Beta Thalassemia Minor: Pathogenesis and clinical findings
Authors: Andrew Brack Yan Yu Huneza Nadeem Reviewers: Wendy Yao Katie Lin Liana Martel Ran (Marissa) Zhang Man-Chiu Poon* Lynn Savoie* * MD at time of publication
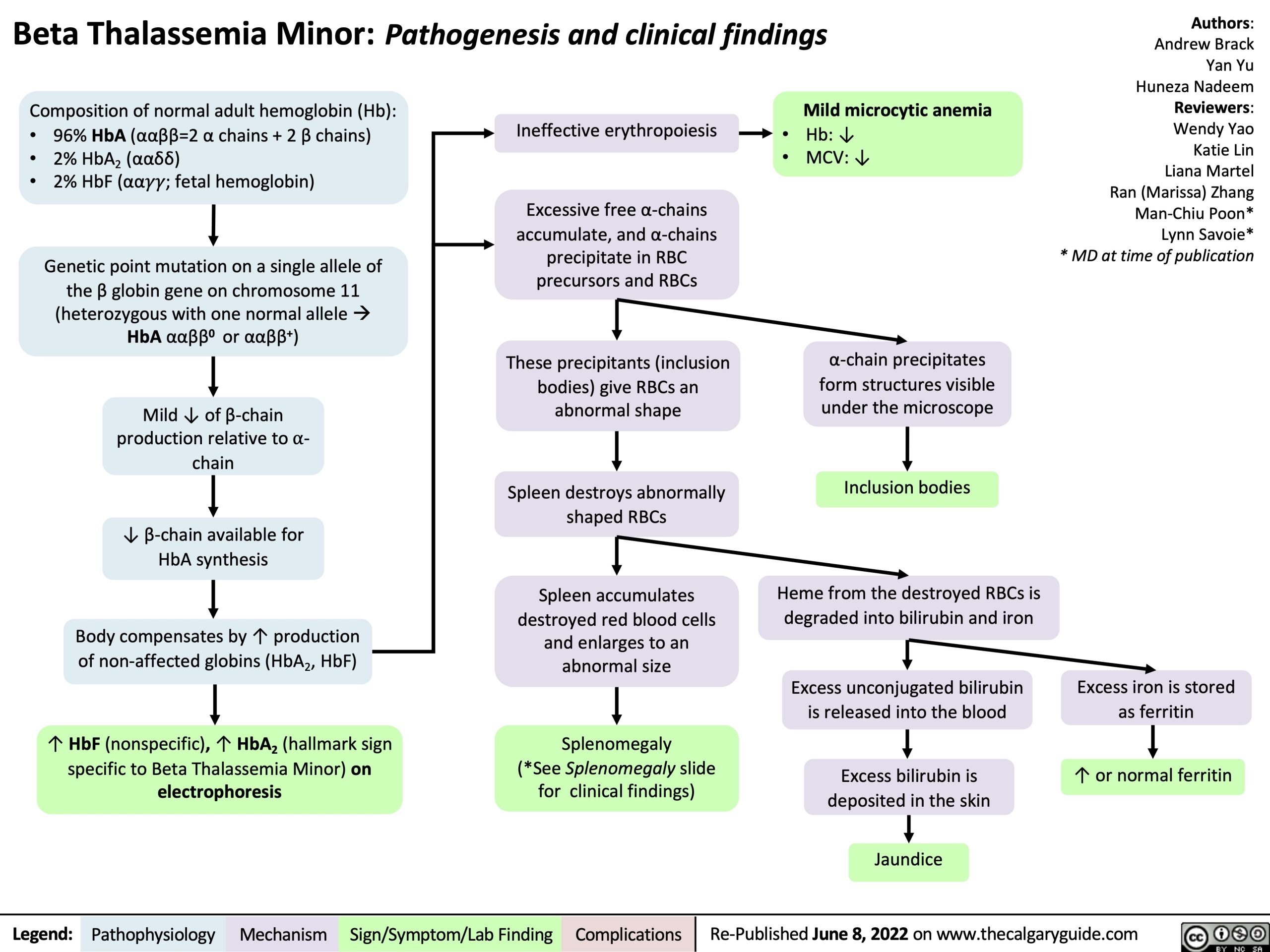
Composition of normal adult hemoglobin (Hb):
Ineffective erythropoiesis
Excessive free α-chains accumulate, and α-chains precipitate in RBC precursors and RBCs
These precipitants (inclusion bodies) give RBCs an abnormal shape
Spleen destroys abnormally shaped RBCs
Spleen accumulates destroyed red blood cells and enlarges to an abnormal size
Splenomegaly (*See Splenomegaly slide for clinical findings)
Mild microcytic anemia
Hb: ↓ MCV: ↓
• • •
96% HbA (ααββ=2 α chains + 2 β chains) 2% HbA2 (ααẟẟ)
2% HbF (αα!!; fetal hemoglobin)
Genetic point mutation on a single allele of the β globin gene on chromosome 11 (heterozygous with one normal alleleà HbA ααββ0 or ααββ+)
Mild ↓ of β-chain production relative to ⍺- chain
↓ β-chain available for HbA synthesis
Body compensates by ↑ production
of non-affected globins (HbA , HbF) 2
↑ HbF (nonspecific), ↑ HbA2 (hallmark sign specific to Beta Thalassemia Minor) on electrophoresis
• •
α-chain precipitates form structures visible under the microscope
Inclusion bodies
Heme from the destroyed RBCs is degraded into bilirubin and iron
Excess unconjugated bilirubin is released into the blood
Excess bilirubin is deposited in the skin
Jaundice
Excess iron is stored as ferritin
↑ or normal ferritin
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Re-Published June 8, 2022 on www.thecalgaryguide.com