SEARCH RESULTS FOR: T cell
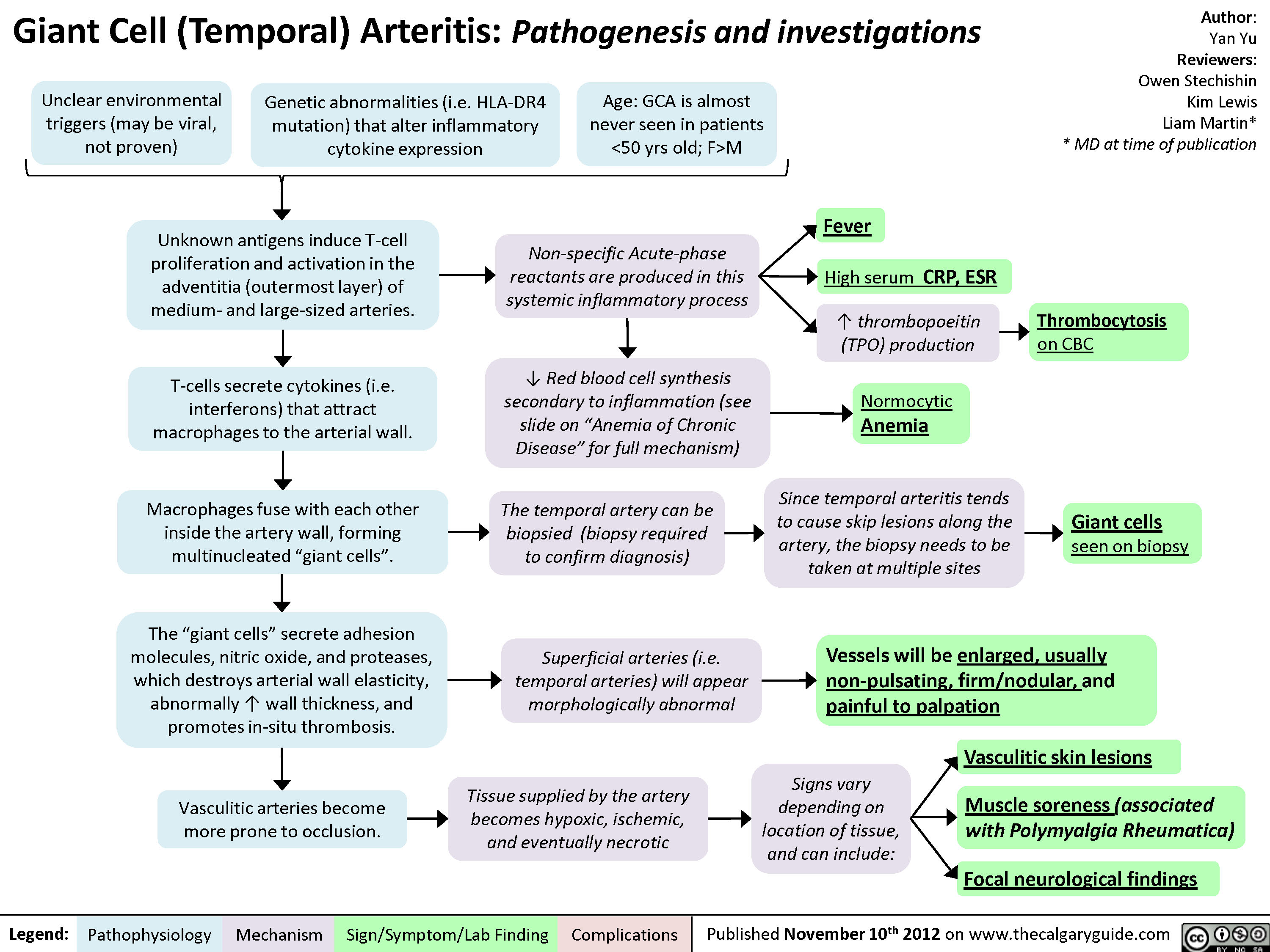
Giant Cell (Temporal) Arteritis: Pathogenesis and investigations

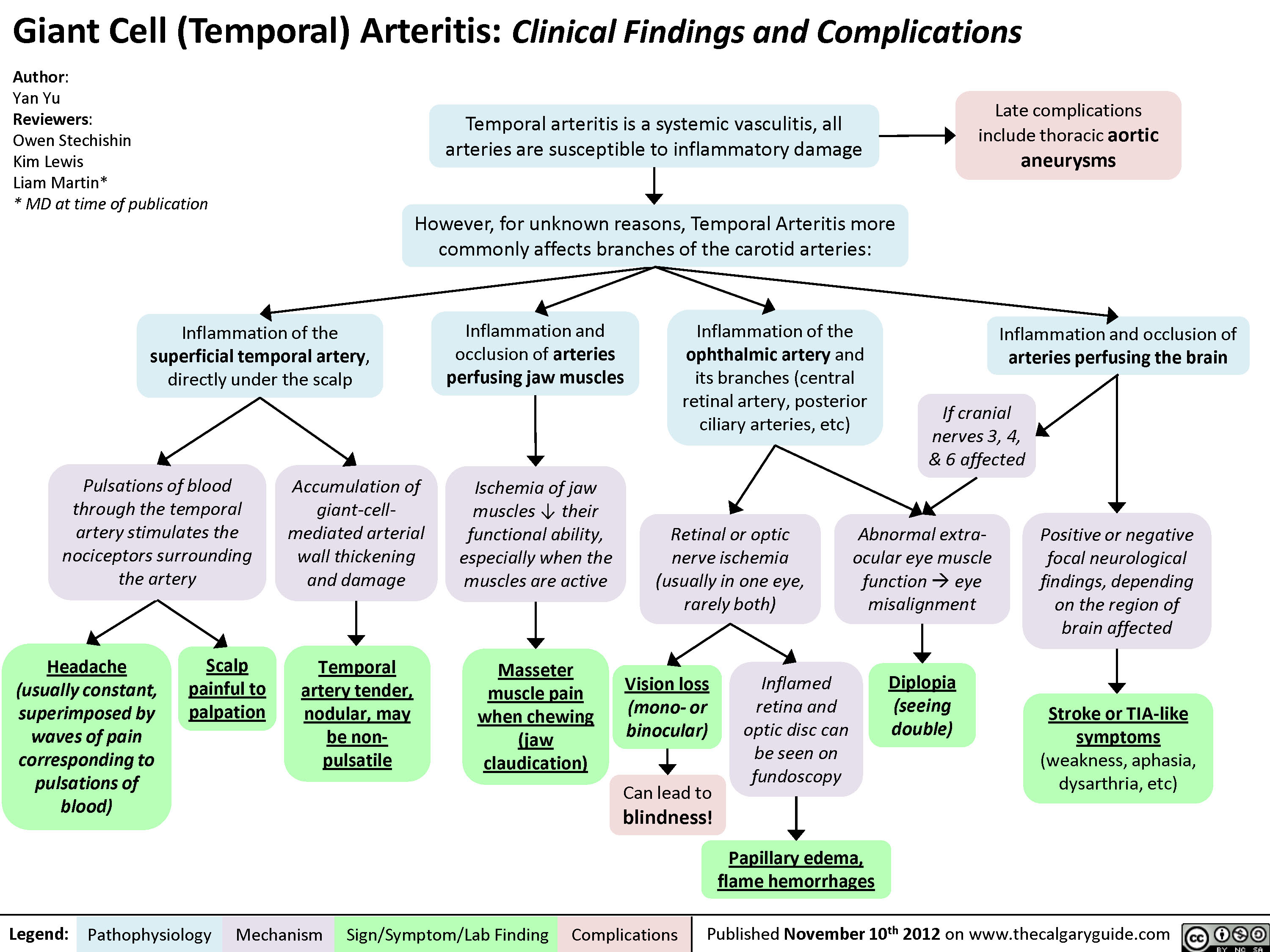
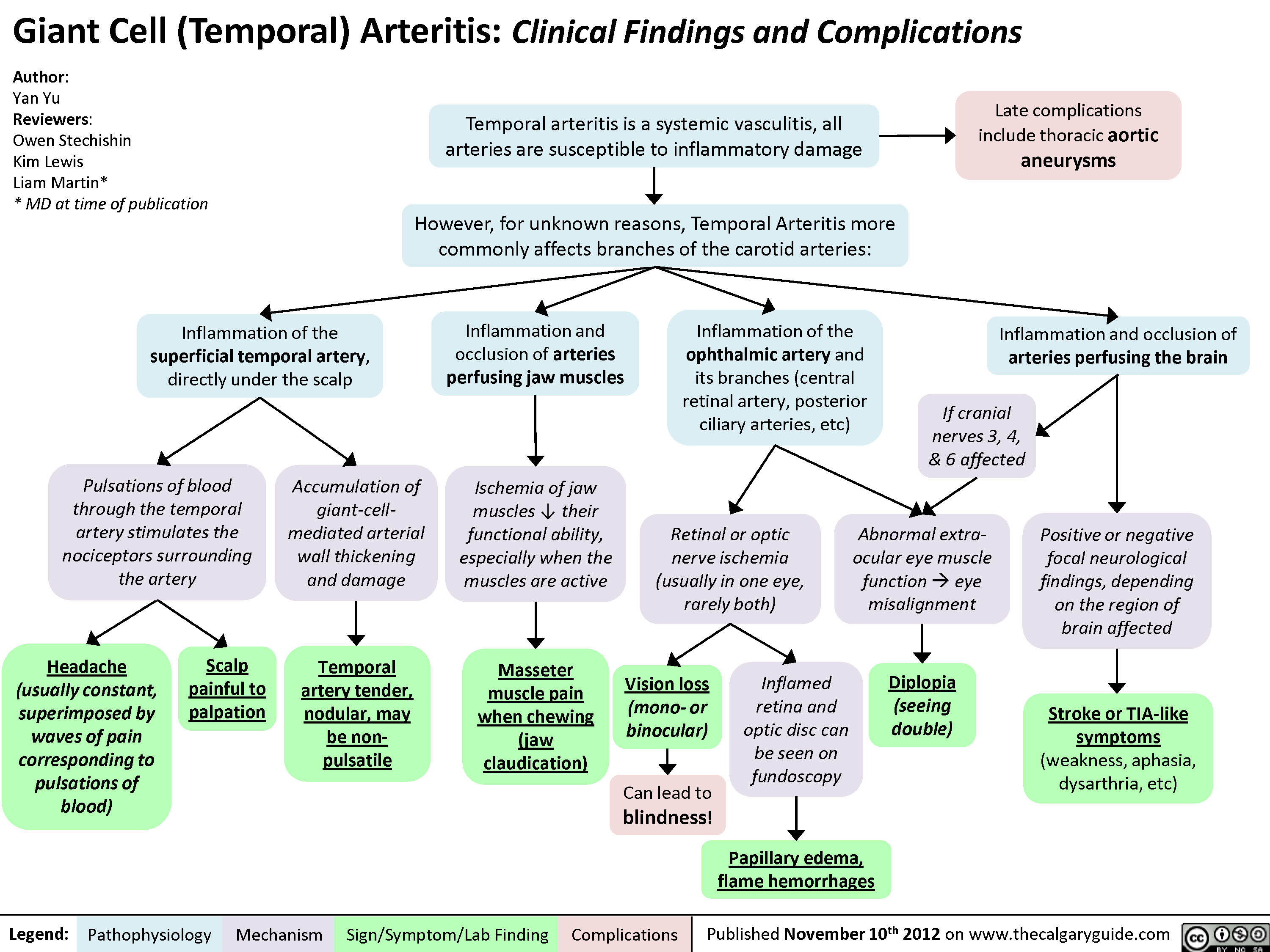
Giant Cell (Temporal) Arteritis: Clinical findings and Complications
Hypokalemia: Clinical Findings
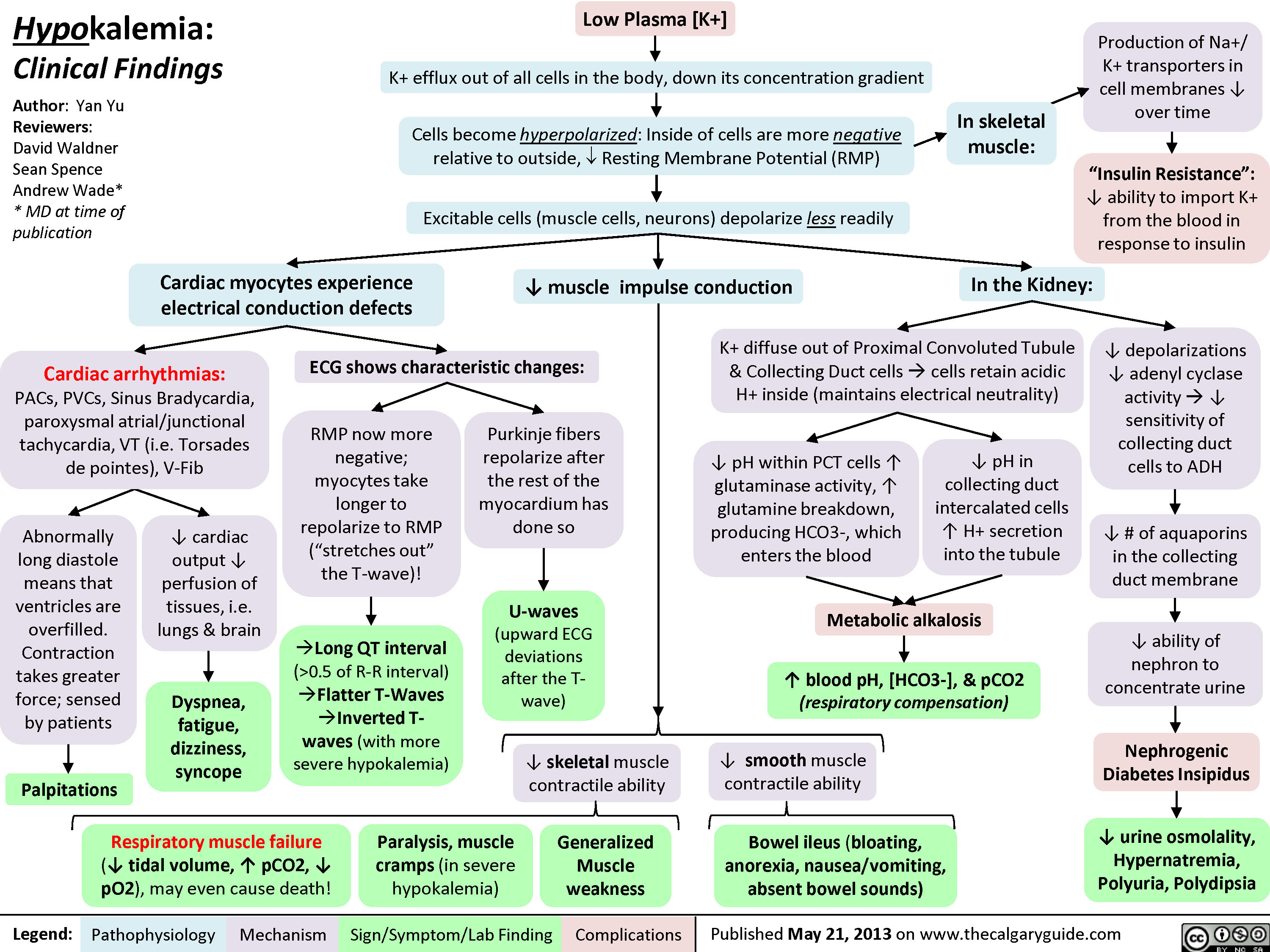 0.5 of R-R interval)?Flatter T-Waves ?Inverted T-waves (with more severe hypokalemia)Purkinje fibers repolarize after the rest of the myocardium has done soU-waves (upward ECG deviations after the T-wave)Cells become hyperpolarized: Inside of cells are more negative relative to outside, ? Resting Membrane Potential (RMP)In the Kidney:Generalized Muscle weaknessK+ diffuse out of Proximal Convoluted Tubule & Collecting Duct cells ? cells retain acidic H+ inside (maintains electrical neutrality)? pH within PCT cells ? glutaminase activity, ? glutamine breakdown, producing HCO3-, which enters the blood? blood pH, [HCO3-], & pCO2 (respiratory compensation)Low Plasma [K+]Abnormally long diastole means that ventricles are overfilled. Contraction takes greater force; sensed by patientsDyspnea, fatigue, dizziness, syncope? cardiac output ? perfusion of tissues, i.e. lungs & brainCardiac arrhythmias: PACs, PVCs, Sinus Bradycardia, paroxysmal atrial/junctional tachycardia, VT (i.e. Torsades de pointes), V-Fib? smooth muscle contractile abilityBowel ileus (bloating, anorexia, nausea/vomiting, absent bowel sounds)? pH in collecting duct intercalated cells ? H+ secretion into the tubuleMetabolic alkalosisParalysis, muscle cramps (in severe hypokalemia)Respiratory muscle failure (? tidal volume, ? pCO2, ? pO2), may even cause death!? depolarizations ? adenyl cyclase activity ? ? sensitivity of collecting duct cells to ADH? ability of nephron to concentrate urineNephrogenic Diabetes Insipidus? urine osmolality, Hypernatremia, Polyuria, Polydipsia? # of aquaporins in the collecting duct membrane"Insulin Resistance": ? ability to import K+ from the blood in response to insulinIn skeletal muscle:
117 kB / 307 word" title="Yu, Yan - Hypokalemia clinical findings - FINAL.pptx
Production of Na+/ K+ transporters in cell membranes ? over timeHypokalemia: Clinical FindingsAuthor: Yan YuReviewers:David WaldnerSean SpenceAndrew Wade** MD at time of publicationLegend:Published May 21, 2013 on www.thecalgaryguide.comMechanismPathophysiologySign/Symptom/Lab FindingComplicationsPalpitationsExcitable cells (muscle cells, neurons) depolarize less readilyK+ efflux out of all cells in the body, down its concentration gradientCardiac myocytes experience electrical conduction defects? muscle impulse conductionECG shows characteristic changes:? skeletal muscle contractile abilityRMP now more negative; myocytes take longer to repolarize to RMP("stretches out" the T-wave)! Long QT interval (>0.5 of R-R interval)?Flatter T-Waves ?Inverted T-waves (with more severe hypokalemia)Purkinje fibers repolarize after the rest of the myocardium has done soU-waves (upward ECG deviations after the T-wave)Cells become hyperpolarized: Inside of cells are more negative relative to outside, ? Resting Membrane Potential (RMP)In the Kidney:Generalized Muscle weaknessK+ diffuse out of Proximal Convoluted Tubule & Collecting Duct cells ? cells retain acidic H+ inside (maintains electrical neutrality)? pH within PCT cells ? glutaminase activity, ? glutamine breakdown, producing HCO3-, which enters the blood? blood pH, [HCO3-], & pCO2 (respiratory compensation)Low Plasma [K+]Abnormally long diastole means that ventricles are overfilled. Contraction takes greater force; sensed by patientsDyspnea, fatigue, dizziness, syncope? cardiac output ? perfusion of tissues, i.e. lungs & brainCardiac arrhythmias: PACs, PVCs, Sinus Bradycardia, paroxysmal atrial/junctional tachycardia, VT (i.e. Torsades de pointes), V-Fib? smooth muscle contractile abilityBowel ileus (bloating, anorexia, nausea/vomiting, absent bowel sounds)? pH in collecting duct intercalated cells ? H+ secretion into the tubuleMetabolic alkalosisParalysis, muscle cramps (in severe hypokalemia)Respiratory muscle failure (? tidal volume, ? pCO2, ? pO2), may even cause death!? depolarizations ? adenyl cyclase activity ? ? sensitivity of collecting duct cells to ADH? ability of nephron to concentrate urineNephrogenic Diabetes Insipidus? urine osmolality, Hypernatremia, Polyuria, Polydipsia? # of aquaporins in the collecting duct membrane"Insulin Resistance": ? ability to import K+ from the blood in response to insulinIn skeletal muscle:
117 kB / 307 word" />
0.5 of R-R interval)?Flatter T-Waves ?Inverted T-waves (with more severe hypokalemia)Purkinje fibers repolarize after the rest of the myocardium has done soU-waves (upward ECG deviations after the T-wave)Cells become hyperpolarized: Inside of cells are more negative relative to outside, ? Resting Membrane Potential (RMP)In the Kidney:Generalized Muscle weaknessK+ diffuse out of Proximal Convoluted Tubule & Collecting Duct cells ? cells retain acidic H+ inside (maintains electrical neutrality)? pH within PCT cells ? glutaminase activity, ? glutamine breakdown, producing HCO3-, which enters the blood? blood pH, [HCO3-], & pCO2 (respiratory compensation)Low Plasma [K+]Abnormally long diastole means that ventricles are overfilled. Contraction takes greater force; sensed by patientsDyspnea, fatigue, dizziness, syncope? cardiac output ? perfusion of tissues, i.e. lungs & brainCardiac arrhythmias: PACs, PVCs, Sinus Bradycardia, paroxysmal atrial/junctional tachycardia, VT (i.e. Torsades de pointes), V-Fib? smooth muscle contractile abilityBowel ileus (bloating, anorexia, nausea/vomiting, absent bowel sounds)? pH in collecting duct intercalated cells ? H+ secretion into the tubuleMetabolic alkalosisParalysis, muscle cramps (in severe hypokalemia)Respiratory muscle failure (? tidal volume, ? pCO2, ? pO2), may even cause death!? depolarizations ? adenyl cyclase activity ? ? sensitivity of collecting duct cells to ADH? ability of nephron to concentrate urineNephrogenic Diabetes Insipidus? urine osmolality, Hypernatremia, Polyuria, Polydipsia? # of aquaporins in the collecting duct membrane"Insulin Resistance": ? ability to import K+ from the blood in response to insulinIn skeletal muscle:
117 kB / 307 word" title="Yu, Yan - Hypokalemia clinical findings - FINAL.pptx
Production of Na+/ K+ transporters in cell membranes ? over timeHypokalemia: Clinical FindingsAuthor: Yan YuReviewers:David WaldnerSean SpenceAndrew Wade** MD at time of publicationLegend:Published May 21, 2013 on www.thecalgaryguide.comMechanismPathophysiologySign/Symptom/Lab FindingComplicationsPalpitationsExcitable cells (muscle cells, neurons) depolarize less readilyK+ efflux out of all cells in the body, down its concentration gradientCardiac myocytes experience electrical conduction defects? muscle impulse conductionECG shows characteristic changes:? skeletal muscle contractile abilityRMP now more negative; myocytes take longer to repolarize to RMP("stretches out" the T-wave)! Long QT interval (>0.5 of R-R interval)?Flatter T-Waves ?Inverted T-waves (with more severe hypokalemia)Purkinje fibers repolarize after the rest of the myocardium has done soU-waves (upward ECG deviations after the T-wave)Cells become hyperpolarized: Inside of cells are more negative relative to outside, ? Resting Membrane Potential (RMP)In the Kidney:Generalized Muscle weaknessK+ diffuse out of Proximal Convoluted Tubule & Collecting Duct cells ? cells retain acidic H+ inside (maintains electrical neutrality)? pH within PCT cells ? glutaminase activity, ? glutamine breakdown, producing HCO3-, which enters the blood? blood pH, [HCO3-], & pCO2 (respiratory compensation)Low Plasma [K+]Abnormally long diastole means that ventricles are overfilled. Contraction takes greater force; sensed by patientsDyspnea, fatigue, dizziness, syncope? cardiac output ? perfusion of tissues, i.e. lungs & brainCardiac arrhythmias: PACs, PVCs, Sinus Bradycardia, paroxysmal atrial/junctional tachycardia, VT (i.e. Torsades de pointes), V-Fib? smooth muscle contractile abilityBowel ileus (bloating, anorexia, nausea/vomiting, absent bowel sounds)? pH in collecting duct intercalated cells ? H+ secretion into the tubuleMetabolic alkalosisParalysis, muscle cramps (in severe hypokalemia)Respiratory muscle failure (? tidal volume, ? pCO2, ? pO2), may even cause death!? depolarizations ? adenyl cyclase activity ? ? sensitivity of collecting duct cells to ADH? ability of nephron to concentrate urineNephrogenic Diabetes Insipidus? urine osmolality, Hypernatremia, Polyuria, Polydipsia? # of aquaporins in the collecting duct membrane"Insulin Resistance": ? ability to import K+ from the blood in response to insulinIn skeletal muscle:
117 kB / 307 word" />
Giant Cell (Temporal) Arteritis - Pathogenesis and investigations
Giant Cell (Temporal) Arteritis - Clinical findings and Complications
Hyperosmolar Hyperglycemic State
![Hyperosmolar Hyperglycemic State (HHS)
Note: HHS is only seen in Type II DM patients!
Note: In patients with either DKA or HHS, always look for an underlying cause (i.e. an infection)
Author: Yan Yu Reviewers:
Peter Vetere
Gill Goobie
Hanan Bassyouni* * MD at time of publication
Alters total body water & ion osmosis
Inadequate insulin production, insulin resistance, non- adherence to insulin Tx
Relative Insulin deficit
Stresses that ↑ Insulin demand: infections, pneumonia, MI, pancreatitis, etc)
Hyperglycemia
(Very high blood [glucose], higher than in DKA)
When blood [glucose] > 12mmol/L, glucose filtration > reabsorption, ↑ urine [glucose]
Glucosuria
Glucose in filtrate promotes osmotic diuresis: large- volume urine output
Polyuria
Dehydration
(↓ JVP, orthostasis: postural hypotension/ postural tachycardia, ↑ resting HR)
Some insulin still present, but not enoughsome glucose is utilized by muscle/fat cells, some remain in the blood
Cells not “starved”, but still need more energy
↑ release of Catabolic hormones: Glucagon, Epinephrine, Cortisol, GH
Body tries to ↑ blood [glucose], to hopefully ↑ cell glucose absorption
Hypothalamic cells sense low intra-cellular glucose, triggering feelings of hunger
Polyphagia
Note: the presence of some insulin directly inhibits lipolysis; thus, in HHS there is no ketone body production, and no subsequent metabolic acidosis and ketouria (unlike in DKA). If ketones are detected in an HHS patient it’s likely secondary to starvation or other mechanisms.
↓ ECF volume, ↑ ECF osmolarity (i.e. hypernatremia)
↑ Gluconeogenesis ↑ Glycogenolysis (in liver)
↓ Protein synthesis, ↑ proteolysis
(in muscle)
↑ Gluconeogenic substrates for liver If the patient doesn’t drink enough
water to replenish lost blood volume If pt is alert and
Electrolyte imbalance
water is accessible
Water osmotically leaves neurons, shrinking them
Neural damage: delirium, lethargy, seizure, stupor, coma
↓ renal perfusion, ↓ GFR
Renal Failure
(pre-renal cause; see relevant slides)
Polydipsia Note: in HHS, body K+ is lost via osmotic diuresis. But diffusion of K+ out of cells
may cause serum [K+] to be falsely normal/elevated. To prevent hypokalemia, give IV KCl along with IV insulin as soon as serum K+ <5.0mmol/L. But ensure patient has good renal function/urine output first, to avoid iatrogenic hyperkalemia!
Note: Electrolyte imbalances (i.e. hyperkalemia, hypernatremia) are worsened by the acute renal failure commonly coexisting with DKA/HHS
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published November 3, 2016 on www.thecalgaryguide.com](http://calgaryguide.ucalgary.ca/wp-content/uploads/2015/05/Hyperosmolar-Hyperglycemic-State-HHS.jpg "Hyperosmolar Hyperglycemic State (HHS)
Note: HHS is only seen in Type II DM patients!
Note: In patients with either DKA or HHS, always look for an underlying cause (i.e. an infection)
Author: Yan Yu Reviewers:
Peter Vetere
Gill Goobie
Hanan Bassyouni* * MD at time of publication
Alters total body water & ion osmosis
Inadequate insulin production, insulin resistance, non- adherence to insulin Tx
Relative Insulin deficit
Stresses that ↑ Insulin demand: infections, pneumonia, MI, pancreatitis, etc)
Hyperglycemia
(Very high blood [glucose], higher than in DKA)
When blood [glucose] > 12mmol/L, glucose filtration > reabsorption, ↑ urine [glucose]
Glucosuria
Glucose in filtrate promotes osmotic diuresis: large- volume urine output
Polyuria
Dehydration
(↓ JVP, orthostasis: postural hypotension/ postural tachycardia, ↑ resting HR)
Some insulin still present, but not enoughsome glucose is utilized by muscle/fat cells, some remain in the blood
Cells not “starved”, but still need more energy
↑ release of Catabolic hormones: Glucagon, Epinephrine, Cortisol, GH
Body tries to ↑ blood [glucose], to hopefully ↑ cell glucose absorption
Hypothalamic cells sense low intra-cellular glucose, triggering feelings of hunger
Polyphagia
Note: the presence of some insulin directly inhibits lipolysis; thus, in HHS there is no ketone body production, and no subsequent metabolic acidosis and ketouria (unlike in DKA). If ketones are detected in an HHS patient it’s likely secondary to starvation or other mechanisms.
↓ ECF volume, ↑ ECF osmolarity (i.e. hypernatremia)
↑ Gluconeogenesis ↑ Glycogenolysis (in liver)
↓ Protein synthesis, ↑ proteolysis
(in muscle)
↑ Gluconeogenic substrates for liver If the patient doesn’t drink enough
water to replenish lost blood volume If pt is alert and
Electrolyte imbalance
water is accessible
Water osmotically leaves neurons, shrinking them
Neural damage: delirium, lethargy, seizure, stupor, coma
↓ renal perfusion, ↓ GFR
Renal Failure
(pre-renal cause; see relevant slides)
Polydipsia Note: in HHS, body K+ is lost via osmotic diuresis. But diffusion of K+ out of cells
may cause serum [K+] to be falsely normal/elevated. To prevent hypokalemia, give IV KCl along with IV insulin as soon as serum K+ <5.0mmol/L. But ensure patient has good renal function/urine output first, to avoid iatrogenic hyperkalemia!
Note: Electrolyte imbalances (i.e. hyperkalemia, hypernatremia) are worsened by the acute renal failure commonly coexisting with DKA/HHS
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published November 3, 2016 on www.thecalgaryguide.com")
central-retinal-artery-occlusion-pathogenesis-and-clinical-findings
 (i.e. Valvular, arrhythmias, congenital defects) (i.e. OCP, Protein C&S deficiency, ATIII deficiency) (i.e. leukemia/lymphoma, sickle cell, polycythemia) Endothelial cell damage Abnormal blood flow 1` coagulation and/or 1 blood viscosity and creates hypercoagulable state causing localized stasis 4, anti-coagulation inflammation
Abbreviations: • GCA — Giant Cell Arteritis • SLE — Systemic Lupus Erythematosus • GPA — granulomatosis with polyangitis • OCP — Oral contraceptive pill • ATIII — Anti-thrombin Ill
Thrombus formation
Blockage of central retinal artery
Central Retinal Artery Occlusion (CRAO)
Authors: Graeme Prosperi-Porta Reviewers: Stephanie Cote Usama Malik Johnathan Wong* * MD at time of publication Carotid Artery Atherosclerosis
Atherosclerotic plaque dislodges from carotid artery
The retina becomes pale 4, perfusion of retinal Slow retinal artery blood Acute retinal edema Ganglion cells and axons from NI, perfusion arterioles due to upstream flow allows for caused by ischemia results death due to ischemia CRAO segmentation of the blood column in a blurred appearance of the retina results in disc pallor seen months after CRAO The choroidal vessels supplying the macula via the posterior ciliary artery become more prominent within a background of retinal pallor")
infectious-esophagitis-pathogenesis-and-clinical-findings

Herpes Simplex Infection of squamous cells and macrophages
Colonization facilitated by use of antacid therapy
Nuclear Large Superficial Squamous Macrophage inclusion bodies esophageal ulceration ulcers cell inclusion bodies aggregation Legend: Pathophysiology Mechanism Sign/Symptom/Lab Finding Complications
Spores and pseudohyphae seen on biopsy
• Invas.on of underlying blood vessels
• White plaques over erythematous base
'1' neutrophils due to inflammatory response")
non-depolarizing-neuromuscular-blocks-ndnmbs
 Eg. pancuronium, rocuronium, atracurium, vecuronium
Abbreviations NDNMBs — Non-Depolarizing Neuromuscular Blockers ACh — Acetylcholine
Quick Facts 1° Indication = Skeletal muscle paralysis to facilitate tracheal intubation, and used during indicated surgeries or mechanical ventilation
Route of Administration = IV
Metabolism & Excretion = Redistribution, hepatic clearance/renal excretion (extent varies greatly by drug). NOT degraded by acetylcholinesterase or pseudocholinesterase
See Acetylcholinesterase Inhibitors slide for reversal of NDN M Bs
Competitive antagonism at post-synaptic nicotinic receptors on muscles
Competitive antagonism at the pre-synaptic nicotinic receptors on neurons
Vagolytic effect (esp. pancuronium)
Anaphylactic/ anaphylactoid reactions
4, Binding sites for ACh at post-synaptic nicotinic receptors on muscles
4, Binding sites for ACh at pre-synaptic nicotinic receptors on neurons
Blockage of vagal muscarinic receptors in sinoatrial nodes
IgE antibodies attach to ammonium ion components of NDNMBs Non-immunologic mast cell degranulation (esp. atracurium)
4, Muscle cell depolarization
4, Positive feedback for continued ACh ► release in response to high frequency stimulation
Skeletal muscle paralysis
Tetanic fade, Train-of-Four fade
4, —• Parasympathetic Tachycardia effects on heart
Release of histamine from mast cells and basophils
Bronchospasm
Hypotension
Legend: Pathophysiology Mechanism Sign/Symptom/Lab Finding Complications I Published March 3, 2018 on www.thecalgaryguide.com
0€3,0 BY NC SA")
non-depolarizing-neuromuscular-blocks-ndnmbs
 Eg. pancuronium, rocuronium, atracurium, vecuronium
Abbreviations NDNMBs — Non-Depolarizing Neuromuscular Blockers ACh — Acetylcholine
Quick Facts 1° Indication = Skeletal muscle paralysis to facilitate tracheal intubation, and used during indicated surgeries or mechanical ventilation
Route of Administration = IV
Metabolism & Excretion = Redistribution, hepatic clearance/renal excretion (extent varies greatly by drug). NOT degraded by acetylcholinesterase or pseudocholinesterase
See Acetylcholinesterase Inhibitors slide for reversal of NDN M Bs
Competitive antagonism at post-synaptic nicotinic receptors on muscles
Competitive antagonism at the pre-synaptic nicotinic receptors on neurons
Vagolytic effect (esp. pancuronium)
Anaphylactic/ anaphylactoid reactions
4, Binding sites for ACh at post-synaptic nicotinic receptors on muscles
4, Binding sites for ACh at pre-synaptic nicotinic receptors on neurons
Blockage of vagal muscarinic receptors in sinoatrial nodes
IgE antibodies attach to ammonium ion components of NDNMBs Non-immunologic mast cell degranulation (esp. atracurium)
4, Muscle cell depolarization
4, Positive feedback for continued ACh ► release in response to high frequency stimulation
Skeletal muscle paralysis
Tetanic fade, Train-of-Four fade
4, —• Parasympathetic Tachycardia effects on heart
Release of histamine from mast cells and basophils
Bronchospasm
Hypotension
Legend: Pathophysiology Mechanism Sign/Symptom/Lab Finding Complications I Published March 3, 2018 on www.thecalgaryguide.com
0€3,0 BY NC SA")
Celiac Disease: Complications
 Carbohydrate Protein Fat Secretory maldigestion maldigestion malabsorption diarrhea
Legend:
Fermentation by gut bacteria 1 Gas production
Bloating
Fat retained in stool
Steatorrhea
Abdominal pain
Pathophysiology Mechanism
Sign/Symptom/Lab Finding
Growth Retardation
Authors: Yoyo Chan Reviewers: Peter Bishay Usama Malik Sylvain Coderre* * MD at time of publication
IgA response
Autoimmune IgA deposits Lymphocyte response in sub-epidermal skin layer against enamel
Dermatitis Herpetiformis (Chronic pruritic blisters)
Nutritional deficiency
Dental enamel hypoplasia
Vitamin D and calcium deficiency
Zinc, selenium Folate Iron Osteoporosis deficiency deficiency deficiency Anemia t Risk of miscarriages")
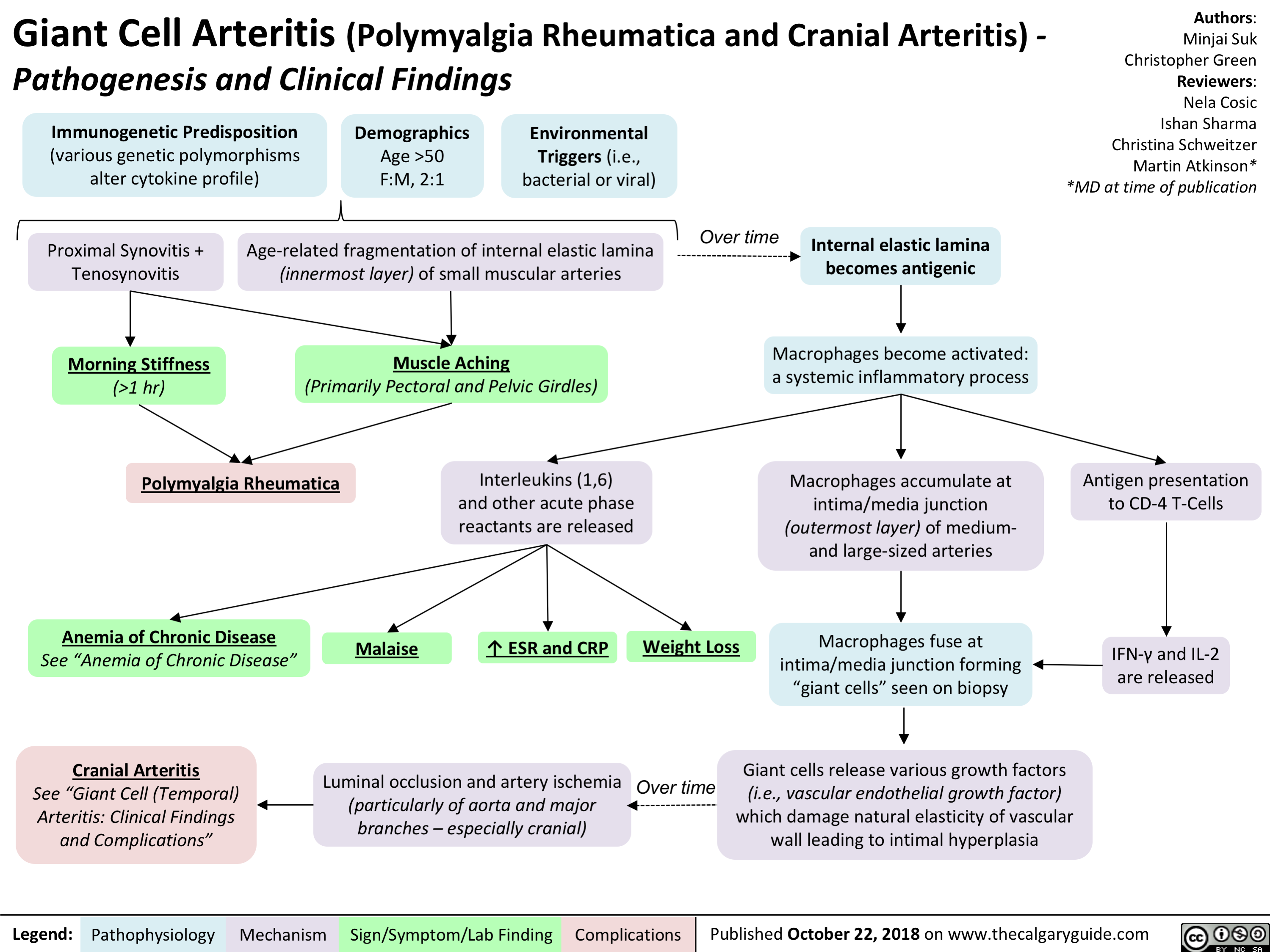
Giant Cell Arteritis: Pathogenesis and Clinical Findings
DiGeorge Syndrome: Pathogenesis and Clinical Findings
, though their role is less well understood.
Abbreviations:
• PA-VSD – pulmonary atresia
with ventricular septal defect
• TBX1 – T-box Protein 1
• VSD – ventricular septal defect
Heterozygous deletion at chromosomal region 22q11.2
The region’s main gene product, TBX1*, exhibits haploinsufficiency: even a heterozygote for this gene product, producing half the normal quantities of TBX1, is insufficient to produce a normal phenotype
Abnormal pharyngeal arch development
Hypoplastic / aplastic thymus ↓ T cells
(lymphopenia)
Craniofacial malformations (e.g. tracheomalacia, horizontal Eustachian tubes, cleft palate)
Impaired ear and sinus drainage
Abnormal conotruncus development
Heart defects
(e.g. interrupted aortic arch, tetralogy of Fallot, PA-VSD/VSD, truncus arteriosus)
Hypoplastic parathyroid glands
Hypocalcemia Seizures
(usually neonatal onset)
Memory Aid:
CATCH-22
Cyanotic congenital heart disease
Abnormal facies
Thymic hypoplasia Cognitive impairment Hypoparathyroidism, hypocalcemia
22q11.2 deletion
Abnormal T cell regulation
and development
Compromised cytotoxic T cells
Susceptibility to intracellular pathogens
Compromised helper T cells
↓ communication with memory B cells
↓immunoglobulins
(progressive hypogammaglobulinemia)
Susceptibility to extracellular pathogens
Autoimmunity and Atopy
Note: There is phenotypic variability in DiGeorge Syndrome. Other features include: thin upper lip, upslanted palpebral fissures, prominent nose, low-set ears, small mouth, hearing impairment, long tapered fingers, scoliosis, vertebral malformations, learning disabilities, and failure to thrive.
Viral Infections
Bacterial Infections
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published April 21, 2019 on www.thecalgaryguide.com")
chronic-myeloid-leukemia
: Pathogenesis and Clinical Presentation
Translocation of a Chr 9 segment onto Chr 22, creating a Philadelphia chromosome (Chr 22) containing the BCR-ABL1 fusion gene
Mutations from ionizing radiation
Other genetic abnormalities
Authors: Yan Yu Katie Lin Reviewers: Jennifer Au Crystal Liu Danielle Chang Lynn Savoie* *Indicates faculty member at time of initial publication
These genetic abnormalities accumulate in the earliest cell of the blood cell differentiation sequence: the pluripotent hematopoietic stem cell
Hematopoietic stem cell division in the bone marrow becomes unregulated
1. Chronic Stage (85% of clinical presentation): Hematopoietic stem cell division/differentiation in the bone marrow results in ↑ production of multiple blood cell lines (detectable on CBC, but patients are usually asymptomatic at this stage)
Acquired ↑ genetic abnormalities
2. Accelerated Stage
More and more immature precursor cells (”blasts”) divide and accumulate in bone marrow (where 10-19% of blood cells are “blasts”.) Blasts start to spill over into the peripheral blood
Acquired ↑ genetic abnormalities
3. Blast crisis (transformation into AML/ALL)
Neoplastic blast cells have filled up the bone marrow (where >20% of blood cells are blasts). More blasts spill out into the peripheral blood.
Multifactorial causes, most Weight loss, malaise, fever/
with unclear mechanisms
Neoplastic division of platelet precursor cells
Neoplastic division of WBC precursor cells, especially neutrophil precursors
Dividing “blasts” limit the space and resources available for RBC synthesis
chills, night sweats Thrombocytosis
Leukocytosis:
· Neutrophilia, basophilia, & eosinophilia
· “Left shift”: ↑ neutrophil & band production
· Disorderly WBC differential: i.e. “myelocyte bulge”
Trapping of WBC’s in the spleen enlarges the spleen
Splenomegaly:
· Left upper quadrant pain · Early satiety (large spleen compresses the stomach)
· Associated hepatomegaly (if spleen is overfilled & WBCs spill over into liver)
Anemia
↓ oxygenation of blood means blood is less red & body tries to compensate
Pallor Dyspnea Tachycardia
High turnover of these cancerous cells → excess cell lysis
Release of intracellular contents (uric acid, K+, LDH) into plasma
· Hyperkalemia · High (LDH)
Hyperuricemia
Gout
Acute Kidney Injury
Expanding marrow pushing on bone
Bone marrow expands into sternum
Bone pain
Sternal tenderness
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Re-Published June 15, 2019 on www.thecalgaryguide.com")
Acute Lymphoblastic Leukemia
![Acute Lymphoblastic Leukemia (ALL): Pathogenesis and Clinical Presentation
Authors: Yan Yu, Katie Lin Reviewers: Crystal Liu, Kara Hawker, Jennifer Au, Lynn Savoie* * MD at time of initial publication
Note: ALL is much rarer than AML and is usually seen in children
Accumulation of genetic abnormalities in immature lymphoid precursor cells (B/T cell precursors)
Neoplastic lymphoid precursor cells (“blasts”) divide and accumulate in bone marrow
Abundance of blasts displaces other blood precursors from marrow, inhibiting their development/differentiati on
After neoplastic blasts fill up bone marrow, they spill out into blood
High turnover of these cancerous cells
Multifactorial causes, most with unclear mechanisms
Expanding marrow pushing on bone
Pancytopenia on CBC
20% of marrow is blasts (on bone marrow aspirate and/or biopsy)
Neoplastic blasts continue to divide and accumulate in lymph
nodes and spleen (can occur, but not that common)
Blasts detected as white blood cells on CBC
High rate of cell lysis
Weight loss, malaise, fever/chills, night sweats
Bone pain (worse than that felt in AML, especially in children)
↓ in neutrophils
↓ in RBCs
↓ in platelets, reduced blood clotting ability
Lymphadenopathy Splenomegaly
May cause leukocytosis, despite pancytopenia
Release of intracellular contents (uric acid, K+, LDH) into plasma
Greater chances of infection
Anemia
Fatigue, shortness of breath, pallor
Easy bruising and petechiae on skin
Hyperuricemia Hyperkalemia High [LDH]
Tumor lysis syndrome
Acute kidney injury
Gout
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Re-Published May 5, 2019 on www.thecalgaryguide.com](http://calgaryguide.ucalgary.ca/wp-content/uploads/2015/05/Acute-Lymphoblastic-Leukemia.jpg "Acute Lymphoblastic Leukemia (ALL): Pathogenesis and Clinical Presentation
Authors: Yan Yu, Katie Lin Reviewers: Crystal Liu, Kara Hawker, Jennifer Au, Lynn Savoie* * MD at time of initial publication
Note: ALL is much rarer than AML and is usually seen in children
Accumulation of genetic abnormalities in immature lymphoid precursor cells (B/T cell precursors)
Neoplastic lymphoid precursor cells (“blasts”) divide and accumulate in bone marrow
Abundance of blasts displaces other blood precursors from marrow, inhibiting their development/differentiati on
After neoplastic blasts fill up bone marrow, they spill out into blood
High turnover of these cancerous cells
Multifactorial causes, most with unclear mechanisms
Expanding marrow pushing on bone
Pancytopenia on CBC
20% of marrow is blasts (on bone marrow aspirate and/or biopsy)
Neoplastic blasts continue to divide and accumulate in lymph
nodes and spleen (can occur, but not that common)
Blasts detected as white blood cells on CBC
High rate of cell lysis
Weight loss, malaise, fever/chills, night sweats
Bone pain (worse than that felt in AML, especially in children)
↓ in neutrophils
↓ in RBCs
↓ in platelets, reduced blood clotting ability
Lymphadenopathy Splenomegaly
May cause leukocytosis, despite pancytopenia
Release of intracellular contents (uric acid, K+, LDH) into plasma
Greater chances of infection
Anemia
Fatigue, shortness of breath, pallor
Easy bruising and petechiae on skin
Hyperuricemia Hyperkalemia High [LDH]
Tumor lysis syndrome
Acute kidney injury
Gout
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Re-Published May 5, 2019 on www.thecalgaryguide.com")
Lyme Disease Pathogenesis and Clinical Findings

Tick bite from Borrelia burgdorferi infected Ixodes ricinus (tick from European countries)
Tick bite from Borrelia burgdorferi infected Ixodes scapularis (Deer tick/black-legged tick in SE Canada and NE United States)
Binding of OspC (a surface protein expressed by B. Burgdorferi) to human plasminogen allowing the spirochete to spread from bite site to other host organs and tissues
B. burgdorferi spreads through skin and other tissues via bloodstream in human host.
If tick bite lasts 36-72 hours or more, this is sufficient time for ticks to transmit the infection. (<36 hours of tick attachment results in a lower rate of infection: 1.2% -1.4%)
Lyme Disease
A vector-borne, infectious multi-system disease with highest risk in late spring and summer by the spirochete Borrelia burgdorferi
Early Disease Stage (<30 days)
Macrophages and T-cells produce ↑ inflammatory (TNF- α, IFN-γ) and ↑ anti-inflammatory cytokines, causing eosinophils to concentrate adjacent to the tick bite site
Early Disseminated Disease Stage (<3 months)
B. burgdorferi attach to host cell integrins
Pro-inflammatory response with production of matrix glycosaminoglycans and extracellular matrix proteins which have an affinity to attack collagen fibrils on the heart, nerves, and joints
1. Multiple erythema migrans 2. Meningitis
3. Meningoradiculoneuritis
4. Cranial nerve palsies
5. Carditis
6. Borrelial lymphocytoma
Late Disease Stage (>3 months)
Ongoing and repeated innate and adaptive host immune response to B. burgdorferi
Chronic inflammatory state results in synovial hypertrophy, vascular
proliferation, and ↑ mononuclear cell infiltrate in large joints
Large joint arthritis (most commonly affecting the knees)
Erythema migrans (a slowly expanding red skin patch with partial central clearing resulting in a “target clearing lesion” appearance) at site of tick bite
Systemic inflammatory response
after dissemination of the spirochete to body tissues and organs
Flu-like symptoms (fever, chills, muscle aches, headache, fatigue, joint aches, swollen lymph nodes)
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published July 24, 2019 on www.thecalgaryguide.com
References
• David A. Wetter and Colin A. Ruff. CMAJ August 09, 2011 183 (11) 1281; DOI: https://doi.org/10.1503/cmaj.101533
• https://www.canada.ca/en/public-health/services/diseases/lyme-disease/causes-lyme-disease.html
• Borrelia burgdorferi Infection-Associated Surface Proteins ErpP, ErpA, and ErpC Bind Human Plasminogen. Catherine A. Brissette, Katrin Haupt, Diana Barthel, Anne E. Cooley, Amy Bowman, Christina Skerka, Reinhard Wallich, Peter F. Zipfel, Peter Kraiczy, Brian Stevenson. Infection and Immunity Dec 2008, 77 (1) 300-306; DOI: 10.1128/IAI.01133-08
• https://www.uptodate.com/contents/what-to-do-after-a-tick-bite-to-prevent-lyme-disease-beyond- the-basics
• Murray, T. S., & Shapiro, E. D. (2010). Lyme disease. Clinics in laboratory medicine, 30(1), 311–328. doi:10.1016/j.cll.2010.01.003
• Weedon, David. Weedon's Skin Pathology E-Book: Expert Consult-Online and Print. Elsevier Health Sciences, 2009.")
Ataxia Telangiectasia Pathogenesis and Clinical Findings
; involuntary muscle contractions, hypotonia, IQ decline, and abnormal eye movement
Loss of ATM leads to mitotic defects and arrest in gamete genetic recombination process
Gonadal dysgenesis and delayed puberty
DNA damage to tumor suppressors such as p53 and BRCA1
Impaired signaling of downstream cell cycle regulators
Impaired genome stability and increased disposition to cancer
↑ Acute Lymphocytic Leukemia of T cell origin (in children) and Chronic Lymphoblastic Leukemia (in adults)
Impaired recombination of DNA in immune cells
Thymic hypoplasia; humoral & cellular immunodeficiency
↓ or absent functional immunoglobulins IgA, IgE, and IgG2 that function to prevent respiratory infections
Respiratory infections with bronchiectasis and pneumonia
Cells less able to undergo apoptosis in response to ionizing radiation
Accumulation of DNA defects in the cells of sun exposed areas such as skin, hair, and conjunctiva
Mucocutaneous telangiectasia on the bulbar conjunctiva and ears between 2-6 years of age
May progress to involve periorbital skin, trunk, extremities, body folds, and other mucosal surfaces
Sterility
DNA damage and genomic instability
Premature melanocyte stem cell differentiation
Premature graying of skin and hair
Abbreviations:
• ATM – Ataxia-telangiectasia
mutated protein
• p53 – Tumor protein 53
• BRCA 1 – Breast cancer
susceptibility protein.
• IgA, IgE, IgG2 – Immunoglobulins
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published August 4, 2019 on www.thecalgaryguide.com")
Erythema Nodosum pathogenesis and clinical findings
 ~28-48% of cases
Medications (Ex. Birth Control Pills, Sulfa drugs) ~3-10% of cases
Malignancy (ex. Lymphoma)
Autoimmune conditions (ex. Sarcoidosis and
Inflammatory Bowel Disease) ~11-25% of cases
Pregnancy ~1-3% of cases
Antigenic Stimuli / Bacteria / Viruses / Chemical Agents all could trigger the following process: Phase 1. Neutrophils Infiltrate the fibrous septa between fat lobules in the subcutaneous fat
Phase 2. Neutrophils release reactive oxygen species, leading to oxidative tissue damage and inflammation
Phase 3. Opening of inter-endothelial junction and the migration of more inflammatory cells into the septal venules, including macrophages, histocytes, and eosinophils
Phase 4. Macrophages secrete inflammatory cytokines, which stimulates the proliferation of more helper T cells (Th1)
Phase 5. Th1 cells secrete more cytokines, leading to the further release of Th1 cytokines and mediating the immune complexes deposition in the septal venules of the subcutaneous fat (panniculitis). The Th1 immune reaction is called Type IV Delayed Hypersensitivity Reaction
Phase 6. Activated macrophages produce hydrolytic enzymes and transform into multi- nucleated giant cells, called Miescher’s Radial Granulomas. These consist of small, well defined aggregations of small histocytes arranged radially around a small cleft of variable shapes in the septal venules of the subcutaneous fat
Phase 1-4. Lesions are red tender nodules, poorly defined, vary in size from 2-6 cm, and usually on shins ( 1st week)
Fat Lobules T lymphocytes
Macrophages
Note: we’ve done extensive research and can’t figure out why erythema nodosum happens mostly on the shins. If you have an answer, please email us!
Phase 5. Lesions become tense, hard, and painful; and they change in color into bluish or livid. (2nd week)
Phase 6. Lesions become fluctuant as in abscess, but do not ulcerate. Lesions fade to a yellowish color
Epidermal layer Dermal-Epidermal Junction
Dermal layer Subcutaneous Fat Layer
Phase 6. Miescher’s Radial Granulomas
Fat Lobules
T lymphocytes
Macrophages
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published August 25, 2019 on www.thecalgaryguide.com")
iga-vasculitis-henoch-scholein-purpura-pathogenesis-and-clinical-findings
 : Pathogenesis and clinical findings
Authors: Mia Koegler Nela Cosic Reviewers: Crystal Liu Yan Yu* Martin Atkinson* * MD at time of publication
Infectious Agents
50% have preceding upper respiratory tract infections, i.e., influenza virus or Group A Strep
Drugs
I.e., antibiotics (penicillin, erythromycin), NSAIDs and biologics (tumor necrosis factor α inhibitors)
Immunogenetic and cellular predisposition
Various genetic polymorphisms alter cell- mediated immune response, IgA levels elevated in 50% of people
↑ Circulating galactose-deficient IgA1 (GD-IgA1). Deficiency in galactosylation of IgAà↓ IgA serum clearanceàadhesion of IgA complexes, which then deposit into the endothelial lining of blood vesselsàattraction of various inflammatory cells to the area:
Formation of Secretion of Interleukin 8 (IL8) - cytokine that induces Neutrophils infiltrate Activation of complement immune complexes neutrophilic chemotaxis and macrophage phagocytosis the tissue site factors (C3, C4)
Leukocytoclastic vasculitis (histopathologic term for small vessels inflamed by neutrophilic autoimmune response)
Inflamed cutaneous vessels become enlarged in clusters
Symmetrical palpable purpura (red/purple, non- blanchable papules) distributed on lower limbs and buttocks areas
Cutaneous small vessel vasculitis (100%)
Inflamed gastric vessels - hemorrhage and edema within bowel wall
Gastrointestinal (85%)
Colicky abdominal pain (commonly in the periumbilical region), nausea, vomiting
Gastrointestinal
GI bleeding (hematemesis, melena), Intussusception
Glomerular mesangial proliferation and inflammation
↑ mast cell deposition in joints
Joints (60-85%)
Arthralgia's (common), arthritis (especially knees and ankles)
Arthralgia often transient. No permanent sequelae
Sympathetic nervous system activation
Glomerulosclerosis, tubulointerstitial and podocyte damage
Renal tissue ischemia
↑ Na sensitivity in renal tubules (↑ Na and water retention)
Renal (10-50%)
Increased renin secretion
HTN, nephrotic/nephritic syndrome, renal insufficiency
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published September 1, 2019 on www.thecalgaryguide.com")
X-linked Severe Combined Immunodeficiency (SCID)
: Pathogenesis & clinical findings
Abbreviations:
• Ig: Immunoglobulin
• IL: Interleukin
• NK: Natural Killer
• Th2: T helper 2 cell
• Treg: T regulatory cell
↓ IL-15 signaling ↓NK cell differentiation
and proliferation
↓ or absent NK cells in the blood, bone marrow and peripheral lymphoid tissues
Impaired innate immune system
Chronic Stress Response
Mutation of the IL2-RG gene encoding the interleukin receptor common gamma chain located on the X-chromosome
↓ IL-2 signaling ↓T cell
proliferation
Sequence analysis shows mutated, duplicated or deleted IL2-RG gene
↓ IL-7 signaling Global ↓ in
lymphocyte survival
Absent thymic shadow on X-ray
↑ susceptibility to fungal infections
Complete defect in cell mediated and humoral immunity
↓ antibody responses to vaccinations
↑ susceptibility to extracellular pathogens, most notably bacteria
Authors: Kyo Farrington Reviewers: Paul Adamiak Jessica Tjong Louis Girard* *MD at time of publication
↓ IL-4 signaling ↓Th2
differentiation
↓T cell help for B cell activation and class switching
Dysfunctional B cells
Genetic Predisposition
↓ Treg development
↑ risk of developing an autoimmune disease
↓ T cell response to mitogens or
anti-CD3 stimulation
↓ or absent T lymphocytes in the blood, bone marrow and peripheral lymphoid tissues
↑ susceptibility to viral infections (often gastrointestinal ones like rotavirus and/or enterovirus)
Chronic diarrhea
Hypoplastic lymphoid tissues (I.e. tonsils, adenoids, lymph nodes)
Hypermetabolic State
Failure to thrive
↓ antibody production in response to antigen exposure
↓ IgA, IgM and IgG serum concentrations
Note: IL2-RG gene encodes the interleukin receptor common gamma chain, which is a sub-unit common to the receptor complexes for IL-2, IL-4, IL-7, IL-9,
IL-15 and IL-21. The bolded/italicized cytokines contribute most to the pathophysiology of X-linked Severe Combine Immunodeficiency (SCID).
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Re-Published October 12, 2019 on www.thecalgaryguide.com")
Innate-Immune-Response

Pathogens overcome physical barriers (e.g., epithelium, cilia)
Trauma
Damage-associated molecular patterns
Pathogen-associated molecular patterns
Examples of tissue-resident macrophages: • Alveolar macrophages – Lung
• Histiocytes – Connective tissue
• Kupffer cells – Liver
Recognition by pattern recognition receptors (e.g., toll-like receptors)
• Mesangial cells – Kidney • Microglial cells – Brain
• Osteoclasts – Bone
Microbe engulfed and exposed to oxidative burst
Microbes destroyed
Pus
Pro-inflammatory chemokines
Recruitment of circulating
granulocytes and monocytes
Pro-inflammatory cytokines (e.g., IL-1β, TNFα, IL-6)
Tissue-resident macrophage activation
Antimicrobial proteins
Unresolved infection/ inflammation
Antigen presented to T cells
Recruitment of adaptive immune response
Enhanced immune responses
Acute phase protein production by liver (i.e., C- reactive protein)
Prostaglandin production in the hypothalamus
Fever
Endothelial tight junctions on vasculature disrupted
Intravascular fluid leak into extravascular space
Edema
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published January 19, 2020 on www.thecalgaryguide.com")
pharmaceuticals-under-investigation-by-who-for-treating-covid-19-proposed-mechanisms
:
Yan Yu*, Stephen Vaughan*
* MD at time of publication
Note: This slide is based on literature available up to 03/30/2020. Medicines shown here are those included in the WHO SOLIDARITY trial, which were selected based on in vitro work and clinical data from MERS and SARS. Mechanisms are preliminary and there is insufficient data to support or refute the use of these agents for COVID- 19. Research is ongoing.
Viral replication is terminated
Fewer new cells infected
↓ activation of inflammatory and immune responses
Improvement or halt in progression of clinical signs of infection*
*See slide on pathophysiology and clinical findings of COVID19
Proposed Mechanisms
COVID-19 Viral Replication Pathway
Virus adheres to Angiotensin Converting Enzyme 2 (ACE-2) receptor on body cells
Endocytosis of virus in clathrin coated vesicles
Vesicles mature through endolysosomal pathway
Virus membrane fuses with mature endolysosome releasing viral RNA into cytosol
Viral RNA uses host cell ribosomes to make new viral proteins like RNA-polymerase
Viral RNA-polymerase incorporates nucleotides from the host cell
New viral RNA is produced
Viral RNA and proteins packaged into new viral particles
Viral particles released from cell
Chloroquine or Hydroxychloroquine (CQ, HCQ)
Weak basicity leads to ↑ pH of endosomes and lysosomes
N-terminal glycosylation of ACE-2 in Golgi is inhibited
Abnormal ACE-2 receptor expressed on cell surface
Viral membrane cannot fuse with immature endosome
Altered virus- ACE-2 interaction impairs entry into host cell
Viral contents are not released
Interferon-β (IFNβ) (Given with LPV/RTV)
Ritonavir (RTV)
(given with LPV)
Inhibition of CYP450, a drug metabolizing enzyme
↓ degradation of Lopinavir (LPV)
↑ plasma half life and duration of action of LPV
Binding to interferon receptor
Impaired maturation of endosomes
Activation of JAK/STAT pathway
Transcription of IFN- regulated genes
↑ expression of antiviral and immunomodulatory proteins
Antiviral effects may ↑ response to LPV/RTV (mechanism uncertain)
Remdesivir (RDV)
RDV is phosphorylated to RDV-triphosphate (RDV-TP)
Lopinavir (LPV)
Inhibition of viral 3- chymotripsin-like protease
Inhibition of viral replication (multiple mechanisms)
RDV-TP competes with ATP for binding to viral RNA polymerase
Viral protein precursors are not cleaved into mature viral proteins
Incorporation of RDV-TP terminates growing RNA
Newly formed viral particles can’t infect new cells
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published March 30, 2020 on www.thecalgaryguide.com")
Humoral-Immunity
 or circulate in the blood
Memory B cells proliferate and differentiate into plasma cells in response to re- exposure to antigen
↑ Rate and amplitude of secondary immune response on repeat exposure
Antigens (Ag) are produced from pathogens (bacteria, viruses, fungi, parasites) or the patient (via trauma, tumor, metabolism), & circulate in plasma, lymph, or other tissue
Clonal expansion
(proliferation of the activated B cells)
↑ WBCs (lymphocytosis) Autoimmune disease if B
cells recognize self-antigen
T cell-dependent Ag:
Ag-presenting cells (such as dendritic cells or macrophages) present Ag to CD4+ helper T cells and activate them. Activated helper-T cells then stimulate B cells
T cell-independent Ag:
Ags such as peptides, carbohydrates and lipids
may be directly recognized by B cells, triggering their activation
Complement:
Circulating serum complement proteins detect and bind Ag. Ags tagged with C3 complement fragment bind B cell co-receptor complex and enhance B cell activation.
Abbreviations:
Ab – Antibody
Ag – Antigen
Ig – Immunoglobulin WBC – White Blood Cell
Naïve B cells
Activated B cells
(in secondary lymphoid organs, such as the spleen or lymph nodes)
Plasma cells first produce IgM
Cytokines and T cells stimulate Ig class switching of B cells (changing the heavy chain constant regions of the Ig molecule)
Ig production switches from IgM to IgG, IgA, IgE, or IgD
IgG is the most common Ig in immune reactions. IgA concentrates at mucosa, IgE degranulates mast cells, IgD helps mature B cells.
Differentiation (into memory B cells or plasma cells)
Plasma cells produce antibodies, which contribute to immunity in 3 ways:
Opsonization: Abs coat pathogens, helping recognition by phagocytes
Neutralization: Abs bind to pathogen surface molecules that are needed to invade host cells, thereby neutralizing them
Activate Complement: Abs activate complement proteins via the classical pathway (see Complement Activation slide)
Clearance of pathogen by adaptive immune response
↑ Serum Ig
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published May 2, 2020 on www.thecalgaryguide.com")
pertussis-pathogenesis-clinical-findings-and-complications
 which damage mucosal cells
Pertussis toxin produces cyclic AMP (cAMP) and disrupts normal intracellular signalling, impairing the immune response initially
Pertussis (“Whooping Cough”) Respiratory syndrome consisting of severe fits of paroxysmal coughing and stridor
Nasopharyngeal swab produces positive culture and/or positive PCR result (either is diagnostic)
Initial immune dampening allows the bacteria to take hold and begin replicating. During this “incubation period”, the bacteria has not yet replicated to the point of causing symptoms.
1. Catarrhal Stage (5-10 days) After a few days, continued
damage to nasopharynx epithelial cells stimulates the immune system to ↑ its response once again
2. Paroxysmal Stage (1-2 weeks)
Tracheal cytotoxin released by B. pertussis impairs normal cilia function and ciliary beating in the trachea
3. Convalescent stage (2 weeks - months) Immune defenses successfully
eliminate the majority of B. pertussis from the respiratory tract
↑ mucus production from goblet cells of the respiratory epithelium
Mucus blocks airway, prevents air entry
Collapsed lung
Rarely, areas of chest or abdominal wall are weakened, allowing contents to bulge out
Hernia
↑ proinflammatory cytokine production
Mild fever
Cold-like symptoms
Mild dry cough, runny nose, sneezing, nasal congestion
↓ fluid clearance from the respiratory tract
Fluid in the trachea narrows tracheal diameter
“Whooping” cough
Severe, rapid and sequential coughing fits, followed by characteristic “whooping” sound on inspiration due to a stridor from a narrower trachea
Fluid build up in the lungs
Environment more susceptible to co-infection
Other bacteria colonize the lungs
Pneumonia
Paroxysmal coughing fits ↓ in frequency and number
Cough may sound louder (mechanism unknown)
but overall symptoms ↓
Some B. pertussis still remain
Residual cough flares
This stage may be prolonged in unvaccinated individuals who eliminate the bacterium more slowly
Intense cough can break ribsàsharp rib ends puncture lungàair leaks out
↑ pressure on bladder
If weak urethral sphincters:
Urinary incontinence
Cough ↑ intra- abdominal pressure
If dripping mucus triggers gag reflex while a cough is contracting abdominal muscles:
Vomiting
Coughing fits disrupt regular inspiration and ↓ oxygenation
Hypoxia
If hypoxia is profound enough to affect brain
Seizures
Abdominal muscles tire from coughing, and coughing fits make it difficult to sleep
Extreme fatigue
Rarely, violent coughing causes trauma to head
Intracranial hemorrhage
Vertebral or carotid dissection
Cerebral ischemia Coma or death
Notes:
• B. pertussis is a Gram- negative strict aerobe
• An effective vaccine exists to prevent infection by B. pertussis
• Pertussis most commonly infects children <18 months prior to completion of scheduled vaccination series, or adolescents with ↓ immunity
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published October 4, 2020 on www.thecalgaryguide.com
Include a sentence!
When sending in the final draft of a slide, please include in the email a sentence that describes the slide that you've produced in detail. This will help us boost the relevance of the slide with search engines.
E.g., Disease X presents with symptom Y due to pathophysiology Z
Pertussis presents with fits of severe paroxysmal coughing due to impaired mucociliary clearance.")
Bronchiolitis-updated
 - but can be others such as rhinovirus,
adenovirus, parainfluenza, influenza, and coronaviruses - initially colonizes the nasopharyngeal mucosa Virus travels via the epithelium to the lower airways to the terminal bronchioles (small airways)
Authors: Nick Baldwin Rebecca Lindsay Reviewers: Kayla Nelson, Yan Yu Timothy Fu, Danielle Nelson* *MD at time of publication
Upper airway mucosal inflammation
Bronchiolitis
(bronchiole inflammation)
Apnea
(cessation of breathing; via unknown mechanism, potentially apnea reflex)
RSV-fusion protein facilitates fusion of the virus to the host cell and directs viral penetration as well as facilitates fusion of the infected cell with its healthy neighbors
Forms syncytia (multinucleated cells)
Cytokines are released into circulation
↑ thermo- regulatory set- point at the hypothalamus
Mild Fever
Copious coryza
(nasal discharge)
Protein and fluid leak into nasopharyngeal interstitium
↑ Capillary permeability
Protein and fluid leak into the bronchiole interstitium, accumulating around airway walls
Airway wall becomes thickened, more readily apparent on x-ray
Peribronchial cuffing
(X-ray finding: bronchi appear like thickened ‘cuffs’ when viewed head-on)
Inflammation stimulates the upregulation of mucous secreting goblet cells
↑ mucous production
Mucous within alveoli ↑ intra- alveolar surface tensionà collapsing alveolar walls
During inspiration, air enters the collapsed alveoli if airway is not yet occluded
↑ intra-alveolar pressure causes the alveoli to suddenly pop open
Inspiratory crackles on auscultation
Syncytia slough off the bronchial epithelium into airways
Airways become narrower and occlude
Disruption of the ciliated epithelial cells (which transport mucous out of the airways and into the pharynx, to be swallowed or evacuated)
↓ mucous clearance from airways
Excess airway mucous triggers cough reflex
Cough
Interstitial edema
Nasal Congestion
Air is absorbed distal to occlusion (gas trapping)
When all air is absorbed, alveoli collapse (resorptive atelectasis)
↓ gas exchange between blood and air in remaining alveoli
↓ O2 saturation & ↑ CO2 content of blood
Narrower airways, especially during expiration, causes audible turbulent airflow
Wheeze on auscultation
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Re-Published November 29, 2020 on www.thecalgaryguide.com")
mrna-vaccines-against-coronavirus-disease-2019-covid-19-production-and-mechanism-of-action
:
Production and Mechanism of Action
Vaccine Production
The “spike protein” is known to be a major viral surface
Authors: Ryan Brenneis, Yan Yu* Reviewers: Davis MacLean, Hannah Yaphe, Timothy Fu, Stephen Vaughan* * MD at time of publication
References
1. ACS Nano 2020, 14, 10, 12522–12537, Publication Date: October 9, 2020, https://doi.org/10.1021/acsnano.0c07197
2. NEJM 2020, Publication Date: December 10, 2020, DOI: 10.1056/NEJMoa2034577
3. Expert Review of Vaccines 2017, 16, 9, 871-881, Publication Date: 2017, DOI: 10.1080/14760584.2017.1355245
4. NEJM 2020, 383, 2439-2450, Publication Date: December 17, 2020, DOI: 10.1056/NEJMoa2027906
5. NEJM 2020, 383, 2427-2438, Publication Date: December 17, 2020, DOI: 10.1056/NEJMoa2028436
6. BMJ 2000, 321, 7271, 1237-1238, Publication Date: November 18, 2000, DOI: 10.1136.bmj.321.7271.1237
Notes:
SARS-CoV-2 (RNA virus causing COVID-19) collected from an infected patient (ie. with a nasopharyngeal swab)
Various reagents are added to the sample containing both human cells and viral constituents
Reagents cause cell/viral membrane lysisàspilling cell contents, viral particles, and viral RNA
Fats, proteins, and carbohydrates removed through various washing reagents, leaving nucleic acids (like RNA)
Reverse transcriptase polymerase chain reaction produces complementary DNA (cDNA) from viral RNA
cDNA library allows for SARS-CoV-2 genome to be mapped through whole-genome sequencing technology
SARS-CoV-2’s spike protein DNA sequence is identified, and is used as a template to create synthetic viral spike protein mRNA
Extra RNA bases are added to this mRNA strand to promote its stabilityàresulting RNA strand is now called “nucleoside-modified RNA” (“modRNA”)
antigen (substance that elicits an immune response) from studies of other coronaviruses (e.g. SARS- CoV-1 and MERS-CoV)
Pfizer/BNT162b2 vaccine contents:
Moderna mRNA-1273 vaccine:
Note: Lipid nano- particles are spherical hollow “balls” made of an outer lipid membrane plus other emulsifiers and membrane stabilizers.
Lipid nanoparticles are capable of engulfing smaller molecules (like RNA) and merging with normal cell membranes
Spike protein modRNA is then isolated (using a series of precipitation, extraction, and chromatography methods)
Final modRNA lipid nanoparticle vaccine is now created and ready for intramuscular injection
The modRNA vaccine is injected intramuscularly into a healthy person 2nd dose after 3-4 weeks needed to strengthen the immune response
(to a level exceeding the immune response in patients recovered from Covid-19), boosting vaccine efficacy especially in older individuals4,5
Lipid nanoparticle fuses with human cells’ phospholipid membranes via endocytosis, releasing modRNA into the cell’s cytosol
modRNA is translated by human ribosomes naturally found in the cell’s cytosol, producing viral spike protein components
•
•
Foreign substance can cause local tissue inflammation
The spike proteins encoded by the modRNA of each of the two vaccines are similar
It is the proprietary lipid nanoparticle formulation (unknown to the public) that is unique to each vaccine
Pain, redness, swelling at injection site (Transient)
Proprietary Pfizer/ BioNTech lipid nanoparticle
The modRNA encodes a
full-length spike protein modified with two proline amino acids (for stability and immunogenicity)2
The modRNA encodes a full-length spike protein modified with two proline amino acids (for stability)1
Proprietary Moderna lipid nanoparticle
Encapsulating this modRNA within Pfizer/ BioNTech’s lipid nanoparticle creates the 162b2 vaccine
This specific formulation requires colder storage temperatures (-700C)
Encapsulating this modRNA within Moderna’s lipid nanoparticle creates the mRNA-1273 vaccine
This specific formulation can be stored at slightly warmer temperatures (-200C)
Muscles are preferred injection sites as they have greater blood supply than other body tissues
Vaccine Action
Able to bring in immune cells faster to process foreign antigens6
Able to drain away foreign vaccine material fasterà minimizing local reactions6
Cell-mediated Immunity
Spike protein degraded by intracellular enzymes into fragments
Humoral Immunity
Natural cellular processes release spike protein components from the cell into the bloodstream
Spike protein components are engulfed by antigen presenting cells (dendritic cells, B cells, macrophages), fragmented, & bound to unique MHC Class II proteins
MHC Class II proteins bring spike protein fragments to the antigen presenting cell’s surface, to present them to circulating naïve CD4+ (helper) T cells
Some naïve helper T cells are able to successfully bind to the spike protein-MHC Class II protein complexes
Binding activates these spike-protein specific helper T cells
Spike protein fragments bound by MHC Class I proteins
MHC Class I proteins bring spike protein fragments to the human cell surface
MHC Class I proteins present spike protein fragments to naïve CD8+ T cell
Naïve CD8+ T cells that able to bind to the spike protein-MHC Class I protein complex become activated, and travel to the lymphatic system to mature
MHC = Major Histocompatability Complex; cell surface proteins key to immune function
CD = Cluster of Differentiation; glycoproteins on T cell surfaces that are co-receptors and facilitate T cell binding to antigens/MHC complexes. They also distinguish the types of T cells.
Some of these T-Cells mature into cytotoxic T cells that now recognize the viral spike protein
Cytotoxic T-cells bind to human cells infected with SARS-CoV-2 expressing spike protein or spike protein fragments
Cytotoxic T cell releases enzymes perforating infected cell, causing cell death to occur
Immune system identifies and destroys human cells infected with SARS-CoV-2, slowing viral spread
Other T cell’s can mature into memory T cells (stimulated by cytokines released by helper T cells)
Memory T cells travel to lymphatic tissue, awaiting activation from future exposure to spike protein
More rapid cell-mediated immune response to future SARS-CoV-2 infection (immunity)
Activated helper T cells specific to the viral spike protein secrete cytokines to stimulate immune activity
Systemic cytokine releaseàsystemic reactions like fever, chills, fatigue, myalgias (Transient)
Some B cells mature into plasma cells that produce IgG antibodies against the viral spike protein
Antibodies to spike protein mark SARS-CoV-2, allowing immune system to destroy virus
Eradication of SARS-CoV-2 in extracellular compartments
Activated helper T cell interacts with
naïve B cells in lymphatic tissue
Some B cells mature into memory B cells specific to SARS-CoV- 2 spike protein
Future exposure to spike protein re-activates memory B cell in lymphatic tissue & creates plasma cells, producing antibodies more rapidly
Rapid humoral immune response to future SARS-CoV-2 infection (immunity)
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Clinical Finding
End Result
Published December 19, 2020 on www.thecalgaryguide.com")
Adenovirus-Vector-Vaccines-Against-COVID19-Production-and-Mechanism-of-Action
, a mild human adenovirus, is isolated
Previous exposure to Ad5 or Ad26 may have sensitized immune system to the adenovirus vector1
Potential for human adenovirus vaccine to fail due to previous exposureàimmunity not built against spike protein
CanSino
Adenovirus type 5 (Ad5), a mild human adenovirus, is isolated
Oxford/AstraZeneca
Chimpanzee adenovirus AZD1222 (ChAdOx1), previously shown to be safe & to elicit an immune response in humans2, is isolated
Vaccine Production
SARS-CoV-2 (virus causing COVID-19) synthetic DNA library sequenced from viral RNA using reverse transcriptase polymerase chain reaction and whole genome sequencing technology
Spike protein DNA sequence isolated from SARS-CoV-2 genome
A promoter sequence is added to the spike protein DNA sequence, allowing human RNA polymerase to recognize and transcribe the spike protein DNA when introduced into human cells
Recombinant genetic technology inserts the modified spike protein DNA into a plasmid: a circular piece of DNA that acts as a shuttle allowing for the insertion of new genes (such as the spike protein gene) into host genomes (like the adenovirus vector DNA genome)
Adenoviral DNA isolated using various lytic & washing reagents (chemicals that break open cell membranes and remove non- nucleic acid cellular materials)
Adenoviral DNA sequenced using whole genome sequencing, then modified as follows:
Chimpanzee virus negates possibility of previous immunity to the viral vector1
Chimpanzee viral vector more likely to successfully
generate immune response to the spike protein
E1 region of adenoviral genome E3 region of adenoviral genome deleted to create deleted to block viral replication3 room for insertion of SARS-CoV-2 spike protein DNA3
Adenovirus used in the final vaccine cannot replicate
within human cells and cannot cause human disease
References
1. ACS Nano 2020, 14, 10, 12522–12537, Publication Date: October 9, 2020, https://doi.org/10.1021/acsnano.0c07197
Modified adenoviral DNA genome is reinserted into The viral vector & the spike protein plasmid are mixed together, and DNA recombination
the adenovirus particle, creating the “viral vector”
technology inserts the spike protein gene from the plasmid into the adenovirus DNA2
Adenovirus containing transcribable SARS-CoV-2 spike protein DNA is introduced into a special cellular culture, allowing the virus to replicate despite its modified DNA2, 3
Authors: Ryan Brenneis, Yan Yu* Reviewers: Davis MacLean, Hannah Yaphe Stephen Vaughan* * MD at time of publication
2. Nature 2020, 586, 578–582, Publication Date: October 20, 2020, https://doi.org/10.1038/s41586- 020-2608-y
3. Frontiers in Immunology 2018, 9, 1963, Publication Date: September 19, 2018, doi: 10.3389/fimmu.2018.01963
4. NPJ Vaccines 2020, 5, 69, Publication Date: July 27, 2020, doi: 10.1038/s41541-020-00221-3
5. The Lancet 2020, Publication Date: Dec. 8, 2020, https://doi.org/10.1016/S0140-6736(20)32623-4
6. BMJ 2000, 321, 7271, 1237-1238, Publication Date: November 18, 2000,
DOI: 10.1136.bmj.321.7271.1237
7. NEJM 2021, Publication Date: Jan. 13, 2021, DOI: 10.1056/NEJMoa2034201
Adenovirus containing transcribable SARS-CoV-2 spike protein DNA is isolated and concentrated to a high enough level for administration as a vaccine
Adenoviruses have an outer protein layer (called a capsid) to protect its DNA
DNA is more stable than mRNA due to deoxyribose sugar backbone and intermolecular bonds between strands
Enhanced stability compared to mRNA lipid nanoparticle vaccines
Can be stored at 2-8°C for up to 3-6 months
Muscles are preferred injection sites as they have greater blood supply than other body tissues
Immune cells arrive faster to The viral vector vaccine is injected intramuscularly into a healthy person process foreign antigens6
Foreign substance can cause local tissue inflammation
Pain, redness, swelling at injection site (Transient)
Note: The Johnson and Johnson vaccine may be 90% effective after a single dose7
Foreign vaccine material drains away fasterà minimizing local reactions6
2nd dose after 28 days recommended to strengthen the immune response (to a level exceeding the immune response in patients recovered from Covid-19), boosting vaccine efficacy especially in older individuals5
Vaccine Action
Cell-mediated Immunity
Spike protein degraded by intracellular enzymes into fragments
Adenovirus surface antigens interact with human cellular receptors, allowing viral entry into human cell via endocytosis3 Adenovirus vector travels to cell nucleus, merges with nuclear envelope and injects its DNA (including the spike protein DNA) into the nucleus
RNA polymerases in the nucleus transcribe the viral DNA, making messenger RNA (mRNA) for SARS-CoV-2 spike protein
mRNA is transported back into the cytosol & translated by ribosomes naturally found there, producing full length SARS-CoV-2 spike protein
Humoral Immunity
Natural cellular processes release spike protein components from the cell into the bloodstream
Spike protein components are engulfed by antigen presenting cells (dendritic cells, B cells, macrophages), fragmented, & bound to unique MHC Class II proteins
MHC Class II proteins bring spike protein fragments to the antigen presenting cell’s surface, to present them to circulating naïve CD4+ (helper) T cells
Some naïve helper T cells are able to successfully bind to the spike protein-MHC Class II protein complexes
Binding activates these spike-protein specific helper T cells
Spike protein fragments are bound by MHC Class I proteins
MHC Class I proteins bring spike protein fragments to the human cell surface MHC Class I proteins present spike protein fragments to naïve CD8+ T cell
Naïve CD8+ T cells that able to bind to the spike protein-MHC Class I protein complex become activated, and travel to the lymphatic system to mature3
MHC = Major Histocompatability Complex; cell surface proteins key to immune function
CD = Cluster of Differentiation; glycoproteins on T cell surfaces that are co-receptors and facilitate T cell binding to antigens/MHC complexes. They also distinguish the types of T cells.
Some of these T cells mature into cytotoxic T cells that now recognize the SARS-CoV-2 spike spike protein
Cytotoxic T cells bind to human cells expressing spike protein or spike protein fragments (e.g. future COVID-19 infection)
Cytotoxic T cells release enzymes perforating infected human cells, causing cell death to occur
Immune system can now more quickly identify & destroy human cells showing signs of COVID-19 infection
Some T cell’s can mature into memory T cells (stimulated by cytokines released by helper T cells)
Memory T cells travel to lymphatic tissue, awaiting activation from exposure to spike protein in the future
More rapid cell-mediated immune response to
future SARS-CoV-2 infection (immunity)
Activated helper T cells specific to the viral spike protein secrete cytokines to stimulate immune activity
Systemic cytokine releaseàsystemic reactions like fever, chills, fatigue, myalgias (Transient)
Note: Duration of cellular/ humoral immunity is unknown
Some B cells mature into plasma cells that produce IgG antibodies against the viral spike protein
Antibodies to spike protein mark SARS-CoV-2, allowing immune system to destroy virus
Eradication of SARS-CoV-2 in extracellular compartments
Activated helper T cell interacts with naïve B cells in lymphatic tissue
Some B cells mature into memory B cells specific to SARS-CoV- 2 spike protein
Future exposure to spike protein re-activates memory B cell in lymphatic tissue & creates plasma cells, producing antibodies more rapidly
Rapid humoral immune response to future SARS-CoV-2 infection (immunity)3
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Clinical Finding
End result
Published February 11, 2021 on www.thecalgaryguide.com")
AAA-Pathogenesis
: Pathogenesis
Different parts of the aorta have different embryologic origins
Atherosclerosis
Hypertension
Age > 65
Progressive deterioration of aorta structural integrity over life span
Connective Tissue Disease
Structurally abnormal protein or protein organization in aorta
Autoimmunity
Infection (e.g Chlamydia, Mycoplasma pneumoniae, Helicobacter pylori, human cytomegalovirus, herpes simplex virus)
Antigens (substance that causes immune response) on virus or bacteria resemble local proteins in abdominal aorta
Antibodies produced in response to infection inappropriately target host cells in the aorta
Antibodies tag cells in the abdominal aorta for destruction by T-lymphocytes
Immune-mediated destruction of aorta
Smoking
Genetics
Unclear mechanisms
Subacute (not clinically detectable) inflammation of aortic tissue
Inflammatory cytokines are released and immune cells are recruited
↑ pressure on aorta and other vessel walls
Infiltration of vessel wall by lymphocytes and macrophages
Production of enzymes that break down elastin & collagen proteins (which provide most tensile strength to aorta)
Aorta susceptible to damage
Degradation of aortic connective tissue
Biomechanical stress on vessels
Authors: Olivia Genereux Davis Maclean Reviewers: Jason Waechter* Amy Bromley* Yan Yu* *MD at time of publication
The exact mechanisms are complex, debatable, and an area of intensive research – the 3 mechanisms and associated pathophysiology presented here are generally thought to be the main causes of abdominal aortic aneurysms
Infrarenal aorta has poorly developed vaso vasorum (dedicated blood supply to vessel wall)
Infrarenal aorta relies solely on nutrient diffusion from aortic blood that crosses abdominal aorta
Infrarenal aortic wall has fewer “lamellar” units (fibromuscular units) than other regions of the aorta
Infrarenal aorta is less elastic & less able to distribute stress
Loss of smooth muscle cells & thinning of tunica media
Destruction of elastin in tunica media
Normal layers of the aortic wall
↓ aortic tensile strength (ability to withstand stretching) Aorta expands and dilates due to internal pressure
Tunica Intima (inner-most tissue layer of aorta)
Tunica Media (layers of elastic
tissue (elastin) and muscle fibers)
Adventitia (thin outermost collagenous layer)
(longitudinal section)
Aortic aneurysms are usually infrarenal (85%)
Abdominal Aortic Aneurysm
Infrarenal aorta more prone to ischemia and has impaired repair potential
Abnormal, irreversible dilation of a focal area of abdominal aorta (area of aorta between diaphragm & aortic bifurcation) to twice the diameter of adjacent normal artery segment
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Re-Published February 27, 2021 on www.thecalgaryguide.com")
Bacterial-Osteomyelitis

Trauma:
Penetrating injury or open fracture introduces bacteria directly to bone
Hematogenous:
Blood carries bacteria from distant site of infection to bone
Contiguous:
Neighboring site of infection seeds bacteria directly to bone
Bacterial Osteomyelitis
Innate Immune cells and local mast cells release vasoactive cytokines
Capillaries dilate to ↑blood flow to affected area and become more permeable to allow for exit of immune cells and plasma contents
Localized Immune response
Poorly understood mechanismsàinnate immune systems unable to clear the bacteria in some individualsàchronic infection
Immune cells produce pyrogenic intermediates (i.e. PGE2, IL-6, and IL-1)
Pyrogenic intermediates travel through the
bloodstream to the hypothalamus and alters the body’s thermal setpoint
Fever
Immune cells produce inflammatory cytokines (i.e. TNFα and IL6)
Systemic inflammatory cytokines result in the production of Non-specific acute- phase reactants
High Serum CRP and ESR
Warmth Erythema
Progressive blood vessel occlusion by immune cellular and bacterial debris causing infarct and necrosis of bone
More permeable capillaries allow plasma to leak into surrounding periosteal tissue
More permeable capillaries allow macrophages and neutrophils to exit the capillaries at the site of infection
Immune cells phagocytize bacteria, producing an of opaque off-white fluid
Pus
Immune cells sequester necrotic bone tissue into a regional abscess within medullary bone
Sequestrum seen as localized opacity on CT or late X-ray imaging
↑ Osteoclast activity to remove damaged medullary bone and ↑ Osteoblast activity to reform new bone
Osteoclasts and osteoblasts remodel and deposit new bone in area surrounding necrotic tissue
Swelling
↑ Periosteal fluid applies ↑ pressure to surrounding nerve endings
Bone Pain manifesting as refusal to use limb
New layer of bone formed
Positive Bone Scan in area of infection
Abbreviations:
• IL – Interleukin
• PGE2 – Prostaglandin E2
• TNFα – Tumor necrosis factor alpha
Involucrum / Periosteal Reaction: seen as regional luminance surrounding sequestrum on CT or late X-ray imaging
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published May 21, 2021 on www.thecalgaryguide.com")
Dry-Eye-Syndrome-Pathogenesis
: Pathogenesis
The Pathophysiology of Dry eye disease is complex and an area of active investigation – The mechanism and causes presented here represented the highest yield causes and mechanism for students
Post laser eye surgery
Disruption of corneal nerves
↓ corneal sensitivity
Damage to trigeminal nerve, the sensory innervation of the eye (due to: Herpes Zoster, tumor, trauma, etc)
Blepharitis (eyelid inflammation)
Many medications can cause dry eye via
multiple mechanisms presented here (e.g. ↓ corneal sensitivity, Meibomian gland dysfunction, lacrimal gland atrophy)
Sex Hormones (e.g. androgens & estrogens) play a complex and poorly understood role in mediating dry eye disease (net effect is that women are more often affected by dry eye)
Obstructed meibomian glands
Eyelid damage Gland atrophy
These items represent general causes of meibomian gland dysfunction – exact causes are numerous, their pathophysiology is beyond the scope of this slide
Contact lens (long term use)
Corneal nerve adaptation to chronic mechanical stimulation
Autoimmune disease (e.g. Sjögren's syndrome)
Chronic inflammatory infiltration of the lacrimal gland (and salivary gland)
Autoimmune Lymphocytic infiltration
Inflammatory cytokine release
Autoantibody production
Cell death and apoptosis
Lacrimal gland degeneration
Meibomian gland dysfunction (Located along the eyelid margins, these glands produce meibum, an oily substance that prevents evaporation of the tear film)
↓ meibum secretion Loss of lipid layer
covering the eye, ↓ the barrier that blocks evaporation of tear film
Tear Film instability
Lifestyle
Extended reading or
TV or electronic device uses
Exposure Keratopathy (any condition causing dryness due to incomplete or inadequate eyelid closure, e.g. Bell’s Palsy)
↓ activity of the afferent portion of
corneal reflex arc (responsible for reflex tearing: tearing in response to irritation of the eye)
Mechanical damage to goblet cells
secrete mucins – a substance that lubricates the eye and preserves tear film
↓ blink rate
↑ time and area
↓ normal reflex tearing
for evaporation
Dry climate Wind exposure
Infiltrative diseases
(e.g. sarcoidosis)
Lacrimal gland infiltration
↓ Lacrimal gland secretion of the the watery aqueous layer of the tear film (Aqueous deficient dry eye)
Deficient or unstable tear film (Evaporative dry eye)
↑ tear evaporation
Direct damage to lacrimal gland (e.g. infection or trauma of the eye)
Authors: Davis Maclean, Yan Yu*, Michael Penny, O.D.
Reviewers: Natalie Arnold, Saleel Jivraj, O.D., Adam Muzychuk*, Victor Penner* *MD at time of publication
Hyperosmolar Tear Film (hyperosmolarity = ↑ solutes and ↓ solvent)
(Further) Tear Film instability
Corneal and conjunctival epithelial
cells dry out, including goblet cells (which secrete mucins – a substance that lubricates the eye)
Inflammatory immune response àRecruitment and activation of CD4+ (Helper) T-Cells, further produce cytokines
Further irritation and damage to ocular surface structures (cornea, conjunctiva and Meibomian glands) and lacrimal glands
See Calgary Guide: “Dry Eye Syndrome
(Kerato- conjunctivitis sicca):Clinical Findings” for signs and symptoms
Dry Eye Syndrome (Keratoconjunctivitis sicca): A multifactorial disease of the ocular surface and tears characterized by loss of tear film homeostasis, tear film hyperosmolality and inflammation
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published August 7, 2021 on www.thecalgaryguide.com")
covid-19-pathophysiology-and-clinical-findings
: Pathophysiology and Clinical Findings
Authors: Ryan Brenneis, Yan Yu* Reviewers: Ciara Hanly, Yonglin Mai ()*, Stephen Vaughan* * MD at time of publication
-Respiratory failure -Septic shock -Multiple organ dysfunction
Critical
Respiratory droplet production (cough, sneeze, talking, breathing) by human host or animal vector infected with SARS-CoV-2
Note: Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2) is the name of the betacoronavirus (a positive sense, single stranded RNA virus). COVID-19 is the name of the disease caused by this virus.
rate, ↓ oxygen saturation -Bilateral interstitial infiltrates on Chest X-ray, progressively worsening
Small droplets (<5μm in diameter) are aerosolized and become airborne
Inhalation of aerosolized particles that are suspended in air
Patient exposed to the virus (SARS-CoV-2)
Droplet contacts the mucus membranes (eyes, nose, mouth) of recipient
Live viral particles can adhere to inanimate objects called fomites (e.g. doorknobs)
Symptoms are on a continuum -Worsening dyspnea: ↑ respiratory
-Fever
-Cough
-Myalgia, fatigue -Nausea/vomiting -Diarrhea
-Loss of taste/smell
Recipient touching infected fomite subsequently touches any of their mucus membranes
Mild
Moderate
Severe
Virus spreads in the body via 1) mucus membrane spread to surrounding cells and 2) entering the blood
Virus adheres to angiotensin-converting enzyme 2 (ACE-2) receptor on body cells, mimics ACE-2, & gains access into cell
COVID-19
Symptomatic infection with SARS-CoV-2
Viral proliferation in cells of tissues with more ACE-2 receptors: lungs (type II pneumocytes); vasculature (endothelial cells), kidneys (proximal tubular epithelium), heart (myocardium), GI tract (enterocytes)
Cell death and ↑ in inflammatory cytokines triggers immune response
Neutrophils move to lungs, release reactive oxygen species and cytokines
Alveolar/capillary damage
Fluid accumulates in interstitium and alveoli
↑ Distance for O2 to diffuse from alveoli to capillaryà ↓ blood O2 saturation
Cytokines induce the hypothalamus to release prostaglandins
↑ body temperature set point to fight infection
Virus disassembles, releasing viral RNA, which uses host cell’s ribosomes to make new viral proteins like RNA-polymerases
Newly made viral RNA-polymerases use cell’s own nucleotides to synthesize new viral RNA
Bilateral ground-glass opacities (CT lung) & interstitial infiltrates on Chest X-ray
Dyspnea
Airway irritation
Cough
Myocardial cell damage
↑ Troponin
↑ skeletal muscle cell damage
Viral RNA & proteins packaged into new viral particles
New virus assembled and released from cell, killing the cell and contributing to disease symptoms
Average incubation period (time from initial infection to symptom onset) is 4-5 days; can be up to 14 days
Heart tries to compensate for
Fever Myalgia
hypoxemiaà↑ cardiac output Arrhythmogenic
and strain on myocardium
state
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published March 22, 2020, updated Aug 18, 2021 on www.thecalgaryguide.com")
asthma-pathogenesis
 Naushad Hirani* * MD at time of publication
Genetic factors
(i.e. HLA gene mutations, defects in bronchial airway epithelium)
Environmental factors
(i.e. excess hygiene, fewer siblings, antibiotics within the first two years)
Asthma:
Defined as airway hyper-responsiveness causing variable and reversible airflow obstruction
Atopy:
predisposition to allergic hyper-sensitivity in airways
First exposure to triggers*
sensitizes helper T cells
Stimulation of B-cells to produce IgE, which binds to mast cell surfaces
Activated Helper-T cells & IgE-sensitized mast cells now line the airways
Triggers of airway hyper- responsiveness include:
Upper respiratory tract infections (URTIs)
Allergens (pollen, animal dander, dust, mold, etc)
Air pollution, cigarette smoke, other chemicals
Drugs (aspirin, NSAIDs, Beta- blockers)
Cold air
Exercise
Early response (0-2 hrs)
Delayed response (3-4 hrs)
Allergens cross-link IgEs on mast cells
Activated mast cells & helper T cells release cytokines
Mast cells release histamines, leukotrienes, and other inflammatory mediators
Induce maturation of granular WBCs like eosinophils
Eosinophils migrate into:
Vascular permeabilityà edema of airway mucosa
Goblet cell hyperplasia à mucus secretion
Bronchial smooth muscle contraction
Airway obstruction
Second exposure to triggers
Asthma Airways Bronchiole constriction
Eyes Conjunctivitis Nose Rhinitis
Note: Delayed response presents within 3-4 hrs, peaks within 6-8 hrs, and resolves within 24 hrs
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published Dec 17, 2012, updated Aug 19, 2021 on www.thecalgaryguide.com")
Pneumoconioses

Inhalation of carbonaceous dust
(Carboconiosis)
Inhalation of metal dust
(Metaloconiosis)
Inhalation of silica dust
(Silicosis)
“-Coniosis”: disease which comes from inhaling dust particles
Internalized asbestos fibers disrupt cellular processes through a complex series of theorized mechanisms
Inhaled dust particles (1-5μm in size) are trapped and deposited in the alveolar airspaces
Alveolar macrophages ingest dust particles, which activates the macrophages and sometimes causes apoptosis
Activated macrophages create an inflammatory microenvironment by releasing pro-inflammatory cytokines, chemokines and reactive oxygen species
Inflammatory products (e.g. reactive oxygen species) damage alveolar epithelial cells, causing activation and release of inflammatory cytokines into the alveolar space
Inhaled dust particles, inflammatory products & other pro- tussive mediators activate airway vagal afferent receptors
Activated receptors stimulate the cough center located in the medulla oblongata
Cough
Alveolar capillaries vasoconstrict in response to hypoxia à↑ Pulmonary vascular resistance
Pulmonary Hypertension
Right heart must pump blood into lungs against higher pressure àcardiomyocyte growth (via sarcomeres formed in parallel within myofibrils)àconcentric hypertrophy of right heart
Cor Pulmonale (right heart failure due to pulmonary hypertension)
Asbestos fibers accumulate in the airspace and translocate to the pleural surface
Macrophage apoptosis: ↓ alveolar macrophages
Fibroblasts are recruited to the alveolar wall and are activated
Activated fibroblasts produce and deposit collagen in the
extracellular space between alveoli
Thickening of tissue between alveoli and capillaries ↑ the diffusion distance of atmospheric and blood gasses
↓ Diffusion of CO2 from blood to alveoli and ↓ diffusion of O2 from alveoli to blood
↑ Respiratory rate to maintain minute ventilation due to ↓ lung volumes and diffusion limitations
Dyspnea and Exertional Hypoxemia
↓ Innate immune response in the lungs
Chest X-Ray: Nodular and reticulonodular opacities are seen in varying lung regions depending on the underlying inhaled dust
Excessive collagen
deposition ↓ lung compliance and ↓ lung expansion
↓ Diffusing capacity for carbon monoxide (DLCO) on pulmonary function test
↓ Arterial oxygen contentà ↑ deoxyhemoglobin and ↓ oxyhemoglobin
↑ Deoxyhemoglobin within the vasculature causes the skin and mucous membranes to appear blue
Cyanosis
↑ Risk of respiratory infections. Mycobacterial infections (e.g. tuberculosis) associated primarily with silicosis
↓ Inhalation volume
↓ Expiratory volume
Hypoxemia
↓Total lung capacity (TLC) and ↓ residual volume (RV) on pulmonary function test
↓Forced vital capacity (FVC) and ↓ forced expiratory volume in 1 second (FEV1) on pulmonary function test
Note:
Forced Vital Capacity: the volume of air that can forcibly be blown out after full inspiration
Forced Expiratory Volume in 1 second: the volume of air that can forcibly be blown out in first 1 second, after full inspiration
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published September 12, 2021 on www.thecalgaryguide.com")
Hypersensitivity-Pneumonitis
*, Kerri Johannson* * MD at time of publication
Farming and Compost
Farmer’s Lung (Common)
Bird and Animal Proteins
Bird Fancier’s Lung (Common)
Water Contamination and Ventilation
Organic antigen identified by dendritic cells
Manufacturing and Chemical Workers
Grain and Flour Processors
Notes:
Immune complex - Production of IgG antibodies (Type III hypersensitivity)
Cell mediated - Sensitization of helper T cells (Type IV hypersensitivity)
• Thereare3typesofhypersensitivity pneumonitis: acute, subacute, chronic
• InacuteHP,removalofincitingantigen results in resolution of symptoms within days.
*Lymphoplasmocytic interstitial infiltrate
with bronchiolocentric distribution on pathology
Epithelial injury (exact mechanism unknown)
*Organizing pneumonia
Dyspnea, tachypnea, and crackles
Abbreviations:
• HP – Hypersensitivity Pneumonitis
• PFT – Pulmonary Function Test
↑ Neutrophils, mast cells, macrophages, CD8+ T cells, & inflammatory cytokines
• *TriadofmainfindingsforsubacuteHP Systemic release of
cytokines disrupts hypothalamic regulation
Fever
Macrophages ingest antigens
*Poorly formed granulomas on pathology
Tissue breakdown from neutrophils activates fibroblasts, which deposit collagen
CT: Normal or diffuse ground- glass opacity (acute)
Chronic deposition of collagen replaces normal lung parenchyma by scar tissues (Chronic Findings)
Neutrophil elastase breaks down lung elastic fibers
Tissue destruction of alveolar walls creates larger air spaces
CT: Emphysema
CT: Honeycombing
(End stage Iung disease)
Pathology: Advanced interstitial fibrosis
PFT: Restrictive pattern ↓FEV1, ↓FVC, and ↓ DLCO
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published September 1, 2016, updated October 5, 2021 on www.thecalgaryguide.com")
Twins Mechanisms and Complications

Allows multiple follicles to mature
IVF (multiple embryo transfer)
Ovulation induction/ superovulation fertility medications
Embryo splits during days 0-3
Embryo splits during days 4-8, after trophoblast cells (outer layer of blastocyst) have already invaded the endometrium and formed part of the placenta
Each embryo forms its own amniotic sac, both fed by 1 placenta
Monochorionic (1 placenta) Diamniotic (2 amniotic sacs) Monozygotic Twins
Embryo splits during days 9-12, when both placental and amniotic sac development have started
The embryos must now develop within a shared amniotic sac & placenta
Monochorionic (1 placenta) Monoamniotic (1 amniotic sac) Monozygotic Twins
Two ova ovulatedàEach ovum fertilized by a separate sperm
Cells still undifferentiated at this stage, allows each embryo to develop it’s own placenta and amniotic sac
Dichorionic (2 placentas) Diamniotic (2 amniotic sacs) Monozygotic Twins
Two embryos develop that implant separately into the endometrium and develop their own placenta and amniotic sac
Dizygotic Twins (Dichorionic, Diamniotic by default)
Complications of all types of twins:
Overdistention of the uterus
Weakened uterine muscles leading to uterine atony
Postpartum Hemorrhage (see relevant slide)
Larger placental mass without ↑ placental blood flow
Placental hypoperfusion (plus other complex mechanisms)
Gestational Hypertension and Pre- Eclampsia
↑ plasma volumeà diluting red blood cells, plus ↑ fetal iron consumption from mother’s stores
Anemia
High β- HCG
Mechanism unknown
Hyperemesis Gravidarum
Operative Delivery
Preterm Birth
Insufficient maternal nutritional supplies
Abnormal placental vascular connections
Fetal Death
Interplacental vascular connections lead to uneven distribution of nutrients and blood flow
Twin-Twin Transfusion Syndrome (growth discordance between twins, see relevant slide)
Note: all complications are more likely in monozygotic twins
Umbilical cords become entangled within shared amniotic sac
Cord Entanglement
Authors: Jemimah Raffé- Devine, Yan Yu* Reviewers: Brianna Ghali Jadine Paw* *MD at the time of publication
Uterine crowding
↑ risk of fetal malpresentation
Less space in uterus for continued growth
Congenital Anomalies
Intrauterine Growth Restriction
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published October 18, 2021 on www.thecalgaryguide.com")
Drug Reaction with Eosinophilia and Systemic Symptoms (DRESS)
:
Authors: Lauren D. Lee, Harry C. Liu* Reviewers: Mehul Gupta, Brian Rankin, Julia Chai, Stephen Williams Yan Yu* Laurie Parsons* *MD at time of publication
Pathogenesis and clinical findings
Genetic susceptibility
Certain HLAs (e.g., HLA-B*58:01, HLA-B*57:01, and HLA-A*31:01)
HLA alleles encode for MHC structure and may influence how specific drugs/drug metabolites interact with T cell receptors and MHC proteins on antigen-presenting cells
Exposure to offending drug
E.g., Aromatic anticonvulsants (lamotrigine, carbamazepine, and phenytoin), allopurinol, and sulfonamides
Drug-specific CD4+ and CD8+ T cells produce tumor necrosis factor-alpha and interferon gamma
↑ activated T-cells 2-6 weeks after drug exposure
Drug Reaction with Eosinophilia and Systemic Symptoms (DRESS)
Latent Viral infection
Latent viruses (HHV-6, HHV- 7, CMV, and EBV) concealed in regulatory T-cells
Reactivation of latent viruses may be contributory or secondary to T cell activation by drugs
Eosinophilia +/- atypical lymphocytes
T helper type 2 cells recruit and activate eosinophils by releasing cytokines
Activated leukocytes create a humoral immune and allergic response
Dysfunction of regulatory T cells, resulting in failure to control unwanted immune responses against “self”
Autoimmune processes with single- or multi-organ involvement
CMV- Cytomegalovirus EBV- Epstein-Barr Virus HHV- Human Herpesvirus HLA- Human Leukocyte Antigens
MHC – Major Histo- compatibility Complex
Endocrine system
mechanism of dysfunction unclear but may be linked to autoreactive T cells
Autoimmune Type 1 thyroiditis diabetes
Lymphadenopathy
Fever
Facial edema
Liver
lobular inflammation, dispersed foci of necrotic hepatocytes, granulomatous infiltrates consisting of eosinophils
Hepatitis
Kidney
Interstitial edema and infiltrates of lymphocytes, histiocytes, eosinophils, and plasma cells
Acute interstitial nephritis
Lung
increased pulmonary infiltrate and edema
Morbilliform Skin Eruption
Spongiosis
Epidermal layer
Dermal- Epidermal Junction Dermal layer
Interface dermatitis
Skin eruption
(morbilliform to diffuse erythema with follicular accentuation)
Interstitial pneumonitis
Pleural effusion
Eosinophilic infiltration
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published October 19, 2021 on www.thecalgaryguide.com")
COPD-发病机制
 45
(on ABGs)
Ventilation- perfusion mismatch
High A-a gradient
(calculated from ABGs)
Low, flat diaphragm, >10 posterior ribs
(on frontal CXR)
High TLC and VC
(on spirometry)
• •
PaO2: partial pressure of O2 in arterial blood PaCO2: partial pressure of CO2 in arterial blood
• In the setting of fever and productive cough, especially if lung field opacifications are seen on CXR: consider sputum gram stain and culture to rule out pneumonia.
Air does not block X-ray beams, will appear black on X-ray film
Chronic hypercapnia makes breathing centers less sensitive to the high PaCO2 stimulus for breathing, & more reliant on the low PaO2 stimulus
(“CO2 retention”)
Give O2 carefully to these patients (high PaO2 may suppress patients’ hypoxic respiratory drive, ↓ their breathing, & ↑↑↑ PaCO2)
↑ retrosternal air space
(on lateral CXR)
Hyper-lucent
(darker) lung fields, ↓ lung markings (on frontal CXR)
• Arterial Blood Gasses (ABGs)
• Chest X-Ray (CXR): frontal and
lateral
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published January 7, 2013 on www.thecalgaryguide.com
COPD: !"#$ 气流阻塞
肺泡通气↓ 呼气时,胸膜腔正压挤压气 道à 阻塞↑
作者: Yan Yu 审稿人: Jason Baserman, Jennifer Au, Naushad Hirani*, Juri Janovcik* 译者:Zihong Xie (谢梓泓) 翻译审稿人: Yonglin Mai (麦泳琳), Zesheng Ye (叶泽生) * 发表时担任临床医生
慢性阻塞性肺疾病 (COPD)
肺组织损伤
没有弹性回缩力将
气体排出肺
肺实质与血管分布减少导 致气体交换面积↓
弥散功能↓ (肺功能检查)
更多的CO2残留 并扩散到血液中
高碳酸血症: PaCO2 > 45
(动脉血气)
血流灌注通气不良的肺泡
时无法获得足够的氧气
总呼气时长较正常长
FEV1/FEV < 0.7
(肺功能检查)
肺无法完全排空
更多空气潴留在肺部
(肺过度充气)
低氧血症: PaO2 < 70mmHg
(动脉血气)
通气-灌注不匹配
肺泡-动脉氧分压差↑ (可通过动脉血气分析计算得出)
横膈低平, 下移至第10肋后端 及以下部位 (胸部正位片)
TLC与VC增大 (肺功能检查)
缩写: • • FEV1: 1秒用 •
VC:肺活量
PaO2: 动脉血 力呼气量 氧分压
空气不会阻挡X射线, 在X光片上呈现为黑色
慢性高碳酸血症使呼吸中枢对PaCO2 刺激呼吸的敏感性下降 & 更依赖于低PaO2的刺激 (“二氧化碳潴留”)
给患者吸氧时需注意(高PaO2
可能会抑制患者低氧时对呼吸的 刺激,使呼吸驱动↓ & PaCO2↑↑↑ )
• FVC: 用力肺 • 活量
• TLC:肺总量 慢阻肺相关检查 :
PaCO2: 动脉 血二氧化碳 分压
胸骨后间隙↑
(胸部侧位片) 肺纹理↓
• 肺功能检查
• 动脉血气分析(Arterial Blood Gasses, ABGs)
• 胸部正侧位片
• 当患者发热和湿咳,特别是胸片上见肺野不清晰时:
肺透亮度↑, (胸部正位片)
考虑进行痰革兰氏染色及痰培养以排除肺炎可能
图注:
病理生理
机制
体征/临床表现/实验室检查
并发症
2013年1月7日发布于 www.thecalgaryguide.com
COPD: Complications Lung inflammation
Chronic Obstructive Pulmonary Disease (COPD)
Airway obstruction ↓ inhaled air in alveoli and terminal bronchioles
Rupture of emphasematous bullae on surface of lung
Inhaled air leaks into pleural cavity and is trapped there
Pneumothorax
Feeling a loss of control over one’s life, and hopelessness for the future
Goblet cell proliferation, ↑ mucus production
Death of airway
epithelium ciliated cells
↓ oxygenation of the blood passing through the lungs
Chronic hypoxemia
Kidneys compensate by ↑ erythropoietin (EPO) production
↑ Hemoglobin and red blood cell synthesis
Polycythemia (secondary)
Hypoxic alveoli cause the pulmonary arterioles perfusing them to reflexively vasoconstrict
Since most alveoli in the lungs are hypoxic, hypoxic vasoconstriction occurs across entire lung
Vasoconstriction ↑ blood pressure within lung vasculature
Pulmonary hypertension
↑ workload of the right ventricle (to pump against higher pressures)
To compensate, the right ventricle progressively hypertrophies and dilates, but over time its output ↓
Cor pulmonale
(Right heart failure in isolation, not due to Left heart failure)
Mucus trapped in airways, serve as nidus for infection
Acute exacerbation of COPD (AECOPD)
Pneumonia
The chronic, systemic inflammation in COPD is a hyper-metabolic state that consumes calories
Macro-nutrient deficiency
Trouble with respiration lead to inactivity and deconditioning
Wasting, muscle atrophy
More inactivity and deconditioning perpetuates the cycle
Depression
Author: Yan Yu Reviewers: Jason Baserman Naushad Hirani* Juri Janovcik* * MD at time of publication
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published January 7, 2013 on www.thecalgaryguide.com
COPD: !"# 肺部炎症
杯状细胞增殖, 气道上皮纤毛 粘液产生↑ 细胞死亡
黏液潴留呼吸道,成为感
染的病灶
慢性阻塞性肺疾病 (COPD) 气道阻塞à 吸入肺泡和终末细
肺大疱破裂
吸入的空气渗入
并潴留于胸腔
气胸
感觉生活失控,对未
来感到绝望
抑郁
作者: Yan Yu 审稿人: Jason Baserman, Naushad Hirani*, Juri Janovcik* 译者: Zihong Xie (谢梓泓) 翻译审稿人: Yonglin Mai (麦泳琳), Zesheng Ye (叶泽生) * 发表时担任临床医生
支气管的空气 ↓
流经肺的血液进行气 缺氧的肺泡à灌注肺泡的肺小动
慢性阻塞性肺疾 病急性加重期 (AECOPD)
肺炎
体交换↓ 慢性低氧血症
肾脏合成促红细胞 生成素进行代偿↑
血红蛋白与红 细胞合成↑
红细胞增多症 (继发性)
脉发生反射性血管收缩
肺大部分肺泡缺氧à整个肺 都出现缺氧性血管收缩
肺血管收缩 à 肺血管压力↑ 肺动脉高压
↑ 右心室负荷(泵血时对抗高压) 为了代偿,右心室逐渐肥大和扩张,
但随着病程进展,右心室输出量 ↓
肺心病 (单独出现右心衰竭,非左心衰)
COPD所致的慢性全身 呼吸困难导致活 性炎症会使机体处于高 动量减少和活动
代谢状态,消耗能量 耐量降低
宏量营养 素缺乏症
消瘦,肌肉萎缩
运动量下降和活动耐量
的降低造成恶性循环
图注:
病理生理
机制
体征/临床表现/实验室检查
并发症
2013年1月7日发布于 www.thecalgaryguide.com
" title="COPD: 发病机制
作者: Yan Yu 审稿人:Jason Baserman, Jennifer Au, Naushad Hirani*, Juri Janovcik* 译者: Zihong Xie (谢梓泓) 翻译审稿人:Yonglin Mai (麦泳琳), Zesheng Ye (叶泽生) * 发表时担任临床医生
/012
(如a1-抗胰蛋白酶缺乏) 阻止肺组织损伤的能力↓
+,-.
(如长期吸烟、环境污染、感染)
肺内产生自由基
34*5
肺抗蛋白酶的失活
↑氧化应激,炎性细胞因子,蛋白酶功能
支气管的持续、反复损伤
炎性细胞浸润, 杯状细胞增殖, 气道上皮纤毛 尤其中性粒细 黏液产生↑ 细胞死亡
气道弹性↓ (弹性回缩
肺实质的蛋白水解破坏↑ 维持气道开放 肺泡永久性异常
的结构支持↓ 扩张
胞 力)
肺气体潴留 气道狭窄与 肺过度 肺大泡
气道黏液潴留,成为感染 狭窄 病灶
塌陷 充气
肺气肿
(容易肺泡 破裂)
气道纤维化和
%&'()*
慢性阻塞性肺疾病(COPD)
临床表现 并发症 (参阅相关幻灯片) (参阅相关幻灯片)
图注:
病理生理
机制
体征/临床表现/实验室检查
并发症
2013年1月7日发布于 www.thecalgaryguide.com
COPD: Clinical Findings Lung tissue
Chronic Obstructive Pulmonary Disease (COPD)
damage
↓ elastic recoil to push air out of lungs on expiration
Lungs don’t fully empty, air is trapped in alveoli (lung hyperinflation)
↑ lung volume means diaphragm is tonically contracted (flatter)
If occurring around airways
Airflow obstruction
↑ mucus production
↓ number of epithelial ciliated cells to clear away the mucus (the cells have been killed by airway inflammation)
Chronic cough with sputum
Author: Yan Yu Reviewers: Jason Baserman Jennifer Au Naushad Hirani* Juri Janovcik* * MD at time of publication
During expiration, positive pleural pressure squeezes on airwaysà↑ obstruction
↓ ventilation of alveoli
↓ oxygenation of blood (hypoxemia)
↓ perfusion of body tissues (i.e. brain, muscle)
Fatigue; ↓ exercise tolerance
Total expiration time takes longer than normal
Prolonged expiration
More effort needed to ventilate larger lungs
Respiratory muscles must work harder to breathe
Turbulent airflow in narrower airways is heard on auscultation
Expiratory Wheeze
Diaphragm can’t flatten much further to generate deep breaths
To breathe, chest wall must expand out more
Dyspnea
Shortness of breath, especially on exertion
Breathes are rapid & shallow
If end-stage:
Chronic fatigue causes deconditioning
Muscle weakness & wasting
Barrel chest
If end-stage: diaphragm will be “flat”. Continued
Patient tries to expire against higher mouth air pressure, forcing airways to open wider
Pursed-lip breathing
Patient breathes with accessory muscles as well as diaphragm to try to improve airflow
inspiratory effort further contracts diaphragmà pull the lower chest wall inwards
Hoover’s sign
(paradoxical shrinking of lower chest during inspiration)
Tripod sitting position (activates pectoral muscles)
Neck (SCM, scalene) muscles contracted
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published January 7, 2013 on www.thecalgaryguide.com
COPD: !"#$
慢性阻塞性肺疾病 (COPD) 如果出现在气道周围 气流阻塞
肺不能完全排空
气体,气体潴留
于肺泡(肺过度
充气)
总呼气时长大于 正常时长
呼气相延长
肺组织损伤
呼气时,将空气排出肺外 的弹性回缩力↓
肺不能完全排空气体,
气体潴留于肺泡内
(肺过度充气)
肺容积↑,膈肌紧张 性收缩(膈肌平坦)
呼气时,胸膜腔正压挤压气道 à 气道阻塞↑
肺泡通气↓ 血液氧合↓ (低
氧血症)
身体组织灌注 量↓ (比如脑、 肌肉)
疲劳; 运动耐量↓
黏液生成↑ 清除黏液的上皮纤
毛细胞数量↓ (受 气道炎症损伤)
慢性咳嗽伴咳 痰
作者: Yan Yu
审稿人: Jason Baserman, Jennifer Au, Naushad Hirani*, Juri Janovcik* 译者: Zihong Xie (谢梓泓) 翻译审稿人: Yonglin Mai (麦泳琳),
Zesheng Ye (叶泽生)
* 发表时担任临床医生
容积较大 的肺需要
更加努力 才能通气
呼吸肌必须
更用力才能 呼吸
听诊闻及狭窄气
道中的湍流气流
呼气喘鸣音
呼吸困难 气促,尤其是劳累
膈肌无法进一步收缩以
产生深呼吸
呼吸浅快
为了呼吸,
胸壁必须延
展得更大
桶状胸
晚期病人:
患者试图在较高的口 慢性疲劳导致 患者动用辅助呼吸肌和膈肌呼吸,
腔内气压下进行呼气, 活动耐量下降 从而使气道更开放
以改善气流
晚期病人:膈肌 “平坦” ,持续吸气进一步压 缩膈肌à 向内拉季肋部胸壁
胡佛征 (吸气时,胸廓下侧季肋部内收)
缩唇呼吸
肌肉无力 & 消瘦
端坐呼吸 (调动胸肌)
颈部肌肉收
缩(胸锁乳
突肌、斜角
肌)
图注:
病理生理
机制
体征/临床表现/实验室检查
并发症
2013年1月7日发布于 www.thecalgaryguide.com
COPD: Findings on Investigations
Chronic Obstructive Pulmonary Disease (COPD)
Author: Yan Yu Reviewers: Jason Baserman Jennifer Au Naushad Hirani* Juri Janovcik* * MD at time of publication
Airflow obstruction
Lung tissue damage
↓ ventilation of alveoli
Blood perfusing ill- ventilated alveoli does not receive normal amounts of oxygen
During expiration, positive pleural pressure squeezes on airwaysà↑ obstruction)
No elastic recoil to push air out of lungs
Loss of lung parenchyma and vasculature ↓ surface area for gas exchange
↓ diffusion capacity
(on spirometry)
Hypoxemia: PaO2 < 70mmHg (on ABGs)
Abbreviations:
• FEV1: Forced expiratory volume in 1 second
• FVC: Forced vital capacity
• TLC: Total lung capacity
• VC: Vital Capacity
Investigations for COPD :
• Spirometry (Pulmonary function test)
Total expiration time takes longer than normal
FEV1/FEV < 0.7
(on spirometry)
Lungs don’t fully empty
More air trapped within lungs (hyperinflation)
More CO2 remains and diffuses into the blood
Hypercapnia: PaCO2 > 45
(on ABGs)
Ventilation- perfusion mismatch
High A-a gradient
(calculated from ABGs)
Low, flat diaphragm, >10 posterior ribs
(on frontal CXR)
High TLC and VC
(on spirometry)
• •
PaO2: partial pressure of O2 in arterial blood PaCO2: partial pressure of CO2 in arterial blood
• In the setting of fever and productive cough, especially if lung field opacifications are seen on CXR: consider sputum gram stain and culture to rule out pneumonia.
Air does not block X-ray beams, will appear black on X-ray film
Chronic hypercapnia makes breathing centers less sensitive to the high PaCO2 stimulus for breathing, & more reliant on the low PaO2 stimulus
(“CO2 retention”)
Give O2 carefully to these patients (high PaO2 may suppress patients’ hypoxic respiratory drive, ↓ their breathing, & ↑↑↑ PaCO2)
↑ retrosternal air space
(on lateral CXR)
Hyper-lucent
(darker) lung fields, ↓ lung markings (on frontal CXR)
• Arterial Blood Gasses (ABGs)
• Chest X-Ray (CXR): frontal and
lateral
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published January 7, 2013 on www.thecalgaryguide.com
COPD: !"#$ 气流阻塞
肺泡通气↓ 呼气时,胸膜腔正压挤压气 道à 阻塞↑
作者: Yan Yu 审稿人: Jason Baserman, Jennifer Au, Naushad Hirani*, Juri Janovcik* 译者:Zihong Xie (谢梓泓) 翻译审稿人: Yonglin Mai (麦泳琳), Zesheng Ye (叶泽生) * 发表时担任临床医生
慢性阻塞性肺疾病 (COPD)
肺组织损伤
没有弹性回缩力将
气体排出肺
肺实质与血管分布减少导 致气体交换面积↓
弥散功能↓ (肺功能检查)
更多的CO2残留 并扩散到血液中
高碳酸血症: PaCO2 > 45
(动脉血气)
血流灌注通气不良的肺泡
时无法获得足够的氧气
总呼气时长较正常长
FEV1/FEV < 0.7
(肺功能检查)
肺无法完全排空
更多空气潴留在肺部
(肺过度充气)
低氧血症: PaO2 < 70mmHg
(动脉血气)
通气-灌注不匹配
肺泡-动脉氧分压差↑ (可通过动脉血气分析计算得出)
横膈低平, 下移至第10肋后端 及以下部位 (胸部正位片)
TLC与VC增大 (肺功能检查)
缩写: • • FEV1: 1秒用 •
VC:肺活量
PaO2: 动脉血 力呼气量 氧分压
空气不会阻挡X射线, 在X光片上呈现为黑色
慢性高碳酸血症使呼吸中枢对PaCO2 刺激呼吸的敏感性下降 & 更依赖于低PaO2的刺激 (“二氧化碳潴留”)
给患者吸氧时需注意(高PaO2
可能会抑制患者低氧时对呼吸的 刺激,使呼吸驱动↓ & PaCO2↑↑↑ )
• FVC: 用力肺 • 活量
• TLC:肺总量 慢阻肺相关检查 :
PaCO2: 动脉 血二氧化碳 分压
胸骨后间隙↑
(胸部侧位片) 肺纹理↓
• 肺功能检查
• 动脉血气分析(Arterial Blood Gasses, ABGs)
• 胸部正侧位片
• 当患者发热和湿咳,特别是胸片上见肺野不清晰时:
肺透亮度↑, (胸部正位片)
考虑进行痰革兰氏染色及痰培养以排除肺炎可能
图注:
病理生理
机制
体征/临床表现/实验室检查
并发症
2013年1月7日发布于 www.thecalgaryguide.com
COPD: Complications Lung inflammation
Chronic Obstructive Pulmonary Disease (COPD)
Airway obstruction ↓ inhaled air in alveoli and terminal bronchioles
Rupture of emphasematous bullae on surface of lung
Inhaled air leaks into pleural cavity and is trapped there
Pneumothorax
Feeling a loss of control over one’s life, and hopelessness for the future
Goblet cell proliferation, ↑ mucus production
Death of airway
epithelium ciliated cells
↓ oxygenation of the blood passing through the lungs
Chronic hypoxemia
Kidneys compensate by ↑ erythropoietin (EPO) production
↑ Hemoglobin and red blood cell synthesis
Polycythemia (secondary)
Hypoxic alveoli cause the pulmonary arterioles perfusing them to reflexively vasoconstrict
Since most alveoli in the lungs are hypoxic, hypoxic vasoconstriction occurs across entire lung
Vasoconstriction ↑ blood pressure within lung vasculature
Pulmonary hypertension
↑ workload of the right ventricle (to pump against higher pressures)
To compensate, the right ventricle progressively hypertrophies and dilates, but over time its output ↓
Cor pulmonale
(Right heart failure in isolation, not due to Left heart failure)
Mucus trapped in airways, serve as nidus for infection
Acute exacerbation of COPD (AECOPD)
Pneumonia
The chronic, systemic inflammation in COPD is a hyper-metabolic state that consumes calories
Macro-nutrient deficiency
Trouble with respiration lead to inactivity and deconditioning
Wasting, muscle atrophy
More inactivity and deconditioning perpetuates the cycle
Depression
Author: Yan Yu Reviewers: Jason Baserman Naushad Hirani* Juri Janovcik* * MD at time of publication
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published January 7, 2013 on www.thecalgaryguide.com
COPD: !"# 肺部炎症
杯状细胞增殖, 气道上皮纤毛 粘液产生↑ 细胞死亡
黏液潴留呼吸道,成为感
染的病灶
慢性阻塞性肺疾病 (COPD) 气道阻塞à 吸入肺泡和终末细
肺大疱破裂
吸入的空气渗入
并潴留于胸腔
气胸
感觉生活失控,对未
来感到绝望
抑郁
作者: Yan Yu 审稿人: Jason Baserman, Naushad Hirani*, Juri Janovcik* 译者: Zihong Xie (谢梓泓) 翻译审稿人: Yonglin Mai (麦泳琳), Zesheng Ye (叶泽生) * 发表时担任临床医生
支气管的空气 ↓
流经肺的血液进行气 缺氧的肺泡à灌注肺泡的肺小动
慢性阻塞性肺疾 病急性加重期 (AECOPD)
肺炎
体交换↓ 慢性低氧血症
肾脏合成促红细胞 生成素进行代偿↑
血红蛋白与红 细胞合成↑
红细胞增多症 (继发性)
脉发生反射性血管收缩
肺大部分肺泡缺氧à整个肺 都出现缺氧性血管收缩
肺血管收缩 à 肺血管压力↑ 肺动脉高压
↑ 右心室负荷(泵血时对抗高压) 为了代偿,右心室逐渐肥大和扩张,
但随着病程进展,右心室输出量 ↓
肺心病 (单独出现右心衰竭,非左心衰)
COPD所致的慢性全身 呼吸困难导致活 性炎症会使机体处于高 动量减少和活动
代谢状态,消耗能量 耐量降低
宏量营养 素缺乏症
消瘦,肌肉萎缩
运动量下降和活动耐量
的降低造成恶性循环
图注:
病理生理
机制
体征/临床表现/实验室检查
并发症
2013年1月7日发布于 www.thecalgaryguide.com
" />
45
(on ABGs)
Ventilation- perfusion mismatch
High A-a gradient
(calculated from ABGs)
Low, flat diaphragm, >10 posterior ribs
(on frontal CXR)
High TLC and VC
(on spirometry)
• •
PaO2: partial pressure of O2 in arterial blood PaCO2: partial pressure of CO2 in arterial blood
• In the setting of fever and productive cough, especially if lung field opacifications are seen on CXR: consider sputum gram stain and culture to rule out pneumonia.
Air does not block X-ray beams, will appear black on X-ray film
Chronic hypercapnia makes breathing centers less sensitive to the high PaCO2 stimulus for breathing, & more reliant on the low PaO2 stimulus
(“CO2 retention”)
Give O2 carefully to these patients (high PaO2 may suppress patients’ hypoxic respiratory drive, ↓ their breathing, & ↑↑↑ PaCO2)
↑ retrosternal air space
(on lateral CXR)
Hyper-lucent
(darker) lung fields, ↓ lung markings (on frontal CXR)
• Arterial Blood Gasses (ABGs)
• Chest X-Ray (CXR): frontal and
lateral
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published January 7, 2013 on www.thecalgaryguide.com
COPD: !"#$ 气流阻塞
肺泡通气↓ 呼气时,胸膜腔正压挤压气 道à 阻塞↑
作者: Yan Yu 审稿人: Jason Baserman, Jennifer Au, Naushad Hirani*, Juri Janovcik* 译者:Zihong Xie (谢梓泓) 翻译审稿人: Yonglin Mai (麦泳琳), Zesheng Ye (叶泽生) * 发表时担任临床医生
慢性阻塞性肺疾病 (COPD)
肺组织损伤
没有弹性回缩力将
气体排出肺
肺实质与血管分布减少导 致气体交换面积↓
弥散功能↓ (肺功能检查)
更多的CO2残留 并扩散到血液中
高碳酸血症: PaCO2 > 45
(动脉血气)
血流灌注通气不良的肺泡
时无法获得足够的氧气
总呼气时长较正常长
FEV1/FEV < 0.7
(肺功能检查)
肺无法完全排空
更多空气潴留在肺部
(肺过度充气)
低氧血症: PaO2 < 70mmHg
(动脉血气)
通气-灌注不匹配
肺泡-动脉氧分压差↑ (可通过动脉血气分析计算得出)
横膈低平, 下移至第10肋后端 及以下部位 (胸部正位片)
TLC与VC增大 (肺功能检查)
缩写: • • FEV1: 1秒用 •
VC:肺活量
PaO2: 动脉血 力呼气量 氧分压
空气不会阻挡X射线, 在X光片上呈现为黑色
慢性高碳酸血症使呼吸中枢对PaCO2 刺激呼吸的敏感性下降 & 更依赖于低PaO2的刺激 (“二氧化碳潴留”)
给患者吸氧时需注意(高PaO2
可能会抑制患者低氧时对呼吸的 刺激,使呼吸驱动↓ & PaCO2↑↑↑ )
• FVC: 用力肺 • 活量
• TLC:肺总量 慢阻肺相关检查 :
PaCO2: 动脉 血二氧化碳 分压
胸骨后间隙↑
(胸部侧位片) 肺纹理↓
• 肺功能检查
• 动脉血气分析(Arterial Blood Gasses, ABGs)
• 胸部正侧位片
• 当患者发热和湿咳,特别是胸片上见肺野不清晰时:
肺透亮度↑, (胸部正位片)
考虑进行痰革兰氏染色及痰培养以排除肺炎可能
图注:
病理生理
机制
体征/临床表现/实验室检查
并发症
2013年1月7日发布于 www.thecalgaryguide.com
COPD: Complications Lung inflammation
Chronic Obstructive Pulmonary Disease (COPD)
Airway obstruction ↓ inhaled air in alveoli and terminal bronchioles
Rupture of emphasematous bullae on surface of lung
Inhaled air leaks into pleural cavity and is trapped there
Pneumothorax
Feeling a loss of control over one’s life, and hopelessness for the future
Goblet cell proliferation, ↑ mucus production
Death of airway
epithelium ciliated cells
↓ oxygenation of the blood passing through the lungs
Chronic hypoxemia
Kidneys compensate by ↑ erythropoietin (EPO) production
↑ Hemoglobin and red blood cell synthesis
Polycythemia (secondary)
Hypoxic alveoli cause the pulmonary arterioles perfusing them to reflexively vasoconstrict
Since most alveoli in the lungs are hypoxic, hypoxic vasoconstriction occurs across entire lung
Vasoconstriction ↑ blood pressure within lung vasculature
Pulmonary hypertension
↑ workload of the right ventricle (to pump against higher pressures)
To compensate, the right ventricle progressively hypertrophies and dilates, but over time its output ↓
Cor pulmonale
(Right heart failure in isolation, not due to Left heart failure)
Mucus trapped in airways, serve as nidus for infection
Acute exacerbation of COPD (AECOPD)
Pneumonia
The chronic, systemic inflammation in COPD is a hyper-metabolic state that consumes calories
Macro-nutrient deficiency
Trouble with respiration lead to inactivity and deconditioning
Wasting, muscle atrophy
More inactivity and deconditioning perpetuates the cycle
Depression
Author: Yan Yu Reviewers: Jason Baserman Naushad Hirani* Juri Janovcik* * MD at time of publication
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published January 7, 2013 on www.thecalgaryguide.com
COPD: !"# 肺部炎症
杯状细胞增殖, 气道上皮纤毛 粘液产生↑ 细胞死亡
黏液潴留呼吸道,成为感
染的病灶
慢性阻塞性肺疾病 (COPD) 气道阻塞à 吸入肺泡和终末细
肺大疱破裂
吸入的空气渗入
并潴留于胸腔
气胸
感觉生活失控,对未
来感到绝望
抑郁
作者: Yan Yu 审稿人: Jason Baserman, Naushad Hirani*, Juri Janovcik* 译者: Zihong Xie (谢梓泓) 翻译审稿人: Yonglin Mai (麦泳琳), Zesheng Ye (叶泽生) * 发表时担任临床医生
支气管的空气 ↓
流经肺的血液进行气 缺氧的肺泡à灌注肺泡的肺小动
慢性阻塞性肺疾 病急性加重期 (AECOPD)
肺炎
体交换↓ 慢性低氧血症
肾脏合成促红细胞 生成素进行代偿↑
血红蛋白与红 细胞合成↑
红细胞增多症 (继发性)
脉发生反射性血管收缩
肺大部分肺泡缺氧à整个肺 都出现缺氧性血管收缩
肺血管收缩 à 肺血管压力↑ 肺动脉高压
↑ 右心室负荷(泵血时对抗高压) 为了代偿,右心室逐渐肥大和扩张,
但随着病程进展,右心室输出量 ↓
肺心病 (单独出现右心衰竭,非左心衰)
COPD所致的慢性全身 呼吸困难导致活 性炎症会使机体处于高 动量减少和活动
代谢状态,消耗能量 耐量降低
宏量营养 素缺乏症
消瘦,肌肉萎缩
运动量下降和活动耐量
的降低造成恶性循环
图注:
病理生理
机制
体征/临床表现/实验室检查
并发症
2013年1月7日发布于 www.thecalgaryguide.com
" title="COPD: 发病机制
作者: Yan Yu 审稿人:Jason Baserman, Jennifer Au, Naushad Hirani*, Juri Janovcik* 译者: Zihong Xie (谢梓泓) 翻译审稿人:Yonglin Mai (麦泳琳), Zesheng Ye (叶泽生) * 发表时担任临床医生
/012
(如a1-抗胰蛋白酶缺乏) 阻止肺组织损伤的能力↓
+,-.
(如长期吸烟、环境污染、感染)
肺内产生自由基
34*5
肺抗蛋白酶的失活
↑氧化应激,炎性细胞因子,蛋白酶功能
支气管的持续、反复损伤
炎性细胞浸润, 杯状细胞增殖, 气道上皮纤毛 尤其中性粒细 黏液产生↑ 细胞死亡
气道弹性↓ (弹性回缩
肺实质的蛋白水解破坏↑ 维持气道开放 肺泡永久性异常
的结构支持↓ 扩张
胞 力)
肺气体潴留 气道狭窄与 肺过度 肺大泡
气道黏液潴留,成为感染 狭窄 病灶
塌陷 充气
肺气肿
(容易肺泡 破裂)
气道纤维化和
%&'()*
慢性阻塞性肺疾病(COPD)
临床表现 并发症 (参阅相关幻灯片) (参阅相关幻灯片)
图注:
病理生理
机制
体征/临床表现/实验室检查
并发症
2013年1月7日发布于 www.thecalgaryguide.com
COPD: Clinical Findings Lung tissue
Chronic Obstructive Pulmonary Disease (COPD)
damage
↓ elastic recoil to push air out of lungs on expiration
Lungs don’t fully empty, air is trapped in alveoli (lung hyperinflation)
↑ lung volume means diaphragm is tonically contracted (flatter)
If occurring around airways
Airflow obstruction
↑ mucus production
↓ number of epithelial ciliated cells to clear away the mucus (the cells have been killed by airway inflammation)
Chronic cough with sputum
Author: Yan Yu Reviewers: Jason Baserman Jennifer Au Naushad Hirani* Juri Janovcik* * MD at time of publication
During expiration, positive pleural pressure squeezes on airwaysà↑ obstruction
↓ ventilation of alveoli
↓ oxygenation of blood (hypoxemia)
↓ perfusion of body tissues (i.e. brain, muscle)
Fatigue; ↓ exercise tolerance
Total expiration time takes longer than normal
Prolonged expiration
More effort needed to ventilate larger lungs
Respiratory muscles must work harder to breathe
Turbulent airflow in narrower airways is heard on auscultation
Expiratory Wheeze
Diaphragm can’t flatten much further to generate deep breaths
To breathe, chest wall must expand out more
Dyspnea
Shortness of breath, especially on exertion
Breathes are rapid & shallow
If end-stage:
Chronic fatigue causes deconditioning
Muscle weakness & wasting
Barrel chest
If end-stage: diaphragm will be “flat”. Continued
Patient tries to expire against higher mouth air pressure, forcing airways to open wider
Pursed-lip breathing
Patient breathes with accessory muscles as well as diaphragm to try to improve airflow
inspiratory effort further contracts diaphragmà pull the lower chest wall inwards
Hoover’s sign
(paradoxical shrinking of lower chest during inspiration)
Tripod sitting position (activates pectoral muscles)
Neck (SCM, scalene) muscles contracted
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published January 7, 2013 on www.thecalgaryguide.com
COPD: !"#$
慢性阻塞性肺疾病 (COPD) 如果出现在气道周围 气流阻塞
肺不能完全排空
气体,气体潴留
于肺泡(肺过度
充气)
总呼气时长大于 正常时长
呼气相延长
肺组织损伤
呼气时,将空气排出肺外 的弹性回缩力↓
肺不能完全排空气体,
气体潴留于肺泡内
(肺过度充气)
肺容积↑,膈肌紧张 性收缩(膈肌平坦)
呼气时,胸膜腔正压挤压气道 à 气道阻塞↑
肺泡通气↓ 血液氧合↓ (低
氧血症)
身体组织灌注 量↓ (比如脑、 肌肉)
疲劳; 运动耐量↓
黏液生成↑ 清除黏液的上皮纤
毛细胞数量↓ (受 气道炎症损伤)
慢性咳嗽伴咳 痰
作者: Yan Yu
审稿人: Jason Baserman, Jennifer Au, Naushad Hirani*, Juri Janovcik* 译者: Zihong Xie (谢梓泓) 翻译审稿人: Yonglin Mai (麦泳琳),
Zesheng Ye (叶泽生)
* 发表时担任临床医生
容积较大 的肺需要
更加努力 才能通气
呼吸肌必须
更用力才能 呼吸
听诊闻及狭窄气
道中的湍流气流
呼气喘鸣音
呼吸困难 气促,尤其是劳累
膈肌无法进一步收缩以
产生深呼吸
呼吸浅快
为了呼吸,
胸壁必须延
展得更大
桶状胸
晚期病人:
患者试图在较高的口 慢性疲劳导致 患者动用辅助呼吸肌和膈肌呼吸,
腔内气压下进行呼气, 活动耐量下降 从而使气道更开放
以改善气流
晚期病人:膈肌 “平坦” ,持续吸气进一步压 缩膈肌à 向内拉季肋部胸壁
胡佛征 (吸气时,胸廓下侧季肋部内收)
缩唇呼吸
肌肉无力 & 消瘦
端坐呼吸 (调动胸肌)
颈部肌肉收
缩(胸锁乳
突肌、斜角
肌)
图注:
病理生理
机制
体征/临床表现/实验室检查
并发症
2013年1月7日发布于 www.thecalgaryguide.com
COPD: Findings on Investigations
Chronic Obstructive Pulmonary Disease (COPD)
Author: Yan Yu Reviewers: Jason Baserman Jennifer Au Naushad Hirani* Juri Janovcik* * MD at time of publication
Airflow obstruction
Lung tissue damage
↓ ventilation of alveoli
Blood perfusing ill- ventilated alveoli does not receive normal amounts of oxygen
During expiration, positive pleural pressure squeezes on airwaysà↑ obstruction)
No elastic recoil to push air out of lungs
Loss of lung parenchyma and vasculature ↓ surface area for gas exchange
↓ diffusion capacity
(on spirometry)
Hypoxemia: PaO2 < 70mmHg (on ABGs)
Abbreviations:
• FEV1: Forced expiratory volume in 1 second
• FVC: Forced vital capacity
• TLC: Total lung capacity
• VC: Vital Capacity
Investigations for COPD :
• Spirometry (Pulmonary function test)
Total expiration time takes longer than normal
FEV1/FEV < 0.7
(on spirometry)
Lungs don’t fully empty
More air trapped within lungs (hyperinflation)
More CO2 remains and diffuses into the blood
Hypercapnia: PaCO2 > 45
(on ABGs)
Ventilation- perfusion mismatch
High A-a gradient
(calculated from ABGs)
Low, flat diaphragm, >10 posterior ribs
(on frontal CXR)
High TLC and VC
(on spirometry)
• •
PaO2: partial pressure of O2 in arterial blood PaCO2: partial pressure of CO2 in arterial blood
• In the setting of fever and productive cough, especially if lung field opacifications are seen on CXR: consider sputum gram stain and culture to rule out pneumonia.
Air does not block X-ray beams, will appear black on X-ray film
Chronic hypercapnia makes breathing centers less sensitive to the high PaCO2 stimulus for breathing, & more reliant on the low PaO2 stimulus
(“CO2 retention”)
Give O2 carefully to these patients (high PaO2 may suppress patients’ hypoxic respiratory drive, ↓ their breathing, & ↑↑↑ PaCO2)
↑ retrosternal air space
(on lateral CXR)
Hyper-lucent
(darker) lung fields, ↓ lung markings (on frontal CXR)
• Arterial Blood Gasses (ABGs)
• Chest X-Ray (CXR): frontal and
lateral
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published January 7, 2013 on www.thecalgaryguide.com
COPD: !"#$ 气流阻塞
肺泡通气↓ 呼气时,胸膜腔正压挤压气 道à 阻塞↑
作者: Yan Yu 审稿人: Jason Baserman, Jennifer Au, Naushad Hirani*, Juri Janovcik* 译者:Zihong Xie (谢梓泓) 翻译审稿人: Yonglin Mai (麦泳琳), Zesheng Ye (叶泽生) * 发表时担任临床医生
慢性阻塞性肺疾病 (COPD)
肺组织损伤
没有弹性回缩力将
气体排出肺
肺实质与血管分布减少导 致气体交换面积↓
弥散功能↓ (肺功能检查)
更多的CO2残留 并扩散到血液中
高碳酸血症: PaCO2 > 45
(动脉血气)
血流灌注通气不良的肺泡
时无法获得足够的氧气
总呼气时长较正常长
FEV1/FEV < 0.7
(肺功能检查)
肺无法完全排空
更多空气潴留在肺部
(肺过度充气)
低氧血症: PaO2 < 70mmHg
(动脉血气)
通气-灌注不匹配
肺泡-动脉氧分压差↑ (可通过动脉血气分析计算得出)
横膈低平, 下移至第10肋后端 及以下部位 (胸部正位片)
TLC与VC增大 (肺功能检查)
缩写: • • FEV1: 1秒用 •
VC:肺活量
PaO2: 动脉血 力呼气量 氧分压
空气不会阻挡X射线, 在X光片上呈现为黑色
慢性高碳酸血症使呼吸中枢对PaCO2 刺激呼吸的敏感性下降 & 更依赖于低PaO2的刺激 (“二氧化碳潴留”)
给患者吸氧时需注意(高PaO2
可能会抑制患者低氧时对呼吸的 刺激,使呼吸驱动↓ & PaCO2↑↑↑ )
• FVC: 用力肺 • 活量
• TLC:肺总量 慢阻肺相关检查 :
PaCO2: 动脉 血二氧化碳 分压
胸骨后间隙↑
(胸部侧位片) 肺纹理↓
• 肺功能检查
• 动脉血气分析(Arterial Blood Gasses, ABGs)
• 胸部正侧位片
• 当患者发热和湿咳,特别是胸片上见肺野不清晰时:
肺透亮度↑, (胸部正位片)
考虑进行痰革兰氏染色及痰培养以排除肺炎可能
图注:
病理生理
机制
体征/临床表现/实验室检查
并发症
2013年1月7日发布于 www.thecalgaryguide.com
COPD: Complications Lung inflammation
Chronic Obstructive Pulmonary Disease (COPD)
Airway obstruction ↓ inhaled air in alveoli and terminal bronchioles
Rupture of emphasematous bullae on surface of lung
Inhaled air leaks into pleural cavity and is trapped there
Pneumothorax
Feeling a loss of control over one’s life, and hopelessness for the future
Goblet cell proliferation, ↑ mucus production
Death of airway
epithelium ciliated cells
↓ oxygenation of the blood passing through the lungs
Chronic hypoxemia
Kidneys compensate by ↑ erythropoietin (EPO) production
↑ Hemoglobin and red blood cell synthesis
Polycythemia (secondary)
Hypoxic alveoli cause the pulmonary arterioles perfusing them to reflexively vasoconstrict
Since most alveoli in the lungs are hypoxic, hypoxic vasoconstriction occurs across entire lung
Vasoconstriction ↑ blood pressure within lung vasculature
Pulmonary hypertension
↑ workload of the right ventricle (to pump against higher pressures)
To compensate, the right ventricle progressively hypertrophies and dilates, but over time its output ↓
Cor pulmonale
(Right heart failure in isolation, not due to Left heart failure)
Mucus trapped in airways, serve as nidus for infection
Acute exacerbation of COPD (AECOPD)
Pneumonia
The chronic, systemic inflammation in COPD is a hyper-metabolic state that consumes calories
Macro-nutrient deficiency
Trouble with respiration lead to inactivity and deconditioning
Wasting, muscle atrophy
More inactivity and deconditioning perpetuates the cycle
Depression
Author: Yan Yu Reviewers: Jason Baserman Naushad Hirani* Juri Janovcik* * MD at time of publication
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published January 7, 2013 on www.thecalgaryguide.com
COPD: !"# 肺部炎症
杯状细胞增殖, 气道上皮纤毛 粘液产生↑ 细胞死亡
黏液潴留呼吸道,成为感
染的病灶
慢性阻塞性肺疾病 (COPD) 气道阻塞à 吸入肺泡和终末细
肺大疱破裂
吸入的空气渗入
并潴留于胸腔
气胸
感觉生活失控,对未
来感到绝望
抑郁
作者: Yan Yu 审稿人: Jason Baserman, Naushad Hirani*, Juri Janovcik* 译者: Zihong Xie (谢梓泓) 翻译审稿人: Yonglin Mai (麦泳琳), Zesheng Ye (叶泽生) * 发表时担任临床医生
支气管的空气 ↓
流经肺的血液进行气 缺氧的肺泡à灌注肺泡的肺小动
慢性阻塞性肺疾 病急性加重期 (AECOPD)
肺炎
体交换↓ 慢性低氧血症
肾脏合成促红细胞 生成素进行代偿↑
血红蛋白与红 细胞合成↑
红细胞增多症 (继发性)
脉发生反射性血管收缩
肺大部分肺泡缺氧à整个肺 都出现缺氧性血管收缩
肺血管收缩 à 肺血管压力↑ 肺动脉高压
↑ 右心室负荷(泵血时对抗高压) 为了代偿,右心室逐渐肥大和扩张,
但随着病程进展,右心室输出量 ↓
肺心病 (单独出现右心衰竭,非左心衰)
COPD所致的慢性全身 呼吸困难导致活 性炎症会使机体处于高 动量减少和活动
代谢状态,消耗能量 耐量降低
宏量营养 素缺乏症
消瘦,肌肉萎缩
运动量下降和活动耐量
的降低造成恶性循环
图注:
病理生理
机制
体征/临床表现/实验室检查
并发症
2013年1月7日发布于 www.thecalgaryguide.com
" />
COPD-临床表现
 45
(on ABGs)
Ventilation- perfusion mismatch
High A-a gradient
(calculated from ABGs)
Low, flat diaphragm, >10 posterior ribs
(on frontal CXR)
High TLC and VC
(on spirometry)
• •
PaO2: partial pressure of O2 in arterial blood PaCO2: partial pressure of CO2 in arterial blood
• In the setting of fever and productive cough, especially if lung field opacifications are seen on CXR: consider sputum gram stain and culture to rule out pneumonia.
Air does not block X-ray beams, will appear black on X-ray film
Chronic hypercapnia makes breathing centers less sensitive to the high PaCO2 stimulus for breathing, & more reliant on the low PaO2 stimulus
(“CO2 retention”)
Give O2 carefully to these patients (high PaO2 may suppress patients’ hypoxic respiratory drive, ↓ their breathing, & ↑↑↑ PaCO2)
↑ retrosternal air space
(on lateral CXR)
Hyper-lucent
(darker) lung fields, ↓ lung markings (on frontal CXR)
• Arterial Blood Gasses (ABGs)
• Chest X-Ray (CXR): frontal and
lateral
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published January 7, 2013 on www.thecalgaryguide.com
COPD: !"#$ 气流阻塞
肺泡通气↓ 呼气时,胸膜腔正压挤压气 道à 阻塞↑
作者: Yan Yu 审稿人: Jason Baserman, Jennifer Au, Naushad Hirani*, Juri Janovcik* 译者:Zihong Xie (谢梓泓) 翻译审稿人: Yonglin Mai (麦泳琳), Zesheng Ye (叶泽生) * 发表时担任临床医生
慢性阻塞性肺疾病 (COPD)
肺组织损伤
没有弹性回缩力将
气体排出肺
肺实质与血管分布减少导 致气体交换面积↓
弥散功能↓ (肺功能检查)
更多的CO2残留 并扩散到血液中
高碳酸血症: PaCO2 > 45
(动脉血气)
血流灌注通气不良的肺泡
时无法获得足够的氧气
总呼气时长较正常长
FEV1/FEV < 0.7
(肺功能检查)
肺无法完全排空
更多空气潴留在肺部
(肺过度充气)
低氧血症: PaO2 < 70mmHg
(动脉血气)
通气-灌注不匹配
肺泡-动脉氧分压差↑ (可通过动脉血气分析计算得出)
横膈低平, 下移至第10肋后端 及以下部位 (胸部正位片)
TLC与VC增大 (肺功能检查)
缩写: • • FEV1: 1秒用 •
VC:肺活量
PaO2: 动脉血 力呼气量 氧分压
空气不会阻挡X射线, 在X光片上呈现为黑色
慢性高碳酸血症使呼吸中枢对PaCO2 刺激呼吸的敏感性下降 & 更依赖于低PaO2的刺激 (“二氧化碳潴留”)
给患者吸氧时需注意(高PaO2
可能会抑制患者低氧时对呼吸的 刺激,使呼吸驱动↓ & PaCO2↑↑↑ )
• FVC: 用力肺 • 活量
• TLC:肺总量 慢阻肺相关检查 :
PaCO2: 动脉 血二氧化碳 分压
胸骨后间隙↑
(胸部侧位片) 肺纹理↓
• 肺功能检查
• 动脉血气分析(Arterial Blood Gasses, ABGs)
• 胸部正侧位片
• 当患者发热和湿咳,特别是胸片上见肺野不清晰时:
肺透亮度↑, (胸部正位片)
考虑进行痰革兰氏染色及痰培养以排除肺炎可能
图注:
病理生理
机制
体征/临床表现/实验室检查
并发症
2013年1月7日发布于 www.thecalgaryguide.com
COPD: Complications Lung inflammation
Chronic Obstructive Pulmonary Disease (COPD)
Airway obstruction ↓ inhaled air in alveoli and terminal bronchioles
Rupture of emphasematous bullae on surface of lung
Inhaled air leaks into pleural cavity and is trapped there
Pneumothorax
Feeling a loss of control over one’s life, and hopelessness for the future
Goblet cell proliferation, ↑ mucus production
Death of airway
epithelium ciliated cells
↓ oxygenation of the blood passing through the lungs
Chronic hypoxemia
Kidneys compensate by ↑ erythropoietin (EPO) production
↑ Hemoglobin and red blood cell synthesis
Polycythemia (secondary)
Hypoxic alveoli cause the pulmonary arterioles perfusing them to reflexively vasoconstrict
Since most alveoli in the lungs are hypoxic, hypoxic vasoconstriction occurs across entire lung
Vasoconstriction ↑ blood pressure within lung vasculature
Pulmonary hypertension
↑ workload of the right ventricle (to pump against higher pressures)
To compensate, the right ventricle progressively hypertrophies and dilates, but over time its output ↓
Cor pulmonale
(Right heart failure in isolation, not due to Left heart failure)
Mucus trapped in airways, serve as nidus for infection
Acute exacerbation of COPD (AECOPD)
Pneumonia
The chronic, systemic inflammation in COPD is a hyper-metabolic state that consumes calories
Macro-nutrient deficiency
Trouble with respiration lead to inactivity and deconditioning
Wasting, muscle atrophy
More inactivity and deconditioning perpetuates the cycle
Depression
Author: Yan Yu Reviewers: Jason Baserman Naushad Hirani* Juri Janovcik* * MD at time of publication
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published January 7, 2013 on www.thecalgaryguide.com
COPD: !"# 肺部炎症
杯状细胞增殖, 气道上皮纤毛 粘液产生↑ 细胞死亡
黏液潴留呼吸道,成为感
染的病灶
慢性阻塞性肺疾病 (COPD) 气道阻塞à 吸入肺泡和终末细
肺大疱破裂
吸入的空气渗入
并潴留于胸腔
气胸
感觉生活失控,对未
来感到绝望
抑郁
作者: Yan Yu 审稿人: Jason Baserman, Naushad Hirani*, Juri Janovcik* 译者: Zihong Xie (谢梓泓) 翻译审稿人: Yonglin Mai (麦泳琳), Zesheng Ye (叶泽生) * 发表时担任临床医生
支气管的空气 ↓
流经肺的血液进行气 缺氧的肺泡à灌注肺泡的肺小动
慢性阻塞性肺疾 病急性加重期 (AECOPD)
肺炎
体交换↓ 慢性低氧血症
肾脏合成促红细胞 生成素进行代偿↑
血红蛋白与红 细胞合成↑
红细胞增多症 (继发性)
脉发生反射性血管收缩
肺大部分肺泡缺氧à整个肺 都出现缺氧性血管收缩
肺血管收缩 à 肺血管压力↑ 肺动脉高压
↑ 右心室负荷(泵血时对抗高压) 为了代偿,右心室逐渐肥大和扩张,
但随着病程进展,右心室输出量 ↓
肺心病 (单独出现右心衰竭,非左心衰)
COPD所致的慢性全身 呼吸困难导致活 性炎症会使机体处于高 动量减少和活动
代谢状态,消耗能量 耐量降低
宏量营养 素缺乏症
消瘦,肌肉萎缩
运动量下降和活动耐量
的降低造成恶性循环
图注:
病理生理
机制
体征/临床表现/实验室检查
并发症
2013年1月7日发布于 www.thecalgaryguide.com
" title="COPD: 临床表现
作者: Yan Yu 审稿人:Jason Baserman, Jennifer Au, Naushad Hirani*, Juri Janovcik* 译者: Zihong Xie (谢梓泓) 翻译审稿人:Yonglin Mai (麦泳琳), Zesheng Ye (叶泽生) * 发表时担任临床医生
/012
(如a1-抗胰蛋白酶缺乏) 阻止肺组织损伤的能力↓
+,-.
(如长期吸烟、环境污染、感染)
肺内产生自由基
34*5
肺抗蛋白酶的失活
↑氧化应激,炎性细胞因子,蛋白酶功能
支气管的持续、反复损伤
炎性细胞浸润, 杯状细胞增殖, 气道上皮纤毛 尤其中性粒细 黏液产生↑ 细胞死亡
气道弹性↓ (弹性回缩
肺实质的蛋白水解破坏↑ 维持气道开放 肺泡永久性异常
的结构支持↓ 扩张
胞 力)
肺气体潴留 气道狭窄与 肺过度 肺大泡
气道黏液潴留,成为感染 狭窄 病灶
塌陷 充气
肺气肿
(容易肺泡 破裂)
气道纤维化和
%&'()*
慢性阻塞性肺疾病(COPD)
临床表现 并发症 (参阅相关幻灯片) (参阅相关幻灯片)
图注:
病理生理
机制
体征/临床表现/实验室检查
并发症
2013年1月7日发布于 www.thecalgaryguide.com
COPD: Clinical Findings Lung tissue
Chronic Obstructive Pulmonary Disease (COPD)
damage
↓ elastic recoil to push air out of lungs on expiration
Lungs don’t fully empty, air is trapped in alveoli (lung hyperinflation)
↑ lung volume means diaphragm is tonically contracted (flatter)
If occurring around airways
Airflow obstruction
↑ mucus production
↓ number of epithelial ciliated cells to clear away the mucus (the cells have been killed by airway inflammation)
Chronic cough with sputum
Author: Yan Yu Reviewers: Jason Baserman Jennifer Au Naushad Hirani* Juri Janovcik* * MD at time of publication
During expiration, positive pleural pressure squeezes on airwaysà↑ obstruction
↓ ventilation of alveoli
↓ oxygenation of blood (hypoxemia)
↓ perfusion of body tissues (i.e. brain, muscle)
Fatigue; ↓ exercise tolerance
Total expiration time takes longer than normal
Prolonged expiration
More effort needed to ventilate larger lungs
Respiratory muscles must work harder to breathe
Turbulent airflow in narrower airways is heard on auscultation
Expiratory Wheeze
Diaphragm can’t flatten much further to generate deep breaths
To breathe, chest wall must expand out more
Dyspnea
Shortness of breath, especially on exertion
Breathes are rapid & shallow
If end-stage:
Chronic fatigue causes deconditioning
Muscle weakness & wasting
Barrel chest
If end-stage: diaphragm will be “flat”. Continued
Patient tries to expire against higher mouth air pressure, forcing airways to open wider
Pursed-lip breathing
Patient breathes with accessory muscles as well as diaphragm to try to improve airflow
inspiratory effort further contracts diaphragmà pull the lower chest wall inwards
Hoover’s sign
(paradoxical shrinking of lower chest during inspiration)
Tripod sitting position (activates pectoral muscles)
Neck (SCM, scalene) muscles contracted
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published January 7, 2013 on www.thecalgaryguide.com
COPD: !"#$
慢性阻塞性肺疾病 (COPD) 如果出现在气道周围 气流阻塞
肺不能完全排空
气体,气体潴留
于肺泡(肺过度
充气)
总呼气时长大于 正常时长
呼气相延长
肺组织损伤
呼气时,将空气排出肺外 的弹性回缩力↓
肺不能完全排空气体,
气体潴留于肺泡内
(肺过度充气)
肺容积↑,膈肌紧张 性收缩(膈肌平坦)
呼气时,胸膜腔正压挤压气道 à 气道阻塞↑
肺泡通气↓ 血液氧合↓ (低
氧血症)
身体组织灌注 量↓ (比如脑、 肌肉)
疲劳; 运动耐量↓
黏液生成↑ 清除黏液的上皮纤
毛细胞数量↓ (受 气道炎症损伤)
慢性咳嗽伴咳 痰
作者: Yan Yu
审稿人: Jason Baserman, Jennifer Au, Naushad Hirani*, Juri Janovcik* 译者: Zihong Xie (谢梓泓) 翻译审稿人: Yonglin Mai (麦泳琳),
Zesheng Ye (叶泽生)
* 发表时担任临床医生
容积较大 的肺需要
更加努力 才能通气
呼吸肌必须
更用力才能 呼吸
听诊闻及狭窄气
道中的湍流气流
呼气喘鸣音
呼吸困难 气促,尤其是劳累
膈肌无法进一步收缩以
产生深呼吸
呼吸浅快
为了呼吸,
胸壁必须延
展得更大
桶状胸
晚期病人:
患者试图在较高的口 慢性疲劳导致 患者动用辅助呼吸肌和膈肌呼吸,
腔内气压下进行呼气, 活动耐量下降 从而使气道更开放
以改善气流
晚期病人:膈肌 “平坦” ,持续吸气进一步压 缩膈肌à 向内拉季肋部胸壁
胡佛征 (吸气时,胸廓下侧季肋部内收)
缩唇呼吸
肌肉无力 & 消瘦
端坐呼吸 (调动胸肌)
颈部肌肉收
缩(胸锁乳
突肌、斜角
肌)
图注:
病理生理
机制
体征/临床表现/实验室检查
并发症
2013年1月7日发布于 www.thecalgaryguide.com
COPD: Findings on Investigations
Chronic Obstructive Pulmonary Disease (COPD)
Author: Yan Yu Reviewers: Jason Baserman Jennifer Au Naushad Hirani* Juri Janovcik* * MD at time of publication
Airflow obstruction
Lung tissue damage
↓ ventilation of alveoli
Blood perfusing ill- ventilated alveoli does not receive normal amounts of oxygen
During expiration, positive pleural pressure squeezes on airwaysà↑ obstruction)
No elastic recoil to push air out of lungs
Loss of lung parenchyma and vasculature ↓ surface area for gas exchange
↓ diffusion capacity
(on spirometry)
Hypoxemia: PaO2 < 70mmHg (on ABGs)
Abbreviations:
• FEV1: Forced expiratory volume in 1 second
• FVC: Forced vital capacity
• TLC: Total lung capacity
• VC: Vital Capacity
Investigations for COPD :
• Spirometry (Pulmonary function test)
Total expiration time takes longer than normal
FEV1/FEV < 0.7
(on spirometry)
Lungs don’t fully empty
More air trapped within lungs (hyperinflation)
More CO2 remains and diffuses into the blood
Hypercapnia: PaCO2 > 45
(on ABGs)
Ventilation- perfusion mismatch
High A-a gradient
(calculated from ABGs)
Low, flat diaphragm, >10 posterior ribs
(on frontal CXR)
High TLC and VC
(on spirometry)
• •
PaO2: partial pressure of O2 in arterial blood PaCO2: partial pressure of CO2 in arterial blood
• In the setting of fever and productive cough, especially if lung field opacifications are seen on CXR: consider sputum gram stain and culture to rule out pneumonia.
Air does not block X-ray beams, will appear black on X-ray film
Chronic hypercapnia makes breathing centers less sensitive to the high PaCO2 stimulus for breathing, & more reliant on the low PaO2 stimulus
(“CO2 retention”)
Give O2 carefully to these patients (high PaO2 may suppress patients’ hypoxic respiratory drive, ↓ their breathing, & ↑↑↑ PaCO2)
↑ retrosternal air space
(on lateral CXR)
Hyper-lucent
(darker) lung fields, ↓ lung markings (on frontal CXR)
• Arterial Blood Gasses (ABGs)
• Chest X-Ray (CXR): frontal and
lateral
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published January 7, 2013 on www.thecalgaryguide.com
COPD: !"#$ 气流阻塞
肺泡通气↓ 呼气时,胸膜腔正压挤压气 道à 阻塞↑
作者: Yan Yu 审稿人: Jason Baserman, Jennifer Au, Naushad Hirani*, Juri Janovcik* 译者:Zihong Xie (谢梓泓) 翻译审稿人: Yonglin Mai (麦泳琳), Zesheng Ye (叶泽生) * 发表时担任临床医生
慢性阻塞性肺疾病 (COPD)
肺组织损伤
没有弹性回缩力将
气体排出肺
肺实质与血管分布减少导 致气体交换面积↓
弥散功能↓ (肺功能检查)
更多的CO2残留 并扩散到血液中
高碳酸血症: PaCO2 > 45
(动脉血气)
血流灌注通气不良的肺泡
时无法获得足够的氧气
总呼气时长较正常长
FEV1/FEV < 0.7
(肺功能检查)
肺无法完全排空
更多空气潴留在肺部
(肺过度充气)
低氧血症: PaO2 < 70mmHg
(动脉血气)
通气-灌注不匹配
肺泡-动脉氧分压差↑ (可通过动脉血气分析计算得出)
横膈低平, 下移至第10肋后端 及以下部位 (胸部正位片)
TLC与VC增大 (肺功能检查)
缩写: • • FEV1: 1秒用 •
VC:肺活量
PaO2: 动脉血 力呼气量 氧分压
空气不会阻挡X射线, 在X光片上呈现为黑色
慢性高碳酸血症使呼吸中枢对PaCO2 刺激呼吸的敏感性下降 & 更依赖于低PaO2的刺激 (“二氧化碳潴留”)
给患者吸氧时需注意(高PaO2
可能会抑制患者低氧时对呼吸的 刺激,使呼吸驱动↓ & PaCO2↑↑↑ )
• FVC: 用力肺 • 活量
• TLC:肺总量 慢阻肺相关检查 :
PaCO2: 动脉 血二氧化碳 分压
胸骨后间隙↑
(胸部侧位片) 肺纹理↓
• 肺功能检查
• 动脉血气分析(Arterial Blood Gasses, ABGs)
• 胸部正侧位片
• 当患者发热和湿咳,特别是胸片上见肺野不清晰时:
肺透亮度↑, (胸部正位片)
考虑进行痰革兰氏染色及痰培养以排除肺炎可能
图注:
病理生理
机制
体征/临床表现/实验室检查
并发症
2013年1月7日发布于 www.thecalgaryguide.com
COPD: Complications Lung inflammation
Chronic Obstructive Pulmonary Disease (COPD)
Airway obstruction ↓ inhaled air in alveoli and terminal bronchioles
Rupture of emphasematous bullae on surface of lung
Inhaled air leaks into pleural cavity and is trapped there
Pneumothorax
Feeling a loss of control over one’s life, and hopelessness for the future
Goblet cell proliferation, ↑ mucus production
Death of airway
epithelium ciliated cells
↓ oxygenation of the blood passing through the lungs
Chronic hypoxemia
Kidneys compensate by ↑ erythropoietin (EPO) production
↑ Hemoglobin and red blood cell synthesis
Polycythemia (secondary)
Hypoxic alveoli cause the pulmonary arterioles perfusing them to reflexively vasoconstrict
Since most alveoli in the lungs are hypoxic, hypoxic vasoconstriction occurs across entire lung
Vasoconstriction ↑ blood pressure within lung vasculature
Pulmonary hypertension
↑ workload of the right ventricle (to pump against higher pressures)
To compensate, the right ventricle progressively hypertrophies and dilates, but over time its output ↓
Cor pulmonale
(Right heart failure in isolation, not due to Left heart failure)
Mucus trapped in airways, serve as nidus for infection
Acute exacerbation of COPD (AECOPD)
Pneumonia
The chronic, systemic inflammation in COPD is a hyper-metabolic state that consumes calories
Macro-nutrient deficiency
Trouble with respiration lead to inactivity and deconditioning
Wasting, muscle atrophy
More inactivity and deconditioning perpetuates the cycle
Depression
Author: Yan Yu Reviewers: Jason Baserman Naushad Hirani* Juri Janovcik* * MD at time of publication
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published January 7, 2013 on www.thecalgaryguide.com
COPD: !"# 肺部炎症
杯状细胞增殖, 气道上皮纤毛 粘液产生↑ 细胞死亡
黏液潴留呼吸道,成为感
染的病灶
慢性阻塞性肺疾病 (COPD) 气道阻塞à 吸入肺泡和终末细
肺大疱破裂
吸入的空气渗入
并潴留于胸腔
气胸
感觉生活失控,对未
来感到绝望
抑郁
作者: Yan Yu 审稿人: Jason Baserman, Naushad Hirani*, Juri Janovcik* 译者: Zihong Xie (谢梓泓) 翻译审稿人: Yonglin Mai (麦泳琳), Zesheng Ye (叶泽生) * 发表时担任临床医生
支气管的空气 ↓
流经肺的血液进行气 缺氧的肺泡à灌注肺泡的肺小动
慢性阻塞性肺疾 病急性加重期 (AECOPD)
肺炎
体交换↓ 慢性低氧血症
肾脏合成促红细胞 生成素进行代偿↑
血红蛋白与红 细胞合成↑
红细胞增多症 (继发性)
脉发生反射性血管收缩
肺大部分肺泡缺氧à整个肺 都出现缺氧性血管收缩
肺血管收缩 à 肺血管压力↑ 肺动脉高压
↑ 右心室负荷(泵血时对抗高压) 为了代偿,右心室逐渐肥大和扩张,
但随着病程进展,右心室输出量 ↓
肺心病 (单独出现右心衰竭,非左心衰)
COPD所致的慢性全身 呼吸困难导致活 性炎症会使机体处于高 动量减少和活动
代谢状态,消耗能量 耐量降低
宏量营养 素缺乏症
消瘦,肌肉萎缩
运动量下降和活动耐量
的降低造成恶性循环
图注:
病理生理
机制
体征/临床表现/实验室检查
并发症
2013年1月7日发布于 www.thecalgaryguide.com
" />
45
(on ABGs)
Ventilation- perfusion mismatch
High A-a gradient
(calculated from ABGs)
Low, flat diaphragm, >10 posterior ribs
(on frontal CXR)
High TLC and VC
(on spirometry)
• •
PaO2: partial pressure of O2 in arterial blood PaCO2: partial pressure of CO2 in arterial blood
• In the setting of fever and productive cough, especially if lung field opacifications are seen on CXR: consider sputum gram stain and culture to rule out pneumonia.
Air does not block X-ray beams, will appear black on X-ray film
Chronic hypercapnia makes breathing centers less sensitive to the high PaCO2 stimulus for breathing, & more reliant on the low PaO2 stimulus
(“CO2 retention”)
Give O2 carefully to these patients (high PaO2 may suppress patients’ hypoxic respiratory drive, ↓ their breathing, & ↑↑↑ PaCO2)
↑ retrosternal air space
(on lateral CXR)
Hyper-lucent
(darker) lung fields, ↓ lung markings (on frontal CXR)
• Arterial Blood Gasses (ABGs)
• Chest X-Ray (CXR): frontal and
lateral
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published January 7, 2013 on www.thecalgaryguide.com
COPD: !"#$ 气流阻塞
肺泡通气↓ 呼气时,胸膜腔正压挤压气 道à 阻塞↑
作者: Yan Yu 审稿人: Jason Baserman, Jennifer Au, Naushad Hirani*, Juri Janovcik* 译者:Zihong Xie (谢梓泓) 翻译审稿人: Yonglin Mai (麦泳琳), Zesheng Ye (叶泽生) * 发表时担任临床医生
慢性阻塞性肺疾病 (COPD)
肺组织损伤
没有弹性回缩力将
气体排出肺
肺实质与血管分布减少导 致气体交换面积↓
弥散功能↓ (肺功能检查)
更多的CO2残留 并扩散到血液中
高碳酸血症: PaCO2 > 45
(动脉血气)
血流灌注通气不良的肺泡
时无法获得足够的氧气
总呼气时长较正常长
FEV1/FEV < 0.7
(肺功能检查)
肺无法完全排空
更多空气潴留在肺部
(肺过度充气)
低氧血症: PaO2 < 70mmHg
(动脉血气)
通气-灌注不匹配
肺泡-动脉氧分压差↑ (可通过动脉血气分析计算得出)
横膈低平, 下移至第10肋后端 及以下部位 (胸部正位片)
TLC与VC增大 (肺功能检查)
缩写: • • FEV1: 1秒用 •
VC:肺活量
PaO2: 动脉血 力呼气量 氧分压
空气不会阻挡X射线, 在X光片上呈现为黑色
慢性高碳酸血症使呼吸中枢对PaCO2 刺激呼吸的敏感性下降 & 更依赖于低PaO2的刺激 (“二氧化碳潴留”)
给患者吸氧时需注意(高PaO2
可能会抑制患者低氧时对呼吸的 刺激,使呼吸驱动↓ & PaCO2↑↑↑ )
• FVC: 用力肺 • 活量
• TLC:肺总量 慢阻肺相关检查 :
PaCO2: 动脉 血二氧化碳 分压
胸骨后间隙↑
(胸部侧位片) 肺纹理↓
• 肺功能检查
• 动脉血气分析(Arterial Blood Gasses, ABGs)
• 胸部正侧位片
• 当患者发热和湿咳,特别是胸片上见肺野不清晰时:
肺透亮度↑, (胸部正位片)
考虑进行痰革兰氏染色及痰培养以排除肺炎可能
图注:
病理生理
机制
体征/临床表现/实验室检查
并发症
2013年1月7日发布于 www.thecalgaryguide.com
COPD: Complications Lung inflammation
Chronic Obstructive Pulmonary Disease (COPD)
Airway obstruction ↓ inhaled air in alveoli and terminal bronchioles
Rupture of emphasematous bullae on surface of lung
Inhaled air leaks into pleural cavity and is trapped there
Pneumothorax
Feeling a loss of control over one’s life, and hopelessness for the future
Goblet cell proliferation, ↑ mucus production
Death of airway
epithelium ciliated cells
↓ oxygenation of the blood passing through the lungs
Chronic hypoxemia
Kidneys compensate by ↑ erythropoietin (EPO) production
↑ Hemoglobin and red blood cell synthesis
Polycythemia (secondary)
Hypoxic alveoli cause the pulmonary arterioles perfusing them to reflexively vasoconstrict
Since most alveoli in the lungs are hypoxic, hypoxic vasoconstriction occurs across entire lung
Vasoconstriction ↑ blood pressure within lung vasculature
Pulmonary hypertension
↑ workload of the right ventricle (to pump against higher pressures)
To compensate, the right ventricle progressively hypertrophies and dilates, but over time its output ↓
Cor pulmonale
(Right heart failure in isolation, not due to Left heart failure)
Mucus trapped in airways, serve as nidus for infection
Acute exacerbation of COPD (AECOPD)
Pneumonia
The chronic, systemic inflammation in COPD is a hyper-metabolic state that consumes calories
Macro-nutrient deficiency
Trouble with respiration lead to inactivity and deconditioning
Wasting, muscle atrophy
More inactivity and deconditioning perpetuates the cycle
Depression
Author: Yan Yu Reviewers: Jason Baserman Naushad Hirani* Juri Janovcik* * MD at time of publication
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published January 7, 2013 on www.thecalgaryguide.com
COPD: !"# 肺部炎症
杯状细胞增殖, 气道上皮纤毛 粘液产生↑ 细胞死亡
黏液潴留呼吸道,成为感
染的病灶
慢性阻塞性肺疾病 (COPD) 气道阻塞à 吸入肺泡和终末细
肺大疱破裂
吸入的空气渗入
并潴留于胸腔
气胸
感觉生活失控,对未
来感到绝望
抑郁
作者: Yan Yu 审稿人: Jason Baserman, Naushad Hirani*, Juri Janovcik* 译者: Zihong Xie (谢梓泓) 翻译审稿人: Yonglin Mai (麦泳琳), Zesheng Ye (叶泽生) * 发表时担任临床医生
支气管的空气 ↓
流经肺的血液进行气 缺氧的肺泡à灌注肺泡的肺小动
慢性阻塞性肺疾 病急性加重期 (AECOPD)
肺炎
体交换↓ 慢性低氧血症
肾脏合成促红细胞 生成素进行代偿↑
血红蛋白与红 细胞合成↑
红细胞增多症 (继发性)
脉发生反射性血管收缩
肺大部分肺泡缺氧à整个肺 都出现缺氧性血管收缩
肺血管收缩 à 肺血管压力↑ 肺动脉高压
↑ 右心室负荷(泵血时对抗高压) 为了代偿,右心室逐渐肥大和扩张,
但随着病程进展,右心室输出量 ↓
肺心病 (单独出现右心衰竭,非左心衰)
COPD所致的慢性全身 呼吸困难导致活 性炎症会使机体处于高 动量减少和活动
代谢状态,消耗能量 耐量降低
宏量营养 素缺乏症
消瘦,肌肉萎缩
运动量下降和活动耐量
的降低造成恶性循环
图注:
病理生理
机制
体征/临床表现/实验室检查
并发症
2013年1月7日发布于 www.thecalgaryguide.com
" title="COPD: 临床表现
作者: Yan Yu 审稿人:Jason Baserman, Jennifer Au, Naushad Hirani*, Juri Janovcik* 译者: Zihong Xie (谢梓泓) 翻译审稿人:Yonglin Mai (麦泳琳), Zesheng Ye (叶泽生) * 发表时担任临床医生
/012
(如a1-抗胰蛋白酶缺乏) 阻止肺组织损伤的能力↓
+,-.
(如长期吸烟、环境污染、感染)
肺内产生自由基
34*5
肺抗蛋白酶的失活
↑氧化应激,炎性细胞因子,蛋白酶功能
支气管的持续、反复损伤
炎性细胞浸润, 杯状细胞增殖, 气道上皮纤毛 尤其中性粒细 黏液产生↑ 细胞死亡
气道弹性↓ (弹性回缩
肺实质的蛋白水解破坏↑ 维持气道开放 肺泡永久性异常
的结构支持↓ 扩张
胞 力)
肺气体潴留 气道狭窄与 肺过度 肺大泡
气道黏液潴留,成为感染 狭窄 病灶
塌陷 充气
肺气肿
(容易肺泡 破裂)
气道纤维化和
%&'()*
慢性阻塞性肺疾病(COPD)
临床表现 并发症 (参阅相关幻灯片) (参阅相关幻灯片)
图注:
病理生理
机制
体征/临床表现/实验室检查
并发症
2013年1月7日发布于 www.thecalgaryguide.com
COPD: Clinical Findings Lung tissue
Chronic Obstructive Pulmonary Disease (COPD)
damage
↓ elastic recoil to push air out of lungs on expiration
Lungs don’t fully empty, air is trapped in alveoli (lung hyperinflation)
↑ lung volume means diaphragm is tonically contracted (flatter)
If occurring around airways
Airflow obstruction
↑ mucus production
↓ number of epithelial ciliated cells to clear away the mucus (the cells have been killed by airway inflammation)
Chronic cough with sputum
Author: Yan Yu Reviewers: Jason Baserman Jennifer Au Naushad Hirani* Juri Janovcik* * MD at time of publication
During expiration, positive pleural pressure squeezes on airwaysà↑ obstruction
↓ ventilation of alveoli
↓ oxygenation of blood (hypoxemia)
↓ perfusion of body tissues (i.e. brain, muscle)
Fatigue; ↓ exercise tolerance
Total expiration time takes longer than normal
Prolonged expiration
More effort needed to ventilate larger lungs
Respiratory muscles must work harder to breathe
Turbulent airflow in narrower airways is heard on auscultation
Expiratory Wheeze
Diaphragm can’t flatten much further to generate deep breaths
To breathe, chest wall must expand out more
Dyspnea
Shortness of breath, especially on exertion
Breathes are rapid & shallow
If end-stage:
Chronic fatigue causes deconditioning
Muscle weakness & wasting
Barrel chest
If end-stage: diaphragm will be “flat”. Continued
Patient tries to expire against higher mouth air pressure, forcing airways to open wider
Pursed-lip breathing
Patient breathes with accessory muscles as well as diaphragm to try to improve airflow
inspiratory effort further contracts diaphragmà pull the lower chest wall inwards
Hoover’s sign
(paradoxical shrinking of lower chest during inspiration)
Tripod sitting position (activates pectoral muscles)
Neck (SCM, scalene) muscles contracted
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published January 7, 2013 on www.thecalgaryguide.com
COPD: !"#$
慢性阻塞性肺疾病 (COPD) 如果出现在气道周围 气流阻塞
肺不能完全排空
气体,气体潴留
于肺泡(肺过度
充气)
总呼气时长大于 正常时长
呼气相延长
肺组织损伤
呼气时,将空气排出肺外 的弹性回缩力↓
肺不能完全排空气体,
气体潴留于肺泡内
(肺过度充气)
肺容积↑,膈肌紧张 性收缩(膈肌平坦)
呼气时,胸膜腔正压挤压气道 à 气道阻塞↑
肺泡通气↓ 血液氧合↓ (低
氧血症)
身体组织灌注 量↓ (比如脑、 肌肉)
疲劳; 运动耐量↓
黏液生成↑ 清除黏液的上皮纤
毛细胞数量↓ (受 气道炎症损伤)
慢性咳嗽伴咳 痰
作者: Yan Yu
审稿人: Jason Baserman, Jennifer Au, Naushad Hirani*, Juri Janovcik* 译者: Zihong Xie (谢梓泓) 翻译审稿人: Yonglin Mai (麦泳琳),
Zesheng Ye (叶泽生)
* 发表时担任临床医生
容积较大 的肺需要
更加努力 才能通气
呼吸肌必须
更用力才能 呼吸
听诊闻及狭窄气
道中的湍流气流
呼气喘鸣音
呼吸困难 气促,尤其是劳累
膈肌无法进一步收缩以
产生深呼吸
呼吸浅快
为了呼吸,
胸壁必须延
展得更大
桶状胸
晚期病人:
患者试图在较高的口 慢性疲劳导致 患者动用辅助呼吸肌和膈肌呼吸,
腔内气压下进行呼气, 活动耐量下降 从而使气道更开放
以改善气流
晚期病人:膈肌 “平坦” ,持续吸气进一步压 缩膈肌à 向内拉季肋部胸壁
胡佛征 (吸气时,胸廓下侧季肋部内收)
缩唇呼吸
肌肉无力 & 消瘦
端坐呼吸 (调动胸肌)
颈部肌肉收
缩(胸锁乳
突肌、斜角
肌)
图注:
病理生理
机制
体征/临床表现/实验室检查
并发症
2013年1月7日发布于 www.thecalgaryguide.com
COPD: Findings on Investigations
Chronic Obstructive Pulmonary Disease (COPD)
Author: Yan Yu Reviewers: Jason Baserman Jennifer Au Naushad Hirani* Juri Janovcik* * MD at time of publication
Airflow obstruction
Lung tissue damage
↓ ventilation of alveoli
Blood perfusing ill- ventilated alveoli does not receive normal amounts of oxygen
During expiration, positive pleural pressure squeezes on airwaysà↑ obstruction)
No elastic recoil to push air out of lungs
Loss of lung parenchyma and vasculature ↓ surface area for gas exchange
↓ diffusion capacity
(on spirometry)
Hypoxemia: PaO2 < 70mmHg (on ABGs)
Abbreviations:
• FEV1: Forced expiratory volume in 1 second
• FVC: Forced vital capacity
• TLC: Total lung capacity
• VC: Vital Capacity
Investigations for COPD :
• Spirometry (Pulmonary function test)
Total expiration time takes longer than normal
FEV1/FEV < 0.7
(on spirometry)
Lungs don’t fully empty
More air trapped within lungs (hyperinflation)
More CO2 remains and diffuses into the blood
Hypercapnia: PaCO2 > 45
(on ABGs)
Ventilation- perfusion mismatch
High A-a gradient
(calculated from ABGs)
Low, flat diaphragm, >10 posterior ribs
(on frontal CXR)
High TLC and VC
(on spirometry)
• •
PaO2: partial pressure of O2 in arterial blood PaCO2: partial pressure of CO2 in arterial blood
• In the setting of fever and productive cough, especially if lung field opacifications are seen on CXR: consider sputum gram stain and culture to rule out pneumonia.
Air does not block X-ray beams, will appear black on X-ray film
Chronic hypercapnia makes breathing centers less sensitive to the high PaCO2 stimulus for breathing, & more reliant on the low PaO2 stimulus
(“CO2 retention”)
Give O2 carefully to these patients (high PaO2 may suppress patients’ hypoxic respiratory drive, ↓ their breathing, & ↑↑↑ PaCO2)
↑ retrosternal air space
(on lateral CXR)
Hyper-lucent
(darker) lung fields, ↓ lung markings (on frontal CXR)
• Arterial Blood Gasses (ABGs)
• Chest X-Ray (CXR): frontal and
lateral
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published January 7, 2013 on www.thecalgaryguide.com
COPD: !"#$ 气流阻塞
肺泡通气↓ 呼气时,胸膜腔正压挤压气 道à 阻塞↑
作者: Yan Yu 审稿人: Jason Baserman, Jennifer Au, Naushad Hirani*, Juri Janovcik* 译者:Zihong Xie (谢梓泓) 翻译审稿人: Yonglin Mai (麦泳琳), Zesheng Ye (叶泽生) * 发表时担任临床医生
慢性阻塞性肺疾病 (COPD)
肺组织损伤
没有弹性回缩力将
气体排出肺
肺实质与血管分布减少导 致气体交换面积↓
弥散功能↓ (肺功能检查)
更多的CO2残留 并扩散到血液中
高碳酸血症: PaCO2 > 45
(动脉血气)
血流灌注通气不良的肺泡
时无法获得足够的氧气
总呼气时长较正常长
FEV1/FEV < 0.7
(肺功能检查)
肺无法完全排空
更多空气潴留在肺部
(肺过度充气)
低氧血症: PaO2 < 70mmHg
(动脉血气)
通气-灌注不匹配
肺泡-动脉氧分压差↑ (可通过动脉血气分析计算得出)
横膈低平, 下移至第10肋后端 及以下部位 (胸部正位片)
TLC与VC增大 (肺功能检查)
缩写: • • FEV1: 1秒用 •
VC:肺活量
PaO2: 动脉血 力呼气量 氧分压
空气不会阻挡X射线, 在X光片上呈现为黑色
慢性高碳酸血症使呼吸中枢对PaCO2 刺激呼吸的敏感性下降 & 更依赖于低PaO2的刺激 (“二氧化碳潴留”)
给患者吸氧时需注意(高PaO2
可能会抑制患者低氧时对呼吸的 刺激,使呼吸驱动↓ & PaCO2↑↑↑ )
• FVC: 用力肺 • 活量
• TLC:肺总量 慢阻肺相关检查 :
PaCO2: 动脉 血二氧化碳 分压
胸骨后间隙↑
(胸部侧位片) 肺纹理↓
• 肺功能检查
• 动脉血气分析(Arterial Blood Gasses, ABGs)
• 胸部正侧位片
• 当患者发热和湿咳,特别是胸片上见肺野不清晰时:
肺透亮度↑, (胸部正位片)
考虑进行痰革兰氏染色及痰培养以排除肺炎可能
图注:
病理生理
机制
体征/临床表现/实验室检查
并发症
2013年1月7日发布于 www.thecalgaryguide.com
COPD: Complications Lung inflammation
Chronic Obstructive Pulmonary Disease (COPD)
Airway obstruction ↓ inhaled air in alveoli and terminal bronchioles
Rupture of emphasematous bullae on surface of lung
Inhaled air leaks into pleural cavity and is trapped there
Pneumothorax
Feeling a loss of control over one’s life, and hopelessness for the future
Goblet cell proliferation, ↑ mucus production
Death of airway
epithelium ciliated cells
↓ oxygenation of the blood passing through the lungs
Chronic hypoxemia
Kidneys compensate by ↑ erythropoietin (EPO) production
↑ Hemoglobin and red blood cell synthesis
Polycythemia (secondary)
Hypoxic alveoli cause the pulmonary arterioles perfusing them to reflexively vasoconstrict
Since most alveoli in the lungs are hypoxic, hypoxic vasoconstriction occurs across entire lung
Vasoconstriction ↑ blood pressure within lung vasculature
Pulmonary hypertension
↑ workload of the right ventricle (to pump against higher pressures)
To compensate, the right ventricle progressively hypertrophies and dilates, but over time its output ↓
Cor pulmonale
(Right heart failure in isolation, not due to Left heart failure)
Mucus trapped in airways, serve as nidus for infection
Acute exacerbation of COPD (AECOPD)
Pneumonia
The chronic, systemic inflammation in COPD is a hyper-metabolic state that consumes calories
Macro-nutrient deficiency
Trouble with respiration lead to inactivity and deconditioning
Wasting, muscle atrophy
More inactivity and deconditioning perpetuates the cycle
Depression
Author: Yan Yu Reviewers: Jason Baserman Naushad Hirani* Juri Janovcik* * MD at time of publication
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published January 7, 2013 on www.thecalgaryguide.com
COPD: !"# 肺部炎症
杯状细胞增殖, 气道上皮纤毛 粘液产生↑ 细胞死亡
黏液潴留呼吸道,成为感
染的病灶
慢性阻塞性肺疾病 (COPD) 气道阻塞à 吸入肺泡和终末细
肺大疱破裂
吸入的空气渗入
并潴留于胸腔
气胸
感觉生活失控,对未
来感到绝望
抑郁
作者: Yan Yu 审稿人: Jason Baserman, Naushad Hirani*, Juri Janovcik* 译者: Zihong Xie (谢梓泓) 翻译审稿人: Yonglin Mai (麦泳琳), Zesheng Ye (叶泽生) * 发表时担任临床医生
支气管的空气 ↓
流经肺的血液进行气 缺氧的肺泡à灌注肺泡的肺小动
慢性阻塞性肺疾 病急性加重期 (AECOPD)
肺炎
体交换↓ 慢性低氧血症
肾脏合成促红细胞 生成素进行代偿↑
血红蛋白与红 细胞合成↑
红细胞增多症 (继发性)
脉发生反射性血管收缩
肺大部分肺泡缺氧à整个肺 都出现缺氧性血管收缩
肺血管收缩 à 肺血管压力↑ 肺动脉高压
↑ 右心室负荷(泵血时对抗高压) 为了代偿,右心室逐渐肥大和扩张,
但随着病程进展,右心室输出量 ↓
肺心病 (单独出现右心衰竭,非左心衰)
COPD所致的慢性全身 呼吸困难导致活 性炎症会使机体处于高 动量减少和活动
代谢状态,消耗能量 耐量降低
宏量营养 素缺乏症
消瘦,肌肉萎缩
运动量下降和活动耐量
的降低造成恶性循环
图注:
病理生理
机制
体征/临床表现/实验室检查
并发症
2013年1月7日发布于 www.thecalgaryguide.com
" />
Asthma clinical findings

Author: Yan Yu Reviewers: Jason Baserman Jennifer Au Yonoglin Mai (麦泳琳) Naushad Hirani* * MD at time of publication
Variable, sporadic airway obstruction in response to triggers
Associated allergic eosinophil response
Eosinophils infiltrate: Skin
If severe:
↓ ventilation of alveoli
↓ oxygenation of blood (hypoxemia)
During expiration, positive pleural pressure squeezes on airwaysà↑↑ airway obstruction
Heart rate to improve
perfusion of tissue
Tachycardia
Respiratory centers rate of breathing to
compensate
Tachypnea
Gas is trapped within alveolià hyperinflates lungs
Ventilating larger lungs needs more effort
Patients need to voluntarily contract
their expiratory muscles faster and more forcefully to effectively expire
Narrower airways àturbulent
airflow, heard on auscultation
Expiratory Wheeze (high-pitched expiratory sound)
Nose
Rhinitis/ sinusitis
Runny nose, sneezing, etc
Atopic dermatitis
Skin rash, hives
Eyes
Conjunctivitis
Red itchy eyes, visual blurring
Episodic
dyspnea
(shortness of breath)
Chest tightness
During severe attacks:
Note: Asthma attacks often have two phases:
• An immediate attack (within 0-2 hours of the trigger, due to acute release of histamine from mast cells)
• A delayed attack (due to eosinophil infiltration of airways, presents within 3-4 hours after exposure to the trigger, peaks within 6-8 hours, and resolves within 24 hours).
Keep the possibility of a delayed attack in mind when treating patients in Emergency!
Note: Symptoms often worse at night or early in the morning.
Note: Asthma should be suspected in children experiencing dyspnea with multiple episodes of Upper Respiratory Tract Infections or Croup.
Patient compensates by activating accessory respiratory muscles to ↑ thoracic volume
Visible contraction of
neck muscles (Scalene, sternocleidomastoids)
↑↑↑ airway obstruction on
expiration, lungs take more time to empty
Prolonged expiratory phase of breathing
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published Dec 17, 2012 and updated Dec 4, 2021 on www.thecalgaryguide.com")
necrotizing fasciitis

Laceration
Recent surgery
Injection
Burn
Blunt force trauma
Childbirth
Lower extremity wounds
Bacteria enters tissue through open wound
Infection of muscle fascia Local immune response
Production of exotoxins by bacteria
Disruptions of protective skin barrier
Bacteria introduced into tissue during injury
Necrotizing Fasciitis
Type I infection: mixed aerobic and anaerobic bacteria Type II infection: group A streptococcus
Type III infection: marine organisms, clostridial infections Type IV infection: fungal organisms
Poor blood supply of muscle fascia allows for progressive spread of infection
Systemic immune response
Pyrogens produced by immune system
Pyrogens travel through
the bloodstream to the hypothalamus and alters the body’s thermal setpoint
Transmission of bacteria from infected tissue to blood
Sepsis
Streptolysin (exotoxin) causes blood clot formation
Blood clots in vessels
Tissue ischemia in epidermis, dermis, subcutaneous fat, muscle fascia, and/or muscle
Stimulation of programmed cell death
Tissue destruction
Pain more severe than clinical findings
↓ blood flow fails to meet tissue’s needs
Tissue death
Build up of gas in subcutaneous
tissue from bacteria metabolism
Crepitus
↑ serum creatinine
kinase from protein breakdown
↑ blood flow to infected tissue
Warmth Erythema
Immune cells release vasoactive cytokines into the blood
Capillary vasodilation
Fluid and proteins shift from cells and capillaries to interstitial space
Blood
vessel dilation
↓ perfusion of vital organs
Organ failure
Hypotension
↑ heart rate to perfuse vital organs
Tachycardia
Bacteria releases toxins which are taken up into the bloodstream
Immune cells produce inflammatory cytokines
Circulating toxins activate T cells, over- activating the systemic immune response
Toxic Shock syndrome
Infection ↑ white blood cell production in bone marrow
↑ white blood cells
Destructionof peripheral nerve endings
Insensitivity to pain
Tissue hypoxia à anaerobic metabolism
Poor perfusion of lungs impairs gas exchange
Tachypnea
Cytokines affect dopamine production in the basal ganglia
Acute malaise
Production of non-specific acute phase reactants
↑ C reactive protein and erythrocyte sedimentation rate
Fluid-filled blisters
Edema
Fever Compartment syndrome (see relevant Calgary Guide slide)
Amputation ↑ serum
lactate
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
First published Nov 20, 2013, updated Dec 19, 2021 on www.thecalgaryguide.com")
Minimal Change Disease
 present in serum
Immunofluorescence test negative
Idiopathic/Primary Minimal Change Disease
No identifiable extraglomerular disease process causes this condition
Secondary Minimal Change Disease
Infections, NSAIDS, neoplasms via unclear mechanisms
Minimal change to glomerulus seen on light microscopy
Podocyte effacement seen on electron microscopy
Abnormal T Cell activation and release of cytokines (sometimes called permeability factors) that are toxic to podocytes
Podocyte foot processes efface (become flattened) or fuse together
Damage to negatively charged foot processes damages the charge barrier of the glomerulus that repels negatively charged molecules
↑ filtration of larger negatively charged molecules, such as low-molecular weight proteins like albumin, from the blood into the renal tubular filtrate
Induces ↑ hepatic lipoprotein synthesis and ↓ lipoprotein catabolism
Hyperlipidemia
(↑ serum LDL, VLDL, and triglycerides)
↑ lipid filtration through glomerulus
Lipiduria (fatty casts)
Hypo-albuminemia
↓ oncotic pressure in capillaries
Fluid leaks into interstitial space
↑ Filtration of Proteins C and S and antithrombin
Hypercoagulable state
Proteinuria
↑ Filtration of immunoglobulins
Immunosuppression
Infections
↑ Filtration of Plasminogen
Plasminogen converted to plasmin in the cortical collecting duct via urokinase- type plasminogen activator
Plasmin activates the epithelial sodium channel
↓ intravascular volume
Hypotension
Pre-Renal Acute Kidney Injury
Underfill edema
(see slide)
Thrombosis Edema (especially peri-
orbital, scrotal, labial, and extremities)
Overfill
edema
(see slide)
↑ Na+ and water reabsorption
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published December 30, 2021 on www.thecalgaryguide.com")
NSAIDs and the Kidney Nephrotoxicity

Authors: Kyle Moxham Mehul Gupta Reviewers: Emily Wildman Yan Yu* Adam Bass* * MD at time of publication
Inhibition of Cyclooxygenase COX-1 (expressed in kidney) and COX-2 (expressed in kidney and sites of inflammation)
NSAID induced nephrotoxicity: associated with chronic NSAID usage independent of dosage
COX inhibition ↑ conversion of arachidonic acid (AA) to leukotrienes, causing systemic T-cell dysfunction (unknown mechanism)
Type IV systemic hypersensitivity (delayed
T helper cell mediated) reaction to drug exposure
T cells release inflammatory cytokines into the bloodstream
(see Calgary Guide slide on NSAIDs and the Kidney: Mechanism of Action and Side Effects)
T cells infiltrate the renal interstitium, sparing the glomeruli and blood vessels
Overproduction of cytokines by T cells causing inflammation,
tissue damage, and cell death, of the renal intersitium
Drug Induced Acute interstitial nephritis (AIN): a type of immune-mediated tubulointerstitial injury
Activated T-cells infiltrate the glomerulus and cause podocyte injury (epithelial cells attached to the glomerular basement membrane)
Membranous nephropathy:
nephrotic syndrome involving autoimmune glomerular basement membrane thickening & complete podocyte effacement (seen on kidney biopsy)
Minimal change disease: nephrotic syndrome caused by autoimmune podocyte effacement (seen on kidney biopsy)
Cytokine mediated activation and
proliferation of immune cells like macrophages and eosinophils
Cytokines travel to hypothalamus,
causing change in the body’s thermal set point
Fever
Podocyte effacement allows for serum proteins to across the glomerulus into the tubular lumen (see Calgary Guide slide on Nephrotic Syndrome for full mechanisms)
Repeated NSAID exposure causing
recurrent unrecognized AIN and damage of the kidney
Chronic interstitial nephritis /analgesic nephropathy
Infiltrating immune cells (predominantly neutrophils) are filtered into/enter the renal tubules, and form clumps (“casts”) within the tubulesà casts are then released into urine
WBC cast on urinalysis
↑ Blood eosinophils
Immune cells infiltrate the
dermis and epidermis of the skin
Rash
Abnormal quantities of protein present in urine
Protein present in the blood is improperly filtered into the filtrate at the glomerulus
Protein- Creatinine
Ratio >3.5mg/mg
Proteinuria >3.5g/d
Hypo- albuminemia
↓ plasma oncotic pressure resulting in fluid extravasation into the interstitium (see Calgary guide slide on Edema for full mechanisms)
Pitting Edema
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published January 13, 2022 on www.thecalgaryguide.com")
Langerhans Cell Histiocytosis
 CD1a+/CD207+ (Langerhans cell phenotype surface receptors) dendritic cells in tissue(s)
Somatic BRAFV600E mutation: BRAF is a kinase in the MAPK pathway
Other somatic (non- reproductive cell) mutations
Idiopathic (unknown cause)
Mutation(s) can occur in one of these precursor cell types*
Hematopoietic stem cell (earliest cell of blood cell differentiation) in bone marrow
Committed dendritic cell (type of myeloid antigen-presenting cell) precursor in bone marrow or blood
Committed dendritic cell precursor in tissue
Constitutive activation of the MAPK pathway (signalling pathway that regulates variety of cellular processes) in one of these precursor cell types
*Note: Based on the “misguided myeloid differentiation” modelàthe earlier the mutation(s) occur in the myeloid cell differentiation pathway, the more severe the disease.
Langerhans Cell Histiocytosis
Accumulation of abnormal CD1a+/CD207+ dendritic cells (Langerhans Cell Histiocytosis cells or LCH cells) with an inflammatory background in one or more organs
Authors:
Ran (Marissa) Zhang Reviewers:
Mehul Gupta
Kiera Pajunen
Yan Yu*
Lynn Savoie*
* MD at time of publication
↑ recruitment & activation of T cells, macrophages, eosinophils in tissue(s) around the body
↓ CCR7 & CXCR4 (chemokine receptors) expression on LCH cellsàinhibits migration of LCH cells to lymph nodes
↑ BCLXL (an apoptosis regulator protein) expression on LCH cellsà inhibits apoptosis of LCH cells
Immune cell infiltration & ↑ pro-inflammatory chemokine/cytokine release à dysregulated local & systemic inflammation
Accumulation of LCH cells in tissue(s) around the body
Inflammatory lesion (an area of abnormal tissue) formation in one or more organs:
In pituitary stalk
Mass effectà
obstruction of antidiuretic hormone (complex mechanisms)
In liver
Invasion & accumulation of cells foreign to liverà expands liver
Chronic local inflammation
Scarring of bile ducts
↓ bilirubin clearance from liveràbuildup into serum
↑ serum bilirubin
Jaundice
In cortical bone Cytokine production
à↑ osteoclast
(cells that break down bone) activity
↑ rate of bone loss
Osteolytic bone lesions
In bone marrow
Unclear mechanism but likely due to macrophage activation
↑ phagocytosis (ingestion & destruction) of blood cells
In spleen
Invasion & accumulation of cells foreign to spleen
Forms aggregates that expand the red pulp (functions as the blood filter in spleen)
Splenomegaly
In skin
Unclear mechanisms
Variable presentations: most commonly pinpoint erythematous (red) papules or erythematous plaques with crusting & scaling
Hepatomegaly • BRAF- B-Raf proto-oncogene, serine/threonine kinase
• CCR7- C-C motif chemokine receptor type 7
• CD1a- Cluster of differentiation 1A
• CD207- C-type lectin domain family 4 member k • CXCR4- C-X-C chemokine receptor type 4
• LCH- Langerhans Cell Histiocytosis
• MAPK- Mitogen-activated protein kinase
Diabetes Insipidus
Abbreviations:
• BCLXL- B-cell lymphoma-extra large
Anemia, thrombocytopenia &/or neutropenia
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published January 13, 2022 on www.thecalgaryguide.com")
Vitiligo Pathogenesis and Clinical Findings
 that, through complex mechanisms, stimulate the immune system’s CD8+ T-cells to destroy melanocytes
Melanocytes enter apoptotic or senescent state
↓ Functional melanocytes
↓ Melanin production
Overall loss of functional melanocytes
Vitiligo
Normal Skin
Pigmented Epidermis Dermal- Epidermal Junction
Dermis
Autoimmune destruction of melanocytes
Depigmented Epidermis
Melanocytes
Immune-mediated destruction of melanocytes (by both neutrophils and CD8+ T cells)
A depigmenting skin disorder characterized by selective loss of melanocytes
Depigmentation in areas of
↑ pressure, friction and/or trauma
Nonsegmental vitiligo
Smooth unpigmented macules or patches in bilateral, often symmetric pattern
Somatic mosaicism (mutation limited to a subset of cells) in zygote during development
Segmental vitiligo
Smooth unpigmented macules or patches in unilateral pattern not crossing midline
Vitiligo
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published February 15, 2022 on www.thecalgaryguide.com")
isotretinoin-systemic-retinoid-mechanisms-and-side-effects
:
Mechanisms and Side Effects
Isotretinoin
Authors: Ayaa Alkhaleefa Reviewers: Mehul Gupta, Ben Campbell Stephen Williams, Lauren Lee, Yan Yu*, Laurie Parsons* * MD at time of publication
↑ Apoptotic signaling in sebocytes
Inhibits androgen nuclear receptors responsible for sebum secretion
↓ Sebaceous lipogenesis
↑ Apoptotic signaling in neural crest cells during embryonic development
Alterations in hindbrain, neural crest, otic anlage, and reduced pharyngeal arch in embryo
Craniofacial, cardiac, thymic, and central nervous system malformations in fetus
Isotretinoin isomerizes to all-trans retinoic acid (ATRA)
ATRA enters cell nucleus and binds retinoic acid receptors and retinoic X receptors
ATRA induces tumour necrosis factor-related apoptosis-inducing ligand
↑ Apoptotic signaling in epidermal keratinocytes
↑ Expression of FoxO1
↑ Expression of p53 (tumour suppressor)
Release of caspases 3, 6, 7, and 9
↑ Cell cycle inhibitors p21 and p27
↓ Pro-survival proteins (Survivin)
Sebaceous gland involution
Sebum suppression
C. acnes unable to break down sebum into pro- inflammatory lipids
↓ Colonization with C. acnes
Teratogenicity
↑ Cornification (death) of epidermal keratinocytes
↓ Corneodesmosomes (main adhesive structures of the stratum corneum)
↓ Cohesion of corneocytes (dead keratinocytes)
↓ Corneocyte buildup in pilosebaceous follicles
C. acnes unable to populate and release cytokines in corneocytes
Epidermis
Dermis
Pilosebaceous follicle
↓ Stratum corneum thickness
↑ Trans-epidermal water loss
Dryness, peeling & inflammation
of lips (cheilitis), skin (dermatitis) & mucosa (mucositis)
↓ Comedogenesis
Sebum
Hair follicle Dermal-
Epidermal Junction
Sebaceous gland (sebocytes)
Death of sebocytes in pilosebaceous follicles
Reduction of number and size of acne lesions
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published March 20, 2022 on www.thecalgaryguide.com")
irritable-bowel-syndrome-ibs-pathogenesis-and-clinical-findings
: Pathogenesis and clinical findings
Authors: Ben Campbell Reviewers: Mehul Gupta Kiana Hampton Yan Yu* Edwin Cheng* * MD at time of publication
Note:
*Several signaling pathways in the gut are believed to play a role in IBS. The serotonin pathway is represented here for illustrative purposes, and it is a common target of therapy.
Pathogenesis is multifactorial, not fully understood, and may arise from any one or a combination of these factors
Genetic factors
are believed to influence many of these pathways
Environmental factors
Gut infection, ischemia, injury, chronic low-level inflammation, altered microbiome
Abnormal gut chemical milieu (cytokines, lymphocytes, mast cells, hormones)
One common pathway: altered quantity or activity of serotonin-releasing enterochromaffin * cells and serotonin reuptake transporters in gut
Serotonin is a key stimulator of gut muscle contraction and sensory signaling
Altered bowel motility
Bowel motility can ↑, ↓ or alternate ↑/↓
Psychosocial factors
Anxiety, depression, adverse experiences, dysregulated stress response
Brain can alter normal reflexes of enteric nervous system (e.g. via hypothalamic stress hormone release)
↑ Activity of sensory receptors in gut + recruitment of formerly ”silent” receptors
Altered visceral bowel sensation
Visceral hypersensitivity
Complex mechanisms
↓ Brain’s ability to modulate pain signals from gut;
↑ vigilance to pain
Irritable Bowel Syndrome
Disorder of brain-gut interaction with gastrointestinal manifestations
Not a predominantly inflammatory condition
During times of ↑ bowel motility:
No lesions in gut mucosa
(Absence of ”red flags” for organic disease)
↑ Impacts of other factors that precipitate diarrhea
Dietary sensitivity
(e.g. to intake of fermentable oligo/di/ monosaccharides and polyols— FODMAPs—which draw water into gut lumen by osmosis)
↑ Impacts of other factors that precipitate constipation
Dietary sensitivity (e.g. to inadequate intake of motility- promoting fibre)
During times of ↓ bowel motility:
↑ Time for water absorption from feces and bacterial fermentation
↑ Hard stool and trapped gas in bowel
↑ Number and sensitivity of active pain receptors (nociceptors) in gut
Hyperalgesia
↑ Response to painful stimulus
Stretch receptors in gut stimulate pain in normally unconscious neural pathways
Allodynia
Pain in response to normal stimulus
↑ Peristalsis
↓ Time for water absorption from feces
↑ Water volume in gut lumen
↑ Stretch / stimulation of visceral afferent nerves in gut
Absence of bloody stool and anemia
Absence of nocturnal symptoms and inflammatory markers
Constipation
Bloating
Diffuse abdominal pain
All symptoms may exacerbate psychosocial factors
Bowel distension
Diarrhea
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published April 30, 2022 on www.thecalgaryguide.com")
hypercalcemia-pathogenesis

Ectopic (outside of kidney) production of 1-α-hydroxylase
1-α-hydroxylase converts calcidiol to calcitriol (aka 1,25-OH Vitamin D or active Vitamin D)
↑ 1-25 OH Vitamin D
↑ Absorption of Ca2+ in the small intestine
↑ Vitamin D Intake
Dietary Vitamin D is converted to calcidiol (aka 25- OH Vitamin D or inactive Vitamin D) in the liver
Primary Hyperparathyroidism
↑ or normal* serum parathyroid hormone (PTH) level
Tumour released Parathyroid Hormone related Peptide (PTHrP) mimics natural PTH (PTH level itself is low)
Tumour produces bone- reabsorbing substances (IL-6, IL-1, RANKL)
Paget’s Disease
Osteoblasts produce abnormally high levels of RANKL
Block Na+/Cl- cotransporter (NCC) on distal convoluted tubule (DCT) cells of the nephron
↓ Na+ and Cl- transport from lumen into DCT cells
↓ Intracellular Na+ in DCT cells
+ To compensate Na ,
↑ Ca-ATPase and 3:Na:Ca exchanger activity on DCT cells (moves 3 Na+ into cell, 1 Ca+ out into peritubular capillary)
↑ Ca+ moving into bloodstream
↓ Mechanical stimulation of osteocytes
↓ Signaling of osteoblasts (cells that form bone)
↓ New bone formation
↓ Ca2+ incorporation
into bone
↑ Ca2+ in plasma
↑ reabsorption of Ca2+ from the distal portion of nephron back into the blood
↑ 1-α-hydroxylase production in the proximal convoluted tubule
Milk Alkali Syndrome
Ca2+ intake exceeding 2000 mg per day leads to unregulated gut Ca2+ absorption into the blood
PTH binds to osteoblasts on the surface of bone
Osteoblasts produce RANKL to stimulate osteoclast function
↑ Osteoclast (cells that breakdown bone) activity
↑ Bone breakdown
Ca2+ within bone is released into the bloodstream
Osteolytic bone metastases
Hyperthyroidism
↑ Catabolic thyroid hormones
↑ RANKL: OP ratio, RANKL promotes bone breakdown by stimulating osteoclast growth while OP (osteogenic protein) promotes bone formation
Hypercalcemia Hypercalcemia
Authors: Alexander Arnold, Peter Vetere, Huneza Nadeem Reviewers: Mark Elliott, Ran (Marissa) Zhang, Yan Yu*, Hanan Bassyouni* * MD at time of publication
See Hypercalcemia: Clinical Findings slide
*Note: A normal PTH value is abnormal in the context of hypercalcemia, since hypercalcemia would normally negatively feed back to suppress PTH production.
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published February 11, 2014, updated Aug 11, 2022 on www.thecalgaryguide.com")
asthma-how-treatments-work-and-common-side-effects

Long-acting Beta Agonists (Controller)
Short-acting Muscarinic Antagonists (Exacerbation)
Long-acting Muscarinic Antagonists (Controller)
Magnesium Sulfate (Exacerbation)
Monoclonal Antibodies (Controller)
Leukotriene Receptor Antagonists (Controller)
Inhaled Corticosteroids (ICS) (Controller)
Systemic Corticosteroids (Exacerbation)
Binding to beta-2 adrenergic receptors and subsequent intracellular signal cascade in bronchial smooth muscle
Off-target binding occurs in other systemic cells
Inhibition of muscarinic acetylcholine receptors in airway muscle cells
↓ Activation of the inositol triphosphate (IP3)
intracellular pathway
(IP3 pathway normally functions to mobilize intracellular Ca2+ stores)
Inhibition of Ca2+ channels on airway smooth muscle surface
↓ Influx of Ca2+
Binding and inactivation of inflammatory signal molecules (IgE, IL-5)
Inhibition of leukotriene receptors in lung and immune cells
Steroid binds to nuclear receptors within cells
↓ Gene expression/synthesis of immune mediators (ex. cytokines)
Stimulation of
Na/K ATPase (which transports K into cells)
Hypokalemia Palpitations Tachycardia Muscle cramps Tremor
Authors: Chunpeng Nie Reviewers: Sravya Kakumanu, Ben Campbell, *Tara Lohmann
* MD at time of publication
↓ Activation of mast cells (by ↓ IgE) and eosinophils (by ↓ IL-5/leukotrienes)
↓ Release of inflammatory cytokines by these cells (↓ Type 2 inflammatory response in airways)
↓ Permeability of airway vasculature
↓ Microvascular leakage into airway
Inadvertent sympathetic nervous system activation
Bronchial smooth muscle cells have ↓ Ca2+ release from intracellular stores (i.e. from sarcoplasmic reticulum)
↓↓ Cytoplasmic Ca2+
Myosin (muscle protein) unable to be activated for muscle contraction
↓ Smooth muscle contraction in bronchioles
Bronchodilation
ICS cause ↓
immune cell activity in the oropharynx
Susceptibility to infection and irritation of the oropharynx from inhaled particles and pathogens
Hoarseness Thrush
Systemic corticosteroids cause ↓ immune cell activity in whole body
↑ Susceptibility to any infection
Many other side effects
Similar effects on other muscles in the body outside bronchi
Dry mouth
Urinary retention
Constipation
↓ Mucosal edema
↓ Airway Mucus
Airflow improvement
↑ Peak flow, ↑ Oxygenation, ↓ Dyspnea
Legend:
Pathophysiology
Mechanism
Treatment Effect
Complications
Published October 9, 2022 on www.thecalgaryguide.com")
chronic-pancreatitis-complications
 from acinar cells in exocrine pancreas
(early in disease course)
Proteins precipitate and form aggregates within pancreatic ducts
Accumulation of protein aggregates and localized fibrosis block pancreatic ducts
Rupture of acinar cells near blocked ducts → release of intracellular enzymes and fluid
Accumulation of enzyme- rich fluid within pancreas
Intra-pancreatic pseudocysts (differ from pseudocysts in acute pancreatitis, which are primarily extra-pancreatic)
Chronic Pancreatitis
Recurrent episodes of acute pancreatitis leading to irreversible fibroinflammatory pancreatic damage
Inflammatory cytokines are
continuously released from damaged pancreas over years
Cytokines damage endothelium of intra- and peri-pancreatic blood vessels (including splenic vein, which runs posteriorly behind pancreas and allows for its venous drainage)
Thin and weakened
vessel walls balloon outwards from pressure of blood flow
Pseudoaneurysms
Venous stasis (low blood flow) → ↑ concentration of clotting factors
Obstruction of peripancreatic ducts
Author: Ashar Memon Reviewers: Yan Yu*, Kiana Hampton, Sylvain Coderre* * MD at time of publication
Exocrine insufficiency
(↓ secretion of digestive enzymes, e.g., Lipase, into gastro-intestinal tract)
↓ digestion of foods and absorption of nutrients (including fats)
↓ absorption of fat-soluble vitamin D
Metabolic bone disease
(a group of disorders of decreased bone mineralization)
Blood vessels dilate and swell from increased blood flow
Gastric varices
Cytokines perpetually activate pancreatic stellate cells (stellate cells produce proteins that remodel extra- cellular matrix)
Pancreatic stellate cells increase amounts of collagen and other extra- cellular matrix molecules in pancreas → Fibrosis
Pancreatic proteolytic enzymes (e.g., trypsin) in fluid-filled pseudocysts digest walls of adjacent blood vessels
Fibrotic tissue and pseudocysts compress peripancreatic structures (including splenic vein)
Cytokines stimulate apoptosis of hormone- producing pancreatic Islet cells
(e.g., beta cells)
Endocrine insufficiency
(↓ production and secretion of pancreatic hormones)
Damaged endothelial cells of splenic vein trigger coagulation cascade
(See Calgary Guide slide on Coagulation Cascade)
Splenic vein thrombosis
↑ resistance to blood flow through splenic vein
Cytokines
stimulate apoptosis of acinar cells in exocrine pancreas
Malnutrition
↓ secretion of insulin
↓ cellular uptake and metabolism of glucose → hyperglycemia
Diabetes mellitus
Collateral blood vessels develop around stomach so blood can circumvent splenic vein and relieve splenic vein hypertension
Duodenal obstruction Biliary obstruction
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published October 18, 2022 on www.thecalgaryguide.com")
thiazide-diuretics-mechanism-of-action-and-adverse-side-effects
 Zhang, Julian Midgley* * MD at time of publication
Thiazides in circulation enter the proximal convoluted tubule (PCT) cells by basolateral Organic Acid Transporter 1 (OAT1)
↑ Intracellular thiazide availability
for apical OAT4 exchangers
OAT4 reabsorb urate in lumen in exchange
for thiazide that is excreted
↑ Urate in serum Hyperuricemia Uric acid build up in
joints Gout
Block the Na-Cl cotransporter (NCC) in the distal convoluted tubule (DCT) of the nephron
Impaired water excretion, vasopressin release, and ↑ water intake (mechanisms unclear)
Hyponatremia
Lack of intracellular Na+ needed to drive the Na+/K+ ATPase (moves 2 K+ into cell, 3 Na+ out into peritubular capillary)
↓ Intracellular Na+ drives Ca-ATPase and 3:Na:Ca exchanger activity (moves 3 Na+ into cell, 1 Ca+2 out into peritubular capillary)
↑ Ca+2 in serum
Hypercalcemia
See Hypercalcemia: Clinical Findings slide
↓ Na+ and Cl- reabsorption into proximal DCT cells
↑ Na+ in DCT filtrate by ∼3-5%
Water follows Na+ to maintain balanced osmotic pressure
↑ Water available for excretion
Mild Diuresis
↓ Blood volume Hypotension
↑ Na+ filtrate delivery to cortical collecting duct (CCD)
Epithelial sodium channels (ENaC)s on principal cells of the CCD transport Na+ from filtrate into the principal cells
↑ Intracellular Na+ drives Na/K+ ATPase activity on principal cells (moves 2 K+ into cell, 3 Na+ out into peritubular capillary)
↑ Intracellular K+ drives H/K+ ATPase activity on
intercalated cells (moves 1 H+ into cell, 1 K+ out into tubular filtrate)
↓ K+ in serum
Hypokalemia
See Hypokalemia: Clinical Findings slide
Acute response to
high dose thiazide diuretics (mechanism unclear)
Hyperlipidemia
Hypokalemia induces hyperpolarization of pancreatic beta cells
↓ Number of voltage gated Ca+2 channels
open given intracellular charge
↓ Ca+2 influx prevents exocytosis mediated insulin release
↓ Glucose uptake in the body
Hyperglycemia
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published October 18, 2022 on www.thecalgaryguide.com")
type-ii-proximal-renal-tubular-acidosis-pathogenesis-and-laboratory-findings
 Zhang, Julian Midgley* * MD at time of publication
Overview of bicarbonate reabsorption in the proximal tubule (PT):
Serum bicarbonate (HCO3-) is filtered by the glomerulus and enters PT lumen
H+ ATPase and sodium- hydrogen exchanger 3
(NHE3) on luminal surface of PT cell secrete H+ into PT lumen
H+ combines with HCO3- in
PT lumen to form carbonic acid (H2CO3)
H2CO3 is broken down to H2O and CO2 by luminal membrane carbonic anhydrase 4
CO2 diffuses into PT cell
CO2 reacts with H2O to form H2CO3 in PT cell, catalyzed by cytosolic carbonic anhydrase 2
H2CO3 rapidly breaks down intracellularly to form HCO3- and H+
HCO3- is reabsorbed into plasma by sodium
bicarbonate transporter (NBCE1) on basolateral surface of PT cell
HCO3- is available for use to buffer plasma H+
H+ is available intracellularly for use by H+ ATPase and NHE3 exchanger
Mutation encoding NHE3 exchanger
↓ H+ secretion into PT lumen
Galactosemia, Wilson disease, cystinosis, tyrosinemia, glycogen storage disorders
Dent disease, Lowe syndrome
Infiltrative disorders
e.g., amyloidosis, multiple myeloma, monoclonal gammopathies
Drugs
e.g., tenofivir, ifosfamide, cisplatin
Carbonic anhydrase inhibitors
↓ H2CO3 breakdown to H2O and CO2
Mutation encoding CAII enzyme, carbonic anhydrase inhibitors
↓ H2CO3 production in PT lumen
↓ CO2 diffusion into PT cell
↓ H2CO3 production in PT cell
↓ HCO3- production in PT cell
↓ HCO3- reabsorption by PT cell into plasma
Type II/Proximal Renal Tubular Acidosis (RTA)
Defective HCO3- reabsorption in proximal tubule
Normal anion gap metabolic acidosis (NAGMA)
See NAGMA slide
Accumulation of toxic metabolites systemically, including in the PT
Disruption of endocytosis and intracellular transport systemically, including in the PT
Accumulation of light chain/amyloid deposits in PT
Interfere with PT’s ability to reabsorb ions/molecules
• • • •
Fanconi Syndrome:
generalized PT dysfunction
Defective tubular reabsorption of other ions/molecules to varying
degrees, including phosphate, glucose, sodium and amino acids
Tubular proteinuria Phosphaturia
Glucosuria
↑ Na+ secretionàhypovolemia
Mutation encoding NBCE1 transporter
↑ HCO3- delivery to distal nephron
K+ in distal nephron lumen binds to HCO3-
K+ is lost through osmotic diuresis
Hypokalemia
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published October 24, 2022 on www.thecalgaryguide.com")
Granulomatosis with Polyangiitis Pathogenesis

Environmental exposures
Silica dust, cigarette smoke, infections (Staphylococcus aureus)
Genetic factors
Alpha-1 antitrypsin deficiency, proteinase 3 gene mutation
↑ Production of cytokines and antineutrophil cytoplasmic antibodies (ANCAs) (mechanism unknown) Cytokines bind to endothelial cells (that line blood vessels) and neutrophils, priming them
Proteinase 3 (PR3), an enzyme that degrades extracellular matrix proteins, migrates from neutrophil granules to neutrophil cell surface
Circulating ANCAs bind to PR3 on neutrophils
PR3 stimulates maturation of dendritic cells in lungs
Dendritic cells present antigen (PR3) to naïve CD4+ T cells in peripheral lymph nodes
T cells differentiate into type 1 and type 17 helper T cells (Th1 and Th17 cells)
Th1 and Th17 cells secrete cytokines (interferon γ (INF-γ) and tumor necrosis factor α (TNF-α)) in lungs
Secreted cytokines trigger macrophage maturation
Formation of granulomas (giant cells with central necrosis surrounded by plasma cells, lymphocytes, and dendritic cells) primarily in lungs and upper airways
ANCA-activated neutrophils release proinflammatory cytokines, attracting more neutrophils to endothelium (blood vessel wall)
ANCA-activated neutrophils undergo firm adhesion to endothelium
ANCAs stimulate ↑ secretion of proteolytic enzymes and reactive oxygen species from neutrophils
Endothelial damage and tissue injury
Granulomatosis with polyangiitis (GPA)
ANCA-associated vasculitis affecting medium and small-sized arteries, associated with necrotizing granulomas
Constitutional symptoms with involvement of multiple organ systems
(see slide on clinical findings)
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published December 4, 2022 on www.thecalgaryguide.com
Granulomatosis with polyangiitis: Clinical findings Inflammation-mediated endothelial damage and granuloma formation
(see slide on pathogenesis)
Granulomatosis with polyangiitis (GPA)
Author:
Oswald Chen
Reviewers:
Ben Campbell
*Liam Martin
* MD at time of publication
ANCA-associated vasculitis affecting medium and small-sized arteries, associated with necrotizing granulomas
Constitutional symptoms
Fever, unintentional weight loss, night sweats, arthralgias
Skin involvement
Inflammation of cutaneous vessels
Systemic inflammation obstructing blood flow, with granulomatous lesions primarily in upper airways and lungs
Ear, nose, and throat involvement
↑ C-reactive protein (CRP) and erythrocyte sedimentation rate (ESR) (markers of inflammation)
Necrotizing granulomas on biopsy of affected tissue
Positive PR3-ANCA/c-ANCA blood test (antibodies present in ~90% of patients, described in GPA Pathogenesis slide)
Renal involvement
Inflammation of renal vessels
Rupture of basement membrane (layer that filters blood from glomerular capillaries into Bowman’s capsule)
Pauci-immune glomerulonephritis (see Nephritic Syndrome slide)
Rapidly progressive glomerulonephritis
Eye involvement
Inflammation of ocular tissue
Conjunctivitis
Scleritis/ episcleritis (painful red eye)
Lower respiratory tract involvement
Inflammation of pulmonary vessels
Vessel occlusion and ischemia
Skin necrosis
Vessels burst and blood pools under skin
Round and retiform (net- like) palpable purpura of lower extremities
Granulomatous destruction of nasal cartilage
Collapse of nasal bridge
Saddle nose deformity
Inflammation of paranasal sinus and nasal cavity vessels
↓ Perfusion of lungs
Dyspnea
Damage to interstitial capillaries
Hemoptysis
Diffuse alveolar hemorrhage
Rhinitis/ sinusitis
Granulomatous obstruction of eustachian tube
Otitis media (see Otitis Media slide)
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published December 4, 2022 on www.thecalgaryguide.com")
Acute and Chronic Gastritis: Pathogenesis and clinical findings

Noninfectious
NSAIDs, alcohol, gastric reflux
Stress
Critical illness, trauma
Gastric ulceration (see Peptic Ulcer Disease slide)
↓ Gastric mucosal defense
(↓ secretion of prostaglandins and mucus)
Acid erodes gastric mucosa
Inflammatory cells (primarily neutrophils) infiltrate site of injury
Acute Gastritis
Acute inflammation of gastric mucosa
Inflammatory cells (primarily lymphocytes and plasma cells) accumulate in gastric mucosa
Autoimmune Metaplastic Environmental Metaplastic Atrophic Gastritis (AMAG) Atrophic Gastritis (EMAG)
Atrophic Gastritis
Chronic inflammation of gastric mucosa
Inflammatory cells destroy gastric glandular epithelial cells
Gastric mucosal atrophy and metaplasia (replacement of gastric mucosal cells with intestinal epithelial cells, commonly goblet cells)
Hypochlorhydria
(parietal cell loss → ↓ hydrochloric acid secretion)
↓ Iron absorption
Iron deficiency anemia
(see Iron Deficiency Anemia slide)
Activation of chemoreceptors and mechanoreceptors
Activation
of visceral nociceptors
Visceral afferents stimulate chemoreceptor trigger zone of medulla
Nausea/ vomiting
Autoimmune
Associated with human leukocyte antigens HLA-B8 and HLA-DR3, which are proteins expressed on immune cell surface
Antibodies destroy parietal cells and intrinsic factor in gastric body and fundus
Epigastric pain (typically burning or gnawing) Dyspepsia (abdominal discomfort after
eating, often with early satiety and bloating)
Parietal cell loss →
↓ intrinsic factor →
↓ vitamin B12 absorption
Vitamin B12 deficiency
(see Vitamin B12 Deficiency slide)
Macrocytic Peripheral anemia neuropathy
↓ Gastric acidity leads to ↓ inhibition of G cells in antrum Hypergastrinemia (↑ gastrin release from G cells)
Gastrin binds to cholecystokinin-B (CCK-B) receptors on parietal and enterochromaffin-like (ECL) cells of gastric body
↑ CCK-B signaling leads to ↑ cell proliferation and ↓ cell death (↓ apoptotic activity)
Hyperplasia (↑ number of cells)
Low-grade dysplasia (disordered growth of epithelium) High-grade dysplasia
Carcinoid tumor (0.7% of cases) Malignancy arising from ECL cells
Gastric adenocarcinoma (0.3% of cases) Malignancy arising from gastric epithelial cells
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published November 15, 2022 on www.thecalgaryguide.com")
Ischemic Stroke: Pathogenesis
 of basal or brainstem penetrating arteries
Large artery atherosclerosis
Cholesterol plaque ↓ diameter of intra- or extracranial vessel
Cardiac embolism
Blood clot in heart breaks free, travels to brain
Other
E.g. volume loss, severe infection
Unknown
E.g. 2 or more mechanisms
Modest ↓ in O2 at penumbra (see figure)
Authors: Mizuki Lopez Andrea Kuczynski Illustrator: Mizuki Lopez Reviewers: Sina Marzoughi Usama Malik Hannah Mathew Ran (Marissa) Zhang Andrew M Demchuk* Gary M. Klein* * MD at time of publication
Significant ↓ in O2 at ischemic core (see figure)
↑ Anaerobic metabolism ↓ ATP
Production
Dysfunction of Na+/K+ ATPase pump (for 1 ATP molecule, 3 Na+ moved out of cell, 2 K+ moved into cell)
H2O influx following Na+ Cerebral edema
Compression of vessels and surrounding tissue damages blood-brain barrier
↑ Permeability of damaged blood-brain barrier
Infiltration by peripheral immune cells
Immune cells release inflammatory cytokines
↓ Cerebral Blood Flow
Penumbra Ischemic core
Metabolic demands are greater than supply of ATP
Cell death
Microglia (resident neural immune cells) activate to clean dead cell debris
Microglia release inflammatory cytokines (TNFα, IFγ, IL-1β)
Cytokines lead to astrocyte activation (support cells for neurons)
Astrocytes release more inflammatory cytokines
Inflammation of brain tissue
↑ Na+, Ca2+ influx, K+ outflux
↓ Glutamate (excitatory neurotransmitter) reuptake by astrocytes (support cells for neurons)
↑ Glutamate in extracellular fluid
Spreading depolarization from core (unclear mechanism)
Activate biochemical pathways including glutamate receptor activation
↑ Glutamate activity
Activate glutamate receptors that conduct Ca2+
↑ Ca2+ influx into neuron
Activation of catabolic proteases, lipases, nucleases in neuron
Dysfunction of neuronal protein synthesis and activity
Neuronal cell death
↑ Volume of dead (infarcted) brain tissue
Neurons depolarize and release glutamate
Reversal of Na+ Dependent Glutamate Reuptake Transporters on astrocytes (normally 3 Na++ 1 H+ + 1 glutamate into cell, for 2 K+ out)
↑ Glutamate in extracellular fluid
Stroke symptoms (e.g. weakness, slurred speech, visual field losses, autonomic dysfunction)
(see Ischemic Stroke: Impairment by Localization stroke slide)
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published November 14, 2017; updated November 6, 2022 on www.thecalgaryguide.com")
Myelodysplastic Syndrome Pathogenesis and clinical findings
: Pathogenesis and clinical findings
Authors: Mao Ding Reviewers: Ashar Memon Man-Chiu Poon Yan Yu* * MD at time of publication
Stem cells differentiateà accumulation of bone marrow cells with aberrant morphology & maturation
Idiopathic (unknown cause)
Treatment related: exposure to ionizing radiation, chemotherapeutic agents (e.g., alkylating agents, anti- metabolite, topoisomerase II inhibitors)
Environmental toxins (e.g., tobacco, benzene & other organic solvents)
Familial predisposition to MDS
Acquired somatic (non- reproductive) gene mutations (DNA alterations)
Inherited
germline (reproductive cells) gene mutations
Recurrent mutations cause alteration(s) in one or more protein functions with/without chromosomal abnormalities in hematopoietic stem cell (earliest cell of blood cell differentiation):
Mutation in the transcription factor gene RUNX1 impairs regulation of normal hematopoietic (blood cell) development
Mutation(s) in epigenetic regulator genes (DNMT3A, TET2) impairs regulation of DNA methylation
Mutation in splicing regulator genes (SF3B1) causes mistakes in splicing mRNA moleculesàaberrant translation (production) of proteins
Chromosomal abnormalities: deletions (chromosome 5, 7, and/or 20), duplications (chromosome 8), structural abnormalities (inversion of a gene segment)
Myelodysplastic Syndrome
Mutation-associated clonal disorders of hematopoietic stem cells, causing dysplasia (abnormal development) of one or more myeloid cell lineages (granulocytes, monocytes, red blood cells & platelets) in the bone marrow
Genetic mutations or chromosomal abnormalities occur in hematopoietic stem cellsàclonal expansion of abnormal cells in bone marrow
Apoptosis (programed cell death) of clonal cells, inhibiting development of granulocytes, monocytes, red blood cells & platelets in the bone marrow
Neoplastic myeloid precursor cells (blasts) accumulate in the bone marrow
Acute Myeloid Leukemia (AML)
> 20% Blasts in the bone marrow
Excessive blasts displace other precursor cells & inhibit differentiation
Pancytopenia
(↓ number of cells of ALL 3 cell lines: platelets, white blood cells & red blood cells) (see Acute Myeloid Leukemia for signs/symptoms/complications)
↓ Number of mature functional blood cells leave the bone marrow to go to peripheral bloodà↓ number of cells of one or more cell lines
Bone marrow’s inability to make sufficient blood cells cause extramedullary hematopoiesis (formation/activation of blood cells outside the medulla of bone) at sites such as liver and/or spleen
Hematopoietic stem cells migrate to the spleen/liver & differentiate into blood cells
Expanding bone marrow physically pushes on bone’s cortex from within, activating nociceptors
Multifactorial causes mostly with unclear mechanisms
Intracellular granules precipitate inside blasts & the precipitate spills into the blood
↑Division & death of cancerous cells → ↑ cell lysis & release of intracellular contents into plasma
Bone pain
B symptoms: Weight
loss, fever, night sweats
Auer Rods (needle- like crystals) seen on blood smear
↑ Serum levels of uric acid, K+, LDH
↓ Red blood cells
Anemia
fatigue, pallor
↓ White blood cells
Leukopenia
Recurrent infections
↓ Platelets
Thrombo- cytopenia
Bruising, bleeding
Accumulation of cells within the spleen/liver increase the size of the organ
Splenomegaly Hepatomegaly
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published March 4, 2023 on www.thecalgaryguide.com")
Primary Hemostasis

Disorders of primary hemostasis:
Collagen and microfibrils (subendothelial elements
that are normally protected) are exposed to circulating blood due to vessel injury
Storage granules in endothelial cells release large multimers of clotting factor von Willebrand (vWF) when damaged
Acquired or inherited reduction in vWF quantity or quality (see vWF deficiency slide)
von Willebrand Disease
Defect in expression of GpIb interferes with platelet ability to bind to vWF
Bernard-Soulier syndrome
Defect in platelet membrane glycoproteins IIb and IIIa
Glanzmann’s Thrombasthenia
Insufficient platelets to form a platelet plug
Thrombocytopenia
(low platelet count)
Disorders of primary hemostasis: problems with formation of a platelet plug
Platelet adhesion: vWF binds to collagen and interacts with the platelet surface receptor glycoprotein Ib (GpIb) allowing platelet adhesion
Platelet activation: mediated by agonists like ADP, thrombin and collagen
Mucocutaneous bleeding (bleeding of the skin and mucous membrane)
↑ Closure time, ↑ bleeding time
Platelets change shape and release substances from their granules
Conformational changes in platelet cell surface glycoprotein IIb/IIIa
↑Affinity for fibrinogen through GpIIb/IIIa
ADP
Fibrinogen, vWF, and other substances
Thromboxane A2 (see note)
Induces aggregation
Epistaxis
Menorrhagia Note:
Petechiae
Platelet aggregation: fibrinogen forms bridges between adjacent platelets by binding to activated glycoprotein IIb/IIIa
Interaction with coagulation factors: platelets provide a scaffold that allows the activation of phospholipid-dependent coagulation factors
Primary hemostasis
(formation of a platelet plug)
Secondary hemostasis (see slide for normal physiology)
Drugs like Aspirin and NSAIDs inhibit Thromboxane A2 synthesis (by inhibiting cyclooxygenase) and thereby inhibit platelet aggregation
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published March 30, 2023 on www.thecalgaryguide.com")
Sarcoidosis pathogenesis and CXR findings
 such as tumor necrosis factor to neighboring cells
Tumor necrosis factor is a pro- inflammatory cytokine promoting cell survival and proliferation, predominantly in the upper/middle lung for unknown causes
More macrophages and helper T cells are activated and accumulate in the mediastinum, peritracheal region, and the upper/middle lung
Macrophages and helper T cells promote fibroblasts (cells that produce collagen) in the upper and middle lungs
Macrophages in the lungs present the antigen to the helper T cells in lymph nodes
Helper T cells become activated and accumulate in the lungs
Image Credit: Radiopaedia
The right peritracheal and bilateral hilar lymph nodes becomes swollen
Inflammatory signals stimulate macrophages to form granulomas (clusters of white blood cells) in the lungs, peritracheal and bilateral hilar lymph nodes
X-ray images show visible white opacities in areas of increased densities, such as areas of macrophage and helper T cell concentration
Enlarged peritracheal lymph node Stage 1 and 2
Enlarged bilateral hilar lymph node Stage 1 and 2
Interstitial Opacities Stage 2 and 3
Bilateral and symmetric lacey lung markings in upper/middle lungs
Decreased lung volume on either sides (higher than normal diaphragm)
Collagen accumulate which causes fibrotic tissue (scarred and thickened), replacing healthy tissue
Pulmonary fibrosis of upper and middle lung Stage 4
The fibrotic tissue is stiffer, reduces amount of air held in lungs, and has a higher density (appearing white)
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published April 7, 2023 on www.thecalgaryguide.com")
Superficial thickness burns

Dermis
Sub-cutaneous tissue
Erythema
Radiation
Sunlight (most common), x-ray, nuclear emission/explosion
Specific to sunlight radiation, UV rays reach keratinocytes in the epidermis
p53 tumor suppressor protein is induced in keratinocytes
Transient cell cycle arrest Apoptotic pathway is
Fire
Contact
Scald
Chemical
Electrical
Direct damage to keratinocytes
Skin erosion and sloughing of skin cells
Direct stimulation of nociceptive nerve endings in the epidermis
DNA repair mechanisms are activated
Mistakes in repair process
Malignancy
(See ‘Basal Cell Carcinoma’ Slide & ‘Squamous Cell Carcinoma’ Slide)
Fluid leaves vasculature and enters interstitial tissues of the skin, causing it to swell
Edema
Triggers release of endothelin A proalgesic protein
Selective excitement of nociceptive nerve ending in epidermis
Pain
initiated in keratinocytes
Keratinocytes apoptose
Prostaglandins, arachidonic acid metabolites, substance P & proinflammatory cytokines are released into surrounding tissue
↑ Vascular permeability
↑ bloodflow carries warmth to body area
Irritation of endothelial cells in the dermal vascular plexus
Release of endothelium derived vasodilators such as nitric oxide
Vasodilation, ↑ blood flow through vessels
↑ blood under skin leads to skin appearing red
Warmth Pressing on skin occludes blood vessels temporarily, making skin Blanchable underneath appear white immediately after pressure is lifted
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published Dec 2, 2013, updated Aug 25, 2023 on www.thecalgaryguide.com")
Sugammadex
) in plasma when administered IV
↓ Concentration of functional nNMBs in plasma
Creates a concentration gradient from muscle tissue (high) to plasma (low)
nNMBs move from muscle compartment to plasma
Sugammadex in plasma encapsulates nNMBs that moved to the plasma
↓ Concentration of functional nNMBs in the plasma
↓ Concentration of nNMBs at the nicotinic acetylcholine receptor within the skeletal neuromuscular junction
Reverses neuromuscular blockade created by nNMBs
Sugammadex
Progesterone is similar in structure to nNMBs
Sugammadex binds progesterone
↓ Progesterone activity in the body
Progesterone is critical for maintenance of early pregnancy
Unknown significance, avoid use in early pregnancy
Sugammadex-nNMB complex is cleared by the kidneys
Higher concentrations of sugammadex facilitate faster nNMB clearance
Unknown mechanisms
Post operative nausea and vomiting
Headache Bradycardia Cardiovascular Collapse
↓ Effectiveness of progesterone-based contraception for 7 days
↓ Clearance in patients with severe renal impairment
Reversal of profoundly deep neuromuscular blockade at higher doses
Binds to IgG or IgE receptors on sensitized basophils/mast cells in allergic reactions
Activation of basophils/mast cells
Degranulation of basophils/mast cells
Release of granulation products
Anaphylaxis Bronchospasm Hypotension
Authors: Arzina Jaffer, Kayleigh Yang Reviewers: Jasleen Brar, Mao Ding Joseph Ahn* * MD at time of publication
Movement of limbs or body during anesthesia
Coughing during anesthesia
Grimacing or suckling on the endotracheal tube
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published January 16, 2024 on www.thecalgaryguide.com")
Avascular Necrosis AVN of the Femoral Head Findings on MRI
 of the Femoral Head: Findings on MRI
Traumatic or atraumatic disruption of blood supply to the proximal femur (for full pathogenesis, see Calgary Guide slide Avascular Necrosis: Pathogenesis and clinical findings)
Ischemia of the femoral head (usually unilateral for traumatic and bilateral for non-traumatic)
Prolonged anoxia (total oxygen deprivation) within the femoral head
Cell death (necrosis) of osteocytes and marrow cells in the femoral head, forming a focal lesion (sequestrum)
Histiocytes and giant cells (immune cells) aggregate around the sequestrum, forming a “reactive zone” around the periphery of the sequestrum
Apoptotic osteocytes in the anoxic reactive zone cannot be phagocytosed leading to dysregulated bone remodeling and osteosclerosis (hardening of bone and ↑ bone mineralization and density due to ↓ resorption and ↑ bone formation)
Femoral head becomes progressively weaker while the mechanical load on it remains the same
Progressive femoral head/subchondral bone collapse Osteoarthritis
Areas with ↑ fluid content appear darker on T1w images
Areas with ↑ fluid content appear brighter on T2w images
Areas with ↑ bone density and ↓ fat content appear darker on T1w images
Areas with ↑ bone density and ↓ fat content appear darker on T2w images
Basic MRI Physiology
Edema and inflammation increases fluid content
Sclerotic areas have ↑ bone density and thus ↓ fat content
Location of Signs
Inflamed reactive zones are darker on T1w images
Inflamed reactive zones are brighter on T2w images
Sclerotic areas are darker on both T1w and T2w images
Authors: George S. Tadros Reviewers: Matthew Hobart, Mao Ding Shahab Marzoughi David Cornell* * MD at time of publication
T1w
Signs on both T1-weighted and T2-weighted images are most commonly seen on the superior anterolateral aspect of the femoral head
Single dark band on T1-weighted MRI
A single band-like crescentic lesion of low signal intensity is seen on T1-weighted MRI images (white arrows). This band represents the edematous reactive zone between the necrotic and normal tissue, and typically extends to the subchondral plate
Double Line Sign on T2-weighted fat-saturated MRI
An outer distal low signal intensity line is seen (white arrows) representing reactive bone sclerosis
Image source: radiologymasterclass
T2w
An inner proximal high signal intensity line is also seen (red arrows) representing vascular and repair tissue at the periphery of the sequestrum
Image source: radiologymasterclass
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published Jan 27, 2024 on www.thecalgaryguide.com")
Acute Wound Healing

Puncture (small piercing caused by sharp object)
Acute injury to the skin
Crush (damage by compression)
Acute Wound Healing:
Pathogenesis and clinical findings
Author: Amanda Eslinger Mina Youakim Reviewers: Heena Singh Shahab Marzoughi Yan Yu Laurie Parsons* * MD at time of publication
8 – 365+ days post-injury
Remodeling
(↓ blood vessels & organized collagen)
Extensively cross-linked type 1 collagen replaces the disorganized
collagen laid down in the proliferative phase
↑ Protein content in collagen
Scarring (fibrotic tissue replaces previously healthy tissue)
Disruption of structure and function of dermis, epidermis and subdermal tissues
Subendothelial and endothelial damage activates the coagulation pathway
Formation of a platelet plug
Bleeding is slowed or stopped by
hemostatic plug (hemostasis)
Clot unifies wound edges
0 – 7 days post-injury
Inflammation
(In disrupted skin layers)
4 - 14 days post-injury
Proliferation of collagen, extracellular matrix & blood vessels
TGF-β attracts fibroblasts to the site of the wound
Fibroblast & macrophage stimulate tissue growth &
angiogenesis which replaces hemostatic plug
Scabbing (protective crust overlying damaged tissue)
Re-epithelialization beneath the scab sloughs it off
Healing (newly replaced tissue replaces damaged one)
In response to irritant, mast cells release histamine
Complement activation causing nearby endothelial cells to release prostaglandins
Vasodilation occurs around the wound area
Localized ↑ vascular supply (reception of blood and fluid from vessels)
↑ Hydrostatic pressure forces fluid from vessels into surrounding tissue
Edema (swelling from fluid buildup)
Erythema (redness)
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
First Published Sept 19, 2013; updated Jan 30, 2024 on www.thecalgaryguide.com")
Chancroid

Micro-abrasions occur in the epidermal tissue of the genital area
Sexual Transmission of Haemophilus ducreyi (H. ducreyi) bacteria from an infected sexual partner
H. ducreyi enters the epidermis through micro abrasions H. ducreyi infects epithelial cells
H. ducreyi secretes Large Supernatant Proteins LspA1 and LspA2 (which inhibit phagocytosis by neutrophils and macrophages)
T cells activate and release cytokines IL-6 and IL-8 Neutrophils are recruited to the site of infection
Local macrophages form a collar around the base of the papule to try to reach and engulf H. ducreyi
↑ Localized buildup of immune cells
While infection persists, H. ducreyi release lipooligosaccharides (LOS)
LOS travels to lymph nodes in the inguinal region Lymph nodes synthesize and proliferate T
cells specific to H. ducreyi antigen Inguinal lymph nodes swell due
to ↑ number of T cells
Inguinal buboes (swollen inguinal lymph nodes)
Neutrophils surround the bacteria and attempt to engulf it
Localized epidermal buildup of neutrophils and H. ducreyi
Papules (small protrusions on the skin)
Continued build-up of H. ducreyi, neutrophils, dead white blood cells (pus), and macrophages
Pustules (pus-filled skin lesion)
↑ Local pressure from growing pustule compresses surrounding vessels
↓ Local blood flow erodes and sloughs off the local epidermal roof
Ulcers (open skin sore)
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published Feb 8, 2024 on www.thecalgaryguide.com")
Neonatal Necrotising Enterocolitis in Premature Neonates
 in Premature Neonates:
Pathogenesis and clinical findings
Prematurity risk factors ↓ Intestinal motility
↑ Intestinal stasis allows bacteria more time to proliferate
Bacterial overgrowth in gut
↓ Goblet cells in intestinal epithelium
↓ Intestinal mucus layer production leads to impaired mechanical defense against pathogenic bacteria
Immature tight junctions in intestinal epithelium
↑ Permeability of intestinal epithelial barrier
↑ Toll-like receptor 4 (TLR4) expression on intestinal epithelial cells
Aberrant bacterial colonization of gut
Impaired gut barrier allows for ↑ bacterial translocation across intestinal epithelium
TLR4 on intestinal mesentery endothelial cells bind lipopolysaccharides (LPS) on Gram-negative gut bacteria
Immune cells release proinflammatory mediators (TNF, IL-12, IL-18)
Cytokines mediate ↑ enterocyte apoptosis (including enteric stem cells) and ↓ enterocyte proliferation
Intestinal mucosa healing is impaired, leading to local inflammation & injury
TLR4 on intestinal epithelial cells binds LPS from Gram-negative gut bacteria
Authors: Rachel Bethune Naima Riaz Reviewers: Nicola Adderley Michelle J. Chen Kamran Yusuf* Jean Mah* * MD at time of publication
Endothelial nitric oxide synthase expression is reduced
Vasoconstriction from ↓ NO reduces blood flow to intestines
Prolonged ↓ in O2 perfusion results in irreversible intestinal mucosal cell death (necrosis)
Gas escapes into abdominal cavity
Leakage of intestinal contents irritates parietal peritoneum
Bacteria enter bloodstream
Pneumo- peritoneum
Abdominal distention
Peritonitis
Sepsis
Blood from tissue damage mixes with intestinal contents
Bloody stool
Intestinal sensory neurons detect damage and send signals to medullary vomiting centre
Bilious vomiting
Damaged intestinal cells are unable to absorb nutrients
Short gut syndrome
Persistent intestinal mucosal injury creates penetrating lesions through intestinal wall
Intestinal perforation
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published May 6, 2019; updated Mar 21, 2024 on www.thecalgaryguide.com")
Lichen Sclerosus
: Pathogenesis and clinical findings
Authors: Mina Youakim Reviewers: Elise Hansen Sunawer Aujla Shahab Marzoughi Jori Hardin* * MD at time of publication
Histamine receptor binding stimulates sensory nerve endings
Pruritus (itching)
Unknown triggers
Genetic predisposition (HLA-DQ7 and HLA-DR12)
Chronic inflammation (i.e. chronic infection, chronic toxin exposure) and trauma
Medications (e.g. carbamazepine, pembrolizumab, nivolumab, ipilimumab)
↑ Activation of CD4+ and CD8+ T cells released from macrophages (white blood cell) in the perineum and genital skin tissue infiltrate into the dermal-epidermal junction
T-cells proliferate in a horizontal linear formation Pro-inflammatory response activation
Fibroblasts (contributes to the formation of connective tissue) proliferate and persist producing altered collagen under the epidermis
Collagen deposits and hyalinizes (transforms into acellular translucent material) beneath the epidermal layer
Sclerotic plaques (localized areas of thickened skin)
T cells release of pro-inflammatory cytokines (interleukins and transforming growth factor β)
↑ Oxidative stress and cell damage
Progressive basal layer degeneration thins the overall skin thickness
Mast cells respond to increased need for immune cell flow to area
Nitric oxide is released when histamine binds to vascular receptors
Localized area appears red
Erythema (reddening of the skin)
Erosion/ulceration
Skin fissures (linear cleavage of skin)
Localized histamine release
Nitric oxide induces localized vasodilation (↑ blood flow)
Normal Skin
Lichen Sclerosus
Epidermal layer
Basal layer
Dermal-Epidermal Junction Dermal layer
Hypopigmented patches (localized, pale areas of skin)
Epidermal atrophy (crinkling paper-type skin appearance)
Thinned skin is weakened and prone to physical stress or trauma
↑ fluid in the extracellular space due to capillary leakage from ↑ blood flow
Superficial dermal edema (swelling)
Fibrotic tissue deposition following healing of damaged tissue
Atrophic Epidermal layer
Hyalinized collagen deposits
Basal layer degeneration Band of T-cell infiltrate
Dermal layer
Scarring
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published Mar 25, 2024 on www.thecalgaryguide.com
Lichen Sclerosus (genital manifestation): Pathogenesis and clinical findings
Authors: Mina Youakim Reviewers: Elise Hansen Sunawer Aujla Shahab Marzoughi Jori Hardin* * MD at time of publication
Histamine receptor binding stimulates sensory nerve endings
Pruritus (itching)
Unknown triggers
Genetic predisposition (HLA-DQ7 and HLA-DR12)
Chronic inflammation (i.e. chronic infection, chronic toxin exposure) and trauma
Medications (e.g. carbamazepine, pembrolizumab, nivolumab, ipilimumab)
↑ Activation of CD4+ and CD8+ T cells released from macrophages (white blood cell) in the perineum and genital skin tissue infiltrate into the dermal-epidermal junction
T-cells proliferate in a horizontal linear formation Pro-inflammatory response activation
Fibroblasts (contributes to the formation of connective tissue) proliferate and persist producing altered collagen under the epidermis
Collagen deposits and hyalinizes (transforms into acellular translucent material) beneath the epidermal layer
Sclerotic plaques (localized areas of thickened skin)
T cells release of pro-inflammatory cytokines (interleukins and transforming growth factor β)
↑ Oxidative stress and cell damage
Mast cells respond to increased need for immune cell flow to area
Nitric oxide is released when histamine binds to vascular receptors
↑ fluid in the extracellular space due to capillary leakage from ↑ blood flow
Superficial dermal edema (swelling)
Epidermal atrophy (crinkling paper- type skin appearance)
Thinned skin is weakened and prone to physical stress or trauma
Localized histamine release
Nitric oxide induces localized vasodilation (↑ blood flow)
Localized area appears red
Erythema (reddening of the skin)
Erosion/ulceration
Skin fissures (linear cleavage of skin)
Normal Skin
Lichen Sclerosus
Epidermal layer
Basal layer
Dermal-Epidermal Junction Dermal layer
Atrophic Epidermal layer
Hyalinized collagen deposits
Basal layer degeneration Band of T-cell infiltrate
Dermal layer
Progressive basal layer degeneration thins the overall skin thickness
Hypopigmented patches (localized, pale areas of skin)
Fibrotic tissue deposition following healing of damaged tissue
Scarring
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published MONTH, DAY, YEAR on www.thecalgaryguide.com
Chronic inflammation (i.e. chronic
Reviewers:
Dermal layer
Unknown triggers
Genetic predisposition (HLA-DQ7 and HLA-DR12)
infection, chronic toxin exposure) and trauma
Medications (e.g. carbamazepine, pembrolizumab, nivolumab, ipilimumab)
Elise Hansen Sunawer Aujla Shahab Marzoughi Jori Hardin* * MD at time of publication
↑ Activation of CD4+ and CD8+ T cells released from macrophages (white blood cell) in the perineum and genital skin tissue infiltrate into the dermal-epidermal junction
T-cells proliferate in a horizontal linear formation
Pro-inflammatory response activation
↑ Expression of MicroRNA-155 (enhances pro-inflammatory response and ↓ expression of tumor suppression genes)
Fibroblasts (contributes to the formation of connective tissue) proliferate and persist producing altered collagen
Collagen deposits and hyalinizes (transforms into acellular translucent material) beneath the epidermal layer
Sclerotic plaques (localized areas of thickened skin)
T cells release of pro-inflammatory cytokines (interleukins and transforming growth factor β)
↑ Oxidative stress and cell damage
Mast cells respond to increased need for immune cell flow to area
Nitric oxide is released when histamine binds to vascular receptors
↑ fluid in the extracellular space due to capillary leakage from ↑ blood flow
Superficial dermal edema (swelling)
Epidermal atrophy (crinkling paper- type skin appearance)
Thinned skin is weakened and prone to physical stress or trauma
Lichen Sclerosus
Localized histamine release
Nitric oxide induces localized vasodilation (↑ blood flow)
Localized area appears red
Erythema (reddening of the skin)
Erosion/ulceration
Skin fissures (linear cleavage of skin)
Atrophic Epidermal layer
Hyalinized collagen deposits
Basal layer degeneration Band of T-cell infiltrate
Histamine receptor binding stimulates sensory nerve endings
Pruritus (itching)
Normal Skin
Progressive basal layer degeneration thins the overall skin thickness
Hypopigmented patches (localized, pale areas of skin)
Epidermal layer
Basal layer
Dermal-Epidermal Junction
Fibrotic tissue deposition following healing of damaged tissue
Scarring
Chronic inflammation (i.e. chronic
Reviewers:
Dermal layer
Unknown triggers
Genetic predisposition (HLA-DQ7 and HLA-DR12)
infection, chronic toxin exposure) and trauma
Medications (e.g. carbamazepine, pembrolizumab, nivolumab, ipilimumab)
Elise Hansen Sunawer Aujla Shahab Marzoughi Jori Hardin* * MD at time of publication
↑ Activation of CD4+ and CD8+ T cells released from macrophages in the perineum and genital skin tissue infiltrate into the dermal-epidermal junction
T-cells proliferate in a horizontal linear formation Pro-inflammatory response activation
↑ Expression of MicroRNA-155 (short segment of RNA which enhances pro- inflammatory response and ↓ expression of tumor suppression genes)
Fibroblasts proliferate and persist producing altered collagen
Collagen deposits and hyalinizes (transforms into acellular translucent material) beneath the epidermal layer
Sclerotic plaques (localized areas of thickened skin)
T cells release of pro-inflammatory cytokines (interleukins and transforming growth factor β)
↑ Oxidative stress and cell damage
Progressive basal layer degeneration thins the overall skin thickness
Hypopigmented patches (localized, pale areas of skin)
Epidermal layer
Basal layer
Dermal-Epidermal Junction
Mast cells respond to increased need for immune cell flow to area
Nitric oxide is released when histamine binds to vascular receptors
Superficial dermal edema (swelling)
Epidermal atrophy (crinkling paper- type skin appearance)
Localized histamine release
Nitric oxide induces localized vasodilation (↑ blood flow)
Localized area appears red
Erythema (reddening of the skin)
Histamine receptor binding stimulates sensory nerve endings
Pruritus (itching)
Normal Skin
Lichen Sclerosus
Skin fissures (linear cleavage of skin)
Erosion/ulceration
Fibrotic tissue deposition following healing of damaged tissue
Scarring
Atrophic Epidermal layer
Hyalinized collagen deposits
Basal layer degeneration Band of T-cell infiltrate
Reviewers:
Dermal layer
Unknown triggers
Genetic predisposition (HLA-DQ7 and HLA-DR12)
Chronic inflammation (i.e. chronic infection, chronic toxin exposure) and trauma
Medications (e.g. carbamazepine, pembrolizumab, nivolumab, ipilimumab)
Elise Hansen Sunawer Aujla Shahab Marzoughi Jori Hardin* * MD at time of publication
↑ Activation and infiltration of CD4+ and CD8+ T cells into the dermal-epidermal junction
T-cells proliferate in a band (horizontal linear) formation Pro-inflammatory response activation
↑ Expression of MicroRNA-155 (short segment of RNA which enhances pro-inflammatory response and ↓ expression of tumor suppression genes)
Fibroblasts proliferate and persist producing altered collagen
Collagen deposits and hyalinizes (transforms into acellular translucent material) beneath the epidermal layer
Sclerotic plaques (localized areas of thickened skin)
T cells release of pro-inflammatory cytokines (interleukins and transforming growth factor β)
↑ Oxidative stress and cell damage
Progressive basal layer degeneration thins the epidermis
Hypopigmented patches (localized, pale areas of skin)
Epidermal layer
Basal layer
Dermal-Epidermal Junction
Mast cells respond to increased need for immune cell flow to area
Nitric oxide is released when histamine binds to vascular receptors
Superficial dermal edema (swelling)
Epidermal atrophy (crinkling paper- type skin appearance)
Localized histamine release
Histamine receptor binding stimulates sensory nerve endings
Pruritus (itching)
Skin fissures (linear cleavage of skin)
Nitric oxide induces localized vasodilation (↑ blood flow)
Localized area appears red
Erythema (reddening of the skin)
Scarring
Atrophic Epidermal layer
Hyalinized collagen deposits
Basal layer degeneration Band of T-cell infiltrate
Erosion/ulceration
Fibrotic tissue deposition following healing of damaged tissue
Normal Skin
Lichen Sclerosus
Chronic inflammation (i.e. chronic
Elise Hansen
Unknown triggers
Genetic predisposition infection, chronic toxin exposure) Medications (e.g. carbamazepine, (HLA-DQ7 and HLA-DR12) and trauma pembrolizumab, nivolumab, ipilimumab)
↑ Activation and infiltration of CD4+ and CD8+ T cells into the dermal-epidermal junction
Sunawer Aujla Shahab Marzoughi Name Name* * MD at time of publication
T-cells proliferate in a band (horizontal linear) formation
Pro-inflammatory response activation (increase in pro-inflammatory cytokines such as Interleukins 1-alpha and 1-beta)
↑ Expression of MicroRNA-155 (short segment of RNA which enhances pro-inflammatory response and ↓ expression of tumor suppression genes)
Fibroblasts proliferate and persist producing altered collagen
T cells release of pro-inflammatory cytokines (interleukins and transforming growth factor β)
↑ Oxidative stress and cell damage
Progressive basal layer degeneration thins the epidermis
Hypopigmented patches (localized, pale areas of skin)
Histamine receptor binding stimulates sensory nerve endings
Localized area appears red
Localized histamine release
Localized vasodilation (↑ blood flow)
Superficial dermal edema (swelling)
Pruritus
Erythema
Collagen deposits and hyalinizes (transforms into acellular translucent material) beneath the epidermal layer
Normal Skin
Sclerotic plaques (localized areas of thickened skin)
Epidermal layer
Basal layer
Dermal-Epidermal
Junction Dermal layer
Skin fissures (linear cleavage of skin)
Bleeding
Erosion/Ulceration
Epidermal atrophy (crinkling paper- type skin appearance)
Lichen Sclerosus
Atrophic Epidermal layer
Hyalinized collagen deposits
Basal layer degeneration
Band of T-cell infiltrate
Dermal layer
Fibrotic tissue deposition following healing of damaged tissue
Scarring
Lichen Sclerosus (genital manifestation): Pathogenesis and clinical findings
Authors: Mina Youakim Reviewers: Elise Hansen Sunawer Aujla Shahab Marzoughi Name Name* * MD at time of publication
Pruritus
Histamine receptor binding stimulates sensory nerve endings
Localized area appears red
Erythema
Unknown triggers
Genetic predisposition (HLA-DQ7 and HLA-DR12)
Chronic inflammation and trauma
Medications (e.g. carbamazepine, pembrolizumab, nivolumab, ipilimumab)
↑ Activation and infiltration of CD4+ and CD8+ T cells into the dermal-epidermal junction
T-cells proliferate in a band formation Pro-inflammatory response activation
↑ Expression of MicroRNA-155 (short segment of RNA which enhances pro-inflammatory response and ↓ expression of tumor suppression genes)
Fibroblasts proliferate and persist producing altered collagen
T cells release of pro-inflammatory cytokines (interleukins and transforming growth factor β)
Localized histamine release
Localized vasodilation (↑ blood flow)
Superficial dermal edema (swelling)
Collagen deposits and hyalinizes beneath the atrophic epidermal layer
Hypopigmented patches (localized, pale areas of skin)
↑ Oxidative stress and cell damage
Progressive basal layer degeneration thins the epidermis
Skin fissures
Lichen Sclerosus
Epidermal atrophy (crinkling paper- type skin appearance)
Sclerotic plaques (localized areas of thickened skin)
Bleeding Scarring
Erosion/Ulceration
Normal Skin
Epidermal layer
Basal layer
Dermal-Epidermal
Junction Dermal layer
Atrophic Epidermal layer
Hyalinized collagen deposits
Basal layer degeneration
Band of T-cell infiltrate
Dermal layer
Legend:
Published MONTH, DAY, YEAR on www.thecalgaryguide.com
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications")
Irritant Contact Dermatitis Pathogenesis and Clinical Findings

Hyperkeratotic skin is less amenable to skin stretching and pressure
Skin fissures (cracks in the skin)
Irritant agents
(abrasives, cleaning solution, oxidative & reducing agents, dust, soils, water)
Acute exposure triggers inflammatory response
Keratinocytes undergo cytotoxic damage with ↑ neutrophil & cytokine release
Common occupational exposures (housekeeping, cleaning, catering, medical/dental, construction)
Risk factors
(atopy, fair skin, low temperature, low humidity)
Stimulation of local nociceptors (free nerve endings extend into the mid epidermis)
Perivascular (around the blood vessel) inflammation causes histamine release from mast cells
Damaged keratinocytes are destroyed via
apoptosis (programmed cell death)
Epidermal Necrosis (death of epidermal tissue)
Shedding of necrotic tissue
Ulceration (deep open wound on skin)
Burning
pain (uncomfortable stinging sensation)
Pruritus (itching)
Histamine causes local blood vessel dilation and ↑ blood flow to the area of skin affected
Erythema (area appears red from ↑ blood flow)
Burning & Itching Spongiosis
Neutrophils Neutrophils
Histamine causes local blood vessel walls to have
↑ permeability, thereby ↑ leakage of fluid
Spongiosis
(↑ fluid between keratinocytes in the epidermis)
Fluid continues to build up from ongoing inflammation
Vesiculation (fluid collections in the epidermis)
Long-term skin scratching causes chronic irritation which eventually hardens the skin
Lichenification
(thick, hardened patches of skin)
↑ Overall keratin production
Hyperkeratosis (thickening of the outermost skin layer)
Further fluid buildup bursts vesicles leaving behind erosions and dried crust on the epidermis
Crust (scaling over the skin)
Lichenification
Erosions (open sore on skin)
Ulcer
Epidermis
Perivascular Inflammation
Hyperkeratosis
Dermis
Dermal-epidermal junction
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published Oct 19, 2016; updated Mar 30, 2024 on www.thecalgaryguide.com")
Febrile Neutropenia Pathogenesis and clinical findings
: Pathogenesis and clinical findings
Administration of cytotoxic chemotherapy for cancer treatment
Acquired aplastic anemia
Autoreactive T cells destroy bone marrow stem cells
Congenital mutations in ELA2 gene (encodes neutrophil elastase)
↑ Neutrophil apoptosis
Chemotherapy eliminates beneficial bacterial species from gut microbiota
New microbiota composition allows for growth of colonizing bacteria
Indwelling catheters inserted to deliver chemotherapy
Skin-colonizing bacteria access tissues through catheters
Bacteria penetrate tissue barriers
Chemotherapy injures gastrointestinal mucosa
Broken mucosal barrier increases susceptibility to infections
Chemotherapy destroys circulating neutrophils
Chemotherapy impairs bone marrow stem cells
↓ Production of neutrophils
Neutropenia (Absolute Neutrophil Count (ANC) < 0.5x109 cells/L
↓ Immune cell ↓ Production of engulfment of microbes inflammatory mediators
Fewer circulating neutrophils blunts the innate immune response
Pathogens enter bloodstream from tissues Systemic infection
Authors: Max Lazar Braxton Phillips Reviewers: Naman Siddique Michelle J. Chen Lynn Savoie* * MD at time of publication
Dormant viral infections reactivate (e.g. cytomegalovirus, herpes simplex virus)
↑ Susceptibility to common bacterial infections
Positive blood bacterial cultures
Fever (T ≥ 38.3 oC or sustained T ≥ 38 oC for 1 hour)
Immune system mounts an excessive inflammatory response that damages tissues and organs
Sepsis
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published Dec 5, 2018; updated Mar 30, 2024 on www.thecalgaryguide.com")
Costochondritis

Infection spreads through blood to ribcage and chest wall
Age-related degeneration of cartilage within ribcage joints
Immune cells present in joints secrete proinflammatory cytokines
Cytokines sensitize nociceptors (pain receptors) in joints
Repetitive trauma or strain to chest wall muscles (e.g. chronic cough, chest wall injury, overexertion of chest wall muscles)
Excessive stretch and tearing of ribcage cartilage and ligaments
Autoimmune disease (e.g. rheumatoid arthritis)
Antigen presenting cells display self-antigens to CD4+ T cells
Mechanical stimuli (e.g. Damaged cells release cytokines Pathogens activate immune cells at site of stretch) activate nociceptors that recruit immune cells inflammation
Costochondritis
Benign inflammation of the ribcage (costal) cartilage, particularly at the costosternal junctions (connection between sternum and cartilage) and the costochondral junctions (connection between the cartilage and rib)
Authors:
Michelle J. Chen Reviewers:
Raafi Ali
Yan Yu*
Gerhard Kiefer*
* MD at time of publication
Cartilage, ligament, or muscle injury
Presence of foreign pathogen in costal cartilage
Injury or pathogen activates inflammatory cascade in the ribcage cartilage
Immune cells proliferate so that they exceed the available space in the cartilage matrix of the ribcage
Cartilage swelling compresses intercostal nerves
Compression acts as a mechanical stimulus to activate nociceptors
Sharp, localized pain reproducible upon palpation over ribcage
Immune cells secrete cytokines
Cytokines activate nociceptors so that they become more sensitive to mechanical stimuli
Muscle or ligament stretch from normal use activates sensitized nociceptors
Pain increases with inspiration or cough
Natural resolution of inflammation over time
Macrophages phagocytose apoptotic cells (immune and cartilage cells) and cellular debris
Immune cells release factors that degrade proinflammatory cytokines
Fewer cytokines reduce immune cell recruitment into costal cartilage
Pain usually self-resolves
Immune cells release lipid molecules that bind to the same cells (autocrine signaling) to inhibit further production of inflammatory cytokines and chemokines
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published Apr 29, 2024 on www.thecalgaryguide.com")
Acute Otitis Media Complications
 accumulates in mastoid air spaces
Untreated middle ear infection allows further bacterial proliferation
Persistent effusion
causes ion channel changes in inner ear
Composition of endolymph and perilymph in inner ear changes
Vestibular/Labyrinth dysfunction
Feelings of Vertigo imbalance
Purulent discharge from middle ear through perforation
Otorrhea (ear discharge)
↑ Pressure in middle ear
Pressure stretches tympanic membrane
Cytokines reach hypothalamus through the bloodstream
Hypothalamus responds to stimulation and ↑ thermoregulatory set-point
High-grade fever
Infection spreads into bloodstream
Sepsis
Cytokines alter metabolism pathways of neurotransmitters in the brain
Cerebral cortex dysfunction
Infection spreads to cranium
Intracranial complications (e.g., meningitis, brain abscess, thrombus)
Author: Jody Platt Stephanie de Waal Reviewers: Yan Yu Elizabeth De Klerk William Kim Annie Pham Michelle J. Chen Danielle Nelson* * MD at time of publication
Helper T cells and macrophages release inflammatory cytokines into the bloodstream
Perforation of tympanic membrane
↓ Conduction of sound waves
Conductive hearing loss
Pressure in middle ear diffuses out of perforation
↓ Tympanic membrane stretching
Otalgia (ear pain) fades
Accumulated debris compresses cranial nerve VII
Facial nerve palsy
Mastoid abscess
Febrile seizures
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published Feb 28, 2013, updated Apr 29, 2024 on www.thecalgaryguide.com")
Wiskott-Aldrich Syndrome
: Pathogenesis and clinical findings X-linked recessive inheritance (mostly males impacted) Spontaneous DNA mutation
Genetic mutations in Wiskott-Aldrich Syndrome Protein (WASP) impair actin cytoskeleton remodeling in hematopoietic cells (immature blood cells)
Authors: Mina Mina Reviewers: Hana Osman Mao Ding Maharshi Gandhi Shahab Marzoughi Louis-Philippe Girard* * MD at time of publication
Megakaryocytes (platelet precursor cells) depend on the cytoskeleton changing shape to form pseudopodia (arm-like projections) which bud off forming cellular fragments (platelets)
Abnormal cytoskeleton reorganization leads to ineffective thrombocytopoiesis (production of platelets)
Microthrombocytopenia (↓ platelet size and quantity)
Issues with formation of a platelet plug due to abnormal platelets (dysfunction of primary hemostasis)
Reduced ability for platelet adhesion, activation, and/or aggregation
Natural killer cells depend on the cytoskeleton reorganization to form immunological synapses (communication) with body cells in order to have effective surveillance
T cells depend on the cytoskeleton remodeling to form pseudopods which allow them to synapse with other cells when a pathogen is encountered
Defective immune synapse formation
↓ Cancer immunosurveillance
↑ Risk of malignancy
Helper T cells cannot activate B cells which generate antibodies (immunoglobulins) to destroy pathogens
Regulatory T cells can not sufficiently downregulate effector cells which typically limit the immune response and prevent autoimmune conditions
↑ Risk of autoimmune diseases
Recurrent opportunistic infections
Immune dysregulation
Impaired immune response
↓ Number and function of T cells
Immune responses contribute to Type 2 immune responses
Atopic dermatitis (AD; chronic inflammation that causes itchy skin)
↑ Immunoglobulin (Ig) A
Epistaxis (nosebleed)
Red blood cells leak from capillaries
Menorrhagia (heavy menstrual bleeding)
Petechiae (small, flat, red spots that appear on the skin)
Inflammation in AD triggers release of interleukins (modulatory proteins during inflammatory and immune responses) leading to Immunoglobulin (Ig) class switching
↑ IgE levels
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published Apr 20, 2024 on www.thecalgaryguide.com")
Scabies pathogenesis and clinical findings

Authors: Heena Singh Amanda Eslinger Nirav Saini Reviewers: Shahab Marzoughi Danny Guo Yan Yu Richard Haber* * MD at time of publication
Mechanical irritation and immunological reaction
Mast cell activation and histamine release
Itch
(> 10 Mites)
Stratum corneum
Stratum lucidum Stratum granulosum
Stratum spinosum Stratum basale
Females lay 2-3 ova/day which hatch in the stratum corneum in pockets after 3 days
The larvae molt and mature for 2 weeks and mate within the pockets of the stratum corneum
Following mating, female mites begin burrowing
Cycle is propagated as new female mites create more burrows in stratum corneum
Superficial, Linear, Tortuous (i.e., Twisted) +/- Scaly Burrows
↑ Exposure to mite antigens such as Sar s 14.3 and Sar s 14.2
Type 2 helper T-cells produce proteins IL-4, IL-5, IL-13
↑ Serum Immunoglobulin G & E ↑ Eosinophil Count in Peripheral Blood Inappropriate T helper-type immune response in skin (mechanism unknown)
Antigens forming from mite feces, ova, or decomposing bodies
Type IV hypersensitivity reaction (delayed immune response 2-4 weeks following initial contact)
Skin barrier dysfunction from inflammation and histamine release
Pre-existing immunosuppression Chronic inflammatory response Allergic reaction to ↑ mite activity at night
Uncontrollable Itch (Worse At Night) + Excoriations (Scratch Marks)
Urticarial (Hive-Like) Crusted Papules Eczematous Plaques
Skin breakdown Opening for bacteria (commonly S. aureus) 2° Bacterial infection Pustules
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published Oct 17, 2013; updated Jun 9, 2024 on www.thecalgaryguide.com")
Acute Otitis Media Pathogenesis and Clinical Findings in Children

Exposure to tobacco smoke
Impaired macrophage function in nasopharynx
Lack of immunizations
Lack of breastfeeding
Infant does not receive antibodies from breast milk
Overcrowding
Age 6 – 16 months
Immune system deficiencies
Close proximity of kids & ↓ sanitation (e.g. daycare)
↑ Infection risk Upper Respiratory Tract Infection (i.e. bacterial (Streptococcus pneumoniae, Haemophilus influenzae, Moraxella catarrhalis), viral)
Immature Eustachian tube anatomy which facilitates pathogen transmission to middle ear
Author: Jody Platt Stephanie de Waal Reviewers: Yan Yu Elizabeth De Klerk William Kim Annie Pham Michelle J. Chen Danielle Nelson* * MD at time of publication
Abnormal anatomical structure (e.g. cleft palate)
↑ Nasopharyngeal streptococcus pneumoniae
Lack of immunity to pathogens
↑ Colonization of nasopharynx with bacterial pathogens
Inflammation & edema of respiratory mucosa including the nose, nasopharynx, and Eustachian tube
Obstruction of the Eustachian tube
Air from middle ear resorbs into circulation which creates a low-pressure environment
Negative pressure gradient pulls viral/bacterial pathogens into middle ear
Degenerating white blood cells, tissue debris, and microorganisms accumulate & develops into purulent effusion
Inflammation & infection of middle ear
Complications of acute otitis media**
↑ Pressure in middle ear
Stretching of tympanic membrane
Helper T cells & macrophages release cytokines into the bloodstream
Cytokines trigger hypothalamus to ↑ thermoregulatory set-point
Effusion behind tympanic membrane
Effusion obstructs visualization of ossicles
Neutrophils infiltrate middle ear & phagocytose pathogens
Pus accumulates behind the tympanic membrane
Blood vessels of the tympanic membrane vasodilate
Bulging tympanic membrane
Otalgia (ear pain)
Discomfort disrupts daily activities in young children
Fever
Irritable Poor feeding
Tympanic membrane erythema
Painful blisters on tympanic membrane (e.g. Bullous myringitis)
Can persist for up to 3 months after infection resolves
Loss of tympanic membrane landmarks (i.e. handle of malleus, light reflex)
** See corresponding Calgary Guide slide
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published Feb 28, 2013; updated Aug 25, 2024 on www.thecalgaryguide.com")
Pityriasis Tinea Versicolor
 Versicolor: Pathogenesis and clinical findings
Authors: Kerry Yang Reviewers: Mina Youakim Shahab Marzoughi Jori Hardin* * MD at time of publication
↑ Loosening of the stratum corneum
Scaling of skin
Hyperhidrosis (↑ sweating)
Poor ↑ Sebaceous nutrition gland secretions
Development of a lipid- rich environment
Use of oils on skin
↑ Environmental heat & humidity
Immunosuppression
Lipophilic Malassezia (normal constituent of skin flora) yeast cells transform into the pathogenic, mycelial (fungal thread) form, commonly occurring on the trunk, neck, and proximal extremities
Malassezia infect the stratum corneum (the outer layer) of the skin
↑ Malassezia – associated ligand – activated transcription factors
Rate of different gene transcriptions modified
↑ Melanin production by melanocytes
↑ In melanin more than the ↓ in melanin
Hyperpigmented macules, patches, and plaques
↑ Production of the enzyme keratinase
↑ Activation of Langerhans cells (macrophages in the skin)
Alteration in the expression of cytokines and chemokines in keratinocytes (epidermal skin cells)
↑ Inflammatory response
Activation of cutaneous nerve fibers
Itch signal transduction
Mild pruritis (itchiness)
Ligand-activated transcription factors produced by Malassezia
↑ Degradation of keratin in the skin
↑ Production of dicarboxylic acids by certain Malassezia species
Competitively inhibits the rate- limiting enzyme in melanin production tyrosinase
↓ Production of melanin
↓ In melanin more than the ↑ in melanin
Hypopigmented macules, patches, and plaques
↑ Production of inflammatory mediators
Dyspigmentation
Malassezia, particularly M. globosa, M. furfur, and M. sympodialis
Loosened Epidermal layer
Dermal-Epidermal Junction
Dermal layer
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published Aug 25, 2024 on www.thecalgaryguide.com")
Secondary hypoglycemia Insulin Mediated

Non-insulinoma pancreatic islet cell disorder
(e.g. Non-insulinoma pancreatogenous hypoglycemia syndrome)
Nesidioblastosis (pancreatic islet tissues formed from pancreatic duct budding) formation
↑ In number & size of abnormal pancreatic beta islet cells throughout the entire pancreas
↑ Insulin production & secretion by abnormal pancreatic islet cells
↑ Glycogen synthesis
Insulin autoimmune syndrome
Autoantibodies bind to insulin molecules released post-meal
Insulin molecules unable to exert effects
Hyperglycemia Promotes insulin release
↓ Plasma glucose concentration
↓ Insulin release & ↓ total insulin levels
Insulin molecules dissociate from autoantibodies
↑ Free insulin levels (inappropriate for the ↓ plasma glucose levels)
↓ Glycogenolysis (glycogen breakdown to glucose)
Hypoglycemia
(serum glucose of <3mmol/L)
Intake of insulin secretagogues (e.g. Sulphonylureas, Meglitinide analogues)
Binds to adenosine triphosphate (ATP) dependant potassium channels on pancreatic islet cells
Inhibit potassium ion efflux through ATP-dependent potassium channels
Pancreatic islet cell depolarization
Opens voltage-gated calcium channel
Calcium influx into pancreatic islet cells
↑ Insulin release
↓ Gluconeogenesis (glucose synthesis from non-carbohydrate compounds)
Exogenous insulin intake
Suppress endogenous insulin production (by beta cells)
Low C-peptide levels
(byproduct of endogenous pro- insulin being converted to insulin)
90% caused by somatic mutations of pancreatic beta islet cells
10% due to genetic changes linked to MEN1(Multipl e endocrine neoplasia type-1) gene mutation
↑ Pancreatic islet cells proliferation
↑ Insulin secretion by hyperplastic (increasing in number) pancreatic islet cells
Neuroglycopenic symptoms (altered mental status, seizures)
Neurogenic symptoms (palpitations, tremulousness, diaphoresis
Authors: Iffat Naeem Run Xuan (Karen) Zeng Reviewers: Gurreet Bhandal Luiza Radu Yan Yu* Samuel Fineblit* * MD at time of publication
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published Sept 29, 2024 on www.thecalgaryguide.com")
Vaccine-Mediated Immunity General Physiology
 phagocytose and process antigen
Adjuvants in vaccines activate local immune response
↑ Production of pro-inflammatory signals (chemokines & cytokines)
Chemokines & cytokines act as signaling molecules that attract APCs to draining lymph nodes
APCs present antigen on MHC I molecules to CD8+ T cells
APCs present antigen on major histocompatibility complex II (MHC-II) molecules to naïve CD4+ T helper cells
Recognition of antigenic material activates naïve CD4+ T helper cells
A subset of naïve B cells differentiate into short-lived plasma cells
Plasma cells produce antibodies
Antibodies cover the pathogen’s surface (neutralization)
Antibody-coated pathogens are phagocytosed by macrophages/neutrophils (opsonization)
↑ Pathogen-specific antibody titers
CD8+ T cells multiply to form an antigen- specific cytotoxic T cell population (clonal expansion)
↑ Lymphocyte counts
Authors: Tanis Orsetti Reviewers: Allesha Eman Michelle J. Chen Dr. Russell Sterrett* Prevention of specific pathogen from causing severe disease upon subsequent exposure * MD at time of publication
Some activated CD4+ T helper cells stimulate naïve B cells
A subset of activated CD4+ T helper cells differentiate into memory CD4+ T cells
A subset of CD8+ T cells differentiate into memory CD8+ T cells
A subset of naïve B cells differentiate into memory B cells
Memory B cells circulate the body and monitor for secondary exposure to the pathogen
Memory T cells circulate the body and monitor for secondary exposure
Memory T cells are activated upon secondary exposure to the pathogen
Activated memory T cells differentiate into effector T cells
Memory B cells differentiate into plasma cells when there is a secondary exposure to the pathogen
Effector CD4+ T helper cells stimulate B lymphocytes
CD8+ cytotoxic T cells induce apoptosis of infected cells
Vaccine-Mediated Immunity
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published Nov 23, 2024 on www.thecalgaryguide.com")
Chronic Inflammatory Demyelinating Polyneuropathy

Systemic illness (e.g., hepatitis B, Hepatitis C, HIV, thyroid Disease, nephrotic syndrome, irritable bowel syndrome)
Authors: Andrea Soumbasis Reviewers: Braxton Phillips Shahab Marzoughi Sina Marzoughi* * MD at time of publication
Autoimmune response initiation
Lumbar Puncture: Cytoalbuminologic Dissociation CSF protein count elevated with normal cell count
Cell-mediated response: Putative antigen (unidentified immune target) presents to auto-reactive T cells
Activated T cells secrete inflammatory mediators (matrix metalloproteinases)
Inflammatory response ↑ peripheral nerve permeability & T cells cross blood-nerve barrier
Humoral Response: Immunoglobulin & complement deposits on compact myelin & nodal regions
Autoantibodies target unknown epitope (antigen molecule recognized by antibody) on outer surface of Schwann cells
IgG4 antibodies target axoglial junction (Contactin-1, Neurofascin 155) that maintain Node of Ranvier of myelinated axons
↑ Permeability of blood- nerve barrier allows large proteins to leak into CSF
CD8+ T cells, CD4+ T cells & macrophages infiltrate peripheral nerves
Macrophages form clusters around endoneurium, release proinflammatory cytokines & strip away myelin via phagocytosis
Abnormal expression & distribution of Contactin-1 & Neurofascin 155 antibodies along axoglial junction
Disrupted ion channel segregation & paranodal structure
↓ Saltatory conduction
Nodopathy & Paranodopothy: Neurofascin 155 – Distal sensorimotor ataxia
Contactin-1 – Sensorimotor ataxia Caspr-1 – Neuropathic pain & nephrotic syndrome
Onion bulb formation: Nerve biopsy showing concentric, circular layers of cells surrounding the axon
Segmental demyelination & remyelination of peripheral nerves
↑ Demyelination
Chronic Inflammatory Demyelinating Polyneuropathy
Progressive weakness & sensory loss over a period of greater than 8 weeks due to autoimmune dysfunction
Peripheral nerve degeneration & conduction velocity
Nerve Conduction Study: Partial nerve conduction block, conduction velocity slowing, prolonged distal motor latencies, delay or disappearance of CMAP (compound muscle action potential)
Lesions of larger myelinated fibers in the dorsal columns for vibration & position sense
Lesions of peripheral motor nerves in non-length dependent pattern (does not only affect longest nerves first)
Motor Deficits
↓ Sensory input and/or ↓ motor output
Abnormal Reflexes
Globally ↓ reflexes
Lesions affecting sympathetic & parasympathetic nerve fibers
Autonomic Deficits
Bladder & bowel dysfunction, heart rate irregularities, blood pressure fluctuation
Sensory Deficits
Gait ataxia
Proximal weakness (muscles closer to the trunk or center of the body): Difficulty climbing stairs, rising from seated position, lifting objects overhead, frequent falls
Distal weakness (muscles farther from the trunk or the extremities): Tripping due to foot drop, difficulty with fine motor tasks
↓ Vibration & proprioception
Paraesthesias (e.g., tingling, numbness)
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published Dec 29, 2024 on www.thecalgaryguide.com")
Barretts Esophagus

Authors: Sophia Khan Reviewers: Claire Song Shahab Marzoughi Sylvain Coderre* * MD at time of publication
Reflux of stomach acid, bile salts + digestive enzymes
through lower esophageal sphincter (located at junction between esophagus and stomach)
Chronic reflux exposure to squamous epithelium (cells lining distal esophagus)
Squamous epithelial cells release inflammatory cytokines: interleukin-8 and interleukin-1beta
Inflammation of squamous epithelium
Adaptive changes of squamous epithelium to prevent damage by acidic reflux
Migration of stem cells in gastric cardia (proximal region of the stomach) into distal esophagus
Replacement of esophageal squamous epithelium by gastric columnar epithelium
Conversion (metaplasia) of normal esophageal squamous epithelium into abnormal gastric columnar epithelium with interspersed goblet cells
Z line (the squamocolumnar junction between the esophagus and stomach) is irregular and displaced proximally on esophagus on endoscopy
Heartburn (retrosternal burning sensation from stomach reflux)
Regurgitation (involuntary expulsion
of stomach content back into esophagus)
Development of goblet cells (intestinal mucus-producing cells) within esophageal epithelium
Abnormal presence of goblet cells on histology of distal esophagus
Abnormal presence of columnar epithelial cells on histology of distal esophagus
↑ Atypical proliferation &
↓ apoptosis of Barrett’s epithelial cells
Dysplasia (disordered growth of cells with the
potential to develop into cancer) of Barrett’s epithelium
Esophageal adenocarcinoma
Improper division of nuclear chromatin (packaged DNA)
Cellular nuclei increase in size due to retained chromatin from improper division
Excess chromatin absorbs more stain during histology
Double- stranded DNA breaks
Nuclear enlargement on histology
Continued acidic reflux causes oxidative DNA damage
Hyperchromatic (darkly stained) nuclei on histology
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published Dec 15, 2024 on www.thecalgaryguide.com")
Malignant Esophagitis

Chronic gastric reflux (acidic partially- digested stomach contents)
Conversion of normal esophageal squamous epithelium to metaplastic columnar epithelium
Gastroesophageal Reflux Disease
Recurrent regurgitation of gastric content
Chronic Irritation & damage to the esophageal mucosa
Lesions and plaques develop in distal esophagus
Malignancy develops in the mucus-producing glandular cells in the lower esophagus
Smoking
Carcinogenic tobacco condensate is swallowed
Irritation of the the esophageal lining
Heavy alcohol consumption
Mutation in alcohol- dehydrogenase, which breaks down ethanol
↓ Alcohol breakdown
Irritation of the esophageal mucosa & lining
↑ Inflammation of the squamous epithelium located in the upper & middle esophagus
Achalasia (Esophageal smooth muscle motility disorder)
Food stasis in the esophagus ↑ Bacteria production
Lactic acid production & fermentation damages esophageal mucosa
Chronic inflammation in the esophageal epithelium
Malignant cells invade the aorta & tracheobronchial region
↑ Spread & growth of cancer in other organ systems
Metastatic invasion into the aorta, lungs, liver, bones, & kidneys through the lymph nodes
↑ Spread & growth of cancer in other organ systems
↑ Metabolic demand
Unintentional weight loss
Generalized chest pain
Hoarseness Cough
Esophageal adenocarcinoma (malignancy of the glandular cells of the esophagus)
Esophageal squamous cell carcinoma (malignancy of the squamous epithelium of the esophagus)
↑ Metabolic demand
↑ Weight loss
Chronic GI bleeding
Iron-deficiency anemia
↑ Ulceration
Exposure to stomach acid
↑ Tumor- induced inflammation
↑ Fibrosis
Irregular stricture of the upper & middle esophagus
Dysphagia
Esophageal wall narrowing
Abnormal reflexes of GI tract
Dysphagia
Indigestion
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published Dec 15, 2024 on www.thecalgaryguide.com")
Constipation in Children

Older Child (>1 years old)
Dietary (e.g., lack of fluids / fiber)
Mechanical (e.g.,. intestinal Atresia, anal atresia)
Congenital malformation (narrowing, absence, or malrotation) of structure of intestine / anus
Interrupted flow of bowel contents
Genetic (e.g., cystic Fibrosis)
Mucous blocks pancreatic duct
Inability of pancreatic enzymes to reach small intestine
↓ Digestion after a meal
Large, thickened stool
Neurologic (e.g., Hirschsprung's disease)
Congenital disruption of the migration of neural crest cells to the distal colon
Affected segment of the colon fails to relax
Progressive secondary dilation of the healthy proximal colon
Systemic (e.g., hypothyroidism)
Thyroid hormone deficiency
Reduction in the stimulation of gut tone & contractility by thyroid hormones
↓ Peristalsis (intestinal contractions) of the bowel
Dietary
(e.g., lack of fluids/ fiber)
Mucosal (e.g., celiac disease)
Inappropriate immune response against gluten
Intestinal mucosal injury
Malabsorpti on of water and other nutrients
Functional (e.g., pain)
Prolonged stool retention in bowel
↑ Time in bowel causing over- absorption of water from stool into the large intestine
Mechanical (e.g., bowel obstruction)
Mechanical obstruction in the intestines
Interrupted flow of bowel contents
Intestinal obstruction
Neurologic (e.g., neglect, physical abuse)
Disturbance in brain-intestine axis
Mechanisms not fully understood
Visceral hyper- sensitivity
(increased pain sensation)
Withholding behaviour
Lack of soluble fiber
↓ Attractive forces between water & stool
Prevention of secretion of water into stool
Lack of insoluble fiber
↓ Stool bulk and laxation
↓ Secretion of water &
mucous into stool
Lack of soluble fiber
↓ Attractive forces between water & stool
Prevention of secretion of water into stool
Lack of insoluble fiber
↓ Stool bulk and laxation
↓ Secretion of water & mucous into stool
Formation of dry & hard stool
Formation of dry & hard stool
Difficulty passing stool
Difficulty passing stool
Authors: Jennifer Wytsma Reviewers: Sophia Khan, Shahab Marzoughi, Sylvain Coderre* * MD at time of publication
Intestinal obstruction
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published Dec 15, 2024 on www.thecalgaryguide.com")
Polycythemia Vera Pathogenesis
: Pathogenesis and Clinical Findings
Authors: Caitlin Bittman Noriyah Al Awadhi Yan Yu Peter Duggan* Reviewers: Maharshi Gandhi Kevin Zhan Michelle J. Chen Paul Ratti Merna Adly Crystal Liu Kareem Jamani* Man-Chiu Poon* Lynn Savoie* * MD at time of publication
Familial PV inherited as autosomal dominant incomplete penetrance (5% of PV cases)
Predisposition to acquire a second somatic mutation
Hematopoietic cells with no known mutations in their DNA
Acquired JAK2 (Janus kinase 2) mutation in a single hematopoietic pluripotent stem cell in the bone marrow
JAK2 mutated cells in the bone marrow ↑ production of hematopoietic cells (RBCs, WBCs, platelets) and cytokines
DNA changes leads to continuous replication of the mutated cell
Abnormal cell becomes the predominant hematopoietic stem cell in the bone marrow
↑ RBC production independent of erythropoietin (EPO)
↑ Mast cell production
↑ Leukocyte production (basophils, neutrophils, eosinophils, monocytes
↑ Platelet production (thrombocytosis)
Overproduced platelets often have abnormal function of activation and aggregation
Hypercellular bone marrow on biopsy with trilineage growth (erythroid, granulocytic, and megakaryocytic cell lines)
↑ Hemoglobin concentration
↑ Hematocrit (proportion of blood volume occupied by RBCs)
↑ RBC leads to down-regulation of EPO by kidneys
↓ Serum EPO
↑ Levels of histamine, which is stored in granules within basophils & mast cells
Contact with water causes degranulation and subsequent histamine release from mast cells & basophils
Aquagenic pruritis (itchy skin after contact with water)
Histamines released from mast cells in the stomach causes hypersecretion of gastric acid
Stomach ulcers
Acquired Von Willebrand syndrome (with platelets >1,000,000/microL)
Despite thrombocytosis, there is an ↑ risk of bleeding due to platelet dysfunction
↑ Risk of hemorrhage & abnormal bleeding (e.g. GI and mucosal bleeding)
Easy bruising, purpura, petechiae, dental bleeds, epistaxis
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published Aug 7, 2012; updated Feb 22, 2025 on www.thecalgaryguide.com")
Tonsillitis Pathogenesis and clinical findings

Group A Streptococci (GAS) (most common bacteria)
Group B, C & G Strep,
Fusobacterium necrophorum
Age 5-15 (tonsils have ↑ role in immune function at this age)
Tonsillitis
Inflammation of the tonsils
Infectious agent exposure
Susceptible host Pathogen colonizes the oropharynx
Acute suppurative disease
Immune cells release proinflammatory cytokines & antibodies
Inflammatory mediators ↑ vascular permeability of tonsils
Leakage of protein & fluid into surrounding tissue
Regional nodes receive ↑ lymph
Enlarged anterior cervical nodes
Sinusitis**
Pharyngitis**
Local spread of pathogen
Acute otitis media**
Pneumonia**
Cervical lymphadenitis
Bacteria spread from
tonsils into lymphatic system & bloodstream
Bacteremia
F. necrophorum
invades lateral pharyngeal space & soft tissue in neck
Thrombosis forms in peritonsillar vein
Thrombosis extends into internal jugular vein
Lemierre’s syndrome
Systemic inflammatory cytokines disrupt hypothalamic regulation
Fever
Additional immune cells are recruited to facilitate immune response
Macrophages phagocytize pathogen
Bacteria invade distant tissue & elicit local inflammatory response
Hepatitis Osteomyelitis
Infective endocarditis
Bacteria illicit systemic response
Sepsis
Tonsillar tissue become swollen & irritated
Tonsillar hypertrophy
Localized collection of pus forms
Immune cells cause inadvertent cellular injury & hemolysis
Palatal petechiae
Meningitis
Products of immune response & cellular debris are deposited into tonsillar tissue
Peritonsillar or Tonsillar retropharyngeal abscess** exudate
** See corresponding Calgary Guide slide
Toxin-mediated disease
Bacteria release exotoxins into bloodstream
Inflammatory mediators & cytokines are overactivated (cytokine storm)
Skin has local inflammatory response
Toxic shock syndrome**
Scarlet fever**
Post-infectious disease
Antibodies to GAS cross react with host tissue
Acute rheumatic fever** Post-strep glomerulonephritis
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published Nov 5, 2018; updated Mar 14, 2025 on www.thecalgaryguide.com")
Polymyalgia Rheumatica
 is s9ll poorly understood. PMR is a rheuma9c condi9on characterized
Author: Anna Bobyn Reviewers: Anika Zaman, Kevin Zhan Luiza Radu, Emily J. Doucette Glen Hazlewood* *MD at time of publication
by pain and s9ffness around the neck, shoulder, and pelvic girdle.
Non-Modifiable factors
Assigned female at birth:
3♀ : 1♂
Environmental factors
Age-Related Changes: PMR almost exclusively affects people >50
Polymorphisms:
(eg. HLA-DR1, Interleukin (IL)-1, IL-6, TNF⍺)
Infectious Triggers: Epstein-Barr virus, Mycoplasma pneumoniae, parvovirus
Link to Vaccina=ons:
COVID-19, influenza, respiratory syncy=al virus
Innate immunity involvement: Macrophages & dendri=c cells recognize & respond to unknown an=gen
Immune cells ↑ production of cytokines (IL-6, IFN-γ, TNF-α)
Adap=ve immunity involvement: No strong evidence of autoan=body role
Early CD4+ T cell ac=va=on & infiltra=on of T cells into vascular =ssue
Subclinical vascular inflammation, particularly of medium & large arteries
↑ Vascular Endothelial Growth Factor & micro-vasculariza=on
Vascular dysfunction, ischemia, edema, & sensitization of pain receptors
Systemic inflamma=on
Local inflamma=on of synovial membrane, joint bursae, & tendon sheaths
B cell dysregula=on (↓ naïve B cells, ↑ memory B cells)
↑ Serum inflammatory
markers (CRP & ESR)
↑ Risk of cardiovascular
events (heart attack, stroke)
Low-grade fever (37.2- 38°C)
↓ Passive & ac>ve range of mo>on
Bilateral morning stiffness lasting >30 minutes
Fa>gue & malaise
Muscle pain in proximal joints
(shoulder & pelvic girdle)
Development of Giant Cell Arteri>s**
(15-20% of PMR pa>ents)
**See corresponding Calgary Guide slide
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complica=ons
Published Mar 23, 2025 on www.thecalgaryguide.com")
HIV Pathogenesis and Clinical Findings

Perinatal transmission during
pregnancy, childbirth or breast feeding
Direct contact of patient bloodstream or mucous membranes with HIV-1 infected blood, semen, rectal fluids, vaginal
discharge or breast milk (HIV-2 is transmitted in similar fashion but is less common & more likely to be seen in West Africa)
Authors:
Taylor Krawec
Ishjot Litt, Naima Riaz
Reviewers:
Candace Chan, Shahab Marzoughi
Emily J. Doucette, Sarah Smith*
* MD at time of publication
Glycoproteins in HIV viral envelope
bind to CD4 & chemokine receptors
on CD4+ T-cells & other immune cells
Viral & host cell
membranes fuse
Viral single-stranded RNA
(ssRNA) genome & enzymes
are released into the host cell
Host CD4+ cells are destroyed via direct cytotoxic
effects & immune cell-mediated cytotoxicity
CD4+ cell count ↓
Virions are disseminated throughout host
↑ Viral load
Viral proteins are transcribed
& assembled into virions
Virions bud off host
cell membrane
ssRNA is transcribed into proviral
DNA by viral reverse transcriptase
using host nucleotides
Infected cells revert to memory
state & establish latent HIV reservoir
Chronic HIV infection
Host is unable to keep up
with loss of CD4+ cells
Proviral DNA enters
nucleus & integrates
into host DNA
Acute HIV infection
Infected immune
cells release
proinflammatory
cytokines
Antibodies to HIV are generated
in attempt to control infection
(2-12 weeks post infection)
Positive antibody-antigen immunoassay
Cytokines disrupt
hypothalamic
temperature
regulation
Systemic immune
system activation
results in ↑
metabolic demand
Cytokines
extravasate
to the dermis
Fever Maculopapular
Chills
Fatigue
rash
Legend: Pathophysiology Mechanism
Sign/Symptom/Lab Finding Viral load ↓ &
stabilizes around
set-point viral load
Oral hairy leukoplakia
(oral infection with
Epstein-Barr Virus)
Complications
Viral load ↑ & CD4+ cell
count ↓ steadily over several
years without treatment
CD4+ cell count ↓
to 200-500 cells/μL
Impaired host immune response
↑ risk of opportunistic infections
Oropharyngeal
or vulvovaginal
candidiasis
Severe manifestations of
common infections (ex.
herpes simplex, HPV)
Published April 22, 2025 on www.thecalgaryguide.com
Virus evades immune
detection to allow
ongoing viral
replication, chronic
immune activation &
↓ thymic function
Vague viral
symptoms (ex.
fatigue, weight loss,
lymphadenopathy)
CD4+ count ↓
to < 200 cells/μL
Acquired
immunodeficiency
syndrome (AIDS)")
Trachoma

Mechanical vectors: Eye-seeking
flies (e.g. Musca sorbens)
Chlamydia trachomatis infection (serotype A, B & C)
Trachoma
Chronic keratoconjunctivitis (inflammation of the conjunctiva & cornea) caused by recurrent infection with C. trachomatis
Active Phase
Bacteria infects the conjunctival epithelial cells
Redness, irritation & mucopurulent discharge
Immune response triggers inflammation of the upper tarsal conjunctiva
Herbert’s pits (small, sunken scars at the limbus (border
between cornea & sclera) from healed follicles
Corneal lesions
Pannus (invasion by superficial vessels) at the limbus
Lymphoid follicles consisting mainly of B & T cells form in the upper tarsal conjunctiva
Trachomatous inflammation - follicular: Presence of five or
more white or yellow follicles, each 0.5 mm or bigger in size
Inflammation leads to papillae formation with dilated blood vessels, infiltration
of lymphocytes, plasma cells & neutrophils & proliferation of epithelial cells
Small, raised bumps with a central blood vessel in the upper conjunctiva
Papillary hypertrophy causes conjunctival thickening & opaqueness Trachomatous inflammation - intense: Papillae obscure deep tarsal blood vessels
Repeated re-infection or untreated infections
Cicatricial (scarring) Phase
Proliferation of fibroblasts & deposition of collagen leads to scarring (fibrosis)
Trachomatous scarring: White, horizontal lines or bands on upper tarsal conjunctiva
Scarring blocks
meibomian gland ducts
Conjunctival epithelium atrophy
& goblet cell destruction Subconjunctival
fibrosis
Entropion (eyelid turns inward)
Cornea ulcers
& scarring
↑ Tear evaporation
↓ Tear production
Scar contraction distorts
upper tarsal plate
Dry eye
Trachomatous trichiasis
(eyelashes grow inward
toward the eye)
Corneal
abrasion
Corneal opacity
& blindness
Published Sept, 2 2015; updated May 7, 2025 on www.thecalgaryguide.com
Legend: Pathophysiology Mechanism
Sign/Symptom/Lab Finding Complications")
Vaccine Mediated Immunity Comparing Vaccine Subtypes
,
Varicella, Rotavirus, Influenza
COVID-19
Serial passage in sub-optimal
culture conditions weaken
pathogen
Pathogen maintains ability to
slowly replicate (attenuated)
Vaccine components (live
attenuated pathogen,
adjuvants, stabilizers,
preservatives) are
formulated & administered
Attenuated pathogen enters
host cells & replicates,
mimicking natural infection
Identify target protein of specific pathogen
RNA polymerase transcribes DNA for target
protein into mRNA (in vitro transcription)
mRNA is purified & concentrated
mRNA is encapsulated in lipid
nanoparticles to prevent degradation
Vaccine components (mRNA, lipid
nanoparticles, adjuvants, preservatives)
are formulated & administered
Host cells take up mRNA & synthesize the
antigenic protein
Diphtheria, Tetanus, Pertussis,
Pneumococcus, Meningococcus,
Haemophilus influenzae type b, Influenza,
Hepatitis B, Human Papilloma Virus
Polio (IPV), Hepatitis A
Antigen-presenting cells (APCs) take up
intracellular & extracellular antigens
Infected cells & APCs present intracellular
antigens to CD8+ T cells via major
histocompatibility complex-I (MHC-I) molecules
Robust cell-mediated immunity
↑ Lymphocyte counts
Legend: Pathophysiology Mechanism
Sign/Symptom/Lab Finding Complications
Authors:
Tanis Orsetti
Reviewers:
Jessica Hammal
Emily J. Doucette
James D. Kellner*
* MD at time of publication
Inactivated Whole Virus Vaccines
Identify target protein of specific
pathogen (antigenic protein)
Pathogen is inactivated using
chemicals/detergents/heat
Large quantity of antigenic protein
produced in a laboratory setting
Pathogen is unable
to replicate
Vaccine components (pathogen, adjuvants, stabilizers,
preservatives) are formulated & administered
APCs take up
extracellular antigens
APCs present extracellular antigens to CD4+
cells via major histocompatibility complex-II
(MHC-II) molecules
Activated CD4+ T helper cells stimulate B lymphocytes
B lymphocytes produce antibodies specific to pathogen
Robust humoral immunity
↑ Pathogen-specific antibody titers
Published May 31, 2025 on www.thecalgaryguide.com")
Heparin-Induced Thrombocytopenia

Platelets aggregate
within the bloodstream
↓ Free unbound
platelets in bloodstream
Mild Thrombocytopenia:
platelet count between
100 x 109/L - 150 x 109/L
Fewer platelets
available for thrombosis
(formation of a
pathological blood clot)
Self limiting with
no thrombotic risk
Legend: Heparin-Induced Thrombocytopenia (HIT): Pathogenesis and clinical findings
Obesity Hypertension Macrovascular disease Hyperlipidemia
Heparin (often unfractionated)
complexes with platelet factor 4 (PF4)
on the platelet surface, due to
heparin’s negative charge & PF4’s
affinity for long-chain polysaccharides
Immune-Mediated:
Type 2 (within 5-15 days
of heparin administration)
Anti-heparin/PF4
Immunoglobulin G
antibodies binds to
the heparin-PF4
complex in the blood
Splenic macrophages
remove platelet-
immunoglobulin G
complexes in the spleen
Thrombocytopenia:
Platelet count < 100 x
109/L (though < 20 x
109/L should prompt
consideration of
other diagnoses)
↑ Vascular inflammation & endothelial
dysfunction activate platelets
Authors:
Kelle Edgar, Hadi Hassan
Sergio F. Sharif
Reviewers:
Haotian Wang, Jessica Asgarpour
Yan Yu*, Lynn Savoie*
Kareem Jamani*
* MD at time of publication
Activated platelets
release PF4
Anti-heparin/PF4
antibodies activate
adjacent platelets
Activated
platelets release
inflammatory
mediators
Circulating anti-
heparin/PF4 antibodies
bind adjacent cellular
targets
Anti-heparin/PF4
antibodies bind
endogenous heparan
sulfate on endothelium
Activated
endothelium
releases
procoagulants
thrombin &
tissue factor
Activated
platelets release
procoagulants
(factors
promoting blood
clotting)
Free platelets are
consumed from
formation of thrombi
Hypercoagulable state
(↑ risk of thrombosis)
↓ Platelets available
to clot at different
area of the body
Thrombosis in
coronary, peripheral
or cerebral arteries
Bleeding (rare)
Myocardial
infarction
Acute
ischemia
Stroke
Inflammatory
mediators disrupt
hypothalamic body
thermoregulation
Fever/
chills
Inflammatory mediators
promote vasodilation of
arterioles under the skin
↑ Blood to
surface of skin
Flushing of skin
Venous thrombosis
in lower leg or lung
Deep vein
thrombosis
Pulmonary
embolism
Microthrombi occlude
vessels surrounding
injection site, limiting
nutrient supply
Necrosis (tissue death)
at heparin injection site
Published Aug 29, 2014; updated Jun 16, 2025 on www.thecalgaryguide.com
Pathophysiology Mechanism
Sign/Symptom/Lab Finding Complications")
Varicella Zoster Virus
 - Chicken Pox: Pathogenesis and clinical findings Exposure to
individual with
VZV infection
VZV enters
respiratory tract
& conjunctiva
VZV enters
dendritic
cells (DC)
DCs travel to
regional
lymph nodes
Primary viremia
Authors:
Morgan Garland,
Sean Spence
VZV replicates
VZV proteins ↑ major
↓ MHCI
Reviewers:
in nasopharynx
histocompatibility
expression
Yan Yu, Danny Guo,
& CD4+ T cells
complex class I
on CD4+ T
Emily J. Doucette,
(MHCI) degradation
cell surface
Laurie Parsons*,
James D. Kellner*
*MD at time of publication
Acute phase
VZV evades the immune system
VZV travels through the blood in CD4+ T cells
Immune cells release
cytokines to fight the
virus (stronger in adults)
Fluid & cellular debris
accumulate between
epidermal layers
Generalized pruritic (itchy) vesicular
rash at different stages of development
(macule, papule, vesicle)
Immune system activates
& releases inflammatory
compounds
VZV replicates in liver,
spleen & lymph nodes
Erythema
(redness) & pain
Scratching lesions
introduces bacteria
Fibroblasts lay down
collagen to heal skin lesions
Resets thermostat
in hypothalamus
Systemic
response
Tissue damage
in oropharynx
Direct
inflammation in
neural tissues
Cerebellar ataxia
(more common
in children)
Bacterial superinfection from S.
aureus & Streptococcus pyogenes
(more common in children)
Scarring
Fever
(prodromal)
Malaise
(prodromal)
Sore throat
(prodromal)
Encephalitis (more
common in adults)
Abscess
Cellulitis
Toxic shock syndrome (TSS)
Secondary viremia
Immune cells release cytokines to
fight infection (stronger in adults)
Inflammation & fluid
build up in lungs
VZV travels through the blood in CD4+ T
cells expressing skin homing receptors
Fluid & cellular
debris accumulate
in damaged alveoli Lungs more susceptible to
secondary bacterial infection
Pneumonia** (more
common in adults)
VZV colonizes
keratinocytes
over several days
VZV colonizes
neurons & glial
cells in the brain
VZV colonizes
alveolar
epithelial cells
Resolution & latency
VZV tracks along
neurons from sites of
infection to dorsal root
ganglia & cranial nerves
↓ MHCI expression
in sensory ganglia
Adaptive
immune system
controls viremia
↓ Viral gene expression
Resolution of VZV
infection &
development of
lifelong immunity
Direct viral toxicity (DNA damage, mitochondrial
dysfunction, & protein misfolds in infected cells)
Apoptosis & dysfunction of infected cells
Latent
infection
Stress or immune suppression may
precipitate reactivation of VZV later in life Shingles**
**See corresponding Calgary Guide slide
Pathophysiology Mechanism
Published Nov 12, 2012; updated Jun 22, 2025 on www.thecalgaryguide.com
Legend: Sign/Symptom/Lab Finding Complications")
GLP-1 Receptor Agonists
 Receptor Agonists: Mechanism of action and side effects
Management of Type
2 Diabetes Mellitus**
Weight
management
Authors: Saania Tariq, Trevor Low
Reviewers: Rafael Sanguinetti
Luiza Radu, Emily J. Doucette
Activated GLP-1 receptors in
Activated GLP-1
Hanan Bassyouni*
* MD at time of publication
the myenteric plexus inhibit
receptors stimulate
gastric motor neurons
pancreatic islet cells
GLP-1 Receptor Agonists
Synthetic analogs (e.g., Semaglutide) of GLP-1 peptide hormone endogenously
produced by intestinal enteroendocrine L cells directly activate GLP-1 receptors
to modulate food intake & glucose metabolism.
↓ Gastric
motility
↑ Pyloric sphincter
tone via disinhibition
↓ Glucagon release
from α-cells
↑ Regeneration,
↑ growth &
↓ apoptosis of β-cells
Slowed gastric emptying
Intramuscular injection or oral ingestion
prolongs GLP-1 receptor activation
↑ β-cell function & glucose-
dependent insulin production
GLP-1 receptor activation on neurons in
the arcuate nucleus of the hypothalamus
modulates appetite & satiety
Activation of GLP-1 receptors
in the gastrointestinal system
regulates metabolism
Prolonged gastric
distension & stasis
activates
chemoreceptors &
mechanoreceptors
Delayed digestion of
gastric contents ↓
glucose spikes after
meals
↑ Glycogen synthesis &
glucose uptake in liver &
skeletal muscles
Neuropeptide Y &
agouti-related protein
expressing neurons are
hyperpolarized
Pro-opiomelanocortin
expressing neurons
become ↑ excitable
Activated GLP-1 receptors
signal to hypothalamus
via vagus nerve afferents
↑ Vagal afferent
signaling to area
postrema
(vomiting centre)
Nausea &
vomiting
↓ Release of
neuropeptide Y &
agouti-related protein
↑ Anorexigenic
signalling
↑ Parasympathetic
output via dorsal motor
nucleus of vagus nerve
GLP-1 agonists directly
activate receptors in
area postrema
↑ Satiety (feeling
of fullness)
↓ Adiposity
(weight loss)
↓ Orexigenic drive
↓ Inflammatory stress in
pancreatic β-cells
↓ Appetite
↓ Caloric intake
↓ Inflammatory
cytokines & lipotoxicity
↓ Systemic
inflammation
**See corresponding Calgary Guide slide
Legend: Sign/Symptom/Lab Finding Pathophysiology Mechanism
Complications
↓ Hepatic glucose
production
(glycogenolysis)
↓ Fasting & postprandial
blood glucose
↓ Red blood cell (RBC)
glycation (glucose
irreversibly binds to
hemoglobin in RBCs)
↓ Production
of glycated
proteins and
lipids
↓ % of glycated
RBCs over time
↑ β-cell function
improves insulin
secretion
↓ Vascular
endothelial
damage and
atherosclerosis
↓ Endothelial
damage to
glomeruli
↑ Insulin sensitivity in
skeletal muscle & liver
↓ Hemoglobin
A1c
↓ Risk of
cardiovascular
complications
↓ Risk of renal
complications
Published Jun 22, 2025 on www.thecalgaryguide.com")