SEARCH RESULTS FOR: shock
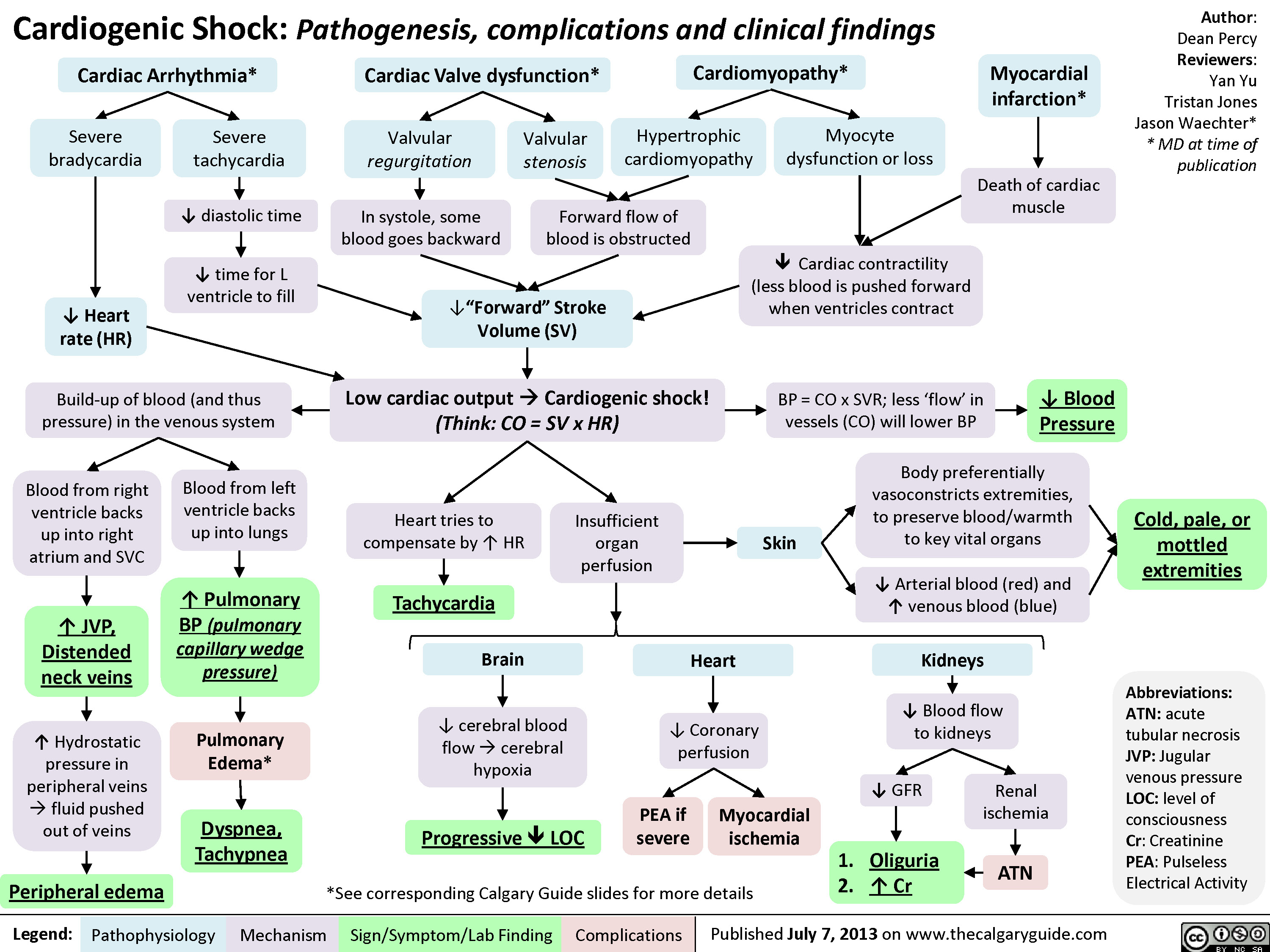
Cardiogenic Shock
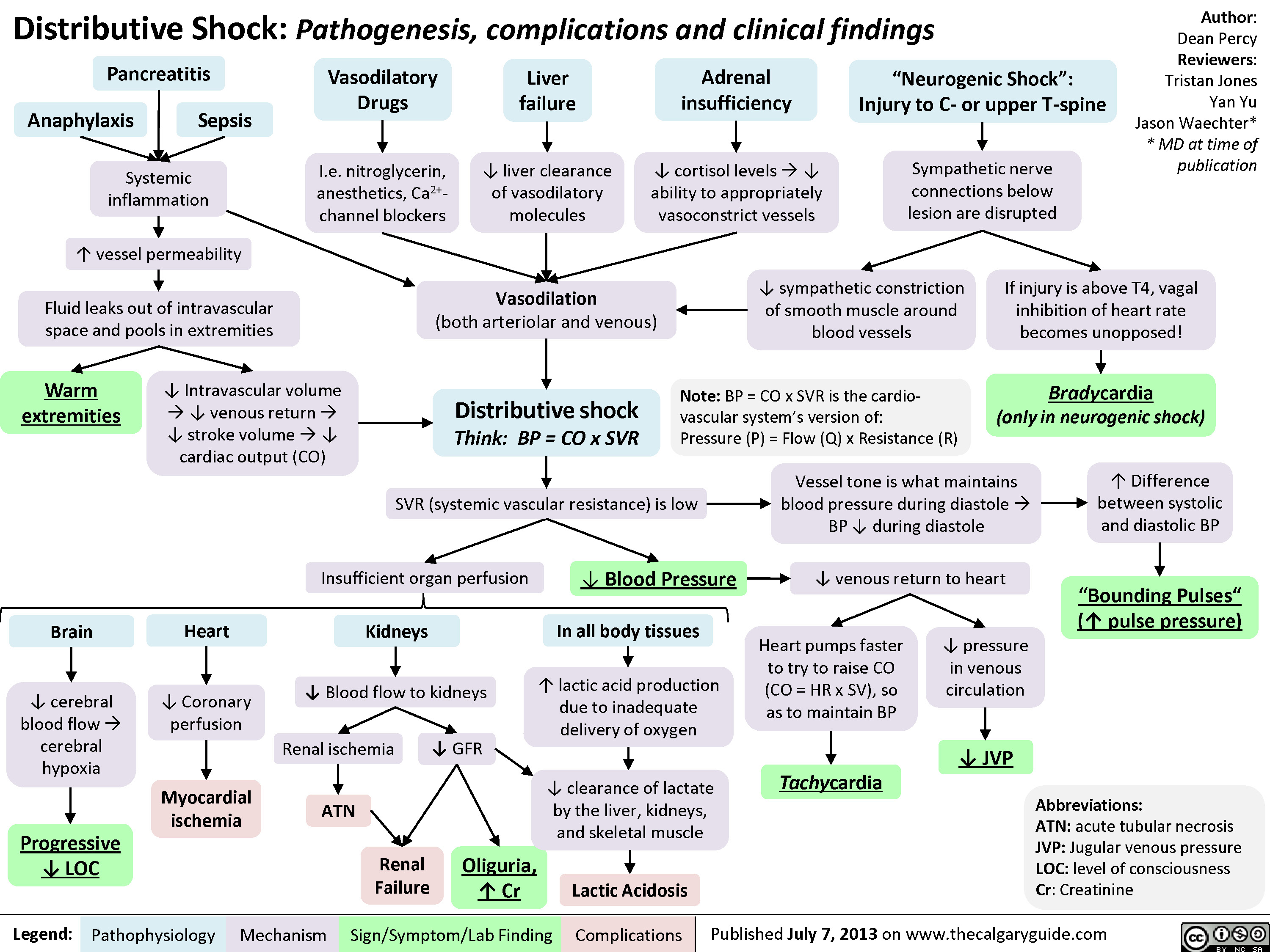
Distributive Shock
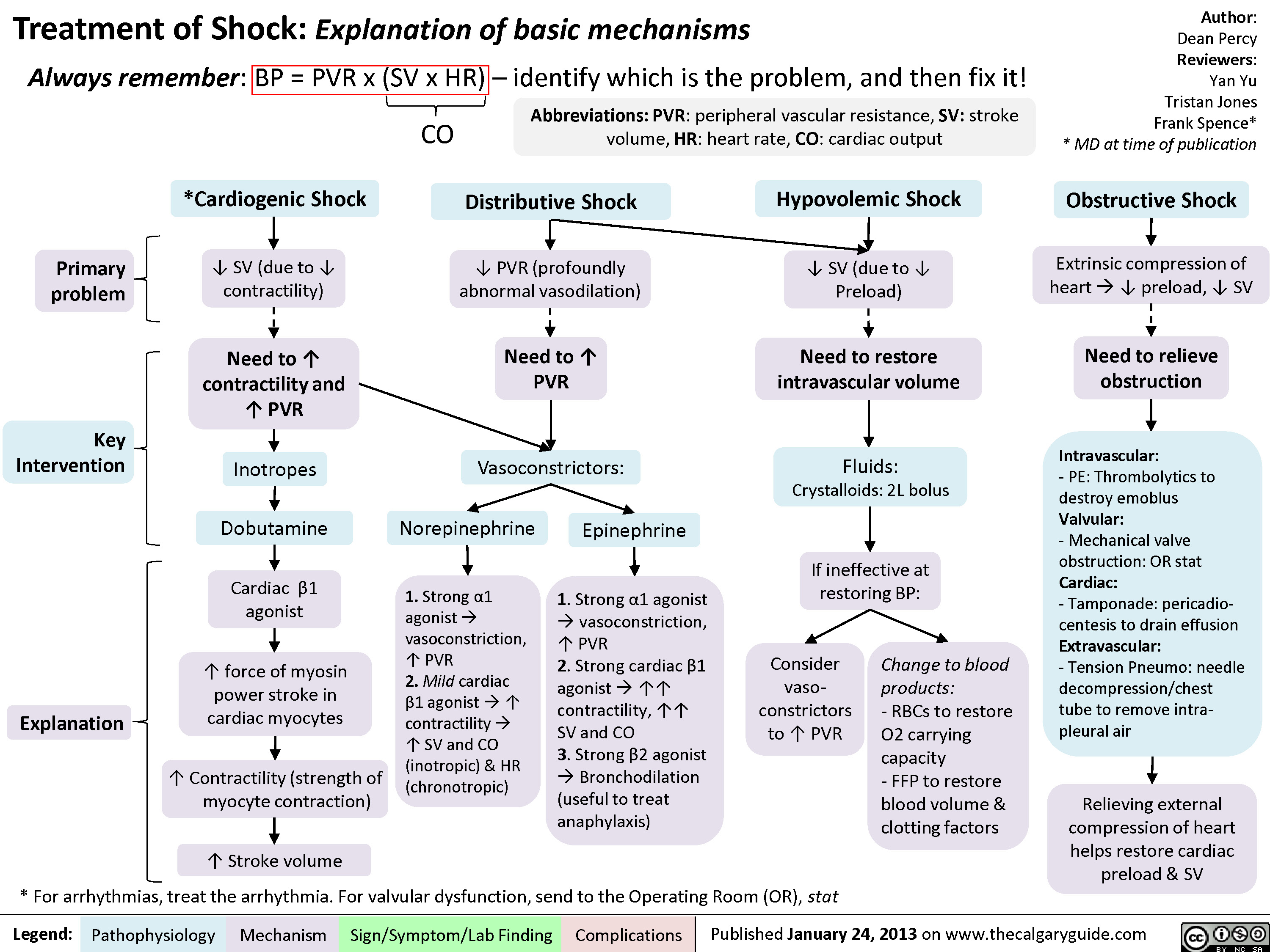
Drugs used to treat shock
Pediatric Uncompensated Shock: pathogenesis and clinical findings

Hypernatremia Physiology
![Hypernatremia: Physiology Unreplaced H2O loss
Hypodipsia
H2O shift into cells
Severe exercise, electroshock induced seizures
Transient ↑ cell osmolality
Na+ overload
Inappropriate IV hypertonic solution, salt poisoning
Abbreviations:
H2O: Water
GI: Gastrointestinal
DM: Diabetes Mellitus
DI: Diabetes Insipidus
Na+: Sodium ion
IV: Intravenous
ADH: Antidiuretic Hormone LOC: Level of Consciousness
Skin
Sweat, burns
GI
Vomiting, bleeding, osmotic diarrhea
Fluid [Na+] < serum [Na+]
↑ H2O loss compared to Na+ loss
Renal
DM, Mannitol, Diuretics
Absent thirst mechanism
Hypothalamic lesion impairs normal drive for H2O intake
Nephrogenic
↑ renal resistance to ADH
H2O Deprivation Test + no AVP response
↓ access to H2O
DI
Central
↓ ADH secretion
H2O Deprivation Test + AVP response
↑ [Na+] 10- 15 mEq/L within a few minutes
Weakness, irritability, seizures, coma
↑ thirst, ↓ urinary frequency and volume
Note:
Hypernatremia
Serum [Na+] > 145 mmol/L
Intracranial hemorrhage
Headache, vomiting, ↓ LOC
• Plasma [Na+] is regulated by water intake/excretion, not by changes in [Na+].
• Effects on plasma [Na+] of IV fluids or loss of bodily fluids is determined by the tonicity of the fluid, not the osmolality.
Authors: Mannat Dhillon Reviewers: Andrea Kuczynski Kevin McLaughlin* * MD at time of publication
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published January 11, 2019 on www.thecalgaryguide.com](http://calgaryguide.ucalgary.ca/wp-content/uploads/2019/01/Hypernatremia-Physiology-.jpg "Hypernatremia: Physiology Unreplaced H2O loss
Hypodipsia
H2O shift into cells
Severe exercise, electroshock induced seizures
Transient ↑ cell osmolality
Na+ overload
Inappropriate IV hypertonic solution, salt poisoning
Abbreviations:
H2O: Water
GI: Gastrointestinal
DM: Diabetes Mellitus
DI: Diabetes Insipidus
Na+: Sodium ion
IV: Intravenous
ADH: Antidiuretic Hormone LOC: Level of Consciousness
Skin
Sweat, burns
GI
Vomiting, bleeding, osmotic diarrhea
Fluid [Na+] < serum [Na+]
↑ H2O loss compared to Na+ loss
Renal
DM, Mannitol, Diuretics
Absent thirst mechanism
Hypothalamic lesion impairs normal drive for H2O intake
Nephrogenic
↑ renal resistance to ADH
H2O Deprivation Test + no AVP response
↓ access to H2O
DI
Central
↓ ADH secretion
H2O Deprivation Test + AVP response
↑ [Na+] 10- 15 mEq/L within a few minutes
Weakness, irritability, seizures, coma
↑ thirst, ↓ urinary frequency and volume
Note:
Hypernatremia
Serum [Na+] > 145 mmol/L
Intracranial hemorrhage
Headache, vomiting, ↓ LOC
• Plasma [Na+] is regulated by water intake/excretion, not by changes in [Na+].
• Effects on plasma [Na+] of IV fluids or loss of bodily fluids is determined by the tonicity of the fluid, not the osmolality.
Authors: Mannat Dhillon Reviewers: Andrea Kuczynski Kevin McLaughlin* * MD at time of publication
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published January 11, 2019 on www.thecalgaryguide.com")
Takotsubo Cardiomyopathy- Pathogenesis and clinical findings

Sepsis, and Septic Shock- Pathogenesis and Clinical Findings

Comorbidities
Immunosuppression or ↑ susceptibility (e.g. splenectomy)
Pathogen virulence
Invasion and host immune avoidance
Vulnerable infection site
↑ likelihood of spread of infection & mortality
Genetics
↑ Sensitivity of innate immune response
Community acquired
Hospital acquired
Infection of host
Innate immune response
Fever, Leukocytosis/ Leukopenia, Left Shift/ Bandemia
Compensatory response
Tachypnea, Altered Level of Consciousness, Hypotension
↓ perfusion and oxygen delivery to organs
Dysregulated Host Response
Pro- and anti-inflammatory response
Life-threatening organ dysfunction caused by a dysregulated host response to infection
The Sequential Organ Failure Assessment (SOFA) or quick
SOFA (qSOFA) Scores may be used to assess mortality risk
Respiratory
↓ PaO2 / FiO2 (mmHg)
Nervous
↓ Level of consciousness
Septic Shock
Cardiovascular
↓ Mean Arterial Pressure
Organ Dysfunction
Liver
↑ Bilirubin
Kidney
↑ Creatinine, Acute oliguria
Coagulation
Thrombocytopenia, ↑ INR or aPTT
Require vasopressors to ↑ mean arterial pressure
Persistent hypotension despite adequate fluid resuscitation
↓ Mean Arterial Pressure (< 65 mmHg), ↑ Lactate (> 2 mmol/L)
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published February 12, 2019 on www.thecalgaryguide.com")
Ischemic Colitis
 supply blood to colon
Surgical repair of aorta
Borders of SMA and IMA collaterals at the splenic flexure and rectosigmoid junction are vulnerable to ischemia (“watershed” areas)
Atherosclerosis and narrowing of mesenteric arteries
Low flow state
(e.g., CHF, hypotension, arrhythmia)
Underlying CAD/PVD
Atrial fibrillation, endocarditis
Embolic arterial occlusion of SMA and/or IMA
Trauma, infection, clotting abnormalities
Mesenteric vein thrombosis
Vascular risk factors (e.g., smoking, hypertension)
Thrombotic arterial occlusion of SMA and/or IMA
Endograft coverage of IMA
Nonocclusive hypoperfusion
Inadequate blood flow to meet the cellular metabolic needs in the colon
Ischemic Colitis
Tachypnea Tachycardia Hyperthermia Hypotension
Ischemic period
Loss of oxygen and nutrients to bowel
Reperfusion period
Influx of O2àreacts to produce more oxygen free radicals
Lipid peroxidation
Systemic inflammatory response syndrome*
Nausea and vomiting
Abdominal pain (generally left sided)
Peritonitis
Leukocytosis
Systemic shock
(inadequate perfusion to tissue)
Author: Audrey Caron Michael Blomfield Reviewers: Tony Gu Yan Yu* Edwin Cheng* * MD at time of publication
Systemic shock
(inadequate perfusion to body tissue)
Hematochezia (Bloody stool)
Gangrene (tissue death)
Hemorrhage
Tissue damage/cell death (starting from mucosa and submucosa going outwards to serosa)
Mucosal ulceration
Colonic inflammation
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published November 10, 2019 on www.thecalgaryguide.com")
Hereditary Hemorrhagic Telangiectasia (Osler-Weber-Rendu disease)
:
Pathogenesis and Clinical Findings
Inherited or de novo mutation in the ACVRL1, ENG, or Smad4 genes
Abnormal signalling within the transforming growth factor ß (TGF-ß) pathway
Unclear mechanismsàInability of vascular mural cells to stabilize and remodel newly formed blood vessels
Excessive proliferation of endothelial cells and ensuing overgrowth of blood vessels
Authors: Tony Gu Reviewers: Brian Rankin Yan Yu* Laurie Parsons* * MD at time of publication
Formation of friable telangiectasias
(small dilated vessels apparent near the surface of skin or mucous membranes)
Formation of Arteriovenous malformations (AVMs):
Direct connection between arteries and veins without intervening capillary bed
Nasal telangiectasias
Epistaxis (nosebleeds)
Gastrointestinal telangiectasias
Gastrointestinal bleeding
Mucocutaneous telangiectasias
Cerebral AVMs
Hepatic AVMs
Left to right shunting of blood
Heart works harder to perfuse tissues
Heart failure
Pulmonary AVMs
Rupture
High flow left to right shunting of blood (the steal effect)
Cerebral ischemia
No oxygenation at capillaries
Hypoxemia
↑ erythropoietin production
Secondary polycythemia
No filtering from capillaries
Hemorrhage, shock, death
Venous emboli enter arteries (paradoxical embolism)
Stroke
Venous bacteria enter arteries
Cerebral abscess
Iron deficiency anemia
↓ serum iron is associated with ↑ coagulation factor VIII levels (mechanism unclear)
Venous thromboembolism
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published July 28, 2020 on www.thecalgaryguide.com")
acute-pancreatitis-complications
:
Interstitial edematous pancreatitis
Local accumulation of fluid in the pancreas
<2 weeks after onset
Acute peripancreatic fluid collection (not encapsulated)
Walled off by fibrous & granulation tissue
>2 weeks after onset
Pancreatic pseudocyst
(completely encapsulated)
Peritoneal irritation à pain
Large cyst can (very rarely) compress surrounding bowel
Acute Pancreatitis
Inflammatory cytokines are released from damaged pancreas
If recurrentàchronic pancreatitis (see relevant slide)
Inflammation damages pancreatic exocrine
cellsàInappropriate release of pancreatic enzymes into surrounding tissue & vasculature àdigesting pancreatic parenchyma
Authors: Nissi Wei, *Yan Yu Reviewers: Dean Percy, Miles Mannas, Varun Suresh, Brandon Hisey, *Kerri Novak, *Sylvain Coderre * MD at time of publication
complete resolution (most cases)
Necrotic tissue is vulnerable to
infection (esp. Gram neg GI bacteria)
inflammation & necrosis activate cytokine cascade
Severe, necrosis (15%): Necrotizing pancreatitis
Local infection
Severe pancreatic inflammation shifts body fluid into retroperitoneal spaceàintravascular volume depletion
Systemic Inflammatory Response Syndrome (SIRS) (see relevant slide)
Organ failure (may be sole feature on presentation)
Stagnant fluid can more easily become infected
Infection spreads to bloodstream
Cardiac failure Hypovolemic shock Renal failure
Local accumulation of fluid & necrosis in the pancreas
< 4 weeks after onset:
Acute necrotic collection (not encapsulated)
Walled off by fibrous & granulation tissue
> 4 weeks after onset
walled-off necrosis
(completely encapsulated)
When treated with excess fluid resuscitation:
Intra- abdominal hypertension
Respiratory failure (ARDS)
Disseminated intravascular coagulation (DIC)
Bowel obstruction Gastric outlet (see relevant slide) obstruction
Infected pancreatic necrosis
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published September 20, 2016, updated September 7, 2020 on www.thecalgaryguide.com")
Beta-Blockers-Mechanism-of-Action-and-Side-Effects
 to these receptors, ↓ their normal adrenergic tone
Beta-2 receptor antagonism Beta-1 receptor antagonism
Lungs Eyes Central nervous system Heart Kidneys ↓ cAMP (intracellular messenger) productionàcomplex, tissue-specific intracellular mechanisms resulting in a variety of effects in different tissues:
Throughout body tissue
Epinephrine (via cAMP) indirectly ↑ the activity of the Na+/K+ pump on cell membranes (a pump that moves 3 Na+ out of cells per 2 K+ moved into cells)
Blocking epinephrine from binding
the beta-2 receptor and producing cAMPà↓ activity of Na+/K+ pump à↓K+ moved into cells
↑ proportion of K+ now resides in extracellular fluid, detectable in serum (total body K+ remains the same)
Hyperkalemia (see Calgary Guide: Hyperkalemia – Clinical findings)
Blocking sympathetic hormonesà↓ relaxation of smooth muscle circumferentially wrapped around airways
↑ resting airway muscle toneà bronchoconstriction
↑ resistance to airflow
Wheezing, dyspnea, chest tightness
Exacerbation of underlying airway disease (e.g. asthma)
↓ ciliary epithelium’s production of aqueous humor (fluid that fills anterior chamber of the eye)
Reduced intraocular pressure
Blocking adrenergic response mediated by epinephrine and norepinephrine (e.g. the physiologic “fight- or-flight” response to stress)
↓ tremor, irritability, anxiety
↓ ability to produce adrenergic symptoms in response to hypoglycemia
Hypoglycemia unawareness
Coronary perfusion pressure = diastolic blood pressure in aorta – LV end diastolic pressure
↓ inotropy (contractility of cardiac muscle)
↓ chronotropy (heart rate and conduction velocity)
↓ renin releaseà↓ creation of angiotensin II & aldosterone
+ ↓ reabsorption of Na
and H2O in nephron
↑ urinary Na+ & H2O loss
↓ total blood volume
Decompensation of acute heart failure
Dizziness and fatigue Hypotension (Blood pressure = cardiac
output x systemic vascular resistance)
↓ O2 demand of myocardial tissue
Bradycardia
Inability to ↑ heart rate in response to stress (e.g. shock, sepsis)
↓ stroke volume
↓ cardiac output
Beta blockers ↓ diastolic blood pressure, & thus may ↓ coronary perfusion pressure
Before giving beta blockers, ensure blood pressure isn’t too low
Otherwise, may worsen acute myocardial ischemia
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published Jan 14, 2021, updated Feb 7, 2021 on www.thecalgaryguide.com")
AAA-Clinical-Findings-and-Complications
: Clinical Findings and Complications AAA = Abnormal, irreversible dilation of a focal area of abdominal aorta (area of aorta between
diaphragm & aortic bifurcation) to twice the diameter of adjacent normal artery segments
Asymptomatic, non-ruptured aneurysm:
Most AAAs are asymptomatic until rupture or days before impending rupture
Asymptomatic AAAs are only detectable on imaging or by palpation
Given their structural weakness, AAAs are at risk of rupture (risk ↑ with ↑ size of aneurysm)
Ruptured AAA: a medical emergency
Aorta lies in between the peritoneal and retroperitoneal space
Symptomatic, non-ruptured aneurysm: rarely, an unruptured AAA can cause symptoms or complications (0.1%-1% of AAAs)
Authors: Olivia Genereux, Davis Maclean, Yan Yu* Reviewers: Jason Waechter*, Amy Bromley*, Sandeep Aggarwal*, Gregory Samis* *MD at time of publication
Posterior aortic wall rupture
Retroperitoneal hemorrhage
Sudden and severe abdominal and/or back pain
Anterior aortic wall rupture
Peritoneal hemorrhage
Peritoneal space is larger and holds larger volumes of blood
Peritoneal hemorrhages are large (entire blood volume can pool in the peritoneal space in minutes)
Prior to rupture, the adventitia (thin outermost collagenous layer of the aorta) may dilate significantly
Adventitia is the only layer of the aorta that contains sensory innervation à nociceptors there can be activated by adventitia dilation
Very rarely (0.1%), areas of stagnant blood flow within the AAA allow for blood & clotting factors to accumulate
Thrombi (blood clots) develop in these aneurysms
Thrombi may dislodge and travel (embolize) to distal vasculature, cutting off blood- flow to these areas
This process (referred to as tamponade) is crucial in preventing catastrophic blood loss, allowing for a window of opportunity for treatment
Compensated Hypovolemic Shock: Low blood pressure &
poor organ perfusion, but blood loss has temporarily stopped. This state of shock is compatible with life if patient is otherwise healthy (e.g. no coronary artery disease)
↓ space for blood to accumulate in the retroperitoneal space (compared to the peritoneal space)
Rapid pressure ↑ in retroperitoneal space overcomes the pressure in aorta
Since blood will only travel from areas of ↑ pressure to areas of ↓ pressure, this pressure gradient prevents further blood loss from ruptured aorta
Massive hemorrhage due to high blood flow volume through aorta
Hypotension and rapid progression to hypovolemic shock
Abdominal and/or àorgan ischemia
back pain
The most common symptom of a symptomatic non- ruptured aneurysm, and may be the first sign of an impending aneurysm rupture
Death within minutes
(rare)
If low blood pressure is now (inappropriately) treated with fluid resuscitation, the blood pressure will ↑
Differential pressure gradient is reversed (aortic blood pressure > pressure in retroperitoneal space)
Bleeding into retroperitoneal space resumes
Decompensation of hypovolemic shock Possible death
Kidney ischemia
Lower limb ischemia
Time exists to transfer patient to tertiary care center for surgical treatment
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Re-Published February 28, 2021 on www.thecalgaryguide.com")
Fat-Embolism-Syndrome

Non-trauma related (rare)
Long bone fracture
Pelvic fracture
Orthopedic Trauma
Intraosseous access
Soft tissue injuries
Chest compressions
Bone marrow transplant
Pancreatitis
Diabetes mellitus
Fat from bone marrow is disrupted and leaks into bloodstream via damaged blood vessels
Fat globules obstruct dermal capillaries
Capillaries rupture
Blood leaks into the skin
Petechial rash
Non-Orthopedic Trauma (less common)
Fat from injured adipose tissue is released from adipocytes into bloodstream
Metabolic disturbance mobilizes stored fat and moves it into circulation
Fat Embolism Syndrome
the presence of fat globules in circulation
Fat globules damage blood vessel walls
Platelets stick to damaged areas Platelet aggregation
↑ circulating free fatty acids
↑ inflammatory cytokines (TNF, IL1, IL6)
↑ serum C Reactive Protein (an acute phase reactant)
C reactive protein binds to lipid vesicles in circulation
↑ formation of lipid complexes in the blood
Obstruction of cerebral vasculature
↓ blood flow and oxygen delivery to brain tissue
Neurological findings: ranging from ↓ level of consciousness to seizures
Notes:
Large quantities of fat globules can obstruct pulmonary vasculature
Blood clots form throughout the body
Disseminated intravascular coagulopathy
Back up of blood into right heart àRight ventricular dysfunction
↓ pulmonary arterial blood flow à↓ gas exchange in the lungs
Higher CO2 & lower O2 levels in blood àdetected by chemoreceptors
Chemoreceptors stimulate respiratory centre in the brain to ↑ rate of respiration
Dyspnea / Tachypnea
Authors: Tabitha Hawes Reviewers: Hannah Koury, Alyssa Federico, Davis MacLean, Mehul Gupta, Yan Yu*, Jeremy Lamothe* * MD at time of publication
• Underlined findings indicate classic triad of symptoms (petechial rash, neurologic findings, dyspnea/tachypnea)
• Clinical presentation of fat embolism syndrome is variable and may present with any or all of these findings
↓ pumping of blood into systemic circulation
Hypotension Obstructive shock
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published July 19, 2021 on www.thecalgaryguide.com")
covid-19-pathophysiology-and-clinical-findings
: Pathophysiology and Clinical Findings
Authors: Ryan Brenneis, Yan Yu* Reviewers: Ciara Hanly, Yonglin Mai ()*, Stephen Vaughan* * MD at time of publication
-Respiratory failure -Septic shock -Multiple organ dysfunction
Critical
Respiratory droplet production (cough, sneeze, talking, breathing) by human host or animal vector infected with SARS-CoV-2
Note: Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2) is the name of the betacoronavirus (a positive sense, single stranded RNA virus). COVID-19 is the name of the disease caused by this virus.
rate, ↓ oxygen saturation -Bilateral interstitial infiltrates on Chest X-ray, progressively worsening
Small droplets (<5μm in diameter) are aerosolized and become airborne
Inhalation of aerosolized particles that are suspended in air
Patient exposed to the virus (SARS-CoV-2)
Droplet contacts the mucus membranes (eyes, nose, mouth) of recipient
Live viral particles can adhere to inanimate objects called fomites (e.g. doorknobs)
Symptoms are on a continuum -Worsening dyspnea: ↑ respiratory
-Fever
-Cough
-Myalgia, fatigue -Nausea/vomiting -Diarrhea
-Loss of taste/smell
Recipient touching infected fomite subsequently touches any of their mucus membranes
Mild
Moderate
Severe
Virus spreads in the body via 1) mucus membrane spread to surrounding cells and 2) entering the blood
Virus adheres to angiotensin-converting enzyme 2 (ACE-2) receptor on body cells, mimics ACE-2, & gains access into cell
COVID-19
Symptomatic infection with SARS-CoV-2
Viral proliferation in cells of tissues with more ACE-2 receptors: lungs (type II pneumocytes); vasculature (endothelial cells), kidneys (proximal tubular epithelium), heart (myocardium), GI tract (enterocytes)
Cell death and ↑ in inflammatory cytokines triggers immune response
Neutrophils move to lungs, release reactive oxygen species and cytokines
Alveolar/capillary damage
Fluid accumulates in interstitium and alveoli
↑ Distance for O2 to diffuse from alveoli to capillaryà ↓ blood O2 saturation
Cytokines induce the hypothalamus to release prostaglandins
↑ body temperature set point to fight infection
Virus disassembles, releasing viral RNA, which uses host cell’s ribosomes to make new viral proteins like RNA-polymerases
Newly made viral RNA-polymerases use cell’s own nucleotides to synthesize new viral RNA
Bilateral ground-glass opacities (CT lung) & interstitial infiltrates on Chest X-ray
Dyspnea
Airway irritation
Cough
Myocardial cell damage
↑ Troponin
↑ skeletal muscle cell damage
Viral RNA & proteins packaged into new viral particles
New virus assembled and released from cell, killing the cell and contributing to disease symptoms
Average incubation period (time from initial infection to symptom onset) is 4-5 days; can be up to 14 days
Heart tries to compensate for
Fever Myalgia
hypoxemiaà↑ cardiac output Arrhythmogenic
and strain on myocardium
state
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published March 22, 2020, updated Aug 18, 2021 on www.thecalgaryguide.com")
Ectopic Pregnancy

Endometriosis
Tubal surgery or disorders
Age >35
Risk factor accumulation over time
Smoking
Impairment in tubal motility; impaired immunity (risk factor for PID)
Tubal scarring leading to adhesions, obstruction, and alteration of tubal function
Ectopic Pregnancy:
Implantation of developing blastocyst outside the uterine cavity, most commonly in fallopian tube (other locations: interstitial > cornual > cervical > ovarian > abdominal)
Embryo releases human chorionic gonadotropin (β-hCG), which supports corpus luteum to continue producing progesterone
On transvaginal ultrasound: Extrauterine gestational sac with a yolk sac or embryo
Embryo & trophoblast deathàloss of hormone support for the decidua (modified endometrial lining)
Progesterone maintains the endometrial lining, preventing it from shedding
Missed period
Penetration of ovum into muscular wall of fallopian tube
Tubal distention àTubal rupture
Intra-abdominal hemorrhage
Pregnancy cannot survive without the uterine endometrium
Maternal blood extrudes through fimbriae of fallopian tubes and into peritoneal cavity
Lower abdominal pain (including peritonitis in cases of hemoperitoneum)
Hemoperitoneum
(blood in the peritoneal cavity)
Sloughing of decidua out of the uterus through the vagina
Vaginal bleeding (usually in first trimester)
Cessation of human chorionic gonadotropin (β-hCG) release from embryo
β-hCG plateaus or decreases
Authors: Jemimah Raffé-Devine Tahsin Khan Yan Yu* Reviewers: Brianna Ghali Bishwas Paudel Mackenzie Grisdale Christina Schweitzer Ron Cusano* Jadine Paw* * MD at time of publication
Syncope
↓ Level of consciousness
Positive β-hCG, but rising <35% over 2 days
Discriminatory zone: β-hCG >2000 + absence of intrauterine pregnancy
Hypotension
Shock
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published Oct 1, 2017, updated Oct 19, 2021 on www.thecalgaryguide.com")
necrotizing fasciitis

Laceration
Recent surgery
Injection
Burn
Blunt force trauma
Childbirth
Lower extremity wounds
Bacteria enters tissue through open wound
Infection of muscle fascia Local immune response
Production of exotoxins by bacteria
Disruptions of protective skin barrier
Bacteria introduced into tissue during injury
Necrotizing Fasciitis
Type I infection: mixed aerobic and anaerobic bacteria Type II infection: group A streptococcus
Type III infection: marine organisms, clostridial infections Type IV infection: fungal organisms
Poor blood supply of muscle fascia allows for progressive spread of infection
Systemic immune response
Pyrogens produced by immune system
Pyrogens travel through
the bloodstream to the hypothalamus and alters the body’s thermal setpoint
Transmission of bacteria from infected tissue to blood
Sepsis
Streptolysin (exotoxin) causes blood clot formation
Blood clots in vessels
Tissue ischemia in epidermis, dermis, subcutaneous fat, muscle fascia, and/or muscle
Stimulation of programmed cell death
Tissue destruction
Pain more severe than clinical findings
↓ blood flow fails to meet tissue’s needs
Tissue death
Build up of gas in subcutaneous
tissue from bacteria metabolism
Crepitus
↑ serum creatinine
kinase from protein breakdown
↑ blood flow to infected tissue
Warmth Erythema
Immune cells release vasoactive cytokines into the blood
Capillary vasodilation
Fluid and proteins shift from cells and capillaries to interstitial space
Blood
vessel dilation
↓ perfusion of vital organs
Organ failure
Hypotension
↑ heart rate to perfuse vital organs
Tachycardia
Bacteria releases toxins which are taken up into the bloodstream
Immune cells produce inflammatory cytokines
Circulating toxins activate T cells, over- activating the systemic immune response
Toxic Shock syndrome
Infection ↑ white blood cell production in bone marrow
↑ white blood cells
Destructionof peripheral nerve endings
Insensitivity to pain
Tissue hypoxia à anaerobic metabolism
Poor perfusion of lungs impairs gas exchange
Tachypnea
Cytokines affect dopamine production in the basal ganglia
Acute malaise
Production of non-specific acute phase reactants
↑ C reactive protein and erythrocyte sedimentation rate
Fluid-filled blisters
Edema
Fever Compartment syndrome (see relevant Calgary Guide slide)
Amputation ↑ serum
lactate
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
First published Nov 20, 2013, updated Dec 19, 2021 on www.thecalgaryguide.com")
sepsis-y-shock-septico-patogenesis-y-hallazgos-clinicos

Vitiligo Pathogenesis and Clinical Findings
 that, through complex mechanisms, stimulate the immune system’s CD8+ T-cells to destroy melanocytes
Melanocytes enter apoptotic or senescent state
↓ Functional melanocytes
↓ Melanin production
Overall loss of functional melanocytes
Vitiligo
Normal Skin
Pigmented Epidermis Dermal- Epidermal Junction
Dermis
Autoimmune destruction of melanocytes
Depigmented Epidermis
Melanocytes
Immune-mediated destruction of melanocytes (by both neutrophils and CD8+ T cells)
A depigmenting skin disorder characterized by selective loss of melanocytes
Depigmentation in areas of
↑ pressure, friction and/or trauma
Nonsegmental vitiligo
Smooth unpigmented macules or patches in bilateral, often symmetric pattern
Somatic mosaicism (mutation limited to a subset of cells) in zygote during development
Segmental vitiligo
Smooth unpigmented macules or patches in unilateral pattern not crossing midline
Vitiligo
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published February 15, 2022 on www.thecalgaryguide.com")
Hypovolämischer Schock: Pathogenese, Komplikationen und klinische Befunde

complications-of-pulmonary-embolism
![Complications of Pulmonary Embolism
Authors:
Sravya Kakumanu, Dean Percy, Yan Yu
Reviewers:
Tristan Jones, Ciara Hanly, Jieling Ma (马杰羚), Ben Campbell, Dr. Man-Chiu Poon*, Dr. Lynn Savoie*, Dr. Tara Lohmann * * MD at time of publication
IF CHRONIC:
Unresolved clot after 2 years leading to fibrosis of pulmonary vasculature
Chronic Thromboembolic Pulmonary Hypertension (CTEPH)
(<5% of PE cases)
Venous Stasis Hypercoagulable state
Vessel Injury
Virchow’s Triad (*See Suspected Deep Vein Thrombosis slide)
Deep Vein Thrombosis
Clot migrates from deep limb veins à femoral àiliac veins
ACUTE/MASSIVE PE:
Clot obstructs pulmonary arterial or arteriolar flow
Lung infarction (tissue death) from ischemia
Inflammatory cells migrate to site and release cytokines
↑ Permeability of blood vessels
Permeability-driven (exudate) fluid leakage into pleural space
Pleural Effusion
Clot migratesàinferior vena cava àright atrium (RA) of heartà right ventricle (RV) à gets lodged in pulmonary arteries/arterioles
Pulmonary Embolism (PE)
↑ RV afterload
↑ RV pressure and expansion
Well-ventilated (V) areas of lung do not receive adequate blood supply (Q)
V/Q Mismatch
Leftward shift of ventricular septum
↓ Left ventricle filling in diastole
↓ Cardiac output
Obstructive Shock
Impaired heart filling
Pulseless Electrical Activity
(ECG activity in absence of palpable pulse)
Back up of pressure in systemic venous system
↑ Pressure in capillaries draining parietal pleura
Pressure-driven (transudate) fluid leakage into pleural space
For signs and symptoms, see the Obstructive Shock slide
For signs and symptoms refer to CTEPH slide
Chronic ↑ RV afterload
↑ Stretching of myocytes causing RV hypertrophy and dilation
↓ RV ejection fraction
Right Heart Failure
“Cor Pulmonale”
For signs and symptoms, see the Right Heart Failure slide
Failure to oxygenate blood
Type I Respiratory Failure
Hypoxemic: patient has ↓ blood [O2]
IF MASSIVE PE (less common):
↑ Alveolar dead space
Failure to ventilate
Type II Respiratory Failure Hypercapnic: patient has ↑ blood [CO2]
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published August 7, 2012, updated Mar 31, 2022 on www.thecalgaryguide.com](https://calgaryguide.ucalgary.ca/wp-content/uploads/2014/09/Complications-of-Pulmonary-Embolism-2022.jpg "Complications of Pulmonary Embolism
Authors:
Sravya Kakumanu, Dean Percy, Yan Yu
Reviewers:
Tristan Jones, Ciara Hanly, Jieling Ma (马杰羚), Ben Campbell, Dr. Man-Chiu Poon*, Dr. Lynn Savoie*, Dr. Tara Lohmann * * MD at time of publication
IF CHRONIC:
Unresolved clot after 2 years leading to fibrosis of pulmonary vasculature
Chronic Thromboembolic Pulmonary Hypertension (CTEPH)
(<5% of PE cases)
Venous Stasis Hypercoagulable state
Vessel Injury
Virchow’s Triad (*See Suspected Deep Vein Thrombosis slide)
Deep Vein Thrombosis
Clot migrates from deep limb veins à femoral àiliac veins
ACUTE/MASSIVE PE:
Clot obstructs pulmonary arterial or arteriolar flow
Lung infarction (tissue death) from ischemia
Inflammatory cells migrate to site and release cytokines
↑ Permeability of blood vessels
Permeability-driven (exudate) fluid leakage into pleural space
Pleural Effusion
Clot migratesàinferior vena cava àright atrium (RA) of heartà right ventricle (RV) à gets lodged in pulmonary arteries/arterioles
Pulmonary Embolism (PE)
↑ RV afterload
↑ RV pressure and expansion
Well-ventilated (V) areas of lung do not receive adequate blood supply (Q)
V/Q Mismatch
Leftward shift of ventricular septum
↓ Left ventricle filling in diastole
↓ Cardiac output
Obstructive Shock
Impaired heart filling
Pulseless Electrical Activity
(ECG activity in absence of palpable pulse)
Back up of pressure in systemic venous system
↑ Pressure in capillaries draining parietal pleura
Pressure-driven (transudate) fluid leakage into pleural space
For signs and symptoms, see the Obstructive Shock slide
For signs and symptoms refer to CTEPH slide
Chronic ↑ RV afterload
↑ Stretching of myocytes causing RV hypertrophy and dilation
↓ RV ejection fraction
Right Heart Failure
“Cor Pulmonale”
For signs and symptoms, see the Right Heart Failure slide
Failure to oxygenate blood
Type I Respiratory Failure
Hypoxemic: patient has ↓ blood [O2]
IF MASSIVE PE (less common):
↑ Alveolar dead space
Failure to ventilate
Type II Respiratory Failure Hypercapnic: patient has ↑ blood [CO2]
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published August 7, 2012, updated Mar 31, 2022 on www.thecalgaryguide.com")
burn-shock-pathogenesis-complications-and-clinical-findings

Endothelial cell lining in blood vessel walls is compromised
↑ Local vessel permeability
Shift of plasma + proteins from vessel into interstitial space
Direct vascular thermal injury (within burn wound)
↑ Production of circulating inflammatory mediators (ex. IL-1, IL-6, TNF-!)
↑ Systemic vessel permeability
↑ Circulating reactive oxygen species
Damage to DNA, proteins, and lipids throughout body, including myocardium (muscle cells of the heart)
↑ Myocardial stress
Myocardial dysfunction
↓ Cardiac contractility ↓ Stroke volume ↓ Cardiac output Cardiogenic
Shock
Refer to Cardiogenic Shock:
Pathogenesis, Complications and Clinical Findings
↓ Protein concentration in vessels causes ↓ intravascular oncotic pressure
Further shift of plasma from vessel into interstitial space (↑ interstitial proteins pull plasma into interstitium)
↓ Intravascular plasma
↑ RBCs per unit volume of plasma
↑ Systemic vasoconstriction to maintain blood pressure (↑ Afterload)
↓ Circulating blood volume leads to less venous return (↓ Preload)
↑ Hematocrit
Pitting edema (burned & unburned tissue)
↓ Circulating blood volume
Hypovolemic Shock
Refer to Hypovolemic Shock: Pathogenesis, Complications and Clinical Findings
Burn Shock: A complication of large burns causing end-organ hypoperfusion with resultant organ dysfunction
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published August 15, 2022 on www.thecalgaryguide.com")
ascending-cholangitis-pathogenesis-clinical-findings

Bacteria ascends into biliary tract from duodenum via Sphincter of Oddi
Gallstone in the common bile duct
Stricture of biliary/hepatic ducts
Biliary / pancreatic duct malignancy
Biliary obstruction (partial bile duct obstruction)
↓ Excretion of bilirubin into
bileà ↑ bilirubin in blood
↑ Serum bilirubin
Deposition of bilirubin in the skin and mucous membranes
Jaundice*†
Parasites in bile duct
(E.g. Clonorchis)
Complication of endoscopic retrograde cholangiopancreatography (ERCP)
Bile accumulates in biliary tract
Bile duct dilation on ultrasound
Sludge (bile precipitants) impact bile ducts worsening obstruction
Obstruction & detergents in bile inflame ductal mucosa. Inflammation then spreads to adjacent structures
Stimulates phrenic (C3-C5) and foregut autonomic nerves (T5-T8)
Dull right upper quadrant pain*† radiating to back and right shoulder
↑ Intra-biliary pressure
Impaired forward flowà
↑ backflow of bile
Impaired bile secretion damages ductal epithelium of the biliary tract
Damaged ductal epithelium leaks ALP and GGT (enzymes) into blood
↑ Serum ALP and GGT
↑ Permeability of bile ductules
Reduced flushing of bile out to duodenum
Inflammatory response triggered
Fever*† ↑ WBCs
Tachycardia Hypotension† Confusion† Oliguria
Bacterial translocation from biliary tract into blood
Bacteremia
Massive systemic inflammatory response
Septic Shock
(see Distributive Shock slide)
* Charcot’s triad ~20% of cases | † Reynolds’ pentad ~7% of cases
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
First published November 18, 2015; Updated August 31, 2022 on www.thecalgaryguide.com")
acute-lower-gi-bleeds-pathogenesis-and-clinical-findings

Intestinal vessels are stretched over the domes of the diverticula
Angiodysplasia
Formation of
dilated, thin- walled vessels in GI tract mucosa/ submucosa
Colorectal Cancer
Infectious Colitis
Invasion of bacteria and/or bacterial toxins into intestinal wall
Cell damage and cell death
Sloughing off of intestinal epithelial cells
Ischemic Colitis
↓ Blood flow to a portion of the colon
Lack of oxygen delivery to colon wall
Necrosis (cell death) in the colon wall
Inflammatory Bowel Diseases (Note: these are chronic diseases and rarely present with acute lower GI bleed)
New, extremely friable blood vessels develop within the tumor
Malignant tissue invades the colon wall and disrupts colonic blood vessels
Crohn Disease
Immune- mediated full thickness inflammation of bowel wall
Ulcer formation and disruption of intestinal vessels
Ulcerative Colitis
Recurrent immune- mediated inflammation of colon mucosa
Authors:
Miranda Schmidt Illustrator:
Mizuki Lopez Reviewers:
Vina Fan,
Ben Campbell, Kerri Novak*
* MD at initial time of publication
Acute Lower GI Bleed
↑ Risk for vessel damage and rupture
Blood travels rapidly through GI tract
Hematochezia
(bright red blood per rectum)
May result in significant blood loss
Blood from the small intestine or right colon travels a longer distance through the GI tract
Bacteria in the GI tract has time to oxidize hemoglobin in the blood
Oxidization makes blood a darker color
Melena (rare in a lower GI bleed) (tarry black stool)
Inferior Vena Cava
Diaphragm
Esophagus
Loss of red
blood cells results in a loss of hemoglobin (a component of red blood cells)
Fluid from the extravascular space moves into the blood vessels to maintain vascular volume
Fluid is administered in a healthcare setting to compensate for blood loss
Hypovolemic Shock (rare in a lower GI bleed) (↓ Oxygen delivery to tissues due to low blood volume)
See “Hypovolemic Shock” slide for signs and symptoms
Lower GI Bleeds
are intra-luminal GI tract bleeds that occur anywhere distal to the ligament of Treitz (transition between duodenum and jejunum)
After 24 hours, the addition of fluid to the intravascular space dilutes hemoglobin in the blood
Normocytic Anemia
Duodenum
Ligament of Treitz
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published August 31, 2022 on www.thecalgaryguide.com")
ventilator-associated-pneumonia-pathogenesis-and-clinical-findings
 for invasive mechanical ventilation to maintain respiration
Paralytics and
sedatives lead to inhibition of cough reflex
Damage to cilia on epithelial cells of trachea during insertion of ETT
↓ Mucociliary clearance
Insertion of nasogastric (NG) tube for feeding
Constant opening by NG tube ↓ esophageal sphincter function
Introduction of oropharyngeal microbes during intubation
Severe acute illness impairs phagocytosis and dysregulates T- cells (mechanism unclear)
Medications, chronic disease, or severe acute illness can weaken immune system
Desensitization of
pharyngoglottal
adduction reflex (PAR) (PAR normally induces closure of epiglottis to protect the airway when swallowing)
↑ Reflux of gastric contents
Accumulation of subglottic secretions containing microbes
Microbial colonization on inside of ETT
Development of biofilm on inside of ETT
Dislodgement of biofilm into lower airway
↑ Age or chronic disease can weaken respiratory function
Micro aspirations of subglottic and gastric contents
Impaired mechanisms to remove microbes from airway Positive pressure pushes microbes down
Fever/rigors ↑ White blood cell count
Introduction of pathogenic microbes to airway Susceptible patient (not required for infection)
Septic shock
See Distributive Shock slide
Sepsis
Microbes descend airway and infect lungs
Ventilator-Associated Pneumonia (VAP)
Occurs > 48-72 hours after intubation
↑ Inflammatory response at infection site promoting immune cell extravasation and cytokine release
Cytokine signalling ↑ permeability of capillaries leading to ↑ fluid leakage into interstitium and alveoli
Authors: Sravya Kakumanu Reviewers: Ben Campbell *Tara Lohmann *Bryan Yipp * MD at time of publication
Acute respiratory distress syndrome
See ARDS pathogenesis slide
Pleural effusion Lung consolidation on chest x-ray (CXR) ↑ Sputum production to clear fluid on CXR (Note: In patients with Acute Respiratory Distress Syndrome (ARDS), look for VAP consolidation in non- within alveoli/airways (may be
Positive microbial culture from sputum
dependent/upper regions of the lung where ARDS consolidation would be unexpected to extend to)
purulent in worse infections)
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published September 2, 2022 on www.thecalgaryguide.com")
syok-luka-bakar-patogenesis-komplikasi-dan-temuan-klinis

syok-hipovolemik-patogenesis-komplikasi-dan-temuan-klinis

Syok Obstruktif: Patogenesis, komplikasi, dan temuan klinis

Syok Kardiogenik: Patogenesis, komplikasi, dan temuan klinis

Syok Distributif: Patogenesis, komplikasi, dan temuan klinis

Acute Liver Failure: Pathogenesis and clinical findings

NAPQI binds hepatocellular proteins
(see Acetaminophen Overdose: pathogenesis and clinical findings slide)
Drug-induced liver injury
Metabolism of drugs by the liver can produce reactive drug metabolites
Intracellular stress, mitochondrial injury, or immune response
Viral Hepatitis (i.e. HAV, HBV, HEV, HSV)
Acute infection or infection flare provokes an immune response against infected hepatocytes
Autoimmune Hepatitis
Autoimmune antibodies attack hepatocytes (see Auto-immune Hepatitis (AIH) slide)
Ischemia (i.e. from shock)
↓ O2 delivery to the liver
Hepatocellular hypoxia
Wilson’s Disease
Heritable mutation in the ATP7B gene
↓ Biliary excretion of copper
Hepatic copper accumulation injures hepatocytes (see Wilson’s Disease slide)
Accelerated rate of hepatocellular necrosis or apoptosis
Hepatocyte death exceeds regeneration such that liver function is compromised within a short amount of time
Acute Liver Failure
An illness of <26 weeks duration in the absence of pre-existing cirrhosis, characterized by INR ≥1.5 and evidence of altered mentation (hepatic encephalopathy)
Injured hepatocytes leak hepatic enzymes (AST, ALT, GGT) into circulation
↑ Liver enzymes
Hepatocellular inflammation
Stimulation of foregut
autonomic nerves
Right upper quadrant pain
↓ Toxin metabolism
Toxins build up and activate microglial cells (brain macrophage)
Oxidative stress and cerebral edema
Hepatic encephalopathy
Characteristic set of neuropsychiatric symptoms (see Hepatic Encephalopathy slide)
↓ Hepatocellular function and number
↓ Complement protein synthesis
↓ Ability to clear immune complexes and activate B cells
Accumulation of pigmented bilirubin
↓ Synthesis of coagulation factors
↑ INR
↓ Conjugation of bilirubin by the liver and ↓ transport into bile for excretion
↑ Serum bilirubin
Jaundice
Infection
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published November 15, 2022 on www.thecalgaryguide.com")
hypovolemic-shock

Inflammation (pancreatitis, cirrhosis, post-operative, etc.)
Inflammatory mediators vessel permeability and fluid leaks out
Trauma
Ruptured vessels leak fluid into potential spaces
Hemorrhagic losses
(GI bleed, postpartum hemorrhage, etc.)
↓ Intravascular volume
↓ Venous return to the heart
↓ Cardiac output (blood pumped from the heart)
Hypovolemic Shock
↓ Oxygen delivery to tissues due to low blood volume
Insufficient organ perfusion
Non-Hemorrhagic losses
(dehydration, GI losses, skin losses / burns, renal losses, etc.)
‘Third Spacing’ of fluid
(fluid located outside the intravascular or intracellular space; large collections can occur in the pelvis, thorax, GI tract, long bones of children, intra-abdominally, retroperitoneally)
P = Q x R; less ‘flow’ in the vessels (Q), with vessels not constricting enough to maintain resistance (R)à pressure (P) will drop
↓ Blood Pressure
Caution: young, healthy individuals can maintain blood pressure during circulatory collapse with cardiac output and vasoconstriction; do not use blood pressure as an indicator of shock severity in children
Carotid sinus baroreceptors sense low blood pressure ↓ Carotid sinus inhibition of sympathetic nervous system Release of sympathetic catecholamines (epinephrine and
↓ Pressure in venous circulation
Brain
Heart
Kidneys
↓ Blood in the right internal jugular vein
↓ Oxygen delivery to the brain
↓ Myocardial contractility (from lactic acidosis)
↓ Blood flow to kidneys
↓ Jugular Venous Pressure
Catecholamines bind to beta-1 receptors in the sinoatrial node of the heart
Beta-1 receptor activation causes ↑ heart rate
Tachycardia
norepinephrine)
Catecholamines bind to and stimulate alpha-1 receptors in peripheral vessels
Vasoconstriction of peripheral vessels
↓ Blood flow to peripheral tissue
Catecholamines bind to and stimulate beta receptors in sweat glands
Diaphoresis
(sweating)
In all body tissues
Inadequate oxygen delivery
↓ ATP production
↑ Anaerobic metabolism
↓ Body temperature
Impaired neurological functioning
Renal ischemia
Activation of the renin-angiotensin aldosterone system
↓ Glomerular filtration rate
↓ Clearance of lactic acid by the kidney
↑ Lactic acid production
↓ Rate of activity of clotting enzymes
Lactic Acidosis
Unknown mechanism
Coagulopathy Hypothermia
Trauma Triad of Death
↑ Capillary Cold, mottled refill time extremities
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published January 24, 2013, updated December 4, 2022 on www.thecalgaryguide.com")
Wstrząs obturacyjny: Patogeneza, powikłania oraz zmiany kliniczne

Wstrząs dystrybucyjny: Patogeneza, powikłania oraz zmiany kliniczne

Wstrząs kardiogenny: Patogeneza, powikłania oraz zmiany kliniczne

Wstrząs oparzeniowy: patogeneza, powikłania i zmiany kliniczne

Leczenie wstrząsu: wyjaśnienie podstawowych mechanizmów

Burns - Full Thickness - Pathogenesis and Clinical Findings 2023

Author and Illustrator: Amanda Eslinger Tracey Rice Reviewers:
Sunawer Aujla Alexander Arnold Duncan Nickerson* Jori Hardin*
* MD at time of publication
Epidermis
Dermis
Sub-cutaneous tissue
Fire
Contact Scald Chemical Electrical
Transfer of heat energy causes direct injury to keratinocytes
Hypoxic injury (lack of oxygen) causes ischemia-related cell death leading to necrosis
Full thickness burn
Non-uniform damage to the epidermal, dermal & subcutaneous layers at varying widths & depths due to unpredictable injury pattern
Degraded
epidermal
& dermal layers cover granulated tissue
Ulceration covered by eschar: a thick, dried, black (necrotic) layer
Long-term risk of ulcers & infections
↑ Vascular permeability in subcutaneous layers
↑ Intravascular fluid leaves capillaries, ↓ uptake by lymph vessels
Edema
Compression of surrounding muscles, nerves & vessels
Ischemia and/or necrosis
↓ Immunologic
response due to impaired epidermal barrier function
↑ Microbial growth creating biofilm & secretion of chemicals that inhibit natural protective process
Irritated,
inflamed wound bed +/- exudate
↑ Risk of infection & septic shock
Destruction
of somatosensory structures
Hypoesthesia (↓ sensation to stimuli)
↑ Risk of
repeated injury due to ↓ response to noxious stimuli
Destruction of cutaneous capillary beds
↓ Healing due to ↓ dermal structures throughout wound
White or leathery appearance
Chronic wounds require surgical interventions
↑ Risk of contractures & skin barrier weakness
Legend:
Mechanism of Injury
Pathophysiology
Sign/Symptom
Complication
Published December 2, 2013, updated June 26, 2023 on www.thecalgaryguide.com")
Burns - Full Thickness - Pathogenesis and Clinical Findings

Author and Illustrator: Amanda Eslinger Tracey Rice Reviewers:
Sunawer Aujla Alexander Arnold Duncan Nickerson* Jori Hardin*
* MD at time of publication
Epidermis
Dermis
Sub-cutaneous tissue
Fire
Contact Scald Chemical Electrical
Transfer of heat energy causes direct injury to keratinocytes
Hypoxic injury (lack of oxygen) causes ischemia-related cell death leading to necrosis
Full thickness burn
Non-uniform damage to the epidermal, dermal & subcutaneous layers at varying widths & depths due to unpredictable injury pattern
Degraded
epidermal
& dermal layers cover granulated tissue
Ulceration covered by eschar: a thick, dried, black (necrotic) layer
Long-term risk of ulcers & infections
↑ Vascular permeability in subcutaneous layers
↑ Intravascular fluid leaves capillaries, ↓ uptake by lymph vessels
Edema
Compression of surrounding muscles, nerves & vessels
Ischemia and/or necrosis
↓ Immunologic
response due to impaired epidermal barrier function
↑ Microbial growth creating biofilm & secretion of chemicals that inhibit natural protective process
Irritated,
inflamed wound bed +/- exudate
↑ Risk of infection & septic shock
Destruction
of somatosensory structures
Hypoesthesia (↓ sensation to stimuli)
↑ Risk of
repeated injury due to ↓ response to noxious stimuli
Destruction of cutaneous capillary beds
↓ Healing due to ↓ dermal structures throughout wound
White or leathery appearance
Chronic wounds require surgical interventions
↑ Risk of contractures & skin barrier weakness
Legend:
Mechanism of Injury
Pathophysiology
Sign/Symptom
Complication
Published December 2, 2013, updated June 26, 2023 on www.thecalgaryguide.com")
Shock por quemaduras

Tatalaksana Syok Penjelasan dari mekanisme dasar

Overview of burns
 Less common Burns from any source can result in 3 damage zones
Common
Fire Scald
Zone of coagulation:
innermost zone; maximum damage through necrosis and irreversible tissue loss
Zone of stasis: middle zone; decreased tissue perfusion, potentially salvageable
Zone of hyperemia:
outermost zone; tissue perfusion is increased, damage is reversible
Epidermis, dermis, and subcutaneous tissue
Full Thickness (3rd degree burn)
Normal Skin
Epidermis only
Superficial Thickness (1st degree burn)
Epidermis and superficial (papillary) dermis
Superficial Partial Thickness (2nd degree burn)
Epidermis and deep (reticular) dermis
Deep Partial Thickness (2nd degree burn)
Skin and deep tissues, muscle, fascia, nerves, blood vessels, bone
Composite tissue injury
(4th degree burn)
Compartment syndrome
Epidermal layer Dermal-Epidermal Junction
Superficial (Papillary) Dermis
Deep (Reticular) Dermis
Note: The total burn surface area (TBSA) can be estimated using the rule of nines (1st degree burns are not included): Head and Neck: 9% total, Chest and Upper Back: 9% each, Arm: 9% each, Leg: 18% each (front and back),
Abdomen and Lower Back: 9% each, Genital Area: 1%
Refer to Complications of Burns
Burn Shock
Refer to Burn Shock: Pathogenesis, Complications, and Clinical Findings
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published November 8, 2023 on www.thecalgaryguide.com")
Death Cardiovascular Respiratory and Neurologic Mechanisms

Hypoxemia (Type I Respiratory Failure): low dissolved oxygen in blood (PaO2)
Lungs can’t oxygenate blood fast enough
Lungs can’t rid blood of CO2 fast enough
Hypercapnia / hypercarbia (Type II Respiratory Failure): elevated dissolved CO2 in blood (PaCO2)
Cerebral vasodilation
Toxins: e.g. cyanide, pesticides, arsenic Severe anemia
Distributive problems:
Systemic inflammation (sepsis, anaphylaxis, pancreatitis), adrenal insufficiency, vasodilatory drugs
Obstructive problems: Cardiac tamponade*, tension pneumothorax* or massive pulmonary embolism*
Hypovolemic* problems (low blood volume): Hemorrhage, dehydration, widespread skin disruption or burns
Cardiac valve dysfunction
Myocardial infarction* or cardiomyopathy
Cardiac arrhythmia or heart block
Disturbed electrical activity in cardiomyocytes
Peripheral metabolic disturbances
Hypokalemia*, Hyperkalemia* Acidosis* (including renal failure) Hypothermia*
Toxins* (e.g. cocaine, beta blockers, tricyclics) Severe thyroid derangement
Inappropriate systemic vasodilation
Adjacent forces impair heart filling
Low cardiac preload
Low stroke volume (SV; depends on valves, contractility, preload)
Decreased systemic vascular resistance (SVR)
Low blood pressure (BP = CO x SVR)
Decreased cardiac output (CO = SV x HR)
Disseminated intravascular coagulationàwidespread thrombi that occlude blood flow (also causes hemorrhage, see relevant box at left)
Methemoglobinemia: some hemoglobin gets stuck in a state that can’t carry O2
Hemoglobin has reduced capacity to carry or release O2
Drugs: e.g. dapsone, nitrates
Carbon monoxide poisoning
Circulatory collapse / shock: inadequate perfusion of tissue with blood
Respiratory collapse: blood has insufficient useable O2 content
Ventricular fibrillation (VF) or pulseless ventricular tachycardia (VT)
Hypoxia*: inadequate O2 delivery or utilization in tissues
Hypoxia creates metabolic disturbances that impair cardiac cells. Alternatively, any of the preceding conditions marked with (*) can directly trigger cardiac arrest first
Pulseless Electrical Activity (PEA): organized activity on ECG with no cardiac output (can be preceded or mimicked by pseudo-PEA, in which there is still some output on ultrasound)
Low atmospheric pressure or oxygen content Severe lung disease
Asthma, COPD, interstitial lung disease, congestive heart failure, pulmonary hypertension, pulmonary embolism, lung collapse / atelectasis
Acute respiratory distress syndrome
Pneumonia, aspiration pneumonitis, inhalational injury, systemic inflammation, drowning
Severe hypoventilation
Respiratory fatigue, advanced COPD, chest wall disorders, neuromuscular disorders, upper airway obstruction, toxins (e.g. opioids, botulism)
Can degenerate at any time
Asystole: no cardiac electrical activity or output
Death
Respiratory arrest: cessation of breathing
Inability to protect airway
Decreased level of consciousness
Note
This is a broad overview of the many scenarios that can result in death. For detailed explanations of the various disease mechanisms, refer to the corresponding slides.
* = reversible causes of cardiac arrest (Hs and Ts)
Author:
Ben Campbell
Reviewers:
Yan Yu*
Huma Ali*
* MD at time of publication
Bradycardia
(low heart rate, HR)
Unopposed parasympathetic stimulation of heart (can also cause vasodilation, see Distributive problems)
Disruption of spinal cord sympathetic control
Injury to cervical or upper thoracic spinal cord
Irreversible cessation of cardiac, respiratory, and brain function
Prolonged seizure initially causes increased cardiovascular activity, until the system fatigues
Disruption of respiratory control center in medulla
Expanding skull contents squeeze brainstem (herniation)
Increased intracranial pressure
Edema from intracranial hemorrhage, trauma, brain mass
Edema, inflammation, hypoxia and/or metabolic derangements cause diffuse neuron dysfunction
Central nervous system infection
Dementia, particularly with delirium
Massive ischemic stroke
Seizure
activity prevents or alters breathing
Metabolic disturbances that affect the central nervous system Hypoglycemia Hypocalcemia, hypercalcemia Hyponatremia, hypernatremia Uremia
Acute liver failure (hyperNH4) Many drugs / toxins Withdrawal (e.g. EtOH)
Status epilepticus
Brainstem lesion (e.g. stroke, neoplasm, inflammatory)
Nervous System Insult
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published November 11, 2023 on www.thecalgaryguide.com
Respiratory System Insult
Cardiovascular System Insult Cardiogenic problems")
Complication of MI - Acute Mitral Regurgitation
 commonly in the posterior descending artery
Plaque ruptures
Exposed plaque contentsàPlatelet adhesion and aggregation
Artery becomes partially or completely occluded
Mitral Regurgitation**
(Back flow of blood from left ventricle to left atrium during systole)
↑ Left atrial pressure
Blood from left atrium backs up into pulmonary venous system
↑ Pulmonary venous pressure
↑ Hydrostatic pressure in alveolar capillaries
↑ Fluid leak from alveolar capillaries to interstitium (pulmonary edema**)
↓ Gas exchange
Attempt at
physiologic compensation à ↑ Respiratory rate
Tachypnea
Holosystolic murmur
Heard loudest over the mitral valve (5th intercostal space, mid-clavicular line), with radiation to the axilla
↑ Volume of blood to left ventricle during diastole
↓ Blood supply to the portion of the ventricle that supports the papillary muscle à↓ Muscle movement
Left ventricle dysfunction
↓ Blood supply to the posterior medial papillary muscle
Cell death (myocardial infarction)
Papillary muscle ischemia (muscle is intact but cannot contract)
↑ Blood in right atriumà↑ Blood in superior vena cava
↑ Blood in internal jugular vein
↑ Jugular venous pressure (JVP)
Redistribution of interstitial fluid when lying flat (reduced effect of gravity)
↓ Forward blood flow from left ventricle to aorta
↓ Stroke volume (SV)
↓ Cardiac output (CO) CO = SV x HR (heart rate)
Sympathetic nervous system attempts to physiologically compensate
↑ Heart rate
Tachycardia
↓ Blood pressure (BP) because BP = CO X SVR (systemic vascular resistance)
Cardiogenic shock**
Papillary muscle rupture
Papillary muscle unable to provide adequate tension on mitral valve
Mitral valve unable to stay closed during systole
Orthopnea
Difficulty breathing
Dyspnea
Paroxysmal nocturnal dyspnea
**See corresponding Calgary Guide slides for more details
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published November 25, 2023 on www.thecalgaryguide.com")
Angioedema Bradykinin Mediated
 & precursors (kininogen), triggered by ↑ systemic estrogen
Rheumatologic disorders & B- cell lymphoproliferative disease
Complement cascade activation results in ↑ C1 protease production
C1 esterase inhibitor is utilized to neutralize C1 protease, with its consumption exceeding its synthesis
Plasma cell proliferation (i.e., dyscrasia/ monoclonal gammopathy)
Immunoglobulin G antibodies act against C1 esterase inhibitor to render it non-functional
Genetic or spontaneous mutation in C1 esterase inhibitor gene
C1 esterase inhibitor deficiency
C1 esterase inhibitor dysfunction
Inhibition of angiotensin converting enzyme, dipeptidyl peptidase-4, or neprilysin induced metabolism of bradykinin
↓ Bradykinin (peptide hormone) degradation in plasma
Misfunctioning C1 esterase inhibiter is unable to inactivate bradykinin cascade members
↓ C1 esterase inhibiter results in inadequate inactivation of bradykinin cascade members
↑ Bradykinin protein production in plasma
Cutaneous Tissue
Mucosal Tissue
Epidermal layer
Dermal-Epidermal Junction
Dermal layer
Subcutaneous layer
Systemic bradykinin excess
Bradykinin-2 receptor binding on endothelial and vascular smooth muscle cells Hyperpermeability pathway activation, with the transcription of some signalling molecules taking hours Released pro-inflammatory mediators act on venules & arterioles in subcutaneous & submucosa tissues
Relaxation of vascular smooth muscle Dissociation of endothelial cell junctions
↑ Capillary blood flow ↑ Vascular permeability
↑ Plasma release into interstitial tissues (specific regions of the body hypothesized to be affected due to local differences in endothelial structure and its response to permeability inducing stimuli)
Dilation & ↑ permeability of vasculature results in fluid release into surrounding tissues
Mucosal Layer
Muscularis mucosae
Submucosal layer
Muscularis externa
Intestinal edema (Fluid buildup in Intestine tissues)
Intestinal swelling (↑ intra-abdominal pressure)
Laryngeal edema (Fluid buildup in larynx)
Swelling in larynx ↓ air flow into & out of lungs
Asphyxia (body is deprived of oxygen) Dyspnea (difficult in breathing)
Author: Aaron Varga Reviewers: Tracey Rice Sunawer Aujla Shahab Marzoughi Maharshi Gandhi Jori Hardin* Yan Yu* * MD at time of publication
Peripheral edema (Fluid buildup in extremities such as the hands, ankles, and feet)
Ascites, bowel obstruction, &/or hypovolemic shock
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published Mar 21, 2024 on www.thecalgaryguide.com")
Choque Hipovolemico

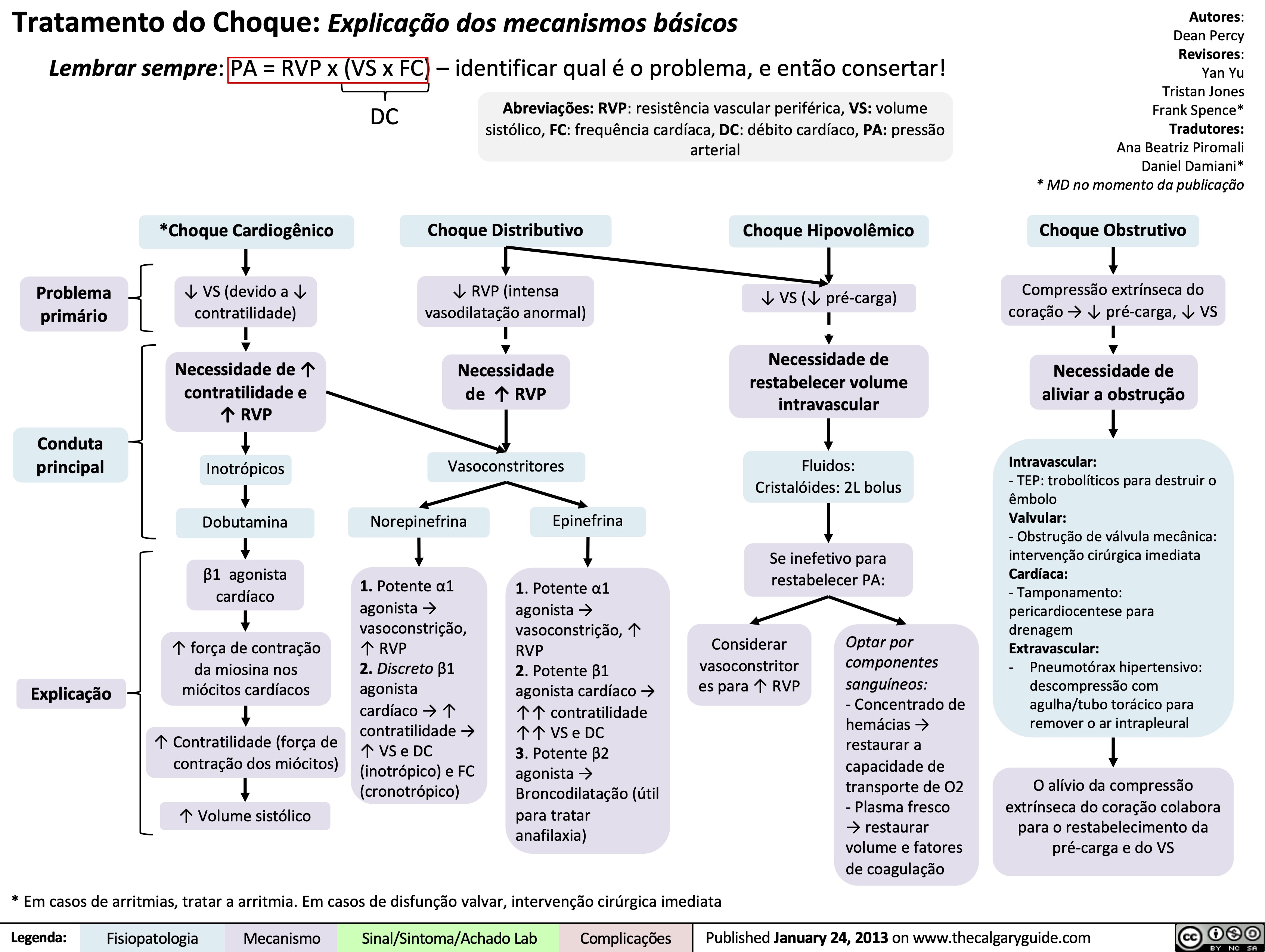
Tratamento do Choque: Explicacao dos mecanismos basicos
Massive Transfusion Protocol
![Massive Transfusion Protocol: Considerations and rationale
Massive transfusion protocol (MTP) is a tool used by clinicians when there is a need to rapidly administer a large amount of blood products, including packed red blood cells (pRBCs), fresh frozen plasma (FFP), and platelets. Complications of MTP are commonly referred to as “The Lethal Triad” referring to hypothermia, acidosis and coagulopathy.
Authors: Kayleigh Yang Arzina Jaffer
Reviewers: Jasleen Brar,
Luiza Radu, Karl Darcus*
* MD at time of publication
Intervention
Indications Initial Response Pathophysiology Transfusion Targets
≥ 3 pRBCs unit transfusion requirement in 1 hour
Shock index (heart rate/systolic blood pressure) > 1
Blood volume loss >50% in ≤3 hours
ABC Score ≥ 3 of: 1. Penetrating mechanism of injury 2. Systolic blood pressure < 90 mmHg 3. Heart rate > 120 beats per minute 4. Evidence of hemoperitoneum or hemopericardium on ultrasound (positive FAST U/S exam)
RABT Score ≥ 2 of: 1. Penetrating mechanism of injury 2. Shock index > 1 3. Positive FAST U/S 4. Known or suspected pelvic fracture
Call for help
Activate institution's MTP protocol
Send for STAT type and screen
Establish large-bore intravenous access
Fluid resuscitation
Collect and send STAT bloodwork including hemoglobin, platelet, INR, fibrinogen, electrolytes, creatinine and arterial blood gas (ABG).
Citrate present in blood products to avoid clotting during storage
Stored pRBCs break down and release potassium due to time mediated degeneration
Temporary accumulation of citrate in patient's blood with rapid use of blood products
Citrate chelates calcium
Less negative cell membrane resting potential
Anaerobic metabolism
Promotes hypocalcaemia
Changes in membrane excitability
Lactic acid buildup
Coagulopathy
(see coagulation cascade slide)
Cardiac dysrhythmias (peaked T-waves, atrial block, “sine wave”, asystolic EKG changes)
Metabolic acidosis
End organ damage
Continued blood loss
Volume overload
Avoid hypocalcemia
Avoid hyperkalemia
pH 7.35-7.45
Bleeding source control
Hemoglobin >70-90
Platelets >50 INR <1.5 Fibrinogen >1.5
Avoid dilutional coagulopathy (clotting factor dilution)
Mean Arterial Pressure (MAP) >60mmHg
Temperature >35.0°C
Slow (over 5-10 minutes) IV calcium administration
Inhaled beta agonists
Insulin/Dextrose
EKG monitoring
Sodium bicarbonate
Increase minute ventilation
Fastest control method to prevent further blood loss (i.e., packing wounds)
Early tranexamic acid administration
Administer pRBCs, FFP, and platelets in a 1:1:1 ratio (fibrinogen replacement indicated if <1.5 despite FFP)
Minimize crystalloid use
Administer crystalloids in a 3:1 ratio to estimated blood loss until blood products available
Administer vasopressors to meet target, do not overshoot
Temperature monitoring Fluid warming
↑ [Potassium] in pRBCs solution
Administration of pRBCs ↑ potassium in patient's blood
Blood loss
↓ Hemoglobin
Tissue hypoperfusion
Tissue hypoxia
↑ Diluent volume
↓ Concentration of clotting factors
Tissue death
↓ Coagulation ability
↑ Transfusion requirements
Early fluid resuscitation
Rapid transfusion of cooled or room-temperature blood products/fluids
↑ Blood pressure
Development of hypothermia
↑ Bleeding and clot dislodgement potential
↓ Enzyme activity in the coagulation cascade
↓ Coagulation ability
Legend:
Pathophysiology
Mechanism
Targets
Intervention
Published Sept 5, 2024 on www.thecalgaryguide.com](https://calgaryguide.ucalgary.ca/wp-content/uploads/2024/09/Massive-Transfusion-Protocol.jpg "Massive Transfusion Protocol: Considerations and rationale
Massive transfusion protocol (MTP) is a tool used by clinicians when there is a need to rapidly administer a large amount of blood products, including packed red blood cells (pRBCs), fresh frozen plasma (FFP), and platelets. Complications of MTP are commonly referred to as “The Lethal Triad” referring to hypothermia, acidosis and coagulopathy.
Authors: Kayleigh Yang Arzina Jaffer
Reviewers: Jasleen Brar,
Luiza Radu, Karl Darcus*
* MD at time of publication
Intervention
Indications Initial Response Pathophysiology Transfusion Targets
≥ 3 pRBCs unit transfusion requirement in 1 hour
Shock index (heart rate/systolic blood pressure) > 1
Blood volume loss >50% in ≤3 hours
ABC Score ≥ 3 of: 1. Penetrating mechanism of injury 2. Systolic blood pressure < 90 mmHg 3. Heart rate > 120 beats per minute 4. Evidence of hemoperitoneum or hemopericardium on ultrasound (positive FAST U/S exam)
RABT Score ≥ 2 of: 1. Penetrating mechanism of injury 2. Shock index > 1 3. Positive FAST U/S 4. Known or suspected pelvic fracture
Call for help
Activate institution's MTP protocol
Send for STAT type and screen
Establish large-bore intravenous access
Fluid resuscitation
Collect and send STAT bloodwork including hemoglobin, platelet, INR, fibrinogen, electrolytes, creatinine and arterial blood gas (ABG).
Citrate present in blood products to avoid clotting during storage
Stored pRBCs break down and release potassium due to time mediated degeneration
Temporary accumulation of citrate in patient's blood with rapid use of blood products
Citrate chelates calcium
Less negative cell membrane resting potential
Anaerobic metabolism
Promotes hypocalcaemia
Changes in membrane excitability
Lactic acid buildup
Coagulopathy
(see coagulation cascade slide)
Cardiac dysrhythmias (peaked T-waves, atrial block, “sine wave”, asystolic EKG changes)
Metabolic acidosis
End organ damage
Continued blood loss
Volume overload
Avoid hypocalcemia
Avoid hyperkalemia
pH 7.35-7.45
Bleeding source control
Hemoglobin >70-90
Platelets >50 INR <1.5 Fibrinogen >1.5
Avoid dilutional coagulopathy (clotting factor dilution)
Mean Arterial Pressure (MAP) >60mmHg
Temperature >35.0°C
Slow (over 5-10 minutes) IV calcium administration
Inhaled beta agonists
Insulin/Dextrose
EKG monitoring
Sodium bicarbonate
Increase minute ventilation
Fastest control method to prevent further blood loss (i.e., packing wounds)
Early tranexamic acid administration
Administer pRBCs, FFP, and platelets in a 1:1:1 ratio (fibrinogen replacement indicated if <1.5 despite FFP)
Minimize crystalloid use
Administer crystalloids in a 3:1 ratio to estimated blood loss until blood products available
Administer vasopressors to meet target, do not overshoot
Temperature monitoring Fluid warming
↑ [Potassium] in pRBCs solution
Administration of pRBCs ↑ potassium in patient's blood
Blood loss
↓ Hemoglobin
Tissue hypoperfusion
Tissue hypoxia
↑ Diluent volume
↓ Concentration of clotting factors
Tissue death
↓ Coagulation ability
↑ Transfusion requirements
Early fluid resuscitation
Rapid transfusion of cooled or room-temperature blood products/fluids
↑ Blood pressure
Development of hypothermia
↑ Bleeding and clot dislodgement potential
↓ Enzyme activity in the coagulation cascade
↓ Coagulation ability
Legend:
Pathophysiology
Mechanism
Targets
Intervention
Published Sept 5, 2024 on www.thecalgaryguide.com")
Rapid sequence induction and intubation
: Indications & considerations
“Full stomach”: ↑ risk of regurgitation, vomiting, aspiration Life-threatening injury or illness requiring immediate or rapid airway control
↓ Gastro- esophageal sphincter competence (elderly, pregnancy, hiatus hernia, obesity)
↑ Intragastric pressure (pregnancy, obesity, bowel obstruction, large abdominal tumors)
Delayed gastric emptying (narcotics, anticholinergics, pregnancy, renal failure, diabetes)
↓ Level of consciousness (drug/alcohol overdose, head injury, trauma or shock state)
Respiratory & ventilatory compromise (i.e., hypoxic or hypercapnic respiratory failure)
Achalasia (esophageal motility disorder resulting in impaired swallowing)
Dynamically deteriorating clinical situation (i.e., trauma)
GI bleed
Impaired airway reflexes
↓ Muscle tone of structures in the airway (i.e., tongue, pharyngeal walls, & soft palate)
Patients who did not stop GLP-1 agonist preoperatively as advised
Impaired clearance of secretions or vomitus
↓ Safe apnea time before hemodynamic decompensation
Unprotected airway
Need for rapidly securing airway while avoiding aspiration & hemodynamic compromise
Rapid sequence intubation (RSI): Simultaneous administration of induction agent (unconsciousness) & neuromuscular blocking agent (paralysis) to achieve intubation conditions (~45-60 seconds after IV push) for rapid control of an emergency airway
Preoxygenation
Deranged physiologic conditions (i.e., hypotension, acidosis, hypoxemia)
Reduced tolerance for
apnea (period with no ventilation or oxygenation)
Pre-oxygenate with high flow O2 (15L) to create a large pulmonary & tissue reservoir of oxygen
↓ Significant oxygen desaturation during apnea
↑ Oxygen saturation on pulse oximetry
Induction
Laryngoscopy & intubation are a potent sympathetic nervous system stimulus
Airway manipulation causes a surge in catecholamines
Paralysis
Visualization & passage of endotracheal tube requires relaxation of vocal cords & surrounding muscles
Neuromuscular blocking agents facilitate paralysis
Rescue
Some induction agents (i.e., propofol) are vasodilators
Hemodynamically unstable or patients in shock
Hypotension
Tachycardia
↑ Intracranial pressure (ICP)
Hypertension
Suppress cough & gag reflex
Prevent laryngospasm (involuntary closure of vocal cords to airway manipulation)
Minimize movement during procedure
Vasoactive agents (i.e., ephedrine, phenylephrine) ↑ systemic vascular resistance
Atropine & glycopyrrolate ↑ heart rate
Lidocaine (Na+ channel blocker) & opioids (μ receptor agonist) ↓ transmission of pain
↓ Sympathetic response, myocardial demand & physiologic stress
Anesthetics (i.e., propofol) achieve unconsciousness for paralysis & intubation
↓ Airway trauma & damage to vocal cords
Bag mask ventilation typically avoided in this step to ↓ gastric insufflation & risk of aspiration
Cricoid pressure (Sellick maneuver): posterior displacement of cricoid ring to compress esophagus against C-spine to prevent passive regurgitation of gastric contents to airway. Applied from start of induction, released when placement of endotracheal tube is confirmed by capnography.
Intubation
↑ Blood pressure and/or cardiac output
Authors: Jen Guo Reviewers: Priyanka Grewa Luiza Radu Leyla Baghirzada* * MD at time of publication
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published November 18, 2024 on www.thecalgaryguide.com")
Pelvic Ring Fractures

Diastasis (separation) of pubic symphysis
Anterior Posterior Compression (APC)
Axial shear force (e.g. fall)
Superior or inferior displacement of hemipelvis
Vertical Shear (VS)
Grade 3: APC 2 with additional disruption of posterior sacroiliac ligaments
Lateral compression force
(e.g. automobile collides with pedestrian)
Internal rotation of hemipelvis
Lateral Compression (LC)
Authors: Meaghan Mackenzie Stephanie de Waal Nojan Mannani Reviewers: Annalise Abbott Usama Malik Sunawer Aujla Mankirat Bhogal Michelle J. Chen Dr. Prism Schneider* Dr. Alyssa Federico* * MD at time of publication
Grade 1: pubic symphysis diastasis < 2.5 cm
Grade 2: pubic symphysis diastasis >2.5 cm, anterior sacroiliac diastasis, disruption of sacrospinous and sacrotuberous ligaments
Grade 1: pubic rami fracture with ipsilateral sacral compression fracture
Grade 2: pubic rami fracture with ipsilateral sacral and ilium fractures
Grade 3: ipsilateral LC 1 or 2 injury and contralateral APC injury (windswept pelvis)
Young-Burgess Classification of Pelvic Ring Fractures
(Combination of any of the three classes can also occur)
Blood vessels surrounding the fracture site rupture during injury
Pelvic has capacity of hold large volume of blood
Supporting ligaments are stretched or ruptured
Damage to adjacent urogenital structures
Gross hematuria
Posterior urethral tear or bladder rupture
Scrotal, labial or perineal hematoma
Displacement of hemipelvis
Limb-length discrepancy
Damage to adjacent neurological structures
Flank hematoma
Potential hemorrhagic shock
Loss of rectal tone or perirectal sensation
L5 or S1 dermatomal paresthesia
Chronic instability
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published Nov 16, 2017; updated Nov 22, 2024 on www.thecalgaryguide.com")
Tonsillitis Pathogenesis and clinical findings

Group A Streptococci (GAS) (most common bacteria)
Group B, C & G Strep,
Fusobacterium necrophorum
Age 5-15 (tonsils have ↑ role in immune function at this age)
Tonsillitis
Inflammation of the tonsils
Infectious agent exposure
Susceptible host Pathogen colonizes the oropharynx
Acute suppurative disease
Immune cells release proinflammatory cytokines & antibodies
Inflammatory mediators ↑ vascular permeability of tonsils
Leakage of protein & fluid into surrounding tissue
Regional nodes receive ↑ lymph
Enlarged anterior cervical nodes
Sinusitis**
Pharyngitis**
Local spread of pathogen
Acute otitis media**
Pneumonia**
Cervical lymphadenitis
Bacteria spread from
tonsils into lymphatic system & bloodstream
Bacteremia
F. necrophorum
invades lateral pharyngeal space & soft tissue in neck
Thrombosis forms in peritonsillar vein
Thrombosis extends into internal jugular vein
Lemierre’s syndrome
Systemic inflammatory cytokines disrupt hypothalamic regulation
Fever
Additional immune cells are recruited to facilitate immune response
Macrophages phagocytize pathogen
Bacteria invade distant tissue & elicit local inflammatory response
Hepatitis Osteomyelitis
Infective endocarditis
Bacteria illicit systemic response
Sepsis
Tonsillar tissue become swollen & irritated
Tonsillar hypertrophy
Localized collection of pus forms
Immune cells cause inadvertent cellular injury & hemolysis
Palatal petechiae
Meningitis
Products of immune response & cellular debris are deposited into tonsillar tissue
Peritonsillar or Tonsillar retropharyngeal abscess** exudate
** See corresponding Calgary Guide slide
Toxin-mediated disease
Bacteria release exotoxins into bloodstream
Inflammatory mediators & cytokines are overactivated (cytokine storm)
Skin has local inflammatory response
Toxic shock syndrome**
Scarlet fever**
Post-infectious disease
Antibodies to GAS cross react with host tissue
Acute rheumatic fever** Post-strep glomerulonephritis
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published Nov 5, 2018; updated Mar 14, 2025 on www.thecalgaryguide.com")
Varicella Zoster Virus
 - Chicken Pox: Pathogenesis and clinical findings Exposure to
individual with
VZV infection
VZV enters
respiratory tract
& conjunctiva
VZV enters
dendritic
cells (DC)
DCs travel to
regional
lymph nodes
Primary viremia
Authors:
Morgan Garland,
Sean Spence
VZV replicates
VZV proteins ↑ major
↓ MHCI
Reviewers:
in nasopharynx
histocompatibility
expression
Yan Yu, Danny Guo,
& CD4+ T cells
complex class I
on CD4+ T
Emily J. Doucette,
(MHCI) degradation
cell surface
Laurie Parsons*,
James D. Kellner*
*MD at time of publication
Acute phase
VZV evades the immune system
VZV travels through the blood in CD4+ T cells
Immune cells release
cytokines to fight the
virus (stronger in adults)
Fluid & cellular debris
accumulate between
epidermal layers
Generalized pruritic (itchy) vesicular
rash at different stages of development
(macule, papule, vesicle)
Immune system activates
& releases inflammatory
compounds
VZV replicates in liver,
spleen & lymph nodes
Erythema
(redness) & pain
Scratching lesions
introduces bacteria
Fibroblasts lay down
collagen to heal skin lesions
Resets thermostat
in hypothalamus
Systemic
response
Tissue damage
in oropharynx
Direct
inflammation in
neural tissues
Cerebellar ataxia
(more common
in children)
Bacterial superinfection from S.
aureus & Streptococcus pyogenes
(more common in children)
Scarring
Fever
(prodromal)
Malaise
(prodromal)
Sore throat
(prodromal)
Encephalitis (more
common in adults)
Abscess
Cellulitis
Toxic shock syndrome (TSS)
Secondary viremia
Immune cells release cytokines to
fight infection (stronger in adults)
Inflammation & fluid
build up in lungs
VZV travels through the blood in CD4+ T
cells expressing skin homing receptors
Fluid & cellular
debris accumulate
in damaged alveoli Lungs more susceptible to
secondary bacterial infection
Pneumonia** (more
common in adults)
VZV colonizes
keratinocytes
over several days
VZV colonizes
neurons & glial
cells in the brain
VZV colonizes
alveolar
epithelial cells
Resolution & latency
VZV tracks along
neurons from sites of
infection to dorsal root
ganglia & cranial nerves
↓ MHCI expression
in sensory ganglia
Adaptive
immune system
controls viremia
↓ Viral gene expression
Resolution of VZV
infection &
development of
lifelong immunity
Direct viral toxicity (DNA damage, mitochondrial
dysfunction, & protein misfolds in infected cells)
Apoptosis & dysfunction of infected cells
Latent
infection
Stress or immune suppression may
precipitate reactivation of VZV later in life Shingles**
**See corresponding Calgary Guide slide
Pathophysiology Mechanism
Published Nov 12, 2012; updated Jun 22, 2025 on www.thecalgaryguide.com
Legend: Sign/Symptom/Lab Finding Complications")
Traitement du choc

Choc cardiogenique

Choc distributif

Hemodynamic Changes in Pregnancy
 formation
↑ Risk of
disseminated
intravascular
coagulation**
Thrombus forms in deep
vein of leg, calf, or pelvis
↑ Plasma
volume
↑ Hydrostatic pressure
in peripheral vessels
pushes fluid into
extravascular spaces
↑ Water in vessels
↓ red blood cell
(RBC) concentration
(↓ hematocrit)
↑ Risk of
blood loss
during
Peripheral edema
↑ Blood flow to uterus
delivery Mild tachycardia
to supply fetus with
oxygen & nutrients
Anemia early in
pregnancy (first
trimester)
Kidneys detect ↓ oxygen levels
from ↓ hematocrit & upregulate
erythropoietin production
Erythropoietin stimulates ↑ RBC
production in bone marrow
Gestational
hypertension
(new onset
hypertension
after 20 weeks
gestation)
↑ Hematocrit later in pregnancy
Deep vein
thrombosis**
Authors:
Orly Aziza
Alam Randhawa
Reviewers:
Riya Prajapati
Michelle J. Chen
Jessica Revington
Rachel Wang*
* MD at time of publication
** See corresponding Calgary Guide slide
Legend: Pathophysiology Mechanism
↑ Risk of developing preeclampsia**
Sign/Symptom/Lab Finding Complications
Pale or
Cold
Dizziness
clammy skin Fatigue
↑ Risk of shock
↑ Risk of
organ damage
↑ Risk of syncope
Published Aug 25, 2025 on www.thecalgaryguide.com
Thrombus travels
through venous
circulation to the
heart & into the
pulmonary arteries
Pulmonary
embolism")
Choc obstructif

Obstructive Shock

↓ Right ventricle stroke volume
(blood volume ejected per beat)
↓ Blood return to left atrium
Underfilled left ventricle
Obstructive shock
↓ systemic blood flow due to a physical
obstruction of the heart’s pumping function
Insufficient
organ
perfusion
Tension pneumothorax**
↑ Pressure in the pleural cavity
Intrathoracic pressure exceeds
venous filling pressures &
compresses heart chambers
↓ Venous return to right
atrium & left atrium
Author:
Ryan Dion
Sergio F. Sharif
Dean Percy
Reviewers:
Yan Yu
Tristan Jones
Jason Waechter*
* MD at time of publication
** See corresponding
Calgary Guide slides
↓ Preload (degree of left ventricular filling)
as left ventricle receives low levels of blood
↓ Cardiac output (volume of
blood ejected from the
↓ Blood pressure (BP)
heart/min)
Baroreceptors in carotid sinus & aortic
arch detect low blood pressure
Skin
Reflexive release of catecholamines
via sympathetic nervous system
Blood supply diverted
away from peripheral
tissues to vital organs
Tachycardia
Hypotension
Cold
extremities
Legend: Pathophysiology Mechanism
Sign/Symptom/Lab Finding Brain
↓ Cerebral blood
flow causes
cerebral hypoxia
Altered consciousness
Heart
↓ Coronary
artery
perfusion
Myocardial
ischemia**
Complications
Blood backs up into
venous system
Kidneys
↓ Blood flow
to kidneys
Prerenal acute kidney
injury **
↑ Pressure distends
veins
↑ Jugular
venous
pressure
Pressurized fluid leaks
out of veins into
tissue
Fluid enters
pulmonary
perivascular spaces
Fluid impedes normal gas
exchange in lungs
Tachypnea (↑
respiratory rate)
Dyspnea
Published July 7, 2013; updated Sept 28, 2025 on www.thecalgaryguide.com")
Acute Tubular Necrosis

Septic shock** Cardiogenic shock**
Cholesterol embolization
syndrome (rare)
Severe hypotension
↓ Mean arterial pressure
reduces renal perfusion pressure
which ↓ renal blood flow
Renal vascular autoregulation
fails to maintain renal perfusion
Tubular cell hypoxia & depletion
of adenosine triphosphate (ATP)
(primary energy carrier in cells)
Cell ion pump failure, cell swelling,
& cell membrane disruption
Ischemic tubular necrosis (renal tubular
cell injury secondary to ↓ blood flow)
Myoglobinuria
Hemoglobinuria
Aminoglycosides
Ethylene glycol
Amphotericin
Cisplatin
↓ Efficiency of kidney reabsorption & excretion
Kidney removes ↓
potassium (K+) from
the blood volume
Hyperkalemia** (high K+
concentration in the blood)
↑ Serum K+ levels
induce abnormal
cardiac electrical
conduction patterns &
excessive cardiac
excitability
Fatal cardiac
dysrhythmia (irregular
heartbeat pattern)
Kidney removes
↓ urea from the
blood volume
Kidney removes ↓
creatinine from the
blood volume
Azotemia (↑ serum
urea & creatinine)
Uremic toxins
systemically
impair
neutrophil
function
Uremic toxins (indoxyl
sulfate or paracresyl
sulfate) induce oxidative
stress in the brain through
accumulation of reactive
oxygen species (ROS)
↑ Risk of infection
ROS damage neuronal membranes & ion channels &
induce dysfunctional mitochondria in the brain
Authors:
Adam Bubelenyi
Reviewers:
Britney Wong
Luiza Radu
Jessica Revington
Veronica Hammer*
Braden Manns*
* MD at time of publication
**See corresponding Calgary Guide slide
Dysfunctional brain mitochondria cannot
properly metabolize purines & urea
Brain cannot use any metabolic or cellular pathways requiring ATP
↓ Cerebral neuronal signaling
Legend: Pathophysiology Mechanism
Sign/Symptom/Lab Finding Intravenous (IV)
administration of
iodinated
contrast medium
Tumor lysis syndrome
Cancer cell apoptosis (cell death)
releases ↑ uric acid & phosphate
Heme-containing
pigments
Medications
Uric acid & calcium phosphate
crystals precipitate in renal tubules
Nephrotoxic substances damage renal tubular cells through unique mechanisms
Lipid peroxidation
(oxidative degradation) of
lipid bilayer weakens renal
tubular cell membranes
Damage to mitochondria in tubular
cells causes depletion of ATP
Formation of oxygen free
radicals in renal tubular cells
Impairment of ATP-
dependent processes
Activation of pro-inflammatory
cytokines & enzymes in the kidney ↑ Renal tubular cell
permeability
Disruption of protein synthesis &
enzyme function in renal tubular cells
Degradation of tubular cell
membrane lipids & proteins
Free radicals
oxidize tubular
cell proteins &
impair structure
& function
Toxic tubular necrosis (renal tubular injury due to nephrotoxic substances)
Acute Tubular Necrosis
Acute kidney injury via renal tubular cell damage & cell death
Denudation (removal of surface tissue layers) &
erosion of the tubular basement membrane
Kidney removes ↓
sodium (Na+) & water
from the blood volume
↓ Bicarbonate
reabsorption from
the proximal tubule
of the kidney
Necrotic tubular cells fall into tubular lumen
Ongoing tubule damage
↓ ability to concentrate
urine solutes
↓ Bicarbonate
neutralization of
acids in the blood
Necrotic
tubular cells
release
intracellular
contents
Obstruction of
tubular lumen
impedes
glomerular
filtration
↓ Urine osmolality
(low urine solute
concentration)
↑ Water in blood
volume dilutes
Na+ serum
concentration
Metabolic acidosis
(blood pH <7.35)
Muddy brown
granular casts
↓ Glomerular
filtration rate (GFR)
Hyponatremia** (low Na+
concentration in the blood)
↑ Na+ retention in
the circulatory blood
volume & ↓ Na+
excretion in the urine
↓ Urine output
Accumulation of ROS activates microglia
& releases pro-inflammatory cytokines
↑ Fluid retention in
the circulatory system
Aggressive IV fluid
rehydration in septic
shock or cardiogenic
shock patients
Cerebral inflammation ↑ oxidative
damage to neurons & glial cells
Fluid overload
Uremic encephalopathy
Pulmonary edema (excess
fluid accumulation in lung
alveoli & interstitium)
Published November 15, 2025 on www.thecalgaryguide.com
Complications
Disruption of
DNA & RNA
synthesis
within renal
tubular cells
Tubular epithelial
cells in urine
Epithelial cell
casts in urine
(cast composed
of epithelial
cells embedded
in a matrix)
↓ Ability to clear
bacteria in urine
↑ Risk of urinary
tract infection")
Heart Transplant Indications and Benefits

Used as bridge to transplant
Failure of treatment with
conventional heart failure
therapies
Cardiac transplant recommended
Major systemic diseases,
including cancer
Assess surgical readiness and
comorbidities with evaluation by
cardiac surgeon & cardiologist
Psychological Instability
Active or recent
substance abuse
Bridge to transplant with
inotropes or mechanical
circulatory support devices
(VAD, ECMO, IABP)
Cardiac Transplant
Surgery to replace dysfunctional
heart with a functional donor heart
**See corresponding Calgary Guide slide(s)
Legend: Pathophysiology Mechanism
Sign/Symptom/Lab Finding Complications
Heart Transplant: Indications & Benefits Indications
Kate Fougere
Authors:
Reviewers:
Emily Kuervers
Sergio F. Sharif
Dr. Robert Miller*
Severe cardiomyopathy
Myocardial dysfunction
* MD at time of publication
Disease of the cardiac
Pump Failure
↓ Stroke volume (SV),
muscle (Dilated,
↓ Forward flow,
↓ cardiac output (CO),
hypertrophic, restrictive
backup into lungs
↓ blood pressure (BP)
cardiomyopathies**)
Acyanotic heart
disease
Normal concentration
of oxygenated Hb
↓ Perfusion
of organs with
↑ pulmonary
congestion
Congenital Heart Disease**
Disruption of normal cardiac
formation that is
unsuccessfully treated with
surgical repair
Cyanotic heart disease
↑ Concentration of
deoxygenated
hemoglobin (Hb)
Cyanosis
(dusky, blue
skin
discoloration)
Cardiac Allograph
Vasculopathy (CAV)
Diffuse concentric narrowing
of coronary arteries
Antibody mediated graft rejection &
fibrosis
Symptoms of
heart failure **
Cardiovascular risk factors exacerbated
by immunosuppressive drugs
↓ BP
Reduced SV with
no response to
medical therapy
Pump Failure
↓ Forward flow,
backup into lungs
↓ Perfusion of organs
↓ Level of consciousness
End organ damage
Abnormal electrical activity does not respond to
pharmacological agents, cardiac ablation or
electrophysiological interventions (ICD)
Refractory arrythmias (ventricular
tachycardia, ventricular fibrillation) &
subsequent cardiac death
Device complications arise
while awaiting
transplantation
Device infection
Clotting or bleeding complications
(stroke or hemorrhage)
Abbreviations:
• ICD - Implantable cardioverter defibrillators
• ECMO- Extracorporeal membrane oxygenation
• IABP - Intra-aortic balloon pump
Low survival benefit from transplant
↓ Capacity to manage post transplant
requirements
Graft rejection
↑ Potential for relapse after transplant
↓ Symptoms of heart failure**
Restoration of cardiac output &
normalization of volume status
↑ Life expectancy
↑ Quality of life
Published Nov 23, 2025 on www.thecalgaryguide.com")
Childhood Immunization Schedule

Spores enter contaminated wounds &
bacteria produce tetanospasmin to
invade central nervous system (CNS)
Muscle rigidity spasms,
hyperreflexia, autonomic
dysfunction, laryngeal spasms
↑ Mortality from
respiratory
obstruction & failure
Bacteria attach to ciliated
respiratory epithelial cells &
release toxins
Prolonged paroxysmal cough,
inspiratory “whoop” sound, emesis,
apnea, cyanosis, leukocytosis
↑ Risk of
pneumonia, seizures
& encephalopathy
Virus invades oropharynx/GI tract &
replicates in lymphoid tissue before
hematogenous spread to motor neurons
↑ Risk of paralytic
poliomyelitis &
respiratory failure
Bacteria colonize the
nasopharynx & invade
the bloodstream & CNS
Virus invades hepatocytes
via specific receptors &
replicates within liver cells
Bacteria colonize the
nasopharynx & may invade the
lungs, bloodstream, or meninges
Virus invades mature
enterocytes in the
small intestine
Bacteria colonize the nasopharynx
& enter the bloodstream to cross
the blood–brain barrier
Virus invades respiratory
epithelium & spreads to regional
lymphoid tissue & bloodstream
Virus infects upper respiratory tract
(URT) & disseminates via viremia to
salivary glands & other organs
Virus invades URT &
enters the bloodstream &
regional lymphoid tissue
Virus infects URT &
lymphoid tissue before
invading neural tissue
Virus infects anogenital
& oropharyngeal basal
layer epithelium tissue
Influenza virus
Virus invades upper
& lower respiratory
epithelium
Severe acute respiratory
syndrome coronavirus 2
(SARS-CoV-2)**
Viral invasion of mucous
membranes & may invade
extrapulmonary tissues
Legend: Muscle weakness,
asymmetric reduction
in tone, quadriplegia
Fever, fatigue, shortness of breath,
nausea, emesis, headache, stiff neck,
altered mental status, otitis media**
Fatigue, anorexia, nausea,
jaundice, arthralgia, right
upper quadrant pain
Otitis media**, sinusitis,
pneumonia**, meningitis**,
bacteremia
Gastroenteritis with
emesis, fever, diarrhea,
malaise & dehydration
Fever, headache, neck
stiffness, altered level of
consciousness, purpuric rash
Koplik spots, conjunctivitis, fever,
rhinorrhea, cough, diarrhea, otitis
media, pneumonia
Parotitis, headache, fever, malaise,
sensorineural hearing loss, orchitis,
mastitis, oophoritis, pancreatitis
Fever, lymphadenopathy, rash,
congenital anomalies such as hearing
loss, cataracts & cardiac defects
↑ Mortality from
meningitis,
pneumonia & sepsis
↑ Risk of cirrhosis,
hepatic malignancy,
& liver failure
↑ Mortality from
respiratory, CNS &
cardiac dysfunction
↑ Mortality from
hypovolemic shock
& CNS infection
↑ Mortality from
sepsis, multiorgan
failure & necrosis
↑ Mortality from
pneumonia &
encephalitis
↑ Mortality
from meningitis
& encephalitis
↑ Mortality
from congenital
rubella
Vesicular & pruritic rash,
fever, malaise, pneumonia,
encephalitis, cellulitis
↑ Morbidity from necrotizing
fasciitis, CNS, soft tissue &
respiratory infections
Often asymptomatic, or
may present with painless
anogenital warts
↑ Risk of anogenital
& head & neck
malignancy
Fever, cough, myalgia, malaise,
cough, headache, emesis, diarrhea,
abdominal pain, febrile seizures
↑ Mortality from
widespread
multiorgan infection
Fever, cough, fatigue, shortness of
breath, anosmia, ageusia,
pneumonia, multiorgan dysfunction
↑ Mortality from
respiratory distress
& multiorgan failure
DTaP-IPV-Hib-HB vaccine
Given at 2, 4 & 6 months old
Protects against
diphtheria, Tetanus,
Pertussis, Polio,
Haemophilus influenzae
type B, & Hepatitis B
DTaP-IPV-Hib vaccine
Given at 18 months old
Protects against
diphtheria, Tetanus,
Pertussis, Polio, &
Haemophilus
influenzae type B
Pneumococcal conjugate vaccine
Given at 2, 4 & 12 months old (additional 4th dose given at
6 months if at ↑ risk of invasive pneumococcal disease)
Authors:
McKayla Kirkpatrick, Stacey Holbrook
Reviewers:
Merry Faye Graff, Emily J. Doucette, Charissa
Chen, Amanda Ang, Danielle Nelson*
* MD at time of publication
Tdap-IPV vaccine
Given at 4 years old
Tdap vaccine
Given in Grade 9
Protects against
diphtheria, Tetanus,
Pertussis, & Polio
Protects against
diphtheria, Tetanus,
& Pertussis
HB vaccine
Given in Grade 6
Protects against
Hepatitis B virus
Protects against
Streptococcus pneumoniae
Rotavirus vaccine
Given at 2 & 4 months old
Protects against rotavirus
Meningococcal conjugate (MenconC)
Given at 4 & 12 months old
MenC-ACYW
(Meningococcal type A,
C, Y, W-135) in Grade 9
Protects against
Neisseria meningitidis
MMR-Var vaccine (e.g., Priorix-Tetra)
Given at 12 & 18 months old
Protects against measles,
mumps, rubella & varicella
Pathophysiology Mechanism
Sign/Symptom/Lab Finding Complications
HPV vaccine
Given in Grade 6 (2-3 doses over 6 months)
Seasonal influenza vaccine
Given annually starting at 6 months or older
COVID-19 vaccine
Given at 6 months or older Published Aug 19, 2015; updated Dec 31, 2025 on www.thecalgaryguide.com
Protects against HPV
Protects against influenza viruses
predicted to circulate each fall & winter
Protects against SARS-CoV2")
Necrosis versus Apoptosis

Hypoxia
(↓ Oxygen
delivered to tissues)
Physical agents
(extreme temperatures,
trauma, radiation)
Bacterial, viral, or
fungal infections
&/or microbe toxins
Necrosis
Uncontrolled & pathologic cell death
Harmful stimuli trigger ↓ ATP (cellular energy)
↓ ATP leads to ion pump failure & ↑ cell permeability (“leakiness”)
↑ Cellular ion influx causes cell to swell & lyse (rupture its plasma membrane)
Intracellular contents spill into the extracellular space
Intracellular contents activate inflammasomes (cytoplasmic multiprotein complexes)
Pro-inflammatory cytokine interleukin-1β is secreted & activates the immune system
Excessive cytokine release provokes a large inflammatory response
Inflammation damages surrounding healthy tissues & organs
Ischemia (↓ Blood
flow) to organs
except brain
↓ Oxygen denatures
enzymes à dead cell
skeletons cannot breakdown
Cells remain
“coagulated”
(firm) for days
Coagulative
necrosis
↓ Blood flow to limb(s)
+/- bacterial invasion
Gangrenous
necrosis
↑ Pain or ↓ feeling in affected limb(s)
Skin color changes (red, purple, black)
Damaged adipocytes (fat cells) release
triglyceride (fat) contents that group into oil cysts
Scars form &
calcify (harden)
Fat necrosis
Damaged endothelium (blood
vessel wall) becomes leaky
Fibrin (blood protein) deposits in
endothelium with immune complexes
Fibrinoid
necrosis
Brain ischemia &/or
infection kills cells
Digestive enzymes transform dead
tissue into creamy liquid pus
Liquefactive necrosis
Coagulative + liquefactive necrosis mechanisms à granular debris
Caseous necrosis
Legend: Pathophysiology Mechanism
Sign/Symptom/Lab Finding Complications
-----------------------------------------------------------------------------------------------------
Authors:
Tyra Fernandes
Catherine R Jarvis
Reviewers:
Mitchell Chorney
Sarah Smith*
* MD at time of publication
Apoptosis
Programmed cell death via an energy-dependent process
Extrinsic pathway: cellular stress or
damage signals in the extracellular
environment trigger apoptosis
Intrinsic pathway: internal
cellular stressors (damage from
chemotherapy, UV radiation,
drugs) trigger apoptosis
Ligands such as TNF-α (tumor
necrosis factor-alpha, produced by
macrophages) & FasL (Fas ligand,
produced by T cells) bind to apoptotic
receptors on the cell’s surface
Pro-apoptotic factors recognize
cellular stressors
Cytochrome C releases from the
intermembrane space of the
mitochondria to the cytoplasm
Ligand binding activates initiator
caspases (protease (protein
breakdown)-like enzymes) 2, 8, 9, 10
Cytochrome C activates Caspase-9
Initiator caspases activate
effector caspases 3, 6, 7
Caspase-9 activates
effector caspases 3 & 7
Effector caspases begin execution pathway
Caspases degrade the cell’s nuclear envelope & DNA
à DNA repair is inhibited & cell shrinks
Cellular shrinkage forms apoptotic bodies
Phagocytes engulf apoptotic bodies to prevent inflammation
Ineffective phagocytosis à inflammation
Published Feb 8, 2026 on www.thecalgaryguide.com
Final Slide is 1st
Necrosis vs Apoptosis: Pathogenesis and clinical findings
Toxic chemicals
(poisons, drug
toxicities)
Hypoxia
(↓ Oxygen
delivered to tissues)
Physical agents
(extreme temperatures,
trauma, radiation)
Toxin release
from bacteria,
viruses, or fungi
Necrosis
Uncontrolled & pathologic cell death
Harmful stimuli trigger ↓ ATP (cellular energy)
↓ ATP leads to ion pump failure & ↑ cell permeability
↑ Cellular ion influx causes cell to swell & lyse (rupture its plasma membrane)
Intracellular contents spill into the extracellular space
Intracellular contents activate inflammasomes (cytoplasmic multiprotein complexes)
Pro-inflammatory cytokine interleukin-1β is secreted and activates the immune system
Excessive cytokine release provokes a large inflammatory response
Inflammation damages surrounding healthy tissues & organs
Ischemia (↓ Blood
flow) to organs
except brain
↓ Oxygen denatures
enzymes à cell skeleton
cannot be broken down
Cell is preserved
in “coagulated”
state for days
Coagulative
necrosis
↓ Blood flow to limbs
+/- bacterial invasion
Gangrenous
necrosis
↑ Pain or ↓ feeling in limb
Skin color changes (red, purple, black)
Damaged adipocytes (fat cells) release
triglycerides (fat) contents that group into oil cysts
Scars form &
calcify (harden)
Fat necrosis
Damaged endothelium (blood
vessel wall) becomes leaky
Fibrin (blood protein) deposits in
endothelium with immune complexes
Fibrinoid
necrosis
Brain ischemia &/or
infection kills cells
Digestive enzymes transform dead
tissue into creamy liquid pus
Liquefactive necrosis
Coagulative & liquefactive necrosis mixture à granular debris
Caseous necrosis
Legend: Pathophysiology Mechanism
Sign/Symptom/Lab Finding -----------------------------------------------------------------------------------------------------
Complications
Authors:
Tyra Fernandes
Catherine R Jarvis
Reviewers:
Mitchell Chorney
Sarah Smith*
* MD at time of publication
Apoptosis
Programmed cell death via an energy-dependent process
Extrinsic pathway: cellular stress or
damage signals in the extracellular
environment trigger apoptosis
Intrinsic pathway: internal
cellular stressors (chemotherapy,
UV radiation, drugs, misfolded
proteins) trigger apoptosis
Ligands such as TNF-α (tumor
necrosis factor-alpha, produced by
macrophages) & FasL (Fas ligand,
produced by T cells) bind to apoptotic
receptors on the cell’s surface
Pro-apoptotic factors recognize
cellular stressors
Cytochrome C releases from the
intermembrane space of the
mitochondria to the cytoplasm
Ligand binding activates initiator
caspases (protease (protein
breakdown)-like enzymes) 2, 8, 9, 10
Cytochrome C activates Caspase-9
Initiator caspases activate
effector caspases 3, 6, 7
Caspase-9 activates
effector caspases 3 & 7
Effector caspases begin execution pathway
Caspases degrade the cell’s nuclear envelope & DNA
à DNA repair is inhibited & cell shrinks
Cellular shrinkage forms apoptotic bodies
Phagocytes engulf apoptotic bodies to prevent inflammation
Ineffective phagocytosis à inflammation
Published MONTH, DAY, YEAR on www.thecalgaryguide.com
Necrosis vs Apoptosis
Toxic chemicals
(poisons, drug
toxicities)
Hypoxia
(↓ Oxygen
delivered to tissues)
Physical agents
(extreme temperatures,
trauma, radiation)
Biological agents
(bacteria,
viruses, fungi)
Necrosis
Non-physiologic & uncontrolled cell death
Noxious (harmful) stimuli trigger increased cell permeability and ion pump failure
Cell swells & ruptures its plasma membrane à cell lysis (breakdown)
Intracellular contents spill into the extracellular space
Intracellular contents activate inflammasomes (cytoplasmic multiprotein complexes)
Pro-inflammatory cytokine interleukin-1β is secreted and activates the immune system
Excessive cytokine release provokes a large inflammatory response
Inflammation damages surrounding healthy tissues & organs
Acute tubular
necrosis
(damaged kidney
tubule cells)
Gangrene
(body tissue death)
Myocardial infarction
(heart attack)
Steatohepatitis
(progressive
liver disease)
↓ Blood flow to a
Myocardium (heart
portion of the body
muscle) is deprived
Necrotic renal
(usually limbs)
of oxygen &
(kidney) tubular
becomes damaged
epithelial cells
deteriorate &
slough off
↑ Extracellular
matrix proteins
(especially
collagen) are
produced and
deposit in liver
Granular casts
on urine
microscopy
↑ Pain
or
↓ Feeling
Skin
color
change
(red,
purple,
black)
↑ Blood
troponin
levels
↓ Heart
function
Liver fibrosis
(scarring)
Legend: Authors:
Tyra Fernandes
Catherine R Jarvis
Reviewers:
-----------------------------------------------------------------------------------------------------
Pathophysiology Mechanism
Sign/Symptom/Lab Finding Complications
Mitchell Chorney
Name Name*
* MD at time of publication
Apoptosis
Programmed cell death via an energy-dependent process
Extrinsic Pathway: Apoptotic signals
from other cells cause initiation
Intrinsic Pathway: Internal cellular
stressors (chemotherapy, UV
radiation, drugs, misfolded
proteins) cause initiation
Ligands such as TNF-α (tumor necrosis
factor-alpha, produced by
macrophages) & FasL (Fas ligand,
produced by T cells) bind to apoptotic
receptors on the cell’s surface
Pro-apoptotic factors recognize
cellular stressors
Cytochrome P releases from the
intermembrane space of the
Ligand binding activates initiator
mitochondria to the cytoplasm
caspases (protease (protein
breakdown)-like enzymes) 2, 8, 9, 10
Cytochrome P activates Caspase-9
Initiator caspases activate
effector caspases 3, 6, 7
Caspase-9 activates caspases
3 & 7
Execution Pathway
Caspases degrade the cell’s nuclear envelope & DNA
à DNA repair is inhibited & cell shrinks
Cellular shrinkage forms apoptotic bodies
Phagocytes engulf apoptotic bodies to prevent inflammation
Ineffective phagocytosis à inflammation
Published MONTH, DAY, YEAR on www.thecalgaryguide.com
Necrosis vs Apoptosis
Hypoxia
Chemical agents
Physical agents Biological agents
Necrosis
Non-physiologic & uncontrolled
cell death
Oncolysis (ion pump failure and
increased cell permeability
cause cell swelling) occurs due
to noxious stimuli
Plasma membrane ruptures and
cell is lysed (broken down)
Intracellular contents spill into
the extracellular space
Authors:
Tyra Fernandes
Reviewers:
Mitchell Chorney
Catherine R Jarvis
Name Name*
* MD at time of publication
Renal tubule is damaged due to
reduced oxygen supply and toxicity
Granular casts on urine microscopy
Acute tubular necrosis (kidney
disorder of the tubule cells
Extracellular matrix proteins accumulate
in liver causing collagen deposition
Liver fibrosis
Steatohepatitis
(progressive liver disease)
Intracellular contents activate
inflammasomes (cytoplasmic
multiprotein complexes)
Myocardium (heart muscle) is
deprived of oxygen
↑ Blood troponin levels
Myocardial infarction
-----------------------------------------------------------------------------------------------------
Apoptosis
Programmed cell death via an
energy-dependent process
Extrinsic Pathway: Apoptotic signals
from other cells cause initiation
Ligands such as TNF-α (tumor necrosis
factor-alpha, produced by
macrophages) and FasL (Fas ligand,
produced by T cells) bind to apoptotic
receptors on the cell’s surface
Ligand binding activates initiator
caspases (protease-like
enzymes) 2, 8, 9, 10
Initiator caspases activate
effector caspases 3, 6, 7
Intrinsic Pathway: Internal cellular
stressors (chemotherapy, UV
radiation, drugs, & misfolded
proteins) cause initiation
Pro-apoptotic factors recognize
cellular stressors
Cytochrome P releases from the
intermembrane space of the
mitochondria to the cytoplasm
Cytochrome P activates Caspase-9
Caspase-9 activates caspases
3 & 7
Pro-inflammatory cytokine
interleukin-1β is secreted and
activates the immune system
Excessive cytokine release
provokes a large inflammatory
response
Legend: Inflammatory
damage to the
surrounding
healthy tissues
Microscopic
Effects:
Pathophysiology Mechanism
Sign/Symptom/Lab Finding Complications
Execution Pathway
Caspases degrade the cell’s nuclear envelope. DNA
fragments inhibit DNA repair, which shrinks the cell
Cellular shrinkage forms apoptotic bodies
Phagocytes engulf apoptotic bodies to prevent inflammation
Ineffective phagocytosis à inflammation
Published MONTH, DAY, YEAR on www.thecalgaryguide.com
Necrosis vs Apoptosis
Authors:
Tyra Fernandes
Reviewers:
Mitchell Chorney
Catherine R Jarvis
Hypoxia, physical agents,
chemical agents, biologic agents
Necrosis (Non-physiologic
& uncontrolled cell death)
Oncolysis (cell swelling due to
the failure of ion pumps and
increased cell permeability)
occurs due to noxious stimuli
Plasma membranes rupture
leading to cell lysis
Intracellular contents spill into
the extracellular space
Inflammasomes (cytoplasmic
multiprotein complexes) are
activated
Pro-inflammatory cytokine
interleukin-1β is secreted and
activates the immune system
Excessive cytokine release
provokes a large inflammatory
response
Legend: Microscopic Effects:
Steatohepatitis
Collagen deposition due
to the accumulation of
extracellular matrix
proteins
Liver fibrosis
Myocardial
Infarction
Ischemic or toxic
injury to the renal
tubule
Granular casts on
urine microscopy
Acute tubular
necrosis
Oxygen supply
deprivation to the
myocardium
Elevated blood
troponin levels
-----------------------------------------------------------------------------------------------------
Extrinsic Pathway: Apoptotic Signals
from other cells cause initiation
Ligands such as TNF-α (tumor necrosis
factor-alpha, produced by
macrophages) and FasL (Fas ligand,
produced by T cells) bind to apoptotic
receptors on the cell’s surface
Ligand binding activates initiator
caspases (protease-like
enzymes) 2, 8, 9, 10
Initiator caspases activate
effector caspases 3, 6, 7
Apoptosis (Programmed
cell death via an energy-
dependent process)
Name Name*
* MD at time of publication
Intrinsic Pathway: Internal cellular
stressors chemotherapy, UV
radiation, drugs, & misfolded
proteins) cause initiation
Pro-apoptotic factors recognize
cellular stressors
Cytochrome P releases from the
intermembrane space of the
mitochondria to the cytoplasm
Cytochrome P activates Caspase-9
Caspase-9 activates caspases 3 and
7
Execution Pathway
Cellular shrinkage
forms apoptotic
bodies
Inflammatory damage to
surrounding healthy tissues à
potential organ failure
Pathophysiology Mechanism
Sign/Symptom/Lab Finding Complications
The nuclear envelope
degrades and DNA
fragments which
inhibits DNA repair
which shrinks the cell
Phagocytes engulf
apoptotic bodies to
prevent inflammation
Ineffective phagocytosis
à inflammation
Published MONTH, DAY, YEAR on www.thecalgaryguide.com
Authors:
Necrosis vs Apoptosis
Tyra Fernandes
Reviewers:
Mitchell Chorney
Necrosis Apoptosis
Catherine R Jarvis
Hypoxia: ischemia, respiratory
insufficiency
Physical agents: trauma, radiation,
Non-physiologic &
temperature, electrical shock
uncontrolled cell death
Biological agents: bacteria, viruses, fungi
Chemical agents: poisons, drugs,
Cell swelling (oncosis) due to
occupational exposures
the failure of ion pumps and
increased cell permeability
Collagen deposition leading
to liver fibrosis
Cell lysis due to the rupture of
the plasma membrane
Steatohepatitis
Microscopic
Spillage of intracellular contents
Effects:
into the extracellular space
Acute tubular
Myocardial
Activation of cytoplasmic
necrosis
Infarction
multiprotein complexes, known
as inflammasomes
Ischemic or toxic
Oxygen supply
injury to the renal
deprivation to the
tubule
myocardium
Secretion of pro-inflammatory
cytokine interleukin-1β, which
-----------------------------------------------------------------------------------------------------
Name Name*
* MD at time of publication
Programmed cell death via
an energy-dependent
process
Extrinsic pathway Intrinsic pathway
The cell receives apoptotic signals
Initiated by internal cellular
from other cells
stressors such as chemotherapy
drugs, UV radiation, and misfolded
Apoptosis is activated via ligands
proteins
binding to apoptotic receptors on
the cell surface
Pro-apoptotic factors recognize
cellular stressors
Ligand binding activates initiator
caspases (2, 8, 9, 10), which are
Cytochrome P releases from the
protease-like enzymes
intermembrane space of the
mitochondria to the cytoplasm
Examples of ligands include: TNF-α
(produced by macrophages) and
Caspase-9 is activated
FasL (produced by T cells)
Activation of downstream effector
Effector caspases (3, 6, 7) are
caspases 3 and 7
activated
activates the immune system
Excessive cytokine release
provokes a large inflammatory
response
Legend: Granular casts on
urine microscopy
Elevated blood
troponin levels
Damage to surrounding
healthy tissues may lead to
organ failure
Prevention of
inflammation
Execution pathway
The nuclear envelope
degrades and DNA fragments
which inhibits DNA repair
Cellular shrinkage
forms apoptotic
bodies
Phagocy
engulfment of
apoptotic bodies
Published MONTH, DAY, YEAR on www.thecalgaryguide.com
Pathophysiology Mechanism
Sign/Symptom/Lab Finding Complications
Necrosis versus apoptosis
Necrosis
Non-physiologic &
uncontrolled cell death due
to noxious stimuli
Cell swelling (oncosis) à cell
lysisà spillage of
intracellular contents à
tissue damage
Inflammatory response,
leading to inflammasome
activation and the secretion of
pro-inflammatory cytokine
interleukin-1β
Macroscopic
Effects:
Hypoxia: ischemia, respiratory insufficency
Physical agents: trauma, radiation,
temperature, electrical shock
Biological agents: bacteria, viruses, fungi
Chemical agents: poisons, drugs,
occupational exposures
Acute tubular
necrosis due to
chemical agents
Myocardial
infarction due to
hypoxia
Microscopic
Effects:
Stroke due to
hypoxia in the brain
Gangrene due to
hypoxia of the limbs
Steatohepatitis due
to chemical agents
-----------------------------------------------------------------------------------------------------
Apoptosis
Programmed cell death via
an energy-dependent
process
Execution phase: pro-& anti-
apoptotic proteins mediate
cell breakdown by caspases
Caspase activity blocks DNA
repair à leading to
fragmented DNA and cell
shrinkage
Apoptosis is
inhibited in cancer
the overexpression
of anti-apoptotic
proteins
Authors:
Tyra Fernandes
Reviewers:
Mitchell Chorney
Name Name*
* MD at time of publication
Extrinsic pathway: immune cell activation
activates executioner caspases; ligands such
as TNF-α & FasL bind to receptors on the cell
membrane
Intrinsic pathway: internal cellular factors
cause pro-apoptotic factors to increase
mitochondrial membrane permeability.
Cytochrome c is released from the inner
mitochondrial membrane, which ultimately
activates caspases
Cellular fragments are
engulfed by immune cells
which prevents an
inflammatory response
Organism death via
necrosis
Menstruation Embryogenesis
Endometrial
shedding of the
inner lining of
the uterus is an
apoptotic
mechanism
The tissue
between the
fingers and toes
degrades via
apoptosis in
utero à
separation of
the digits
Legend: Pathophysiology Mechanism
Sign/Symptom/Lab Finding Abbreviations:
• IL-1β– interleukin-1β
• TNF-α– tumour necrosis factor - alpha
• FasL– CD95 ligand; mediates apoptosis
Published MONTH, DAY, YEAR on www.thecalgaryguide.com
Complications
References
• https://next.amboss.com/us/article/VP0GdT?q=apoptosis#Y0d79977cce8fc2c548ac720f8060d667
• https://www.ncbi.nlm.nih.gov/books/NBK557627/
• https://www.ncbi.nlm.nih.gov/books/NBK499821/
• https://pmc.ncbi.nlm.nih.gov/articles/PMC5855670/
• https://www.sciencedirect.com/topics/pharmacology-toxicology-and-pharmaceutical-science/oncosis
• https://www.nature.com/articles/s41421-020-0167-x
• https://pmc.ncbi.nlm.nih.gov/articles/PMC3714593/
• https://www.ncbi.nlm.nih.gov/books/NBK537076/
Hypoxia
Physical agents
Biological agents
Chemical agents")
Shock da ustione
