Content
Collaboration
About Us
Contact Us
SEARCH RESULTS FOR:
Thalassemia
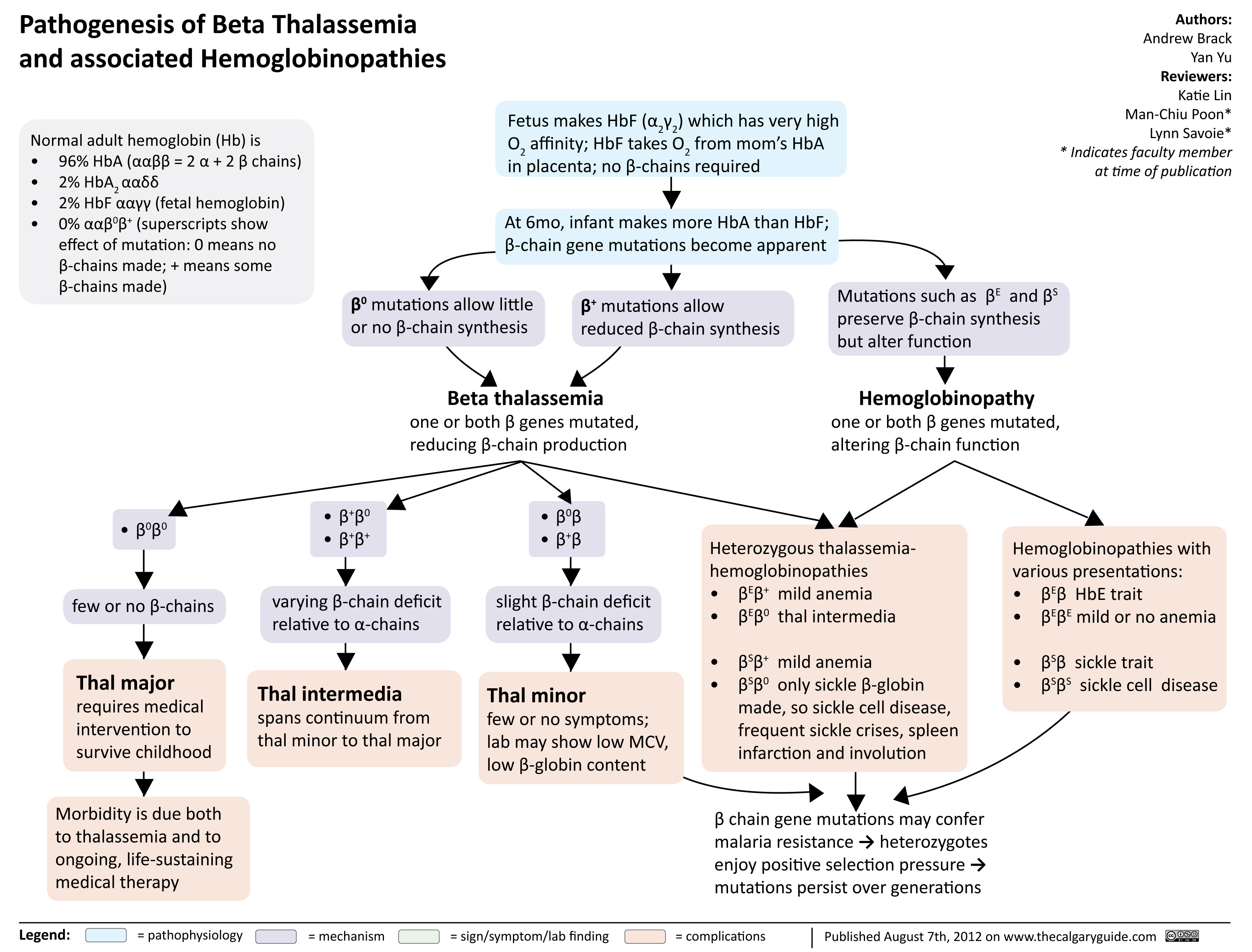
Pathogenesis of Beta Thalassemia
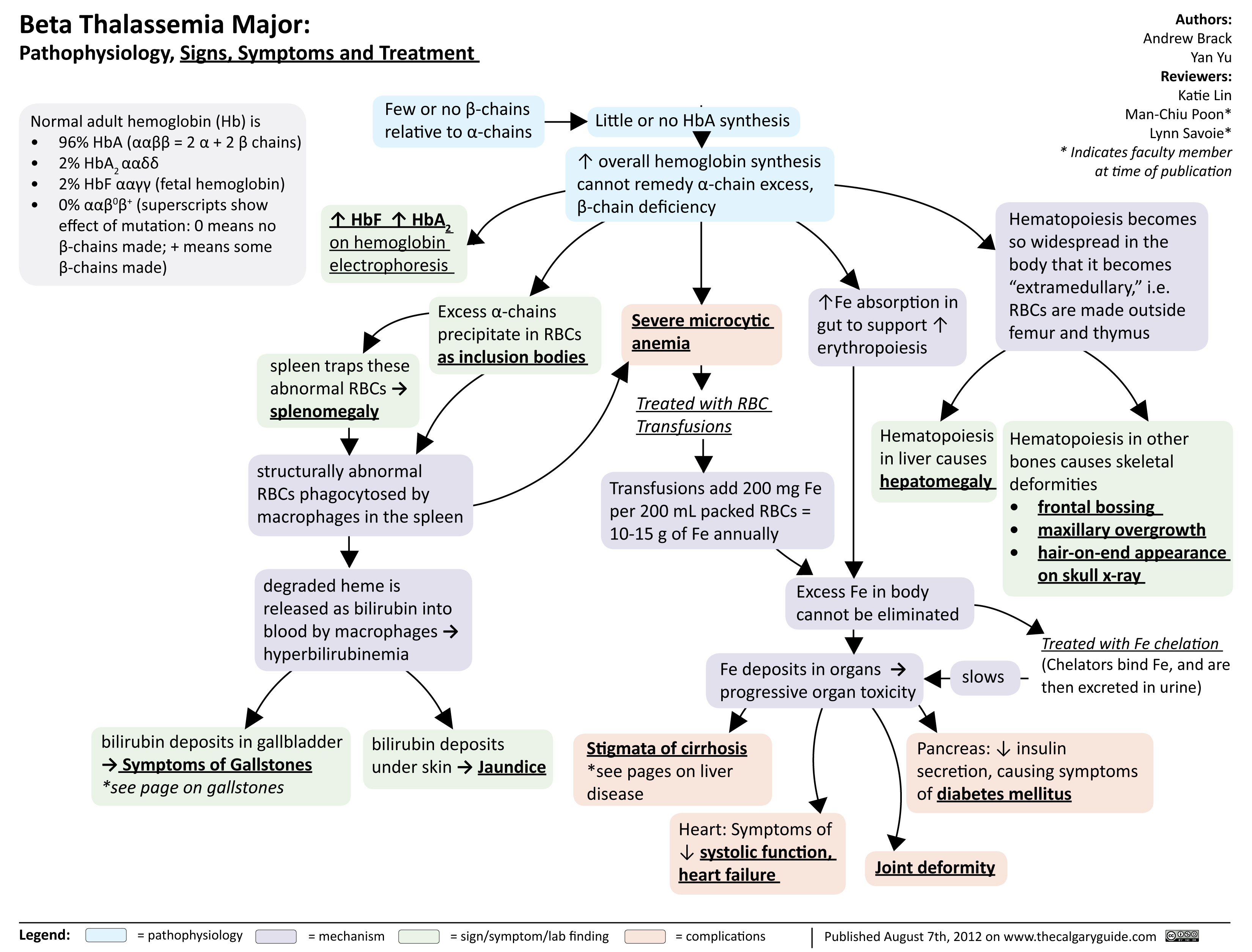
Beta Thalassemia Signs Symptoms Treatment
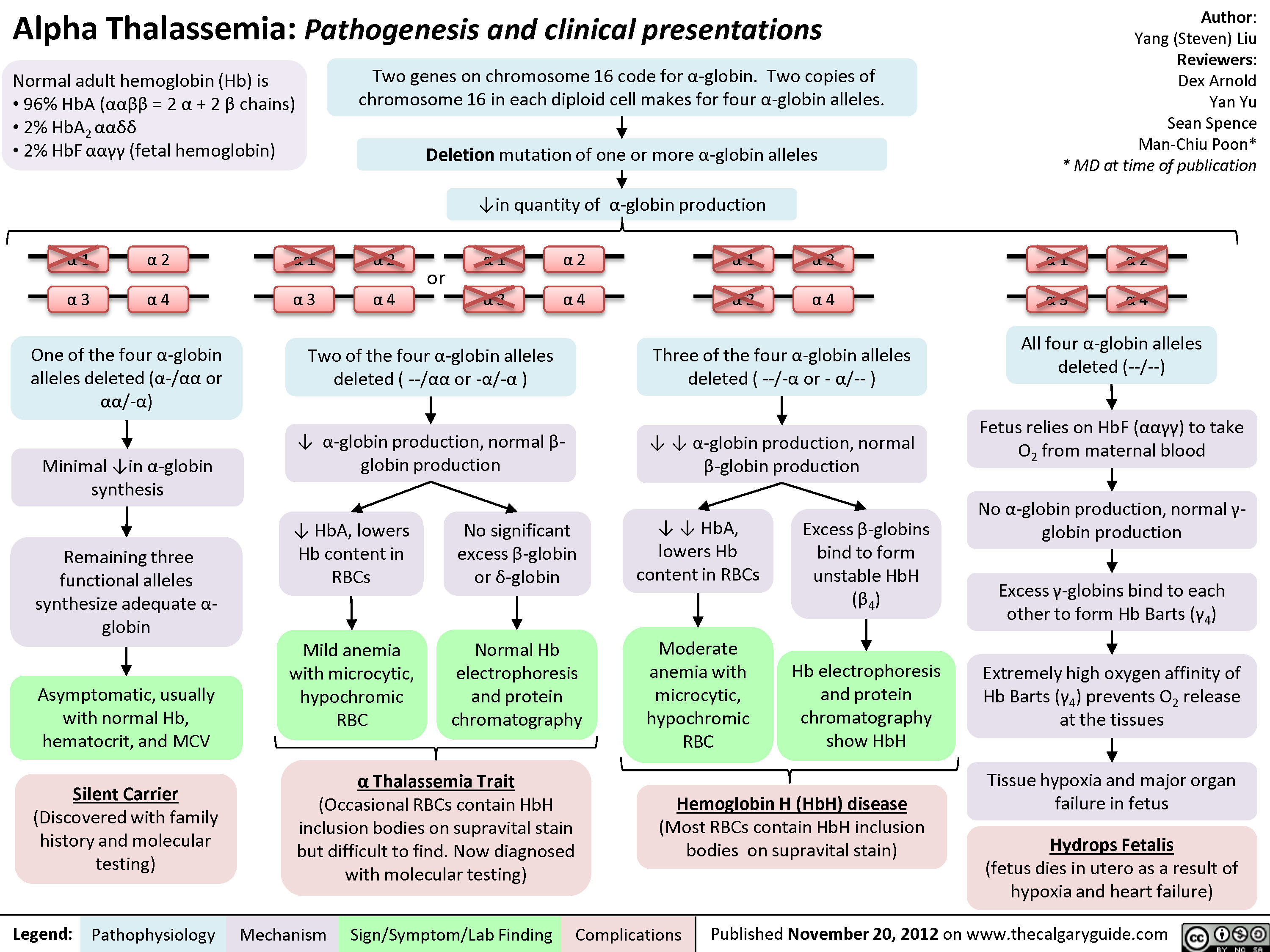
Alpha Thalassemia Pathogenesis
Hemolytic Anemia - Pathophysiology
beta-thalassemia-minor
Unconjugated Hyperbilirubinemia
 may result in slightly macrocytic anemia (because reticulocytes have larger volumes than RBCs)
Defects in the RBC’s environment
Defects in RBC membranes
Ex. Hereditary Spherocytosis:
mutation causing deficiency of RBC structural proteins like ankyrin or spectrin
RBC membranes become weakened and form blebs that break off
↓ RBC surface area while volume remains constantà RBC becomes spherical
Spherocytes in spleen trapped and phagocytosed by splenic macrophages (extravascular hemolysis)
Defects in RBC internal contents (thalassemia, hemoglobinopathies, and metabolic defects)
Ex. Sickle Cell Disease: point mutation in hemoglobin (Hgb) structure (GluàVal)
Inappropriate Hgb polymerization in low oxygen environments due to mutationàRBC becomes rigid, forms a sickle shape
Inflexible RBCs become trapped in the spleen’s sinusoid membranes àphagocytosed by splenic macrophages (extravascular hemolysis)
Infection triggers immune system activation
Autoimmune processes
TTP/HUS (abnormal platelet aggregation blocking blood vessels)
Production of abnormal
antibodies and immune complexes targeted against RBC surface antigens
Immunoglobulin-bound RBCs are marked for
destruction by the immune system (by either the cell- mediated or complement- mediated pathways)
DIC (fibrin deposition blocking blood vessels)
Artificial heart valve
RBCs are sheared when they flow past an abnormal surface
Rate of hemolytic RBC destruction > rate of bone marrow RBC synthesis (reticulocytosis)
↓ total number of RBCs in the body (despite normal RBC production/volume)
Normocytic anemia
Authors: Yan Yu Katie Lin Man-Chiu Poon* Reviewers: Andrew Brack Julia Heighton JoyAnne Krupa Lynn Savoie* * MD at time of publication
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Re-Published June 15, 2019 on www.thecalgaryguide.com")
 Zhang Man-Chiu Poon* Lynn Savoie* * MD at time of publication
Composition of normal adult hemoglobin (Hb):
Ineffective erythropoiesis
Excessive free α-chains accumulate, and α-chains precipitate in RBC precursors and RBCs
These precipitants (inclusion bodies) give RBCs an abnormal shape
Spleen destroys abnormally shaped RBCs
Spleen accumulates destroyed red blood cells and enlarges to an abnormal size
Splenomegaly (*See Splenomegaly slide for clinical findings)
Mild microcytic anemia
Hb: ↓ MCV: ↓
• • •
96% HbA (ααββ=2 α chains + 2 β chains) 2% HbA2 (ααẟẟ)
2% HbF (αα!!; fetal hemoglobin)
Genetic point mutation on a single allele of the β globin gene on chromosome 11 (heterozygous with one normal alleleà HbA ααββ0 or ααββ+)
Mild ↓ of β-chain production relative to ⍺- chain
↓ β-chain available for HbA synthesis
Body compensates by ↑ production
of non-affected globins (HbA , HbF) 2
↑ HbF (nonspecific), ↑ HbA2 (hallmark sign specific to Beta Thalassemia Minor) on electrophoresis
• •
α-chain precipitates form structures visible under the microscope
Inclusion bodies
Heme from the destroyed RBCs is degraded into bilirubin and iron
Excess unconjugated bilirubin is released into the blood
Excess bilirubin is deposited in the skin
Jaundice
Excess iron is stored as ferritin
↑ or normal ferritin
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Re-Published June 8, 2022 on www.thecalgaryguide.com")
 factor
incompatibility
Maternal sensitization
against Rh-positive fetal cells
Maternal antibodies against Rh
factor attack fetal RBCs
ABO incompatibility
Native maternal antibodies against non-
native blood types attack fetal RBCs
Sepsis** Widespread systemic inflammation Cytokines & complement factors damage RBC membranes
Disseminated intravascular
coagulation** (clotting
proteins become overactive)
Clots form in
systemic circulation
Small clots shear & damage
RBCs in circulation
Red blood cell
(RBC) enzyme
defects
Glucose-6 phosphatase
dehydrogenase (G6PD)
deficiency**
G6PD protects RBCs against
oxidative damage
↑ RBC sensitivity to oxidative
stress (during acute stressors)
Pyruvate kinase
deficiency (PKD)
Abnormal glycolysis & cellular
energy production
↓ RBCs life spans
RBC membrane
defects
Hereditary spherocytosis
RBCs are abnormally round (spherocytes)
RBC
membranes
easily damaged
in circulation
Bilverdin reductase
Hereditary elliptocytosis
RBCs are abnormally elongated or oval
converts bilverdin to
Sickle cell
Abnormal hemoglobin (HbS)
RBCs become abnormally
unconjugated bilirubin
disease**
polymerizes under ↓ O2
sickle shaped
Hemoglobinopathies
Thalassemia's**
Defective hemoglobin
chains in RBCs
Irregularly
shaped RBCs
trapped in
spleen
RBC
sequestration
↑ Production of RBCs (Polycythemia**)
Accumulation of blood (i.e.
Cephalohematoma, hemorrhage)
↑ RBC load &
turnover
Causes of ↓
hepatocellular
bilirubin clearance
Genetic defects in
uridine diphosphate
glucuronosyltransfe
rase (UGT) enzyme
(conjugates
bilirubin in liver)
Gilbert syndrome
Unconjugated bilirubin
↓ UGT production
remains insoluble & cannot
Crigler–Najjar
be excreted in bile
syndrome Type II
Crigler–Najjar
syndrome Type I
No UGT production
Breast milk jaundice Breast milk contains β-glucuronidase
Causes of ↑
entero-hepatic
bilirubin circulation
Intestinal obstruction
Obstruction blocks bile flow from liver to intestines Breastfeeding
jaundice
Inadequate milk intake (volume
depletion/dehydration)
↑ Reabsorption of bilirubin
in the intestines
**See corresponding Calgary Guide slide
Legend: Macrophages engulf
old or damaged RBCs
in spleen & liver
↑ Hemolysis (RBC
breakdown)
RBCs release cellular
contents including heme
(from hemoglobin)
Heme oxygenase
converts heme to
bilverdin
↑ Unconjugated
(indirect) bilirubin
accumulation in blood
↑ Total serum
bilirubin levels
Excess bilirubin
deposition into elastin-
rich tissues (eg. skin,
sclera) due to ↑
affinity for elastin
Pathologic neonatal jaundice
(yellow discoloration of skin
within first 24 hours of life)
Pathophysiology Mechanism
Sign/Symptom/Lab Finding Complications
Published July 13, 2025 on www.thecalgaryguide.com
Authors:
Merry Faye Graff
Khushi Arora
Reviewers:
Annie Pham
Emily J. Doucette
Danielle Nelson*
* MD at time of publication
Hemolytic
anemia**
↓ Bilirubin
conjugation
β-glucuronidase deconjugates bilirubin
Bilirubin builds up
in hepatic system
Unconjugated
bilirubin crosses
blood-brain barrier
Bilirubin-induced
neurologic
dysfunction (BIND)
Kernicterus (bilirubin-
induced neurological
damage)
Scleral icterus
(yellowing of
sclera in eyes)")