SEARCH RESULTS FOR: diabetes
lower-urinary-tract-infections-complications
, stagnant
urine (anatomical variant, obstruction,
neurogenic bladder, urinary reflux)
Bacterial entry (Less Common):
Indwelling catheter, surgical inoculation,
hematogenousspread, trauma
(Staphylococcus, Enterococcus, Candida)
Fecal bacteria access urethra
(E. coli, Proteus, Klebsiella)
Impairment of body's natural defense
systems, or stagnant urine, allow for
bacterial accumulation
Portal of entry bypasses body's physical
defenses (gravity and repetitive outward
urine flow)
Bacterial fimbriae and pili allow
them to ascend urethra and
adhere to epithelium
Lower Urinary Tract Infection (LUTI): Pathogenesis and clinical findings
Suprapubic
Tenderness
Bacterial colony irritates
urinary epithelium
Urgency:
Sensation of need to urinate
quickly or impending
incontinence
Stimulation of
inflammatory
response
Stimulation of urinary reflex
Pathogens use
enzymes to reduce
nitrate to nitrite
Delirium in Elderly
Frequency:
Repetitive need to
urinate
Unique response of altered fluid
status, electrolytes and mental
status, likely as a result of
increased inflammatory cytokines
Lower Urinary Tract Infection (“Cystitis”):
Infection of bladder or distal tract by capable bacteria
colonizing epithelium and causing symptoms
Legend: Pathophysiology Mechanism Sign/Symptom/Lab Finding Complications Published March 16, 2014 on www.thecalgaryguide.com
Author:
Brett Edwards
Reviewers:
Riley Hartmann
Jan Rudzinski
Haotian Wang
Steve Vaughan*
* MD at time of publication
Usual Pathogens (“KEEPS”):
K – Klebsiella
E – E. coli (90%)
E – Enterococcus, Enterobacteriaceae
P – Proteus, Pseudomonas
S – Staph. saprophyticus, Serratia
Urine Findings:
↑ Colony Count (>107 CFU/L)
↑ WBC (>10 WBC/μL)
(+) Bacterial culture
(+) Nitrites, Leukocyte Esterase
(+) Foul, turbid urine
+/- Hematuria (rare)")
lower-urinary-tract-infection-pathogenesis-and-clinical-findings
, stagnant
urine (anatomical variant, obstruction,
neurogenic bladder, urinary reflux)
Bacterial entry (Less Common):
Indwelling catheter, surgical inoculation,
hematogenousspread, trauma
(Staphylococcus, Enterococcus, Candida)
Fecal bacteria access urethra
(E. coli, Proteus, Klebsiella)
Impairment of body's natural defense
systems, or stagnant urine, allow for
bacterial accumulation
Portal of entry bypasses body's physical
defenses (gravity and repetitive outward
urine flow)
Bacterial fimbriae and pili allow
them to ascend urethra and
adhere to epithelium
Lower Urinary Tract Infection (LUTI): Pathogenesis and clinical findings
Suprapubic
Tenderness
Bacterial colony irritates
urinary epithelium
Urgency:
Sensation of need to urinate
quickly or impending
incontinence
Stimulation of
inflammatory
response
Stimulation of urinary reflex
Pathogens use
enzymes to reduce
nitrate to nitrite
Delirium in Elderly
Frequency:
Repetitive need to
urinate
Unique response of altered fluid
status, electrolytes and mental
status, likely as a result of
increased inflammatory cytokines
Lower Urinary Tract Infection (“Cystitis”):
Infection of bladder or distal tract by capable bacteria
colonizing epithelium and causing symptoms
Legend: Pathophysiology Mechanism Sign/Symptom/Lab Finding Complications Published March 16, 2014 on www.thecalgaryguide.com
Author:
Brett Edwards
Reviewers:
Riley Hartmann
Jan Rudzinski
Haotian Wang
Steve Vaughan*
* MD at time of publication
Usual Pathogens (“KEEPS”):
K – Klebsiella
E – E. coli (90%)
E – Enterococcus, Enterobacteriaceae
P – Proteus, Pseudomonas
S – Staph. saprophyticus, Serratia
Urine Findings:
↑ Colony Count (>107 CFU/L)
↑ WBC (>10 WBC/μL)
(+) Bacterial culture
(+) Nitrites, Leukocyte Esterase
(+) Foul, turbid urine
+/- Hematuria (rare)
WBCs onsite
release enzymes
Cytokines released
systemically
Fever, Malaise,
↑WBC
(>11 x 109 cells/L)
(Rare in LUTI)")
Hypokalemia: Clinical Findings
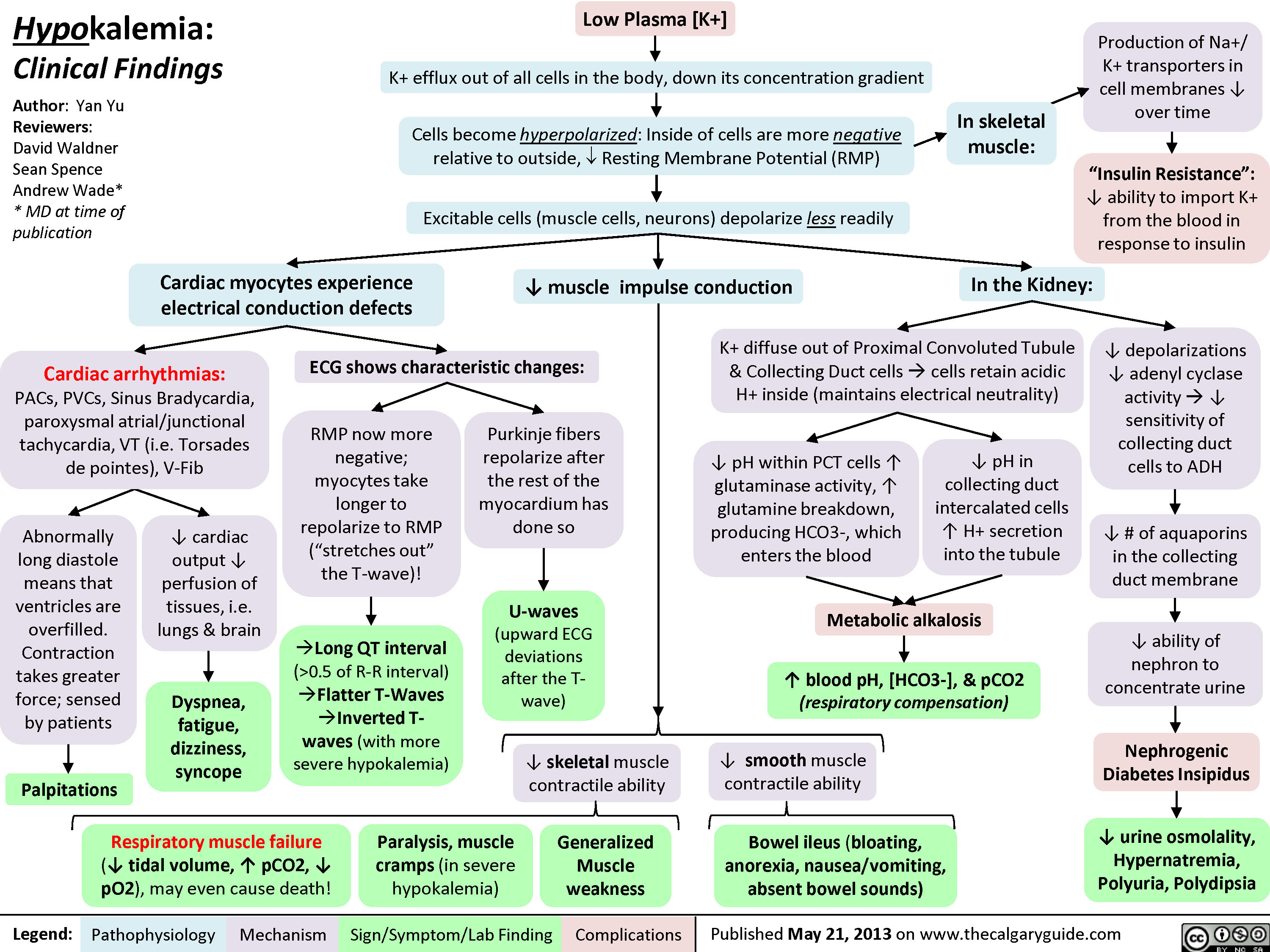 0.5 of R-R interval)?Flatter T-Waves ?Inverted T-waves (with more severe hypokalemia)Purkinje fibers repolarize after the rest of the myocardium has done soU-waves (upward ECG deviations after the T-wave)Cells become hyperpolarized: Inside of cells are more negative relative to outside, ? Resting Membrane Potential (RMP)In the Kidney:Generalized Muscle weaknessK+ diffuse out of Proximal Convoluted Tubule & Collecting Duct cells ? cells retain acidic H+ inside (maintains electrical neutrality)? pH within PCT cells ? glutaminase activity, ? glutamine breakdown, producing HCO3-, which enters the blood? blood pH, [HCO3-], & pCO2 (respiratory compensation)Low Plasma [K+]Abnormally long diastole means that ventricles are overfilled. Contraction takes greater force; sensed by patientsDyspnea, fatigue, dizziness, syncope? cardiac output ? perfusion of tissues, i.e. lungs & brainCardiac arrhythmias: PACs, PVCs, Sinus Bradycardia, paroxysmal atrial/junctional tachycardia, VT (i.e. Torsades de pointes), V-Fib? smooth muscle contractile abilityBowel ileus (bloating, anorexia, nausea/vomiting, absent bowel sounds)? pH in collecting duct intercalated cells ? H+ secretion into the tubuleMetabolic alkalosisParalysis, muscle cramps (in severe hypokalemia)Respiratory muscle failure (? tidal volume, ? pCO2, ? pO2), may even cause death!? depolarizations ? adenyl cyclase activity ? ? sensitivity of collecting duct cells to ADH? ability of nephron to concentrate urineNephrogenic Diabetes Insipidus? urine osmolality, Hypernatremia, Polyuria, Polydipsia? # of aquaporins in the collecting duct membrane"Insulin Resistance": ? ability to import K+ from the blood in response to insulinIn skeletal muscle:
117 kB / 307 word" title="Yu, Yan - Hypokalemia clinical findings - FINAL.pptx
Production of Na+/ K+ transporters in cell membranes ? over timeHypokalemia: Clinical FindingsAuthor: Yan YuReviewers:David WaldnerSean SpenceAndrew Wade** MD at time of publicationLegend:Published May 21, 2013 on www.thecalgaryguide.comMechanismPathophysiologySign/Symptom/Lab FindingComplicationsPalpitationsExcitable cells (muscle cells, neurons) depolarize less readilyK+ efflux out of all cells in the body, down its concentration gradientCardiac myocytes experience electrical conduction defects? muscle impulse conductionECG shows characteristic changes:? skeletal muscle contractile abilityRMP now more negative; myocytes take longer to repolarize to RMP("stretches out" the T-wave)! Long QT interval (>0.5 of R-R interval)?Flatter T-Waves ?Inverted T-waves (with more severe hypokalemia)Purkinje fibers repolarize after the rest of the myocardium has done soU-waves (upward ECG deviations after the T-wave)Cells become hyperpolarized: Inside of cells are more negative relative to outside, ? Resting Membrane Potential (RMP)In the Kidney:Generalized Muscle weaknessK+ diffuse out of Proximal Convoluted Tubule & Collecting Duct cells ? cells retain acidic H+ inside (maintains electrical neutrality)? pH within PCT cells ? glutaminase activity, ? glutamine breakdown, producing HCO3-, which enters the blood? blood pH, [HCO3-], & pCO2 (respiratory compensation)Low Plasma [K+]Abnormally long diastole means that ventricles are overfilled. Contraction takes greater force; sensed by patientsDyspnea, fatigue, dizziness, syncope? cardiac output ? perfusion of tissues, i.e. lungs & brainCardiac arrhythmias: PACs, PVCs, Sinus Bradycardia, paroxysmal atrial/junctional tachycardia, VT (i.e. Torsades de pointes), V-Fib? smooth muscle contractile abilityBowel ileus (bloating, anorexia, nausea/vomiting, absent bowel sounds)? pH in collecting duct intercalated cells ? H+ secretion into the tubuleMetabolic alkalosisParalysis, muscle cramps (in severe hypokalemia)Respiratory muscle failure (? tidal volume, ? pCO2, ? pO2), may even cause death!? depolarizations ? adenyl cyclase activity ? ? sensitivity of collecting duct cells to ADH? ability of nephron to concentrate urineNephrogenic Diabetes Insipidus? urine osmolality, Hypernatremia, Polyuria, Polydipsia? # of aquaporins in the collecting duct membrane"Insulin Resistance": ? ability to import K+ from the blood in response to insulinIn skeletal muscle:
117 kB / 307 word" />
0.5 of R-R interval)?Flatter T-Waves ?Inverted T-waves (with more severe hypokalemia)Purkinje fibers repolarize after the rest of the myocardium has done soU-waves (upward ECG deviations after the T-wave)Cells become hyperpolarized: Inside of cells are more negative relative to outside, ? Resting Membrane Potential (RMP)In the Kidney:Generalized Muscle weaknessK+ diffuse out of Proximal Convoluted Tubule & Collecting Duct cells ? cells retain acidic H+ inside (maintains electrical neutrality)? pH within PCT cells ? glutaminase activity, ? glutamine breakdown, producing HCO3-, which enters the blood? blood pH, [HCO3-], & pCO2 (respiratory compensation)Low Plasma [K+]Abnormally long diastole means that ventricles are overfilled. Contraction takes greater force; sensed by patientsDyspnea, fatigue, dizziness, syncope? cardiac output ? perfusion of tissues, i.e. lungs & brainCardiac arrhythmias: PACs, PVCs, Sinus Bradycardia, paroxysmal atrial/junctional tachycardia, VT (i.e. Torsades de pointes), V-Fib? smooth muscle contractile abilityBowel ileus (bloating, anorexia, nausea/vomiting, absent bowel sounds)? pH in collecting duct intercalated cells ? H+ secretion into the tubuleMetabolic alkalosisParalysis, muscle cramps (in severe hypokalemia)Respiratory muscle failure (? tidal volume, ? pCO2, ? pO2), may even cause death!? depolarizations ? adenyl cyclase activity ? ? sensitivity of collecting duct cells to ADH? ability of nephron to concentrate urineNephrogenic Diabetes Insipidus? urine osmolality, Hypernatremia, Polyuria, Polydipsia? # of aquaporins in the collecting duct membrane"Insulin Resistance": ? ability to import K+ from the blood in response to insulinIn skeletal muscle:
117 kB / 307 word" title="Yu, Yan - Hypokalemia clinical findings - FINAL.pptx
Production of Na+/ K+ transporters in cell membranes ? over timeHypokalemia: Clinical FindingsAuthor: Yan YuReviewers:David WaldnerSean SpenceAndrew Wade** MD at time of publicationLegend:Published May 21, 2013 on www.thecalgaryguide.comMechanismPathophysiologySign/Symptom/Lab FindingComplicationsPalpitationsExcitable cells (muscle cells, neurons) depolarize less readilyK+ efflux out of all cells in the body, down its concentration gradientCardiac myocytes experience electrical conduction defects? muscle impulse conductionECG shows characteristic changes:? skeletal muscle contractile abilityRMP now more negative; myocytes take longer to repolarize to RMP("stretches out" the T-wave)! Long QT interval (>0.5 of R-R interval)?Flatter T-Waves ?Inverted T-waves (with more severe hypokalemia)Purkinje fibers repolarize after the rest of the myocardium has done soU-waves (upward ECG deviations after the T-wave)Cells become hyperpolarized: Inside of cells are more negative relative to outside, ? Resting Membrane Potential (RMP)In the Kidney:Generalized Muscle weaknessK+ diffuse out of Proximal Convoluted Tubule & Collecting Duct cells ? cells retain acidic H+ inside (maintains electrical neutrality)? pH within PCT cells ? glutaminase activity, ? glutamine breakdown, producing HCO3-, which enters the blood? blood pH, [HCO3-], & pCO2 (respiratory compensation)Low Plasma [K+]Abnormally long diastole means that ventricles are overfilled. Contraction takes greater force; sensed by patientsDyspnea, fatigue, dizziness, syncope? cardiac output ? perfusion of tissues, i.e. lungs & brainCardiac arrhythmias: PACs, PVCs, Sinus Bradycardia, paroxysmal atrial/junctional tachycardia, VT (i.e. Torsades de pointes), V-Fib? smooth muscle contractile abilityBowel ileus (bloating, anorexia, nausea/vomiting, absent bowel sounds)? pH in collecting duct intercalated cells ? H+ secretion into the tubuleMetabolic alkalosisParalysis, muscle cramps (in severe hypokalemia)Respiratory muscle failure (? tidal volume, ? pCO2, ? pO2), may even cause death!? depolarizations ? adenyl cyclase activity ? ? sensitivity of collecting duct cells to ADH? ability of nephron to concentrate urineNephrogenic Diabetes Insipidus? urine osmolality, Hypernatremia, Polyuria, Polydipsia? # of aquaporins in the collecting duct membrane"Insulin Resistance": ? ability to import K+ from the blood in response to insulinIn skeletal muscle:
117 kB / 307 word" />
Nephrotic Syndrome: Pathogenesis and Clinical Findings
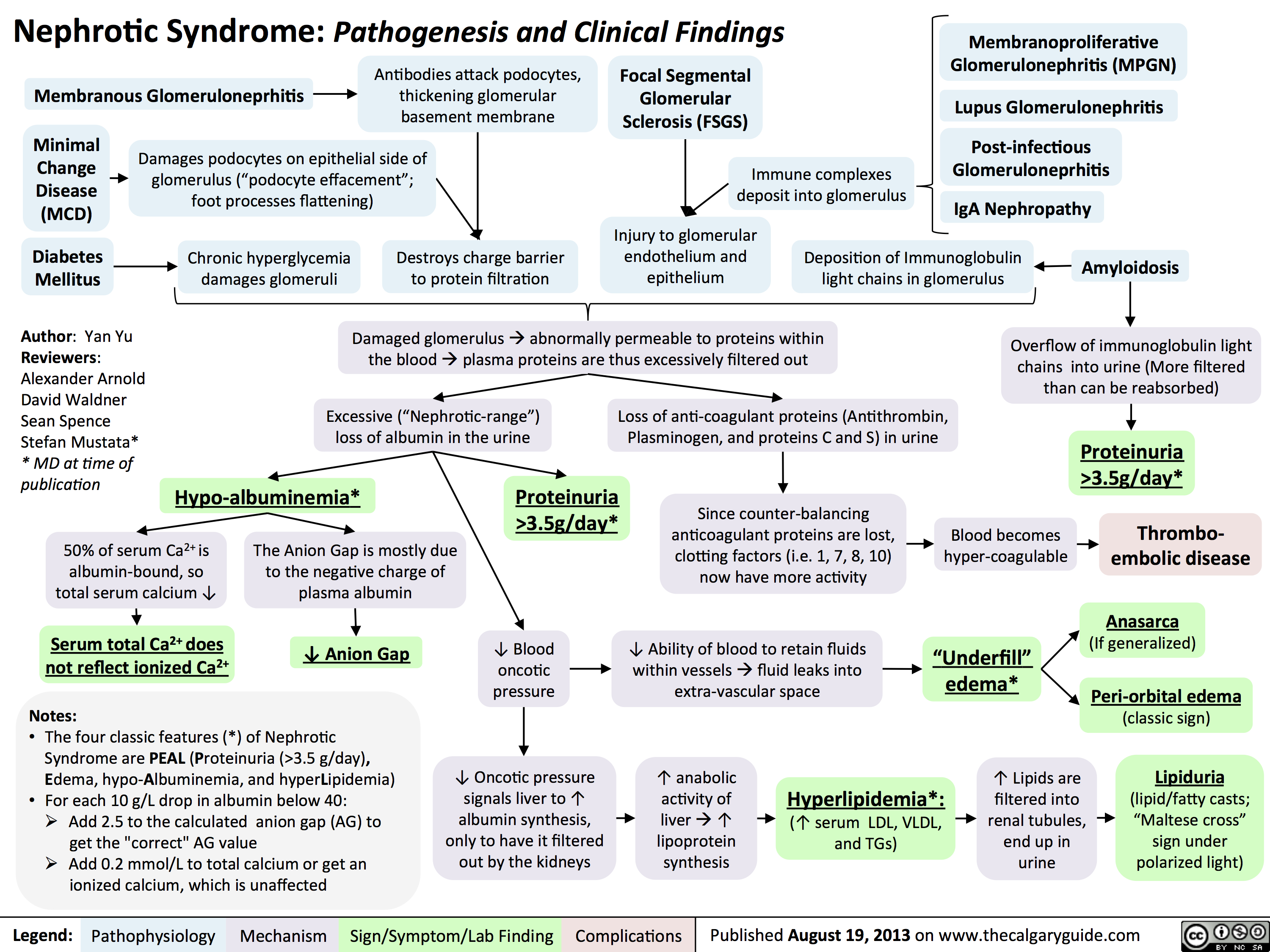 3.5g/day*? Ability of blood to retain fluids within vessels ? fluid leaks into extra-vascular spaceInjury to glomerular endothelium and epitheliumImmune complexes deposit into glomerulusDamaged glomerulus ? abnormally permeable to proteins within the blood ? plasma proteins are thus excessively filtered out? Oncotic pressure signals liver to ? albumin synthesis, only to have it filtered out by the kidneys? anabolic activity of liver ? ? lipoprotein synthesisHyperlipidemia*:(? serum LDL, VLDL, and TGs)Lipiduria(lipid/fatty casts; "Maltese cross" sign under polarized light)Since counter-balancing anticoagulant proteins are lost, clotting factors (i.e. 1, 7, 8, 10) now have more activityThrombo-embolic diseaseBlood becomes hyper-coagulable? Lipids are filtered into renal tubules, end up in urineMembranoproliferative Glomerulonephritis (MPGN)Lupus Glomerulonephritis Post-infectious GlomeruloneprhitisIgA NephropathyDamages podocytes on epithelial side of glomerulus ("podocyte effacement"; foot processes flattening)Diabetes MellitusChronic hyperglycemia damages glomeruliDeposition of Immunoglobulin light chains in glomerulusAmyloidosisAnasarca(If generalized)Peri-orbital edema (classic sign)Focal Segmental Glomerular Sclerosis (FSGS)Membranous GlomeruloneprhitisAntibodies attack podocytes, thickening glomerular basement membraneOverflow of immunoglobulin light chains into urine (More filtered than can be reabsorbed)Proteinuria >3.5g/day*The Anion Gap is mostly due to the negative charge of plasma albumin? Anion GapNotes: The four classic features (*) of Nephrotic Syndrome are PEAL (Proteinuria (>3.5 g/day), Edema, hypo-Albuminemia, and hyperLipidemia)For each 10 g/L drop in albumin below 40:Add 2.5 to the calculated anion gap (AG) to get the "correct" AG valueAdd 0.2 mmol/L to total calcium or get an ionized calcium, which is unaffected50% of serum Ca2+ is albumin-bound, so total serum calcium ? Serum total Ca2+ does not reflect ionized Ca2+ ? Blood oncotic pressure" title="Destroys charge barrier to protein filtrationNephrotic Syndrome: Pathogenesis and Clinical FindingsAuthor: Yan YuReviewers:Alexander ArnoldDavid WaldnerSean SpenceStefan Mustata** MD at time of publicationLegend:Published August 19, 2013 on www.thecalgaryguide.comMechanismPathophysiologySign/Symptom/Lab FindingComplicationsExcessive ("Nephrotic-range") loss of albumin in the urineHypo-albuminemia*Loss of anti-coagulant proteins (Antithrombin, Plasminogen, and proteins C and S) in urineMinimal Change Disease (MCD)"Underfill" edema*Proteinuria >3.5g/day*? Ability of blood to retain fluids within vessels ? fluid leaks into extra-vascular spaceInjury to glomerular endothelium and epitheliumImmune complexes deposit into glomerulusDamaged glomerulus ? abnormally permeable to proteins within the blood ? plasma proteins are thus excessively filtered out? Oncotic pressure signals liver to ? albumin synthesis, only to have it filtered out by the kidneys? anabolic activity of liver ? ? lipoprotein synthesisHyperlipidemia*:(? serum LDL, VLDL, and TGs)Lipiduria(lipid/fatty casts; "Maltese cross" sign under polarized light)Since counter-balancing anticoagulant proteins are lost, clotting factors (i.e. 1, 7, 8, 10) now have more activityThrombo-embolic diseaseBlood becomes hyper-coagulable? Lipids are filtered into renal tubules, end up in urineMembranoproliferative Glomerulonephritis (MPGN)Lupus Glomerulonephritis Post-infectious GlomeruloneprhitisIgA NephropathyDamages podocytes on epithelial side of glomerulus ("podocyte effacement"; foot processes flattening)Diabetes MellitusChronic hyperglycemia damages glomeruliDeposition of Immunoglobulin light chains in glomerulusAmyloidosisAnasarca(If generalized)Peri-orbital edema (classic sign)Focal Segmental Glomerular Sclerosis (FSGS)Membranous GlomeruloneprhitisAntibodies attack podocytes, thickening glomerular basement membraneOverflow of immunoglobulin light chains into urine (More filtered than can be reabsorbed)Proteinuria >3.5g/day*The Anion Gap is mostly due to the negative charge of plasma albumin? Anion GapNotes: The four classic features (*) of Nephrotic Syndrome are PEAL (Proteinuria (>3.5 g/day), Edema, hypo-Albuminemia, and hyperLipidemia)For each 10 g/L drop in albumin below 40:Add 2.5 to the calculated anion gap (AG) to get the "correct" AG valueAdd 0.2 mmol/L to total calcium or get an ionized calcium, which is unaffected50% of serum Ca2+ is albumin-bound, so total serum calcium ? Serum total Ca2+ does not reflect ionized Ca2+ ? Blood oncotic pressure" />
3.5g/day*? Ability of blood to retain fluids within vessels ? fluid leaks into extra-vascular spaceInjury to glomerular endothelium and epitheliumImmune complexes deposit into glomerulusDamaged glomerulus ? abnormally permeable to proteins within the blood ? plasma proteins are thus excessively filtered out? Oncotic pressure signals liver to ? albumin synthesis, only to have it filtered out by the kidneys? anabolic activity of liver ? ? lipoprotein synthesisHyperlipidemia*:(? serum LDL, VLDL, and TGs)Lipiduria(lipid/fatty casts; "Maltese cross" sign under polarized light)Since counter-balancing anticoagulant proteins are lost, clotting factors (i.e. 1, 7, 8, 10) now have more activityThrombo-embolic diseaseBlood becomes hyper-coagulable? Lipids are filtered into renal tubules, end up in urineMembranoproliferative Glomerulonephritis (MPGN)Lupus Glomerulonephritis Post-infectious GlomeruloneprhitisIgA NephropathyDamages podocytes on epithelial side of glomerulus ("podocyte effacement"; foot processes flattening)Diabetes MellitusChronic hyperglycemia damages glomeruliDeposition of Immunoglobulin light chains in glomerulusAmyloidosisAnasarca(If generalized)Peri-orbital edema (classic sign)Focal Segmental Glomerular Sclerosis (FSGS)Membranous GlomeruloneprhitisAntibodies attack podocytes, thickening glomerular basement membraneOverflow of immunoglobulin light chains into urine (More filtered than can be reabsorbed)Proteinuria >3.5g/day*The Anion Gap is mostly due to the negative charge of plasma albumin? Anion GapNotes: The four classic features (*) of Nephrotic Syndrome are PEAL (Proteinuria (>3.5 g/day), Edema, hypo-Albuminemia, and hyperLipidemia)For each 10 g/L drop in albumin below 40:Add 2.5 to the calculated anion gap (AG) to get the "correct" AG valueAdd 0.2 mmol/L to total calcium or get an ionized calcium, which is unaffected50% of serum Ca2+ is albumin-bound, so total serum calcium ? Serum total Ca2+ does not reflect ionized Ca2+ ? Blood oncotic pressure" title="Destroys charge barrier to protein filtrationNephrotic Syndrome: Pathogenesis and Clinical FindingsAuthor: Yan YuReviewers:Alexander ArnoldDavid WaldnerSean SpenceStefan Mustata** MD at time of publicationLegend:Published August 19, 2013 on www.thecalgaryguide.comMechanismPathophysiologySign/Symptom/Lab FindingComplicationsExcessive ("Nephrotic-range") loss of albumin in the urineHypo-albuminemia*Loss of anti-coagulant proteins (Antithrombin, Plasminogen, and proteins C and S) in urineMinimal Change Disease (MCD)"Underfill" edema*Proteinuria >3.5g/day*? Ability of blood to retain fluids within vessels ? fluid leaks into extra-vascular spaceInjury to glomerular endothelium and epitheliumImmune complexes deposit into glomerulusDamaged glomerulus ? abnormally permeable to proteins within the blood ? plasma proteins are thus excessively filtered out? Oncotic pressure signals liver to ? albumin synthesis, only to have it filtered out by the kidneys? anabolic activity of liver ? ? lipoprotein synthesisHyperlipidemia*:(? serum LDL, VLDL, and TGs)Lipiduria(lipid/fatty casts; "Maltese cross" sign under polarized light)Since counter-balancing anticoagulant proteins are lost, clotting factors (i.e. 1, 7, 8, 10) now have more activityThrombo-embolic diseaseBlood becomes hyper-coagulable? Lipids are filtered into renal tubules, end up in urineMembranoproliferative Glomerulonephritis (MPGN)Lupus Glomerulonephritis Post-infectious GlomeruloneprhitisIgA NephropathyDamages podocytes on epithelial side of glomerulus ("podocyte effacement"; foot processes flattening)Diabetes MellitusChronic hyperglycemia damages glomeruliDeposition of Immunoglobulin light chains in glomerulusAmyloidosisAnasarca(If generalized)Peri-orbital edema (classic sign)Focal Segmental Glomerular Sclerosis (FSGS)Membranous GlomeruloneprhitisAntibodies attack podocytes, thickening glomerular basement membraneOverflow of immunoglobulin light chains into urine (More filtered than can be reabsorbed)Proteinuria >3.5g/day*The Anion Gap is mostly due to the negative charge of plasma albumin? Anion GapNotes: The four classic features (*) of Nephrotic Syndrome are PEAL (Proteinuria (>3.5 g/day), Edema, hypo-Albuminemia, and hyperLipidemia)For each 10 g/L drop in albumin below 40:Add 2.5 to the calculated anion gap (AG) to get the "correct" AG valueAdd 0.2 mmol/L to total calcium or get an ionized calcium, which is unaffected50% of serum Ca2+ is albumin-bound, so total serum calcium ? Serum total Ca2+ does not reflect ionized Ca2+ ? Blood oncotic pressure" />
Pathogenesis of Diabetes mellitus DM), Type II

Diabetic Hypoglycemia
![Yu, Yan - Diabetic Hypoglycemia - Clinical Findings - FINAL.pptx
? Epinephrine(Released within seconds as [glucose] falls further) Growth hormone, ? Cortisol (if hypoglycemia persists for minutes)Glucagon should ? when [glucose] falls. But here, glucagon release is inhibited by 1) diabetic auto-immune destruction of Alpha cells & 2) the high insulin.43210Plasma Glucose concentration (mmol/L)Liver should ? glycogenolysis & gluconeogenesisPeripheral vaso-constrictionPlasma [glucose] stays lowActivation of sympathetic (adrenergic) receptors across body, triggering Neurogenic symptomsPlasma [glucose] ?Excess subcutaneous insulin or insulin-secretagogue ?? [insulin] in the bloodOver time: [insulin] in the DM patient depends only on how much was injected or how much secretagogue was consumed; not on the body's physiological state.[Insulin] stays high in excessively-treated DM patientsPlasma [glucose] normally ?, but...High insulin transports plasma glucose into cells!In pts with existing diabetic autonomic neuropathy, epi-nephrine secretion will already be ?Brain does not get enough glucose, ? neuron function ? Neuroglycopenic symptomsTx: glucose intake![Glucose] returns to normalIf no glucose intake:Hypoglycemia-unawareness: No autonomic Sx felt so hypoglycemia not treated early ? pts present later on with more severe hypoglycemia and neuroglycopenic sxBrain cells kept chronically euglycemic due to GLUT1 receptor over-expression (despite rest of body being hypoglycemic)With many hypoglycemic events over time:Brain feels no need to ? glucose, so it ? autonomic epinephrine secretion!This is the normal sequence of hormone responses to ?ing plasma glucose levels.But this normal hormonal response will be blunted over time if there is 1) continued hypoglycemia dampening the sympathetic nervous system, and 2) long-standing diabetic neuropathy! (To be explained later in this flow chart)Abbreviations: [ ] = concentrationTx = TreatmentDM = Diabetes mellitusDiabetic Hypoglycemia: Pathogenesis and Clinical FindingsConfusionCan't concentrateWeaknessSlurred speech? coordination (staggering, etc)SeizuresComa, deathAdrenergic symptoms (epinephrine-mediated):Anxiety, irritability, trembling, pallor (skin vasoconstriction), palpitations, ? systolic BP, tachycardia Cholinergic symptoms(Acetylcholine-mediated):Sweating, hunger, tingling, blurry visionNote: In pts w/out DM, endogenous insulin secretion normally stops when blood [glucose] drops to <4mmol/LAuthor: Yan YuReviewers: Peter Vetere, Gillian Goobie, Hanan Bassyouni** MD at time of publicationLegend:Published June 14, 2013 on www.thecalgaryguide.comMechanismPathophysiologySign/Symptom/Lab FindingComplicationsMany hypoglycemic events over time blunt epinephrine secretion further.Hypoglycemia unawareness can be reversedIf pt stays hypoglycemia-free for >6 weeks, brain restores its ability to detect low glucose levels? peripheral glucose delivery and uptake (saving more glucose for the brain)Lack of glucagon effect reinforces hypoglycemia
124 kB / 361 words](http://calgaryguide.ucalgary.ca/wp-content/uploads/2015/05/Diabetic-Hypoglycemia-Clinical-Findings.jpg "Yu, Yan - Diabetic Hypoglycemia - Clinical Findings - FINAL.pptx
? Epinephrine(Released within seconds as [glucose] falls further) Growth hormone, ? Cortisol (if hypoglycemia persists for minutes)Glucagon should ? when [glucose] falls. But here, glucagon release is inhibited by 1) diabetic auto-immune destruction of Alpha cells & 2) the high insulin.43210Plasma Glucose concentration (mmol/L)Liver should ? glycogenolysis & gluconeogenesisPeripheral vaso-constrictionPlasma [glucose] stays lowActivation of sympathetic (adrenergic) receptors across body, triggering Neurogenic symptomsPlasma [glucose] ?Excess subcutaneous insulin or insulin-secretagogue ?? [insulin] in the bloodOver time: [insulin] in the DM patient depends only on how much was injected or how much secretagogue was consumed; not on the body's physiological state.[Insulin] stays high in excessively-treated DM patientsPlasma [glucose] normally ?, but...High insulin transports plasma glucose into cells!In pts with existing diabetic autonomic neuropathy, epi-nephrine secretion will already be ?Brain does not get enough glucose, ? neuron function ? Neuroglycopenic symptomsTx: glucose intake![Glucose] returns to normalIf no glucose intake:Hypoglycemia-unawareness: No autonomic Sx felt so hypoglycemia not treated early ? pts present later on with more severe hypoglycemia and neuroglycopenic sxBrain cells kept chronically euglycemic due to GLUT1 receptor over-expression (despite rest of body being hypoglycemic)With many hypoglycemic events over time:Brain feels no need to ? glucose, so it ? autonomic epinephrine secretion!This is the normal sequence of hormone responses to ?ing plasma glucose levels.But this normal hormonal response will be blunted over time if there is 1) continued hypoglycemia dampening the sympathetic nervous system, and 2) long-standing diabetic neuropathy! (To be explained later in this flow chart)Abbreviations: [ ] = concentrationTx = TreatmentDM = Diabetes mellitusDiabetic Hypoglycemia: Pathogenesis and Clinical FindingsConfusionCan't concentrateWeaknessSlurred speech? coordination (staggering, etc)SeizuresComa, deathAdrenergic symptoms (epinephrine-mediated):Anxiety, irritability, trembling, pallor (skin vasoconstriction), palpitations, ? systolic BP, tachycardia Cholinergic symptoms(Acetylcholine-mediated):Sweating, hunger, tingling, blurry visionNote: In pts w/out DM, endogenous insulin secretion normally stops when blood [glucose] drops to <4mmol/LAuthor: Yan YuReviewers: Peter Vetere, Gillian Goobie, Hanan Bassyouni** MD at time of publicationLegend:Published June 14, 2013 on www.thecalgaryguide.comMechanismPathophysiologySign/Symptom/Lab FindingComplicationsMany hypoglycemic events over time blunt epinephrine secretion further.Hypoglycemia unawareness can be reversedIf pt stays hypoglycemia-free for >6 weeks, brain restores its ability to detect low glucose levels? peripheral glucose delivery and uptake (saving more glucose for the brain)Lack of glucagon effect reinforces hypoglycemia
124 kB / 361 words")
chronic-hypertensive-retinopathy-pathogenesis-and-clinical-findings
 • 1° HTN
Retinal Detachment
Vitreous Hemorrhage
Central/Branch Retinal Artery/Vein Occlusions
Risk Factors for 2° HTN (ex. Hyperaldosterone, Cushing's, Acromegaly, Chronic Kidney Disease, Obstructive Sleep Apnea, Diabetes Mellitus, Hypo/Hyper-thyroid, Adrenal Hyperplasia, Renal Artery Stenosis)
2° HTN
Ophthalmic Artery Hypertension ,17
Stage 1: Mild/vasoconstrictive
Stage 2: Moderate/sclerotic
Stage 3: Severe/exudative
Stage 4: Malignant
Abbreviations: • HTN — Hypertension • BRB — Blood-retinal barrier • RPE — Retinal pigment epithelium
Legend:
Pathophysiology
Mechanism
Acute and chronic vasospasm
Authors: Graeme Prosperi-Porta Reviewers: Stephanie Cote Usama Malik Johnathan Wong* * MD at time of publication
Diffuse and focal arterial narrowing and vascular tortuosity
Atherosclerosis and hyalinization causes arteriolar wall thickening resulting in a diffuse light reflex appearing red-brown coloured
Thickening of the arteriolar wall and/or sclerotic thickening at the arteriole/venule crossing compresses the underlying venule
BRB breakdown causes dot/blot hemorrhages in the inner retina and flame hemorrhages in the nerve fiber layer
Serum proteins and lipids leakage from damaged BRB appears as white or yellow areas with sharp margins
Occlusion of the terminal retinal arterioles causes fluffy white ischemic lesions in the inner retinal nerve fiber layer
Hyper-pigmented patches surrounded by a hypo-pigmented ring due to RPE clumping around atrophic areas in the choroid
Sign/Symptom/Lab Finding
lschemia of optic disc arterioles causes optic nerve swelling and blurred disc margins. Leakage of optic disc arterioles causes hemorrhage and disc edema.
Complications
Copper Wiring
AV nicking
Retinal Hemorrhages
Yellow Hard Exudates
Cotton-wool Spots
Elschnig's Spots
Papilledema")
2nd gen antipsychotics (Slovenian translation) - FINAL VERSION
: primeri: klozapin, olanzapin, kvetiapin, risperidon, paliperidon itd.
antagonisti dopaminskih D2 receptorjev zavirajo delovanje DA na D2 receptorjih po celotnih mo2ganih
antagonisti serotoninskih 2A receptorjev zavirajo delovanje 5-HT na 5-HT2A receptorjih po celotnih mo2ganih
antagonisti serotoninskih 2C receptorjev klozapin, olanzapin, kvetiapin
antagonisti ACh M1 receptorjev zavirajo delovanje ACh po telesu (v ustih, prebavilih, o6eh, mo2ganih)
antagonisti aradrenoceptorjev v oiilju dilatacija gladkih migic v stenah arteriol
antagonisti histaminskih H1 receptorjev zavirajo delovanje histamina po telesu
sano-presnovni kink' mehanizem neznan
Legenda:
patofiziologija mehanizem
Pomni: posledica velikih razlik v afiniteti za receptorska vezavna mesta so svojevrstni terapevtski in varnostni profili atipicnih a nti psi hoti kov. Kloza pi n med vsemi velja za najud nkovitejSega, a i ma tudi najve6 neZelenih uC'inkov, vkljUuja agranulocitozo (0,5-2 %). Tako predstavlja terapijo drugega izbora, potrebno je red no spremljanje bolnikove krvne slike.
mezolimbiEna pot DA blokada > 5-HT blokado
nigrostriatna pot 5-HT blokada > DA blokado
4, pozitivnih simptomov terapevtski ueinek
blokada 5-HT vodi v sprokanje DA v striatumu
tubero- blokada 5-HT zavre sprokanje infundibularna pot prolaktina v adenohipofizi
mezokortikalna pot
blokada 5-HT2c receptorjev stimulira sprokanje DA in NA v prefrontalni skorji
zamegljen vid
kognitivna upolasnjenost
sposobnost vzdrievanja krvnega tlaka
omotica
apetit
T tel. tee
ortostatska hipotenzija
tveganje za debelost
trigliceridi na tee
inzulinska odpornost —■ diabetes tipa 2
znak/simptom/laboratorijska najdba
4, halucinacii
blodeni
avtorica: Sara Meunier pregledala: Yan Yu, Aaron Mackie*
* dr. med. ob objavi
prevedel in priredil: Jan KejZar, dr. med., specializant psihiatrije pregledala: doc. dr. Brigita Novak Sarotar, dr. med., spec. psih.
4, ekstrapiramidnih simptomov (EPS)*
4, hiperprolaktinemije*
prokognitivni in antidepresivni ainki
4, kognitivnih simptomov* 4, negativnih simptomov*
* Glede na anti-psihotike prve generacije, glej ustreznogradivo!
okrajgave: 5-HT - serotonin DA - dopamin NA - noradrenalin ACh - acetilholin
visoko presnovno tveganje: klozapin, olanzapin zmerno presnovno tveganje: risperidon, kvetiapin nizko presnovnotveganje:antipsihotikitretjegeneracije
1 tveganje za diabeti6no ketoacidozo pri bolnikih z visokim tveganjem tveganje za srbo-iilne dogodke tveganje za prezgodnjo smrt
zaplet Objavljeno 15. junija 2017 na www.thecalgaryguide.com.")
Pituitary Mass Effects
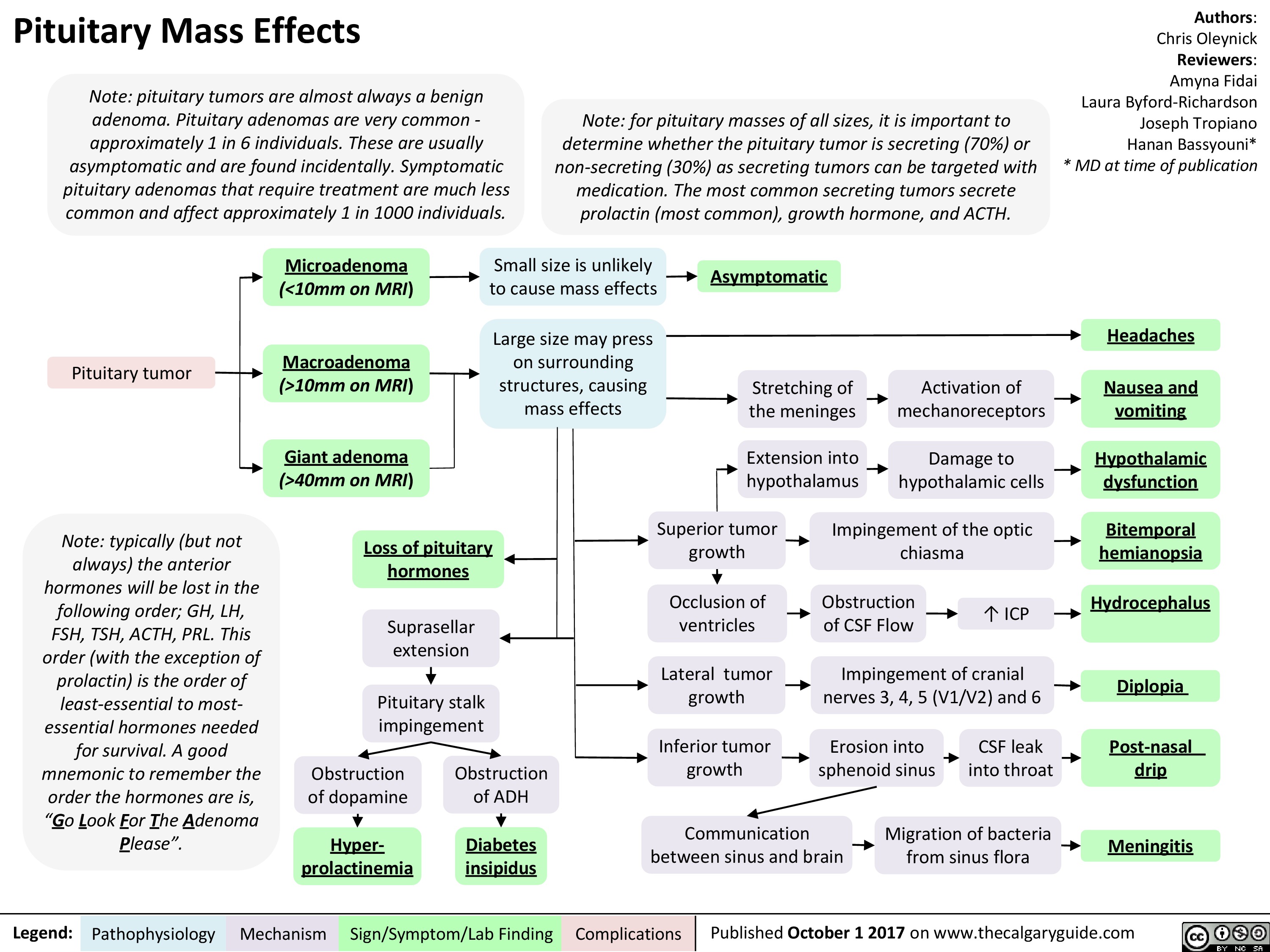 10mm on MRI) vomiting Giant adenoma Extension into hypothalamus —1■• Damage to hypothalamic cells Hypothalamic (>40mm on MRI) dysfunction Obstruction of dopamine Superior tumor growth Impingement of the optic chiasma Bitemporal Loss of pituitary hemianopsia hormones ICP Suprasellar extension Occlusion of ventricles Obstruction of CSF Flow Hydrocephalus Lateral tumor growth Impingement of cranial nerves 3, 4, 5 (V1/V2) and 6 4 Pituitary stalk impingement Diplopia Inferior tumor growth Erosion into sphenoid sinus CSF leak into throat Post-nasal Obstruction of ADH drip Communication between sinus and brain Migration of bacteria from sinus flora Hyper-Diabetes Meningitis prolactinemia insipidus
Pathophysiology Mechanism
Sign/Symptom/Lab Finding
Complications
Published October 1 2017 on www.thecalgaryguide.com
" title="Pituitary Mass Effects
Note: pituitary tumors are almost always a benign adenoma. Pituitary adenomas are very common -approximately 1 in 6 individuals. These are usually asymptomatic and are found incidentally. Symptomatic pituitary adenomas that require treatment are much less common and affect approximately 1 in 1000 individuals.
Pituitary tumor
Note: typically (but not always) the anterior hormones will be lost in the following order; GH, LH, FSH, TSH, ACTH, PRL. This order (with the exception of prolactin) is the order of least-essential to most-essential hormones needed for survival. A good mnemonic to remember the order the hormones are is, "Go Look For The Adenoma Please".
Legend:
Note: for pituitary masses of all sizes, it is important to determine whether the pituitary tumor is secreting (70%) or non-secreting (30%) as secreting tumors can be targeted with medication. The most common secreting tumors secrete prolactin (most common), growth hormone, and ACTH.
Authors: Chris Oleynick Reviewers: Amyna Fidai Laura Byford-Richardson Joseph Tropiano Hanan Bassyouni* * MD at time of publication
Microadenoma Small size is unlikely to cause mass effects (<10mm on MRI) Asymptomatic Macroadenoma Large size may press on surrounding structures, causing mass effects Headaches Stretching of the meninges Activation of mechanoreceptors Nausea and (>10mm on MRI) vomiting Giant adenoma Extension into hypothalamus —1■• Damage to hypothalamic cells Hypothalamic (>40mm on MRI) dysfunction Obstruction of dopamine Superior tumor growth Impingement of the optic chiasma Bitemporal Loss of pituitary hemianopsia hormones ICP Suprasellar extension Occlusion of ventricles Obstruction of CSF Flow Hydrocephalus Lateral tumor growth Impingement of cranial nerves 3, 4, 5 (V1/V2) and 6 4 Pituitary stalk impingement Diplopia Inferior tumor growth Erosion into sphenoid sinus CSF leak into throat Post-nasal Obstruction of ADH drip Communication between sinus and brain Migration of bacteria from sinus flora Hyper-Diabetes Meningitis prolactinemia insipidus
Pathophysiology Mechanism
Sign/Symptom/Lab Finding
Complications
Published October 1 2017 on www.thecalgaryguide.com
" />
10mm on MRI) vomiting Giant adenoma Extension into hypothalamus —1■• Damage to hypothalamic cells Hypothalamic (>40mm on MRI) dysfunction Obstruction of dopamine Superior tumor growth Impingement of the optic chiasma Bitemporal Loss of pituitary hemianopsia hormones ICP Suprasellar extension Occlusion of ventricles Obstruction of CSF Flow Hydrocephalus Lateral tumor growth Impingement of cranial nerves 3, 4, 5 (V1/V2) and 6 4 Pituitary stalk impingement Diplopia Inferior tumor growth Erosion into sphenoid sinus CSF leak into throat Post-nasal Obstruction of ADH drip Communication between sinus and brain Migration of bacteria from sinus flora Hyper-Diabetes Meningitis prolactinemia insipidus
Pathophysiology Mechanism
Sign/Symptom/Lab Finding
Complications
Published October 1 2017 on www.thecalgaryguide.com
" title="Pituitary Mass Effects
Note: pituitary tumors are almost always a benign adenoma. Pituitary adenomas are very common -approximately 1 in 6 individuals. These are usually asymptomatic and are found incidentally. Symptomatic pituitary adenomas that require treatment are much less common and affect approximately 1 in 1000 individuals.
Pituitary tumor
Note: typically (but not always) the anterior hormones will be lost in the following order; GH, LH, FSH, TSH, ACTH, PRL. This order (with the exception of prolactin) is the order of least-essential to most-essential hormones needed for survival. A good mnemonic to remember the order the hormones are is, "Go Look For The Adenoma Please".
Legend:
Note: for pituitary masses of all sizes, it is important to determine whether the pituitary tumor is secreting (70%) or non-secreting (30%) as secreting tumors can be targeted with medication. The most common secreting tumors secrete prolactin (most common), growth hormone, and ACTH.
Authors: Chris Oleynick Reviewers: Amyna Fidai Laura Byford-Richardson Joseph Tropiano Hanan Bassyouni* * MD at time of publication
Microadenoma Small size is unlikely to cause mass effects (<10mm on MRI) Asymptomatic Macroadenoma Large size may press on surrounding structures, causing mass effects Headaches Stretching of the meninges Activation of mechanoreceptors Nausea and (>10mm on MRI) vomiting Giant adenoma Extension into hypothalamus —1■• Damage to hypothalamic cells Hypothalamic (>40mm on MRI) dysfunction Obstruction of dopamine Superior tumor growth Impingement of the optic chiasma Bitemporal Loss of pituitary hemianopsia hormones ICP Suprasellar extension Occlusion of ventricles Obstruction of CSF Flow Hydrocephalus Lateral tumor growth Impingement of cranial nerves 3, 4, 5 (V1/V2) and 6 4 Pituitary stalk impingement Diplopia Inferior tumor growth Erosion into sphenoid sinus CSF leak into throat Post-nasal Obstruction of ADH drip Communication between sinus and brain Migration of bacteria from sinus flora Hyper-Diabetes Meningitis prolactinemia insipidus
Pathophysiology Mechanism
Sign/Symptom/Lab Finding
Complications
Published October 1 2017 on www.thecalgaryguide.com
" />
Pressure Ulcers Pathogenesis and clinical findings

Must remove slough/eschar to determine stage
-110.
Kennedy terminal ulcer (often precedes death)
—1111.
Pressure Ulcers: Pathogenesis and clinical findings
Bed, wheelchair, stretcher, car seat
External physical compression
Involuntary muscle movement, passive repositioning of torso Shear forces (dermis/epidermis fixed through contact with a surface while deeper tissues are moved; vessels angulate and thrombose, creating undermining of ulcer)
Inability to move well, aging skin (loss of elasticity, blood flow, and subcutaneous fat) Friction (person dragged across surface, damaging stratum corneum)
Bowel/bladder incontinence, diaphoresis, wound drainage Moisture (skin maceration)
4, in movement (coma, neuro injury, post-surgery, etc.) Limited mobility
Unrelieved pressure greater than arterial capillary pressure (>32 mmHg, with more rapid ulcer formation at higher pressures; normal range 12-32 mmHg) Disrupts blood supply and deprives tissues of oxygen and nutrients Pressure Ulcer (local injury to skin and/or underlying tissues, often over bony prominence)
Authors: Rebecca (Becky) Phillips Reviewers: Gurleen Chahal Usama Malik Laurie M. Parsons* * MD at time of publication
Notes: • More common in ages 65+ • Risk factors: diabetes, peripheral arterial disease, immunodeficiency, steroid therapy, smoking, dementia, poor nutrition, sensory deficit, circulatory disturbance, prolonged immobility • Grading system from National Pressure Ulcer Advisory Panel
Pear- or butterfly-shaped sacral ulcer
Stage I (non-blanchable erythema of intact skin; heralds impending ulcer)
May be warmer, painful, edematous, indurated, or discolored compared to surrounding tissue
Legend:
Stage II (partial thickness skin loss)
Erosion, serum-filled blister, or shallow ulcer with red-pink wound bed
Pathophysiology Mechanism
Stage III (full thickness skin loss; damage to subcutaneous tissue but not underlying fascia)
*
Exposed subcutaneous fat. May have slough, undermining, or tunneling (nose bridge, ear, occiput, and malleolar ulcers will appear shallow due to absence of subcutaneous tissue)
Sign/Symptom/Lab Finding
Stage IV (full thickness tissue loss)
Bone, tendon, or muscle exposed. Slough or eschar may be present. Often have undermining or tunneling
Complications
Bacterial invasion via contiguous spread (commonly S. Aureus and coagulase-negative staphylococci)
Osteomyelitis")
Erectile Dysfunction: Pathogenesis

1. Assess CVD Disease risk* a. I% Blood pressure b. I% Fasting glucose or HbA1c c. TG's & cholesterol 1. Penile duplex sonography 2. Cavernosometry
Legend:
Endocrinologic Erectile Dysfunction
Hypogonadism, hyperprolactinemia, hyperthyroidism, alcoholism, iatrogenic
.J, circulating free testosterone
•
1. 4, 7 AM free testosterone* 2. l• Thyroid Stimulating Hormone 3. l• Prolactin 4. l• Follicle Stimulating Hormone 5. l• Luteinizing Hormone
4, release of NO and cGMP levels within corpora cavernosa and smooth muscle relaxation
Pathophysiology Mechanism
Neurogenic Erectile Dysfunction
Neurologic disease, trauma, iatrogenic, diabetes mellitus
Central (cerebral or spinal cord); peripheral (afferent/sensory neuropathy) or efferent (autonomic neuropathy)
4, parasympathetic nerve firing
4, NO release
Psychogenic Erectile Dysfunction
•
Sudden onset, sporadic (circumstantial), younger, nocturnal/AM erection present
•
Anxiety, depression, strained relationship, lack of sexual arousal, psychological disorder
Possible mechanisms include an imbalance of central neurotransmitters, over inhibition of spinal erection center by the brain, and sympathetic overactivity
1. Abnormal Nocturnal penile 1. Normal Nocturnal penile tumescence and rigidity* tumescence and rigidity*
Erectile Dysfunction -• (persistent or recurrent inability to achieve an erection sufficient to achieve desired sexual performance)
Sign/Symptoni/Lab Finding
Complications
Authors: Braden Milian Reviewers: Alex Tang Usama Malik Jay C. Lee* * MD at time of publication")
Mixed Urinary Incontinence Pathogenesis and clinical findings
 4, Urinary leakage preceded by a sudden, strong urge to void
Overflow Incontinence vir Overfilling of the bladder from obstruction; BOO (tumour, stone, BPH, urethral or bladder neck stricture)
Detrusor Overactivity Ilr OAB (idiopathic), CNS lesion (neurogenic), inflammation/ infection (cystitis, UTI), diabetes mellitus
4. Bladder Wall Compliance
Progressive t in intravesicle pressure during bladder filling pushing urine from the bladder
Authors: Braden Millan Reviewers: Alex Tang Usama Malik Jay C. Lee* * MD at time of publication
Stress Urinary Incontinence (SUI) + Episodic involuntary urinary leakage with sudden l• in intra-abdominal pressure
4.
Urethral hypermobility, intrinsic sphincter deficiency, or a poorly coapting urethra
4,
4, Pelvic floor muscle and ligament strength causing 4. tone of vesicoureteral sphincter unit; 4, urethral strength and associated striated and smooth muscle; iatrogenic
Legend:
Failure to Void Weak Stream (+ dribbling), Intermittent, Straining, '1` PVR if a complication of urinary retention; obstruction visible on cystoscopy
Failure to Store Frequency, Urgency, Nocturia, Dysuria if SUI or UUI not caused by obstruction
Pathophysiology Mechanism
Urodynamic Studies SUI — 4, urethral closure pressure with 11` IAP/Bladder Volume and urinary leakage UUI — involuntary detrusor contraction and/or detrusor sphincter dyssynergia
Incontinence, 4, Quality of Life, UTI's")
Penyembuhan Fraktur: Tahapan dan Faktor Pengganggu
 (osifikasi intramembran)
Penyembuhan tulang tahap inflamasi, kalus halus, and kalus keras
Penulis: Spencer Montgomery Penyunting: Yan Yu Dr. Gerhard Kiefer* Penerjemah: M Harmen Reza S* * MD (dokter) pada saat publikasi
Legenda:
Fraktur
Catatan: Penyembuhan fraktur melibatkan campuran antara jalur penyembuhan primer dan sekunder
Tahap Inflamasi (0-7Hari)
Stabilitas relatif pada lokasi fraktur: (pergerakan pada ujung tulang) - e.g. bidai, paku intramedular, traksi
Penyembuhan tulang sekunder (indirek) (osifikasi endokondral)
Kerusakan pembuluh darah lokal 4 hematoma 4 4, perfusi/02 ke tulang 4 osteonekrosis pada garis fraktur 4 terbentuk inflamasi lokal
Pergerakan pada lokasi fraktur
++ nyeri
Tahap Kalus Halus (mgg 1— 3)
Tahap Kalus Keras (mgg 3 — bin 3)
Remodeling (bin — thn)
Patofisiologi Mekanisme
• 1Kondrosit menyusun tulang rawan di lokasi hematoma, menjembatani kedua ujung tulang 4 nyeri berkurang
1
Osteoblas mengendapkan Ca3(PO4)2 ke matriks tulang rawan, membentuk kalus,1` stabilitas lokasi fraktur
Faktor yang dapat mengganggu penyembuhan fraktur
Tembakau Memperlama waktu penyembuhan, mekanisme belum jelas. Tiga hipotesis: 1) Nikotin: 4, aliran darah, dapat bersifat toksik pada osteoblas 2) Karbon Monoksida: 4, 02 ke lokasi fraktur 3) Hidrogen Sianida: menginhibisi metabolisme oksidatif pada tingkat sel
Penyalahgunaan alkohol Memperlama waktu penyembuhan, mekanisme tidak diketahui
Kortikosteroid & AINS jangka panjang Menghalangi respon inflamasi yang membatu penyembuhan
Remodeling tulang oleh pasangan Osteoklas-osteoblas : mengikir kalus agar tulang dapat mencapai bentuk efisien, sepanjang jalur gaya mekanisnya
Tanda/Gejala/Penunjang
Komplikasi
Kuinolon Menyebabkan pembentukan kalus imatur
Defisiensi Vitamin C Mengurangi pembentukan kolagen (Vit C adalah kofaktor kunci sintesis kolagen)
Diabetes Produksi kalus lemah (studi hewan)
Rifampicin & gentamycin topikal Toksik terhadap osteoblas
Hipotiroidisme Menginhibisi osifikasi endokondral (studi hewan)
Defisiensi Vitamin D Kurangnya absorpsi Ca2+ & Fosfat dari saluran cerna, 4, mineralisasi tulang.")
intrauterine-growth-restriction-iugr-pathogenesis
: Pathogenesis
Authors: Ricki Hagen Reviewers: Jaimie Bird Sarah McQuillan* * MD at time of publication
Abbreviations:
• DM: diabetes mellitus
• HTN: hypertension • IUGR: intrauterine growth restriction
• SGA: small for gestational age
• SLE: systemic lupus erythematosus
• TORCH: Toxoplasmosis, Others, Rubella, CMV, HSV
Maternal Factors
Maternal-Fetal Factors Placental malformations
(Ex. previa, accreta, infarction, abnormal implantation, ischemia)
Gestational HTN/ Preeclampsia
Multiple gestation Gestational DM
Fetal Factors Structural anomalies
(often comorbid with cytogenetic disorders)
Congenital infections
(Ex. TORCH)
Inborn errors of metabolism
Chromosomal disorders/ genetic syndromes
Multiple unclear intrinsic fetal mechanisms
Note:
Teratogenic medications (Ex. Warafin, Valproic Acid, Folic Acid Antagonists)
High altitude living
Smoking, ETOH and/or drug use
Malnutrition/ Low pre-gestational weight
Multiple unclear extrinsic fetal mechanisms
Medical conditions
(Ex: chronic HTN, cyanotic heart disease, severe chronic anemia, kidney disease)
Autoimmune conditions (Ex. Type 1 DM, SLE)
Decreased uteroplacental blood flow
Nutrient supply to fetus compromised
Reduction of total body mass, bone
mineral content, and muscle mass
Blood flow redirected away from vital organs to brain, placenta, heart and adrenal glands
Reduction of overall fetal size to increase survival
IUGR
Failure to reach genetically determined growth potential
• IUGR is not synonymous with SGA • Constitutional SGA is due to
paternal and maternal factors such as height, weight, ethnicity, and parity; it is not associated with increased risk for infant mortality or morbidity
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published October, 30, 2018 on www.thecalgaryguide.com")
Benzodiazepine (BZD) withdrawal: clinical findings and complications
 withdrawal: clinical findings and complications
Abrupt cessation of chronic ingestion of BZDs
Administration of BZD antagonist (flumazenil) on patients who have developed -* tolerance/dependence to BZD
Withdrawal Seizure
Negative physiological reactions BZD intake inhibition a mygd to f, • of a la Withdrawal symptoms Benzodiazepine Withdrawal GABA receptor activity (less inhibition alleviated by ingesting BZD Tolerance GABA BZD intake Conformational changes in the GABA receptor 1, receptor's Withdrawal Insomnia Pro-excitatory 4— state of excitatory neurotransmitters) 4— to the agent activity affinity for the agent
A
Activation of ACC and OFC
Feelings of fear
Activation of PAG
Behavioural response of fight or flight
Legend: Pathophysiology Mechanism
Activation of hypothalamus '1` Cortisol CAD, T2DM, Stroke
Sign/Symptom/Lab Finding
Activation of PBN
V
t RR, SOB, Asthma, or a sense of being smothered
Activation of LC
t Sympathetic Activity
t BP, t HR variability, tremor, and diaphoresis
Authors: Usama Malik Reviewers: Sina Marzoughi Aaron Mackie* * MD at time of publication
Notes: • The onset of withdrawal can vary according to the half-life of the BZD involved. Symptoms may be delayed up to three weeks in BZDs with long half-lives, but may appear as early as 24 to 48 hours after cessation of BZDs with short half-lives.
Abbreviations: • ACC: Anterior Cingulate Cortex • BP: Blood Pressure • CAD: Coronary Artery Disease • HR: Heart Rate • LC: Locus Coeruleus • MI: Myocardial Infarction • OFC: Orbitofrontal Cortex • PAG: Periaqueductal Gray • PBN: Parabrachial Nucleus • RR: Respiratory Rate • SOB: Shortness of Breath • T2DM: Type 2 Diabetes
I` atherosclerosis, cardiac ischemia, MI, or sudden death")
Hypernatremia Physiology
![Hypernatremia: Physiology Unreplaced H2O loss
Hypodipsia
H2O shift into cells
Severe exercise, electroshock induced seizures
Transient ↑ cell osmolality
Na+ overload
Inappropriate IV hypertonic solution, salt poisoning
Abbreviations:
H2O: Water
GI: Gastrointestinal
DM: Diabetes Mellitus
DI: Diabetes Insipidus
Na+: Sodium ion
IV: Intravenous
ADH: Antidiuretic Hormone LOC: Level of Consciousness
Skin
Sweat, burns
GI
Vomiting, bleeding, osmotic diarrhea
Fluid [Na+] < serum [Na+]
↑ H2O loss compared to Na+ loss
Renal
DM, Mannitol, Diuretics
Absent thirst mechanism
Hypothalamic lesion impairs normal drive for H2O intake
Nephrogenic
↑ renal resistance to ADH
H2O Deprivation Test + no AVP response
↓ access to H2O
DI
Central
↓ ADH secretion
H2O Deprivation Test + AVP response
↑ [Na+] 10- 15 mEq/L within a few minutes
Weakness, irritability, seizures, coma
↑ thirst, ↓ urinary frequency and volume
Note:
Hypernatremia
Serum [Na+] > 145 mmol/L
Intracranial hemorrhage
Headache, vomiting, ↓ LOC
• Plasma [Na+] is regulated by water intake/excretion, not by changes in [Na+].
• Effects on plasma [Na+] of IV fluids or loss of bodily fluids is determined by the tonicity of the fluid, not the osmolality.
Authors: Mannat Dhillon Reviewers: Andrea Kuczynski Kevin McLaughlin* * MD at time of publication
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published January 11, 2019 on www.thecalgaryguide.com](http://calgaryguide.ucalgary.ca/wp-content/uploads/2019/01/Hypernatremia-Physiology-.jpg "Hypernatremia: Physiology Unreplaced H2O loss
Hypodipsia
H2O shift into cells
Severe exercise, electroshock induced seizures
Transient ↑ cell osmolality
Na+ overload
Inappropriate IV hypertonic solution, salt poisoning
Abbreviations:
H2O: Water
GI: Gastrointestinal
DM: Diabetes Mellitus
DI: Diabetes Insipidus
Na+: Sodium ion
IV: Intravenous
ADH: Antidiuretic Hormone LOC: Level of Consciousness
Skin
Sweat, burns
GI
Vomiting, bleeding, osmotic diarrhea
Fluid [Na+] < serum [Na+]
↑ H2O loss compared to Na+ loss
Renal
DM, Mannitol, Diuretics
Absent thirst mechanism
Hypothalamic lesion impairs normal drive for H2O intake
Nephrogenic
↑ renal resistance to ADH
H2O Deprivation Test + no AVP response
↓ access to H2O
DI
Central
↓ ADH secretion
H2O Deprivation Test + AVP response
↑ [Na+] 10- 15 mEq/L within a few minutes
Weakness, irritability, seizures, coma
↑ thirst, ↓ urinary frequency and volume
Note:
Hypernatremia
Serum [Na+] > 145 mmol/L
Intracranial hemorrhage
Headache, vomiting, ↓ LOC
• Plasma [Na+] is regulated by water intake/excretion, not by changes in [Na+].
• Effects on plasma [Na+] of IV fluids or loss of bodily fluids is determined by the tonicity of the fluid, not the osmolality.
Authors: Mannat Dhillon Reviewers: Andrea Kuczynski Kevin McLaughlin* * MD at time of publication
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published January 11, 2019 on www.thecalgaryguide.com")
Meralgia paresthetica- Pathogenesis and Clinical Findings

Shoulder Dystocia: Complications

Previous
shoulder
dystocia
Size discrepancy between fetal
shoulders and maternal pelvis
Multiparity
Maternal
diabetes
Inadequate
uterine tone (over
distension of
uterus, prolonged
2nd stage) and
birth canal trauma
from complicated
delivery
**Attempts to
disimpact and/or
deliver a
macrosomic fetus
Traction to head
can lead to
stretching and
tearing of
brachial plexus
nerves
Intentional or
incidental
fracture of the
fetus’:
Dysfunctional
or prolonged
labour and/or
contractions
↓ oxygenation
to fetus
Authors:
Danielle Hubbert
Risk Factors: *Up to 50% have no risk factors = Obstetrical Emergency that is challenging to predict Mark Diaz
Fetal death
Legend: Pathophysiology Mechanism Sign/Symptom/Lab Finding Complications Published March 12, 2019 on www.thecalgaryguide.com
Prolonged
2nd stage of
labour
Maternal
obesity
Operative
vaginal
delivery
Fetal anterior shoulder becomes impacted
against maternal pubis symphysis and
fails to deliver spontaneously with normal efforts
Clavicular fracture
Erbs palsy (C5-6)
Klumpke palsy (C8-T1)
(rarely permanent)
Episiotomy or
3rd-4th degree
perineal tears Postpartum
hemorrhage
Hypoxia/asphyxia (see PPH slide)
Turtle sign – fetal head
retracts tight against
perineum
Uterine
rupture
Antepartum Risks Intrapartum Risks
Cord
Compression
Weakened and
distended
musculature
Fetal Complications Maternal Complications
Clavicle, to
↓ diameter
of shoulders
Humerus, when
sweeping
posterior arm
across chest
Humeral
fracture
Reviewers:
Dalynne Peters
Angela Deane
Ingrid Kristensen*
*MD at time of publication")
Small Bowel Infarction
 versus the colon’s dual blood supply (SMA and IMA)
• With decreased perfusion colonic tissue tends to suffer from ischemia rather than more serious infarction
• Colonic ischemia presents with pain, diarrhea, and rectal bleeding
Abbreviations
• SMA - Superior mesenteric
artery
• IMA - Inferior mesenteric artery
Atrial fibrillation
Blood stasis in left atria of heart more
prone to coagulation
Embolism occluding SMA
Hypertension, dyslipidemia, smoking, diabetes, + family hx
Atherosclerosis (of SMA)
Thrombosis in the superior mesenteric artery
↓ Arterial perfusion of the small intestine (↓ O2 delivery to bowel tissue)
Ischemia of bowels Infarction of bowels
Venous trauma
↓ blood flow and endothelial injury
hypercoagulable state
Mesenteric venous thrombosis, backing up arterial blood
Food in the
intestine ↑ demand for blood in gut
Death of cells under visceral peritoneum stimulates autonomic nerves
Post-prandial abdominal pain
Severe central abdominal pain
Small bowel infarction from
SMA occlusion is commonly pain progressive, and out of proportion with the patient’s physical exam findings
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Re-Published June 15, 2019 on www.thecalgaryguide.com")
acanthosis-nigricans-pathogenesis-and-clinical-findings

Macrosomia-Fetal-Complications

Size discrepancy between fetal shoulders and maternal pelvic inlet
Anterior shoulder becomes impacted behind the symphysis pubis during delivery
Shoulder Dystocia
(↑risk in infants of diabetic mothers – see slide on Gestational Diabetes)
Injuries acquired as a result of the birthing process in an infant with shoulder dystocia
↑ incidence of preterm birth
Surfactant deficiency
Respiratory Distress Syndrome
Umbilical cord compression
↓ delivery of oxygenated blood to fetus
Hypoxia/Asphyxia
Infant gasping
Perinatal aspiration of stained amniotic fluid
Meconium Aspiration Syndrome
↑ incidence of cesarean deliveries
↓ duration/absence of labour
↓ release of maternal epinephrine and glucocorticoids
↓ activation of epithelial sodium channels on type II pneumocytes
Delayed resorption of fetal lung fluid
Transient Tachypnea of the Newborn
Notes
↑ oxygen demands
Fetal hypoxia
↑ production of erythropoetin
Polycythemia Neonatal Jaundice
Maternal diabetes
↑ intrauterine exposure to
excessive nutrients and glucose
2Fetal Hyperinsulinism
↑ glucose utilization and suppression of hepatic glucose production
Termination of the maternal glucose supply at delivery
Hypoglycemia
Brachial Plexus Injury
Clavicular Fracture
Humeral Fracture
1. Complications of macrosomia ↑ with birth weight. Risk of stillbirth ↑ above 5 kg 2. Most common in the setting of poorly controlled maternal diabetes; however,
hyperinsulinism may be absent if macrosomia is secondary to a different etiology (e.g. post-term fetus, genetic conditions).
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published September 22, 2019 on www.thecalgaryguide.com")
Incisional-Hernia
 that ↑ risk of infection
Post-op wound Infection
Obesity
Chronic constipation
Chronic cough Pregnancy
↓ clotting factors
Vigorous cough
Severe Hypertension
Seroma
Post-op hematoma Bulging fluid separates High risk
Sutures unsuitable Poor surgical for tension technique
↑ intraabdominal pressure
Fascial Incision separates
Notes:
fascial incision
surgeries* High Risk Surgeries*
Connective tissue disorder
Suboptimal fascial closure
• • •
Emergency surgeries Midline incisions
Acute abdominal surgeries
↓ wound healing/collagen synthesis
Fascial defect at previous incision site
Incisional Hernia:
Protrusion of tissues through prior fascial incision
• Deep wound infection = most common cause of incisional hernias
• Diagnosis on physical exam +/- CT scan if patient is obese
• Treatment = surgery
Bulge at prior incision site
Palpable fascial defect
Bowel and other abdominal contents protrude through defect
Mechanical bowel obstruction (see relevant slide)
Constipation /obstipation
Contents unable to be pushed back through defect (incarceration)
Vascular supply is compromised to herniated contents
Contents become ischemic (strangulated)
Prolonged pressure on skin & bowel over time
Ulceration & ischemia
↓ blood flow to skin layers
Discoloration of skin
Bulge ↑ with coughing/straining
Ulcers extend through bowel wall
Authors: Karly Nikkel Meaghan Ryan Reviewers Michael Blomfield Tony Gu Yan Yu* Edwin Cheng* *MD at time of publication
Colo-enteric fistula
Bowel Perforation
Abdominal Pain
Abdominal Distension
Nausea/ Vomiting
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published November 13, 2019 on www.thecalgaryguide.com")
vomiting-pathogenesis
 Center
Toxins circulating in bloodstream: Chemotherapy, Opioids
Offending substance travels through circulation and binds to receptors in the CTZ, outside the blood brain barrier
Abbreviations:
GERD: Gastroesophageal Reflux Disease PUD: Peptic Ulcer Disease
IBD: Inflammatory Bowel Disease
CTZ: Chemoreceptor Trigger Zone
CNX: Cranial Nerve Ten
H1: Histamine Receptor
M1: Muscarinic Receptor
Disrupted inner ear balance: Motion Sickness
Activation of H1 & M1 receptors in vestibular center traveling via Cerebellum
Stimulates Solitary Tract Nucleus (Medulla)
(Medulla)
Vagus Nerve (CNX) and enteric nervous system activation, resulting in:
Gastric relaxation, ↓ pylorus tone, retrograde duodenal peristalsis
Downward diaphragm contraction, abdominal & chest wall muscles contract: ↑ intra-gastric pressure
Vomiting
(Forceful expulsion of material from stomach and intestines)
Upper and lower esophageal sphincter relaxation and glottis closure
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Re-published February 16, 2020 on www.thecalgaryguide.com")
Fecal-Incontinence

↓ anal resting tone
Aging:
Degene- ration of muscle fibers
Movement disorders (e.g. arthritis, Parkinson’s); aging is a risk factorà ↓ mobility
↓ timely access to bathrooms
Inflammation
of colon (e.g., Ulcerative colitis, Radiation proctitis)
↓ capacity of
rectal smooth muscle to stretch
↓ capacity to store stool
↑ urgency of defecation
↑ reflex relaxation of internal anal sphincter
Chronic diarrhea, diarrhea- predominant irritable bowel syndrome, laxatives
Yan Yu* Erika Dempsey* * MD at time of publication
Stretch injury of Pelvic surgery
Chronic constipation
Build up of solid, immobile mass of stool in the rectum
Loose stool is able to flow around impacted stool, exiting anal canal (overflow diarrhea)
Sensory neuro- pathy (e.g. Diabetes)
Altered
mental conditions (e.g. stroke, dementia)
pudendal nerve (innervating the pelvic muscles and external anal sphincter)
Local neuronal damage
Impaired pelvic muscle and external anal sphincter motor control
Pelvic trauma
Rectal prolapse
Direct external anal sphincter impairment
↑ Stool volume
↑ Loose stools
Rectal hyposensitivity (↓ perception of rectal distension)
Patient fails to sense rectal fullness and voluntarily releases their external anal sphincter
Voluntary external anal sphincter contraction is no longer sufficient in closing the anus
Loose stool is more prone to escape through anal canal compared to solid stool
Continence mechanisms are impaired
Fecal Incontinence: The unintentional loss of solid or liquid stool
Skin Skin
Continence mechanisms are intact, but overwhelmed or ignored
infection Skin erythema
erosion
Inability to control what is widely considered a basic, fundamental bodily process
↑ caretaker burden Social stigma
↑ skin contact with acidic irritant (stool)
↑ rate of institutionalization, (e.g., admission into long-term care)
↓ confidence, sense of agency
↑ stress, anxiety
Skin inflammation
↓ social activity, work ↓ help-seeking ↓ treatment
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published May 2, 2020 on www.thecalgaryguide.com")
Diabetic-Nephropathy

↑ Intrarenal angiotensin II
↑ Advanced glycation end products (AGE)
Shunting of glucose through non- glycolytic pathways (e.g. polyol)
Activation of Protein Kinase C (PKC) pathway
Excessive production and accumulation of glycolytic intermediates (e.g. sorbitol, hexosamine, succinate)
hyperglycemia
Succinate via GPR91
↑ Free radical production (oxidative stress)
Activation of cellular signalling, transcription factors and cytokines (e.g. TGF-β-Smad-MAPK, IGF-1, NF-κB)
↑NADPH oxidase activity
↑Blood volume ↑ Blood pressure ↑ Renal perfusion
Relative afferent arteriole dilatation, efferent arteriole constriction
Initial ↑ in glomerular filtration rate (GFR)
Podocyte loss/injury
Authors: Steven Chen Shannon Gui Yan Yu* Reviewers: Julia Heighton Ryan Brenneis Sophia Chou* * MD at time of publication
Initial glomerular hyperfiltration at time of diagnosis
↓ Production of matrix metallo- proteinases
Aberrant extracellular matrix (ECM) protein expression and accumulation
Sheer stress to glomeruli à pressure-induced damage
↑ Glomerular basement membrane permeability to proteins like albumin
↓ Extracellular matrix regulation
“Metabolic Pathway”
Mesangial matrix expansion
Kimmelstiel-
Wilson lesions (pink
hyaline nodules due to accumulation of damaged proteins)
Tubular fibrosis
Scarred glomeruli are less able to effectively filter blood
↓ in glomerular filtration rate (GFR)
Albuminuria
Usually occurs after ~5 years from time of diagnosis in T1DM; can occur at time of diagnosis in T2DM
Abbreviations
IGF: Insulin-like growth factor
MAPK: Mitogen-activated protein kinases NADPH: Nicotinamide adenine dinucleotide phosphate
NF-κB: Nuclear factor kappa-light-chain- enhancer of activated B cells
TGF-β: Transforming growth factor-β
Protein endocytosis into tubular cells causing inflammation
“Hemodynamic Pathway”
Diabetic Nephropathy
Overt diabetic nephropathy may take upwards of 15-25 years to develop
Note: The mechanisms presented here have been simplified. The cross- talk and signaling between the metabolic and hemodynamic factors do not manifest in a step-wise fashion, but rather occur in parallel.
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published May 3, 2020 on www.thecalgaryguide.com")
Cellulitis
 and entry of pathogen
Risk Factors: Immunocompromised Host: -Diabetes mellitus+ -Lymphedema -Malnourishment
-Older patient+
-Obesity+
-Peripheral vascular disease General Infection Risk: -History of cellulitis+ +highest risk factors
Risk Factors for MRSA Cellulitis: Increased exposure to MRSA: -Contact sports
-Crowded living conditions -Health care workers -Indigenous descent
-Sharing towels, equipment
Increased susceptibility:
-Immunodeficiency -Young age
Direct inoculation (e.g. trauma) Organism virulence overwhelms host defense mechanisms (related to risk factors)
Cellulitis: A bacterial infection in which pathogens penetrate deep dermis and/or subcutaneous fat
Cytokines activate immune response
Accumulation of pus (bacteria, white blood cells, dead skin)
Abscess formation
Infection spreads to nearby lymph nodes
Lymphadenitis
Infection spreads through lymph vessels
Ascending lymphangitis
Local inflammatory response in skin
Pain Warmth Edema Erythema (redness)
with indistinct margins
Vesicles and bullae
Organisms penetrate blood vessels
Bacteremia (presence of bacteria in blood)
Systemic inflammation
Distant spread to bone
Osteomyelitis
Distant spread to endocardium (inner lining of heart chambers and valves)
Endocarditis
Fever Malaise
Chills
Sepsis
(rarely)
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published September 27, 2020 on www.thecalgaryguide.com")
Cubital-Tunnel-Syndrome-Ulnar-Neuropathy
: Pathogenesis and clinical findings
Osteoarthritis
Rheumatoid arthritis
Diabetes mellitus (see Calgary Guide slide on Diabetic Neuropathy)
Degenerative bone & joint changes
Autoimmune damage to joints
Abnormal bone structure/lesions
Idiopathic
Hyperglycemia causes nerve damage (complex mechanisms)
Elbow trauma
Repetitive elbow flexion
Authors: Chris Oleynick Alexandros Mouratidis Yan Yu* Reviewers: Annalise Abbott Sean Crooks Davis Maclean Hannah Koury Jeremy LaMothe* Ian Auld* * MD at time of publication
Inflammation or edema in cubital tunnel (space between medial epicondyle of humerus and olecranon of ulna) where ulnar nerve is found
↓ size of the cubital tunnelà↑ pressure on internal contents (e.g. ulnar nerve)
Axonal conduction is interrupted
Myelin sheath is damaged
↓ blood supply to nerve
Weakness of adductor pollicus longus (works to adduct thumb)
↑ compensatory activity of flexor
pollicis longus with pinching
↓ activity of hypothenar muscles (which move the 5th digit)
↓ activity of 5th digit palmar interosseus muscles (which adduct the 5th digit)
Cubital Tunnel syndrome: compression neuropathy
Paresthesia
Abnormal sensations of skin (“pins & needles”, tingling, burning, and/or numbness) in ulnar nerve sensory distribution
ulnar nerve
↓ activity of muscles innervated by ulnar nerve (medial forearm flexors, hypothenar muscles, interossei muscles, adductor pollicis longus)
+ Tinel sign
over cubital tunnel (tapping at medial elbow causes discomfort and/or paresthesia)
Weak pinch & ↓ grip strength
+ Froment’s sign
(thumb hyperflexion compensates for weak pinch)
Hypothenar atrophy
+ Wartenburg’s sign
(involuntary, excess abduction of 5th digit)
of the
Altered sensation in areas innervated by ulnar nerve (medial forearm and wrist, 5th digit, and medial half of 4th digit)
+ elbow flexion test
(flexing elbow & extending wrist further ↓ volume of the cubital tunnelà paresthesia in ulnar nerve sensory distribution
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
First published January 12, 2017, re-published February 28, 2021 on www.thecalgaryguide.com")
Corneal-Abrasion

Trichiasis
(Misdirected eyelashes directly abrading the ocular surface)
↓ tear production (See Calgary Guide - Dry eye pathogenesis)
↓ quantity or quality of tear film
Any condition causing incomplete or inadequate eyelid closureà↑ exposure of eye surface to atmosphere (Bell’s palsy, proptosis/ exophthalmos)
↑ Tear evaporation
↓ lubrication of eye surface
Dryness and desiccative damage to ocular surface structures
↓ protective barriers against mechanical or foreign body damage
Infection of epithelial defect by pathogens
Damage deepens to inner layers of the cornea
Immune response causes white blood cells to accumulate
Pathogen and immune cells obstruct blood flow, causing tissue necrosis
Infectious corneal ulcer
Any condition affecting the trigeminal nerve and/or peripheral corneal nerves (e.g. Herpes zoster/simplex infection, diabetes, medications, surgery)
Tight lens
↓ O2 reaching cornea
Corneal epithelium hypoxiaà cellular damage
Contact Lens use
Extended use Lens dehydrates
Corneal epithelium adheres to lens and is removed with the lens
Neurotrophic keratopathy
(corneal damage secondary to loss of innervation)
↓ stimulation of the cornea by neurotransmitters (complex and multifactorial)
↓ Blinking (if bilateral)
Area of defect stained bright green under cobalt-blue filtered light
Impaired sensory innervation of the
corneaàImpaired Reflex Tearing
↓ adhesion between corneal epithelium and Bowman’s layer
Trauma from insertion or removal of contact lens
Previous traumatic abrasion with damage to Bowman’s layer or inadequate epithelial adherence (e.g. corneal dystrophy)
Spontaneous erosion (occurs without antecedent injury or foreign body)
↓ structural integrity of corneal epithelium
Fluorescein dyes the exposed Bowman’s layer (inner corneal layer between epithelium and stroma)
Corneal Epithelial Damage (the transparent portion of the eye that covers the anterior portion of the eye and covers the pupil iris and anterior chamber)
Corneal Abrasion: Focal area of epithelial loss - outermost layer of cornea (damage may extend to the bowman's layer below as well)
Abrasions that overlay the pupil
Epithelium (outermost portion on cornea)
Bowman’s layer
Stroma
Descemet’s membrane
Endothelium
Cornea (cross section)
Layers of the cornea
Concurrent damage to other anterior
segment structures in trauma
Can induce traumatic uveitis
Irritation and spasms of the iris/ciliary body muscle complex
Light stimulus induces further movement of
irritated/inflamed structures
Damaged tissues & ensuing inflammatory responseà Inflammatory mediator release
Conjunctival injection (vasodilation of vessels in the conjunctiva)
Red eye
Damage to corneal nerves stimulates corneal nociceptors
Pain/ Foreign body sensation
prevent light entry through the pupil
Acute vision loss
Hyperalgesia (lowered peripheral nerve
threshold for firing while damaged tissues are healing, during which normally non- noxious stimuli like light, wind or temperature change - can induce pain)
Nociceptors stimulate afferent neurons in trigeminal nerve, which then activates efferent neurons in the facial nerve
Stimulation of the lacrimal gland
Tearing
Anterior chamber of the eye
Photophobia (Light sensitivity or light-induced pain/ discomfort) – Pathophysiology is complex and multifactorial
Authors: Yejun Hong, Davis Maclean Reviewers: Mehul Gupta, Adam Muzychuk*, Victor Penner*, Yan Yu* *MD at time of publication
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published July 19, 2021 on www.thecalgaryguide.com")
Fat-Embolism-Syndrome

Non-trauma related (rare)
Long bone fracture
Pelvic fracture
Orthopedic Trauma
Intraosseous access
Soft tissue injuries
Chest compressions
Bone marrow transplant
Pancreatitis
Diabetes mellitus
Fat from bone marrow is disrupted and leaks into bloodstream via damaged blood vessels
Fat globules obstruct dermal capillaries
Capillaries rupture
Blood leaks into the skin
Petechial rash
Non-Orthopedic Trauma (less common)
Fat from injured adipose tissue is released from adipocytes into bloodstream
Metabolic disturbance mobilizes stored fat and moves it into circulation
Fat Embolism Syndrome
the presence of fat globules in circulation
Fat globules damage blood vessel walls
Platelets stick to damaged areas Platelet aggregation
↑ circulating free fatty acids
↑ inflammatory cytokines (TNF, IL1, IL6)
↑ serum C Reactive Protein (an acute phase reactant)
C reactive protein binds to lipid vesicles in circulation
↑ formation of lipid complexes in the blood
Obstruction of cerebral vasculature
↓ blood flow and oxygen delivery to brain tissue
Neurological findings: ranging from ↓ level of consciousness to seizures
Notes:
Large quantities of fat globules can obstruct pulmonary vasculature
Blood clots form throughout the body
Disseminated intravascular coagulopathy
Back up of blood into right heart àRight ventricular dysfunction
↓ pulmonary arterial blood flow à↓ gas exchange in the lungs
Higher CO2 & lower O2 levels in blood àdetected by chemoreceptors
Chemoreceptors stimulate respiratory centre in the brain to ↑ rate of respiration
Dyspnea / Tachypnea
Authors: Tabitha Hawes Reviewers: Hannah Koury, Alyssa Federico, Davis MacLean, Mehul Gupta, Yan Yu*, Jeremy Lamothe* * MD at time of publication
• Underlined findings indicate classic triad of symptoms (petechial rash, neurologic findings, dyspnea/tachypnea)
• Clinical presentation of fat embolism syndrome is variable and may present with any or all of these findings
↓ pumping of blood into systemic circulation
Hypotension Obstructive shock
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published July 19, 2021 on www.thecalgaryguide.com")
Pathogenese des Diabetes Mellitus (DM), Typ II
, Typ II
Genetische Prädisposition:
Poly- oder monogenetische Faktoren (z.B. „Maturity- onset diabetes of the young“ (MODY)) können eine Insulinresistenz prädispositionieren
Altern: Betazellen verlieren im Rahmen des Alterungsprozesses an Masse, wodurch sich, bei bereits bestehender Pädisposition, im Alter ein DM Typ II entwickeln kann .
Medikamente: z.B. Cortison, Psychopharmaka, hochaktive antiretrovirale Therapie(HAART), Minipille (nur auf Progesteron basierende Kontrazeptiva)
Blutzucker bleibt im Normbereich
Zuckervergiftung: Hyperglykämien wirken toxisch auf Betazellen
Hyperglykämie
Beachte: Es besteht eine beträchtliche genetische Komponente: bei familiärer Vorbelastung ist die Erkrankungswahrscheinlichkeit sehr hoch ( bei eineiigen Zwillingen bis zu 90%). Bei Verwandtschaftsgrad ersten Grades ist das Risiko einer Erkrankung 5-10 Mal höher
Ungesunder Lebensstil (z.B. Übergewicht, Bewegungsmangel)
Intraabdominell akkumuliert „Viszeralfett“ (Bauchfett) welches als endokrines Organ funktioniert:
Beachte: „Adipokine“ sind von Fettgewebe produzierte Entzündungsmediatoren (z.B. TNFalpha). Je mehr Fettgewebe ein Mensch besitzt, desto mehr Adipokine werden produziert.
Entzündungsm ediatoren
Freie Fettsäuren
Adipokine
Komplexe, teils unklare Interaktionen mit dem Gewebe
Insulinresistenz
(Die Wirkung des Insulins auf Leber, Muskel und Fettgewebe ist vermindert, Glucose kann deshalb nicht als Energiequelle genutzt werden )
Zunächst kompensieren Betazellen des Pankreas die verminderte Insulinwirkung durch vermehrte Produktion
Fettvergiftung:
Längerfristig nimmt die Kompensationsfähigkeit der Betazellen ab, Insulinproduktion ↓ (relativer Insulinmangel)
Nach Jahren nimmt die Leistungsfähigkeit der Betazellen soweit ab, dass kein Insulin mehr produziert wird (absoluter Insulinmangel)
Diabetes Mellitus Typ II
Freie Fettsäuren blockieren den GLUT2 der Betazellen, Glucoseimport ↓
Betazellen erkennen den erhöhten Blutzucker nicht, Insulinproduktion sinkt
Da der Körper keine Glukose nutzen kann, werden Triglyceride zu freien Fettsäuren umgewandelt damit diese als Energiequelle dienen können
Autor: Yan Yu Rezensenten: Peter Vetere Gillian Goobie Doreen Rabi* Übersetzerin: Sarah Schwarz Übersetzungsprüfer: Gesche Tallen* * MD zum Zeitpunkt der Veröffentlichung
Legende:
Pathophysiologie
Mechanismen
Symptome/Klinische Befunde
Komplikationen
Veröffentlicht 11. Juli 2013 auf www.thecalgaryguide.com")
Pathogenese des Diabetes Mellitus (DM), Typ I
, Typ I
Genetische Prädisposition
IDDM1 (HLA) Mutation IDDM2 (Insulingene) Mutationen
Diet (Kuhmilch, Nitrosamine)
Viren (Röteln, Coxsackie-Virus, Masern)
Drogen/Toxine
(Pyrinuron,Alloxan, Streptozocin, Pentamidine)
Direkter Schaden an Betazellen,
sodass deren Antigene dem
Immunsystem ausgesetzt werden
Stress (häufige Infektionen, Operationen, Pupertät)
Externe Risikofaktoren
Reifende T-Zellen im Thymus sind nicht in der Lage, die Fähigkeit der Erkennung von Insulingenen zu entwickeln T-Zellen greifen Insulin produzierende Betazellen an
Mutationen des HLA (MCH) Gens können die Bindung von T-Zellen an Betazellen fördern aber auch verhindern
Körperfremde Antigene imitieren Betazellantigene (Molekulare Mimiky), sodass sich die Immunantwort gegen diese Antigene auch gegen Betazellen richtet
Beeinträchtigung der Toleranz des Immunsystem gegen körpereigene pankreatische Betazellen
Autoimmunreaktion gegen Betazellen
(sowohl durch angeborenes, als auch durch erworbenes Immunsystem: Infiltration der Langerhans-Inseln durch Monozyten und Zerstörung durch T-Zellen, auch als Insulitis bezeichnet )
Atrophie der Langerhans-Inseln auf die Hälfte ihrer Ausgangsmasse
Längerfristig sind nur noch <10% der Betazellen funktionstüchtig
Beachte: Die Anzahl an Autoantikörpern im Körper korrelieren mit der Wahrscheinlichkeit einen DM Typ I zu entwickeln
Autor: Yan Yu
Rezensenten:
Peter Vetere
Gillian Goobie
*Doreen Rabi
Übersetzerin: Sarah Schwarz Übersetzungsprüfer: Gesche Tallen*
* MD zum Zeitpunkt der Veröffentlichung
Produktion von Autoantikörper gegen Betazellen
“Prä-diabetes”
Diabetes Mellitus Typ I Absoluter Insulinmangel
Anti-Insulin & Anti-GAD65 nachweisbar im Serum
Noch asymptomatisch Hoher postprandialer Blutzucker
Abgeschwächte Insulinreaktion nach i.v. Glucosegabe
Legende:
Pathophysiologie
Mechanismen
Symptome/Klinische Befunde
Komplikationen
Veröffentlicht: 11. Juli 2013 auf www.thecalgaryguide.com")
Drug Reaction with Eosinophilia and Systemic Symptoms (DRESS)
:
Authors: Lauren D. Lee, Harry C. Liu* Reviewers: Mehul Gupta, Brian Rankin, Julia Chai, Stephen Williams Yan Yu* Laurie Parsons* *MD at time of publication
Pathogenesis and clinical findings
Genetic susceptibility
Certain HLAs (e.g., HLA-B*58:01, HLA-B*57:01, and HLA-A*31:01)
HLA alleles encode for MHC structure and may influence how specific drugs/drug metabolites interact with T cell receptors and MHC proteins on antigen-presenting cells
Exposure to offending drug
E.g., Aromatic anticonvulsants (lamotrigine, carbamazepine, and phenytoin), allopurinol, and sulfonamides
Drug-specific CD4+ and CD8+ T cells produce tumor necrosis factor-alpha and interferon gamma
↑ activated T-cells 2-6 weeks after drug exposure
Drug Reaction with Eosinophilia and Systemic Symptoms (DRESS)
Latent Viral infection
Latent viruses (HHV-6, HHV- 7, CMV, and EBV) concealed in regulatory T-cells
Reactivation of latent viruses may be contributory or secondary to T cell activation by drugs
Eosinophilia +/- atypical lymphocytes
T helper type 2 cells recruit and activate eosinophils by releasing cytokines
Activated leukocytes create a humoral immune and allergic response
Dysfunction of regulatory T cells, resulting in failure to control unwanted immune responses against “self”
Autoimmune processes with single- or multi-organ involvement
CMV- Cytomegalovirus EBV- Epstein-Barr Virus HHV- Human Herpesvirus HLA- Human Leukocyte Antigens
MHC – Major Histo- compatibility Complex
Endocrine system
mechanism of dysfunction unclear but may be linked to autoreactive T cells
Autoimmune Type 1 thyroiditis diabetes
Lymphadenopathy
Fever
Facial edema
Liver
lobular inflammation, dispersed foci of necrotic hepatocytes, granulomatous infiltrates consisting of eosinophils
Hepatitis
Kidney
Interstitial edema and infiltrates of lymphocytes, histiocytes, eosinophils, and plasma cells
Acute interstitial nephritis
Lung
increased pulmonary infiltrate and edema
Morbilliform Skin Eruption
Spongiosis
Epidermal layer
Dermal- Epidermal Junction Dermal layer
Interface dermatitis
Skin eruption
(morbilliform to diffuse erythema with follicular accentuation)
Interstitial pneumonitis
Pleural effusion
Eosinophilic infiltration
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published October 19, 2021 on www.thecalgaryguide.com")
Diabetische Ketoazidose (DKA)

Betrifft hauptsächlich Patienten mit Diabetes Mellitus Typ 1: Bestimmte Situationen (z.B. Infektionen) erhöhen den Insulinbedarf, welcher durch fehlende Produktion/unzureichende Substitution nicht gedeckt werden kann
Autor: Yan Yu Rezensenten: Peter Vetere, Gill Goobie, Sean Spence, Hanan Bassyouni* Übersetzung: Sarah Schwarz Übersetzungsprüfung: Dr. Gesche Tallen * MD zum Zeitpunkt der Veröffentlichung
Hyperglykämie
(erhöhter Blutzucker)
Bei >12mmol/L, Glukosefiltration > - resorption, Glukose verbleibt im Urin
Glukosurie
Glukose ist osmotisch wirksam und zieht große Mengen Wasser mit sich (Osmotische Diurese)
Polyurie
Schwere Dehydrierung(bis zu 4-5L)
(ZVD↓, Orthostase: posturale Hypotension/ posturale Tachykardie, Ruhepuls ↑ )
Insulinmangel enthemmt die Lipolyse, sodass der Körper Energie aus Triglyceriden produziert
Freisetzung von freien Fettsäuren aus Fettgewebe
Absoluter
Insulinmangel
Hypothalamische Zellen induzieren bei geringem intrazellulären Glukosespiegel ein starkes Hungergefühl
Glukose bleibt im Blut & kann nicht durch Muskel- /Fettgewebe verstoffwechselt werden
“Ausgehungerte” Zellen triggern die Freisetzung kataboler Hormone: Glucagon, Katecholamine, Cortisol, Somatotropin
Damit intrazellulärer Glukosespiegel ↑, erhöht der Körper den Blutzucker
Hydrolyse von freien Fettsäure in
der Leber (Ketogenese) Polyphagie
↓ Proteinsynthese, ↑ Proteolyse (in Muskelgewebe)
↑ Gluconeogenese, ↑ Glykogenolyse (in der Leber)
Acetyl Co-A
Energiequelle für “ausgehungerte” Zellen
Beachte: Neben Glukose sind Ketonkörper die einzigen Energieträger die von Nervenzellen metabolisiert werden können. Deshalb werden sie vom Körper bei geringem Glukoseangebot produziert.
Ketonkörper
( β-Hydroxybutyrat, Acetoacetat, Aceton, akkumulieren im Blut)
Ketonurie
Metabolische Azidose
(↑Anionenlücke: Ketonkörper verbrauchen den HCO3--Puffer)
Substrate der Gluconeogenese↑
Durch ↓ Extrazellulärflüssigkeit, werden Ketonkörper konzentriert → Azidose
Stört das enterische Nervensystem, Magenentleerung ↓, Ileus
Bauchschmerzen, Übelkeit, Erbrechen
(Dehydration↑)
Abatmen von CO2 um Azidose auszugleichen
Kussmaul- Atmung (Tiefe, schnelle Züge)
Abatmen von Keton- körpern
Azeton- geruch der Atemluft
Beeinträchtigung elektrischer Signale zum Gehirn, Rücken- mark und Nervenzellen
Perfusion des Gehirns, Rückenmark, Nervenzellen↓
Verschiebung des Wasser- und Elektrolythaushalts
Falls Trinkwasser vorhanden
Polydipsie Beachte: Während der DKA verliert der Körper K+ durch
Elektrolytstörung
Schwäche, Delirium +/- Koma
osmotische Diurese und Erbrechen. Da gleichzeitig allerdings K+ aus den Zellen diffundiert, kann das Serumkalium unauffällig/erhöht sein. Um Hypokaliämien zu verhindern sollte bei K+ <5.0mmol/L KCl zusammen mit Insulin i.v. verabreicht werden. Cave: gute Nierenfunktion des Patienten
Therapie: 1) +++ Flüssigkeit. 2) Insulin + KCl. 3) Auffüllen der Anionenlücke. 4) Auslösende Faktoren identifizieren. 5) Wenig PO4 (typischerweise wenige Stunden bis einen Tag nach der Ketoazidose durch ↑ ATP-Produktion.)
Legende:
Pathophysiologie
Mechanismen
Symptome/Klinische Befunde
Komplikationen
Publikationsdatum: 17 Juni 2013 auf www.thecalgaryguide.com
Hyperosmolares/ Hyperglykämisches Koma (HHS)
Beachte: betrifft nur Patienten mit Diabetes mellitus Typ II
Beachte: Es sollte stets nach auslösenden Faktoren z.B. einer Infektion gesucht werden
Autor: Yan Yu Rezensenten:
Peter Vetere
Gill Goobie
Hanan Bassyouni* Übersetzung:
Sarah Schwarz Übersetzungsprüfung: Dr. Gesche Tallen
* MD zum Zeitpunkt der Veröffentlichung
Verschiebung des Wasser- und Elektrolythaushalts
Elektrolytstörung
Unzureichende Insulin- produktion, Insulinresistenz, Nichtansprechen auf Insulintherapie
Relativer Insulinmangel
Situationen mit ↑Insulinbedarf: Infekt, Pneumonie, Herzinfarkt, Pancreatitis, etc.)
Hyperglykämie
(Stark erhöhter Blutzucker, höher als bei der DKA)
Bei >12mmol/L, Glukosefiltration > - resorption, Glukose verbleibt im Urin
Glukosurie
Glukose ist osmotisch wirksam und zieht große Mengen Wasser mit sich (Osmotische Diurese)
Polyurie Dehydration
(ZVD↓, Orthostase: posturale Hypotension/posturale Tachykardie, Ruhepuls ↑ )
Durch das wenige noch vorhandene Insulin, wird ein Teil der Glukose von Muskel- /Fettgewebe verstoffwechselt, ein Teil verbleibt im Blut
Körperzellen brauchen eine weitere Energiequelle
Freisetzung kataboler Hormone: Glucagon, Katecholamine, Cortisol, Somatrotropin
Damit intrazellulärer Glukosespiegel ↑, erhöht der Körper den Blutzucker
Hypothalamische Zellen induzieren bei geringem intrazellulären Glukosespiegel ein starkes Hungergefühl
Polyphagie
Beachte: Durch das wenige noch
vorhandene Insulin wird die Lipolyse gehemmt und werden keine Ketonkörper produziert. Es kommt nicht zur Azidose und Ketonurie (Gegensatz zur DKA). Sollte es doch zur Ketourie kommen, ist das meist Folge von Hungerzuständen oder anderen Mechanismen
Extrazellulärflüssigkeit ↓ mit erhöhter Osmolarität (z.B. Hypernatriämie)
↑ Gluconeogenese, ↑ Glykogenolyse (in der Leber)
↓ Proteinsynthese, ↑ Proteolyse (in Muskelgewebe)
Substrate der Gluconeogenese ↑
Sollte der Patient nicht ausreichend trinken um das Volumendefizit auszugleichen
Falls Trinkwasser vorhanden
Polydipsie
Wasser verlässt Zellen entlang des osmotischen Gradienten, Neuronen schrumpfen
Neuronaler Schaden: Delirium, Krampfanfall, Benommenheit, Koma
Renale Perfusion↓, GFR ↓
Nierenversagen
(prä-renal, siehe entsprechende Folie)
Beachte: Während der DKA verliert der Körper K+ durch osmotische Diurese. Da gleichzeitig allerdings K+ aus den Zellen diffundiert, kann das Serumkalium unauffällig/erhöht sein. Um Hypokaliämien zu verhindern sollte bei K+ <5.0mmol/L KCl zusammen mit Insulin i.v. verabreicht werden.
Cave: gute Nierenfunktion des Patienten
Beachte: Elektrolytstörungen (z.B. Hyperkaliämien, Hypernatriämien) verschlechtern sich bei akutem Nierenversagen, welches bei der DKA &HK häufig auftritt
Legende:
Pathophysiologie
Mechanismen
Symptome/Klinische Befunde
Komplikationen
Veröffentlicht: 3. November 2016 auf www.thecalgaryguide.com")
Hyperosmolares/ Hyperglykämisches Koma (HHS)

Betrifft hauptsächlich Patienten mit Diabetes Mellitus Typ 1: Bestimmte Situationen (z.B. Infektionen) erhöhen den Insulinbedarf, welcher durch fehlende Produktion/unzureichende Substitution nicht gedeckt werden kann
Autor: Yan Yu Rezensenten: Peter Vetere, Gill Goobie, Sean Spence, Hanan Bassyouni* Übersetzung: Sarah Schwarz Übersetzungsprüfung: Dr. Gesche Tallen * MD zum Zeitpunkt der Veröffentlichung
Hyperglykämie
(erhöhter Blutzucker)
Bei >12mmol/L, Glukosefiltration > - resorption, Glukose verbleibt im Urin
Glukosurie
Glukose ist osmotisch wirksam und zieht große Mengen Wasser mit sich (Osmotische Diurese)
Polyurie
Schwere Dehydrierung(bis zu 4-5L)
(ZVD↓, Orthostase: posturale Hypotension/ posturale Tachykardie, Ruhepuls ↑ )
Insulinmangel enthemmt die Lipolyse, sodass der Körper Energie aus Triglyceriden produziert
Freisetzung von freien Fettsäuren aus Fettgewebe
Absoluter
Insulinmangel
Hypothalamische Zellen induzieren bei geringem intrazellulären Glukosespiegel ein starkes Hungergefühl
Glukose bleibt im Blut & kann nicht durch Muskel- /Fettgewebe verstoffwechselt werden
“Ausgehungerte” Zellen triggern die Freisetzung kataboler Hormone: Glucagon, Katecholamine, Cortisol, Somatotropin
Damit intrazellulärer Glukosespiegel ↑, erhöht der Körper den Blutzucker
Hydrolyse von freien Fettsäure in
der Leber (Ketogenese) Polyphagie
↓ Proteinsynthese, ↑ Proteolyse (in Muskelgewebe)
↑ Gluconeogenese, ↑ Glykogenolyse (in der Leber)
Acetyl Co-A
Energiequelle für “ausgehungerte” Zellen
Beachte: Neben Glukose sind Ketonkörper die einzigen Energieträger die von Nervenzellen metabolisiert werden können. Deshalb werden sie vom Körper bei geringem Glukoseangebot produziert.
Ketonkörper
( β-Hydroxybutyrat, Acetoacetat, Aceton, akkumulieren im Blut)
Ketonurie
Metabolische Azidose
(↑Anionenlücke: Ketonkörper verbrauchen den HCO3--Puffer)
Substrate der Gluconeogenese↑
Durch ↓ Extrazellulärflüssigkeit, werden Ketonkörper konzentriert → Azidose
Stört das enterische Nervensystem, Magenentleerung ↓, Ileus
Bauchschmerzen, Übelkeit, Erbrechen
(Dehydration↑)
Abatmen von CO2 um Azidose auszugleichen
Kussmaul- Atmung (Tiefe, schnelle Züge)
Abatmen von Keton- körpern
Azeton- geruch der Atemluft
Beeinträchtigung elektrischer Signale zum Gehirn, Rücken- mark und Nervenzellen
Perfusion des Gehirns, Rückenmark, Nervenzellen↓
Verschiebung des Wasser- und Elektrolythaushalts
Falls Trinkwasser vorhanden
Polydipsie Beachte: Während der DKA verliert der Körper K+ durch
Elektrolytstörung
Schwäche, Delirium +/- Koma
osmotische Diurese und Erbrechen. Da gleichzeitig allerdings K+ aus den Zellen diffundiert, kann das Serumkalium unauffällig/erhöht sein. Um Hypokaliämien zu verhindern sollte bei K+ <5.0mmol/L KCl zusammen mit Insulin i.v. verabreicht werden. Cave: gute Nierenfunktion des Patienten
Therapie: 1) +++ Flüssigkeit. 2) Insulin + KCl. 3) Auffüllen der Anionenlücke. 4) Auslösende Faktoren identifizieren. 5) Wenig PO4 (typischerweise wenige Stunden bis einen Tag nach der Ketoazidose durch ↑ ATP-Produktion.)
Legende:
Pathophysiologie
Mechanismen
Symptome/Klinische Befunde
Komplikationen
Publikationsdatum: 17 Juni 2013 auf www.thecalgaryguide.com
Hyperosmolares/ Hyperglykämisches Koma (HHS)
Beachte: betrifft nur Patienten mit Diabetes mellitus Typ II
Beachte: Es sollte stets nach auslösenden Faktoren z.B. einer Infektion gesucht werden
Autor: Yan Yu Rezensenten:
Peter Vetere
Gill Goobie
Hanan Bassyouni* Übersetzung:
Sarah Schwarz Übersetzungsprüfung: Dr. Gesche Tallen
* MD zum Zeitpunkt der Veröffentlichung
Verschiebung des Wasser- und Elektrolythaushalts
Elektrolytstörung
Unzureichende Insulin- produktion, Insulinresistenz, Nichtansprechen auf Insulintherapie
Relativer Insulinmangel
Situationen mit ↑Insulinbedarf: Infekt, Pneumonie, Herzinfarkt, Pancreatitis, etc.)
Hyperglykämie
(Stark erhöhter Blutzucker, höher als bei der DKA)
Bei >12mmol/L, Glukosefiltration > - resorption, Glukose verbleibt im Urin
Glukosurie
Glukose ist osmotisch wirksam und zieht große Mengen Wasser mit sich (Osmotische Diurese)
Polyurie Dehydration
(ZVD↓, Orthostase: posturale Hypotension/posturale Tachykardie, Ruhepuls ↑ )
Durch das wenige noch vorhandene Insulin, wird ein Teil der Glukose von Muskel- /Fettgewebe verstoffwechselt, ein Teil verbleibt im Blut
Körperzellen brauchen eine weitere Energiequelle
Freisetzung kataboler Hormone: Glucagon, Katecholamine, Cortisol, Somatrotropin
Damit intrazellulärer Glukosespiegel ↑, erhöht der Körper den Blutzucker
Hypothalamische Zellen induzieren bei geringem intrazellulären Glukosespiegel ein starkes Hungergefühl
Polyphagie
Beachte: Durch das wenige noch
vorhandene Insulin wird die Lipolyse gehemmt und werden keine Ketonkörper produziert. Es kommt nicht zur Azidose und Ketonurie (Gegensatz zur DKA). Sollte es doch zur Ketourie kommen, ist das meist Folge von Hungerzuständen oder anderen Mechanismen
Extrazellulärflüssigkeit ↓ mit erhöhter Osmolarität (z.B. Hypernatriämie)
↑ Gluconeogenese, ↑ Glykogenolyse (in der Leber)
↓ Proteinsynthese, ↑ Proteolyse (in Muskelgewebe)
Substrate der Gluconeogenese ↑
Sollte der Patient nicht ausreichend trinken um das Volumendefizit auszugleichen
Falls Trinkwasser vorhanden
Polydipsie
Wasser verlässt Zellen entlang des osmotischen Gradienten, Neuronen schrumpfen
Neuronaler Schaden: Delirium, Krampfanfall, Benommenheit, Koma
Renale Perfusion↓, GFR ↓
Nierenversagen
(prä-renal, siehe entsprechende Folie)
Beachte: Während der DKA verliert der Körper K+ durch osmotische Diurese. Da gleichzeitig allerdings K+ aus den Zellen diffundiert, kann das Serumkalium unauffällig/erhöht sein. Um Hypokaliämien zu verhindern sollte bei K+ <5.0mmol/L KCl zusammen mit Insulin i.v. verabreicht werden.
Cave: gute Nierenfunktion des Patienten
Beachte: Elektrolytstörungen (z.B. Hyperkaliämien, Hypernatriämien) verschlechtern sich bei akutem Nierenversagen, welches bei der DKA &HK häufig auftritt
Legende:
Pathophysiologie
Mechanismen
Symptome/Klinische Befunde
Komplikationen
Veröffentlicht: 3. November 2016 auf www.thecalgaryguide.com")
Nephrotisches Syndrom: Pathogenese und klinische Befunde

Lupus-Nephritis
Postinfektiöse Glomeruloneprhitis
IgA Nephropathie
Membranöse Glomerulonephritis
Antikörper greifen Podozyten an, Verdickung der glomerulären Basalmembran
Fokal-segmentale Glomerulosklerose (FSSGN)
Schädigung des glomerulären Endo- und Epithels
Minimal Change Glomerulo- nephritis (MCNG)
Diabetes mellitus
Autor: Yan Yu Rezensenten: Alexander Arnold David Waldner
Sean Spence
Stefan Mustata* Übersetzung:
Sarah Schwarz Übersetzungsprüfung: Gesche Tallen*
* MD zum Zeitpunkt der Veröffentlichung
Podozyten-Schädigung auf der epithelialen Seite des Glomerulums (Abflachung der Podozytenfortsätze)
Glomeruläre Immunkomplex- ablagerungen
Chronische Hyperglykämien schädigen das Glomerulum
Geschädigter Proteinfilter (v.a. für geladene Proteine)
Ablagerungen von Immunglobulin-Leichtketten im Glomerulum
Amyloidose
Geschädigte Glomeruli --> gestörte Filterbarriere v.a. für Proteine --> übermäßige Filtration von Plasmaproteinen
Vermehrte renale Ausscheidung von Immunglobulin-Leichtketten (Filtration>Resorption)
Hypoalbuminämie*
Übermäßiger Verlust an Albumin über den Urin
Proteinurie >3.5g/Tag*
Verlust an Antikoagulationsproteinen (Antithrombin, Plasminogen, Protein C & S) über den Urin
Koagulations- /Gerinnungsfaktoren (z.B. 1,7,8, 10) sind in Überzahl
Proteinurie >3.5g/Tag*
Thrombophilie
Anasarka
(Generalisiertes Ödem)
Lidödem
(klassisches Frühzeichen)
Lipidurie
(zeigt unter
gekreuztem polarisiertem Licht eine Malteserkreuz- form)
50% des Serumkalziums sind an Albumin gebunden, sodass Serumkalzium- spiegel ↓
Serum- Ca2+ repräsentiert nicht mehr das Gesamt-Ca2+
Beachte:
• DerklassischeSymptomkomplex(*)desnephrotischen Syndroms besteht aus: Proteinurie (>3,5g/Tag), Ödemen, Hypoalbuminämie,Hyperlipidämie
• Fürjede10g/LmitAlbumin<40:
➔ Addiere 2.5 zur errechneten Anionenlücke um
dessen “wahren” Wert zu bekommen
➔ Addiere 0,2mmol/L zum Gesamt-Ca um den
Wert des ionisierten Kalziums zu errechnen
Blut neigt zur Bildung von Thromben
Ödeme*
↑ Renale Filtrationder Lipide und Ausscheidung über den Urin
Die Anionenlücke ergibt sich hauptsächlich aus negativ geladenem Serumalbumin
Anionenlücke↓
Kolloid- osmotischer Druck ↓
Flüssigkeit kann nicht mehr in den Blutgefäßen gehalten werden und diffundiert ins Gewebe
Signalisiert der Leber die Albuminproduktion zuerhöhen,Albumin wird aber weiterhin über die Nieren verloren
Synthese- arbeit der Leber↑, auch ↑ Lipoprotein- synthese
Hyperlipidämie*:
(Serum-LDL, -VLDL, - TG ↑ )
Legende:
Pathophysiologie
Mechanismen
Symptome/Klinische Befunde
Komplikationen
Veröffentlicht: 19. August 2013 auf www.thecalgaryguide.com")
Celulitis

Organismos penetran los vasos sanguíneos
Bacteremia (presencia de bacterias en sangre)
Piel normal
Capa epidérmica
Unión dérmica- epidérmica Capa dérmica
Grasa subcutánea
Flora cutánea residente: Staphylococcus coagulasa negativos*
Flora cutánea transitoria:
Staphylococcus aureus*
Streptococcus pyogenes
Bacterias gram negativas Hongos
Patógeno en dermis profunda y grasa subcutánea
*patógenos más comunes
Rotura de la barrera cutánea (puede que no sea evidente) y entrada de patógenos
Virulencia del organismo supera los mecanismos de defensa del huésped (asociado a los factores de riesgo)
Celulitis: Una infección bacteriana en la que los patógenos penetran la dermis profunda y/o la grasa subcutánea
Citocinas activan la respuesta inmune
Acumulación de pus (bacterias, glóbulos blancos, piel muerta)
Formación de abscesos
Factores de riesgo:
Huésped inmunodeprimido: -Diabetes mellitus+
-Linfedema
-Desnutrición
-Paciente adulto mayor+ -Obesidad+
-Enfermedad vascular periférica Riesgo de infección general: -Historia de celulitis+
+factores de riesgo mayores
Factores de riesgo para celulitis por SARM:
Mayor exposición a SARM: -Deportes de contacto -Hacinamiento
-Trabajadores de la salud -Ascendencia indígena -Compartir toallas, equipos Mayor susceptibilidad: -Inmunodeficiencia
-Edad temprana
Infección se propaga a los ganglios linfáticos cercanos
Linfadenitis
infección se propaga a través de los vasos linfáticos
Linfangitis ascendente
Respuesta inflamatoria local en la piel
Diseminación a distancia en endocardio (revestimiento interno de las cámaras y válvulas del corazón)
Endocarditis SARM: Staphylococcus aureus resistente a meticilina
Dolor
Calor
Fiebre Malestar
Diseminación a distancia en el hueso
Osteomielitis
Escalofríos
Inflamación sistémica
Edema Eritema (enrojecimiento)
con márgenes indefinidos
Vesículas y ampollas
(poco frecuente)
Sepsis Abreviaturas:
Leyenda: Patofisiología
Mecanismo
Signos/Síntomas/Hallazgos de Laboratorio
Complicaciones
Publicado el 27 Septiembre, 2020 en www.thecalgaryguide.com")
chronic-hypertension-complications
 ≥ 135/85 (on ambulatory or home blood pressure measurement) in patients without diabetes, or BP ≥ 130/80 in patients with diabetes
Authors: Samin Dolatabadi, Yan Yu* Reviewers: Meena Assad, Jessica Krahn Juliya Hemmett* * MD at time of publication
↑ Afterloadà ↑ Resistance to left ventricle ejection
To overcome resistance and preserve cardiac
output, the myocardium undergoes structural and functional changes
Left ventricular hypertrophy and fibrosis
Stiff ventricle
↓ Contractility of the left ventricle
Impaired forward flow of blood from heart
Chronic stress on the endothelium of systemic blood vessels
↑ Blood pressure in retinal circulation
Hypertensive Retinopathy (See
slide on Chronic Hypertensive Retinopathy: Pathogenesis and Clinic Findings)
Smooth muscle of kidney’s
afferent arterioles constricts to prevent transmission of ↑ blood pressure to glomerulus
Overtime, smooth muscles of afferent arterioles hypertrophy from prolonged vasoconstriction
Chronic stress and trauma on endothelial and smooth muscle cells of kidney
Injury leads to excretion of cytokines and extracellular matrix such as fibrin and collagen into subendothelial layer
Endothelial dysfunction (See slide on Atherosclerosis: Pathogenesis)
Atherosclerosis
Loss of normal arterial architecture in the brain due to stress of ↑ blood pressure
Weakening of cerebral arteries
Formation and rupture of microaneurysms
Intracerebral Hemorrhage
Accumulation of plaques in
the walls of cerebral arteries
↓ Cerebral blood flow
Ischemic Stroke
Accumulation of plaques in
the walls of coronary arteries
↓ Myocardial blood flow
Oxygen supply- demand mismatch
Coronary Artery Disease
Thickening of arteriolar wall and narrowing of afferent arterioles
↓ Glomerular blood flow
Glomerular and tubular ischemia
Glomerular sclerosis and tubular atrophy
Blood backs up into lungs
↓ perfusion of blood throughout the bodyà inability of the heart to meet metabolic demands
Congestive Heart Failure
Hypertensive Nephrosclerosis
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published December 4, 2021 on www.thecalgaryguide.com")
Primary Aldosteronism Pathogenesis
à↑ aldosterone
Overproduction of aldosterone, independent from renin-angiotensin- aldosterone system (RAAS)
Ectopic aldosterone secreting tumor
adrenocortical tissue outside of the adrenal glands produce aldosterone
Familial hyperaldosteronism (FH) Type I
Rare mutation causing ACTH- sensitive aldosterone production in the zona fasciculata
Aldosterone binds its receptor on the principal cells located in the collecting duct of the kidney
↑ Na+ and K+ channel insertion on luminal surface of the principal cell and ↑ Na/K ATPase activity on basolateral surface
Na+ follows the concentration gradient and moves into the principal cell cytoplasmàK+ moves into the collecting duct to maintain electroneutrality
Aldosterone binds its receptor on the alpha intercalated cells located in the late distal tubule and collecting duct of the kidney
↑ H+ ATPase and Na/H+ ATPase activity on the luminal surface of alpha intercalated cellsà↑ H+ excretion
H+ loss permits HCO3- to move down the electrochemical gradient across the luminal surfaceà↑ HCO3- resorption into peritubular capillaries
Metabolic alkalosis
K+ efflux (or hypokalemia of any etiology) is counterbalanced by influx of H+ into tubular cells to maintain electroneutrality
↓ pH in tubular cells activates glutaminase (a pH dependent enzyme) generating glutamate from glutamine
Glutamate within renal tubules dissociates into NH4+, HCO3- Intracellular acidosis within tubular cells
Disrupted cellular signaling within the collecting duct results in reduced APQ2 translocation to luminal surface
Authors: Kyle Moxham Reviewers: Emily Wildman Austin Laing Yan Yu* Matthew Harding* * MD at time of publication
Hypertension
↑ Na+ reabsorption
H2O follows active reabsorption
of solute into circulationà ↑ effective arterial blood volume (EABV)
Chronic EABV expansion resets hypothalamic osmotic sensitive ADH releaseàdecreased ADH production
↑ K+ excretion
Hypokalemia (see our Hypokalemia: Clinical Findings Slide)
↓ Na+ delivery to macula densa in the distal convoluted tubule
↓Endogenous renin secretion by juxtaglomerular apparatus while aldosterone continues to be independently produced
Polydipsia & Polyuria
Mild hypernatremia
↓ Renin: Aldosterone ratio
Nephrogenic diabetes insipidus: ↓ urinary concentrating ability
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published December 4, 2021 on www.thecalgaryguide.com")
necrotizing fasciitis

Laceration
Recent surgery
Injection
Burn
Blunt force trauma
Childbirth
Lower extremity wounds
Bacteria enters tissue through open wound
Infection of muscle fascia Local immune response
Production of exotoxins by bacteria
Disruptions of protective skin barrier
Bacteria introduced into tissue during injury
Necrotizing Fasciitis
Type I infection: mixed aerobic and anaerobic bacteria Type II infection: group A streptococcus
Type III infection: marine organisms, clostridial infections Type IV infection: fungal organisms
Poor blood supply of muscle fascia allows for progressive spread of infection
Systemic immune response
Pyrogens produced by immune system
Pyrogens travel through
the bloodstream to the hypothalamus and alters the body’s thermal setpoint
Transmission of bacteria from infected tissue to blood
Sepsis
Streptolysin (exotoxin) causes blood clot formation
Blood clots in vessels
Tissue ischemia in epidermis, dermis, subcutaneous fat, muscle fascia, and/or muscle
Stimulation of programmed cell death
Tissue destruction
Pain more severe than clinical findings
↓ blood flow fails to meet tissue’s needs
Tissue death
Build up of gas in subcutaneous
tissue from bacteria metabolism
Crepitus
↑ serum creatinine
kinase from protein breakdown
↑ blood flow to infected tissue
Warmth Erythema
Immune cells release vasoactive cytokines into the blood
Capillary vasodilation
Fluid and proteins shift from cells and capillaries to interstitial space
Blood
vessel dilation
↓ perfusion of vital organs
Organ failure
Hypotension
↑ heart rate to perfuse vital organs
Tachycardia
Bacteria releases toxins which are taken up into the bloodstream
Immune cells produce inflammatory cytokines
Circulating toxins activate T cells, over- activating the systemic immune response
Toxic Shock syndrome
Infection ↑ white blood cell production in bone marrow
↑ white blood cells
Destructionof peripheral nerve endings
Insensitivity to pain
Tissue hypoxia à anaerobic metabolism
Poor perfusion of lungs impairs gas exchange
Tachypnea
Cytokines affect dopamine production in the basal ganglia
Acute malaise
Production of non-specific acute phase reactants
↑ C reactive protein and erythrocyte sedimentation rate
Fluid-filled blisters
Edema
Fever Compartment syndrome (see relevant Calgary Guide slide)
Amputation ↑ serum
lactate
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
First published Nov 20, 2013, updated Dec 19, 2021 on www.thecalgaryguide.com")
Langerhans Cell Histiocytosis
 CD1a+/CD207+ (Langerhans cell phenotype surface receptors) dendritic cells in tissue(s)
Somatic BRAFV600E mutation: BRAF is a kinase in the MAPK pathway
Other somatic (non- reproductive cell) mutations
Idiopathic (unknown cause)
Mutation(s) can occur in one of these precursor cell types*
Hematopoietic stem cell (earliest cell of blood cell differentiation) in bone marrow
Committed dendritic cell (type of myeloid antigen-presenting cell) precursor in bone marrow or blood
Committed dendritic cell precursor in tissue
Constitutive activation of the MAPK pathway (signalling pathway that regulates variety of cellular processes) in one of these precursor cell types
*Note: Based on the “misguided myeloid differentiation” modelàthe earlier the mutation(s) occur in the myeloid cell differentiation pathway, the more severe the disease.
Langerhans Cell Histiocytosis
Accumulation of abnormal CD1a+/CD207+ dendritic cells (Langerhans Cell Histiocytosis cells or LCH cells) with an inflammatory background in one or more organs
Authors:
Ran (Marissa) Zhang Reviewers:
Mehul Gupta
Kiera Pajunen
Yan Yu*
Lynn Savoie*
* MD at time of publication
↑ recruitment & activation of T cells, macrophages, eosinophils in tissue(s) around the body
↓ CCR7 & CXCR4 (chemokine receptors) expression on LCH cellsàinhibits migration of LCH cells to lymph nodes
↑ BCLXL (an apoptosis regulator protein) expression on LCH cellsà inhibits apoptosis of LCH cells
Immune cell infiltration & ↑ pro-inflammatory chemokine/cytokine release à dysregulated local & systemic inflammation
Accumulation of LCH cells in tissue(s) around the body
Inflammatory lesion (an area of abnormal tissue) formation in one or more organs:
In pituitary stalk
Mass effectà
obstruction of antidiuretic hormone (complex mechanisms)
In liver
Invasion & accumulation of cells foreign to liverà expands liver
Chronic local inflammation
Scarring of bile ducts
↓ bilirubin clearance from liveràbuildup into serum
↑ serum bilirubin
Jaundice
In cortical bone Cytokine production
à↑ osteoclast
(cells that break down bone) activity
↑ rate of bone loss
Osteolytic bone lesions
In bone marrow
Unclear mechanism but likely due to macrophage activation
↑ phagocytosis (ingestion & destruction) of blood cells
In spleen
Invasion & accumulation of cells foreign to spleen
Forms aggregates that expand the red pulp (functions as the blood filter in spleen)
Splenomegaly
In skin
Unclear mechanisms
Variable presentations: most commonly pinpoint erythematous (red) papules or erythematous plaques with crusting & scaling
Hepatomegaly • BRAF- B-Raf proto-oncogene, serine/threonine kinase
• CCR7- C-C motif chemokine receptor type 7
• CD1a- Cluster of differentiation 1A
• CD207- C-type lectin domain family 4 member k • CXCR4- C-X-C chemokine receptor type 4
• LCH- Langerhans Cell Histiocytosis
• MAPK- Mitogen-activated protein kinase
Diabetes Insipidus
Abbreviations:
• BCLXL- B-cell lymphoma-extra large
Anemia, thrombocytopenia &/or neutropenia
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published January 13, 2022 on www.thecalgaryguide.com")
hyperkalemia-pathophysiology-intracellular-shift-and-intake
![Hyperkalemia (intracellular shift and ↑ intake): Pathophysiology
↑ K+ dietary intake (rarely causative)
β2 receptor inhibition (i.e. beta blockers)
Digoxin
α1 receptor stimulation (i.e. epinephrine, norepinephrine)
Insulin deficiency or resistance (i.e. diabetes mellitus)
↓ Stimulation of NHE1 (moves 1 Na+ into cell, 1 H+ out of cell) throughout body
Normal Anion- Gap Metabolic Acidosis (NAGMA)
Excess serum H+ results in ↓ NHE1 activity (See NAGMA: Pathogenesis and Laboratory Findings slide)
↑ Serum osmolarity (i.e. hyperglycemia)
Osmotic movement of water from cells to serum
Cell lysis (i.e. tumour lysis syndrome, rhabdomyolysis, hemolytic anemia)
↑ K+ release from lysed cells into the serum
Na+/K+ ATPase (moves 3K+ into cell, 2 Na+ out) activity inhibited on cells throughout body
↓ Activity of Na+/K+ ATPase on cells throughout the body
↓ Amount of K+ entering cells
↓ NHE1 activity prevents Na+ from entering the cell
Lack of high intracellular [Na+] needed to drive the Na+/K+ ATPase on cells
Loss of water from cells ↑ intracellular [K+]
K+ moves down concentration gradient from cell into serum
↑ K+ available for absorption
Since K+ is dissolved in water, some K+ is carried by water as water osmotically moves through water channels into the serum (phenomenon known as “solvent drag”)
See Hyperkalemia: Clinical Findings slide
Authors:
Mannat Dhillon, Joshua Low, Emily Wildman Reviewers:
Huneza Nadeem, Marissa (Ran) Zhang, Andrea Kuczynski, Yan Yu*, Kevin McLaughlin*, Adam Bass*
* MD at time of publication
↑ K+ in serum
Hyperkalemia
Serum [K+] > 5.1 mmol/L
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published July 17, 2022 on www.thecalgaryguide.com](https://calgaryguide.ucalgary.ca/wp-content/uploads/2022/07/Hyperkalemia-shift-1.jpg "Hyperkalemia (intracellular shift and ↑ intake): Pathophysiology
↑ K+ dietary intake (rarely causative)
β2 receptor inhibition (i.e. beta blockers)
Digoxin
α1 receptor stimulation (i.e. epinephrine, norepinephrine)
Insulin deficiency or resistance (i.e. diabetes mellitus)
↓ Stimulation of NHE1 (moves 1 Na+ into cell, 1 H+ out of cell) throughout body
Normal Anion- Gap Metabolic Acidosis (NAGMA)
Excess serum H+ results in ↓ NHE1 activity (See NAGMA: Pathogenesis and Laboratory Findings slide)
↑ Serum osmolarity (i.e. hyperglycemia)
Osmotic movement of water from cells to serum
Cell lysis (i.e. tumour lysis syndrome, rhabdomyolysis, hemolytic anemia)
↑ K+ release from lysed cells into the serum
Na+/K+ ATPase (moves 3K+ into cell, 2 Na+ out) activity inhibited on cells throughout body
↓ Activity of Na+/K+ ATPase on cells throughout the body
↓ Amount of K+ entering cells
↓ NHE1 activity prevents Na+ from entering the cell
Lack of high intracellular [Na+] needed to drive the Na+/K+ ATPase on cells
Loss of water from cells ↑ intracellular [K+]
K+ moves down concentration gradient from cell into serum
↑ K+ available for absorption
Since K+ is dissolved in water, some K+ is carried by water as water osmotically moves through water channels into the serum (phenomenon known as “solvent drag”)
See Hyperkalemia: Clinical Findings slide
Authors:
Mannat Dhillon, Joshua Low, Emily Wildman Reviewers:
Huneza Nadeem, Marissa (Ran) Zhang, Andrea Kuczynski, Yan Yu*, Kevin McLaughlin*, Adam Bass*
* MD at time of publication
↑ K+ in serum
Hyperkalemia
Serum [K+] > 5.1 mmol/L
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published July 17, 2022 on www.thecalgaryguide.com")
patellar-tendon-rupture-pathogenesis-and-clinical-findings

Repetitive activities involving rapid knee flexion and extension
Tendon inflammation and micro-tearing
Fluoroquinolone (antibiotic class) use
Smoking
Immobilization of knee
↓ Muscle and tendon flexibility and strength
Steroid use (including intraarticular injections)
Changes in collagen fibrils composing tendon (exact mechanism unknown)
Disrupted blood supply to tendon and poor healing
Compromised integrity and strength of the tendon
Sudden heavy load on flexed knee while foot is planted
Quadriceps muscles contract while lengthening (eccentric contraction)àforce exceeds tendon strength
Patellar tendon innervated by common peroneal (fibular) nerve
Nociceptors (pain receptors) within tendon activated by tendon rupture
Pain and tenderness below the patella
Collagen fibres in tendon tear
Tearing or popping sensation at time of injury
Patellar Tendon Rupture
Tendon/Ligament detaches from bony attachment on patella or tibial tubercle No connection of quadriceps to tibia via patellar tendon
↑ Force on tibial tubercle during tendon detachment
Avulsion fracture of tibial tubercle
Trauma to tendon ruptures surrounding blood vessels
Coagulation cascade and inflammatory mediators released at rupture site
Quadriceps muscles unable to stabilize patellar tracking
Instability of knee joint
Quadriceps muscle tension pulls patella upwards with no resistance from patellar tendon
Patellar tendon cannot use distal attachment site for leverage
Knee extensor muscles unable to effectively contract
Knee buckling
Abnormal gait
Patella alta
(high sitting patella) on X-ray
Palpable gap below the patella
Bruising below the patella
Swelling below the patella
↑ Risk of falls
Loss of patellar reflex
Inability to perform a straight leg raise (hip flexes and knee cannot remain straight)
Hematoma (blood collection under the skin)
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published August 28, 2022 on www.thecalgaryguide.com")
diabetes-insipidus-pathogenesis-and-clinical-findings
![Diabetes Insipidus: Pathogenesis and clinical findings
Hereditary
Autoimmune/ Idiopathic
Auto-antibodies destroy neurons that release antidiuretic hormone (ADH)
Mass Effect/ Tumor Invasion
Mass pressing on hypothalamus or pituitary
Electrolyte Imbalance
(mechanism unclear)
Hereditary
Lithium (Li)
(mechanism unclear)
Li enters principal cells of collecting ducts via ENaCs
Li inhibits GSK3β, reducing adenylyl cyclase activity
↓ cAMP- dependent phosphorylation of aquaporin-2
↑ Serum [Ca2+]
Activation of
CaSR in thick ascending limb of Loop of Henle
↓ NaCl reabsorption in thick ascending limb
↓ Generation of medullary osmotic gradient
↓ Serum [K+]
↑ Degradation of aquaporin-2 channels in collecting duct
↓ Aquaporin- 2 channels transporting water across apical membrane of collecting duct
Mutation of AVPR2 gene on X chromosome
Antidiuretic hormone (ADH) receptor cannot reach basolateral surface of principal cells of collecting duct
Mutation of aquaporin-2 gene on chromosome 12
↓ Fusion of aquaporins with apical membrane of collecting duct
Mutation of WFS1 gene on chromosome 4 (Wolfram syndrome)
↓ Processing of antidiuretic hormone (ADH) precursors and ↓ADH-releasing neurons
Surgery/ Trauma
Injury to hypothalamus or pituitary stalk
Mutation of PCSK1 gene on chromosome 5
Deficiency in PC1/3 (encoded by PCSK1)
↓ Processing of ADH by PC1/3
Aquaporin dysfunction
↓ Kidney response to ADH, which mediates reabsorption of water down its osmotic gradient through aquaporins
↓ Production of ADH by hypothalamus or ↓ secretion from ADH-releasing neurons in posterior pituitary (depending on location of lesion)
Central Diabetes Insipidus
Nephrogenic Diabetes Insipidus
Abbreviations:
AVPR2: arginine vasopressin receptor 2 CaSR: calcium-sensing receptor
ENaC: epithelial sodium channel
GSK3β: glycogen synthase kinase type 3 beta PC1/3: proprotein convertase
Diabetes Insipidus
Decreased ability of kidneys to concentrate urine
↓ Reabsorption of water from collecting duct into vasculature
Author:
Oswald Chen
Reviewers:
Huneza Nadeem,
Ran (Marissa) Zhang,
Yan Yu*
Sam Fineblit*
* MD at time of publication
Urine becomes more dilute
↓ Urine osmolality
↑ Urine output
↓ Blood volume
Blood becomes more concentrated
Occurs during late sleep period
Nocturia
Polyuria
(>3 L/day)
↑ Serum osmolality
Activation of hypothalamic osmoreceptors
Hypernatremia
(Serum [Na+] >145 mEq/L)
Polydipsia
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published September 25, 2022 on www.thecalgaryguide.com](https://calgaryguide.ucalgary.ca/wp-content/uploads/2022/09/Diabetes-Insipidus.jpg "Diabetes Insipidus: Pathogenesis and clinical findings
Hereditary
Autoimmune/ Idiopathic
Auto-antibodies destroy neurons that release antidiuretic hormone (ADH)
Mass Effect/ Tumor Invasion
Mass pressing on hypothalamus or pituitary
Electrolyte Imbalance
(mechanism unclear)
Hereditary
Lithium (Li)
(mechanism unclear)
Li enters principal cells of collecting ducts via ENaCs
Li inhibits GSK3β, reducing adenylyl cyclase activity
↓ cAMP- dependent phosphorylation of aquaporin-2
↑ Serum [Ca2+]
Activation of
CaSR in thick ascending limb of Loop of Henle
↓ NaCl reabsorption in thick ascending limb
↓ Generation of medullary osmotic gradient
↓ Serum [K+]
↑ Degradation of aquaporin-2 channels in collecting duct
↓ Aquaporin- 2 channels transporting water across apical membrane of collecting duct
Mutation of AVPR2 gene on X chromosome
Antidiuretic hormone (ADH) receptor cannot reach basolateral surface of principal cells of collecting duct
Mutation of aquaporin-2 gene on chromosome 12
↓ Fusion of aquaporins with apical membrane of collecting duct
Mutation of WFS1 gene on chromosome 4 (Wolfram syndrome)
↓ Processing of antidiuretic hormone (ADH) precursors and ↓ADH-releasing neurons
Surgery/ Trauma
Injury to hypothalamus or pituitary stalk
Mutation of PCSK1 gene on chromosome 5
Deficiency in PC1/3 (encoded by PCSK1)
↓ Processing of ADH by PC1/3
Aquaporin dysfunction
↓ Kidney response to ADH, which mediates reabsorption of water down its osmotic gradient through aquaporins
↓ Production of ADH by hypothalamus or ↓ secretion from ADH-releasing neurons in posterior pituitary (depending on location of lesion)
Central Diabetes Insipidus
Nephrogenic Diabetes Insipidus
Abbreviations:
AVPR2: arginine vasopressin receptor 2 CaSR: calcium-sensing receptor
ENaC: epithelial sodium channel
GSK3β: glycogen synthase kinase type 3 beta PC1/3: proprotein convertase
Diabetes Insipidus
Decreased ability of kidneys to concentrate urine
↓ Reabsorption of water from collecting duct into vasculature
Author:
Oswald Chen
Reviewers:
Huneza Nadeem,
Ran (Marissa) Zhang,
Yan Yu*
Sam Fineblit*
* MD at time of publication
Urine becomes more dilute
↓ Urine osmolality
↑ Urine output
↓ Blood volume
Blood becomes more concentrated
Occurs during late sleep period
Nocturia
Polyuria
(>3 L/day)
↑ Serum osmolality
Activation of hypothalamic osmoreceptors
Hypernatremia
(Serum [Na+] >145 mEq/L)
Polydipsia
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published September 25, 2022 on www.thecalgaryguide.com")
quadriceps-tendon-rupture-pathogenesis-and-clinical-findings

Quadricep muscles atrophy
↓ Quadricep muscle strength
Smoking
Chronic disease (e.g. renal failure, gout, rheumatoid arthritis, and diabetes)
Fluoroquinolone (antibiotic class) use
Changes in collagen fibrils composing tendonàimpaired tendon strength (exact mechanism unknown)
Quadriceps tendon shortens
↓ Quadricep tendon flexibility
Disrupted blood supply to quadriceps tendon and poor healing
Compromised integrity of the quadriceps tendon and ↓ strength
Sudden heavy load on flexed knee while foot is plantedàquadriceps muscles contract while lengthening (eccentric contraction)
Force on quadriceps tendon exceeds its strength
Quadriceps Tendon Rupture
Tendon detaches from bony attachment on patella
↑ Force on patella during quadriceps tendon detachment
Collagen fibres in quadriceps tendon tear
No connection of quadriceps to tibia via quadriceps tendon
Patellar tendon tension pulls patella downwards with no resistance from quadriceps muscles
Quadriceps tendon innervated by common peroneal (fibular) nerve
Nociceptors (pain receptors) within tendon activated by tendon rupture
Pain and tenderness above the patella
Trauma to tendon ruptures blood vessels
Coagulation cascade and inflammatory mediators released at rupture site
Quadriceps muscles unable to stabilize patellar tracking
Knee joint instability
Patella baja (low sitting patella) on X-ray
Palpable gap above the patella
Loss of patellar reflex
Patellar tendon cannot use proximal attachment site for leverage
Knee extensor muscles unable to effectively contract
Inability to perform a straight leg raise (hip flexes while knee cannot remain straight)
Bruising above the patella
Swelling above the patella
Avulsion fracture of patella
Tearing or popping
sensation at time of injury
Knee buckling
Abnormal gait
↑ Risk of falls
Hematoma (blood collection under the skin)
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published October 2, 2022 on www.thecalgaryguide.com")
chronic-pancreatitis-complications
 from acinar cells in exocrine pancreas
(early in disease course)
Proteins precipitate and form aggregates within pancreatic ducts
Accumulation of protein aggregates and localized fibrosis block pancreatic ducts
Rupture of acinar cells near blocked ducts → release of intracellular enzymes and fluid
Accumulation of enzyme- rich fluid within pancreas
Intra-pancreatic pseudocysts (differ from pseudocysts in acute pancreatitis, which are primarily extra-pancreatic)
Chronic Pancreatitis
Recurrent episodes of acute pancreatitis leading to irreversible fibroinflammatory pancreatic damage
Inflammatory cytokines are
continuously released from damaged pancreas over years
Cytokines damage endothelium of intra- and peri-pancreatic blood vessels (including splenic vein, which runs posteriorly behind pancreas and allows for its venous drainage)
Thin and weakened
vessel walls balloon outwards from pressure of blood flow
Pseudoaneurysms
Venous stasis (low blood flow) → ↑ concentration of clotting factors
Obstruction of peripancreatic ducts
Author: Ashar Memon Reviewers: Yan Yu*, Kiana Hampton, Sylvain Coderre* * MD at time of publication
Exocrine insufficiency
(↓ secretion of digestive enzymes, e.g., Lipase, into gastro-intestinal tract)
↓ digestion of foods and absorption of nutrients (including fats)
↓ absorption of fat-soluble vitamin D
Metabolic bone disease
(a group of disorders of decreased bone mineralization)
Blood vessels dilate and swell from increased blood flow
Gastric varices
Cytokines perpetually activate pancreatic stellate cells (stellate cells produce proteins that remodel extra- cellular matrix)
Pancreatic stellate cells increase amounts of collagen and other extra- cellular matrix molecules in pancreas → Fibrosis
Pancreatic proteolytic enzymes (e.g., trypsin) in fluid-filled pseudocysts digest walls of adjacent blood vessels
Fibrotic tissue and pseudocysts compress peripancreatic structures (including splenic vein)
Cytokines stimulate apoptosis of hormone- producing pancreatic Islet cells
(e.g., beta cells)
Endocrine insufficiency
(↓ production and secretion of pancreatic hormones)
Damaged endothelial cells of splenic vein trigger coagulation cascade
(See Calgary Guide slide on Coagulation Cascade)
Splenic vein thrombosis
↑ resistance to blood flow through splenic vein
Cytokines
stimulate apoptosis of acinar cells in exocrine pancreas
Malnutrition
↓ secretion of insulin
↓ cellular uptake and metabolism of glucose → hyperglycemia
Diabetes mellitus
Collateral blood vessels develop around stomach so blood can circumvent splenic vein and relieve splenic vein hypertension
Duodenal obstruction Biliary obstruction
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published October 18, 2022 on www.thecalgaryguide.com")
Achalasia: Findings on Fluoroscopy with Barium Swallow
 tract during fluoroscopy)
Primary Achalasia
(idiopathic)
Secondary (Pseudo) Achalasia
(due to another disease process such as tumor, Chaga’s disease, diabetes mellitus)
Achalasia
Secondary disease processes can cause denervation or nerve dysfunction of esophageal myenteric plexus (EMP). Tumors can obstruct the lumen and infiltrate the EMP.
Visual differences from primary achalasia
Look for fixed abnormalities and/or mucosal irregularities and shouldering in the setting of a tumor
Nerves controlling the lower esophagus are damaged, making it difficult for food and liquids to pass the esophagus
See “Achalasia: Pathogenesis and Clinical Findings” for full pathogenesis of primary and secondary achalasia
Inflammation and degeneration of nerves in the wall of esophagus
Dysfunction of the esophageal myenteric plexus (network of nerves in the GI tract responsible for peristalsis and sphincter relaxation)
Incomplete relaxation of the Loss of peristalsis in distal lower esophageal sphincter (LES) esophagus
Barium has difficulty passing through the LES to the stomach
Barium outlines the GI tract, following esophageal abnormalities
Normal findings
Esophageal Dilatation
Occurs upstream of narrow LES
Bird Beak Sign / Rat Tail Sign
Smooth narrowing of the distal esophagus
Incomplete LES relaxation
Barium Swallow may be normal in some patients with achalasia. Esophageal manometry (gold standard for diagnosis) or upper endoscopy can be considered instead.
Image Source: Radiopaedia
Legend:
Pathophysiology
Mechanism
Radiographic Findings
Complications
Published November 9, 2022 on www.thecalgaryguide.com")
Ischemic Stroke Impairment by Localization
 in respective blood vessels
Ischemia: ↓ blood flow
(See Ischemic Stroke: Pathogenesis slide)
Left hemisphere damage
Right hemisphere damage
Motor and sensory cortices of upper limb and face damage
Urinary incontinence Aphasia (inability to comprehend or produce
Ischemia in
the middle cerebral artery (MCA)
MCA divides into segments
language) (See Aphasia slide)
Left sided agnosia (visual perceptual deficits)
Contralateral hemiparesis (weakness on side of body opposite to injury) & sensory deficits, visual field deficits, aphasia, agnosia (inability to process sensory information), apraxia (motor planning deficits) & agraphia (inability to communicate by writing)
M1-MCA (sphenoidal segment)
M2-MCA (insular segment)
Ischemia in the posterior cerebral artery
Spares the lower extremity, affects the upper extremity and face
Lesion to frontal lobe (Broca area) Infarction of occipital cortex
Lesion to superior temporal gyrus of temporal lobe (Wernicke area)
No homonymous hemianopsia (one-sided visual field loss) Expressive Broca’s/motor aphasia (inability to produce language)
Contralateral homonymous hemianopsia
(visual field loss on opposite side)
Receptive Wernicke’s/sensory aphasia
(inability to comprehend language)
Sensory loss, memory loss, contralateral homonymous hemianopsia & alexia (reading difficulty)
Ischemia of the occipital lobe, posteromedial temporal lobes, midbrain & thalamus
Ischemia in the vertebral basilar artery
Ischemia in the basilar artery
Ischemia of brainstem & medulla
Ischemia of midbrain, thalami, inferior temporal & occipital lobes
Cranial nerve disorders: dysarthria (slurred/slowed speech) (IX, X), diplopia (double vision), facial numbness or paresthesia (VII), Foville’s syndrome (ipsilateral cerebellar ataxia), Horner's syndrome, (paresis of conjugate gaze and contralateral hemiparesis, facial palsy, pain & thermal hypoesthesia)
Motor deficits: Millard-Gubler syndrome (pons lesion), Raymond’s syndrome (ipsilateral abducens impairment, contralateral central facial paresis & contralateral hemiparesis), Wallenburg syndrome (sensory deficits in the contralateral limb, ipsilateral face), ataxia (abnormal gait), unilateral or bilateral sensory loss of position & vibration
Cranial nerve disorders: dysconjugate gaze (unpaired eye movements) (III, IV, VI), ipsilateral facial hypoalgesia (↓ pain sensitivity) (V), unilateral lower motor neuron face paralysis (VII), vertigo (spinning sensation), dysarthria (weak speech muscles) (IX, X)
Motor deficits: contralateral hemiparesis, quadriplegia (paralysis of all 4 limbs), contralateral limb hypoalgesia
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
First published February 3, 2018, updated February 28, 2023 on www.thecalgaryguide.com")
Central Retinal Vein Occlusion

↓ Anticoagulation (e.g., Protein C or S def) and/or ↑ coagulation (e.g., malignancy, Polycythemia Vera)
Non-ischemic (perfused) CRVO
Glaucoma
(a disease characterized by ↑ intraocular pressure)
Optic disc drusen
(calcified nodules located within the optic disk – the most anterior part of the optic nerve)
Diabetes
Dyslipidemia
Hypertension
Vasculitis
(inflammation of blood vessels; e.g., sarcoid, Systemic Lupus Erythematosus)
Endothelial damage
Pupillary responses do not vary between both eyes when a light is shone in one eye at a time
Relative afferent pupillary defect (RAPD) mild or absent
Lost vascular wall integrity
Normal retinal electrical response to a light stimuli
Normal
electro- retinography (ERG) with b- wave >60%
Few scattered Intra-retinal hemorrhages
↓ In retinal electrical response to a light stimuli
Pupils respond differently to a light stimulus shone in one eye at a time
Severe ↓ in visual acuity (Snellen acuity of <20/400)
Visual field deficits
↓ b-wave amplitude (<60%) on ERG
RAPD present
↑ Pressure compromises retinal vein outflow
Central retinal atherosclerosis (build-up of fat & cholesterol plaque in arteries) & hardening
Atherosclerotic changes in the central retinal artery compress the central retinal vein (since they are both held together in region of the optic disc)
Venous stasis (stagnation of blood flow) ↑ Likelihood of thrombus formation
Central Retinal Vein Occlusion (CRVO)
A common retinal vascular disease characterized by blockage of the main vein that drains blood from the retina
CRVO classified based on the degree of perfusion (as seen through retinal angiography)
Ischemic (nonperfused) CRVO
↑ Intraluminal venous pressure
Dilated tortuous retinal veins
Normal visual fields
Vascular wall integrity lost
Four-quadrant hemorrhages described as “blood and thunder” on fundus exam
Obstruction due to thrombus
↓ Retinal capillary perfusion causes ischemia
Hypoxia in the retinal tissues
↑ Release of vascular endothelial growth factor to revascularize diseased tissue & overcome hypoxic conditions
Angiogenesis (formation of new blood vessels)
Intraretinal infarcts
“Cotton wool spots” on fundus exam
Venous capillary fluid/protein leakage
Mild retina, macula and disc edema
Moderate ↓in visual acuity often (Snellen acuity >20/400)
New vessel proliferation can occur in the anterior chamber of the eye
This change can block the outflow path of the aqueous humor in the eye
Neovascular glaucoma
Neovascular vessels are structurally different in comparison to regular retinal vasculature
Neovascular vessels are more fragile
↑ Likelihood of rupture Vitreous hemorrhage
Neovascular vessels lack tight junctions
↑ Fluid leakage
Retina, macula and disc edema
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
First Published June 28, 2017, updated March 4, 2023 on www.thecalgaryguide.com")
Carpal Tunnel Syndrome

↑ Blood sugar: deposition of advanced glycation end (AGE) products (proteins or lipids glycated when exposed to sugar)
AGE attaches to and prevents tendons from moving properly
Authors: Amanda Eslinger Yvette Ysabel Yao Reviewers: Matthew Harding Owen Stechishin Mao Ding, Cory Toth* * MD at time of publication
Wrist trauma (distal radius fractures, carpal/metacarpal fractures, tendon ruptures)
Repetitive Strain Injury (repetitive hand & wrist movements)
Irritation, swelling & thickening of tendons in carpal tunnel
Calcium deposits
Calcifica tion
Deposition Amyloidosis
Amyloid
(protein aggregates) deposition
Gout
(See Gout Slide)
Uric acid crystal deposition
Pregnancy
↑ Concentration of hormones & uterine pressure on inferior vena cava
Backup of blood into systemic circulation
Autoimmune
(i.e. Rheumatoid arthritis, scleroderma, lupus, Sjogren’s syndrome)
↑ Inflammatory cytokines causing inflammation
Hypo- thyroidism
Myxedema (swelling of skin and underlying tissues) in carpal tunnel
Idiopathic or Congenital
Edema
Narrowed carpal tunnel leads to ↑ internal pressure
Carpal Tunnel Syndrome
Vascular: median artery thrombosis in carpal tunnel
Median nerve compression inside the carpal tunnel Mechanical disruption of median nerve
Compression exacerbated with flexed wrists (i.e. sleep, driving, holding phone/cup)
Disruption of daily activities and sleep
Ischemia (↓ blood supply) to median nerve
Hypoxia (↓ oxygenated blood flow)
Metabolic conduction block (impaired axonal transport due to ischemia)
Nerve conduction study
(show sensory nerve impulses slowing across the wrist, followed by mild / moderate / severe loss of sensory nerve amplitude
Damage to the myelin sheath
↓ Saltatory conduction (action potential propagation along myelinated axons)
Neuropraxia (nerve compression blocks conduction)
Interruption in axonal continuity
Axonotmesis (endoneural tube stays intact but myelin & distal axon degenerates)
Recovery possible
Full disruption of myelin, axon & nerve sheath
Neurotmesis (axons no longer have an endoneural tube to guide regrowth)
Recovery impossible
↓ Ability to contract and use abductor pollicis brevis muscle
↓ Signals through median nerve
Interference with signals to the brain causes unusual sensations
Hypoalgesia (↓ pain Dysesthesia (tingling, burning, or sensitivity at 1st 3 1⁄2 digits) painful sensation at 1st 3 1⁄2 digits)
Thenar muscle wasting
Reduced hand dexterity
Weak thumb abduction
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published December 2nd, 2013, updated March 22, 2023 on www.thecalgaryguide.com")
Coronary Artery Bypass Graft CABG Indications
: Indications
Author: Breanne Gordulic Reviewers: Miranda Schmidt Ben Campbell Sunawer Aujla Angela Kealey* * MD at time of publication
Symptomatic multivessel (≥ 3 vessels) coronary artery disease (MVCAD) or complex MVCAD
Acute coronary syndrome (ACS)
Left main coronary artery disease
Multivessel (≥ 3 vessels) CAD and diabetes
Cardiac surgery required for other pathology
Multivessel CAD, LV dysfunction and congestive heart failure (CHF)
Complex CAD includes stenosed vein grafts, bifurcation lesions, calcified lesions, total occlusions, ostial lesions
STEMI initial treatment is PCI/thrombolysis
CABG outcomes compared to percutaneous coronary intervention (PCI) in MVCAD include ↑ survival in diabetes, ↑ survival with LV dysfunction, ↓ repeat revascularization, ↓ myocardial infarction, ↓ stroke
↑ Risk of failure in complex CAD with PCI
Rapid reperfusion to myocardium most important in STEMI to decrease myocardial damage
CABG can be considered for residual stenoses 6-8 weeks later
NSTEMI or unstable angina (UA) with MVCAD involving at least three vessels including the proximal left anterior descending (LAD)
Left main coronary artery divides into left anterior descending (LAD) and left circumflex (LCx) which supplies 2/3 of myocardium
↑ Survival Myocardial infarction from left main artery occlusion Death
Left main stenosis
Ventricular dysrhythmias
Ongoing ischemia
LV dysfunction Hemodynamic instability
↑ risk of PCI
CABG has mortality benefit
↓ Number of operations
↑ Risk of cardiovascular disease in diabetes
Revascularization indicated along with other cardiac surgery
Multivessel CAD with >90% stenosis and CHF
LV ejection fraction <35%
↑ Risk of atherosclerosis from hyperglycemia and dyslipidemia
CABG bypasses several atherosclerotic plaques in coronary arteries
↑ Durability
↑ Complete perfusion
Valve stenosis or regurgitation Septal defect
Aortic root or arch pathology
Combination procedure
Evidence of ischemia at rest
Evidence of
impaired LV function at rest
↓ All-cause mortality in CABG vs medical management
Chronic obstructive pulmonary disease
Abbreviations:
• ACS- acute coronary syndrome. Acute reduction in
blood flow to heart muscle resulting in cell death. • CAD- coronary artery disease. Narrowing or blockage of the coronary arteries by plaque
• NSTEMI- myocardial infarction (heart attack) with no ST elevation on electrocardiogram
• PCI- percutaneous coronary intervention. A balloon tipped catheter is used to open blocked coronary arteries; a stent may be placed.
• STEMI- myocardial infarction with ST segment elevation on electrocardiogram
Consider revascularization (restore blood flow to blocked or narrowed blood vessels) of coronary arteries to increase perfusion to myocardium (heart muscle)
Coronary artery bypass graft recommended
Assess surgical risk and comorbidities with evaluation by heart team that includes both a cardiac surgeon and interventional cardiologist (SYNTAX Trial)
Individual management plan for patients with comorbidities that increase mortality
Frailty
Chronic Renal Failure
↑ Inflammation and deregulated angiogenesis affects all organ systems
↓ Physiologic reserve and ↓ ability to recover from acute stress
↑ Pneumonia
↑ Respiratory and Renal Failure
↑ Stroke
↑ In hospital mortality
↓ Survival two years after surgery
Use Society of Thoracic Surgeons Score, EuroSCORE, or SYNTAX II Score to predict patient outcome with anatomy, disease severity, and preoperative characteristics
Coronary Artery Bypass Graft
Surgery to take healthy blood vessels from the body and connect them proximally and distally to blocked coronary arteries
Cardiopulmonary bypass, fluid overload, ↑ renal vasoconstriction and ↓ renal oxygenation from rewarming
Kidney injury
↑ End stage kidney disease
Blood flow restored to
ischemic myocardium
↓ Angina
↑ Quality of life ↑ LV function ↑ Survival
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published April 12, 2023 on www.thecalgaryguide.com")
Diabetic Retinopathy

Ethnicity: White youth, African American, Hispanic, Asian-Pacific Islanders, and American Indigenous
Type 1 Diabetes Mellitus (T1DM) (see Diabetes Mellitus, Type I for pathogenesis)
Type 2 Diabetes Mellitus (T2DM) (see Diabetes Mellitus, Type II for pathogenesis)
Polycystic Ovarian Syndrome
Family history of T2DM
History of Gestational Diabetes
↑ Body Mass Index
↑ Age
Ethnicity: Indigenous Americans, African American, Hispanic
Poor glycemic control
Chronic high blood sugaràinflammatory response
↑ cytokines and growth factors including vascular endothelial growth factor (VEGF)
Vascular permeability Retinal neovascularization
Vascular endothelial dysfunction: basement membrane thickening, vascular cell death, vascular occlusion from platelet aggregation
Retinal hypoxia
Outpouchings of the weakened capillary walls or endothelial buds attempting to re-vascularize the ischemic retina
Weakened blood-retinal barrier (BRB) allows for rupture into the deeper retinal layers
Lipid deposits with sharp margins due to lipoproteins & other proteins leaking through the damaged blood-retinal barrier
Nerve fiber layer infarcts from occlusions of the precapillary arterioles
Retinal hemorrhages occur in the more superficial nerve layer
Focal areas of saccular venous bulges due to significant retinal ischemia & endothelial wall weakening and damage
Diabetic Retinopathy (DR)
A complication of diabetes due to chronic hyperglycemia resulting in abnormal permeability and ischemia of retinal vessels
Micro-aneurysms
Dot/blot hemorrhages Hard exudates (yellow
opaque solids on retina)
Cotton-wool spots (cloudy translucent patches on retina)
Flame hemorrhages
(multiple opaque red patches)
Venous beading (veins with areas of narrowing forming bead-like segments)
Traction retinal detachment
Bleeding into the vitreous humor
Pericyte death, breakdown of endothelial tight junctions & basement membrane thickening damages blood-retinal barrier
Damaged blood- retinal border leaks fluid into the retinal tissues
Macular edema
(swelling of macula)
Mild Non-proliferative DR
Moderate Non-proliferative DR
Severe Non-proliferative DR
Proliferative DR
Presence of neovascularization
Localized retinal ischemia causes upregulation of vascular endothelial growth factor causing fine, irregular & easily friable neovascularization in the disc, macula, and/or retina
Neovascularization
(fine loop networks of new blood vessels)
Neovascular membranes in vitreous gel form vitreoretinal adhesions and contract
Fragile vessels extending into the vitreous humor
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published June 28, 2017, updated May 18, 2023 on www.thecalgaryguide.com")
Physiology of Anti-diuretic hormone
/Arginine Vasopressin (AVP)
Authors: Manaswi Yerrabattini Reviewers: Parker Lieb Mao Ding Shyla Bharadia Laura Hinz* * MD at time of publication
Limbic activation (e.g., pain, nausea)
Placental production of vasopressinase during pregnancy catalyzes the breakdown of ADH, leading to a temporary Diabetes Insipidus state (see Diabetes Insipidus: Pathogenesis and Clinical Findings slide)
Systemic arteriole vasoconstriction
Hypovolemia/Hypotension
Hyperosmolar state (i.e., extracellular fluid osmolarity above a certain threshold, most commonly due to hypernatremia)
Sensed by osmoreceptors in hypothalamus
Angiotensin II synthesized through activation of Renin- Angiotensin-Aldosterone System (RAAS) (see Physiology of RAAS slide)
Binds to receptors located in the hypothalamus
↓ pressure sensed by baroreceptors in the heart (left atrium and large veins)
Receptors transmit signals to brain via the vagus nerve
↓ Arterial baroreceptor firing
↑ Sympathetic activity of nerves innervating afferent arterioles
↑ Hypothalamic secretion of ADH (peptide hormone), transportation to posterior pituitary, and release from posterior pituitary into blood circulation
Blood Vessels (Minor role)
Kidneys (Main role)
ADH binds to to Vasopressin-2 receptors on basolateral side of principal cells in kidneys
↑ Insertion of aquaporin II channels onto apical membrane of late distal tubule and collecting ducts
↑ Water reabsorption
↓ Urine Output and Maintains narrow range of serum osmolarity ↑ Urine Osmolarity and preserves sodium homeostasis
ADH binds to Vasopressin-1 receptors on smooth muscle of blood vessels
ADH activates calcium signaling pathway
↑ Blood Pressure
Maintains overall fluid volume status
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published May 20, 2023 on www.thecalgaryguide.com")
Knee Osteoarthritis

Articular trauma
Inflammatory disease or infection (e.g. Rheumatoid or septic arthritis)
Obesity and ↑leptin ↑ Chondrocytes,
inflammatory mediators, and metalloproteinases
Extracellular matrix degradation
↑ Knee joint loading forces
Metabolic syndromes (e.g. diabetes mellitus)
↑ Oxidative stress and insulin resistance
Low-grade systemic inflammation
↓ Elasticity and ↑ degradation of cartilage
↑ Friction in knee joint with movement
↓ Cartilage along femoral groove and posterior surface of patella
Pain, catching, and crepitus (crackling/clicking sound) in the patellofemoral joint
Inability/difficulty with kneeling or climbing stairs
Abnormal distribution of forces accumulate and stress articular surface
↑ Damage/laxity to soft tissue structures stabilizing knee joint
Knee Osteoarthritis
(Multifactorial entity characterized by cartilage breakdown, deterioration of connective tissue, and bone deformities)
↓ Cartilage between distal femur and proximal tibia ↓ Joint spaceàto articular dysfunction
Radiographic changes
See Osteoarthritis (OA): X-Ray Features slide
Repeated attempts to repair cartilage and joint disruption
Subchondral bone thickening (sclerosis) under joint cartilage and bone spur (osteophyte) formation around joint line
Rotational/antero-posterior instability and ↑ external adduction moments during walking
Alterations in proteoglycans, fiber arrangement, and collagen composition in soft tissue structures within/around knee joint
↑ Shear forces and medial compartment narrowing erode and pinch soft tissue structures within the knee joint
Cruciate ligament degeneration
Weakened passive stabilizers of the knee joint
Knee giving way and instability (falls)
Meniscal tears, if large àprevents knee extension/flexion
Locking of the knee
Joint line tenderness:
Patient points to area of tenderness/pain reproducible upon palpation
Anatomical axis of hip, knee, and ankle joints ↑ loading medially
Medial > lateral joint line tenderness
↑ Joint friction activating nociceptors in the surrounding anatomical tissues
Injury and inflammation ↑ nociceptive responses in soft tissue structures and subchondral bone within knee joint
Nociceptive feedback to brain inhibits activity of motor cortex neurons controlling muscles around the knee
↓ Motor output and muscle activation over time
↓ Muscle strength/endurance, lower limb muscle use, functional ability (walking, stairs, etc.)
Joint inflammationà accumulation of fluid within joint
Stiffness, swelling, redness, and pain
Limited joint space reduces range of motion for femur to roll/slide on tibia
↓ Knee flexion and extension
Flexion contracture and antalgic gait
Reduced weight acceptance of the joint and surrounding muscles/tendons
↓ Mobility and physical dysfunction
Muscular atrophy
Reduced function of active stabilizers of the knee joint (quadriceps, adductors, hamstrings)
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published July 30, 2023 on www.thecalgaryguide.com")
Pharmacotherapy for Dyslipidemia Overview

Bile-acid sequestrants
Bind bile acids in intestinal lumen to prevent reabsorption by enterohepatic (gut-liver) circulation
↑ Excretion of bile acids and cholesterol in stool
↓ LDL in blood
Side effects: GI disturbances, commonly interact with other drugs by interfering with absorption
See Calgary guide slide on “Bile-acid sequestrants: Mechanisms of action & side effects” for complete description of mechanism and side effects
(clinical imbalance of lipids)
Hypertriglyceridemia (if VLDL mediated & in need of treatment for pancreatitis prevention)
Hypercholesteremia
(↑ LDL in blood)
Ezetimibe
Inhibits cholesterol absorption via NPC1L1 transporter
↑ Hepatic (liver) LDL receptor expression
↑ LDL clearance from blood
↓ LDL in blood
Avoid in pregnancy
See Calgary guide slide on “Ezetimibe: Mechanisms of action & side effects” for complete description of mechanism and side effects
Combined hyperlipidemia (↑ Triglycerides and ↑ cholesterol)
Statins (ex. rosuvastatin, atorvastatin, simvastatin, pravastatin)
Competitive inhibitors of HMG-CoA reductase (rate-limiting enzyme in cholesterol synthesis)
Fibrates (ex. fenofibrate, gemfibrozil)
Activate PPAR! (nuclear receptor)
↑ Lipolysis (breakdown of lipids) and free fatty acid oxidation
↓ Triglycerides in blood
Side effects: GI discomfort, rash, pruritis
Contraindicated in pregnancy, renal failure, liver & gallbladder disease
See Calgary guide slide on “Fibrates: Mechanisms of action & side effects” for complete description of mechanism and side effects
PCSK9 inhibitors (ex. evolocumab and alirocumab which are monoclonal antibodies)
Inhibit PCSK9 (holds the LDL:LDL receptor complex together as it is internalized into the cell for destruction of LDL)
LDL receptor returns to surface without being destroyed
↑ LDL receptor expression
↑ LDL clearance from blood
↓ LDL in blood
See Calgary guide slide on “PCSK9 Inhibitors: Mechanisms of action & side effects” for complete description of mechanism and side effects
↓ Cholesterol synthesis in liver
↑ LDL receptor expression in liver
LDL receptor recognizes apoB100 (structural protein on LDL) and apoE (structural protein found on chylomicron, VLDL, IDL)
↑ Clearance of LDL cholesterol from bloodstream
↓ LDL cholesterol in blood ↑ HDL in blood
↓ Triglycerides in blood
↓ Atherosclerosis (plaque along walls of blood vessels)
Abbreviations: HDL – High density lipoprotein; HMG-CoA – Hydroxymethylglutaryl- CoA; LDL – Low-density lipoprotein; PCSK9 – Proprotein convertase subtilisin/kexin type 9; PPAR! – Peroxisome proliferator-activated receptor alpha; NPC1L1 – Niemann-Pick C1-Like 1; VLDL – Very low-density lipoprotein
Side effects: Myalgias (muscular aches), rhabdomyolysis (muscle breakdown), transaminitis (liver inflammation), liver failure, ↑ risk of diabetes mellitus
Contraindicated in pregnancy
See Calgary guide slide on “Statins: Mechanisms of action & side effects” for complete description of mechanism and side effects
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published Nov 6, 2023 on www.thecalgaryguide.com")
Non-Alcoholic Fatty Liver Disease
)
Steatohepatitis: chronic inflammatory and apoptotic climate in the hepatocytes (in the absence of alcohol consumption, termed Non-Alcoholic Steatohepatitis (NASH))
Fibrosis of the Liver: excessive scarring of liver tissue resulting from chronic inflammation, although liver architecture is largely intact
Fat droplets form and grow in the hepatocytes
Hepatic mitochondria increase their workload in attempt to break down the excess free fatty acids through beta-oxidation
↑ in cellular workload creates more reactive oxygen speciesà Inflammation and apoptosis of hepatocytes
On-going inflammation damages hepatic stellate cells (the primary extracellular matrix–producing cells of the liver) causing the release of fibrinogenic cytokines
Cirrhosis of the liver: normal lobular structure distorts and is replaced by regenerating nodules and bridging septa, disrupting normal liver blood flow
Deposition of fibrotic
material and collagen within the perisinusoidal spaces of the liver
Decompensated Cirrhosis Hepatocellular carcinoma
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published November 25, 2023 on www.thecalgaryguide.com")
Gestational Diabetes Risk factors and pathogenesis

Authors:
Amyna Fidai
Maharshi Gandhi Reviewers:
Laura Byford-Richardson Shahab Marzoughi
Yan Yu*
Hanan Bassyouni*
* MD at time of publication
High risk population (Aboriginal, Hispanic, South Asian, Asian, African)
Previous or current macrosomia (>4000g) or polyhydramnios
Other conditions associated with Diabetes Mellitus such as polycystic ovarian syndrome, hypertension, metabolic syndrome
Placental counter regulatory hormones (particularly Human Placental Growth Hormone) oppose the action of insulin
↑ Insulin resistance (liver, muscle, adipose tissues become less responsive to insulin)
↑ Fetal demands after 18 weeks gestation
(fetus requires 80% of its energy from maternal glucose)
↑ Carbohydrate intake to keep up with the demands
Previous history of gestational diabetes or glucose intolerance
Family history of diabetes
Advanced maternal age
Obesity
Previous unexplained stillbirth
Multiples (larger placental mass and activity)
Corticosteroid use
↑ Risk for pregnancy induced glucose intolerance (mechanism unclear and/or complex)
↑ Insulin requirements Initially pancreatic beta cells work
overtime to keep up with the ↑ insulin demands
Eventually insulin demands are not met due to the exhaustion of pancreatic beta cells
Plasma glucose rises (Fasting plasma glucose ≥5.3 mmol/L)
Gestational Diabetes (or exacerbation of pre-existing DM)
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published Jan 28, 2017, updated Feb 24, 2024 on www.thecalgaryguide.com")
Onychomycosis

Proximal Subungual
White Superficial
Tinea infection (e.g. Tinea Pedis, Corporis, Capitis)
Infection spreads to distal hyponychial space
Dermatophytes colonize local tissue in nail plate and nail bed
Dermatophytes feed on keratinized tissue
General Symptoms (All Subtypes)
Spongiosis (Intercellular edema)
Acanthosis (Thickening of stratum spinosum layer of epidermis)
Hyperkeratosis (Thickening of stratum corneum In effort to rid infection)
Papillomatosis (Projections of dermal papillae)
Secondary damage to nail matrix
Loss of nail
Keratinocytes produce an acute, low-grade inflammatory cytokine response
Onychomycosis
Dermatophytic infection of the nail bed
Inflammation promotes ↑ fluid to tissues for ↑ immune cell delivery
Widespread inflammation thickens parts of the epidermis in efforts to shed the infection
Inflammation and epidermal hyperplasia (↑ growth of cells) influence local dermal papillae (group of cells just beneath the hair follicle) to proliferate and project above the skin
Distal Subungual
Superinfecting bacteria or other fungi proliferate beneath the compromised nail imparting a yellowish appearance
Distal Subungual Subtype
(Thick yellow nails, keratin and debris accumulate distally underneath nail plate)
Dermatophytes invade the proximal end of the nail plate
Dermatophytes penetrate through the cuticle to the newly forming nail plate moving distally
Proximal Subungual Subtype (Whitish discolouration of nail plate that begins proximally and moves distally, indicative of immunosuppression)
Fungi predominantly invade various areas of the superficial nail plate layers eventually joining together
White Superficial Subtype (Chalky white scale that spreads slowly beneath nail plate, well-defined “white islands” that coalesce as disease progresses)
The entirety of the nail plate is infected by the dermatophytes
Widespread inflammation thickens the nail plate as well as beneath the nail (subungual hyperkeratosis) in efforts to shed the infection
Total Dystrophic Subtype (End-stage nail disease, entire nail becomes thick and dystrophic)
Local spread of infection Dermatophytes spread causing cracks in the skin deeper into toe
Abnormal keratinization in hyponychium
Keratin accumulates between nail plate and hyponychium
Fissure (splits in the skin)
Bacteria enters lymphatics and bloodstream
Cellulitis Sepsis
Onycholysis (nail plate separates from nail bed)
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published Mar 13, 2024 on www.thecalgaryguide.com
Dermatophyte Onychomycosis: Pathogenesis and clinical findings
Authors: Holly Zahary Loreman Reviewers: Mina Youakim Elise Hansen Shahab Marzoughi Jodi Hardin* * MD at time of publication
Host Risk Factors
Environmental Risk Factors
Immuno- compromised
↓ Immune response to infection
Older age
Peripheral vascular disease
Reduced blood circulation
Diabetes
Pre-existing nail dystrophy
Previous nail trauma
Integrity of nail unit is compromised
Micro- traumatic pressure on nail
Dark, warm shoe environment
Optimal conditions for fugal growth
Exposure to tinea pedis or onychomycosis
Direct spread of infection to nail unit
High blood sugar favoring infection
Dermatophytes invade corneocytes on stratum corneum, the uppermost non-living layer of keratinized skin
Compromise/breaking of hyponychial seal or cuticle (connection between hyponychium and nail plate)
Proximal Subungual
White Superficial
Distal Subungual
Superinfecting bacteria or other fungi proliferate beneath the compromised nail imparting a yellowish appearance
Distal Subungual Subtype
(Thick yellow nails, keratin and debris accumulate distally underneath nail plate)
Infection spreads to distal hyponychial space
Dermatophytes colonize local tissue in nail plate and nail bed
Dermatophytes feed on keratinized tissue
Keratinocytes produce an acute, low-grade inflammatory cytokine response
Onychomycosis
Dermatophytic infection of the nail bed
Inflammation promotes ↑ fluid to tissues for ↑ immune cell delivery
Widespread inflammation thickens parts of the epidermis in efforts to shed the infection
Inflammation and epidermal hyperplasia (↑ growth of cells) influence local dermal papillae (group of cells just beneath the hair follicle) to proliferate and project above the skin
General Symptoms (All Subtypes)
Spongiosis (Intercellular edema)
Acanthosis (Thickening of stratum spinosum layer of epidermis)
Hyperkeratosis (Thickening of stratum corneum In effort to rid infection)
Papillomatosis (Projections of dermal papillae)
Secondary damage to nail matrix
Loss of nail
Tinea infection (e.g. Tinea Pedis, Corporis, Capitis)
Dermatophytes invade the proximal end of the nail plate
Dermatophytes penetrate through the cuticle to the newly forming nail plate moving distally
Proximal Subungual Subtype (Whitish discolouration of nail plate that begins proximally and moves distally, indicative of immunosuppression)
Fungi predominantly invade various areas of the superficial nail plate layers eventually joining together
White Superficial Subtype (Chalky white scale that spreads slowly beneath nail plate, well-defined “white islands” that coalesce as disease progresses)
The entirety of the nail plate is infected by the dermatophytes
Widespread inflammation thickens the nail plate as well as beneath the nail (subungual hyperkeratosis) in efforts to shed the infection
Total Dystrophic Subtype (End-stage nail disease, entire nail becomes thick and dystrophic)
Dermatophytes spread deeper into toe
Bacteria enters lymphatics and bloodstream
Abnormal keratinization in hyponychium
Keratin accumulates between nail plate and hyponychium
Local spread of infection causing cracks in the skin
Fissure (splits in the skin)
Cellulitis Sepsis
Onycholysis (nail plate separates from nail bed)
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published MONTH, DAY, YEAR on www.thecalgaryguide.com
Dermatophyte Onychomycosis: Pathogenesis and clinical findings
Authors: Holly Zahary Loreman Reviewers: Mina Youakim Elise Hansen Shahab Marzoughi Jodi Hardin* * MD at time of publication
Host Risk Factors
Environmental Risk Factors
Immuno- Older compromised age
↓ Immune response to infection
Peripheral vascular disease
Reduced blood circulation
Diabetes
Pre-existing nail dystrophy
Previous nail trauma
Integrity of nail unit is compromised
Micro- traumatic pressure on nail
Dark, warm shoe environment
Optimal conditions for fugal growth
Exposure to tinea pedis or onychomycosis
Direct spread of infection to nail unit
High blood sugar favoring infection
Dermatophytes invade corneocytes on stratum corneum, the uppermost non-living layer of keratinized skin
Compromise/breaking of hyponychial seal or cuticle (connection between hyponychium and nail plate)
Tinea infection (e.g. Tinea Pedis, Corporis, Capitis)
Infection spreads to distal hyponychial space
Dermatophytes colonize local tissue in nail plate and nail bed
Dermatophytes feed on keratinized tissue
Keratinocytes produce an acute, low-grade inflammatory cytokine response
Onychomycosis
Dermatophytic infection of the nail bed
General Symptoms (All Subtypes)
Spongiosis (Intercellular edema)
Acanthosis (Thickening of stratum spinosum layer of epidermis)
Papillomatosis
(Projections of dermal papillae)
Hyperkeratosis (Thickening of stratum corneum In effort to rid infection)
Secondary damage to nail matrix
Loss of nail
Proximal Subungual
White Superficial
Distal Subungual
Distal Subungual Subtype
(Thick yellow nails, keratin and debris accumulate distally underneath nail plate)
Proximal Subungual Subtype (Whitish discolouration of nail plate that begins proximally and moves distally, indicative of immunosuppression)
White Superficial Subtype (Chalky white scale that spreads slowly beneath nail plate, well-defined “white islands” that coalesce as disease progresses)
Total Dystrophic Subtype (End-stage nail disease, entire nail becomes thick and dystrophic)
Local spread of infection causing cracks in the skin
Dermatophytes spread deeper into toe
Abnormal keratinization in hyponychium
Keratin accumulates between nail plate and hyponychium
Onycholysis (nail plate separates from nail bed)
Fissure (splits in the skin)
Bacteria enters lymphatics and bloodstream
Cellulitis Sepsis
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Legend:
Published MONTH, DAY, YEAR on www.thecalgaryguide.com
Dermatophyte Onychomycosis: Pathogenesis and clinical findings
Authors: Holly Zahary Loreman Reviewers: Mina Youakim Elise Hansen Shahab Marzoughi Jodi Hardin* * MD at time of publication
Host Risk Factors
Environmental Risk Factors
Immuno- Older compromised age
↓ Immune response to infection
Peripheral vascular disease
Reduced blood circulation
Diabetes
Pre-existing nail dystrophy
Previous nail trauma
Integrity of nail unit is compromised
Micro- traumatic pressure on nail
Dark, warm shoe environment
Optimal conditions for fugal growth
Exposure to tinea pedis or onychomycosis
Direct spread of infection to nail unit
High blood sugar favoring infection
Dermatophytes invade corneocytes on stratum corneum, the uppermost non-living layer of keratinized skin
Compromise/breaking of hyponychial seal or cuticle (connection between hyponychium and nail plate)
Tinea infection (e.g. Tinea Pedis, Corporis, Capitis)
Infection spreads to distal hyponychial space
Dermatophytes colonize local tissue in nail plate and nail bed
Dermatophytes feed on keratinized tissue
Keratinocytes produce an acute, low-grade inflammatory cytokine response
Onychomycosis
Dermatophytic infection of the nail bed
Proximal Subungual
White Superficial
General Symptoms (All Subtypes)
Spongiosis (Intercellular edema)
Acanthosis (Thickening of stratum spinosum layer of epidermis)
Papillomatosis
(Projections of dermal papillae)
Hyperkeratosis (Thickening of stratum corneum In effort to rid infection)
Secondary damage to nail matrix
Loss of nail
Distal Subungual
Distal Subungual Subtype
(Thick yellow nails, keratin and debris accumulate distally underneath nail plate)
Proximal Subungual Subtype (Whitish discolouration of nail plate that begins proximally and moves distally, indicative of immunosuppression)
White Superficial Subtype (Chalky white scale that spreads slowly beneath nail plate, well- defined “white islands” that coalesce as disease progresses)
Total Dystrophic Subtype (End-stage nail disease, entire nail becomes thick and dystrophic)
Local spread of infection causing cracks in the skin
Dermatophytes spread deeper into toe
Abnormal keratinization in hyponychium
Keratin accumulates between nail plate and hyponychium
Onycholysis (nail plate separates from nail bed)
Bacteria enters lymphatics and bloodstream
Fissure (splits in the skin)
Cellulitis
Sepsis
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Legend:
Published MONTH, DAY, YEAR on www.thecalgaryguide.com
Dermatophyte Onychomycosis (Tinea Unguium): Pathogenesis, clinical findings,
Authors: Holly Zahary Loreman Reviewers: Elise Hansen Name Name* * MD at time of publication
and complications
Host Risk Factors
Environmental Risk Factors
Immuno- compromised
↓ immune response to infection
Older age
Peripheral vascular disease
Diabetes
Pre-existing nail dystrophy
Previous Nail Trauma
Integrity of nail unit is compromised
Micro-traumatic pressure on nail
Dark, warm shoe environment
Optimal conditions for fugal growth
Exposure to tinea pedis or onychomycosis
Direct spread of infection to nail unit
Reduced blood circulation
High blood sugar, favoring infection
Tinea pedis infection (see ‘Tinea Capitis, Tinea Corporis, and Tinea Pedis’)
Infection spreads to distal hyponychial space Dermatophytes colonize local tissue in nail plate and nail bed Dermatophytes feed on keratinized tissue
Proximal Subungual
White Superficial
Dermatophytes invade corneocytes on stratum corneum, the uppermost non-living layer of keratinized skin
Compromise/breaking of hyponychial seal or cuticle (connection between hyponychium and nail plate)
Keratinocytes produce an acute, low-grade inflammatory cytokine response
Onychomycosis (Tinea Unguium)
(dermatophytic infection of the nail bed)
Distal Subungual
General Symptoms (All Subtypes)
Spongiosis
Intercellular edema
Acanthosis
Thickening of stratum spinosum layer of epidermis
Papillomatosis
Projections of dermal papillae
Hyperkeratosis
Thickening of stratum corneum In effort to rid infection
Secondary damage to nail matrix
Loss of nail
Distal Subungual Subtype
Thick yellow nails, keratin and debris accumulate distally underneath nail plate
Proximal Subungual Subtype
Whitish discolouration of nail plate that begins proximally and moves distally, indicative of immunosuppression
White Superficial Subtype
Chalky white scale that spreads slowly beneath nail plate, well-defined “white islands” that coalesce as disease progresses
Total Dystrophic Subtype End-stage nail disease, entire nail becomes thick and dystrophic
Local spread of infection causing cracks in the skin
Dermatophytes spread deeper into toe
Abnormal keratinization in hyponychium
Keratin accumulates between nail plate and hyponychium
Onycholysis (nail plate separates from nail bed)
Tissue Damage
Cellulitis
Sepsis
Bacteria enters lymphatics and bloodstream
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Legend:
Published MONTH, DAY, YEAR on www.thecalgaryguide.com")
Macrosomia Pathogenesis and Complications

Genes encoding cellular growth are mutated and induce ↑ cell proliferation
Fetus with XY chromosomes
Y chromosome predisposes fetus to excess growth factor
Pregnancy longer than 42 weeks
Birth weight ↑ as gestational age ↑
Pregnant parent with BMI > 30 kg/m2
↑ Central adipose tissue release insulin- desensitizing factors
↑ Insulin resistance promotes hepatic glucose production
Macrosomia
Parent has previously had ≥ 2 births
Average birth weight ↑ with each successive pregnancy
Pregnant parent with type 2 diabetes or gestational diabetes (1-hour 50 g glucose challenge test >140 mg/dL at 24-28 weeks gestation)
Parent’s glucose-rich blood is carried to the fetus through the placenta
↑ Levels of glucose present in fetal circulation promotes excessive growth
(Fetus grows beyond absolute birth weight (> 4000 g) regardless of gestational age)
Fetal dysregulation of glucose and fetal programming of later adiposity
Metabolic syndromes (e.g. hypoglycemia, hyperinsulinemia)
↑ Insulin levels delay pulmonary maturation
Respiratory distress
Large fetal size in the uterus
Cardiac mass ↑ in proportion to body size
Fetal cardiac remodeling (e.g. ↑ left ventricular mass)
Uterine muscle wall stretched beyond optimal range
Uterine rupture
Parent pushes fetus into birth canal
Maternal nutrition
supply is unable to meet fetus’ increased metabolic demands
↑ Uterine distension prevents uterine muscles from contracting (uterine atony)
Fetus takes longer to descend through the birth canal
Large fetal size overstretches pelvic structures
Perianal trauma (e.g. lacerations to pelvic floor, vagina, rectum)
Less space in birth canal prevents the parent from delivering the anterior fetal shoulder after the fetal head
Shoulder dystocia (baby’s shoulder stuck during birth)
Arrested labour (slow cervical dilation)
Insufficient space in birth canal to deliver fetus
Assisted vaginal birth/ cesarian section
Protracted labour (slow fetal descent)
Stillbirth
Fetal distress (↓ heart rate)
Lack of mechanical contraction of the spiral arteries, normally provided by uterine muscles
Blood loss (≥ 500-1000 mL 24 hours post birth)
Postpartum hemorrhage
Authors:
Akaya Blair
Reviewers:
Dasha Mori
Michelle J. Chen
Dr. Ian Mitchell*
* MD at time of publication
↑ Frequency/ prolonged admission (≥ 3 days) to neonatal intensive care unit
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published Mar 21, 2024 on www.thecalgaryguide.com")
Operative Vaginal Delivery Indications
: Indications
Authors: Taylor Pigott Akaya Blair Reviewers: Michelle J. Chen Sylvie Bowden* Stephanie Cooper* Sarah Glaze* * MD at time of publication
Abnormal fetal heart rate/fetal distress
↓ Risk of maternal and fetal morbidity/ morality
Rupture of membranes without fetal head adequately applied to maternal pelvis
Small pelvic outlet Narrow vaginal canal
Cardiovascular disease
Diabetes Hypertension Abdominal trauma
Short umbilical cord
Large for gestational age (LGA) fetus
Neurological/ muscular disease
↑ Pain during labor
Maternal contraindication to Valsalva maneuver (e.g., cardiac disease, cystic lung)
Rush of fluid past the fetal head out through cervix
Umbilical cord prolapse
Umbilical vasoconstriction
Umbilical cord compression
↓ Blood flow through umbilical cord
Fetal presenting part applies pressure on umbilical cord
Impaired vascular function and narrowed vessels ↓ blood flow around the body
↓ Oxygen transfer across the placenta
Fetal oxygen deprivation and hypoxia
Operative vaginal delivery with forceps or vacuum expedites delivery
Placenta partially or completely separates from uterine wall (abruption)
Increased tension of umbilical cord on placenta
Fetal shoulder is lodged behind maternal pubic symphysis
Delivery of the body is delayed
Inability to adequately bear down to push
Myometrium runs out of energy from repeated contractions and becomes less able to contract (↓ uterine tone)
Uterine spiral arteries dependent on contractions for vasoconstriction remain dilated, allowing blood flow
Postpartum hemorrhage
Uterine rupture
↑ Risk of future pelvic organ prolapse
↑ Risk of future incontinence
Prolonged labour
Repeated contraction/ relaxation of the uterus without progress tears and damages uterine muscles
↓ Maternal endurance
Inadequate fluid and food intake prior to/ during labor
Multiples
↑ Sympathetic nervous system activation during labor
Maternal exhaustion
Uterine muscles stretch to accommodate multiple growing fetuses
↑ Pushing against pelvic floor and perineum
Pushing force damages pelvic floor muscles and nerves
↓ Myometrial contractility
↑ Need for vaginal exams to check on labour progress
Bacterial overgrowth in vagina and/or uterus
Maternal infection
Fetal infection
↓ Strength of contractions
Epidural anesthetic
Inability to ambulate to urinate
↑ Urinary catheterizations
↑ Epinephrine secretion from adrenal cortex
Blood is shunted away from uterus/internal organs and towards heart and skeletal muscle
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published 03, ##, 2023 on www.thecalgaryguide.com")
Diabetes Mellitus Pathophysiology Behind Lab Findings

↓ Glucose uptake into cells
Autoimmune destruction of pancreatic beta cells (type 1 diabetes mellitus)
Insulin resistance (type 2 diabetes mellitus)
Other causes of diabetes mellitus (genetic, drug- induced, pregnancy, etc.)
↑ Lipolysis (fat breakdown) in adipose tissue
↑ Free fatty acids & glycerol in bloodstream
↑ Oxidation of fatty acids in liver to form acetyl CoA
↑ Conversion of acetyl CoA to ketone bodies (ketogenesis) to be used as fuel for the brain
Relative or absolute insulin deficiency
↓ Activation of adipose (fat tissue) & muscle cell transmembrane insulin receptors
↓ Activation of intracellular insulin signaling pathways (PI3K & MAP kinase)
Cells cannot use glucose as a source of energy; thus body reacts as if it is starving
↑ Protein breakdown in muscle tissue ↑ Amino acids & lactate in bloodstream
↑ Substrates (glycerol, amino acids & lactate) available to produce glucose in the liver
↑ Glycogenolysis (breakdown of stored glucose - glycogen) & gluconeogenesis (production of glucose) in liver
Glucose in bloodstream glycates (coats) hemoglobin in red blood cells; thus ↑ blood glucose leads to ↑ glycated hemoglobin
↑ Hemoglobin A1C
↑ Fasting blood glucose level ↑ Random blood glucose level
↑ Blood glucose following oral glucose tolerance test
Diabetic ketoacidosis (DKA) in absolute insulin deficiency *See DKA slide
Filtered ketone bodies exceed the reabsorption capacity of renal tubules
Ketonuria (↑ ketones in urine)
↑ Glucose in bloodstream
Build-up of glycation end products causes damage to glomerular tissue
Transient glomerular hyperfiltration followed by long-term ↓ in glomerular filtration rate
Albuminuria (↑ albumin in urine)
Diabetic nephropathy *See slide
Filtered glucose exceeds reabsorption capacity of renal tubules in the kidney
Glucosuria (↑ glucose in urine)
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published July 8, 2024 on www.thecalgaryguide.com")
Neonatal Hypoglycemia Pathogenesis
, glycogenolysis (breakdown of glycogen into glucose), and fatty acid oxidation (conversation of fatty acids to ATP)
Low glucose levels stimulate the neonate’s appetite, allowing them to adapt to intermittent feeds
Infant feeding regularly on a diet with sufficient carbohydrates creates a consistent plasma glucose concentration
Pregnant Parent Causes
Impaired Glucose Production
Inadequate Glucose Supply
Pregnant parent using β-blockers
β-blockers prevent epinephrine from binding to adrenergic receptors in skeletal muscle and liver
Blocked sympathetic signaling in these organs prevents glycogenolysis
↓ Breakdown of glycogen into glucose
Infant of a parent with diabetes
Parent has a high level of glucose in their blood
The fetus receives a glucose-rich blood supply
Fetal pancreatic β cells ↑ insulin production
Post-birth, glucose levels in the infant’s circulation ↓ but insulin levels
remain high
Excessive glucose uptake into cells
Fetus born with a metabolism disorder (e.g. organic amino acid disorder, disorder of glycogen metabolism, disorder of gluconeogenesis)
Genes that make proteins or hormones responsible for production, breakdown and regulation of glycogen are mutated
Fetus born with an endocrine disorder (e.g. congenital adrenal hyperplasia, hypopituitarism, Turner Syndrome)
Pituitary and/or adrenal gland dysfunction ↓ release of hormones that regulate glucose balance
Fetal growth is restricted, fetus is small for gestational age, or is premature (<37 weeks gestation)
Glycogen is deposited during the 3rd trimester of pregnancy. Infants born early have fewer stores; smaller infants born at term have ↓ stores, ↑ insulin sensitivity, & poorly coordinated counter- regulatory hormones
↑ Glucose demand for the transition to life out of the womb
Glycogen stores used up quickly
Perinatal stress (e.g. sepsis, asphyxia)
Stress stimulates fetal adrenal glands to ↑ epinephrine secretion
Fetal pancreatic ⍺- cells ↑ glucagon secretion
↑ Breakdown of muscles and fat for glucose- building blocks
Glycogen stores used up quickly
↑ Glucose usage as fetal metabolic demands ↑ to manage stress
Fetal β-cells secrete inappropriately high levels of insulin despite hypoglycemic state; this hyperinsulinism state can last for months & resolve spontaneously
↓ Serum glucose Neonatal hypoglycemia
Authors: Dasha Mori Reviewers: Michelle J. Chen *Dr. Danielle Nelson *MD at time of publication
Blood glucose < 2.6 mmol/L in term & preterm infants within 72 hours of birth or < 3.3 mmol/L after 72 hours of birth
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published July 19, 2024 on www.thecalgaryguide.com")
Diabete Gestazionale fattori di rischio e patogenesi

Obesity Pathogenesis
 Zeng Reviewers: Gurreet Bhandal, Raafi Ali, Mizuki Lopez
Energy homeostasis dysregulation (Peptide hormones regulating hunger/satiety that influence the hypothalamic control of eating behaviours)
Luiza Radu, Dr. Sam Fineblit*
* MD at time of publication
Endocrine dysregulation
Neurological (↓ caloric reserve activates the neurological system)
Adiposity-related
Hypothalamus (hemostatic area)
↑ Production of ghrelin (peptide hormone)
↑ Activation of agouti-related protein (A Neuropeptide Y (NPY) neuron clusters in the hypothalamus via vagus nerve
↑ Appetite
Mesolimbic area (hedonic area)
Reward Driven Eating (meso-limbic region of brain)
↑ Food consumption
↑ Dopamine & endogenous opioid signals
↑ Pleasure & desire to consume more food
Prefrontal cortex (executive functioning)
↓ Glucagon-like peptide-1 (GLP-1) & pancreatic peptide YY (PYY) secretion post- meal
↓ Stimulation of Proopiomelanocortin (POMC) neuron clusters in hypothalamus
↓ Beta-melanocyte stimulating hormone (MSH) (endogenous peptide) production
Defect in leptin receptor gene
↓ Circulating
soluble leptin receptors (SLR)
↑ Serum leptin levels
Leptin resistance
Inactive leptin gene
↓ Adipocyte leptin production & secretion
↓ Leptin
transport across blood brain barrier
↓ Hypothalamus suppression of hunger
↓ Satiety sensation
↑ Caloric intake & weight gain
↑ Dietary carbohydrate intake
↑ Insulin production & secretion
↓ Cellular response to insulin
↑ Insulin resistance
↑ Insulin production & secretion to compensate for insulin resistance
Impaired glycemic control
Metabolic syndrome, type 2 diabetes mellitus and cardiovascular disease **
Metabolic adaptation to reduced-caloric intake efforts
↑ Body energy conservation
↓ Baseline energy expenditure
↓ Resting metabolic rate (the amount of energy body requires to function while at rest)
↓ Ability to lose weight & fat mass
↑ Hunger
↑ Caloric intake & weight gain
Obesity (A combination of metrics including ↑ BMI, ↑ weight circumference, ↑ waist-to-hip ratio, skinfold thickness, and other standardized measures varied by equipment available at various institutions resulting in a negative impact on the health of the individual)
**See slide on Pathogenesis of Type II Diabetes Mellitus
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published Aug 13, 2024 on www.thecalgaryguide.com")
Transient Tachypnea of the Newborn

↑ Work of breathing
Maternal factors leading to impaired fetal lung development
Lack of labour-induced hormone changes (i.e. cortisol, catecholamines)
↓ Alveolar fluid reabsorption through epithelial aquaporin channels
↑ Alveolar fluid in lungs Disruption of laminar flow in airways
↑ Resistance to airflow
↑ Lung expansion to compensate
Limited surface area for gas exchange in the alveoli
↓ Ventilation
Fluid buildup in the major bronchi in the perihilar region
Prominent perihilar streaking on chest x-ray
↑ Intrathoracic pressure pushes the diaphragm down
Blunting of costophrenic angle on chest x-ray
Hyperinflation of lungs
Expanded lung fields on chest x-ray
↑ Deoxygenated hemoglobin in the bloodstream
Blue/purple discoloration of the skin particularly around the lips & fingers (cyanosis)
↓ Oxygenated hemoglobin in the bloodstream
↑ Nasal passage size allows for more air to enter the lungs with each breath
Nasal flaring
Scalene/intercostal/ sternocleidomastoid /abdominal muscle activation to assist with breathing
Accessory muscle use
↓Oxygen saturation (SpO2)
Neonate takes more breaths to compensate for limited oxygenation
Respiratory rate > 60 breaths/minute (tachypnea)
Transient Tachypnea of the Newborn
Temporary respiratory condition in newborns characterized by impaired lung function and rapid breathing
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published Oct 4, 2024 on www.thecalgaryguide.com")
Neonatal Hypoglycemia Clinical Presentation
 or was born to pregnant person with diabetes
Fetus was exposed to high levels of glucose through placental circulation
Fetus adapted to high blood glucose by producing high amounts of insulin
After birth, placental glucose supply is stopped but insulin remains high
↑ Insulin promotes inappropriately ↑ uptake of glucose into cells
put them at high risk for hypoglycemia
Cells cannot produce enough ATP from glucose to power physiological functions
Immature nervous system, use of fatty acids or proteins as an alternate ATP source, or adaptation to low glucose in utero can mask symptoms of hypoglycemia
Asymptomatic hypoglycemia
Neonates with low blood glucose often don’t show any symptoms and are detected by screening infants who are at a high risk for hypoglycemia
min are at risk for hypoglycemia
Infant is small for gestational age or experienced fetal growth restriction
Smaller neonates have smaller glycogen stores
Preterm infants born < 37 weeks
Glycogen is stored during the 3rd trimester; preterm infants did not have opportunity to build up stores
Loss of glucose source from placental circulation after birth puts preterm neonate at risk for low blood sugar
Body’s sympathetic system detects low glucose and triggers physical symptoms (neurogenic symptoms)
Symptomatic hypoglycemia
Neurons in brain are unable to produce enough ATP and thus function properly, triggering symptoms related to low sugar in the CNS (neuroglycopenic symptoms)
Non-specific symptoms are not exclusive to hypoglycemia and warrant further investigation to exclude other differential diagnosis (sepsis, inborn errors of metabolism, neonatal abstinence syndrome, neonatal encephalopathy, perinatal asphyxiation)
Jitteriness or tremors
Sweating Irritability Tachypnea Pallor
Pathologic poor feeding
Weak or a high- pitched cry
Change in level of consciousness (lethargy or coma)
Seizures
Pathologic hypotonia for gestational age
Apnea Bradycardia Cyanosis Hypothermia
Neonates with perinatal stress (e.g. birth asphyxia, meconium aspiration)
Body uses more glucose to produce ATP to manage condition causing physiological stress
Glucose stores are used up more quickly
Born to pregnant person with beta-blocker use
Body in stress releases epinephrine to promote sympathetic responses including glycogen breakdown into glucose
Beta blockers from placental circulation prevent epinephrine from binding to its receptor in infant’s body
Neonatal Hypoglycemia: Asymptomatic and symptomatic hypoglycemia satisfy the criteria of blood glucose < 2.6 mmol/L in both term and preterm infants within 72 hours of birth, and after 72 hours of age glucose < 3.3 mmol/L
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published Oct 4, 2024 on www.thecalgaryguide.com")
Rapid sequence induction and intubation
: Indications & considerations
“Full stomach”: ↑ risk of regurgitation, vomiting, aspiration Life-threatening injury or illness requiring immediate or rapid airway control
↓ Gastro- esophageal sphincter competence (elderly, pregnancy, hiatus hernia, obesity)
↑ Intragastric pressure (pregnancy, obesity, bowel obstruction, large abdominal tumors)
Delayed gastric emptying (narcotics, anticholinergics, pregnancy, renal failure, diabetes)
↓ Level of consciousness (drug/alcohol overdose, head injury, trauma or shock state)
Respiratory & ventilatory compromise (i.e., hypoxic or hypercapnic respiratory failure)
Achalasia (esophageal motility disorder resulting in impaired swallowing)
Dynamically deteriorating clinical situation (i.e., trauma)
GI bleed
Impaired airway reflexes
↓ Muscle tone of structures in the airway (i.e., tongue, pharyngeal walls, & soft palate)
Patients who did not stop GLP-1 agonist preoperatively as advised
Impaired clearance of secretions or vomitus
↓ Safe apnea time before hemodynamic decompensation
Unprotected airway
Need for rapidly securing airway while avoiding aspiration & hemodynamic compromise
Rapid sequence intubation (RSI): Simultaneous administration of induction agent (unconsciousness) & neuromuscular blocking agent (paralysis) to achieve intubation conditions (~45-60 seconds after IV push) for rapid control of an emergency airway
Preoxygenation
Deranged physiologic conditions (i.e., hypotension, acidosis, hypoxemia)
Reduced tolerance for
apnea (period with no ventilation or oxygenation)
Pre-oxygenate with high flow O2 (15L) to create a large pulmonary & tissue reservoir of oxygen
↓ Significant oxygen desaturation during apnea
↑ Oxygen saturation on pulse oximetry
Induction
Laryngoscopy & intubation are a potent sympathetic nervous system stimulus
Airway manipulation causes a surge in catecholamines
Paralysis
Visualization & passage of endotracheal tube requires relaxation of vocal cords & surrounding muscles
Neuromuscular blocking agents facilitate paralysis
Rescue
Some induction agents (i.e., propofol) are vasodilators
Hemodynamically unstable or patients in shock
Hypotension
Tachycardia
↑ Intracranial pressure (ICP)
Hypertension
Suppress cough & gag reflex
Prevent laryngospasm (involuntary closure of vocal cords to airway manipulation)
Minimize movement during procedure
Vasoactive agents (i.e., ephedrine, phenylephrine) ↑ systemic vascular resistance
Atropine & glycopyrrolate ↑ heart rate
Lidocaine (Na+ channel blocker) & opioids (μ receptor agonist) ↓ transmission of pain
↓ Sympathetic response, myocardial demand & physiologic stress
Anesthetics (i.e., propofol) achieve unconsciousness for paralysis & intubation
↓ Airway trauma & damage to vocal cords
Bag mask ventilation typically avoided in this step to ↓ gastric insufflation & risk of aspiration
Cricoid pressure (Sellick maneuver): posterior displacement of cricoid ring to compress esophagus against C-spine to prevent passive regurgitation of gastric contents to airway. Applied from start of induction, released when placement of endotracheal tube is confirmed by capnography.
Intubation
↑ Blood pressure and/or cardiac output
Authors: Jen Guo Reviewers: Priyanka Grewa Luiza Radu Leyla Baghirzada* * MD at time of publication
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published November 18, 2024 on www.thecalgaryguide.com")
Pre-Eclampsia Pathogenesis

Primigravida (1st pregnancy)
Advanced maternal age
Genetic factors (e.g. sFLT1)
Collagen vascular disease
Previous preeclamptic pregnancies
Multiple gestation
Other unclear causes
Dysregulated immune and non-immune decidual cells (specialized endometrial cells) promote abnormal placental trophoblast invasion and uterine spiral artery formation (exact mechanism unclear)
Multiple unclear, complicated mechanisms
Abnormal association of placental vasculature with uterine vasculature early in pregnancy (utero-placental mismatch) disrupts adequate blood perfusion from placenta to fetus (< 20 weeks gestation or early-onset)
Fetus experiences under- perfusion, hypoxia, ischemia, and oxidative stress
Placental cells release molecules toxic to vascular endothelium into the maternal circulation
Damaged maternal blood vessels are less able to perfuse the placenta
Systemic dysfunction of maternal blood vessel endothelium (endothelial cell activation) (>20 weeks gestation or late-onset)
Inflammatory cells retain memory postpartum which increases risk for developing preeclampsia in subsequent pregnancies
Hypoxia leads to placental cell death, which results in release of fetal antigens
Authors:
Yan Yu
Jasmine Nguyen Reviewers:
Kayla Nelson
Radhmila Parmar
Sean Spence
Monica Kidd*
Katrina Krakowski* Maryam Nasr-Esfahani* Riya Prajapati
Michelle J. Chen
Jadine Paw*
* MD at time of publication
Placental ischemia & injury
Pre-existing maternal diseases damage endothelial cells (e.g. vasculitis, diabetes, renal diseases, chronic hypertension)
Placenta is unable to supply sufficient O2 and nutrients to meet demands of growing fetus
Maternal Clinical Findings and Complications**
Fetal Clinical
Findings and Complications**
**See corresponding Calgary Guide slides
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published Feb 10, 2017; updated Nov 22, 2024 on www.thecalgaryguide.com")
Polycystic Ovarian Syndrome
: Pathogenesis and clinical findings
Genetic Susceptibility:
↑ Expression of LH receptors in granulosa cells, and anterior pituitary ↑ production of luteinizing hormone (LH)
↑ Serum LH compared to FSHà higher levels of LH increasingly activate thecal cells
Excess nutrients (from overeating and sedentary behavior) is stored as visceral fat
↑ Central adiposity (accumulation of fat around abdominal area)
Adipose tissue ↑ secretion of estrogen, inflammatory mediators, adipokines, free fatty acids
̄ Hepatic synthesis of sex hormone-binding globulin
Acne
Hair loss on scalp, and less commonly eyebrows and eyelashes (alopecia)
Excessive hair growth around mouth, chin, chest, abdomen, upper arms, thighs, and upper & lower back (hirsutism)
Metabolic syndrome develops, including ↑ insulin resistance, obesity, dyslipidemia, liver disease, & cardiovascular disease
Theca cells in ovaries ↑ androgen secretion
↑ Serum androgens
State of hyperandrogenism
Androgen and estrogen levels are elevated too early in menstrual cycle (estrogen has negative feedback inhibition on anterior pituitary hormone production)
Early suppression of FSH Limited proliferation of follicles
Relative reduction in secretion of progesterone Imbalance between progesterone and estrogen
Unpredictable ovulation
↑ Circulating insulin- like growth factor
Growth factor promote keratinocyte and dermal fibroblast proliferation
Velvety, darkening of the skin fold areas such as back of neck, armpit area, groin (acanthosis nigricans)
↑ Risk of type 2 diabetes mellitus
Since granulosa cells normally convert testosterone to estrogen, ̄ granulosa cell function means more testosterone exists
Since follicle stimulating hormone (FSH) activates granulosa cells, ̄ FSH means ̄ granulosa cell function.
Follicles arrested in development accumulate fluid, becoming cysts
Polycystic ovaries visible on ultrasound
Infertility
Unpredictable uterine bleeding
↑ Risk of endometrial hyperplasia & endometrial cancer
Irregular menstruation (amenorrhea/oligomenorrhea)
Authors: Lauren Standerwick Claire Song Reviewers: Mackenzie Grisdale Amy Fowler Christina Schweitzer Michelle J. Chen Dr. Yan Yu* Dr. Bernard Corenblum* Dr. Sylvie Bowden* * MD at time of publication
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published Nov 20, 2017; updated Dec 15, 2024 on www.thecalgaryguide.com")
Pituitary Tumour Classification and Clinical Outcomes
 (lack of hormone-producing cells or due to gene mutations)
Authors: Chris Oleynick Caroline Kokorudz Reviewers: Amyna Fidai Laura Byford- Richardson Joseph Tropiano Julia Gospodinov Luiza Radu Hanan Bassyouni* * MD at time of publication
hormones (70%)
Relative abundance & predisposition for lactotroph (prolactin-producing), somatotroph (growth hormone – producing), thyrotroph (TSH producing) & corticotroph (adrenocorticotropic hormone – producing) cells to form adenomas (epithelial cell tumours)
↑ Lactotroph cells
↑ Prolactin (PRL) (most common)
Hyper- prolactinemia (high prolactin blood levels)
↑ Somatotroph cells
↑ Growth hormone (GH)
Acromegaly* (excess tissue growth & metabolic dysfunction)
↑ Corticotroph cells
↑ Adrenocortico- tropic hormone (ACTH)
Cushing disease* (prolonged exposure to excess cortisol)
↑ Thyrotroph cells
↑ Thyroid stimulating hormone (TSH)
Central hyper- thyroidism (↑ T4 & T3)
Large size (macro) (>10mm on MRI) may cause mass effect (tissue compression from mass)
Small size (micro) (<10mm on MRI) unlikely to cause mass effect (compression to surrounding structures)
Asymptomatic
Headache
Nausea & vomiting
Compresses pituitary gland & impairs blood supply & function of pituitary cells
Impinges pituitary stalk & disrupts hormone transport from hypothalamus to the anterior and posterior pituitary
Compresses surrounding structures
↑ Pressure & stretching of dural mater
Stretching of the meninges activates mechanoreceptors (stretch receptors) in GI tract
Pituitary hormone deficiency (typically in left-right order with mass effect):
Dopamine release obstruction
↓ Inhibition of prolactin secretion
Antidiuretic hormone (ADH) obstruction
Inferior tumour growth
Growth into sphenoid sinus
Lateral growth of the tumor compresses surrounding cranial nerves
Superior tumour growth
↓ GH
↓ Protein production & muscle cell proliferation
↓ Luteinizing hormone (LH) (triggers ovulation & sex hormone synthesis) & follicle stimulating hormone (FSH) (stimulates ovarian follicles & sperm growth)
↓ (TSH)
↓ ACTH (stimulates cortisol & androgen release)
Cranial nerve (CN) II (optic)
↓ Visual acuity
Diplopia (double vision)
CN III, (oculomotor) IV (trochlear), or VI (abducens)
Ptosis* (drooping upper eyelid)
CN V (trigeminal) branches V1 & V2
Facial numbness
Ophthalmoplegia (eye muscle paralysis)
Compresses optic chiasma
Bitemporal hemianopsia (decreased lateral peripheral vision)
Tumour extends into hypothalamus
Hypothalamic dysfunction due to damage to hypothalamic cells
Disruption of multiple regulatory systems (i.e. sleep-wake cycles, appetite, temperature)
Tumour occludes ventricles
Tumour obstructs CSF flow
↑ Intracranial pressure
Hydrocephalus* (abnormal buildup of CSF in ventricles of the brain) and papilledema
Bacteria migrates from sinus flora into sphenoid sinus
Cerebrospinal fluid (CSF) leaks into throat
Post-nasal drip and nasopharyngeal mass
Stunted growth and short stature in children
↓ Muscle mass & fatigue
Diabetes insipidus* (excessive urination & extreme thirst)
Central hypothyroidism* (↓ T4 & T3)
Hyperprolactinemia
Meningitis* (inflammation of the meningeal layers of central nervous system)
*See relevant Calgary Guide slide
Hypogonadotropic hypogonadism (↓ Sex hormones)
Adrenal insufficiency* (↓ 8 AM cortisol)
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published Oct 1, 2027; updated Jan 5, 2025 on www.thecalgaryguide.com")
Chronic Mesenteric Ischemia

HLA-B52
allele &
other genetic
factors
Dyslipidemia
(abnormal blood
lipid levels)
Various etiologies (trauma,
coagulopathies, inflammatory
bowel disease, paraneoplastic
syndrome, surgery, portal
hypertension, cardiac)
High blood
pressure
Fibromuscular
dysplasia
(systemic vascular
disease)
Takayasu’s
arteritis (systemic
inflammatory
artery disease)
Median arcuate
ligament syndrome
Atherosclerosis
(plaque deposition in
the subendothelium;
cause of 90% of chronic
mesenteric ischemia
cases)
Arterial wall
thickening
Growth & hardening of
arterial wall
Compression of
celiac trunk by
the median
arcuate ligament
Thromboembolism
(clot formation)
Turbulent blood flow at
the level of the lesion
Mesenteric artery
stenosis (narrowing)
Abdominal bruit
(audible whoosh due to
artery narrowing)
Mucosal and
submucosal injury
& inflammation
Insufficient blood supply to the
gastrointestinal tract to meet
demand (i.e., “intestinal angina”)
Diarrhea ±
constipation
Gut motility
dysfunction
Activation of the
vomiting centre
in the brain
Chronic Mesenteric Ischemia
Chronic hypoperfusion of the bowel due to
(typically) multivessel mesenteric artery
stenosis or occlusion for >3 months
Aortic
dissection
(tear)
Radiation
exposure
Vascular injury
Exposure of
subendothelium
& disruption of
blood flow
Mesenteric artery
occlusion (blockage)
Compensation by the
mesenteric vasculature
through expansion of
collateral circulation
Majority of
patients are
asymptomatic
Nausea &
vomiting
Detectable
stenosis/occlusion of
mesenteric arteries
Intake of food increases metabolic
demand of bowel beyond levels
accommodated by strained blood supply
Mesenteric artery stenosis/
occlusion with medical
imaging (ie. CT Angiogram)
Postprandial abdominal discomfort
(typically epigastric pain ~30 minutes
following a meal, lasting for 1-4 hours)
Published May 19, 2025 on www.thecalgaryguide.com
Author: Liam Fitzgerald
Reviewers: Sophia Khan
Shahab Marzoughi
Sergio F. Sharif
Sylvain P. Coderre*
* MD at time of publication
Tobacco use
Buerger’s disease
(inflammatory
artery disease)
Formation of focal
inflammatory blood
clots
Weight loss
(>20 lbs)
Food aversion, smaller
meals, avoidance of fatty
foods (caloric restriction)
Legend: Pathophysiology Mechanism
Sign/Symptom/Lab Finding Complications")
Tetralogy of Fallot

Maternal exposure
to alcohol or
anticonvulsants
Maternal infections (e.g., hepatitis
B virus, coxsackievirus-B, human
cytomegalovirus virus, rubella)
Variable interaction between genetic associations and environmental risk factors
Antero-cephalad deviation of conal septum (muscular structure that forms the boundary
between right & left ventricular outflow tract during embryological development)
Tetralogy of Fallot
Cyanotic congenital heart disease with a combination of four cardiac defects
Ventricular septal
defect (VSD)**
Overriding aortic root
(aorta above VSD rather than left ventricle)
Right ventricular outflow
tract obstruction (RVOTO)
Right Ventricular
Hypertrophy
Hole in septum
Absent or reduced
Stressors (eg., crying,
between ventricles Right ventricle of
Oxygenated blood from the left
pulmonary valve
defecation, fever,
heart pumps
ventricle and deoxygenated blood
closure (P2)
dehydration) trigger acute
harder to push
from right ventricle flows into aorta
↑ Left ventricular
pressure
infundibular spasm (spasm of
blood through
conus arteriosus)
RVOTO
Chronic volume
overload
L-R ventricular
shunting
Single, loud S2
Spasm ↓ blood flow through
right ventricular outflow tract
Aortic
Dilated
aortic root
regurgitation** Hypercyanois or “tet” spells
Turbulent blood
flow from left
ventricle through
the hole in septum
to right ventricle
Turbulent diastolic
backflow through
incompetent aortic
valve
Deoxygenated
blood flows
through aorta to
systemic circulation
↓ O2 Saturation
Chemoreceptors in the carotid & aortic bodies
stimulate respiratory centre in the medulla oblongata
Harsh, holosystolic
murmur
Diastolic
decrescendo
murmur
↑ Deoxygenated blood
entering systemic circulation
↑ Catecholamine release from adrenal
medulla stimulating ↑ contractility of heart
↓ Blood flow
across VSD
↑ contraction against outflow obstruction
↑ pulmonary vascular resistance
↓ Intensity or
disappearance
of holosystolic
murmur
Peripheral & central
chemoreceptors detect ↓ arterial
O2 content, triggering brainstem
respiratory centre to stimulate ↑
work & rate of breathing
↓ Pulmonary blood flow
↑ Turbulence of blood
flow through RVOTO
↑ R-L shunting
↑ Deoxygenated hemoglobin (dark
red) relative to oxygenated hemoglobin
(bright red), reflecting more blue light
Cyanosis (blue discoloration of the
mucous membranes, nail beds, skin)
Paroxysmal
hyperpnea (rapid
and deep
respirations)
Irritability
and crying
Dyspnea
(shortness of
breath)
Poor feeding
Accentuation of systolic
crescendo/decrescendo
murmur with harsh ejection
**See corresponding
Calgary Guide slides
Legend: Mechanism
Published Jul 8, 2015; updated Jun 16, 2025 on www.thecalgaryguide.com
Pathophysiology Sign/Symptom/Lab Finding Complications")
Obesity Hypoventilation Syndrome
![Obesity Hypoventilation Syndrome: Pathogenesis and clinical findings
Obesity (BMI ≥ 30 kg/m2) risk factors: Poor eating patterns, sedentary lifestyle, genetic predisposition,
hypothyroidism, Cushing’s syndrome, socio-economic factors, age
Sleep-disordered breathing risk factors: Family history, tonsillar or adenoidal
hypertrophy, ↑ neck circumference, type 2 diabetes, HTN
Authors: Mohammad Omer
Mujtaba Siddique
Reviewers:
Ali Babwani
Luiza Radu
Jonathan Liu*
MD at time of publication*
↑ Adipose deposition
in abdomen
Abdominal fat pushes
against diaphragm
↑ Diaphragmatic
displacement
↑ Resistance to chest
wall expansion
↑Leptin resistance
High pressure
Pharyngeal
on upper airway
dilations unable
Secondary depression
↓ Chest wall
↓ Leptins ability to stimulate
↑ lung
to compensate
Narrowing of
(compromised function) of
Poor ventilation to
expansion
ventilation (mechanism unknown)
collapsibility
for weight
upper airways
respiratory system
lower lobes of lungs
↓ Tidal volume (air
that moves in/out of
lungs in a respiratory
cycle)
↑ Respiratory rate
↑ Chest wall thickness ↑ Leptin (a hormone released by
adipose tissues that controls hunger by
signaling fullness)
↑ Adipose
deposition near
upper airways
↑ Buildup of
edema in lower
extremities
↑ Respiratory
workload
↓ Chest wall
compliance (ability to
stretch)
↓ Leptin receptor
expression
↓ Leptin through
blood-brain barrier
↓ Pharyngeal space
Respiratory system is
unable to compensate to ↑
Fluid shifts from
demands
legs to neck during
sleep
Hypoventilation in sleep
↓ Ventilation (air exchange in lungs)
↑ PaCO₂ (partial pressure of arterial carbon
dioxide)
↑ Serum [H+]
↑ Serum [HCO3
-] by renal
reabsorption buffers [H+] rise
↓ PaO₂
(partial pressure of arterial oxygen)
Hypoxia (low
O₂ in tissue)
Higher PaCO₂ required to
reduce pH
↓ O₂ levels in alveoli triggers pulmonary
vessel vasoconstriction
PaCO₂ > 45
mmHG
Respiratory
acidosis
↓ response to CO₂ in central
chemoreceptors in brain
Pulmonary hypertension (high pressure in
pulmonary arteries)
↓ Neural drive
↓ Ventilatory responsiveness
) Right heart pumps against higher
pulmonary pressure leading to
cardiomyocyte hypertrophy
Cor pulmonale
(right-sided heart
failure)
Fatigue
Chronic hypercapnia
(↑ CO2 retention)
Pathophysiology Legend: Mechanism
Sign/Symptom/Lab Finding Complications
Morning headaches
Daytime lethargy
Published Jun 16, 2025 on www.thecalgaryguide.com](https://calgaryguide.ucalgary.ca/wp-content/uploads/2025/06/Obesity-Hypoventilation-Syndrome.jpg "Obesity Hypoventilation Syndrome: Pathogenesis and clinical findings
Obesity (BMI ≥ 30 kg/m2) risk factors: Poor eating patterns, sedentary lifestyle, genetic predisposition,
hypothyroidism, Cushing’s syndrome, socio-economic factors, age
Sleep-disordered breathing risk factors: Family history, tonsillar or adenoidal
hypertrophy, ↑ neck circumference, type 2 diabetes, HTN
Authors: Mohammad Omer
Mujtaba Siddique
Reviewers:
Ali Babwani
Luiza Radu
Jonathan Liu*
MD at time of publication*
↑ Adipose deposition
in abdomen
Abdominal fat pushes
against diaphragm
↑ Diaphragmatic
displacement
↑ Resistance to chest
wall expansion
↑Leptin resistance
High pressure
Pharyngeal
on upper airway
dilations unable
Secondary depression
↓ Chest wall
↓ Leptins ability to stimulate
↑ lung
to compensate
Narrowing of
(compromised function) of
Poor ventilation to
expansion
ventilation (mechanism unknown)
collapsibility
for weight
upper airways
respiratory system
lower lobes of lungs
↓ Tidal volume (air
that moves in/out of
lungs in a respiratory
cycle)
↑ Respiratory rate
↑ Chest wall thickness ↑ Leptin (a hormone released by
adipose tissues that controls hunger by
signaling fullness)
↑ Adipose
deposition near
upper airways
↑ Buildup of
edema in lower
extremities
↑ Respiratory
workload
↓ Chest wall
compliance (ability to
stretch)
↓ Leptin receptor
expression
↓ Leptin through
blood-brain barrier
↓ Pharyngeal space
Respiratory system is
unable to compensate to ↑
Fluid shifts from
demands
legs to neck during
sleep
Hypoventilation in sleep
↓ Ventilation (air exchange in lungs)
↑ PaCO₂ (partial pressure of arterial carbon
dioxide)
↑ Serum [H+]
↑ Serum [HCO3
-] by renal
reabsorption buffers [H+] rise
↓ PaO₂
(partial pressure of arterial oxygen)
Hypoxia (low
O₂ in tissue)
Higher PaCO₂ required to
reduce pH
↓ O₂ levels in alveoli triggers pulmonary
vessel vasoconstriction
PaCO₂ > 45
mmHG
Respiratory
acidosis
↓ response to CO₂ in central
chemoreceptors in brain
Pulmonary hypertension (high pressure in
pulmonary arteries)
↓ Neural drive
↓ Ventilatory responsiveness
) Right heart pumps against higher
pulmonary pressure leading to
cardiomyocyte hypertrophy
Cor pulmonale
(right-sided heart
failure)
Fatigue
Chronic hypercapnia
(↑ CO2 retention)
Pathophysiology Legend: Mechanism
Sign/Symptom/Lab Finding Complications
Morning headaches
Daytime lethargy
Published Jun 16, 2025 on www.thecalgaryguide.com")
GLP-1 Receptor Agonists
 Receptor Agonists: Mechanism of action and side effects
Management of Type
2 Diabetes Mellitus**
Weight
management
Authors: Saania Tariq, Trevor Low
Reviewers: Rafael Sanguinetti
Luiza Radu, Emily J. Doucette
Activated GLP-1 receptors in
Activated GLP-1
Hanan Bassyouni*
* MD at time of publication
the myenteric plexus inhibit
receptors stimulate
gastric motor neurons
pancreatic islet cells
GLP-1 Receptor Agonists
Synthetic analogs (e.g., Semaglutide) of GLP-1 peptide hormone endogenously
produced by intestinal enteroendocrine L cells directly activate GLP-1 receptors
to modulate food intake & glucose metabolism.
↓ Gastric
motility
↑ Pyloric sphincter
tone via disinhibition
↓ Glucagon release
from α-cells
↑ Regeneration,
↑ growth &
↓ apoptosis of β-cells
Slowed gastric emptying
Intramuscular injection or oral ingestion
prolongs GLP-1 receptor activation
↑ β-cell function & glucose-
dependent insulin production
GLP-1 receptor activation on neurons in
the arcuate nucleus of the hypothalamus
modulates appetite & satiety
Activation of GLP-1 receptors
in the gastrointestinal system
regulates metabolism
Prolonged gastric
distension & stasis
activates
chemoreceptors &
mechanoreceptors
Delayed digestion of
gastric contents ↓
glucose spikes after
meals
↑ Glycogen synthesis &
glucose uptake in liver &
skeletal muscles
Neuropeptide Y &
agouti-related protein
expressing neurons are
hyperpolarized
Pro-opiomelanocortin
expressing neurons
become ↑ excitable
Activated GLP-1 receptors
signal to hypothalamus
via vagus nerve afferents
↑ Vagal afferent
signaling to area
postrema
(vomiting centre)
Nausea &
vomiting
↓ Release of
neuropeptide Y &
agouti-related protein
↑ Anorexigenic
signalling
↑ Parasympathetic
output via dorsal motor
nucleus of vagus nerve
GLP-1 agonists directly
activate receptors in
area postrema
↑ Satiety (feeling
of fullness)
↓ Adiposity
(weight loss)
↓ Orexigenic drive
↓ Inflammatory stress in
pancreatic β-cells
↓ Appetite
↓ Caloric intake
↓ Inflammatory
cytokines & lipotoxicity
↓ Systemic
inflammation
**See corresponding Calgary Guide slide
Legend: Sign/Symptom/Lab Finding Pathophysiology Mechanism
Complications
↓ Hepatic glucose
production
(glycogenolysis)
↓ Fasting & postprandial
blood glucose
↓ Red blood cell (RBC)
glycation (glucose
irreversibly binds to
hemoglobin in RBCs)
↓ Production
of glycated
proteins and
lipids
↓ % of glycated
RBCs over time
↑ β-cell function
improves insulin
secretion
↓ Vascular
endothelial
damage and
atherosclerosis
↓ Endothelial
damage to
glomeruli
↑ Insulin sensitivity in
skeletal muscle & liver
↓ Hemoglobin
A1c
↓ Risk of
cardiovascular
complications
↓ Risk of renal
complications
Published Jun 22, 2025 on www.thecalgaryguide.com")