
Seizure
Products from blood breakdown in cerebral spinal fluid
Irritation of meninges (membranes surrounding the brain)
Aseptic meningitis (non-infectious inflammation)
Meningismus
(neck pain + rigidity)
Cerebral infarction (death of tissue)
Obstructs cerebral spinal fluid flow and absorption at subarachnoid granulations
Hydrocephalus (fluid build up in ventricles)
↓ Level of consciousness
Reduced cerebral blood flow
Dilation of cranial vessels to ↑ blood flow
Rapid ↑ internal carotid artery intracranial pressure
Refer to Increased Intracranial Pressure: Clinical Findings slide
Internal carotid artery
Pituitary ischemia
Hypopituitarism
[underactive pituitary gland, failing to produce 1+ pituitary hormone(s)]
Refer to hypopituitarism slides
Myocardial disruption
Left ventricle dysfunction
↑ Pressure in left heart
Blood forced backwards into pulmonary veins
↑ Pulmonary blood pressure
Fluid from blood vessels leaks into lungs
Dysrhythmias (disturbance in rate/rhythm of heart) causing ↓ cardiac output
Syncope
(loss of consciousness due to ↓ blood flow to the brain)
Pulmonary edema
(excess accumulation of fluid in lung)
Cerebral hypoperfusion
Sudden ↑in blood volume
Vessels and meninges suddenly stretch
Thunderclap Headache (worst headache of patient's life)
Shortness of breath
Reactive cerebral hyperemia (excess blood in vessels supplying the brain)
Artery specific findings:
Rapid ↑ internal carotid artery intracranial pressure
Middle cerebral artery
Posterior communicating artery
Compression of outer CN3 Compression of inner CN3
Anterior communicating artery
Nonreactive pupil
Gaze palsy
(eye deviates down and out)
Diplopia
(double vision)
Ptosis
(drooping of upper eyelid)
Frontal lobe ischemia
Avolition
(complete lack of motivation)
Ischemia of motor strip pertaining to the legs
Bilateral leg weakness
Motor strip ischemia
Hemiparesis
(weakness/ inability to move one side of the body)
Ischemia of parietal association areas (brain regions integral for motor control of the eyes, the extremities and spatial cognition)
Aphasia
(impaired ability to speak and/or understand language)/ neglect
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published July 1, 2014, updated August 10, 2022 on www.thecalgaryguide.com")
patellar-tendon-rupture-pathogenesis-and-clinical-findings

Repetitive activities involving rapid knee flexion and extension
Tendon inflammation and micro-tearing
Fluoroquinolone (antibiotic class) use
Smoking
Immobilization of knee
↓ Muscle and tendon flexibility and strength
Steroid use (including intraarticular injections)
Changes in collagen fibrils composing tendon (exact mechanism unknown)
Disrupted blood supply to tendon and poor healing
Compromised integrity and strength of the tendon
Sudden heavy load on flexed knee while foot is planted
Quadriceps muscles contract while lengthening (eccentric contraction)àforce exceeds tendon strength
Patellar tendon innervated by common peroneal (fibular) nerve
Nociceptors (pain receptors) within tendon activated by tendon rupture
Pain and tenderness below the patella
Collagen fibres in tendon tear
Tearing or popping sensation at time of injury
Patellar Tendon Rupture
Tendon/Ligament detaches from bony attachment on patella or tibial tubercle No connection of quadriceps to tibia via patellar tendon
↑ Force on tibial tubercle during tendon detachment
Avulsion fracture of tibial tubercle
Trauma to tendon ruptures surrounding blood vessels
Coagulation cascade and inflammatory mediators released at rupture site
Quadriceps muscles unable to stabilize patellar tracking
Instability of knee joint
Quadriceps muscle tension pulls patella upwards with no resistance from patellar tendon
Patellar tendon cannot use distal attachment site for leverage
Knee extensor muscles unable to effectively contract
Knee buckling
Abnormal gait
Patella alta
(high sitting patella) on X-ray
Palpable gap below the patella
Bruising below the patella
Swelling below the patella
↑ Risk of falls
Loss of patellar reflex
Inability to perform a straight leg raise (hip flexes and knee cannot remain straight)
Hematoma (blood collection under the skin)
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published August 28, 2022 on www.thecalgaryguide.com")
quadriceps-tendon-rupture-pathogenesis-and-clinical-findings

Quadricep muscles atrophy
↓ Quadricep muscle strength
Smoking
Chronic disease (e.g. renal failure, gout, rheumatoid arthritis, and diabetes)
Fluoroquinolone (antibiotic class) use
Changes in collagen fibrils composing tendonàimpaired tendon strength (exact mechanism unknown)
Quadriceps tendon shortens
↓ Quadricep tendon flexibility
Disrupted blood supply to quadriceps tendon and poor healing
Compromised integrity of the quadriceps tendon and ↓ strength
Sudden heavy load on flexed knee while foot is plantedàquadriceps muscles contract while lengthening (eccentric contraction)
Force on quadriceps tendon exceeds its strength
Quadriceps Tendon Rupture
Tendon detaches from bony attachment on patella
↑ Force on patella during quadriceps tendon detachment
Collagen fibres in quadriceps tendon tear
No connection of quadriceps to tibia via quadriceps tendon
Patellar tendon tension pulls patella downwards with no resistance from quadriceps muscles
Quadriceps tendon innervated by common peroneal (fibular) nerve
Nociceptors (pain receptors) within tendon activated by tendon rupture
Pain and tenderness above the patella
Trauma to tendon ruptures blood vessels
Coagulation cascade and inflammatory mediators released at rupture site
Quadriceps muscles unable to stabilize patellar tracking
Knee joint instability
Patella baja (low sitting patella) on X-ray
Palpable gap above the patella
Loss of patellar reflex
Patellar tendon cannot use proximal attachment site for leverage
Knee extensor muscles unable to effectively contract
Inability to perform a straight leg raise (hip flexes while knee cannot remain straight)
Bruising above the patella
Swelling above the patella
Avulsion fracture of patella
Tearing or popping
sensation at time of injury
Knee buckling
Abnormal gait
↑ Risk of falls
Hematoma (blood collection under the skin)
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published October 2, 2022 on www.thecalgaryguide.com")
unstable-angina-pathogenesis-and-clinical-findings
![Unstable Angina/Unstable Angina Pectoris: Pathogenesis and clinical findings Primary cause:
Secondary causes:
Coronary artery vasospasm - primary or drug induced (Ex: cocaine, triptans)
Coagulopathy
(Ex: antiphospholipid antibody syndrome)
Vasculitic syndromes (Ex: Takayasu arteritis)
Authors: Marisa Vigna Ryan Wilkie Yan Yu* Reviewers: Julena Foglia Davis Maclean Mehul Gupta Andrew Grant* * MD at time of publication
Atherosclerosis
Fatty plaque accumulates inside the intimal walls of arteries Coronary arterial atherosclerotic plaque rupture or erosion
Plaque disruption exposes subendothelial components of damaged vessel wall to platelets, initiating the coagulation cascade and platelet adhesion
Aggregation of platelets results in the formation of a thrombus Thrombus partially occludes blood flow through a coronary artery âmyocardial blood supply
Congenital anomalies (Ex: myocardial bridge, anomalous coronary)
Spontaneous coronary artery dissection
Increased blood viscosity (Ex: polycythemia, thrombocytopenia)
Factors thatámyocardial (cardiac muscle) oxygen demand (Ex: tachycardia, hypotension, hypertension, anemia, exertion, stress)
Coronary embolism (Ex: A. Fib, endocarditis, prosthetic valve thrombus)
áheart rate, contractility, and/or wall tension ámyocardial oxygen demand
Myocardial ischemia due to imbalance between blood supply and oxygen demand (insufficient blood/oxygen supply)
Unstable Angina/Unstable Angina Pectoris
Can be new onset angina; typically progressive in frequency, severity, or duration; can occur at rest
Subtotal occlusion of a coronary arteryà
reduced, but continued, myocardial blood supply
Maintained perfusion means cardiomyocytes are still alive and thus do not leak troponin into bloodstream
Normal serum troponin
Diaphoresis
(sweating)
Since bloodflow occurs from epicardium to endocardium, myocardial ischemia is more
pronounced in the subendocardium (region furthest away from heart’s external surface)
Sufficient blood flow is maintained in regions superficial to the subendocardium, resulting in non-transmural (partial thickness) heart wall ischemia
Non-inferior wall ischemia triggers a predominantáin sympathetic nervous system activity, given the proximity of cardiac sympathetic nerve innervation
Ischemiaâ cardiomyocyte resting membrane potential andâ action potential duration
Voltage gradient between normal and subendocardial ischemic zones creates injury currents, shifting the ST- vector on ECG
ECG: ST depression
and/or T wave inversion
Cardiac sensory nerve fibres mix with somatic sensory nerve
fibres and enter the spinal cord via the T1-T4 nerve roots
Brain perceives increased cardiac sensory nerve signaling as nerve pain coming from the skin of T1-T4 dermatomes (“Referred Pain”)
Myocardial ischemia causes hypoxic stress on cardiomyocytesàâaerobic (requiring oxygen) metabolism,áanaerobic (not requiring oxygen) metabolism
áanerobic respirationálactic acid production,á[H+], andâcellular pH which impairs cardiomyocyte function
Cardiomyocyte dysfunction impairs myocardial relaxation in diastole and/orâ left ventricular contractility in systole
âleft ventricular cardiac output àbackup of blood in the left ventricle, atrium, and pulmonary vasculature
ápulmonary capillary pressures pushes fluid out of the capillaries into the alveoli in the lungs
Fluid filled alveoliâgas exchange andâ oxygenation, triggering harder and faster breathing in order to compensate
Dyspnea
Activation of sweat glands via acetylcholine release
Hormones bind to cardiac β1 receptors
Tachycardia
(áheart rate)
Epinephrine/ Norepinephrine hormone release from the adrenal medulla
Hormones bind to arterial smooth muscle α1 receptors ávascular tone (vasoconstriction)
Hypertension
The Vagus nerve sits in close physical proximity to the inferior wall of the heart àinferior wall ischemia triggers involuntary Vagus nerve activation
Since the Vagus nerve coordinates parasympathetic activity,áVagus nerve activity leads to a variety of parasympathetic nervous system responses:
Retrosternal discomfort: May present as pain, heaviness, tightness, aching, pressure, burning or squeezing
Pain radiation to T1-T4 dermatomes:
Left shoulder and arm, lower jaw, neck, abdomen, upper back
Syncope
(fainting)
Bradycardia
Nausea Hypotension
(âheart rate)
(âblood pressure)
(áblood pressure)
(shortness of breath)
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Findings
Complications
Published Oct 18, 2015, updated Aug 29, 2021 on www.thecalgaryguide.com](https://calgaryguide.ucalgary.ca/wp-content/uploads/2015/10/Unstable-Angina-2021.jpg "Unstable Angina/Unstable Angina Pectoris: Pathogenesis and clinical findings Primary cause:
Secondary causes:
Coronary artery vasospasm - primary or drug induced (Ex: cocaine, triptans)
Coagulopathy
(Ex: antiphospholipid antibody syndrome)
Vasculitic syndromes (Ex: Takayasu arteritis)
Authors: Marisa Vigna Ryan Wilkie Yan Yu* Reviewers: Julena Foglia Davis Maclean Mehul Gupta Andrew Grant* * MD at time of publication
Atherosclerosis
Fatty plaque accumulates inside the intimal walls of arteries Coronary arterial atherosclerotic plaque rupture or erosion
Plaque disruption exposes subendothelial components of damaged vessel wall to platelets, initiating the coagulation cascade and platelet adhesion
Aggregation of platelets results in the formation of a thrombus Thrombus partially occludes blood flow through a coronary artery âmyocardial blood supply
Congenital anomalies (Ex: myocardial bridge, anomalous coronary)
Spontaneous coronary artery dissection
Increased blood viscosity (Ex: polycythemia, thrombocytopenia)
Factors thatámyocardial (cardiac muscle) oxygen demand (Ex: tachycardia, hypotension, hypertension, anemia, exertion, stress)
Coronary embolism (Ex: A. Fib, endocarditis, prosthetic valve thrombus)
áheart rate, contractility, and/or wall tension ámyocardial oxygen demand
Myocardial ischemia due to imbalance between blood supply and oxygen demand (insufficient blood/oxygen supply)
Unstable Angina/Unstable Angina Pectoris
Can be new onset angina; typically progressive in frequency, severity, or duration; can occur at rest
Subtotal occlusion of a coronary arteryà
reduced, but continued, myocardial blood supply
Maintained perfusion means cardiomyocytes are still alive and thus do not leak troponin into bloodstream
Normal serum troponin
Diaphoresis
(sweating)
Since bloodflow occurs from epicardium to endocardium, myocardial ischemia is more
pronounced in the subendocardium (region furthest away from heart’s external surface)
Sufficient blood flow is maintained in regions superficial to the subendocardium, resulting in non-transmural (partial thickness) heart wall ischemia
Non-inferior wall ischemia triggers a predominantáin sympathetic nervous system activity, given the proximity of cardiac sympathetic nerve innervation
Ischemiaâ cardiomyocyte resting membrane potential andâ action potential duration
Voltage gradient between normal and subendocardial ischemic zones creates injury currents, shifting the ST- vector on ECG
ECG: ST depression
and/or T wave inversion
Cardiac sensory nerve fibres mix with somatic sensory nerve
fibres and enter the spinal cord via the T1-T4 nerve roots
Brain perceives increased cardiac sensory nerve signaling as nerve pain coming from the skin of T1-T4 dermatomes (“Referred Pain”)
Myocardial ischemia causes hypoxic stress on cardiomyocytesàâaerobic (requiring oxygen) metabolism,áanaerobic (not requiring oxygen) metabolism
áanerobic respirationálactic acid production,á[H+], andâcellular pH which impairs cardiomyocyte function
Cardiomyocyte dysfunction impairs myocardial relaxation in diastole and/orâ left ventricular contractility in systole
âleft ventricular cardiac output àbackup of blood in the left ventricle, atrium, and pulmonary vasculature
ápulmonary capillary pressures pushes fluid out of the capillaries into the alveoli in the lungs
Fluid filled alveoliâgas exchange andâ oxygenation, triggering harder and faster breathing in order to compensate
Dyspnea
Activation of sweat glands via acetylcholine release
Hormones bind to cardiac β1 receptors
Tachycardia
(áheart rate)
Epinephrine/ Norepinephrine hormone release from the adrenal medulla
Hormones bind to arterial smooth muscle α1 receptors ávascular tone (vasoconstriction)
Hypertension
The Vagus nerve sits in close physical proximity to the inferior wall of the heart àinferior wall ischemia triggers involuntary Vagus nerve activation
Since the Vagus nerve coordinates parasympathetic activity,áVagus nerve activity leads to a variety of parasympathetic nervous system responses:
Retrosternal discomfort: May present as pain, heaviness, tightness, aching, pressure, burning or squeezing
Pain radiation to T1-T4 dermatomes:
Left shoulder and arm, lower jaw, neck, abdomen, upper back
Syncope
(fainting)
Bradycardia
Nausea Hypotension
(âheart rate)
(âblood pressure)
(áblood pressure)
(shortness of breath)
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Findings
Complications
Published Oct 18, 2015, updated Aug 29, 2021 on www.thecalgaryguide.com")
type-ii-proximal-renal-tubular-acidosis-pathogenesis-and-laboratory-findings
 Zhang, Julian Midgley* * MD at time of publication
Overview of bicarbonate reabsorption in the proximal tubule (PT):
Serum bicarbonate (HCO3-) is filtered by the glomerulus and enters PT lumen
H+ ATPase and sodium- hydrogen exchanger 3
(NHE3) on luminal surface of PT cell secrete H+ into PT lumen
H+ combines with HCO3- in
PT lumen to form carbonic acid (H2CO3)
H2CO3 is broken down to H2O and CO2 by luminal membrane carbonic anhydrase 4
CO2 diffuses into PT cell
CO2 reacts with H2O to form H2CO3 in PT cell, catalyzed by cytosolic carbonic anhydrase 2
H2CO3 rapidly breaks down intracellularly to form HCO3- and H+
HCO3- is reabsorbed into plasma by sodium
bicarbonate transporter (NBCE1) on basolateral surface of PT cell
HCO3- is available for use to buffer plasma H+
H+ is available intracellularly for use by H+ ATPase and NHE3 exchanger
Mutation encoding NHE3 exchanger
↓ H+ secretion into PT lumen
Galactosemia, Wilson disease, cystinosis, tyrosinemia, glycogen storage disorders
Dent disease, Lowe syndrome
Infiltrative disorders
e.g., amyloidosis, multiple myeloma, monoclonal gammopathies
Drugs
e.g., tenofivir, ifosfamide, cisplatin
Carbonic anhydrase inhibitors
↓ H2CO3 breakdown to H2O and CO2
Mutation encoding CAII enzyme, carbonic anhydrase inhibitors
↓ H2CO3 production in PT lumen
↓ CO2 diffusion into PT cell
↓ H2CO3 production in PT cell
↓ HCO3- production in PT cell
↓ HCO3- reabsorption by PT cell into plasma
Type II/Proximal Renal Tubular Acidosis (RTA)
Defective HCO3- reabsorption in proximal tubule
Normal anion gap metabolic acidosis (NAGMA)
See NAGMA slide
Accumulation of toxic metabolites systemically, including in the PT
Disruption of endocytosis and intracellular transport systemically, including in the PT
Accumulation of light chain/amyloid deposits in PT
Interfere with PT’s ability to reabsorb ions/molecules
• • • •
Fanconi Syndrome:
generalized PT dysfunction
Defective tubular reabsorption of other ions/molecules to varying
degrees, including phosphate, glucose, sodium and amino acids
Tubular proteinuria Phosphaturia
Glucosuria
↑ Na+ secretionàhypovolemia
Mutation encoding NBCE1 transporter
↑ HCO3- delivery to distal nephron
K+ in distal nephron lumen binds to HCO3-
K+ is lost through osmotic diuresis
Hypokalemia
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published October 24, 2022 on www.thecalgaryguide.com")
Granulomatosis with Polyangiitis Pathogenesis

Environmental exposures
Silica dust, cigarette smoke, infections (Staphylococcus aureus)
Genetic factors
Alpha-1 antitrypsin deficiency, proteinase 3 gene mutation
↑ Production of cytokines and antineutrophil cytoplasmic antibodies (ANCAs) (mechanism unknown) Cytokines bind to endothelial cells (that line blood vessels) and neutrophils, priming them
Proteinase 3 (PR3), an enzyme that degrades extracellular matrix proteins, migrates from neutrophil granules to neutrophil cell surface
Circulating ANCAs bind to PR3 on neutrophils
PR3 stimulates maturation of dendritic cells in lungs
Dendritic cells present antigen (PR3) to naïve CD4+ T cells in peripheral lymph nodes
T cells differentiate into type 1 and type 17 helper T cells (Th1 and Th17 cells)
Th1 and Th17 cells secrete cytokines (interferon γ (INF-γ) and tumor necrosis factor α (TNF-α)) in lungs
Secreted cytokines trigger macrophage maturation
Formation of granulomas (giant cells with central necrosis surrounded by plasma cells, lymphocytes, and dendritic cells) primarily in lungs and upper airways
ANCA-activated neutrophils release proinflammatory cytokines, attracting more neutrophils to endothelium (blood vessel wall)
ANCA-activated neutrophils undergo firm adhesion to endothelium
ANCAs stimulate ↑ secretion of proteolytic enzymes and reactive oxygen species from neutrophils
Endothelial damage and tissue injury
Granulomatosis with polyangiitis (GPA)
ANCA-associated vasculitis affecting medium and small-sized arteries, associated with necrotizing granulomas
Constitutional symptoms with involvement of multiple organ systems
(see slide on clinical findings)
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published December 4, 2022 on www.thecalgaryguide.com
Granulomatosis with polyangiitis: Clinical findings Inflammation-mediated endothelial damage and granuloma formation
(see slide on pathogenesis)
Granulomatosis with polyangiitis (GPA)
Author:
Oswald Chen
Reviewers:
Ben Campbell
*Liam Martin
* MD at time of publication
ANCA-associated vasculitis affecting medium and small-sized arteries, associated with necrotizing granulomas
Constitutional symptoms
Fever, unintentional weight loss, night sweats, arthralgias
Skin involvement
Inflammation of cutaneous vessels
Systemic inflammation obstructing blood flow, with granulomatous lesions primarily in upper airways and lungs
Ear, nose, and throat involvement
↑ C-reactive protein (CRP) and erythrocyte sedimentation rate (ESR) (markers of inflammation)
Necrotizing granulomas on biopsy of affected tissue
Positive PR3-ANCA/c-ANCA blood test (antibodies present in ~90% of patients, described in GPA Pathogenesis slide)
Renal involvement
Inflammation of renal vessels
Rupture of basement membrane (layer that filters blood from glomerular capillaries into Bowman’s capsule)
Pauci-immune glomerulonephritis (see Nephritic Syndrome slide)
Rapidly progressive glomerulonephritis
Eye involvement
Inflammation of ocular tissue
Conjunctivitis
Scleritis/ episcleritis (painful red eye)
Lower respiratory tract involvement
Inflammation of pulmonary vessels
Vessel occlusion and ischemia
Skin necrosis
Vessels burst and blood pools under skin
Round and retiform (net- like) palpable purpura of lower extremities
Granulomatous destruction of nasal cartilage
Collapse of nasal bridge
Saddle nose deformity
Inflammation of paranasal sinus and nasal cavity vessels
↓ Perfusion of lungs
Dyspnea
Damage to interstitial capillaries
Hemoptysis
Diffuse alveolar hemorrhage
Rhinitis/ sinusitis
Granulomatous obstruction of eustachian tube
Otitis media (see Otitis Media slide)
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published December 4, 2022 on www.thecalgaryguide.com")
Granulomatosis with polyangiitis: Clinical findings

Granulomatosis with polyangiitis (GPA)
Author:
Oswald Chen
Reviewers:
Ben Campbell
*Liam Martin
* MD at time of publication
ANCA-associated vasculitis affecting medium and small-sized arteries, associated with necrotizing granulomas
Constitutional symptoms
Fever, unintentional weight loss, night sweats, arthralgias
Skin involvement
Inflammation of cutaneous vessels
Systemic inflammation obstructing blood flow, with granulomatous lesions primarily in upper airways and lungs
Ear, nose, and throat involvement
↑ C-reactive protein (CRP) and erythrocyte sedimentation rate (ESR) (markers of inflammation)
Necrotizing granulomas on biopsy of affected tissue
Positive PR3-ANCA/c-ANCA blood test (antibodies present in ~90% of patients, described in GPA Pathogenesis slide)
Renal involvement
Inflammation of renal vessels
Rupture of basement membrane (layer that filters blood from glomerular capillaries into Bowman’s capsule)
Pauci-immune glomerulonephritis (see Nephritic Syndrome slide)
Rapidly progressive glomerulonephritis
Eye involvement
Inflammation of ocular tissue
Conjunctivitis
Scleritis/ episcleritis (painful red eye)
Lower respiratory tract involvement
Inflammation of pulmonary vessels
Vessel occlusion and ischemia
Skin necrosis
Vessels burst and blood pools under skin
Round and retiform (net- like) palpable purpura of lower extremities
Granulomatous destruction of nasal cartilage
Collapse of nasal bridge
Saddle nose deformity
Inflammation of paranasal sinus and nasal cavity vessels
↓ Perfusion of lungs
Dyspnea
Damage to interstitial capillaries
Hemoptysis
Diffuse alveolar hemorrhage
Rhinitis/ sinusitis
Granulomatous obstruction of eustachian tube
Otitis media (see Otitis Media slide)
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published December 4, 2022 on www.thecalgaryguide.com")
hypovolemic-shock

Inflammation (pancreatitis, cirrhosis, post-operative, etc.)
Inflammatory mediators vessel permeability and fluid leaks out
Trauma
Ruptured vessels leak fluid into potential spaces
Hemorrhagic losses
(GI bleed, postpartum hemorrhage, etc.)
↓ Intravascular volume
↓ Venous return to the heart
↓ Cardiac output (blood pumped from the heart)
Hypovolemic Shock
↓ Oxygen delivery to tissues due to low blood volume
Insufficient organ perfusion
Non-Hemorrhagic losses
(dehydration, GI losses, skin losses / burns, renal losses, etc.)
‘Third Spacing’ of fluid
(fluid located outside the intravascular or intracellular space; large collections can occur in the pelvis, thorax, GI tract, long bones of children, intra-abdominally, retroperitoneally)
P = Q x R; less ‘flow’ in the vessels (Q), with vessels not constricting enough to maintain resistance (R)à pressure (P) will drop
↓ Blood Pressure
Caution: young, healthy individuals can maintain blood pressure during circulatory collapse with cardiac output and vasoconstriction; do not use blood pressure as an indicator of shock severity in children
Carotid sinus baroreceptors sense low blood pressure ↓ Carotid sinus inhibition of sympathetic nervous system Release of sympathetic catecholamines (epinephrine and
↓ Pressure in venous circulation
Brain
Heart
Kidneys
↓ Blood in the right internal jugular vein
↓ Oxygen delivery to the brain
↓ Myocardial contractility (from lactic acidosis)
↓ Blood flow to kidneys
↓ Jugular Venous Pressure
Catecholamines bind to beta-1 receptors in the sinoatrial node of the heart
Beta-1 receptor activation causes ↑ heart rate
Tachycardia
norepinephrine)
Catecholamines bind to and stimulate alpha-1 receptors in peripheral vessels
Vasoconstriction of peripheral vessels
↓ Blood flow to peripheral tissue
Catecholamines bind to and stimulate beta receptors in sweat glands
Diaphoresis
(sweating)
In all body tissues
Inadequate oxygen delivery
↓ ATP production
↑ Anaerobic metabolism
↓ Body temperature
Impaired neurological functioning
Renal ischemia
Activation of the renin-angiotensin aldosterone system
↓ Glomerular filtration rate
↓ Clearance of lactic acid by the kidney
↑ Lactic acid production
↓ Rate of activity of clotting enzymes
Lactic Acidosis
Unknown mechanism
Coagulopathy Hypothermia
Trauma Triad of Death
↑ Capillary Cold, mottled refill time extremities
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published January 24, 2013, updated December 4, 2022 on www.thecalgaryguide.com")
Approach to Arterial Blood Gases ABGs

Normal pO2 should be FiO2 x 4-5
(FiO2 in room air= 21%)
pH >7.40 = Alkalemia
1. Adequate oxygenation?
2. Define the acid-base disturbance
Diarrhea (Loss of electrolytes not reabsorbed through bowel)
Normal ABG on Room Air = 7.40 / 40 / 90 / 24
pO2 ↓ than expected
Possible hypoxemia
See slide on
Hypoxemia: Pathogenesis and Clinical Findings
(pH)
(Loss of acidic fluids from stomach)
(HCO3-)
(pCO2)
(pO2)
Renal Tubular Acidosis (RTA)
pH <7.40 = Acidemia
Metabolic derangement (Due to poisons, infection, or ketones)
Gain of acid (H+)
Brain unable to promote respiratory drive
(ex. drugs, trauma)
Inability for chest wall to expand (ex. obesity, neuromuscular weakness, pleural or chest wall abnormalities)
Vomiting or
nasogastric
suction
Impaired tubular transport of H+
(Due to loop/thiazide diuretics, hypomagnesemia, congenital abnormalities)
1o Hyperaldosteronism =
↑ H+ ion secretion at
distal tubule
(Due to tumours, congenital abnormalities , malignant hypertension, etc)
Type II RTA = HCO3- not reabsorbed
Type I or IV RTA = H+ not secreted
GI or renal loss of HCO3-
Hypoventilation
↑ CO2 from ↓ exhalation Respiratory Acidosis Acute= ↑10:↑1, Chronic= ↑10:↑3
GI or renal loss of acid (H+)
↑ HCO3- from ↓ H+ to bind with Metabolic Alkalosis ↑7:↑10
↑ Ventilation from stress, trauma, infection
↓ CO2 from ↑ exhalation Respiratory Alkalosis Acute= ↓10:↓2, Chronic= ↓10:↓4
3. Identify the primary (1o) process
4. Compensatory mechanisms in lungs and kidneys attempt to lose/retain CO2 or HCO3- to maintain normal pH. Determine if compensation is appropriate (approximate normal ratio of change in pCO2 : HCO3- is shown in green boxes). If inappropriate, there is a secondary (2o) process occurring
HCO3- binds with extra H+
↓ HCO3- Metabolic Acidosis ↓12 :↓10
Less CO2 than expected, due to ↑ exhalation = Less acid in serum
2o Respiratory alkalosis
More CO2 than expected, due to ↓ exhalation = More acid in serum
2o Respiratory acidosis
Less HCO3- than expected, due to 2o gain of H+ or loss of HCO3-
2o Metabolic acidosis
More HCO3- than expected, due to 2o loss of H+
2o Metabolic alkalosis
Less CO2 than expected, due to ↑ exhalation = Less acid in serum
2o Respiratory alkalosis
↑ CO2 than expected, due to ↓ exhalation = More acid in serum
2o Respiratory acidosis
Less HCO3- than expected, due to 2o gain of H+ or loss of HCO3-
2o Metabolic acidosis
More HCO3- than expected, due to 2o loss of H+
2o Metabolic alkalosis
5. Calculate anion gap (AG) to determine the presence and type of metabolic acidosis. This is done regardlessofthe1o or2oprocess, as there may also be a hidden metabolic acidosis
6. HAGMA only: In normal blood buffer system, acid gain should match bicarbonate lost. If not, identify if another process is causing an inappropriate loss or gain of HCO3-
Gain of acid (∆AG) = AG-12 LossofHCO3- (∆HCO3-)=24-HCO3-
7. Calculate Osmolar Gap (for HAGMA) or Urine net charge (for NAGMA) to narrow the etiology
Authors:
Sravya Kakumanu
Reviewers:
Huneza Nadeem, Ben Campbell *Adam Bass, *Yan Yu
* MD at time of publication
(normal is ~12)
Calculate AG = Na+- Cl- - HCO3-
If a metabolic acidosis is present, ↓ HCO3- is
compensatedby↑otherserumanionstomaintain neutral charge
If no metabolic acidosis was identified in steps 2-4:
No Metabolic acidosis present
Classically, the compensating anion is Cl-,maintaininganormalAG
AG ≤ 12
If a 1o or 2o metabolic acidosis was identified in steps 2-4:
Normal Anion Gap Metabolic Acidosis (NAGMA)
In this scenario, a normal distal nephron should be excreting excess acid
Excess acid is normally excreted as ammonium (NH +) with Cl- 4
The more Cl- in the urine relative to cations, the more acid is being excreted Calculate urine (U) net charge to determine cause of NAGMA = UNa+ + UK+ - UCl-
When the cause is addition of an abnormal acid, HCO3- is used up asabuffer,andthecompensatinganionistheabnormalacid's conjugate base – which is not measured in the AG calculation
↓ HCO3- is compensated by unmeasured anionsà the AG ↑ AG>12
High Anion Gap Metabolic Acidosis (HAGMA)
Calculate ∆AG = AG-12
Calculate ∆HCO3- = 24-HCO3-
∆HCO3- > ∆AG
Loss of HCO3- > Gain of acid
Additional process causing further loss of HCO3-
HAGMA + NAGMA
∆HCO3- = ∆AG
Loss of HCO3- = Gain of acid
Only acid gain from HAGMA causing HCO3- loss
HAGMA only
∆HCO3- < ∆AG
Loss of HCO3- < Gain of acid
Additional process causing gain of HCO3- despite losses due to HAGMA
HAGMA + Metabolic alkalosis
See slide on
Metabolic Alkalosis: Pathogenesis
-ve U net charge Appropriately large Cl- excretionà
Toxic alcohol ingestion ↑ serum osmolality (and H+) from metabolites produced, so osmolality helps identify etiology
Calculate osmolar gap to determine cause of HAGMA
= (measured serum osm) – (expected serum osm)
= (measured serum osm) – (2(Na+) + urea + serum glucose)
+ve U net charge
Inappropriately small Cl- excretionà impaired acid excretion is the issue
Type IV or Type I RTA See slide on Normal Anion Gap Metabolic Acidosis: Pathogenesis and Laboratory Findings
HCO3
GI losses or Type II RTA
-
loss is the issue
Osmolar gap > 10 (extra osmoles present)
Toxic alcohol poisoning
Osmolar gap ≤10 (normal)
Test for other causes
See slide on High Anion Gap Metabolic Acidosis: Pathogenesis and Laboratory Findings
Legend:
Disease State
Mechanism
Lab Finding/Calculation
Published January 29, 2023 on www.thecalgaryguide.com")
Coronary Artery Bypass Graft CABG Indications
: Indications
Author: Breanne Gordulic Reviewers: Miranda Schmidt Ben Campbell Sunawer Aujla Angela Kealey* * MD at time of publication
Symptomatic multivessel (≥ 3 vessels) coronary artery disease (MVCAD) or complex MVCAD
Acute coronary syndrome (ACS)
Left main coronary artery disease
Multivessel (≥ 3 vessels) CAD and diabetes
Cardiac surgery required for other pathology
Multivessel CAD, LV dysfunction and congestive heart failure (CHF)
Complex CAD includes stenosed vein grafts, bifurcation lesions, calcified lesions, total occlusions, ostial lesions
STEMI initial treatment is PCI/thrombolysis
CABG outcomes compared to percutaneous coronary intervention (PCI) in MVCAD include ↑ survival in diabetes, ↑ survival with LV dysfunction, ↓ repeat revascularization, ↓ myocardial infarction, ↓ stroke
↑ Risk of failure in complex CAD with PCI
Rapid reperfusion to myocardium most important in STEMI to decrease myocardial damage
CABG can be considered for residual stenoses 6-8 weeks later
NSTEMI or unstable angina (UA) with MVCAD involving at least three vessels including the proximal left anterior descending (LAD)
Left main coronary artery divides into left anterior descending (LAD) and left circumflex (LCx) which supplies 2/3 of myocardium
↑ Survival Myocardial infarction from left main artery occlusion Death
Left main stenosis
Ventricular dysrhythmias
Ongoing ischemia
LV dysfunction Hemodynamic instability
↑ risk of PCI
CABG has mortality benefit
↓ Number of operations
↑ Risk of cardiovascular disease in diabetes
Revascularization indicated along with other cardiac surgery
Multivessel CAD with >90% stenosis and CHF
LV ejection fraction <35%
↑ Risk of atherosclerosis from hyperglycemia and dyslipidemia
CABG bypasses several atherosclerotic plaques in coronary arteries
↑ Durability
↑ Complete perfusion
Valve stenosis or regurgitation Septal defect
Aortic root or arch pathology
Combination procedure
Evidence of ischemia at rest
Evidence of
impaired LV function at rest
↓ All-cause mortality in CABG vs medical management
Chronic obstructive pulmonary disease
Abbreviations:
• ACS- acute coronary syndrome. Acute reduction in
blood flow to heart muscle resulting in cell death. • CAD- coronary artery disease. Narrowing or blockage of the coronary arteries by plaque
• NSTEMI- myocardial infarction (heart attack) with no ST elevation on electrocardiogram
• PCI- percutaneous coronary intervention. A balloon tipped catheter is used to open blocked coronary arteries; a stent may be placed.
• STEMI- myocardial infarction with ST segment elevation on electrocardiogram
Consider revascularization (restore blood flow to blocked or narrowed blood vessels) of coronary arteries to increase perfusion to myocardium (heart muscle)
Coronary artery bypass graft recommended
Assess surgical risk and comorbidities with evaluation by heart team that includes both a cardiac surgeon and interventional cardiologist (SYNTAX Trial)
Individual management plan for patients with comorbidities that increase mortality
Frailty
Chronic Renal Failure
↑ Inflammation and deregulated angiogenesis affects all organ systems
↓ Physiologic reserve and ↓ ability to recover from acute stress
↑ Pneumonia
↑ Respiratory and Renal Failure
↑ Stroke
↑ In hospital mortality
↓ Survival two years after surgery
Use Society of Thoracic Surgeons Score, EuroSCORE, or SYNTAX II Score to predict patient outcome with anatomy, disease severity, and preoperative characteristics
Coronary Artery Bypass Graft
Surgery to take healthy blood vessels from the body and connect them proximally and distally to blocked coronary arteries
Cardiopulmonary bypass, fluid overload, ↑ renal vasoconstriction and ↓ renal oxygenation from rewarming
Kidney injury
↑ End stage kidney disease
Blood flow restored to
ischemic myocardium
↓ Angina
↑ Quality of life ↑ LV function ↑ Survival
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published April 12, 2023 on www.thecalgaryguide.com")
normal neonatal changes pathogenesis and clinical findings
.
Authors: Erin Auld, Dasha Mori Reviewers: Kayla Feragen Mao Ding Danielle Nelson* *MD at time of publication
Birth weight: Loss of birth weight up to 10%; should be re-gained within 10-14 days (30 g/day)
Stool: transitions from meconium (first stool of neonate that is black, tarry and sticky) to normal (green/brown or yellow mustard) within 2-3 days
Meconium passage: within 24 hours of birth
Growth
• Height: 2.5 cm/month
• Head Circumference: Average 1 cm/month in the first year with greatest growth in the first month • Weight: 20-30 g/day for the first 3 months
Urination: within the first 24 hours
Mother receives intravenous fluids during delivery
Colostrum (first milk secretion that contains antibodies) produced in first 2-3 days of lactation post-partum (Lactogenesis I)
GI tract maturation
Adequate dietary intake
Maturation of urinary tract
Adequate fluid intake
↑ maternal blood volume
Fluid moves between fetus and mother through placenta
Fetus’s fluid volume ↑
Infant’s urine output ↑ in first 24 hours post-partum
Low breast milk intake in first 2-3 days
↓ progesterone, ↑ prolactin in mother at birth
Infant ingests colostrum
Mostly water loss, some fat loss
↑ Breast Milk Production (Lactogenesis II)
Stomach stretches
↑ in gastrointestinal tract motility (gastrocolic reflex)
Ingestion of milk post-partum further stimulates GI maturation
110-120 kilocalories/kg/day
Gastrointestinal tract formation begins when the fetus is 4 weeks old. Maturation continues into infancy.
Normal gestational age (38-42 weeks)
Fetus ingests amniotic fluid in utero
Stimulates structural changes, enzymatic activity, and metabolic activity of GI tract
Mature detrusor sphincter complex, appropriate bladder capacity, proper renal perfusion, and regular arousal of the neonate
Breast milk: Feeding when infant demands it, approximately every 2-3 hours Formula: Feeding when infant demands it, approximately every 4 hours
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published Oct 24, 2015, updated Sept 28, 2023 on www.thecalgaryguide.com")
Alcohol Withdrawal Syndrome
: Pathophysiology & clinical findings
Authors: Rupali Manek Gurreet Bhandal Erika Russell Reviewers: Harjot Atwal Yvette Ysabel Yao Mao Ding Nureen Pirbhai* * MD at time of publication
Chronic alcohol use
↓Autonomic adrenergic systems
↓ Dopamine in the nucleus accumbens
↑ EtOH depressant effects on brain
↓ Glutamate- induced excitation
↑ GABA-induced inhibition
↑ Glutamate receptors in attempt to maintain normal arousal state
GABA insensitivity (↑ GABA needed to maintain a constant inhibitory tone)
Long-term physical dependence
Abrupt alcohol cessation àabrupt ↓ in blood EtOH concentration
Alcohol withdrawal Syndrome
Symptoms that occur when patients stop drinking or significantly decrease their alcohol intake after long-term dependence
↓ GABA-induced inhibition &↑ glutamate-induced excitation relative to chronic alcohol use
Central nervous system overactivity
Withdrawal seizures: Generalized tonic-clonic convulsions 24-48 hours after alcohol cessation
Fluid and electrolyte abnormalities
↑ Autonomic adrenergic systems (rebound over-activity of the brain and noradrenergic systems)
↑ Sympathetic activity: ↑ Heart rate (HR), ↑respiratory rate (RR) ↑blood pressure (BP), tremor & diaphoresis (↑sweating)
Early symptoms: Insomnia, tremulousness, anxiety, digestive upset, anorexia, headache, sweating, palpitations 6-12 hours after alcohol cessation
↑ Dopamine in nucleus accumbens
Alcoholic hallucinosis: Usually visual (but can be auditory or tactile), normal vitals 12-24 hours after alcohol cessation, typically resolve within 24-48 hours
Alcohol withdrawal delirium (delirium tremens or DT):
Hallucinations (mostly visual), disorientation, tachycardia, hypertension, hyperthermia, agitation, and diaphoresis 48-96 hours after alcohol cessation and lasts 1-5 days
Hypovolemia
(from diaphoresis, hyperthermia, vomiting, ↑RR & ↓oral intake)
Metabolic acidosis
(from hypoperfusion, infection, alcoholic ketoacidosis, or ↓
thiamine & other B vitamins)
↓ Potassium (K) (renal & extrarenal K losses, alterations in aldosterone concentrations, and changes in K distribution across the cell membrane)
↓ Phosphate
(from malnutrition)
Cardiac failure, rhabdomyolysis or muscle breakdown
↓ Phosphate available to make ATP
↓ ATP
↓ Magnesium (common in patients with DT)
Impaired Na-K ATPase function
Dysrhythmias
↑ Glutamate-activated depolarization in the brain
↑ Neuronal excitability
Seizures
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published Apr 15, 2017, updated Oct 18, 2023 on www.thecalgaryguide.com
Alcohol Withdrawal Syndrome (AWS): Pathophysiology & clinical findings
Authors: Rupali Manek Gurreet Bhandal Erika Russell Reviewers: Harjot Atwal Yvette Ysabel Yao Mao Ding Nureen Pirbhai* * MD at time of publication
Chronic alcohol use
↓Autonomic adrenergic systems
↓ Dopamine in the nucleus accumbens
↑ EtOH depressant effects on brain
↓ Glutamate- induced excitation
↑ GABA-induced inhibition
↑ Glutamate receptors in attempt to maintain normal arousal state
GABA insensitivity (↑ GABA needed to maintain a constant inhibitory tone)
Long-term physical dependence
Abrupt alcohol cessation àabrupt ↓ in blood EtOH concentration
Alcohol withdrawal Syndrome
Symptoms that occur when patients stop drinking or significantly decrease their alcohol intake after long-term dependence
↓ GABA-induced inhibition &↑ glutamate-induced excitation relative to chronic alcohol use
Central nervous system overactivity
Withdrawal seizures: Generalized tonic-clonic convulsions 24-48 hours after alcohol cessation
Fluid and electrolyte abnormalities
↑ Autonomic adrenergic systems (rebound over-activity of the brain and noradrenergic systems)
↑ Sympathetic activity: ↑ Heart rate (HR), ↑respiratory rate (RR) ↑blood pressure (BP), tremor & diaphoresis (↑sweating)
Early symptoms: Insomnia, tremulousness, anxiety, digestive upset, anorexia, headache, sweating, palpitations 6-12 hours after alcohol cessation
↑ Dopamine in nucleus accumbens
Alcoholic hallucinosis: Usually visual (but can be auditory or tactile), normal vitals 12-24 hours after alcohol cessation, typically resolve within 24-48 hours
Alcohol withdrawal delirium (delirium tremens or DT):
Hallucinations (mostly visual), disorientation, tachycardia, hypertension, hyperthermia, agitation, and diaphoresis 48-96 hours after alcohol cessation and lasts 1-5 days
Hypovolemia
(from diaphoresis, hyperthermia, vomiting, ↑RR & ↓oral intake)
Metabolic acidosis
(from hypoperfusion, infection, alcoholic ketoacidosis, or ↓ thiamine & other B vitamins)
↓ Potassium (K) (renal & extrarenal K losses, alterations in aldosterone concentrations, and changes in K distribution across the cell membrane)
↓ Magnesium (common in patients with DT)
Dysrhythmias, seizures
↓ Phosphate
(from malnutrition)
↓ Phosphate available to make ATP
↓ ATP
Cardiac failure, rhabdomyolysis or muscle breakdown
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published April 28th, 2014 on www.thecalgaryguide.com
Alcohol Withdrawal: Pathophysiology & clinical findings
Authors: Erika Russell Rupali Manek Gurreet Bhandal Reviewers: Yvette Ysabel Yao Harjot Atwal Nureen Pirbhai* * MD at time of publication
↓Autonomic adrenergic systems
↓ Glutamate-induced excitation
Chronic alcohol use
↑ EtOH depressant effects on brain
↓ Dopamine in the nucleus accumbens
↑ GABA-induced inhibition
↑ Glutamate receptors in attempt to maintain normal arousal state GABA insensitivity (↑ GABA needed to maintain a constant inhibitory tone)
↑ Autonomic adrenergic systems (rebound over- activity of the brain and noradrenergic systems)
↑ Sympathetic activity: ↑ Heart rate (HR), ↑respiratory rate (RR) ↑blood pressure (BP), tremor & diaphoresis (↑sweating)
Physical dependence due to chronic alcohol use
Abrupt alcohol cessation
Abrupt ↓ in blood EtOH concentration
Alcohol withdrawal
↓ GABA-induced inhibition &↑ glutamate-induced excitation relative to chronic alcohol use
Central nervous system overactivity
Withdrawal seizures: Generalized tonic-clonic convulsions 24-48 hours after alcohol cessation
Fluid and electrolyte abnormalities
↑ Dopamine in nucleus accumbens
Alcoholic hallucinosis: Usually visual (but can be auditory or tactile), normal vitals
12-24 hours after alcohol cessation, typically resolve within 24-48 hours
Early symptoms: Insomnia, tremulousness, anxiety, digestive upset, anorexia, headache, sweating, palpitations 6-12 hours after alcohol cessation
Alcohol withdrawal delirium (delirium tremens or DT):
Hallucinations (mostly visual), disorientation, tachycardia, hypertension, hyperthermia, agitation, and diaphoresis 48-96 hours after alcohol cessation and lasts 1-5 days
Hypovolemia (from diaphoresis, hyperthermia, vomiting,
↑RR & ↓oral intake)
Metabolic acidosis (from
hypoperfusion, infection, alcoholic ketoacidosis, or ↓ thiamine & other B vitamins)
↓ Potassium (K) (renal & extrarenal K losses, alterations in aldosterone concentrations, and changes in K distribution across the cell membrane)
↓ Magnesium (common in patients with DT)
Dysrhythmias, seizures
↓ Phosphate (from malnutrition)
↓ Phosphate available to make ATP
↓ ATP
Cardiac failure, rhabdomyolysis or muscle breakdown
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published April 28th, 2014 on www.thecalgaryguide.com
Authors: Erika Russell Rupali Manek Gurreet Bhandal Reviewers: ↓Autonomic adrenergic systems ↓ Dopamine in the nucleus accumbens Yvette Ysabel Yao Harjot Atwal Nureen Pirbhai* * MD at time of publication
Alcohol Withdrawal: Pathophysiology & clinical findings Chronic alcohol use
↑ EtOH depressant effects on brain
↓ Glutamate-induced excitation ↑ GABA-induced inhibition
↑ Glutamate receptors in attempt to maintain normal arousal state GABA insensitivity (↑ GABA needed to maintain a constant inhibitory tone)
↑ Autonomic adrenergic systems (rebound over- activity of the brain and noradrenergic systems)
↑ Sympathetic activity: ↑ Heart rate (HR), ↑respiratory rate (RR) ↑blood pressure (BP), tremor & diaphoresis (↑sweating)
Physical dependence due to chronic alcohol use
Abrupt alcohol cessation
Abrupt ↓ in blood EtOH concentration Alcohol withdrawal
↓ GABA-induced inhibition &↑ Glutamate-induced excitation relative to chronic alcohol use
Central nervous system overactivity
Withdrawal seizures: Generalized tonic-clonic convulsions 24-48 hours after alcohol cessation
Fluid and electrolyte abnormalities
↓ Potassium (K) (renal & extrarenal K losses, alterations in aldosterone concentrations, and changes in K distribution across the cell membrane)
↑ Dopamine in nucleus accumbens
Alcoholic hallucinosis: Usually visual (but can be auditory or tactile), normal vitals
12-24 hours after alcohol cessation, typically resolve within 24-48 hours
Alcohol withdrawal delirium (delirium tremens or DT):
Hallucinations (mostly visual), disorientation, tachycardia, hypertension, hyperthermia, agitation, and diaphoresis 48-96 hours after alcohol cessation and lasts 1-5 days
Early symptoms: Insomnia, tremulousness, anxiety, digestive upset, anorexia, headache, sweating, palpitations 6-12 hours after alcohol cessation
Hypovolemia (from diaphoresis, hyperthermia, vomiting, ↑RR & ↓oral intake)
Metabolic acidosis (from
hypoperfusion, infection, alcoholic ketoacidosis, or ↓ thiamine & other B vitamins)
↓ Magnesium (common in patients with DT)
Dysrhythmias, seizures
↓ Phosphate (from malnutrition) Cardiac failure, rhabdomyolysis
or muscle breakdown
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published April 28th, 2014 on www.thecalgaryguide.com
Alcohol Withdrawal: Pathophysiology & clinical findings Chronic alcohol use
↓ Dopamine in the nucleus accumbens ↓Autonomic adrenergic systems
Authors: Erika Russell Rupali Manek Gurreet Bhandal Reviewers: Yvette Ysabel Yao Harjot Atwal ↑ GABA-induced inhibition Nureen Pirbhai* * MD at time of publication
↑ EtOH depressant effects on brain
↓ Glutamate-induced excitation
↑ Glutamate receptors in attempt to maintain normal arousal state
GABA insensitivity (↑ GABA needed to maintain a constant inhibitory tone)
↑ Autonomic adrenergic systems (rebound over- activity of the brain and noradrenergic systems)
↑ Sympathetic activity: ↑Heart rate (HR), ↑respiratory rate (RR) ↑blood pressure (BP), tremor & diaphoresis (↑sweating)
Physical dependence due to chronic alcohol use
Abrupt alcohol cessation
Abrupt ↓ in blood EtOH concentration Alcohol withdrawal
↓ GABA-induced inhibition &↑ Glutamate-induced excitation relative to chronic alcohol use
Central nervous system overactivity
Withdrawal seizures: Generalized tonic-clonic convulsions 24-48 hours after alcohol cessation
Fluid and electrolyte abnormalities
↑ Dopamine in nucleus accumbens
Alcoholic hallucinosis: Usually visual (but can be auditory or tactile), normal vitals
12-24 hours after alcohol cessation, typically resolve within 24-48 hours
Alcohol withdrawal delirium (delirium tremens or DT):
Hallucinations (mostly visual), disorientation, tachycardia, hypertension, hyperthermia, agitation, and diaphoresis 48-96 hours after alcohol cessation and lasts 1-5 days
Early symptoms: Insomnia, tremulousness, anxiety, digestive upset, anorexia, headache, sweating, palpitations 6-12 hours after alcohol cessation
Hypovolemia (from diaphoresis, hyperthermia, vomiting, ↑RR & ↓oral intake)
Metabolic acidosis (from
hypoperfusion, infection, alcoholic ketoacidosis, or ↓ thiamine & other B vitamins)
↓ Potassium (K) (renal & extrarenal K losses, alterations in aldosterone concentrations, and changes in K distribution across the cell membrane)
↓ Magnesium
(common in patients with DT)
Dysrhythmias, seizures
↓ Phosphate (from malnutrition)
Cardiac failure, rhabdomyolysis or muscle breakdown
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published April 28th, 2014 on www.thecalgaryguide.com
Alcohol Withdrawal: Pathophysiology & clinical findings
Authors: Erika Russell Rupali Manek Gurreet Bhandal Reviewers: Yvette Ysabel Yao Harjot Atwal Nureen Pirbhai* * MD at time of publication
↑ EtOH depressant effects on brain
↓ Glutamate-induced excitation
↑ Glutamate receptors in attempt to maintain normal arousal state
Chronic alcohol use
↑ GABA-induced inhibition
GABA insensitivity (↑ GABA needed to maintain a constant inhibitory tone)
↓ Dopamine in the nucleus accumbens ↓Autonomic adrenergic systems
Abrupt alcohol cessation
Physical dependence due to chronic alcohol use
Alcohol withdrawal
↓ GABA-induced inhibition &↑ Glutamate-induced excitation relative to chronic alcohol use
Central nervous system overactivity
Withdrawal seizures: Generalized tonic-clonic convulsions 24-48 hours after alcohol cessation
↑ Autonomic adrenergic systems (rebound over-activity of the brain and noradrenergic systems)
↑ Sympathetic activity: ↑HR, ↑RR ↑BP, tremor & diaphoresis (↑sweating)
Early symptoms: Insomnia, tremulousness, anxiety, digestive upset, anorexia, headache, sweating, palpitations
6-12 hours after alcohol cessation
↑ Dopamine in nucleus accumbens
Alcoholic hallucinosis: Usually visual (but can be auditory or tactile), normal vitals
12-24 hours after alcohol cessation, typically resolve within 24-48 hours
Alcohol withdrawal delirium (delirium tremens or DT):
Hallucinations (predominately visual), disorientation, tachycardia, hypertension, hyperthermia, agitation, and diaphoresis 48-96 hours after alcohol cessation and lasts 1-5 days
Fluid and electrolyte abnormalities
Hypovolemia (from diaphoresis, hyperthermia, vomiting, ↑ RR & ↓
oral intake)
Metabolic acidosis (from
hypoperfusion, infection, alcoholic ketoacidosis, or ↓ thiamine & other B vitamins)
↓ Potassium (K) (renal & extrarenal K losses, alterations in aldosterone concentrations, and changes in K distribution across the cell membrane)
↓ Magnesium
(common in patients with DT)
Dysrhythmias, seizures
↓ Phosphate (from malnutrition)
Cardiac failure, rhabdomyolysis or muscle breakdown
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published April 28th, 2014 on www.thecalgaryguide.com
Alcohol Withdrawal: Pathophysiology & clinical findings
Authors: Erika Russell Rupali Manek Gurreet Bhandal Reviewers: Yvette Ysabel Yao Harjot Atwal Nureen Pirbhai* * MD at time of publication
↓ GABA-induced inhibition &↑ Glutamate-induced excitation relative to chronic alcohol use
CNS overactivity
Alcohol withdrawal delirium (delirium tremens or DT): Hallucinations (predominately visual), disorientation, tachycardia, hypertension, hyperthermia, agitation, and diaphoresis
48-96 hours after alcohol cessation and lasts 1-5 days
Alcohol cessation ↓ Dopamine
Alcohol withdrawal
Chronic alcohol use
↑ EtOH depressant effects on brain
in the NAc
↓Autonomic adrenergic systems
↑ Dopamine in nucleus accumbens
(NAc) relative to chronic alcohol use
Alcoholic hallucinosis: Usually visual (but can be auditory or tactile), normal vitals 12-24 hours after alcohol cessation, typically resolve within 24-48 hours
↑ Autonomic adrenergic systems relative to chronic alcohol use
↑ Sympathetic activity: ↑HR, ↑RR ↑BP, tremor & diaphoresis (↑sweating)
↓ Glutamate- induced excitation
↑ Glutamate receptors in attempt to maintain normal arousal state
↑ GABA-induced inhibition
GABA insensitivity (↑ GABA needed to maintain a constant inhibitory tone)
Early symptoms: Insomnia, tremulousness, anxiety, digestive upset, anorexia, headache, sweating, palpitations
6-12 hours after alcohol cessation
Withdrawal seizures: Generalized tonic- clonic convulsions 24-48 hours after alcohol cessation
Fluid and electrolyte abnormalities
Abbreviations:
CNS – Central nervous system K – Potassium
DT – Delirium tremens
Mg – Magnesium
NAc – Nucleus accumbens NMDA – N-methyl-D-aspartate EtOH – Alcohol
NT – Neurotransmitter
Hypovolemia (from diaphoresis, hyperthermia, vomiting, ↑ RR & ↓
oral intake)
Metabolic acidosis (from hypoperfusion, infection, alcoholic ketoacidosis, or ↓
thiamine & other B vitamins)
↓ K (renal & extrarenal K losses, alterations in aldosterone concentrations, and changes in K distribution across the cell membrane)
↓ Mg (common in patients with DT)
Dysrhythmias, seizures
↓ Phosphate (from malnutrition)
Cardiac failure, rhabdomyolysis or muscle breakdown
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published April 28th, 2014 on www.thecalgaryguide.com
Alcohol Withdrawal: Clinical Findings and Complications
Authors: Erika Russell Reviewers: Harjot Atwal Nureen Pirbhai* * MD at time of publication
Note: *The onset of alcohol withdrawal generally begins 6-24 hours after the last drink with symptoms peaking between 24-36 hours after and gradually lessening.
Symptoms typically progress from early symptoms and increased sympathetic activityà hallucinationsàseizures àpotentially Delirium Tremens.
Alcohol withdrawal is mild-moderate in severity for 90% of patients. Those who progressively worsen however can enter Delirium Tremens (DT) which has a mortality rate of up to 20%. More likely to have DT if had DT before, age >30 years, concurrent illness, >2 days after EtOH cessation before seeking help, and history of sustained drinking.
Long term, heavy alcohol use that leads to physical dependence
Abrupt ↓ in blood EtOH concentration
Negative physiological reactions to ↓ alcohol intake
Adaptive suppression of GABA activity from chronic alcohol enhancement
Alcohol Withdrawal*
Withdrawal symptoms alleviated by ingesting alcohol
Alcohol taken to relieve withdrawal AND/OR
Social and internal relapse cues trigger urge to use alcohol
Blood EtOH levels >600 mg% can lead to lethal respiratory depression by suppressing the respiratory centers in the brainstem
Upregulated autonomic adrenergic systems from chronic alcohol inhibition
Discontinuation of alcohol leads to rebound over-
activity of the brain and noradrenergic systems
Increased Sympathetic Activity Tachycardia, hypertension, tremor and diaphoresis
Generalized Tonic-Clonic Seizures
Usually begin within 8-24 hours of alcohol cessation and peak after 24 hours. Risk of having seizures ↑ with repeated withdrawals. 1/3 of people can progress to DT if seizures left untreated.
Discontinuation of alcohol causes a sudden relative
deficiency in inhibitory GABA activity
Reduction in dopamine in the nucleus accumbens
(NAc) from chronic alcohol exposure
Discontinuation of alcohol causes a increase in dopamine levels in NAc
Hallucinations
Commonly visual (but can be auditory or tactile), develop 12-24 hours after alcohol cessation.
Early Symptoms
Anxiety, insomnia, vivid dreams, anorexia, nausea, headache and psychomotor agitation
Delirium Tremens (DT)
Life-threatening state of greatly exaggerated withdrawal symptoms (severe tachycardia, diaphoresis etc.) with confusion/disorientation and hallucinations that generally appears 72-96 hours after the last drink and lasts 2-3 days.
Legend:
Published MONTH, DAY, YEAR on www.thecalgaryguide.com
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications")
Pharmacotherapy for Dyslipidemia Overview

Bile-acid sequestrants
Bind bile acids in intestinal lumen to prevent reabsorption by enterohepatic (gut-liver) circulation
↑ Excretion of bile acids and cholesterol in stool
↓ LDL in blood
Side effects: GI disturbances, commonly interact with other drugs by interfering with absorption
See Calgary guide slide on “Bile-acid sequestrants: Mechanisms of action & side effects” for complete description of mechanism and side effects
(clinical imbalance of lipids)
Hypertriglyceridemia (if VLDL mediated & in need of treatment for pancreatitis prevention)
Hypercholesteremia
(↑ LDL in blood)
Ezetimibe
Inhibits cholesterol absorption via NPC1L1 transporter
↑ Hepatic (liver) LDL receptor expression
↑ LDL clearance from blood
↓ LDL in blood
Avoid in pregnancy
See Calgary guide slide on “Ezetimibe: Mechanisms of action & side effects” for complete description of mechanism and side effects
Combined hyperlipidemia (↑ Triglycerides and ↑ cholesterol)
Statins (ex. rosuvastatin, atorvastatin, simvastatin, pravastatin)
Competitive inhibitors of HMG-CoA reductase (rate-limiting enzyme in cholesterol synthesis)
Fibrates (ex. fenofibrate, gemfibrozil)
Activate PPAR! (nuclear receptor)
↑ Lipolysis (breakdown of lipids) and free fatty acid oxidation
↓ Triglycerides in blood
Side effects: GI discomfort, rash, pruritis
Contraindicated in pregnancy, renal failure, liver & gallbladder disease
See Calgary guide slide on “Fibrates: Mechanisms of action & side effects” for complete description of mechanism and side effects
PCSK9 inhibitors (ex. evolocumab and alirocumab which are monoclonal antibodies)
Inhibit PCSK9 (holds the LDL:LDL receptor complex together as it is internalized into the cell for destruction of LDL)
LDL receptor returns to surface without being destroyed
↑ LDL receptor expression
↑ LDL clearance from blood
↓ LDL in blood
See Calgary guide slide on “PCSK9 Inhibitors: Mechanisms of action & side effects” for complete description of mechanism and side effects
↓ Cholesterol synthesis in liver
↑ LDL receptor expression in liver
LDL receptor recognizes apoB100 (structural protein on LDL) and apoE (structural protein found on chylomicron, VLDL, IDL)
↑ Clearance of LDL cholesterol from bloodstream
↓ LDL cholesterol in blood ↑ HDL in blood
↓ Triglycerides in blood
↓ Atherosclerosis (plaque along walls of blood vessels)
Abbreviations: HDL – High density lipoprotein; HMG-CoA – Hydroxymethylglutaryl- CoA; LDL – Low-density lipoprotein; PCSK9 – Proprotein convertase subtilisin/kexin type 9; PPAR! – Peroxisome proliferator-activated receptor alpha; NPC1L1 – Niemann-Pick C1-Like 1; VLDL – Very low-density lipoprotein
Side effects: Myalgias (muscular aches), rhabdomyolysis (muscle breakdown), transaminitis (liver inflammation), liver failure, ↑ risk of diabetes mellitus
Contraindicated in pregnancy
See Calgary guide slide on “Statins: Mechanisms of action & side effects” for complete description of mechanism and side effects
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published Nov 6, 2023 on www.thecalgaryguide.com")
Death Cardiovascular Respiratory and Neurologic Mechanisms

Hypoxemia (Type I Respiratory Failure): low dissolved oxygen in blood (PaO2)
Lungs can’t oxygenate blood fast enough
Lungs can’t rid blood of CO2 fast enough
Hypercapnia / hypercarbia (Type II Respiratory Failure): elevated dissolved CO2 in blood (PaCO2)
Cerebral vasodilation
Toxins: e.g. cyanide, pesticides, arsenic Severe anemia
Distributive problems:
Systemic inflammation (sepsis, anaphylaxis, pancreatitis), adrenal insufficiency, vasodilatory drugs
Obstructive problems: Cardiac tamponade*, tension pneumothorax* or massive pulmonary embolism*
Hypovolemic* problems (low blood volume): Hemorrhage, dehydration, widespread skin disruption or burns
Cardiac valve dysfunction
Myocardial infarction* or cardiomyopathy
Cardiac arrhythmia or heart block
Disturbed electrical activity in cardiomyocytes
Peripheral metabolic disturbances
Hypokalemia*, Hyperkalemia* Acidosis* (including renal failure) Hypothermia*
Toxins* (e.g. cocaine, beta blockers, tricyclics) Severe thyroid derangement
Inappropriate systemic vasodilation
Adjacent forces impair heart filling
Low cardiac preload
Low stroke volume (SV; depends on valves, contractility, preload)
Decreased systemic vascular resistance (SVR)
Low blood pressure (BP = CO x SVR)
Decreased cardiac output (CO = SV x HR)
Disseminated intravascular coagulationàwidespread thrombi that occlude blood flow (also causes hemorrhage, see relevant box at left)
Methemoglobinemia: some hemoglobin gets stuck in a state that can’t carry O2
Hemoglobin has reduced capacity to carry or release O2
Drugs: e.g. dapsone, nitrates
Carbon monoxide poisoning
Circulatory collapse / shock: inadequate perfusion of tissue with blood
Respiratory collapse: blood has insufficient useable O2 content
Ventricular fibrillation (VF) or pulseless ventricular tachycardia (VT)
Hypoxia*: inadequate O2 delivery or utilization in tissues
Hypoxia creates metabolic disturbances that impair cardiac cells. Alternatively, any of the preceding conditions marked with (*) can directly trigger cardiac arrest first
Pulseless Electrical Activity (PEA): organized activity on ECG with no cardiac output (can be preceded or mimicked by pseudo-PEA, in which there is still some output on ultrasound)
Low atmospheric pressure or oxygen content Severe lung disease
Asthma, COPD, interstitial lung disease, congestive heart failure, pulmonary hypertension, pulmonary embolism, lung collapse / atelectasis
Acute respiratory distress syndrome
Pneumonia, aspiration pneumonitis, inhalational injury, systemic inflammation, drowning
Severe hypoventilation
Respiratory fatigue, advanced COPD, chest wall disorders, neuromuscular disorders, upper airway obstruction, toxins (e.g. opioids, botulism)
Can degenerate at any time
Asystole: no cardiac electrical activity or output
Death
Respiratory arrest: cessation of breathing
Inability to protect airway
Decreased level of consciousness
Note
This is a broad overview of the many scenarios that can result in death. For detailed explanations of the various disease mechanisms, refer to the corresponding slides.
* = reversible causes of cardiac arrest (Hs and Ts)
Author:
Ben Campbell
Reviewers:
Yan Yu*
Huma Ali*
* MD at time of publication
Bradycardia
(low heart rate, HR)
Unopposed parasympathetic stimulation of heart (can also cause vasodilation, see Distributive problems)
Disruption of spinal cord sympathetic control
Injury to cervical or upper thoracic spinal cord
Irreversible cessation of cardiac, respiratory, and brain function
Prolonged seizure initially causes increased cardiovascular activity, until the system fatigues
Disruption of respiratory control center in medulla
Expanding skull contents squeeze brainstem (herniation)
Increased intracranial pressure
Edema from intracranial hemorrhage, trauma, brain mass
Edema, inflammation, hypoxia and/or metabolic derangements cause diffuse neuron dysfunction
Central nervous system infection
Dementia, particularly with delirium
Massive ischemic stroke
Seizure
activity prevents or alters breathing
Metabolic disturbances that affect the central nervous system Hypoglycemia Hypocalcemia, hypercalcemia Hyponatremia, hypernatremia Uremia
Acute liver failure (hyperNH4) Many drugs / toxins Withdrawal (e.g. EtOH)
Status epilepticus
Brainstem lesion (e.g. stroke, neoplasm, inflammatory)
Nervous System Insult
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published November 11, 2023 on www.thecalgaryguide.com
Respiratory System Insult
Cardiovascular System Insult Cardiogenic problems")
Turner Syndrome Pathogenesis and Clinical Findings

Partial or complete absence of second sex chromosome, leaving only one normal X-chromosome
Possible Chromosomal Profiles
Other meiotic error → deletion or misdivision of X-chromosomal material
Complete loss of one X- chromosome in all cells (45,X) (45%)
Skeletal Abnormalities
↓ Expression of SHOX gene (present on X- and Y-chromosomes)
↓ Cellular proliferation in growth plates of bones in extremities during embryonic development
Short stature
High-arched palate
Genu valgum (knock knees)
Micrognathia (smaller lower jaw)
Broad chest
Cubitus valgus (forearm angled outward)
Mosaicism (complete loss of one X-chromosome in some cells (e.g., 45,X/46,XXX, etc)) (50%)
Congenital Heart Defects (most serious)
Single copy of TIMP1 gene and presence of risk TIMP3 allele, and differential expression of KDM6A gene
Bicuspid aortic valve
Aortic coarctation
Aortic dilation (worsened by hypertension)
One X-chromosome and presence of Y-chromosomal material in some or all cells (e.g., 45,X/46,XY)
Presence of an
X- isochromosome (most commonly i(Xq))
Other structural abnormalities of X-chromsome (e.g., Ring Chromosome X, partial deletion of X, X or Y marker chromosome)
Endocrine Disorders
Turner Syndrome
The most common sex chromosomal abnormality in females (affects 1/2000-3000)
Results in deletion or non- functioning of one X chromosome
Clinical presentations vary depending on chromosome profile
Aortic dissection
Renal disease
Neurocognitive Deficits
Mechanism unknown
Deficit in social skills
Specific (non-verbal) learning disorder (otherwise normal intelligence)
↓ Executive function skills
↓ Visuospatial skills ↓ Attention
Accelerated follicular apoptosis (i.e., loss of oocytes from ovaries) → streak gonads
Ovaries are unable to respond to high gonadotropins (FSH, LH)
Hypergonadotropic hypogonadism
Premature ovarian insufficiency
↓ Expression of immune- associated genes on X- chromosome
↑ Autoimmunity
Autoimmune diseases (celiac, thyroiditis, IBD, metabolic abnormalities)
↓ Estrogen levels
Lack of breast development
↑ Liver enzymes (Additional mechanisms likely)
↓ Bone mineral density
Other Dysmorphic Features
Lymphedema Webbed (buildup of lymph neck
fluidàswelling)
Primary amenorrhea
Infertility
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published November 25, 2023 on www.thecalgaryguide.com")
Sugammadex
) in plasma when administered IV
↓ Concentration of functional nNMBs in plasma
Creates a concentration gradient from muscle tissue (high) to plasma (low)
nNMBs move from muscle compartment to plasma
Sugammadex in plasma encapsulates nNMBs that moved to the plasma
↓ Concentration of functional nNMBs in the plasma
↓ Concentration of nNMBs at the nicotinic acetylcholine receptor within the skeletal neuromuscular junction
Reverses neuromuscular blockade created by nNMBs
Sugammadex
Progesterone is similar in structure to nNMBs
Sugammadex binds progesterone
↓ Progesterone activity in the body
Progesterone is critical for maintenance of early pregnancy
Unknown significance, avoid use in early pregnancy
Sugammadex-nNMB complex is cleared by the kidneys
Higher concentrations of sugammadex facilitate faster nNMB clearance
Unknown mechanisms
Post operative nausea and vomiting
Headache Bradycardia Cardiovascular Collapse
↓ Effectiveness of progesterone-based contraception for 7 days
↓ Clearance in patients with severe renal impairment
Reversal of profoundly deep neuromuscular blockade at higher doses
Binds to IgG or IgE receptors on sensitized basophils/mast cells in allergic reactions
Activation of basophils/mast cells
Degranulation of basophils/mast cells
Release of granulation products
Anaphylaxis Bronchospasm Hypotension
Authors: Arzina Jaffer, Kayleigh Yang Reviewers: Jasleen Brar, Mao Ding Joseph Ahn* * MD at time of publication
Movement of limbs or body during anesthesia
Coughing during anesthesia
Grimacing or suckling on the endotracheal tube
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published January 16, 2024 on www.thecalgaryguide.com")
Bacterial Infections from Transfusion
 recognize bacteria
Immune cells release pyrogens (either bacterial components or signalling molecules) upon recognition
Pyrogens bind to receptors on the hypothalamus
Hypothalamus ↑ body temperature set point
Body generates and conserves heat to reach set point
Immune cells release pro- inflammatory cytokines (messenger protein)
Cytokines activate sympathetic nervous system via hypothalamus
Adrenal glands release stress hormones (epinephrine and norepinephrine) into bloodstream
Stress hormones bind to receptors on cardiomyocytes (heart muscle cells)
↑ Heart contractility
↑ Heart rate
Cytokines circulate throughout the body
Cytokines interact with endothelial cells of the blood vessels
↑ Nitric oxide production
Relaxes smooth muscle cells of blood vessel walls
Vasodilation (↑ blood vessel diameter)
Hypotension
Systemic inflammation
Stimulate nerve endings in muscles
Stimulate nerve endings in gastrointestinal tract’s lining
Blood brain barrier’s integrity is compromised
Nitric oxide reacts with oxygen to form reactive nitrogen species
Tissue damage
Fatigue
Weakness Muscle aches
Nausea and vomiting
Immune and inflammatory cells enter the brain
Authors:
Arzina Jaffer
Kayleigh Yang
Reviewers:
Nimaya De Silva
Raafi Ali
Michelle J. Chen
Yan Yu*
Kareem Jamani*
* MD at time of publication
Inflammation in the brain
Activates pain processing centers in the brain
Headache
Damages neurons and disrupts cell communication
Confusion and altered mental state
Shivering
Fever
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published January 30, 2024 on www.thecalgaryguide.com")
Carbonic Anhydrase Inhibitor Diuretics

Inhibition of carbonic anhydrase on the apical surface of the brush border cells in the proximal convoluted tubule (PCT)
Activation of the Renin- Angiotensin-Aldosterone Systemfromvolume depletion
Activation of principle cell
Epithelial sodium channels (ENaC) on principal cells of the CCD reabsorb ↑ Na+ and waste K+
↓ K+ in serum
Hypokalemia
See Hypokalemia: Clinical
Findings slide
↑ Na+ delivery to the cortical collecting duct (CCD)
H2O follows Na+ into the CCD to maintain a balanced osmotic pressure
↑ H2O available for excretion
Mild diuresis (increase in frequencyandvolumeof urine)
↓ Blood volume
Hypotension
↓ Na+ and HCO3- reabsorption in the PCT
↑ HCO3- delivery to cortical collecting duct
Urine alkalization (increased pH)
Chronic urine alkalization
↓Solubilityof citrate
↓ Urinary citrate
↓ Citrate binding with Ca2+à↑ Ca2+ complexing with oxalate
↑ Spontaneous nucleation, growth and agglomeration of calcium oxalate crystals
Formation of calcium oxalate renal calculi
↑ HCO3- is lost in the urine ↓ pH of the blood
Type II Renal Tubular Acidosis
See Type II/Proximal Renal Tubular Acidosis slide
CAI prevents the up- regulationofglutamine transporters in the PCT
Inability to correct the metabolic acidosis and impaired urinary NH3 excretion
Hyperammonemia (↑ serum NH3 )
↑ Risk of hepatic encephalopathy in individuals with cirrhosis
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published Feb 3, 2024 on www.thecalgaryguide.com
Carbonic Anhydrase Inhibitor Diuretics: Renal Mechanism and Side Effects Carbonic Anhydrase Inhibitors (CAI)
Inhibition of carbonic anhydrase on the apical surface of the brush border cells of the proximal convoluted tubule (PCT)
Authors: Stephanie Happ Reviewers: Matt Hobart Name Name* * MD at time of publication
↓ Na+ and HCO3- reabsorption in the PCT
↑ Na+ delivery to the cortical collecting duct (CCD)
H2O follows Na+ into the CCD to maintain a balanced osmotic pressure
↑ H O available for 2
excretion
Mild diuresis
↓ Blood volume Hypotension
↑ HCO3- delivery to cortical collecting duct
Epithelial sodium channels (ENaC) on principal cells of the CCD reabsorb ↑ Na+
↑ Intracellular Na+ drives Na+/K+ ATPase activity on the principal cells (moving 2 K+ into cell and 3 Na+ out into the peritubular capillary)
↑ Intracellular K+ drives H+/K+ ATPase activity on the intercalated cells (moving 1 H+ into cell and 1 K+ out into the tubular filtrate)
↓ K+ in serum
Hypokalemia
See Hypokalemia: Clinical Findings slide
Urine alkalization
↑ HCO3- is lost in the urine, leading to ↓ pH of the blood
Renal Tubular Acidosis Type II
See Type II/Proximal Renal Tubular Acidosis slide
CAI inhibit the up-regulation of glutamine transporters in the PCT
Inability to correct the metabolic acidosis and
impaired urinary NH3 excretion
Hyperammonemia
↑ Risk of hepatic encephalopathy in individuals with cirrhosis
Chronic urine alkalization leads to marked ↓ in urinary citrate
↓ Ability of citrate to bind to Ca2+ and calcium oxalate stones
↓ Inhibition of spontaneous nucleation
↓ Prevention of growth and agglomeration of crystals
Formation of calcium oxalate renal calculi
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published MONTH, DAY, YEAR on www.thecalgaryguide.com")
Stable Angina

↓ Blood vessel lumen diameter
↓ Volume of blood is supplied to the heart
Predictable period of physical activity or emotional stress
↑ Heart rate
↓ Time for coronary arteries to fill heart with blood (diastole)
↑ Heart contractility
↑ Oxygen demand of heart muscle tissue (myocardium)
↓ Myocardial blood supply
Imbalance between blood supply & oxygen demand causes myocardial ischemia
Angina Pectoris/Stable Angina
Myocardial ischemia causes cardiac muscle cells (cardiomyocytes) to switch from oxygen-dependent (aerobic) to oxygen-absent (anaerobic) metabolism
Anaerobic metabolism produces metabolites that stimulate cardiac spinal afferent nerves
Myocardial visceral afferent & somatic sensory nerve fibers mix & enter the spinal cord via T1-T4 nerve roots
Brain interprets ↑ nerve signaling as nerve pain coming from the skin of T1-T4 dermatomes (referred pain)
↑ lactic acid production & ↓ cellular pH impairs cardiomyocytes’ function
Damaged cardiomyocytes impair myocardial relaxation & cause ↓ left ventricular contractility & cardiac output
Blood backs up into left ventricle, atrium, & pulmonary vasculature
↑ Pulmonary capillary pressures pushes fluid out & into the lung’s alveoli
↓ Gas exchange & oxygenation
↑ Respiratory rate & Dyspnea (shortness of breath)
Blood flow begins at the epicardium (outer heart layer) & ends at endocardium (inner layer)
Subendocardium (innermost heart layer) receives the least blood flow causing non-transmural (partial thickness) heart wall ischemia
Anterior/septal & lateral wall ischemia triggers ↑ sympathetic nervous system (SNS) activity given the proximity of cardiac SNS innervation
Inferior wall ischemia triggers involuntary ↑ in Vagus nerve activity given the nerve’s proximity
Bradycardia (↓ heart rate)
Nausea
Adrenal medulla releases Norepinephrine hormone
Activation of sweat glands via SNS acetylcholine neurotransmitter release
Hypotension (↓ blood pressure)
Pain radiation to left arm, jaw, abdomen & upper back
Chest pain, pressure, or discomfort
Unstable Angina (unpredictable & worsening chest pain)
See relevant Calgary Guide slide on Unstable Angina
Binds arterial smooth muscle α1 receptors
↑ Coronary arteries’ vascular tone (vasoconstriction)
Hypertension (↑ blood pressure)
Activates β1 receptors in the heart
Tachycardia (↑ heart rate)
Diaphoresis (↑ sweating)
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published Aug 8, 2013; updated Feb 5, 2024 on www.thecalgaryguide.com")
Gestational Diabetes Risk factors and pathogenesis

Authors:
Amyna Fidai
Maharshi Gandhi Reviewers:
Laura Byford-Richardson Shahab Marzoughi
Yan Yu*
Hanan Bassyouni*
* MD at time of publication
High risk population (Aboriginal, Hispanic, South Asian, Asian, African)
Previous or current macrosomia (>4000g) or polyhydramnios
Other conditions associated with Diabetes Mellitus such as polycystic ovarian syndrome, hypertension, metabolic syndrome
Placental counter regulatory hormones (particularly Human Placental Growth Hormone) oppose the action of insulin
↑ Insulin resistance (liver, muscle, adipose tissues become less responsive to insulin)
↑ Fetal demands after 18 weeks gestation
(fetus requires 80% of its energy from maternal glucose)
↑ Carbohydrate intake to keep up with the demands
Previous history of gestational diabetes or glucose intolerance
Family history of diabetes
Advanced maternal age
Obesity
Previous unexplained stillbirth
Multiples (larger placental mass and activity)
Corticosteroid use
↑ Risk for pregnancy induced glucose intolerance (mechanism unclear and/or complex)
↑ Insulin requirements Initially pancreatic beta cells work
overtime to keep up with the ↑ insulin demands
Eventually insulin demands are not met due to the exhaustion of pancreatic beta cells
Plasma glucose rises (Fasting plasma glucose ≥5.3 mmol/L)
Gestational Diabetes (or exacerbation of pre-existing DM)
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published Jan 28, 2017, updated Feb 24, 2024 on www.thecalgaryguide.com")
Infectious Small Bowel Diarrhea

May produce Shiga toxins (a specific family of toxins that can lead to complications)
Authors: Noriyah Al Awadhi Yan Yu Sara Cho Reviewers: Paul Ratti Jason Baserman Shahab Marzoughi Kerri Novak* * Indicates MD at time of publication
Y. enterocolitica
(also milk, cheese)
Shellfish, undercooked seafood
V. parahaemolyticus V. cholerae
Enterohemorrhagic E. coli (EHEC)
Daycare centers/nurseries
Drinking/swimming in bad water — mountains/wells
S. aureus
B. cereus
Noroviruses
Rotavirus
G. lamblia
C. parvum
May produce emetic toxins (toxins that cause vomiting)
Adequate amount of organism and/or toxin is ingested
Organism adheres to the intestinal liningàorganism colonizes the small intestine Organism releases enterotoxin (a toxin that affects the intestines)
Binds to and disrupts intestinal transporters used in secretion and absorption of water and electrolytes
Toxin enters systemic vasculature
Shiga toxins inhibit ADAMTS13 (cleaving enzyme)
Failure to cleave von Willebrand Factor (vWF) multimers
Accumulation of vWF multimers
Platelets and thrombi accumulate in microvasculature
Hemolytic uremic syndrome (hemolytic anemia, thrombocytopenia, and acute renal damage)
↑ Water and electrolyte secretion
↓ Fluid absorption
Chemoreceptor trigger zone in medulla detects circulating emetic toxins
Nausea & vomiting
Triggers release of inflammatory mediators and cytokines that travel to the central nervous system
Prostaglandin is synthesized and released
Neurotransmitter cyclic AMP (cAMP) is released
cAMP ↑ hypothalamic thermoregulation set point
Fever
Large volume profuse diarrhea
Loss of water
Distention (swelling) of intestines stimulates visceral sensory pain fibers
Visceral pain fibers crosstalk with somatic nerves from the same spinal cord level
Occasional referred pain/cramping (typically diffuse pain around the abdomen)
Loss of bicarbonate, sodium, potassium, magnesium, and chloride
Dehydration
Electrolyte deficiency
Metabolic acidosis (pH < 7.4 & serum bicarbonate < 24)
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published Aug 7, 2012; updated Mar 25, 2024 on www.thecalgaryguide.com")
Post-Renal Acute Kidney Injury AKI
: Pathogenesis and clinical findings
Blood clot or cellular debris
Foreign body
Neurogenic bladder
Obstruction intrinsic to urine excretion system
Nephrolithiasis (kidney stones)
Anatomical defect(s)
Intra- abdominal adhesions
Retroperitoneal fibrosis (scar-like tissue)
Benign or malignant masses
Prostate cancer
Benign prostatic hyperplasia
↓ Urine flow across point of obstruction Obstruction extrinsic to urine excretion system
Urine buildup distends urine collecting system (hydronephrosis)
Compression of renal vasculature due to mass effect
↑ Volume/pressure proximal to obstruction
↑ Intratubular pressure
↓ Pressure gradient between glomerular afferent arteriole and Bowman’s space
Casts occlude tubules
↓ Filtration of plasma into nephrons
↓ Glomerular Filtration Rate (GFR)
Obstruction is relieved ↓ Intratubular pressure Rapid GFR ↑
Rapid diuresis of fluid and electrolytes
Dilated pelvicalyceal system on ultrasound
↓ Urine output
↓ Venous drainage
and arterial supply
Local ischemia and inflammation of kidney
Impaired resorption, excretion, and fine tuning by tubules
Acute Tubular Necrosis (ATN) with granular casts
↓ Urine output
↓ Clearance of free water and solutes
↑ Intravascular volume
↑ Serum creatinine
↓ Medullary solute concentration, ischemia, ↓ response of collecting ducts to antidiuretic hormone
Lasts > 24hrs
Post-obstructive diuresis causes hypovolemia and electrolyte derangements
Authors: David Campbell, Matthew Hobart Reviewers: Raafi Ali, Luiza Radu Huneza Nadeem, Marissa Zhang, Julian Midgley* * MD at time of publication
Resolves < 24hrs in euvolemia
Physiologic post- obstructive diuresis
↑ Na+ and Cl- delivery to distal convoluted tubule is sensed by macula densa
Secretion of adenosine by macula densa
Adenosine constricts afferent arterioles
↓ GFR
↓ Renal clearance of drugs and waste products
↑ Venous hydrostatic pressure
↑ Volume in arterial system overwhelms pressure regulation mechanisms
Hypertension
Fluid extravasation from veins and capillaries
Generalized Edema
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published Mar 25, 2024 on www.thecalgaryguide.com")
Operative Vaginal Delivery Indications
: Indications
Authors: Taylor Pigott Akaya Blair Reviewers: Michelle J. Chen Sylvie Bowden* Stephanie Cooper* Sarah Glaze* * MD at time of publication
Abnormal fetal heart rate/fetal distress
↓ Risk of maternal and fetal morbidity/ morality
Rupture of membranes without fetal head adequately applied to maternal pelvis
Small pelvic outlet Narrow vaginal canal
Cardiovascular disease
Diabetes Hypertension Abdominal trauma
Short umbilical cord
Large for gestational age (LGA) fetus
Neurological/ muscular disease
↑ Pain during labor
Maternal contraindication to Valsalva maneuver (e.g., cardiac disease, cystic lung)
Rush of fluid past the fetal head out through cervix
Umbilical cord prolapse
Umbilical vasoconstriction
Umbilical cord compression
↓ Blood flow through umbilical cord
Fetal presenting part applies pressure on umbilical cord
Impaired vascular function and narrowed vessels ↓ blood flow around the body
↓ Oxygen transfer across the placenta
Fetal oxygen deprivation and hypoxia
Operative vaginal delivery with forceps or vacuum expedites delivery
Placenta partially or completely separates from uterine wall (abruption)
Increased tension of umbilical cord on placenta
Fetal shoulder is lodged behind maternal pubic symphysis
Delivery of the body is delayed
Inability to adequately bear down to push
Myometrium runs out of energy from repeated contractions and becomes less able to contract (↓ uterine tone)
Uterine spiral arteries dependent on contractions for vasoconstriction remain dilated, allowing blood flow
Postpartum hemorrhage
Uterine rupture
↑ Risk of future pelvic organ prolapse
↑ Risk of future incontinence
Prolonged labour
Repeated contraction/ relaxation of the uterus without progress tears and damages uterine muscles
↓ Maternal endurance
Inadequate fluid and food intake prior to/ during labor
Multiples
↑ Sympathetic nervous system activation during labor
Maternal exhaustion
Uterine muscles stretch to accommodate multiple growing fetuses
↑ Pushing against pelvic floor and perineum
Pushing force damages pelvic floor muscles and nerves
↓ Myometrial contractility
↑ Need for vaginal exams to check on labour progress
Bacterial overgrowth in vagina and/or uterus
Maternal infection
Fetal infection
↓ Strength of contractions
Epidural anesthetic
Inability to ambulate to urinate
↑ Urinary catheterizations
↑ Epinephrine secretion from adrenal cortex
Blood is shunted away from uterus/internal organs and towards heart and skeletal muscle
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published 03, ##, 2023 on www.thecalgaryguide.com")
Sickle Cell Disease Pathogenesis Clinical Findings and Complications
 Liu Priyanka Grewal Reviewers: Alexander Arnold Luiza Radu JoyAnne Krupa Yan Yu* Lynn Savoie* * MD at time of publication
Hemoglobin S (HbS) variant formed instead of normal Hemoglobin A (HbA)
Hb electrophoresis shows approximately 45% HbS, 52% HbA (ααββ), 2% HbA
Hb electrophoresis shows approximately 90% HbS, 8% HbF, & 2% HbA .
No HbA present
(ααδδ), & 1% HbF (ααγγ;2 fetal hemoglobin)
Heterozygous: point mutation in one of the two chromosomes (Hb AS)
Sickle cell trait
(asymptomatic unless severely hypoxic)
Homozygous: point mutation in both chromosomes (Hb SS)
Sickle cell disease
An inherited blood disorder characterized by defective hemoglobin that leads to red blood cells sickling
2
Dehydration Hypoxemia
Acidosis
↓ Volume of RBC cytoplasm
↓ O2 Saturation of Hb
O morereadily 2
released from Hb in low pH environment
↑ Concentration of deoxygenated HbSinred blood cells (RBCs)
Hydrophilic glutamate→ hydrophobic valine substitution makes HbS less soluble in the cytoplasm & more prone to polymerization & precipitation in its deoxygenated state
↑ Concentration of deoxygenated Hb in RBC leads to ↑ polymerization rate Polymerized & precipitated HbS forms long needle-like fibers
RBC shape becomes sickled
Sickle cells on peripheral blood smear
Vaso-occlusion
(sickled RBCs lodge in small vessels, blocking bloodflow to organs & tissues)
Blockage of venous outflow
Occlusion of vessels in lungs ↑ pulmonary blood pressure
Fluid extravasates into interstitial tissue leading to pulmonary edema
Acute chest syndrome (chest pain, hypoxemia (↓blood oxygen), etc.)**
to the penis:
to the spleen:
Priapism (persistent, painful erection)
Splenic Sequestration (blood pools in spleenà splenomegaly & hypotension)
Extravascular hemolysis (macrophages in the spleen phagocytose sickled RBCs)
Normocytic anemia
↑Marrow erythropoiesis (RBC production) to compensate for hemolysis
Infarction of bone
Pain crises
If occurring in hands
Dactylitis (inflammation of digits)
Blockage of arteries ↓ oxygenation of organs & tissues
Vaso-occlusion of the splenic artery
Splenic infarction (↓ blood supply leads to tissue death)
RBC inclusions (structures found in RBCs) not removed by spleen
Howell-Jolly bodies (RBC DNA remnants) on blood smear
Vaso-occlusion of other arteries (cranial, renal, etc)
Stroke, renal failure
↑ RBC breakdown
↑ Unconjugated bilirubin released from RBC breakdown
RBC precursors (reticulocytes) are released into the blood stream
Reticulocytosis (increased number of immature red blood cells) on peripheral blood smear
Patient’s RBC level becomes dependent on increased marrow activity
Bone marrow infarction or viral infections (i.e. Parvovirus B19) suppresses bone marrow activity
Aplastic crises (profound anemia)
** See corresponding Calgary Guide slide(s)
↑ Serum level of unconjugated bilirubin
Some of the circulating unconjugated bilirubin deposits in the skin
Jaundice (yellowing of skin)
↑ Conjugation of bilirubin in liver
↑Amounts of conjugated bilirubin released into bile
Gallstone formation
Cholelithiasis (presence of gallstones in the gallbladder)
Spleen releases invasive encapsulated bacteria (eg. Haemophilus influenzae, Streptococcus pneumoniae, Neisseria meningitidis) into circulation, which causes infections
Meningitis
Sepsis
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published Sept 16, 2013, updated Apr 29, 2024 on www.thecalgaryguide.com")
Gynecomastia

Hyperthyroidism
Klinefelter Syndrome (males with > 1 X chromosome)
Liver cirrhosis
Certain tumors (e.g., germ cell, adrenal, Leydig cell, Sertoli cell)
Anabolic steroid usage (containing testosterone)
Finasteride (treatment for benign prostate hyperplasia and male pattern baldness)
Cimetidine - inhibits stomach acid production
Spironolactone (diuretic
used to treat high blood pressure and heart failure)
Ketoconazole (antifungal)
Cytotoxic agents (e.g. alkylating agents, vincristine, methotrexate)
Imbalance between estrogens and androgens
Estrogen stimulates breast tissue growth in newborn
Changes in metabolic rate ↑ fat production
Unclear mechanism
↑ Proinflammatory mediators and cytokines (e.g. prostaglandin E2, TNF⍺, IL-1, IL-6, cyclooxygenase-2)
Prostaglandin E2 and IL- 6 upregulate aromatase enzyme expression
Available estrogen is higher than available testosterone
↑ Aromatase enzyme activity, converting androgens to estrogen
↓ Testosterone release from the testes
↓ Testosterone
↑ Serum sex hormone binding globulin (SHBG)
SHBG binds estrogen with less affinity to testosterone
Thyroid hormone stimulates liver to express more sex hormone binding globulin
Thyroid hormone stimulates aromatase activity
Overexpression of aromatase enzyme
Seminiferous tubules in the testes hyalinize and fibrose
Suppression of the hypothalamic pituitary thyroid axis through an unclear mechanism
Tumor may produce estradiol
Tumor produces β- human chorionic gonadotropin (β-HCG)
↑ serum testosterone
Inhibits 5-α reductase
Blocks binding of 5-DHT to androgen receptors
↓ 2-hydroxylation of estradiol
Mimics structures of testosterone
Inhibits 17,20 desmolase and 17α-hydroxylase
Damage to Leydig cells in testes
↑ Estrogen to androgen ratio
Pathological causes
Impaired spermatogenesis and testosterone production
↓ GnRH secretion from hypothalamus
↓ Testosterone
↓ Luteinizing hormone (LH) release from anterior pituitary
↓ 5-DHT and/or testosterone binding to androgen receptors in chest tissue
↓ inhibition of breast development
Normal or increased estrogen acts on estrogen receptor on chest tissue
Estrogen receptors stimulate breast development
Estradiol negatively feedbacks on luteinizing hormone
β-HCG stimulates LH receptors on Leydig cells in the testes
Aromatase enzyme converts excess testosterone into estrogen and estradiol
↓ conversion of testosterone to 5- dihydrotestosterone (5-DHT), a more potent form of testosterone
Glandular proliferation in male breasts
Gynecomastia
(development of breast tissue in males)
Drug side- effects
↓ Metabolism of estradiol
Competitively binds to androgen receptors
↑ Serum estradiol levels
Exhibits physical attributes that do not align with gender identity
Psychological distress
In some cases, hormones stabilize
Involution and atrophy of ducts
Gynecomastia resolves
↓ Steroid synthesis
↓ Androstenedione produced (testosterone precursors)
↓ Serum testosterone levels
↓ Testosterone production
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published Jun 9, 2024 on www.thecalgaryguide.com")
Anesthetic Considerations in Pregnancy

↑ Progesterone serum levels
↓ Esophageal tone & sphincter pressure
↑ Aspiration risk >20 weeks gestation & during labor
Aspiration prophylaxis
Rapid sequence induction
Nonparticulate antacid prophylaxis (e.g., sodium citrate, famotidine)
Pre-operative fasting & gastric ultrasound to assess volume
Breathing
↑ Minute ventilation
↓ PaCO2 at baseline
Uterine artery vasoconstriction with maternal PCO2 levels exceeding 32 mmHg
Impaired uteroplacental perfusion
Maintain physiologic alkalosis, avoid maternal hypercapnia
Maintain
maternal arterial PCO2 28–32 mmHg
Avoid permissive hypercapnia
↑ Progesterone serum levels
Disproportionate ↑ in plasma volume
↓ Serum colloid osmotic pressure
Drug-specific alterations in absorption, distribution, metabolism & excretion (e.g. ↑ distribution of acidic drugs due to ↓ albumin; ↑ clearance of renally- metabolized drugs)
Optimize anesthetic dosing for altered pharmacokinetics
Adjust dosages based on drug recommendations
Circulation
↑ Maternal blood volume
Peripheral vasodilation
↓In systemic vascular resistance by 25–30%
↓ Blood pressure
↑Abdominal pressure
Compression of the aorta & inferior vena cava
Supine hypotension
Impaired uteroplacental perfusion
Fetal ischemia
Optimize
placental blood flow
Position patient on left side to maintain uterine displacement
Diaphragmatic elevation & lung compression
↓ Functional residual capacity
↑ Risk of rapid desaturation
Fetal hypoxemia
Maintain maternal & fetal oxygenation
Optimize positioning (e.g., patient on left side to maintain uterine displacement)
& supplement oxygen as needed
Heart rate ↑ 15-25%
↑ Cardiac output
Manifestations of significant blood loss are delayed
Avoid hypotension
↑ Stroke volume
Avoid fluid overload
Treat abnormal variation in blood pressure at lower values
Fetus
Requirement to assess & treat two patients
Achieve optimal anesthetic dosing for mother while maintaining fetus safety
Maintain oxygenation of both mother & fetus & ensure adequate uteroplacental perfusion
Consider fetal-drug transfer & adjust agent & dose accordingly
Monitor maternal vital sign & fetal heart rate
Utilize a multidisciplinary team (e.g., pharmacy, nursing, physical & occupational therapy)
Legend:
Pathophysiology
Mechanism
Goal
Management
Published July 5, 2024 on www.thecalgaryguide.com")
Diabetes Mellitus Pathophysiology Behind Lab Findings

↓ Glucose uptake into cells
Autoimmune destruction of pancreatic beta cells (type 1 diabetes mellitus)
Insulin resistance (type 2 diabetes mellitus)
Other causes of diabetes mellitus (genetic, drug- induced, pregnancy, etc.)
↑ Lipolysis (fat breakdown) in adipose tissue
↑ Free fatty acids & glycerol in bloodstream
↑ Oxidation of fatty acids in liver to form acetyl CoA
↑ Conversion of acetyl CoA to ketone bodies (ketogenesis) to be used as fuel for the brain
Relative or absolute insulin deficiency
↓ Activation of adipose (fat tissue) & muscle cell transmembrane insulin receptors
↓ Activation of intracellular insulin signaling pathways (PI3K & MAP kinase)
Cells cannot use glucose as a source of energy; thus body reacts as if it is starving
↑ Protein breakdown in muscle tissue ↑ Amino acids & lactate in bloodstream
↑ Substrates (glycerol, amino acids & lactate) available to produce glucose in the liver
↑ Glycogenolysis (breakdown of stored glucose - glycogen) & gluconeogenesis (production of glucose) in liver
Glucose in bloodstream glycates (coats) hemoglobin in red blood cells; thus ↑ blood glucose leads to ↑ glycated hemoglobin
↑ Hemoglobin A1C
↑ Fasting blood glucose level ↑ Random blood glucose level
↑ Blood glucose following oral glucose tolerance test
Diabetic ketoacidosis (DKA) in absolute insulin deficiency *See DKA slide
Filtered ketone bodies exceed the reabsorption capacity of renal tubules
Ketonuria (↑ ketones in urine)
↑ Glucose in bloodstream
Build-up of glycation end products causes damage to glomerular tissue
Transient glomerular hyperfiltration followed by long-term ↓ in glomerular filtration rate
Albuminuria (↑ albumin in urine)
Diabetic nephropathy *See slide
Filtered glucose exceeds reabsorption capacity of renal tubules in the kidney
Glucosuria (↑ glucose in urine)
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published July 8, 2024 on www.thecalgaryguide.com")
Neonatal Hypoglycemia Pathogenesis
, glycogenolysis (breakdown of glycogen into glucose), and fatty acid oxidation (conversation of fatty acids to ATP)
Low glucose levels stimulate the neonate’s appetite, allowing them to adapt to intermittent feeds
Infant feeding regularly on a diet with sufficient carbohydrates creates a consistent plasma glucose concentration
Pregnant Parent Causes
Impaired Glucose Production
Inadequate Glucose Supply
Pregnant parent using β-blockers
β-blockers prevent epinephrine from binding to adrenergic receptors in skeletal muscle and liver
Blocked sympathetic signaling in these organs prevents glycogenolysis
↓ Breakdown of glycogen into glucose
Infant of a parent with diabetes
Parent has a high level of glucose in their blood
The fetus receives a glucose-rich blood supply
Fetal pancreatic β cells ↑ insulin production
Post-birth, glucose levels in the infant’s circulation ↓ but insulin levels
remain high
Excessive glucose uptake into cells
Fetus born with a metabolism disorder (e.g. organic amino acid disorder, disorder of glycogen metabolism, disorder of gluconeogenesis)
Genes that make proteins or hormones responsible for production, breakdown and regulation of glycogen are mutated
Fetus born with an endocrine disorder (e.g. congenital adrenal hyperplasia, hypopituitarism, Turner Syndrome)
Pituitary and/or adrenal gland dysfunction ↓ release of hormones that regulate glucose balance
Fetal growth is restricted, fetus is small for gestational age, or is premature (<37 weeks gestation)
Glycogen is deposited during the 3rd trimester of pregnancy. Infants born early have fewer stores; smaller infants born at term have ↓ stores, ↑ insulin sensitivity, & poorly coordinated counter- regulatory hormones
↑ Glucose demand for the transition to life out of the womb
Glycogen stores used up quickly
Perinatal stress (e.g. sepsis, asphyxia)
Stress stimulates fetal adrenal glands to ↑ epinephrine secretion
Fetal pancreatic ⍺- cells ↑ glucagon secretion
↑ Breakdown of muscles and fat for glucose- building blocks
Glycogen stores used up quickly
↑ Glucose usage as fetal metabolic demands ↑ to manage stress
Fetal β-cells secrete inappropriately high levels of insulin despite hypoglycemic state; this hyperinsulinism state can last for months & resolve spontaneously
↓ Serum glucose Neonatal hypoglycemia
Authors: Dasha Mori Reviewers: Michelle J. Chen *Dr. Danielle Nelson *MD at time of publication
Blood glucose < 2.6 mmol/L in term & preterm infants within 72 hours of birth or < 3.3 mmol/L after 72 hours of birth
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published July 19, 2024 on www.thecalgaryguide.com")
Generalized Anxiety Disorder GAD
: Pathogenesis and clinical findings
Authors: Keira Britto Reviewers: Sara Cho, Luiza Radu, *Margaret Oakander *MD at time of publication
Altered cognition
Scan environment for perceived threats
Genetic predisposition & family history: polygenic, >30% heritability
Adverse childhood & life experiences: e.g., abuse, neglect, divorce, bereavement
Morbidity & stressors from chronic medical conditions
Hypersensitivity & reactivity to stimuli
áHPA axis activity
Neurobiological differences
Trait neuroticism (emotional instability & nervousness)
Reacts to unfamiliarity with withdrawal & fear
áLevel of uninhibited excitatory signaling
Psychomotor agitation
Frequent sympathetic nervous system (fight or flight) activation to perceived threats
Cycles of epinephrine surges
Inadequate recovery
State of hyperarousal
Disturbed sleep
Ongoing sleep deprivation
Easily fatigable
áACTH released from hypothalamus
áCortisol released from adrenal glands
áSerotonin uptake
âSerotonin Impaired
neuroplasticity &âcomplexity of synaptic connections in amygdala & hippocampus
âAbility to deal with emotional stressors
Impaired engagement of prefrontal cortex
âRegulation of emotional reactions
áAnticipatory emotional responses
âInhibition from GABA
áAmygdala volume & activity
áFear & anxiety circuits
Attentional focus directed towards worries & perceived threats
Divert blood to muscles
Skeletal muscle activation
Muscle tension
Psychomotor agitation (excessive motor activity associated with a feeling of inner tension)
áEmotional intensity
Chronic state of high physiological stress
âHippocampal BDNF (brain derived neurotrophic factor)
Hippocampal atrophy &â hippocampal neurogenesis
â Inhibitory control of the hippocampus over the HPA axis
áAlertness & wakefulness
Disturbed sleep
Strengthening of memories rooted in fear, to prevent encounters with future threats
Behavioural & cognitive changes to facilitate coping with threats
Irritability
**Excessive, persistent
Bottom up, stimulus driven attention
áDistractibility
Difficulty concentrating
anxiety & worry that is distressing &/or impairs functioning
Irritability
Generalized Anxiety Disorder:
**occurring more days than not for ≥ 6 months; **difficult to control; presence of ≥ 3 remaining symptoms
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published July 21, 2024 on www.thecalgaryguide.com")
Underfill Edema Pathogenesis

Vasodilatory medications
Various mechanisms
Right-sided heart failure
Compromised right heart function ↓ forward flow
↓ Hepatic albumin synthesis
Blood is unable to pass through hepatic vessels disrupted by cirrhosis and backs up in portal vein
↑ Blood pressure in portal vein (portal hypertension)
Less blood volume in hepatic veins and vena cava (underfilling)
Pregnancy
↑ Estrogen, progesterone and relaxin
Vasodilation
Gravity causes fluid accumulation in peripheral veins
↑ Capillary hydrostatic pressure
↑ Net fluid movement into interstitial space
↓ Serum albumin
↓ Capillary oncotic pressure
Fluid extravasation into interstitial space
More blood in portal vein ↑ capillary hydrostatic pressure in venous system
Pressure creates net fluid
movement from vascular space into interstitial space
Less blood volume in arteries (underfilling)
↓ Effective arterial blood volume (EABV)
↓ Renal blood flow activates the renin-angiotensin-aldosterone system (RAAS)
Angiotensin and aldosterone ↑ Anti-diuretic hormone released by tubular Na+ and H2O resorption posterior pituitary ↑ H2O resorption
↑ Fluid in circulation, worsening existing venous congestion
↑ Hydrostatic capillary pressure and fluid extravasation into interstitial space Underfill edema (edema worsened by activation of RAAS)
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published Aug 19, 2015; updated Aug 5, 2024 on www.thecalgaryguide.com")
IgA Nephropathy
 created by mucosa- bound IgA1 plasma cells is secreted into plasma instead of onto mucosal surface
IgA1 plasma cells hyper- responsive to triggers (eg. URTIs, gastroenteritis) ↑ synthesis of GD-IgA1 → spill-over into plasma
Immunoglobulin A1 (IgA1) plasma cells destined to reside in mucosa (eg. gut or respiratory tract) travel to and
reside in inappropriate site(s) (eg. bone marrow) releasing GD-IgA1 into plasma
GD-IgA1 is not cleared from plasma as quickly as IgA1 → ↑ plasma GD-IgA1 levels
Hit 3:
GD-IgA1-IgG complexes deposit in mesangium
C3 predominant complement activation amplifies inflammatory response
Renal biopsy:
IgA deposits in mesangium (100% sensitive)
Renal biopsy: Complement in mesangium (C3 predominant) (90-95% sensitive)
A cascade of multiple immunologic hits is initiated
Hit 1: ↑ Serum levels of GD-IgA1 multiple immunologic hits
GD-IgA1 hinge region is structurally distinct from IgA1 that would normally circulate in plasma (lack of galactosyl groups)
GD-IgA1 hinge region may mimic pathogens (ex. bacteria and viruses) or other antigens
Cross reactivity of IgG against GD-IgA1, or synthesis of anti-GD-IgA1 IgG antibodies
Immunoglobulin G (IgG) binds GD-IgA1 hinge region Hit 2: GD-IgA1-IgG immune complex formation
Circulating GD-IgA1-IgG complexes have high affinity for glomerular endothelial cells where they damage the glycocalyx → ↑ permeability of immunoglobulins into the mesangium
↑ Production of chemokines, cytokines and complement → ↑ mesangial cell proliferation and matrix expansion
Leukocyte recruitment and activation damages glomerulus and mesangium
Hit 4:
Inflammatory response to GD-IgA1 complexes in mesangium induce glomerular structure disruption (endothelium, basement membrane, podocytes, mesangium)
and impaired glomerular function
Loss of barrier functions of glomerulus allows for extravasation of blood & proteins into Bowman’s space and subsequently through tubules
Renal biopsy: Glomerulosclerosis, tubulointerstitial fibrosis, glomerular vasculitis, podocyte damage
Eventual end-Stage Renal Disease (ESRD)
Progressive ↓ of filtration surface area within glomeruli and ↓ number of functional glomeruli
Proteinuria
Synpharyngitic hematuria (hematuria with dysmorphic red cells co-occurring with pharyngitis)
↓ Glomerular Filtration Rate (GFR)
Nephrotic Syndrome
↑ Serum creatinine
Chronic kidney disease and eventually ESRD
IgAN is an autoimmune disease where IgA deposition in the glomerulus leads to an inflammatory cascade, endothelial dysfunction and mesangial expansion that damages glomeruli causing kidneys to leak blood and protein into urine and decreased kidney function. IgA nephropathy is a multifactorial disease requiring multiple immunologic hits
IgA Nephropathy (IgAN)
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published Sept 5, 2024 on www.thecalgaryguide.com")
Major Depressive Disorder 2024
: Pathogenesis and clinical findings
Self blaming tendency
Major stressor(s)
↑ Emotional reactivity Monoamine deficiency
Regional brain area alterations
↓ Behavioral activation
(engagement in meaningful activities to improve mood)
Hunger interoception (perception & awareness of hunger) changes
Attributes the occurrence of negative events to internal/personal factors
Expectation that future events are uncontrollable & internally caused
Feelings of worthlessness or excessive guilt
Recurrent thoughts of death or suicide
Insomnia (melancholic MDD)
Dysregulation of negative feedback control on the hypothalamus–pituitary– adrenal (HPA) axis
↓ Serotonin & norepinephrine signaling
↑ HPA axis sensitivity
Heightened responses to stressors
Major Depressive
Disorder
≥5 symptoms/complications during the same 2-week period; at least 1 of **symptoms; causing clinically significant distress or impairment in social, occupational, or other important areas of functioning
↑ Amygdala activity Changes to neuronal activity
Altered prefrontal cortex & limbic functioning
Depressed mood**
Mechanism unknown; can
precede MDD or be a symptom
Psychomotor agitation (restlessness)
↓ Dopaminergic transmission in ventral striatum
Overactivation of the insula (brain region involved in integrating activity in reward & interoception circuitry)
Under activation of the insula
Mechanism unknown
↓ Activation of mesolimbic system (reward system)
↓ Motivation & anticipation for rewards
↑ Activation of mesolimbic system
Psychomotor
retardation (slowing) Poor concentration
↑ Motivation & anticipation for rewards
Anhedonia (loss of interest/pleasure)**
Anergia (lack of energy)
↑ Eating (particularly high- sugar foods)
Hypersomnia (atypical MDD)
↓ or ↑ Appetite &/or weight
Authors: Keira Britto Sara Cho JoAnna Fay Reviewers: Luiza Radu Sara Meunier Jojo Jiang Alexander Arnold Brienne McLane* Phillip Stokes* *MD at time of publication
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
First published Mar 10, 2014, updated Sept 29, 2024 on www.thecalgaryguide.com")
Hypomagnesemia
![Hypomagnesemia: Physiology
Hyperglycemia
An increased amount of glucose enters renal tubules as glomerulus performs blood filtration
↑ [Solute] in renal tubules from ↑ glucose content exerts osmotic force that pulls water & electrolytes, including Mg2+, into renal tubules
↑ Urinary Mg2+ excretion
Lack of insulin
Lack of insulin receptor signaling in distal convoluted tubule (DCT) ↓ glucose uptake from renal tubules
Hypercalcemia
Ca2+ binds to Ca2+ sensing receptors on thick ascending limb (TAL) of loop of Henle, where resorption of Ca2+ & Mg2+ occurs
Receptor activation ↓ Na-K- 2Cl (NKCC) transporter activity which maintains electro- chemical gradient in TAL
Passive paracellular resorption of Ca2+ and Mg2+, dependent on electrochemical gradient, ↓
↑ Extra- cellular fluid
↓ Resorption of Na+ & H2O from renal tubules
Genetic disorders (e.g. Bartter syndrome, familial hypomagnesemia)
Medications (e.g. loop & thiazide diuretics, certain antibiotics, calcineurin inhibitors)
Some metabolic byproducts of these drugs are nephrotoxic
Inability to absorb free fatty acids (FFAs)
Mg2+, which associates with FFAs, is not absorbed through the gut
Steatorrhea (fat in the stool)
Mal- absorption (often due to inflammation or infection) & diarrhea
Acute pancreatitis
↓ Lipase secretion from pancreas ↑ levels of undigested fats in small intestine
↓ Passive Mg resorption from
tubules
Mg saponification in necrotic fat
2+
Varying mechanisms causing defective Mg2+ re-absorption (e.g. impacts to PCT, TAL, DCT disrupting transporters and ion shifting; ↓ gut resorption of Mg2+)
Nutrients & 2+
electrolytes are lost in stool
undergoes
Renal loss of magnesium
Gastrointestinal loss of magnesium
Hypomagnesemia
Serum [Mg2+] < 0.7 mmol/L
Impairs production and release of parathyroid hormone responsible for ↑ blood Ca2+ Hypocalcemia
Muscle cells are unable to activate Mg2+ dependent ATP hydrolysis
Impairs muscle relaxation and reduces the ability to stop muscular contraction
Lack of Ca2+ disrupts neurotransmitter release and neuronal signaling
Impairs rapid depolarization and repolarization during muscle contraction
Neuromuscular excitability (large, rapid change in membrane voltage due to small stimulus)
Delirium
Apathy
QRS widening and peaking of T waves on ECG
Torsade de Pointes
Constant muscle contraction compresses blood vessels
Reduced blood supply to hands, wrists, feet, and ankles
Trousseau sign (carpopedal spasm with inflation of BP cuff)
Authors: Caroline Kokorudz Reviewers: Shyla Bharadia Allesha Eman Michelle J. Chen Dr. Adam Bass* * MD at time of publication
Chvostek sign (facial muscle twitch with cheek touch)
Seizures
Tetany
Weakness
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published Oct 4, 2024 on www.thecalgaryguide.com](https://calgaryguide.ucalgary.ca/wp-content/uploads/2024/10/Hypomagnesia.jpg "Hypomagnesemia: Physiology
Hyperglycemia
An increased amount of glucose enters renal tubules as glomerulus performs blood filtration
↑ [Solute] in renal tubules from ↑ glucose content exerts osmotic force that pulls water & electrolytes, including Mg2+, into renal tubules
↑ Urinary Mg2+ excretion
Lack of insulin
Lack of insulin receptor signaling in distal convoluted tubule (DCT) ↓ glucose uptake from renal tubules
Hypercalcemia
Ca2+ binds to Ca2+ sensing receptors on thick ascending limb (TAL) of loop of Henle, where resorption of Ca2+ & Mg2+ occurs
Receptor activation ↓ Na-K- 2Cl (NKCC) transporter activity which maintains electro- chemical gradient in TAL
Passive paracellular resorption of Ca2+ and Mg2+, dependent on electrochemical gradient, ↓
↑ Extra- cellular fluid
↓ Resorption of Na+ & H2O from renal tubules
Genetic disorders (e.g. Bartter syndrome, familial hypomagnesemia)
Medications (e.g. loop & thiazide diuretics, certain antibiotics, calcineurin inhibitors)
Some metabolic byproducts of these drugs are nephrotoxic
Inability to absorb free fatty acids (FFAs)
Mg2+, which associates with FFAs, is not absorbed through the gut
Steatorrhea (fat in the stool)
Mal- absorption (often due to inflammation or infection) & diarrhea
Acute pancreatitis
↓ Lipase secretion from pancreas ↑ levels of undigested fats in small intestine
↓ Passive Mg resorption from
tubules
Mg saponification in necrotic fat
2+
Varying mechanisms causing defective Mg2+ re-absorption (e.g. impacts to PCT, TAL, DCT disrupting transporters and ion shifting; ↓ gut resorption of Mg2+)
Nutrients & 2+
electrolytes are lost in stool
undergoes
Renal loss of magnesium
Gastrointestinal loss of magnesium
Hypomagnesemia
Serum [Mg2+] < 0.7 mmol/L
Impairs production and release of parathyroid hormone responsible for ↑ blood Ca2+ Hypocalcemia
Muscle cells are unable to activate Mg2+ dependent ATP hydrolysis
Impairs muscle relaxation and reduces the ability to stop muscular contraction
Lack of Ca2+ disrupts neurotransmitter release and neuronal signaling
Impairs rapid depolarization and repolarization during muscle contraction
Neuromuscular excitability (large, rapid change in membrane voltage due to small stimulus)
Delirium
Apathy
QRS widening and peaking of T waves on ECG
Torsade de Pointes
Constant muscle contraction compresses blood vessels
Reduced blood supply to hands, wrists, feet, and ankles
Trousseau sign (carpopedal spasm with inflation of BP cuff)
Authors: Caroline Kokorudz Reviewers: Shyla Bharadia Allesha Eman Michelle J. Chen Dr. Adam Bass* * MD at time of publication
Chvostek sign (facial muscle twitch with cheek touch)
Seizures
Tetany
Weakness
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published Oct 4, 2024 on www.thecalgaryguide.com")
Precocious Puberty
 > chronological age
Gonadotropin releasing hormone (GnRH)-dependent causes involve activation of the hypothalamic-pituitary-gonadal (HPG) axis. Also known as central precocious puberty
GnRH-independent causes of precocious puberty are related to increased amounts of hormone in the body. Also known as peripheral precocious puberty
Neurogenic (previous CNS insult) and CNS tumors (e.g. hypothalamic hamartoma, germinoma, astrocytoma)
Idiopathic
Tumours causing ↑ hormone levels
Exogenous hormones causing ↑ hormone levels
Genetics causing ↑ hormone levels
21-hydroxylase enzyme deficiency
Growth of ovarian cysts/tumors (ex. germinoma, teratoma, choriocarcinoma)
Growth of β-HCG-secreting tumours (e.g. germinomas, dysgerminomas, hepatomas
Growth of benign or malignant adrenal tumours (e.g. adenoma, carcinoma)
↑ Estrogen and/or progesterone
↑ Beta-HCG stimulates ↑ testosterone production
↑ Cortisol, androgens, and/or aldosterone
Exposure to exogenous sex steroids (e.g. anabolic steroids, topical testosterone, oral contraceptives)
Steroid dependent features (e.g. exposure to estrogen creams in males may lead to breast development)
Missense mutation in LH receptor gene (male limited)
↑ Activation of LH receptors
↑ Testosterone production
Café au lait lesions Polycystic fibrous dysplasia
Puberty onset ages 1-4
↑ Activation and mutation of GNAS1 gene (McCune Albright Syndrome)
Various endocrinopathies
Growth acceleration
Congenital adrenal hyperplasia
↓ Cortisol, ↑ ACTH
Bone age > chronological age Absence of testicular enlargement
Female ambiguous genitalia
Authors: MacKenzie Horn Reviewers: Dasha Mori Michelle J. Chen Dr. Jean Mah* * MD at time of publication
Normal variations in hormone production
↑ Sensitivity to estrogen
Early adrenal androgen secretion
Idiopathic premature thelarche
Idiopathic premature adrenarche
Unilateral or bilateral breast development No other secondary sex characteristics Bone age = chronological age Development of pubic hair ± axillary hair No other secondary sex characteristics Bone age = chronological age
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published Oct 4, 2024 on www.thecalgaryguide.com")
Bone Remodeling Physiology
 activity, ↑ osteoclast (bone cells that break down bone) activity
Hyperparathyroidism ↓ Blood Ca2+
↑ Parathyroid hormone (serum Ca2+ concentration increasing hormone)
Trauma
Osteoblasts detect breach in matrix integrity
Hyperthyroidism
↑ Triiodothyronine (T3)
↑ Bone turnover
Puberty
↑ Growth hormone
↑ Blood calcium (Ca2+)
↑ Calcitonin
(reduces serum Ca2+ by ↓ renal Ca2+ reabsorption & ↑ osteoclast activity)
Net bone resorption process (bone tissue released from bones)
Surveillance osteoblasts produce receptor activator of nuclear activator kappa beta (RANKL; osteoclast stimulating protein)
Monocytes fuse into osteoclasts (cells for bone breakdown)
Osteoclast actions
Secrete HCl to dissolve hydroxyapatite (bone matrix forming inorganic mineral)
Serum markers of bone resorption: C-Telopeptide (CTX) P-Telopeptide (PTX)
Net bone formation process
Hormones activate osteoblast
Osteoblast actions
Secrete osteoprotegerin that binds RANKL
Secrete osteoid seam (a new layer of unmineralized organic bone matrix)
Osteoblasts deposit hydroxyapatite on seam
Phagocytose osteocytes within bone matrix
Authors: Andrew Wu, Jason Kreutz Reviewers:Mizuki Lopez,
Gurreet Bhandal, Luiza Radu *Samuel Fineblit
* MD at time of publication
Secrete collagenase enzyme to help digest collagen
↓ Osteoclast activity
Osteoblasts become osteocytes (mature cells) within bone
Serum markers of bone formation:
↑ Alkaline phosphatase (ALP)
↑ Bone-specific alkaline phosphatase (BSAP; an enzyme produced by osteoblasts during mineralization of bone)
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published Oct 4, 2024 on www.thecalgaryguide.com")
Newborn Disorders of Sexual Development
 & Swyer syndrome (complete gonadal dysgenesis)
Lack or inactivation of sex determining region of the Y chromosome (SRY gene – responsible for testicular differentiation)
Testes produce ↓ testosterone & variable amounts (either ↓/ ↑) of anti- müllerian hormone (AMH – hormone produced by Sertoli cells in male fetuses that signals the regression of the müllerian ducts)
17-beta-hydroxysteroid dehydrogenase 3 deficiency (functional testes with variants in testosterone production)
Variant in any of 5 enzymes involved in the conversion of cholesterol to testosterone
Testes produce insufficient testosterone with no change to AMH production
Wolffian & Müllerian ducts regress
Development of female at birth external genitalia (clitoris, labia majora) or male at birth external genitalia (micropenis, hypospadias)
Puberty may develop secondary sexual characteristics typical of males at birth (deep voice, male pattern facial/body hair)
5-alpha reductase deficiency
Variation in the SRD5A2 gene that codes for 5-alpha-RD2 enzyme (responsible for differentiation of gonads into male reproductive system)
5-alpha-RD2 enzyme is impaired & unable to convert testosterone into dihydrotestosterone (DHT)
DHT unable to block formation of female at birth genitalia & promote development of penis & scrotum
Development of ambiguous external genitalia resembling female at birth (unfused labioscrotal folds, undescended testes)
Puberty may develop secondary sexual characteristics typical of males at birth
Complete androgen insensitivity
Impairment of androgen receptors causes unresponsiveness of testicles to testosterone & no change to AMH production
Testes unable to respond to exogenous testosterone treatment
Wolffian
& Müllerian ducts regress
Development of female at birth external genitalia
Puberty may develop secondary sexual characteristics typical of females at birth (breast enlargement, menarche)
Aromatase deficiency
Variations in the CYP19A1 gene that codes for aromatase enzyme
Aromatase enzyme is impaired & unable to convert androgens (testosterone) into estrogen in the placenta
↑ Testosterone levels & absence of anti-Mullerian hormone
Regression of Wolffian ducts & Mullerian ducts differentiate into fallopian tubes & uterus
Development of normal female at birth internal reproductive structures & ambiguous genitalia that is not clearly female or male at birth
Hypergonadotropic hypogonadism at puberty (↓ function of gonads & do not develop secondary sexual characteristics)
46XX Genotype
XX Ovotesticular disorder of sex development
Functional SOX9 gene that codes for testicular Sertoli cells differentiation despite lack of SRY gene
Fetus exposed to only X chromosome (responsible for female gonadal development) due to absence of testicular determinant (SRY)
Prescence of estradiol in developing ovarian follicles inhibits spermatogenesis (sperm production) & no exposure to AMH
Mullerian ducts differentiate into oviduct, uterus, cervix, upper vagina & regression of Wolffian ducts
Development of ambiguous external genitalia (labioscrotal fusion, hypospadias)
& bilateral ovotestes (both ovarian & testicular tissue) or combination of a unilateral ovary or testis with ovotestis on contralateral side
Puberty may develop secondary sexual characteristics typical of femalea at birth (breast enlargement, menarche)
*Samuel Fineblit *MD at time of publication
Ovaries with 21-hydroxylase deficiency
Congenital adrenal hyperplasia (↑ adrenal tissue cell production due to disruption in cortisol synthesis pathway)
↓ Cortisol/aldosterone production from adrenal glands & ↑ Adrenocorticotropic hormone (ACTH) to compensate
Incomplete development of Wolffian ducts (precursor to male internal genitalia) & Müllerian ducts (precursor to female reproductive tract)
Development of ambiguous external genitalia (one testicle undescended; hypospadias where the urethra position is atypical; clitoromegaly)
Puberty may develop secondary sexual characteristics typical of males or females at birth
Swyer syndrome specific: Wolffian & Müllerian ducts regress
Development of normal female internal reproductive structures (uterus & fallopian tubes) & gonadal streaks (underdeveloped ovaries replaced with fibrous scar tissue)
Puberty is stalled unless treated with hormone replacement therapy
ACTH stimulates adrenal gland to ↑ hormone production (both testosterone & estrogen)
Despite ↑ testosterone production, the levels are not sufficient for development of Wolffian ducts & ovaries cannot produce AMH
Wolffian ducts regress & Müllerian ducts develop
Male typical genitalia (male XY) with microorchidism, hyperpigmented scrotum, & enlarged penis
Signs of salt-wasting: low sodium, low potassium, low blood pressure, failure to thrive
Ambiguous external genitalia (female XX: fusion of labial fold, clitoromegaly, penile urethra, urogenital sinus abnormality, hyperpigmentation)
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published Nov 11, 2024 on www.thecalgaryguide.com")
Suboxone & Methadone

Methadone (a long-acting, full agonist) acts at mu-opioid receptors
Buprenorphine (a long-acting, partial agonist with high affinity properties) displaces full-agonists at mu-opioid receptors (e.g., fentanyl).
Naloxone (a mu-opioid receptor antagonist) displaces buprenorphine at mu-opioid receptors
↓ Effects of buprenorphine in intramuscular and intranasal forms
↓ Potential for abuse of buprenorphine
Repeated intake of increasing doses of methadone
Accumulation of methadone
↑ Potency compared to partial agonist (e.g., buprenorphine)
↓ Inhibitory GABAergic neuronal activity
Disinhibition of dopamine at nucleus accumbens
↑ Dopamine levels at the nucleus accumbens and reward pathways
Euphoria
↑ Adherence
↑ Adherence especially in those with severe opioid dependence
Maintained stimulation of opioid receptors
↑ Buprenorphine required to occupy receptors to produce the same level of effect compared to full-agonists
Saturation of mu- opioid receptors (ceiling effect)
Further intake of buprenorphine does not ↑ stimulation of mu-opioid receptors at the locus coeruleus in the brainstem
Locus coeruleus maintains sensitivity to carbon dioxide concentrations
Ventilation is maintained
↓ Risk of overdose and respiratory depression
↓ Stimulation of mu-opioid receptors
↓ Analgesia
↑ Withdrawal
Potassium channels open
Post-synaptic hyperpolarization of cells
Presynaptic inhibition of neurotransmitter (glutamate and substance P) release
Inhibition of locus coeruleus which is in a hyperexcitable state from chronic opioid use
↓ Noradrenaline release from locus coeruleus
↓ Autonomic excitability throughout brain, spinal cord, peripheral nerves and GI tract.
↓ Withdrawal symptoms (restlessness, anxiety, lacrimation, nausea, vomiting, diarrhea)
↓ Euphoria
Methadone blocks channels involved in repolarization of myocytes
Prolonged repolarization of myocytes
Prolonged QTc interval & cardiac arrythmias
Inhibition of locus coeruleus’ ability to sense carbon dioxide and maintain ventilation
↑ Risk of overdose & respiratory depression
↓ Pain signal transmission
Analgesia
Opioid agonist therapy
Further intake of buprenorphine does not ↓ inhibitory GABAergic neuronal activity
GABAergic neurons maintain inhibitory effect on dopamine at the nucleus accumbens
No ↑ in euphoric effects
↓ Adherence to therapy especially in those with severe opioid dependence
(↓ cravings and withdrawal symptoms thereby reducing risk of relapse)
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published Nov 11, 2024 on www.thecalgaryguide.com")
Cannabis Use Disorder
 in cannabis binds human cannabinoid receptor 1 (CB1) receptors in ventral tegmental area
Biological factors: earlier age of cannabis use onset, male gender, genetic predisposition
Psychosocial factors:
stress, aggression, depression, anxiety, parent/peer cannabis or other substance use, ↓ SES, family dysfunction, bullying, occupational difficulties
Disinhibits dopaminergic signaling and reward response
↑ Motivation to continue cannabis use
Cannabis Use Disorder
Cannabis use that may lead to significant distress or impairment in psychological, physical, or social functioning
Cannabis (CB1 receptor agonist), especially with ↑ THC, acutely causes burst firing of ventral tegmental area
↑ Dopamine in striatal and prefrontal areas
↑ Dopaminergic transmission in the mesolimbic pathway (neuronal network involved in mediation of psychosis)
Delusions, hallucinations, & paranoia occurring with acute or chronic use
Cannabis induced psychotic disorder
↑ THC activates CB1 receptors on GABAergic terminals & ↓ THC activates CB1 receptors on glutaminergic terminals
GABA & glutamate dysregulation and imbalance (can occur with acute or chronic use)
Repeated exposures to ↑THC results in serotonin (5- HT) receptor upregulation
↑ Neuro-
endocrine responses of stress hormones, which may ↑ serotonin reuptake
Sedative action of low dose THC on CB1 receptors causes ↓ sleep latency and ↑ slow wave sleep with acute cannabis use
Tolerance with chronic usage causes reversal of acute effects
↑ Sleep latency & ↓ slow wave sleep
↓ Sleep time & quality; ↓ cerebral restoration & recovery
Cannabis induced sleep disorder
Chronic cannabinoid receptor overstimulation
Receptor dysregulation, desensitization, and downregulation
Hypothalamic pituitary adrenal (HPA) axis dysregulation
Inhibited gastrointestinal motility and digestion
Nausea & vomiting & abdominal pain after cannabis use
Cannabinoid hyperemesis syndrome (cyclic vomiting with cannabis use)
Down regulation of CB1 receptors due to long term cannabis usage, paired with sudden discontinuation of cannabis use
↓ Cannabinoid in ↑CB1 receptors disrupts emotional processing, sleep and appetite homeostasis within the endocannabinoid system
Anxiety, irritability, anger, depression, sleep disruption, appetite loss.
Cannabinoid withdrawal
Mood changes, anxiety and/or panic attacks
Cannabis induced mood and anxiety disorders
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published Nov 11, 2024 on www.thecalgaryguide.com
Sympathetic nervous system dysregulation")
Renal manifestations of SLE
: Renal Manifestations
Authors: Madison Turk Reviewers: Modhawi Alqanaie Mao Ding Luiza Radu Glen Hazlewood* * MD at time of publication
Genetic susceptibility and potential environmental triggers (smoking, silica, Epstein-Barr Virus, hormones, ect)
↓Clearance of dead cell debris in the body
(exact mechanisms unknown; See slide on pathogenesis of SLE)
Extracellular exposure of nuclear proteins (which bind DNA and regulate gene expression)
Immune cells respond to nuclear proteins as if they are non-self, as they usually don’t ‘see’ them
Systemic Lupus Erythematosus
An autoimmune disease characterized by anti-nuclear-antibody production resulting in widespread inflammation and tissue damage in varying affected organs, including one, or a combination of, joints, skin, brain, lungs, kidneys, and blood vessels
Production of auto-antibodies against self nuclear proteins
IC deposition in renal vessels
Activation of inflammatory cells against IC’s causing vascular injury/inflammation
↑Clot formation in vessels leading to ↓vessel lumen diameter and ↓blood flow
↑Reninàcleavage of angiotensinogen to angiotensin Ià cleavage by angiotensin converting enzyme into angiotensin II
Vessel constriction to ↑blood pressure (to ↑ renal blood flow)
IC deposition in tubule basement membrane of the kidney
Formation of immune complexes (IC) (collections of antigen(s), antibodies, and/or complement proteins bound together)
Tubulointerstitial nephritis: (Inflammation of the renal tubules and interstitium, sparing the glomeruli)
Renal ischemia
IC deposition in the glomerulus
Activation of complement, initiating an inflammatory response
Recruitment of myeloid cells (monocytes/ neutrophils)
Production of reactive oxygen species, cytokines and release of cytotoxic granules
Tubular inflammation and damage resulting in ↓sodium reabsorption
Renal release of reninà ↑ angiotensin I/II and resulting ↑ aldosterone
↑Sodium reabsorption, and potassium excretion in the collecting duct
↓Potassium secretion from the Distal Convoluted Tubule
Hyperkalemia Hypokalemia
⍺-intercalated cell damage
↓Acid excretion
Metabolic acidosis
End stage kidney disease
(all manifestations can result in this)
Hypertension
Tissue damage from the inflammatory response
Glomerular nephritis (inflammation/damage of the filtering part of the kidneys)
Fibrosis (thickening or scarring of tissue)
Mesangial and parietal cell proliferation
Podocyte injury
↑blood and protein excretion due to damage to the glomerular filtration membrane
↓protein in blood Edema
Proteinuria Hematuria
↓Functional renal tissue to filter creatinine from blood
↓ Glomerular filtration rate ↑ Plasma creatinine
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published Nov 11, 2024 on www.thecalgaryguide.com")
Rapid sequence induction and intubation
: Indications & considerations
“Full stomach”: ↑ risk of regurgitation, vomiting, aspiration Life-threatening injury or illness requiring immediate or rapid airway control
↓ Gastro- esophageal sphincter competence (elderly, pregnancy, hiatus hernia, obesity)
↑ Intragastric pressure (pregnancy, obesity, bowel obstruction, large abdominal tumors)
Delayed gastric emptying (narcotics, anticholinergics, pregnancy, renal failure, diabetes)
↓ Level of consciousness (drug/alcohol overdose, head injury, trauma or shock state)
Respiratory & ventilatory compromise (i.e., hypoxic or hypercapnic respiratory failure)
Achalasia (esophageal motility disorder resulting in impaired swallowing)
Dynamically deteriorating clinical situation (i.e., trauma)
GI bleed
Impaired airway reflexes
↓ Muscle tone of structures in the airway (i.e., tongue, pharyngeal walls, & soft palate)
Patients who did not stop GLP-1 agonist preoperatively as advised
Impaired clearance of secretions or vomitus
↓ Safe apnea time before hemodynamic decompensation
Unprotected airway
Need for rapidly securing airway while avoiding aspiration & hemodynamic compromise
Rapid sequence intubation (RSI): Simultaneous administration of induction agent (unconsciousness) & neuromuscular blocking agent (paralysis) to achieve intubation conditions (~45-60 seconds after IV push) for rapid control of an emergency airway
Preoxygenation
Deranged physiologic conditions (i.e., hypotension, acidosis, hypoxemia)
Reduced tolerance for
apnea (period with no ventilation or oxygenation)
Pre-oxygenate with high flow O2 (15L) to create a large pulmonary & tissue reservoir of oxygen
↓ Significant oxygen desaturation during apnea
↑ Oxygen saturation on pulse oximetry
Induction
Laryngoscopy & intubation are a potent sympathetic nervous system stimulus
Airway manipulation causes a surge in catecholamines
Paralysis
Visualization & passage of endotracheal tube requires relaxation of vocal cords & surrounding muscles
Neuromuscular blocking agents facilitate paralysis
Rescue
Some induction agents (i.e., propofol) are vasodilators
Hemodynamically unstable or patients in shock
Hypotension
Tachycardia
↑ Intracranial pressure (ICP)
Hypertension
Suppress cough & gag reflex
Prevent laryngospasm (involuntary closure of vocal cords to airway manipulation)
Minimize movement during procedure
Vasoactive agents (i.e., ephedrine, phenylephrine) ↑ systemic vascular resistance
Atropine & glycopyrrolate ↑ heart rate
Lidocaine (Na+ channel blocker) & opioids (μ receptor agonist) ↓ transmission of pain
↓ Sympathetic response, myocardial demand & physiologic stress
Anesthetics (i.e., propofol) achieve unconsciousness for paralysis & intubation
↓ Airway trauma & damage to vocal cords
Bag mask ventilation typically avoided in this step to ↓ gastric insufflation & risk of aspiration
Cricoid pressure (Sellick maneuver): posterior displacement of cricoid ring to compress esophagus against C-spine to prevent passive regurgitation of gastric contents to airway. Applied from start of induction, released when placement of endotracheal tube is confirmed by capnography.
Intubation
↑ Blood pressure and/or cardiac output
Authors: Jen Guo Reviewers: Priyanka Grewa Luiza Radu Leyla Baghirzada* * MD at time of publication
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published November 18, 2024 on www.thecalgaryguide.com")
Calcium Channel Blockers
![Calcium Channel Blockers: Mechanisms & side effects
Authors:
Caroline Kokorudz Reviewers:
Rafael Sanguinetti Andrew Wu
Luiza Radu
Timothy Pollak*
* MD at time of publication
Calcium channel blocker medications
inhibit Ca2+ channels in smooth muscle
Reduction of Ca2+ influx into smooth muscle cells
Inhibits calcium-dependent aldosterone synthesis reducing Na+ & H2O resorption in renal distal tubules
Negative feedback to pituitary gland causing ↑ ACTH (adrenocorticotropic hormone)
↑ Androgens (testosterone)
Testosterone acts on gingival cells (multiple cell types that support teeth) & connective tissue matrix
Gingival hyperplasia (gum overgrowth)
Non-dihydropyridines:
(Phenylalkylamines [verapamil], Benzothiazepines [diltiazem]) less potent vasodilators & selective for heart muscle
Prevents smooth muscle contraction
Dihydropyridines:
(amlodipine, felodipine, nifedipine) vasodilate vascular smooth muscle
↓ Arterial resistance and blood pressure in coronary & peripheral arteries
Coronary artery vasodilation
↓ Pressure in coronary arteries
↑ Blood flow through coronary arteries
Reduced
ischemia relieves angina
Inhibits L-type Ca2+ channels, preventing rapid nodal depolarization
Reduces excitation of sinoatrial (SA) & atrioventricular (AV) nodal tissues
↓ Conduction speed of electrical impulses
↓ Contractile strength of cardiomyocytes (heart muscle cell)
↓ Systemic vascular resistance & cardiac
afterload (heart pumping resistance)
↑ Blood volume flowing into significantly smaller vessels
↑ Capillary blood pressure
↑ Circulation to face
Flushes (red & warm)
↓ Cardiac output
↓ Tissue perfusion & attempt to ↑ cardiac output
Worsens heart failure
↓ Oxygen demand of heart muscle
More favorable oxygen supply to demand ratio
Relieves angina
↓ Blood pressure
↓ Cerebral perfusion
Syncope (fainting)
Relieves angina
Capillary fluid leak increased to interstitial space
Peripheral edema
↑ Intracranial pressure Compresses nerve endings Headache
↓ Heart rate Bradycardia
Suppresses dysrhythmias (abnormal heart rhythm)
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published Nov 21, 2024 on www.thecalgaryguide.com](https://calgaryguide.ucalgary.ca/wp-content/uploads/2024/11/Calcium-Channel-Blockers.jpg "Calcium Channel Blockers: Mechanisms & side effects
Authors:
Caroline Kokorudz Reviewers:
Rafael Sanguinetti Andrew Wu
Luiza Radu
Timothy Pollak*
* MD at time of publication
Calcium channel blocker medications
inhibit Ca2+ channels in smooth muscle
Reduction of Ca2+ influx into smooth muscle cells
Inhibits calcium-dependent aldosterone synthesis reducing Na+ & H2O resorption in renal distal tubules
Negative feedback to pituitary gland causing ↑ ACTH (adrenocorticotropic hormone)
↑ Androgens (testosterone)
Testosterone acts on gingival cells (multiple cell types that support teeth) & connective tissue matrix
Gingival hyperplasia (gum overgrowth)
Non-dihydropyridines:
(Phenylalkylamines [verapamil], Benzothiazepines [diltiazem]) less potent vasodilators & selective for heart muscle
Prevents smooth muscle contraction
Dihydropyridines:
(amlodipine, felodipine, nifedipine) vasodilate vascular smooth muscle
↓ Arterial resistance and blood pressure in coronary & peripheral arteries
Coronary artery vasodilation
↓ Pressure in coronary arteries
↑ Blood flow through coronary arteries
Reduced
ischemia relieves angina
Inhibits L-type Ca2+ channels, preventing rapid nodal depolarization
Reduces excitation of sinoatrial (SA) & atrioventricular (AV) nodal tissues
↓ Conduction speed of electrical impulses
↓ Contractile strength of cardiomyocytes (heart muscle cell)
↓ Systemic vascular resistance & cardiac
afterload (heart pumping resistance)
↑ Blood volume flowing into significantly smaller vessels
↑ Capillary blood pressure
↑ Circulation to face
Flushes (red & warm)
↓ Cardiac output
↓ Tissue perfusion & attempt to ↑ cardiac output
Worsens heart failure
↓ Oxygen demand of heart muscle
More favorable oxygen supply to demand ratio
Relieves angina
↓ Blood pressure
↓ Cerebral perfusion
Syncope (fainting)
Relieves angina
Capillary fluid leak increased to interstitial space
Peripheral edema
↑ Intracranial pressure Compresses nerve endings Headache
↓ Heart rate Bradycardia
Suppresses dysrhythmias (abnormal heart rhythm)
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published Nov 21, 2024 on www.thecalgaryguide.com")
Pre-Eclampsia Pathogenesis

Primigravida (1st pregnancy)
Advanced maternal age
Genetic factors (e.g. sFLT1)
Collagen vascular disease
Previous preeclamptic pregnancies
Multiple gestation
Other unclear causes
Dysregulated immune and non-immune decidual cells (specialized endometrial cells) promote abnormal placental trophoblast invasion and uterine spiral artery formation (exact mechanism unclear)
Multiple unclear, complicated mechanisms
Abnormal association of placental vasculature with uterine vasculature early in pregnancy (utero-placental mismatch) disrupts adequate blood perfusion from placenta to fetus (< 20 weeks gestation or early-onset)
Fetus experiences under- perfusion, hypoxia, ischemia, and oxidative stress
Placental cells release molecules toxic to vascular endothelium into the maternal circulation
Damaged maternal blood vessels are less able to perfuse the placenta
Systemic dysfunction of maternal blood vessel endothelium (endothelial cell activation) (>20 weeks gestation or late-onset)
Inflammatory cells retain memory postpartum which increases risk for developing preeclampsia in subsequent pregnancies
Hypoxia leads to placental cell death, which results in release of fetal antigens
Authors:
Yan Yu
Jasmine Nguyen Reviewers:
Kayla Nelson
Radhmila Parmar
Sean Spence
Monica Kidd*
Katrina Krakowski* Maryam Nasr-Esfahani* Riya Prajapati
Michelle J. Chen
Jadine Paw*
* MD at time of publication
Placental ischemia & injury
Pre-existing maternal diseases damage endothelial cells (e.g. vasculitis, diabetes, renal diseases, chronic hypertension)
Placenta is unable to supply sufficient O2 and nutrients to meet demands of growing fetus
Maternal Clinical Findings and Complications**
Fetal Clinical
Findings and Complications**
**See corresponding Calgary Guide slides
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published Feb 10, 2017; updated Nov 22, 2024 on www.thecalgaryguide.com")
Loop diuretics
 symporter at thick ascending limb of Loop of Henle (aLOH)
Authors: Andrew Wu, Rafael Sanguinetti Reviewers: Luiza Radu, Adam Bass* * MD at time of publication
Over-inhibition of ion symporter in ear at high concentrations
Damage to tight junctions in the blood vessel epithelium in the ear
Disruption of the blood- cochlear barrier
Hearing loss
Opening of ATP-sensitive K+ channels on pancreatic β- cells causes efflux of K+
↓ Intracellular K+ causes a negative intracellular charge
↓ Ability for pancreatic β- cells to depolarize & release insulin
Hyperglycemia (rare)
Stimulation of prostaglandin E2 (PGE2) synthesis in the cortical & medullary aLOH
Stimulation of endothelial nitric
oxide synthase releases nitric oxide
Systemic venodilation (dilation of veins)
↓ Systemic vascular resistance
↓ Blood pressure
↓ Na+ in renal interstitium
↓ Water reabsorption (water follows Na+)
Interstitial washout (↑ NaCl & water wasting in interstitium)
↓ Sodium (Na+) reabsorption
↑ Positive luminal charge at thick aLOH
↓ Electrochemical gradient reduces cellular transport of cations
↓ Ca2+ & Mg2+ reabsorption in the aLOH
Hypocalcaemia & hypomagnesemia
↑ Na+ delivery to the distal convoluted tubule (DCT)
↑ Intracellular Na+ at collecting duct
↑ K+ efflux via renal outer medullary potassium channel (ROMK)
↓ Fluid status
↓ Serum Na+
↓Serum Cl-
↑ Luminal K+ at alpha intercalated cell
Chemical gradient ↑ K+/H+ antiporter activity
↑ K+ excretion
Hypokalemia
Chemical gradient shifts K+ into cells creating a charge gradient to shift H+ out
Hyponatremia Hypovolemia Hypochloremia
↓ Volume status activates angiotensin II, which plays a role in sodium and volume retention
↑ Na+/H+ antiporter 3 (NHE3) activity in the proximal tubule
Kidneys excrete & conserve ions to preserve intracellular charge gradient
↑ H+ loss in the urine ↓ Serum H+
↑ Proximal Na+ reabsorption causes ↑ urate reabsorption via urate-hydroxyl exchangers
Hyperuricemia
Contraction alkalosis (loss of Na+-rich, HCO3- low fluid) & hypokalemia
↑ Serum HCO3-
Metabolic alkalosis (systemic pH > 7.45 due to metabolic process)
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published Nov 18, 2024 on www.thecalgaryguide.com")
Hypocalcemia Pathogenesis

Autoimmune (e.g., autoimmune polyglandular syndrome)
Infiltrative (e.g., hemochro matosis, Wilson’s disease)
Congenital (e.g., DiGeorge)
Hypomagnesemia
Magnesium is needed to produce PTH
Malabsorption
↓ Small bowel surface area
↓ Absorption of 25-OH Vitamin D (calcidiol)
↓ Conversion of calcidiol to 1-25 (OH)2 Vitamin D (calcitriol) by 1-α- hydroxylase
↓ Calcitriol
(impacts GI absorption of calcium)
Chronic Renal Failure
↓ Renal parenchymal mass (site of 1-α- hydroxylase production)
Vitamin D Dependent Type 1 Rickets
Mutation in the gene that encodes 1-α- hydroxylase
↓ Sun exposure
↓ 1-α-hydroxylase Pancreatitis
Destruction or atrophy of parathyroid gland
↓ Parathyroid hormone (PTH)
Calcitriol resistance (e.g., Vitamin D Type 2 rickets)
Inflammation of pancreas
↑ Release of lipase enzyme into serum
↑ Breakdown of fat by lipase
↑ Free fatty acids in serum
↑ Ca2+ binding to negatively charged tail of free fatty acids
↑ Parathyroid hormone (PTH)
PTH resistance
Genetic mutation causes body to not respond to PTH
↓ Renal calcium reabsorption
↑ Calcium excretion
↓ Osteoclast activity
↓ Calcium release from bone
↓ Action of PTH on intestinal cells
↓ Absorption of calcium in the small intestine
↑ Serum phosphate (PO4) (e.g. tumor lysis, rhabdomyolysis)
↑ Ca2+
binding to
negatively charged PO4
↑ Serum pH
↓ H+ in serum binding to proteins
↑ Ca2+ binding to proteins
Calcium sequestration
↓ Absorption of calcium in the small intestine
Hypocalcemia
Author: Breanna Fang Reviewers: Gurreet Bhandal, Raafi Ali, Luiza Radu Samuel Fineblit* * MD at time of publication
Serum calcium levels below 2.10mmol/L
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published Nov 22, 2024 on www.thecalgaryguide.com")
Anesthetic Considerations Laparoscopic Abdominal Surgery

Cardiovascular Effects
Positional
Pneumoperitoneum (introduction of gas eg. CO2 in the abdominal cavity for easier access to organs during laparoscopy)
Systemic vasodilation
↓ Systemic vascular resistance
↑ Systemic blood flow & oxygen delivery
Respiratory acidosis
Acidosis leads to ↓ cardiac contractility
Arrhythmia
Cardiac arrest
Reverse Trendelenburg positioning of patient (patient supine with head of OR table ↑)
↑ Venous pooling (accumulation of blood in veins)
↓ Venous return (blood returning to the heart)
↓ Preload
↓ Blood pressure
Trendelenburg positioning of patient (patient supine with head of OR table ↓)
Low fluid status
Compression of large abdominal vessels
↓ Preload
↓ Blood pressure
High fluid status
↑ Venous return (blood returning to the heart)
↑ Preload (ventricle filling pressure prior to contraction)
↑ Blood pressure
↑ Venous return
↑ Preload
↑ Blood pressure
Vagus nerve stimulation
Bradyarrhythmia (slow and irregular heart rate)
Asystole (absence of electrical activity and contractility of the heart)
↑ Cerebral blood flow
Cephalad (upward) displacement of diaphragm & compression of thoracic cavity
↓ Functional residual capacity (volume remaining after expiration) & pulmonary compliance
↓ Tidal volumes (air inhaled per breath)
Atelectasis (partial or full collapse of lung)
V/Q mismatch
(lung ventilation & perfusion imbalance)
Respiratory Effects
↑ Intraocular pressure (e.g. papilledema)
Ophthalmic compromise (visual system impairment)
↑ Intracranial pressure
Compression of renal vessels à↓ Renal blood flow
Release of catecholamines (norepinephrine & dopamine)
Renin angiotensin aldosterone system activation
Aldosterone & vasopressin release
↑ Blood pressure Renal System Effects
Neurologic Effects
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published Nov 25, 2024on www.thecalgaryguide.com")
Ewing Sarcoma
(q24;q12) detectable with a genetic molecular study called FISH
Authors: Curtis Ostertag Nojan Mannani Reviewers: Mankirat Bhogal Michelle J. Chen Dr. Michael Monument* * MD at time of publication
Well-defined histopathology
Biopsy: sheets of monotonous small round blue cells with increased nuclear to cytoplasmic ratio
Immunohistochemistry staining positive for CD99 (80% of cases)
Diagnostic confirmation by histopathology
Diagnostic confirmation by molecular genetics
X-Ray findings: moth-eaten
lesions, Codman triangle of elevated periosteum, or onion- skin periosteal reaction
MRI findings: solid mass in bone, low T1 intensity, high T2, gadolinium enhancement, surrounding edema & soft tissue mass
Malignant small round cell tumor; the second most common malignant bone tumor in adolescents and young adults. Most common in the axial skeleton (pelvis, shoulder girdle, and ribs) and diaphysis of long bones.
Rapid cell turnover
Tumor expansion in bone, disrupting normal tissue
Possible metastasis to surrounding soft tissue
Possible metastasis to lung/pleura
Nodule/malignant pleural effusion
Absent breath sounds, pleural signs, crackles/rales
Possible metastasis to bone marrow
Replacement of normal hematopoietic tissue, including megakaryocytes (source of platelets)
Thrombocytopenia
Petechiae/purpura
Peritumoral edema
Swelling Stiffness
Invasion of cancer cells to distant tissues via bloodstream
Tumor expands toward skin surface
Palpable lump
Tumor secretes ↑ vascular endothelial growth factor (VEGF)
↑ Generation of immature blood vessels which have ↑ permeability
↑ Expression of inflammatory cytokines (i.e. IL-6)
Massive metabolic demand from cancer cells
Energy starved state
Systemic constitutional symptoms including fevers, chills, weight loss, night sweats, fatigue
↓ Distractions at night brings ↑ attention to pain
Adrenal glands naturally release ↓ cortisol throughout the day with the lowest levels at night. Lack of anti- inflammatory properties from cortisol ↑ pain
Potential gravity- related ↑ in tumor- related pressure & ↑ in active bone remodeling
Tumour growth stretches periosteum (connective tissue around bone) causing osteolysis, inflammation, nerve infiltration, edema, structural weakening (microfractures)
Pain that worsens at night
Limp
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published Dec 30, 2024 on www.thecalgaryguide.com")
Pituitary Tumour Classification and Clinical Outcomes
 (lack of hormone-producing cells or due to gene mutations)
Authors: Chris Oleynick Caroline Kokorudz Reviewers: Amyna Fidai Laura Byford- Richardson Joseph Tropiano Julia Gospodinov Luiza Radu Hanan Bassyouni* * MD at time of publication
hormones (70%)
Relative abundance & predisposition for lactotroph (prolactin-producing), somatotroph (growth hormone – producing), thyrotroph (TSH producing) & corticotroph (adrenocorticotropic hormone – producing) cells to form adenomas (epithelial cell tumours)
↑ Lactotroph cells
↑ Prolactin (PRL) (most common)
Hyper- prolactinemia (high prolactin blood levels)
↑ Somatotroph cells
↑ Growth hormone (GH)
Acromegaly* (excess tissue growth & metabolic dysfunction)
↑ Corticotroph cells
↑ Adrenocortico- tropic hormone (ACTH)
Cushing disease* (prolonged exposure to excess cortisol)
↑ Thyrotroph cells
↑ Thyroid stimulating hormone (TSH)
Central hyper- thyroidism (↑ T4 & T3)
Large size (macro) (>10mm on MRI) may cause mass effect (tissue compression from mass)
Small size (micro) (<10mm on MRI) unlikely to cause mass effect (compression to surrounding structures)
Asymptomatic
Headache
Nausea & vomiting
Compresses pituitary gland & impairs blood supply & function of pituitary cells
Impinges pituitary stalk & disrupts hormone transport from hypothalamus to the anterior and posterior pituitary
Compresses surrounding structures
↑ Pressure & stretching of dural mater
Stretching of the meninges activates mechanoreceptors (stretch receptors) in GI tract
Pituitary hormone deficiency (typically in left-right order with mass effect):
Dopamine release obstruction
↓ Inhibition of prolactin secretion
Antidiuretic hormone (ADH) obstruction
Inferior tumour growth
Growth into sphenoid sinus
Lateral growth of the tumor compresses surrounding cranial nerves
Superior tumour growth
↓ GH
↓ Protein production & muscle cell proliferation
↓ Luteinizing hormone (LH) (triggers ovulation & sex hormone synthesis) & follicle stimulating hormone (FSH) (stimulates ovarian follicles & sperm growth)
↓ (TSH)
↓ ACTH (stimulates cortisol & androgen release)
Cranial nerve (CN) II (optic)
↓ Visual acuity
Diplopia (double vision)
CN III, (oculomotor) IV (trochlear), or VI (abducens)
Ptosis* (drooping upper eyelid)
CN V (trigeminal) branches V1 & V2
Facial numbness
Ophthalmoplegia (eye muscle paralysis)
Compresses optic chiasma
Bitemporal hemianopsia (decreased lateral peripheral vision)
Tumour extends into hypothalamus
Hypothalamic dysfunction due to damage to hypothalamic cells
Disruption of multiple regulatory systems (i.e. sleep-wake cycles, appetite, temperature)
Tumour occludes ventricles
Tumour obstructs CSF flow
↑ Intracranial pressure
Hydrocephalus* (abnormal buildup of CSF in ventricles of the brain) and papilledema
Bacteria migrates from sinus flora into sphenoid sinus
Cerebrospinal fluid (CSF) leaks into throat
Post-nasal drip and nasopharyngeal mass
Stunted growth and short stature in children
↓ Muscle mass & fatigue
Diabetes insipidus* (excessive urination & extreme thirst)
Central hypothyroidism* (↓ T4 & T3)
Hyperprolactinemia
Meningitis* (inflammation of the meningeal layers of central nervous system)
*See relevant Calgary Guide slide
Hypogonadotropic hypogonadism (↓ Sex hormones)
Adrenal insufficiency* (↓ 8 AM cortisol)
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published Oct 1, 2027; updated Jan 5, 2025 on www.thecalgaryguide.com")
Insuficiencia Renal Aguda Post-Renal
: Patogenia y hallazgos clínicos")
Central Precocious Puberty
, trauma, infection, ischemia
Damage to hypothalamic & pituitary structures (i.e. infarct or traumatic sheering forces often causing inflammation and ultimately cell death)
Septo-optic dysplasia (early brain development disorder)
Abnormal pituitary & hypothalamus development (unknown pathogenesis)
Psychosocial stress
International adoption from developing countries
Rapid increase in nutrition
Rapid catch-up growth
Hormonal and metabolic changes
Endocrine disrupting chemicals (ex. Phthalates, bisphenol A)
Hypothalamic inflammation
Suppression of inhibitory and activation of stimulatory elements of the GnRH production pathway
Idiopathic familial central precocious puberty from different genetic mutations
primarily impacting tissues derived from ectoderm)
Makorin RING- finger protein 3( MKRN3) mutation (repressor of GnRH secretion)
Kisspeptin protein and/or receptor mutation (regulates puberty & reproductive function)
Extended Kisspeptin protein availability and/or ↑ production
Kisspeptin receptors signal directly to gonadotropin releasing hormone (GnRH) neurons to release GnRH into portal system
Early ↑ or inappropriate GnRH pulses
Neurofibromatos is T1 (NF1) (causes benign tumors to grow in the brain along nerves)*
Tumor development involving the optic chiasm (>15% of individuals with NF1) may impact hypothalamic function
Damage to and/or disruption of normal hypothalamic & pituitary function
Tuberous sclerosis autosomal dominant disorder (rare genetic disease that causes growth of benign tumors in brain & body)
Dysregulated cell division & growth
Hamartomas form as benign mass composed of cells similar to those surrounding it, occurs in many areas including hypothalamic hamartoma (HH)
HH acts as an accessory, unregulated hypothalamus
Autonomous (unregulated) pulsatile GnRH release
Sturge-Weber Syndrome (vascular disorder causing capillary-venus malformations impacting the face, brain, eyes)
Vascular malformation throughout the brain including the hypothalamus
Impaired blood flow to the hypothalamus
Hypothalamic ischemia
Hypothalamus dysregulation and premature activation of GnRH release
Disruption of excitation and inhibition balance in the CNS
Hypothalamic- pituitary dysregulation
Reduced inhibition of GnRH secretion
Increased corticotropin- releasing hormone
Adrenal gland production & release of androgens by unknown mechanism that play a role in maturation of the HPG axis
Adrenarche (start of androgen production). Including: acne, body odor, & pubarche (pubic hair growth)
Dysregulated GnRH pulses
Dysregulated luteinizing hormone (LH) & follicle simulating hormone (FSH) release
Early GnRH release
Early FSH secretion from pituitary gland
↑ Uterine volume & growth of ovarian follicles
Cyclical ovulation
Premature menarche (first menses < 7 years)
Early LH secretion from pituitary
↑ Gonadal androgen production
↑ Estrogen release from ovaries
gland
**See Calgary Guide slide - “Brain Neoplasms”
↑ Skeletal growth
↑ Bone mineral density
Thelarche (breast development)
Authors: Joanna Keough Reviewers: Run Xuan (Karen) Zeng Luiza Radu Samuel Fineblit* *MD at time of publication
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published Jan 28, 2025 on www.thecalgaryguide.com")
PROP1-Related Combined Pituitary Hormone Deficiency
 from
lactotrophs
Impaired mammary
gland stimulation
prevents adequate
milk production
PROP1-Related Combined Pituitary Hormone Deficiency: Pathogenesis and clinical findings
Consanguinity
Inherited PROP1
mutation
Family history of
PROP1 mutation
Sporadic PROP1
mutation
Autosomal recessive or sporadic
mutation of PROP1 on chromosome
5q35 (various mutations identified;
ex. frameshift, insertion, deletion)
activation ability of pituitary-
specific transcription factor
encoded by PROP1 gene
PROP1-Related Combined Pituitary Hormone Deficiency
Genetic disorder resulting in combined pituitary hormone deficiency (CPHD) characterized by a
deficiency in growth hormone (GH) & ≥ 1 additional anterior pituitary hormone
↓ Follicle stimulating
hormone (FSH) from
somatotrophs
↓Luteinizing
hormone (LH) from
gonadotrophs
↓ GH from
gonadotrophs
↓ Thyroid stimulating
hormone (TSH) from
thyrotrophs
↓ Adrenocorticotropic
hormone (ACTH) from
corticotrophs
↓ Gonad
stimulation
↓ Sex hormone
production
Hypogonadotropic hypogonadism
(hypogonadism due to problem at
level of hypothalamus or pituitary)
↓ Secretion
of insulin-like
growth factor
1 (primarily
↓ Stimulation
of growth at
↓ Thyroid
stimulation
↓ Stimulation of
adrenal glands
epiphyseal
plate of long
bones
↓ Thyroid
by liver) ↓ Serum
hormone levels
cortisol
Absent secondary
sexual characteristics
Delayed or
absent puberty
Short stature
Secondary/central
hypothyroidism**
Secondary/central
adrenal insufficiency**
Postpartum
lactation failure
Authors:
Juliette Eshleman
Taylor Krawec
Reviewers:
Annie Pham
Emily J. Doucette
Danielle Nelson*
*MD at time of publication
Impaired glucose regulation
(↓ gluconeogenesis, ↓
glycogenolysis & ↓ lipolysis)
Altered stress
response to
illness or injury
↓ Vascular tone &
catecholamine response
(fight-or-flight response)
Impaired
glucose
metabolism
Neonatal hypoglycemia**
Adrenal crisis
Hypotension
Weakness
Fatigue
**See corresponding Calgary Guide slide
Legend: Pathophysiology Published May 19, 2025 on www.thecalgaryguide.com
Infertility
Mechanism
Sign/Symptom/Lab Finding Complications")
Tetralogy of Fallot

Maternal exposure
to alcohol or
anticonvulsants
Maternal infections (e.g., hepatitis
B virus, coxsackievirus-B, human
cytomegalovirus virus, rubella)
Variable interaction between genetic associations and environmental risk factors
Antero-cephalad deviation of conal septum (muscular structure that forms the boundary
between right & left ventricular outflow tract during embryological development)
Tetralogy of Fallot
Cyanotic congenital heart disease with a combination of four cardiac defects
Ventricular septal
defect (VSD)**
Overriding aortic root
(aorta above VSD rather than left ventricle)
Right ventricular outflow
tract obstruction (RVOTO)
Right Ventricular
Hypertrophy
Hole in septum
Absent or reduced
Stressors (eg., crying,
between ventricles Right ventricle of
Oxygenated blood from the left
pulmonary valve
defecation, fever,
heart pumps
ventricle and deoxygenated blood
closure (P2)
dehydration) trigger acute
harder to push
from right ventricle flows into aorta
↑ Left ventricular
pressure
infundibular spasm (spasm of
blood through
conus arteriosus)
RVOTO
Chronic volume
overload
L-R ventricular
shunting
Single, loud S2
Spasm ↓ blood flow through
right ventricular outflow tract
Aortic
Dilated
aortic root
regurgitation** Hypercyanois or “tet” spells
Turbulent blood
flow from left
ventricle through
the hole in septum
to right ventricle
Turbulent diastolic
backflow through
incompetent aortic
valve
Deoxygenated
blood flows
through aorta to
systemic circulation
↓ O2 Saturation
Chemoreceptors in the carotid & aortic bodies
stimulate respiratory centre in the medulla oblongata
Harsh, holosystolic
murmur
Diastolic
decrescendo
murmur
↑ Deoxygenated blood
entering systemic circulation
↑ Catecholamine release from adrenal
medulla stimulating ↑ contractility of heart
↓ Blood flow
across VSD
↑ contraction against outflow obstruction
↑ pulmonary vascular resistance
↓ Intensity or
disappearance
of holosystolic
murmur
Peripheral & central
chemoreceptors detect ↓ arterial
O2 content, triggering brainstem
respiratory centre to stimulate ↑
work & rate of breathing
↓ Pulmonary blood flow
↑ Turbulence of blood
flow through RVOTO
↑ R-L shunting
↑ Deoxygenated hemoglobin (dark
red) relative to oxygenated hemoglobin
(bright red), reflecting more blue light
Cyanosis (blue discoloration of the
mucous membranes, nail beds, skin)
Paroxysmal
hyperpnea (rapid
and deep
respirations)
Irritability
and crying
Dyspnea
(shortness of
breath)
Poor feeding
Accentuation of systolic
crescendo/decrescendo
murmur with harsh ejection
**See corresponding
Calgary Guide slides
Legend: Mechanism
Published Jul 8, 2015; updated Jun 16, 2025 on www.thecalgaryguide.com
Pathophysiology Sign/Symptom/Lab Finding Complications")
Obesity Hypoventilation Syndrome
![Obesity Hypoventilation Syndrome: Pathogenesis and clinical findings
Obesity (BMI ≥ 30 kg/m2) risk factors: Poor eating patterns, sedentary lifestyle, genetic predisposition,
hypothyroidism, Cushing’s syndrome, socio-economic factors, age
Sleep-disordered breathing risk factors: Family history, tonsillar or adenoidal
hypertrophy, ↑ neck circumference, type 2 diabetes, HTN
Authors: Mohammad Omer
Mujtaba Siddique
Reviewers:
Ali Babwani
Luiza Radu
Jonathan Liu*
MD at time of publication*
↑ Adipose deposition
in abdomen
Abdominal fat pushes
against diaphragm
↑ Diaphragmatic
displacement
↑ Resistance to chest
wall expansion
↑Leptin resistance
High pressure
Pharyngeal
on upper airway
dilations unable
Secondary depression
↓ Chest wall
↓ Leptins ability to stimulate
↑ lung
to compensate
Narrowing of
(compromised function) of
Poor ventilation to
expansion
ventilation (mechanism unknown)
collapsibility
for weight
upper airways
respiratory system
lower lobes of lungs
↓ Tidal volume (air
that moves in/out of
lungs in a respiratory
cycle)
↑ Respiratory rate
↑ Chest wall thickness ↑ Leptin (a hormone released by
adipose tissues that controls hunger by
signaling fullness)
↑ Adipose
deposition near
upper airways
↑ Buildup of
edema in lower
extremities
↑ Respiratory
workload
↓ Chest wall
compliance (ability to
stretch)
↓ Leptin receptor
expression
↓ Leptin through
blood-brain barrier
↓ Pharyngeal space
Respiratory system is
unable to compensate to ↑
Fluid shifts from
demands
legs to neck during
sleep
Hypoventilation in sleep
↓ Ventilation (air exchange in lungs)
↑ PaCO₂ (partial pressure of arterial carbon
dioxide)
↑ Serum [H+]
↑ Serum [HCO3
-] by renal
reabsorption buffers [H+] rise
↓ PaO₂
(partial pressure of arterial oxygen)
Hypoxia (low
O₂ in tissue)
Higher PaCO₂ required to
reduce pH
↓ O₂ levels in alveoli triggers pulmonary
vessel vasoconstriction
PaCO₂ > 45
mmHG
Respiratory
acidosis
↓ response to CO₂ in central
chemoreceptors in brain
Pulmonary hypertension (high pressure in
pulmonary arteries)
↓ Neural drive
↓ Ventilatory responsiveness
) Right heart pumps against higher
pulmonary pressure leading to
cardiomyocyte hypertrophy
Cor pulmonale
(right-sided heart
failure)
Fatigue
Chronic hypercapnia
(↑ CO2 retention)
Pathophysiology Legend: Mechanism
Sign/Symptom/Lab Finding Complications
Morning headaches
Daytime lethargy
Published Jun 16, 2025 on www.thecalgaryguide.com](https://calgaryguide.ucalgary.ca/wp-content/uploads/2025/06/Obesity-Hypoventilation-Syndrome.jpg "Obesity Hypoventilation Syndrome: Pathogenesis and clinical findings
Obesity (BMI ≥ 30 kg/m2) risk factors: Poor eating patterns, sedentary lifestyle, genetic predisposition,
hypothyroidism, Cushing’s syndrome, socio-economic factors, age
Sleep-disordered breathing risk factors: Family history, tonsillar or adenoidal
hypertrophy, ↑ neck circumference, type 2 diabetes, HTN
Authors: Mohammad Omer
Mujtaba Siddique
Reviewers:
Ali Babwani
Luiza Radu
Jonathan Liu*
MD at time of publication*
↑ Adipose deposition
in abdomen
Abdominal fat pushes
against diaphragm
↑ Diaphragmatic
displacement
↑ Resistance to chest
wall expansion
↑Leptin resistance
High pressure
Pharyngeal
on upper airway
dilations unable
Secondary depression
↓ Chest wall
↓ Leptins ability to stimulate
↑ lung
to compensate
Narrowing of
(compromised function) of
Poor ventilation to
expansion
ventilation (mechanism unknown)
collapsibility
for weight
upper airways
respiratory system
lower lobes of lungs
↓ Tidal volume (air
that moves in/out of
lungs in a respiratory
cycle)
↑ Respiratory rate
↑ Chest wall thickness ↑ Leptin (a hormone released by
adipose tissues that controls hunger by
signaling fullness)
↑ Adipose
deposition near
upper airways
↑ Buildup of
edema in lower
extremities
↑ Respiratory
workload
↓ Chest wall
compliance (ability to
stretch)
↓ Leptin receptor
expression
↓ Leptin through
blood-brain barrier
↓ Pharyngeal space
Respiratory system is
unable to compensate to ↑
Fluid shifts from
demands
legs to neck during
sleep
Hypoventilation in sleep
↓ Ventilation (air exchange in lungs)
↑ PaCO₂ (partial pressure of arterial carbon
dioxide)
↑ Serum [H+]
↑ Serum [HCO3
-] by renal
reabsorption buffers [H+] rise
↓ PaO₂
(partial pressure of arterial oxygen)
Hypoxia (low
O₂ in tissue)
Higher PaCO₂ required to
reduce pH
↓ O₂ levels in alveoli triggers pulmonary
vessel vasoconstriction
PaCO₂ > 45
mmHG
Respiratory
acidosis
↓ response to CO₂ in central
chemoreceptors in brain
Pulmonary hypertension (high pressure in
pulmonary arteries)
↓ Neural drive
↓ Ventilatory responsiveness
) Right heart pumps against higher
pulmonary pressure leading to
cardiomyocyte hypertrophy
Cor pulmonale
(right-sided heart
failure)
Fatigue
Chronic hypercapnia
(↑ CO2 retention)
Pathophysiology Legend: Mechanism
Sign/Symptom/Lab Finding Complications
Morning headaches
Daytime lethargy
Published Jun 16, 2025 on www.thecalgaryguide.com")
Neuroanatomy and Physiology of Fear

Olfaction: olfactory bulb sends direct input
to subcortical & cortical pathways
Sight, sound, touch, & taste: thalamus† pre-processes &
relays sensory information to subcortical & cortical pathways
Authors:
Taryn Stokowski
Andrea Moir
Reviewers:
Erika Russell
Usama Malik
Emily J. Doucette
Brienne McLane*
* MD at time of publication
Subcortical pathway (12 ms) Amygdala† initiates defensive response
Nucleus accumbens (ventral basal ganglia) integrates input to bias selection
of active (fight/flight) or passive (freeze) responses via ventral pallidum
Periaqueductal gray
(PAG) coordinates
motor response
Hypothalamus† coordinates the corresponding
autonomic & hormonal responses
Somatic motor system
Medulla oblongata
relays stimulation
Endocrine system
PAG activates
brainstem motor
nuclei
& spinal cord
Autonomic system
Active only:
hypothalamus†
secretes corticotropin-
releasing hormone
Pituitary gland
If active or
releases
If active
early passive
If passive
adrenocorticotropic
(dorsal
If passive
(sympathetic):
(para-
hormone**
PAG):
(ventral
adrenal gland
sympathetic):
rapid,
PAG):
releases
vagus nerve
muscle
whole-
adrenaline
releases
Adrenal cortex
releases cortisol
activation
body
acetylcholine
muscle
tension
Energy sustained
(e.g, ↑ blood glucose)
Defensive movement
(e.g, startle or flee)
Systemic
physical response
(e.g, ↑ heart rate)
Cortical pathway (300 ms)
Sensory cortices analyze the stimulus in detail
(e.g., visual cortex determines that a coiled object is in view)
Association areas in the parietal, temporal, and frontal
lobes categorize the object based on previous knowledge
(e.g., could this be a garden snake?)
Insular cortex detects
interoceptive information
(e.g., awareness of racing
heart)
Hippocampus† uses memory
to assign contextual meaning
(e.g., has this type of snake
harmed me before?)
Prefrontal cortex (PFC) integrates
interoceptive, sensory, & memory
information to respond to stimulus
Ventromedial PFC &
anterior cingulate cortex†
assess emotional salience
& modulate subcortical
response (e.g., ↓ arousal)
Subjective experience of fear
(e.g., I feel scared by that
garden snake!)
Emotion is regulated
(e.g., body relaxes)
†Part of the Limbic System
**See corresponding Calgary Guide slide
Note: This slide is based on the Two-System Framework by LeDoux & Pine (2016)
Legend: Complications
Published July 19, 2025 on www.thecalgaryguide.com
Pathophysiology Mechanism
Sign/Symptom/Lab Finding")
Hypocalcemia Physiology
![Hypocalcemia: Physiology
Hypomagnesemia**
Pseudohypoparathyroidism
(genetic resistance to PTH)
Sepsis** or severe illness
Authors:
Serra Thai,
Ryan Dion
Reviewers:
Jessica Hammal,
Michelle J. Chen,
Emily J. Doucette,
Hanan Bassyouni*
* MD at time of
publication
Parathyroid gland hypofunction
from surgical removal,
autoimmune disease, or congenital
disease (e.g. DiGeorge Syndrome)
Impaired Mg-dependent
generation of cyclic
adenosine monophosphate
↑ Systemic
inflammation
↑ Calcium
sequestration into
cells (mechanism of
action unknown)
↓ Liver function &
albumin synthesis
↓ Or inappropriately normal
parathyroid hormone (PTH)
in circulation
↓ PTH receptor (PTHR) sensitivity
↓ Albumin-bound
calcium in blood
↓ PTHR signaling in kidneys
↓ PTHR signaling in
osteoblastic lineage
Vitamin D deficiency** (e.g.
cells within bones
↓ intake, malabsorption)
False hypocalcemia (↓
total serum calcium with
normal Ca2+ levels)
Acute pancreatitis**
↓ Sodium-hydrogen
exchanger 3 (NHE3)
activity & expression
in proximal tubule
Chronic
kidney
disease
(CKD)**
↓ 1-⍺ hydroxylase
enzyme (converts
inactive vitamin D to
active form) activity
in kidneys
↑ Claudin14 (tight
junction membrane
proteins) expression in
thick ascending limb (TAL)
of the loop of Henle
↓ Transcription of
calcium
transporter genes
(TRPV5, calbindin
D28K, NCX) in
distal convoluted
tubule
↓ Nuclear factor
kappa B ligand
(RANKL) expression
& binding to
receptors on
osteoclast
precursor cells
Pancreatic enzymes are
prematurely activated
↓ Sodium
reabsorption
triggers
electrochemical
gradient
changes
↓ Glomerular
filtration due
to ↓ kidney
function
↓ Activated
vitamin D
(calcitriol)
synthesis
Claudin14 binds cation
channels composed of
Claudin16 & 19 in TAL
tight junctions
Lipase released from
pancreatic
autodigestion breaks
down peripancreatic fat
↓ Renal phosphate
filtration from
blood
↓ Binding of vitamin
D regulated calcium
transporters in
duodenum & jejunum
Binding blocks
paracellular Ca & Mg
transport from tubule
into vasculature
through tight junctions
↓ Probability
of calcium
transport
channels
opening
↓ Osteoclast
differentiation
(responsible
for breaking
down bone)
Free fatty acids bind
ionized Ca2+ to form
insoluble calcium
soaps (saponification)
↓ Circulating ionized
calcium in blood
↑ Phosphate-calcium
crystal formation
↓ Gastrointestinal
↓ Renal reabsorption
↓ Bone resorption
of calcium
of calcium
reabsorption of calcium Hypocalcemia**
(serum [Ca2+]
<2.1mmol/L+)
**See corresponding Calgary Guide slide
Legend: Pathophysiology Mechanism
Sign/Symptom/Lab Finding Complications
Published Aug 23, 2025 on www.thecalgaryguide.com
Hypocalcemia: Physiology
Authors:
Serra Thai
Ryan Dion
Reviewers:
Jessica Hammal
Michelle J. Chen
Emily J. Doucette
Hanan Bassyouni*
* MD at time of
publication
Parathyroid gland dysfunction
from surgical removal,
autoimmune disease, or
congenital disease (e.g. DiGeorge)
↓ Parathyroid hormone (PTH) in circulation
↓ PTHR signaling in kidneys
Chronic kidney
disease (CKD)
↓ Renal
blood flow
↓ Glomerular
filtration
↓ Sodium-hydrogen
exchanger 3 (NHE3)
activity & expression
in proximal tubule
↓ Sodium
reabsorption
Electrochemical
gradient changes
↓ Phosphate
excretion
↑ Phosphate-calcium
complex formation
**See corresponding Calgary Guide slide
Legend: Pathophysiology Mechanism
Hypomagnesemia**
Pseudohypoparathyroidism
(genetic resistance to PTH)
Sepsis** or severe illness
Impaired Mg-dependent
generation of cyclic
adenosine monophosphate
↑ Inflammation
↓ PTH receptor (PTHR) sensitivity
↑ Calcium
sequestration
into cells
(unknown
mechanism of
action)
↓ Liver
function
Vitamin D deficiency (e.g.
↓ intake, malabsorption)
↓ 1-⍺ hydroxylase
enzyme (converts
inactive vitamin D to
active form) activity in
kidneys
↑ Claudin14 (tight
junction membrane
protein) proteins
are expressed in
the thick ascending
loop of Henle
↓ Transcription of
calcium transporter
genes (TRPV5,
calbindin D28K,
NCX) in the distal
convoluted tubule
↓ Synthesis of activated
vitamin D (calcitriol)
Claudin14 binds to
cation channels
composed of
Claudin16 & 19 found
↓ Binding of vitamin D
in TAL tight junctions
regulated calcium
transporters (TRPV6,
calbindin9k, PMCa2B,
NCX2) in the duodenum
& jejunum
Binding blocks paracellular
Ca & Mg transport from
tubule into vasculature
through tight junctions
between cells
↓ Probability
of calcium
transport
channels
opening
↓ Gastrointestinal
reabsorption of Ca
↓ Renal reabsorption of calcium
↓ Synthesis
of albumin
↓ PTHR signaling in
osteoblastic lineage
cells in bones
↓ Nuclear factor kappa
B ligand (RANKL)
expression & binding to
receptors on osteoclast
precursor cells
↓ Osteoclast
differentiation
(responsible for
breaking down bone)
↓ Bone resorption
↓ Albumin-
bound calcium
with normal free
(biologically
active) Ca levels
False
hypocalcemia
Acute pancreatitis
↓ Lipase secretion
from pancreas
↑ Undigested fats in
small intestine
Free fatty acids
percipitate calcium
Hypocalcemia**
(serum [Ca2+]
<2.1mmol/L+)
Published MONTH, DAY, YEAR on www.thecalgaryguide.com
Sign/Symptom/Lab Finding Complications
Hypomagnesemia*
Impaired magnesium-
dependent generation
of cyclic adenosine
monophosphate
↓ PTH receptor (PTHR) sensitivity
Hypocalcemia: Physiology
Authors:
Serra Thai
Ryan Dion
Reviewers:
Jessica Hammal
Michelle J. Chen
* MD at time of publication
Parathyroid gland dysfunction
from surgical removal,
autoimmune disease, or
congenital disease (e.g. DiGeorge)
Pseudohypoparathyroidism
(genetic resistance to PTH)
Sepsis or severe illness
↓ Parathyroid hormone (PTH) in circulation
↓ PTHR signaling in kidneys
Vitamin D deficiency (e.g.
↓ intake, malabsorption)
↓ Sodium-hydrogen
exchanger 3 (NHE3)
activity & expression in
proximal tubule
↓ 1-⍺ hydroxylase
enzyme (converts
inactive vitamin D to its
active form) activity in
the kidneys
↑ Claudin14 (tight
junction membrane
protein) proteins
are expressed in
the thick ascending
loop of Henle
↓ Transcription of
calcium transporter
genes (TRPV5,
calbindin D28K,
NCX) in the distal
convoluted tubule
Chronic kidney
disease
↓ Sodium
reabsorption
↓ Synthesis of activated
vitamin D (calcitriol)
↓ Renal
blood flow
Electrochemical
gradient changes
Claudin14 binds to
cation channels
composed of
Claudin16 & 19 found
in TAL tight junctions
↓ Glomerular
filtration
↓ Phosphate
excretion
↓ Binding of vitamin D
regulated calcium
transporters (TRPV6,
calbindin9k, PMCa2B,
NCX2) in the duodenum
& jejunum
Binding blocks paracellular
Ca (and Mg) transport
from the tubule into
vasculature through tight
junctions between cells
↓ Probability
of calcium
transport
channels
opening
↑ Phosphate-calcium
complex formation
↓ Gastrointestinal
reabsorption of Ca
↓ Renal reabsorption of calcium
*See corresponding Calgary Guide slide: “Hypomagnesemia: Physiology”
** See corresponding Calgary Guide slide: “Hypoca;cemia: Clinical Findings”
Legend: ↑ Inflammation
↑ Ca
sequestration
into cells
(unknown
mechanism of
action)
↓ Liver
function
↓ Synthesis
of albumin
↓ PTHR signaling in
osteoblastic lineage
cells in bones
↓ Nuclear factor kappa
B ligand (RANKL)
expression and binding
to its receptors on
osteoclast precursor
cells
↓ Differentiation of
osteoclasts which are
responsible for
breaking down
(resorbing) bone
↓ Bone resorption
↓ Albumin-
bound calcium
with normal
free
(biologically
active) Ca
levels
False
hypocalcemia
Acute pancreatitis
↓ Lipase secretion
from pancreas
↑ Undigested fats in
small intestine
Free fatty acids
percipitate calcium
Hypocalcemia**
(serum [Ca2+]
<2.1mmol/L+)
Complications
Published MONTH, DAY, YEAR on www.thecalgaryguide.com
Pathophysiology Mechanism
Sign/Symptom/Lab Finding
Hypocalcemia: Physiology
Authors:
Serra Thai
Reviewers:
Jessica Hammal
Michelle J. Chen
* MD at time of publication
Parathyroid gland dysfunction
from surgical removal,
autoimmune disease, or
congenital disease (e.g. DiGeorge)
Hypomagnesemia**
Pseudohypoparathyroidism
(genetic resistance to PTH)
Sepsis or severe illness
Impaired magnesium-
dependent generation
of cyclic adenosine
monophosphate
↑ Inflammation
↓ Parathyroid hormone (PTH) in circulation
↓ PTH receptor (PTHR) sensitivity
↑ Ca
sequestration
into cells
(unknown
mechanism of
action)
↓ Liver
function
↓ Synthesis
of albumin
↓ PTHR signaling in kidneys
Vitamin D deficiency (e.g.
↓ intake, malabsorption)
↓ Sodium-hydrogen
exchanger 3 (NHE3)
activity & expression in
proximal tubule
↓ 1-⍺ hydroxylase
enzyme (converts
inactive vitamin D to its
active form) activity in
the kidneys
↑ Claudin14 (tight
junction membrane
protein) proteins
are expressed in
the thick ascending
loop of Henle
↓ Transcription of
calcium transporter
genes (TRPV5,
calbindin D28K,
NCX) in the distal
convoluted tubule
Chronic kidney
disease
↓ Sodium
reabsorption
↓ Synthesis of activated
vitamin D (calcitriol)
↓ Renal
blood flow
Electrochemical
gradient changes
Claudin14 binds to
cation channels
composed of
Claudin16 & 19 found
in TAL tight junctions
↓ Glomerular
filtration
↓ Phosphate
excretion
↓ Binding of vitamin D
regulated calcium
transporters (TRPV6,
calbindin9k, PMCa2B,
NCX2) in the duodenum
& jejunum
Binding blocks paracellular
Ca (and Mg) transport
from the tubule into
vasculature through tight
junctions between cells
↓ Probability
of calcium
transport
channels
opening
↑ Phosphate-calcium
complex formation
↓ Gastrointestinal
reabsorption of Ca
↓ Renal reabsorption of calcium
↓ PTHR signaling in
osteoblastic lineage
cells in bones
↓ Nuclear factor kappa
B ligand (RANKL)
expression and binding
to its receptors on
osteoclast precursor
cells
↓ Differentiation of
osteoclasts which are
responsible for
breaking down
(resorbing) bone
↓ Bone resorption
↓ Albumin-
bound calcium
with normal
free Ca levels
due to
homeostatic
mechanisms
False
hypocalcemia
Acute pancreatitis
↓ Lipase secretion
from pancreas
↑ Undigested fats in
small intestine
Free fatty acids
percipitate calcium
Hypocalcemia**
(serum [Ca2+]
<2.1mmol/L+)
Published MONTH, DAY, YEAR on www.thecalgaryguide.com
**See corresponding Calgary Guide slide: “Hypomagnesemia: Physiology”
Legend: Pathophysiology Mechanism
Sign/Symptom/Lab Finding Complications
Hypocalcemia: Physiology
Hypomagnesemia**
Authors:
Serra Thai
Reviewers:
Jessica Hammal
* MD at time of publication
Parathyroid gland
dysfunction: removal,
autoimmune,
congenital (DiGeorge)
Vitamin D
deficiency:
↓intake,
malabsorption
Pseudohypoparathyroidism
Sepsis/severe illness
↓PTHR
activation in
kidneys
CKD
↓ Renal
blood flow
↓ Glomerular
filtration
↓ Sodium-
hydrogen
exchanger 3
(NHE3) activity &
expression in
proximal tubule
↓ Sodium
reabsorption
Electrochemical
gradient changes
↓ Phosphate
excretion
↑ Phosphate calcium
complex formation
**See corresponding Calgary Guide slide(s)
Legend: ↓ 1-alpha
hydroxylase
enzyme activity
↓ Synthesis of
activated vitamin
D (calcitriol)
↓ Binding of vitamin
D regulated calcium
transporters (TRPV6,
calbindin9k, PMCa2B,
NCX2) in the
duodenum & jejunum
↓ Gastrointestinal
reabsorption of
calcium
Pathophysiology Mechanism
Sign/Symptom/Lab Finding Impaired magnesium
dependent generation
of cyclic adenosine
monophosphate ↑ Inflammation
↓ Parathyroid
hormone (PTH)
↑ Claudin14 (tight
junction membrane
protein) activity in
the thick ascending
loop of Henle
↑ Binding of
claudin14 to
claudin16
Inhibition of claudin
16 & 19 from
forming cation
permeable pores in
the tight junctions
↑ Calcium
sequestration
↓ Liver
function
into cells
(mechanism
of action
↓ Synthesis
of albumin
↓ PTH receptor
remains
(PTHR) sensitivity ↓ Albumin-
unknown)
bound calcium
with normal
↓ PTHR
activation in
bones
free Ca levels
due to
homeostatic
mechanisms
↓ Transcription
of calcium
transporter
genes (TRPV5,
calbindin D28K,
NCX) in the distal
convoluted
tubule
↓ Probability
of calcium
transport
channels
opening
↓ Renal reabsorption
of calcium
↓ Osteoblast
stimulation
↓ Nuclear factor
kappa b ligand
(RANKL) secretion
↓ Nuclear factor
kappa b receptor
(RANK) binding on
osteoclast
precursors cells
↓Osteoclast
production
↓ Bone
resorption
False
Hypocalcemia
Acute pancreatitis
↓ Lipase secretion
from pancreas
↑ Undigested fats in
small intestine
Free fatty acids
percipitate calcium
Hypocalcemia**
(serum [Ca2
<2.1mmol/L+])
Published MONTH, DAY, YEAR on www.thecalgaryguide.com
Complications
Hypocalcemia: Physiology
Hypomagnesemia**
Authors:
Serra Thai
Pseudohypoparathyroidism
Reviewers:
Jessica Hammal
* MD at time of publication
Vitamin D
deficiency: ↓
intake,
malabsorption
↓ 1-alpha
hydroxylase
enzyme activity
↓ Synthesis of
activated vitamin
D (calcitriol)
↓ Binding of
vitamin D regulated
calcium transporters
(TRPV6, calbindin9k,
PMCa2B, NCX2) in
the duodenum &
jejunum
↓ Gastrointestinal
reabsorption of
calcium
Parathyroid gland
dysfunction: removal,
autoimmune,
congenital (DiGeorge)
CKD
↓ Renal
blood flow
↓ Glomerular
filtration
**See corresponding Calgary Guide slide(s)
Legend: Sepsis/severe illness
Impaired magnesium
dependent generation of cyclic
adenosine monophosphate ↑ Inflammation
↓ Signaling proteins
↓ Parathyroid
hormone (PTH)
↓ PTH receptor
(PTHR) sensitivity
↑ Calcium
sequestration
into cells
(mechanism
of action
remains
unknown)
Pathophysiology Mechanism
↓PTHR1 activation
in kidneys
↓ Activation of G-
coupled protein
signaling pathways
↑ Expression of
sodium-phosphate
co-transporters
(NaPiIIa & NaPIIc)
in proximal tubule
↓ Expression &
phosphorylation of
transient receptor
potential vanilloid
(TRPV5, calcium
transporter channel) in
distal convoluted tubule
↓ PTHR1 activation
in bones
↓ Osteoblast
↑ Claudin14 (tight
stimulation
junction membrane
protein) activity in
the thick ascending
loop of Henle
↓ Nuclear
factor kappa b
ligand (RANKL)
secretion
↑ Binding of
claudin14 to
claudin16
↓ Nuclear
factor kappa b
receptor
Inhibition of
(RANK) binding
on osteoclast
↓ Phosphate
excretion
Electrochemical
↓ Amount of
claudin 16 & 19
gradient
TRPV5 & ↓
from forming
precursors cells
changes
probability of
cation-permeable
channels
pores in the tight
↑ Phosphate
opening
junctions
calcium
↓Osteoclast
production
complex
formation
↓ Paracellular
transport of
calcium ↓ Renal reabsorption
of calcium
↓ Bone
resorption
Published MONTH, DAY, YEAR on www.thecalgaryguide.com
↓ Liver
function
↓ Synthesis
of albumin
↓ Albumin-
bound calcium
with normal
free calcium
levels due to
homeostatic
mechanisms
False
Hypocalcemia
Acute
pancreatitis
↓ Lipase
secretion
from pancreas
↑ Undigested
fats in small
intestine
Free fatty acids
percipitate
calcium
Hypocalcemia**
(serum [Ca2
<2.1mmol/L+])
Sign/Symptom/Lab Finding Complications
Hypocalcemia: Physiology
Parathyroid gland
dysfunction: removal,
autoimmune,
congenital (DiGeorge)
Hypomagnesemia**
Impaired magnesium
dependent generation
of cyclic adenosine
monophosphate
↓ Parathyroid
hormone (PTH)
↓ PTH
sensitivity
Vitamin D
deficiency:
↓intake,
malabsorption
↓ Enzyme 1-alpha
hydroxylase activity
↓ Synthesis of
activated vitamin
D (calcitriol)
↓ Binding of
vitamin D
regulated calcium
transporters in
the duodenum &
jejunum
↓ Gastrointestinal
reabsorption of
calcium
Legend: ↓PTH receptor
activation in
kidneys
↑ Claudin14
activity in the
thick ascending
loop of Henle
Inhibition of
claudin 16 & 19
from forming
cation permeable
pores in the tight
junctions
↓ Transcription
of calcium
transporter
genes in the
distal
convoluted
tubule
↓ Probability
of calcium
transport
channels
opening
↓ Renal reabsorption
of calcium
↓ Sodium-hydrogen
exchanger 3 (NHE3)
activity & expression in
proximal tubule
↓ Sodium
reabsorption
Electrochemical
gradient changes
↓ Phosphate excretion
↑ Phosphate calcium
complex formation
Pathophysiology Mechanism
Sign/Symptom/Lab Finding Complications
↑ Inflammation
Authors:
Serra Thai
Reviewers:
Jessica Hammal
* MD at time of publication
Sepsis/severe illness
↑ Calcium
sequestration
into cells
↓ Liver
synthesis of
albumin
↓ Albumin-
calcium
binding
False
Hypocalcemia
Acute pancreatitis
↓ Lipase
secretion from
pancreas
↑ Levels of
undigested
fats in small
intestine
Free fatty
acids
percipitate
calcium
↓ PTH receptor
activation in
bones
↓ Osteoblast
stimulation
↓ RANKL
ligand
secretion
↓ RAANK
receptor
binding
↓Osteoclast
production
↓ Bone
resorption
Hypocalcemia
(serum [Ca2+]
<2.1mmol/L)
Published MONTH, DAY, YEAR on www.thecalgaryguide.com
Hypocalcemia: Physiology
Vitamin D
deficiency:
↓intake,
malabsorption
Parathyroid gland
dysfunction: removal,
autoimmune,
congenital (DiGeorge)
Hypomagnesemia**
Impaired magnesium
dependent generation
of cyclic adenosine
monophosphate
↓ Parathyroid
hormone (PTH)
↓ PTH
sensitivity
↓ Enzyme 1-alpha
hydroxylase activity
↓ Synthesis of
activated vitamin
D (calcitriol)
↓ Binding of
vitamin D
regulated calcium
transporters in
the duodenum &
jejunum
↓ Gastrointestinal
reabsorption of calcium
Legend: ↓PTH receptor
activation in
kidneys
↑ Claudin14
activity in the
thick ascending
loop of Henle
Inhibition of
claudin 16 & 19
from forming
cation permeable
pores in the tight
junctions
↓ Transcription
of calcium
transporter
genes in the
distal
convoluted
tubule
↓ Probability
of calcium
transport
channels
opening
↓ Renal reabsorption
of calcium
↓ Sodium-hydrogen
exchanger 3 (NHE3)
activity & expression in
proximal tubule
↓ Sodium
reabsorption
Electrochemical
gradient changes
↓ Phosphate excretion
↑ Phosphate calcium
complex formation
Pathophysiology Mechanism
Sign/Symptom/Lab Finding Complications
↑ Inflammation
Sepsis/severe illness
↑ Calcium
sequestration
into cells
↓ Liver
synthesis of
albumin
↓ Albumin-
calcium
binding
False
Hypocalcemia
Acute pancreatitis
↓ Lipase
secretion from
pancreas
↑ Levels of
undigested
fats in small
intestine
Free fatty
acids
percipitate
calcium
↓ PTH receptor
activation in
bones
↓ Osteoblast
stimulation
↓ RANKL
ligand
secretion
↓ RAANK
receptor
binding
↓Osteoclast
production
↓ Bone
resorption
Hypocalcemia
(serum [Ca2+] <2.1mmol/L)
Authors:
Name Name
Name Name*
Reviewers:
Name Name
Name Name*
* MD at time of publication
Published MONTH, DAY, YEAR on www.thecalgaryguide.com
Hypocalcemia: Physiology
Chronic kidney
disease
↓ Renal
blood flow
↓ Glomerular
filtration
Parathyroid gland
dysfunction: removal,
autoimmune,
congenital (DiGeorge)
Vitamin D deficiency:
↓intake, malabsorption
Sepsis/severe illness
↓ NHE3 activity and
expression in
proximal tubule
↓PTH receptor
activation in
kidneys
↑ Claudin14
activity in the
thick ascending
loop of Henle
↓ Parathyroid
hormone (PTH)
↓ Transcription of
calcium transporter
genes (TRPV5, calbindin
D28K, NCX) in the distal
convoluted tubule
↓ Enzyme 1-alpha
hydroxylase activity
↓ PTH receptor
activation in
bones
↓ osteoblast
stimulation
↓ RANKL ligand
secretion
↑ Inflammation ↓ PTH sensitivity
↓ Glomerular
filtration
↓ Liver
synthesis of
serum albumin
False
Hypocalcemia
↑ Calcium
sequestration
into cells**
↓ Lipase secretion
from pancreas
Acute pancreatitis
Legend: Pathophysiology Mechanism
Sign/Symptom/Lab Finding ↓ Albumin-
bound calcium
Current mechanism
is unknown
↑ levels of undigested
fats in small intestine
Complications
Hypomagnesemia Multiple blood
transfusions
↓ Sodium
reabsorption
Electrochemical
gradient changes
↓ Phosphate
excretion
↑ Phosphate
calcium complex
formation
Inhibits claudin16 and 19
from forming cation
permeable pores in the
tight junctions
↓Probability of
calcium transport
channels opening
↓ Synthesis of activated
vitamin D (calcitriol)
↓ RAANK
receptor
binding
↓ Calcium
reabsorption
↓ Binding of vitamin D
regulated calcium
transporters (TRPV6,
calbindin9K, PMCa2B,
NCX2) in the duodenum
and jejunum
↓osteoclast
production
↓ Bone
resorption
↓ Serum calcium
Hypocalcemia
<2.1 mmol/L
↑ Precipitation of calcium
by free fatty acids
Authors:
Name Name
Name Name*
Reviewers:
Name Name
Name Name*
* MD at time of publication
Published MONTH, DAY, YEAR on www.thecalgaryguide.com
https://journals.physiology.org/doi/full/10.1152/physrev.00003.2004?rfr_dat=cr_pub++0pubmed&url_ver=Z39.88-
2003&rfr_id=ori%3Arid%3Acrossref.org – Calcium Absoprtion across Epithelia
https://academic.oup.com/endo/article-abstract/137/1/13/2498579 - PTH and Calcium Signaling Pathways
https://www.pnas.org/doi/10.1073/pnas.1616733114 - Information on Claudin 14
https://onlinelibrary-wiley-com.ezproxy.lib.ucalgary.ca/doi/full/10.1111/apha.13959 - effects of PTH on renal calcium and
phosphate handling
https://www.ncbi.nlm.nih.gov/books/NBK430912/ - Hypocalcemia overview + causes + pathophys
https://www.orthobullets.com/basic-science/9010/bone-signaling-and-rankl - information about bone signaling
https://www.sciencedirect.com/science/article/abs/pii/S0889852921000682?via%3Dihub – Calcium homeostasis article
https://pubmed.ncbi.nlm.nih.gov/3012979/#:~:text=Abstract,intestine%2C%20require%20the%20parathyroid%20hormone.
vitamin D metabolism and function
–
https://pmc.ncbi.nlm.nih.gov/articles/PMC2669834/#:~:text=Calcium%20is%20actively%20absorbed%20from,for%20proper
%20mineralization%20of%20bone.
– more vitamin D metabolism and specific receptors
https://jidc.org/index.php/journal/article/view/32903236/2331 - sepsis + hypocalcemia](https://calgaryguide.ucalgary.ca/wp-content/uploads/2025/08/Hypocalcemia-Pathogenesis.jpg "Hypocalcemia: Physiology
Hypomagnesemia**
Pseudohypoparathyroidism
(genetic resistance to PTH)
Sepsis** or severe illness
Authors:
Serra Thai,
Ryan Dion
Reviewers:
Jessica Hammal,
Michelle J. Chen,
Emily J. Doucette,
Hanan Bassyouni*
* MD at time of
publication
Parathyroid gland hypofunction
from surgical removal,
autoimmune disease, or congenital
disease (e.g. DiGeorge Syndrome)
Impaired Mg-dependent
generation of cyclic
adenosine monophosphate
↑ Systemic
inflammation
↑ Calcium
sequestration into
cells (mechanism of
action unknown)
↓ Liver function &
albumin synthesis
↓ Or inappropriately normal
parathyroid hormone (PTH)
in circulation
↓ PTH receptor (PTHR) sensitivity
↓ Albumin-bound
calcium in blood
↓ PTHR signaling in kidneys
↓ PTHR signaling in
osteoblastic lineage
Vitamin D deficiency** (e.g.
cells within bones
↓ intake, malabsorption)
False hypocalcemia (↓
total serum calcium with
normal Ca2+ levels)
Acute pancreatitis**
↓ Sodium-hydrogen
exchanger 3 (NHE3)
activity & expression
in proximal tubule
Chronic
kidney
disease
(CKD)**
↓ 1-⍺ hydroxylase
enzyme (converts
inactive vitamin D to
active form) activity
in kidneys
↑ Claudin14 (tight
junction membrane
proteins) expression in
thick ascending limb (TAL)
of the loop of Henle
↓ Transcription of
calcium
transporter genes
(TRPV5, calbindin
D28K, NCX) in
distal convoluted
tubule
↓ Nuclear factor
kappa B ligand
(RANKL) expression
& binding to
receptors on
osteoclast
precursor cells
Pancreatic enzymes are
prematurely activated
↓ Sodium
reabsorption
triggers
electrochemical
gradient
changes
↓ Glomerular
filtration due
to ↓ kidney
function
↓ Activated
vitamin D
(calcitriol)
synthesis
Claudin14 binds cation
channels composed of
Claudin16 & 19 in TAL
tight junctions
Lipase released from
pancreatic
autodigestion breaks
down peripancreatic fat
↓ Renal phosphate
filtration from
blood
↓ Binding of vitamin
D regulated calcium
transporters in
duodenum & jejunum
Binding blocks
paracellular Ca & Mg
transport from tubule
into vasculature
through tight junctions
↓ Probability
of calcium
transport
channels
opening
↓ Osteoclast
differentiation
(responsible
for breaking
down bone)
Free fatty acids bind
ionized Ca2+ to form
insoluble calcium
soaps (saponification)
↓ Circulating ionized
calcium in blood
↑ Phosphate-calcium
crystal formation
↓ Gastrointestinal
↓ Renal reabsorption
↓ Bone resorption
of calcium
of calcium
reabsorption of calcium Hypocalcemia**
(serum [Ca2+]
<2.1mmol/L+)
**See corresponding Calgary Guide slide
Legend: Pathophysiology Mechanism
Sign/Symptom/Lab Finding Complications
Published Aug 23, 2025 on www.thecalgaryguide.com
Hypocalcemia: Physiology
Authors:
Serra Thai
Ryan Dion
Reviewers:
Jessica Hammal
Michelle J. Chen
Emily J. Doucette
Hanan Bassyouni*
* MD at time of
publication
Parathyroid gland dysfunction
from surgical removal,
autoimmune disease, or
congenital disease (e.g. DiGeorge)
↓ Parathyroid hormone (PTH) in circulation
↓ PTHR signaling in kidneys
Chronic kidney
disease (CKD)
↓ Renal
blood flow
↓ Glomerular
filtration
↓ Sodium-hydrogen
exchanger 3 (NHE3)
activity & expression
in proximal tubule
↓ Sodium
reabsorption
Electrochemical
gradient changes
↓ Phosphate
excretion
↑ Phosphate-calcium
complex formation
**See corresponding Calgary Guide slide
Legend: Pathophysiology Mechanism
Hypomagnesemia**
Pseudohypoparathyroidism
(genetic resistance to PTH)
Sepsis** or severe illness
Impaired Mg-dependent
generation of cyclic
adenosine monophosphate
↑ Inflammation
↓ PTH receptor (PTHR) sensitivity
↑ Calcium
sequestration
into cells
(unknown
mechanism of
action)
↓ Liver
function
Vitamin D deficiency (e.g.
↓ intake, malabsorption)
↓ 1-⍺ hydroxylase
enzyme (converts
inactive vitamin D to
active form) activity in
kidneys
↑ Claudin14 (tight
junction membrane
protein) proteins
are expressed in
the thick ascending
loop of Henle
↓ Transcription of
calcium transporter
genes (TRPV5,
calbindin D28K,
NCX) in the distal
convoluted tubule
↓ Synthesis of activated
vitamin D (calcitriol)
Claudin14 binds to
cation channels
composed of
Claudin16 & 19 found
↓ Binding of vitamin D
in TAL tight junctions
regulated calcium
transporters (TRPV6,
calbindin9k, PMCa2B,
NCX2) in the duodenum
& jejunum
Binding blocks paracellular
Ca & Mg transport from
tubule into vasculature
through tight junctions
between cells
↓ Probability
of calcium
transport
channels
opening
↓ Gastrointestinal
reabsorption of Ca
↓ Renal reabsorption of calcium
↓ Synthesis
of albumin
↓ PTHR signaling in
osteoblastic lineage
cells in bones
↓ Nuclear factor kappa
B ligand (RANKL)
expression & binding to
receptors on osteoclast
precursor cells
↓ Osteoclast
differentiation
(responsible for
breaking down bone)
↓ Bone resorption
↓ Albumin-
bound calcium
with normal free
(biologically
active) Ca levels
False
hypocalcemia
Acute pancreatitis
↓ Lipase secretion
from pancreas
↑ Undigested fats in
small intestine
Free fatty acids
percipitate calcium
Hypocalcemia**
(serum [Ca2+]
<2.1mmol/L+)
Published MONTH, DAY, YEAR on www.thecalgaryguide.com
Sign/Symptom/Lab Finding Complications
Hypomagnesemia*
Impaired magnesium-
dependent generation
of cyclic adenosine
monophosphate
↓ PTH receptor (PTHR) sensitivity
Hypocalcemia: Physiology
Authors:
Serra Thai
Ryan Dion
Reviewers:
Jessica Hammal
Michelle J. Chen
* MD at time of publication
Parathyroid gland dysfunction
from surgical removal,
autoimmune disease, or
congenital disease (e.g. DiGeorge)
Pseudohypoparathyroidism
(genetic resistance to PTH)
Sepsis or severe illness
↓ Parathyroid hormone (PTH) in circulation
↓ PTHR signaling in kidneys
Vitamin D deficiency (e.g.
↓ intake, malabsorption)
↓ Sodium-hydrogen
exchanger 3 (NHE3)
activity & expression in
proximal tubule
↓ 1-⍺ hydroxylase
enzyme (converts
inactive vitamin D to its
active form) activity in
the kidneys
↑ Claudin14 (tight
junction membrane
protein) proteins
are expressed in
the thick ascending
loop of Henle
↓ Transcription of
calcium transporter
genes (TRPV5,
calbindin D28K,
NCX) in the distal
convoluted tubule
Chronic kidney
disease
↓ Sodium
reabsorption
↓ Synthesis of activated
vitamin D (calcitriol)
↓ Renal
blood flow
Electrochemical
gradient changes
Claudin14 binds to
cation channels
composed of
Claudin16 & 19 found
in TAL tight junctions
↓ Glomerular
filtration
↓ Phosphate
excretion
↓ Binding of vitamin D
regulated calcium
transporters (TRPV6,
calbindin9k, PMCa2B,
NCX2) in the duodenum
& jejunum
Binding blocks paracellular
Ca (and Mg) transport
from the tubule into
vasculature through tight
junctions between cells
↓ Probability
of calcium
transport
channels
opening
↑ Phosphate-calcium
complex formation
↓ Gastrointestinal
reabsorption of Ca
↓ Renal reabsorption of calcium
*See corresponding Calgary Guide slide: “Hypomagnesemia: Physiology”
** See corresponding Calgary Guide slide: “Hypoca;cemia: Clinical Findings”
Legend: ↑ Inflammation
↑ Ca
sequestration
into cells
(unknown
mechanism of
action)
↓ Liver
function
↓ Synthesis
of albumin
↓ PTHR signaling in
osteoblastic lineage
cells in bones
↓ Nuclear factor kappa
B ligand (RANKL)
expression and binding
to its receptors on
osteoclast precursor
cells
↓ Differentiation of
osteoclasts which are
responsible for
breaking down
(resorbing) bone
↓ Bone resorption
↓ Albumin-
bound calcium
with normal
free
(biologically
active) Ca
levels
False
hypocalcemia
Acute pancreatitis
↓ Lipase secretion
from pancreas
↑ Undigested fats in
small intestine
Free fatty acids
percipitate calcium
Hypocalcemia**
(serum [Ca2+]
<2.1mmol/L+)
Complications
Published MONTH, DAY, YEAR on www.thecalgaryguide.com
Pathophysiology Mechanism
Sign/Symptom/Lab Finding
Hypocalcemia: Physiology
Authors:
Serra Thai
Reviewers:
Jessica Hammal
Michelle J. Chen
* MD at time of publication
Parathyroid gland dysfunction
from surgical removal,
autoimmune disease, or
congenital disease (e.g. DiGeorge)
Hypomagnesemia**
Pseudohypoparathyroidism
(genetic resistance to PTH)
Sepsis or severe illness
Impaired magnesium-
dependent generation
of cyclic adenosine
monophosphate
↑ Inflammation
↓ Parathyroid hormone (PTH) in circulation
↓ PTH receptor (PTHR) sensitivity
↑ Ca
sequestration
into cells
(unknown
mechanism of
action)
↓ Liver
function
↓ Synthesis
of albumin
↓ PTHR signaling in kidneys
Vitamin D deficiency (e.g.
↓ intake, malabsorption)
↓ Sodium-hydrogen
exchanger 3 (NHE3)
activity & expression in
proximal tubule
↓ 1-⍺ hydroxylase
enzyme (converts
inactive vitamin D to its
active form) activity in
the kidneys
↑ Claudin14 (tight
junction membrane
protein) proteins
are expressed in
the thick ascending
loop of Henle
↓ Transcription of
calcium transporter
genes (TRPV5,
calbindin D28K,
NCX) in the distal
convoluted tubule
Chronic kidney
disease
↓ Sodium
reabsorption
↓ Synthesis of activated
vitamin D (calcitriol)
↓ Renal
blood flow
Electrochemical
gradient changes
Claudin14 binds to
cation channels
composed of
Claudin16 & 19 found
in TAL tight junctions
↓ Glomerular
filtration
↓ Phosphate
excretion
↓ Binding of vitamin D
regulated calcium
transporters (TRPV6,
calbindin9k, PMCa2B,
NCX2) in the duodenum
& jejunum
Binding blocks paracellular
Ca (and Mg) transport
from the tubule into
vasculature through tight
junctions between cells
↓ Probability
of calcium
transport
channels
opening
↑ Phosphate-calcium
complex formation
↓ Gastrointestinal
reabsorption of Ca
↓ Renal reabsorption of calcium
↓ PTHR signaling in
osteoblastic lineage
cells in bones
↓ Nuclear factor kappa
B ligand (RANKL)
expression and binding
to its receptors on
osteoclast precursor
cells
↓ Differentiation of
osteoclasts which are
responsible for
breaking down
(resorbing) bone
↓ Bone resorption
↓ Albumin-
bound calcium
with normal
free Ca levels
due to
homeostatic
mechanisms
False
hypocalcemia
Acute pancreatitis
↓ Lipase secretion
from pancreas
↑ Undigested fats in
small intestine
Free fatty acids
percipitate calcium
Hypocalcemia**
(serum [Ca2+]
<2.1mmol/L+)
Published MONTH, DAY, YEAR on www.thecalgaryguide.com
**See corresponding Calgary Guide slide: “Hypomagnesemia: Physiology”
Legend: Pathophysiology Mechanism
Sign/Symptom/Lab Finding Complications
Hypocalcemia: Physiology
Hypomagnesemia**
Authors:
Serra Thai
Reviewers:
Jessica Hammal
* MD at time of publication
Parathyroid gland
dysfunction: removal,
autoimmune,
congenital (DiGeorge)
Vitamin D
deficiency:
↓intake,
malabsorption
Pseudohypoparathyroidism
Sepsis/severe illness
↓PTHR
activation in
kidneys
CKD
↓ Renal
blood flow
↓ Glomerular
filtration
↓ Sodium-
hydrogen
exchanger 3
(NHE3) activity &
expression in
proximal tubule
↓ Sodium
reabsorption
Electrochemical
gradient changes
↓ Phosphate
excretion
↑ Phosphate calcium
complex formation
**See corresponding Calgary Guide slide(s)
Legend: ↓ 1-alpha
hydroxylase
enzyme activity
↓ Synthesis of
activated vitamin
D (calcitriol)
↓ Binding of vitamin
D regulated calcium
transporters (TRPV6,
calbindin9k, PMCa2B,
NCX2) in the
duodenum & jejunum
↓ Gastrointestinal
reabsorption of
calcium
Pathophysiology Mechanism
Sign/Symptom/Lab Finding Impaired magnesium
dependent generation
of cyclic adenosine
monophosphate ↑ Inflammation
↓ Parathyroid
hormone (PTH)
↑ Claudin14 (tight
junction membrane
protein) activity in
the thick ascending
loop of Henle
↑ Binding of
claudin14 to
claudin16
Inhibition of claudin
16 & 19 from
forming cation
permeable pores in
the tight junctions
↑ Calcium
sequestration
↓ Liver
function
into cells
(mechanism
of action
↓ Synthesis
of albumin
↓ PTH receptor
remains
(PTHR) sensitivity ↓ Albumin-
unknown)
bound calcium
with normal
↓ PTHR
activation in
bones
free Ca levels
due to
homeostatic
mechanisms
↓ Transcription
of calcium
transporter
genes (TRPV5,
calbindin D28K,
NCX) in the distal
convoluted
tubule
↓ Probability
of calcium
transport
channels
opening
↓ Renal reabsorption
of calcium
↓ Osteoblast
stimulation
↓ Nuclear factor
kappa b ligand
(RANKL) secretion
↓ Nuclear factor
kappa b receptor
(RANK) binding on
osteoclast
precursors cells
↓Osteoclast
production
↓ Bone
resorption
False
Hypocalcemia
Acute pancreatitis
↓ Lipase secretion
from pancreas
↑ Undigested fats in
small intestine
Free fatty acids
percipitate calcium
Hypocalcemia**
(serum [Ca2
<2.1mmol/L+])
Published MONTH, DAY, YEAR on www.thecalgaryguide.com
Complications
Hypocalcemia: Physiology
Hypomagnesemia**
Authors:
Serra Thai
Pseudohypoparathyroidism
Reviewers:
Jessica Hammal
* MD at time of publication
Vitamin D
deficiency: ↓
intake,
malabsorption
↓ 1-alpha
hydroxylase
enzyme activity
↓ Synthesis of
activated vitamin
D (calcitriol)
↓ Binding of
vitamin D regulated
calcium transporters
(TRPV6, calbindin9k,
PMCa2B, NCX2) in
the duodenum &
jejunum
↓ Gastrointestinal
reabsorption of
calcium
Parathyroid gland
dysfunction: removal,
autoimmune,
congenital (DiGeorge)
CKD
↓ Renal
blood flow
↓ Glomerular
filtration
**See corresponding Calgary Guide slide(s)
Legend: Sepsis/severe illness
Impaired magnesium
dependent generation of cyclic
adenosine monophosphate ↑ Inflammation
↓ Signaling proteins
↓ Parathyroid
hormone (PTH)
↓ PTH receptor
(PTHR) sensitivity
↑ Calcium
sequestration
into cells
(mechanism
of action
remains
unknown)
Pathophysiology Mechanism
↓PTHR1 activation
in kidneys
↓ Activation of G-
coupled protein
signaling pathways
↑ Expression of
sodium-phosphate
co-transporters
(NaPiIIa & NaPIIc)
in proximal tubule
↓ Expression &
phosphorylation of
transient receptor
potential vanilloid
(TRPV5, calcium
transporter channel) in
distal convoluted tubule
↓ PTHR1 activation
in bones
↓ Osteoblast
↑ Claudin14 (tight
stimulation
junction membrane
protein) activity in
the thick ascending
loop of Henle
↓ Nuclear
factor kappa b
ligand (RANKL)
secretion
↑ Binding of
claudin14 to
claudin16
↓ Nuclear
factor kappa b
receptor
Inhibition of
(RANK) binding
on osteoclast
↓ Phosphate
excretion
Electrochemical
↓ Amount of
claudin 16 & 19
gradient
TRPV5 & ↓
from forming
precursors cells
changes
probability of
cation-permeable
channels
pores in the tight
↑ Phosphate
opening
junctions
calcium
↓Osteoclast
production
complex
formation
↓ Paracellular
transport of
calcium ↓ Renal reabsorption
of calcium
↓ Bone
resorption
Published MONTH, DAY, YEAR on www.thecalgaryguide.com
↓ Liver
function
↓ Synthesis
of albumin
↓ Albumin-
bound calcium
with normal
free calcium
levels due to
homeostatic
mechanisms
False
Hypocalcemia
Acute
pancreatitis
↓ Lipase
secretion
from pancreas
↑ Undigested
fats in small
intestine
Free fatty acids
percipitate
calcium
Hypocalcemia**
(serum [Ca2
<2.1mmol/L+])
Sign/Symptom/Lab Finding Complications
Hypocalcemia: Physiology
Parathyroid gland
dysfunction: removal,
autoimmune,
congenital (DiGeorge)
Hypomagnesemia**
Impaired magnesium
dependent generation
of cyclic adenosine
monophosphate
↓ Parathyroid
hormone (PTH)
↓ PTH
sensitivity
Vitamin D
deficiency:
↓intake,
malabsorption
↓ Enzyme 1-alpha
hydroxylase activity
↓ Synthesis of
activated vitamin
D (calcitriol)
↓ Binding of
vitamin D
regulated calcium
transporters in
the duodenum &
jejunum
↓ Gastrointestinal
reabsorption of
calcium
Legend: ↓PTH receptor
activation in
kidneys
↑ Claudin14
activity in the
thick ascending
loop of Henle
Inhibition of
claudin 16 & 19
from forming
cation permeable
pores in the tight
junctions
↓ Transcription
of calcium
transporter
genes in the
distal
convoluted
tubule
↓ Probability
of calcium
transport
channels
opening
↓ Renal reabsorption
of calcium
↓ Sodium-hydrogen
exchanger 3 (NHE3)
activity & expression in
proximal tubule
↓ Sodium
reabsorption
Electrochemical
gradient changes
↓ Phosphate excretion
↑ Phosphate calcium
complex formation
Pathophysiology Mechanism
Sign/Symptom/Lab Finding Complications
↑ Inflammation
Authors:
Serra Thai
Reviewers:
Jessica Hammal
* MD at time of publication
Sepsis/severe illness
↑ Calcium
sequestration
into cells
↓ Liver
synthesis of
albumin
↓ Albumin-
calcium
binding
False
Hypocalcemia
Acute pancreatitis
↓ Lipase
secretion from
pancreas
↑ Levels of
undigested
fats in small
intestine
Free fatty
acids
percipitate
calcium
↓ PTH receptor
activation in
bones
↓ Osteoblast
stimulation
↓ RANKL
ligand
secretion
↓ RAANK
receptor
binding
↓Osteoclast
production
↓ Bone
resorption
Hypocalcemia
(serum [Ca2+]
<2.1mmol/L)
Published MONTH, DAY, YEAR on www.thecalgaryguide.com
Hypocalcemia: Physiology
Vitamin D
deficiency:
↓intake,
malabsorption
Parathyroid gland
dysfunction: removal,
autoimmune,
congenital (DiGeorge)
Hypomagnesemia**
Impaired magnesium
dependent generation
of cyclic adenosine
monophosphate
↓ Parathyroid
hormone (PTH)
↓ PTH
sensitivity
↓ Enzyme 1-alpha
hydroxylase activity
↓ Synthesis of
activated vitamin
D (calcitriol)
↓ Binding of
vitamin D
regulated calcium
transporters in
the duodenum &
jejunum
↓ Gastrointestinal
reabsorption of calcium
Legend: ↓PTH receptor
activation in
kidneys
↑ Claudin14
activity in the
thick ascending
loop of Henle
Inhibition of
claudin 16 & 19
from forming
cation permeable
pores in the tight
junctions
↓ Transcription
of calcium
transporter
genes in the
distal
convoluted
tubule
↓ Probability
of calcium
transport
channels
opening
↓ Renal reabsorption
of calcium
↓ Sodium-hydrogen
exchanger 3 (NHE3)
activity & expression in
proximal tubule
↓ Sodium
reabsorption
Electrochemical
gradient changes
↓ Phosphate excretion
↑ Phosphate calcium
complex formation
Pathophysiology Mechanism
Sign/Symptom/Lab Finding Complications
↑ Inflammation
Sepsis/severe illness
↑ Calcium
sequestration
into cells
↓ Liver
synthesis of
albumin
↓ Albumin-
calcium
binding
False
Hypocalcemia
Acute pancreatitis
↓ Lipase
secretion from
pancreas
↑ Levels of
undigested
fats in small
intestine
Free fatty
acids
percipitate
calcium
↓ PTH receptor
activation in
bones
↓ Osteoblast
stimulation
↓ RANKL
ligand
secretion
↓ RAANK
receptor
binding
↓Osteoclast
production
↓ Bone
resorption
Hypocalcemia
(serum [Ca2+] <2.1mmol/L)
Authors:
Name Name
Name Name*
Reviewers:
Name Name
Name Name*
* MD at time of publication
Published MONTH, DAY, YEAR on www.thecalgaryguide.com
Hypocalcemia: Physiology
Chronic kidney
disease
↓ Renal
blood flow
↓ Glomerular
filtration
Parathyroid gland
dysfunction: removal,
autoimmune,
congenital (DiGeorge)
Vitamin D deficiency:
↓intake, malabsorption
Sepsis/severe illness
↓ NHE3 activity and
expression in
proximal tubule
↓PTH receptor
activation in
kidneys
↑ Claudin14
activity in the
thick ascending
loop of Henle
↓ Parathyroid
hormone (PTH)
↓ Transcription of
calcium transporter
genes (TRPV5, calbindin
D28K, NCX) in the distal
convoluted tubule
↓ Enzyme 1-alpha
hydroxylase activity
↓ PTH receptor
activation in
bones
↓ osteoblast
stimulation
↓ RANKL ligand
secretion
↑ Inflammation ↓ PTH sensitivity
↓ Glomerular
filtration
↓ Liver
synthesis of
serum albumin
False
Hypocalcemia
↑ Calcium
sequestration
into cells**
↓ Lipase secretion
from pancreas
Acute pancreatitis
Legend: Pathophysiology Mechanism
Sign/Symptom/Lab Finding ↓ Albumin-
bound calcium
Current mechanism
is unknown
↑ levels of undigested
fats in small intestine
Complications
Hypomagnesemia Multiple blood
transfusions
↓ Sodium
reabsorption
Electrochemical
gradient changes
↓ Phosphate
excretion
↑ Phosphate
calcium complex
formation
Inhibits claudin16 and 19
from forming cation
permeable pores in the
tight junctions
↓Probability of
calcium transport
channels opening
↓ Synthesis of activated
vitamin D (calcitriol)
↓ RAANK
receptor
binding
↓ Calcium
reabsorption
↓ Binding of vitamin D
regulated calcium
transporters (TRPV6,
calbindin9K, PMCa2B,
NCX2) in the duodenum
and jejunum
↓osteoclast
production
↓ Bone
resorption
↓ Serum calcium
Hypocalcemia
<2.1 mmol/L
↑ Precipitation of calcium
by free fatty acids
Authors:
Name Name
Name Name*
Reviewers:
Name Name
Name Name*
* MD at time of publication
Published MONTH, DAY, YEAR on www.thecalgaryguide.com
https://journals.physiology.org/doi/full/10.1152/physrev.00003.2004?rfr_dat=cr_pub++0pubmed&url_ver=Z39.88-
2003&rfr_id=ori%3Arid%3Acrossref.org – Calcium Absoprtion across Epithelia
https://academic.oup.com/endo/article-abstract/137/1/13/2498579 - PTH and Calcium Signaling Pathways
https://www.pnas.org/doi/10.1073/pnas.1616733114 - Information on Claudin 14
https://onlinelibrary-wiley-com.ezproxy.lib.ucalgary.ca/doi/full/10.1111/apha.13959 - effects of PTH on renal calcium and
phosphate handling
https://www.ncbi.nlm.nih.gov/books/NBK430912/ - Hypocalcemia overview + causes + pathophys
https://www.orthobullets.com/basic-science/9010/bone-signaling-and-rankl - information about bone signaling
https://www.sciencedirect.com/science/article/abs/pii/S0889852921000682?via%3Dihub – Calcium homeostasis article
https://pubmed.ncbi.nlm.nih.gov/3012979/#:~:text=Abstract,intestine%2C%20require%20the%20parathyroid%20hormone.
vitamin D metabolism and function
–
https://pmc.ncbi.nlm.nih.gov/articles/PMC2669834/#:~:text=Calcium%20is%20actively%20absorbed%20from,for%20proper
%20mineralization%20of%20bone.
– more vitamin D metabolism and specific receptors
https://jidc.org/index.php/journal/article/view/32903236/2331 - sepsis + hypocalcemia")
Malignant Renal Mass
 &
prevents flow of urine
↑ Leptin & vasfatin promote
cellular metabolism to attempt to
meet energy demands of
proliferating cells ↑ IL-15 &
VEGF promote
inflammation
↑ VEGF in response to energy
demands causes ↑ angiogenesis
(blood vessel growth)
causing cellular
damage & ↑
angiogenesis
Cadmium & arsenic
in renal tissue
disrupt metal & ion
homeostasis causing
cell damage which
initiates injury-
repair cycle
Generation of
reactive oxygen
species causes
oxidative damage to
genes regulating cell
cycle
↓ Oxidative
phosphorylation
promotes glycolysis/
survival in hypoxic
conditions typical of
tumor
microenvironment
Altered amino
acid
metabolism in
cells enhances
biosynthetic
capabilities &
promotes cell
proliferation
↑ Pressure causes hypoxic
conditions as tubular
electrolyte load is ↑ &
↑ Atypical
energy demand on renal
epithelial cell
tubular cells is ↑
proliferation
promotes tumor
suppressor gene
mutations
Hypoxia stimulates VEGF
to ↑ angiogenesis
↑ Blood flow
damages
endothelial cells
& initiates the
injury-repair
cycle which ↑
the risk of cell
mutations
↑ Angiogenesis ↑ Cellular metabolism ↓ Fidelity of DNA repair mechanisms
↑ Cellular damage results in DNA mutations Altered metabolism promotes survival in tumor microenvironment
↑ Proliferation of cancer cells (typically from proximal tubular epithelial cells, as in clear cell renal cell carcinoma)
Malignant Renal Mass
Metastasis to distal sites (most
commonly bone, lungs, liver, brain)
Cancer cells release proteolytic
(protein degrading) enzymes
including MMPs, cathepsins, & uPA
Degradation of healthy renal tissue
& vasculature surrounding
collecting system of kidneys
impairing flow of urine and causing
blood loss into collecting system
Inactivation of VHL gene ↑
VEGF expression & causes
↑ angiogenesis
↑ Vascular permeability
due to uninhibited growth
↑ Muscle & fat
wasting from ↑
inflammatory
cytokines activates
JAK/STAT & NF-κB
pathways
Gain of function
mutations to
PI3K/AKT/mTOR
cause ↑ glucose
uptake & drive insulin
resistance without
hyperinsulinemia (↑
blood insulin
Cancer cells release of
adipose triglyceride
lipase & hormone-
sensitive lipase
Tumor-generated
↑Secretion of
Tumor-
cell death releases
parathyroid
generated cell
proinflammatory
hormone-
death ↑ IL-6
cytokines & ↓ renal
related protein
levels &
cell production of
(PTHrP)
initiates ↑
erythropoietin (EPO)
production of
↑ lipolysis & browning
(protein regulating
PTHrP is
thrombopoietin
of white adipose tissue
red blood cell
Weaker vessels form
(conversion to
homologous to
(protein
production)
Hydronephrosis
within the kidney
metabolically active
parathyroid
regulating
↑ Creatinine
(hydrostatic
collecting system &
state by ↑ UCP1
hormone (PTH)
platelet
dilation of the
subsequently rupture
expression)
& competes
production)
renal pelvis &
↓ Erythropoiesis (red
with PTH to
calyces)
blood cell production)
bind to PTH
from ↓ EPO
receptors
Thrombocytosis
(↑ platelets)
Compresses
posterior
abdominal
wall nerves
Imbalance of anabolic
& catabolic
metabolism, reducing
body mass due to ↑
catabolism
Brown adipose tissue
thermogenic properties
↑ body temperature
Flank
pain
Hematuria
(presence of
blood in
urine)
Anemia
Bleeding at site of metastasis
Weight loss Drenching night sweats
PTH receptor activation
promotes ↑ bone
resorption & renal Ca2+
reabsorption
↑ Ca2+
Published Aug 31, 2025 on www.thecalgaryguide.com
Familial Renal
Carcinoma Syndrome
Genetic mutations
conferred from birth
Authors:
Kaiden Jobin
Sergio F. Sharif
Reviewers:
Jessica Revington
Nimira Alimohamed*
*MD at time of publication
Gain of function
for proto-
oncogenes
Loss of function
for tumor
suppressor genes
Compresses
stomach/
esophagus
Compresses
bladder
Compresses
diaphragm
↑ Urinary
Early satiety Cough
frequency
Legend: Malignant
renal cells
produce ↑
EPO &
subsequently
↑
erythropoiesis
Polycythemia
(↑ red blood
cells)
Neoplastic
proximal
tubule
cells
secrete ↑
renin
Intrarenal
ischemia
(reduced
blood flow)
due to
tumor
growth
compressing
renal blood
supply
Hypertension
↑ Tumor cell
secretion of
hepatotoxins,
lysosomal enzymes,
& IL-6
Secreted products
hepatocellular injury,
releasing cell contents
IL-6 triggers immune
response against
hepatocellular cell
contents in response
to injury
↑ ALT ↑ AST ↑ ALP ↑ Prothrombin time ↑ Bilirubin
Pathophysiology Mechanism
Sign/Symptom/Lab Finding Complications")
Obstructive Shock

↓ Right ventricle stroke volume
(blood volume ejected per beat)
↓ Blood return to left atrium
Underfilled left ventricle
Obstructive shock
↓ systemic blood flow due to a physical
obstruction of the heart’s pumping function
Insufficient
organ
perfusion
Tension pneumothorax**
↑ Pressure in the pleural cavity
Intrathoracic pressure exceeds
venous filling pressures &
compresses heart chambers
↓ Venous return to right
atrium & left atrium
Author:
Ryan Dion
Sergio F. Sharif
Dean Percy
Reviewers:
Yan Yu
Tristan Jones
Jason Waechter*
* MD at time of publication
** See corresponding
Calgary Guide slides
↓ Preload (degree of left ventricular filling)
as left ventricle receives low levels of blood
↓ Cardiac output (volume of
blood ejected from the
↓ Blood pressure (BP)
heart/min)
Baroreceptors in carotid sinus & aortic
arch detect low blood pressure
Skin
Reflexive release of catecholamines
via sympathetic nervous system
Blood supply diverted
away from peripheral
tissues to vital organs
Tachycardia
Hypotension
Cold
extremities
Legend: Pathophysiology Mechanism
Sign/Symptom/Lab Finding Brain
↓ Cerebral blood
flow causes
cerebral hypoxia
Altered consciousness
Heart
↓ Coronary
artery
perfusion
Myocardial
ischemia**
Complications
Blood backs up into
venous system
Kidneys
↓ Blood flow
to kidneys
Prerenal acute kidney
injury **
↑ Pressure distends
veins
↑ Jugular
venous
pressure
Pressurized fluid leaks
out of veins into
tissue
Fluid enters
pulmonary
perivascular spaces
Fluid impedes normal gas
exchange in lungs
Tachypnea (↑
respiratory rate)
Dyspnea
Published July 7, 2013; updated Sept 28, 2025 on www.thecalgaryguide.com")
GLP-1 Receptor Agonists
 Receptor Agonists: Mechanism of action and side effects
Management of Type
2 Diabetes Mellitus**
Weight
management
Authors: Saania Tariq, Trevor Low
Reviewers: Rafael Sanguinetti
Luiza Radu, Emily J. Doucette
Activated GLP-1 receptors in
Activated GLP-1
Hanan Bassyouni*
* MD at time of publication
the myenteric plexus inhibit
receptors stimulate
gastric motor neurons
pancreatic islet cells
GLP-1 Receptor Agonists
Synthetic analogs (e.g., Semaglutide) of GLP-1 peptide hormone endogenously
produced by intestinal enteroendocrine L cells directly activate GLP-1 receptors
to modulate food intake & glucose metabolism
↓ Gastric
motility
↑ Pyloric sphincter
tone via disinhibition
↓ Glucagon release
from α-cells
↑ Regeneration,
↑ growth &
↓ apoptosis of β-cells
Slowed gastric emptying
Subcutaneous injection or oral ingestion
prolongs GLP-1 receptor activation
↑ β-cell function & glucose-
dependent insulin production
GLP-1 receptor activation on neurons in
the arcuate nucleus of the hypothalamus
modulates appetite & satiety
Activation of GLP-1 receptors
in the gastrointestinal system
regulates metabolism
Prolonged gastric
distension & stasis
activates
chemoreceptors &
mechanoreceptors
Delayed digestion of
gastric contents ↓
glucose spikes after
meals
↑ Glycogen synthesis &
glucose uptake in liver &
skeletal muscles
Neuropeptide Y &
agouti-related protein
expressing neurons are
hyperpolarized
Pro-opiomelanocortin
expressing neurons
become ↑ excitable
Activated GLP-1 receptors
signal to hypothalamus
via vagus nerve afferents
↑ Vagal afferent
signaling to area
postrema
(vomiting centre)
Nausea &
vomiting
↓ Release of
neuropeptide Y &
agouti-related protein
↑ Anorexigenic
signalling
↑ Parasympathetic
output via dorsal motor
nucleus of vagus nerve
GLP-1 agonists directly
activate receptors in
area postrema
↑ Satiety (feeling
of fullness)
↓ Adiposity
(weight loss)
↓ Orexigenic drive
↓ Inflammatory stress in
pancreatic β-cells
↑ β-cell function
improves insulin
secretion
↓ Appetite
↓ Caloric intake
↓ Inflammatory
cytokines & lipotoxicity
↓ Systemic
inflammation
↑ Insulin sensitivity in
skeletal muscle & liver
**See corresponding Calgary Guide slide
Legend: Sign/Symptom/Lab Finding Pathophysiology Mechanism
Complications
↓ Hepatic glucose
production
(glycogenolysis)
↓ Fasting & postprandial
blood glucose
↓ Red blood cell (RBC)
glycation (glucose
irreversibly binds to
hemoglobin in RBCs)
↓ Production
of glycated
proteins and
lipids
↓ % of glycated
RBCs over time
↓ Vascular
endothelial
damage and
atherosclerosis
↓ Endothelial
damage to
glomeruli
↓ Hemoglobin
A1c
↓ Risk of
cardiovascular
complications
↓ Risk of renal
complications
Published Jun 22, 2025 on www.thecalgaryguide.com")
Rickets and Osteomalacia Pathogenesis and Clinical Findings
)
Medications (i.e.,
bisphosphonates)
Hereditary
hypophosphatasia
Medication act as
pyrophosphate
(PPi) analogue
Renal tubular
defects (e.g.,
Faconi
syndrome,
distal renal
tubular
acidosis)
Chronic use of
Tumor-
phosphate binders
induced
(e.g., calcium
osteomalacia
carbonate)
Hereditary
hypophosphatemic
rickets
Calcipenic rickets (insufficient calcium (Ca2+))
↓ Sunlight
exposure
↓ Intestinal
absorption of fat-
soluble vitamins (A,
D, E & K) (e.g., in GI
or liver disease)
↓ Intake
through
food
Vitamin D-
dependent
rickets type 1
Vitamin D-
dependent
rickets type 2
Osteoblasts
secrete ↓ alkaline
phosphatase (ALP)
(cleaves PPi to
create sites
forCa2+ & PO₄³⁻
deposition)
↑ Fibroblast growth
factor 23 (FGF23)
Mutation in
CYP27B1 or
CYP2R1 gene
Mutation in
vitamin D
receptor gene
↑ PPi binds to
growing
hydroxyapatite
crystals in bone &
prevents further
deposition of Ca2+
& PO₄³⁻
↑ Renal
losses of
PO₄³⁻
FGF23 ↑ breakdown of
FGF23 ↑ 24-
1α-hydroxylase (converts
Hydroxylase
Vitamin D
deficiency (most
common cause)
calcidiol (inactive vitamin
activity
D) to calcitriol (active
(catabolizes
calcitriol)
vitamin D) ↓ Calcidiol
Mutations prevent
conversion of
calcidiol to calcitriol
End-organ
resistance
to calcitriol
↓ Cleavage
of PPi
↓ Calcitriol
↓ PO₄³⁻ released
from PPi
↓ PO₄³⁻ absorption
in intestines
↓ Ca2+ absorption from
intestines & kidneys
Hypophosphatemia (↓ Serum PO₄³⁻)
Hypocalcemia** (↓ Serum Ca2+)
Ca2+ deficiency
↓ Availability of PO₄³⁻ & Ca2+ impairs formation of hydroxyapatite
((Ca10(PO4)6(OH)2) crystals (deposition of crystals hardens bone)
Parathyroid gland releases PTH (secondary
hyperparathyroidism) to maintain serum Ca2+
Impaired mineralization of osteoid
↑ Parathyroid hormone** (PTH)
Rickets
Defective mineralization of new bone formation at growth
plate (only occurs in children with open growth plates)
Defective mineralization
of preformed osteoid
Osteomalacia
Defective mineralization of existing bone (can occur
in individuals with open or closed growth plates)
Unmineralized
cartilage
overgrows at
growth plates
↓ Mineralization
impairs structural
integrity of bone
↓ Calcitriol &
PO₄³⁻ impairs
dentin & enamel
mineralization
Unmineralized
cartilage overgrows
at costochondral
junctions
↓ Mineralization
of skull bones
↓ Structural integrity
& ↓ bone density
↓ PO₄³⁻
disrupts ATP
production
in muscles
Osteoblasts
secrete ↑ ALP to
compensate for
↓ mineralization
↑ Diameter of
growth plate &
metaphysis
Shear forces bend
osteopenic bone
during ambulation
Delayed dental
eruption & enamel
hypoplasia
Rachitic rosary
(palpable bead-like
nodules along ribs)
↑ Stress on poorly
mineralized bone
↑ Stimulation
of periosteal
nociceptors
Muscle weakness
&/or poor tone
↑ALP
Metaphyseal
cupping on
X-ray
Widened
wrists &
ankles
Progressive
lateral bowing
of femur & tibia
Abnormal gait
Genu varum/valgum
Delayed
fontanelle
closure
Craniotabes
(softening of
skull bones)
Fractures &/or
pseudofractures
Localized
bone pain or
tenderness
↓ Bone
mineral
density
Authors:
Olivia Yung, Payam Pournazari
Reviewers:
Merry Faye Graff, Yan Yu, Spencer
Montgomery, Emily J. Doucette,
David Hanley*, Danielle Nelson*
* MD at time of publication
Sign/Symptom/Lab Finding Complications
Published Nov 26, 2012; updated Nov 2, 2025 on www.thecalgaryguide.com
↓ Mineralization
impairs growth
plate fusion
↓ Linear
bone
growth
Short
stature
Legend: Pathophysiology Mechanism")
Acute Tubular Necrosis

Septic shock** Cardiogenic shock**
Cholesterol embolization
syndrome (rare)
Severe hypotension
↓ Mean arterial pressure
reduces renal perfusion pressure
which ↓ renal blood flow
Renal vascular autoregulation
fails to maintain renal perfusion
Tubular cell hypoxia & depletion
of adenosine triphosphate (ATP)
(primary energy carrier in cells)
Cell ion pump failure, cell swelling,
& cell membrane disruption
Ischemic tubular necrosis (renal tubular
cell injury secondary to ↓ blood flow)
Myoglobinuria
Hemoglobinuria
Aminoglycosides
Ethylene glycol
Amphotericin
Cisplatin
↓ Efficiency of kidney reabsorption & excretion
Kidney removes ↓
potassium (K+) from
the blood volume
Hyperkalemia** (high K+
concentration in the blood)
↑ Serum K+ levels
induce abnormal
cardiac electrical
conduction patterns &
excessive cardiac
excitability
Fatal cardiac
dysrhythmia (irregular
heartbeat pattern)
Kidney removes
↓ urea from the
blood volume
Kidney removes ↓
creatinine from the
blood volume
Azotemia (↑ serum
urea & creatinine)
Uremic toxins
systemically
impair
neutrophil
function
Uremic toxins (indoxyl
sulfate or paracresyl
sulfate) induce oxidative
stress in the brain through
accumulation of reactive
oxygen species (ROS)
↑ Risk of infection
ROS damage neuronal membranes & ion channels &
induce dysfunctional mitochondria in the brain
Authors:
Adam Bubelenyi
Reviewers:
Britney Wong
Luiza Radu
Jessica Revington
Veronica Hammer*
Braden Manns*
* MD at time of publication
**See corresponding Calgary Guide slide
Dysfunctional brain mitochondria cannot
properly metabolize purines & urea
Brain cannot use any metabolic or cellular pathways requiring ATP
↓ Cerebral neuronal signaling
Legend: Pathophysiology Mechanism
Sign/Symptom/Lab Finding Intravenous (IV)
administration of
iodinated
contrast medium
Tumor lysis syndrome
Cancer cell apoptosis (cell death)
releases ↑ uric acid & phosphate
Heme-containing
pigments
Medications
Uric acid & calcium phosphate
crystals precipitate in renal tubules
Nephrotoxic substances damage renal tubular cells through unique mechanisms
Lipid peroxidation
(oxidative degradation) of
lipid bilayer weakens renal
tubular cell membranes
Damage to mitochondria in tubular
cells causes depletion of ATP
Formation of oxygen free
radicals in renal tubular cells
Impairment of ATP-
dependent processes
Activation of pro-inflammatory
cytokines & enzymes in the kidney ↑ Renal tubular cell
permeability
Disruption of protein synthesis &
enzyme function in renal tubular cells
Degradation of tubular cell
membrane lipids & proteins
Free radicals
oxidize tubular
cell proteins &
impair structure
& function
Toxic tubular necrosis (renal tubular injury due to nephrotoxic substances)
Acute Tubular Necrosis
Acute kidney injury via renal tubular cell damage & cell death
Denudation (removal of surface tissue layers) &
erosion of the tubular basement membrane
Kidney removes ↓
sodium (Na+) & water
from the blood volume
↓ Bicarbonate
reabsorption from
the proximal tubule
of the kidney
Necrotic tubular cells fall into tubular lumen
Ongoing tubule damage
↓ ability to concentrate
urine solutes
↓ Bicarbonate
neutralization of
acids in the blood
Necrotic
tubular cells
release
intracellular
contents
Obstruction of
tubular lumen
impedes
glomerular
filtration
↓ Urine osmolality
(low urine solute
concentration)
↑ Water in blood
volume dilutes
Na+ serum
concentration
Metabolic acidosis
(blood pH <7.35)
Muddy brown
granular casts
↓ Glomerular
filtration rate (GFR)
Hyponatremia** (low Na+
concentration in the blood)
↑ Na+ retention in
the circulatory blood
volume & ↓ Na+
excretion in the urine
↓ Urine output
Accumulation of ROS activates microglia
& releases pro-inflammatory cytokines
↑ Fluid retention in
the circulatory system
Aggressive IV fluid
rehydration in septic
shock or cardiogenic
shock patients
Cerebral inflammation ↑ oxidative
damage to neurons & glial cells
Fluid overload
Uremic encephalopathy
Pulmonary edema (excess
fluid accumulation in lung
alveoli & interstitium)
Published November 15, 2025 on www.thecalgaryguide.com
Complications
Disruption of
DNA & RNA
synthesis
within renal
tubular cells
Tubular epithelial
cells in urine
Epithelial cell
casts in urine
(cast composed
of epithelial
cells embedded
in a matrix)
↓ Ability to clear
bacteria in urine
↑ Risk of urinary
tract infection")
Infection-Related Glomerulonephritis
 trigger the inflammatory cascade
Common infection sites include pharyngitis (infection of the mucous membranes in the oropharynx) &
endocarditis (inflammation of the endocardium) but bacterial infections can originate anywhere in the body
In streptococcal infections, bacteria release nephritis (inflammation of the kidney) associated plasmin receptor antigens (NAPlr) & streptococcal
pyrogenic exotoxin B antigens (SPeB) into the systemic circulation from the infection site. Other bacterial infections may have different pathophysiology
Immune response generated against circulating NAPlr & SPeB antigens includes
↑ production of IgG antibodies (critical for immune response & memory)
IgG antibodies bind with self-antigens (molecules
recognized by the immune system as belonging to the
host) including plasmin & glomerular proteins (proteins
found in the kidney’s filtering unit, the glomerulus)
IgG antibodies activate plasmin
(enzyme responsible for breaking
down protein in the kidneys)
Plasmin degrades
protein in the kidneys
IgG antibodies bind to
glomerular proteins in
the kidney & form
immune complexes in
glomeruli basement
membrane & sub-
epithelial podocytes
(highly specialized
cells in the glomerular
filtration barrier)
Degradation of key
glomerulus components,
including the glomerular
basement membrane
(GBM) & mesangial matrix
Damage to key
glomerulus
components leads to
abnormal regulation
of blood filtration
↓ Selective
permeability of the
glomerular membrane
IgG antibodies bind with NAPIr & SPeB antigens &
form immune complexes (antibody & antigen
combinations involved in immune system signalling)
Immune complexes circulate & deposit in glomeruli
basement membrane & sub-epithelial podocytes
RBCs escape through
the glomerulus into
renal tubules &
progress into the urine
Dysmorphic
hematuria (blood
in the urine
characterized by
misshapen RBCs)
Author:
Joanna Keough
Reviewers:
Britney Wong
Luiza Radu
Jessica Revington
Veronica Hammer*
Louis Girard*
*MD at time of publication
**See corresponding Calgary Guide slide
Legend: Pathophysiology Mechanism
Immune complexes activate
complement (C3, oxidizing
agents, proteases)
Complement activation helps ↑
proliferation of glomerular mesangial
cells (cells with specialized proteins
capable of producing motile forces)
Mesangial cells produce
extracellular matrix in
the glomerulus
Excess
extracellular
matrix
blocks
glomerular
capillaries
Mesangial cells release chemoattractants
(signalling molecules) into the glomerulus
& attract various immune cells
Lymphocytes follow
chemoattractants to the
glomerulus & activate
Neutrophils follow chemoattractants to the
glomerulus & release lysosomal enzymes
Lymphocytes accumulate in the glomerulus
& obstruct glomerular capillaries
Impaired blood flow through glomerular capillaries
Capillary injury Podocyte injury or death
Infection-Related Glomerulonephritis
Glomerular kidney damage sustained after a bacterial or viral infection
↓ Glomerular
filtration rate (GFR)
Creatinine builds
up in the blood
↑ Glomerular membrane permeability allows
large molecules to pass through the membrane
(e.g., red blood cells (RBCs), protein)
↓ GFR impairs the kidney’s
ability to filter & excrete fluid
↓ Estimated GFR
Prolonged
(eGFR)
renal damage
Protein escapes into renal tubules
& progresses into the urine
Chronic kidney disease
Proteinuria (loss of
protein through the urine)
↑ Fluid retention
in systemic
circulation & ↓
renal blood flow
Impaired
potassium
excretion
↓ Urine
output
Impaired
acid waste
product
excretion
Urine contains ↑ large
proteins (e.g., albumin)
Activation of the
intrarenal renin-
angiotensin-aldosterone
system (RAAS)**
Hyperkalemia
(↑ serum
potassium)
Oliguria
(↓ urine
volume)
Metabolic
acidosis**
Albuminuria (loss of
albumin through the urine)
Intrarenal RAAS activation
leads to ↑ angiotensin II
(Ang II) production
↑ Sodium & water retention in systemic
circulation & ↓ excretion in urine (↓ GFR)
Hypertension**
Edema (swelling due to ↑ fluid in body tissues)
Circulating blood volume exerts ↑ pressure on blood vessels & forces the heart to pump harder
Left ventricle thickens & enlarges over time & demonstrates
progressive cardiac injury (e.g., fibrosis, loss of cardiac muscle cells)
Impaired cardiac output
Heart failure
Sign/Symptom/Lab Finding Complications
Published November 15, 2025 on www.thecalgaryguide.com")
Pyelonephritis

Obstruction of bladder outlet
(congenital/acquired)
High blood glucose damages
nerves controlling the bladder,
as well as blood vessels
supplying bladder nerves
Glycosuria (elevated levels
of glucose in urine) create
nutritive environment for
bacterial growth
Excessive urine buildup
increases pressure exerted
against the bladder, pushing
urine from bladder up to ureters
Authors:
Sergio F. Sharif
Reviewers:
Michelle J. Chen
Jessica Revington
Yan Yu*
Juliya Hemmett*
Brandon Christensen*
* MD at time of publication
Shorter urethra
allows for fecal
microbes to more
readily enter the
urinary tract
Catheter inoculates deep
urinary tract with bacteria
& facilitates organisms to
enter bladder
Neurogenic bladder (decreased
bladder control due to nerve damage)
is unable to appropriately release
urine, leading to urinary retention &
failure to clear residing bacteria
Bacteremia
(bacterial infection
in bloodstream)
Bacteria initially colonize the lower urinary tract, then migrate up the tract towards
one or both kidneys (most commonly Gram-negative rods, e.g., Escherichia coli)
Bacteria, such as
Staphylococcus species,
spread from another site
of infection to kidneys
via bloodstream
Acute Pyelonephritis
Inflammation of the kidney interstitium due to upper urinary tract infection, usually bacterial
Collective contribution of
mechanisms stated below, if severe
Sepsis**
Concurrent cystitis
irritates urinary
tract (see lower
UTI slide)**
Bacteria expressing virulence factors (e.g., P fimbriae in Escherichia coli) exploit
host susceptibility & infect upper urinary tract (ureters & kidneys)
Positive urine culture
Dysuria (pain & burning
while urinating)
Bacteria trigger IL-8 release by local
immune cells in the upper urinary tract,
triggering recruitment of neutrophils to
renal tubules & interstitium
Neutrophils degranulate in the infected
tissue, phagocytose bacteria & form
sacrificial neutrophil extracellular traps
(resulting in neutrophil death) in
attempt to clear infection
Neutrophil-mediated
immune response
damages surrounding
kidney tissue (tubular
injury)
Acute
kidney
injury**
Immune cells at the site of infection release cytokines
(e.g., IL-1, TNF), which disseminate systemically
Cytokines act in the brain in
concert with unclear
mechanisms relating to tissue
injury & inflammation
triggered by infection (see
Neurotransmitters &
Pharmacology behind Nausea
and Vomiting slide**)
Cytokines bind
receptors in the
anterior hypothalamus,
triggering pathways
ultimately leading to
heat-generating
responses (e.g.,
shivering)
Cytokines promote
the growth &
release of white
blood cells from
bone marrow into
bloodstream
Dead neutrophils collect
in renal tubules near
infection & attach to
assemblies of
mucoprotein (a renal
tubular epithelium protein
secreted into tubules)
Pyuria (WBCs in
urine)
Nausea/vomiting
Fever
Leukocytosis
(↑ WBCs)
Urine white blood cell
(WBC) casts (cylindrical
particles with WBCs
seen on microscopy)
Costovertebral
Flank pain CT KUB -
angle tenderness
findings may
include focal
or diffuse
involvement,
fat stranding,
gas formation,
&/or other
complications
including
renal scarring
**See corresponding
and abscesses
Calgary Guide slide(s)
Complications
Published November 23, 2025 on www.thecalgaryguide.com
Unclear mechanism
involving systemic
inflammation
worsens mental
status in susceptible
individuals
Altered level of
consciousness
(especially in elderly)
Legend: Pathophysiology Mechanism
Sign/Symptom/Lab Finding")
Hypothyroidism

Iodine
deficiency
Infiltrative disease affecting
thyroid gland cells
Iatrogenic or medication-
related thyroid dysfunction
Hypothyroid phase of thyroiditis
(inflammation of the thyroid)
Pituitary gland dysfunction
(central hypothyroidism)
Progressive autoimmune
destruction of thyroid gland cells
↓ T3/T4 levels
activate the
hypothalamus-
pituitary-thyroid
axis & ↑ TSH
release by the
pituitary gland
Primary
hypothyroidism
(↑ TSH and ↓
T3/T4)
↓ T3/T4 levels
cause ↓ expression
of TH-dependent
myocardial enzymes
essential for cardiac
contractility
Cardiac myocytes
(cardiac muscle
cells) experience ↓
contractility
↓ Heart rate
↑ Bleeding
Impaired function of the thyroid gland & ↓ thyroid hormone (TH) secretion
Pituitary gland secretes ↓ levels of
thyroid stimulating hormone (TSH)
Hypothyroidism
Low or significantly ↓ secretion of triiodothyronine (T3) & thyroxine (T4) from the thyroid gland
↓ T3/T4 levels ↓
production of nitric oxide
(vasodilator) & cause
systemic vasoconstriction
of blood vessels
Systemic vaso-
constriction ↑
systemic
vascular
resistance
↑ Systemic vascular
resistance à ↑
intrarenal vascular
resistance & contributes
to ↓ renal perfusion
↓ T3/T4 levels reduce the rate of metabolic reactions in the body
including ↓ rates of carbohydrate, fat, & protein metabolism
↓ T3 levels allow fibroblasts
(connective tissue cells) to synthesize ↑
glycosaminoglycans (GAGs – structural
cell molecules capable of attracting
large amounts of water)
↓ Basal metabolic rate (base energy
required to sustain bodily functions)
↑ Diastolic blood pressure
(diastolic hypertension)
↓ Renal blood
flow à ↓
glomerular
filtration rate
(GFR)
↓ GFR
impairs renal
clearance of
excess GAGs
↑ GAG deposits
& water retention
in body tissues
↓ Energy to support physiological function
Narrowed pulse
pressure (↓
difference
between systolic
& diastolic blood
pressure)
Compensatory
mechanisms to
manage blood
volume &
circulation during
↓ T3/T4 levels
are overwhelmed
in the presence
of an infection or
systemic trauma
(e.g., heart
attack, stroke)
↓ GFR impairs
removal of excess free
water from the blood
volume by the kidneys
↑ GAG
deposits &
swelling in
skin tissue
↑ GAG deposits
in the larynx
swell & stiffen
vocal cords
↓ Skin gland
activity & ↓
sebum (oil) &
sweat
production
↑
Fatigue
↓ Motility of the
gastrointestinal
(GI) tract causes
↓ movement of
digested food &
↓ rate of waste
excretion
↓ TH levels & ↓
metabolic rate
impair neurologic
function (e.g.,
neurotransmitter
signaling & neuron
functioning)
Thickened skin
Hoarse speech
Dry skin
↑ Sodium (Na+) & water
retention in blood volume
↓ Perspiration
(sweating)
Constipation
Neurologic
symptoms (e.g.
depression,
somnolence)
Abnormally heavy menses
Excessive water retention
dilutes serum Na+ levels in
severe hypothyroidism
↓ Caloric expenditure at rest & with activity
↑ Risk of infertility
Myxedema coma**
(extreme manifestation
of hypothyroidism)
Hyponatremia
(↓ Na+)
↑ Fluid
retention in
body tissues
Weight gain
Musculoskeletal
symptoms (e.g.,
muscle cramps,
joint pain & ↓
muscle tone)
Published June 19, 2013, revised November 23, 2025 on www.thecalgaryguide.com
Normal or ↓
TSH levels
when T3/T4
levels are low
Authors:
Sophia Khan, Ayden Hansen, Jessica
Revington, Rupali Manek, Jaye
Platnich
Reviewers:
Shahab Marzoughi, Gurreet
Bhandal, Raafi Ali, Matthew
Harding, Mark Elliott, Hanan
Bassyouni*, Breanna McSweeney*
* MD at time of publication
**See corresponding Calgary Guide
slides
↓ T3 levels lead to
↓ lipoprotein lipase
(lipid metabolism
enzyme) activity
↑ Triglyceride
levels in blood
↓ TH levels & ↓
metabolic rate
impair cold
tolerance &
thermogenesis
(heat production)
mechanisms
Cold
extremities
(e.g. hands,
feet)
↓ Cold
tolerance
↓ Activation
of the T3-
mediated low-
density
lipoprotein
(LDL) receptor
gene
↓ LDL
receptor
expression
for LDL
clearance
↑ LDL
cholesterol
levels in
the blood
↓ T3/T4 levels ↓
coagulation factor
synthesis by the liver
↓ T3/T4 levels disrupt
the menstrual cycle &
cause anovulation
(absence of ovulation)
Legend: Hyperlipidemia
Pathophysiology Mechanism
Sign/Symptom/Lab Finding Complications")
Diverticulosis and Angiodysplasia

from idiopathic
causes
von Willebrand
Deficiency (lack of
vWF) aggravates
bleeding tendency
Aging weakens the
circular muscles
strutting the colon
High intracolonic pressure
primarily in the sigmoid
colon (e.g., from peristalsis
pushing against colonic
waste that is low in fiber &
harder to move)
Muscular hypertrophy of the colon
in response to repeated high-
pressure contractions of the
smooth muscle as a compensatory
mechanism to move stool in high
resistance areas
Arterial-venous malformations (AVMs;
abnormal connections between
arteries & veins) bypass capillary beds
and rise to the mucosa of the lower GI
tract
Flat cherry-red vessels
originating from a central
artery as seen on upper GI
endoscopy or colonoscopy
Colon wall forms little
outpouchings (diverticuli) that
span around 3-10mm
Gradual & repetitive
expansion thins the
diverticular wall
Stretching of colonic
serosa stimulates somatic
sensory nerves innervating
the colon
Colonic diverticuli trap
feces
Venous rupture of
submucosal veins in the
High pressure arterial blood flows
colon
directly into veins, causing venous
dilation & weakening of vascular
walls
Overlap with other
vascular lesions such as
Dieulafoy’s
Capillaries within
diverticuli burst and
leak blood into the
colon lumen
Stretching of nociceptors
that are primarily sensitive to
mechanical stimuli such as
distension of the colonic wall
Bacteria have more
time to metabolize the
undigested materials,
producing gas
Irregular defecation
(constipation,
increased frequency)
GI tract lumen bleeding
due to irritation of these
malformations
Large
volume
blood loss
Iron
deficiency
anemia
Lower GI bleed
(usually stops by
itself)
Bloating & cramping
(most often painless)
Flatulence
(accumulation of gas)
Lower GI bleed
(occult, slow,
asymptomatic)
Author: Yan Yu
Ali Babwani
Reviewers:
Jason Baserman
Jennifer Au
Paige Sheleme
Tony Gu
Luiza Radu
Sergio F. Sharif
Kerri Novak*
Sylvain Coderre*
* MD at time of publication
Legend: Re-Published Aug 4, 2019 and Nov 23, 2025 on www.thecalgaryguide.com
Pathophysiology Mechanism
Sign/Symptom/Lab Finding Complications")
Vancomycin
, coagulase-negative Staphylococcus spp., Streptococcus spp., Enterococcus spp. (including E. faecium),
& Clostridium difficile (when given orally). Often requires therapeutic drug monitoring due to risk of toxicity.
Blocks cross-linking of
peptidoglycan strands
Drug binds to D-alanyl-D-
alanine (D-Ala-D-Ala) terminal
of peptidoglycan precursors
Acquisition
of van gene
clusters
(vanA/vanB)
Transglycosylation
& transpeptidation
reactions are
inhibited
Target site changes
from D-Ala-D-Ala to
D-Ala-D-Lac or
D-Ala-D-Ser
Bacterial cell
wall becomes
osmotically
unstable
↓ Binding affinity
of drug to bacteria
Bacterial
cell lysis
Development
of vancomycin
resistance
Bacterial
clearance on
microbiological
culture
Legend: Primarily eliminated by
glomerular filtration
Presence of renal
dysfunction impairs
drug clearance
↑ Serum drug levels
Oxidative stress
& mitochondrial
dysfunction of
proximal tubular
epithelium
Direct proximal
tubular cell injury
Acute tubular
necrosis** or acute
interstitial nephritis
↓ Kidney function
↑ Serum creatinine
& ↓ glomerular
filtration rate (GFR)
Prolonged therapy
or prior exposure
Rapid infusion, high dose or co-
administration with anesthetics
Possible direct
damage to
auditory branch of
cranial nerve VIII
(mechanism not
well understood)
Sensorineural
hearing loss,
tinnitus**,
&/or balance
disturbance
Vancomycin-
dependent
antibodies are
produced
Antibodies bind
to platelets &/or
neutrophils
Reversible
cytopenia +/-
bleeding
Platelet &/or
neutrophil
destruction
↓ Platelets &/or
↓ neutrophils
Activation of mast
cells & basophils
(non-IgE)
Self-limited
infusion reaction
(not an allergy)
Mast cell degranulation
releases histamine & other
vasoactive mediators
Erythema of
face, neck &
torso
↓ Blood
pressure
Pruritus
(itching)
Author:
Oleksandr Baran
Reviewers:
Trevor Low,
Emily J. Doucette,
Brandon Christensen*
*MD at time of publication
**See corresponding Calgary Guide slide
Published Dec 7, 2025 on www.thecalgaryguide.com
Pathophysiology Mechanism
Pharmacologic Effects Side Effects")
Diphtheria
 is
incomplete, absent, or waning
Poor hygiene
environments
Exposure to respiratory droplets or
direct contact with infected surface
Colonization of the pharynx or cutaneous sites
with corynebacterium diphtheriae (C. diphtheriae)
C. diphtheriae proliferate locally
& secrete diphtheria exotoxin
Exotoxin enters nearby host cells &
inhibits host cell protein synthesis
Local epithelial tissue necrosis &
activation of inflammatory response
Accumulation of dead
cells, fibrin, bacteria
& inflammatory cells
Lymphatic & hematogenous spread of exotoxin to distant
tissues (preferentially tissues with ↑ metabolic activity)
Author: Julia Fox
Reviewers:
Steven Quan,
Emily J. Doucette,
James D. Kellner*
*MD at time of publication
**See corresponding Calgary Guide slide
Cutaneous Diphtheria
Cutaneous skin
breakdown
Vesicle/pustule
formation
Superficial, non-healing ulcers
with grey pseudomembrane
Respiratory Diphtheria
Presence of inflammatory
mediators in the pharynx
Mucosal edema
& sensitization
of nociceptors
Dense necrotic pseudomembrane forms &
adheres to larynx, pharynx, or tonsils
Pseudomembrane dislodgment
or extension into the airway
Diphtheria antigens drain
to cervical lymph nodes
Cervical lymphadenitis
Pain & inflammatory
response at infection
site (eg. pharyngitis,
tonsillitis, etc.)
Diphtheritic membrane
(grey mucous membranes)
Localized airway obstruction
(severe laryngeal cases)
Bull neck
appearance
Systemic Manifestations
Protein synthesis inhibition
in myocardial cells
Protein synthesis inhibition in
neurons & Schwann cells
Focal myocardiocyte necrosis,
inflammation, & fibrosis
Demyelination, ↓ myelin
regeneration, & axonal injury
Myocarditis**
Damaged myocardium
disrupts conduction
pathways & contractility
Cranial nerve conduction impaired first (especially
CN IX, X, III) due to ↓ length, ↑ metabolic activity
& proximity to mucosal infection sites
ECG changes (arrhythmias
& heart block)
Palatal &
pharyngeal paralysis
Oculomotor
palsy
Dysphonia, dysphagia,
& loss of gag reflex
Protein synthesis inhibition in glomeruli
& renal tubular epithelial cells
Tubular necrosis &
interstitial inflammation
Renal failure
Endothelial injury ↑
glomerular permeability
Long peripheral nerve conduction
impaired later (2-6 weeks after
initial infection)
Ascending
muscle weakness
Paresthesia
& numbness
Acute
tubular
necrosis
Granular
casts in urine
Proteinuria
Microscopic
hematuria
Legend: Pathophysiology Mechanism
Sign/Symptom/Lab Finding Complications
Published Dec 18, 2025 on www.thecalgaryguide.com")
Necrosis versus Apoptosis

Hypoxia
(↓ Oxygen
delivered to tissues)
Physical agents
(extreme temperatures,
trauma, radiation)
Bacterial, viral, or
fungal infections
&/or microbe toxins
Necrosis
Uncontrolled & pathologic cell death
Harmful stimuli trigger ↓ ATP (cellular energy)
↓ ATP leads to ion pump failure & ↑ cell permeability (“leakiness”)
↑ Cellular ion influx causes cell to swell & lyse (rupture its plasma membrane)
Intracellular contents spill into the extracellular space
Intracellular contents activate inflammasomes (cytoplasmic multiprotein complexes)
Pro-inflammatory cytokine interleukin-1β is secreted & activates the immune system
Excessive cytokine release provokes a large inflammatory response
Inflammation damages surrounding healthy tissues & organs
Ischemia (↓ Blood
flow) to organs
except brain
↓ Oxygen denatures
enzymes à dead cell
skeletons cannot breakdown
Cells remain
“coagulated”
(firm) for days
Coagulative
necrosis
↓ Blood flow to limb(s)
+/- bacterial invasion
Gangrenous
necrosis
↑ Pain or ↓ feeling in affected limb(s)
Skin color changes (red, purple, black)
Damaged adipocytes (fat cells) release
triglyceride (fat) contents that group into oil cysts
Scars form &
calcify (harden)
Fat necrosis
Damaged endothelium (blood
vessel wall) becomes leaky
Fibrin (blood protein) deposits in
endothelium with immune complexes
Fibrinoid
necrosis
Brain ischemia &/or
infection kills cells
Digestive enzymes transform dead
tissue into creamy liquid pus
Liquefactive necrosis
Coagulative + liquefactive necrosis mechanisms à granular debris
Caseous necrosis
Legend: Pathophysiology Mechanism
Sign/Symptom/Lab Finding Complications
-----------------------------------------------------------------------------------------------------
Authors:
Tyra Fernandes
Catherine R Jarvis
Reviewers:
Mitchell Chorney
Sarah Smith*
* MD at time of publication
Apoptosis
Programmed cell death via an energy-dependent process
Extrinsic pathway: cellular stress or
damage signals in the extracellular
environment trigger apoptosis
Intrinsic pathway: internal
cellular stressors (damage from
chemotherapy, UV radiation,
drugs) trigger apoptosis
Ligands such as TNF-α (tumor
necrosis factor-alpha, produced by
macrophages) & FasL (Fas ligand,
produced by T cells) bind to apoptotic
receptors on the cell’s surface
Pro-apoptotic factors recognize
cellular stressors
Cytochrome C releases from the
intermembrane space of the
mitochondria to the cytoplasm
Ligand binding activates initiator
caspases (protease (protein
breakdown)-like enzymes) 2, 8, 9, 10
Cytochrome C activates Caspase-9
Initiator caspases activate
effector caspases 3, 6, 7
Caspase-9 activates
effector caspases 3 & 7
Effector caspases begin execution pathway
Caspases degrade the cell’s nuclear envelope & DNA
à DNA repair is inhibited & cell shrinks
Cellular shrinkage forms apoptotic bodies
Phagocytes engulf apoptotic bodies to prevent inflammation
Ineffective phagocytosis à inflammation
Published Feb 8, 2026 on www.thecalgaryguide.com
Final Slide is 1st
Necrosis vs Apoptosis: Pathogenesis and clinical findings
Toxic chemicals
(poisons, drug
toxicities)
Hypoxia
(↓ Oxygen
delivered to tissues)
Physical agents
(extreme temperatures,
trauma, radiation)
Toxin release
from bacteria,
viruses, or fungi
Necrosis
Uncontrolled & pathologic cell death
Harmful stimuli trigger ↓ ATP (cellular energy)
↓ ATP leads to ion pump failure & ↑ cell permeability
↑ Cellular ion influx causes cell to swell & lyse (rupture its plasma membrane)
Intracellular contents spill into the extracellular space
Intracellular contents activate inflammasomes (cytoplasmic multiprotein complexes)
Pro-inflammatory cytokine interleukin-1β is secreted and activates the immune system
Excessive cytokine release provokes a large inflammatory response
Inflammation damages surrounding healthy tissues & organs
Ischemia (↓ Blood
flow) to organs
except brain
↓ Oxygen denatures
enzymes à cell skeleton
cannot be broken down
Cell is preserved
in “coagulated”
state for days
Coagulative
necrosis
↓ Blood flow to limbs
+/- bacterial invasion
Gangrenous
necrosis
↑ Pain or ↓ feeling in limb
Skin color changes (red, purple, black)
Damaged adipocytes (fat cells) release
triglycerides (fat) contents that group into oil cysts
Scars form &
calcify (harden)
Fat necrosis
Damaged endothelium (blood
vessel wall) becomes leaky
Fibrin (blood protein) deposits in
endothelium with immune complexes
Fibrinoid
necrosis
Brain ischemia &/or
infection kills cells
Digestive enzymes transform dead
tissue into creamy liquid pus
Liquefactive necrosis
Coagulative & liquefactive necrosis mixture à granular debris
Caseous necrosis
Legend: Pathophysiology Mechanism
Sign/Symptom/Lab Finding -----------------------------------------------------------------------------------------------------
Complications
Authors:
Tyra Fernandes
Catherine R Jarvis
Reviewers:
Mitchell Chorney
Sarah Smith*
* MD at time of publication
Apoptosis
Programmed cell death via an energy-dependent process
Extrinsic pathway: cellular stress or
damage signals in the extracellular
environment trigger apoptosis
Intrinsic pathway: internal
cellular stressors (chemotherapy,
UV radiation, drugs, misfolded
proteins) trigger apoptosis
Ligands such as TNF-α (tumor
necrosis factor-alpha, produced by
macrophages) & FasL (Fas ligand,
produced by T cells) bind to apoptotic
receptors on the cell’s surface
Pro-apoptotic factors recognize
cellular stressors
Cytochrome C releases from the
intermembrane space of the
mitochondria to the cytoplasm
Ligand binding activates initiator
caspases (protease (protein
breakdown)-like enzymes) 2, 8, 9, 10
Cytochrome C activates Caspase-9
Initiator caspases activate
effector caspases 3, 6, 7
Caspase-9 activates
effector caspases 3 & 7
Effector caspases begin execution pathway
Caspases degrade the cell’s nuclear envelope & DNA
à DNA repair is inhibited & cell shrinks
Cellular shrinkage forms apoptotic bodies
Phagocytes engulf apoptotic bodies to prevent inflammation
Ineffective phagocytosis à inflammation
Published MONTH, DAY, YEAR on www.thecalgaryguide.com
Necrosis vs Apoptosis
Toxic chemicals
(poisons, drug
toxicities)
Hypoxia
(↓ Oxygen
delivered to tissues)
Physical agents
(extreme temperatures,
trauma, radiation)
Biological agents
(bacteria,
viruses, fungi)
Necrosis
Non-physiologic & uncontrolled cell death
Noxious (harmful) stimuli trigger increased cell permeability and ion pump failure
Cell swells & ruptures its plasma membrane à cell lysis (breakdown)
Intracellular contents spill into the extracellular space
Intracellular contents activate inflammasomes (cytoplasmic multiprotein complexes)
Pro-inflammatory cytokine interleukin-1β is secreted and activates the immune system
Excessive cytokine release provokes a large inflammatory response
Inflammation damages surrounding healthy tissues & organs
Acute tubular
necrosis
(damaged kidney
tubule cells)
Gangrene
(body tissue death)
Myocardial infarction
(heart attack)
Steatohepatitis
(progressive
liver disease)
↓ Blood flow to a
Myocardium (heart
portion of the body
muscle) is deprived
Necrotic renal
(usually limbs)
of oxygen &
(kidney) tubular
becomes damaged
epithelial cells
deteriorate &
slough off
↑ Extracellular
matrix proteins
(especially
collagen) are
produced and
deposit in liver
Granular casts
on urine
microscopy
↑ Pain
or
↓ Feeling
Skin
color
change
(red,
purple,
black)
↑ Blood
troponin
levels
↓ Heart
function
Liver fibrosis
(scarring)
Legend: Authors:
Tyra Fernandes
Catherine R Jarvis
Reviewers:
-----------------------------------------------------------------------------------------------------
Pathophysiology Mechanism
Sign/Symptom/Lab Finding Complications
Mitchell Chorney
Name Name*
* MD at time of publication
Apoptosis
Programmed cell death via an energy-dependent process
Extrinsic Pathway: Apoptotic signals
from other cells cause initiation
Intrinsic Pathway: Internal cellular
stressors (chemotherapy, UV
radiation, drugs, misfolded
proteins) cause initiation
Ligands such as TNF-α (tumor necrosis
factor-alpha, produced by
macrophages) & FasL (Fas ligand,
produced by T cells) bind to apoptotic
receptors on the cell’s surface
Pro-apoptotic factors recognize
cellular stressors
Cytochrome P releases from the
intermembrane space of the
Ligand binding activates initiator
mitochondria to the cytoplasm
caspases (protease (protein
breakdown)-like enzymes) 2, 8, 9, 10
Cytochrome P activates Caspase-9
Initiator caspases activate
effector caspases 3, 6, 7
Caspase-9 activates caspases
3 & 7
Execution Pathway
Caspases degrade the cell’s nuclear envelope & DNA
à DNA repair is inhibited & cell shrinks
Cellular shrinkage forms apoptotic bodies
Phagocytes engulf apoptotic bodies to prevent inflammation
Ineffective phagocytosis à inflammation
Published MONTH, DAY, YEAR on www.thecalgaryguide.com
Necrosis vs Apoptosis
Hypoxia
Chemical agents
Physical agents Biological agents
Necrosis
Non-physiologic & uncontrolled
cell death
Oncolysis (ion pump failure and
increased cell permeability
cause cell swelling) occurs due
to noxious stimuli
Plasma membrane ruptures and
cell is lysed (broken down)
Intracellular contents spill into
the extracellular space
Authors:
Tyra Fernandes
Reviewers:
Mitchell Chorney
Catherine R Jarvis
Name Name*
* MD at time of publication
Renal tubule is damaged due to
reduced oxygen supply and toxicity
Granular casts on urine microscopy
Acute tubular necrosis (kidney
disorder of the tubule cells
Extracellular matrix proteins accumulate
in liver causing collagen deposition
Liver fibrosis
Steatohepatitis
(progressive liver disease)
Intracellular contents activate
inflammasomes (cytoplasmic
multiprotein complexes)
Myocardium (heart muscle) is
deprived of oxygen
↑ Blood troponin levels
Myocardial infarction
-----------------------------------------------------------------------------------------------------
Apoptosis
Programmed cell death via an
energy-dependent process
Extrinsic Pathway: Apoptotic signals
from other cells cause initiation
Ligands such as TNF-α (tumor necrosis
factor-alpha, produced by
macrophages) and FasL (Fas ligand,
produced by T cells) bind to apoptotic
receptors on the cell’s surface
Ligand binding activates initiator
caspases (protease-like
enzymes) 2, 8, 9, 10
Initiator caspases activate
effector caspases 3, 6, 7
Intrinsic Pathway: Internal cellular
stressors (chemotherapy, UV
radiation, drugs, & misfolded
proteins) cause initiation
Pro-apoptotic factors recognize
cellular stressors
Cytochrome P releases from the
intermembrane space of the
mitochondria to the cytoplasm
Cytochrome P activates Caspase-9
Caspase-9 activates caspases
3 & 7
Pro-inflammatory cytokine
interleukin-1β is secreted and
activates the immune system
Excessive cytokine release
provokes a large inflammatory
response
Legend: Inflammatory
damage to the
surrounding
healthy tissues
Microscopic
Effects:
Pathophysiology Mechanism
Sign/Symptom/Lab Finding Complications
Execution Pathway
Caspases degrade the cell’s nuclear envelope. DNA
fragments inhibit DNA repair, which shrinks the cell
Cellular shrinkage forms apoptotic bodies
Phagocytes engulf apoptotic bodies to prevent inflammation
Ineffective phagocytosis à inflammation
Published MONTH, DAY, YEAR on www.thecalgaryguide.com
Necrosis vs Apoptosis
Authors:
Tyra Fernandes
Reviewers:
Mitchell Chorney
Catherine R Jarvis
Hypoxia, physical agents,
chemical agents, biologic agents
Necrosis (Non-physiologic
& uncontrolled cell death)
Oncolysis (cell swelling due to
the failure of ion pumps and
increased cell permeability)
occurs due to noxious stimuli
Plasma membranes rupture
leading to cell lysis
Intracellular contents spill into
the extracellular space
Inflammasomes (cytoplasmic
multiprotein complexes) are
activated
Pro-inflammatory cytokine
interleukin-1β is secreted and
activates the immune system
Excessive cytokine release
provokes a large inflammatory
response
Legend: Microscopic Effects:
Steatohepatitis
Collagen deposition due
to the accumulation of
extracellular matrix
proteins
Liver fibrosis
Myocardial
Infarction
Ischemic or toxic
injury to the renal
tubule
Granular casts on
urine microscopy
Acute tubular
necrosis
Oxygen supply
deprivation to the
myocardium
Elevated blood
troponin levels
-----------------------------------------------------------------------------------------------------
Extrinsic Pathway: Apoptotic Signals
from other cells cause initiation
Ligands such as TNF-α (tumor necrosis
factor-alpha, produced by
macrophages) and FasL (Fas ligand,
produced by T cells) bind to apoptotic
receptors on the cell’s surface
Ligand binding activates initiator
caspases (protease-like
enzymes) 2, 8, 9, 10
Initiator caspases activate
effector caspases 3, 6, 7
Apoptosis (Programmed
cell death via an energy-
dependent process)
Name Name*
* MD at time of publication
Intrinsic Pathway: Internal cellular
stressors chemotherapy, UV
radiation, drugs, & misfolded
proteins) cause initiation
Pro-apoptotic factors recognize
cellular stressors
Cytochrome P releases from the
intermembrane space of the
mitochondria to the cytoplasm
Cytochrome P activates Caspase-9
Caspase-9 activates caspases 3 and
7
Execution Pathway
Cellular shrinkage
forms apoptotic
bodies
Inflammatory damage to
surrounding healthy tissues à
potential organ failure
Pathophysiology Mechanism
Sign/Symptom/Lab Finding Complications
The nuclear envelope
degrades and DNA
fragments which
inhibits DNA repair
which shrinks the cell
Phagocytes engulf
apoptotic bodies to
prevent inflammation
Ineffective phagocytosis
à inflammation
Published MONTH, DAY, YEAR on www.thecalgaryguide.com
Authors:
Necrosis vs Apoptosis
Tyra Fernandes
Reviewers:
Mitchell Chorney
Necrosis Apoptosis
Catherine R Jarvis
Hypoxia: ischemia, respiratory
insufficiency
Physical agents: trauma, radiation,
Non-physiologic &
temperature, electrical shock
uncontrolled cell death
Biological agents: bacteria, viruses, fungi
Chemical agents: poisons, drugs,
Cell swelling (oncosis) due to
occupational exposures
the failure of ion pumps and
increased cell permeability
Collagen deposition leading
to liver fibrosis
Cell lysis due to the rupture of
the plasma membrane
Steatohepatitis
Microscopic
Spillage of intracellular contents
Effects:
into the extracellular space
Acute tubular
Myocardial
Activation of cytoplasmic
necrosis
Infarction
multiprotein complexes, known
as inflammasomes
Ischemic or toxic
Oxygen supply
injury to the renal
deprivation to the
tubule
myocardium
Secretion of pro-inflammatory
cytokine interleukin-1β, which
-----------------------------------------------------------------------------------------------------
Name Name*
* MD at time of publication
Programmed cell death via
an energy-dependent
process
Extrinsic pathway Intrinsic pathway
The cell receives apoptotic signals
Initiated by internal cellular
from other cells
stressors such as chemotherapy
drugs, UV radiation, and misfolded
Apoptosis is activated via ligands
proteins
binding to apoptotic receptors on
the cell surface
Pro-apoptotic factors recognize
cellular stressors
Ligand binding activates initiator
caspases (2, 8, 9, 10), which are
Cytochrome P releases from the
protease-like enzymes
intermembrane space of the
mitochondria to the cytoplasm
Examples of ligands include: TNF-α
(produced by macrophages) and
Caspase-9 is activated
FasL (produced by T cells)
Activation of downstream effector
Effector caspases (3, 6, 7) are
caspases 3 and 7
activated
activates the immune system
Excessive cytokine release
provokes a large inflammatory
response
Legend: Granular casts on
urine microscopy
Elevated blood
troponin levels
Damage to surrounding
healthy tissues may lead to
organ failure
Prevention of
inflammation
Execution pathway
The nuclear envelope
degrades and DNA fragments
which inhibits DNA repair
Cellular shrinkage
forms apoptotic
bodies
Phagocy
engulfment of
apoptotic bodies
Published MONTH, DAY, YEAR on www.thecalgaryguide.com
Pathophysiology Mechanism
Sign/Symptom/Lab Finding Complications
Necrosis versus apoptosis
Necrosis
Non-physiologic &
uncontrolled cell death due
to noxious stimuli
Cell swelling (oncosis) à cell
lysisà spillage of
intracellular contents à
tissue damage
Inflammatory response,
leading to inflammasome
activation and the secretion of
pro-inflammatory cytokine
interleukin-1β
Macroscopic
Effects:
Hypoxia: ischemia, respiratory insufficency
Physical agents: trauma, radiation,
temperature, electrical shock
Biological agents: bacteria, viruses, fungi
Chemical agents: poisons, drugs,
occupational exposures
Acute tubular
necrosis due to
chemical agents
Myocardial
infarction due to
hypoxia
Microscopic
Effects:
Stroke due to
hypoxia in the brain
Gangrene due to
hypoxia of the limbs
Steatohepatitis due
to chemical agents
-----------------------------------------------------------------------------------------------------
Apoptosis
Programmed cell death via
an energy-dependent
process
Execution phase: pro-& anti-
apoptotic proteins mediate
cell breakdown by caspases
Caspase activity blocks DNA
repair à leading to
fragmented DNA and cell
shrinkage
Apoptosis is
inhibited in cancer
the overexpression
of anti-apoptotic
proteins
Authors:
Tyra Fernandes
Reviewers:
Mitchell Chorney
Name Name*
* MD at time of publication
Extrinsic pathway: immune cell activation
activates executioner caspases; ligands such
as TNF-α & FasL bind to receptors on the cell
membrane
Intrinsic pathway: internal cellular factors
cause pro-apoptotic factors to increase
mitochondrial membrane permeability.
Cytochrome c is released from the inner
mitochondrial membrane, which ultimately
activates caspases
Cellular fragments are
engulfed by immune cells
which prevents an
inflammatory response
Organism death via
necrosis
Menstruation Embryogenesis
Endometrial
shedding of the
inner lining of
the uterus is an
apoptotic
mechanism
The tissue
between the
fingers and toes
degrades via
apoptosis in
utero à
separation of
the digits
Legend: Pathophysiology Mechanism
Sign/Symptom/Lab Finding Abbreviations:
• IL-1β– interleukin-1β
• TNF-α– tumour necrosis factor - alpha
• FasL– CD95 ligand; mediates apoptosis
Published MONTH, DAY, YEAR on www.thecalgaryguide.com
Complications
References
• https://next.amboss.com/us/article/VP0GdT?q=apoptosis#Y0d79977cce8fc2c548ac720f8060d667
• https://www.ncbi.nlm.nih.gov/books/NBK557627/
• https://www.ncbi.nlm.nih.gov/books/NBK499821/
• https://pmc.ncbi.nlm.nih.gov/articles/PMC5855670/
• https://www.sciencedirect.com/topics/pharmacology-toxicology-and-pharmaceutical-science/oncosis
• https://www.nature.com/articles/s41421-020-0167-x
• https://pmc.ncbi.nlm.nih.gov/articles/PMC3714593/
• https://www.ncbi.nlm.nih.gov/books/NBK537076/
Hypoxia
Physical agents
Biological agents
Chemical agents")
Neuroleptic Malignant Syndrome
: Pathogenesis and clinical findings
Initiation or ↑ dose of dopamine receptor (D2)
antagonist medication (typical antipsychotics more
common than atypical; antiemetics eg. metoclopramide)
Medication blocks dopamine D2
receptors in the central nervous system
Genetic risk factors including DRD2
A1 allele (TaqI A1 polymorphisms)
& CYP2D6 deficiency
Sudden ↓ in central dopamine activity
Rapid withdrawal of dopaminergic
medications (eg. Levodopa)
Abrupt withdrawal of D2 receptor
stimulation in the central nervous system
Depolarization blockade of the nigrostriatal pathway
& basal ganglia (mechanism not fully understood)
Disinhibition of striatal indirect pathway Hyperactive subthalamic nuclei drives
excitation of globus pallidus internus (GPi)
Overactive GPi strongly inhibits
thalamus & ↓ cortical motor output
↓ Cortical motor output produces
sustained muscle contraction
↑Peripheral calcium release from
myocyte sarcoplasmic reticulum
Continuous depolarization & sustained contraction
↑ Metabolic demand & heat production
Dopamine depletion in mesolimbic
& neocortical pathways
(mechanism not fully understood)
Altered mental status
(e.g. delirium)
Hypothalamic dysregulation & autonomic
instability (mechanism not fully understood)
Disinhibition of posterior hypothalamus
sympathetic premotor neurons
Dysregulated thermogenesis &
impaired cooling mechanisms
in the anterior pre-optic area
Strong neuronal excitation in the
paraventricular nucleus amplifies signals
along descending autonomic tracts
Persistent sympathetic tone &
suppressed parasympathetic tone
Diaphoresis Tachycardia
Fluctuating blood
pressure
Severe “lead-pipe”
muscle rigidity
↑ Serum
creatine kinase
Myoglobinuria
Hyperthermia
(fever ≥40◦C)
Acute renal failure
Prolonged illness promotes skeletal muscle damage & breakdown, immobilization, & dehydration
Rhabdomyolysis
Author:
Amena Thraya
Reviewers:
Emily J. Doucette,
Rupang Pandya*
* MD at time of publication
Legend: Pathophysiology Mechanism
Published Feb 24, 2026 on www.thecalgaryguide.com
Sign/Symptom/Lab Finding Complications")
Central Adrenal Insufficiency
 produced in the
hypothalamus
Exogenous
steroids &
narcotics suppress
the pituitary
Tumor or
inflammation
infiltrate the
pituitary
Surgery,
radiation, or
trauma injure
the pituitary
↓ Adrenocorticotropic
hormone (ACTH) produced in
the pituitary
Adrenal zona reticularis
↓ androgen (sex
hormone) production
↓ ACTH causes ↓
melanocyte-
stimulating
hormone expressed
on the same gene
↓ Dehydroepiandrosterone
as ACTH
(DHEAS; a testosterone
precursor)
Aldosterone
produced in the
adrenal zona
glomerulosa is
unaffected as it is
predominantly
controlled by the
renin-angiotensin-
aldosterone system
Adrenal zona
fasciculata ↓
cortisol production
Adrenal crisis**
↓ Testosterone levels in
women (where adrenals
are the primary source)
Melanocytes ↓
melanin (pigment)
production
Skin
hypopigmentation
No change in
aldosterone levels
↓ Libido
(sex drive)
↓ Pubic &
axillary
(armpit)
hair
growth
**See corresponding Calgary Guide slide
Legend: ↓ Cortisol-mediated inhibition
leads to ↑ anti-diuretic
hormone (ADH) secretion
↓ Counter-regulatory hormone
response to ↓ blood sugar
↓ Cortisol-mediated lymphocyte
apoptosis (cell death)
↓ Cortisol-mediated
vasoconstriction ↑ vasodilation Adrenal medulla ↓
catecholamine (epinephrine &
norepinephrine) production
Multifactorial mechanisms
Author:
Jaye Platnich
Samantha McIntosh
Reviewers:
Mark Elliott
Matthew Harding
Alexander Arnold
Catherine R Jarvis
Hanan Bassyouni*
David Campbell*
* MD at time of publication
Hyponatremia
(↓ Serum sodium)
Hypoglycemia
(↓ Glucose)
Lymphocytosis
(↑ Lymphocytes)
↓ Blood pressure
Postural dizziness
Fatigue
Nausea
Vomiting
Weight loss
Musculoskeletal pain
Anorexia (↓ Appetite)
Pathophysiology Mechanism
Sign/Symptom/Lab Finding Complications
Published Oct 21, 2013; updated Mar 21, 2026 on www.thecalgaryguide.com")