SEARCH RESULTS FOR: Nephrotic
Nephrotic Syndrome: Pathogenesis and Clinical Findings
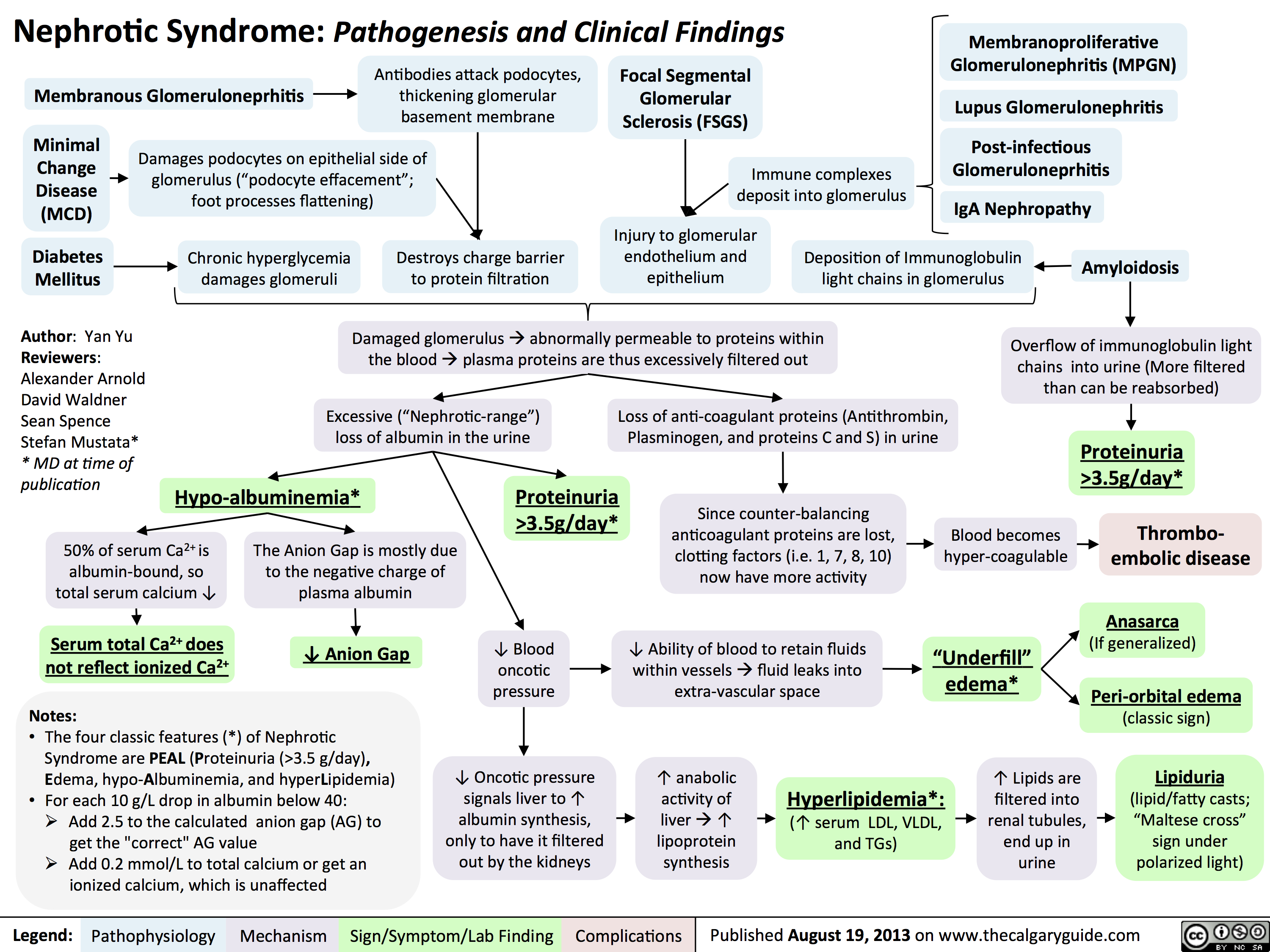 3.5g/day*? Ability of blood to retain fluids within vessels ? fluid leaks into extra-vascular spaceInjury to glomerular endothelium and epitheliumImmune complexes deposit into glomerulusDamaged glomerulus ? abnormally permeable to proteins within the blood ? plasma proteins are thus excessively filtered out? Oncotic pressure signals liver to ? albumin synthesis, only to have it filtered out by the kidneys? anabolic activity of liver ? ? lipoprotein synthesisHyperlipidemia*:(? serum LDL, VLDL, and TGs)Lipiduria(lipid/fatty casts; "Maltese cross" sign under polarized light)Since counter-balancing anticoagulant proteins are lost, clotting factors (i.e. 1, 7, 8, 10) now have more activityThrombo-embolic diseaseBlood becomes hyper-coagulable? Lipids are filtered into renal tubules, end up in urineMembranoproliferative Glomerulonephritis (MPGN)Lupus Glomerulonephritis Post-infectious GlomeruloneprhitisIgA NephropathyDamages podocytes on epithelial side of glomerulus ("podocyte effacement"; foot processes flattening)Diabetes MellitusChronic hyperglycemia damages glomeruliDeposition of Immunoglobulin light chains in glomerulusAmyloidosisAnasarca(If generalized)Peri-orbital edema (classic sign)Focal Segmental Glomerular Sclerosis (FSGS)Membranous GlomeruloneprhitisAntibodies attack podocytes, thickening glomerular basement membraneOverflow of immunoglobulin light chains into urine (More filtered than can be reabsorbed)Proteinuria >3.5g/day*The Anion Gap is mostly due to the negative charge of plasma albumin? Anion GapNotes: The four classic features (*) of Nephrotic Syndrome are PEAL (Proteinuria (>3.5 g/day), Edema, hypo-Albuminemia, and hyperLipidemia)For each 10 g/L drop in albumin below 40:Add 2.5 to the calculated anion gap (AG) to get the "correct" AG valueAdd 0.2 mmol/L to total calcium or get an ionized calcium, which is unaffected50% of serum Ca2+ is albumin-bound, so total serum calcium ? Serum total Ca2+ does not reflect ionized Ca2+ ? Blood oncotic pressure" title="Destroys charge barrier to protein filtrationNephrotic Syndrome: Pathogenesis and Clinical FindingsAuthor: Yan YuReviewers:Alexander ArnoldDavid WaldnerSean SpenceStefan Mustata** MD at time of publicationLegend:Published August 19, 2013 on www.thecalgaryguide.comMechanismPathophysiologySign/Symptom/Lab FindingComplicationsExcessive ("Nephrotic-range") loss of albumin in the urineHypo-albuminemia*Loss of anti-coagulant proteins (Antithrombin, Plasminogen, and proteins C and S) in urineMinimal Change Disease (MCD)"Underfill" edema*Proteinuria >3.5g/day*? Ability of blood to retain fluids within vessels ? fluid leaks into extra-vascular spaceInjury to glomerular endothelium and epitheliumImmune complexes deposit into glomerulusDamaged glomerulus ? abnormally permeable to proteins within the blood ? plasma proteins are thus excessively filtered out? Oncotic pressure signals liver to ? albumin synthesis, only to have it filtered out by the kidneys? anabolic activity of liver ? ? lipoprotein synthesisHyperlipidemia*:(? serum LDL, VLDL, and TGs)Lipiduria(lipid/fatty casts; "Maltese cross" sign under polarized light)Since counter-balancing anticoagulant proteins are lost, clotting factors (i.e. 1, 7, 8, 10) now have more activityThrombo-embolic diseaseBlood becomes hyper-coagulable? Lipids are filtered into renal tubules, end up in urineMembranoproliferative Glomerulonephritis (MPGN)Lupus Glomerulonephritis Post-infectious GlomeruloneprhitisIgA NephropathyDamages podocytes on epithelial side of glomerulus ("podocyte effacement"; foot processes flattening)Diabetes MellitusChronic hyperglycemia damages glomeruliDeposition of Immunoglobulin light chains in glomerulusAmyloidosisAnasarca(If generalized)Peri-orbital edema (classic sign)Focal Segmental Glomerular Sclerosis (FSGS)Membranous GlomeruloneprhitisAntibodies attack podocytes, thickening glomerular basement membraneOverflow of immunoglobulin light chains into urine (More filtered than can be reabsorbed)Proteinuria >3.5g/day*The Anion Gap is mostly due to the negative charge of plasma albumin? Anion GapNotes: The four classic features (*) of Nephrotic Syndrome are PEAL (Proteinuria (>3.5 g/day), Edema, hypo-Albuminemia, and hyperLipidemia)For each 10 g/L drop in albumin below 40:Add 2.5 to the calculated anion gap (AG) to get the "correct" AG valueAdd 0.2 mmol/L to total calcium or get an ionized calcium, which is unaffected50% of serum Ca2+ is albumin-bound, so total serum calcium ? Serum total Ca2+ does not reflect ionized Ca2+ ? Blood oncotic pressure" />
3.5g/day*? Ability of blood to retain fluids within vessels ? fluid leaks into extra-vascular spaceInjury to glomerular endothelium and epitheliumImmune complexes deposit into glomerulusDamaged glomerulus ? abnormally permeable to proteins within the blood ? plasma proteins are thus excessively filtered out? Oncotic pressure signals liver to ? albumin synthesis, only to have it filtered out by the kidneys? anabolic activity of liver ? ? lipoprotein synthesisHyperlipidemia*:(? serum LDL, VLDL, and TGs)Lipiduria(lipid/fatty casts; "Maltese cross" sign under polarized light)Since counter-balancing anticoagulant proteins are lost, clotting factors (i.e. 1, 7, 8, 10) now have more activityThrombo-embolic diseaseBlood becomes hyper-coagulable? Lipids are filtered into renal tubules, end up in urineMembranoproliferative Glomerulonephritis (MPGN)Lupus Glomerulonephritis Post-infectious GlomeruloneprhitisIgA NephropathyDamages podocytes on epithelial side of glomerulus ("podocyte effacement"; foot processes flattening)Diabetes MellitusChronic hyperglycemia damages glomeruliDeposition of Immunoglobulin light chains in glomerulusAmyloidosisAnasarca(If generalized)Peri-orbital edema (classic sign)Focal Segmental Glomerular Sclerosis (FSGS)Membranous GlomeruloneprhitisAntibodies attack podocytes, thickening glomerular basement membraneOverflow of immunoglobulin light chains into urine (More filtered than can be reabsorbed)Proteinuria >3.5g/day*The Anion Gap is mostly due to the negative charge of plasma albumin? Anion GapNotes: The four classic features (*) of Nephrotic Syndrome are PEAL (Proteinuria (>3.5 g/day), Edema, hypo-Albuminemia, and hyperLipidemia)For each 10 g/L drop in albumin below 40:Add 2.5 to the calculated anion gap (AG) to get the "correct" AG valueAdd 0.2 mmol/L to total calcium or get an ionized calcium, which is unaffected50% of serum Ca2+ is albumin-bound, so total serum calcium ? Serum total Ca2+ does not reflect ionized Ca2+ ? Blood oncotic pressure" title="Destroys charge barrier to protein filtrationNephrotic Syndrome: Pathogenesis and Clinical FindingsAuthor: Yan YuReviewers:Alexander ArnoldDavid WaldnerSean SpenceStefan Mustata** MD at time of publicationLegend:Published August 19, 2013 on www.thecalgaryguide.comMechanismPathophysiologySign/Symptom/Lab FindingComplicationsExcessive ("Nephrotic-range") loss of albumin in the urineHypo-albuminemia*Loss of anti-coagulant proteins (Antithrombin, Plasminogen, and proteins C and S) in urineMinimal Change Disease (MCD)"Underfill" edema*Proteinuria >3.5g/day*? Ability of blood to retain fluids within vessels ? fluid leaks into extra-vascular spaceInjury to glomerular endothelium and epitheliumImmune complexes deposit into glomerulusDamaged glomerulus ? abnormally permeable to proteins within the blood ? plasma proteins are thus excessively filtered out? Oncotic pressure signals liver to ? albumin synthesis, only to have it filtered out by the kidneys? anabolic activity of liver ? ? lipoprotein synthesisHyperlipidemia*:(? serum LDL, VLDL, and TGs)Lipiduria(lipid/fatty casts; "Maltese cross" sign under polarized light)Since counter-balancing anticoagulant proteins are lost, clotting factors (i.e. 1, 7, 8, 10) now have more activityThrombo-embolic diseaseBlood becomes hyper-coagulable? Lipids are filtered into renal tubules, end up in urineMembranoproliferative Glomerulonephritis (MPGN)Lupus Glomerulonephritis Post-infectious GlomeruloneprhitisIgA NephropathyDamages podocytes on epithelial side of glomerulus ("podocyte effacement"; foot processes flattening)Diabetes MellitusChronic hyperglycemia damages glomeruliDeposition of Immunoglobulin light chains in glomerulusAmyloidosisAnasarca(If generalized)Peri-orbital edema (classic sign)Focal Segmental Glomerular Sclerosis (FSGS)Membranous GlomeruloneprhitisAntibodies attack podocytes, thickening glomerular basement membraneOverflow of immunoglobulin light chains into urine (More filtered than can be reabsorbed)Proteinuria >3.5g/day*The Anion Gap is mostly due to the negative charge of plasma albumin? Anion GapNotes: The four classic features (*) of Nephrotic Syndrome are PEAL (Proteinuria (>3.5 g/day), Edema, hypo-Albuminemia, and hyperLipidemia)For each 10 g/L drop in albumin below 40:Add 2.5 to the calculated anion gap (AG) to get the "correct" AG valueAdd 0.2 mmol/L to total calcium or get an ionized calcium, which is unaffected50% of serum Ca2+ is albumin-bound, so total serum calcium ? Serum total Ca2+ does not reflect ionized Ca2+ ? Blood oncotic pressure" />
iga-vasculitis-henoch-scholein-purpura-pathogenesis-and-clinical-findings
 : Pathogenesis and clinical findings
Authors: Mia Koegler Nela Cosic Reviewers: Crystal Liu Yan Yu* Martin Atkinson* * MD at time of publication
Infectious Agents
50% have preceding upper respiratory tract infections, i.e., influenza virus or Group A Strep
Drugs
I.e., antibiotics (penicillin, erythromycin), NSAIDs and biologics (tumor necrosis factor α inhibitors)
Immunogenetic and cellular predisposition
Various genetic polymorphisms alter cell- mediated immune response, IgA levels elevated in 50% of people
↑ Circulating galactose-deficient IgA1 (GD-IgA1). Deficiency in galactosylation of IgAà↓ IgA serum clearanceàadhesion of IgA complexes, which then deposit into the endothelial lining of blood vesselsàattraction of various inflammatory cells to the area:
Formation of Secretion of Interleukin 8 (IL8) - cytokine that induces Neutrophils infiltrate Activation of complement immune complexes neutrophilic chemotaxis and macrophage phagocytosis the tissue site factors (C3, C4)
Leukocytoclastic vasculitis (histopathologic term for small vessels inflamed by neutrophilic autoimmune response)
Inflamed cutaneous vessels become enlarged in clusters
Symmetrical palpable purpura (red/purple, non- blanchable papules) distributed on lower limbs and buttocks areas
Cutaneous small vessel vasculitis (100%)
Inflamed gastric vessels - hemorrhage and edema within bowel wall
Gastrointestinal (85%)
Colicky abdominal pain (commonly in the periumbilical region), nausea, vomiting
Gastrointestinal
GI bleeding (hematemesis, melena), Intussusception
Glomerular mesangial proliferation and inflammation
↑ mast cell deposition in joints
Joints (60-85%)
Arthralgia's (common), arthritis (especially knees and ankles)
Arthralgia often transient. No permanent sequelae
Sympathetic nervous system activation
Glomerulosclerosis, tubulointerstitial and podocyte damage
Renal tissue ischemia
↑ Na sensitivity in renal tubules (↑ Na and water retention)
Renal (10-50%)
Increased renin secretion
HTN, nephrotic/nephritic syndrome, renal insufficiency
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published September 1, 2019 on www.thecalgaryguide.com")
Nephrotisches Syndrom: Pathogenese und klinische Befunde

Lupus-Nephritis
Postinfektiöse Glomeruloneprhitis
IgA Nephropathie
Membranöse Glomerulonephritis
Antikörper greifen Podozyten an, Verdickung der glomerulären Basalmembran
Fokal-segmentale Glomerulosklerose (FSSGN)
Schädigung des glomerulären Endo- und Epithels
Minimal Change Glomerulo- nephritis (MCNG)
Diabetes mellitus
Autor: Yan Yu Rezensenten: Alexander Arnold David Waldner
Sean Spence
Stefan Mustata* Übersetzung:
Sarah Schwarz Übersetzungsprüfung: Gesche Tallen*
* MD zum Zeitpunkt der Veröffentlichung
Podozyten-Schädigung auf der epithelialen Seite des Glomerulums (Abflachung der Podozytenfortsätze)
Glomeruläre Immunkomplex- ablagerungen
Chronische Hyperglykämien schädigen das Glomerulum
Geschädigter Proteinfilter (v.a. für geladene Proteine)
Ablagerungen von Immunglobulin-Leichtketten im Glomerulum
Amyloidose
Geschädigte Glomeruli --> gestörte Filterbarriere v.a. für Proteine --> übermäßige Filtration von Plasmaproteinen
Vermehrte renale Ausscheidung von Immunglobulin-Leichtketten (Filtration>Resorption)
Hypoalbuminämie*
Übermäßiger Verlust an Albumin über den Urin
Proteinurie >3.5g/Tag*
Verlust an Antikoagulationsproteinen (Antithrombin, Plasminogen, Protein C & S) über den Urin
Koagulations- /Gerinnungsfaktoren (z.B. 1,7,8, 10) sind in Überzahl
Proteinurie >3.5g/Tag*
Thrombophilie
Anasarka
(Generalisiertes Ödem)
Lidödem
(klassisches Frühzeichen)
Lipidurie
(zeigt unter
gekreuztem polarisiertem Licht eine Malteserkreuz- form)
50% des Serumkalziums sind an Albumin gebunden, sodass Serumkalzium- spiegel ↓
Serum- Ca2+ repräsentiert nicht mehr das Gesamt-Ca2+
Beachte:
• DerklassischeSymptomkomplex(*)desnephrotischen Syndroms besteht aus: Proteinurie (>3,5g/Tag), Ödemen, Hypoalbuminämie,Hyperlipidämie
• Fürjede10g/LmitAlbumin<40:
➔ Addiere 2.5 zur errechneten Anionenlücke um
dessen “wahren” Wert zu bekommen
➔ Addiere 0,2mmol/L zum Gesamt-Ca um den
Wert des ionisierten Kalziums zu errechnen
Blut neigt zur Bildung von Thromben
Ödeme*
↑ Renale Filtrationder Lipide und Ausscheidung über den Urin
Die Anionenlücke ergibt sich hauptsächlich aus negativ geladenem Serumalbumin
Anionenlücke↓
Kolloid- osmotischer Druck ↓
Flüssigkeit kann nicht mehr in den Blutgefäßen gehalten werden und diffundiert ins Gewebe
Signalisiert der Leber die Albuminproduktion zuerhöhen,Albumin wird aber weiterhin über die Nieren verloren
Synthese- arbeit der Leber↑, auch ↑ Lipoprotein- synthese
Hyperlipidämie*:
(Serum-LDL, -VLDL, - TG ↑ )
Legende:
Pathophysiologie
Mechanismen
Symptome/Klinische Befunde
Komplikationen
Veröffentlicht: 19. August 2013 auf www.thecalgaryguide.com")
NSAIDs and the Kidney Nephrotoxicity

Authors: Kyle Moxham Mehul Gupta Reviewers: Emily Wildman Yan Yu* Adam Bass* * MD at time of publication
Inhibition of Cyclooxygenase COX-1 (expressed in kidney) and COX-2 (expressed in kidney and sites of inflammation)
NSAID induced nephrotoxicity: associated with chronic NSAID usage independent of dosage
COX inhibition ↑ conversion of arachidonic acid (AA) to leukotrienes, causing systemic T-cell dysfunction (unknown mechanism)
Type IV systemic hypersensitivity (delayed
T helper cell mediated) reaction to drug exposure
T cells release inflammatory cytokines into the bloodstream
(see Calgary Guide slide on NSAIDs and the Kidney: Mechanism of Action and Side Effects)
T cells infiltrate the renal interstitium, sparing the glomeruli and blood vessels
Overproduction of cytokines by T cells causing inflammation,
tissue damage, and cell death, of the renal intersitium
Drug Induced Acute interstitial nephritis (AIN): a type of immune-mediated tubulointerstitial injury
Activated T-cells infiltrate the glomerulus and cause podocyte injury (epithelial cells attached to the glomerular basement membrane)
Membranous nephropathy:
nephrotic syndrome involving autoimmune glomerular basement membrane thickening & complete podocyte effacement (seen on kidney biopsy)
Minimal change disease: nephrotic syndrome caused by autoimmune podocyte effacement (seen on kidney biopsy)
Cytokine mediated activation and
proliferation of immune cells like macrophages and eosinophils
Cytokines travel to hypothalamus,
causing change in the body’s thermal set point
Fever
Podocyte effacement allows for serum proteins to across the glomerulus into the tubular lumen (see Calgary Guide slide on Nephrotic Syndrome for full mechanisms)
Repeated NSAID exposure causing
recurrent unrecognized AIN and damage of the kidney
Chronic interstitial nephritis /analgesic nephropathy
Infiltrating immune cells (predominantly neutrophils) are filtered into/enter the renal tubules, and form clumps (“casts”) within the tubulesà casts are then released into urine
WBC cast on urinalysis
↑ Blood eosinophils
Immune cells infiltrate the
dermis and epidermis of the skin
Rash
Abnormal quantities of protein present in urine
Protein present in the blood is improperly filtered into the filtrate at the glomerulus
Protein- Creatinine
Ratio >3.5mg/mg
Proteinuria >3.5g/d
Hypo- albuminemia
↓ plasma oncotic pressure resulting in fluid extravasation into the interstitium (see Calgary guide slide on Edema for full mechanisms)
Pitting Edema
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published January 13, 2022 on www.thecalgaryguide.com")
Overfill Edema Pathogenesis

Aberrant filtration of proteins including plasminogen
(glycoprotein in the systemic circulation involved in the dissolution of fibrin blood clots)
↑ Plasminogen concentration in tubular fluid
Conversion of plasminogenà plasmin by urokinase-type plasminogen activator in the cortical collecting tubule
Plasmin activates epithelium
sodium channels (involved in Na+ reabsorption) in principal cells of the cortical collecting duct
Nonsteroidal Anti-inflammatory Drugs
Inhibition of cyclooxygenase throughout body (enzyme that converts arachidonic acidàprostaglandins)
↓ Prostaglandins throughout body
Thiazolidines
↓ Prostaglandin- mediated inhibition of Na+ and Cl- transport in ascending loop of Henle and collecting ducts
↑ Na+ reabsorption from kidney tubules back into blood
↓ Prostaglandin-mediated vasodilation in kidneys
↓ Pressure of blood perfusing kidneys → ↓ pressure gradient between glomerulus and bowman’s capsule
↓ Glomerular filtration rate
↓ Renal excretion of salt & water
↑ Salt and water retention
↑ Effective arterial blood volume
↑ blood pressure in veins
Unclear mechanism but possible theories include upregulation of epithelium sodium channels in the cortical collecting tubule and involvement of other transporters in the proximal tubule and cortical collecting tubule
Acute Renal Failure Chronic Renal Failure
↑ Hydrostatic pressure within capillariesàexceeds hydrostatic pressure within interstitial spaceàfluid moves from capillaries into interstitial space
Overfill Edema
Abnormal accumulation of fluid in the interstitial space where urine Na+ >40meq/L
Authors: Samin Dolatabadi Reviewers: Meena Assad, Jessica Krahn, Brooke Fallis, Ran (Marissa) Zhang, Yan Yu*, Juliya Hemmett* * MD at time of publication
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published January 23, 2022 on www.thecalgaryguide.com")
transudative-pleural-effusions-pathogenesis-and-lab-findings

Left ventricle unable to pump sufficient blood into systemic circulation
Backup of blood in pulmonary veins
↑ Hydrostatic pressure
in pulmonary veins
Pulmonary embolism
R ventricle unable to pump blood due to clot in pulmonary artery
Backup of blood in systemic veins
↑ Hydrostatic pressure
in veins draining parietal pleura
Nephrotic syndrome
Damaged glomerulus has ↑ permeability to plasma proteins in blood
↑ Loss of proteins through urine
↓ Oncotic pressure
in systemic capillaries (including within parietal pleura)
Normally, permeable pleural capillaries do not allow protein leakage into the pleural space
↑ Interstitial fluid leakage across intact pulmonary or pleural capillaries into pleural space
Transudative Pleural Effusion
Absence of bacteria and inflammatory cells in pleural space
No increase in cellular activity in pleural space
Normal levels of glucose metabolism in pleural space = low lactate dehydrogenase (LDH) (LDH increases when glucose metabolism, particularly glycolysis, increases to maintain supply of NAD+)
Large accumulation of pleural fluid (PF) pressing against lung tissue and mediastinum
Lung atelectasis (lung collapse)
See Pleural Effusions: X- ray Findings and Physical Exam Findings of Lung Diseases slides
PF/serum protein ratio < 0.5
PF LDH < 2/3 upper limit of normal
Light’s Criteria: All three criteria must be met to be a transudative pleural effusion
PF/serum LDH ratio < 0.6
See Hypoxemia: Pathogenesis and Clinical Findings slide for pathophysiology and signs of hypoxemia
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published August 9, 2022 on www.thecalgaryguide.com")
Underfill Edema Pathogenesis

Vasodilatory medications
Various mechanisms
Right-sided heart failure
Compromised right heart function ↓ forward flow
↓ Hepatic albumin synthesis
Blood is unable to pass through hepatic vessels disrupted by cirrhosis and backs up in portal vein
↑ Blood pressure in portal vein (portal hypertension)
Less blood volume in hepatic veins and vena cava (underfilling)
Pregnancy
↑ Estrogen, progesterone and relaxin
Vasodilation
Gravity causes fluid accumulation in peripheral veins
↑ Capillary hydrostatic pressure
↑ Net fluid movement into interstitial space
↓ Serum albumin
↓ Capillary oncotic pressure
Fluid extravasation into interstitial space
More blood in portal vein ↑ capillary hydrostatic pressure in venous system
Pressure creates net fluid
movement from vascular space into interstitial space
Less blood volume in arteries (underfilling)
↓ Effective arterial blood volume (EABV)
↓ Renal blood flow activates the renin-angiotensin-aldosterone system (RAAS)
Angiotensin and aldosterone ↑ Anti-diuretic hormone released by tubular Na+ and H2O resorption posterior pituitary ↑ H2O resorption
↑ Fluid in circulation, worsening existing venous congestion
↑ Hydrostatic capillary pressure and fluid extravasation into interstitial space Underfill edema (edema worsened by activation of RAAS)
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published Aug 19, 2015; updated Aug 5, 2024 on www.thecalgaryguide.com")
IgA Nephropathy
 created by mucosa- bound IgA1 plasma cells is secreted into plasma instead of onto mucosal surface
IgA1 plasma cells hyper- responsive to triggers (eg. URTIs, gastroenteritis) ↑ synthesis of GD-IgA1 → spill-over into plasma
Immunoglobulin A1 (IgA1) plasma cells destined to reside in mucosa (eg. gut or respiratory tract) travel to and
reside in inappropriate site(s) (eg. bone marrow) releasing GD-IgA1 into plasma
GD-IgA1 is not cleared from plasma as quickly as IgA1 → ↑ plasma GD-IgA1 levels
Hit 3:
GD-IgA1-IgG complexes deposit in mesangium
C3 predominant complement activation amplifies inflammatory response
Renal biopsy:
IgA deposits in mesangium (100% sensitive)
Renal biopsy: Complement in mesangium (C3 predominant) (90-95% sensitive)
A cascade of multiple immunologic hits is initiated
Hit 1: ↑ Serum levels of GD-IgA1 multiple immunologic hits
GD-IgA1 hinge region is structurally distinct from IgA1 that would normally circulate in plasma (lack of galactosyl groups)
GD-IgA1 hinge region may mimic pathogens (ex. bacteria and viruses) or other antigens
Cross reactivity of IgG against GD-IgA1, or synthesis of anti-GD-IgA1 IgG antibodies
Immunoglobulin G (IgG) binds GD-IgA1 hinge region Hit 2: GD-IgA1-IgG immune complex formation
Circulating GD-IgA1-IgG complexes have high affinity for glomerular endothelial cells where they damage the glycocalyx → ↑ permeability of immunoglobulins into the mesangium
↑ Production of chemokines, cytokines and complement → ↑ mesangial cell proliferation and matrix expansion
Leukocyte recruitment and activation damages glomerulus and mesangium
Hit 4:
Inflammatory response to GD-IgA1 complexes in mesangium induce glomerular structure disruption (endothelium, basement membrane, podocytes, mesangium)
and impaired glomerular function
Loss of barrier functions of glomerulus allows for extravasation of blood & proteins into Bowman’s space and subsequently through tubules
Renal biopsy: Glomerulosclerosis, tubulointerstitial fibrosis, glomerular vasculitis, podocyte damage
Eventual end-Stage Renal Disease (ESRD)
Progressive ↓ of filtration surface area within glomeruli and ↓ number of functional glomeruli
Proteinuria
Synpharyngitic hematuria (hematuria with dysmorphic red cells co-occurring with pharyngitis)
↓ Glomerular Filtration Rate (GFR)
Nephrotic Syndrome
↑ Serum creatinine
Chronic kidney disease and eventually ESRD
IgAN is an autoimmune disease where IgA deposition in the glomerulus leads to an inflammatory cascade, endothelial dysfunction and mesangial expansion that damages glomeruli causing kidneys to leak blood and protein into urine and decreased kidney function. IgA nephropathy is a multifactorial disease requiring multiple immunologic hits
IgA Nephropathy (IgAN)
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published Sept 5, 2024 on www.thecalgaryguide.com")
Chronic Inflammatory Demyelinating Polyneuropathy

Systemic illness (e.g., hepatitis B, Hepatitis C, HIV, thyroid Disease, nephrotic syndrome, irritable bowel syndrome)
Authors: Andrea Soumbasis Reviewers: Braxton Phillips Shahab Marzoughi Sina Marzoughi* * MD at time of publication
Autoimmune response initiation
Lumbar Puncture: Cytoalbuminologic Dissociation CSF protein count elevated with normal cell count
Cell-mediated response: Putative antigen (unidentified immune target) presents to auto-reactive T cells
Activated T cells secrete inflammatory mediators (matrix metalloproteinases)
Inflammatory response ↑ peripheral nerve permeability & T cells cross blood-nerve barrier
Humoral Response: Immunoglobulin & complement deposits on compact myelin & nodal regions
Autoantibodies target unknown epitope (antigen molecule recognized by antibody) on outer surface of Schwann cells
IgG4 antibodies target axoglial junction (Contactin-1, Neurofascin 155) that maintain Node of Ranvier of myelinated axons
↑ Permeability of blood- nerve barrier allows large proteins to leak into CSF
CD8+ T cells, CD4+ T cells & macrophages infiltrate peripheral nerves
Macrophages form clusters around endoneurium, release proinflammatory cytokines & strip away myelin via phagocytosis
Abnormal expression & distribution of Contactin-1 & Neurofascin 155 antibodies along axoglial junction
Disrupted ion channel segregation & paranodal structure
↓ Saltatory conduction
Nodopathy & Paranodopothy: Neurofascin 155 – Distal sensorimotor ataxia
Contactin-1 – Sensorimotor ataxia Caspr-1 – Neuropathic pain & nephrotic syndrome
Onion bulb formation: Nerve biopsy showing concentric, circular layers of cells surrounding the axon
Segmental demyelination & remyelination of peripheral nerves
↑ Demyelination
Chronic Inflammatory Demyelinating Polyneuropathy
Progressive weakness & sensory loss over a period of greater than 8 weeks due to autoimmune dysfunction
Peripheral nerve degeneration & conduction velocity
Nerve Conduction Study: Partial nerve conduction block, conduction velocity slowing, prolonged distal motor latencies, delay or disappearance of CMAP (compound muscle action potential)
Lesions of larger myelinated fibers in the dorsal columns for vibration & position sense
Lesions of peripheral motor nerves in non-length dependent pattern (does not only affect longest nerves first)
Motor Deficits
↓ Sensory input and/or ↓ motor output
Abnormal Reflexes
Globally ↓ reflexes
Lesions affecting sympathetic & parasympathetic nerve fibers
Autonomic Deficits
Bladder & bowel dysfunction, heart rate irregularities, blood pressure fluctuation
Sensory Deficits
Gait ataxia
Proximal weakness (muscles closer to the trunk or center of the body): Difficulty climbing stairs, rising from seated position, lifting objects overhead, frequent falls
Distal weakness (muscles farther from the trunk or the extremities): Tripping due to foot drop, difficulty with fine motor tasks
↓ Vibration & proprioception
Paraesthesias (e.g., tingling, numbness)
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published Dec 29, 2024 on www.thecalgaryguide.com")