SEARCH RESULTS FOR: diabetes mellitus
Nephrotic Syndrome: Pathogenesis and Clinical Findings
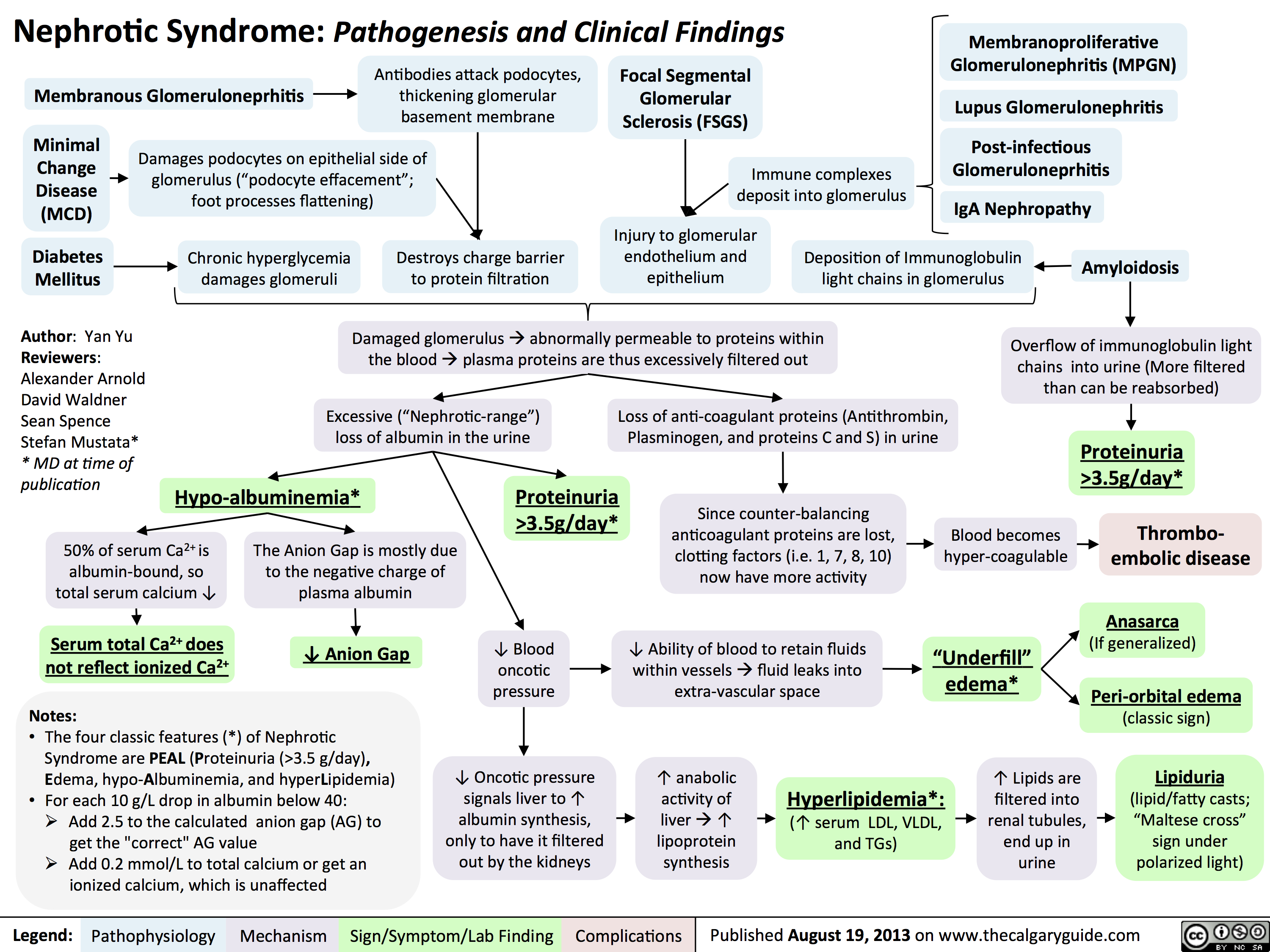 3.5g/day*? Ability of blood to retain fluids within vessels ? fluid leaks into extra-vascular spaceInjury to glomerular endothelium and epitheliumImmune complexes deposit into glomerulusDamaged glomerulus ? abnormally permeable to proteins within the blood ? plasma proteins are thus excessively filtered out? Oncotic pressure signals liver to ? albumin synthesis, only to have it filtered out by the kidneys? anabolic activity of liver ? ? lipoprotein synthesisHyperlipidemia*:(? serum LDL, VLDL, and TGs)Lipiduria(lipid/fatty casts; "Maltese cross" sign under polarized light)Since counter-balancing anticoagulant proteins are lost, clotting factors (i.e. 1, 7, 8, 10) now have more activityThrombo-embolic diseaseBlood becomes hyper-coagulable? Lipids are filtered into renal tubules, end up in urineMembranoproliferative Glomerulonephritis (MPGN)Lupus Glomerulonephritis Post-infectious GlomeruloneprhitisIgA NephropathyDamages podocytes on epithelial side of glomerulus ("podocyte effacement"; foot processes flattening)Diabetes MellitusChronic hyperglycemia damages glomeruliDeposition of Immunoglobulin light chains in glomerulusAmyloidosisAnasarca(If generalized)Peri-orbital edema (classic sign)Focal Segmental Glomerular Sclerosis (FSGS)Membranous GlomeruloneprhitisAntibodies attack podocytes, thickening glomerular basement membraneOverflow of immunoglobulin light chains into urine (More filtered than can be reabsorbed)Proteinuria >3.5g/day*The Anion Gap is mostly due to the negative charge of plasma albumin? Anion GapNotes: The four classic features (*) of Nephrotic Syndrome are PEAL (Proteinuria (>3.5 g/day), Edema, hypo-Albuminemia, and hyperLipidemia)For each 10 g/L drop in albumin below 40:Add 2.5 to the calculated anion gap (AG) to get the "correct" AG valueAdd 0.2 mmol/L to total calcium or get an ionized calcium, which is unaffected50% of serum Ca2+ is albumin-bound, so total serum calcium ? Serum total Ca2+ does not reflect ionized Ca2+ ? Blood oncotic pressure" title="Destroys charge barrier to protein filtrationNephrotic Syndrome: Pathogenesis and Clinical FindingsAuthor: Yan YuReviewers:Alexander ArnoldDavid WaldnerSean SpenceStefan Mustata** MD at time of publicationLegend:Published August 19, 2013 on www.thecalgaryguide.comMechanismPathophysiologySign/Symptom/Lab FindingComplicationsExcessive ("Nephrotic-range") loss of albumin in the urineHypo-albuminemia*Loss of anti-coagulant proteins (Antithrombin, Plasminogen, and proteins C and S) in urineMinimal Change Disease (MCD)"Underfill" edema*Proteinuria >3.5g/day*? Ability of blood to retain fluids within vessels ? fluid leaks into extra-vascular spaceInjury to glomerular endothelium and epitheliumImmune complexes deposit into glomerulusDamaged glomerulus ? abnormally permeable to proteins within the blood ? plasma proteins are thus excessively filtered out? Oncotic pressure signals liver to ? albumin synthesis, only to have it filtered out by the kidneys? anabolic activity of liver ? ? lipoprotein synthesisHyperlipidemia*:(? serum LDL, VLDL, and TGs)Lipiduria(lipid/fatty casts; "Maltese cross" sign under polarized light)Since counter-balancing anticoagulant proteins are lost, clotting factors (i.e. 1, 7, 8, 10) now have more activityThrombo-embolic diseaseBlood becomes hyper-coagulable? Lipids are filtered into renal tubules, end up in urineMembranoproliferative Glomerulonephritis (MPGN)Lupus Glomerulonephritis Post-infectious GlomeruloneprhitisIgA NephropathyDamages podocytes on epithelial side of glomerulus ("podocyte effacement"; foot processes flattening)Diabetes MellitusChronic hyperglycemia damages glomeruliDeposition of Immunoglobulin light chains in glomerulusAmyloidosisAnasarca(If generalized)Peri-orbital edema (classic sign)Focal Segmental Glomerular Sclerosis (FSGS)Membranous GlomeruloneprhitisAntibodies attack podocytes, thickening glomerular basement membraneOverflow of immunoglobulin light chains into urine (More filtered than can be reabsorbed)Proteinuria >3.5g/day*The Anion Gap is mostly due to the negative charge of plasma albumin? Anion GapNotes: The four classic features (*) of Nephrotic Syndrome are PEAL (Proteinuria (>3.5 g/day), Edema, hypo-Albuminemia, and hyperLipidemia)For each 10 g/L drop in albumin below 40:Add 2.5 to the calculated anion gap (AG) to get the "correct" AG valueAdd 0.2 mmol/L to total calcium or get an ionized calcium, which is unaffected50% of serum Ca2+ is albumin-bound, so total serum calcium ? Serum total Ca2+ does not reflect ionized Ca2+ ? Blood oncotic pressure" />
3.5g/day*? Ability of blood to retain fluids within vessels ? fluid leaks into extra-vascular spaceInjury to glomerular endothelium and epitheliumImmune complexes deposit into glomerulusDamaged glomerulus ? abnormally permeable to proteins within the blood ? plasma proteins are thus excessively filtered out? Oncotic pressure signals liver to ? albumin synthesis, only to have it filtered out by the kidneys? anabolic activity of liver ? ? lipoprotein synthesisHyperlipidemia*:(? serum LDL, VLDL, and TGs)Lipiduria(lipid/fatty casts; "Maltese cross" sign under polarized light)Since counter-balancing anticoagulant proteins are lost, clotting factors (i.e. 1, 7, 8, 10) now have more activityThrombo-embolic diseaseBlood becomes hyper-coagulable? Lipids are filtered into renal tubules, end up in urineMembranoproliferative Glomerulonephritis (MPGN)Lupus Glomerulonephritis Post-infectious GlomeruloneprhitisIgA NephropathyDamages podocytes on epithelial side of glomerulus ("podocyte effacement"; foot processes flattening)Diabetes MellitusChronic hyperglycemia damages glomeruliDeposition of Immunoglobulin light chains in glomerulusAmyloidosisAnasarca(If generalized)Peri-orbital edema (classic sign)Focal Segmental Glomerular Sclerosis (FSGS)Membranous GlomeruloneprhitisAntibodies attack podocytes, thickening glomerular basement membraneOverflow of immunoglobulin light chains into urine (More filtered than can be reabsorbed)Proteinuria >3.5g/day*The Anion Gap is mostly due to the negative charge of plasma albumin? Anion GapNotes: The four classic features (*) of Nephrotic Syndrome are PEAL (Proteinuria (>3.5 g/day), Edema, hypo-Albuminemia, and hyperLipidemia)For each 10 g/L drop in albumin below 40:Add 2.5 to the calculated anion gap (AG) to get the "correct" AG valueAdd 0.2 mmol/L to total calcium or get an ionized calcium, which is unaffected50% of serum Ca2+ is albumin-bound, so total serum calcium ? Serum total Ca2+ does not reflect ionized Ca2+ ? Blood oncotic pressure" title="Destroys charge barrier to protein filtrationNephrotic Syndrome: Pathogenesis and Clinical FindingsAuthor: Yan YuReviewers:Alexander ArnoldDavid WaldnerSean SpenceStefan Mustata** MD at time of publicationLegend:Published August 19, 2013 on www.thecalgaryguide.comMechanismPathophysiologySign/Symptom/Lab FindingComplicationsExcessive ("Nephrotic-range") loss of albumin in the urineHypo-albuminemia*Loss of anti-coagulant proteins (Antithrombin, Plasminogen, and proteins C and S) in urineMinimal Change Disease (MCD)"Underfill" edema*Proteinuria >3.5g/day*? Ability of blood to retain fluids within vessels ? fluid leaks into extra-vascular spaceInjury to glomerular endothelium and epitheliumImmune complexes deposit into glomerulusDamaged glomerulus ? abnormally permeable to proteins within the blood ? plasma proteins are thus excessively filtered out? Oncotic pressure signals liver to ? albumin synthesis, only to have it filtered out by the kidneys? anabolic activity of liver ? ? lipoprotein synthesisHyperlipidemia*:(? serum LDL, VLDL, and TGs)Lipiduria(lipid/fatty casts; "Maltese cross" sign under polarized light)Since counter-balancing anticoagulant proteins are lost, clotting factors (i.e. 1, 7, 8, 10) now have more activityThrombo-embolic diseaseBlood becomes hyper-coagulable? Lipids are filtered into renal tubules, end up in urineMembranoproliferative Glomerulonephritis (MPGN)Lupus Glomerulonephritis Post-infectious GlomeruloneprhitisIgA NephropathyDamages podocytes on epithelial side of glomerulus ("podocyte effacement"; foot processes flattening)Diabetes MellitusChronic hyperglycemia damages glomeruliDeposition of Immunoglobulin light chains in glomerulusAmyloidosisAnasarca(If generalized)Peri-orbital edema (classic sign)Focal Segmental Glomerular Sclerosis (FSGS)Membranous GlomeruloneprhitisAntibodies attack podocytes, thickening glomerular basement membraneOverflow of immunoglobulin light chains into urine (More filtered than can be reabsorbed)Proteinuria >3.5g/day*The Anion Gap is mostly due to the negative charge of plasma albumin? Anion GapNotes: The four classic features (*) of Nephrotic Syndrome are PEAL (Proteinuria (>3.5 g/day), Edema, hypo-Albuminemia, and hyperLipidemia)For each 10 g/L drop in albumin below 40:Add 2.5 to the calculated anion gap (AG) to get the "correct" AG valueAdd 0.2 mmol/L to total calcium or get an ionized calcium, which is unaffected50% of serum Ca2+ is albumin-bound, so total serum calcium ? Serum total Ca2+ does not reflect ionized Ca2+ ? Blood oncotic pressure" />
Pathogenesis of Diabetes mellitus DM), Type II

Diabetic Hypoglycemia
![Yu, Yan - Diabetic Hypoglycemia - Clinical Findings - FINAL.pptx
? Epinephrine(Released within seconds as [glucose] falls further) Growth hormone, ? Cortisol (if hypoglycemia persists for minutes)Glucagon should ? when [glucose] falls. But here, glucagon release is inhibited by 1) diabetic auto-immune destruction of Alpha cells & 2) the high insulin.43210Plasma Glucose concentration (mmol/L)Liver should ? glycogenolysis & gluconeogenesisPeripheral vaso-constrictionPlasma [glucose] stays lowActivation of sympathetic (adrenergic) receptors across body, triggering Neurogenic symptomsPlasma [glucose] ?Excess subcutaneous insulin or insulin-secretagogue ?? [insulin] in the bloodOver time: [insulin] in the DM patient depends only on how much was injected or how much secretagogue was consumed; not on the body's physiological state.[Insulin] stays high in excessively-treated DM patientsPlasma [glucose] normally ?, but...High insulin transports plasma glucose into cells!In pts with existing diabetic autonomic neuropathy, epi-nephrine secretion will already be ?Brain does not get enough glucose, ? neuron function ? Neuroglycopenic symptomsTx: glucose intake![Glucose] returns to normalIf no glucose intake:Hypoglycemia-unawareness: No autonomic Sx felt so hypoglycemia not treated early ? pts present later on with more severe hypoglycemia and neuroglycopenic sxBrain cells kept chronically euglycemic due to GLUT1 receptor over-expression (despite rest of body being hypoglycemic)With many hypoglycemic events over time:Brain feels no need to ? glucose, so it ? autonomic epinephrine secretion!This is the normal sequence of hormone responses to ?ing plasma glucose levels.But this normal hormonal response will be blunted over time if there is 1) continued hypoglycemia dampening the sympathetic nervous system, and 2) long-standing diabetic neuropathy! (To be explained later in this flow chart)Abbreviations: [ ] = concentrationTx = TreatmentDM = Diabetes mellitusDiabetic Hypoglycemia: Pathogenesis and Clinical FindingsConfusionCan't concentrateWeaknessSlurred speech? coordination (staggering, etc)SeizuresComa, deathAdrenergic symptoms (epinephrine-mediated):Anxiety, irritability, trembling, pallor (skin vasoconstriction), palpitations, ? systolic BP, tachycardia Cholinergic symptoms(Acetylcholine-mediated):Sweating, hunger, tingling, blurry visionNote: In pts w/out DM, endogenous insulin secretion normally stops when blood [glucose] drops to <4mmol/LAuthor: Yan YuReviewers: Peter Vetere, Gillian Goobie, Hanan Bassyouni** MD at time of publicationLegend:Published June 14, 2013 on www.thecalgaryguide.comMechanismPathophysiologySign/Symptom/Lab FindingComplicationsMany hypoglycemic events over time blunt epinephrine secretion further.Hypoglycemia unawareness can be reversedIf pt stays hypoglycemia-free for >6 weeks, brain restores its ability to detect low glucose levels? peripheral glucose delivery and uptake (saving more glucose for the brain)Lack of glucagon effect reinforces hypoglycemia
124 kB / 361 words](http://calgaryguide.ucalgary.ca/wp-content/uploads/2015/05/Diabetic-Hypoglycemia-Clinical-Findings.jpg "Yu, Yan - Diabetic Hypoglycemia - Clinical Findings - FINAL.pptx
? Epinephrine(Released within seconds as [glucose] falls further) Growth hormone, ? Cortisol (if hypoglycemia persists for minutes)Glucagon should ? when [glucose] falls. But here, glucagon release is inhibited by 1) diabetic auto-immune destruction of Alpha cells & 2) the high insulin.43210Plasma Glucose concentration (mmol/L)Liver should ? glycogenolysis & gluconeogenesisPeripheral vaso-constrictionPlasma [glucose] stays lowActivation of sympathetic (adrenergic) receptors across body, triggering Neurogenic symptomsPlasma [glucose] ?Excess subcutaneous insulin or insulin-secretagogue ?? [insulin] in the bloodOver time: [insulin] in the DM patient depends only on how much was injected or how much secretagogue was consumed; not on the body's physiological state.[Insulin] stays high in excessively-treated DM patientsPlasma [glucose] normally ?, but...High insulin transports plasma glucose into cells!In pts with existing diabetic autonomic neuropathy, epi-nephrine secretion will already be ?Brain does not get enough glucose, ? neuron function ? Neuroglycopenic symptomsTx: glucose intake![Glucose] returns to normalIf no glucose intake:Hypoglycemia-unawareness: No autonomic Sx felt so hypoglycemia not treated early ? pts present later on with more severe hypoglycemia and neuroglycopenic sxBrain cells kept chronically euglycemic due to GLUT1 receptor over-expression (despite rest of body being hypoglycemic)With many hypoglycemic events over time:Brain feels no need to ? glucose, so it ? autonomic epinephrine secretion!This is the normal sequence of hormone responses to ?ing plasma glucose levels.But this normal hormonal response will be blunted over time if there is 1) continued hypoglycemia dampening the sympathetic nervous system, and 2) long-standing diabetic neuropathy! (To be explained later in this flow chart)Abbreviations: [ ] = concentrationTx = TreatmentDM = Diabetes mellitusDiabetic Hypoglycemia: Pathogenesis and Clinical FindingsConfusionCan't concentrateWeaknessSlurred speech? coordination (staggering, etc)SeizuresComa, deathAdrenergic symptoms (epinephrine-mediated):Anxiety, irritability, trembling, pallor (skin vasoconstriction), palpitations, ? systolic BP, tachycardia Cholinergic symptoms(Acetylcholine-mediated):Sweating, hunger, tingling, blurry visionNote: In pts w/out DM, endogenous insulin secretion normally stops when blood [glucose] drops to <4mmol/LAuthor: Yan YuReviewers: Peter Vetere, Gillian Goobie, Hanan Bassyouni** MD at time of publicationLegend:Published June 14, 2013 on www.thecalgaryguide.comMechanismPathophysiologySign/Symptom/Lab FindingComplicationsMany hypoglycemic events over time blunt epinephrine secretion further.Hypoglycemia unawareness can be reversedIf pt stays hypoglycemia-free for >6 weeks, brain restores its ability to detect low glucose levels? peripheral glucose delivery and uptake (saving more glucose for the brain)Lack of glucagon effect reinforces hypoglycemia
124 kB / 361 words")
chronic-hypertensive-retinopathy-pathogenesis-and-clinical-findings
 • 1° HTN
Retinal Detachment
Vitreous Hemorrhage
Central/Branch Retinal Artery/Vein Occlusions
Risk Factors for 2° HTN (ex. Hyperaldosterone, Cushing's, Acromegaly, Chronic Kidney Disease, Obstructive Sleep Apnea, Diabetes Mellitus, Hypo/Hyper-thyroid, Adrenal Hyperplasia, Renal Artery Stenosis)
2° HTN
Ophthalmic Artery Hypertension ,17
Stage 1: Mild/vasoconstrictive
Stage 2: Moderate/sclerotic
Stage 3: Severe/exudative
Stage 4: Malignant
Abbreviations: • HTN — Hypertension • BRB — Blood-retinal barrier • RPE — Retinal pigment epithelium
Legend:
Pathophysiology
Mechanism
Acute and chronic vasospasm
Authors: Graeme Prosperi-Porta Reviewers: Stephanie Cote Usama Malik Johnathan Wong* * MD at time of publication
Diffuse and focal arterial narrowing and vascular tortuosity
Atherosclerosis and hyalinization causes arteriolar wall thickening resulting in a diffuse light reflex appearing red-brown coloured
Thickening of the arteriolar wall and/or sclerotic thickening at the arteriole/venule crossing compresses the underlying venule
BRB breakdown causes dot/blot hemorrhages in the inner retina and flame hemorrhages in the nerve fiber layer
Serum proteins and lipids leakage from damaged BRB appears as white or yellow areas with sharp margins
Occlusion of the terminal retinal arterioles causes fluffy white ischemic lesions in the inner retinal nerve fiber layer
Hyper-pigmented patches surrounded by a hypo-pigmented ring due to RPE clumping around atrophic areas in the choroid
Sign/Symptom/Lab Finding
lschemia of optic disc arterioles causes optic nerve swelling and blurred disc margins. Leakage of optic disc arterioles causes hemorrhage and disc edema.
Complications
Copper Wiring
AV nicking
Retinal Hemorrhages
Yellow Hard Exudates
Cotton-wool Spots
Elschnig's Spots
Papilledema")
Erectile Dysfunction: Pathogenesis

1. Assess CVD Disease risk* a. I% Blood pressure b. I% Fasting glucose or HbA1c c. TG's & cholesterol 1. Penile duplex sonography 2. Cavernosometry
Legend:
Endocrinologic Erectile Dysfunction
Hypogonadism, hyperprolactinemia, hyperthyroidism, alcoholism, iatrogenic
.J, circulating free testosterone
•
1. 4, 7 AM free testosterone* 2. l• Thyroid Stimulating Hormone 3. l• Prolactin 4. l• Follicle Stimulating Hormone 5. l• Luteinizing Hormone
4, release of NO and cGMP levels within corpora cavernosa and smooth muscle relaxation
Pathophysiology Mechanism
Neurogenic Erectile Dysfunction
Neurologic disease, trauma, iatrogenic, diabetes mellitus
Central (cerebral or spinal cord); peripheral (afferent/sensory neuropathy) or efferent (autonomic neuropathy)
4, parasympathetic nerve firing
4, NO release
Psychogenic Erectile Dysfunction
•
Sudden onset, sporadic (circumstantial), younger, nocturnal/AM erection present
•
Anxiety, depression, strained relationship, lack of sexual arousal, psychological disorder
Possible mechanisms include an imbalance of central neurotransmitters, over inhibition of spinal erection center by the brain, and sympathetic overactivity
1. Abnormal Nocturnal penile 1. Normal Nocturnal penile tumescence and rigidity* tumescence and rigidity*
Erectile Dysfunction -• (persistent or recurrent inability to achieve an erection sufficient to achieve desired sexual performance)
Sign/Symptoni/Lab Finding
Complications
Authors: Braden Milian Reviewers: Alex Tang Usama Malik Jay C. Lee* * MD at time of publication")
Mixed Urinary Incontinence Pathogenesis and clinical findings
 4, Urinary leakage preceded by a sudden, strong urge to void
Overflow Incontinence vir Overfilling of the bladder from obstruction; BOO (tumour, stone, BPH, urethral or bladder neck stricture)
Detrusor Overactivity Ilr OAB (idiopathic), CNS lesion (neurogenic), inflammation/ infection (cystitis, UTI), diabetes mellitus
4. Bladder Wall Compliance
Progressive t in intravesicle pressure during bladder filling pushing urine from the bladder
Authors: Braden Millan Reviewers: Alex Tang Usama Malik Jay C. Lee* * MD at time of publication
Stress Urinary Incontinence (SUI) + Episodic involuntary urinary leakage with sudden l• in intra-abdominal pressure
4.
Urethral hypermobility, intrinsic sphincter deficiency, or a poorly coapting urethra
4,
4, Pelvic floor muscle and ligament strength causing 4. tone of vesicoureteral sphincter unit; 4, urethral strength and associated striated and smooth muscle; iatrogenic
Legend:
Failure to Void Weak Stream (+ dribbling), Intermittent, Straining, '1` PVR if a complication of urinary retention; obstruction visible on cystoscopy
Failure to Store Frequency, Urgency, Nocturia, Dysuria if SUI or UUI not caused by obstruction
Pathophysiology Mechanism
Urodynamic Studies SUI — 4, urethral closure pressure with 11` IAP/Bladder Volume and urinary leakage UUI — involuntary detrusor contraction and/or detrusor sphincter dyssynergia
Incontinence, 4, Quality of Life, UTI's")
intrauterine-growth-restriction-iugr-pathogenesis
: Pathogenesis
Authors: Ricki Hagen Reviewers: Jaimie Bird Sarah McQuillan* * MD at time of publication
Abbreviations:
• DM: diabetes mellitus
• HTN: hypertension • IUGR: intrauterine growth restriction
• SGA: small for gestational age
• SLE: systemic lupus erythematosus
• TORCH: Toxoplasmosis, Others, Rubella, CMV, HSV
Maternal Factors
Maternal-Fetal Factors Placental malformations
(Ex. previa, accreta, infarction, abnormal implantation, ischemia)
Gestational HTN/ Preeclampsia
Multiple gestation Gestational DM
Fetal Factors Structural anomalies
(often comorbid with cytogenetic disorders)
Congenital infections
(Ex. TORCH)
Inborn errors of metabolism
Chromosomal disorders/ genetic syndromes
Multiple unclear intrinsic fetal mechanisms
Note:
Teratogenic medications (Ex. Warafin, Valproic Acid, Folic Acid Antagonists)
High altitude living
Smoking, ETOH and/or drug use
Malnutrition/ Low pre-gestational weight
Multiple unclear extrinsic fetal mechanisms
Medical conditions
(Ex: chronic HTN, cyanotic heart disease, severe chronic anemia, kidney disease)
Autoimmune conditions (Ex. Type 1 DM, SLE)
Decreased uteroplacental blood flow
Nutrient supply to fetus compromised
Reduction of total body mass, bone
mineral content, and muscle mass
Blood flow redirected away from vital organs to brain, placenta, heart and adrenal glands
Reduction of overall fetal size to increase survival
IUGR
Failure to reach genetically determined growth potential
• IUGR is not synonymous with SGA • Constitutional SGA is due to
paternal and maternal factors such as height, weight, ethnicity, and parity; it is not associated with increased risk for infant mortality or morbidity
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published October, 30, 2018 on www.thecalgaryguide.com")
Hypernatremia Physiology
![Hypernatremia: Physiology Unreplaced H2O loss
Hypodipsia
H2O shift into cells
Severe exercise, electroshock induced seizures
Transient ↑ cell osmolality
Na+ overload
Inappropriate IV hypertonic solution, salt poisoning
Abbreviations:
H2O: Water
GI: Gastrointestinal
DM: Diabetes Mellitus
DI: Diabetes Insipidus
Na+: Sodium ion
IV: Intravenous
ADH: Antidiuretic Hormone LOC: Level of Consciousness
Skin
Sweat, burns
GI
Vomiting, bleeding, osmotic diarrhea
Fluid [Na+] < serum [Na+]
↑ H2O loss compared to Na+ loss
Renal
DM, Mannitol, Diuretics
Absent thirst mechanism
Hypothalamic lesion impairs normal drive for H2O intake
Nephrogenic
↑ renal resistance to ADH
H2O Deprivation Test + no AVP response
↓ access to H2O
DI
Central
↓ ADH secretion
H2O Deprivation Test + AVP response
↑ [Na+] 10- 15 mEq/L within a few minutes
Weakness, irritability, seizures, coma
↑ thirst, ↓ urinary frequency and volume
Note:
Hypernatremia
Serum [Na+] > 145 mmol/L
Intracranial hemorrhage
Headache, vomiting, ↓ LOC
• Plasma [Na+] is regulated by water intake/excretion, not by changes in [Na+].
• Effects on plasma [Na+] of IV fluids or loss of bodily fluids is determined by the tonicity of the fluid, not the osmolality.
Authors: Mannat Dhillon Reviewers: Andrea Kuczynski Kevin McLaughlin* * MD at time of publication
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published January 11, 2019 on www.thecalgaryguide.com](http://calgaryguide.ucalgary.ca/wp-content/uploads/2019/01/Hypernatremia-Physiology-.jpg "Hypernatremia: Physiology Unreplaced H2O loss
Hypodipsia
H2O shift into cells
Severe exercise, electroshock induced seizures
Transient ↑ cell osmolality
Na+ overload
Inappropriate IV hypertonic solution, salt poisoning
Abbreviations:
H2O: Water
GI: Gastrointestinal
DM: Diabetes Mellitus
DI: Diabetes Insipidus
Na+: Sodium ion
IV: Intravenous
ADH: Antidiuretic Hormone LOC: Level of Consciousness
Skin
Sweat, burns
GI
Vomiting, bleeding, osmotic diarrhea
Fluid [Na+] < serum [Na+]
↑ H2O loss compared to Na+ loss
Renal
DM, Mannitol, Diuretics
Absent thirst mechanism
Hypothalamic lesion impairs normal drive for H2O intake
Nephrogenic
↑ renal resistance to ADH
H2O Deprivation Test + no AVP response
↓ access to H2O
DI
Central
↓ ADH secretion
H2O Deprivation Test + AVP response
↑ [Na+] 10- 15 mEq/L within a few minutes
Weakness, irritability, seizures, coma
↑ thirst, ↓ urinary frequency and volume
Note:
Hypernatremia
Serum [Na+] > 145 mmol/L
Intracranial hemorrhage
Headache, vomiting, ↓ LOC
• Plasma [Na+] is regulated by water intake/excretion, not by changes in [Na+].
• Effects on plasma [Na+] of IV fluids or loss of bodily fluids is determined by the tonicity of the fluid, not the osmolality.
Authors: Mannat Dhillon Reviewers: Andrea Kuczynski Kevin McLaughlin* * MD at time of publication
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published January 11, 2019 on www.thecalgaryguide.com")
acanthosis-nigricans-pathogenesis-and-clinical-findings

Cellulitis
 and entry of pathogen
Risk Factors: Immunocompromised Host: -Diabetes mellitus+ -Lymphedema -Malnourishment
-Older patient+
-Obesity+
-Peripheral vascular disease General Infection Risk: -History of cellulitis+ +highest risk factors
Risk Factors for MRSA Cellulitis: Increased exposure to MRSA: -Contact sports
-Crowded living conditions -Health care workers -Indigenous descent
-Sharing towels, equipment
Increased susceptibility:
-Immunodeficiency -Young age
Direct inoculation (e.g. trauma) Organism virulence overwhelms host defense mechanisms (related to risk factors)
Cellulitis: A bacterial infection in which pathogens penetrate deep dermis and/or subcutaneous fat
Cytokines activate immune response
Accumulation of pus (bacteria, white blood cells, dead skin)
Abscess formation
Infection spreads to nearby lymph nodes
Lymphadenitis
Infection spreads through lymph vessels
Ascending lymphangitis
Local inflammatory response in skin
Pain Warmth Edema Erythema (redness)
with indistinct margins
Vesicles and bullae
Organisms penetrate blood vessels
Bacteremia (presence of bacteria in blood)
Systemic inflammation
Distant spread to bone
Osteomyelitis
Distant spread to endocardium (inner lining of heart chambers and valves)
Endocarditis
Fever Malaise
Chills
Sepsis
(rarely)
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published September 27, 2020 on www.thecalgaryguide.com")
Cubital-Tunnel-Syndrome-Ulnar-Neuropathy
: Pathogenesis and clinical findings
Osteoarthritis
Rheumatoid arthritis
Diabetes mellitus (see Calgary Guide slide on Diabetic Neuropathy)
Degenerative bone & joint changes
Autoimmune damage to joints
Abnormal bone structure/lesions
Idiopathic
Hyperglycemia causes nerve damage (complex mechanisms)
Elbow trauma
Repetitive elbow flexion
Authors: Chris Oleynick Alexandros Mouratidis Yan Yu* Reviewers: Annalise Abbott Sean Crooks Davis Maclean Hannah Koury Jeremy LaMothe* Ian Auld* * MD at time of publication
Inflammation or edema in cubital tunnel (space between medial epicondyle of humerus and olecranon of ulna) where ulnar nerve is found
↓ size of the cubital tunnelà↑ pressure on internal contents (e.g. ulnar nerve)
Axonal conduction is interrupted
Myelin sheath is damaged
↓ blood supply to nerve
Weakness of adductor pollicus longus (works to adduct thumb)
↑ compensatory activity of flexor
pollicis longus with pinching
↓ activity of hypothenar muscles (which move the 5th digit)
↓ activity of 5th digit palmar interosseus muscles (which adduct the 5th digit)
Cubital Tunnel syndrome: compression neuropathy
Paresthesia
Abnormal sensations of skin (“pins & needles”, tingling, burning, and/or numbness) in ulnar nerve sensory distribution
ulnar nerve
↓ activity of muscles innervated by ulnar nerve (medial forearm flexors, hypothenar muscles, interossei muscles, adductor pollicis longus)
+ Tinel sign
over cubital tunnel (tapping at medial elbow causes discomfort and/or paresthesia)
Weak pinch & ↓ grip strength
+ Froment’s sign
(thumb hyperflexion compensates for weak pinch)
Hypothenar atrophy
+ Wartenburg’s sign
(involuntary, excess abduction of 5th digit)
of the
Altered sensation in areas innervated by ulnar nerve (medial forearm and wrist, 5th digit, and medial half of 4th digit)
+ elbow flexion test
(flexing elbow & extending wrist further ↓ volume of the cubital tunnelà paresthesia in ulnar nerve sensory distribution
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
First published January 12, 2017, re-published February 28, 2021 on www.thecalgaryguide.com")
Fat-Embolism-Syndrome

Non-trauma related (rare)
Long bone fracture
Pelvic fracture
Orthopedic Trauma
Intraosseous access
Soft tissue injuries
Chest compressions
Bone marrow transplant
Pancreatitis
Diabetes mellitus
Fat from bone marrow is disrupted and leaks into bloodstream via damaged blood vessels
Fat globules obstruct dermal capillaries
Capillaries rupture
Blood leaks into the skin
Petechial rash
Non-Orthopedic Trauma (less common)
Fat from injured adipose tissue is released from adipocytes into bloodstream
Metabolic disturbance mobilizes stored fat and moves it into circulation
Fat Embolism Syndrome
the presence of fat globules in circulation
Fat globules damage blood vessel walls
Platelets stick to damaged areas Platelet aggregation
↑ circulating free fatty acids
↑ inflammatory cytokines (TNF, IL1, IL6)
↑ serum C Reactive Protein (an acute phase reactant)
C reactive protein binds to lipid vesicles in circulation
↑ formation of lipid complexes in the blood
Obstruction of cerebral vasculature
↓ blood flow and oxygen delivery to brain tissue
Neurological findings: ranging from ↓ level of consciousness to seizures
Notes:
Large quantities of fat globules can obstruct pulmonary vasculature
Blood clots form throughout the body
Disseminated intravascular coagulopathy
Back up of blood into right heart àRight ventricular dysfunction
↓ pulmonary arterial blood flow à↓ gas exchange in the lungs
Higher CO2 & lower O2 levels in blood àdetected by chemoreceptors
Chemoreceptors stimulate respiratory centre in the brain to ↑ rate of respiration
Dyspnea / Tachypnea
Authors: Tabitha Hawes Reviewers: Hannah Koury, Alyssa Federico, Davis MacLean, Mehul Gupta, Yan Yu*, Jeremy Lamothe* * MD at time of publication
• Underlined findings indicate classic triad of symptoms (petechial rash, neurologic findings, dyspnea/tachypnea)
• Clinical presentation of fat embolism syndrome is variable and may present with any or all of these findings
↓ pumping of blood into systemic circulation
Hypotension Obstructive shock
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published July 19, 2021 on www.thecalgaryguide.com")
Pathogenese des Diabetes Mellitus (DM), Typ II
, Typ II
Genetische Prädisposition:
Poly- oder monogenetische Faktoren (z.B. „Maturity- onset diabetes of the young“ (MODY)) können eine Insulinresistenz prädispositionieren
Altern: Betazellen verlieren im Rahmen des Alterungsprozesses an Masse, wodurch sich, bei bereits bestehender Pädisposition, im Alter ein DM Typ II entwickeln kann .
Medikamente: z.B. Cortison, Psychopharmaka, hochaktive antiretrovirale Therapie(HAART), Minipille (nur auf Progesteron basierende Kontrazeptiva)
Blutzucker bleibt im Normbereich
Zuckervergiftung: Hyperglykämien wirken toxisch auf Betazellen
Hyperglykämie
Beachte: Es besteht eine beträchtliche genetische Komponente: bei familiärer Vorbelastung ist die Erkrankungswahrscheinlichkeit sehr hoch ( bei eineiigen Zwillingen bis zu 90%). Bei Verwandtschaftsgrad ersten Grades ist das Risiko einer Erkrankung 5-10 Mal höher
Ungesunder Lebensstil (z.B. Übergewicht, Bewegungsmangel)
Intraabdominell akkumuliert „Viszeralfett“ (Bauchfett) welches als endokrines Organ funktioniert:
Beachte: „Adipokine“ sind von Fettgewebe produzierte Entzündungsmediatoren (z.B. TNFalpha). Je mehr Fettgewebe ein Mensch besitzt, desto mehr Adipokine werden produziert.
Entzündungsm ediatoren
Freie Fettsäuren
Adipokine
Komplexe, teils unklare Interaktionen mit dem Gewebe
Insulinresistenz
(Die Wirkung des Insulins auf Leber, Muskel und Fettgewebe ist vermindert, Glucose kann deshalb nicht als Energiequelle genutzt werden )
Zunächst kompensieren Betazellen des Pankreas die verminderte Insulinwirkung durch vermehrte Produktion
Fettvergiftung:
Längerfristig nimmt die Kompensationsfähigkeit der Betazellen ab, Insulinproduktion ↓ (relativer Insulinmangel)
Nach Jahren nimmt die Leistungsfähigkeit der Betazellen soweit ab, dass kein Insulin mehr produziert wird (absoluter Insulinmangel)
Diabetes Mellitus Typ II
Freie Fettsäuren blockieren den GLUT2 der Betazellen, Glucoseimport ↓
Betazellen erkennen den erhöhten Blutzucker nicht, Insulinproduktion sinkt
Da der Körper keine Glukose nutzen kann, werden Triglyceride zu freien Fettsäuren umgewandelt damit diese als Energiequelle dienen können
Autor: Yan Yu Rezensenten: Peter Vetere Gillian Goobie Doreen Rabi* Übersetzerin: Sarah Schwarz Übersetzungsprüfer: Gesche Tallen* * MD zum Zeitpunkt der Veröffentlichung
Legende:
Pathophysiologie
Mechanismen
Symptome/Klinische Befunde
Komplikationen
Veröffentlicht 11. Juli 2013 auf www.thecalgaryguide.com")
Pathogenese des Diabetes Mellitus (DM), Typ I
, Typ I
Genetische Prädisposition
IDDM1 (HLA) Mutation IDDM2 (Insulingene) Mutationen
Diet (Kuhmilch, Nitrosamine)
Viren (Röteln, Coxsackie-Virus, Masern)
Drogen/Toxine
(Pyrinuron,Alloxan, Streptozocin, Pentamidine)
Direkter Schaden an Betazellen,
sodass deren Antigene dem
Immunsystem ausgesetzt werden
Stress (häufige Infektionen, Operationen, Pupertät)
Externe Risikofaktoren
Reifende T-Zellen im Thymus sind nicht in der Lage, die Fähigkeit der Erkennung von Insulingenen zu entwickeln T-Zellen greifen Insulin produzierende Betazellen an
Mutationen des HLA (MCH) Gens können die Bindung von T-Zellen an Betazellen fördern aber auch verhindern
Körperfremde Antigene imitieren Betazellantigene (Molekulare Mimiky), sodass sich die Immunantwort gegen diese Antigene auch gegen Betazellen richtet
Beeinträchtigung der Toleranz des Immunsystem gegen körpereigene pankreatische Betazellen
Autoimmunreaktion gegen Betazellen
(sowohl durch angeborenes, als auch durch erworbenes Immunsystem: Infiltration der Langerhans-Inseln durch Monozyten und Zerstörung durch T-Zellen, auch als Insulitis bezeichnet )
Atrophie der Langerhans-Inseln auf die Hälfte ihrer Ausgangsmasse
Längerfristig sind nur noch <10% der Betazellen funktionstüchtig
Beachte: Die Anzahl an Autoantikörpern im Körper korrelieren mit der Wahrscheinlichkeit einen DM Typ I zu entwickeln
Autor: Yan Yu
Rezensenten:
Peter Vetere
Gillian Goobie
*Doreen Rabi
Übersetzerin: Sarah Schwarz Übersetzungsprüfer: Gesche Tallen*
* MD zum Zeitpunkt der Veröffentlichung
Produktion von Autoantikörper gegen Betazellen
“Prä-diabetes”
Diabetes Mellitus Typ I Absoluter Insulinmangel
Anti-Insulin & Anti-GAD65 nachweisbar im Serum
Noch asymptomatisch Hoher postprandialer Blutzucker
Abgeschwächte Insulinreaktion nach i.v. Glucosegabe
Legende:
Pathophysiologie
Mechanismen
Symptome/Klinische Befunde
Komplikationen
Veröffentlicht: 11. Juli 2013 auf www.thecalgaryguide.com")
Diabetische Ketoazidose (DKA)

Betrifft hauptsächlich Patienten mit Diabetes Mellitus Typ 1: Bestimmte Situationen (z.B. Infektionen) erhöhen den Insulinbedarf, welcher durch fehlende Produktion/unzureichende Substitution nicht gedeckt werden kann
Autor: Yan Yu Rezensenten: Peter Vetere, Gill Goobie, Sean Spence, Hanan Bassyouni* Übersetzung: Sarah Schwarz Übersetzungsprüfung: Dr. Gesche Tallen * MD zum Zeitpunkt der Veröffentlichung
Hyperglykämie
(erhöhter Blutzucker)
Bei >12mmol/L, Glukosefiltration > - resorption, Glukose verbleibt im Urin
Glukosurie
Glukose ist osmotisch wirksam und zieht große Mengen Wasser mit sich (Osmotische Diurese)
Polyurie
Schwere Dehydrierung(bis zu 4-5L)
(ZVD↓, Orthostase: posturale Hypotension/ posturale Tachykardie, Ruhepuls ↑ )
Insulinmangel enthemmt die Lipolyse, sodass der Körper Energie aus Triglyceriden produziert
Freisetzung von freien Fettsäuren aus Fettgewebe
Absoluter
Insulinmangel
Hypothalamische Zellen induzieren bei geringem intrazellulären Glukosespiegel ein starkes Hungergefühl
Glukose bleibt im Blut & kann nicht durch Muskel- /Fettgewebe verstoffwechselt werden
“Ausgehungerte” Zellen triggern die Freisetzung kataboler Hormone: Glucagon, Katecholamine, Cortisol, Somatotropin
Damit intrazellulärer Glukosespiegel ↑, erhöht der Körper den Blutzucker
Hydrolyse von freien Fettsäure in
der Leber (Ketogenese) Polyphagie
↓ Proteinsynthese, ↑ Proteolyse (in Muskelgewebe)
↑ Gluconeogenese, ↑ Glykogenolyse (in der Leber)
Acetyl Co-A
Energiequelle für “ausgehungerte” Zellen
Beachte: Neben Glukose sind Ketonkörper die einzigen Energieträger die von Nervenzellen metabolisiert werden können. Deshalb werden sie vom Körper bei geringem Glukoseangebot produziert.
Ketonkörper
( β-Hydroxybutyrat, Acetoacetat, Aceton, akkumulieren im Blut)
Ketonurie
Metabolische Azidose
(↑Anionenlücke: Ketonkörper verbrauchen den HCO3--Puffer)
Substrate der Gluconeogenese↑
Durch ↓ Extrazellulärflüssigkeit, werden Ketonkörper konzentriert → Azidose
Stört das enterische Nervensystem, Magenentleerung ↓, Ileus
Bauchschmerzen, Übelkeit, Erbrechen
(Dehydration↑)
Abatmen von CO2 um Azidose auszugleichen
Kussmaul- Atmung (Tiefe, schnelle Züge)
Abatmen von Keton- körpern
Azeton- geruch der Atemluft
Beeinträchtigung elektrischer Signale zum Gehirn, Rücken- mark und Nervenzellen
Perfusion des Gehirns, Rückenmark, Nervenzellen↓
Verschiebung des Wasser- und Elektrolythaushalts
Falls Trinkwasser vorhanden
Polydipsie Beachte: Während der DKA verliert der Körper K+ durch
Elektrolytstörung
Schwäche, Delirium +/- Koma
osmotische Diurese und Erbrechen. Da gleichzeitig allerdings K+ aus den Zellen diffundiert, kann das Serumkalium unauffällig/erhöht sein. Um Hypokaliämien zu verhindern sollte bei K+ <5.0mmol/L KCl zusammen mit Insulin i.v. verabreicht werden. Cave: gute Nierenfunktion des Patienten
Therapie: 1) +++ Flüssigkeit. 2) Insulin + KCl. 3) Auffüllen der Anionenlücke. 4) Auslösende Faktoren identifizieren. 5) Wenig PO4 (typischerweise wenige Stunden bis einen Tag nach der Ketoazidose durch ↑ ATP-Produktion.)
Legende:
Pathophysiologie
Mechanismen
Symptome/Klinische Befunde
Komplikationen
Publikationsdatum: 17 Juni 2013 auf www.thecalgaryguide.com
Hyperosmolares/ Hyperglykämisches Koma (HHS)
Beachte: betrifft nur Patienten mit Diabetes mellitus Typ II
Beachte: Es sollte stets nach auslösenden Faktoren z.B. einer Infektion gesucht werden
Autor: Yan Yu Rezensenten:
Peter Vetere
Gill Goobie
Hanan Bassyouni* Übersetzung:
Sarah Schwarz Übersetzungsprüfung: Dr. Gesche Tallen
* MD zum Zeitpunkt der Veröffentlichung
Verschiebung des Wasser- und Elektrolythaushalts
Elektrolytstörung
Unzureichende Insulin- produktion, Insulinresistenz, Nichtansprechen auf Insulintherapie
Relativer Insulinmangel
Situationen mit ↑Insulinbedarf: Infekt, Pneumonie, Herzinfarkt, Pancreatitis, etc.)
Hyperglykämie
(Stark erhöhter Blutzucker, höher als bei der DKA)
Bei >12mmol/L, Glukosefiltration > - resorption, Glukose verbleibt im Urin
Glukosurie
Glukose ist osmotisch wirksam und zieht große Mengen Wasser mit sich (Osmotische Diurese)
Polyurie Dehydration
(ZVD↓, Orthostase: posturale Hypotension/posturale Tachykardie, Ruhepuls ↑ )
Durch das wenige noch vorhandene Insulin, wird ein Teil der Glukose von Muskel- /Fettgewebe verstoffwechselt, ein Teil verbleibt im Blut
Körperzellen brauchen eine weitere Energiequelle
Freisetzung kataboler Hormone: Glucagon, Katecholamine, Cortisol, Somatrotropin
Damit intrazellulärer Glukosespiegel ↑, erhöht der Körper den Blutzucker
Hypothalamische Zellen induzieren bei geringem intrazellulären Glukosespiegel ein starkes Hungergefühl
Polyphagie
Beachte: Durch das wenige noch
vorhandene Insulin wird die Lipolyse gehemmt und werden keine Ketonkörper produziert. Es kommt nicht zur Azidose und Ketonurie (Gegensatz zur DKA). Sollte es doch zur Ketourie kommen, ist das meist Folge von Hungerzuständen oder anderen Mechanismen
Extrazellulärflüssigkeit ↓ mit erhöhter Osmolarität (z.B. Hypernatriämie)
↑ Gluconeogenese, ↑ Glykogenolyse (in der Leber)
↓ Proteinsynthese, ↑ Proteolyse (in Muskelgewebe)
Substrate der Gluconeogenese ↑
Sollte der Patient nicht ausreichend trinken um das Volumendefizit auszugleichen
Falls Trinkwasser vorhanden
Polydipsie
Wasser verlässt Zellen entlang des osmotischen Gradienten, Neuronen schrumpfen
Neuronaler Schaden: Delirium, Krampfanfall, Benommenheit, Koma
Renale Perfusion↓, GFR ↓
Nierenversagen
(prä-renal, siehe entsprechende Folie)
Beachte: Während der DKA verliert der Körper K+ durch osmotische Diurese. Da gleichzeitig allerdings K+ aus den Zellen diffundiert, kann das Serumkalium unauffällig/erhöht sein. Um Hypokaliämien zu verhindern sollte bei K+ <5.0mmol/L KCl zusammen mit Insulin i.v. verabreicht werden.
Cave: gute Nierenfunktion des Patienten
Beachte: Elektrolytstörungen (z.B. Hyperkaliämien, Hypernatriämien) verschlechtern sich bei akutem Nierenversagen, welches bei der DKA &HK häufig auftritt
Legende:
Pathophysiologie
Mechanismen
Symptome/Klinische Befunde
Komplikationen
Veröffentlicht: 3. November 2016 auf www.thecalgaryguide.com")
Hyperosmolares/ Hyperglykämisches Koma (HHS)

Betrifft hauptsächlich Patienten mit Diabetes Mellitus Typ 1: Bestimmte Situationen (z.B. Infektionen) erhöhen den Insulinbedarf, welcher durch fehlende Produktion/unzureichende Substitution nicht gedeckt werden kann
Autor: Yan Yu Rezensenten: Peter Vetere, Gill Goobie, Sean Spence, Hanan Bassyouni* Übersetzung: Sarah Schwarz Übersetzungsprüfung: Dr. Gesche Tallen * MD zum Zeitpunkt der Veröffentlichung
Hyperglykämie
(erhöhter Blutzucker)
Bei >12mmol/L, Glukosefiltration > - resorption, Glukose verbleibt im Urin
Glukosurie
Glukose ist osmotisch wirksam und zieht große Mengen Wasser mit sich (Osmotische Diurese)
Polyurie
Schwere Dehydrierung(bis zu 4-5L)
(ZVD↓, Orthostase: posturale Hypotension/ posturale Tachykardie, Ruhepuls ↑ )
Insulinmangel enthemmt die Lipolyse, sodass der Körper Energie aus Triglyceriden produziert
Freisetzung von freien Fettsäuren aus Fettgewebe
Absoluter
Insulinmangel
Hypothalamische Zellen induzieren bei geringem intrazellulären Glukosespiegel ein starkes Hungergefühl
Glukose bleibt im Blut & kann nicht durch Muskel- /Fettgewebe verstoffwechselt werden
“Ausgehungerte” Zellen triggern die Freisetzung kataboler Hormone: Glucagon, Katecholamine, Cortisol, Somatotropin
Damit intrazellulärer Glukosespiegel ↑, erhöht der Körper den Blutzucker
Hydrolyse von freien Fettsäure in
der Leber (Ketogenese) Polyphagie
↓ Proteinsynthese, ↑ Proteolyse (in Muskelgewebe)
↑ Gluconeogenese, ↑ Glykogenolyse (in der Leber)
Acetyl Co-A
Energiequelle für “ausgehungerte” Zellen
Beachte: Neben Glukose sind Ketonkörper die einzigen Energieträger die von Nervenzellen metabolisiert werden können. Deshalb werden sie vom Körper bei geringem Glukoseangebot produziert.
Ketonkörper
( β-Hydroxybutyrat, Acetoacetat, Aceton, akkumulieren im Blut)
Ketonurie
Metabolische Azidose
(↑Anionenlücke: Ketonkörper verbrauchen den HCO3--Puffer)
Substrate der Gluconeogenese↑
Durch ↓ Extrazellulärflüssigkeit, werden Ketonkörper konzentriert → Azidose
Stört das enterische Nervensystem, Magenentleerung ↓, Ileus
Bauchschmerzen, Übelkeit, Erbrechen
(Dehydration↑)
Abatmen von CO2 um Azidose auszugleichen
Kussmaul- Atmung (Tiefe, schnelle Züge)
Abatmen von Keton- körpern
Azeton- geruch der Atemluft
Beeinträchtigung elektrischer Signale zum Gehirn, Rücken- mark und Nervenzellen
Perfusion des Gehirns, Rückenmark, Nervenzellen↓
Verschiebung des Wasser- und Elektrolythaushalts
Falls Trinkwasser vorhanden
Polydipsie Beachte: Während der DKA verliert der Körper K+ durch
Elektrolytstörung
Schwäche, Delirium +/- Koma
osmotische Diurese und Erbrechen. Da gleichzeitig allerdings K+ aus den Zellen diffundiert, kann das Serumkalium unauffällig/erhöht sein. Um Hypokaliämien zu verhindern sollte bei K+ <5.0mmol/L KCl zusammen mit Insulin i.v. verabreicht werden. Cave: gute Nierenfunktion des Patienten
Therapie: 1) +++ Flüssigkeit. 2) Insulin + KCl. 3) Auffüllen der Anionenlücke. 4) Auslösende Faktoren identifizieren. 5) Wenig PO4 (typischerweise wenige Stunden bis einen Tag nach der Ketoazidose durch ↑ ATP-Produktion.)
Legende:
Pathophysiologie
Mechanismen
Symptome/Klinische Befunde
Komplikationen
Publikationsdatum: 17 Juni 2013 auf www.thecalgaryguide.com
Hyperosmolares/ Hyperglykämisches Koma (HHS)
Beachte: betrifft nur Patienten mit Diabetes mellitus Typ II
Beachte: Es sollte stets nach auslösenden Faktoren z.B. einer Infektion gesucht werden
Autor: Yan Yu Rezensenten:
Peter Vetere
Gill Goobie
Hanan Bassyouni* Übersetzung:
Sarah Schwarz Übersetzungsprüfung: Dr. Gesche Tallen
* MD zum Zeitpunkt der Veröffentlichung
Verschiebung des Wasser- und Elektrolythaushalts
Elektrolytstörung
Unzureichende Insulin- produktion, Insulinresistenz, Nichtansprechen auf Insulintherapie
Relativer Insulinmangel
Situationen mit ↑Insulinbedarf: Infekt, Pneumonie, Herzinfarkt, Pancreatitis, etc.)
Hyperglykämie
(Stark erhöhter Blutzucker, höher als bei der DKA)
Bei >12mmol/L, Glukosefiltration > - resorption, Glukose verbleibt im Urin
Glukosurie
Glukose ist osmotisch wirksam und zieht große Mengen Wasser mit sich (Osmotische Diurese)
Polyurie Dehydration
(ZVD↓, Orthostase: posturale Hypotension/posturale Tachykardie, Ruhepuls ↑ )
Durch das wenige noch vorhandene Insulin, wird ein Teil der Glukose von Muskel- /Fettgewebe verstoffwechselt, ein Teil verbleibt im Blut
Körperzellen brauchen eine weitere Energiequelle
Freisetzung kataboler Hormone: Glucagon, Katecholamine, Cortisol, Somatrotropin
Damit intrazellulärer Glukosespiegel ↑, erhöht der Körper den Blutzucker
Hypothalamische Zellen induzieren bei geringem intrazellulären Glukosespiegel ein starkes Hungergefühl
Polyphagie
Beachte: Durch das wenige noch
vorhandene Insulin wird die Lipolyse gehemmt und werden keine Ketonkörper produziert. Es kommt nicht zur Azidose und Ketonurie (Gegensatz zur DKA). Sollte es doch zur Ketourie kommen, ist das meist Folge von Hungerzuständen oder anderen Mechanismen
Extrazellulärflüssigkeit ↓ mit erhöhter Osmolarität (z.B. Hypernatriämie)
↑ Gluconeogenese, ↑ Glykogenolyse (in der Leber)
↓ Proteinsynthese, ↑ Proteolyse (in Muskelgewebe)
Substrate der Gluconeogenese ↑
Sollte der Patient nicht ausreichend trinken um das Volumendefizit auszugleichen
Falls Trinkwasser vorhanden
Polydipsie
Wasser verlässt Zellen entlang des osmotischen Gradienten, Neuronen schrumpfen
Neuronaler Schaden: Delirium, Krampfanfall, Benommenheit, Koma
Renale Perfusion↓, GFR ↓
Nierenversagen
(prä-renal, siehe entsprechende Folie)
Beachte: Während der DKA verliert der Körper K+ durch osmotische Diurese. Da gleichzeitig allerdings K+ aus den Zellen diffundiert, kann das Serumkalium unauffällig/erhöht sein. Um Hypokaliämien zu verhindern sollte bei K+ <5.0mmol/L KCl zusammen mit Insulin i.v. verabreicht werden.
Cave: gute Nierenfunktion des Patienten
Beachte: Elektrolytstörungen (z.B. Hyperkaliämien, Hypernatriämien) verschlechtern sich bei akutem Nierenversagen, welches bei der DKA &HK häufig auftritt
Legende:
Pathophysiologie
Mechanismen
Symptome/Klinische Befunde
Komplikationen
Veröffentlicht: 3. November 2016 auf www.thecalgaryguide.com")
Nephrotisches Syndrom: Pathogenese und klinische Befunde

Lupus-Nephritis
Postinfektiöse Glomeruloneprhitis
IgA Nephropathie
Membranöse Glomerulonephritis
Antikörper greifen Podozyten an, Verdickung der glomerulären Basalmembran
Fokal-segmentale Glomerulosklerose (FSSGN)
Schädigung des glomerulären Endo- und Epithels
Minimal Change Glomerulo- nephritis (MCNG)
Diabetes mellitus
Autor: Yan Yu Rezensenten: Alexander Arnold David Waldner
Sean Spence
Stefan Mustata* Übersetzung:
Sarah Schwarz Übersetzungsprüfung: Gesche Tallen*
* MD zum Zeitpunkt der Veröffentlichung
Podozyten-Schädigung auf der epithelialen Seite des Glomerulums (Abflachung der Podozytenfortsätze)
Glomeruläre Immunkomplex- ablagerungen
Chronische Hyperglykämien schädigen das Glomerulum
Geschädigter Proteinfilter (v.a. für geladene Proteine)
Ablagerungen von Immunglobulin-Leichtketten im Glomerulum
Amyloidose
Geschädigte Glomeruli --> gestörte Filterbarriere v.a. für Proteine --> übermäßige Filtration von Plasmaproteinen
Vermehrte renale Ausscheidung von Immunglobulin-Leichtketten (Filtration>Resorption)
Hypoalbuminämie*
Übermäßiger Verlust an Albumin über den Urin
Proteinurie >3.5g/Tag*
Verlust an Antikoagulationsproteinen (Antithrombin, Plasminogen, Protein C & S) über den Urin
Koagulations- /Gerinnungsfaktoren (z.B. 1,7,8, 10) sind in Überzahl
Proteinurie >3.5g/Tag*
Thrombophilie
Anasarka
(Generalisiertes Ödem)
Lidödem
(klassisches Frühzeichen)
Lipidurie
(zeigt unter
gekreuztem polarisiertem Licht eine Malteserkreuz- form)
50% des Serumkalziums sind an Albumin gebunden, sodass Serumkalzium- spiegel ↓
Serum- Ca2+ repräsentiert nicht mehr das Gesamt-Ca2+
Beachte:
• DerklassischeSymptomkomplex(*)desnephrotischen Syndroms besteht aus: Proteinurie (>3,5g/Tag), Ödemen, Hypoalbuminämie,Hyperlipidämie
• Fürjede10g/LmitAlbumin<40:
➔ Addiere 2.5 zur errechneten Anionenlücke um
dessen “wahren” Wert zu bekommen
➔ Addiere 0,2mmol/L zum Gesamt-Ca um den
Wert des ionisierten Kalziums zu errechnen
Blut neigt zur Bildung von Thromben
Ödeme*
↑ Renale Filtrationder Lipide und Ausscheidung über den Urin
Die Anionenlücke ergibt sich hauptsächlich aus negativ geladenem Serumalbumin
Anionenlücke↓
Kolloid- osmotischer Druck ↓
Flüssigkeit kann nicht mehr in den Blutgefäßen gehalten werden und diffundiert ins Gewebe
Signalisiert der Leber die Albuminproduktion zuerhöhen,Albumin wird aber weiterhin über die Nieren verloren
Synthese- arbeit der Leber↑, auch ↑ Lipoprotein- synthese
Hyperlipidämie*:
(Serum-LDL, -VLDL, - TG ↑ )
Legende:
Pathophysiologie
Mechanismen
Symptome/Klinische Befunde
Komplikationen
Veröffentlicht: 19. August 2013 auf www.thecalgaryguide.com")
Celulitis

Organismos penetran los vasos sanguíneos
Bacteremia (presencia de bacterias en sangre)
Piel normal
Capa epidérmica
Unión dérmica- epidérmica Capa dérmica
Grasa subcutánea
Flora cutánea residente: Staphylococcus coagulasa negativos*
Flora cutánea transitoria:
Staphylococcus aureus*
Streptococcus pyogenes
Bacterias gram negativas Hongos
Patógeno en dermis profunda y grasa subcutánea
*patógenos más comunes
Rotura de la barrera cutánea (puede que no sea evidente) y entrada de patógenos
Virulencia del organismo supera los mecanismos de defensa del huésped (asociado a los factores de riesgo)
Celulitis: Una infección bacteriana en la que los patógenos penetran la dermis profunda y/o la grasa subcutánea
Citocinas activan la respuesta inmune
Acumulación de pus (bacterias, glóbulos blancos, piel muerta)
Formación de abscesos
Factores de riesgo:
Huésped inmunodeprimido: -Diabetes mellitus+
-Linfedema
-Desnutrición
-Paciente adulto mayor+ -Obesidad+
-Enfermedad vascular periférica Riesgo de infección general: -Historia de celulitis+
+factores de riesgo mayores
Factores de riesgo para celulitis por SARM:
Mayor exposición a SARM: -Deportes de contacto -Hacinamiento
-Trabajadores de la salud -Ascendencia indígena -Compartir toallas, equipos Mayor susceptibilidad: -Inmunodeficiencia
-Edad temprana
Infección se propaga a los ganglios linfáticos cercanos
Linfadenitis
infección se propaga a través de los vasos linfáticos
Linfangitis ascendente
Respuesta inflamatoria local en la piel
Diseminación a distancia en endocardio (revestimiento interno de las cámaras y válvulas del corazón)
Endocarditis SARM: Staphylococcus aureus resistente a meticilina
Dolor
Calor
Fiebre Malestar
Diseminación a distancia en el hueso
Osteomielitis
Escalofríos
Inflamación sistémica
Edema Eritema (enrojecimiento)
con márgenes indefinidos
Vesículas y ampollas
(poco frecuente)
Sepsis Abreviaturas:
Leyenda: Patofisiología
Mecanismo
Signos/Síntomas/Hallazgos de Laboratorio
Complicaciones
Publicado el 27 Septiembre, 2020 en www.thecalgaryguide.com")
hyperkalemia-pathophysiology-intracellular-shift-and-intake
![Hyperkalemia (intracellular shift and ↑ intake): Pathophysiology
↑ K+ dietary intake (rarely causative)
β2 receptor inhibition (i.e. beta blockers)
Digoxin
α1 receptor stimulation (i.e. epinephrine, norepinephrine)
Insulin deficiency or resistance (i.e. diabetes mellitus)
↓ Stimulation of NHE1 (moves 1 Na+ into cell, 1 H+ out of cell) throughout body
Normal Anion- Gap Metabolic Acidosis (NAGMA)
Excess serum H+ results in ↓ NHE1 activity (See NAGMA: Pathogenesis and Laboratory Findings slide)
↑ Serum osmolarity (i.e. hyperglycemia)
Osmotic movement of water from cells to serum
Cell lysis (i.e. tumour lysis syndrome, rhabdomyolysis, hemolytic anemia)
↑ K+ release from lysed cells into the serum
Na+/K+ ATPase (moves 3K+ into cell, 2 Na+ out) activity inhibited on cells throughout body
↓ Activity of Na+/K+ ATPase on cells throughout the body
↓ Amount of K+ entering cells
↓ NHE1 activity prevents Na+ from entering the cell
Lack of high intracellular [Na+] needed to drive the Na+/K+ ATPase on cells
Loss of water from cells ↑ intracellular [K+]
K+ moves down concentration gradient from cell into serum
↑ K+ available for absorption
Since K+ is dissolved in water, some K+ is carried by water as water osmotically moves through water channels into the serum (phenomenon known as “solvent drag”)
See Hyperkalemia: Clinical Findings slide
Authors:
Mannat Dhillon, Joshua Low, Emily Wildman Reviewers:
Huneza Nadeem, Marissa (Ran) Zhang, Andrea Kuczynski, Yan Yu*, Kevin McLaughlin*, Adam Bass*
* MD at time of publication
↑ K+ in serum
Hyperkalemia
Serum [K+] > 5.1 mmol/L
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published July 17, 2022 on www.thecalgaryguide.com](https://calgaryguide.ucalgary.ca/wp-content/uploads/2022/07/Hyperkalemia-shift-1.jpg "Hyperkalemia (intracellular shift and ↑ intake): Pathophysiology
↑ K+ dietary intake (rarely causative)
β2 receptor inhibition (i.e. beta blockers)
Digoxin
α1 receptor stimulation (i.e. epinephrine, norepinephrine)
Insulin deficiency or resistance (i.e. diabetes mellitus)
↓ Stimulation of NHE1 (moves 1 Na+ into cell, 1 H+ out of cell) throughout body
Normal Anion- Gap Metabolic Acidosis (NAGMA)
Excess serum H+ results in ↓ NHE1 activity (See NAGMA: Pathogenesis and Laboratory Findings slide)
↑ Serum osmolarity (i.e. hyperglycemia)
Osmotic movement of water from cells to serum
Cell lysis (i.e. tumour lysis syndrome, rhabdomyolysis, hemolytic anemia)
↑ K+ release from lysed cells into the serum
Na+/K+ ATPase (moves 3K+ into cell, 2 Na+ out) activity inhibited on cells throughout body
↓ Activity of Na+/K+ ATPase on cells throughout the body
↓ Amount of K+ entering cells
↓ NHE1 activity prevents Na+ from entering the cell
Lack of high intracellular [Na+] needed to drive the Na+/K+ ATPase on cells
Loss of water from cells ↑ intracellular [K+]
K+ moves down concentration gradient from cell into serum
↑ K+ available for absorption
Since K+ is dissolved in water, some K+ is carried by water as water osmotically moves through water channels into the serum (phenomenon known as “solvent drag”)
See Hyperkalemia: Clinical Findings slide
Authors:
Mannat Dhillon, Joshua Low, Emily Wildman Reviewers:
Huneza Nadeem, Marissa (Ran) Zhang, Andrea Kuczynski, Yan Yu*, Kevin McLaughlin*, Adam Bass*
* MD at time of publication
↑ K+ in serum
Hyperkalemia
Serum [K+] > 5.1 mmol/L
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published July 17, 2022 on www.thecalgaryguide.com")
chronic-pancreatitis-complications
 from acinar cells in exocrine pancreas
(early in disease course)
Proteins precipitate and form aggregates within pancreatic ducts
Accumulation of protein aggregates and localized fibrosis block pancreatic ducts
Rupture of acinar cells near blocked ducts → release of intracellular enzymes and fluid
Accumulation of enzyme- rich fluid within pancreas
Intra-pancreatic pseudocysts (differ from pseudocysts in acute pancreatitis, which are primarily extra-pancreatic)
Chronic Pancreatitis
Recurrent episodes of acute pancreatitis leading to irreversible fibroinflammatory pancreatic damage
Inflammatory cytokines are
continuously released from damaged pancreas over years
Cytokines damage endothelium of intra- and peri-pancreatic blood vessels (including splenic vein, which runs posteriorly behind pancreas and allows for its venous drainage)
Thin and weakened
vessel walls balloon outwards from pressure of blood flow
Pseudoaneurysms
Venous stasis (low blood flow) → ↑ concentration of clotting factors
Obstruction of peripancreatic ducts
Author: Ashar Memon Reviewers: Yan Yu*, Kiana Hampton, Sylvain Coderre* * MD at time of publication
Exocrine insufficiency
(↓ secretion of digestive enzymes, e.g., Lipase, into gastro-intestinal tract)
↓ digestion of foods and absorption of nutrients (including fats)
↓ absorption of fat-soluble vitamin D
Metabolic bone disease
(a group of disorders of decreased bone mineralization)
Blood vessels dilate and swell from increased blood flow
Gastric varices
Cytokines perpetually activate pancreatic stellate cells (stellate cells produce proteins that remodel extra- cellular matrix)
Pancreatic stellate cells increase amounts of collagen and other extra- cellular matrix molecules in pancreas → Fibrosis
Pancreatic proteolytic enzymes (e.g., trypsin) in fluid-filled pseudocysts digest walls of adjacent blood vessels
Fibrotic tissue and pseudocysts compress peripancreatic structures (including splenic vein)
Cytokines stimulate apoptosis of hormone- producing pancreatic Islet cells
(e.g., beta cells)
Endocrine insufficiency
(↓ production and secretion of pancreatic hormones)
Damaged endothelial cells of splenic vein trigger coagulation cascade
(See Calgary Guide slide on Coagulation Cascade)
Splenic vein thrombosis
↑ resistance to blood flow through splenic vein
Cytokines
stimulate apoptosis of acinar cells in exocrine pancreas
Malnutrition
↓ secretion of insulin
↓ cellular uptake and metabolism of glucose → hyperglycemia
Diabetes mellitus
Collateral blood vessels develop around stomach so blood can circumvent splenic vein and relieve splenic vein hypertension
Duodenal obstruction Biliary obstruction
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published October 18, 2022 on www.thecalgaryguide.com")
Achalasia: Findings on Fluoroscopy with Barium Swallow
 tract during fluoroscopy)
Primary Achalasia
(idiopathic)
Secondary (Pseudo) Achalasia
(due to another disease process such as tumor, Chaga’s disease, diabetes mellitus)
Achalasia
Secondary disease processes can cause denervation or nerve dysfunction of esophageal myenteric plexus (EMP). Tumors can obstruct the lumen and infiltrate the EMP.
Visual differences from primary achalasia
Look for fixed abnormalities and/or mucosal irregularities and shouldering in the setting of a tumor
Nerves controlling the lower esophagus are damaged, making it difficult for food and liquids to pass the esophagus
See “Achalasia: Pathogenesis and Clinical Findings” for full pathogenesis of primary and secondary achalasia
Inflammation and degeneration of nerves in the wall of esophagus
Dysfunction of the esophageal myenteric plexus (network of nerves in the GI tract responsible for peristalsis and sphincter relaxation)
Incomplete relaxation of the Loss of peristalsis in distal lower esophageal sphincter (LES) esophagus
Barium has difficulty passing through the LES to the stomach
Barium outlines the GI tract, following esophageal abnormalities
Normal findings
Esophageal Dilatation
Occurs upstream of narrow LES
Bird Beak Sign / Rat Tail Sign
Smooth narrowing of the distal esophagus
Incomplete LES relaxation
Barium Swallow may be normal in some patients with achalasia. Esophageal manometry (gold standard for diagnosis) or upper endoscopy can be considered instead.
Image Source: Radiopaedia
Legend:
Pathophysiology
Mechanism
Radiographic Findings
Complications
Published November 9, 2022 on www.thecalgaryguide.com")
Carpal Tunnel Syndrome

↑ Blood sugar: deposition of advanced glycation end (AGE) products (proteins or lipids glycated when exposed to sugar)
AGE attaches to and prevents tendons from moving properly
Authors: Amanda Eslinger Yvette Ysabel Yao Reviewers: Matthew Harding Owen Stechishin Mao Ding, Cory Toth* * MD at time of publication
Wrist trauma (distal radius fractures, carpal/metacarpal fractures, tendon ruptures)
Repetitive Strain Injury (repetitive hand & wrist movements)
Irritation, swelling & thickening of tendons in carpal tunnel
Calcium deposits
Calcifica tion
Deposition Amyloidosis
Amyloid
(protein aggregates) deposition
Gout
(See Gout Slide)
Uric acid crystal deposition
Pregnancy
↑ Concentration of hormones & uterine pressure on inferior vena cava
Backup of blood into systemic circulation
Autoimmune
(i.e. Rheumatoid arthritis, scleroderma, lupus, Sjogren’s syndrome)
↑ Inflammatory cytokines causing inflammation
Hypo- thyroidism
Myxedema (swelling of skin and underlying tissues) in carpal tunnel
Idiopathic or Congenital
Edema
Narrowed carpal tunnel leads to ↑ internal pressure
Carpal Tunnel Syndrome
Vascular: median artery thrombosis in carpal tunnel
Median nerve compression inside the carpal tunnel Mechanical disruption of median nerve
Compression exacerbated with flexed wrists (i.e. sleep, driving, holding phone/cup)
Disruption of daily activities and sleep
Ischemia (↓ blood supply) to median nerve
Hypoxia (↓ oxygenated blood flow)
Metabolic conduction block (impaired axonal transport due to ischemia)
Nerve conduction study
(show sensory nerve impulses slowing across the wrist, followed by mild / moderate / severe loss of sensory nerve amplitude
Damage to the myelin sheath
↓ Saltatory conduction (action potential propagation along myelinated axons)
Neuropraxia (nerve compression blocks conduction)
Interruption in axonal continuity
Axonotmesis (endoneural tube stays intact but myelin & distal axon degenerates)
Recovery possible
Full disruption of myelin, axon & nerve sheath
Neurotmesis (axons no longer have an endoneural tube to guide regrowth)
Recovery impossible
↓ Ability to contract and use abductor pollicis brevis muscle
↓ Signals through median nerve
Interference with signals to the brain causes unusual sensations
Hypoalgesia (↓ pain Dysesthesia (tingling, burning, or sensitivity at 1st 3 1⁄2 digits) painful sensation at 1st 3 1⁄2 digits)
Thenar muscle wasting
Reduced hand dexterity
Weak thumb abduction
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published December 2nd, 2013, updated March 22, 2023 on www.thecalgaryguide.com")
Diabetic Retinopathy

Ethnicity: White youth, African American, Hispanic, Asian-Pacific Islanders, and American Indigenous
Type 1 Diabetes Mellitus (T1DM) (see Diabetes Mellitus, Type I for pathogenesis)
Type 2 Diabetes Mellitus (T2DM) (see Diabetes Mellitus, Type II for pathogenesis)
Polycystic Ovarian Syndrome
Family history of T2DM
History of Gestational Diabetes
↑ Body Mass Index
↑ Age
Ethnicity: Indigenous Americans, African American, Hispanic
Poor glycemic control
Chronic high blood sugaràinflammatory response
↑ cytokines and growth factors including vascular endothelial growth factor (VEGF)
Vascular permeability Retinal neovascularization
Vascular endothelial dysfunction: basement membrane thickening, vascular cell death, vascular occlusion from platelet aggregation
Retinal hypoxia
Outpouchings of the weakened capillary walls or endothelial buds attempting to re-vascularize the ischemic retina
Weakened blood-retinal barrier (BRB) allows for rupture into the deeper retinal layers
Lipid deposits with sharp margins due to lipoproteins & other proteins leaking through the damaged blood-retinal barrier
Nerve fiber layer infarcts from occlusions of the precapillary arterioles
Retinal hemorrhages occur in the more superficial nerve layer
Focal areas of saccular venous bulges due to significant retinal ischemia & endothelial wall weakening and damage
Diabetic Retinopathy (DR)
A complication of diabetes due to chronic hyperglycemia resulting in abnormal permeability and ischemia of retinal vessels
Micro-aneurysms
Dot/blot hemorrhages Hard exudates (yellow
opaque solids on retina)
Cotton-wool spots (cloudy translucent patches on retina)
Flame hemorrhages
(multiple opaque red patches)
Venous beading (veins with areas of narrowing forming bead-like segments)
Traction retinal detachment
Bleeding into the vitreous humor
Pericyte death, breakdown of endothelial tight junctions & basement membrane thickening damages blood-retinal barrier
Damaged blood- retinal border leaks fluid into the retinal tissues
Macular edema
(swelling of macula)
Mild Non-proliferative DR
Moderate Non-proliferative DR
Severe Non-proliferative DR
Proliferative DR
Presence of neovascularization
Localized retinal ischemia causes upregulation of vascular endothelial growth factor causing fine, irregular & easily friable neovascularization in the disc, macula, and/or retina
Neovascularization
(fine loop networks of new blood vessels)
Neovascular membranes in vitreous gel form vitreoretinal adhesions and contract
Fragile vessels extending into the vitreous humor
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published June 28, 2017, updated May 18, 2023 on www.thecalgaryguide.com")
Knee Osteoarthritis

Articular trauma
Inflammatory disease or infection (e.g. Rheumatoid or septic arthritis)
Obesity and ↑leptin ↑ Chondrocytes,
inflammatory mediators, and metalloproteinases
Extracellular matrix degradation
↑ Knee joint loading forces
Metabolic syndromes (e.g. diabetes mellitus)
↑ Oxidative stress and insulin resistance
Low-grade systemic inflammation
↓ Elasticity and ↑ degradation of cartilage
↑ Friction in knee joint with movement
↓ Cartilage along femoral groove and posterior surface of patella
Pain, catching, and crepitus (crackling/clicking sound) in the patellofemoral joint
Inability/difficulty with kneeling or climbing stairs
Abnormal distribution of forces accumulate and stress articular surface
↑ Damage/laxity to soft tissue structures stabilizing knee joint
Knee Osteoarthritis
(Multifactorial entity characterized by cartilage breakdown, deterioration of connective tissue, and bone deformities)
↓ Cartilage between distal femur and proximal tibia ↓ Joint spaceàto articular dysfunction
Radiographic changes
See Osteoarthritis (OA): X-Ray Features slide
Repeated attempts to repair cartilage and joint disruption
Subchondral bone thickening (sclerosis) under joint cartilage and bone spur (osteophyte) formation around joint line
Rotational/antero-posterior instability and ↑ external adduction moments during walking
Alterations in proteoglycans, fiber arrangement, and collagen composition in soft tissue structures within/around knee joint
↑ Shear forces and medial compartment narrowing erode and pinch soft tissue structures within the knee joint
Cruciate ligament degeneration
Weakened passive stabilizers of the knee joint
Knee giving way and instability (falls)
Meniscal tears, if large àprevents knee extension/flexion
Locking of the knee
Joint line tenderness:
Patient points to area of tenderness/pain reproducible upon palpation
Anatomical axis of hip, knee, and ankle joints ↑ loading medially
Medial > lateral joint line tenderness
↑ Joint friction activating nociceptors in the surrounding anatomical tissues
Injury and inflammation ↑ nociceptive responses in soft tissue structures and subchondral bone within knee joint
Nociceptive feedback to brain inhibits activity of motor cortex neurons controlling muscles around the knee
↓ Motor output and muscle activation over time
↓ Muscle strength/endurance, lower limb muscle use, functional ability (walking, stairs, etc.)
Joint inflammationà accumulation of fluid within joint
Stiffness, swelling, redness, and pain
Limited joint space reduces range of motion for femur to roll/slide on tibia
↓ Knee flexion and extension
Flexion contracture and antalgic gait
Reduced weight acceptance of the joint and surrounding muscles/tendons
↓ Mobility and physical dysfunction
Muscular atrophy
Reduced function of active stabilizers of the knee joint (quadriceps, adductors, hamstrings)
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published July 30, 2023 on www.thecalgaryguide.com")
Pharmacotherapy for Dyslipidemia Overview

Bile-acid sequestrants
Bind bile acids in intestinal lumen to prevent reabsorption by enterohepatic (gut-liver) circulation
↑ Excretion of bile acids and cholesterol in stool
↓ LDL in blood
Side effects: GI disturbances, commonly interact with other drugs by interfering with absorption
See Calgary guide slide on “Bile-acid sequestrants: Mechanisms of action & side effects” for complete description of mechanism and side effects
(clinical imbalance of lipids)
Hypertriglyceridemia (if VLDL mediated & in need of treatment for pancreatitis prevention)
Hypercholesteremia
(↑ LDL in blood)
Ezetimibe
Inhibits cholesterol absorption via NPC1L1 transporter
↑ Hepatic (liver) LDL receptor expression
↑ LDL clearance from blood
↓ LDL in blood
Avoid in pregnancy
See Calgary guide slide on “Ezetimibe: Mechanisms of action & side effects” for complete description of mechanism and side effects
Combined hyperlipidemia (↑ Triglycerides and ↑ cholesterol)
Statins (ex. rosuvastatin, atorvastatin, simvastatin, pravastatin)
Competitive inhibitors of HMG-CoA reductase (rate-limiting enzyme in cholesterol synthesis)
Fibrates (ex. fenofibrate, gemfibrozil)
Activate PPAR! (nuclear receptor)
↑ Lipolysis (breakdown of lipids) and free fatty acid oxidation
↓ Triglycerides in blood
Side effects: GI discomfort, rash, pruritis
Contraindicated in pregnancy, renal failure, liver & gallbladder disease
See Calgary guide slide on “Fibrates: Mechanisms of action & side effects” for complete description of mechanism and side effects
PCSK9 inhibitors (ex. evolocumab and alirocumab which are monoclonal antibodies)
Inhibit PCSK9 (holds the LDL:LDL receptor complex together as it is internalized into the cell for destruction of LDL)
LDL receptor returns to surface without being destroyed
↑ LDL receptor expression
↑ LDL clearance from blood
↓ LDL in blood
See Calgary guide slide on “PCSK9 Inhibitors: Mechanisms of action & side effects” for complete description of mechanism and side effects
↓ Cholesterol synthesis in liver
↑ LDL receptor expression in liver
LDL receptor recognizes apoB100 (structural protein on LDL) and apoE (structural protein found on chylomicron, VLDL, IDL)
↑ Clearance of LDL cholesterol from bloodstream
↓ LDL cholesterol in blood ↑ HDL in blood
↓ Triglycerides in blood
↓ Atherosclerosis (plaque along walls of blood vessels)
Abbreviations: HDL – High density lipoprotein; HMG-CoA – Hydroxymethylglutaryl- CoA; LDL – Low-density lipoprotein; PCSK9 – Proprotein convertase subtilisin/kexin type 9; PPAR! – Peroxisome proliferator-activated receptor alpha; NPC1L1 – Niemann-Pick C1-Like 1; VLDL – Very low-density lipoprotein
Side effects: Myalgias (muscular aches), rhabdomyolysis (muscle breakdown), transaminitis (liver inflammation), liver failure, ↑ risk of diabetes mellitus
Contraindicated in pregnancy
See Calgary guide slide on “Statins: Mechanisms of action & side effects” for complete description of mechanism and side effects
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published Nov 6, 2023 on www.thecalgaryguide.com")
Gestational Diabetes Risk factors and pathogenesis

Authors:
Amyna Fidai
Maharshi Gandhi Reviewers:
Laura Byford-Richardson Shahab Marzoughi
Yan Yu*
Hanan Bassyouni*
* MD at time of publication
High risk population (Aboriginal, Hispanic, South Asian, Asian, African)
Previous or current macrosomia (>4000g) or polyhydramnios
Other conditions associated with Diabetes Mellitus such as polycystic ovarian syndrome, hypertension, metabolic syndrome
Placental counter regulatory hormones (particularly Human Placental Growth Hormone) oppose the action of insulin
↑ Insulin resistance (liver, muscle, adipose tissues become less responsive to insulin)
↑ Fetal demands after 18 weeks gestation
(fetus requires 80% of its energy from maternal glucose)
↑ Carbohydrate intake to keep up with the demands
Previous history of gestational diabetes or glucose intolerance
Family history of diabetes
Advanced maternal age
Obesity
Previous unexplained stillbirth
Multiples (larger placental mass and activity)
Corticosteroid use
↑ Risk for pregnancy induced glucose intolerance (mechanism unclear and/or complex)
↑ Insulin requirements Initially pancreatic beta cells work
overtime to keep up with the ↑ insulin demands
Eventually insulin demands are not met due to the exhaustion of pancreatic beta cells
Plasma glucose rises (Fasting plasma glucose ≥5.3 mmol/L)
Gestational Diabetes (or exacerbation of pre-existing DM)
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published Jan 28, 2017, updated Feb 24, 2024 on www.thecalgaryguide.com")
Diabetes Mellitus Pathophysiology Behind Lab Findings

↓ Glucose uptake into cells
Autoimmune destruction of pancreatic beta cells (type 1 diabetes mellitus)
Insulin resistance (type 2 diabetes mellitus)
Other causes of diabetes mellitus (genetic, drug- induced, pregnancy, etc.)
↑ Lipolysis (fat breakdown) in adipose tissue
↑ Free fatty acids & glycerol in bloodstream
↑ Oxidation of fatty acids in liver to form acetyl CoA
↑ Conversion of acetyl CoA to ketone bodies (ketogenesis) to be used as fuel for the brain
Relative or absolute insulin deficiency
↓ Activation of adipose (fat tissue) & muscle cell transmembrane insulin receptors
↓ Activation of intracellular insulin signaling pathways (PI3K & MAP kinase)
Cells cannot use glucose as a source of energy; thus body reacts as if it is starving
↑ Protein breakdown in muscle tissue ↑ Amino acids & lactate in bloodstream
↑ Substrates (glycerol, amino acids & lactate) available to produce glucose in the liver
↑ Glycogenolysis (breakdown of stored glucose - glycogen) & gluconeogenesis (production of glucose) in liver
Glucose in bloodstream glycates (coats) hemoglobin in red blood cells; thus ↑ blood glucose leads to ↑ glycated hemoglobin
↑ Hemoglobin A1C
↑ Fasting blood glucose level ↑ Random blood glucose level
↑ Blood glucose following oral glucose tolerance test
Diabetic ketoacidosis (DKA) in absolute insulin deficiency *See DKA slide
Filtered ketone bodies exceed the reabsorption capacity of renal tubules
Ketonuria (↑ ketones in urine)
↑ Glucose in bloodstream
Build-up of glycation end products causes damage to glomerular tissue
Transient glomerular hyperfiltration followed by long-term ↓ in glomerular filtration rate
Albuminuria (↑ albumin in urine)
Diabetic nephropathy *See slide
Filtered glucose exceeds reabsorption capacity of renal tubules in the kidney
Glucosuria (↑ glucose in urine)
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published July 8, 2024 on www.thecalgaryguide.com")
Obesity Pathogenesis
 Zeng Reviewers: Gurreet Bhandal, Raafi Ali, Mizuki Lopez
Energy homeostasis dysregulation (Peptide hormones regulating hunger/satiety that influence the hypothalamic control of eating behaviours)
Luiza Radu, Dr. Sam Fineblit*
* MD at time of publication
Endocrine dysregulation
Neurological (↓ caloric reserve activates the neurological system)
Adiposity-related
Hypothalamus (hemostatic area)
↑ Production of ghrelin (peptide hormone)
↑ Activation of agouti-related protein (A Neuropeptide Y (NPY) neuron clusters in the hypothalamus via vagus nerve
↑ Appetite
Mesolimbic area (hedonic area)
Reward Driven Eating (meso-limbic region of brain)
↑ Food consumption
↑ Dopamine & endogenous opioid signals
↑ Pleasure & desire to consume more food
Prefrontal cortex (executive functioning)
↓ Glucagon-like peptide-1 (GLP-1) & pancreatic peptide YY (PYY) secretion post- meal
↓ Stimulation of Proopiomelanocortin (POMC) neuron clusters in hypothalamus
↓ Beta-melanocyte stimulating hormone (MSH) (endogenous peptide) production
Defect in leptin receptor gene
↓ Circulating
soluble leptin receptors (SLR)
↑ Serum leptin levels
Leptin resistance
Inactive leptin gene
↓ Adipocyte leptin production & secretion
↓ Leptin
transport across blood brain barrier
↓ Hypothalamus suppression of hunger
↓ Satiety sensation
↑ Caloric intake & weight gain
↑ Dietary carbohydrate intake
↑ Insulin production & secretion
↓ Cellular response to insulin
↑ Insulin resistance
↑ Insulin production & secretion to compensate for insulin resistance
Impaired glycemic control
Metabolic syndrome, type 2 diabetes mellitus and cardiovascular disease **
Metabolic adaptation to reduced-caloric intake efforts
↑ Body energy conservation
↓ Baseline energy expenditure
↓ Resting metabolic rate (the amount of energy body requires to function while at rest)
↓ Ability to lose weight & fat mass
↑ Hunger
↑ Caloric intake & weight gain
Obesity (A combination of metrics including ↑ BMI, ↑ weight circumference, ↑ waist-to-hip ratio, skinfold thickness, and other standardized measures varied by equipment available at various institutions resulting in a negative impact on the health of the individual)
**See slide on Pathogenesis of Type II Diabetes Mellitus
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published Aug 13, 2024 on www.thecalgaryguide.com")
Polycystic Ovarian Syndrome
: Pathogenesis and clinical findings
Genetic Susceptibility:
↑ Expression of LH receptors in granulosa cells, and anterior pituitary ↑ production of luteinizing hormone (LH)
↑ Serum LH compared to FSHà higher levels of LH increasingly activate thecal cells
Excess nutrients (from overeating and sedentary behavior) is stored as visceral fat
↑ Central adiposity (accumulation of fat around abdominal area)
Adipose tissue ↑ secretion of estrogen, inflammatory mediators, adipokines, free fatty acids
̄ Hepatic synthesis of sex hormone-binding globulin
Acne
Hair loss on scalp, and less commonly eyebrows and eyelashes (alopecia)
Excessive hair growth around mouth, chin, chest, abdomen, upper arms, thighs, and upper & lower back (hirsutism)
Metabolic syndrome develops, including ↑ insulin resistance, obesity, dyslipidemia, liver disease, & cardiovascular disease
Theca cells in ovaries ↑ androgen secretion
↑ Serum androgens
State of hyperandrogenism
Androgen and estrogen levels are elevated too early in menstrual cycle (estrogen has negative feedback inhibition on anterior pituitary hormone production)
Early suppression of FSH Limited proliferation of follicles
Relative reduction in secretion of progesterone Imbalance between progesterone and estrogen
Unpredictable ovulation
↑ Circulating insulin- like growth factor
Growth factor promote keratinocyte and dermal fibroblast proliferation
Velvety, darkening of the skin fold areas such as back of neck, armpit area, groin (acanthosis nigricans)
↑ Risk of type 2 diabetes mellitus
Since granulosa cells normally convert testosterone to estrogen, ̄ granulosa cell function means more testosterone exists
Since follicle stimulating hormone (FSH) activates granulosa cells, ̄ FSH means ̄ granulosa cell function.
Follicles arrested in development accumulate fluid, becoming cysts
Polycystic ovaries visible on ultrasound
Infertility
Unpredictable uterine bleeding
↑ Risk of endometrial hyperplasia & endometrial cancer
Irregular menstruation (amenorrhea/oligomenorrhea)
Authors: Lauren Standerwick Claire Song Reviewers: Mackenzie Grisdale Amy Fowler Christina Schweitzer Michelle J. Chen Dr. Yan Yu* Dr. Bernard Corenblum* Dr. Sylvie Bowden* * MD at time of publication
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published Nov 20, 2017; updated Dec 15, 2024 on www.thecalgaryguide.com")
GLP-1 Receptor Agonists
 Receptor Agonists: Mechanism of action and side effects
Management of Type
2 Diabetes Mellitus**
Weight
management
Authors: Saania Tariq, Trevor Low
Reviewers: Rafael Sanguinetti
Luiza Radu, Emily J. Doucette
Activated GLP-1 receptors in
Activated GLP-1
Hanan Bassyouni*
* MD at time of publication
the myenteric plexus inhibit
receptors stimulate
gastric motor neurons
pancreatic islet cells
GLP-1 Receptor Agonists
Synthetic analogs (e.g., Semaglutide) of GLP-1 peptide hormone endogenously
produced by intestinal enteroendocrine L cells directly activate GLP-1 receptors
to modulate food intake & glucose metabolism.
↓ Gastric
motility
↑ Pyloric sphincter
tone via disinhibition
↓ Glucagon release
from α-cells
↑ Regeneration,
↑ growth &
↓ apoptosis of β-cells
Slowed gastric emptying
Intramuscular injection or oral ingestion
prolongs GLP-1 receptor activation
↑ β-cell function & glucose-
dependent insulin production
GLP-1 receptor activation on neurons in
the arcuate nucleus of the hypothalamus
modulates appetite & satiety
Activation of GLP-1 receptors
in the gastrointestinal system
regulates metabolism
Prolonged gastric
distension & stasis
activates
chemoreceptors &
mechanoreceptors
Delayed digestion of
gastric contents ↓
glucose spikes after
meals
↑ Glycogen synthesis &
glucose uptake in liver &
skeletal muscles
Neuropeptide Y &
agouti-related protein
expressing neurons are
hyperpolarized
Pro-opiomelanocortin
expressing neurons
become ↑ excitable
Activated GLP-1 receptors
signal to hypothalamus
via vagus nerve afferents
↑ Vagal afferent
signaling to area
postrema
(vomiting centre)
Nausea &
vomiting
↓ Release of
neuropeptide Y &
agouti-related protein
↑ Anorexigenic
signalling
↑ Parasympathetic
output via dorsal motor
nucleus of vagus nerve
GLP-1 agonists directly
activate receptors in
area postrema
↑ Satiety (feeling
of fullness)
↓ Adiposity
(weight loss)
↓ Orexigenic drive
↓ Inflammatory stress in
pancreatic β-cells
↓ Appetite
↓ Caloric intake
↓ Inflammatory
cytokines & lipotoxicity
↓ Systemic
inflammation
**See corresponding Calgary Guide slide
Legend: Sign/Symptom/Lab Finding Pathophysiology Mechanism
Complications
↓ Hepatic glucose
production
(glycogenolysis)
↓ Fasting & postprandial
blood glucose
↓ Red blood cell (RBC)
glycation (glucose
irreversibly binds to
hemoglobin in RBCs)
↓ Production
of glycated
proteins and
lipids
↓ % of glycated
RBCs over time
↑ β-cell function
improves insulin
secretion
↓ Vascular
endothelial
damage and
atherosclerosis
↓ Endothelial
damage to
glomeruli
↑ Insulin sensitivity in
skeletal muscle & liver
↓ Hemoglobin
A1c
↓ Risk of
cardiovascular
complications
↓ Risk of renal
complications
Published Jun 22, 2025 on www.thecalgaryguide.com")