α1AT Deficiency: Pathophysiology and Clinical Findings
Authors: Sean Spence Reviewers: Danny Guo Yan Yu Merna Adly Crystal Liu Sam Lee* * MD at time of publication
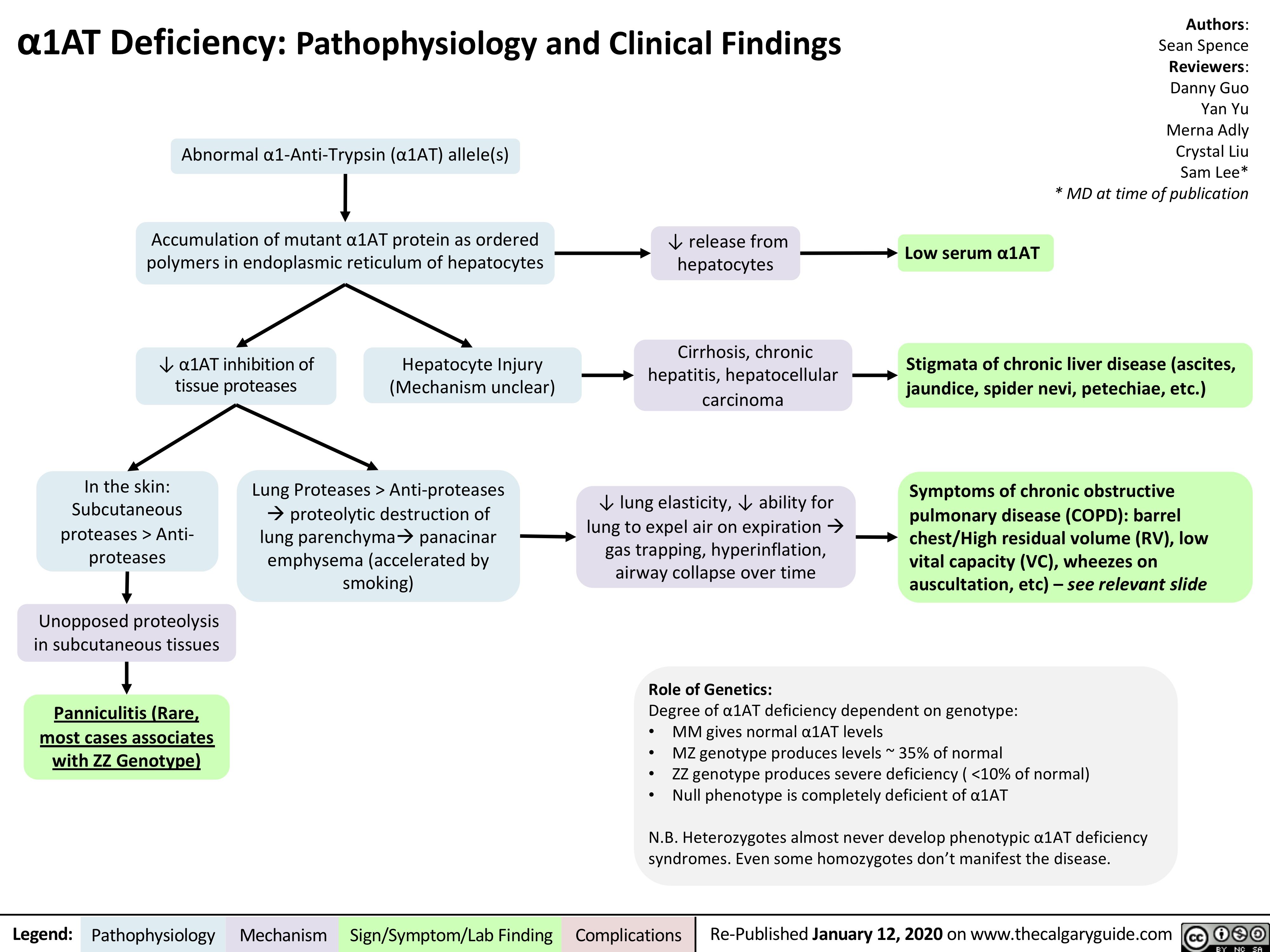
Abnormal α1-Anti-Trypsin (α1AT) allele(s)
Accumulation of mutant α1AT protein as ordered polymers in endoplasmic reticulum of hepatocytes
↓ α1AT inhibition of Hepatocyte Injury tissue proteases (Mechanism unclear)
↓ release from hepatocytes
Cirrhosis, chronic hepatitis, hepatocellular carcinoma
↓ lung elasticity, ↓ ability for lung to expel air on expirationà gas trapping, hyperinflation, airway collapse over time
Role of Genetics:
Low serum α1AT
In the skin: Subcutaneous proteases > Anti- proteases
Unopposed proteolysis in subcutaneous tissues
Panniculitis (Rare, most cases associates with ZZ Genotype)
Lung Proteases > Anti-proteases àproteolytic destruction of lung parenchymaàpanacinar emphysema (accelerated by smoking)
Stigmata of chronic liver disease (ascites, jaundice, spider nevi, petechiae, etc.)
Symptoms of chronic obstructive pulmonary disease (COPD): barrel chest/High residual volume (RV), low vital capacity (VC), wheezes on auscultation, etc) – see relevant slide
Degree of α1AT deficiency dependent on genotype:
• MM gives normal α1AT levels
• MZ genotype produces levels ~ 35% of normal
• ZZ genotype produces severe deficiency ( <10% of normal)
• Null phenotype is completely deficient of α1AT
N.B. Heterozygotes almost never develop phenotypic α1AT deficiency syndromes. Even some homozygotes don’t manifest the disease.
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Re-Published January 12, 2020 on www.thecalgaryguide.com