SEARCH RESULTS FOR:
Myocardial Infarction: Findings on History
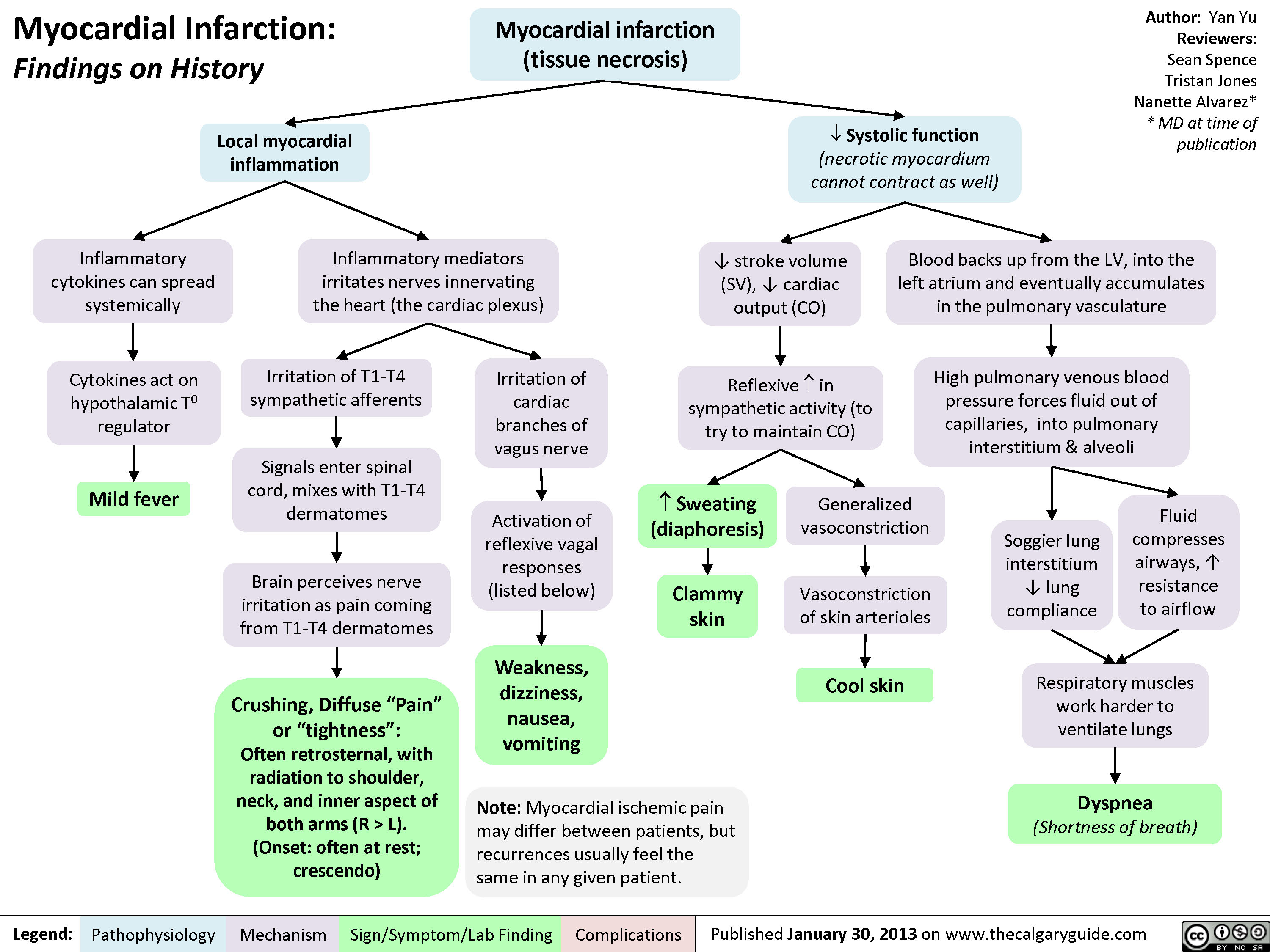 L).(Onset: often at rest; crescendo)Activation of reflexive vagal responses (listed below)Weakness, dizziness, nausea, vomitingInflammatory mediators irritates nerves innervating the heart (the cardiac plexus)Cytokines act on hypothalamic T0 regulatorMild fever? Sweating (diaphoresis)Inflammatory cytokines can spread systemicallyBrain perceives nerve irritation as pain coming from T1-T4 dermatomesBlood backs up from the LV, into the left atrium and eventually accumulates in the pulmonary vasculatureHigh pulmonary venous blood pressure forces fluid out of capillaries, into pulmonary interstitium & alveoliRespiratory muscles work harder to ventilate lungsSoggier lung interstitium ? lung complianceDyspnea(Shortness of breath)Fluid compresses airways, ? resistance to airflow
102 kB / 204 words" title="Yu Yan - MI Findings on History - FINAL.pptx -
Myocardial Infarction: Findings on HistoryLegend:Published January 30, 2013 on www.thecalgaryguide.comMechanismPathophysiologySign/Symptom/Lab FindingComplicationsAuthor: Yan YuReviewers:Sean SpenceTristan JonesNanette Alvarez** MD at time of publication Systolic function(necrotic myocardium cannot contract as well)Reflexive ? in sympathetic activity (to try to maintain CO)Clammy skin? stroke volume (SV), ? cardiac output (CO)Myocardial infarction (tissue necrosis)Note: Myocardial ischemic pain may differ between patients, but recurrences usually feel the same in any given patient.Generalized vasoconstrictionVasoconstriction of skin arteriolesCool skinLocal myocardial inflammationIrritation of T1-T4 sympathetic afferentsIrritation of cardiac branches of vagus nerveSignals enter spinal cord, mixes with T1-T4 dermatomesCrushing, Diffuse "Pain" or "tightness": Often retrosternal, with radiation to shoulder, neck, and inner aspect of both arms (R > L).(Onset: often at rest; crescendo)Activation of reflexive vagal responses (listed below)Weakness, dizziness, nausea, vomitingInflammatory mediators irritates nerves innervating the heart (the cardiac plexus)Cytokines act on hypothalamic T0 regulatorMild fever? Sweating (diaphoresis)Inflammatory cytokines can spread systemicallyBrain perceives nerve irritation as pain coming from T1-T4 dermatomesBlood backs up from the LV, into the left atrium and eventually accumulates in the pulmonary vasculatureHigh pulmonary venous blood pressure forces fluid out of capillaries, into pulmonary interstitium & alveoliRespiratory muscles work harder to ventilate lungsSoggier lung interstitium ? lung complianceDyspnea(Shortness of breath)Fluid compresses airways, ? resistance to airflow
102 kB / 204 words" />
L).(Onset: often at rest; crescendo)Activation of reflexive vagal responses (listed below)Weakness, dizziness, nausea, vomitingInflammatory mediators irritates nerves innervating the heart (the cardiac plexus)Cytokines act on hypothalamic T0 regulatorMild fever? Sweating (diaphoresis)Inflammatory cytokines can spread systemicallyBrain perceives nerve irritation as pain coming from T1-T4 dermatomesBlood backs up from the LV, into the left atrium and eventually accumulates in the pulmonary vasculatureHigh pulmonary venous blood pressure forces fluid out of capillaries, into pulmonary interstitium & alveoliRespiratory muscles work harder to ventilate lungsSoggier lung interstitium ? lung complianceDyspnea(Shortness of breath)Fluid compresses airways, ? resistance to airflow
102 kB / 204 words" title="Yu Yan - MI Findings on History - FINAL.pptx -
Myocardial Infarction: Findings on HistoryLegend:Published January 30, 2013 on www.thecalgaryguide.comMechanismPathophysiologySign/Symptom/Lab FindingComplicationsAuthor: Yan YuReviewers:Sean SpenceTristan JonesNanette Alvarez** MD at time of publication Systolic function(necrotic myocardium cannot contract as well)Reflexive ? in sympathetic activity (to try to maintain CO)Clammy skin? stroke volume (SV), ? cardiac output (CO)Myocardial infarction (tissue necrosis)Note: Myocardial ischemic pain may differ between patients, but recurrences usually feel the same in any given patient.Generalized vasoconstrictionVasoconstriction of skin arteriolesCool skinLocal myocardial inflammationIrritation of T1-T4 sympathetic afferentsIrritation of cardiac branches of vagus nerveSignals enter spinal cord, mixes with T1-T4 dermatomesCrushing, Diffuse "Pain" or "tightness": Often retrosternal, with radiation to shoulder, neck, and inner aspect of both arms (R > L).(Onset: often at rest; crescendo)Activation of reflexive vagal responses (listed below)Weakness, dizziness, nausea, vomitingInflammatory mediators irritates nerves innervating the heart (the cardiac plexus)Cytokines act on hypothalamic T0 regulatorMild fever? Sweating (diaphoresis)Inflammatory cytokines can spread systemicallyBrain perceives nerve irritation as pain coming from T1-T4 dermatomesBlood backs up from the LV, into the left atrium and eventually accumulates in the pulmonary vasculatureHigh pulmonary venous blood pressure forces fluid out of capillaries, into pulmonary interstitium & alveoliRespiratory muscles work harder to ventilate lungsSoggier lung interstitium ? lung complianceDyspnea(Shortness of breath)Fluid compresses airways, ? resistance to airflow
102 kB / 204 words" />
process
lower-urinary-tract-infections-complications
, stagnant
urine (anatomical variant, obstruction,
neurogenic bladder, urinary reflux)
Bacterial entry (Less Common):
Indwelling catheter, surgical inoculation,
hematogenousspread, trauma
(Staphylococcus, Enterococcus, Candida)
Fecal bacteria access urethra
(E. coli, Proteus, Klebsiella)
Impairment of body's natural defense
systems, or stagnant urine, allow for
bacterial accumulation
Portal of entry bypasses body's physical
defenses (gravity and repetitive outward
urine flow)
Bacterial fimbriae and pili allow
them to ascend urethra and
adhere to epithelium
Lower Urinary Tract Infection (LUTI): Pathogenesis and clinical findings
Suprapubic
Tenderness
Bacterial colony irritates
urinary epithelium
Urgency:
Sensation of need to urinate
quickly or impending
incontinence
Stimulation of
inflammatory
response
Stimulation of urinary reflex
Pathogens use
enzymes to reduce
nitrate to nitrite
Delirium in Elderly
Frequency:
Repetitive need to
urinate
Unique response of altered fluid
status, electrolytes and mental
status, likely as a result of
increased inflammatory cytokines
Lower Urinary Tract Infection (“Cystitis”):
Infection of bladder or distal tract by capable bacteria
colonizing epithelium and causing symptoms
Legend: Pathophysiology Mechanism Sign/Symptom/Lab Finding Complications Published March 16, 2014 on www.thecalgaryguide.com
Author:
Brett Edwards
Reviewers:
Riley Hartmann
Jan Rudzinski
Haotian Wang
Steve Vaughan*
* MD at time of publication
Usual Pathogens (“KEEPS”):
K – Klebsiella
E – E. coli (90%)
E – Enterococcus, Enterobacteriaceae
P – Proteus, Pseudomonas
S – Staph. saprophyticus, Serratia
Urine Findings:
↑ Colony Count (>107 CFU/L)
↑ WBC (>10 WBC/μL)
(+) Bacterial culture
(+) Nitrites, Leukocyte Esterase
(+) Foul, turbid urine
+/- Hematuria (rare)")
lower-urinary-tract-infection-pathogenesis-and-clinical-findings
, stagnant
urine (anatomical variant, obstruction,
neurogenic bladder, urinary reflux)
Bacterial entry (Less Common):
Indwelling catheter, surgical inoculation,
hematogenousspread, trauma
(Staphylococcus, Enterococcus, Candida)
Fecal bacteria access urethra
(E. coli, Proteus, Klebsiella)
Impairment of body's natural defense
systems, or stagnant urine, allow for
bacterial accumulation
Portal of entry bypasses body's physical
defenses (gravity and repetitive outward
urine flow)
Bacterial fimbriae and pili allow
them to ascend urethra and
adhere to epithelium
Lower Urinary Tract Infection (LUTI): Pathogenesis and clinical findings
Suprapubic
Tenderness
Bacterial colony irritates
urinary epithelium
Urgency:
Sensation of need to urinate
quickly or impending
incontinence
Stimulation of
inflammatory
response
Stimulation of urinary reflex
Pathogens use
enzymes to reduce
nitrate to nitrite
Delirium in Elderly
Frequency:
Repetitive need to
urinate
Unique response of altered fluid
status, electrolytes and mental
status, likely as a result of
increased inflammatory cytokines
Lower Urinary Tract Infection (“Cystitis”):
Infection of bladder or distal tract by capable bacteria
colonizing epithelium and causing symptoms
Legend: Pathophysiology Mechanism Sign/Symptom/Lab Finding Complications Published March 16, 2014 on www.thecalgaryguide.com
Author:
Brett Edwards
Reviewers:
Riley Hartmann
Jan Rudzinski
Haotian Wang
Steve Vaughan*
* MD at time of publication
Usual Pathogens (“KEEPS”):
K – Klebsiella
E – E. coli (90%)
E – Enterococcus, Enterobacteriaceae
P – Proteus, Pseudomonas
S – Staph. saprophyticus, Serratia
Urine Findings:
↑ Colony Count (>107 CFU/L)
↑ WBC (>10 WBC/μL)
(+) Bacterial culture
(+) Nitrites, Leukocyte Esterase
(+) Foul, turbid urine
+/- Hematuria (rare)
WBCs onsite
release enzymes
Cytokines released
systemically
Fever, Malaise,
↑WBC
(>11 x 109 cells/L)
(Rare in LUTI)")
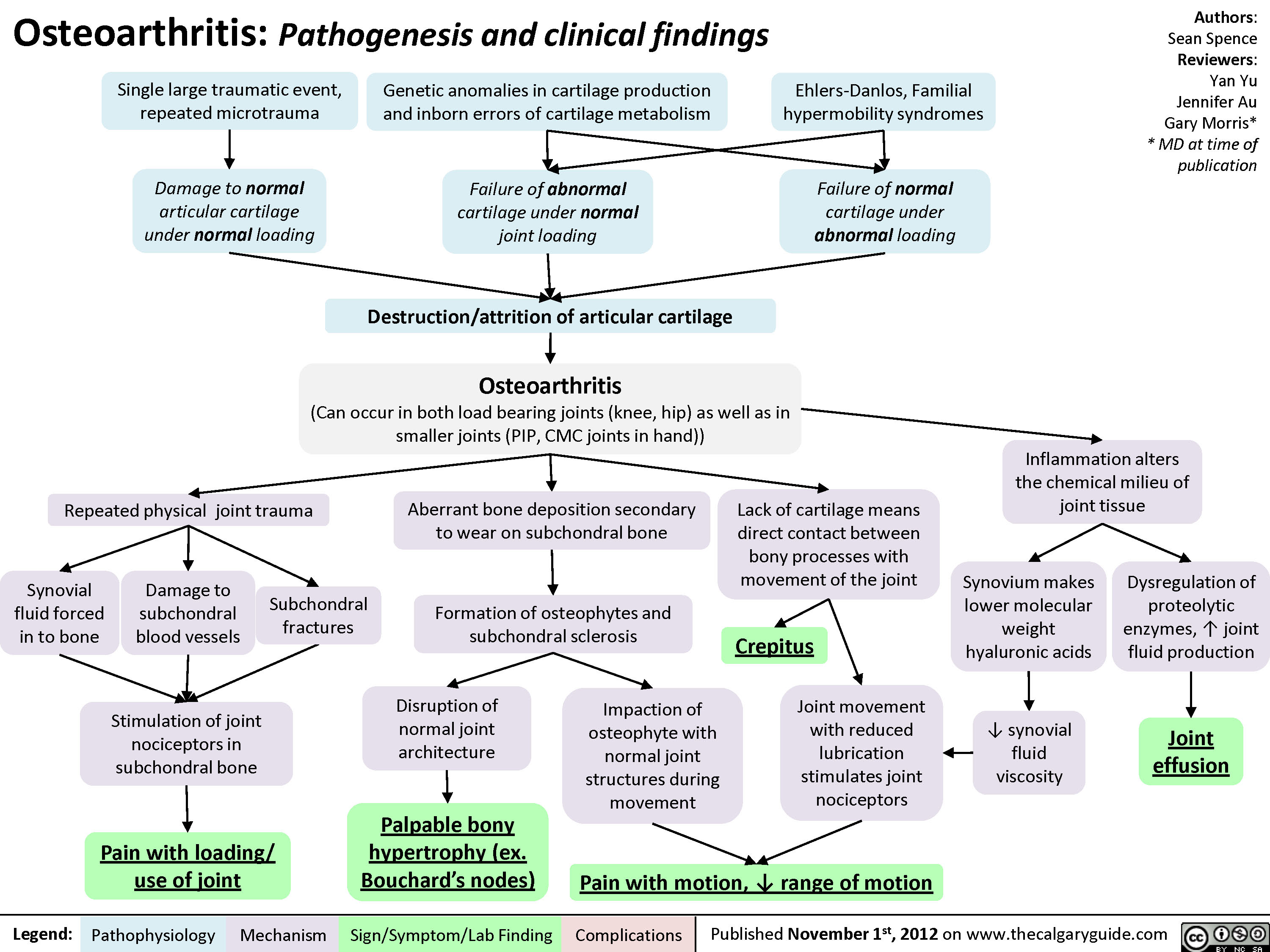
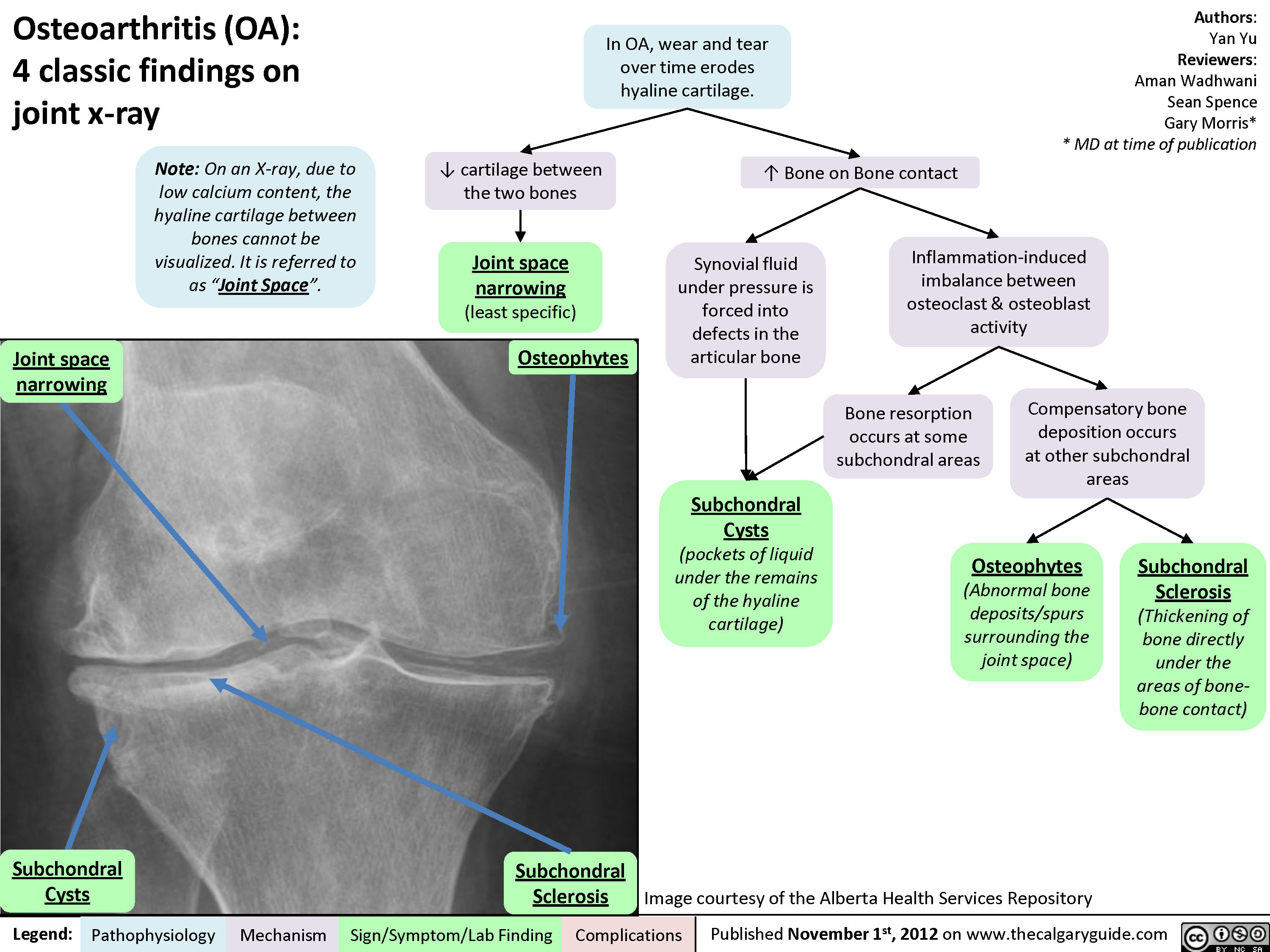
Osteoarthritis (OA): Clinical findings
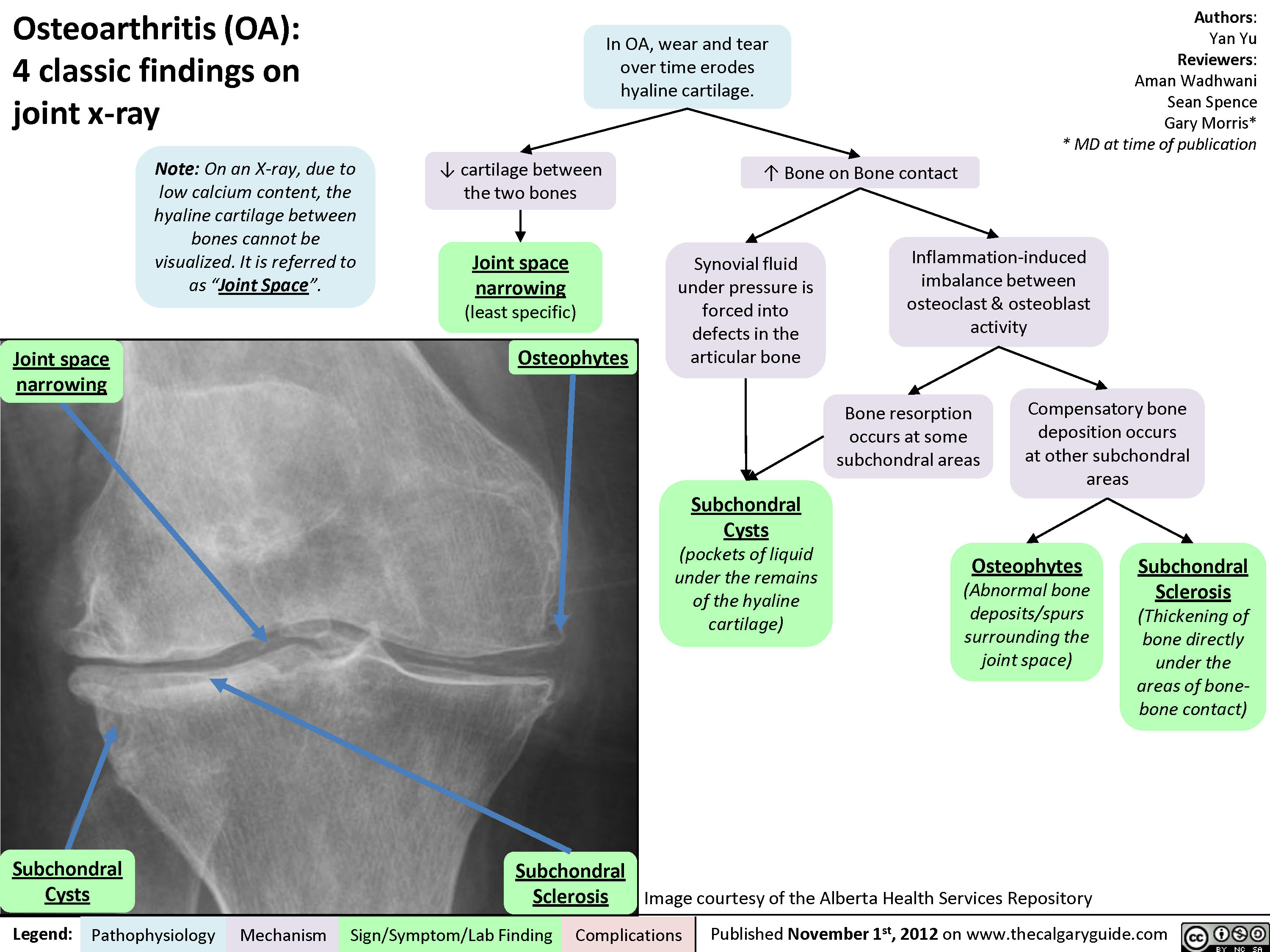
Osteoarthritis (OA): X-ray features
Diffuse Systemic Sclerosis (Scleroderma)
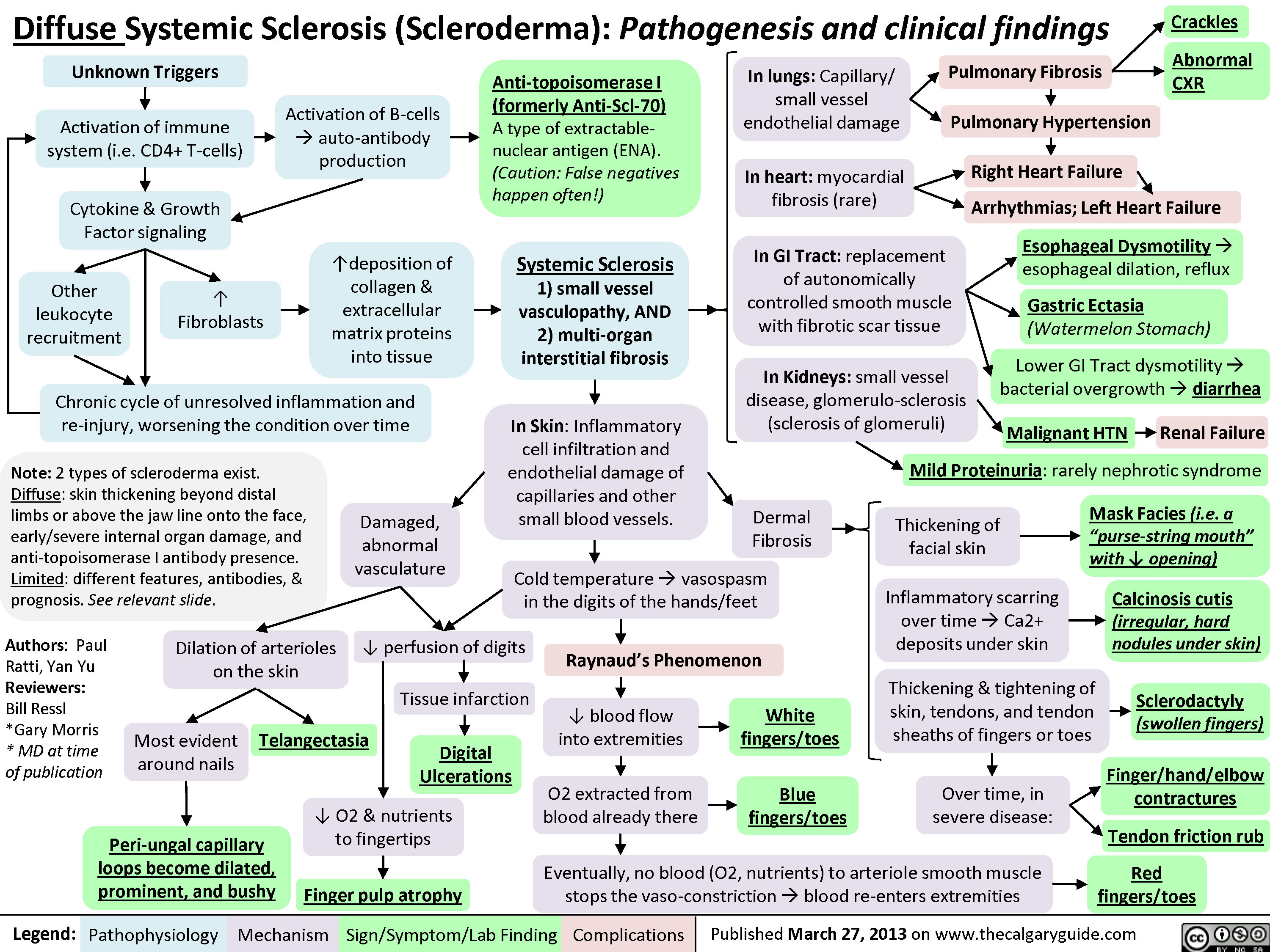
Takayasu's Arteritis: Pathogenesis and clinical findings
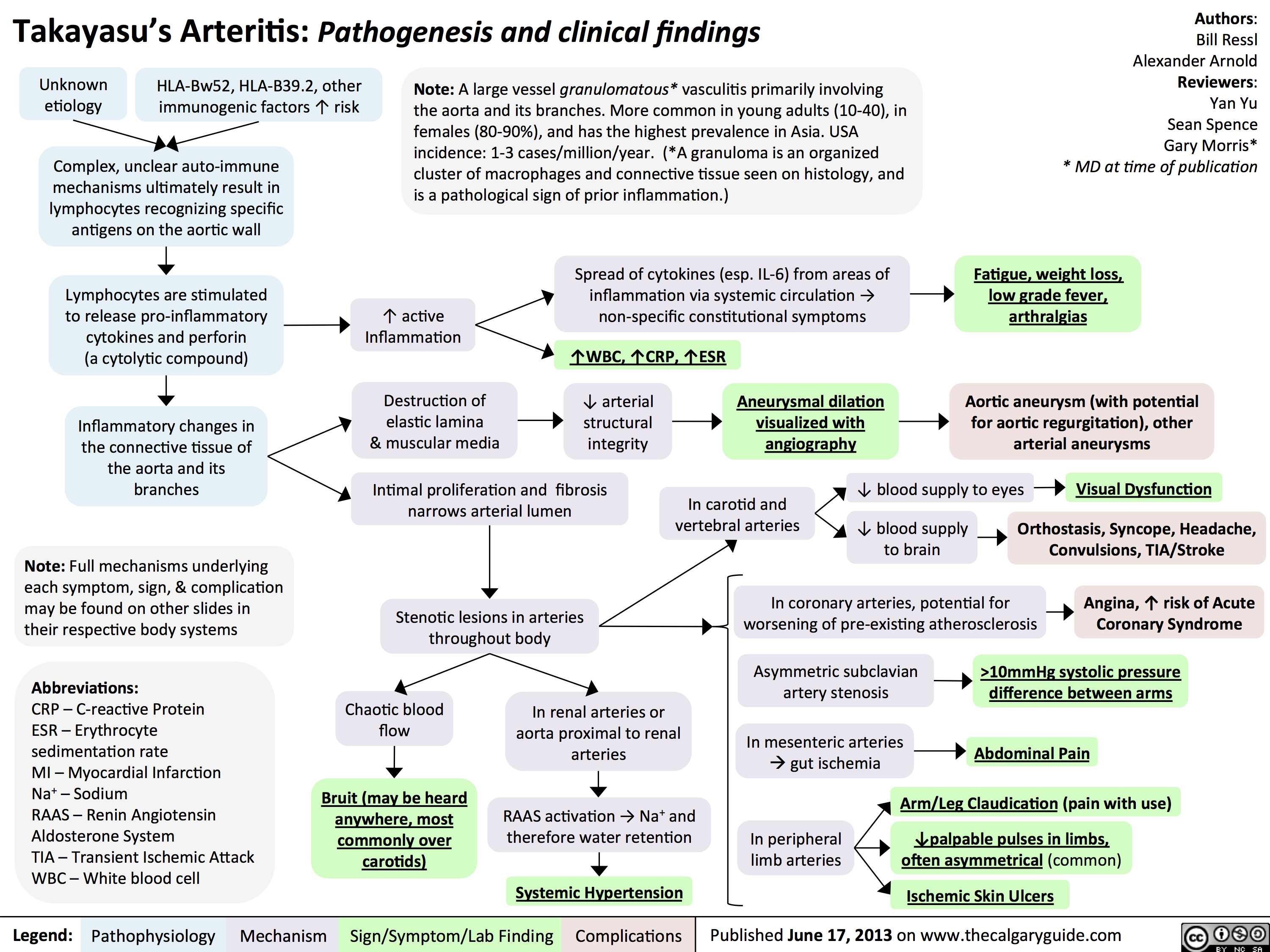
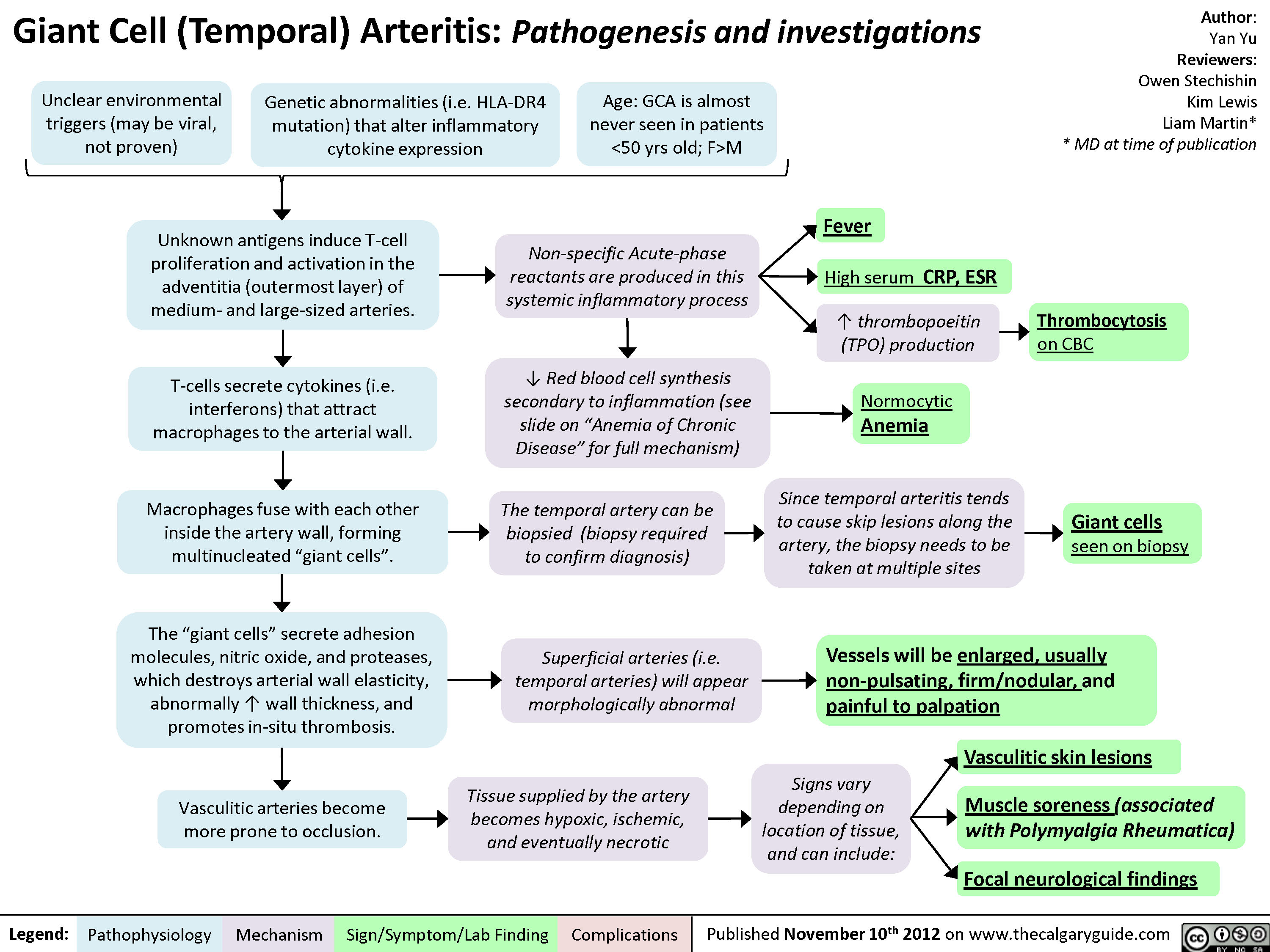
Giant Cell (Temporal) Arteritis: Pathogenesis and investigations

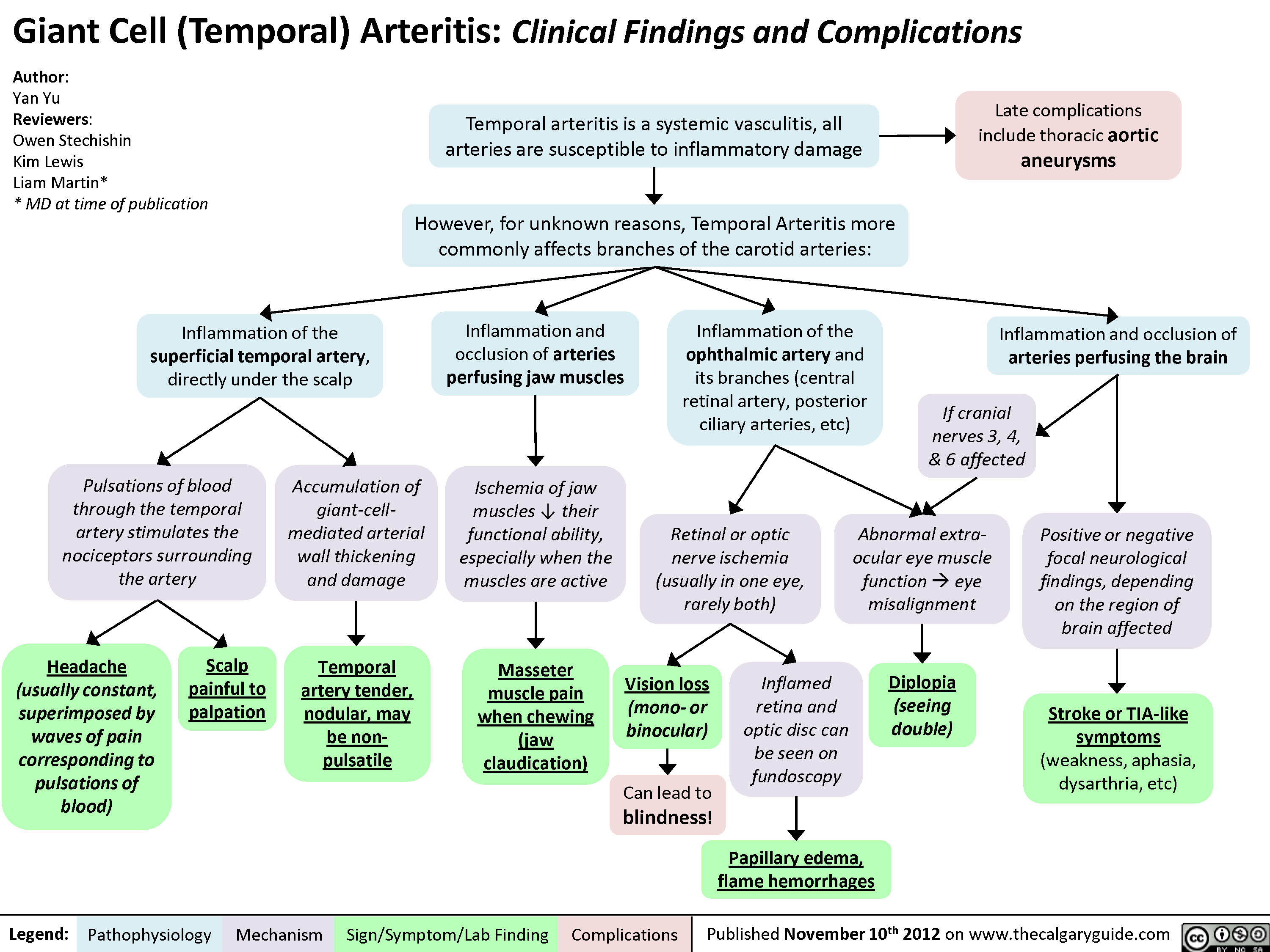
Giant Cell (Temporal) Arteritis: Clinical findings and Complications
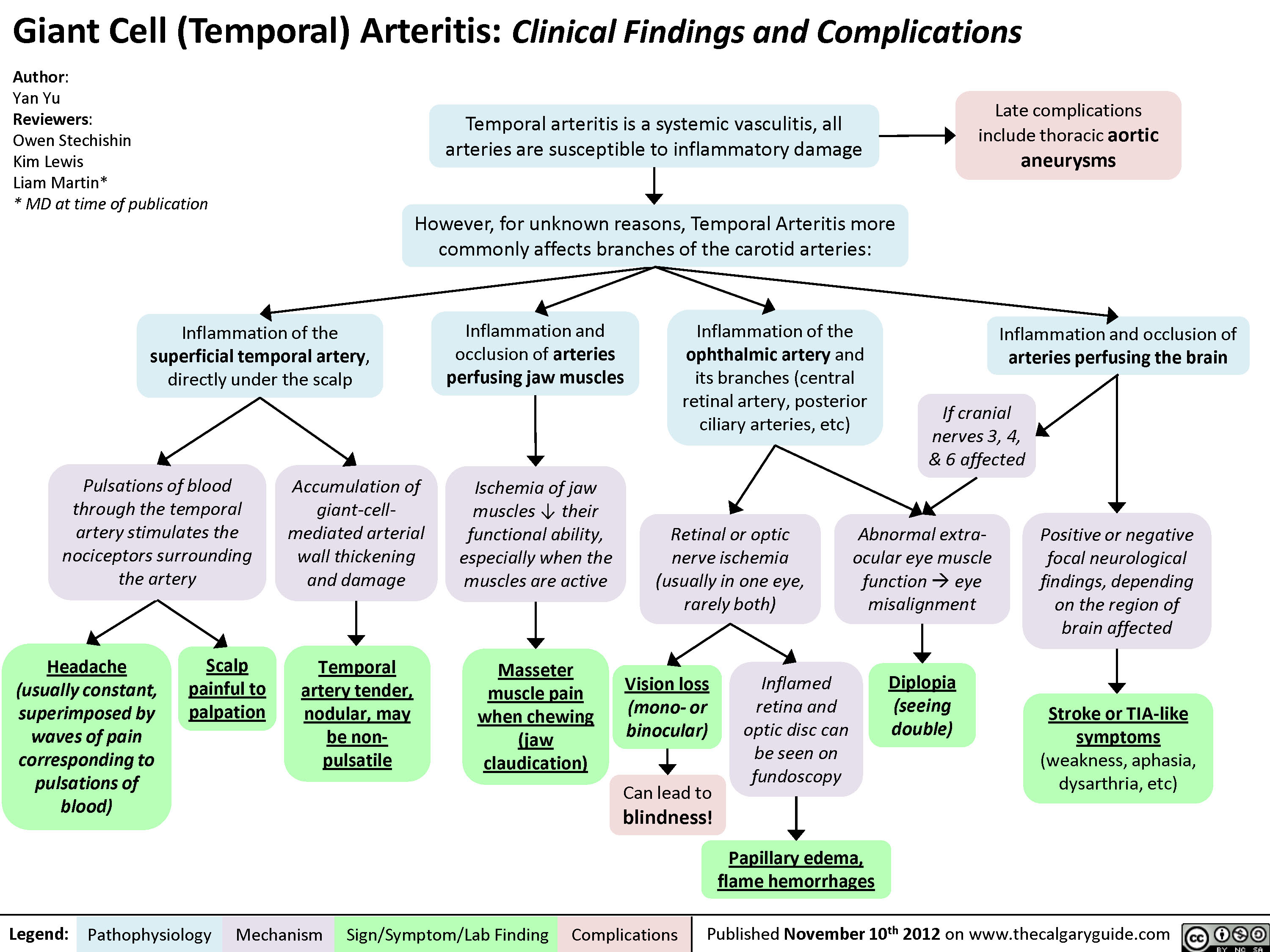
Hyperuricemia Pathogenesis and Complications
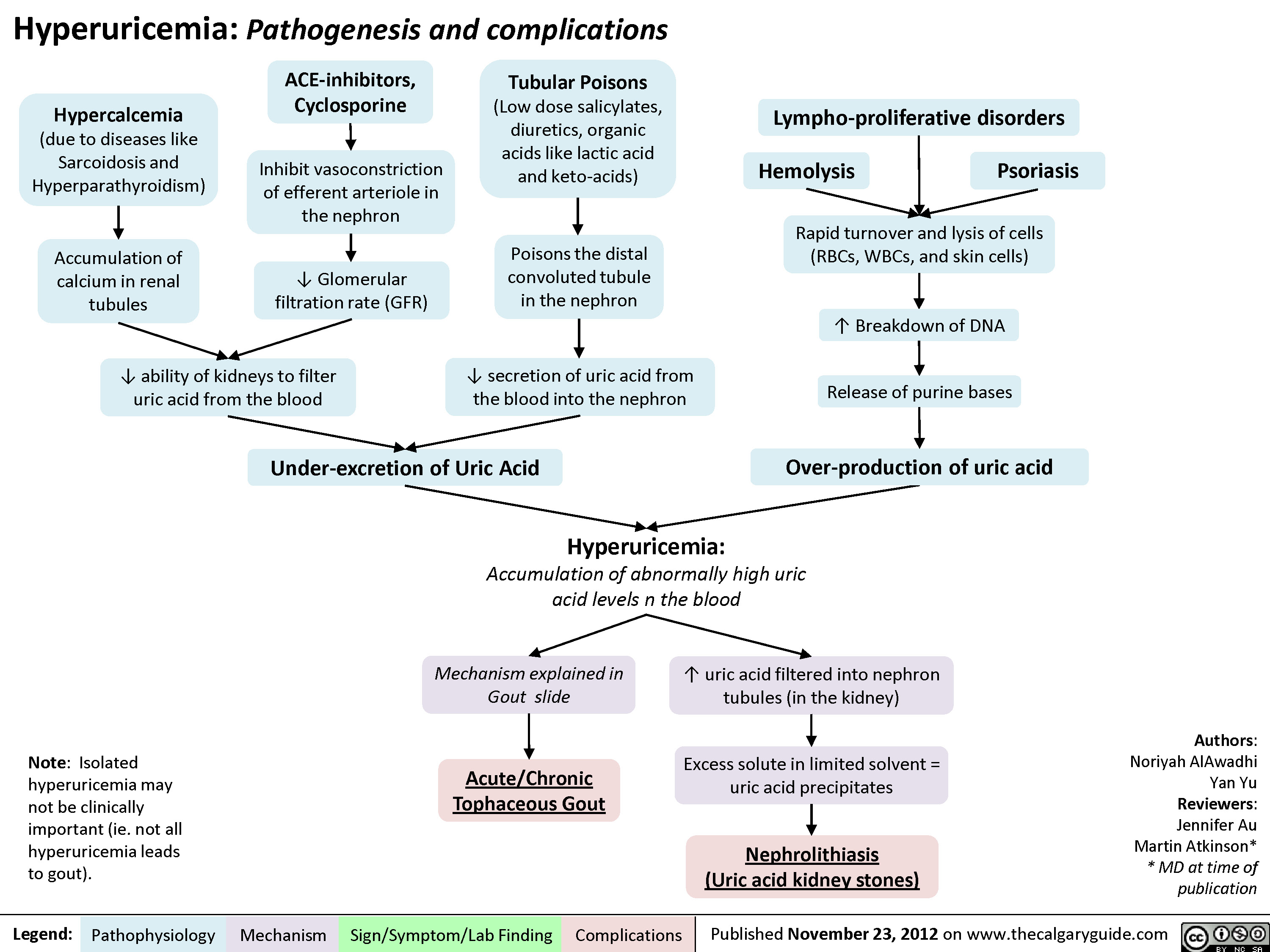
Gout Pathogenesis and Clinical Findings
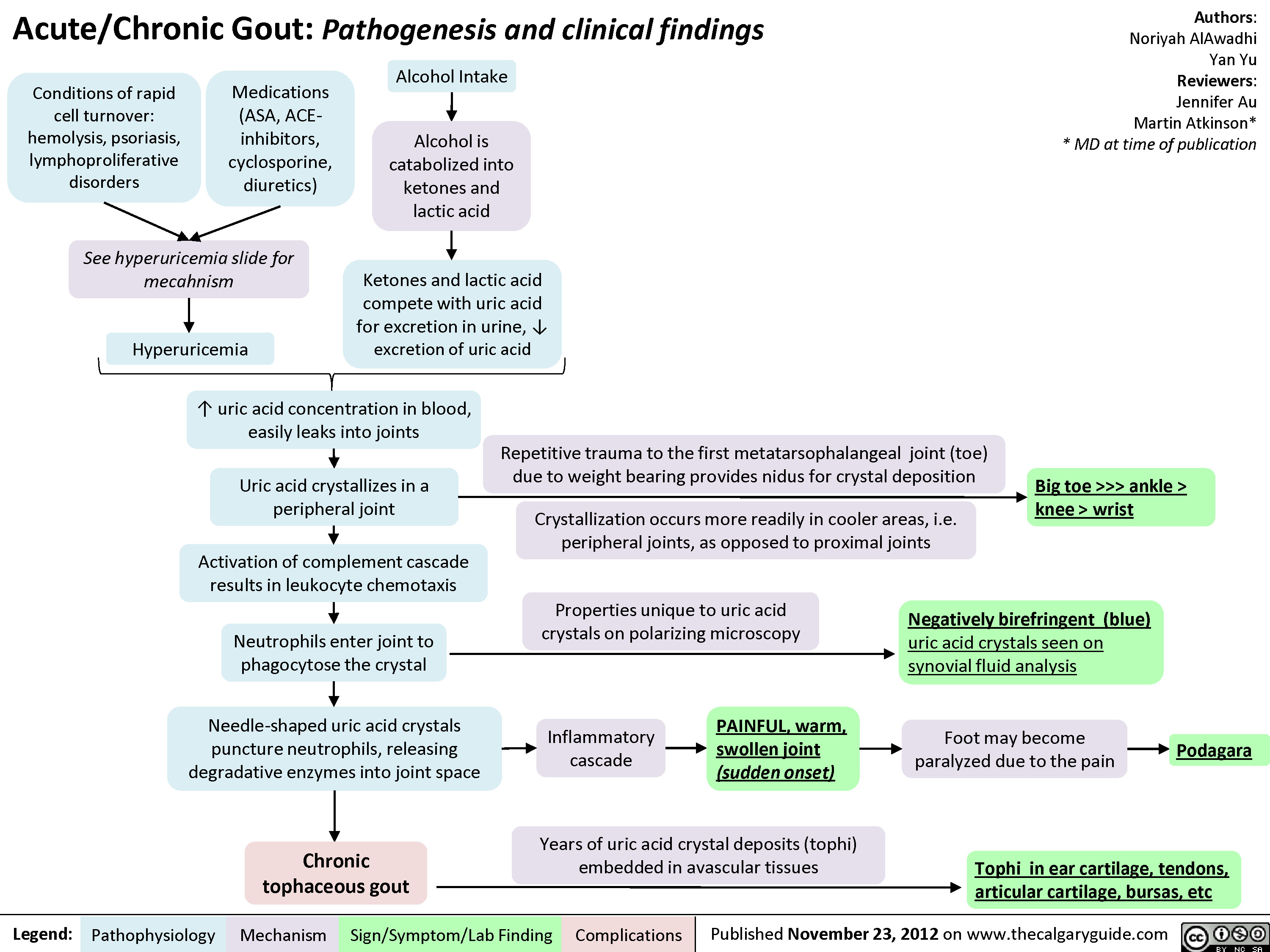
Reactive Arthritis
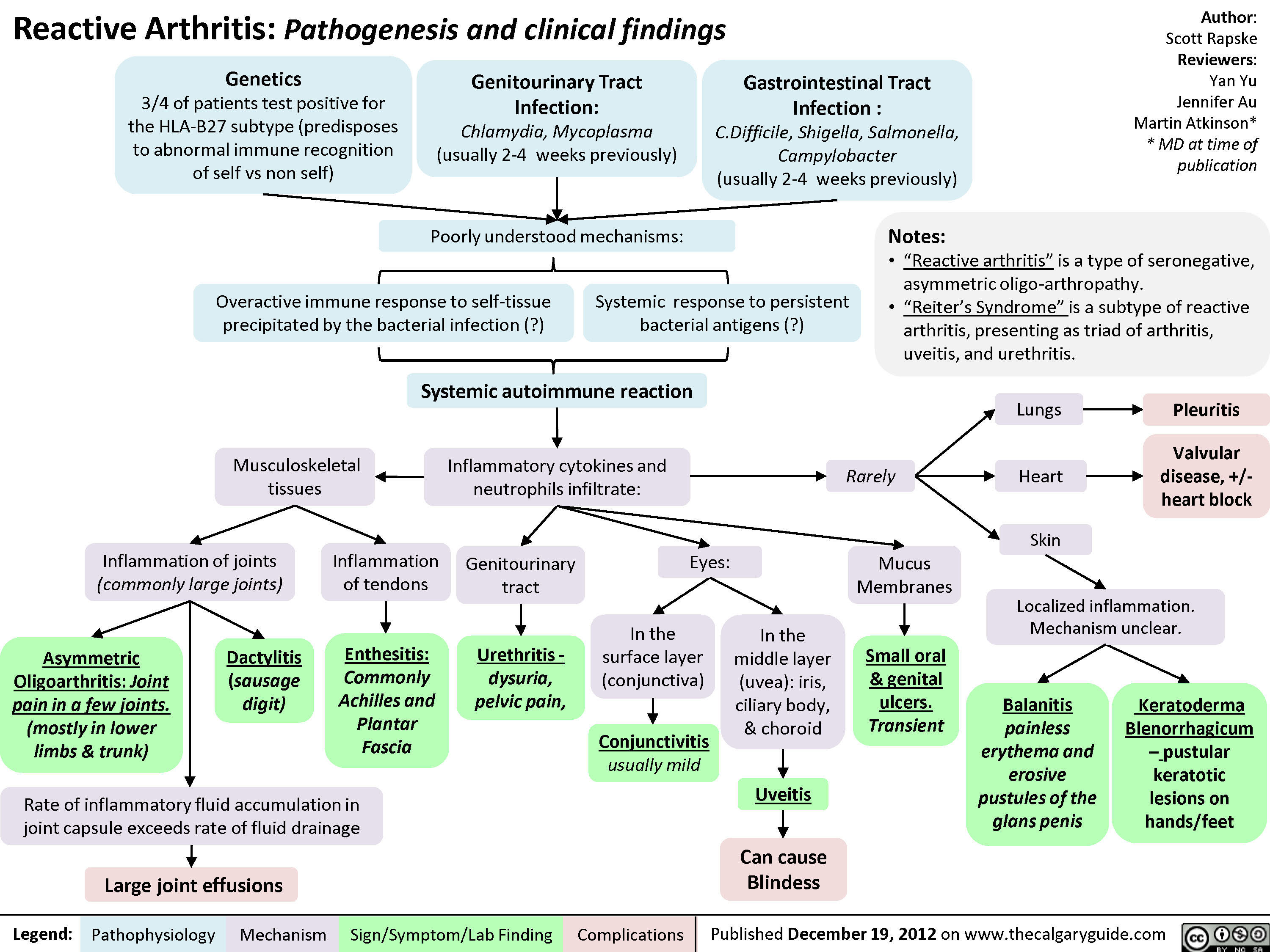
Psoriatic Arthritis: Complications
Psoriatic Arthritis - Pathogenesis and Clinical findings

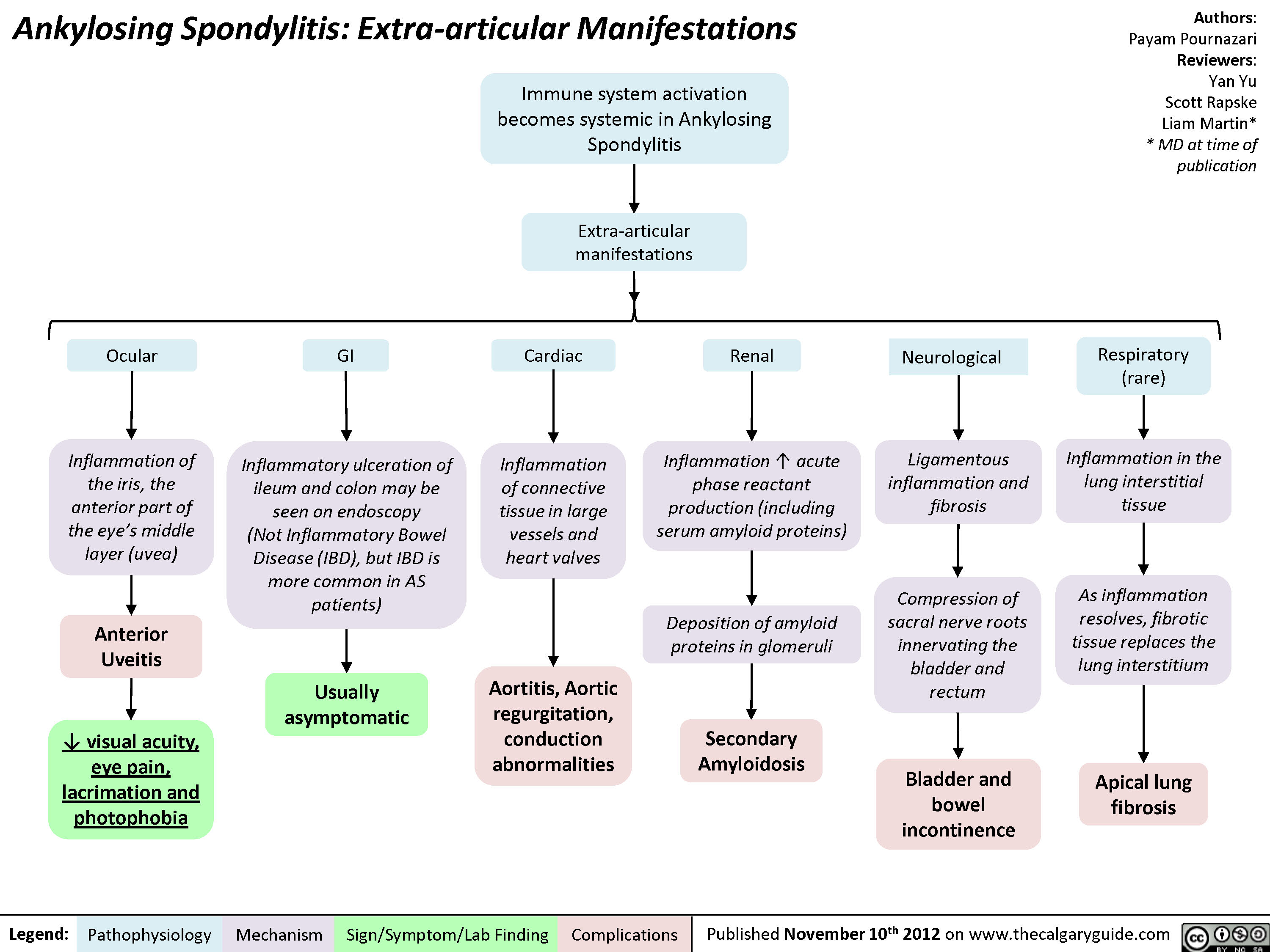
Ankylosing Spondylitis: Extra-articular Manifestations
Ankylosing Spondylitis: Pathogenesis and Clinical findings
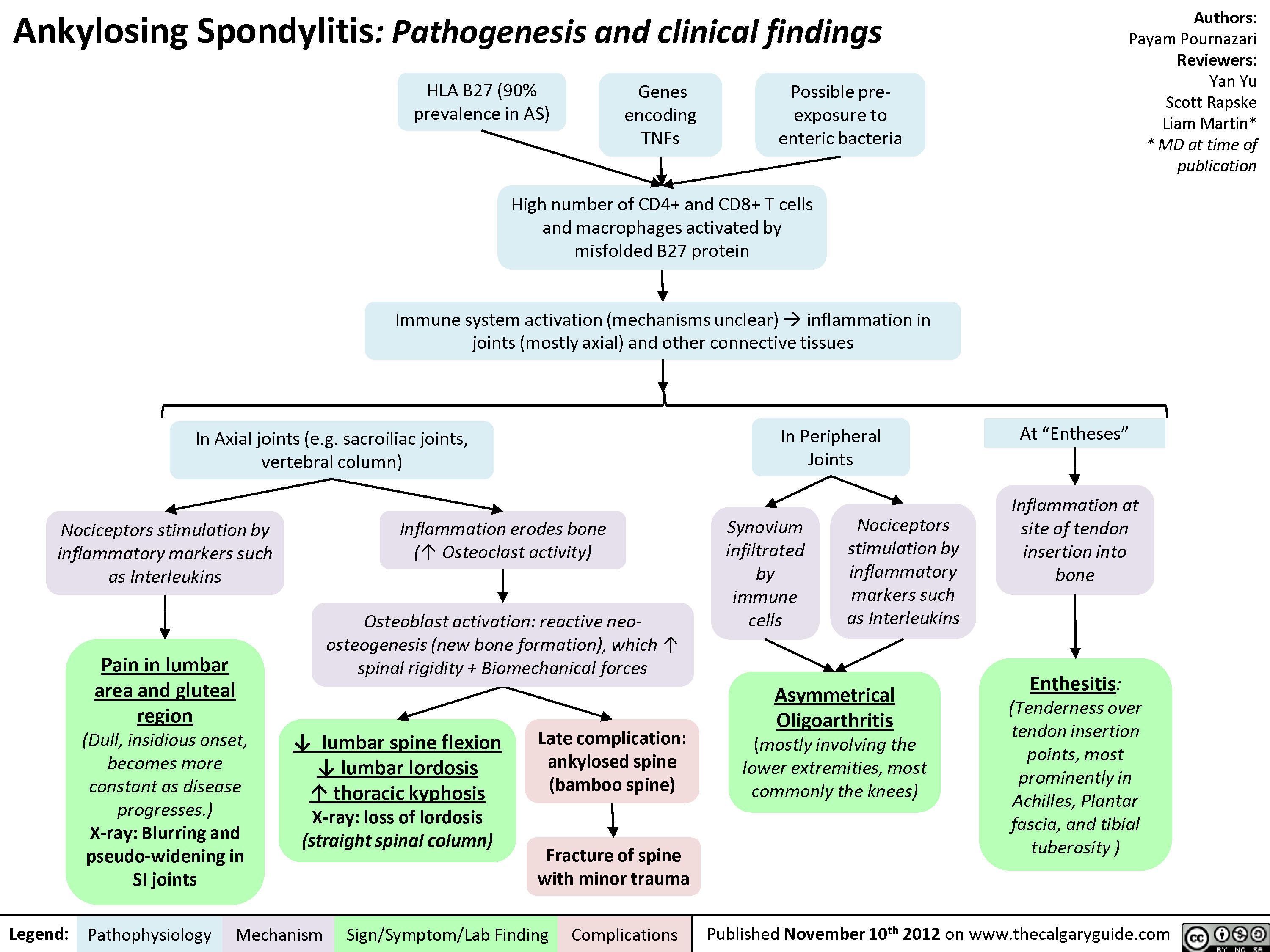
Lupus: Muco-cutaneous manifestations
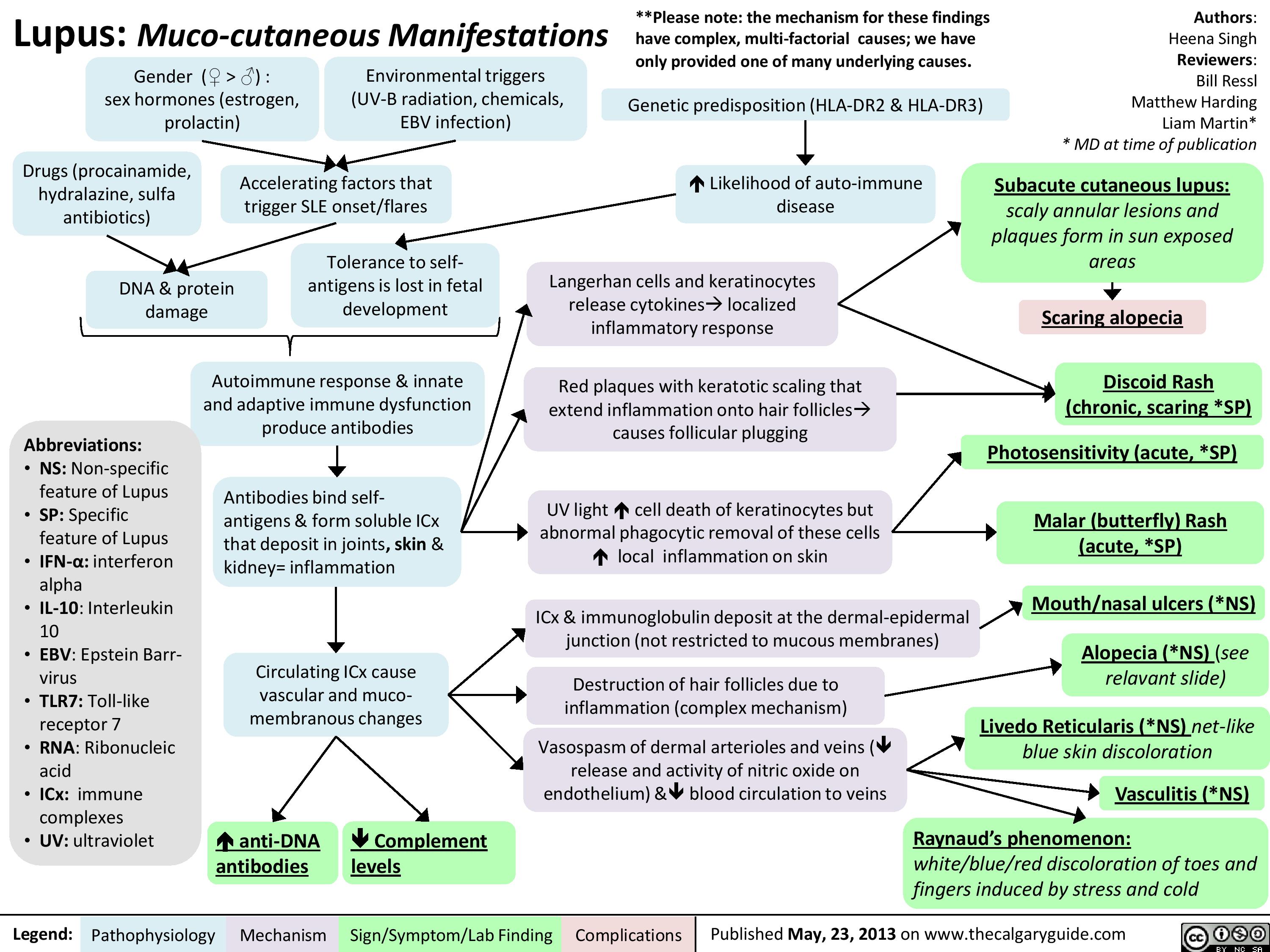
Pathogenesis of Lupus
Rheumatoid arthritis (RA): X-ray features
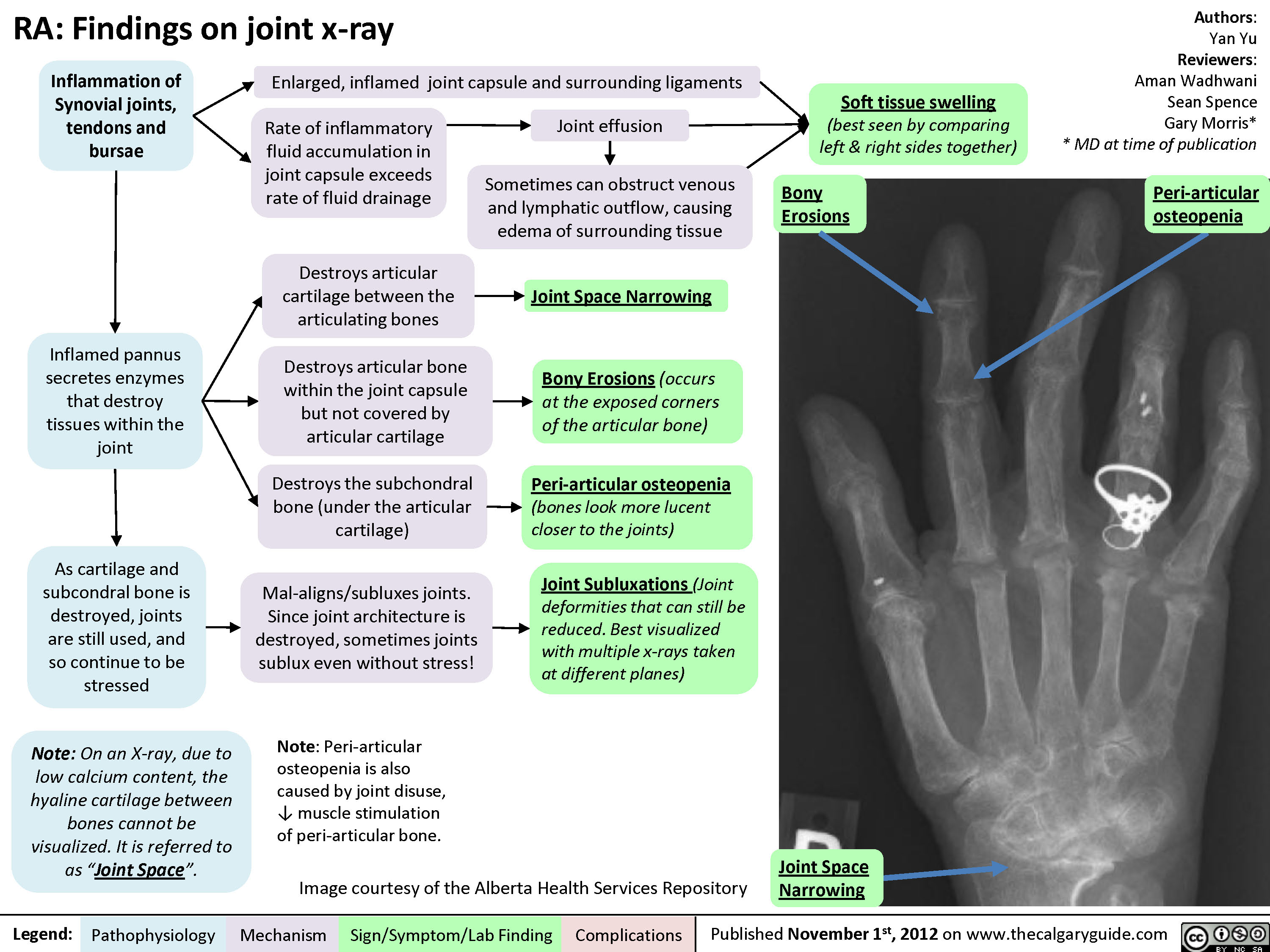
Rheumatoid arthritis (RA): Extra-articular manifestations
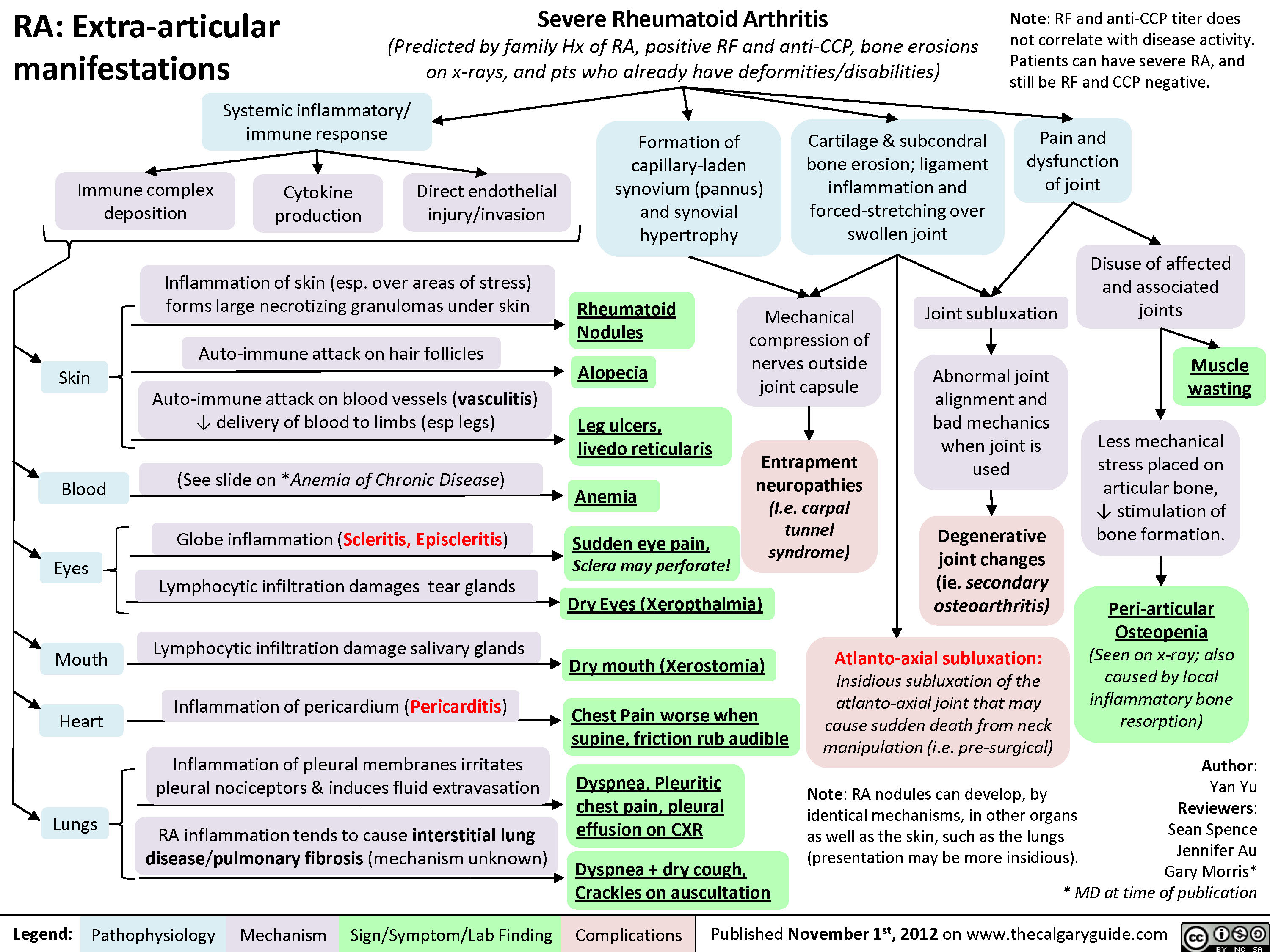
Rheumatoid arthritis (RA): Pathogenesis and Joint diseases features
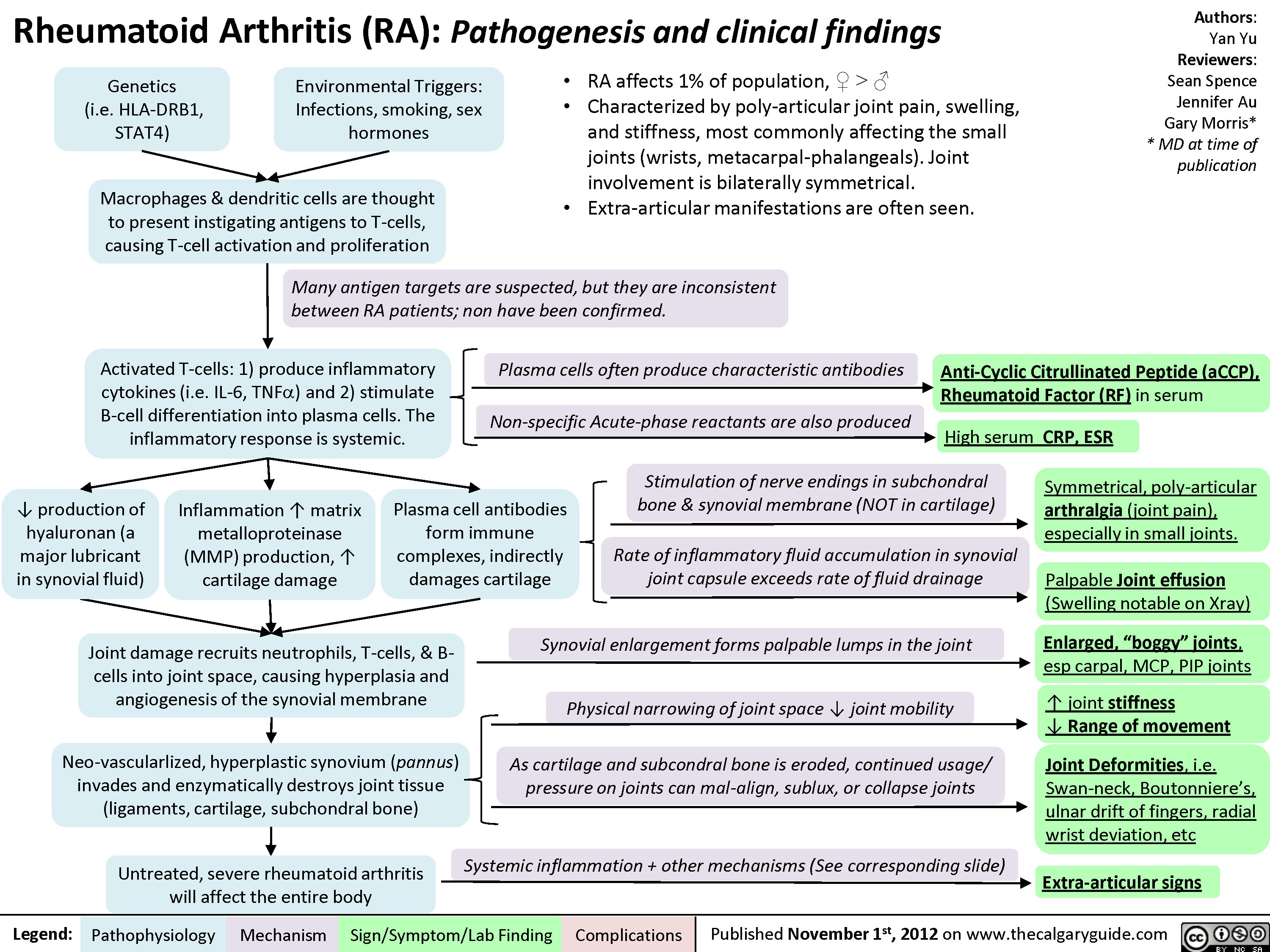
Charcot Joint: Pathogenesis and Clinical findings
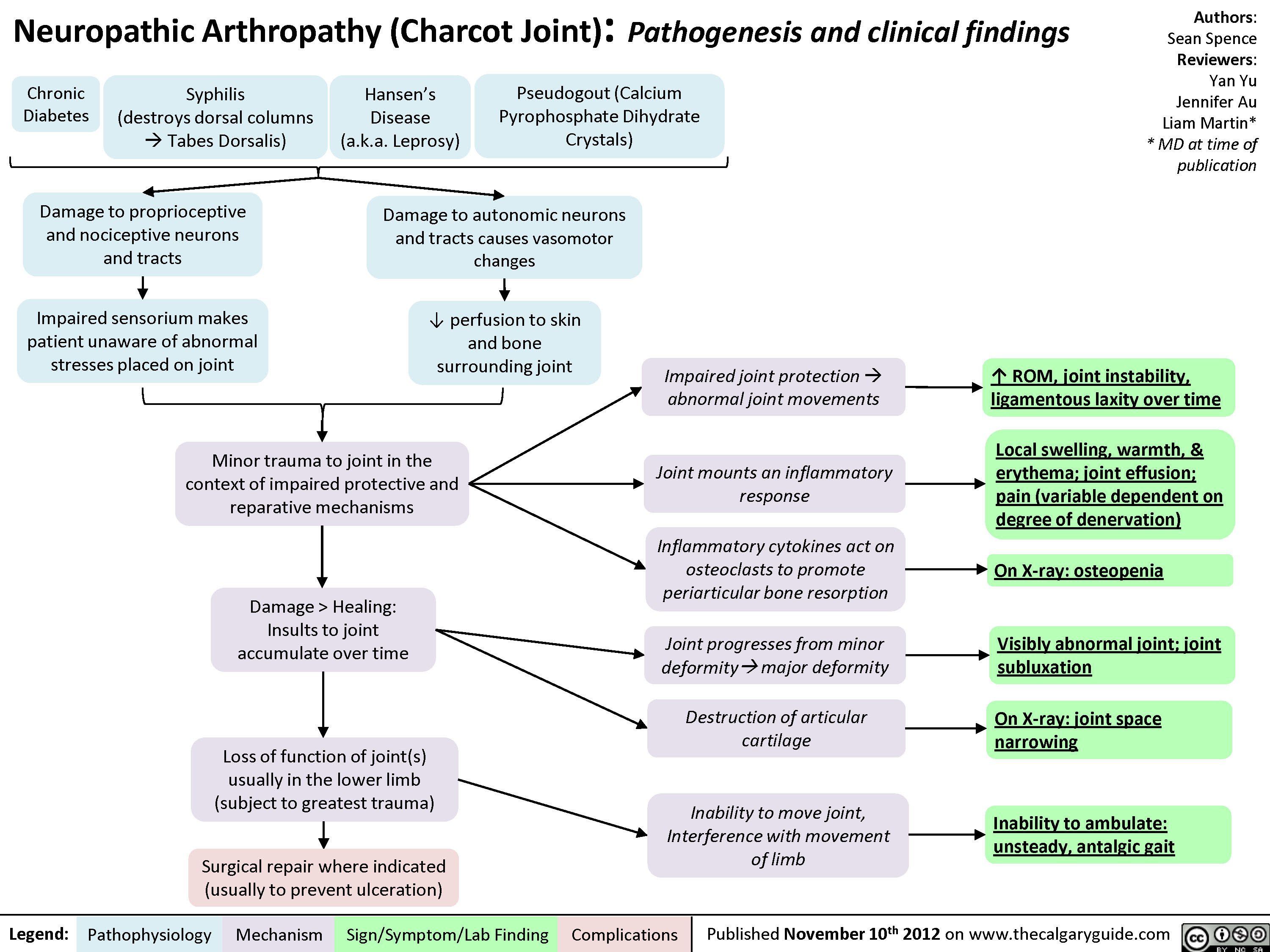
Osteoarthritis (OA): X-ray features
Degenerative Vs Inflammatory Joint Disease
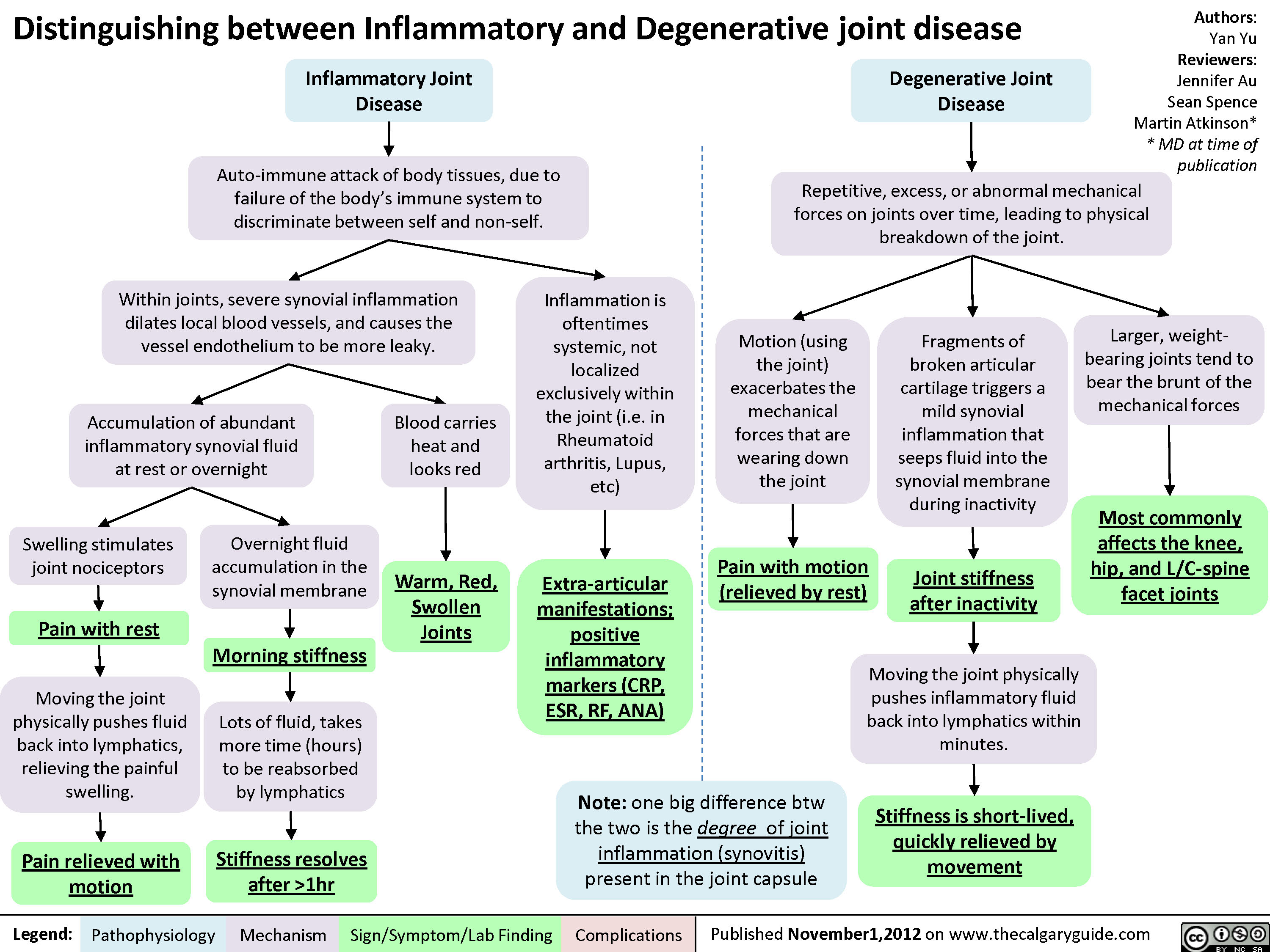
Hypersensitivity: Definitions
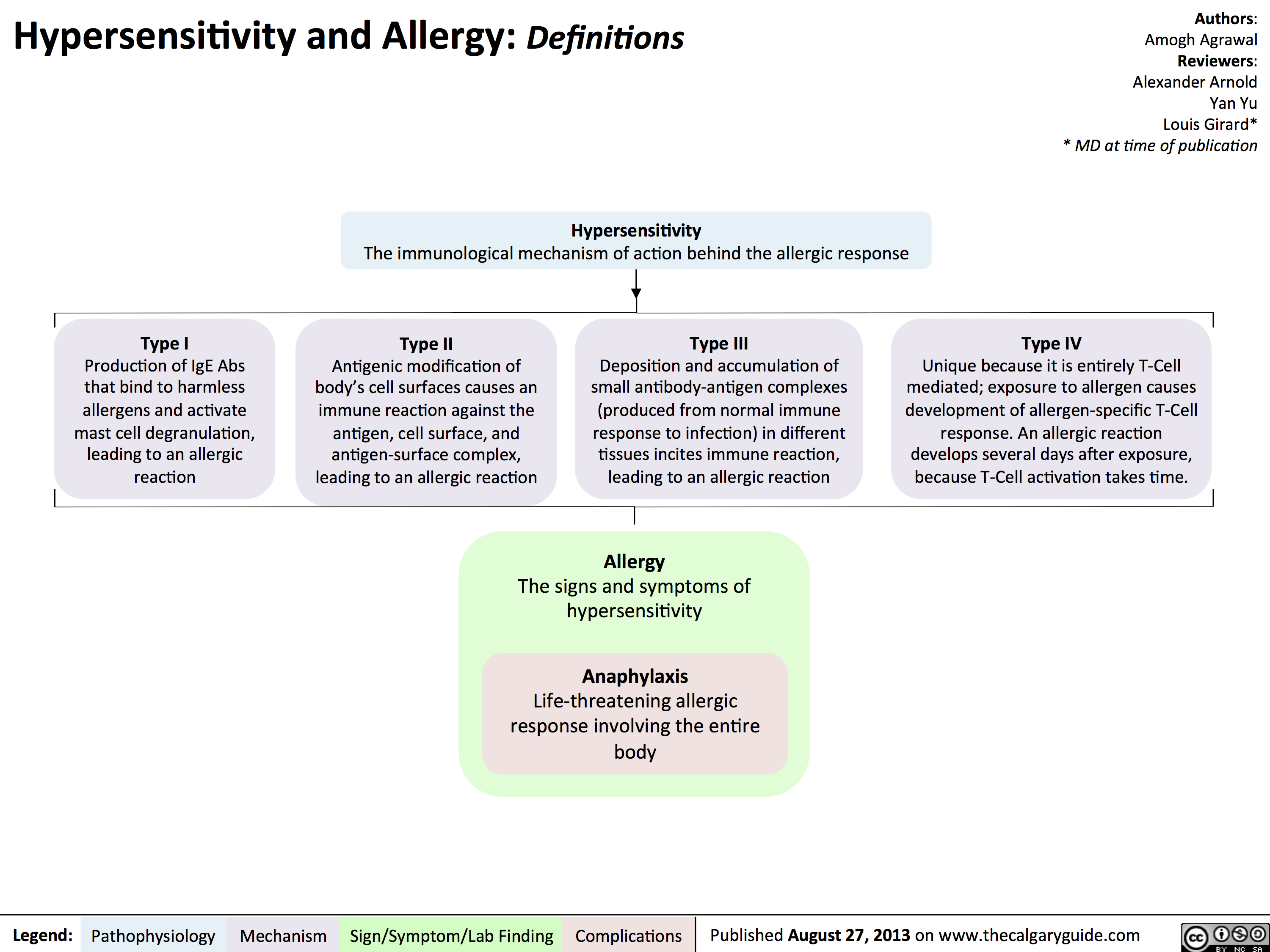
Type I Hypersensitivity: Pathogenesis and clinical findings
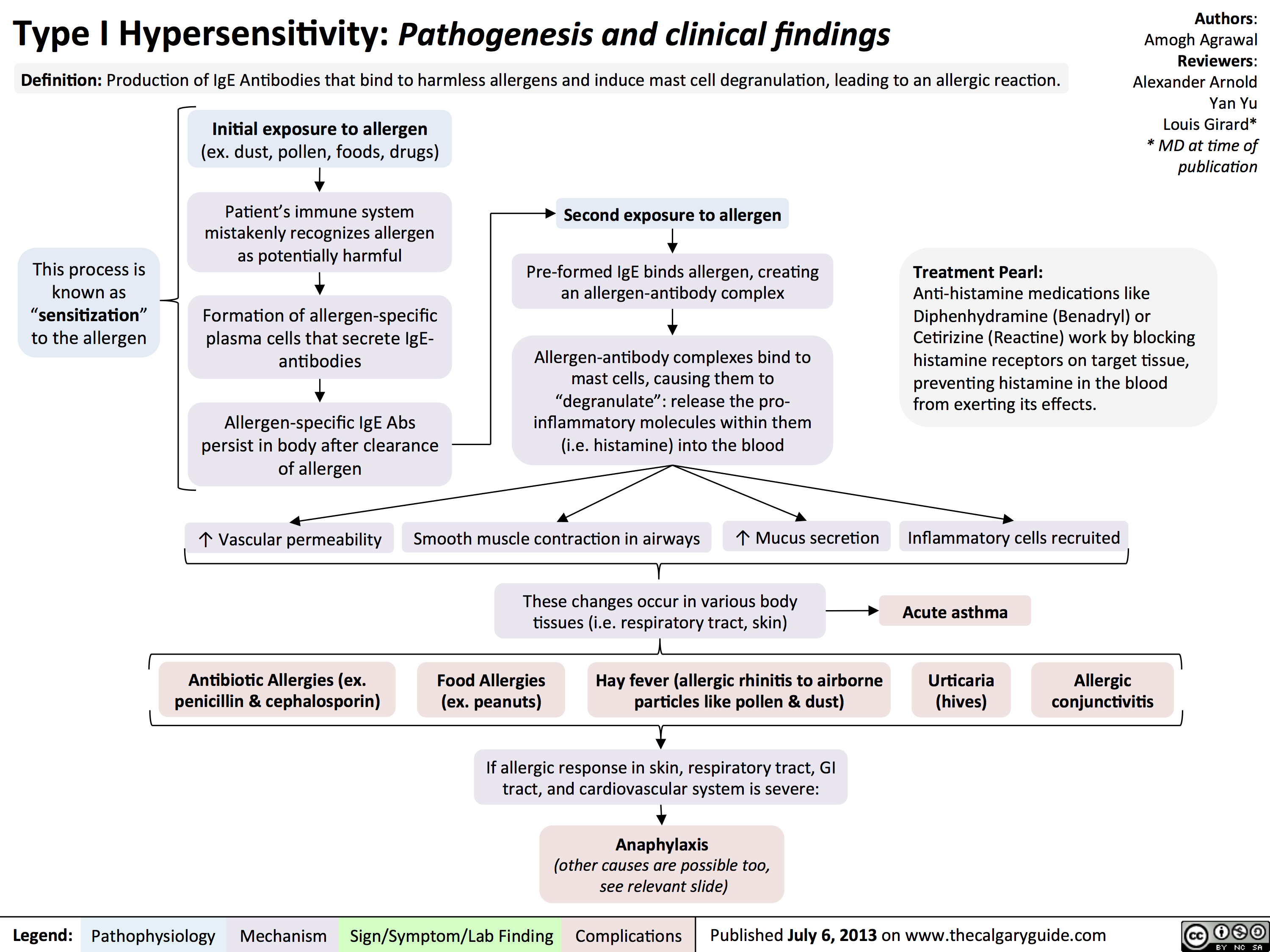
Type II Hypersensitivity: Pathogenesis and clinical findings
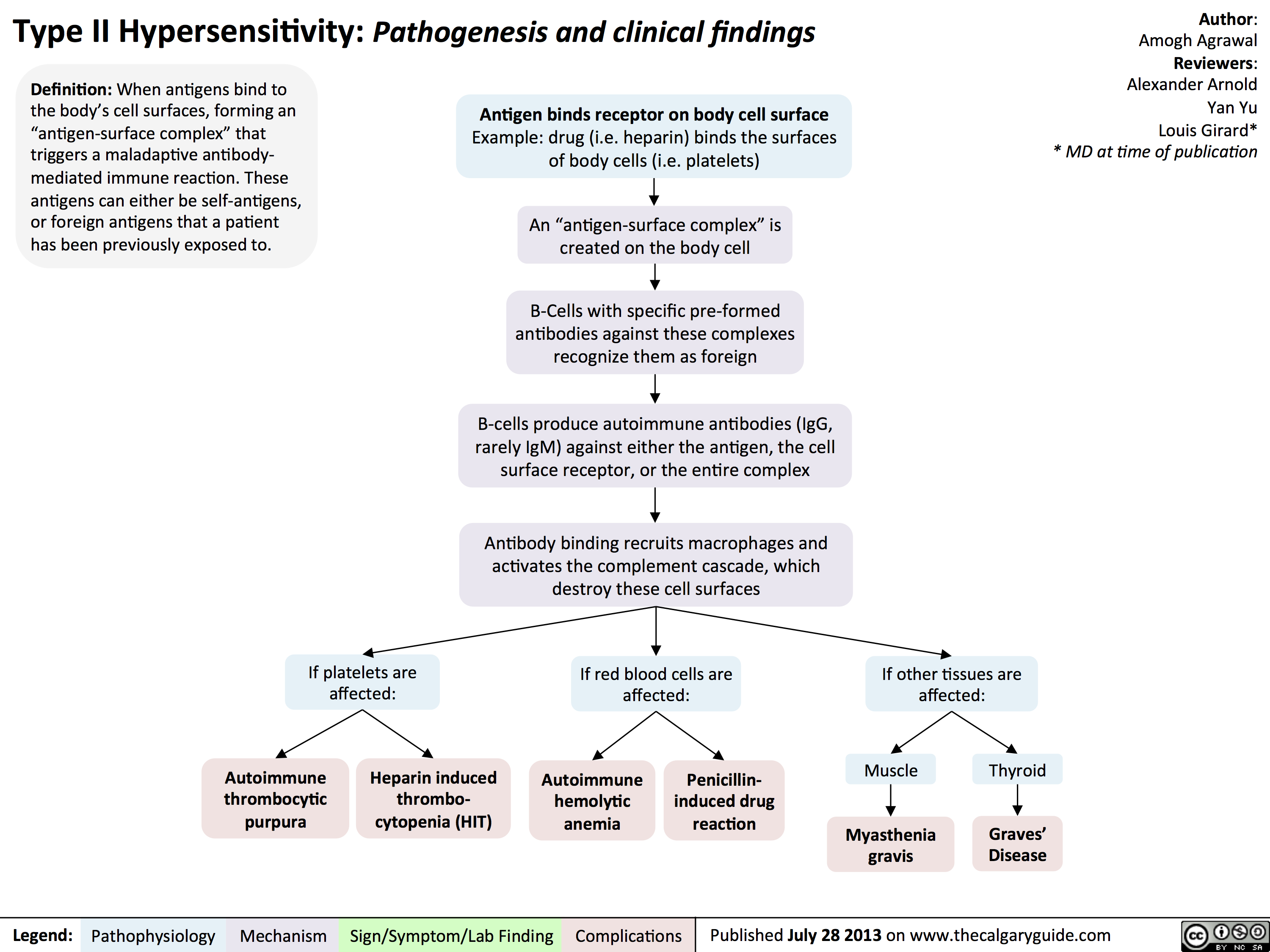
Type III Hypersensitivity: Pathogenesis and clinical findings
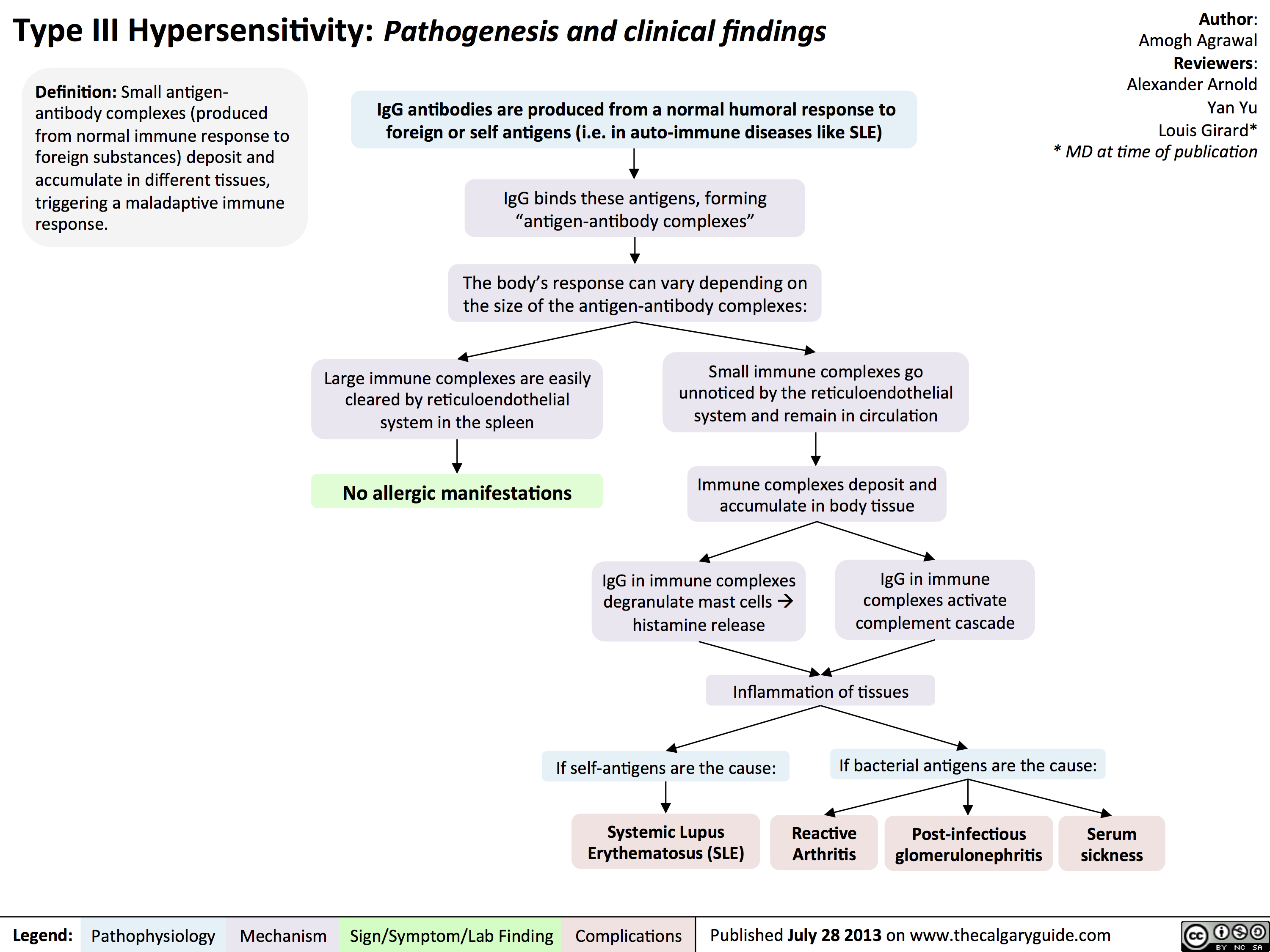
Type IV Hypersensitivity: Pathogenesis and clinical findings
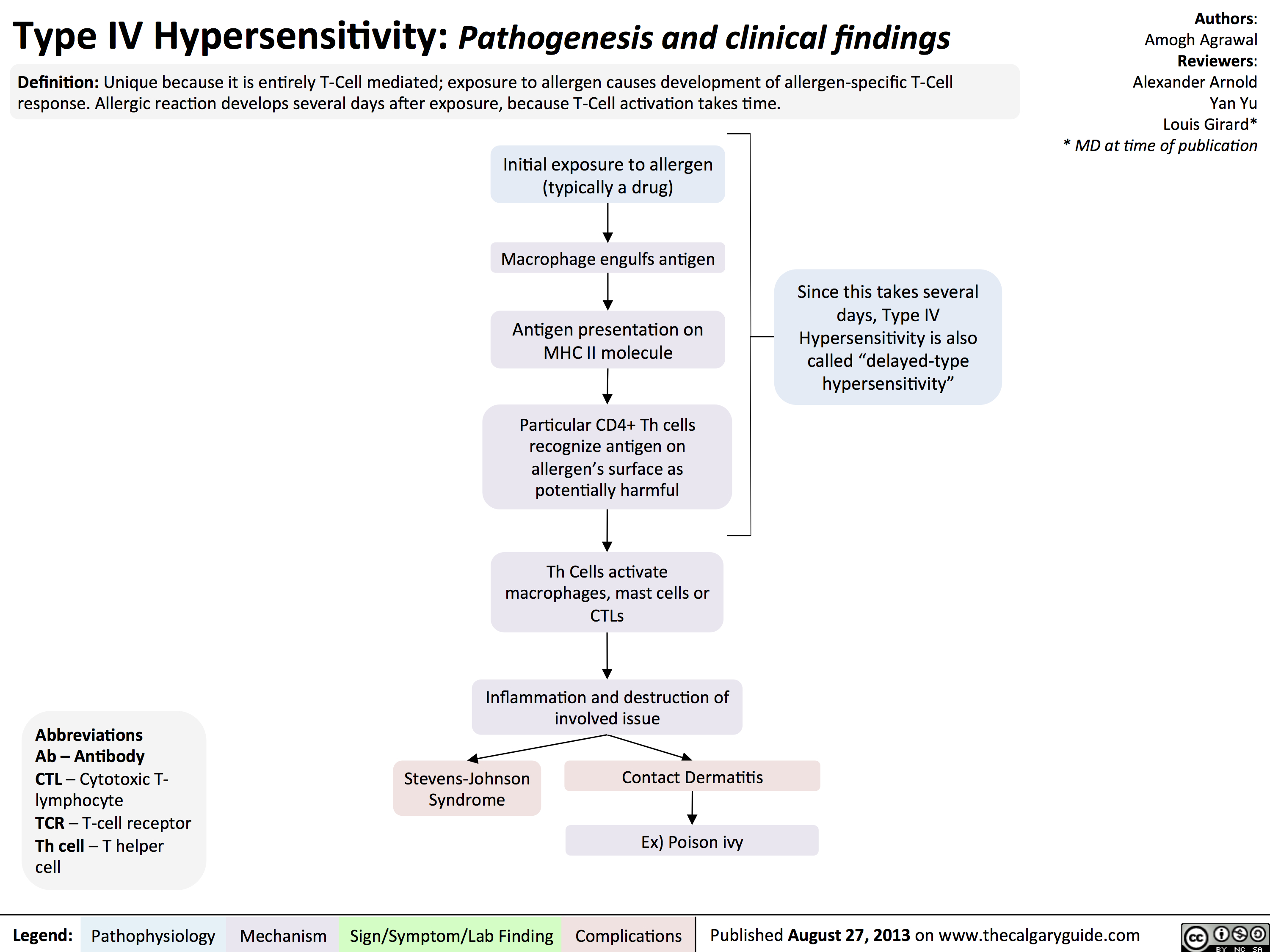
Hypersensitivity Summary
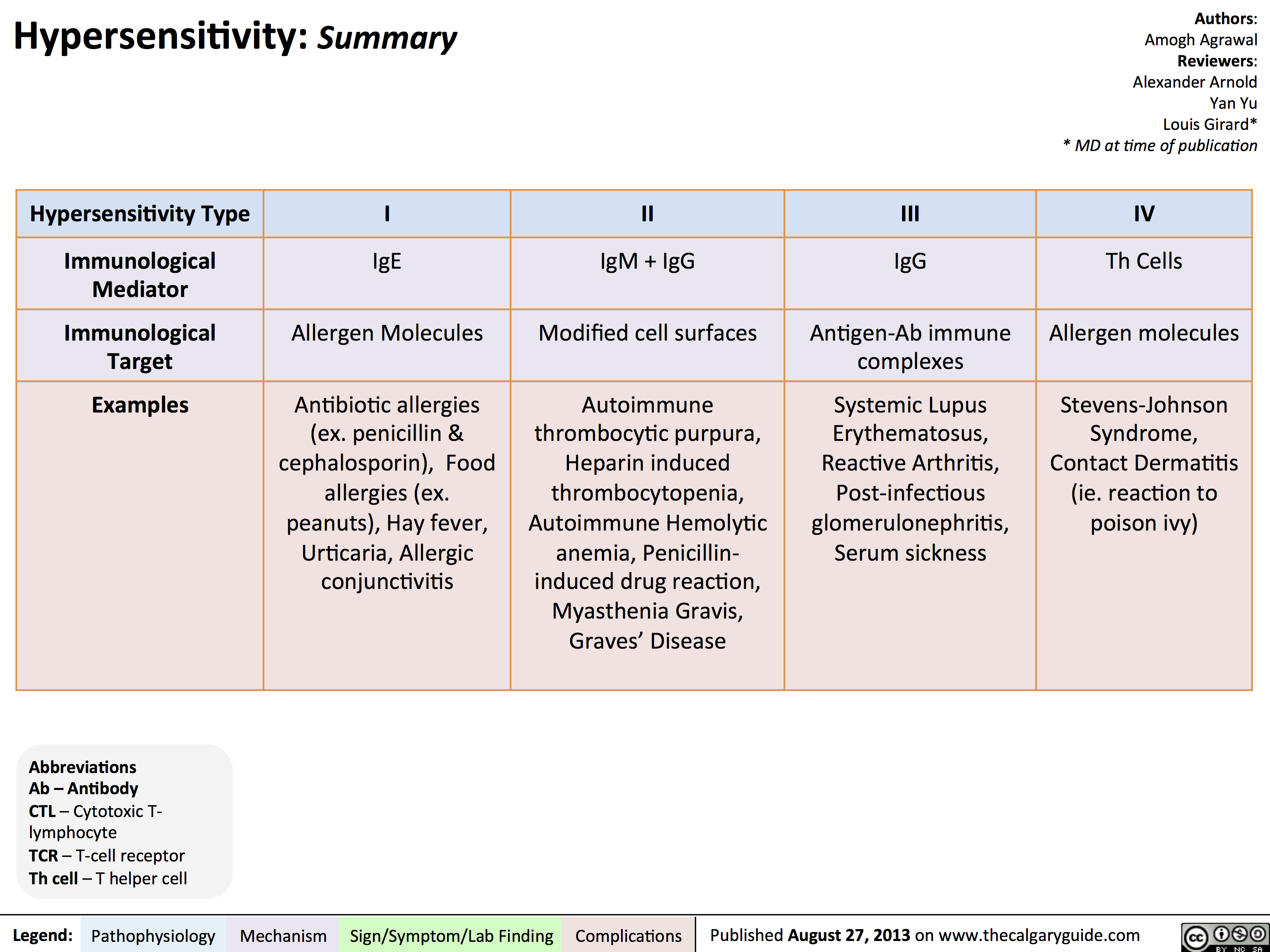
Agammaglobulinemia: Pathogenesis and clinical findings
Endometriosis: Pathogenesis and Complications
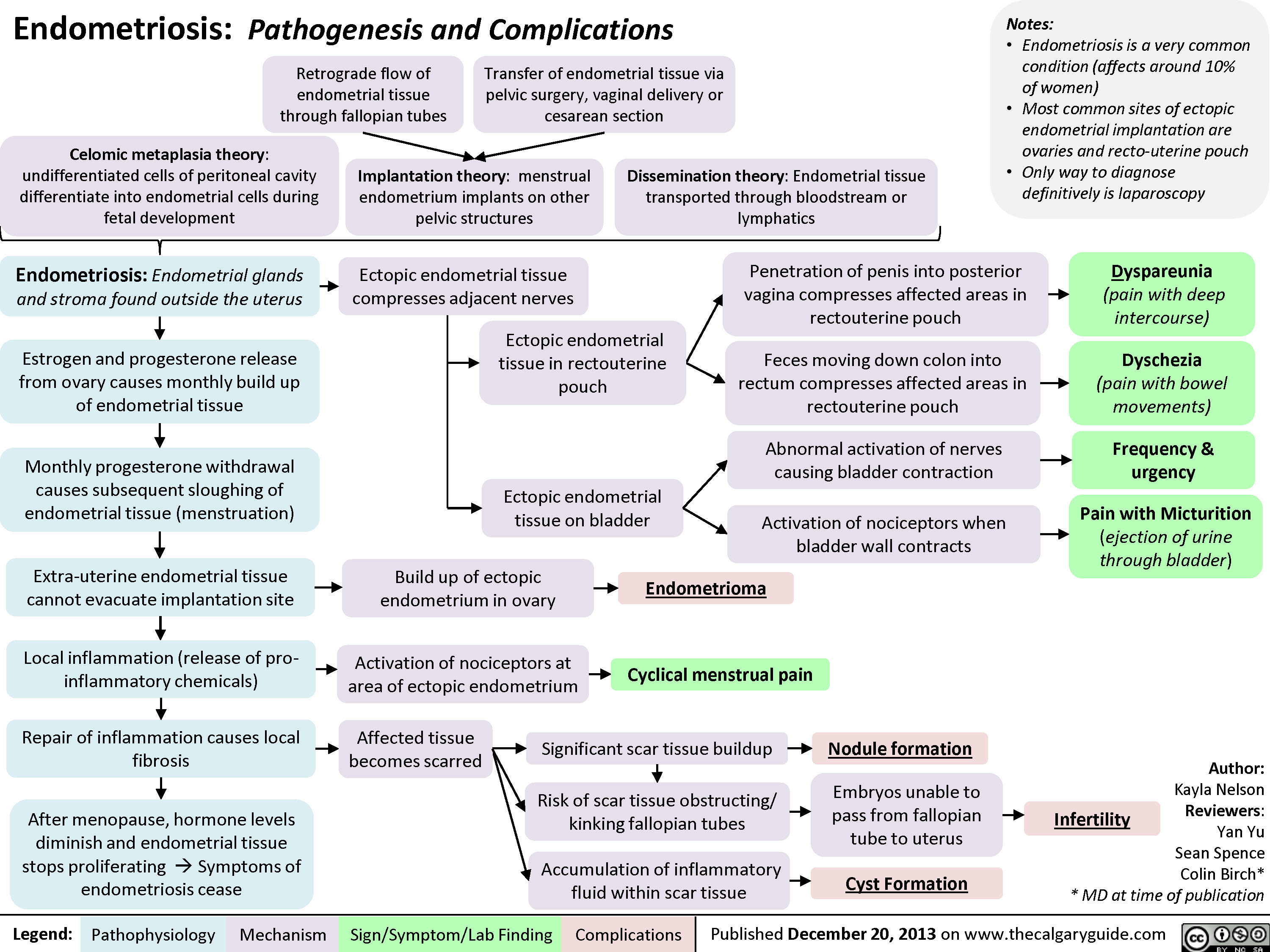
Physiology of the Renin-Angiotensin-Aldosterone System (RAAS)
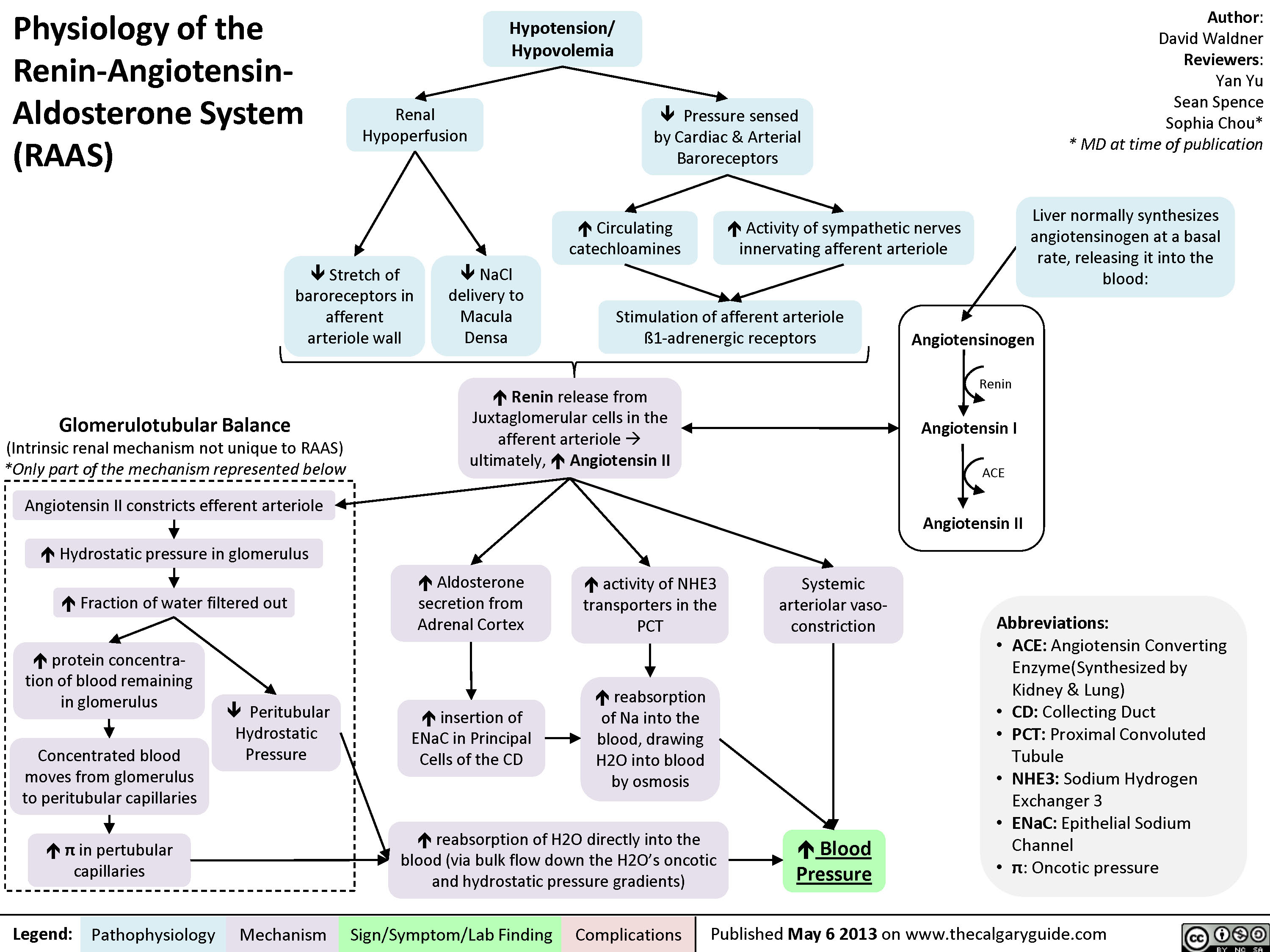
Hypokalemia: Clinical Findings
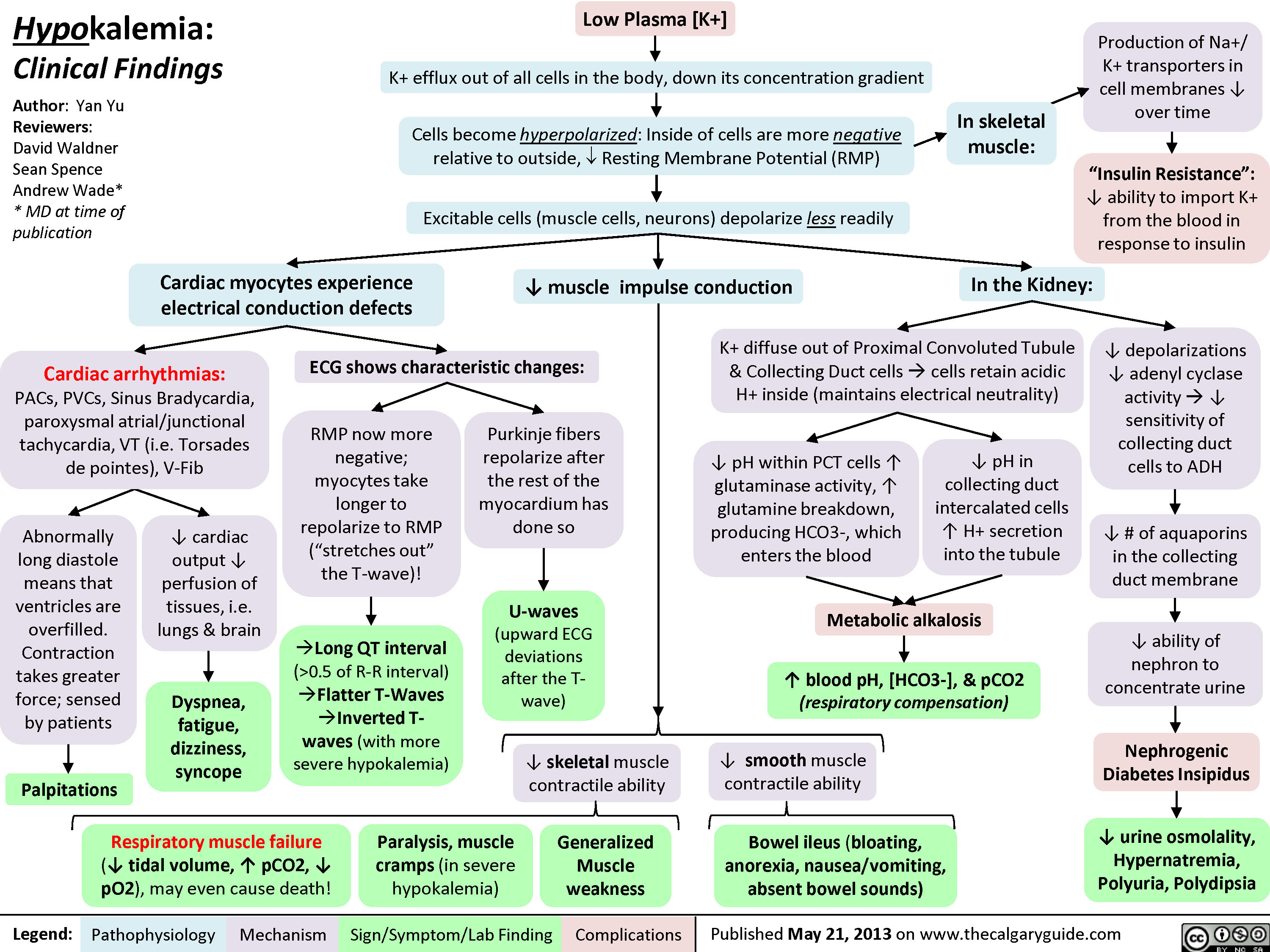 0.5 of R-R interval)?Flatter T-Waves ?Inverted T-waves (with more severe hypokalemia)Purkinje fibers repolarize after the rest of the myocardium has done soU-waves (upward ECG deviations after the T-wave)Cells become hyperpolarized: Inside of cells are more negative relative to outside, ? Resting Membrane Potential (RMP)In the Kidney:Generalized Muscle weaknessK+ diffuse out of Proximal Convoluted Tubule & Collecting Duct cells ? cells retain acidic H+ inside (maintains electrical neutrality)? pH within PCT cells ? glutaminase activity, ? glutamine breakdown, producing HCO3-, which enters the blood? blood pH, [HCO3-], & pCO2 (respiratory compensation)Low Plasma [K+]Abnormally long diastole means that ventricles are overfilled. Contraction takes greater force; sensed by patientsDyspnea, fatigue, dizziness, syncope? cardiac output ? perfusion of tissues, i.e. lungs & brainCardiac arrhythmias: PACs, PVCs, Sinus Bradycardia, paroxysmal atrial/junctional tachycardia, VT (i.e. Torsades de pointes), V-Fib? smooth muscle contractile abilityBowel ileus (bloating, anorexia, nausea/vomiting, absent bowel sounds)? pH in collecting duct intercalated cells ? H+ secretion into the tubuleMetabolic alkalosisParalysis, muscle cramps (in severe hypokalemia)Respiratory muscle failure (? tidal volume, ? pCO2, ? pO2), may even cause death!? depolarizations ? adenyl cyclase activity ? ? sensitivity of collecting duct cells to ADH? ability of nephron to concentrate urineNephrogenic Diabetes Insipidus? urine osmolality, Hypernatremia, Polyuria, Polydipsia? # of aquaporins in the collecting duct membrane"Insulin Resistance": ? ability to import K+ from the blood in response to insulinIn skeletal muscle:
117 kB / 307 word" title="Yu, Yan - Hypokalemia clinical findings - FINAL.pptx
Production of Na+/ K+ transporters in cell membranes ? over timeHypokalemia: Clinical FindingsAuthor: Yan YuReviewers:David WaldnerSean SpenceAndrew Wade** MD at time of publicationLegend:Published May 21, 2013 on www.thecalgaryguide.comMechanismPathophysiologySign/Symptom/Lab FindingComplicationsPalpitationsExcitable cells (muscle cells, neurons) depolarize less readilyK+ efflux out of all cells in the body, down its concentration gradientCardiac myocytes experience electrical conduction defects? muscle impulse conductionECG shows characteristic changes:? skeletal muscle contractile abilityRMP now more negative; myocytes take longer to repolarize to RMP("stretches out" the T-wave)! Long QT interval (>0.5 of R-R interval)?Flatter T-Waves ?Inverted T-waves (with more severe hypokalemia)Purkinje fibers repolarize after the rest of the myocardium has done soU-waves (upward ECG deviations after the T-wave)Cells become hyperpolarized: Inside of cells are more negative relative to outside, ? Resting Membrane Potential (RMP)In the Kidney:Generalized Muscle weaknessK+ diffuse out of Proximal Convoluted Tubule & Collecting Duct cells ? cells retain acidic H+ inside (maintains electrical neutrality)? pH within PCT cells ? glutaminase activity, ? glutamine breakdown, producing HCO3-, which enters the blood? blood pH, [HCO3-], & pCO2 (respiratory compensation)Low Plasma [K+]Abnormally long diastole means that ventricles are overfilled. Contraction takes greater force; sensed by patientsDyspnea, fatigue, dizziness, syncope? cardiac output ? perfusion of tissues, i.e. lungs & brainCardiac arrhythmias: PACs, PVCs, Sinus Bradycardia, paroxysmal atrial/junctional tachycardia, VT (i.e. Torsades de pointes), V-Fib? smooth muscle contractile abilityBowel ileus (bloating, anorexia, nausea/vomiting, absent bowel sounds)? pH in collecting duct intercalated cells ? H+ secretion into the tubuleMetabolic alkalosisParalysis, muscle cramps (in severe hypokalemia)Respiratory muscle failure (? tidal volume, ? pCO2, ? pO2), may even cause death!? depolarizations ? adenyl cyclase activity ? ? sensitivity of collecting duct cells to ADH? ability of nephron to concentrate urineNephrogenic Diabetes Insipidus? urine osmolality, Hypernatremia, Polyuria, Polydipsia? # of aquaporins in the collecting duct membrane"Insulin Resistance": ? ability to import K+ from the blood in response to insulinIn skeletal muscle:
117 kB / 307 word" />
0.5 of R-R interval)?Flatter T-Waves ?Inverted T-waves (with more severe hypokalemia)Purkinje fibers repolarize after the rest of the myocardium has done soU-waves (upward ECG deviations after the T-wave)Cells become hyperpolarized: Inside of cells are more negative relative to outside, ? Resting Membrane Potential (RMP)In the Kidney:Generalized Muscle weaknessK+ diffuse out of Proximal Convoluted Tubule & Collecting Duct cells ? cells retain acidic H+ inside (maintains electrical neutrality)? pH within PCT cells ? glutaminase activity, ? glutamine breakdown, producing HCO3-, which enters the blood? blood pH, [HCO3-], & pCO2 (respiratory compensation)Low Plasma [K+]Abnormally long diastole means that ventricles are overfilled. Contraction takes greater force; sensed by patientsDyspnea, fatigue, dizziness, syncope? cardiac output ? perfusion of tissues, i.e. lungs & brainCardiac arrhythmias: PACs, PVCs, Sinus Bradycardia, paroxysmal atrial/junctional tachycardia, VT (i.e. Torsades de pointes), V-Fib? smooth muscle contractile abilityBowel ileus (bloating, anorexia, nausea/vomiting, absent bowel sounds)? pH in collecting duct intercalated cells ? H+ secretion into the tubuleMetabolic alkalosisParalysis, muscle cramps (in severe hypokalemia)Respiratory muscle failure (? tidal volume, ? pCO2, ? pO2), may even cause death!? depolarizations ? adenyl cyclase activity ? ? sensitivity of collecting duct cells to ADH? ability of nephron to concentrate urineNephrogenic Diabetes Insipidus? urine osmolality, Hypernatremia, Polyuria, Polydipsia? # of aquaporins in the collecting duct membrane"Insulin Resistance": ? ability to import K+ from the blood in response to insulinIn skeletal muscle:
117 kB / 307 word" title="Yu, Yan - Hypokalemia clinical findings - FINAL.pptx
Production of Na+/ K+ transporters in cell membranes ? over timeHypokalemia: Clinical FindingsAuthor: Yan YuReviewers:David WaldnerSean SpenceAndrew Wade** MD at time of publicationLegend:Published May 21, 2013 on www.thecalgaryguide.comMechanismPathophysiologySign/Symptom/Lab FindingComplicationsPalpitationsExcitable cells (muscle cells, neurons) depolarize less readilyK+ efflux out of all cells in the body, down its concentration gradientCardiac myocytes experience electrical conduction defects? muscle impulse conductionECG shows characteristic changes:? skeletal muscle contractile abilityRMP now more negative; myocytes take longer to repolarize to RMP("stretches out" the T-wave)! Long QT interval (>0.5 of R-R interval)?Flatter T-Waves ?Inverted T-waves (with more severe hypokalemia)Purkinje fibers repolarize after the rest of the myocardium has done soU-waves (upward ECG deviations after the T-wave)Cells become hyperpolarized: Inside of cells are more negative relative to outside, ? Resting Membrane Potential (RMP)In the Kidney:Generalized Muscle weaknessK+ diffuse out of Proximal Convoluted Tubule & Collecting Duct cells ? cells retain acidic H+ inside (maintains electrical neutrality)? pH within PCT cells ? glutaminase activity, ? glutamine breakdown, producing HCO3-, which enters the blood? blood pH, [HCO3-], & pCO2 (respiratory compensation)Low Plasma [K+]Abnormally long diastole means that ventricles are overfilled. Contraction takes greater force; sensed by patientsDyspnea, fatigue, dizziness, syncope? cardiac output ? perfusion of tissues, i.e. lungs & brainCardiac arrhythmias: PACs, PVCs, Sinus Bradycardia, paroxysmal atrial/junctional tachycardia, VT (i.e. Torsades de pointes), V-Fib? smooth muscle contractile abilityBowel ileus (bloating, anorexia, nausea/vomiting, absent bowel sounds)? pH in collecting duct intercalated cells ? H+ secretion into the tubuleMetabolic alkalosisParalysis, muscle cramps (in severe hypokalemia)Respiratory muscle failure (? tidal volume, ? pCO2, ? pO2), may even cause death!? depolarizations ? adenyl cyclase activity ? ? sensitivity of collecting duct cells to ADH? ability of nephron to concentrate urineNephrogenic Diabetes Insipidus? urine osmolality, Hypernatremia, Polyuria, Polydipsia? # of aquaporins in the collecting duct membrane"Insulin Resistance": ? ability to import K+ from the blood in response to insulinIn skeletal muscle:
117 kB / 307 word" />
Hyperkalemia: Clinical Findings
![Yu, Yan - Hyperkalemia clinical findings - Published.pptx
Hyperkalemia: Clinical FindingsAuthor: Yan YuReviewers:Alexander ArnoldDavid WaldnerSean SpenceAndrew Wade** MD at time of publicationLegend:Published September 9, 2013 on www.thecalgaryguide.comMechanismPathophysiologySign/Symptom/Lab FindingComplicationsPalpitationsNotes: Symptoms usually manifest when plasma [K+] > 7.0 mmol/L, but can occur at lower [K+]s when hyperkalemia is acute.ECG changes can, but don't necessarily, correlate with a particular [K+].Initially: Excitable cells (muscle cells, neurons) undergo action potentials more readily? [K+ ] gradient between cells and the blood (K+ tends to stay inside cells, less K+ diffuses out)In the Heart:In Skeletal Muscle:[K+] >5.5 mmol/L :faster myocardial repolarization(](http://calgaryguide.ucalgary.ca/wp-content/uploads/2014/09/hyperkalemiaclinicalfindings.jpg) 6.5 mmol/L:? atrial conduction; slow signal transmission from SA to AV nodeCells become slightly depolarized: Resting Membrane Potential (RMP) is brought closer to thresholdIn the Kidney:Muscle weakness and even paralysis (respiratory muscle weakness is rare)? reabsorption of Na+ from Cortical Collecting Duct (CCD)CCD lumen remains more positively chargedMetabolic Acidosis(normal anion gap)Over time (when patients become symptomatic): Chronic membrane depolarization desensitizes voltage-gated Na+ channels (slows their opening) ? ? membrane excitability ? ? action potential generation[K+] > 7.0 mmol/L:? ventricular conductionBradycardiaProlonged, abnormal QRSAV blocks[K+] > 9.0 mmol/L:more conduction abnormalitiesPEA with bizarre wide-QRS rhythmV-fibAsystole? urinary H+ secretion by alpha-intercalated cellsHIGH Plasma [K+] (potassium ion concentration)Dyspnea, fatigue, dizziness, syncope? cardiac output ? ? perfusion of tissues, i.e. lungs & brainCardiac arrhythmias: Conduction blocks (AV block, Bundle branch blocks), VT , V-Fib, Bradycardia, Asystole.?? PR interval ?P-wave flattens, eventually disappearsIf severe, QRS & T-waves fuse:Sine-WavesThe higher the [K+], the slower the voltage-gated Na+ channels open, reflected by distinctive ECG changes:If the K+ is due to ? aldosterone effect ? principal cell dysfunctionHigh pH ? glutamate deamination, which normally produces NH4+? NH4+ reaches the thick ascending limb to be converted to NH3Less NH3 diffuses into the collecting duct to be converted to NH4+ through binding with H+ ? ? NH4+ and therefore ? H+ is excretedK+ moves into proximal tubule cells, causing H+ to diffuse out ? Intracellular alkalosis Irregular force and rhythm of cardiac muscle contraction is sensed by the patient? contraction impulse is conductedDefective electrical conduction through cardiac myocytesMore acid (H=) is retained in the body
118 kB / 357 words" title="Yu, Yan - Hyperkalemia clinical findings - Published.pptx
Hyperkalemia: Clinical FindingsAuthor: Yan YuReviewers:Alexander ArnoldDavid WaldnerSean SpenceAndrew Wade** MD at time of publicationLegend:Published September 9, 2013 on www.thecalgaryguide.comMechanismPathophysiologySign/Symptom/Lab FindingComplicationsPalpitationsNotes: Symptoms usually manifest when plasma [K+] > 7.0 mmol/L, but can occur at lower [K+]s when hyperkalemia is acute.ECG changes can, but don't necessarily, correlate with a particular [K+].Initially: Excitable cells (muscle cells, neurons) undergo action potentials more readily? [K+ ] gradient between cells and the blood (K+ tends to stay inside cells, less K+ diffuses out)In the Heart:In Skeletal Muscle:[K+] >5.5 mmol/L :faster myocardial repolarization("squeezes up" T-wave)Tall, peaked T-Waves Short QT interval (<0.5 of RR interval)[K+] > 6.5 mmol/L:? atrial conduction; slow signal transmission from SA to AV nodeCells become slightly depolarized: Resting Membrane Potential (RMP) is brought closer to thresholdIn the Kidney:Muscle weakness and even paralysis (respiratory muscle weakness is rare)? reabsorption of Na+ from Cortical Collecting Duct (CCD)CCD lumen remains more positively chargedMetabolic Acidosis(normal anion gap)Over time (when patients become symptomatic): Chronic membrane depolarization desensitizes voltage-gated Na+ channels (slows their opening) ? ? membrane excitability ? ? action potential generation[K+] > 7.0 mmol/L:? ventricular conductionBradycardiaProlonged, abnormal QRSAV blocks[K+] > 9.0 mmol/L:more conduction abnormalitiesPEA with bizarre wide-QRS rhythmV-fibAsystole? urinary H+ secretion by alpha-intercalated cellsHIGH Plasma [K+] (potassium ion concentration)Dyspnea, fatigue, dizziness, syncope? cardiac output ? ? perfusion of tissues, i.e. lungs & brainCardiac arrhythmias: Conduction blocks (AV block, Bundle branch blocks), VT , V-Fib, Bradycardia, Asystole.?? PR interval ?P-wave flattens, eventually disappearsIf severe, QRS & T-waves fuse:Sine-WavesThe higher the [K+], the slower the voltage-gated Na+ channels open, reflected by distinctive ECG changes:If the K+ is due to ? aldosterone effect ? principal cell dysfunctionHigh pH ? glutamate deamination, which normally produces NH4+? NH4+ reaches the thick ascending limb to be converted to NH3Less NH3 diffuses into the collecting duct to be converted to NH4+ through binding with H+ ? ? NH4+ and therefore ? H+ is excretedK+ moves into proximal tubule cells, causing H+ to diffuse out ? Intracellular alkalosis Irregular force and rhythm of cardiac muscle contraction is sensed by the patient? contraction impulse is conductedDefective electrical conduction through cardiac myocytesMore acid (H=) is retained in the body
118 kB / 357 words" />
6.5 mmol/L:? atrial conduction; slow signal transmission from SA to AV nodeCells become slightly depolarized: Resting Membrane Potential (RMP) is brought closer to thresholdIn the Kidney:Muscle weakness and even paralysis (respiratory muscle weakness is rare)? reabsorption of Na+ from Cortical Collecting Duct (CCD)CCD lumen remains more positively chargedMetabolic Acidosis(normal anion gap)Over time (when patients become symptomatic): Chronic membrane depolarization desensitizes voltage-gated Na+ channels (slows their opening) ? ? membrane excitability ? ? action potential generation[K+] > 7.0 mmol/L:? ventricular conductionBradycardiaProlonged, abnormal QRSAV blocks[K+] > 9.0 mmol/L:more conduction abnormalitiesPEA with bizarre wide-QRS rhythmV-fibAsystole? urinary H+ secretion by alpha-intercalated cellsHIGH Plasma [K+] (potassium ion concentration)Dyspnea, fatigue, dizziness, syncope? cardiac output ? ? perfusion of tissues, i.e. lungs & brainCardiac arrhythmias: Conduction blocks (AV block, Bundle branch blocks), VT , V-Fib, Bradycardia, Asystole.?? PR interval ?P-wave flattens, eventually disappearsIf severe, QRS & T-waves fuse:Sine-WavesThe higher the [K+], the slower the voltage-gated Na+ channels open, reflected by distinctive ECG changes:If the K+ is due to ? aldosterone effect ? principal cell dysfunctionHigh pH ? glutamate deamination, which normally produces NH4+? NH4+ reaches the thick ascending limb to be converted to NH3Less NH3 diffuses into the collecting duct to be converted to NH4+ through binding with H+ ? ? NH4+ and therefore ? H+ is excretedK+ moves into proximal tubule cells, causing H+ to diffuse out ? Intracellular alkalosis Irregular force and rhythm of cardiac muscle contraction is sensed by the patient? contraction impulse is conductedDefective electrical conduction through cardiac myocytesMore acid (H=) is retained in the body
118 kB / 357 words" title="Yu, Yan - Hyperkalemia clinical findings - Published.pptx
Hyperkalemia: Clinical FindingsAuthor: Yan YuReviewers:Alexander ArnoldDavid WaldnerSean SpenceAndrew Wade** MD at time of publicationLegend:Published September 9, 2013 on www.thecalgaryguide.comMechanismPathophysiologySign/Symptom/Lab FindingComplicationsPalpitationsNotes: Symptoms usually manifest when plasma [K+] > 7.0 mmol/L, but can occur at lower [K+]s when hyperkalemia is acute.ECG changes can, but don't necessarily, correlate with a particular [K+].Initially: Excitable cells (muscle cells, neurons) undergo action potentials more readily? [K+ ] gradient between cells and the blood (K+ tends to stay inside cells, less K+ diffuses out)In the Heart:In Skeletal Muscle:[K+] >5.5 mmol/L :faster myocardial repolarization("squeezes up" T-wave)Tall, peaked T-Waves Short QT interval (<0.5 of RR interval)[K+] > 6.5 mmol/L:? atrial conduction; slow signal transmission from SA to AV nodeCells become slightly depolarized: Resting Membrane Potential (RMP) is brought closer to thresholdIn the Kidney:Muscle weakness and even paralysis (respiratory muscle weakness is rare)? reabsorption of Na+ from Cortical Collecting Duct (CCD)CCD lumen remains more positively chargedMetabolic Acidosis(normal anion gap)Over time (when patients become symptomatic): Chronic membrane depolarization desensitizes voltage-gated Na+ channels (slows their opening) ? ? membrane excitability ? ? action potential generation[K+] > 7.0 mmol/L:? ventricular conductionBradycardiaProlonged, abnormal QRSAV blocks[K+] > 9.0 mmol/L:more conduction abnormalitiesPEA with bizarre wide-QRS rhythmV-fibAsystole? urinary H+ secretion by alpha-intercalated cellsHIGH Plasma [K+] (potassium ion concentration)Dyspnea, fatigue, dizziness, syncope? cardiac output ? ? perfusion of tissues, i.e. lungs & brainCardiac arrhythmias: Conduction blocks (AV block, Bundle branch blocks), VT , V-Fib, Bradycardia, Asystole.?? PR interval ?P-wave flattens, eventually disappearsIf severe, QRS & T-waves fuse:Sine-WavesThe higher the [K+], the slower the voltage-gated Na+ channels open, reflected by distinctive ECG changes:If the K+ is due to ? aldosterone effect ? principal cell dysfunctionHigh pH ? glutamate deamination, which normally produces NH4+? NH4+ reaches the thick ascending limb to be converted to NH3Less NH3 diffuses into the collecting duct to be converted to NH4+ through binding with H+ ? ? NH4+ and therefore ? H+ is excretedK+ moves into proximal tubule cells, causing H+ to diffuse out ? Intracellular alkalosis Irregular force and rhythm of cardiac muscle contraction is sensed by the patient? contraction impulse is conductedDefective electrical conduction through cardiac myocytesMore acid (H=) is retained in the body
118 kB / 357 words" />
Hypocalcemia: Clinical Findings
![Yu, Yan - Hypocalcemia - Clinical Findings - FINAL.pptx
Hypocalcemia: Clinical FindingsAuthor: Yan YuReviewers:David WaldnerSean SpenceGreg Kline** MD at time of publicationLegend:Published May 7, 2013 on www.thecalgaryguide.comMechanismPathophysiologySign/Symptom/Lab FindingComplicationsHypocalcemia(serum [Ca2+] <2.1mmol/L)Altered sensory ability of peripheral nervesLess Ca2+ outside cells, with no change in + charges inside cellsPeripheral paraesthesia? Neuronal](http://calgaryguide.ucalgary.ca/wp-content/uploads/2014/09/Hypocalcemia-Clinical-Findings.jpg "Yu, Yan - Hypocalcemia - Clinical Findings - FINAL.pptx
Hypocalcemia: Clinical FindingsAuthor: Yan YuReviewers:David WaldnerSean SpenceGreg Kline** MD at time of publicationLegend:Published May 7, 2013 on www.thecalgaryguide.comMechanismPathophysiologySign/Symptom/Lab FindingComplicationsHypocalcemia(serum [Ca2+] <2.1mmol/L)Altered sensory ability of peripheral nervesLess Ca2+ outside cells, with no change in + charges inside cellsPeripheral paraesthesia? Neuronal")
Hypercalcemia: Clinical Findings
Nephrotic Syndrome: Pathogenesis and Clinical Findings
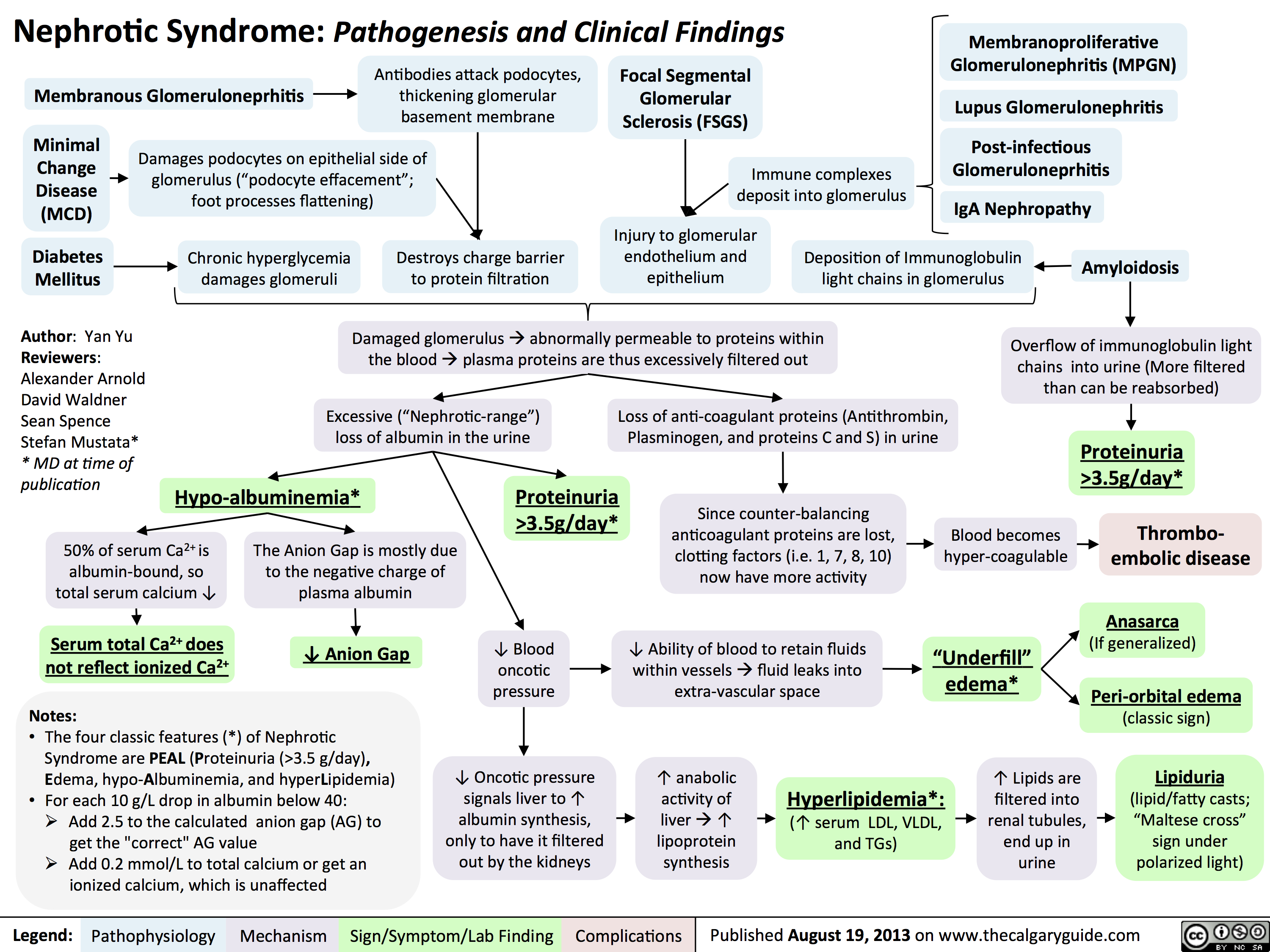 3.5g/day*? Ability of blood to retain fluids within vessels ? fluid leaks into extra-vascular spaceInjury to glomerular endothelium and epitheliumImmune complexes deposit into glomerulusDamaged glomerulus ? abnormally permeable to proteins within the blood ? plasma proteins are thus excessively filtered out? Oncotic pressure signals liver to ? albumin synthesis, only to have it filtered out by the kidneys? anabolic activity of liver ? ? lipoprotein synthesisHyperlipidemia*:(? serum LDL, VLDL, and TGs)Lipiduria(lipid/fatty casts; "Maltese cross" sign under polarized light)Since counter-balancing anticoagulant proteins are lost, clotting factors (i.e. 1, 7, 8, 10) now have more activityThrombo-embolic diseaseBlood becomes hyper-coagulable? Lipids are filtered into renal tubules, end up in urineMembranoproliferative Glomerulonephritis (MPGN)Lupus Glomerulonephritis Post-infectious GlomeruloneprhitisIgA NephropathyDamages podocytes on epithelial side of glomerulus ("podocyte effacement"; foot processes flattening)Diabetes MellitusChronic hyperglycemia damages glomeruliDeposition of Immunoglobulin light chains in glomerulusAmyloidosisAnasarca(If generalized)Peri-orbital edema (classic sign)Focal Segmental Glomerular Sclerosis (FSGS)Membranous GlomeruloneprhitisAntibodies attack podocytes, thickening glomerular basement membraneOverflow of immunoglobulin light chains into urine (More filtered than can be reabsorbed)Proteinuria >3.5g/day*The Anion Gap is mostly due to the negative charge of plasma albumin? Anion GapNotes: The four classic features (*) of Nephrotic Syndrome are PEAL (Proteinuria (>3.5 g/day), Edema, hypo-Albuminemia, and hyperLipidemia)For each 10 g/L drop in albumin below 40:Add 2.5 to the calculated anion gap (AG) to get the "correct" AG valueAdd 0.2 mmol/L to total calcium or get an ionized calcium, which is unaffected50% of serum Ca2+ is albumin-bound, so total serum calcium ? Serum total Ca2+ does not reflect ionized Ca2+ ? Blood oncotic pressure" title="Destroys charge barrier to protein filtrationNephrotic Syndrome: Pathogenesis and Clinical FindingsAuthor: Yan YuReviewers:Alexander ArnoldDavid WaldnerSean SpenceStefan Mustata** MD at time of publicationLegend:Published August 19, 2013 on www.thecalgaryguide.comMechanismPathophysiologySign/Symptom/Lab FindingComplicationsExcessive ("Nephrotic-range") loss of albumin in the urineHypo-albuminemia*Loss of anti-coagulant proteins (Antithrombin, Plasminogen, and proteins C and S) in urineMinimal Change Disease (MCD)"Underfill" edema*Proteinuria >3.5g/day*? Ability of blood to retain fluids within vessels ? fluid leaks into extra-vascular spaceInjury to glomerular endothelium and epitheliumImmune complexes deposit into glomerulusDamaged glomerulus ? abnormally permeable to proteins within the blood ? plasma proteins are thus excessively filtered out? Oncotic pressure signals liver to ? albumin synthesis, only to have it filtered out by the kidneys? anabolic activity of liver ? ? lipoprotein synthesisHyperlipidemia*:(? serum LDL, VLDL, and TGs)Lipiduria(lipid/fatty casts; "Maltese cross" sign under polarized light)Since counter-balancing anticoagulant proteins are lost, clotting factors (i.e. 1, 7, 8, 10) now have more activityThrombo-embolic diseaseBlood becomes hyper-coagulable? Lipids are filtered into renal tubules, end up in urineMembranoproliferative Glomerulonephritis (MPGN)Lupus Glomerulonephritis Post-infectious GlomeruloneprhitisIgA NephropathyDamages podocytes on epithelial side of glomerulus ("podocyte effacement"; foot processes flattening)Diabetes MellitusChronic hyperglycemia damages glomeruliDeposition of Immunoglobulin light chains in glomerulusAmyloidosisAnasarca(If generalized)Peri-orbital edema (classic sign)Focal Segmental Glomerular Sclerosis (FSGS)Membranous GlomeruloneprhitisAntibodies attack podocytes, thickening glomerular basement membraneOverflow of immunoglobulin light chains into urine (More filtered than can be reabsorbed)Proteinuria >3.5g/day*The Anion Gap is mostly due to the negative charge of plasma albumin? Anion GapNotes: The four classic features (*) of Nephrotic Syndrome are PEAL (Proteinuria (>3.5 g/day), Edema, hypo-Albuminemia, and hyperLipidemia)For each 10 g/L drop in albumin below 40:Add 2.5 to the calculated anion gap (AG) to get the "correct" AG valueAdd 0.2 mmol/L to total calcium or get an ionized calcium, which is unaffected50% of serum Ca2+ is albumin-bound, so total serum calcium ? Serum total Ca2+ does not reflect ionized Ca2+ ? Blood oncotic pressure" />
3.5g/day*? Ability of blood to retain fluids within vessels ? fluid leaks into extra-vascular spaceInjury to glomerular endothelium and epitheliumImmune complexes deposit into glomerulusDamaged glomerulus ? abnormally permeable to proteins within the blood ? plasma proteins are thus excessively filtered out? Oncotic pressure signals liver to ? albumin synthesis, only to have it filtered out by the kidneys? anabolic activity of liver ? ? lipoprotein synthesisHyperlipidemia*:(? serum LDL, VLDL, and TGs)Lipiduria(lipid/fatty casts; "Maltese cross" sign under polarized light)Since counter-balancing anticoagulant proteins are lost, clotting factors (i.e. 1, 7, 8, 10) now have more activityThrombo-embolic diseaseBlood becomes hyper-coagulable? Lipids are filtered into renal tubules, end up in urineMembranoproliferative Glomerulonephritis (MPGN)Lupus Glomerulonephritis Post-infectious GlomeruloneprhitisIgA NephropathyDamages podocytes on epithelial side of glomerulus ("podocyte effacement"; foot processes flattening)Diabetes MellitusChronic hyperglycemia damages glomeruliDeposition of Immunoglobulin light chains in glomerulusAmyloidosisAnasarca(If generalized)Peri-orbital edema (classic sign)Focal Segmental Glomerular Sclerosis (FSGS)Membranous GlomeruloneprhitisAntibodies attack podocytes, thickening glomerular basement membraneOverflow of immunoglobulin light chains into urine (More filtered than can be reabsorbed)Proteinuria >3.5g/day*The Anion Gap is mostly due to the negative charge of plasma albumin? Anion GapNotes: The four classic features (*) of Nephrotic Syndrome are PEAL (Proteinuria (>3.5 g/day), Edema, hypo-Albuminemia, and hyperLipidemia)For each 10 g/L drop in albumin below 40:Add 2.5 to the calculated anion gap (AG) to get the "correct" AG valueAdd 0.2 mmol/L to total calcium or get an ionized calcium, which is unaffected50% of serum Ca2+ is albumin-bound, so total serum calcium ? Serum total Ca2+ does not reflect ionized Ca2+ ? Blood oncotic pressure" title="Destroys charge barrier to protein filtrationNephrotic Syndrome: Pathogenesis and Clinical FindingsAuthor: Yan YuReviewers:Alexander ArnoldDavid WaldnerSean SpenceStefan Mustata** MD at time of publicationLegend:Published August 19, 2013 on www.thecalgaryguide.comMechanismPathophysiologySign/Symptom/Lab FindingComplicationsExcessive ("Nephrotic-range") loss of albumin in the urineHypo-albuminemia*Loss of anti-coagulant proteins (Antithrombin, Plasminogen, and proteins C and S) in urineMinimal Change Disease (MCD)"Underfill" edema*Proteinuria >3.5g/day*? Ability of blood to retain fluids within vessels ? fluid leaks into extra-vascular spaceInjury to glomerular endothelium and epitheliumImmune complexes deposit into glomerulusDamaged glomerulus ? abnormally permeable to proteins within the blood ? plasma proteins are thus excessively filtered out? Oncotic pressure signals liver to ? albumin synthesis, only to have it filtered out by the kidneys? anabolic activity of liver ? ? lipoprotein synthesisHyperlipidemia*:(? serum LDL, VLDL, and TGs)Lipiduria(lipid/fatty casts; "Maltese cross" sign under polarized light)Since counter-balancing anticoagulant proteins are lost, clotting factors (i.e. 1, 7, 8, 10) now have more activityThrombo-embolic diseaseBlood becomes hyper-coagulable? Lipids are filtered into renal tubules, end up in urineMembranoproliferative Glomerulonephritis (MPGN)Lupus Glomerulonephritis Post-infectious GlomeruloneprhitisIgA NephropathyDamages podocytes on epithelial side of glomerulus ("podocyte effacement"; foot processes flattening)Diabetes MellitusChronic hyperglycemia damages glomeruliDeposition of Immunoglobulin light chains in glomerulusAmyloidosisAnasarca(If generalized)Peri-orbital edema (classic sign)Focal Segmental Glomerular Sclerosis (FSGS)Membranous GlomeruloneprhitisAntibodies attack podocytes, thickening glomerular basement membraneOverflow of immunoglobulin light chains into urine (More filtered than can be reabsorbed)Proteinuria >3.5g/day*The Anion Gap is mostly due to the negative charge of plasma albumin? Anion GapNotes: The four classic features (*) of Nephrotic Syndrome are PEAL (Proteinuria (>3.5 g/day), Edema, hypo-Albuminemia, and hyperLipidemia)For each 10 g/L drop in albumin below 40:Add 2.5 to the calculated anion gap (AG) to get the "correct" AG valueAdd 0.2 mmol/L to total calcium or get an ionized calcium, which is unaffected50% of serum Ca2+ is albumin-bound, so total serum calcium ? Serum total Ca2+ does not reflect ionized Ca2+ ? Blood oncotic pressure" />
Signs and Symptoms of Hypovolemia
Placental Abruption
Contraindications to Inducing Vaginal Delivery
Active Phase Problems: Pathogenesis and Management
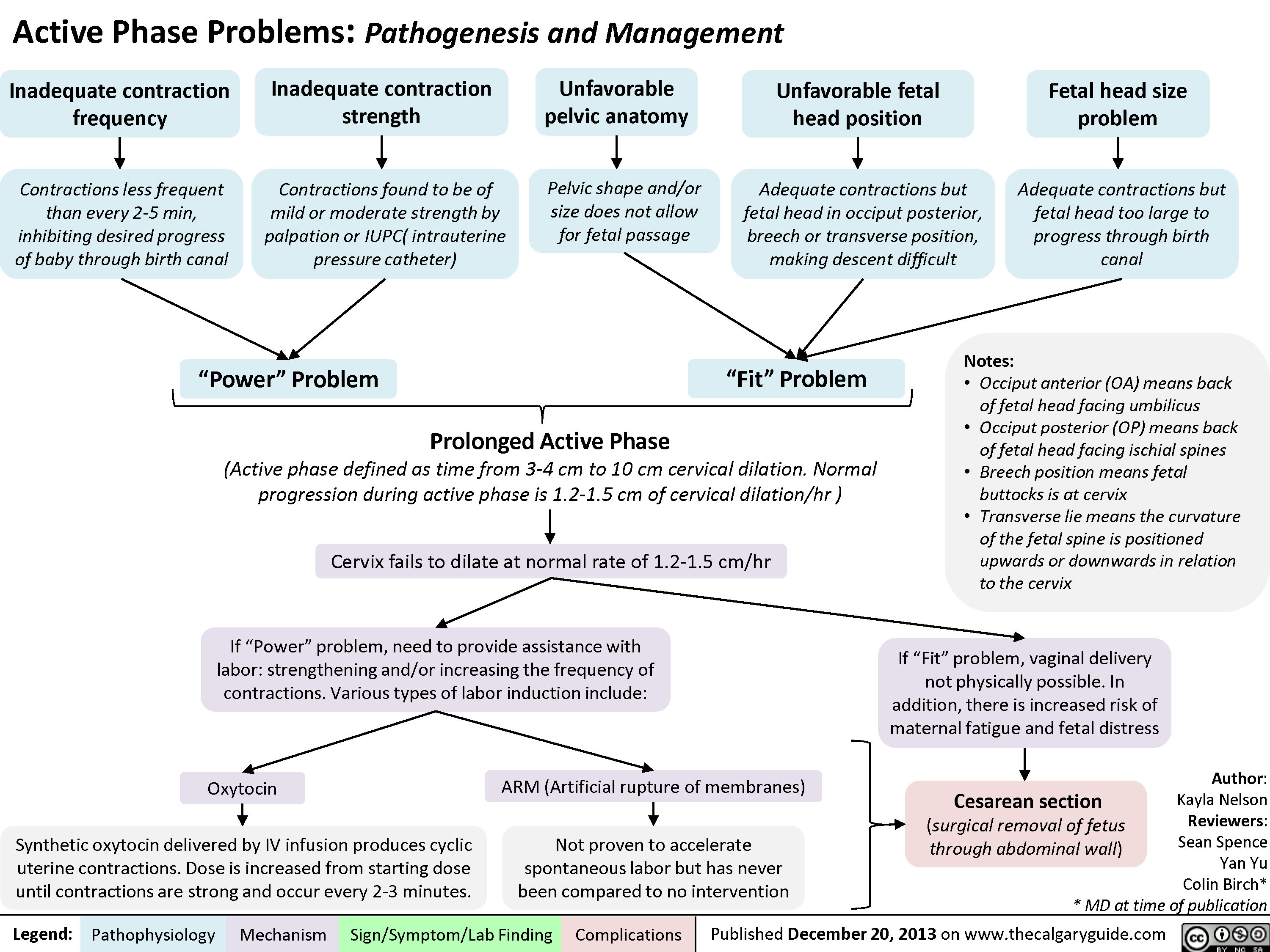
Normal Signs of Placental Detachment

Methods to Improve Fetal Oxygenation
 pattern (i.e. variable decelerations, mild tachycardia, mild ? in HR variability)? Mechanical stress on the fetus, to allow better perfusion of fetusImproved perfusion of maternal organs, including the uterus/fetusNote: if these methods do not restore normal FHR, perform scalp stimulation and scalp pH to better assess fetal hypoxia)Normal FHR pattern
91 kB / 187 words")
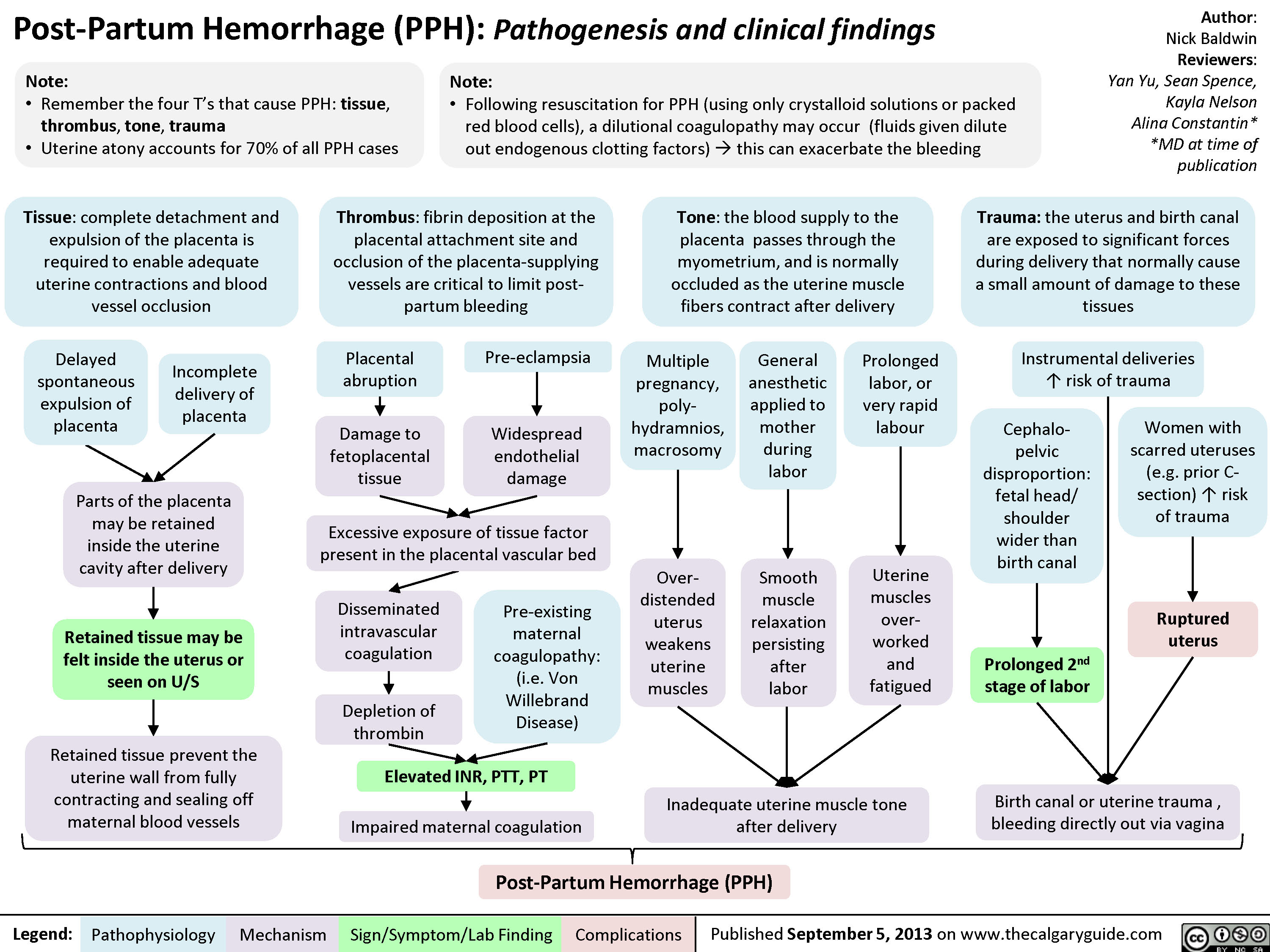
Post-Partum Hemorrhage
Upper Urinary Tract infection (UUTI): Pathogenesis and Clinical Findings

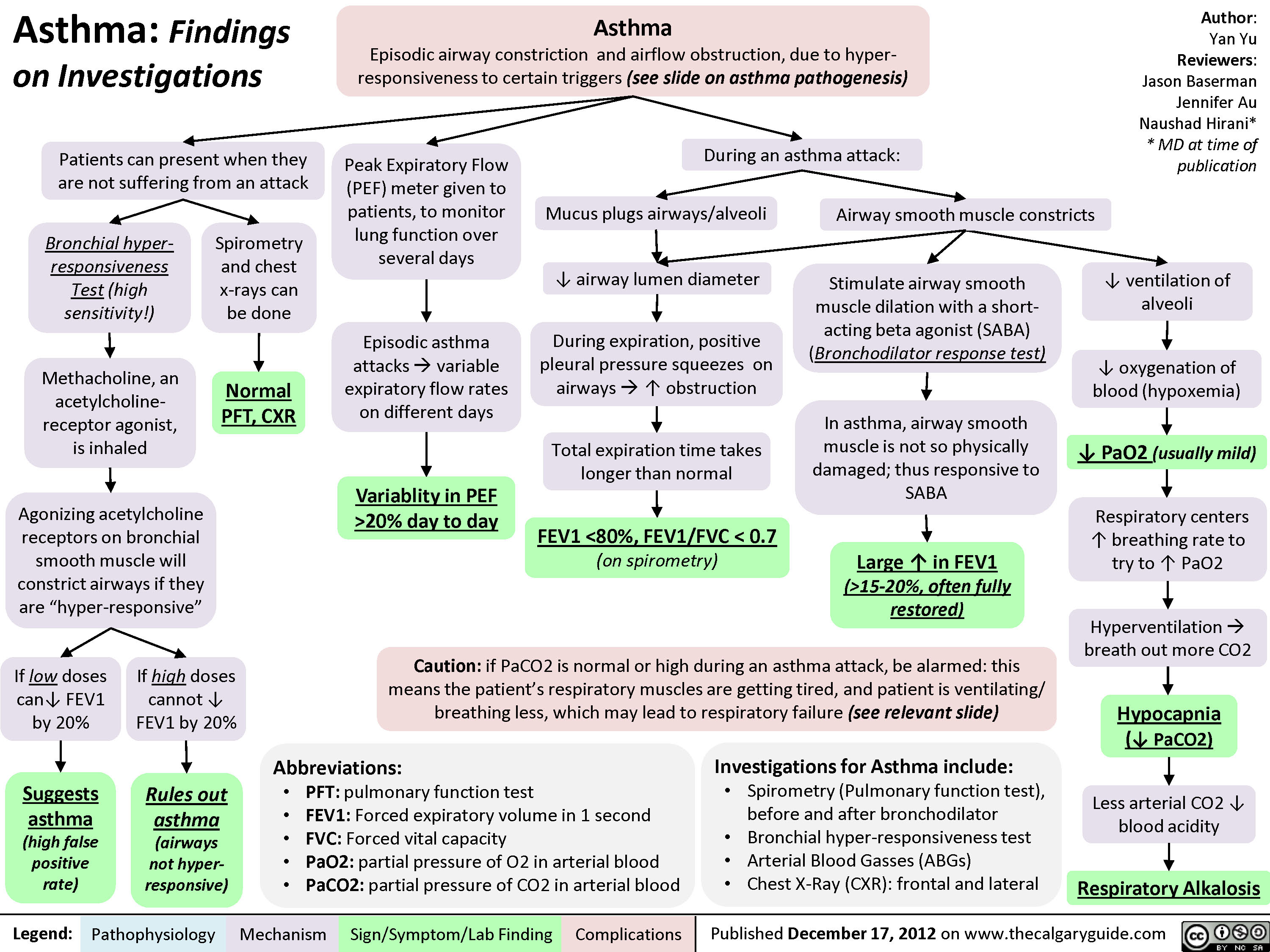
Asthma: Findings on Investigations
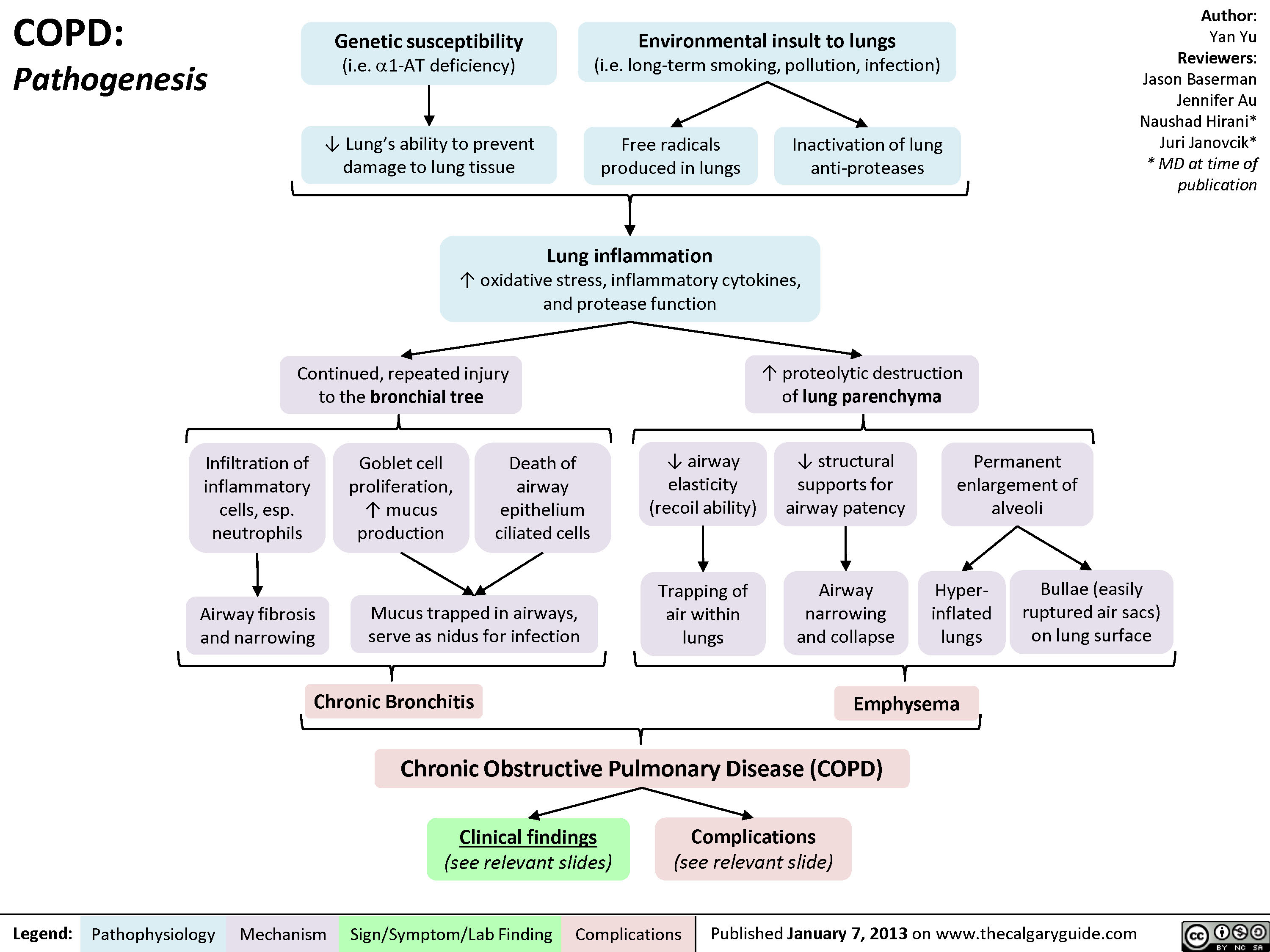
COPD: Pathogenesis
COPD: Clinical Findings
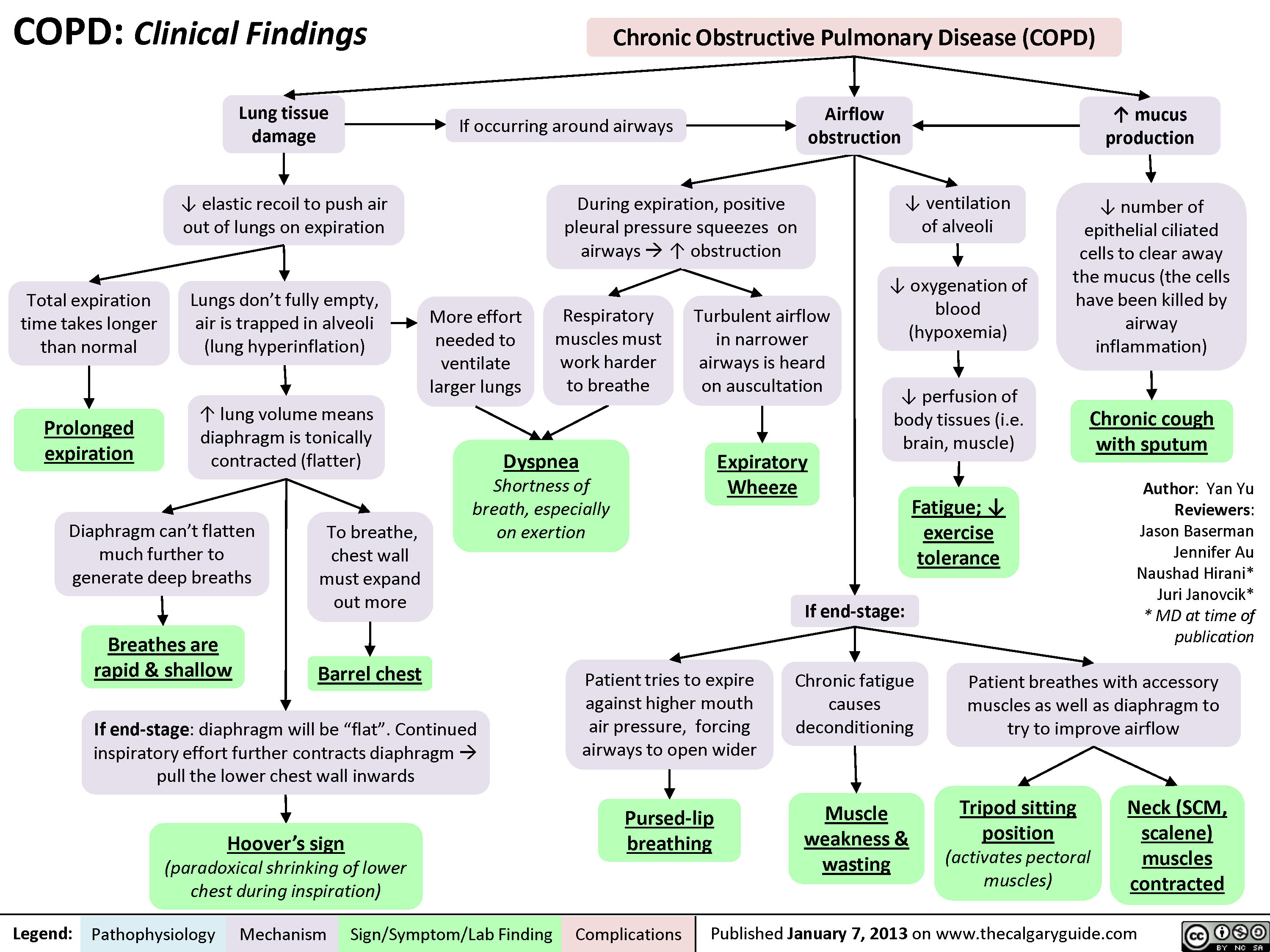
COPD: Findings on Investigations
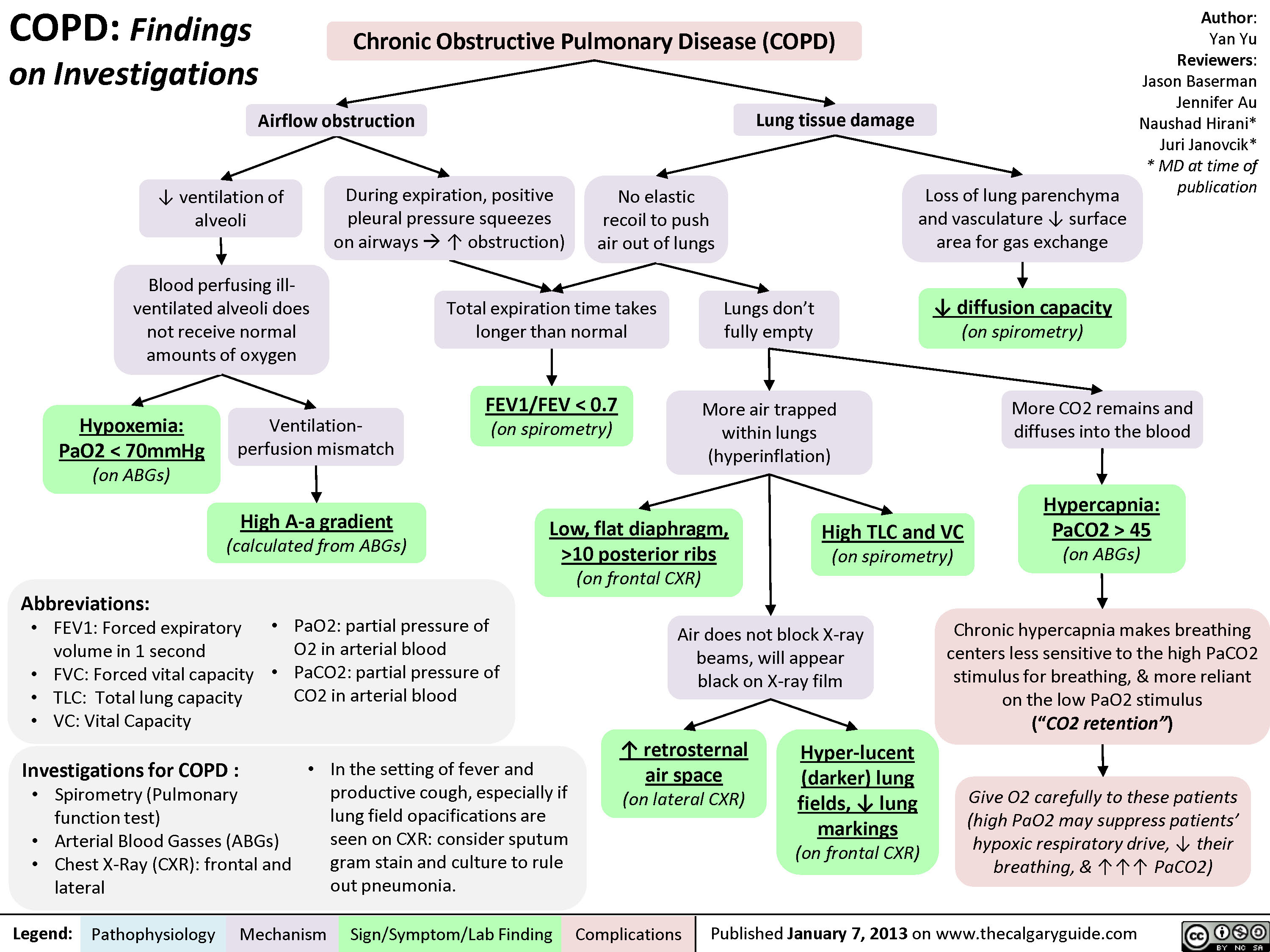
COPD: Complications
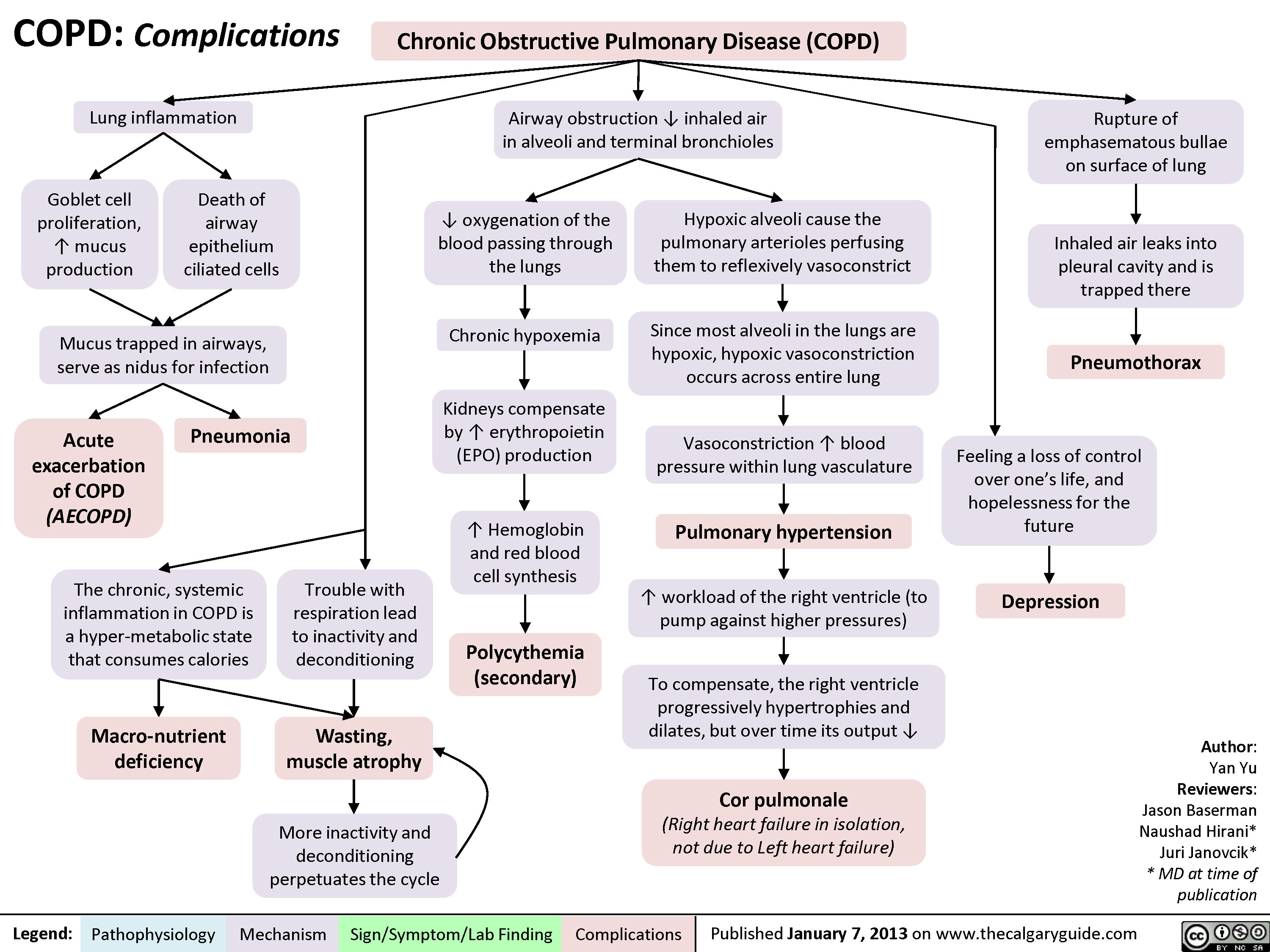
Bronchiectasis: Findings on Chest X-Ray and CT Scan
Cystic Fibrosis
Posterior-Anterior Chest X-Ray
Lateral Chest X-Ray
Coronal CT
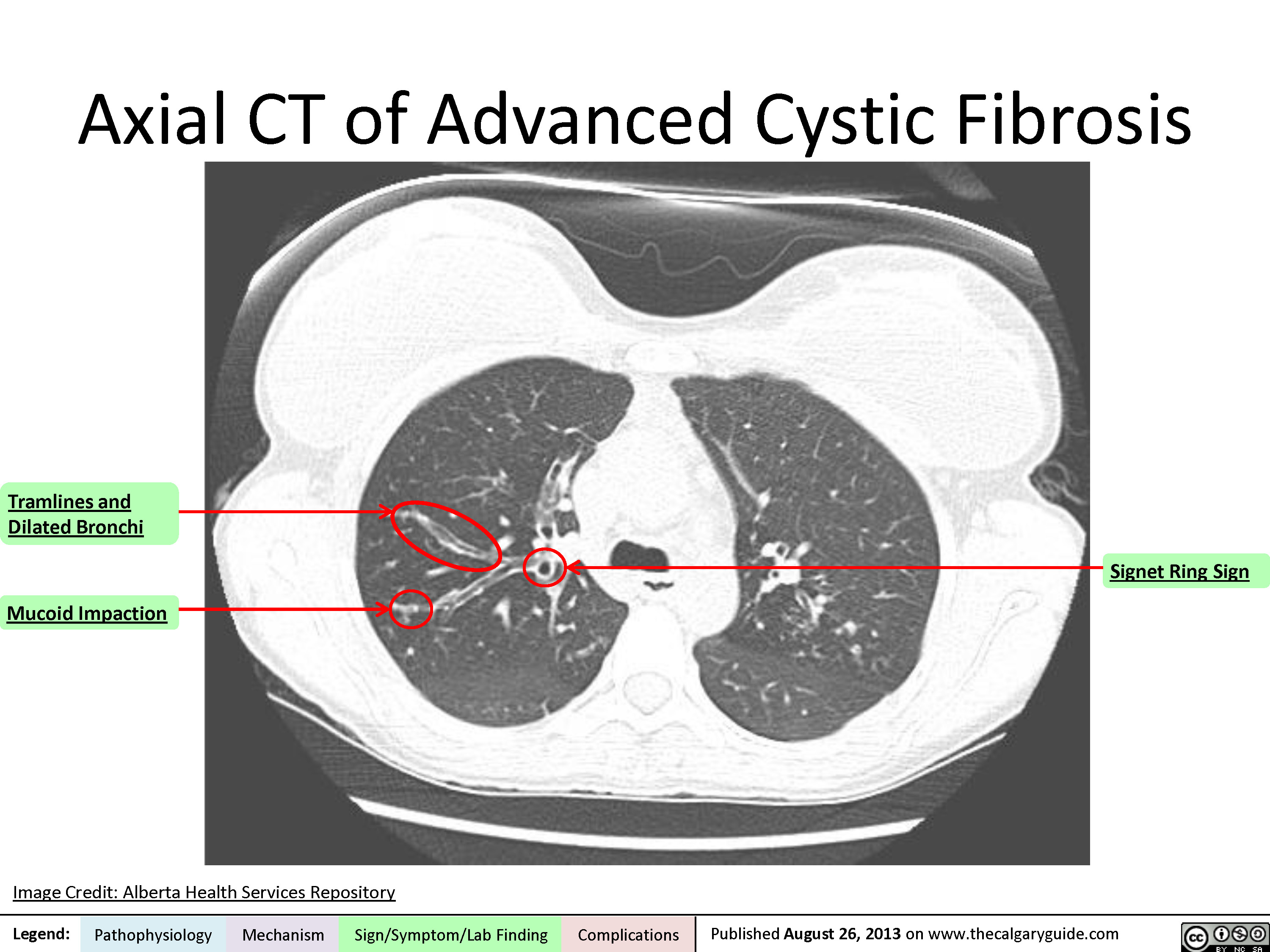
Axial CT
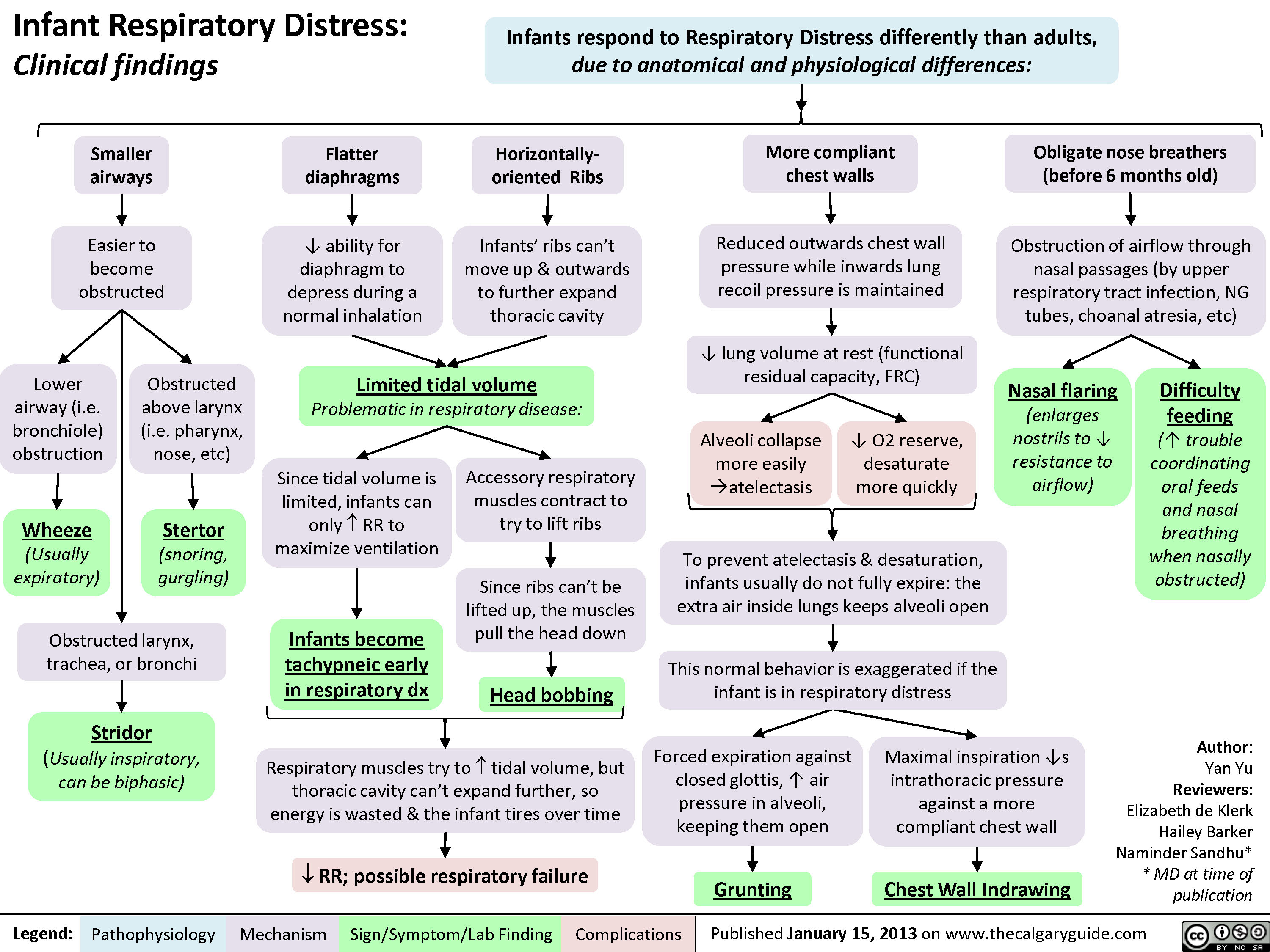
Infant Respiratory Distress: Clinical findings
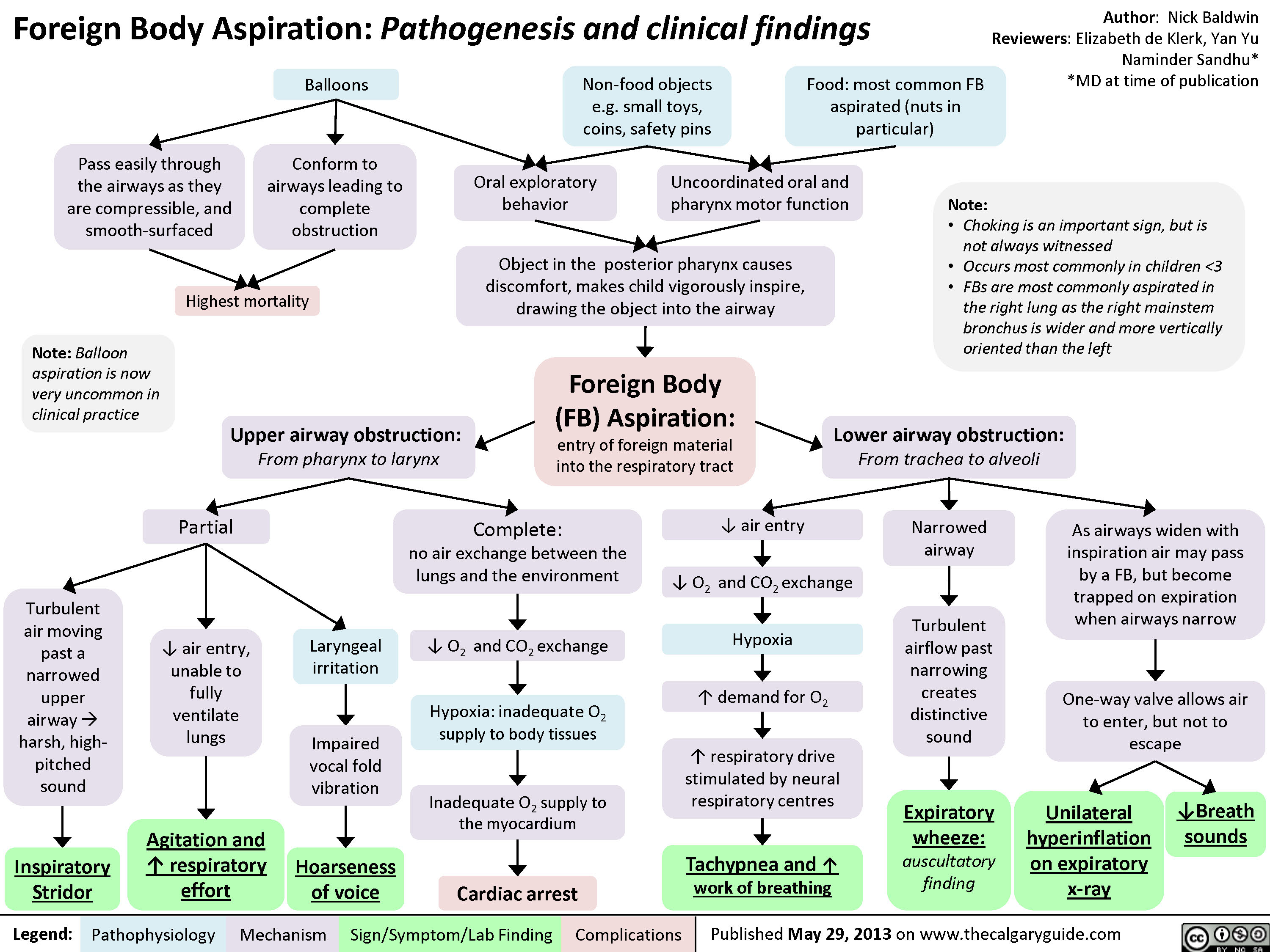
Foreign Body Aspiration
Types of Burns - Summary of Causes and Clinical Findings

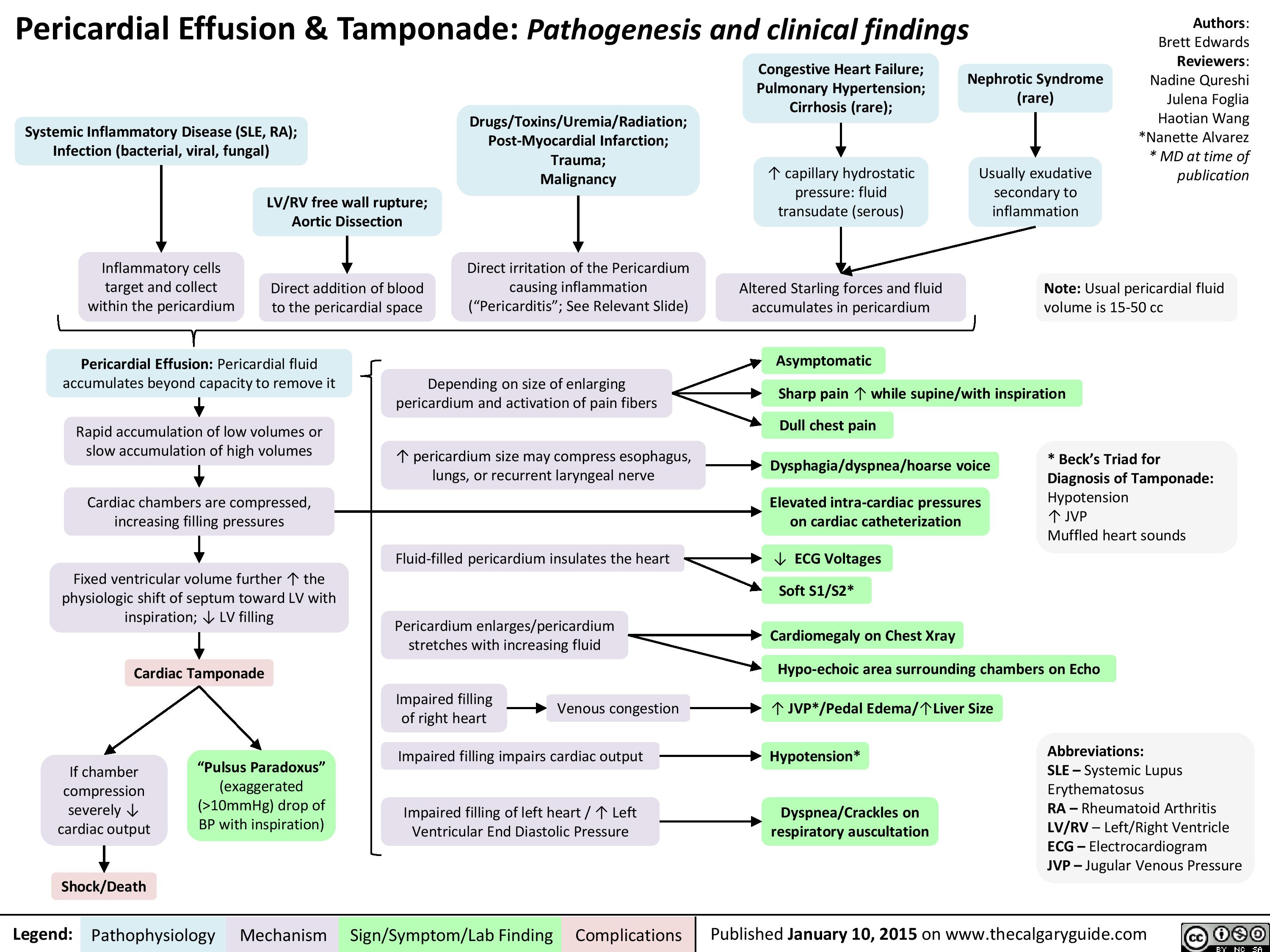
Pericardial Effusion and Tamponade: Pathogenesis and Clinical Findings
Atherosclerosis - Pathogenesis
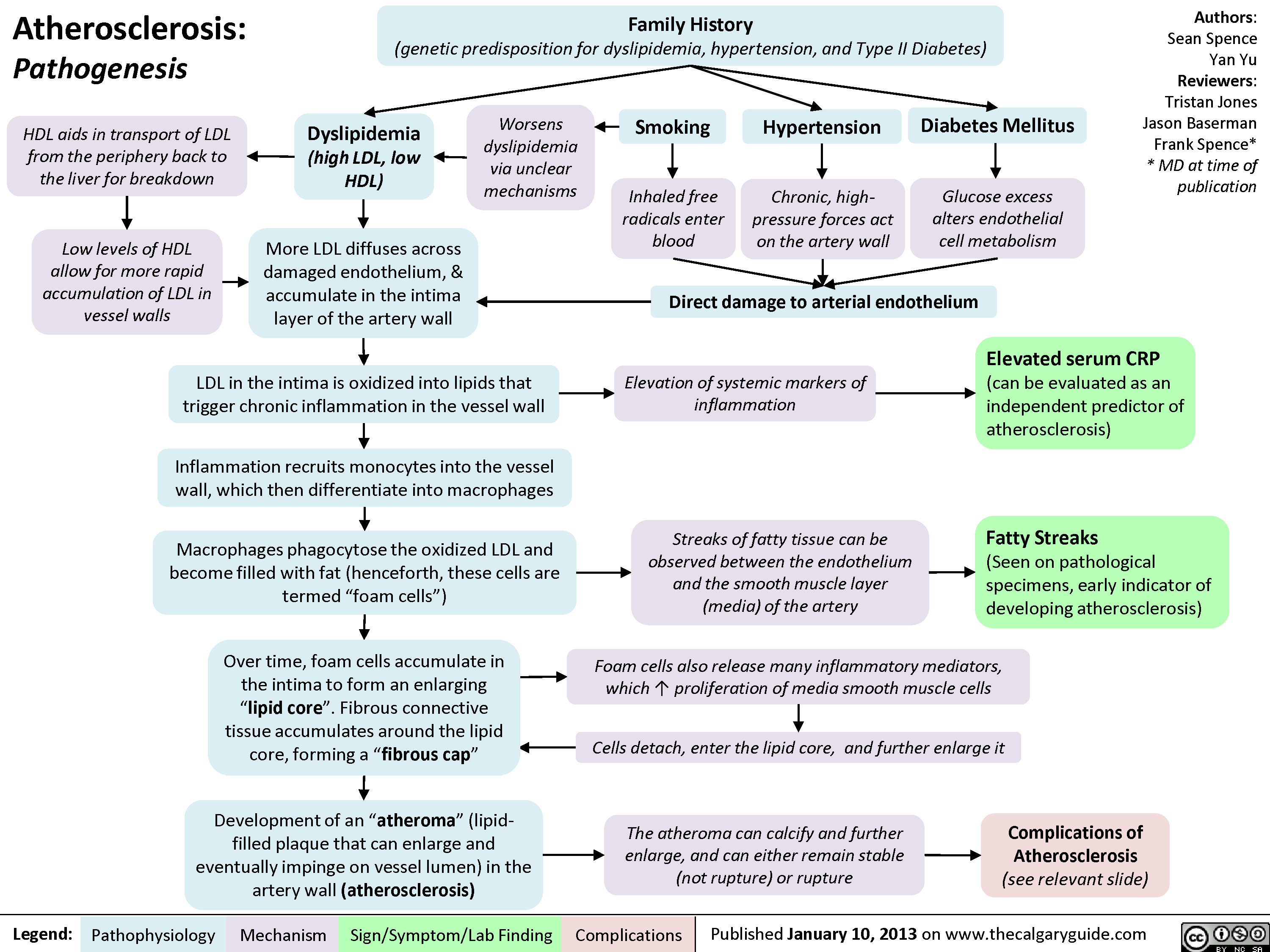
Atherosclerosis - Complications
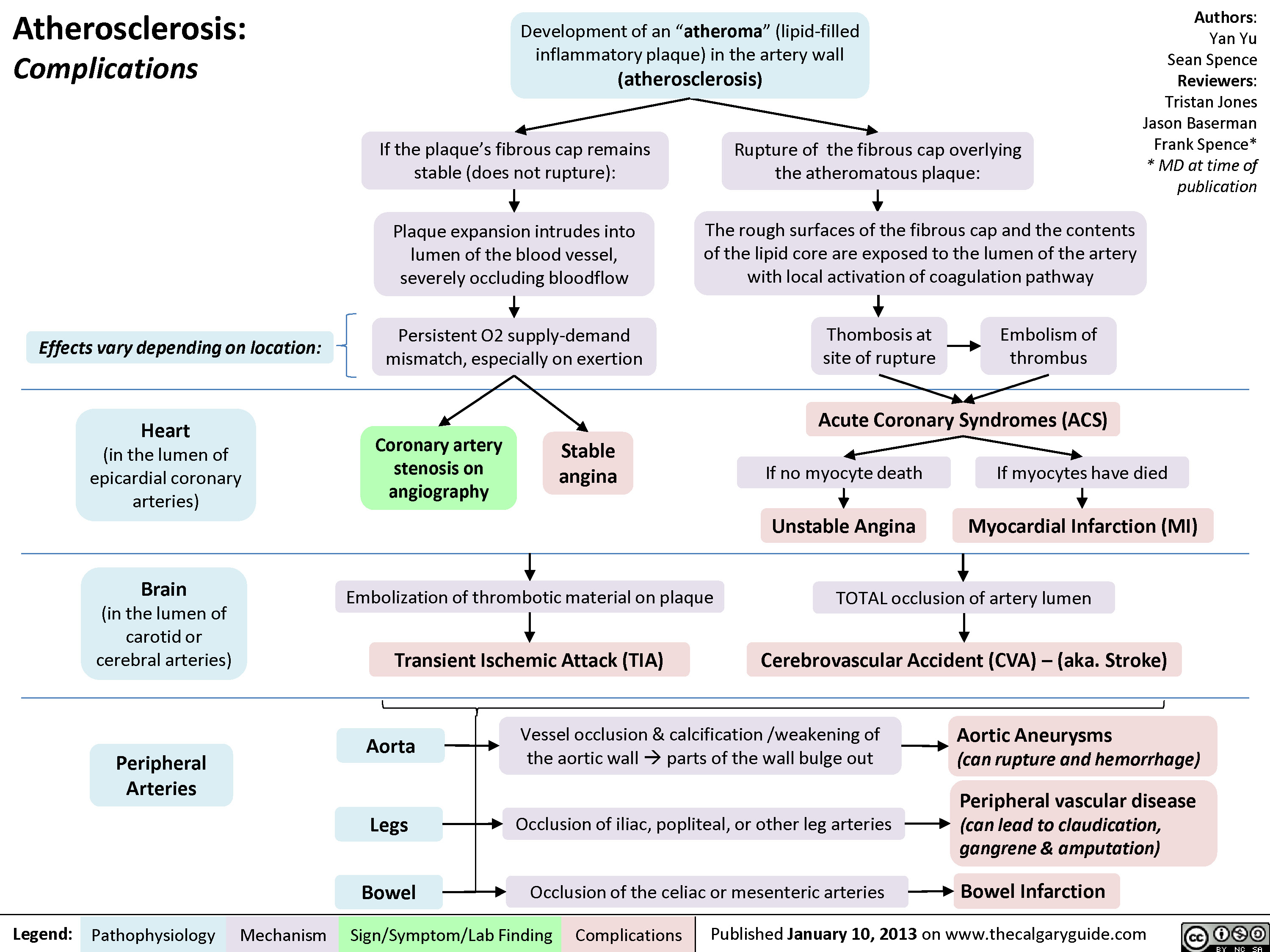
Complications of Myocardial Infarction
myocardial-infarction-findings-on-investigations
Acute, trans-mural myocardial ischemiaIschemia of sub-endocardial myocardiumMyocardial infarctionNote: Both types of ST-segment changes are non-specific: they can indicate Myocardial Infarctions , but can also be false positives (i.e. caused by left ventricular hypertrophy, bundle branch blocks, and other non-myocardial ischemic causes)If ischemia progresses to tissue infarctionPathologic Q-waves (localizes to site of ischemia)Tissue necrosis ? Local myocardial inflammation2-4 hours after MI: troponin proteins released into blood3-8 hours after MI:Creatinine-kinase MB-isozymes released into blood? serum Cardiac Troponins: cTnT, cTnI(Sensitive and most specific serum marker for myocardial necrosis)Relatively faster clearance from circulation? serum CK-MB(less sensitive and specific for myocardial necrosis than Troponins)ST-segment depression(non-localizing)Inflammatory cytokines can spread systemicallyStimulation of neutrophil and monocyte migration towards area of inflammation? WBC count (on CBC)? C-Reactive Protein (CRP)Dead, damaged cardiac myocytes release inner contents into the bloodRelatively slower clearance from circulationSerum CK-MB levels normalize within 3 daysSerum Troponin levels normalize within 14 daysNote: Measuring both CK-MB and Troponins gives a timeline to the MI. For instance, if CK-MB is normal but Troponins are high, it means the MI happened >3 days but <14 days ago.ST-segment elevation(localizes to site of ischemia)Legend:Published January 30, 2013 on www.thecalgaryguide.comMechanismPathophysiologySign/Symptom/Lab FindingComplications
104 kB / 212 words")
myocardial-infarction-findings-on-physical-exam
 Diastolic compliance (necrotic myocardium does not relax as well to accommodate blood)Necrosis of papillary muscles:S4(4th heart sound)? Force of ventricular contractionsMitral valve regurgitationBlood")
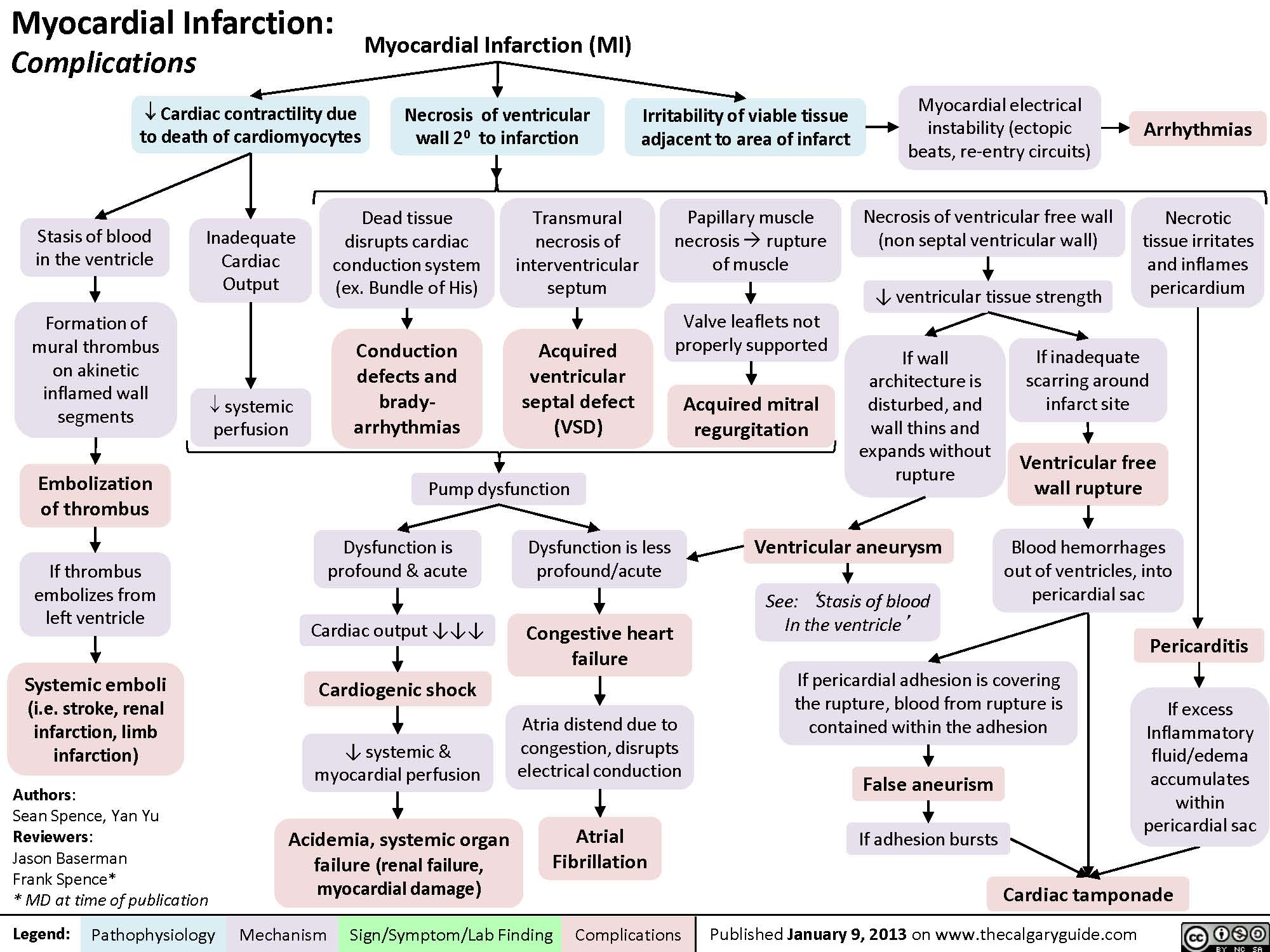
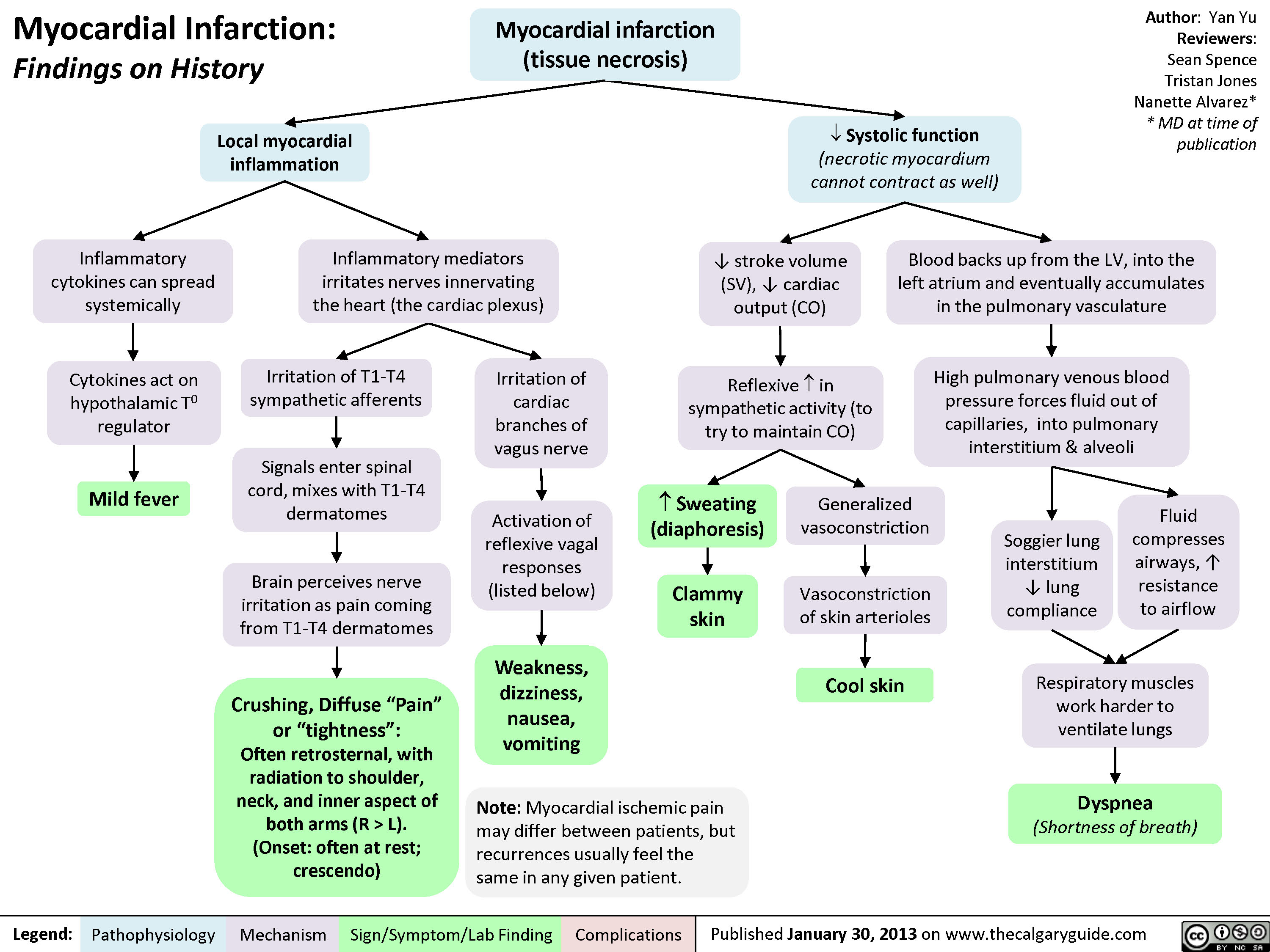
MI Findings on History
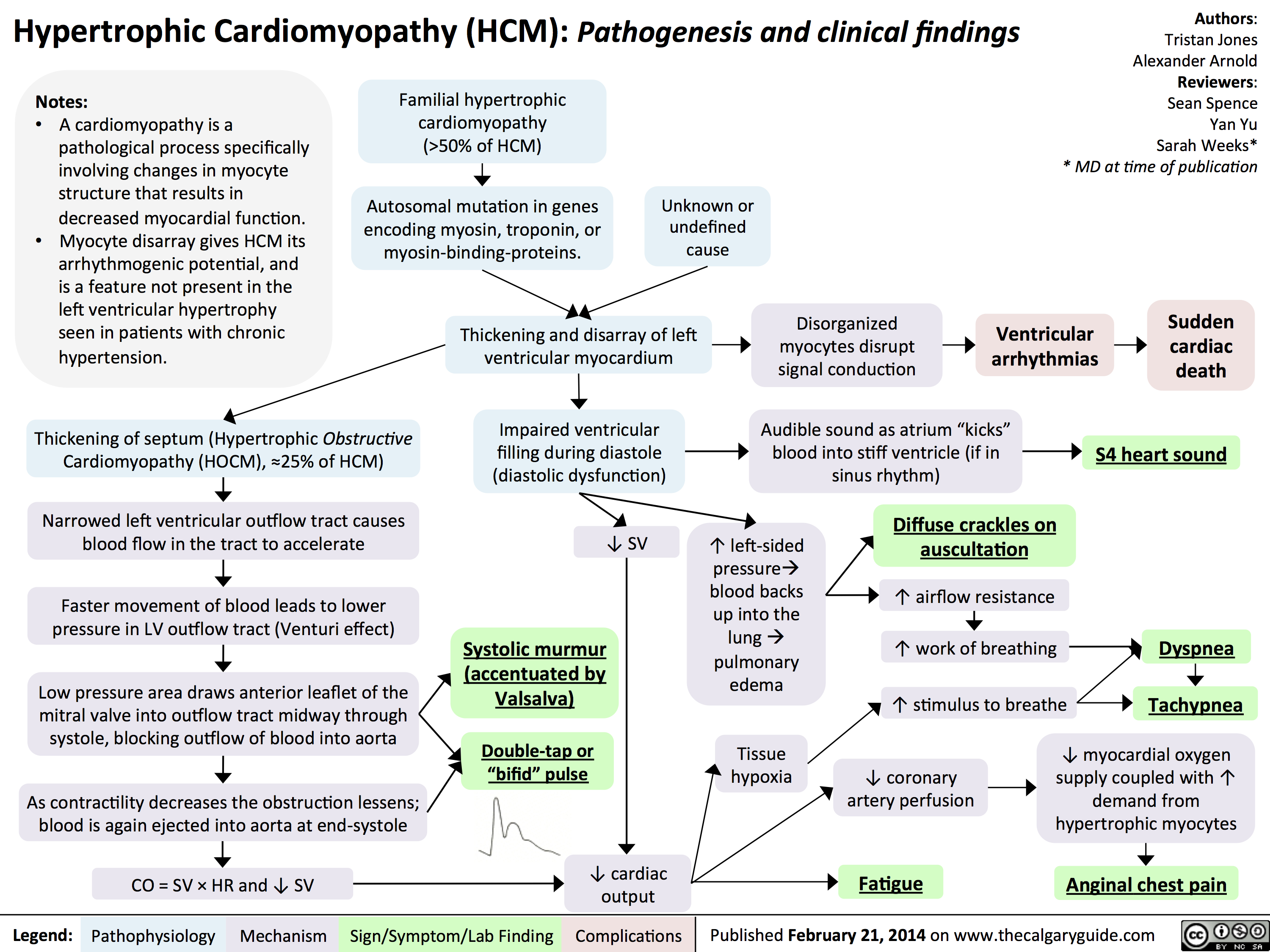
Dilated Cardiomyopathy
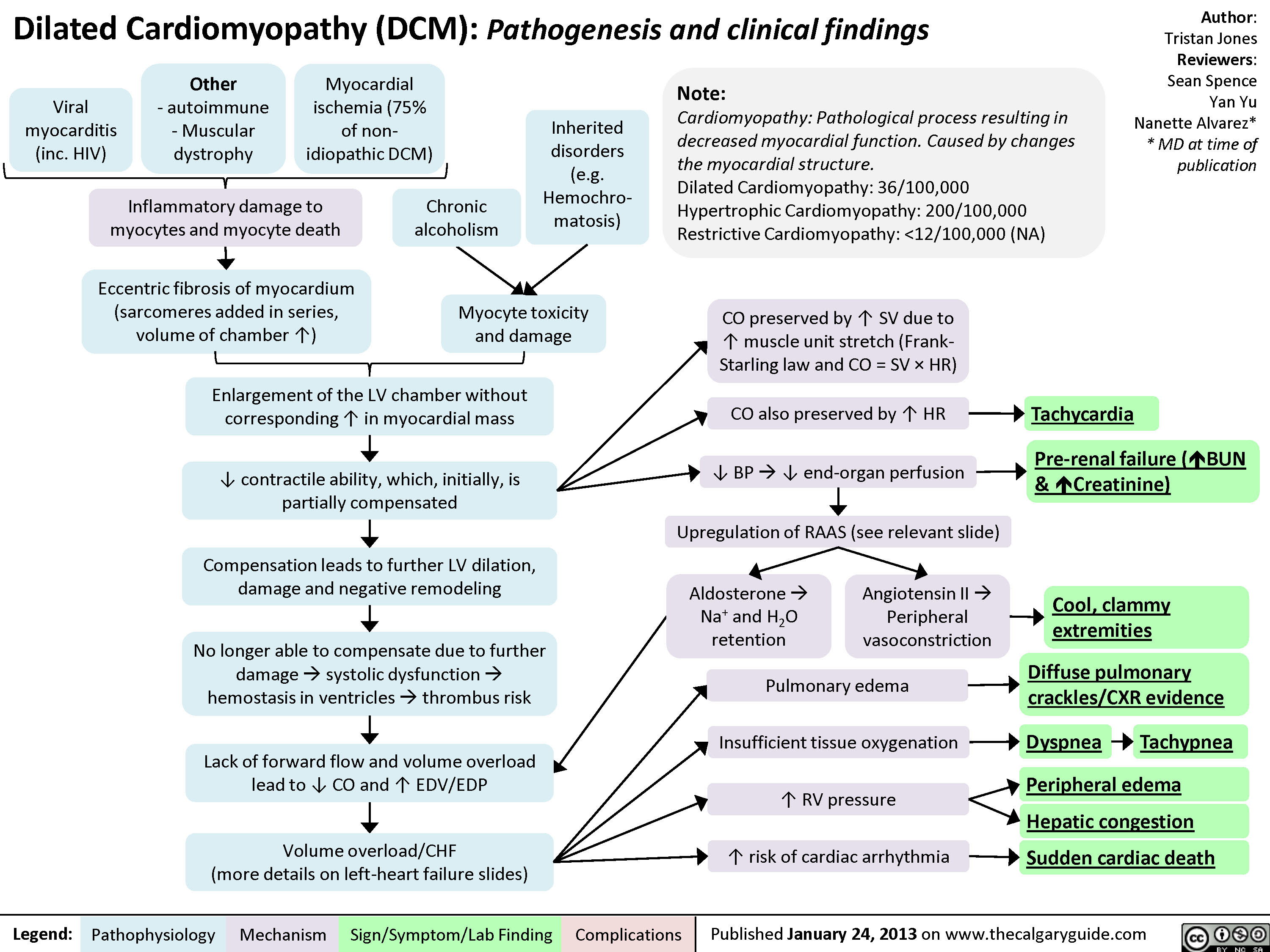
Myocarditis
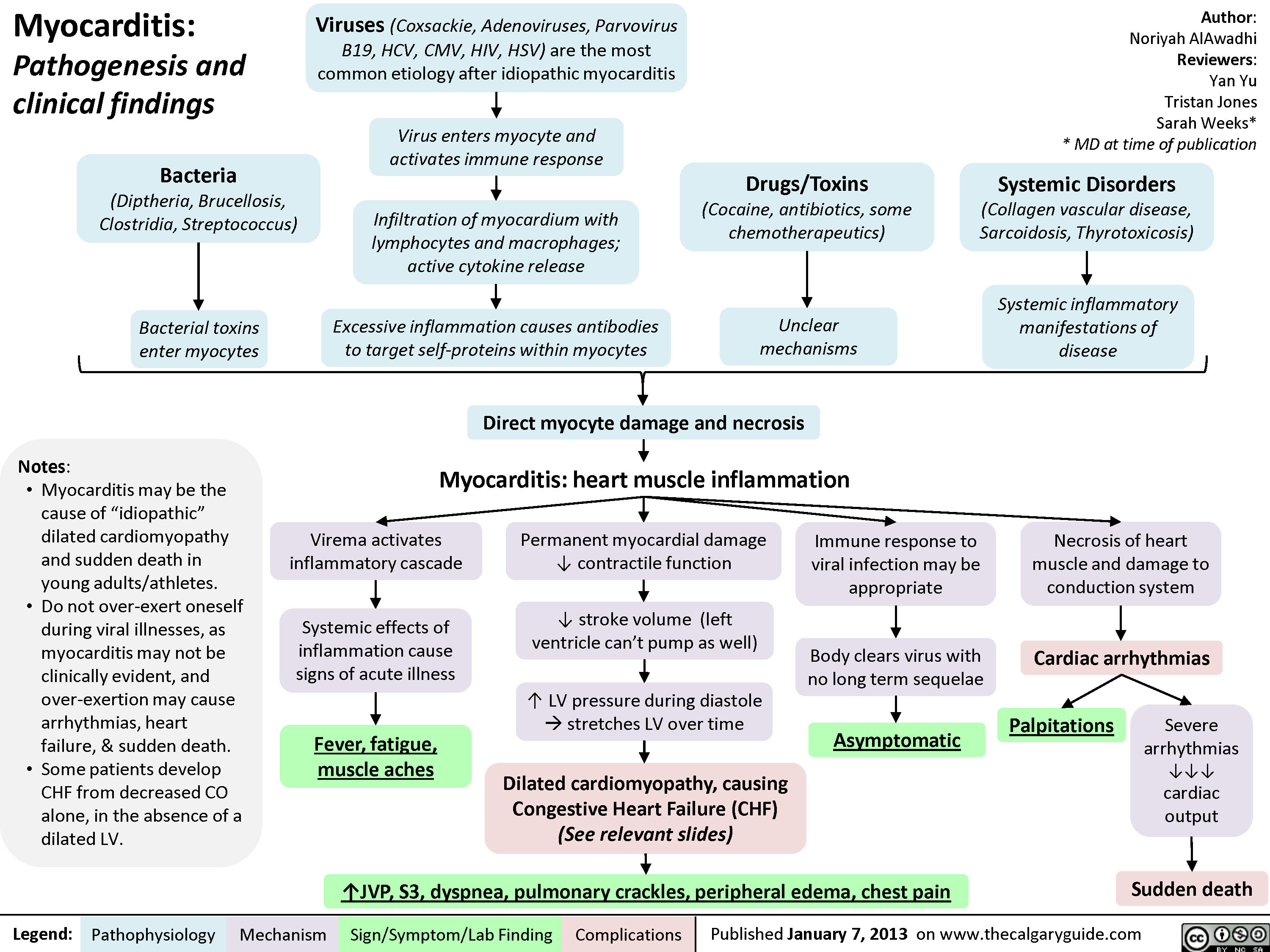
Restrictive Cardiomyopathy
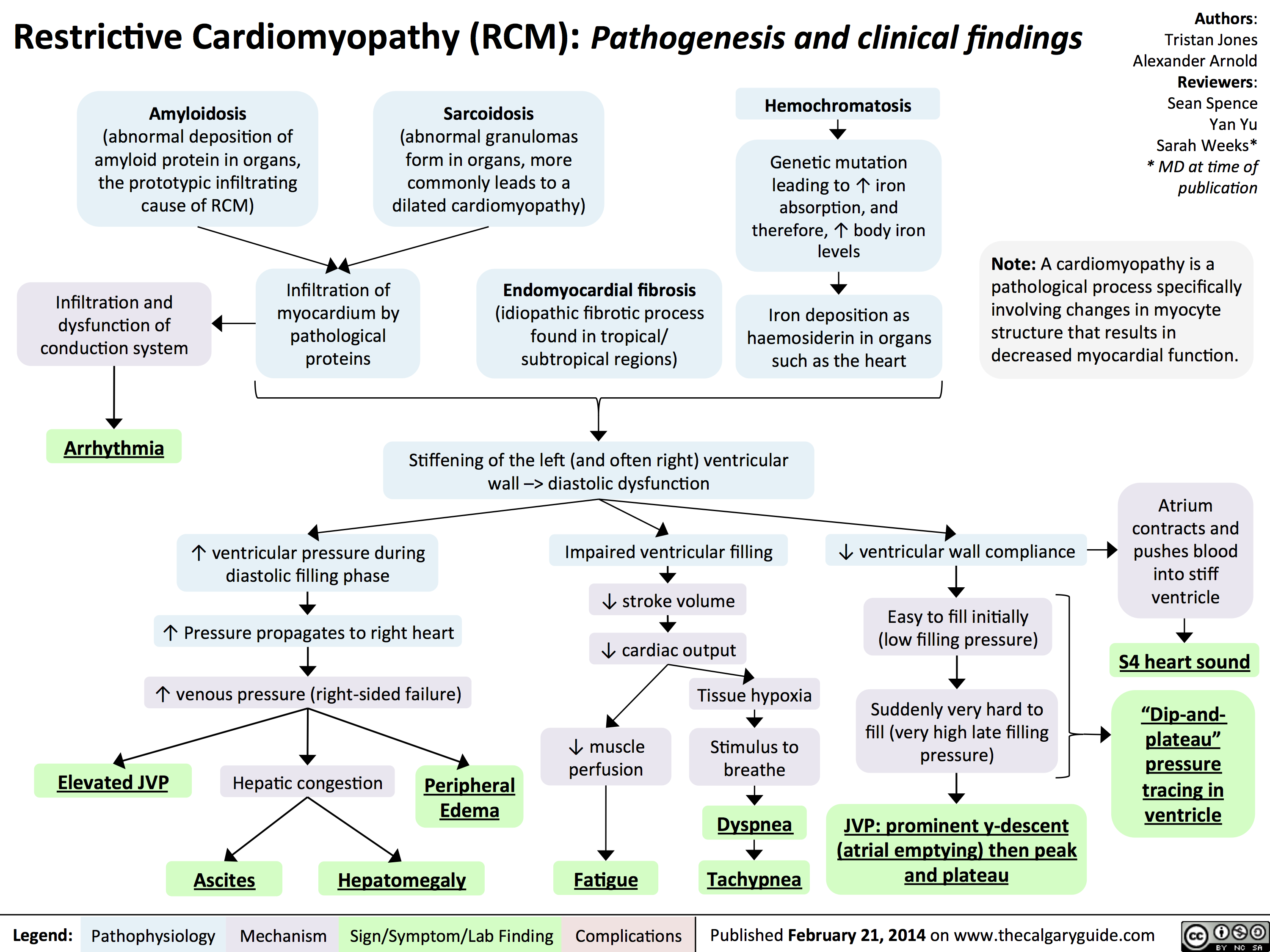
Hypertrophic Cardiomyopathy
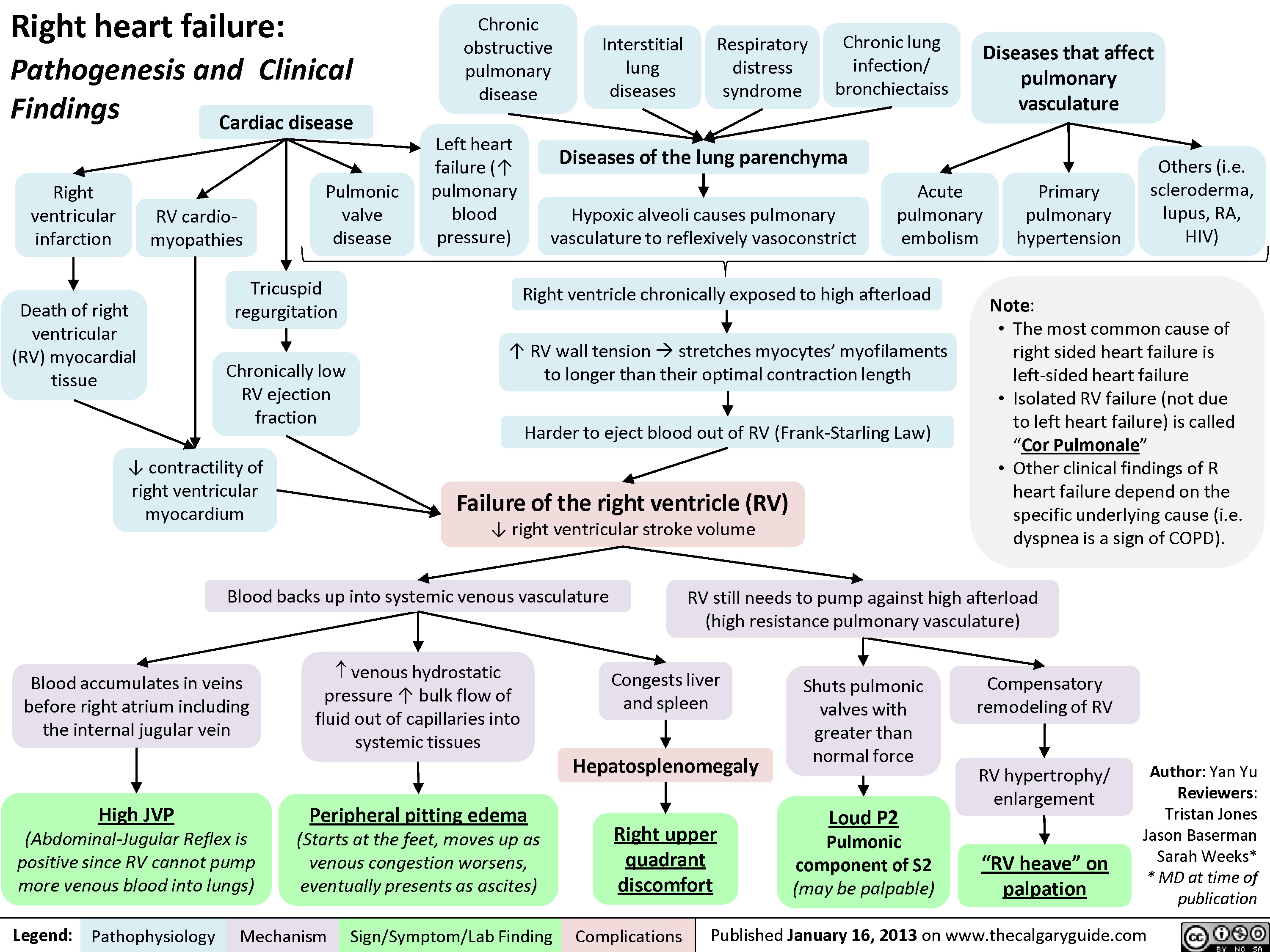
Left Heart Failure - Pathogenesis
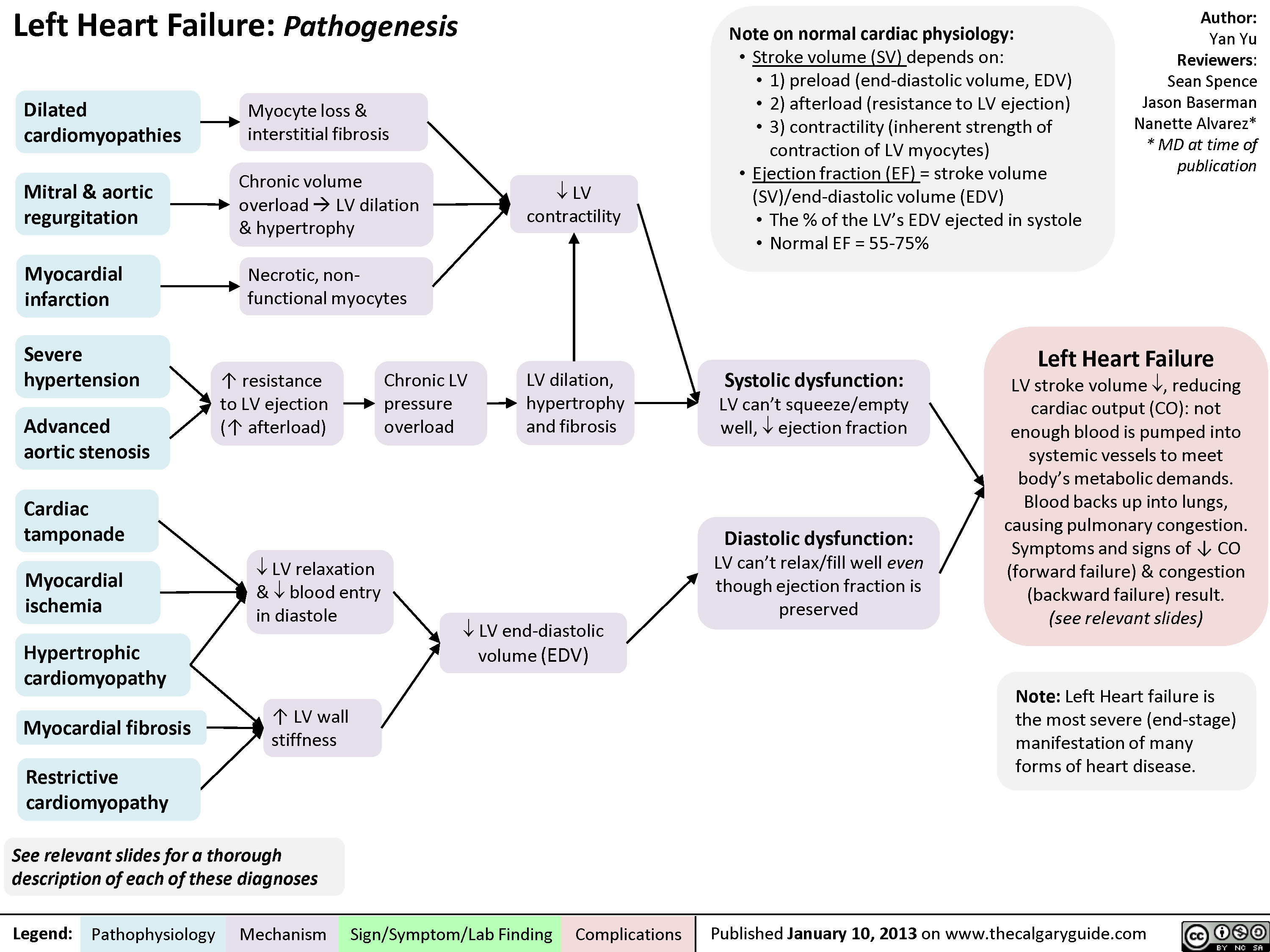
Right Heart Failure
Left Heart Failure
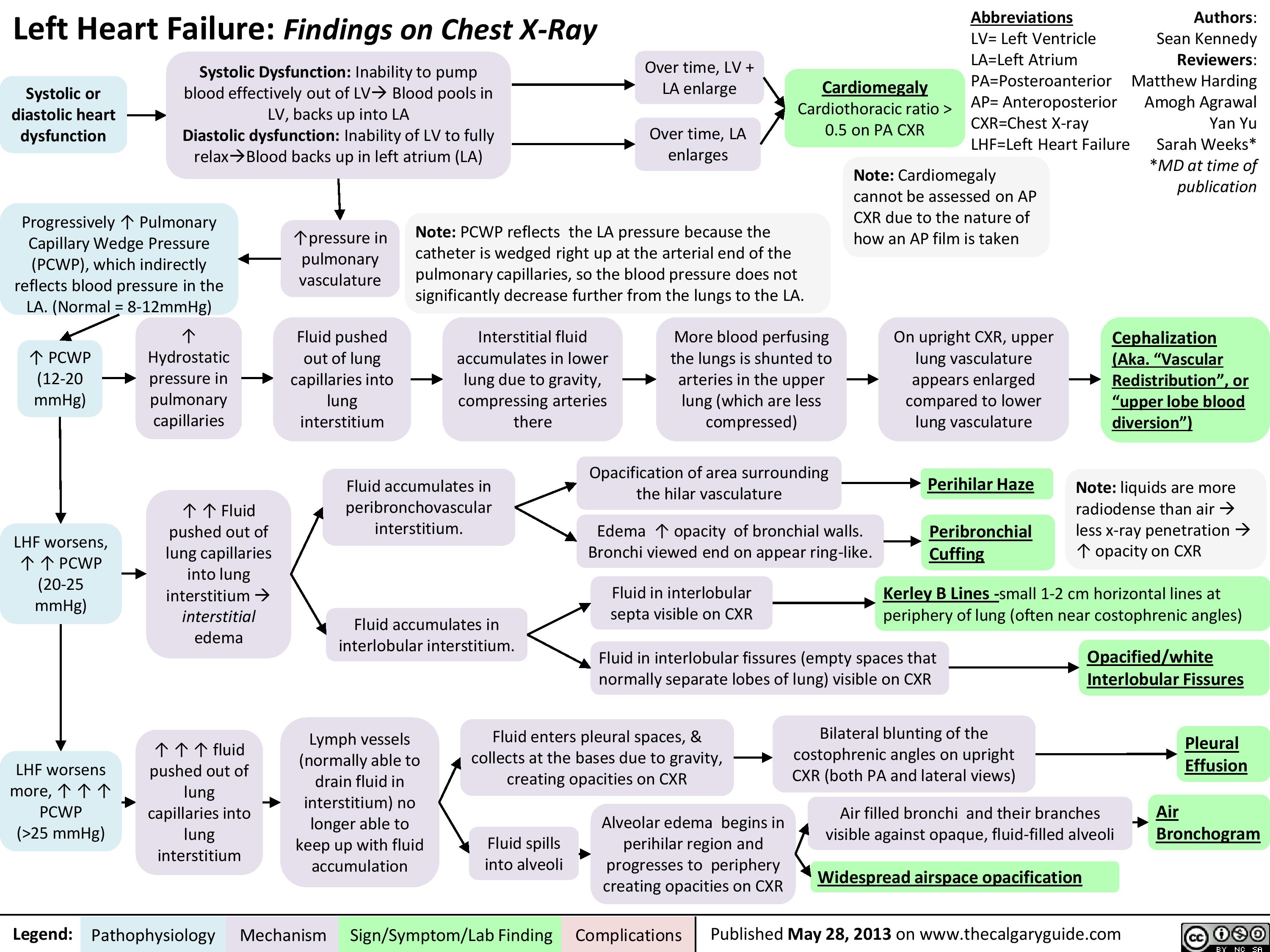
Left Heart Failure - Physical Exam Findings
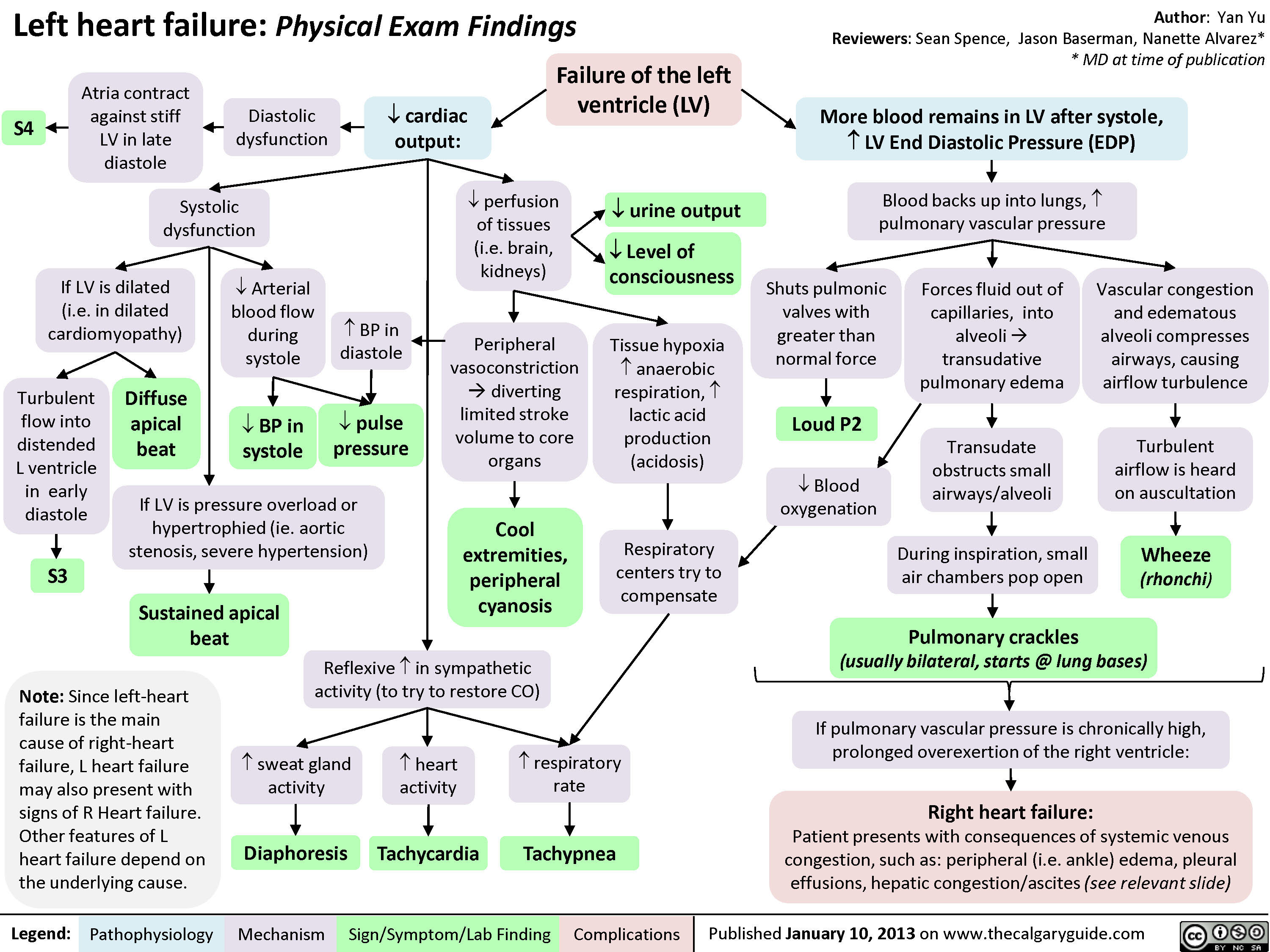
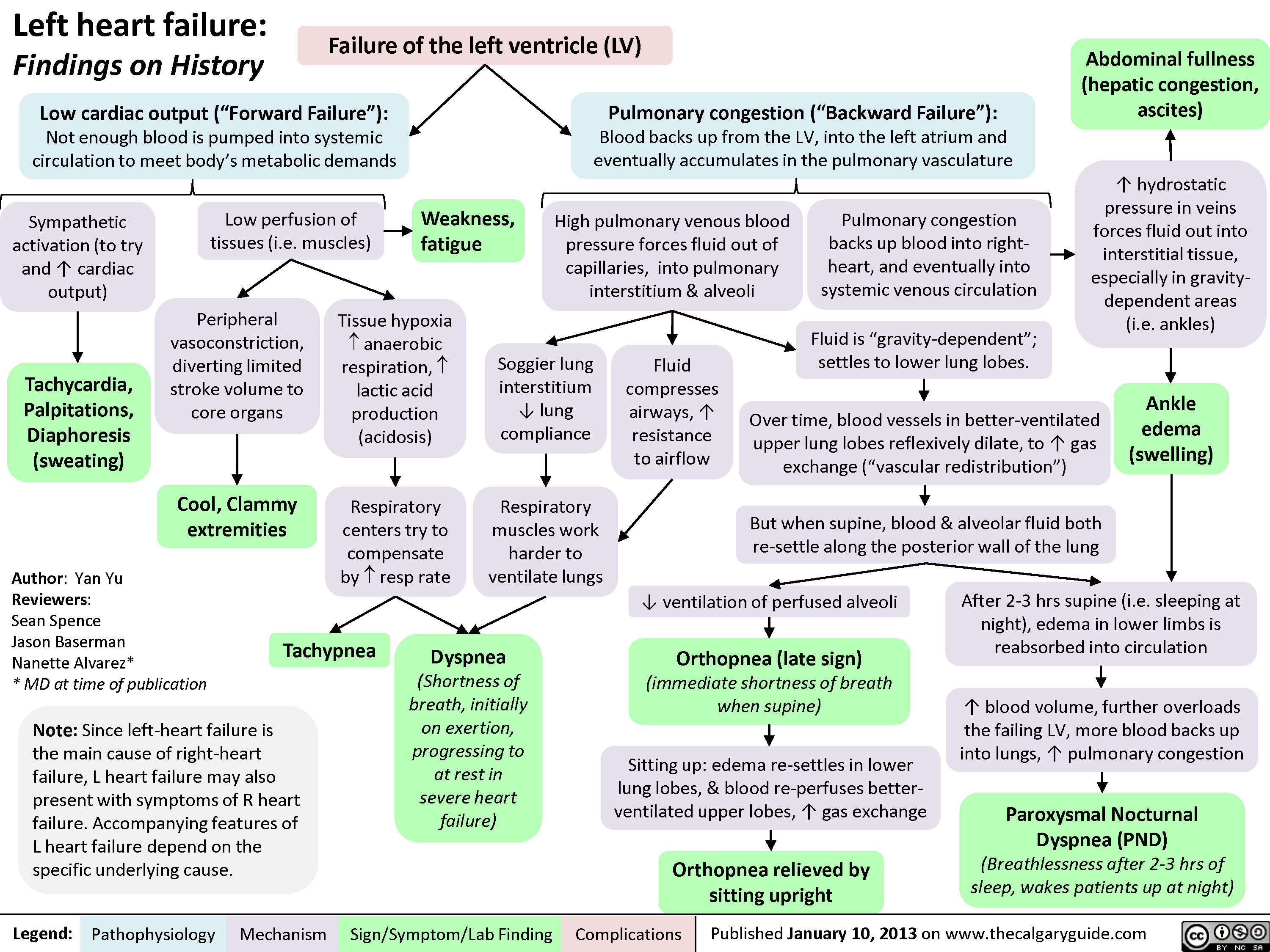
Left Heart Failure - Findings on History
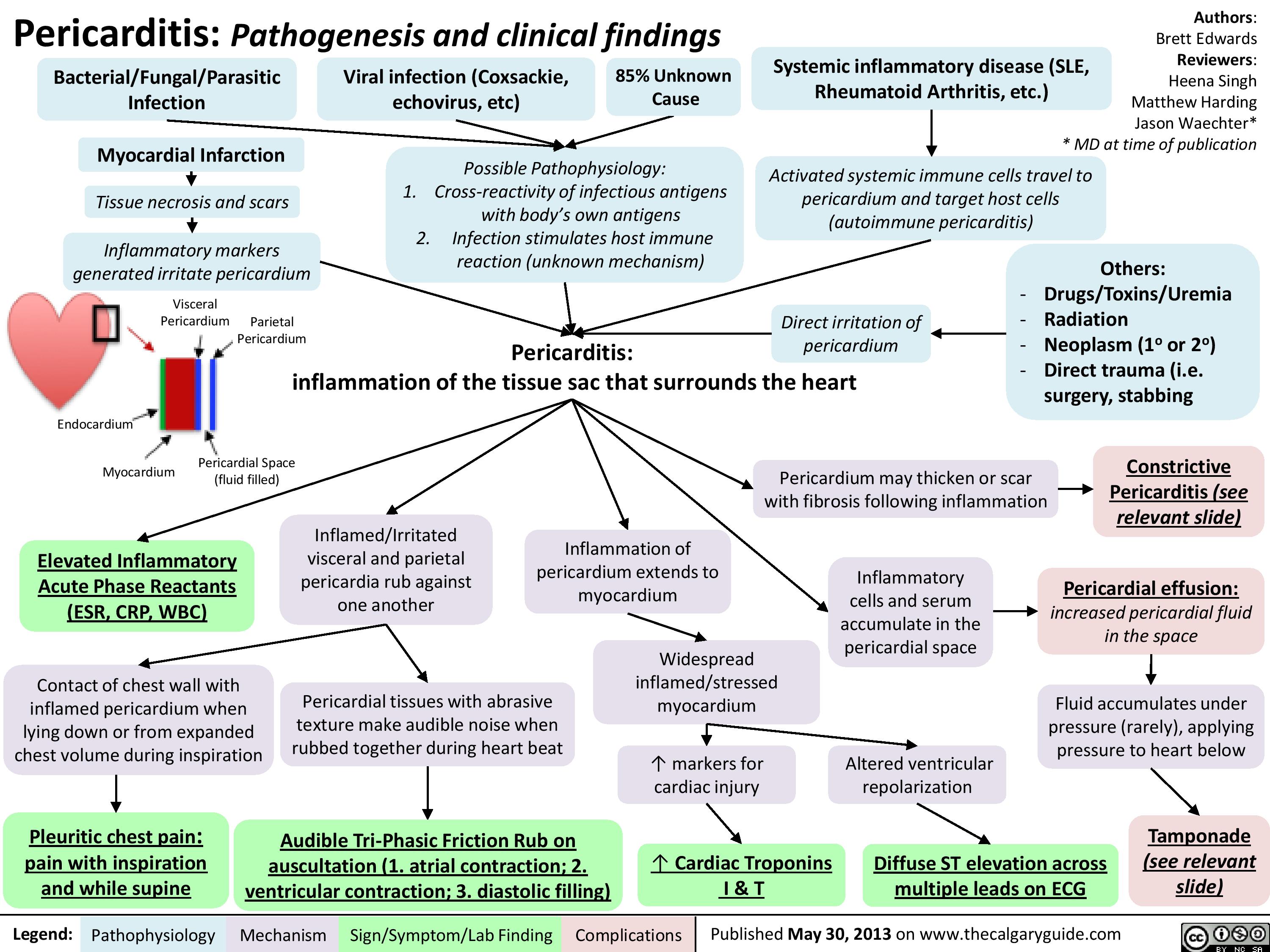
pericarditis
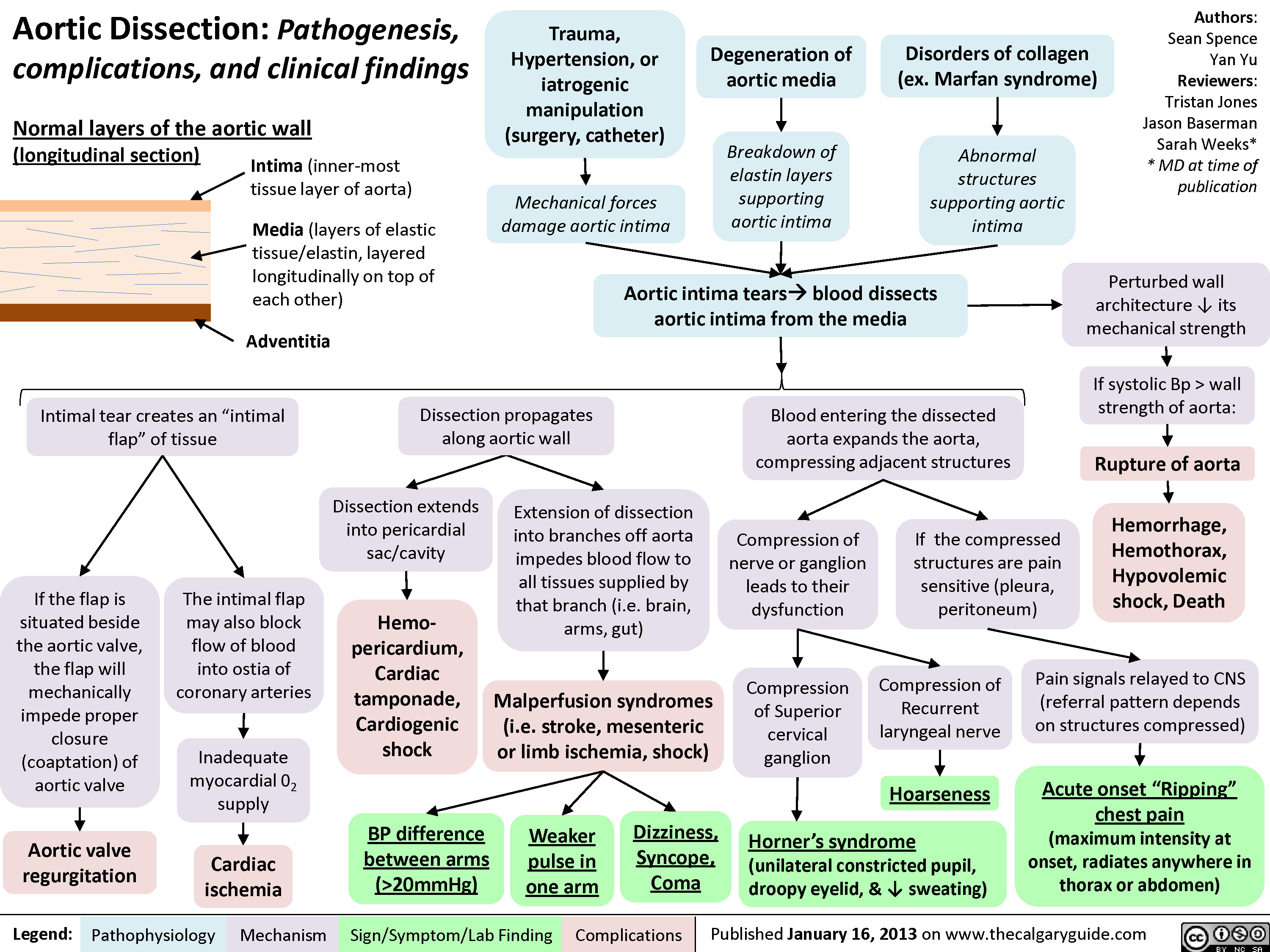
Aortic Dissection
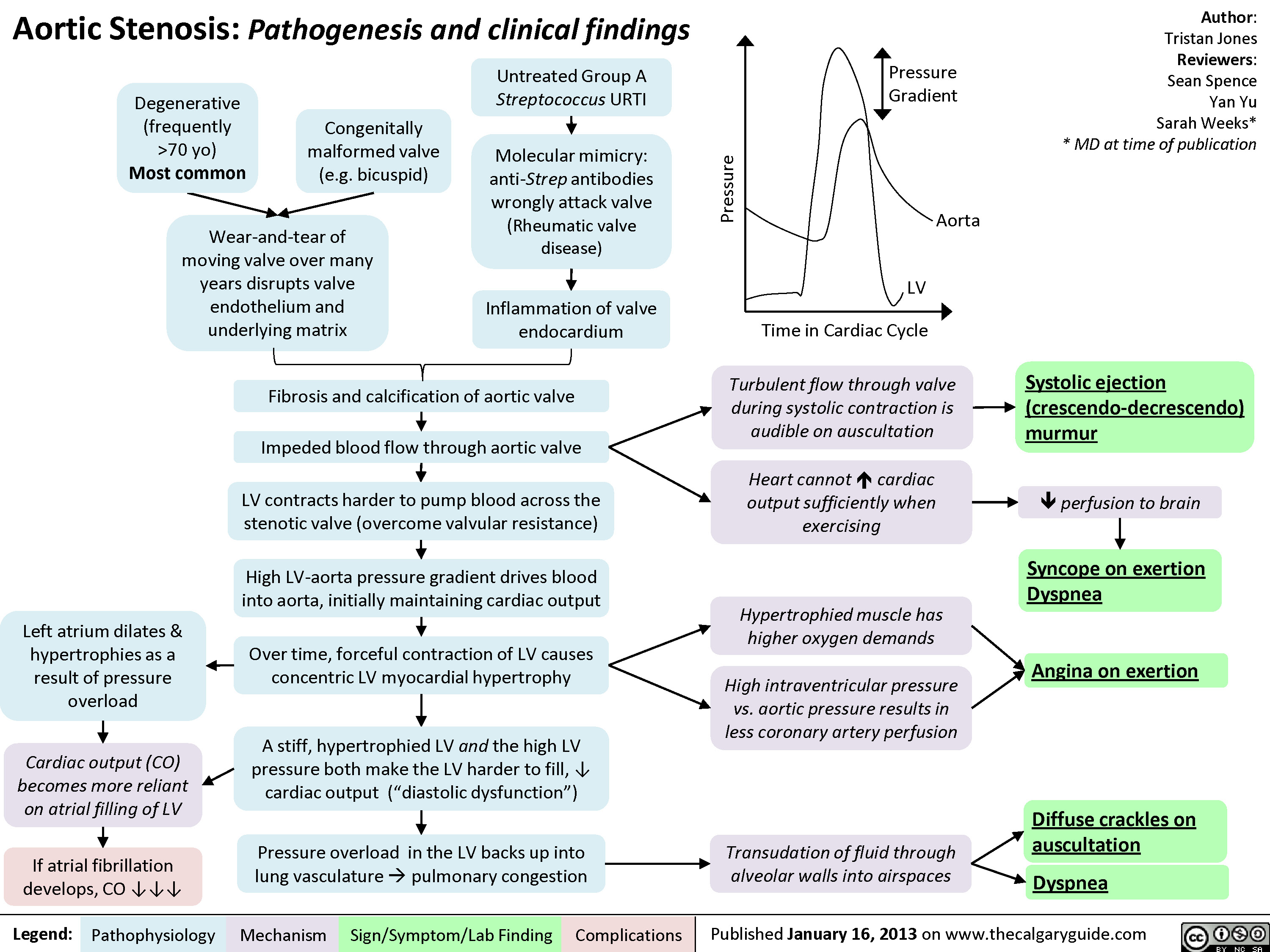
Aortic Stenosis - Pathogenesis and Clinical Findings
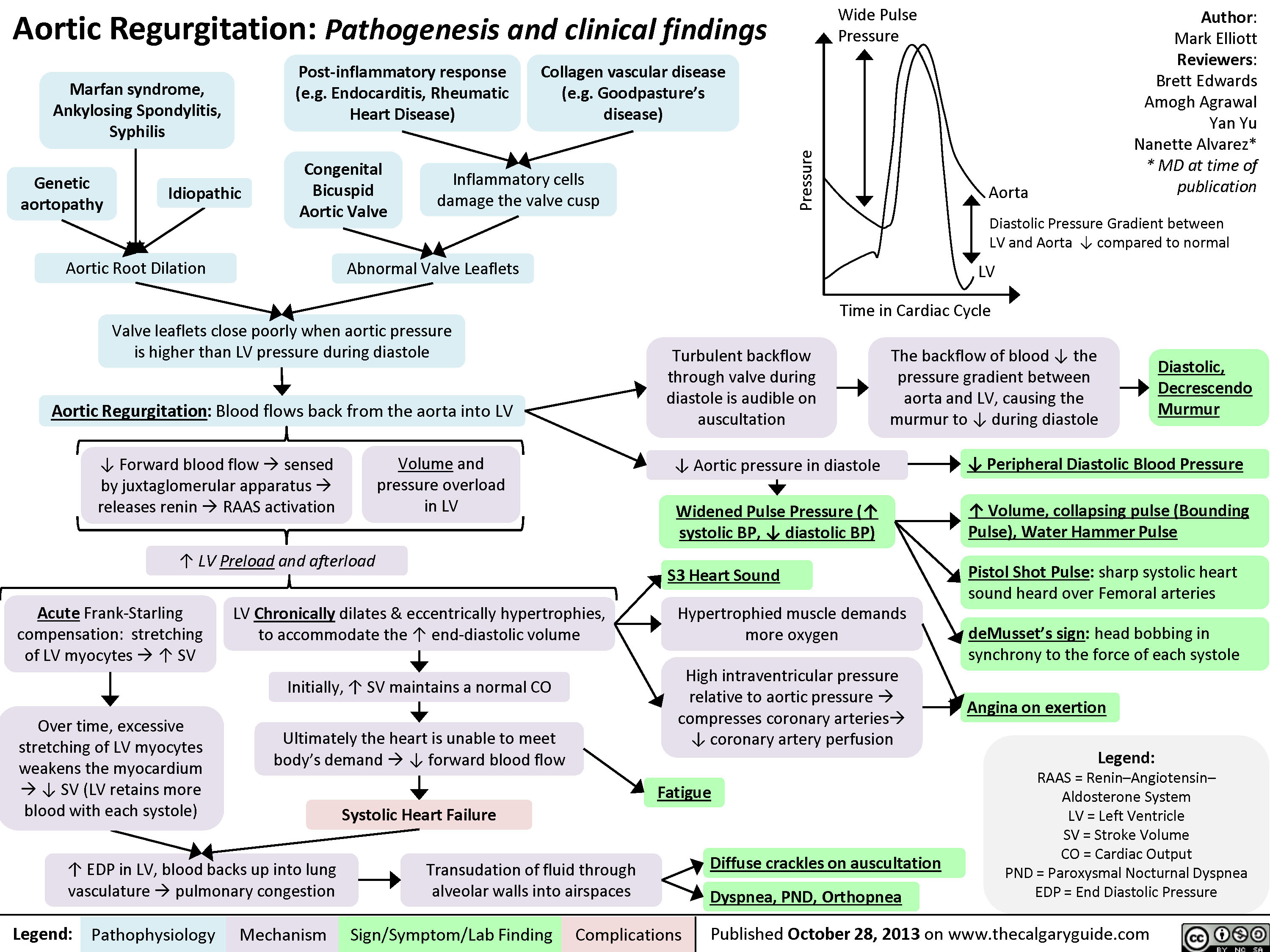
Aortic Regurgitation
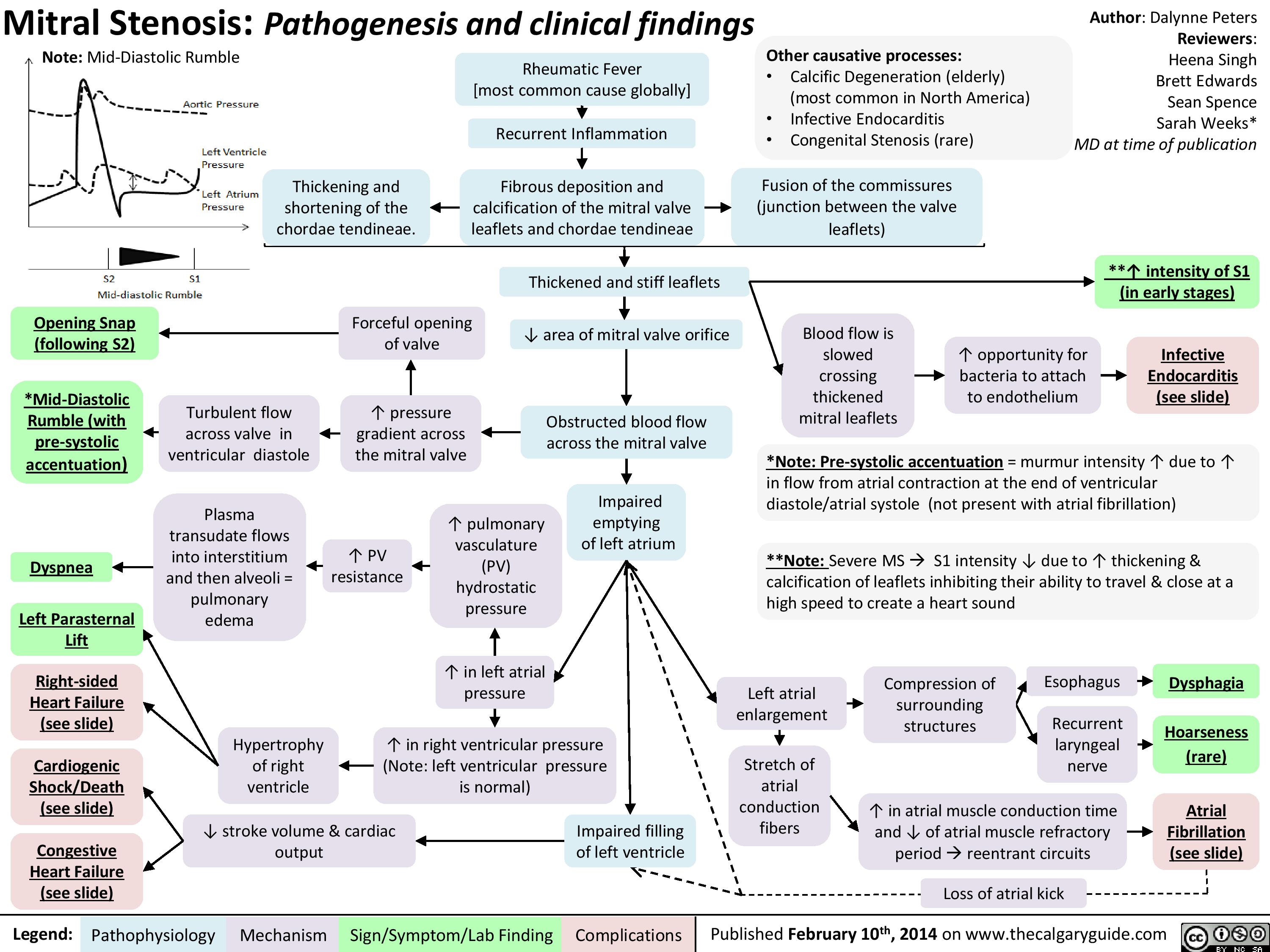
Mitral Stenosis
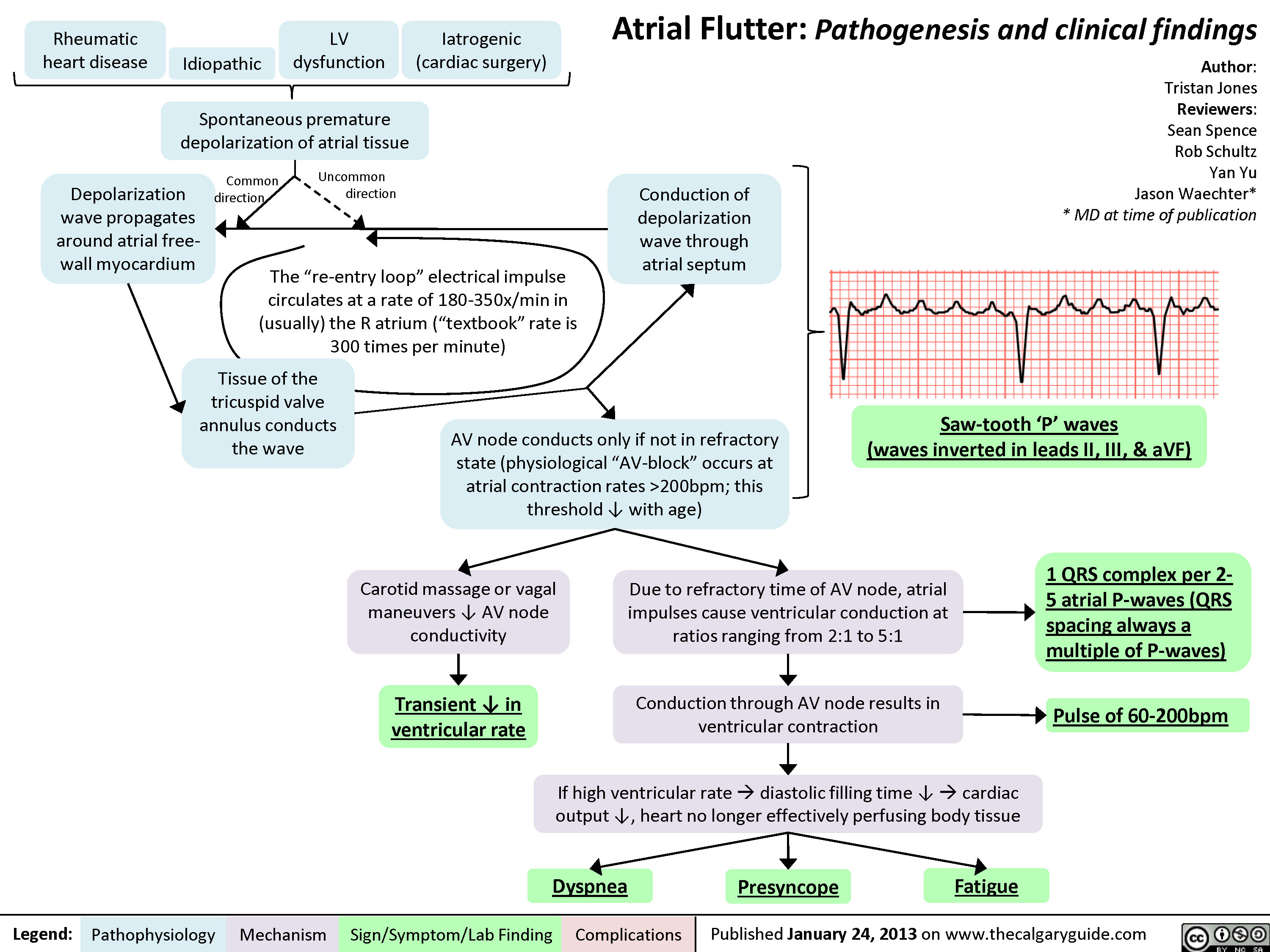
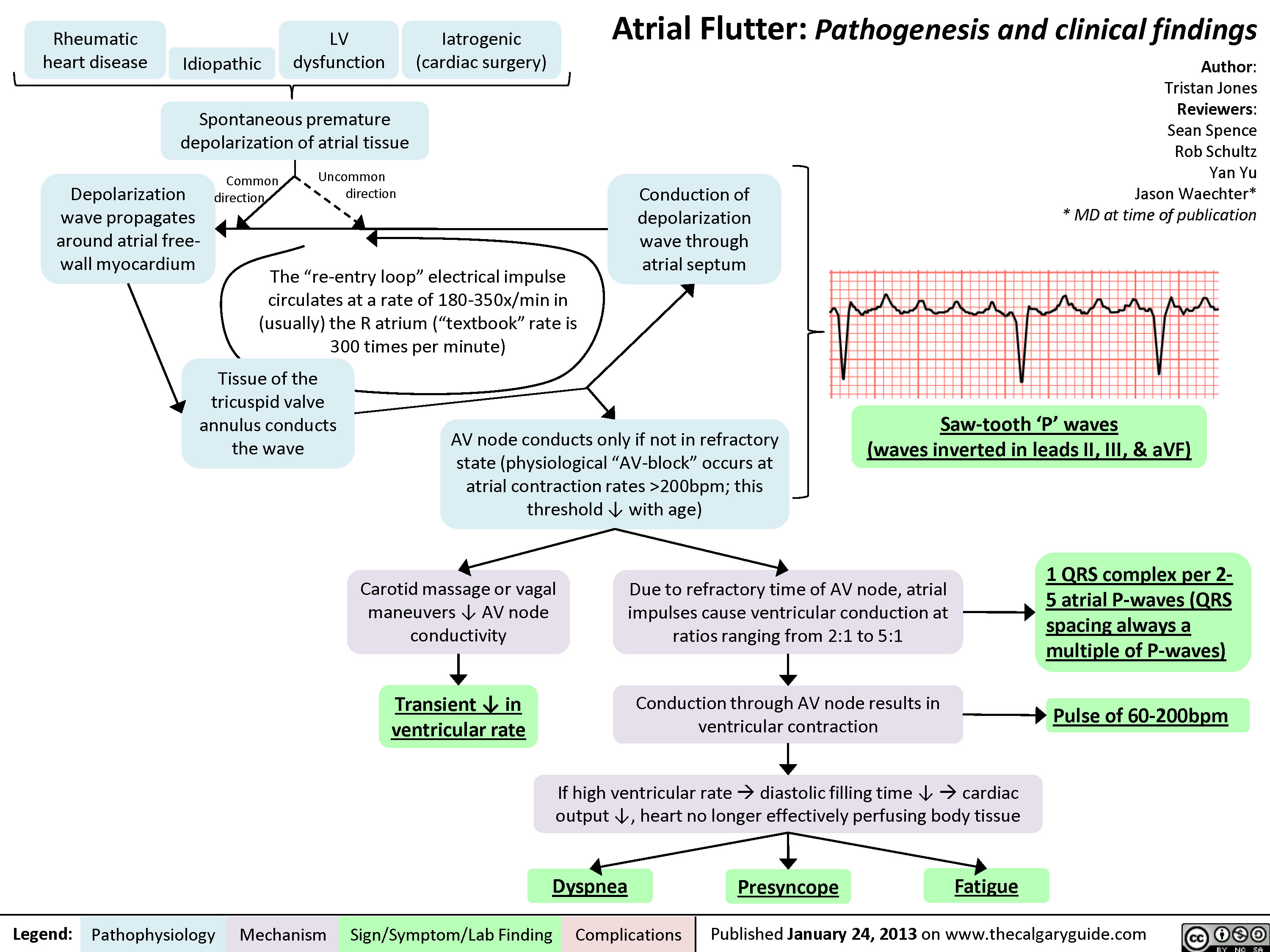
Atrial Flutter
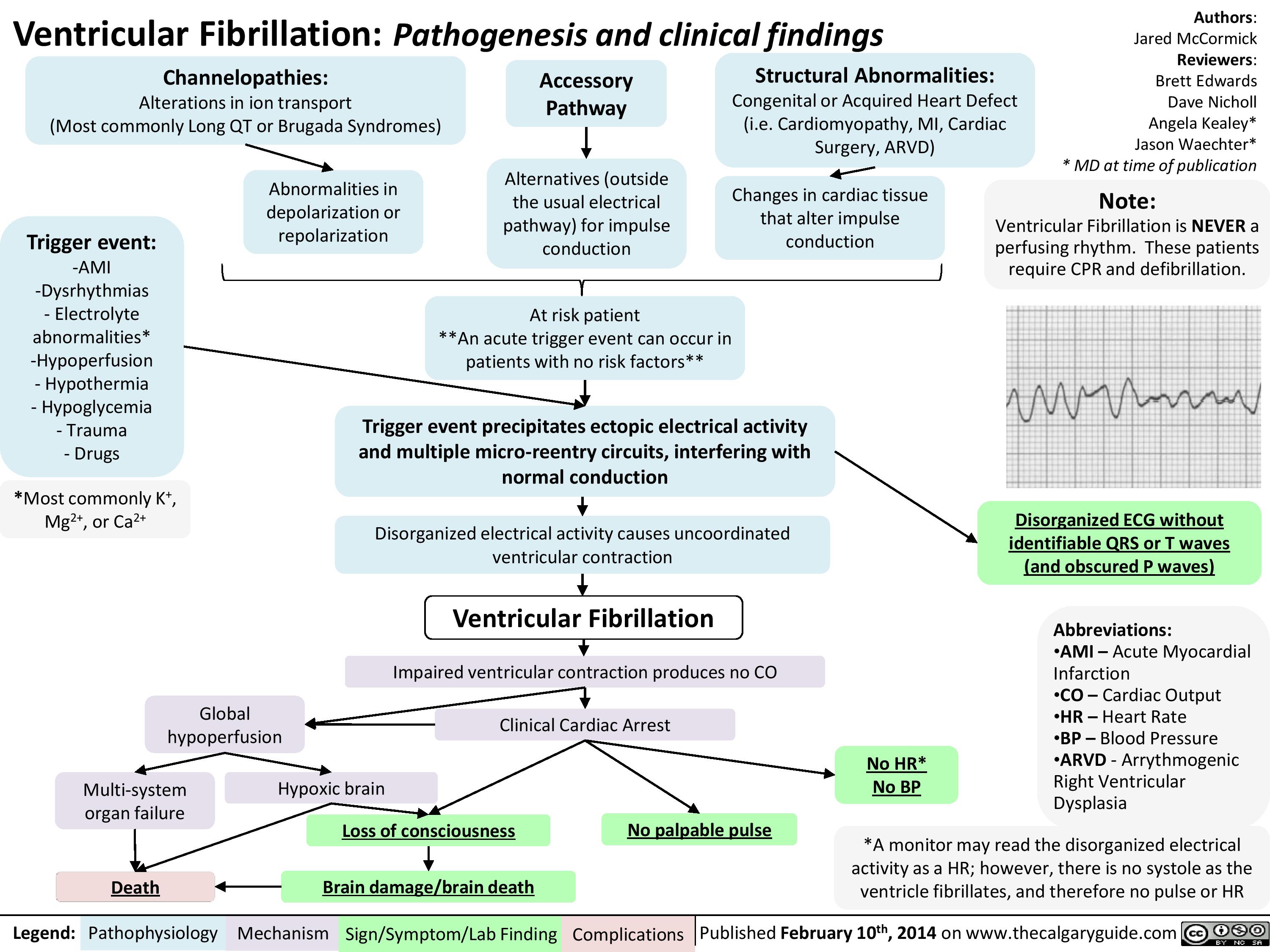
Ventricular fibrillation
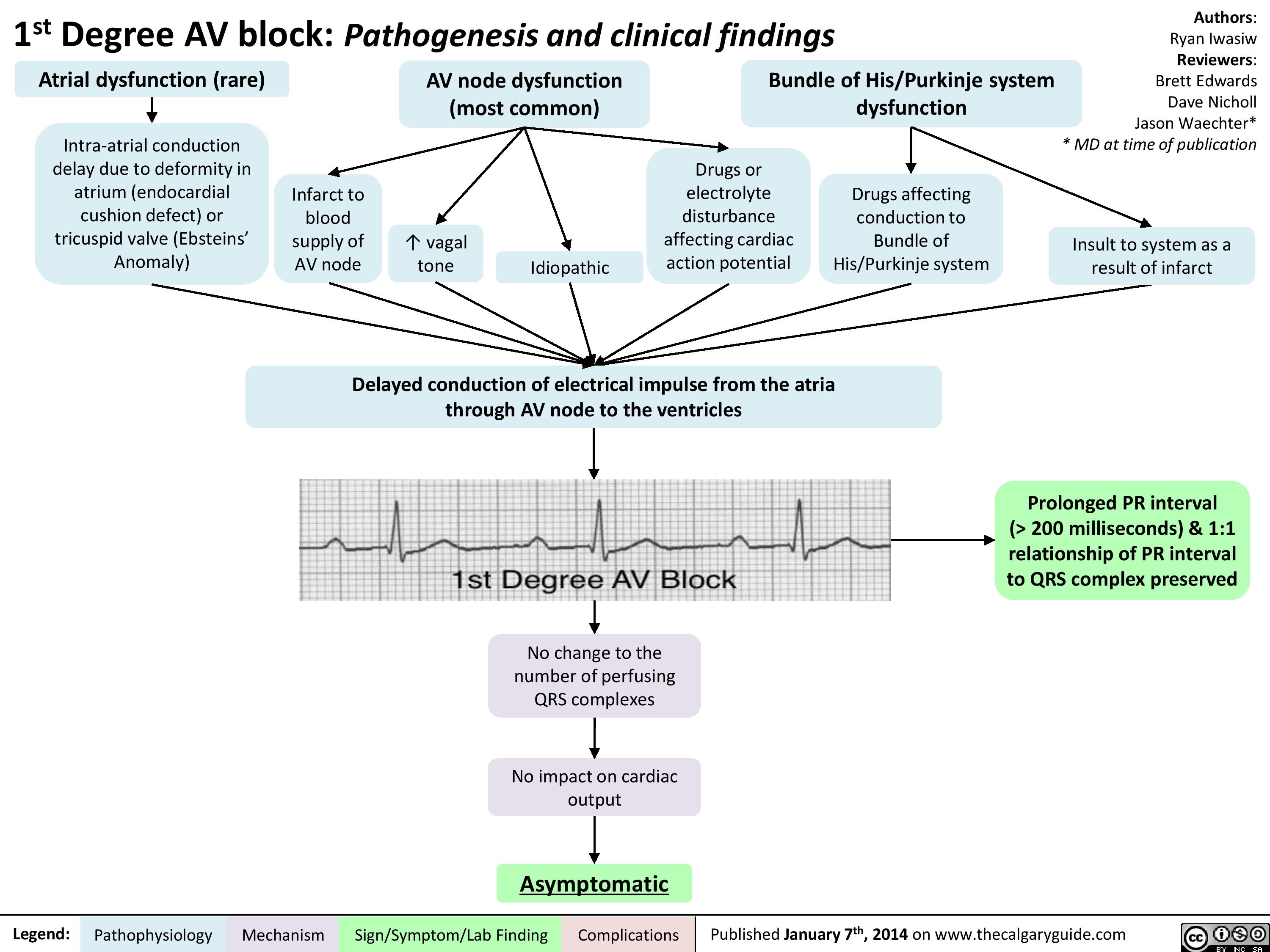
1st Degree AV block
Second Degree Heart Block - Mobitz Type II - Pathogenesis and clinical findings

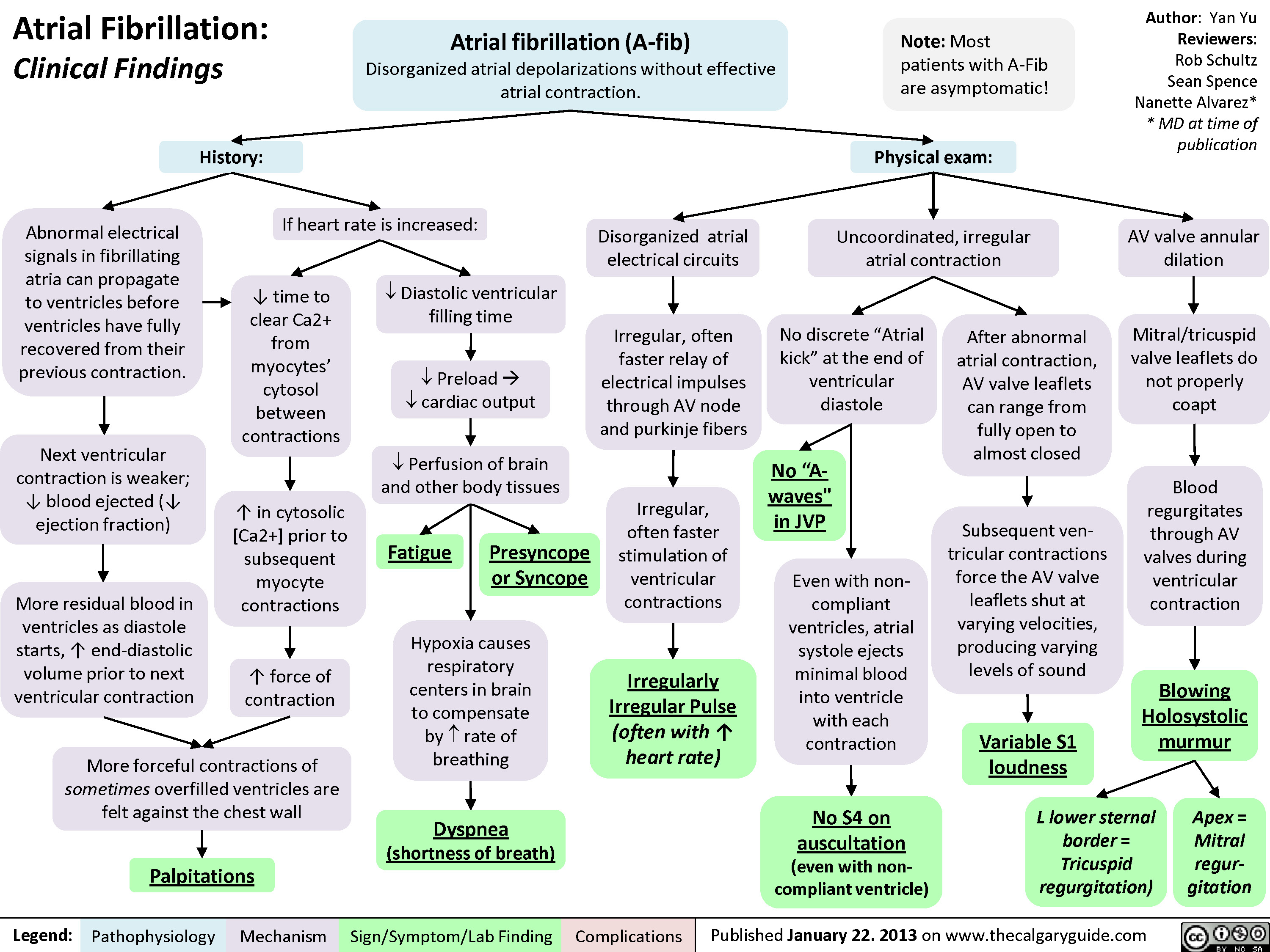
Atrial Fibrillation - Clinical Findings
atrial-fibrillation-complications

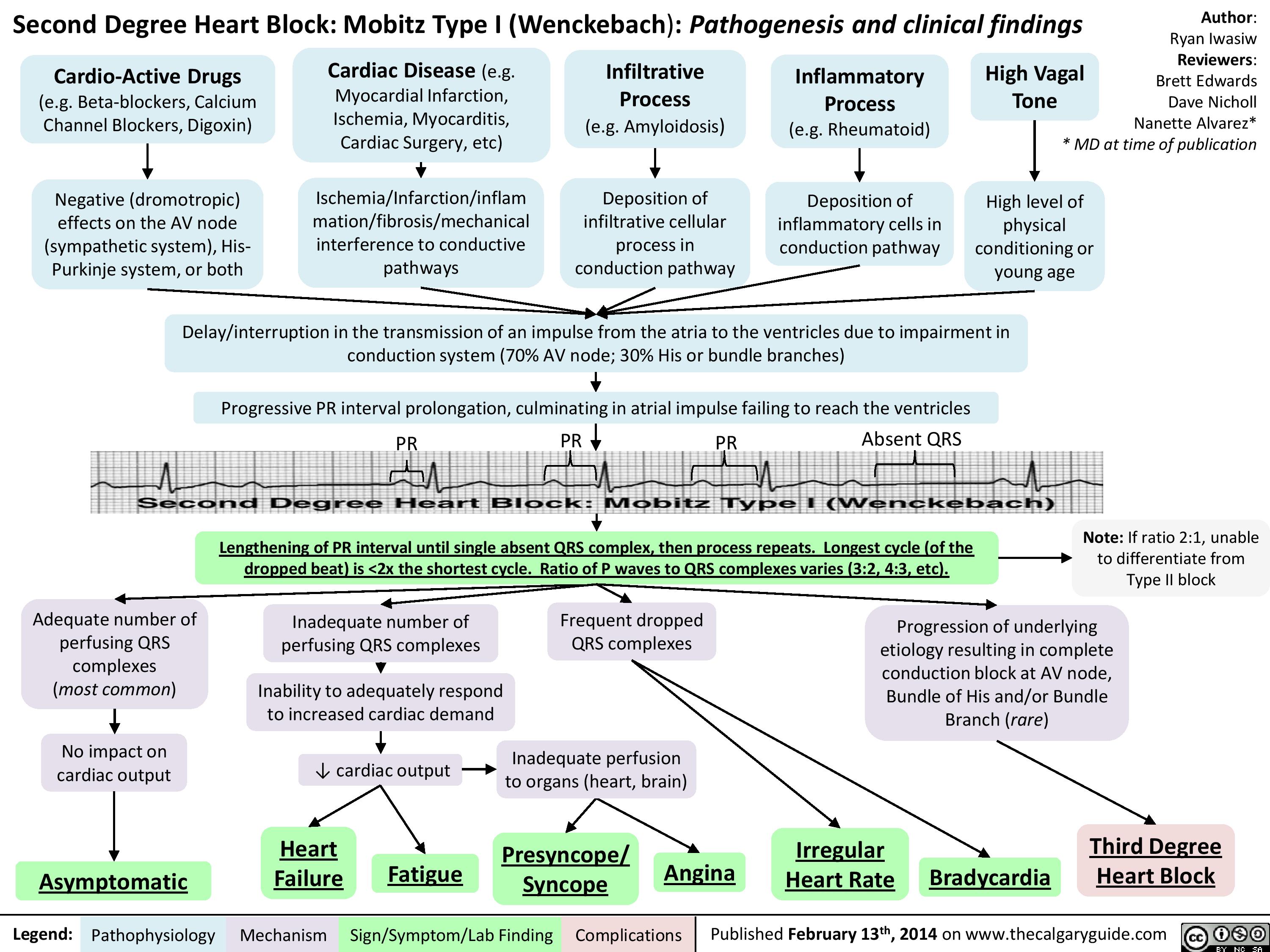
Second Degree Heart Block - Mobitz Type I (Wenckebach) - Pathogenesis and Clinical Findings
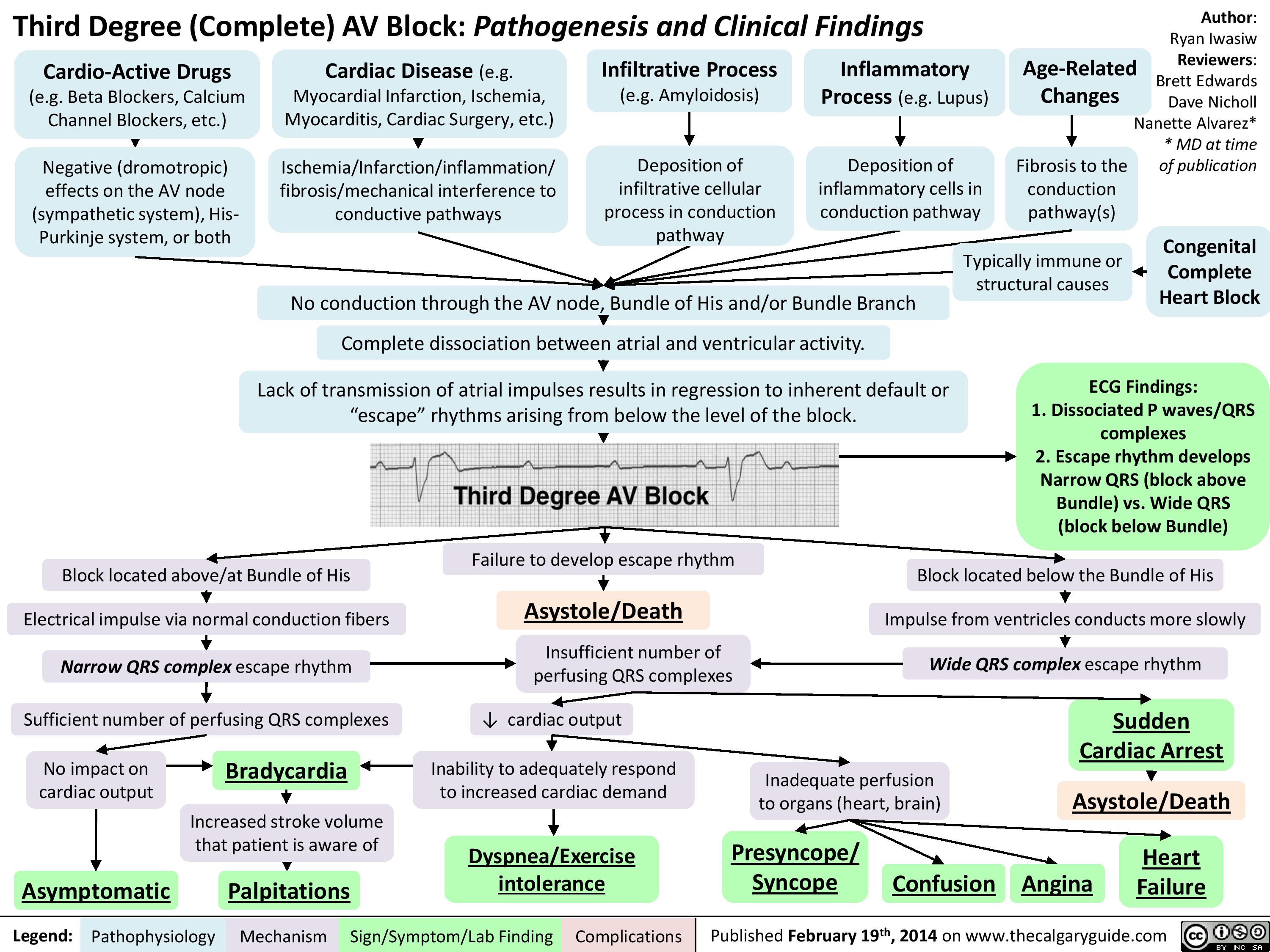
Third Degree (Complete) AV Block - Pathogenesis and Clinical Findings
Atrial Flutter (1)
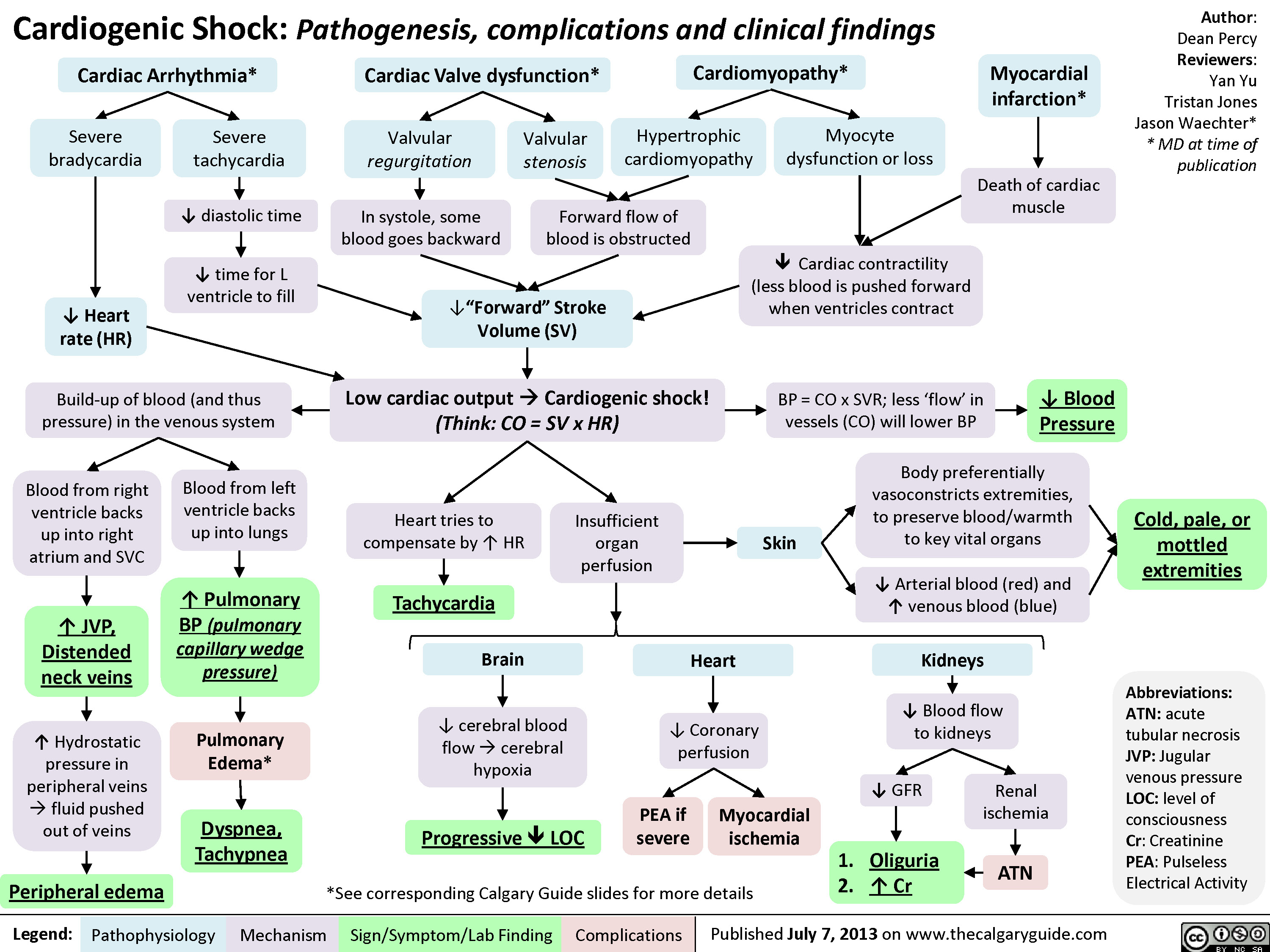
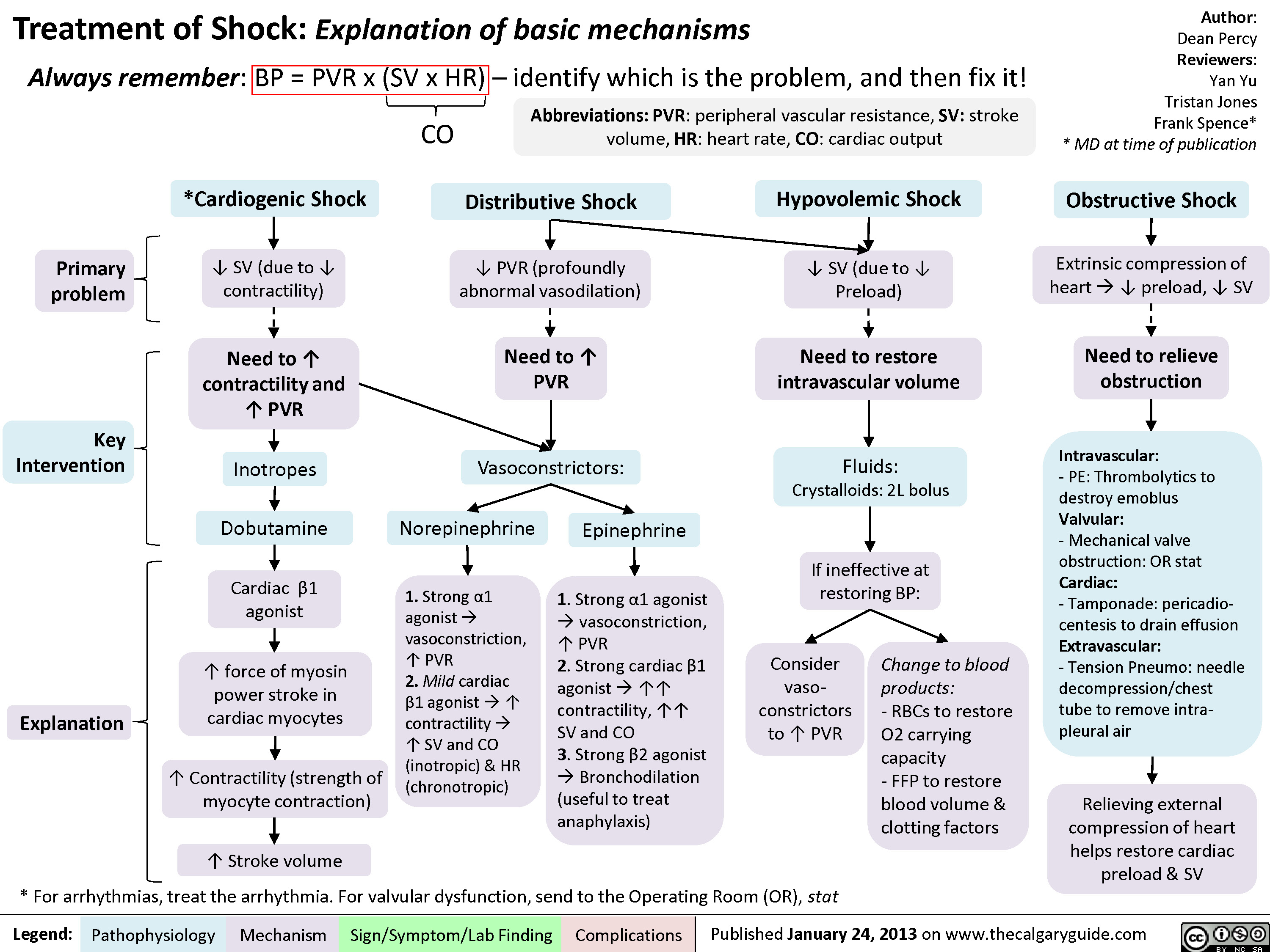
Cardiogenic Shock
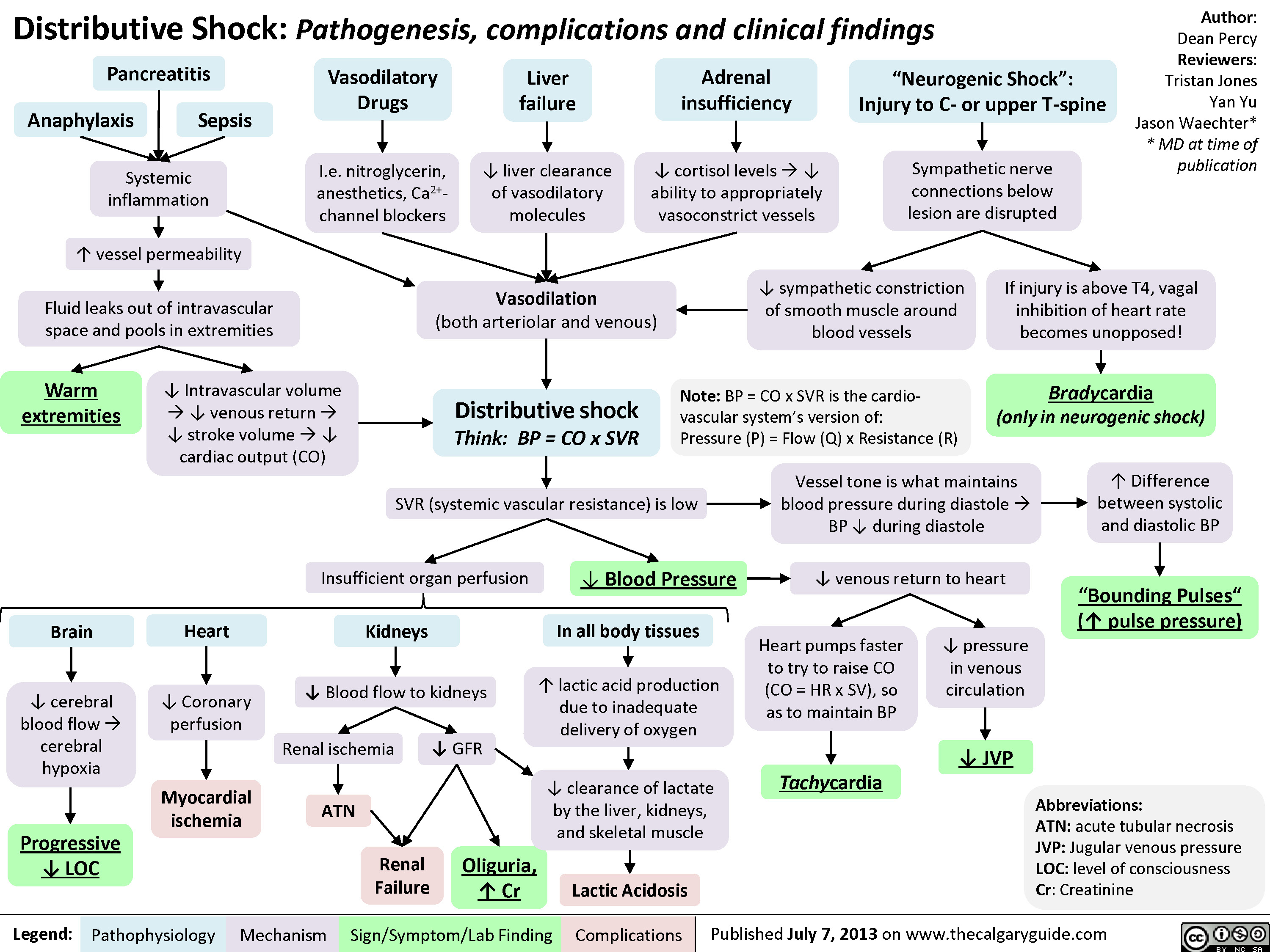
Distributive Shock
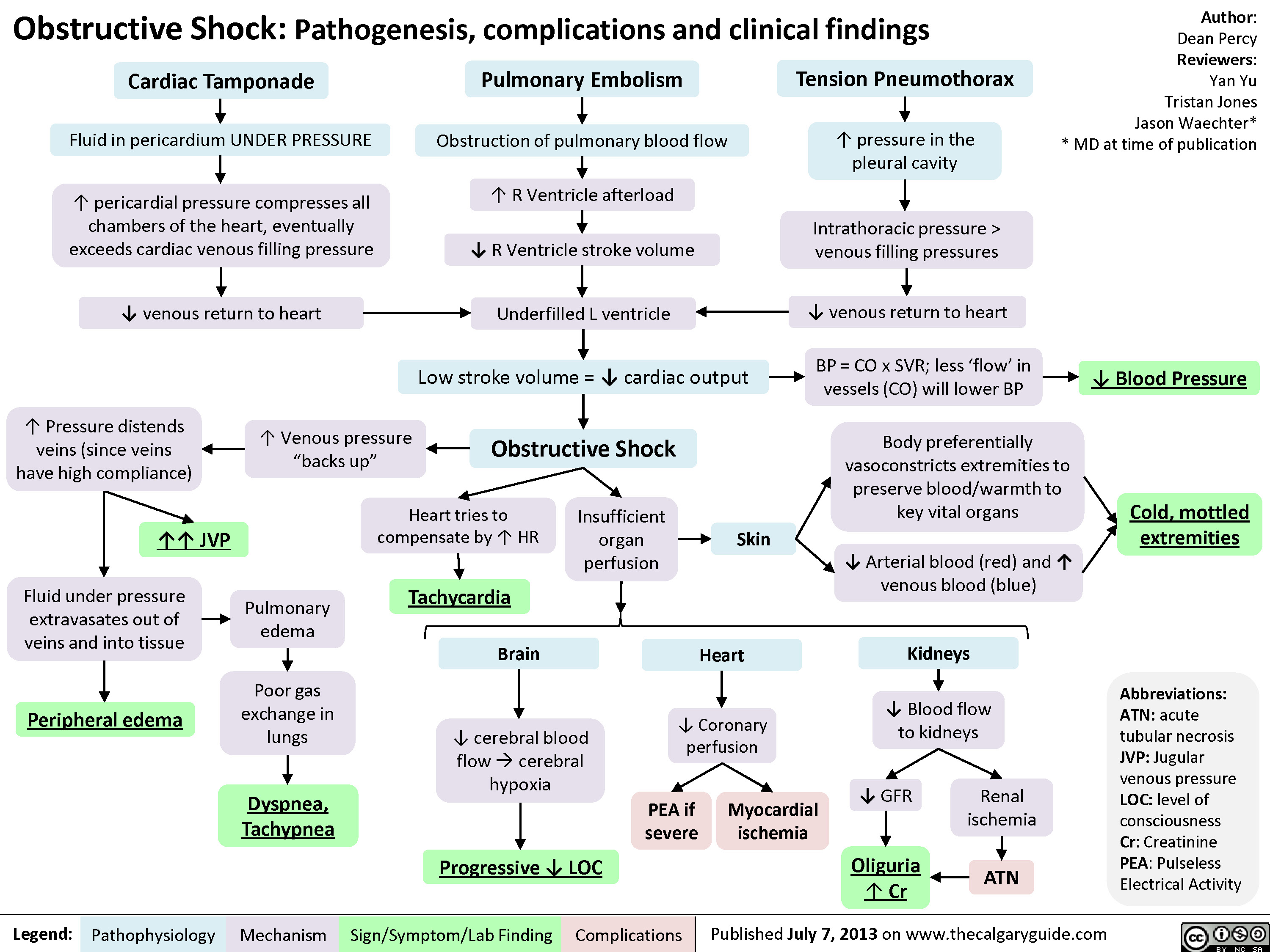
Obstructive Shock
Drugs used to treat shock
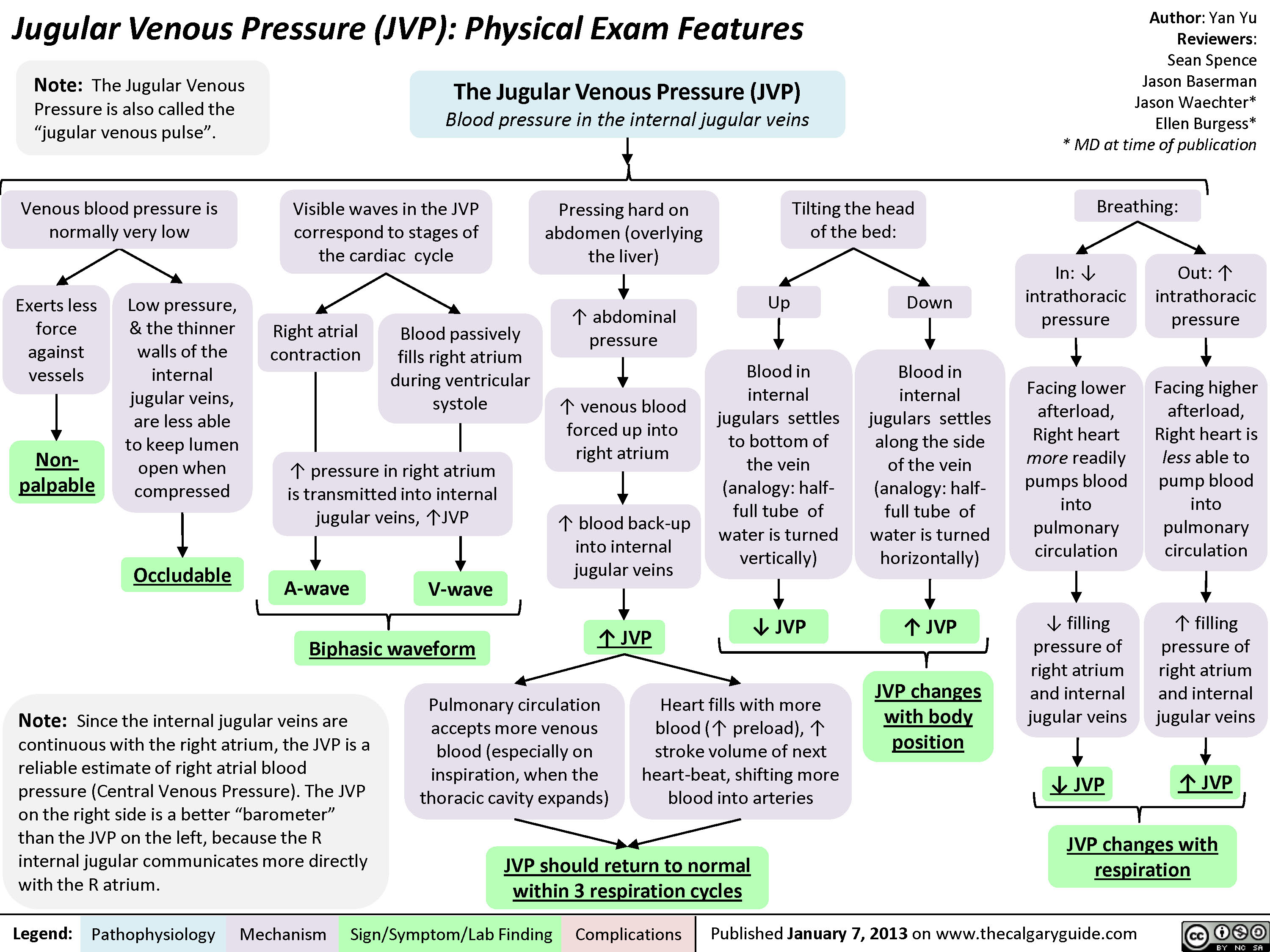
JVP-Physical Exam Features
jvp-kussmals-sign-explained
: Kussmal's Sign explainedExcessive pericardial fluid compresses heart walls on all sidesLegend:Published January 7, 2013 on www.thecalgaryguide.comMechanismPathophysiologySign/Symptom/Lab FindingComplicationsAuthor: Yan YuReviewers:Sean SpenceJason BasermanJason Waechter** MD at time of publication? Right ventricle wall complianceConstrictive pericarditisRight ventricle prevented from fully expanding ? ability of the right ventricle to accommodate higher venous returnBackup of venous blood into right atrium and preceding internal jugular veinsRestrictive cardiomyopathyInflamed, fibrotic pericardium restricts expansion of heartRight ventricle myocardial infarct Cardiac tamponade (rare)InspirationMore venous blood tries to enter the low-pressure thoracic cavity via the right ventricle? pressure in thoracic cavity
JVP should return to normal within 3 respiration cyclesJugular Venous Pressure (JVP): Physical Exam FeaturesExerts less force against vesselsTilting the head of the bed:Low pressure, & the thinner walls of the internal jugular veins, are less able to keep lumen open when compressedLegend:Published January 7, 2013 on www.thecalgaryguide.comMechanismPathophysiologySign/Symptom/Lab FindingComplicationsAuthor: Yan YuReviewers:Sean SpenceJason BasermanJason Waechter** MD at time of publicationBlood in internal jugulars settles to bottom of the vein (analogy: half-full tube of water is turned vertically)Visible waves in the JVP correspond to stages of the cardiac cycleNon-palpableBiphasic waveform? JVP The Jugular Venous Pressure (JVP) Blood pressure in the internal jugular veinsV-waveA-waveRight atrial contractionBlood passively fills right atrium during ventricular systole? JVP? JVPIn: ? intrathoracic pressurePressing hard on abdomen (overlying the liver), or doing a valsalvaFacing lower afterload, Right heart more readily pumps blood into pulmonary circulation? abdominal pressure? venous blood forced up into right atriumVenous blood pressure is normally very lowOccludableNote: Since the internal jugular veins are continuous with the right atrium, the JVP is a reliable estimate of right atrial blood pressure (Central Venous Pressure). The JVP on the right side is a better")
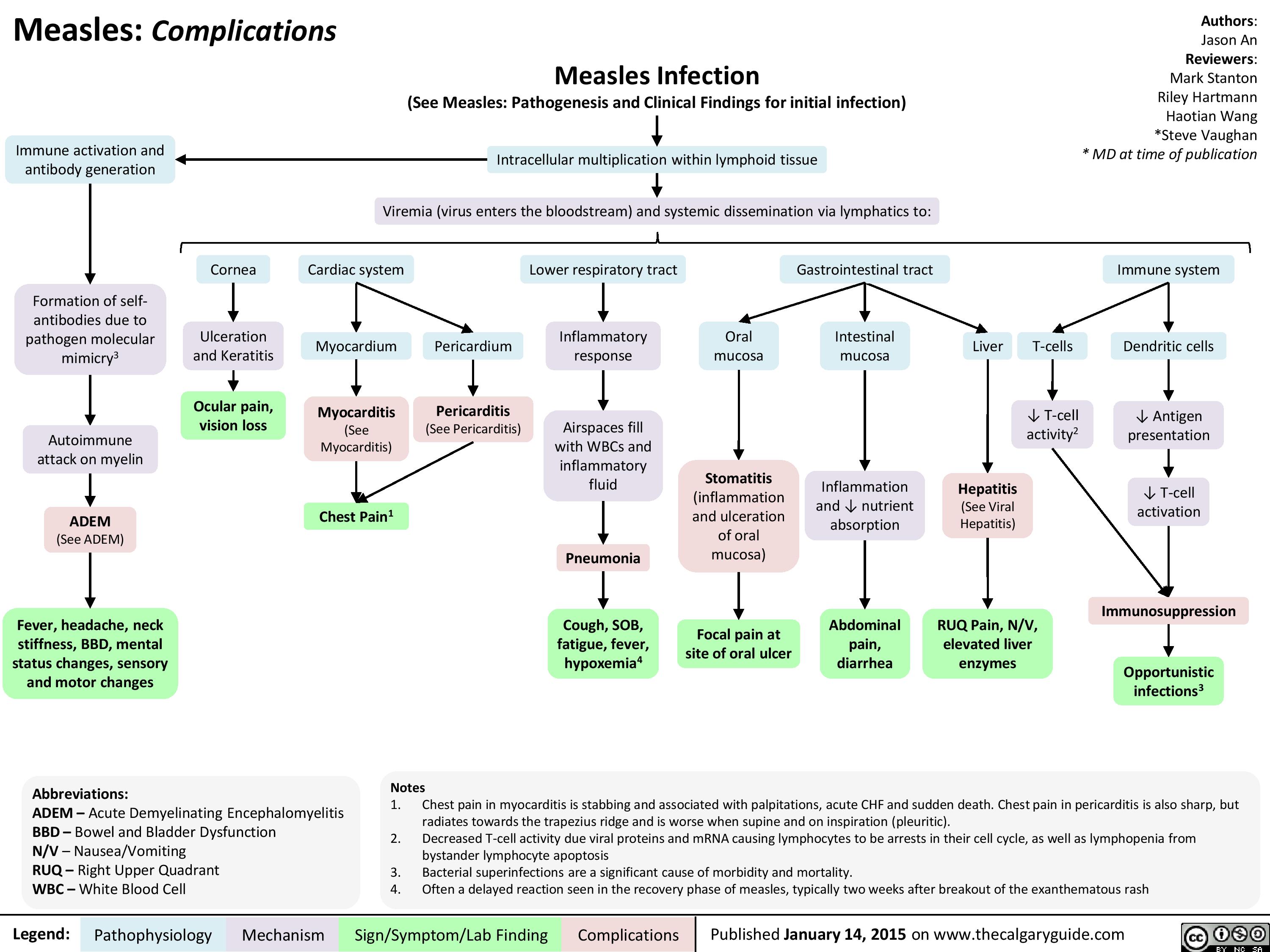
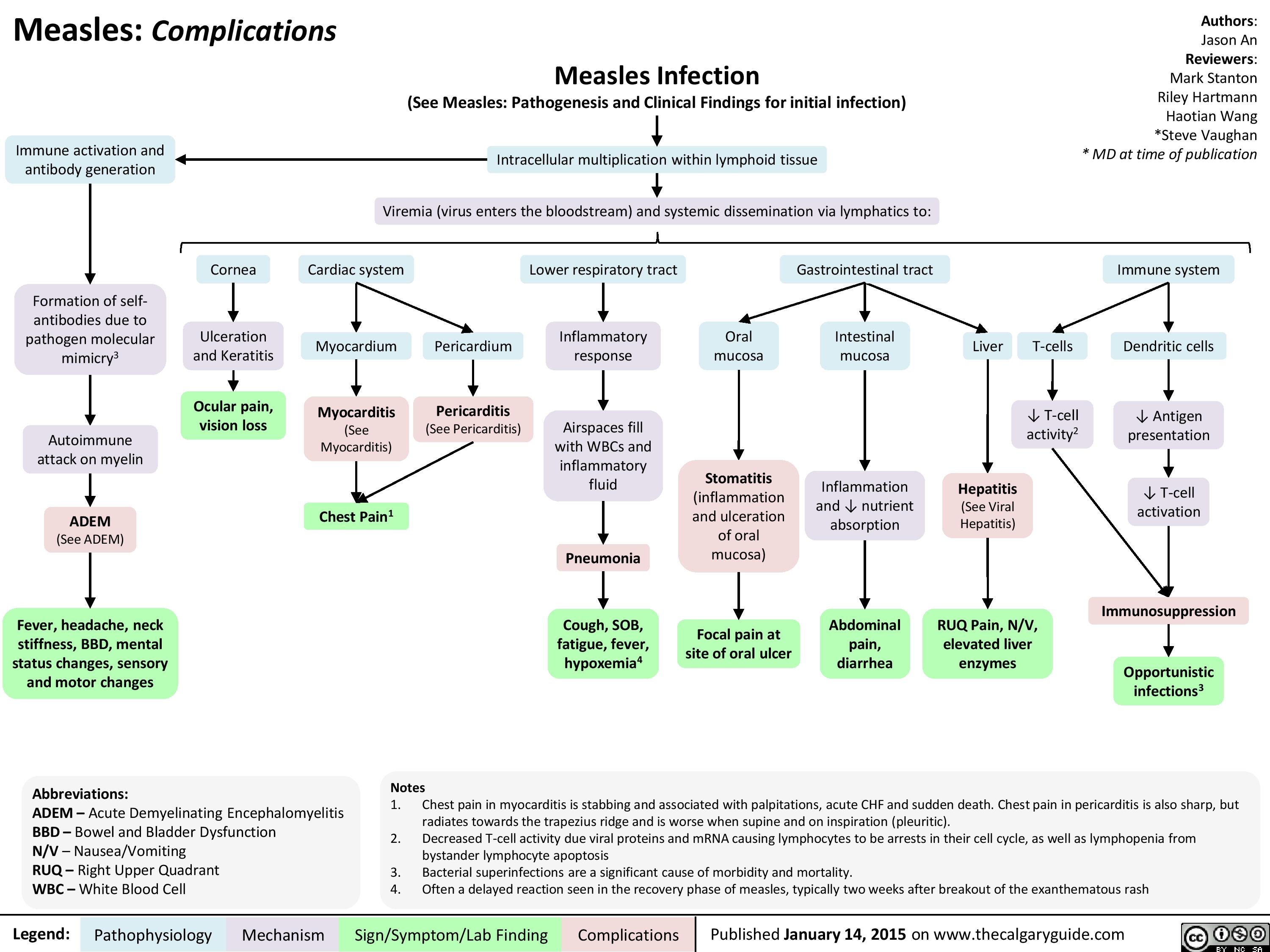
Complications of Measles Pathogenesis and Clinical Findings
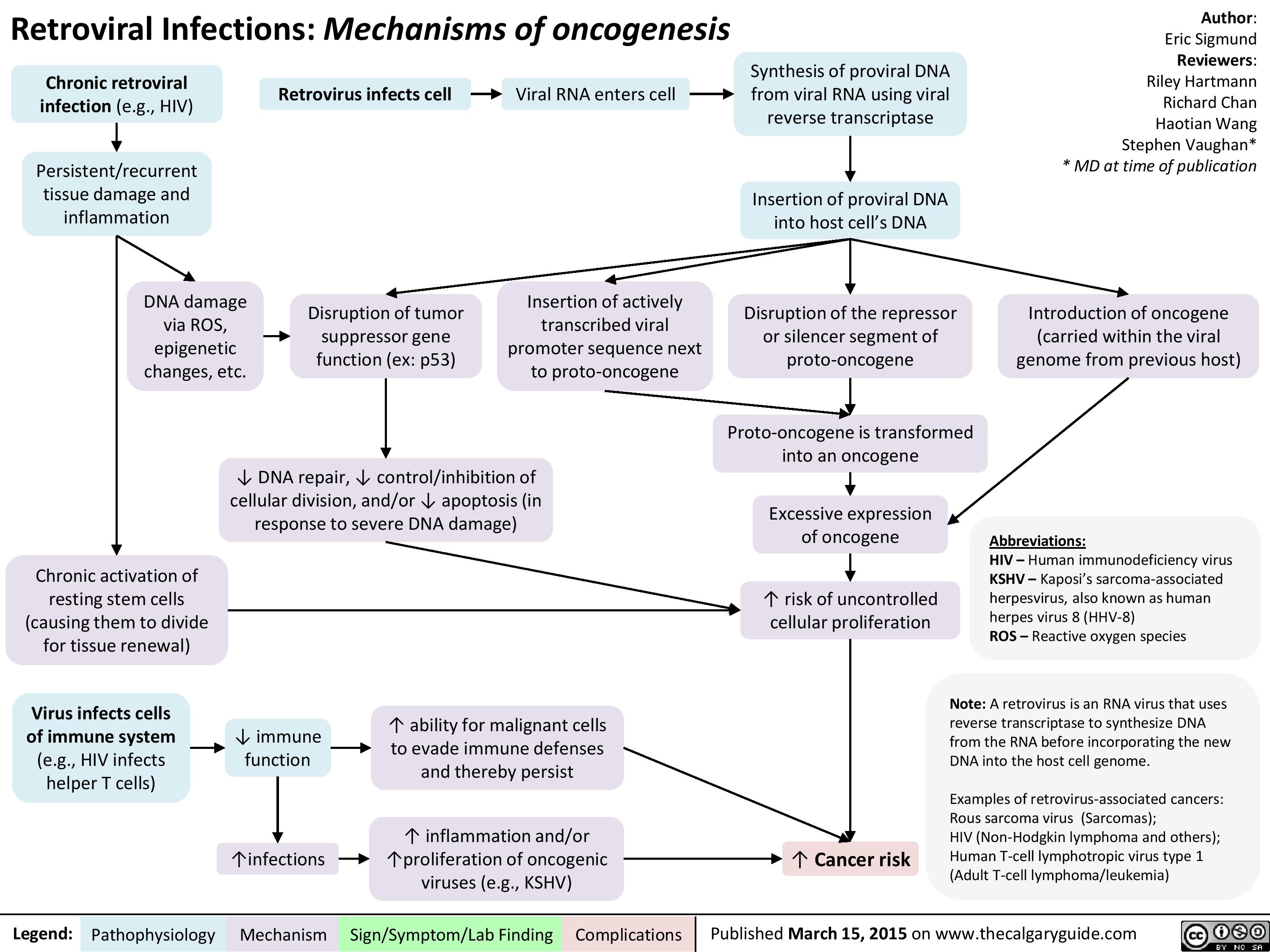
Retroviral Infections Mechanisms of oncogenesis
Superficial Partial Thickness Burns - Pathogenesis and Clinical Findings
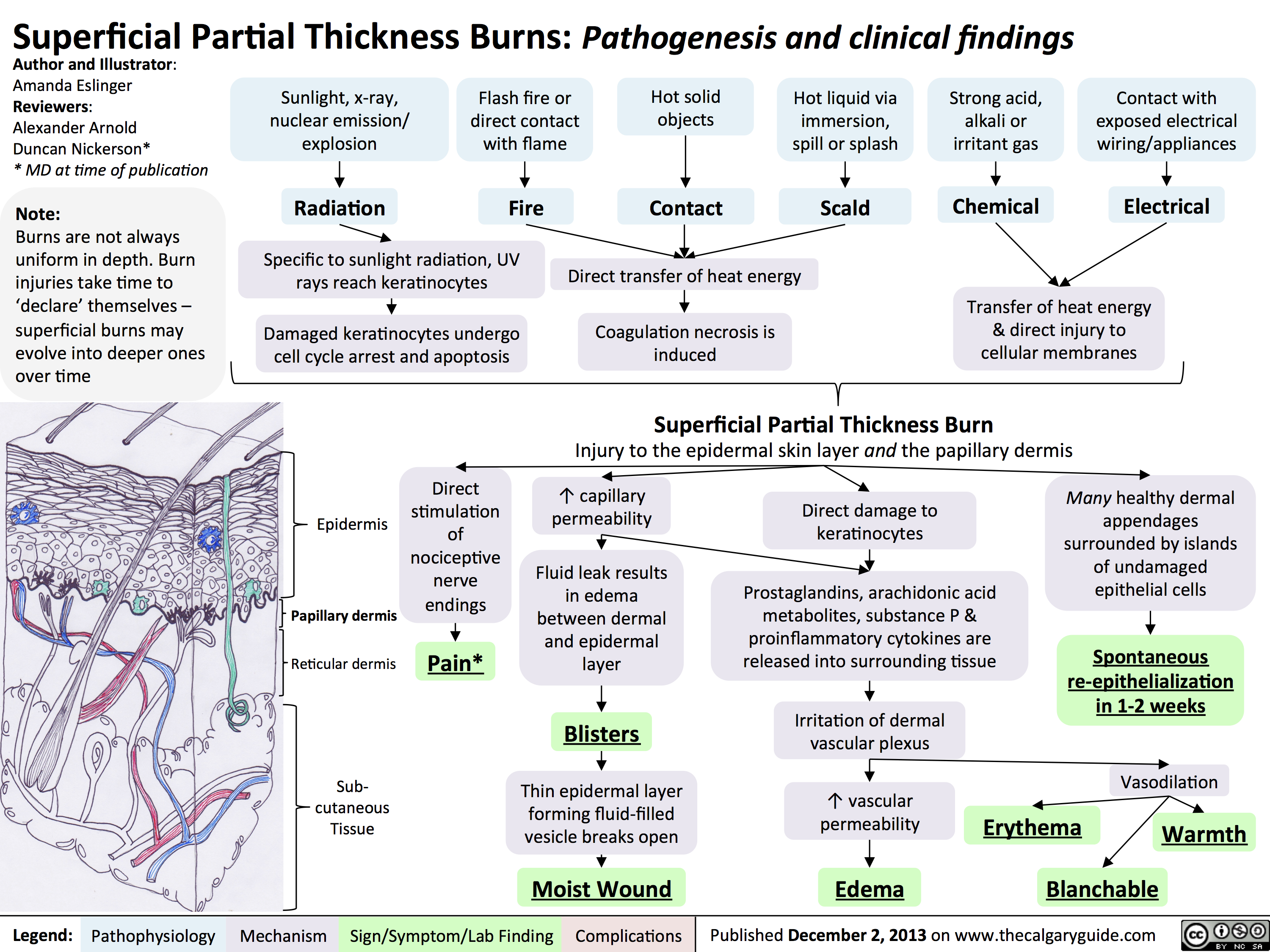
Complications of Burns
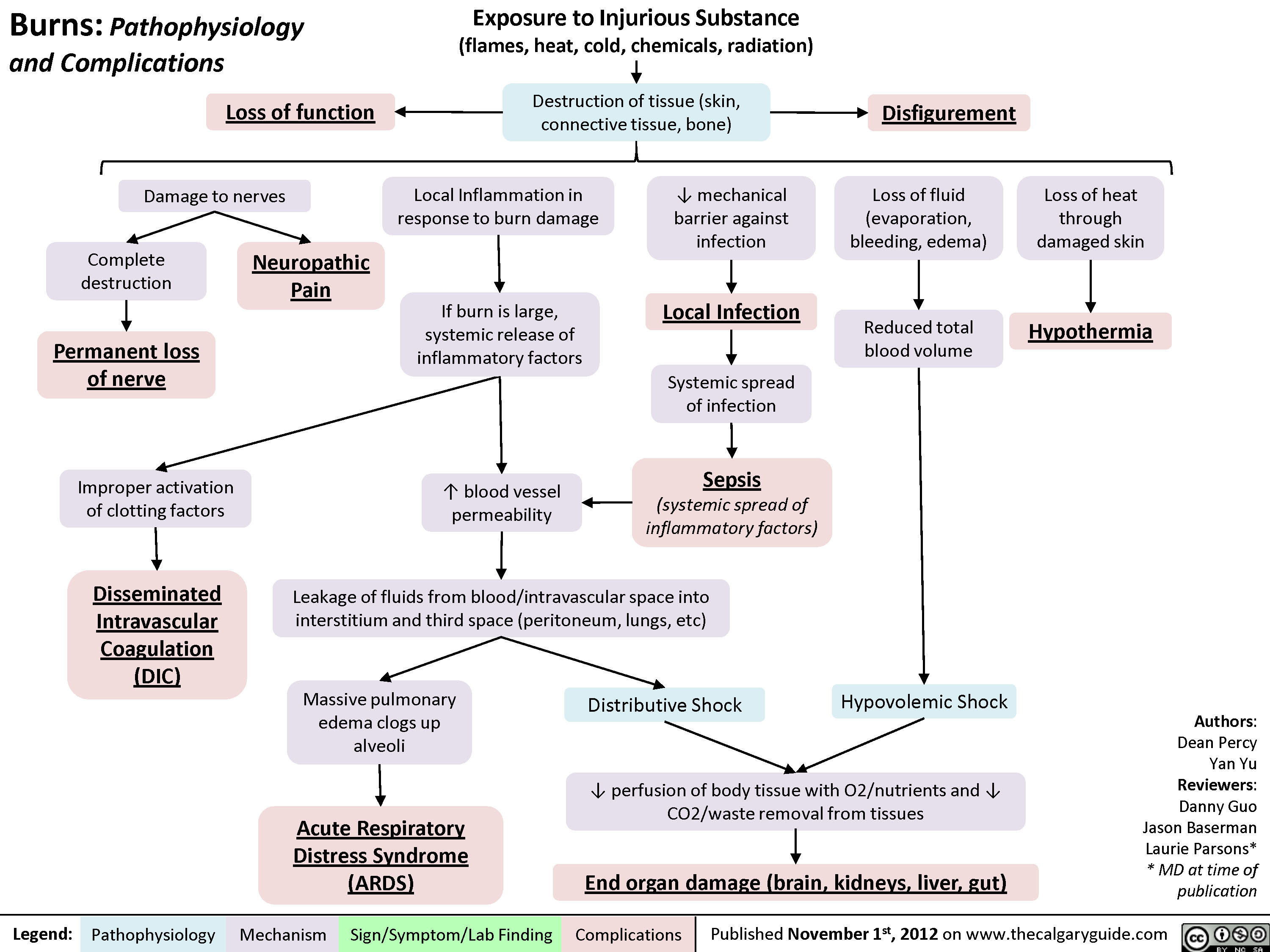
Acne Vulgaris
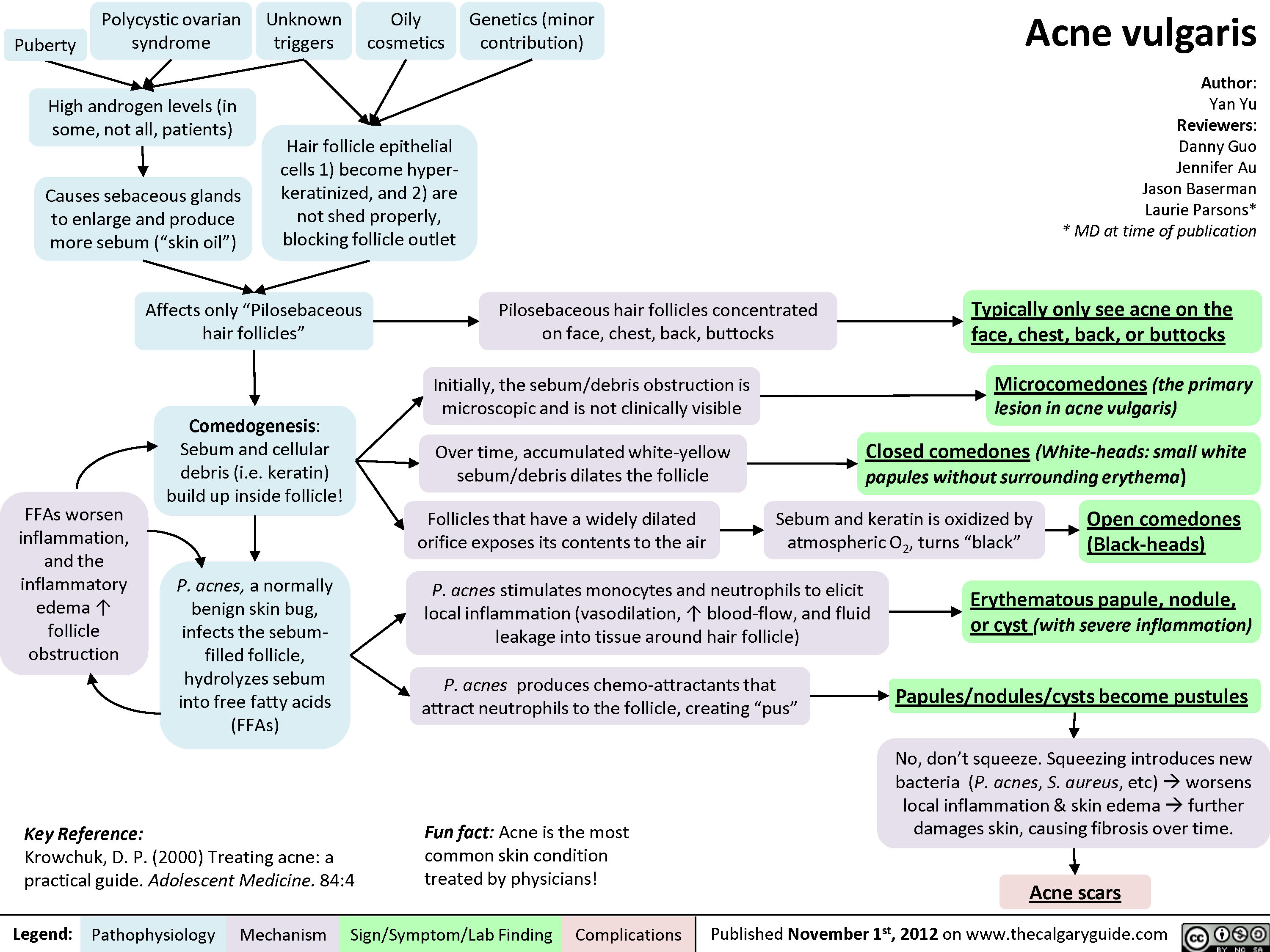
Androgenic Alopecia
Distinguishing between Benign and Malignant Pigmented Lesions
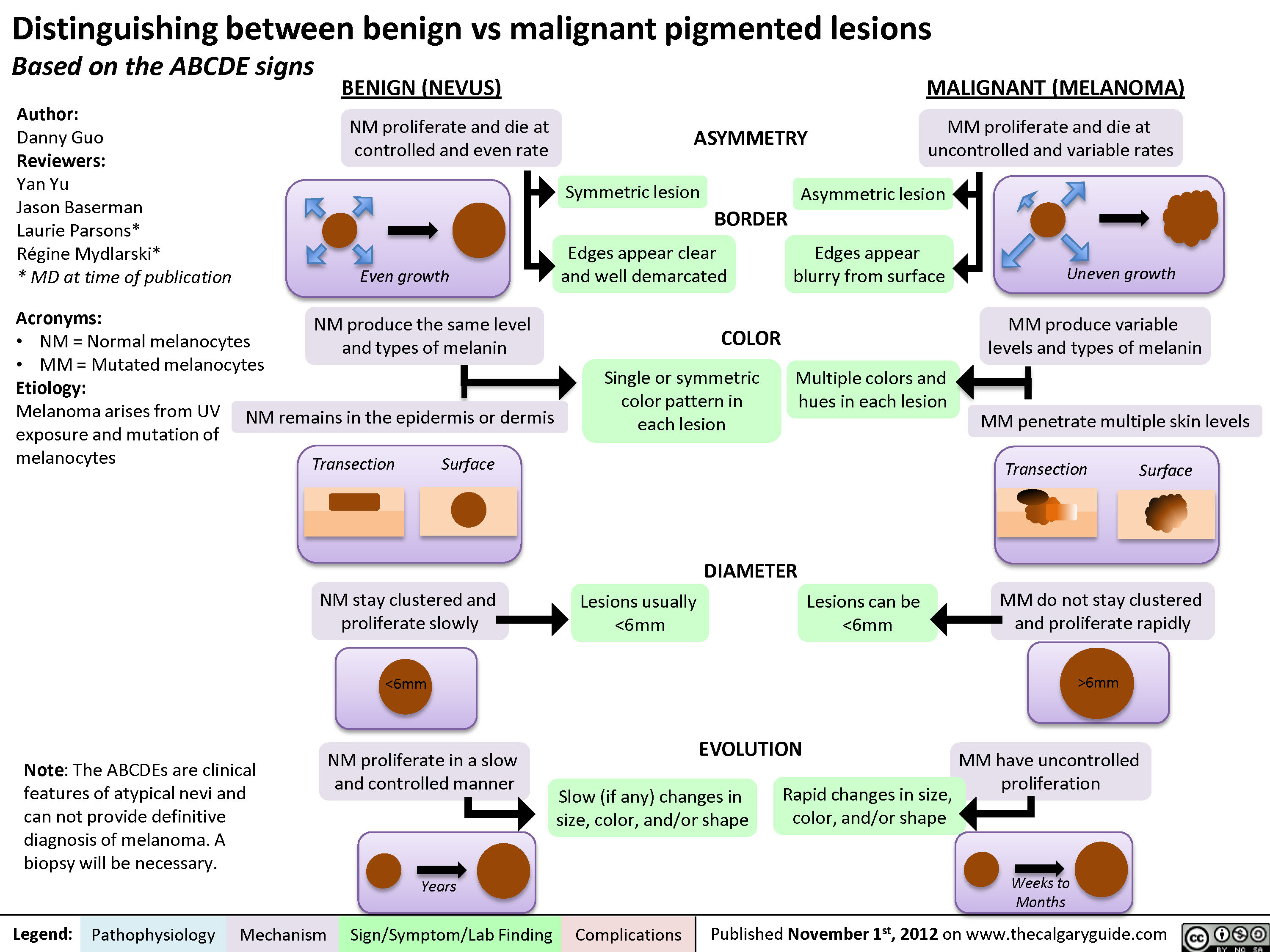
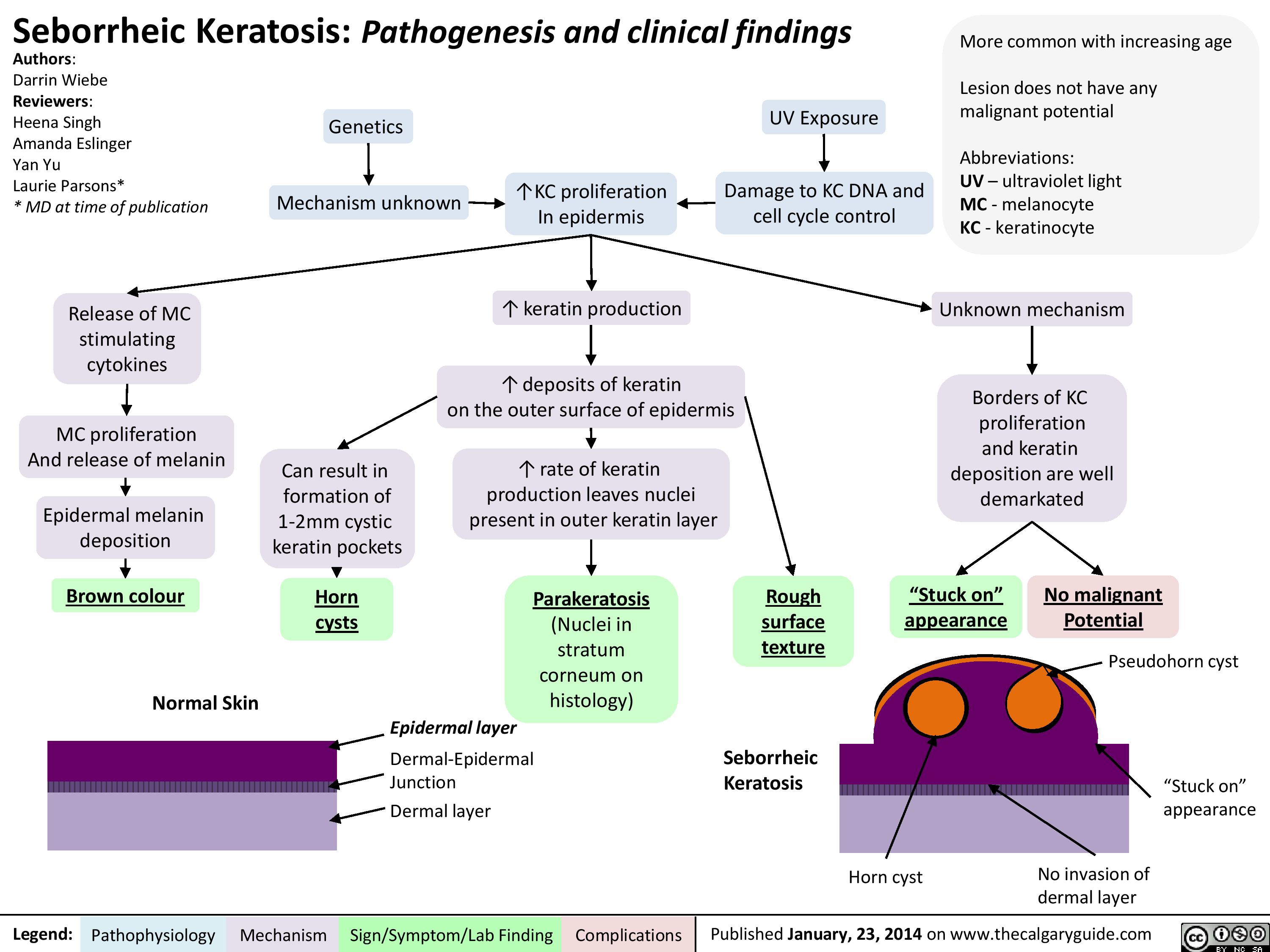
Seborrheic Keratosis
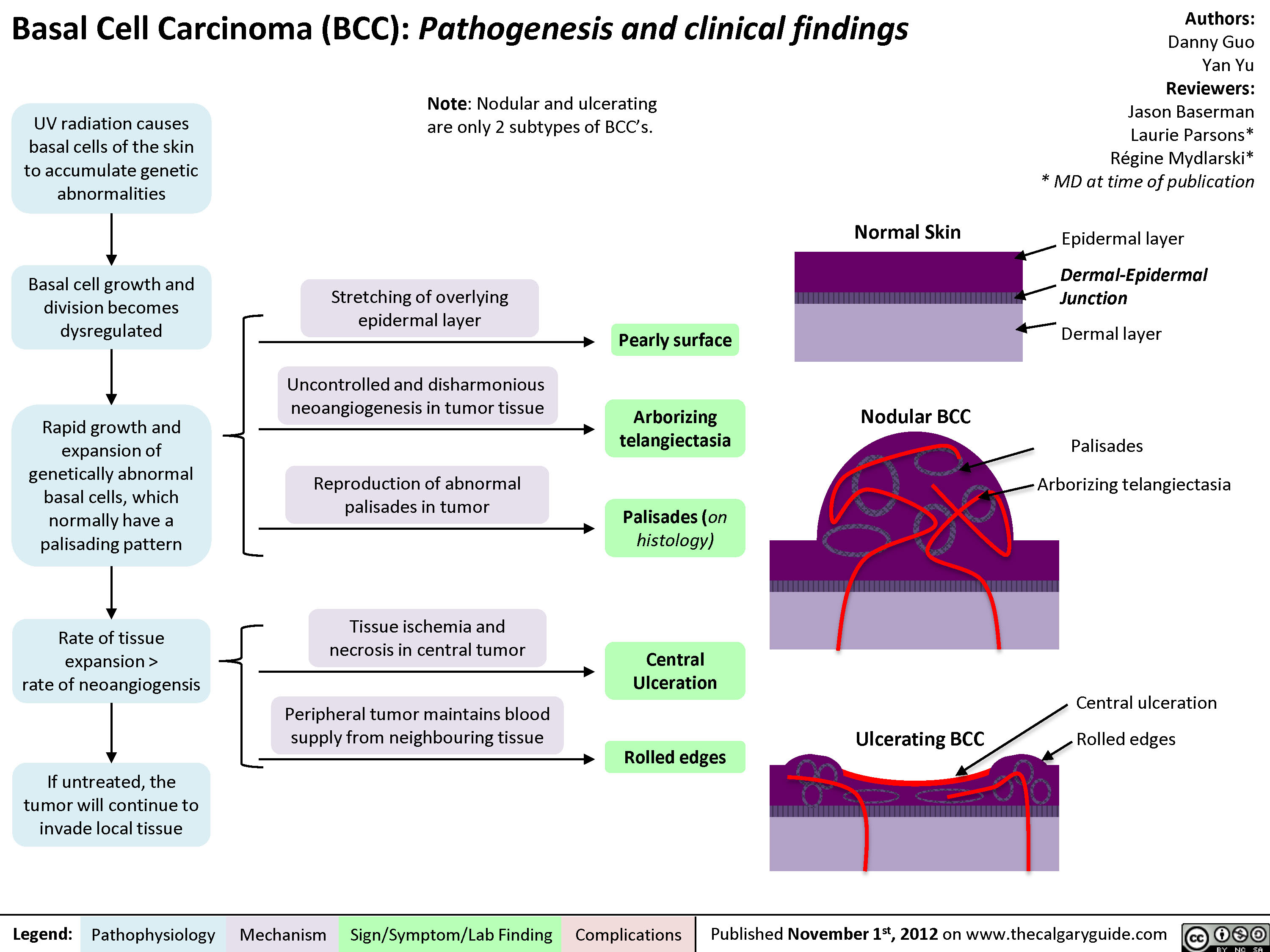
Basal Cell Carcinoma (BCC)
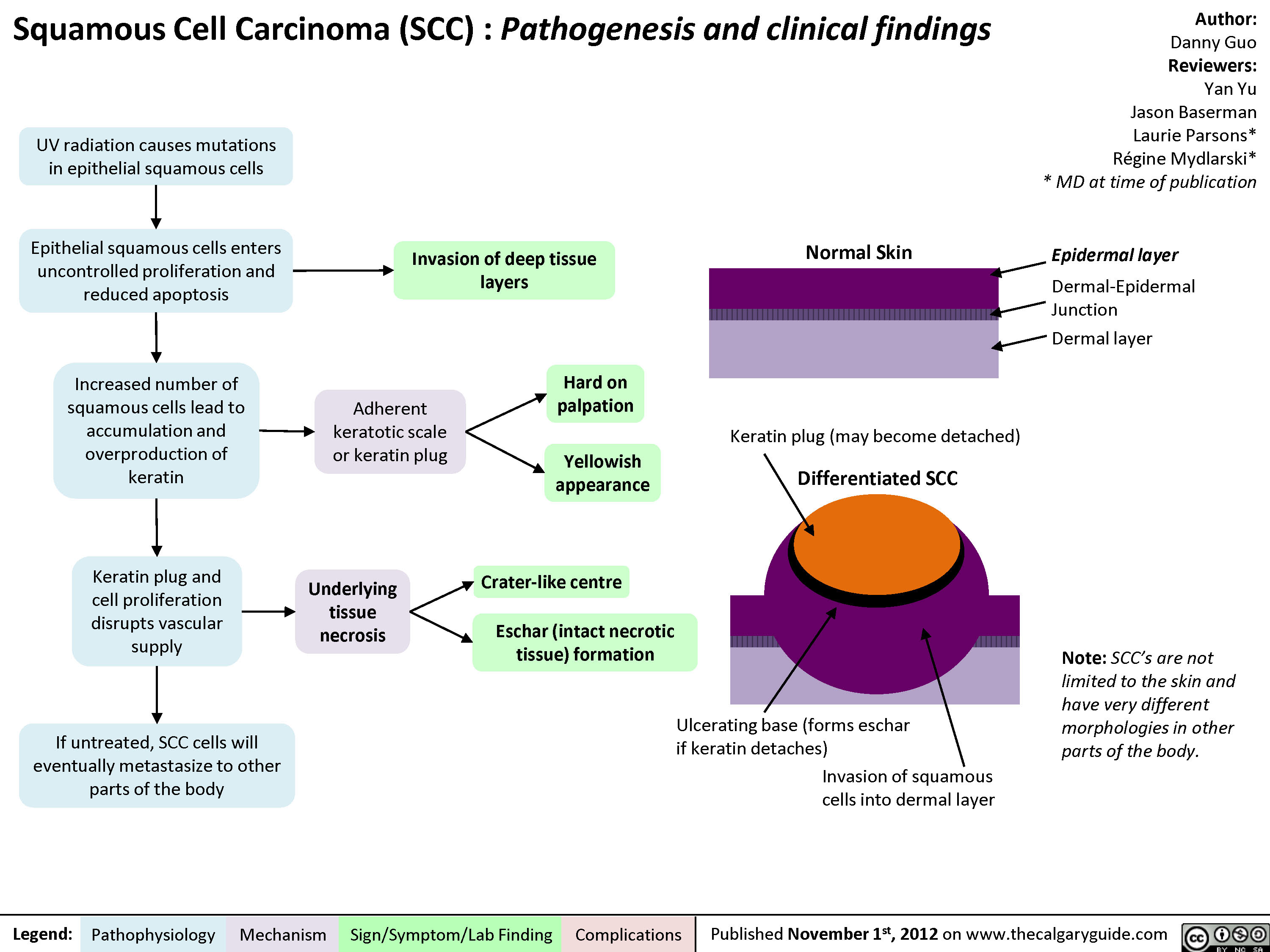
Squamous Cell Carcinoma (SCC)
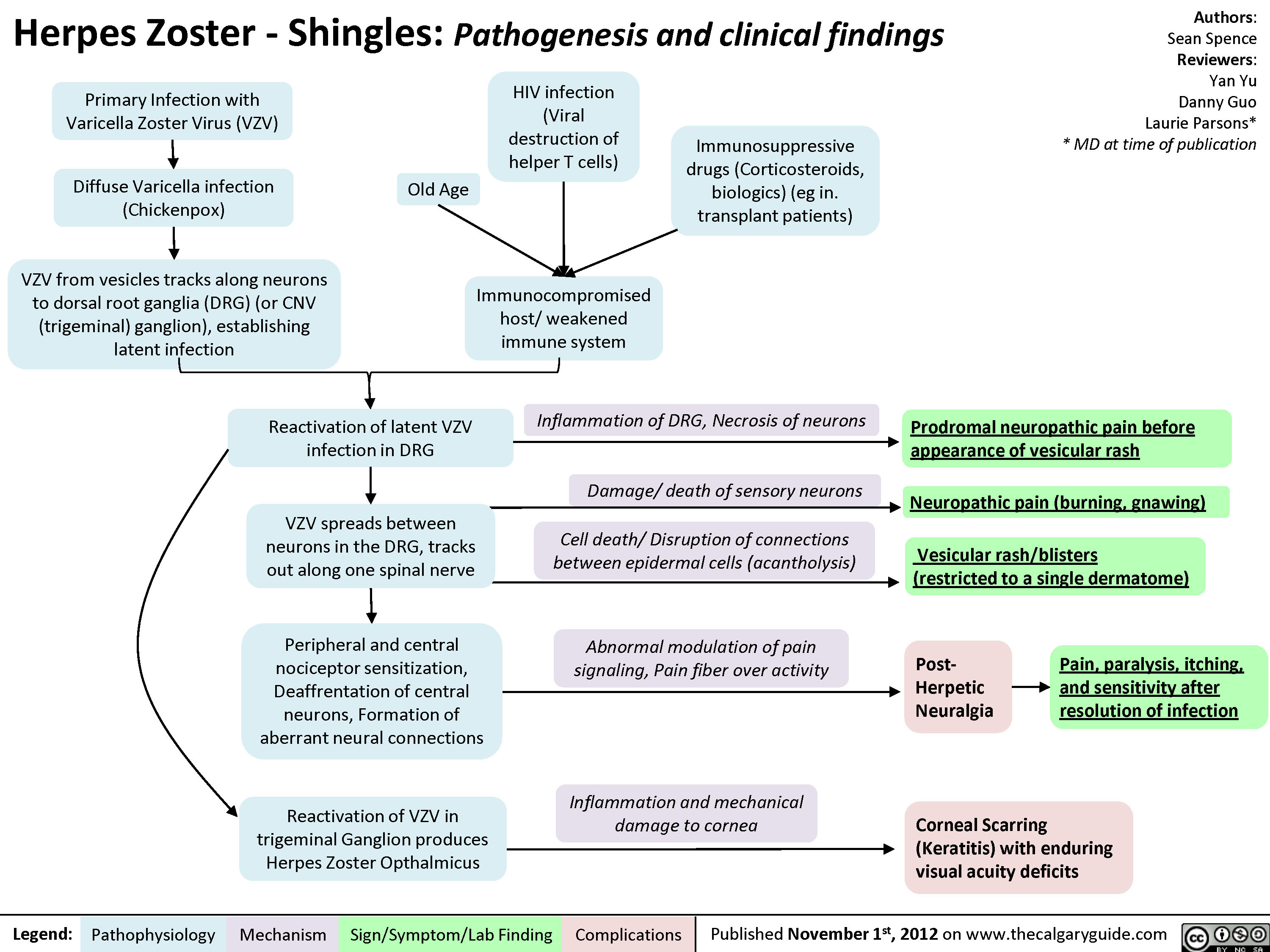
Herpes Zoster (Shingles)
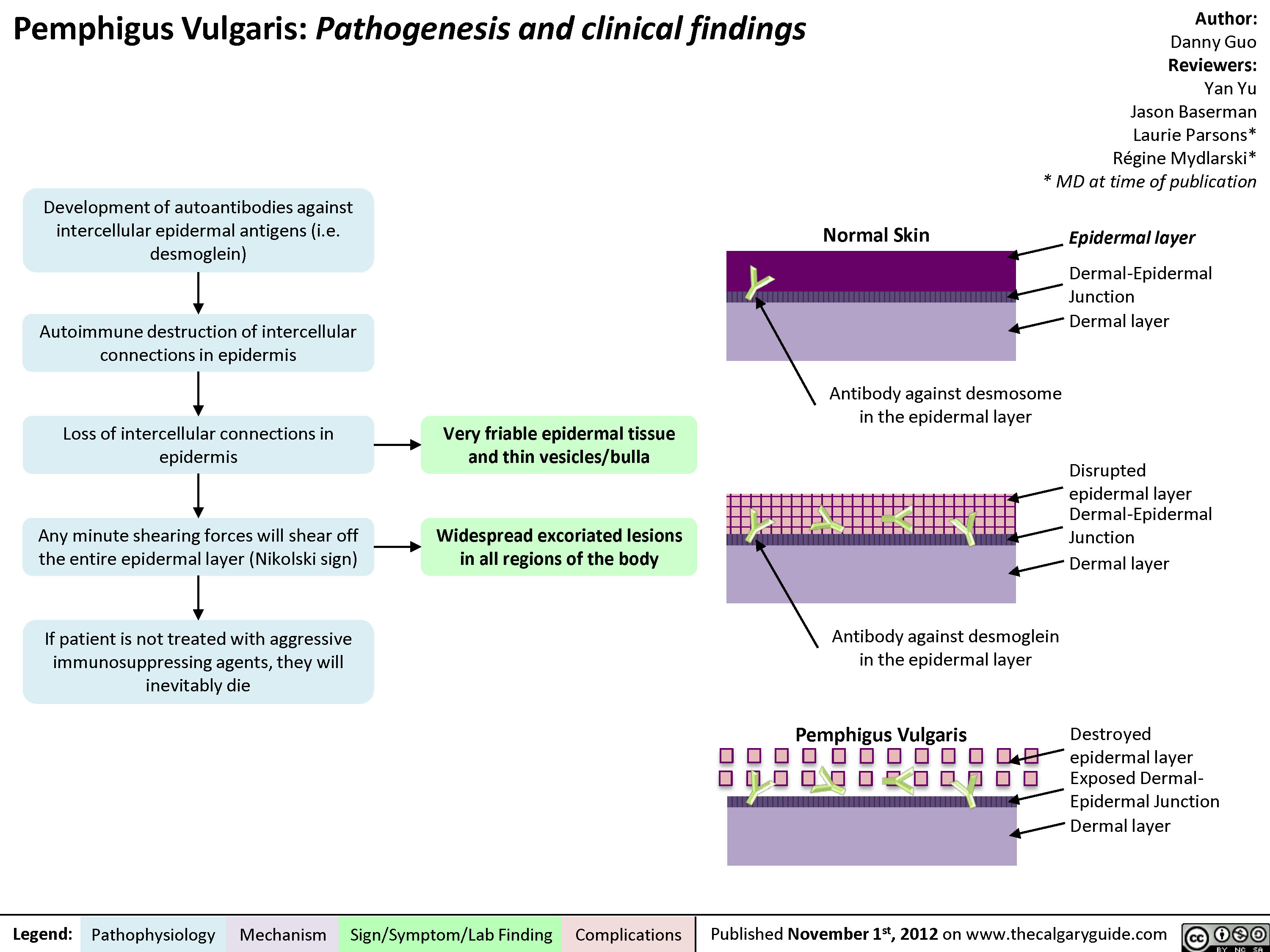
Pemphigus Vulgaris
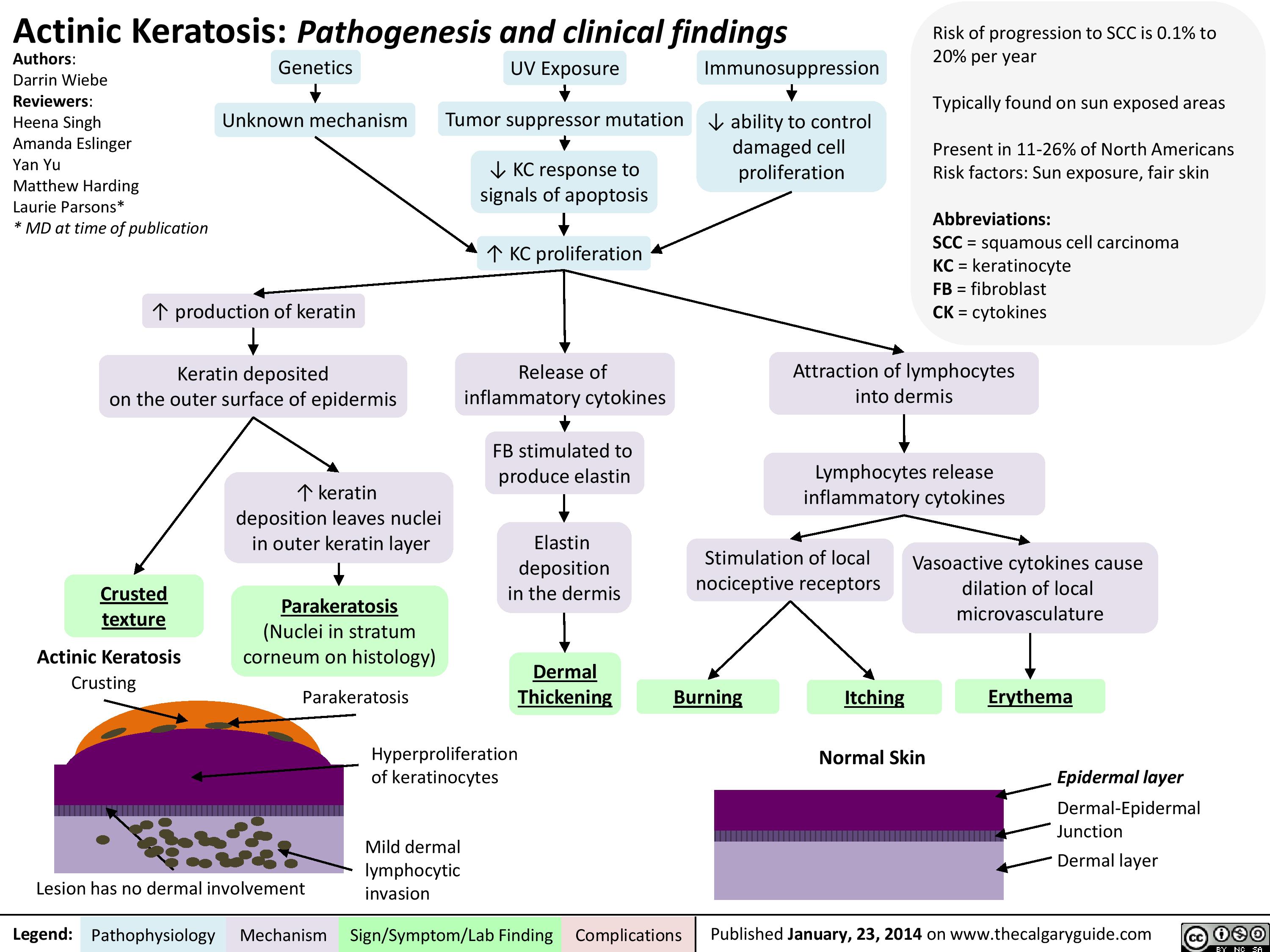
Actinic Keratosis
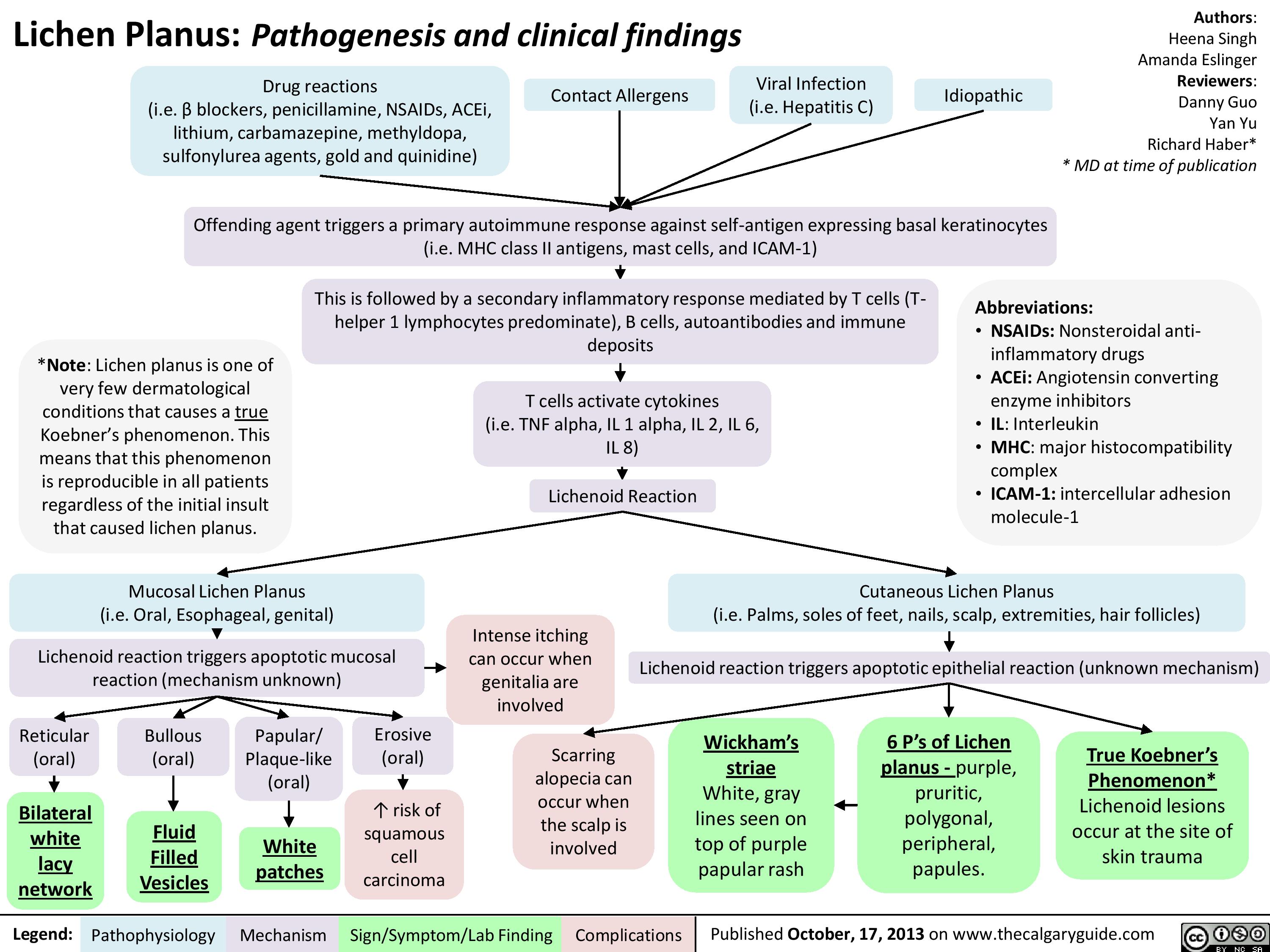
Lichen Planus
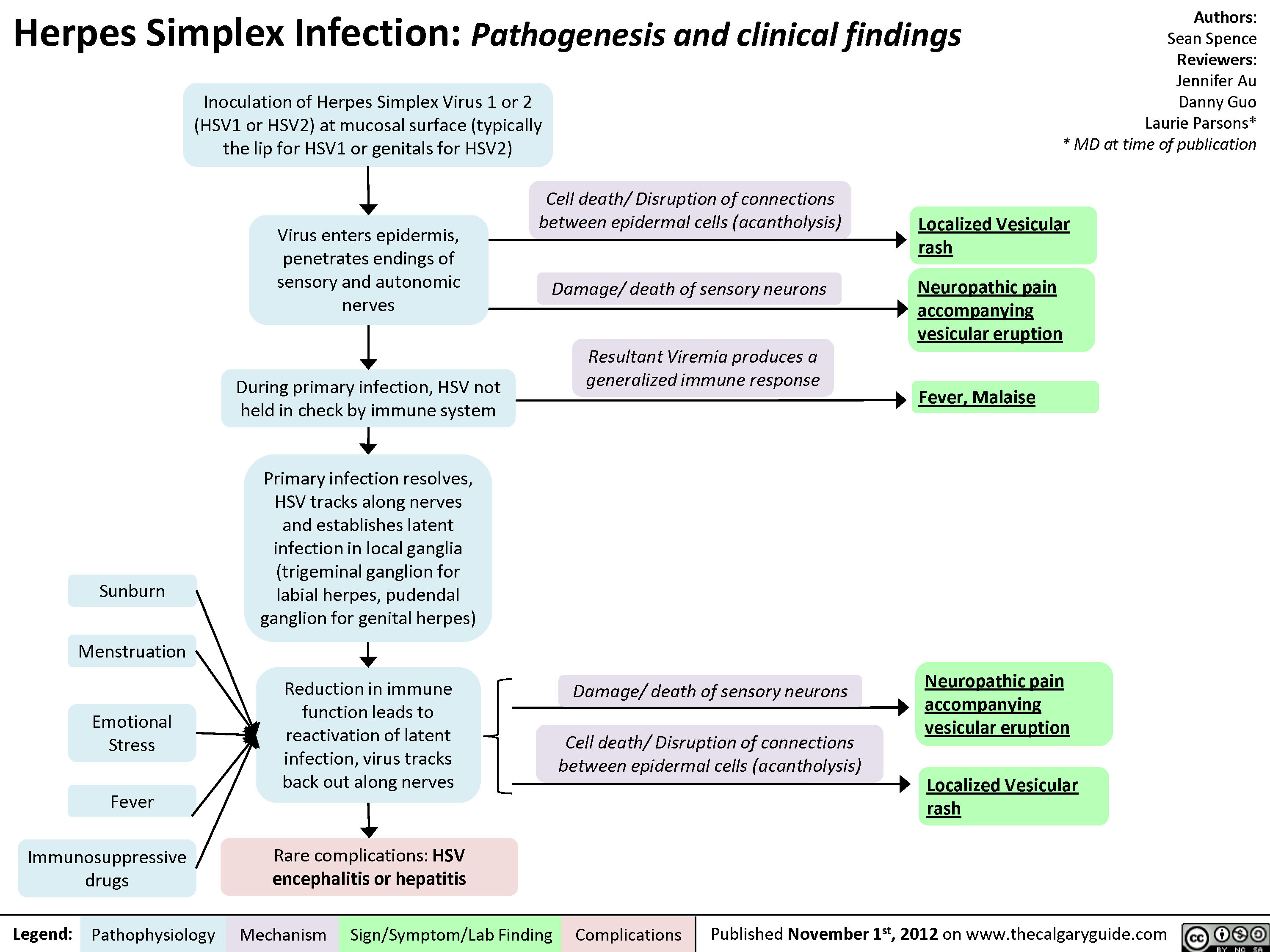
Herpes Simplex Virus (HSV)
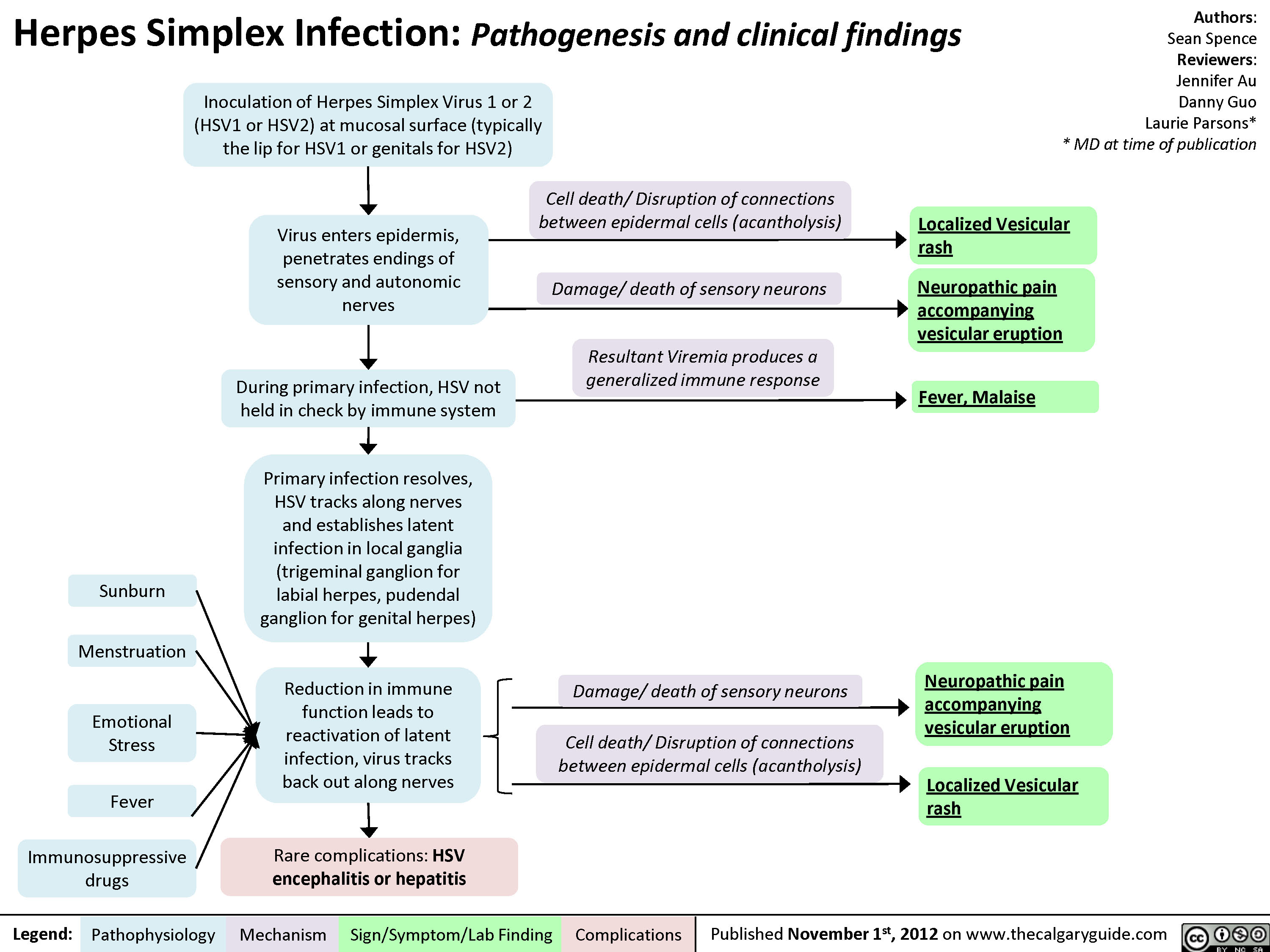
Herpes Simplex Virus (HSV)
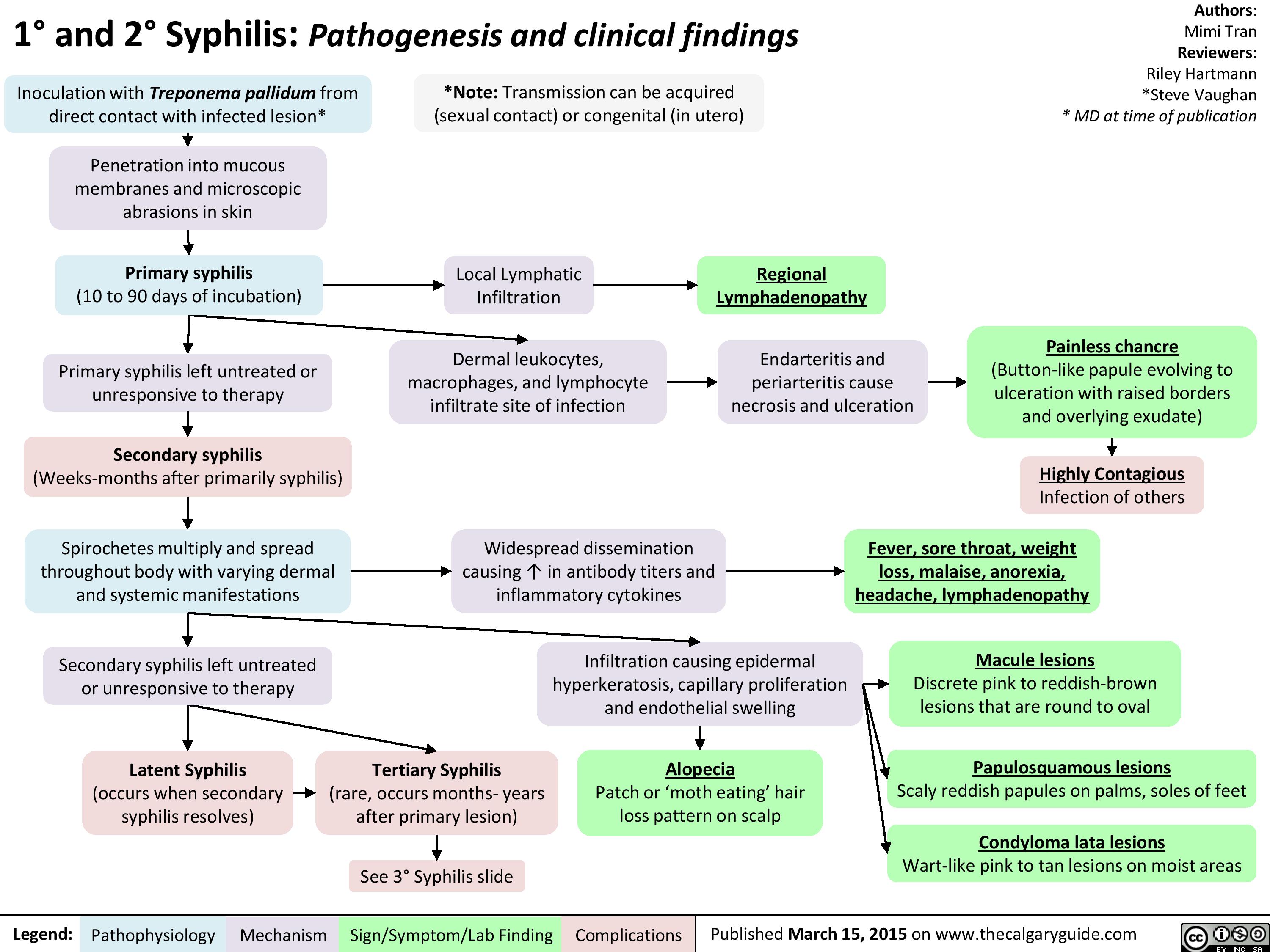
1-and-2-syphilis-pathogenesis-and-clinical-findings
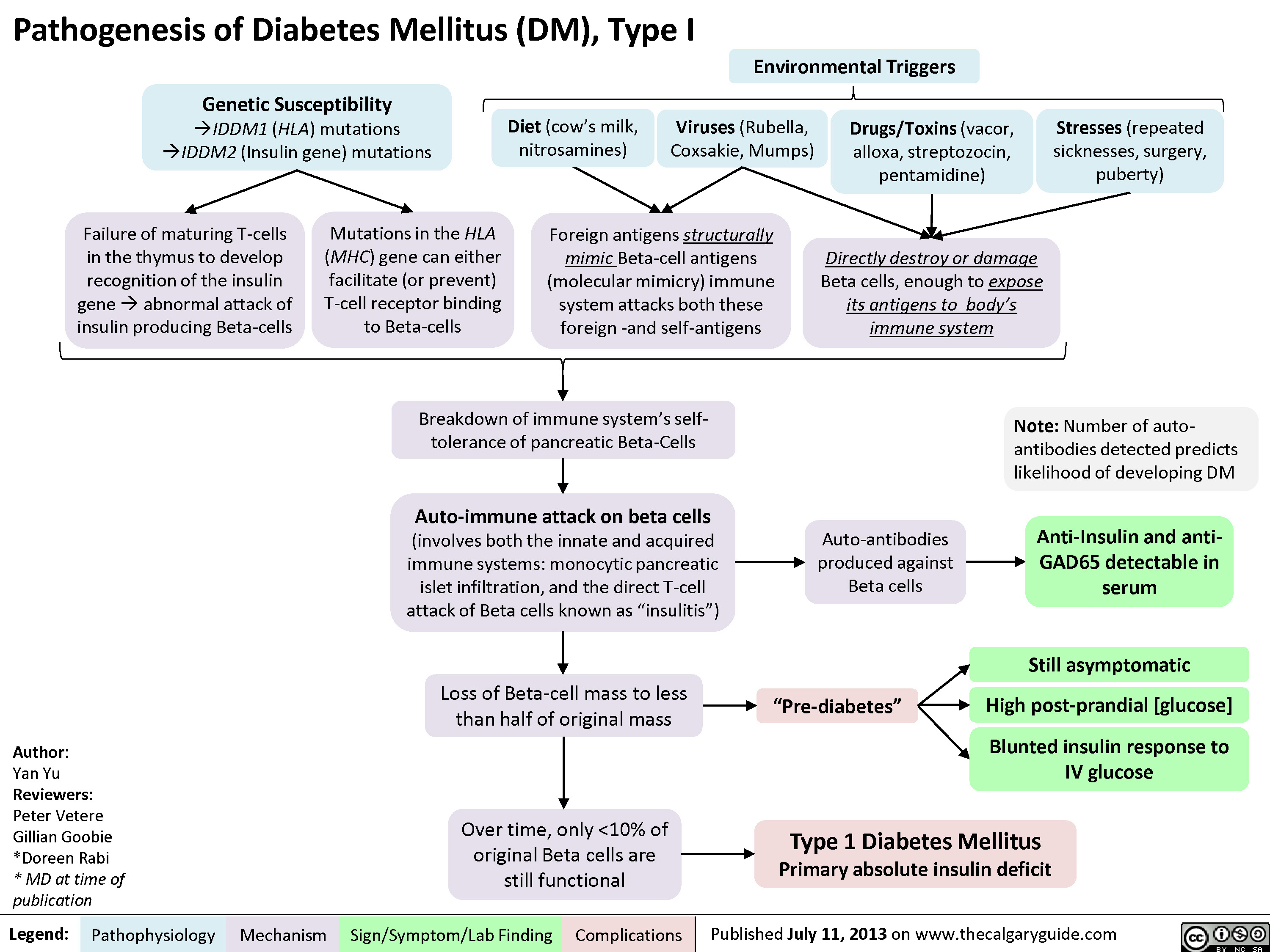
DM I pathogenesis
Pathogenesis of Diabetes mellitus DM), Type II

Diabetic Ketoacidosis

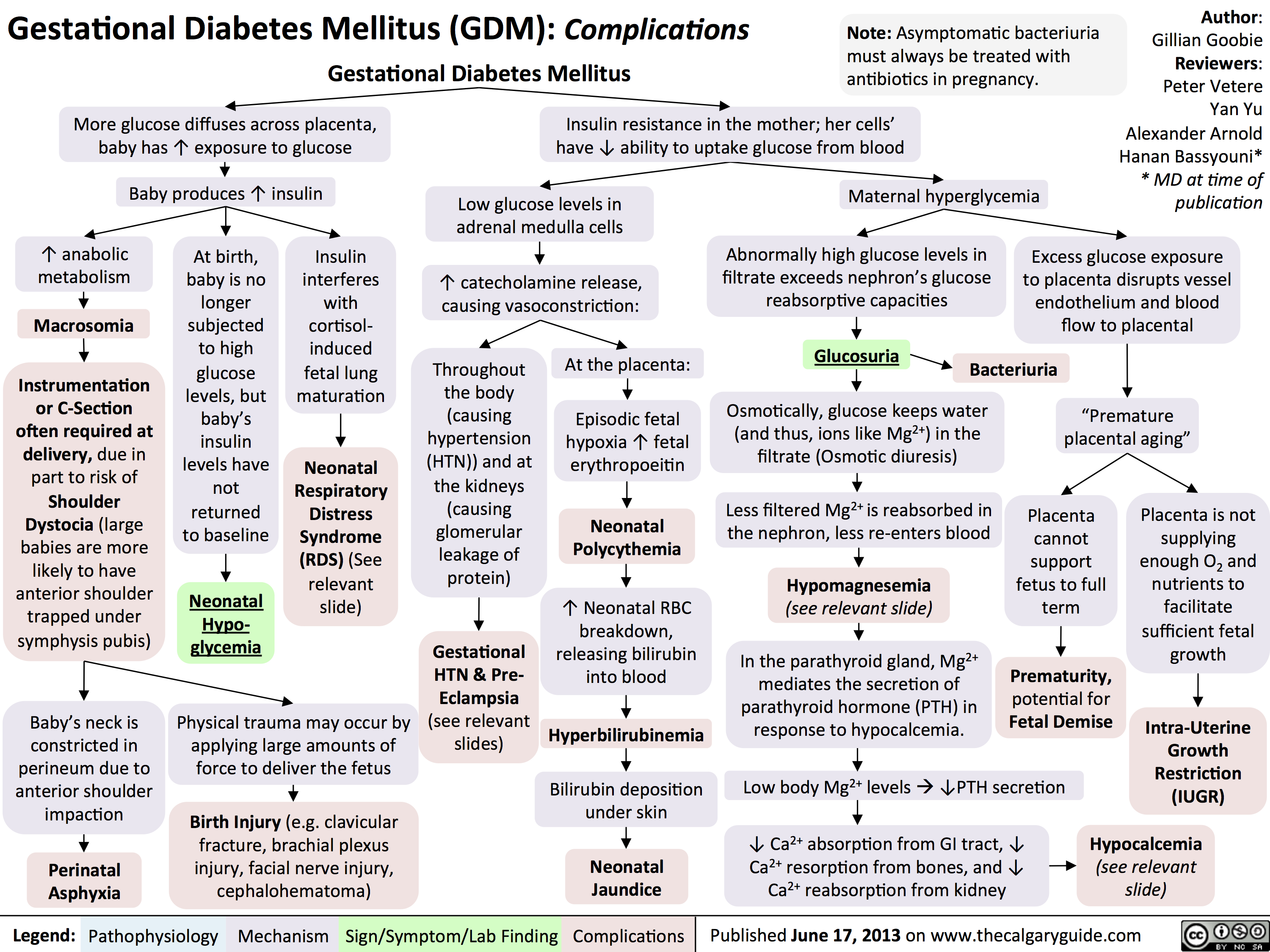
GDM Complications
Hypoglycemia - Pathogenesis

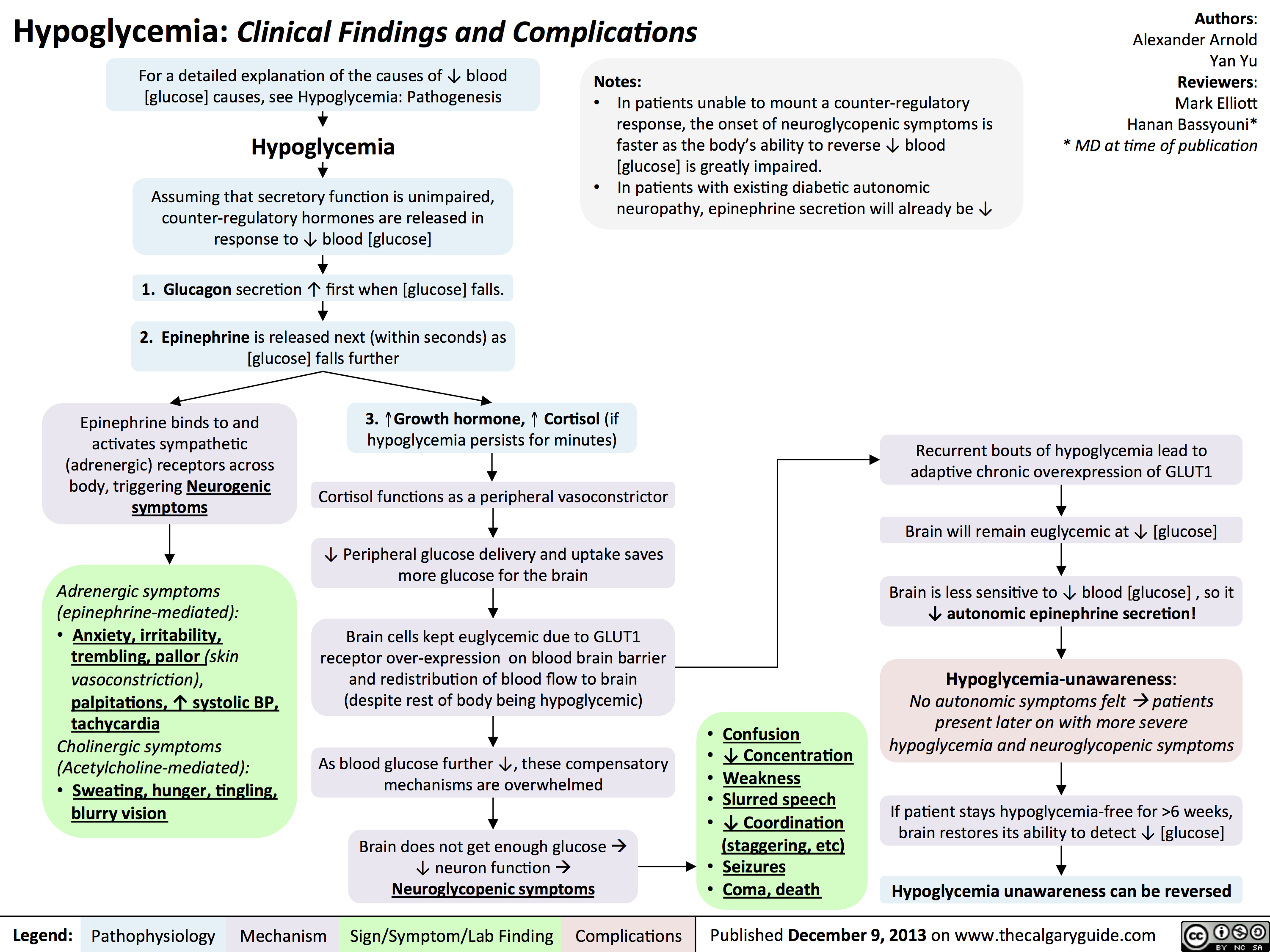
Hypoglycemia - Clinical Findings and Complications
Diabetic Hypoglycemia
![Yu, Yan - Diabetic Hypoglycemia - Clinical Findings - FINAL.pptx
? Epinephrine(Released within seconds as [glucose] falls further) Growth hormone, ? Cortisol (if hypoglycemia persists for minutes)Glucagon should ? when [glucose] falls. But here, glucagon release is inhibited by 1) diabetic auto-immune destruction of Alpha cells & 2) the high insulin.43210Plasma Glucose concentration (mmol/L)Liver should ? glycogenolysis & gluconeogenesisPeripheral vaso-constrictionPlasma [glucose] stays lowActivation of sympathetic (adrenergic) receptors across body, triggering Neurogenic symptomsPlasma [glucose] ?Excess subcutaneous insulin or insulin-secretagogue ?? [insulin] in the bloodOver time: [insulin] in the DM patient depends only on how much was injected or how much secretagogue was consumed; not on the body's physiological state.[Insulin] stays high in excessively-treated DM patientsPlasma [glucose] normally ?, but...High insulin transports plasma glucose into cells!In pts with existing diabetic autonomic neuropathy, epi-nephrine secretion will already be ?Brain does not get enough glucose, ? neuron function ? Neuroglycopenic symptomsTx: glucose intake![Glucose] returns to normalIf no glucose intake:Hypoglycemia-unawareness: No autonomic Sx felt so hypoglycemia not treated early ? pts present later on with more severe hypoglycemia and neuroglycopenic sxBrain cells kept chronically euglycemic due to GLUT1 receptor over-expression (despite rest of body being hypoglycemic)With many hypoglycemic events over time:Brain feels no need to ? glucose, so it ? autonomic epinephrine secretion!This is the normal sequence of hormone responses to ?ing plasma glucose levels.But this normal hormonal response will be blunted over time if there is 1) continued hypoglycemia dampening the sympathetic nervous system, and 2) long-standing diabetic neuropathy! (To be explained later in this flow chart)Abbreviations: [ ] = concentrationTx = TreatmentDM = Diabetes mellitusDiabetic Hypoglycemia: Pathogenesis and Clinical FindingsConfusionCan't concentrateWeaknessSlurred speech? coordination (staggering, etc)SeizuresComa, deathAdrenergic symptoms (epinephrine-mediated):Anxiety, irritability, trembling, pallor (skin vasoconstriction), palpitations, ? systolic BP, tachycardia Cholinergic symptoms(Acetylcholine-mediated):Sweating, hunger, tingling, blurry visionNote: In pts w/out DM, endogenous insulin secretion normally stops when blood [glucose] drops to <4mmol/LAuthor: Yan YuReviewers: Peter Vetere, Gillian Goobie, Hanan Bassyouni** MD at time of publicationLegend:Published June 14, 2013 on www.thecalgaryguide.comMechanismPathophysiologySign/Symptom/Lab FindingComplicationsMany hypoglycemic events over time blunt epinephrine secretion further.Hypoglycemia unawareness can be reversedIf pt stays hypoglycemia-free for >6 weeks, brain restores its ability to detect low glucose levels? peripheral glucose delivery and uptake (saving more glucose for the brain)Lack of glucagon effect reinforces hypoglycemia
124 kB / 361 words](http://calgaryguide.ucalgary.ca/wp-content/uploads/2015/05/Diabetic-Hypoglycemia-Clinical-Findings.jpg "Yu, Yan - Diabetic Hypoglycemia - Clinical Findings - FINAL.pptx
? Epinephrine(Released within seconds as [glucose] falls further) Growth hormone, ? Cortisol (if hypoglycemia persists for minutes)Glucagon should ? when [glucose] falls. But here, glucagon release is inhibited by 1) diabetic auto-immune destruction of Alpha cells & 2) the high insulin.43210Plasma Glucose concentration (mmol/L)Liver should ? glycogenolysis & gluconeogenesisPeripheral vaso-constrictionPlasma [glucose] stays lowActivation of sympathetic (adrenergic) receptors across body, triggering Neurogenic symptomsPlasma [glucose] ?Excess subcutaneous insulin or insulin-secretagogue ?? [insulin] in the bloodOver time: [insulin] in the DM patient depends only on how much was injected or how much secretagogue was consumed; not on the body's physiological state.[Insulin] stays high in excessively-treated DM patientsPlasma [glucose] normally ?, but...High insulin transports plasma glucose into cells!In pts with existing diabetic autonomic neuropathy, epi-nephrine secretion will already be ?Brain does not get enough glucose, ? neuron function ? Neuroglycopenic symptomsTx: glucose intake![Glucose] returns to normalIf no glucose intake:Hypoglycemia-unawareness: No autonomic Sx felt so hypoglycemia not treated early ? pts present later on with more severe hypoglycemia and neuroglycopenic sxBrain cells kept chronically euglycemic due to GLUT1 receptor over-expression (despite rest of body being hypoglycemic)With many hypoglycemic events over time:Brain feels no need to ? glucose, so it ? autonomic epinephrine secretion!This is the normal sequence of hormone responses to ?ing plasma glucose levels.But this normal hormonal response will be blunted over time if there is 1) continued hypoglycemia dampening the sympathetic nervous system, and 2) long-standing diabetic neuropathy! (To be explained later in this flow chart)Abbreviations: [ ] = concentrationTx = TreatmentDM = Diabetes mellitusDiabetic Hypoglycemia: Pathogenesis and Clinical FindingsConfusionCan't concentrateWeaknessSlurred speech? coordination (staggering, etc)SeizuresComa, deathAdrenergic symptoms (epinephrine-mediated):Anxiety, irritability, trembling, pallor (skin vasoconstriction), palpitations, ? systolic BP, tachycardia Cholinergic symptoms(Acetylcholine-mediated):Sweating, hunger, tingling, blurry visionNote: In pts w/out DM, endogenous insulin secretion normally stops when blood [glucose] drops to <4mmol/LAuthor: Yan YuReviewers: Peter Vetere, Gillian Goobie, Hanan Bassyouni** MD at time of publicationLegend:Published June 14, 2013 on www.thecalgaryguide.comMechanismPathophysiologySign/Symptom/Lab FindingComplicationsMany hypoglycemic events over time blunt epinephrine secretion further.Hypoglycemia unawareness can be reversedIf pt stays hypoglycemia-free for >6 weeks, brain restores its ability to detect low glucose levels? peripheral glucose delivery and uptake (saving more glucose for the brain)Lack of glucagon effect reinforces hypoglycemia
124 kB / 361 words")
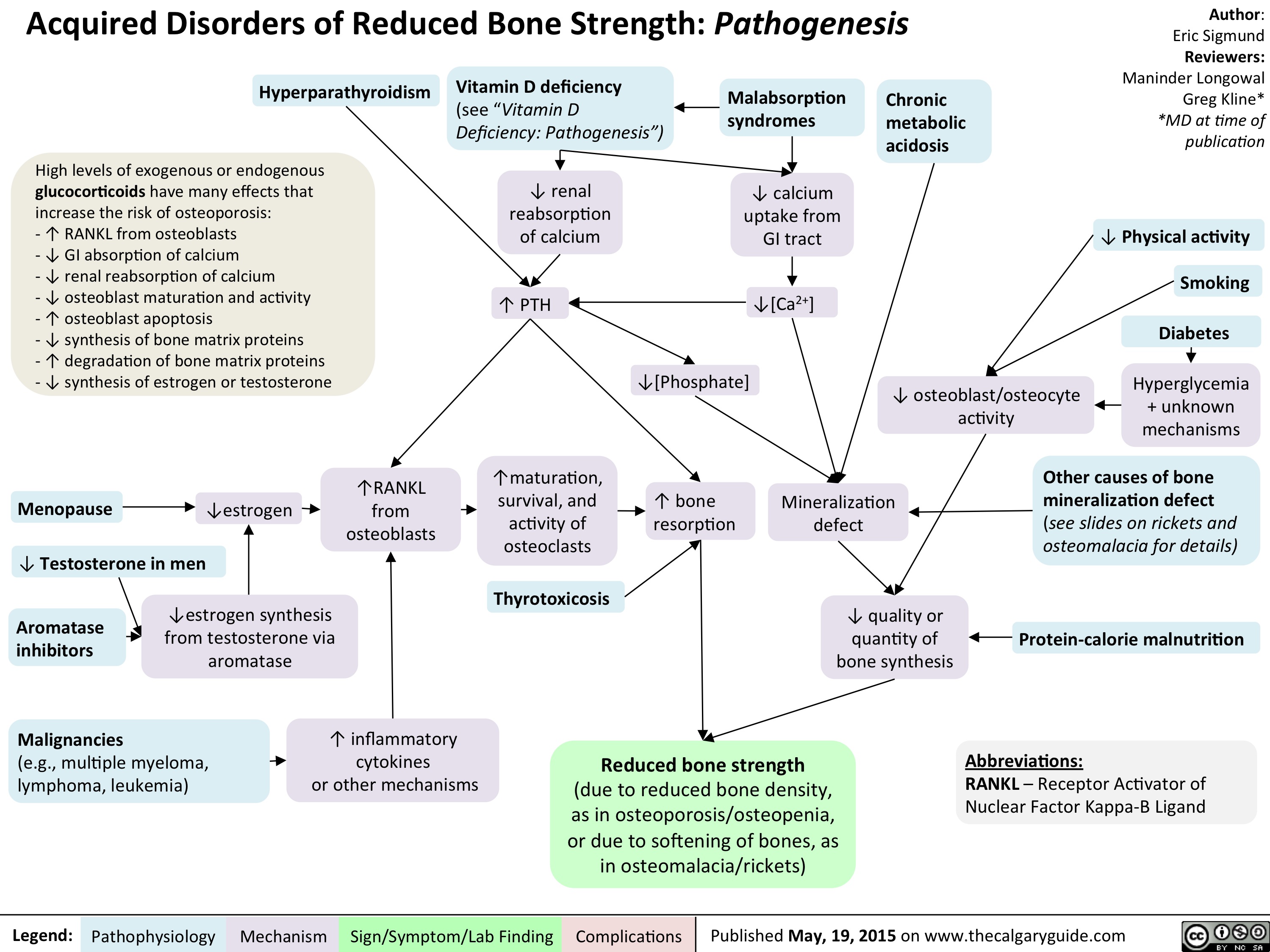
Acquired Disorders of Reduced Bone Strength - Pathogenesis
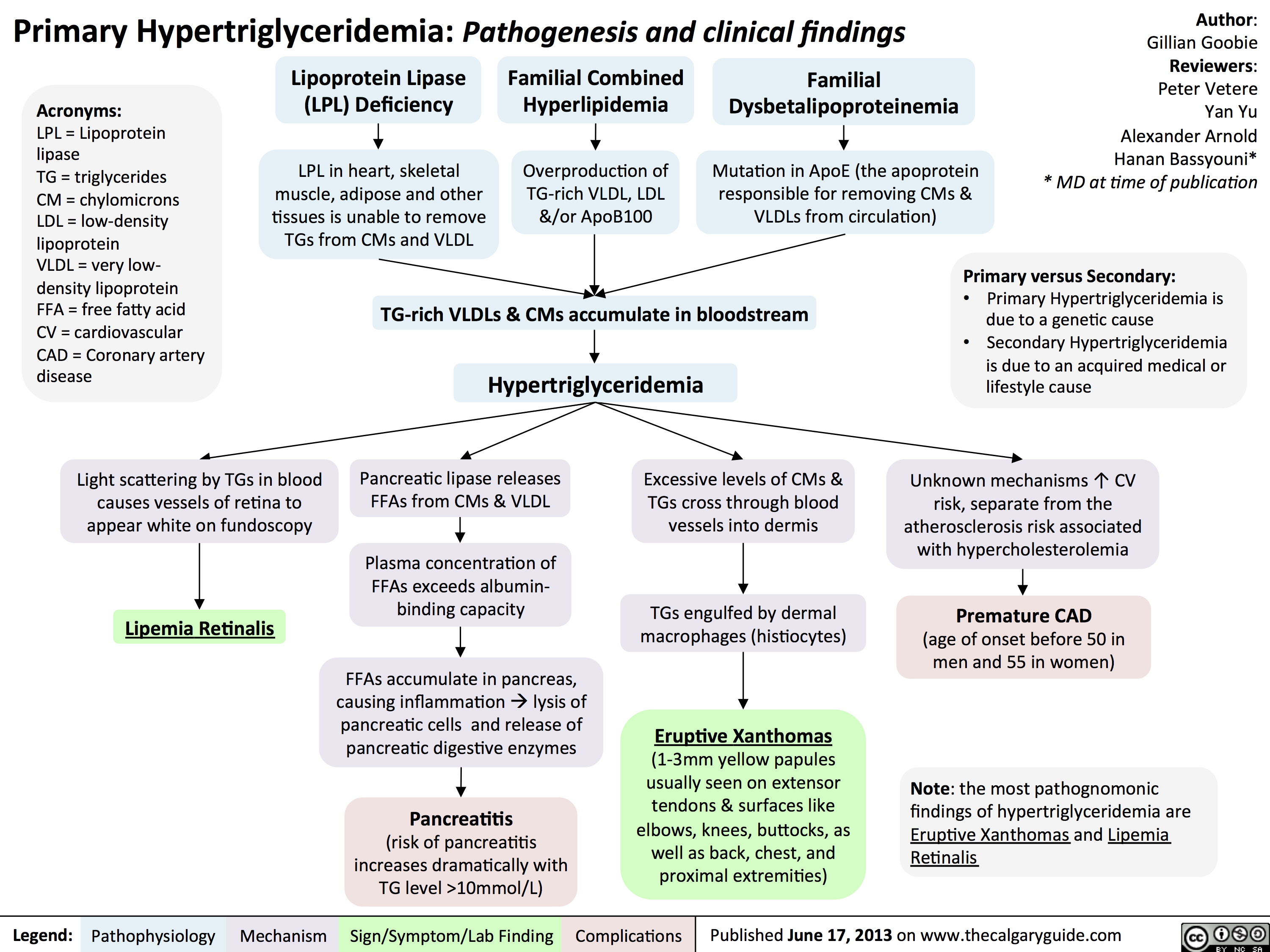
Primary Hypertriglyceridemia
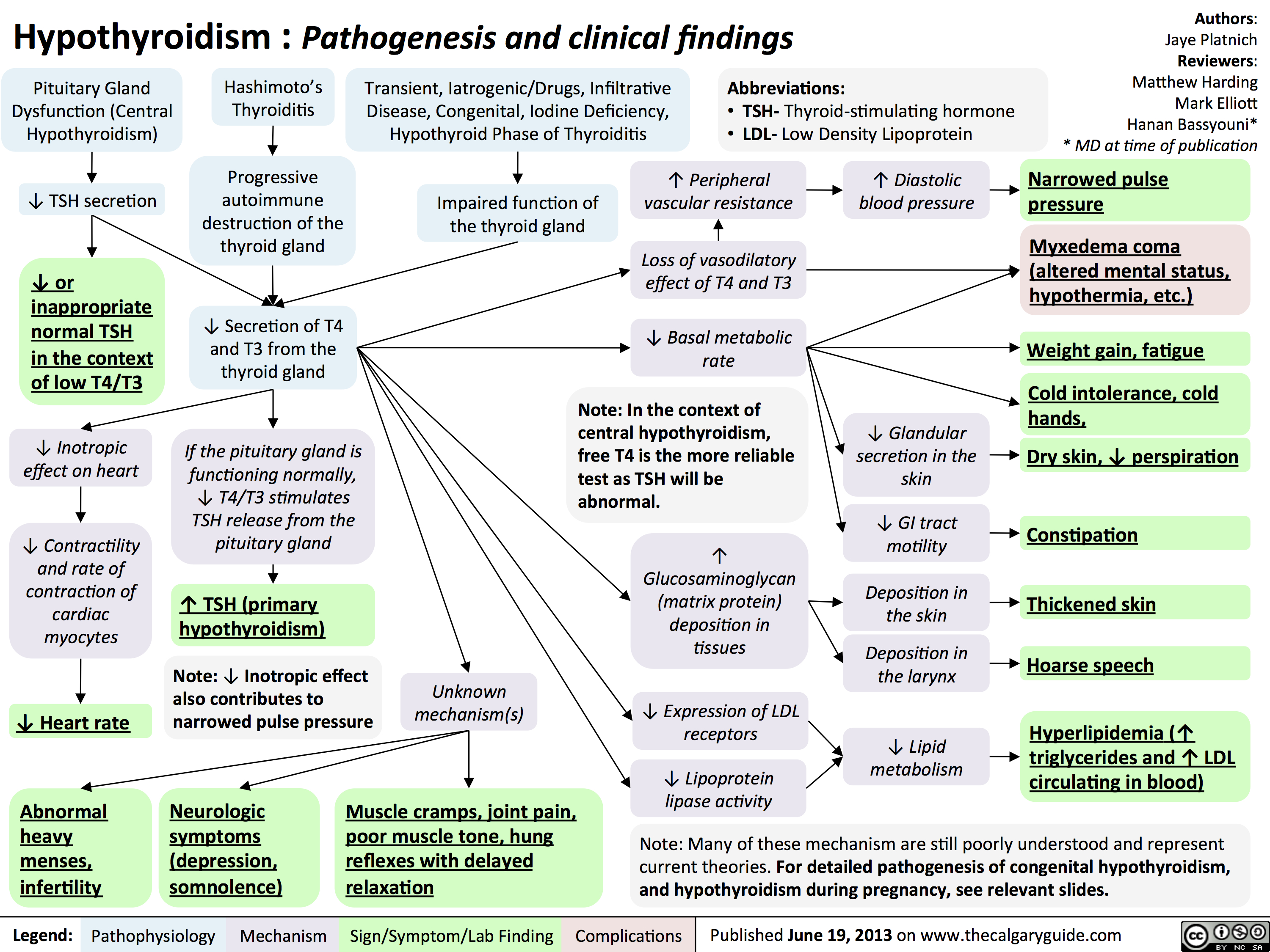
Hypothyroidism
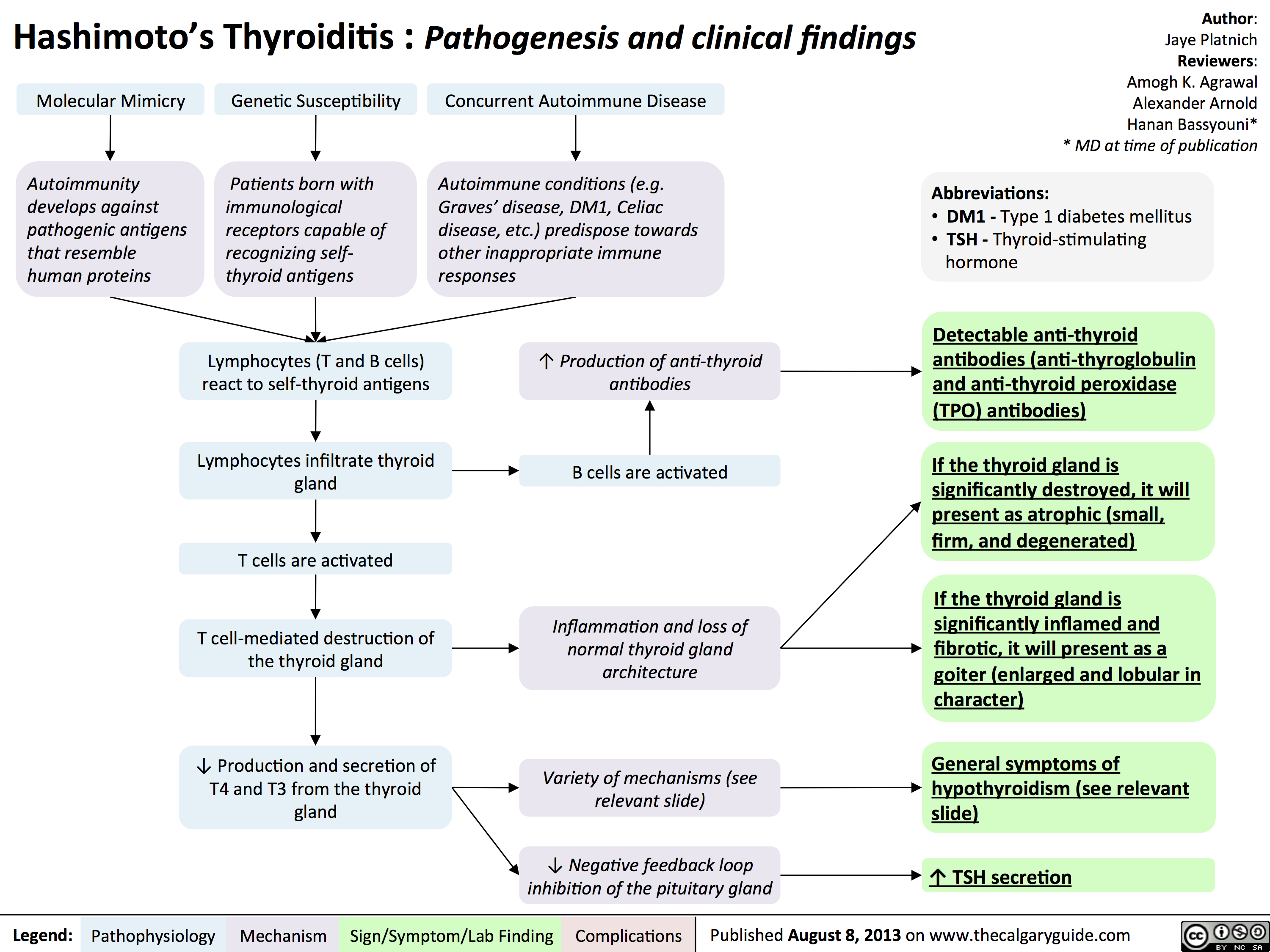
Hashimoto
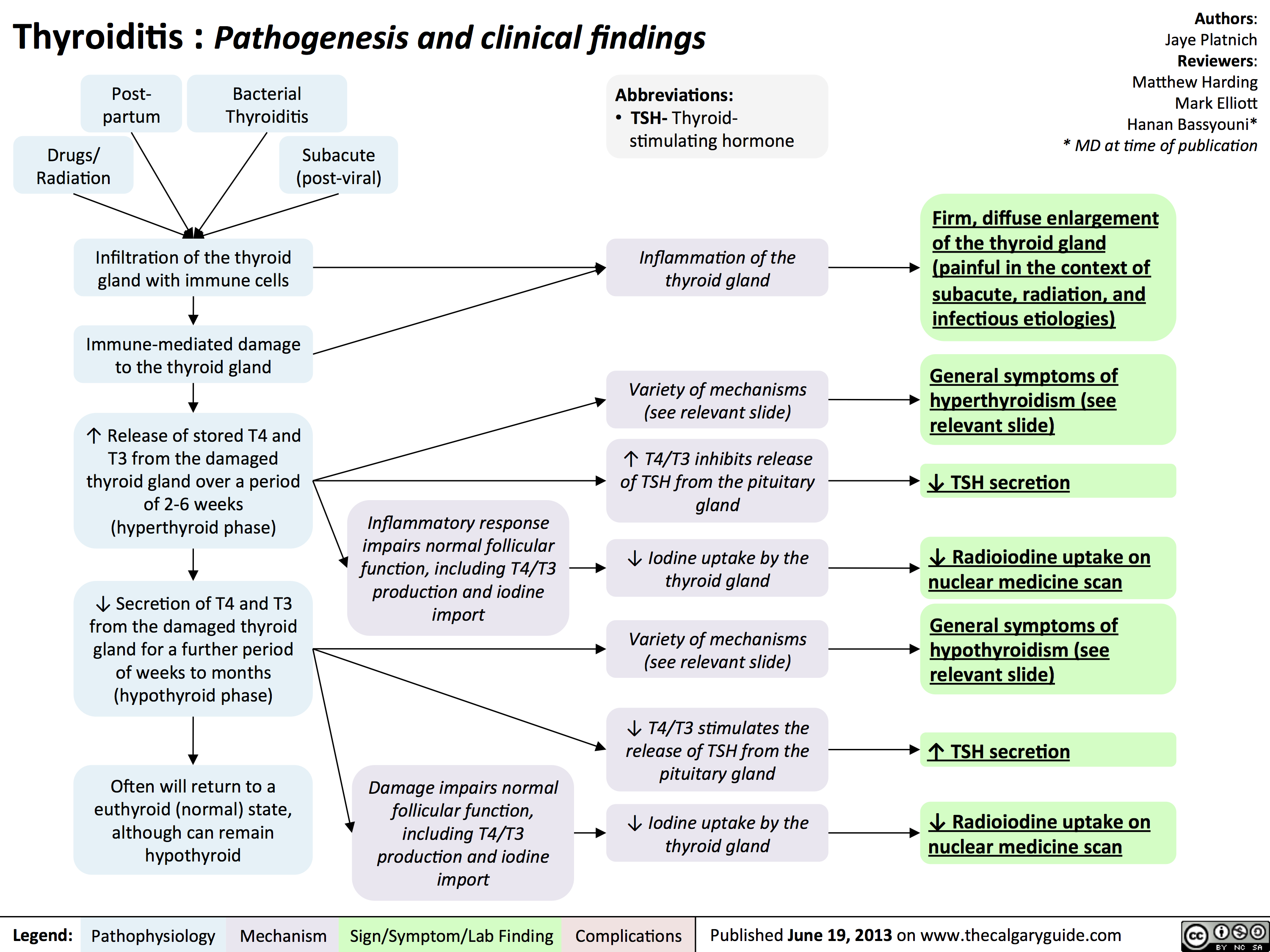
Thyroiditis
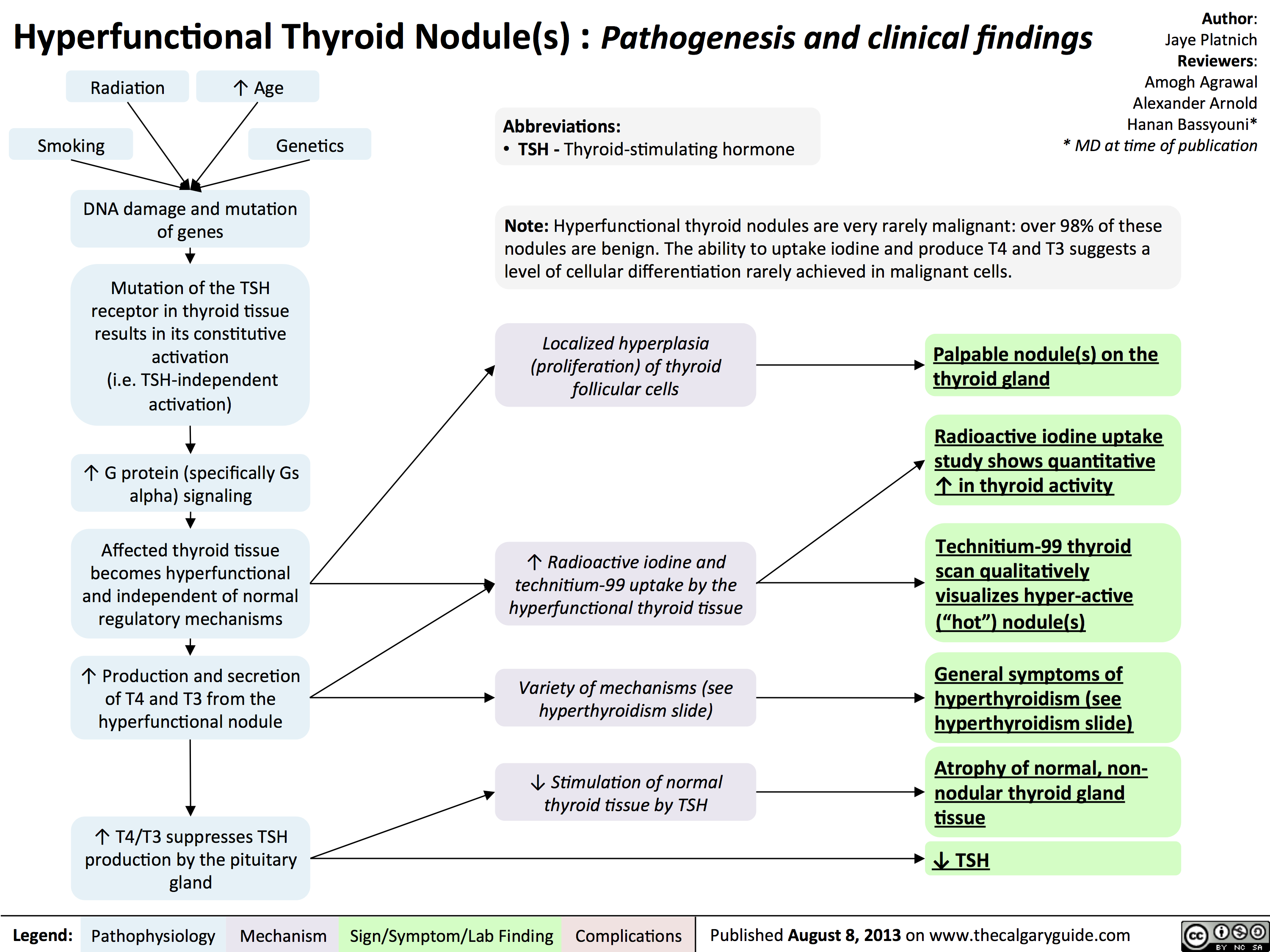
Hyperfunctional
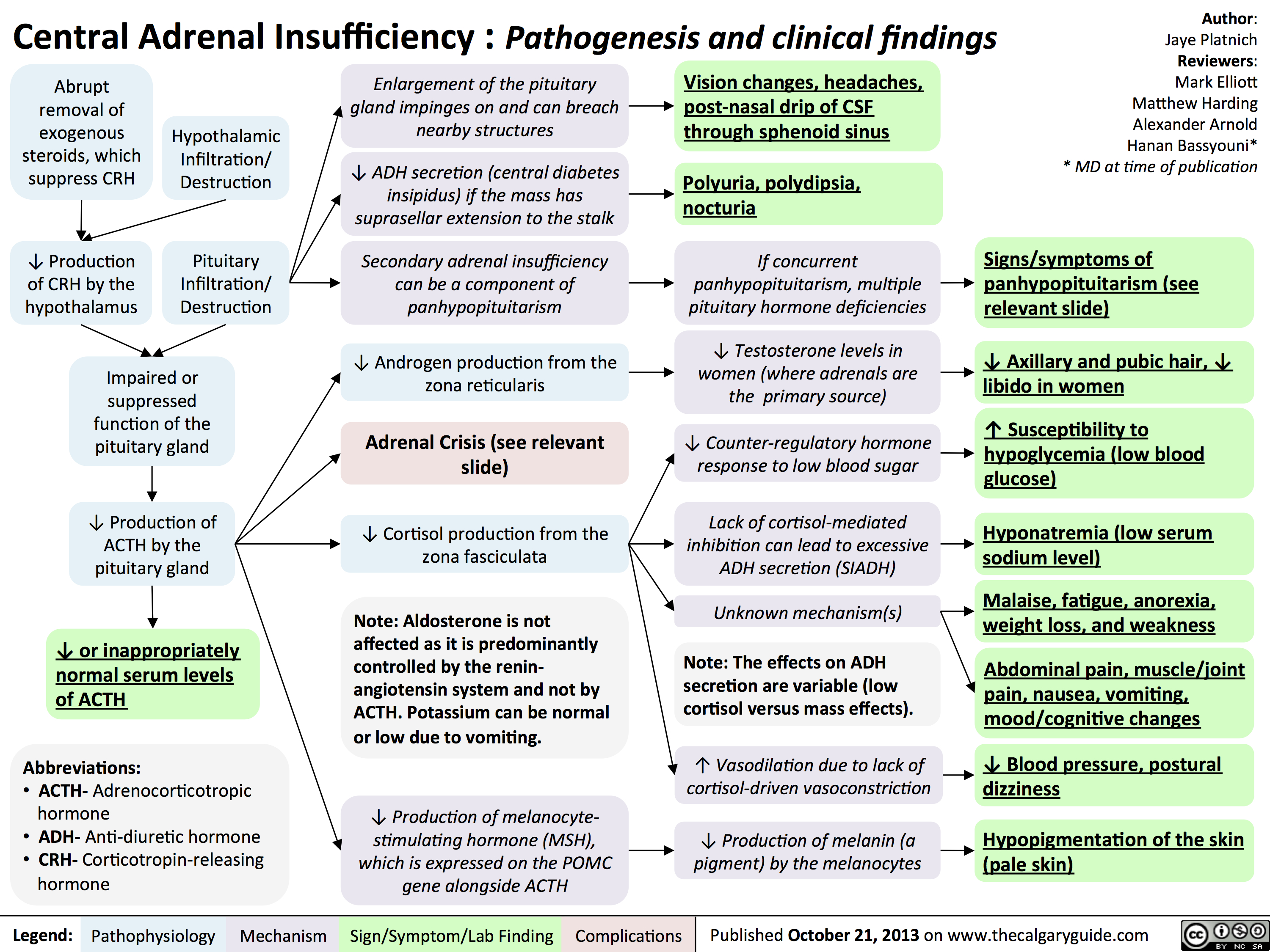
Central Adrenal Insufficiency - Pathogenesis and Clinical Findings
Clinical Findings of Androgen Deficiency
![Yu, Yan - Androgen Deficiency - FINAL.pptx
Hypogonadism in Males:Clinical Findings of Androgen Deficiency? secretion volume from seminal vesicle and prostateAuthor: Yan YuReviewers:Peter VetereGillian GoobieHanan Bassyouni** MD at time of publicationLegend:Published June 18, 2013 on www.thecalgaryguide.comMechanismPathophysiologySign/Symptom/Lab FindingComplications? effect of testosterone on the brain? Libido(sensitive, but less specific)? [testosterone] : [estrogen] ratio at the male breast? ejaculate volume(a sensitive and specific sign)Gynecomastia (palpable breast tissue, not fat, directly under nipple)Fatigue,low mood, irrtabilityHot flashes, sweats(Can be nocturnal; occur only when hypogonadism is severe)Vasomotor neural response of unknown causeFewer spontaneous erections (i.e. in the morning)Lack of androgens (i.e. testosterone, DHT) in men past the age of pubertyIn advanced stages of the disease, after years of hypogonadism:(thus, less commonly seen)Low Bone Mass Density (BMD)Less testosterone to be converted into estrogen in bone? muscle bulk and strengthSmall, soft testicles(<4cm long on orchidometer)Lack of hormones to stimulate and maintain testicular hyperplasia/growthLoss of androgenic hair (on face, midline, and pubic area)Vertebral fracture (height loss), or other fragility fracturesIf sexual development is incomplete from puberty:Note: These clinical findings apply to many disorders, including:-Andropause-Hypopituitarism (suspect if other hormone abnormalities & Sx of mass lesion like visual field loss, diplopia, and headache exist)-Testicular Failure (if Hx of chemo, radiation, excess alcohol, and chronic liver disease)-Klinefelter's (if assoc. tall and eunuchoid stature, breast enlargement and cognitive deficiency - XXY)-Kallman's (if assoc. anosmia, and tall/eunuchoid stature)-Drugs (e.g. ketoconazole, anabolic steroids, spironolactone, digoxin, marijuana)Testosterone's inhibitory effect on estrogen is not enough to prevent breast growthDeficiency in testosterone during puberty delays fusion of epiphysesTall, eunuchoid statureNote: any disease involving an increase in aromatase activity (hyperthyroidism, cirrhosis, HCG-secreting tumors) will also cause relative estrogen excess & subsequent gynecomastia.
111 kB / 272 words](http://calgaryguide.ucalgary.ca/wp-content/uploads/2015/05/Clinical-Findings-of-Androgen-Deficiency.jpg "Yu, Yan - Androgen Deficiency - FINAL.pptx
Hypogonadism in Males:Clinical Findings of Androgen Deficiency? secretion volume from seminal vesicle and prostateAuthor: Yan YuReviewers:Peter VetereGillian GoobieHanan Bassyouni** MD at time of publicationLegend:Published June 18, 2013 on www.thecalgaryguide.comMechanismPathophysiologySign/Symptom/Lab FindingComplications? effect of testosterone on the brain? Libido(sensitive, but less specific)? [testosterone] : [estrogen] ratio at the male breast? ejaculate volume(a sensitive and specific sign)Gynecomastia (palpable breast tissue, not fat, directly under nipple)Fatigue,low mood, irrtabilityHot flashes, sweats(Can be nocturnal; occur only when hypogonadism is severe)Vasomotor neural response of unknown causeFewer spontaneous erections (i.e. in the morning)Lack of androgens (i.e. testosterone, DHT) in men past the age of pubertyIn advanced stages of the disease, after years of hypogonadism:(thus, less commonly seen)Low Bone Mass Density (BMD)Less testosterone to be converted into estrogen in bone? muscle bulk and strengthSmall, soft testicles(<4cm long on orchidometer)Lack of hormones to stimulate and maintain testicular hyperplasia/growthLoss of androgenic hair (on face, midline, and pubic area)Vertebral fracture (height loss), or other fragility fracturesIf sexual development is incomplete from puberty:Note: These clinical findings apply to many disorders, including:-Andropause-Hypopituitarism (suspect if other hormone abnormalities & Sx of mass lesion like visual field loss, diplopia, and headache exist)-Testicular Failure (if Hx of chemo, radiation, excess alcohol, and chronic liver disease)-Klinefelter's (if assoc. tall and eunuchoid stature, breast enlargement and cognitive deficiency - XXY)-Kallman's (if assoc. anosmia, and tall/eunuchoid stature)-Drugs (e.g. ketoconazole, anabolic steroids, spironolactone, digoxin, marijuana)Testosterone's inhibitory effect on estrogen is not enough to prevent breast growthDeficiency in testosterone during puberty delays fusion of epiphysesTall, eunuchoid statureNote: any disease involving an increase in aromatase activity (hyperthyroidism, cirrhosis, HCG-secreting tumors) will also cause relative estrogen excess & subsequent gynecomastia.
111 kB / 272 words")
Hypocalcemia - Clinical Findings
![Yu, Yan - Hypocalcemia - Clinical Findings - FINAL.pptx
Hypocalcemia: Clinical FindingsAuthor: Yan YuReviewers:David WaldnerSean SpenceGreg Kline** MD at time of publicationLegend:Published May 7, 2013 on www.thecalgaryguide.comMechanismPathophysiologySign/Symptom/Lab FindingComplicationsHypocalcemia(serum [Ca2+] <2.1mmol/L)Altered sensory ability of peripheral nervesLess Ca2+ outside cells, with no change in + charges inside cellsPeripheral paraesthesia? Neuronal](http://calgaryguide.ucalgary.ca/wp-content/uploads/2015/05/Hypocalcemia-Clinical-Findings.jpg "Yu, Yan - Hypocalcemia - Clinical Findings - FINAL.pptx
Hypocalcemia: Clinical FindingsAuthor: Yan YuReviewers:David WaldnerSean SpenceGreg Kline** MD at time of publicationLegend:Published May 7, 2013 on www.thecalgaryguide.comMechanismPathophysiologySign/Symptom/Lab FindingComplicationsHypocalcemia(serum [Ca2+] <2.1mmol/L)Altered sensory ability of peripheral nervesLess Ca2+ outside cells, with no change in + charges inside cellsPeripheral paraesthesia? Neuronal")
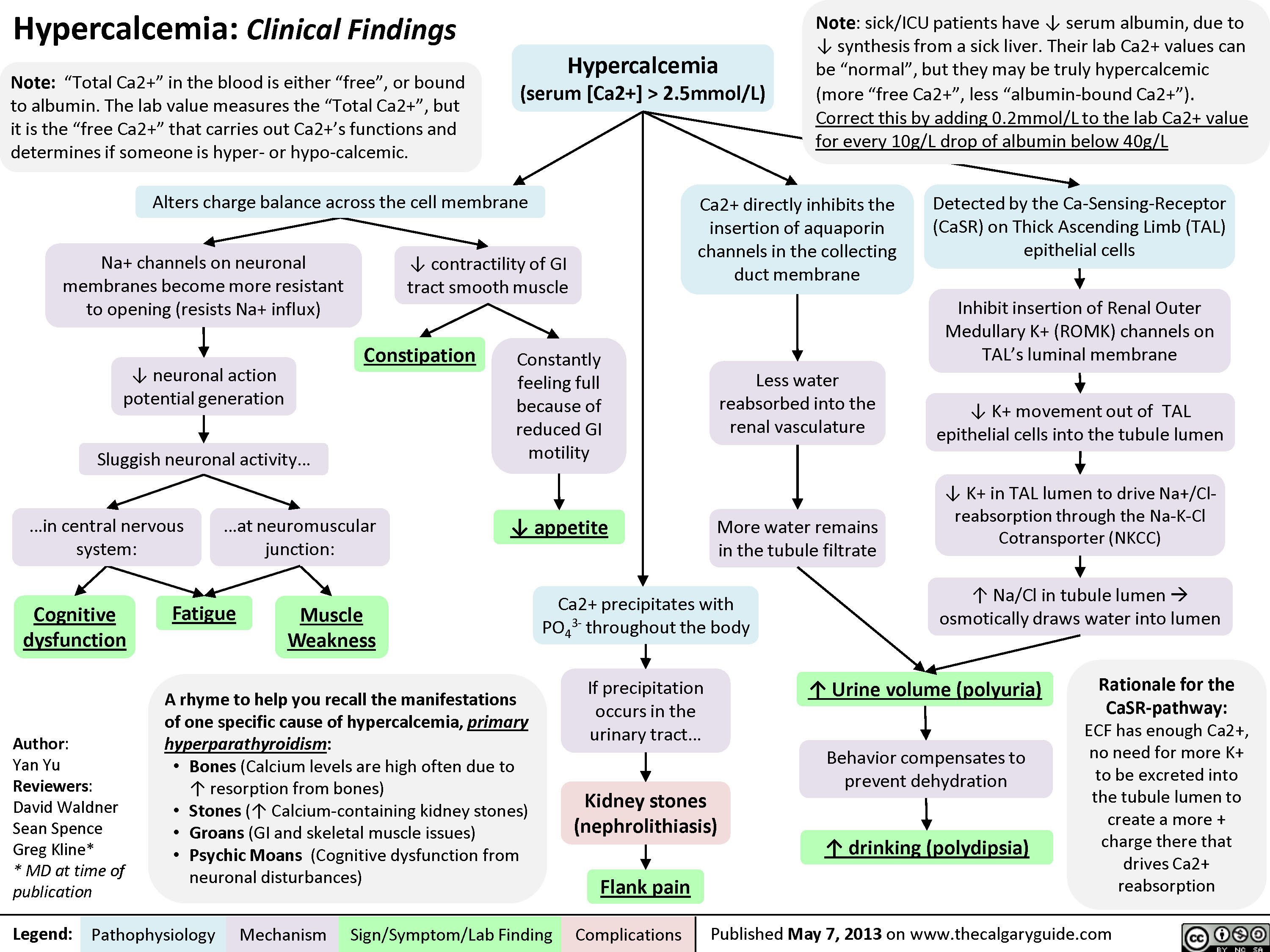
Hypercalcemia - Clinical Findings
![Yu, Yan - Hypercalcemia - Clinical Findings - FINAL.pptx
Hypercalcemia: Clinical FindingsAuthor: Yan YuReviewers:David WaldnerSean SpenceGreg Kline** MD at time of publicationLegend:Published May 7, 2013 on www.thecalgaryguide.comMechanismPathophysiologySign/Symptom/Lab FindingComplicationsHypercalcemia(serum [Ca2+] > 2.5mmol/L)Na+ channels on neuronal membranes become more resistant to opening (resists Na+ influx)Cognitive dysfunctionIf precipitation occurs in the urinary tract...Fatigue? contractility of GI tract smooth muscle? K+ movement out of TAL epithelial cells into the tubule lumen Alters charge balance across the cell membraneCa2+ precipitates with PO43- throughout the bodyDetected by the Ca-Sensing-Receptor (CaSR) on Thick Ascending Limb (TAL) epithelial cells? neuronal action potential generationSluggish neuronal activity...? appetiteConstipationFlank painInhibit insertion of Renal Outer Medullary K+ (ROMK) channels on TAL's luminal membrane? K+ in TAL lumen to drive Na+/Cl- reabsorption through the Na-K-Cl Cotransporter (NKCC)? Na/Cl in tubule lumen ? osmotically draws water into lumen? drinking (polydipsia)? Urine volume (polyuria)Rationale for the CaSR-pathway: ECF has enough Ca2+, no need for more K+ to be excreted into the tubule lumen to create a more + charge there that drives Ca2+ reabsorptionBehavior compensates to prevent dehydrationKidney stones (nephrolithiasis)Constantly feeling full because of reduced GI motilityCa2+ directly inhibits the insertion of aquaporin channels in the collecting duct membraneLess water reabsorbed into the renal vasculatureMore water remains in the tubule filtrateMuscle Weakness...in central nervous system:...at neuromuscular junction:A rhyme to help you recall the manifestations of one specific cause of hypercalcemia, primary hyperparathyroidism:Bones (Calcium levels are high often due to ? resorption from bones)Stones (? Calcium-containing kidney stones)Groans (GI and skeletal muscle issues) Psychic Moans (Cognitive dysfunction from neuronal disturbances)Note: sick/ICU patients have ? serum albumin, due to ? synthesis from a sick liver. Their lab Ca2+ values can be](http://calgaryguide.ucalgary.ca/wp-content/uploads/2015/05/Hypercalcemia-Clinical-Findings.jpg "Yu, Yan - Hypercalcemia - Clinical Findings - FINAL.pptx
Hypercalcemia: Clinical FindingsAuthor: Yan YuReviewers:David WaldnerSean SpenceGreg Kline** MD at time of publicationLegend:Published May 7, 2013 on www.thecalgaryguide.comMechanismPathophysiologySign/Symptom/Lab FindingComplicationsHypercalcemia(serum [Ca2+] > 2.5mmol/L)Na+ channels on neuronal membranes become more resistant to opening (resists Na+ influx)Cognitive dysfunctionIf precipitation occurs in the urinary tract...Fatigue? contractility of GI tract smooth muscle? K+ movement out of TAL epithelial cells into the tubule lumen Alters charge balance across the cell membraneCa2+ precipitates with PO43- throughout the bodyDetected by the Ca-Sensing-Receptor (CaSR) on Thick Ascending Limb (TAL) epithelial cells? neuronal action potential generationSluggish neuronal activity...? appetiteConstipationFlank painInhibit insertion of Renal Outer Medullary K+ (ROMK) channels on TAL's luminal membrane? K+ in TAL lumen to drive Na+/Cl- reabsorption through the Na-K-Cl Cotransporter (NKCC)? Na/Cl in tubule lumen ? osmotically draws water into lumen? drinking (polydipsia)? Urine volume (polyuria)Rationale for the CaSR-pathway: ECF has enough Ca2+, no need for more K+ to be excreted into the tubule lumen to create a more + charge there that drives Ca2+ reabsorptionBehavior compensates to prevent dehydrationKidney stones (nephrolithiasis)Constantly feeling full because of reduced GI motilityCa2+ directly inhibits the insertion of aquaporin channels in the collecting duct membraneLess water reabsorbed into the renal vasculatureMore water remains in the tubule filtrateMuscle Weakness...in central nervous system:...at neuromuscular junction:A rhyme to help you recall the manifestations of one specific cause of hypercalcemia, primary hyperparathyroidism:Bones (Calcium levels are high often due to ? resorption from bones)Stones (? Calcium-containing kidney stones)Groans (GI and skeletal muscle issues) Psychic Moans (Cognitive dysfunction from neuronal disturbances)Note: sick/ICU patients have ? serum albumin, due to ? synthesis from a sick liver. Their lab Ca2+ values can be")
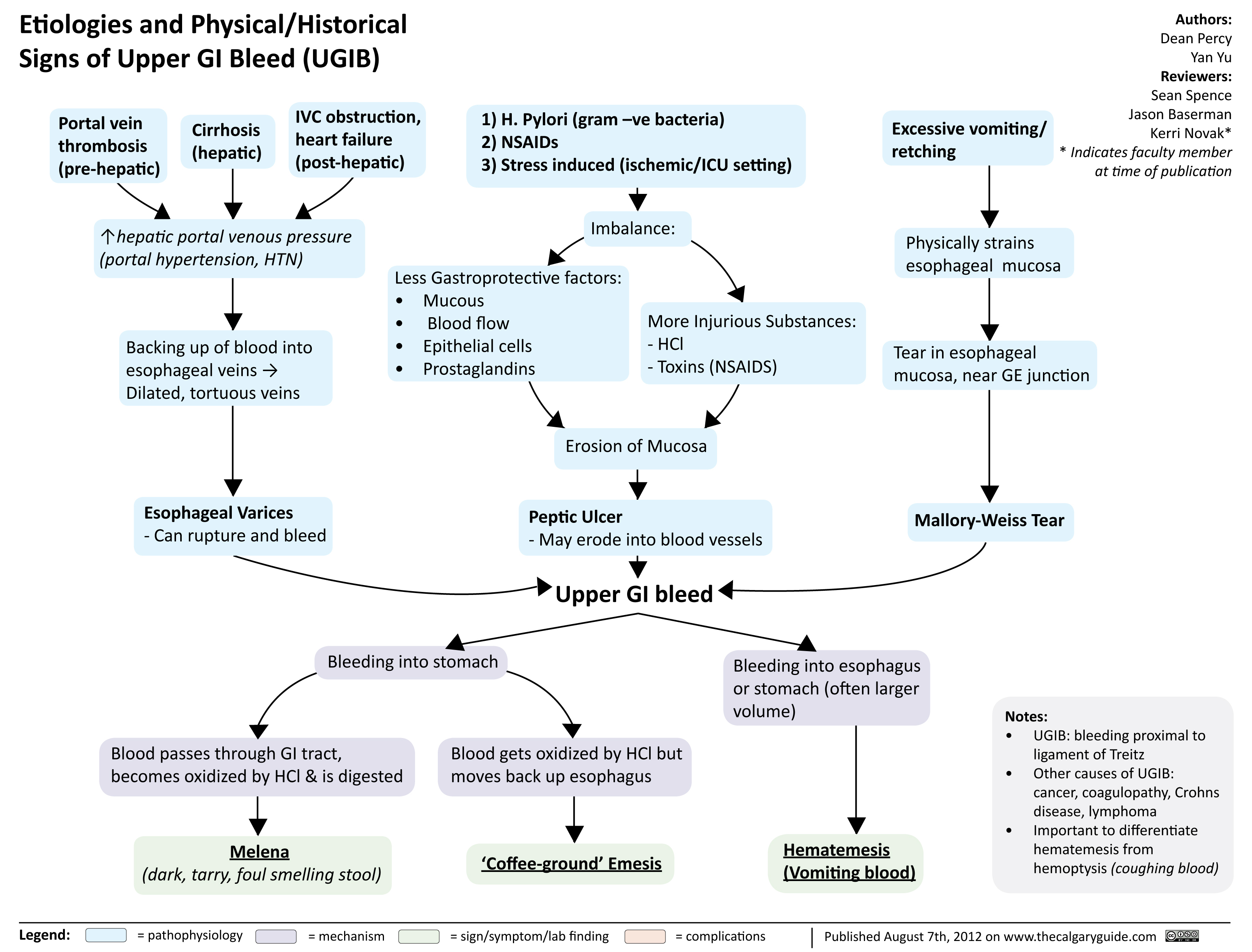
Etiologies and Physical Historical Signs of Upper GI Bleed
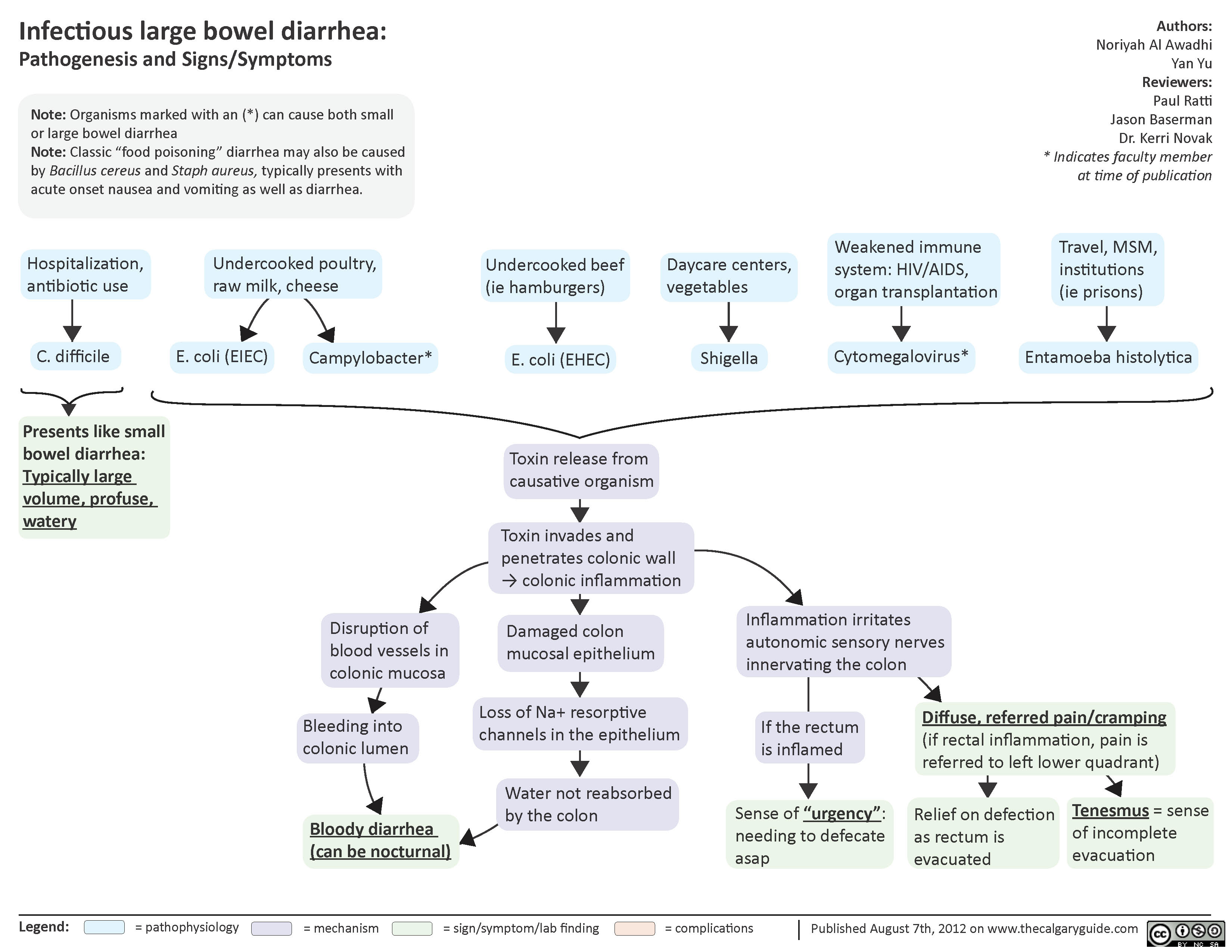
Infectious Large Bowel Diarrhea
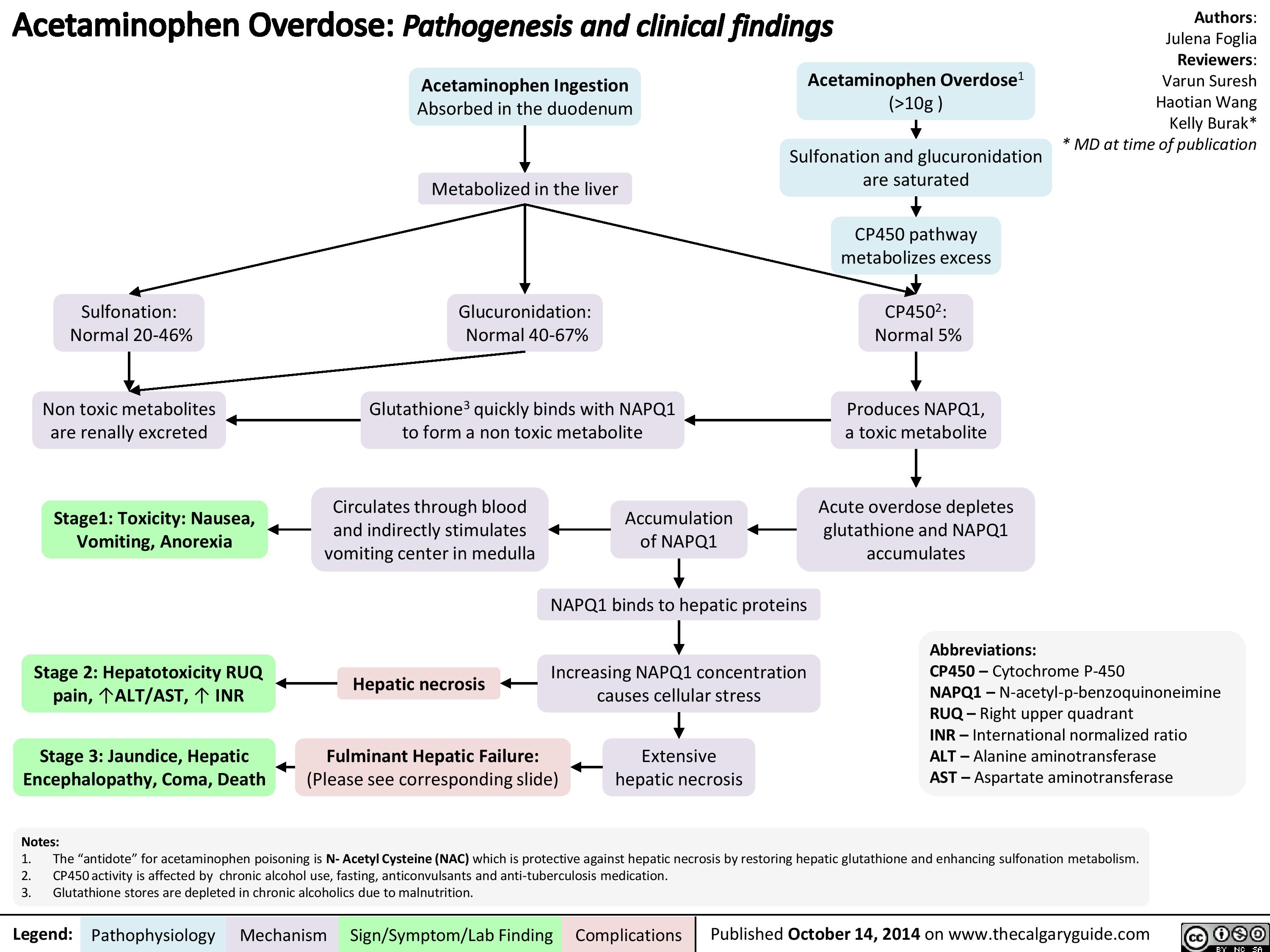
Acetaminophen Overdose
Gall Bladder Disorders
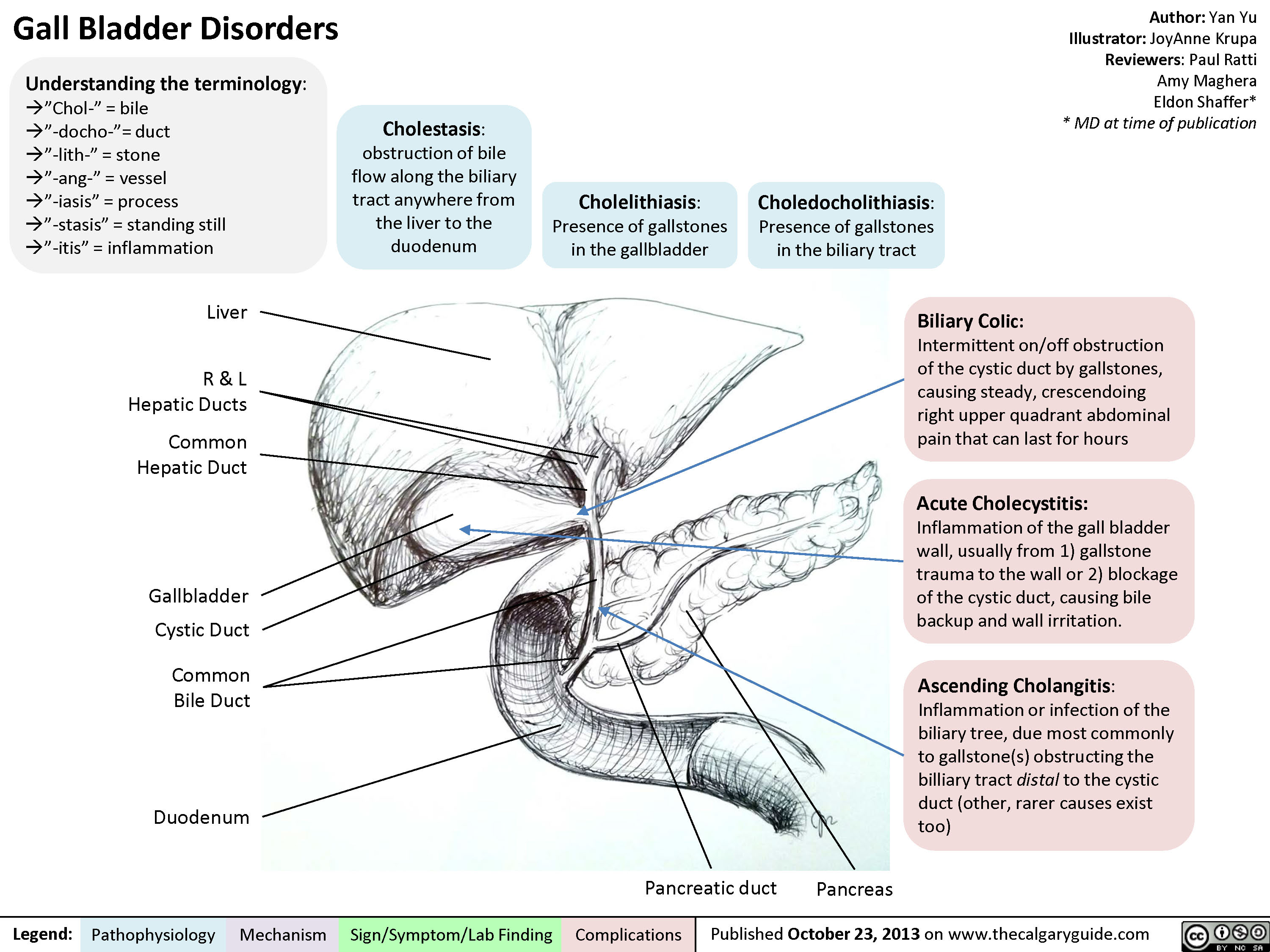
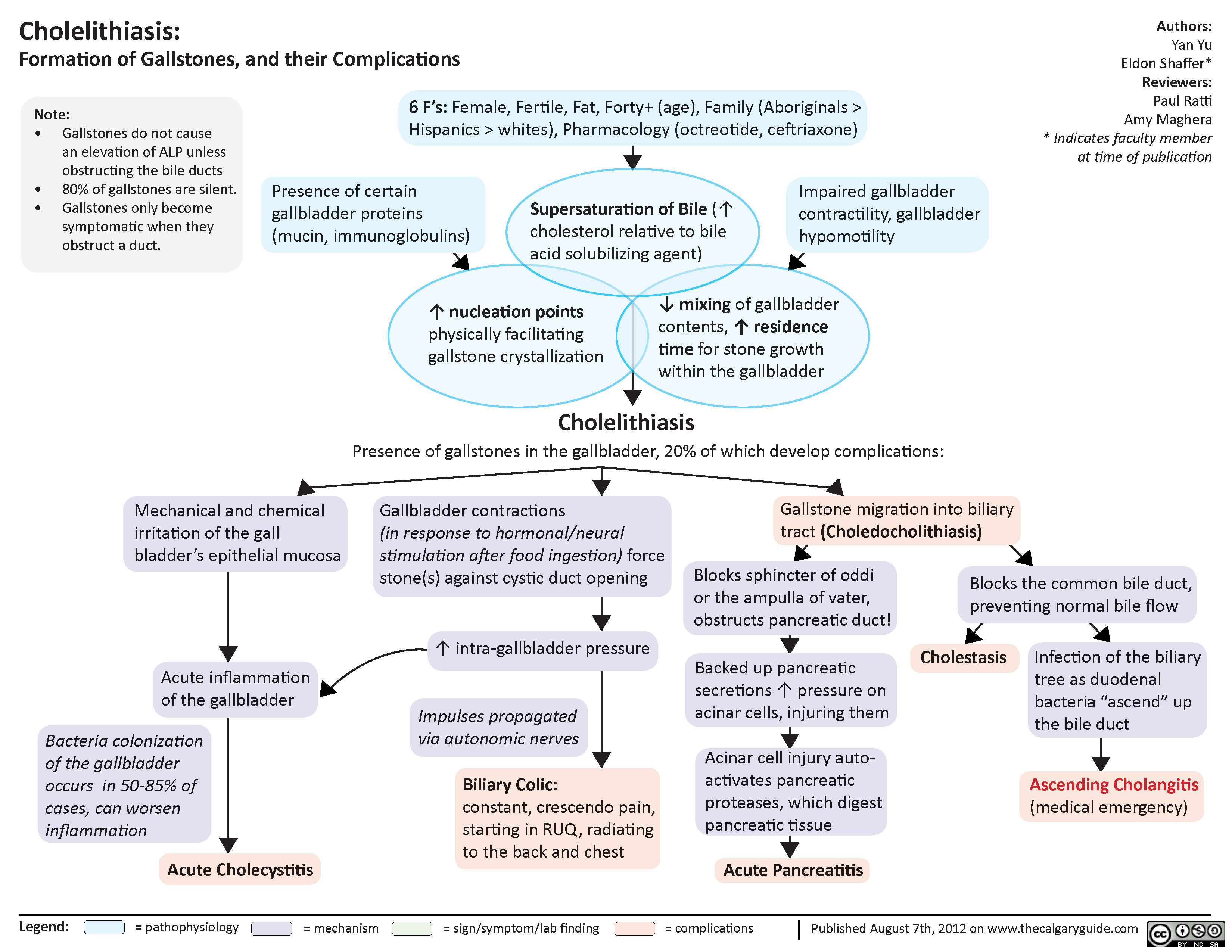
Cholelithiasis
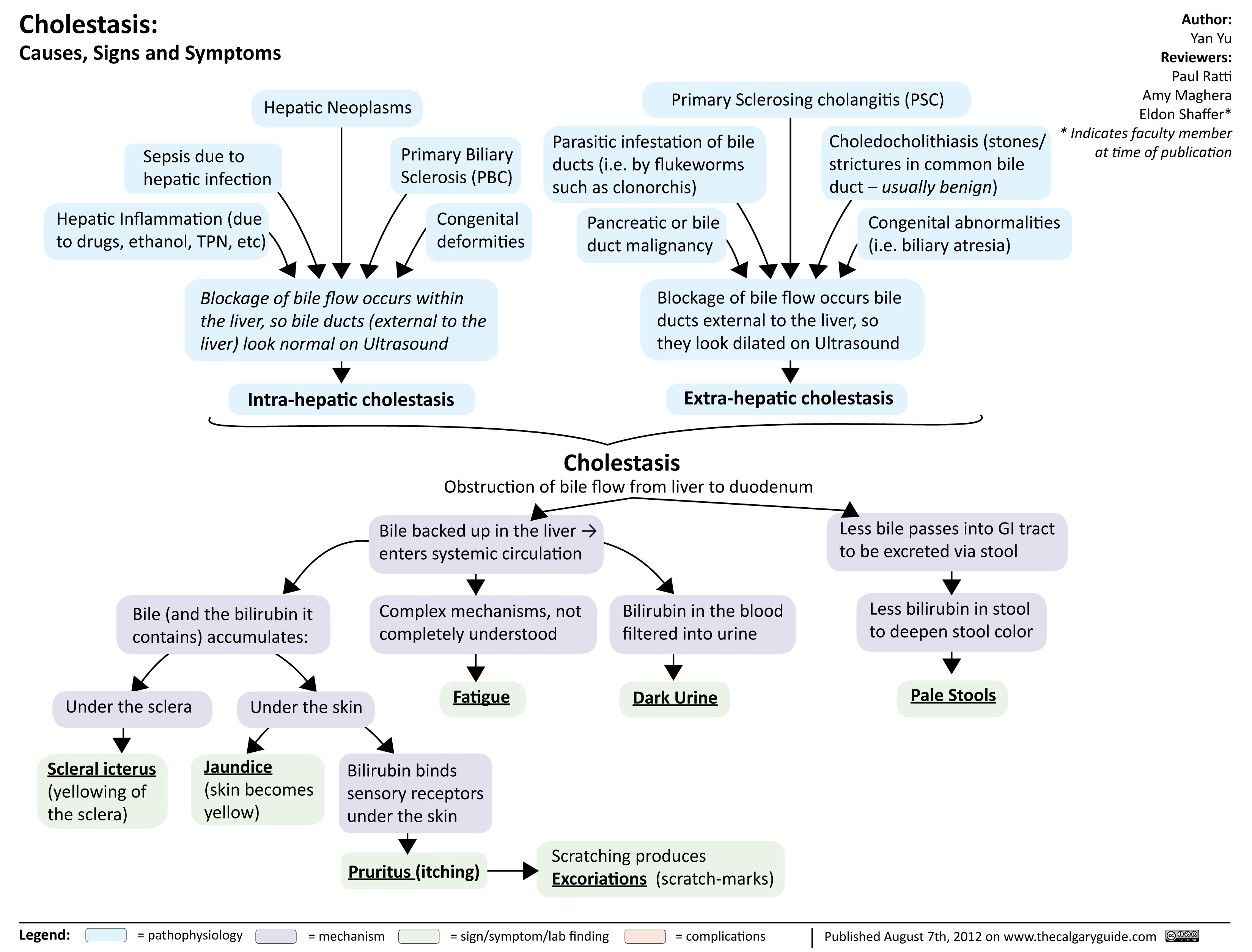
Cholestasis
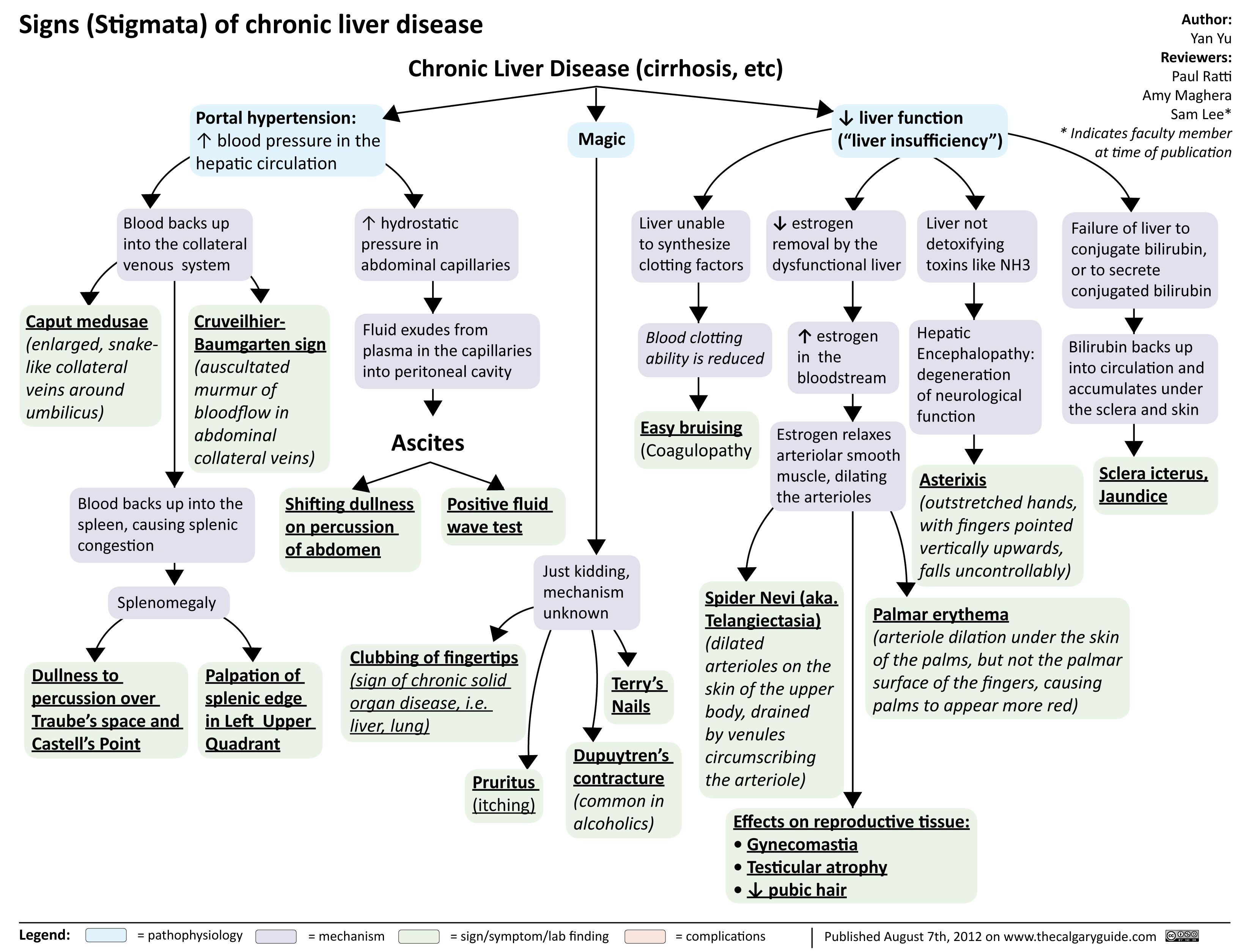
signs of chronic liver disease
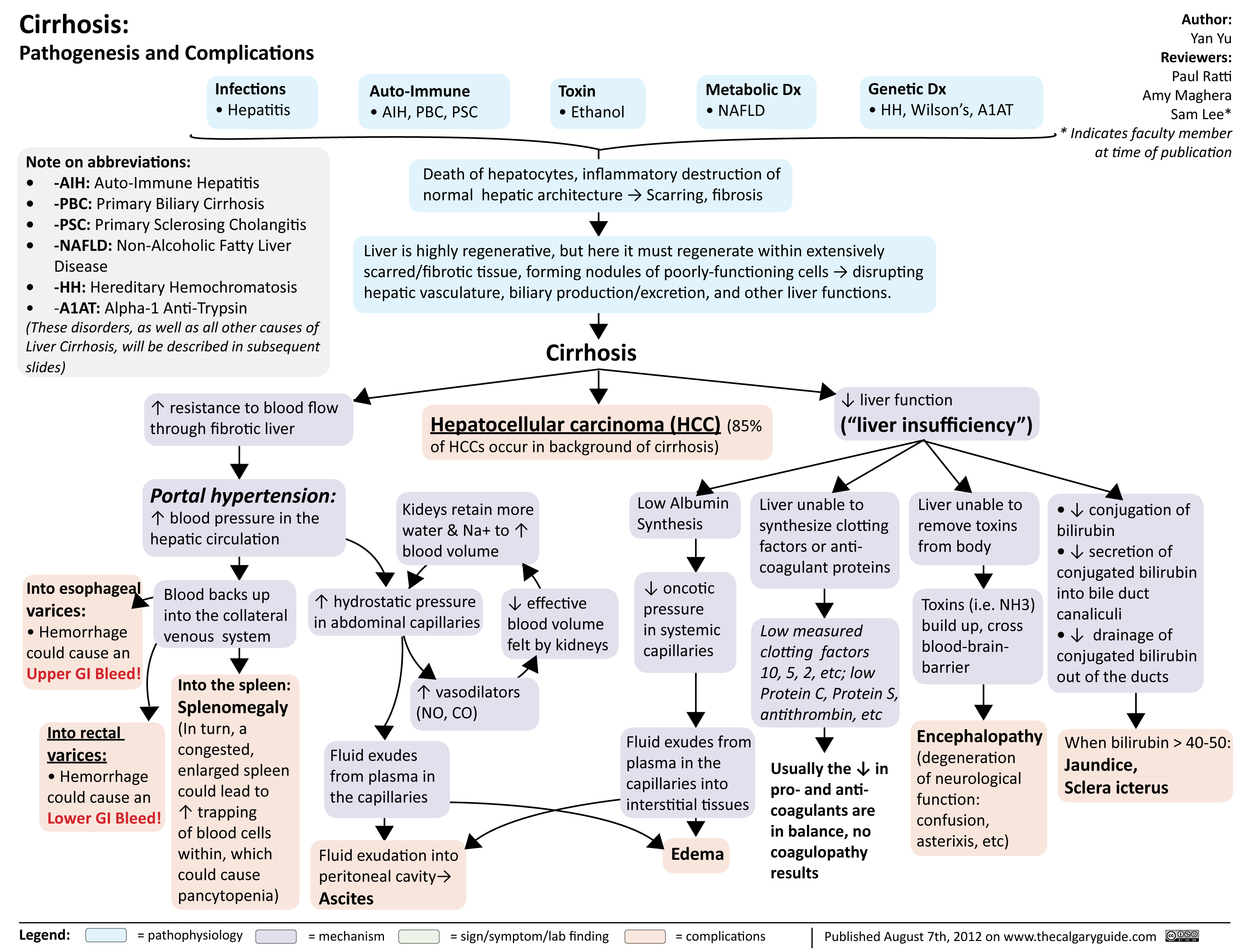
cirrhosis
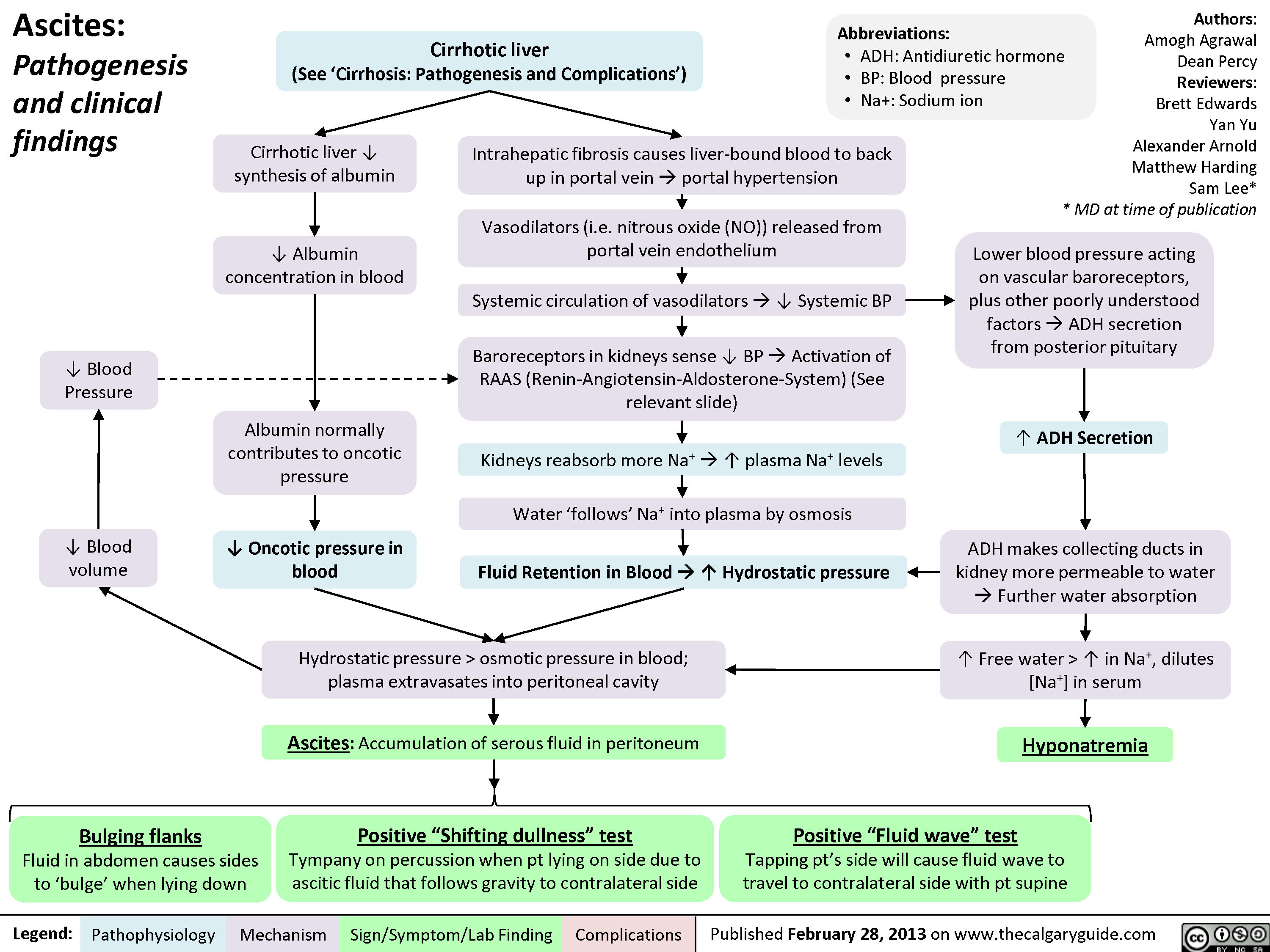
Ascites Pathogenesis and Clinical Findings
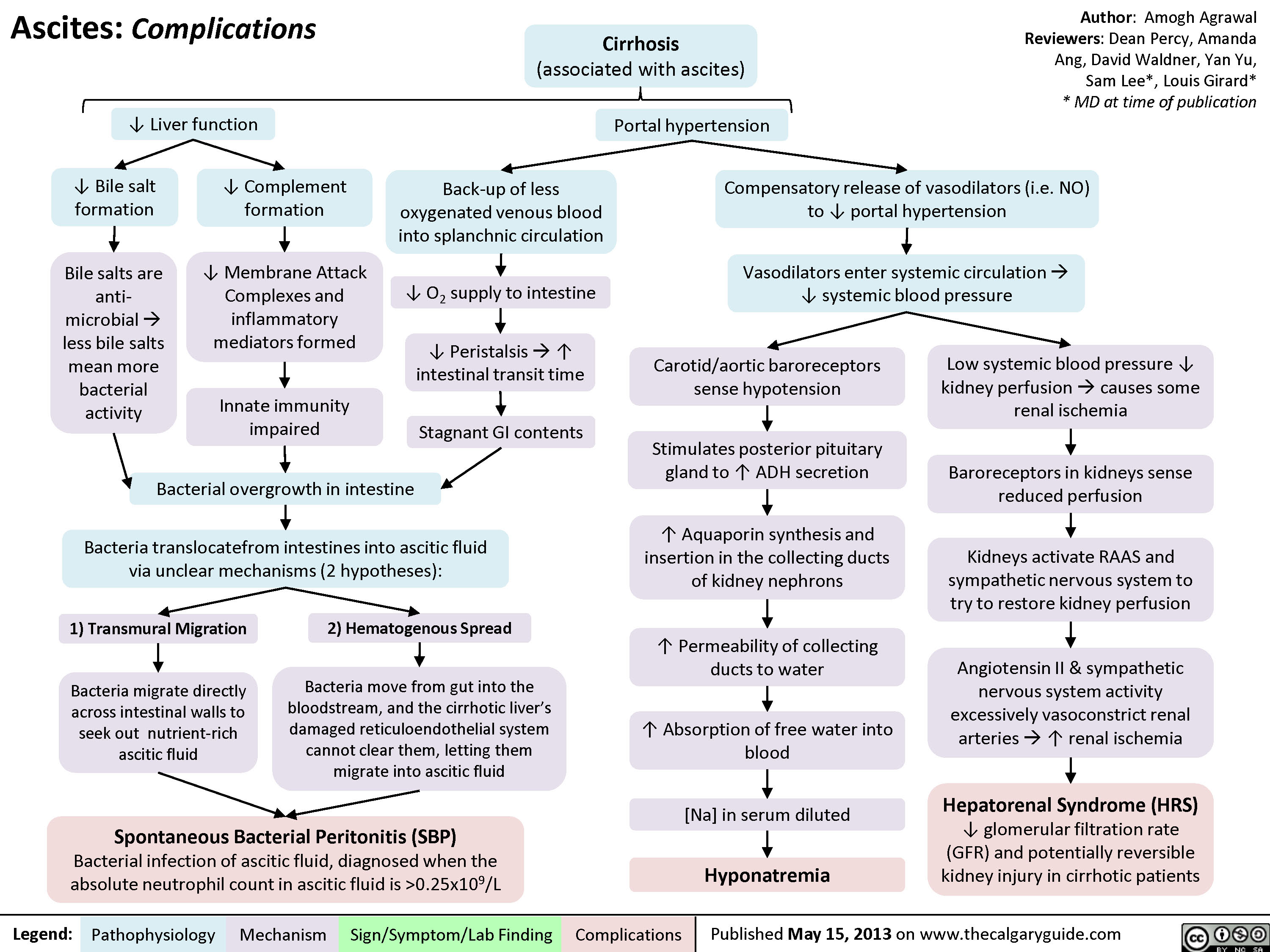
Ascites Complications
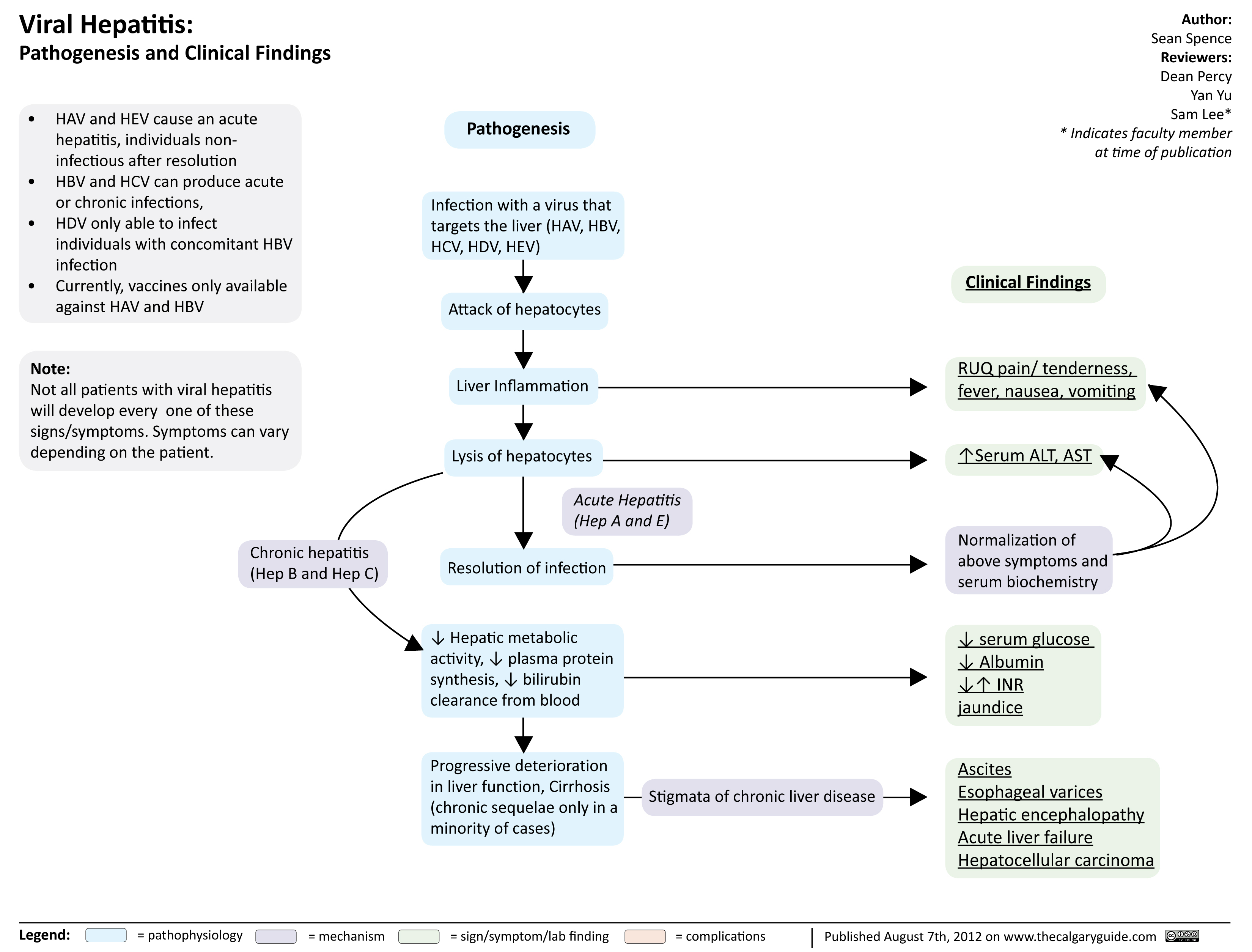
viral hepatits
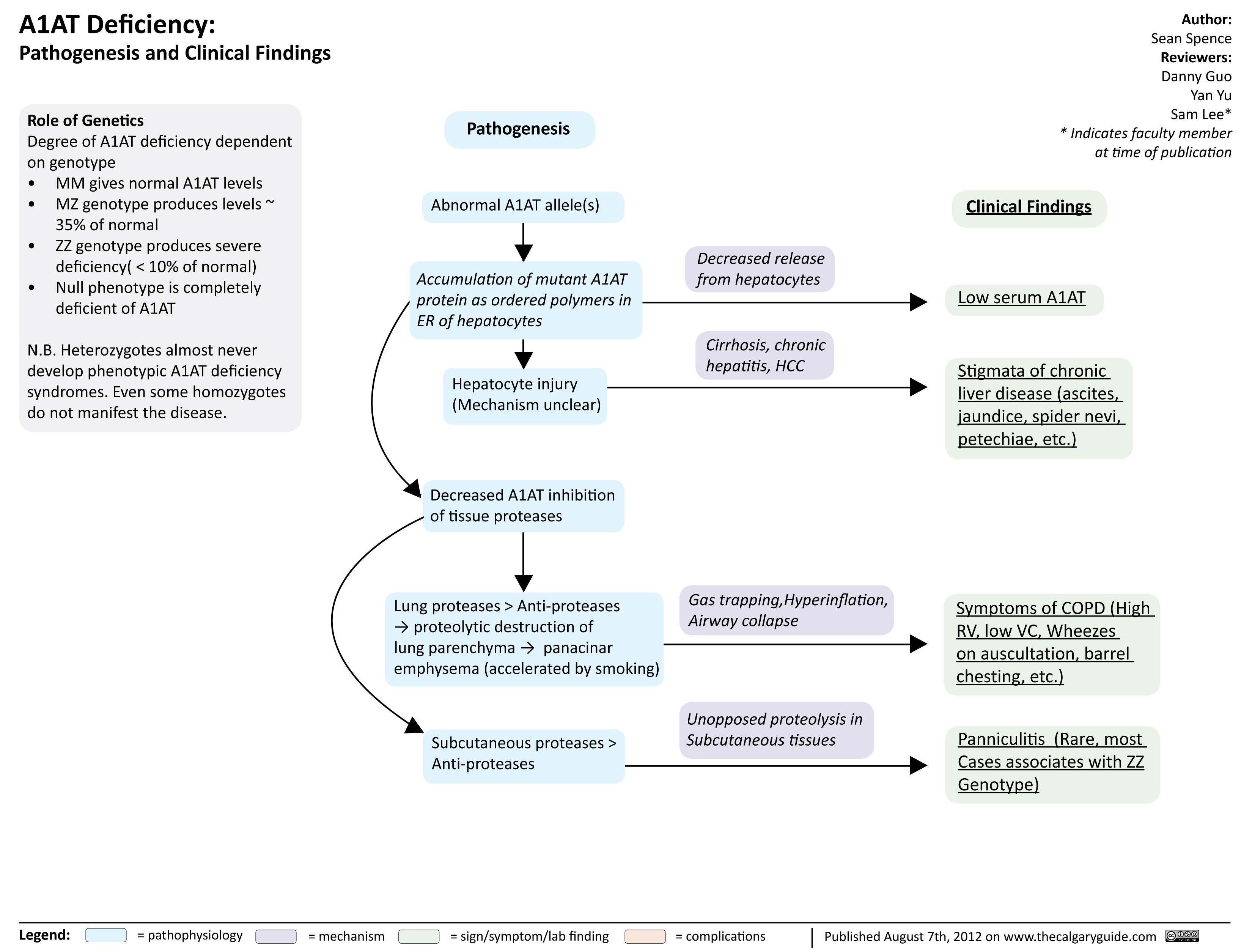
A1ATDeficiency
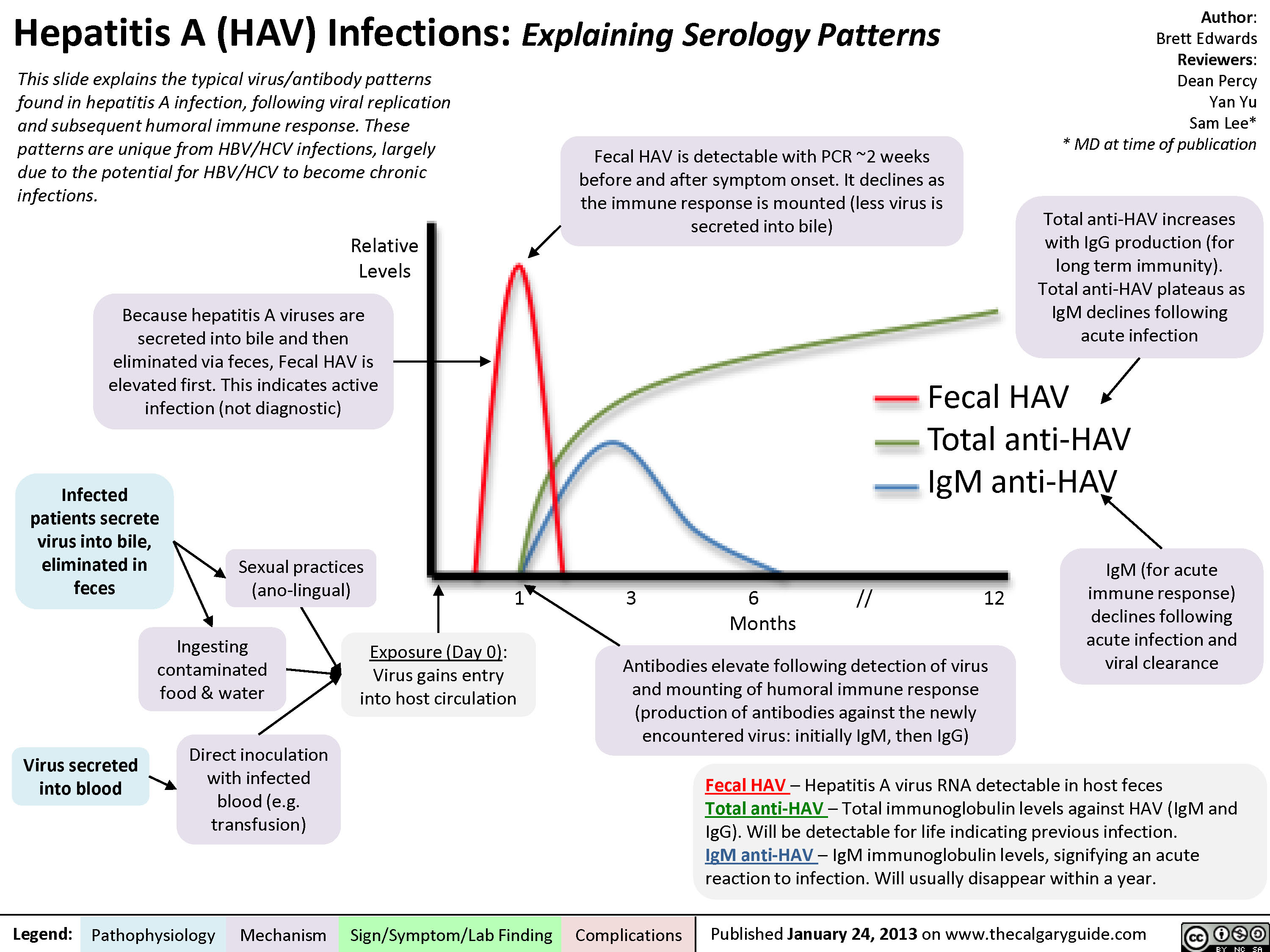
Hepatitis A (HAV) Infections
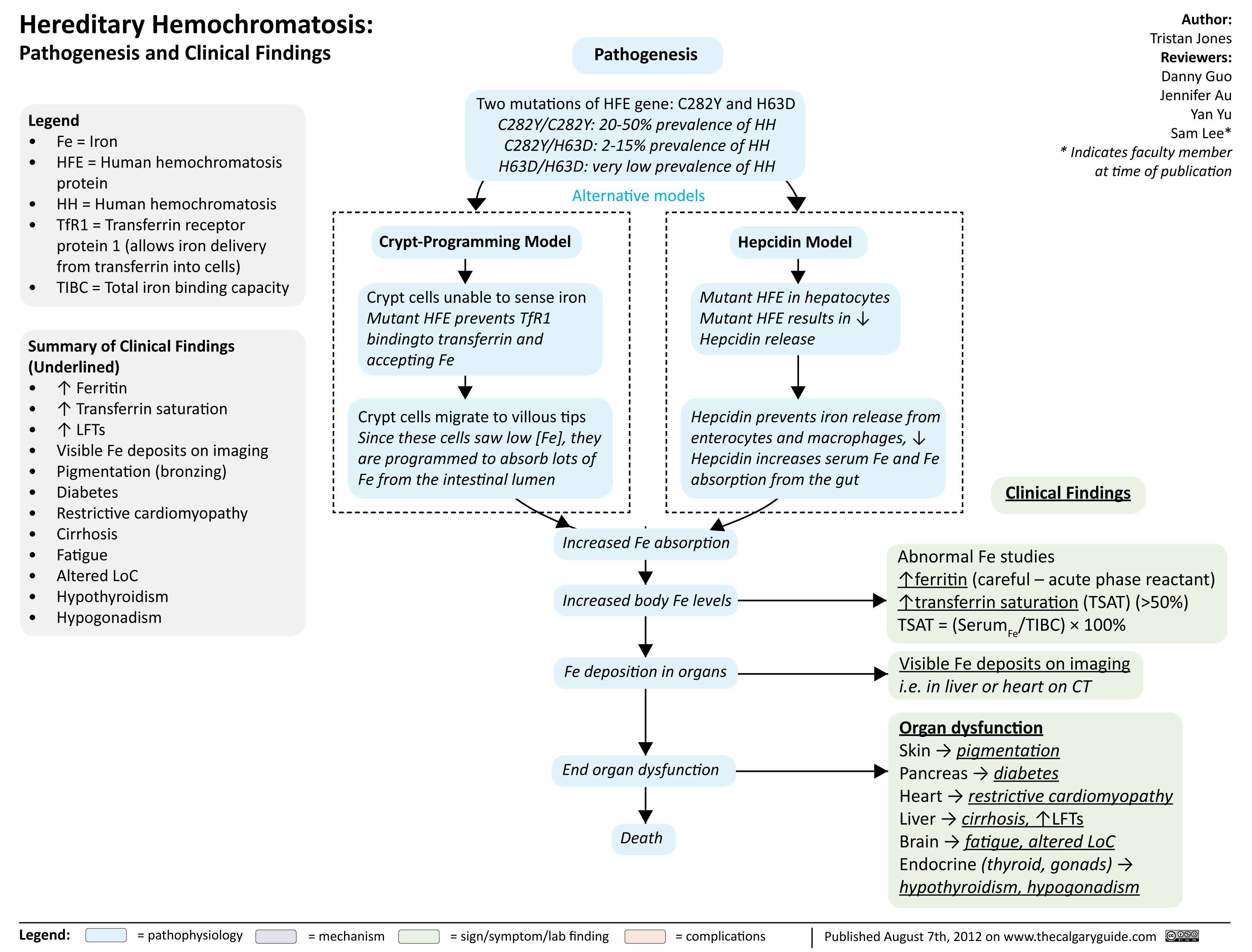
Hereditary hemochromatosis
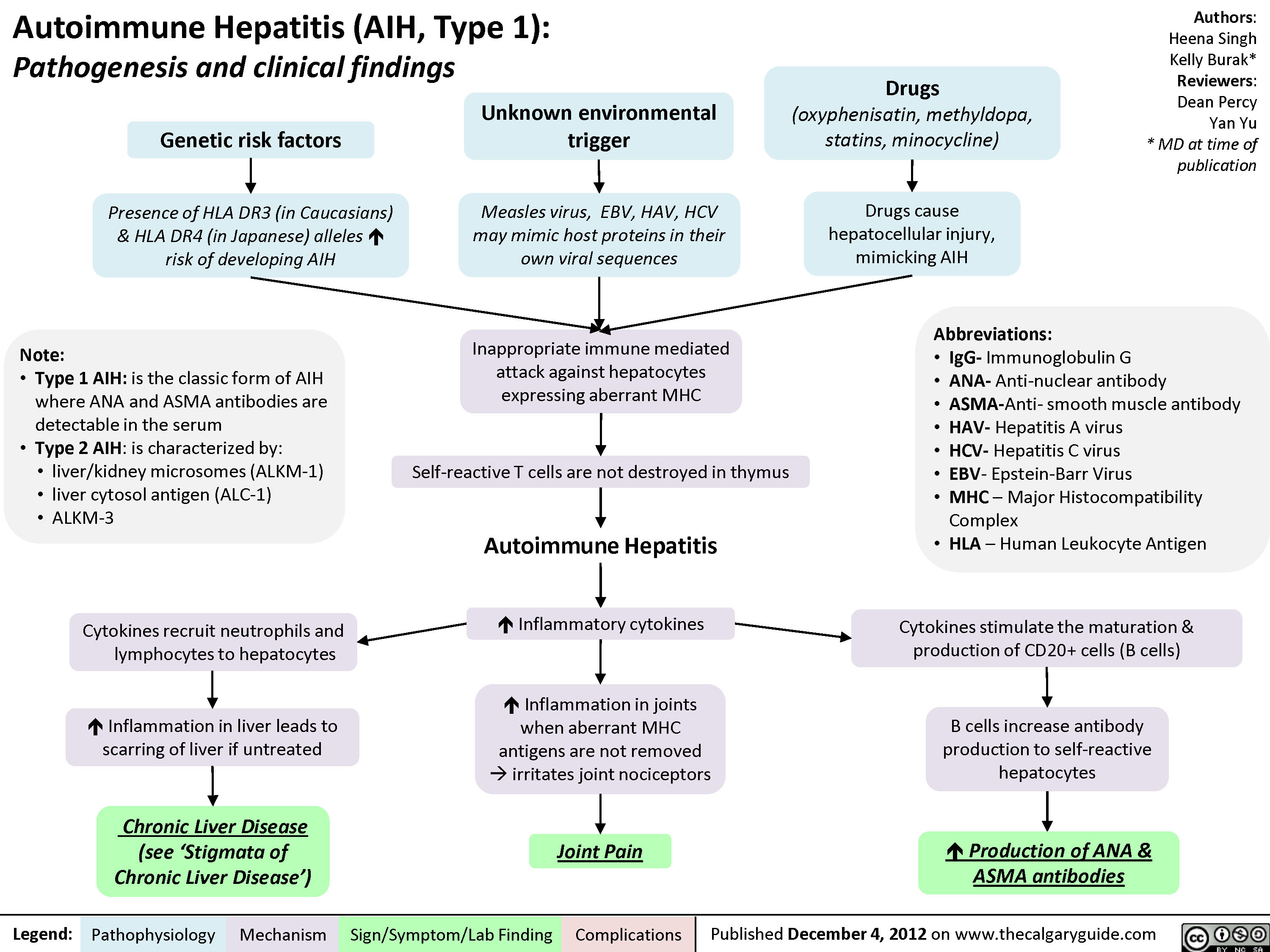
Auto-immune Hepatitis
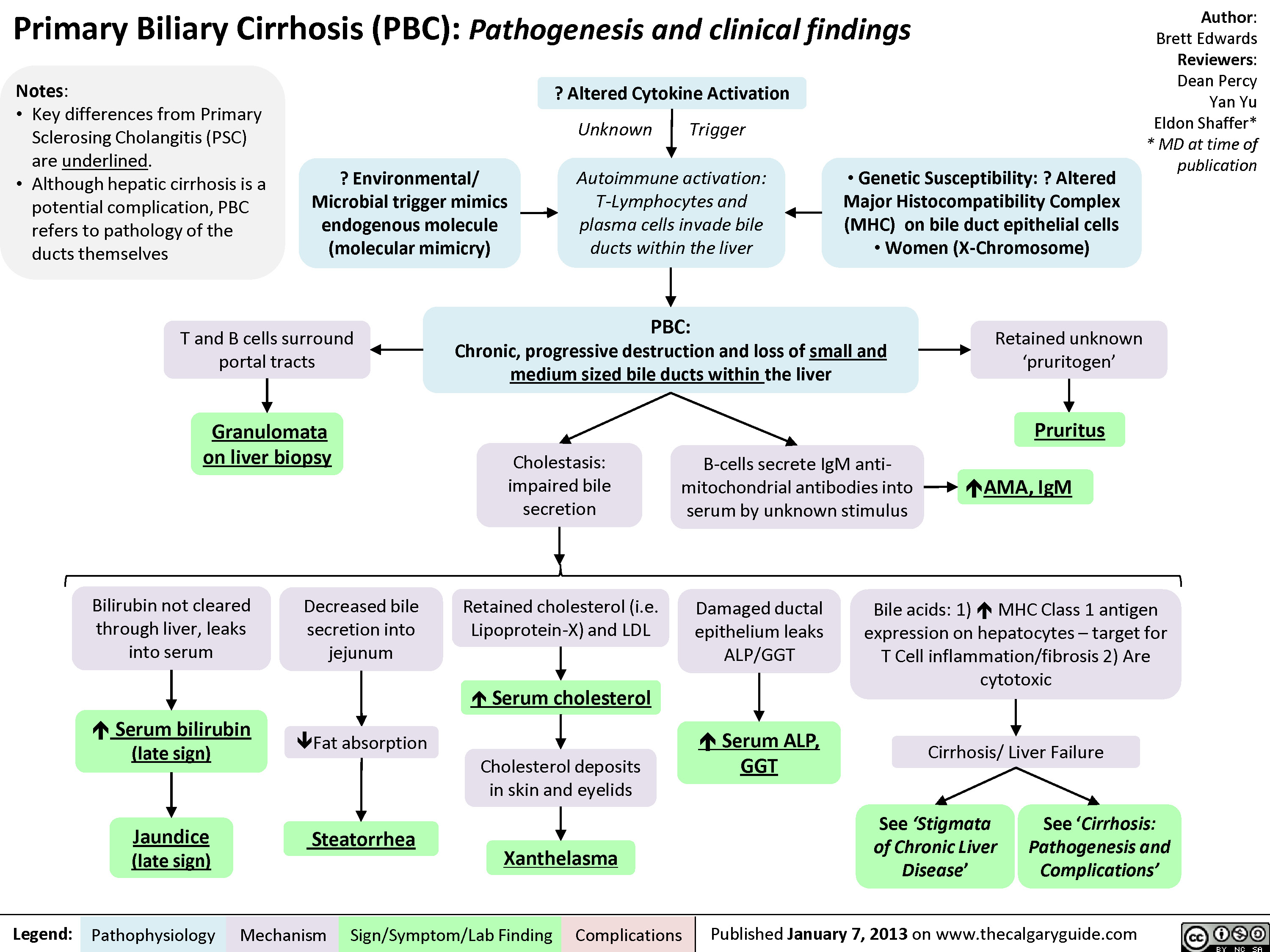
Primary Biliary Cirrhosis (PBC)
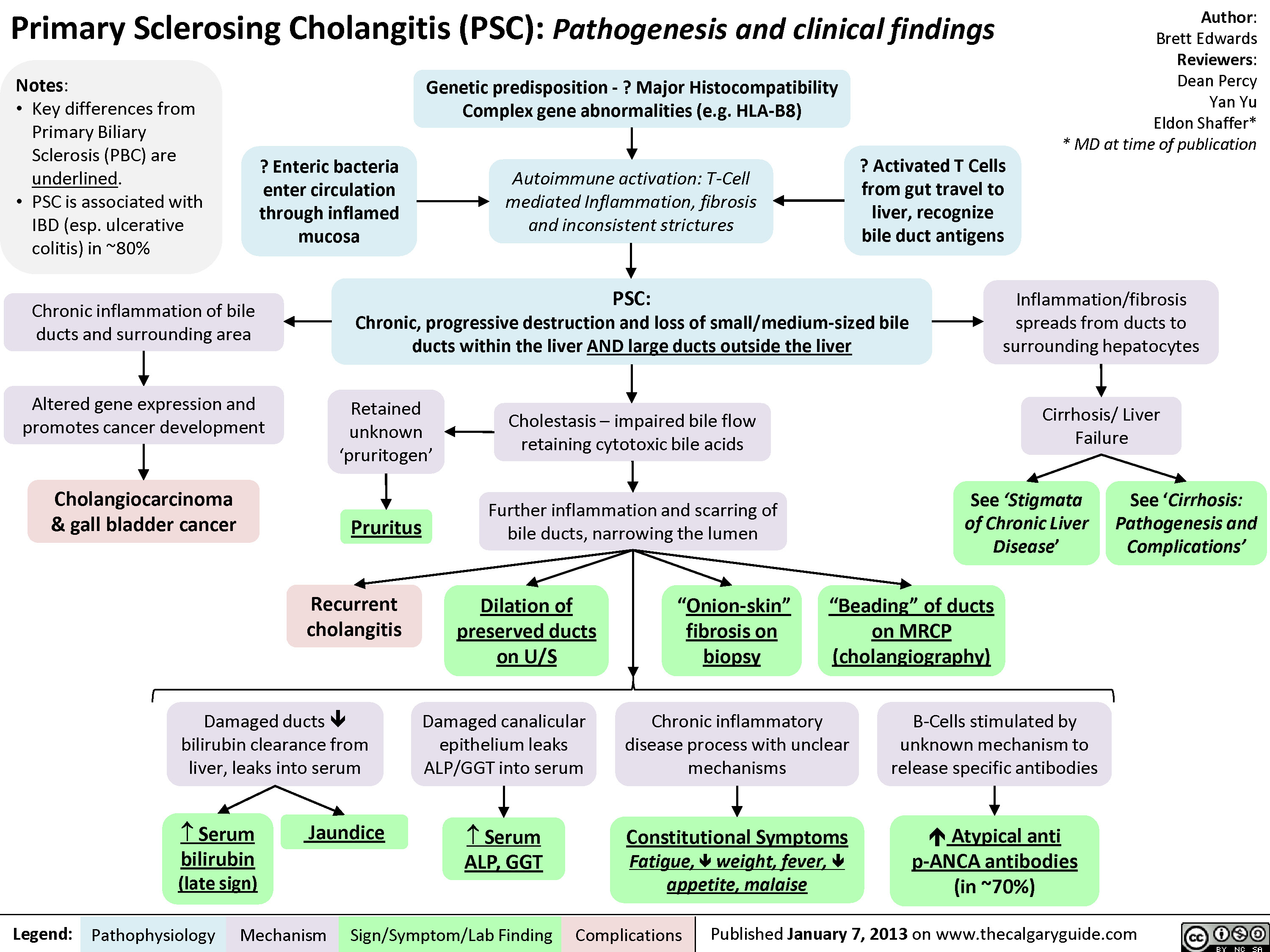
Primary Sclerosing Cholangitis (PSC)
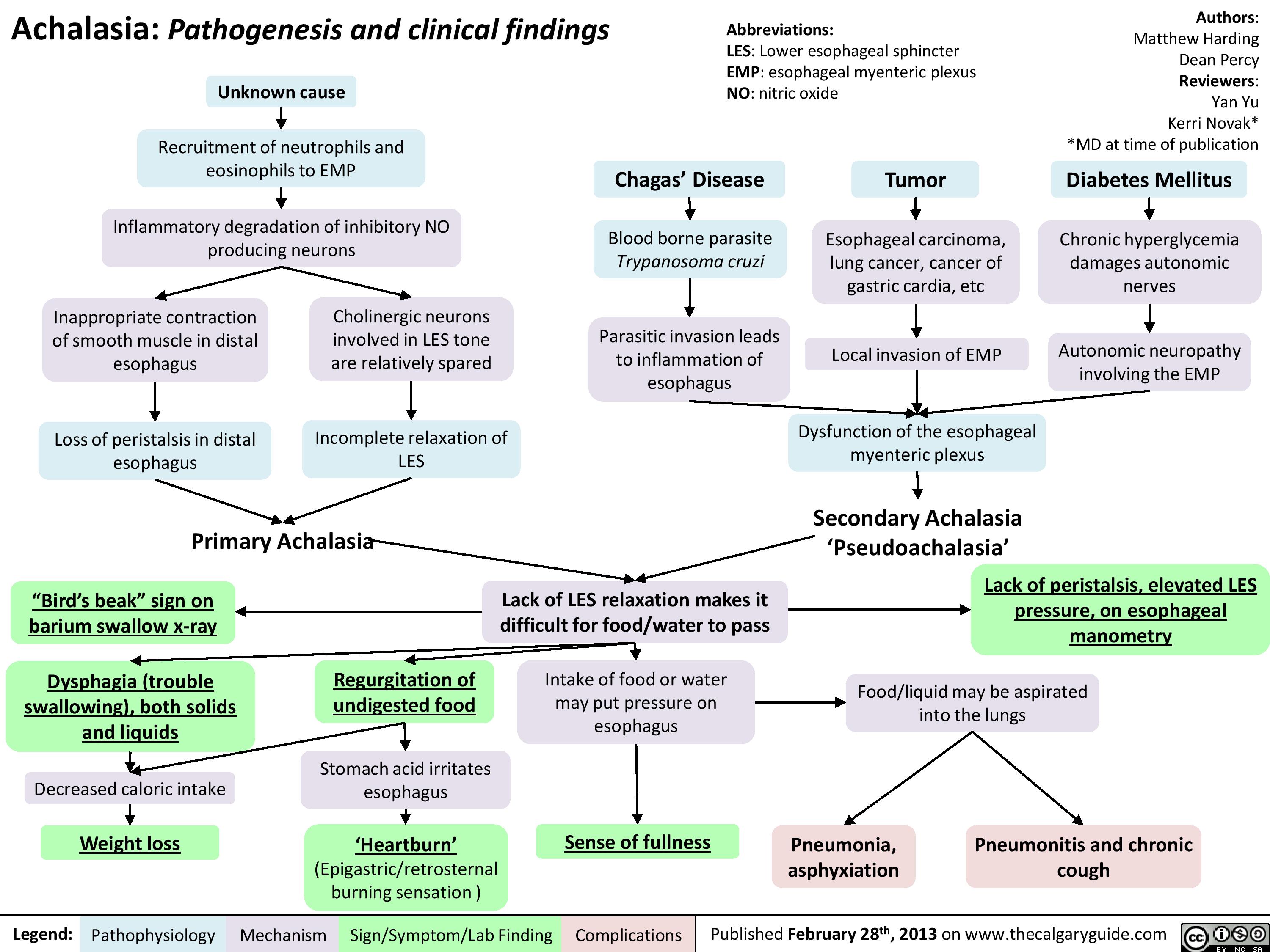
Achalasia Pathogenesis and clinical findings
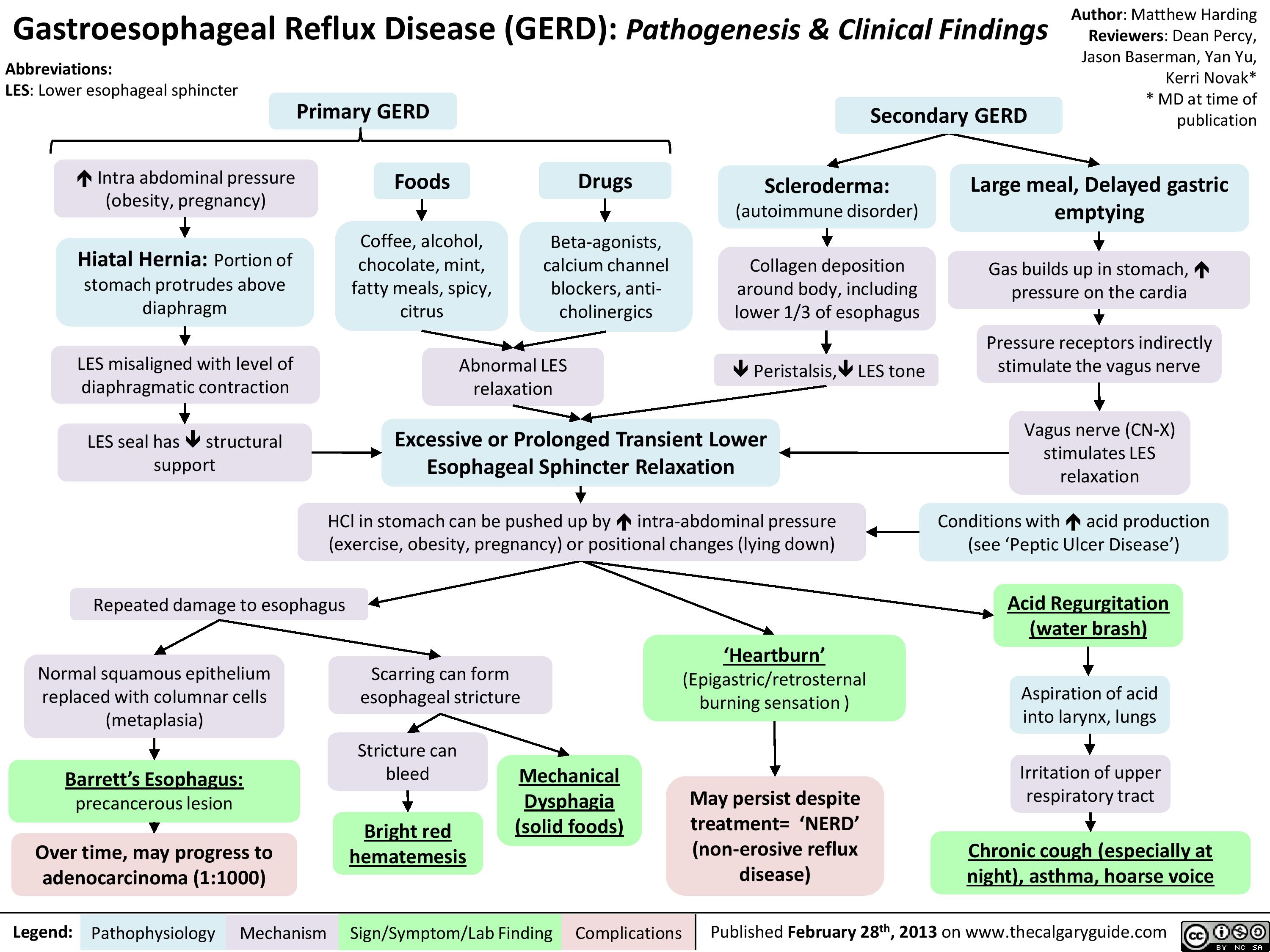
Gastroesophageal Reflux Disease (GERD) Pathogenesis and Clinical Findings
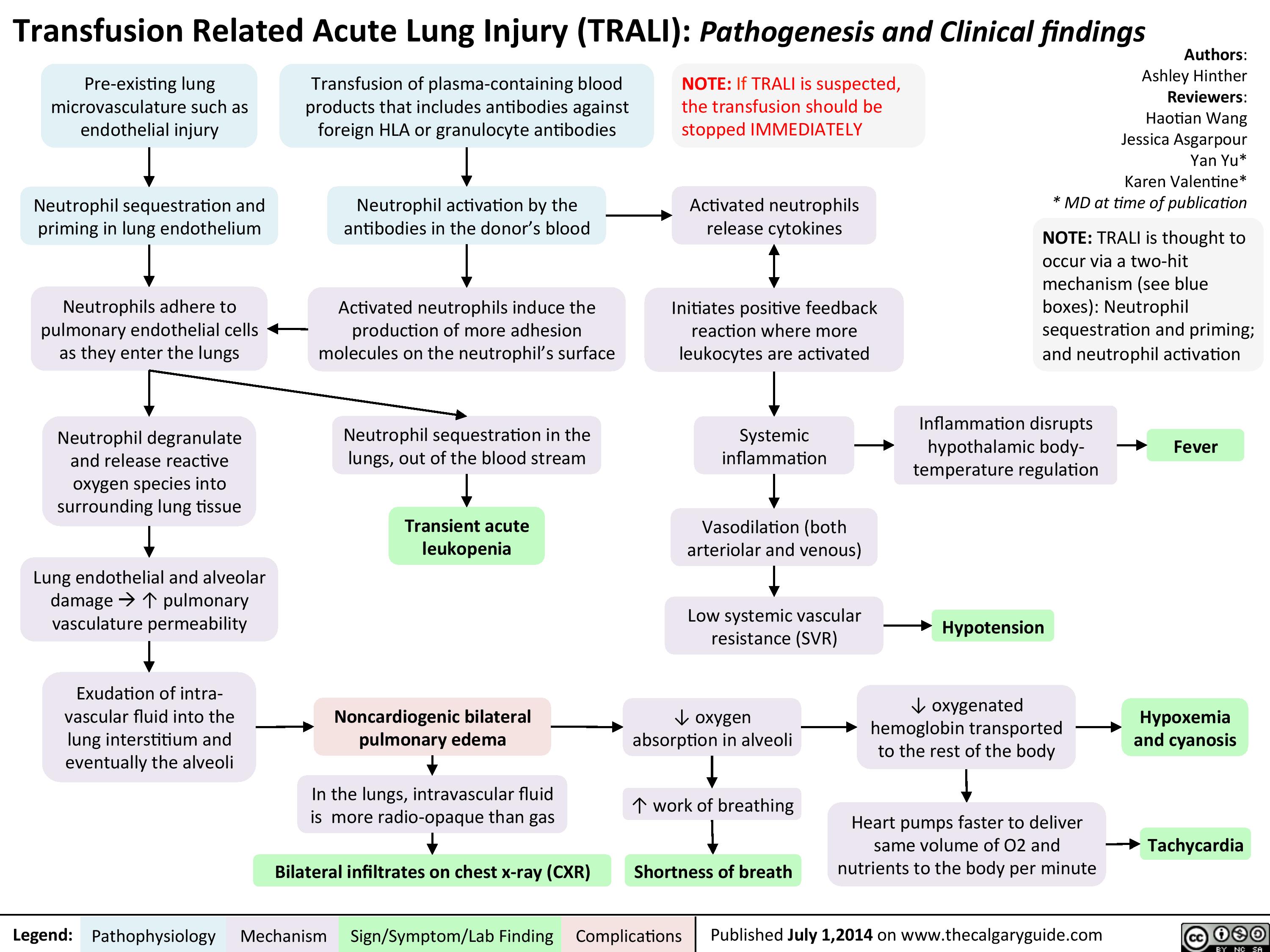
TRALI
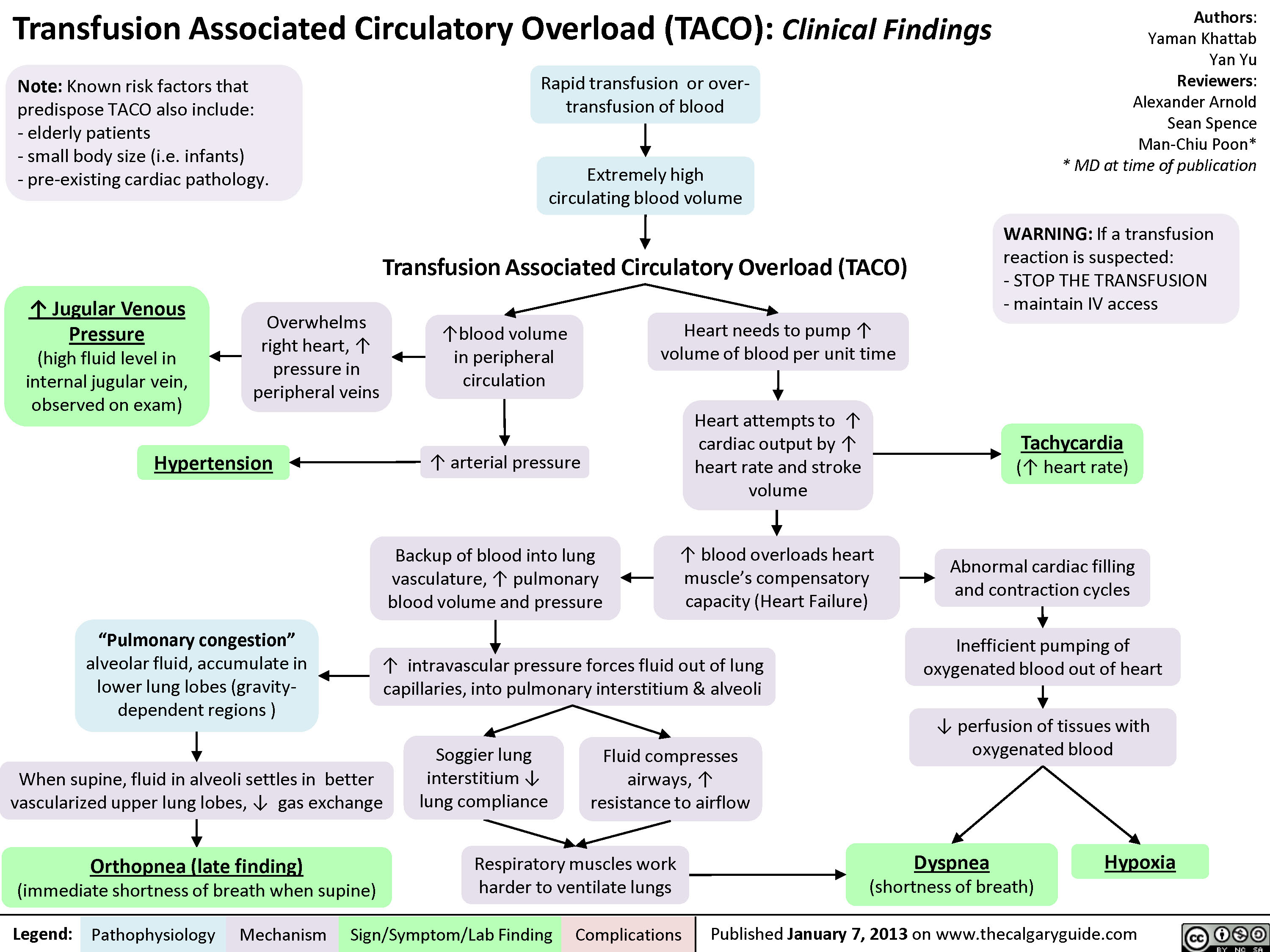
Transfusion Associated Circulatory Overload (TACO)
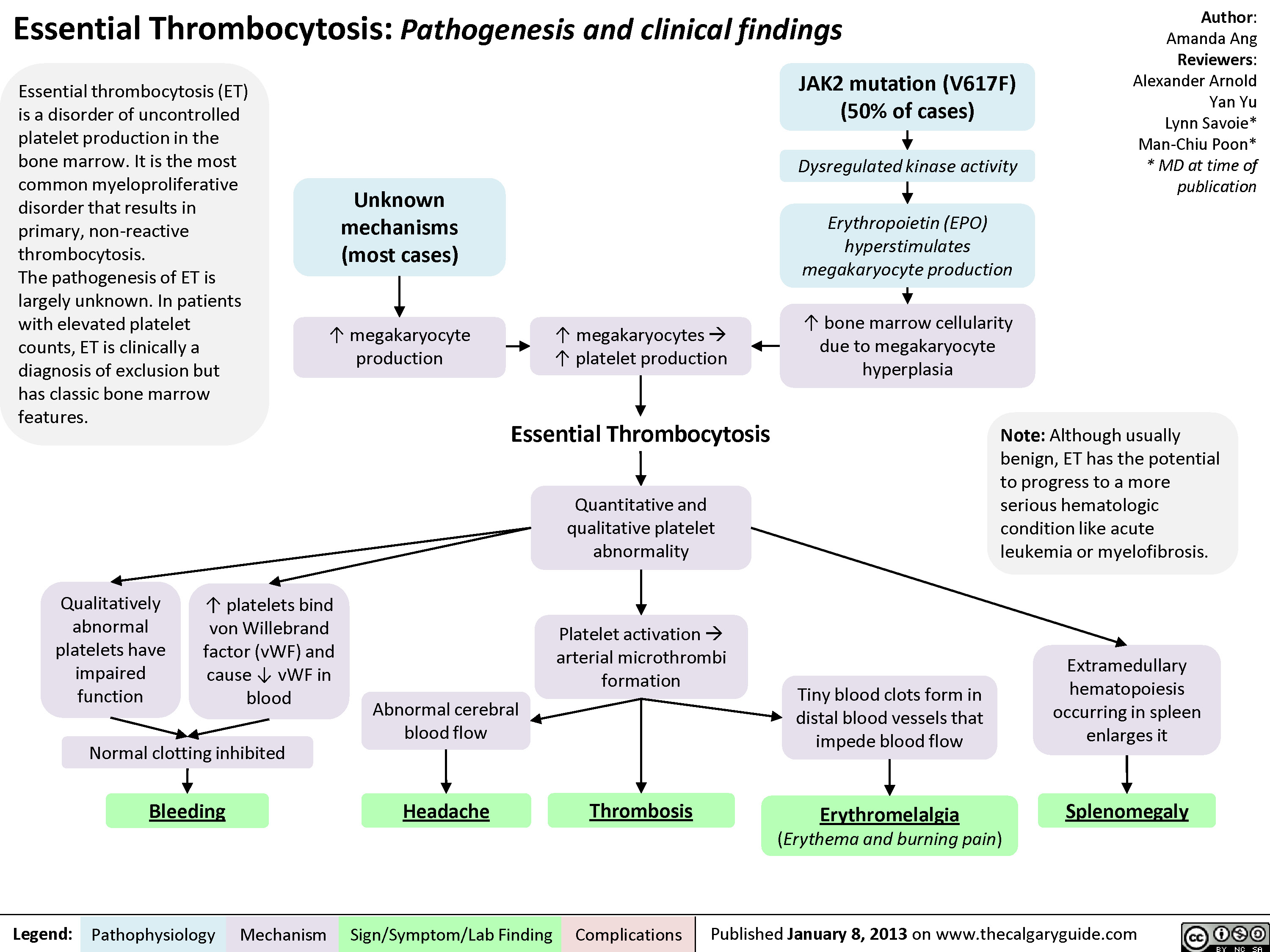
Essential Thrombocytosis (ET)
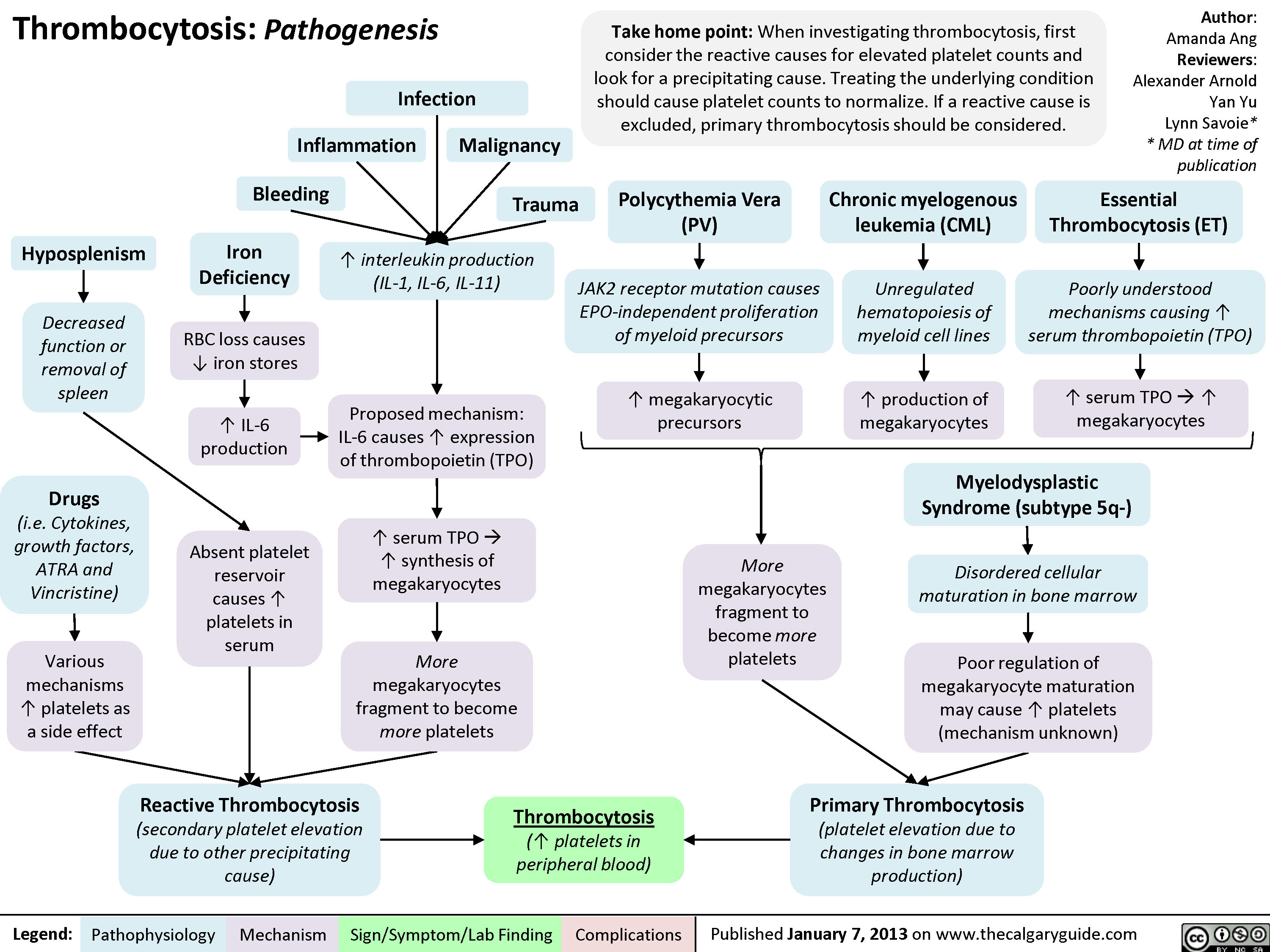
Pathogenesis of thrombocytosis
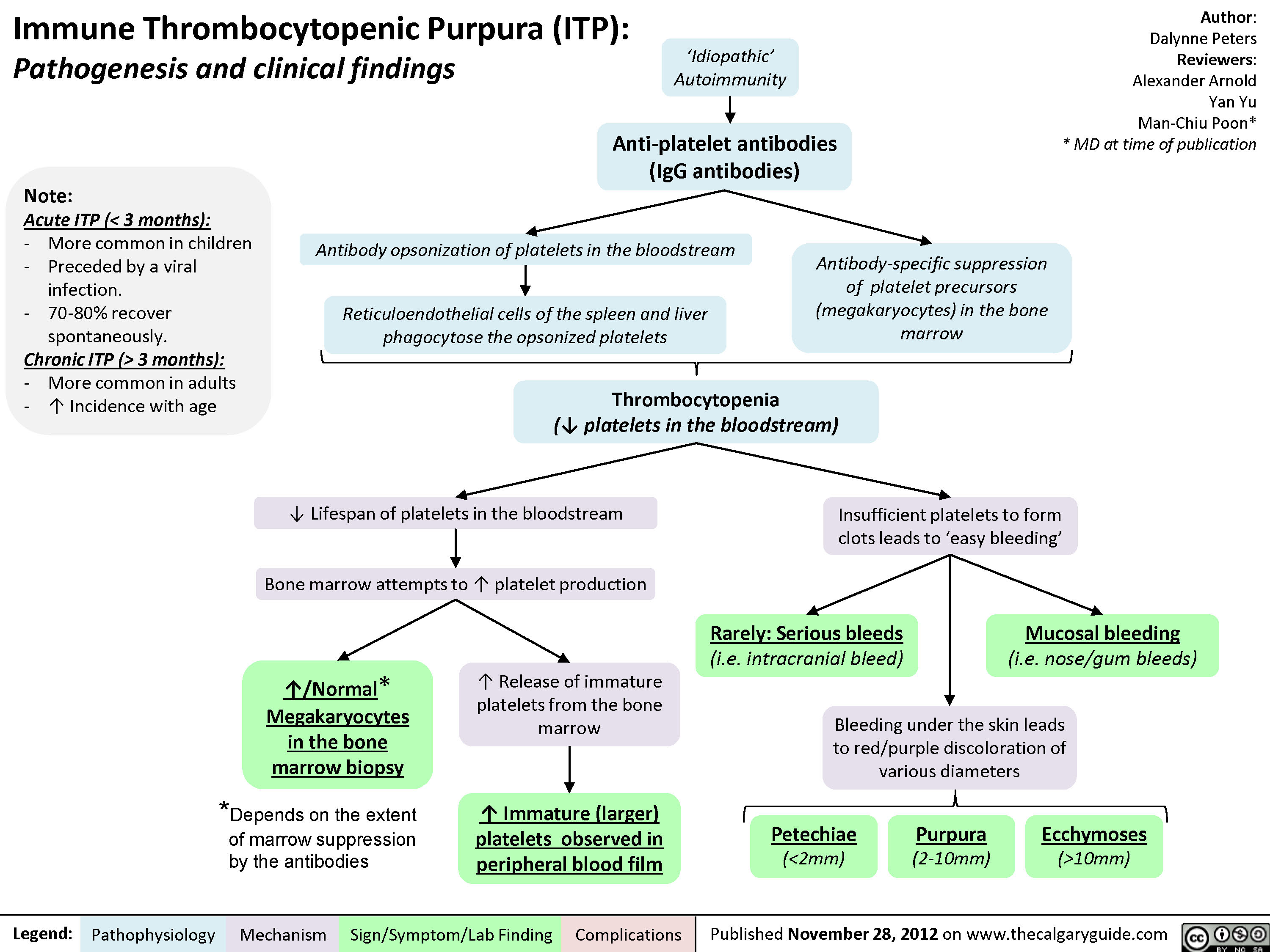
Immune thrombocytopenic purpura
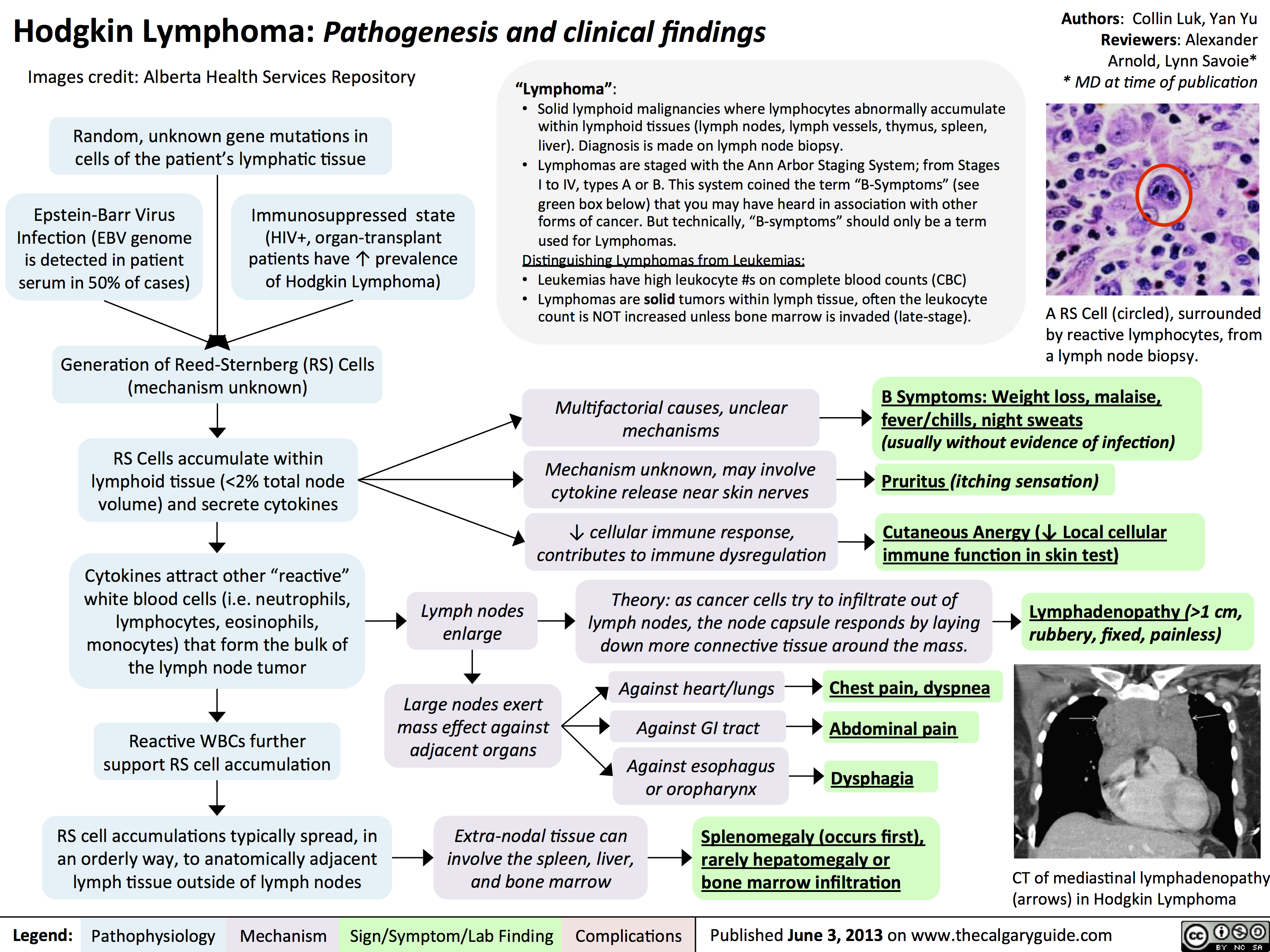
hodgkin lymphoma - pathogenesis and clinical findings
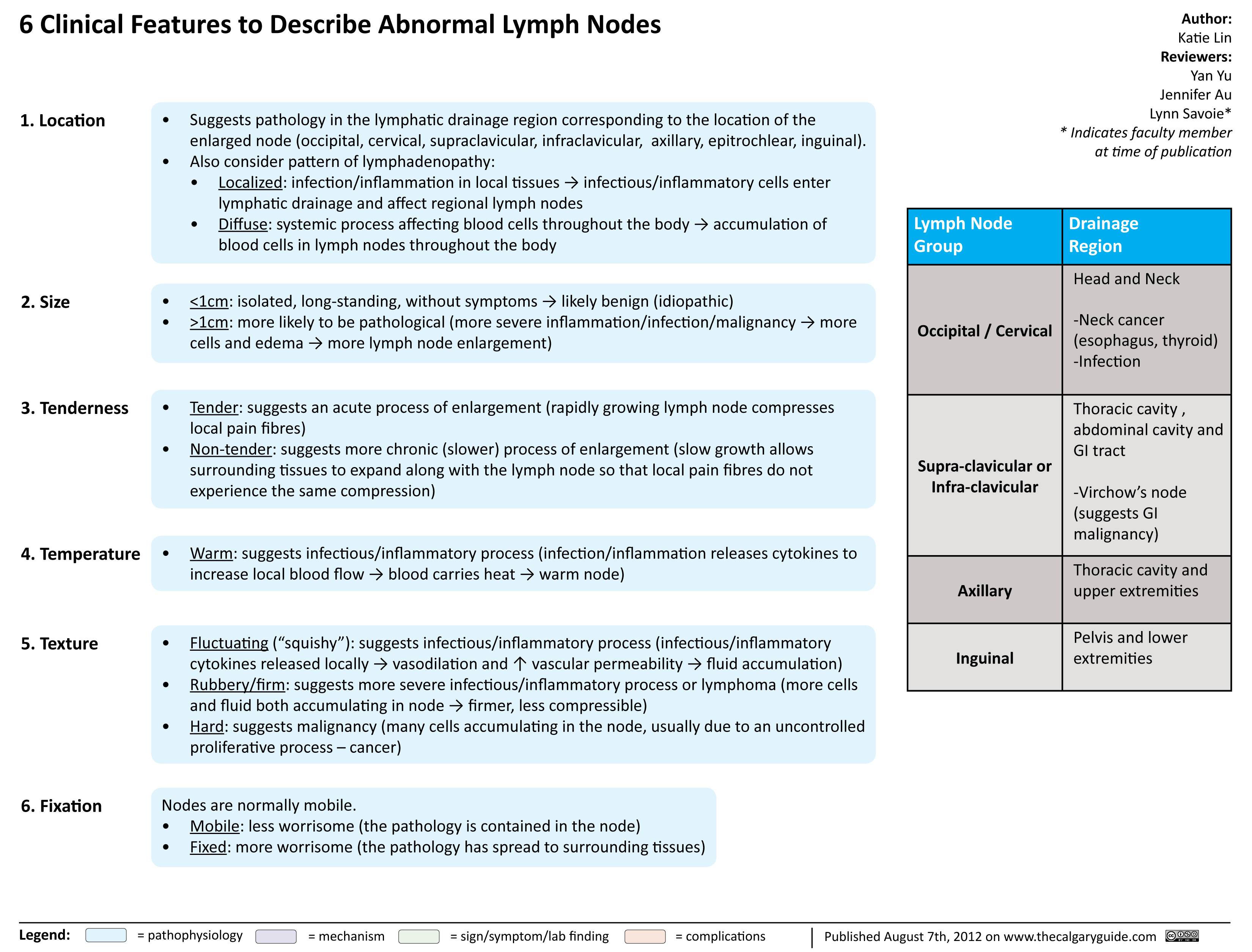
Clinical Features to Describe Abnormal Lymph Nodes
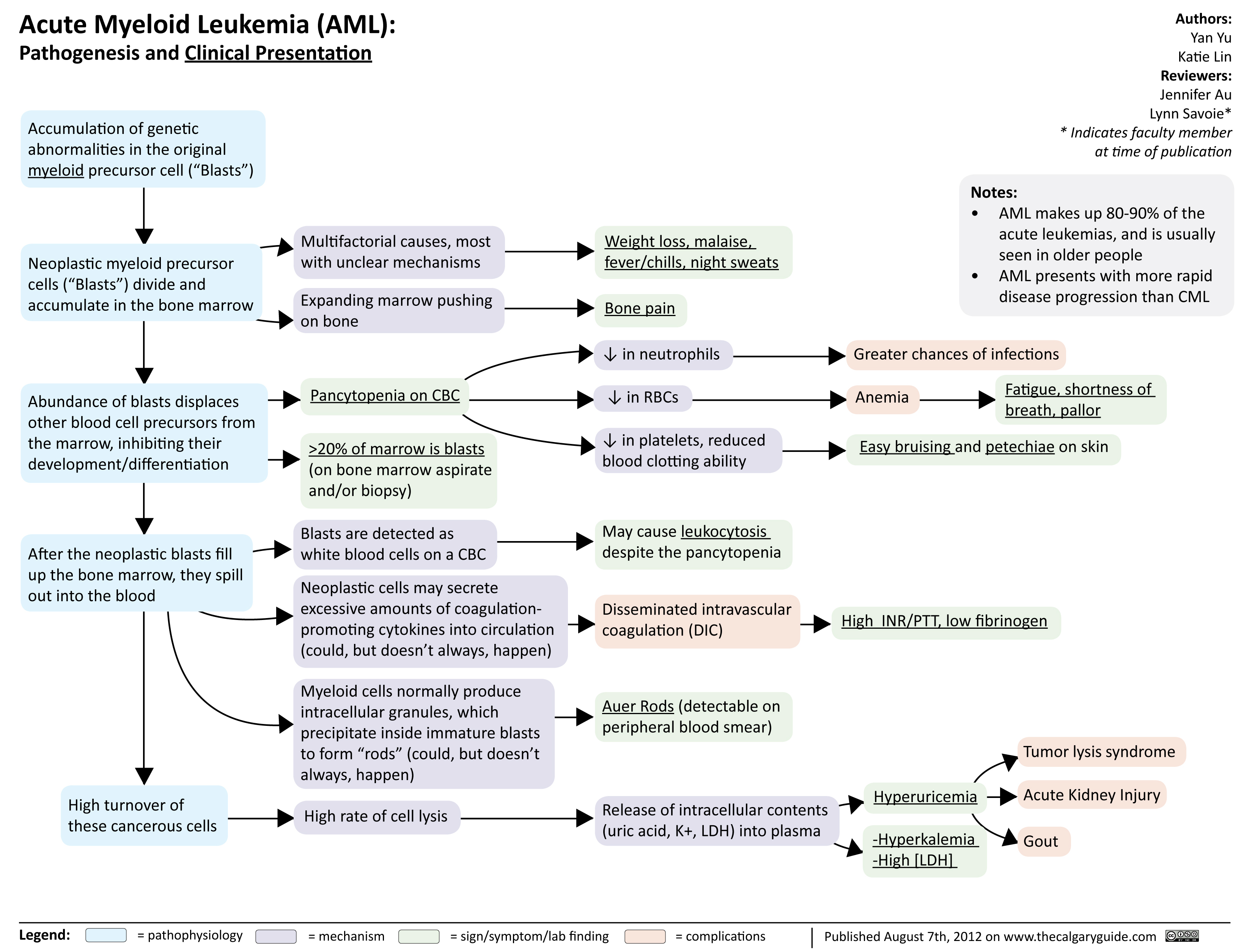
Acute Myeloid Leukemia
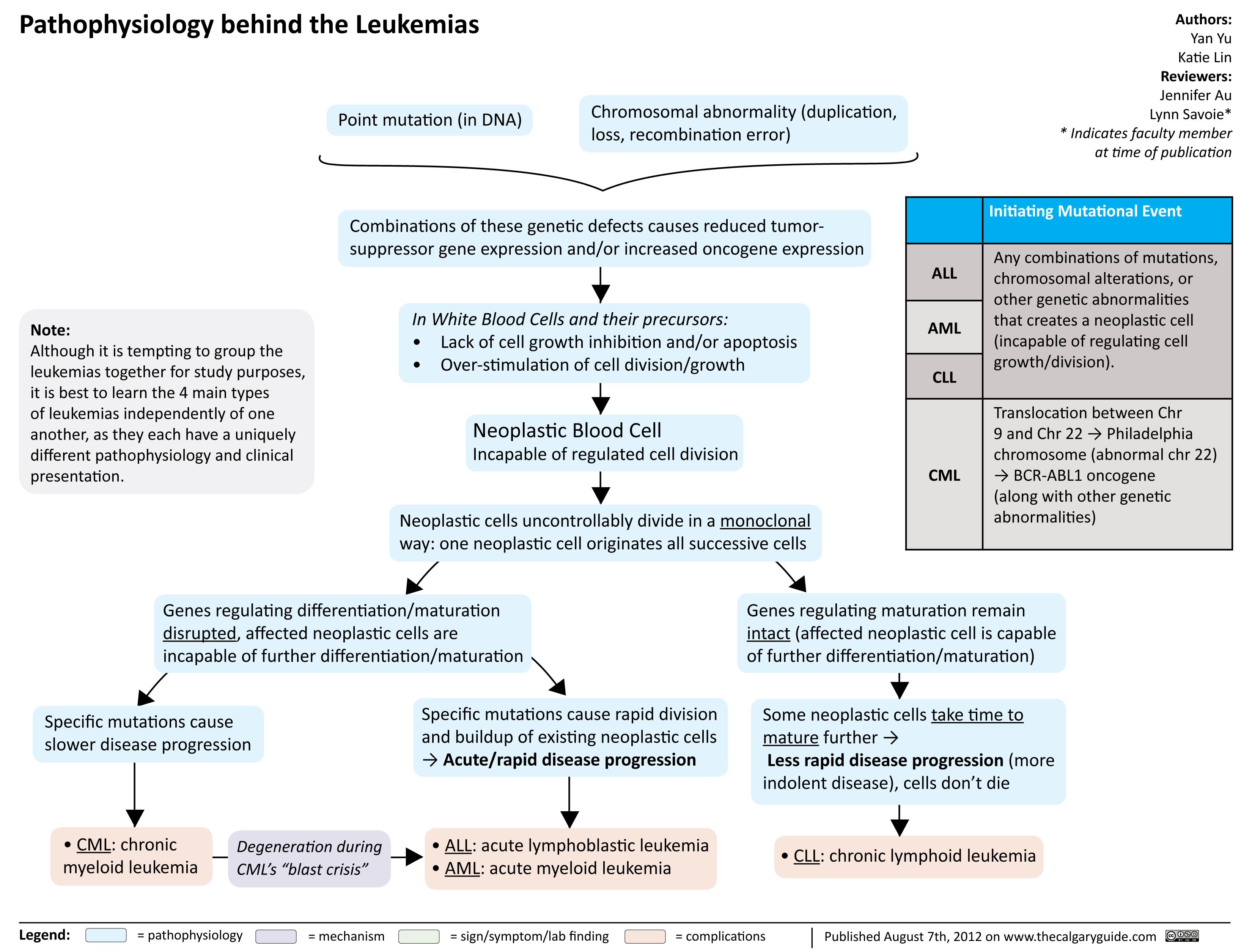
Pathophysiology behind the leukemias
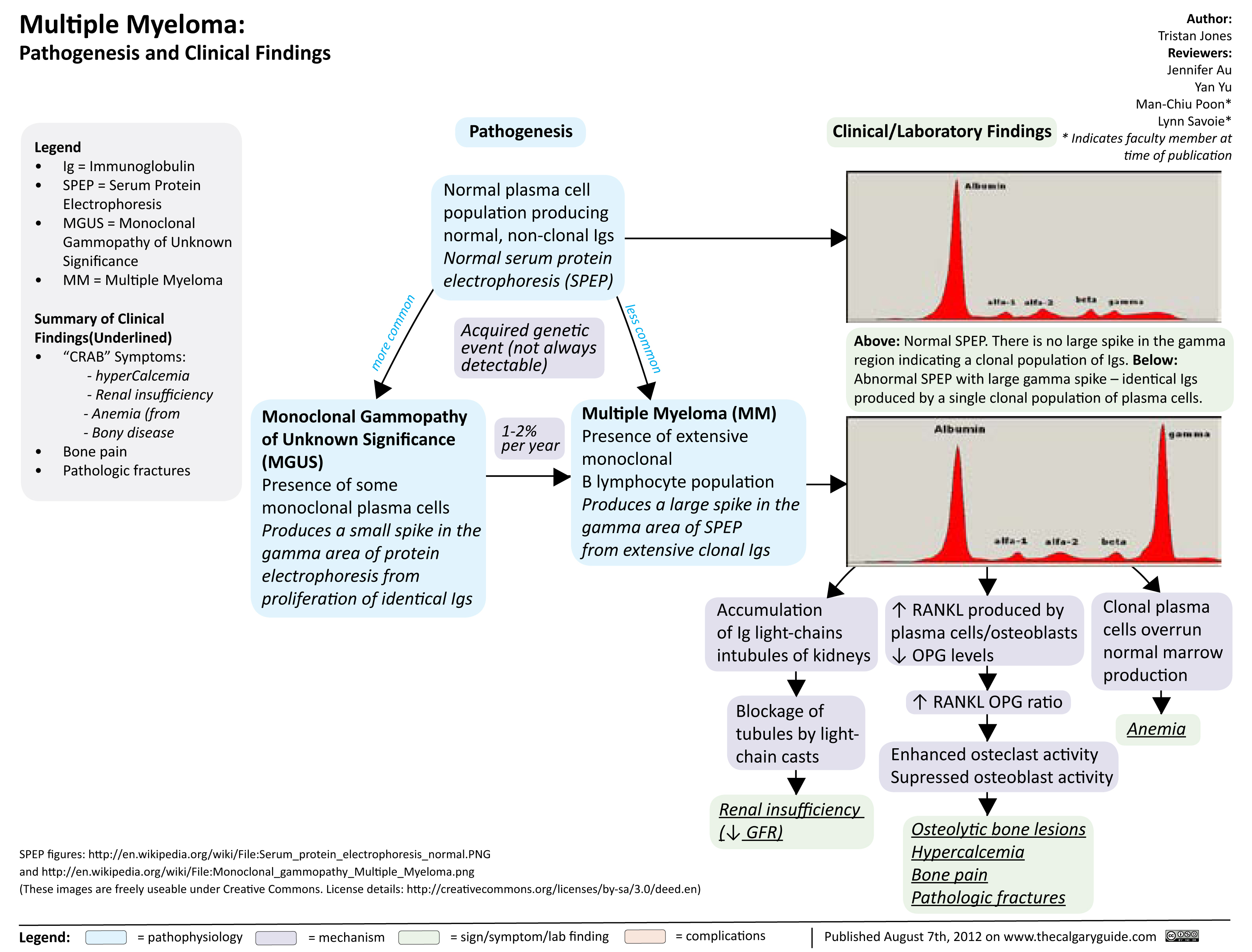
Multiple Myeloma
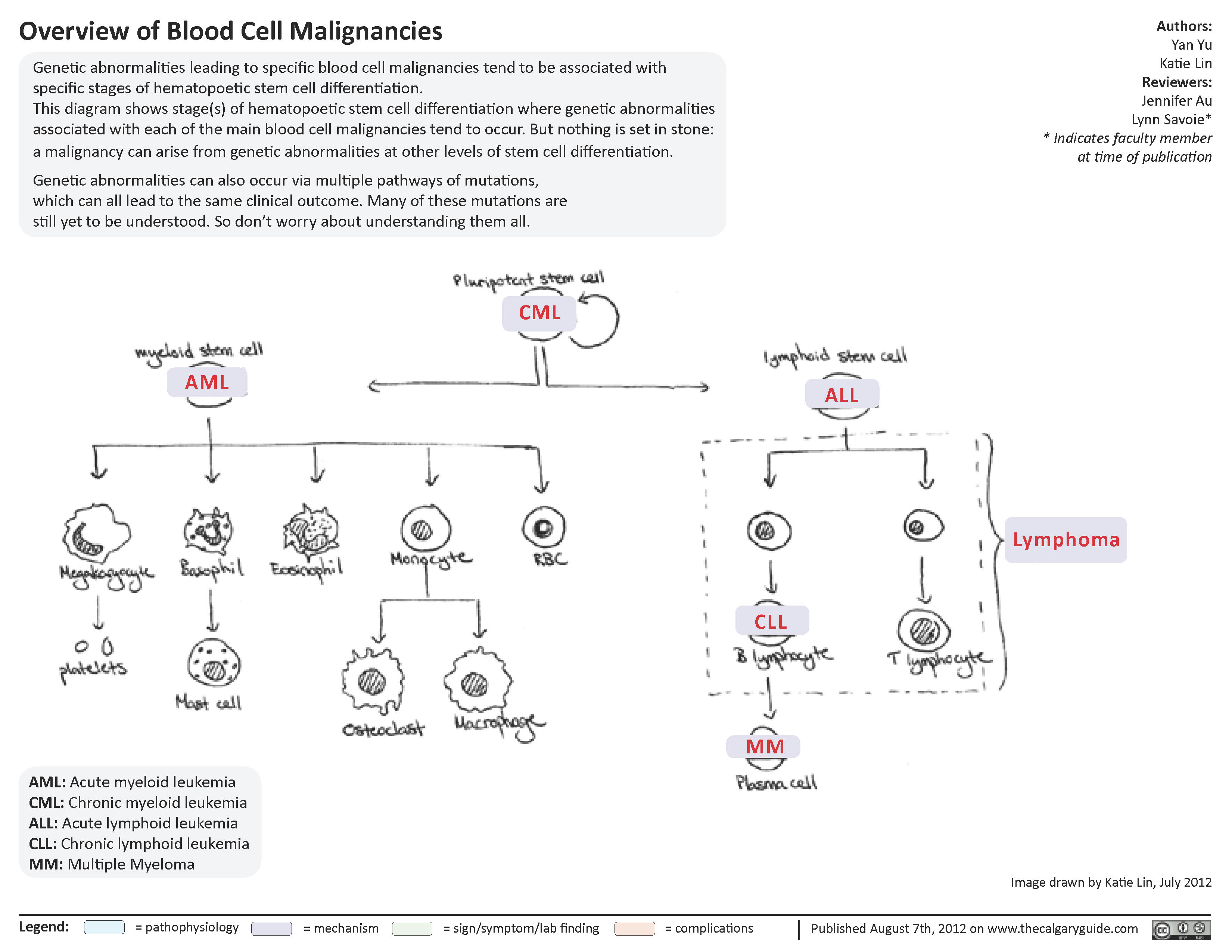
Overview of blood cell malignancies
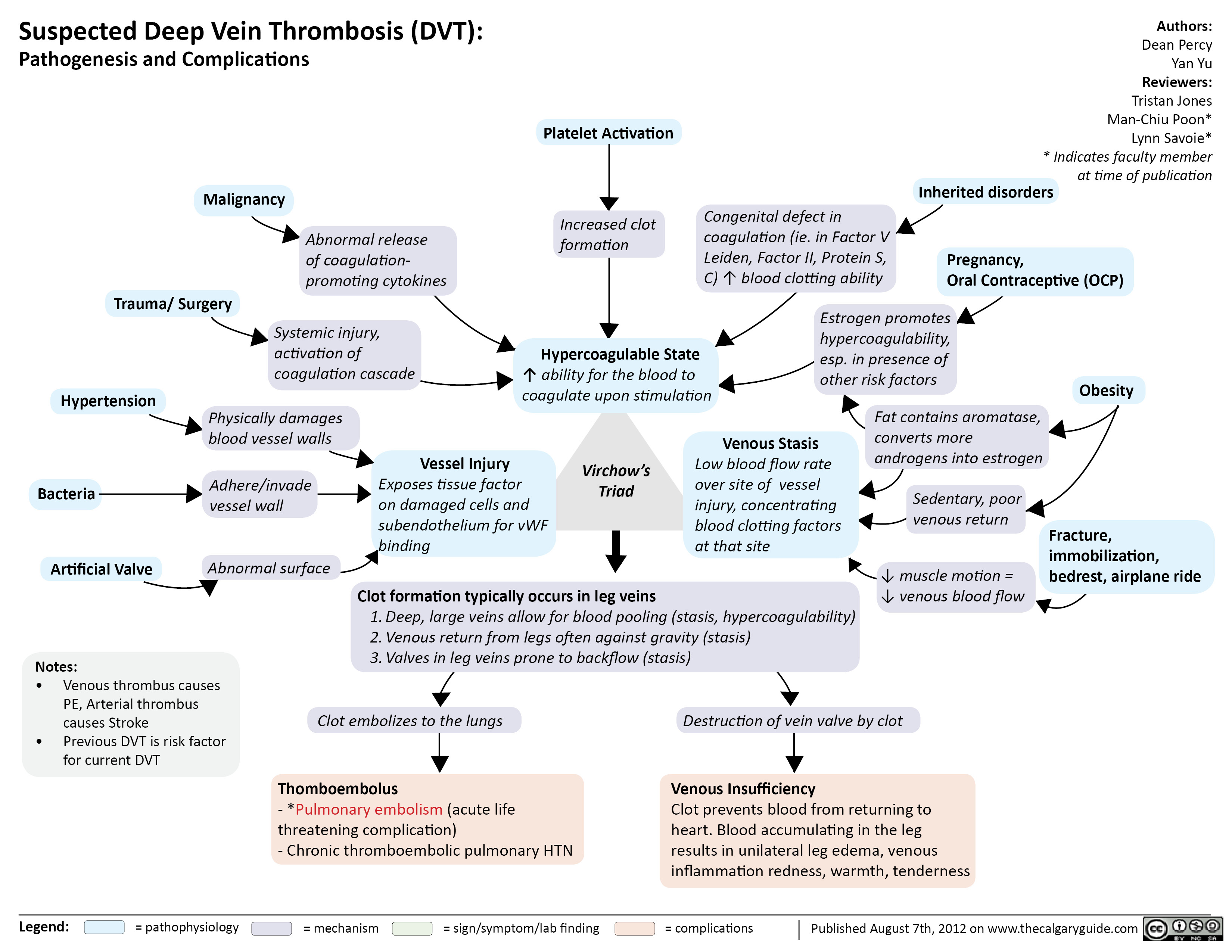
Suspected Deep Vein Thrombosis
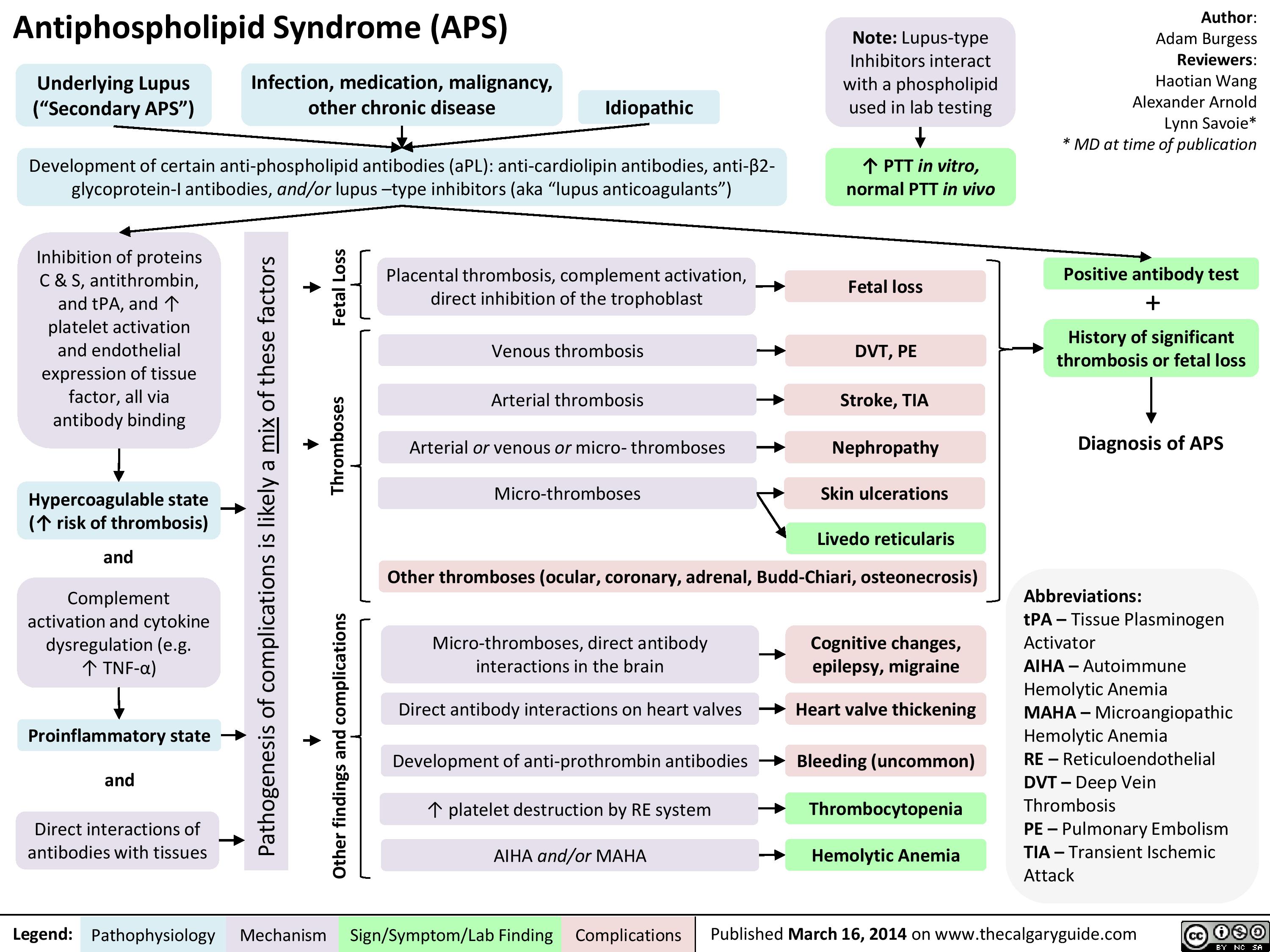
APS
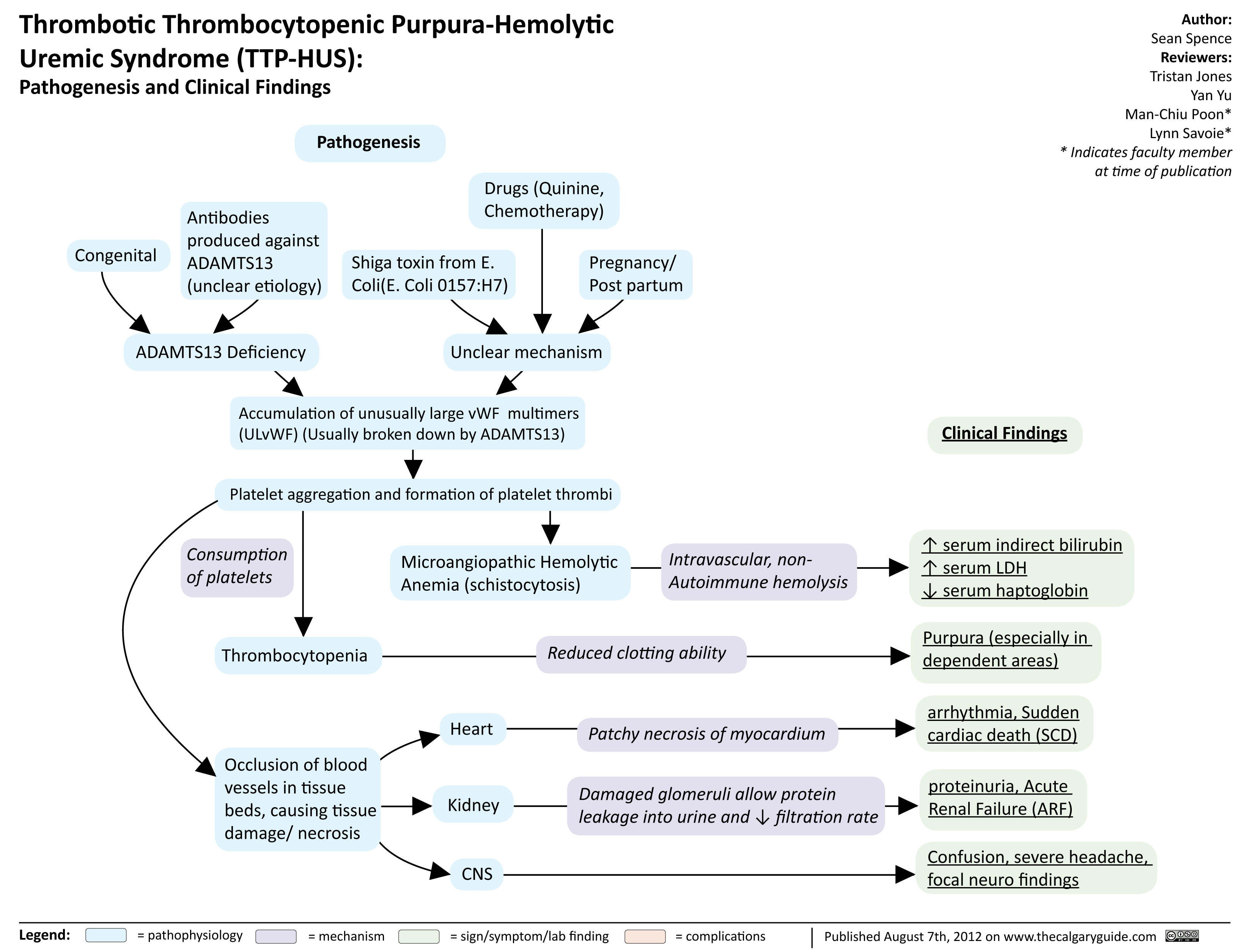
TTP HUS
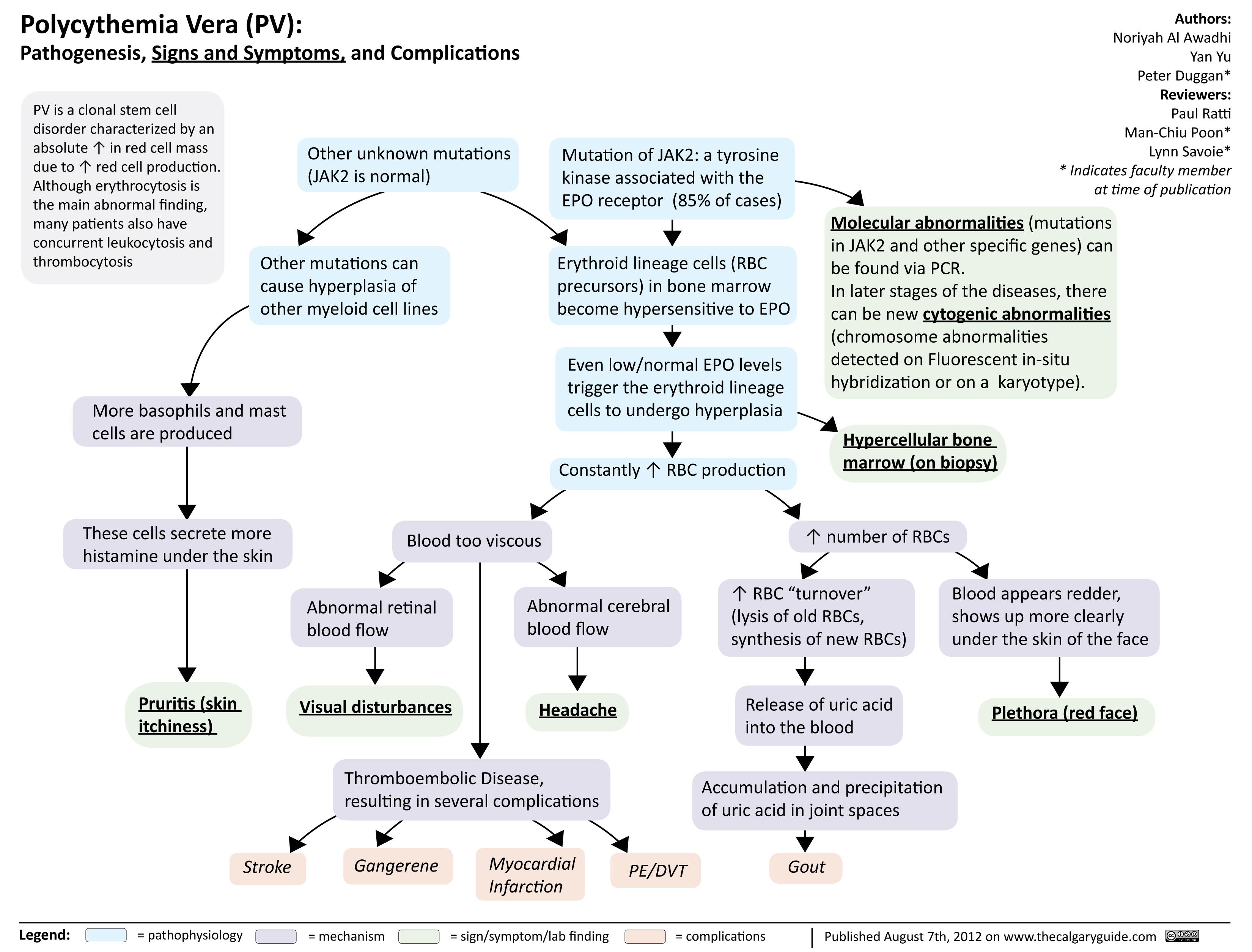
Polycythmia Vera
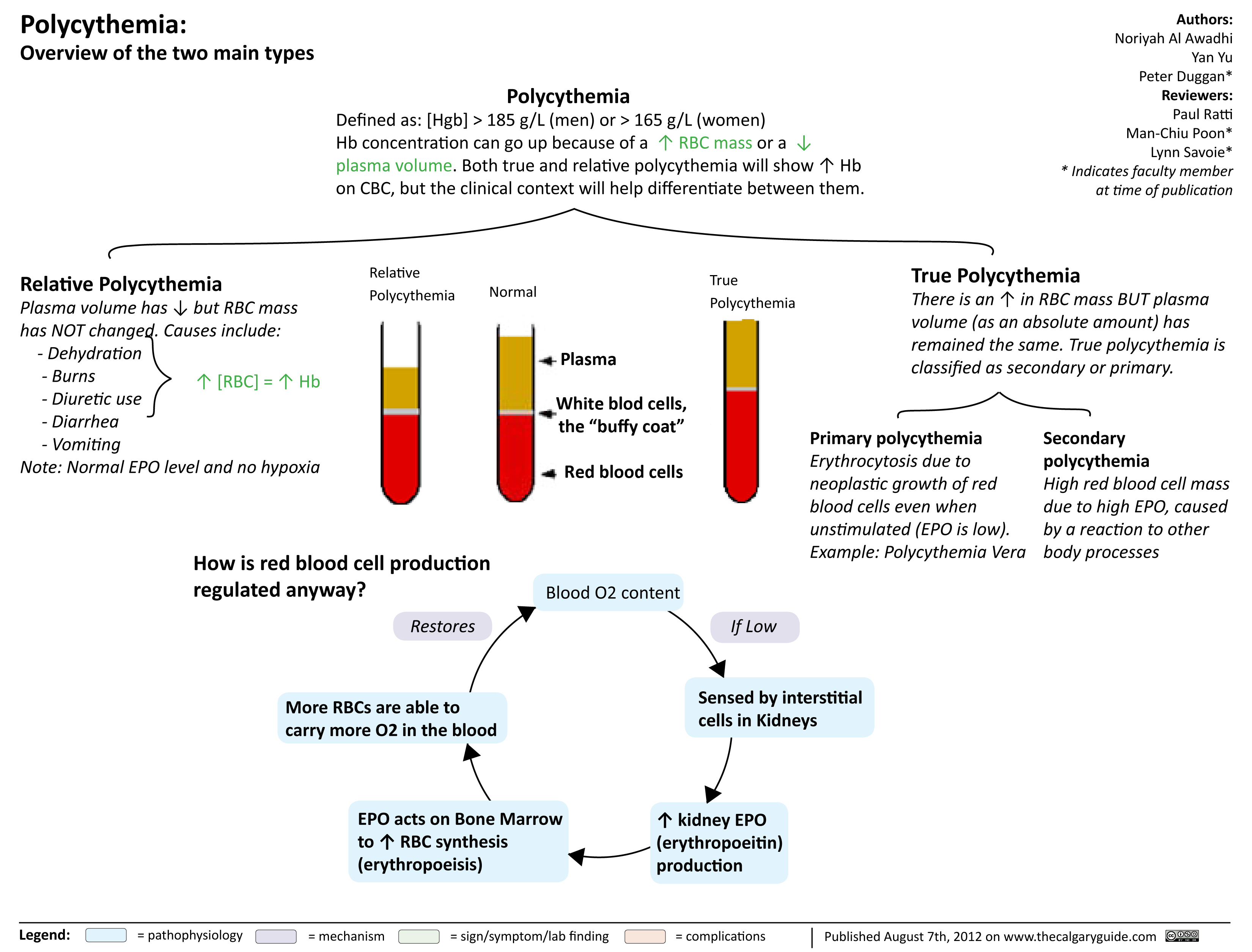
Polycythmia Overview
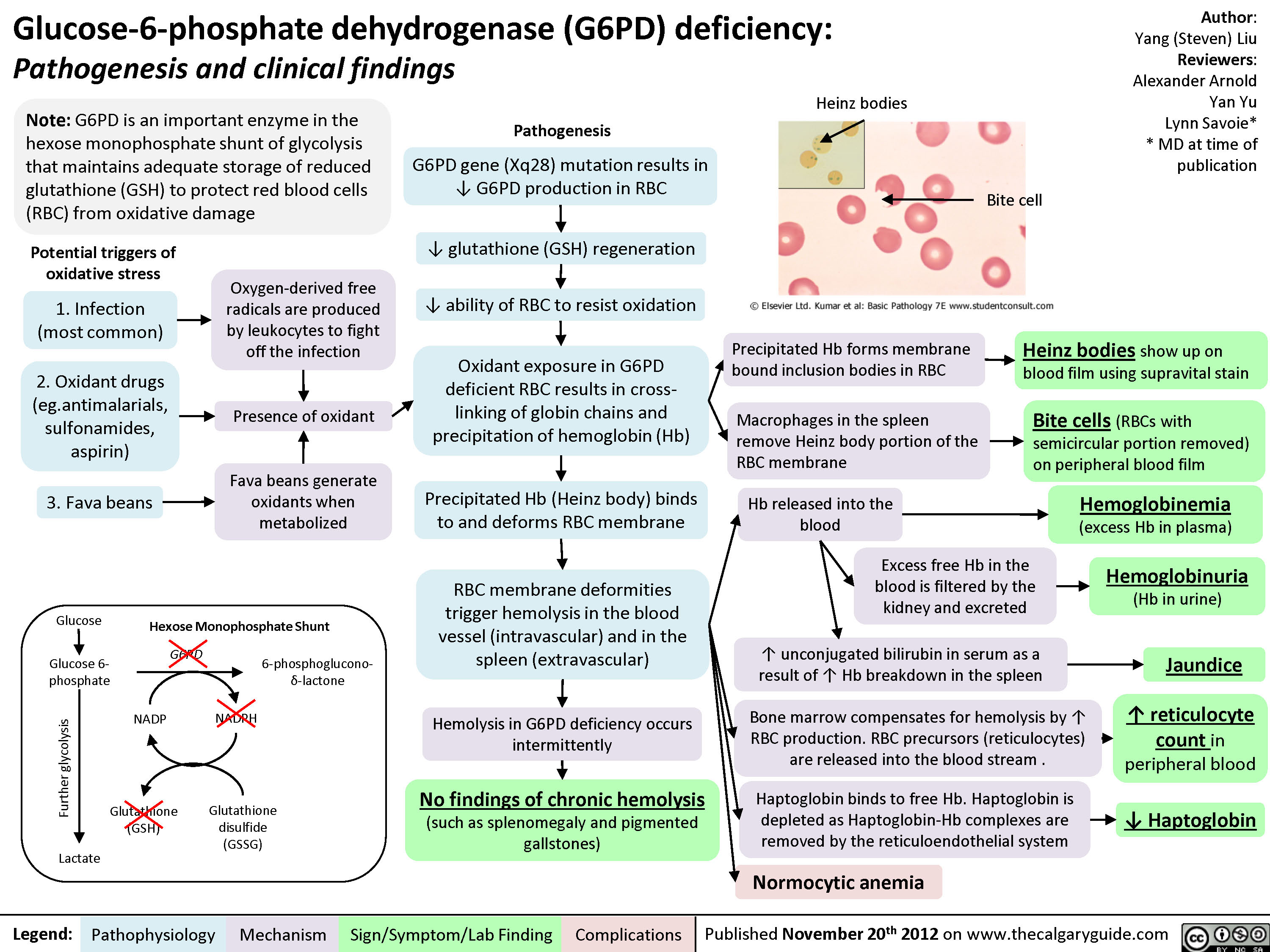
G6PD Deficiency
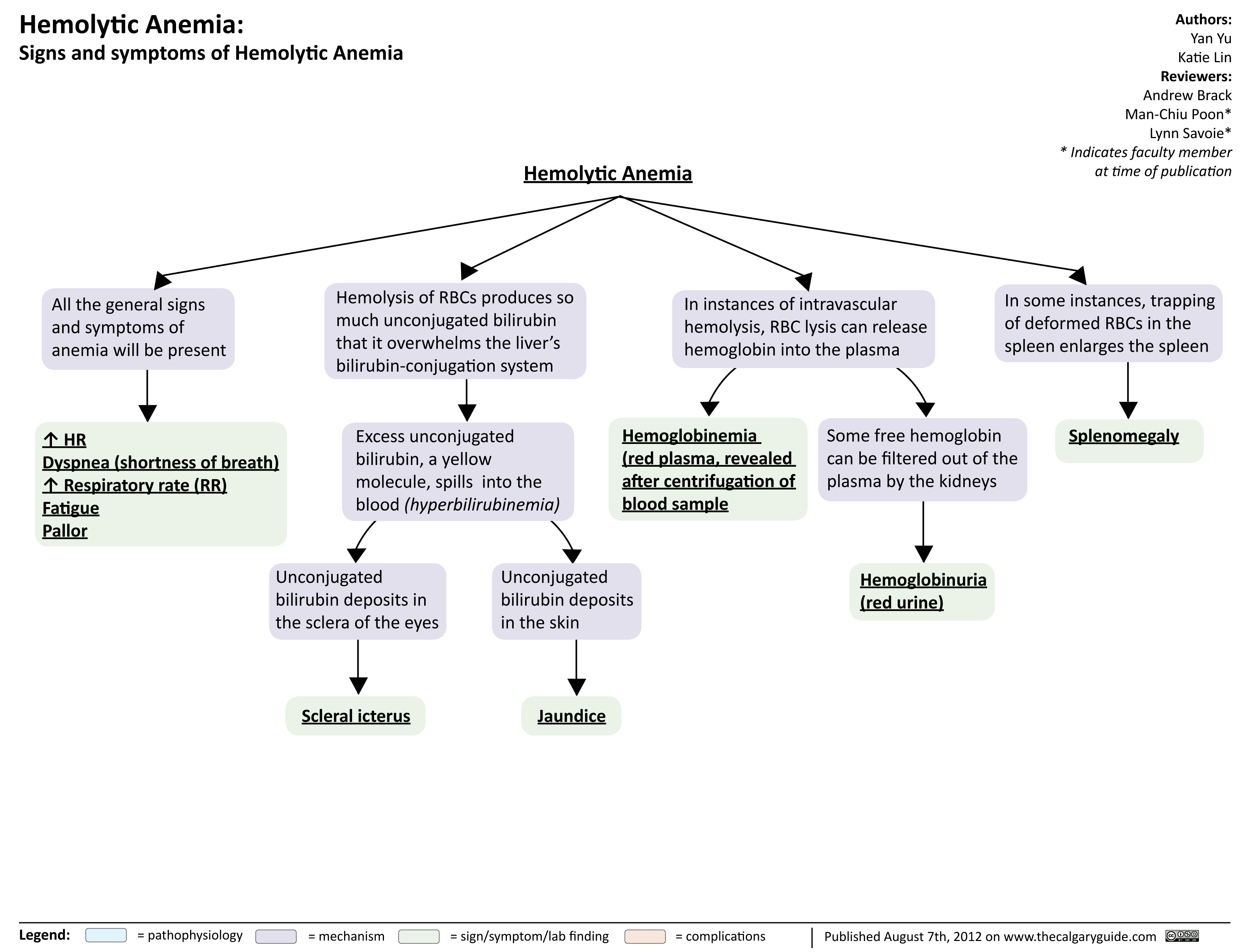
Hemolytic Anemia Signs and Symptoms
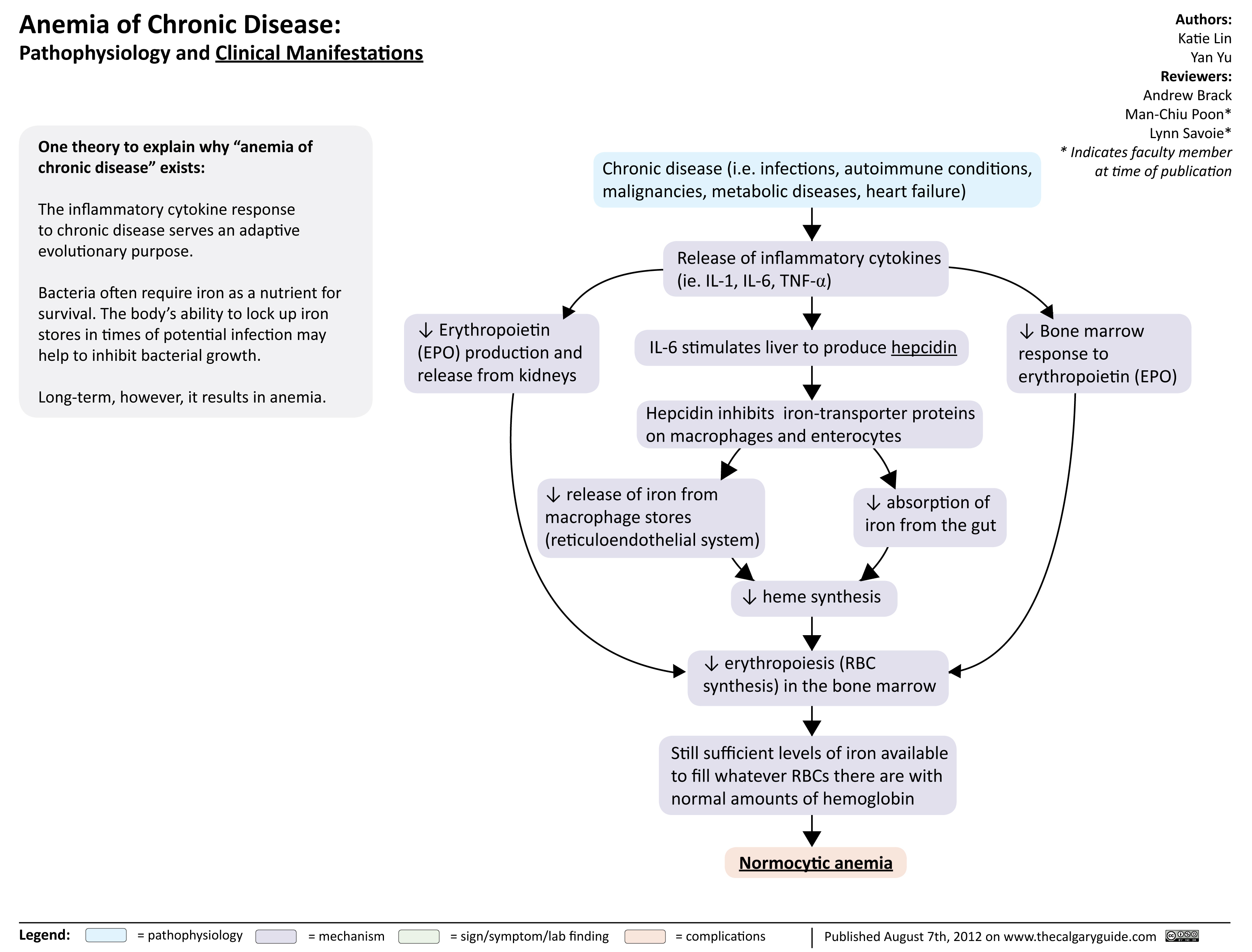
Anemia of Chronic Disease
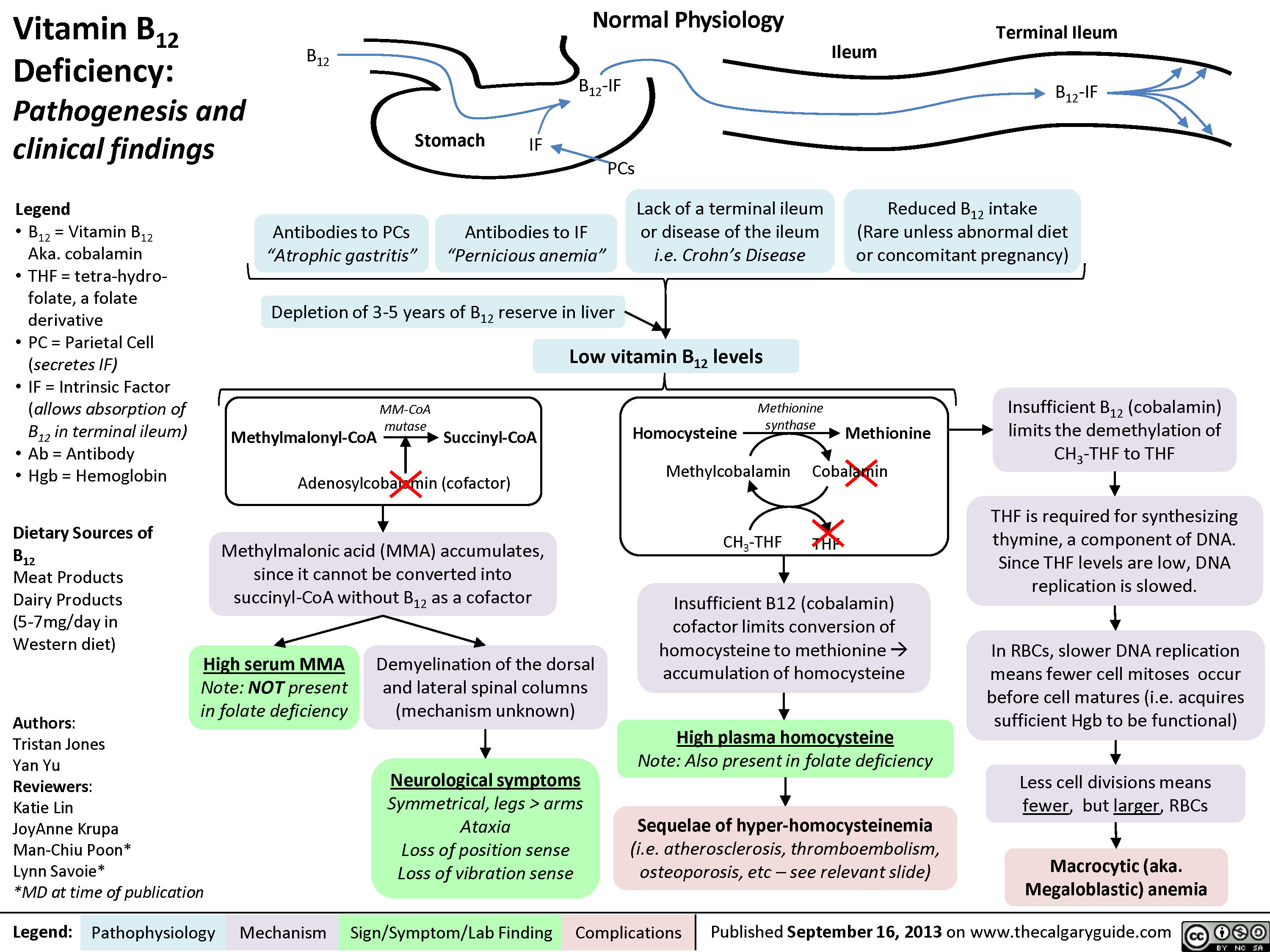
Vitamin B12 Deficiency
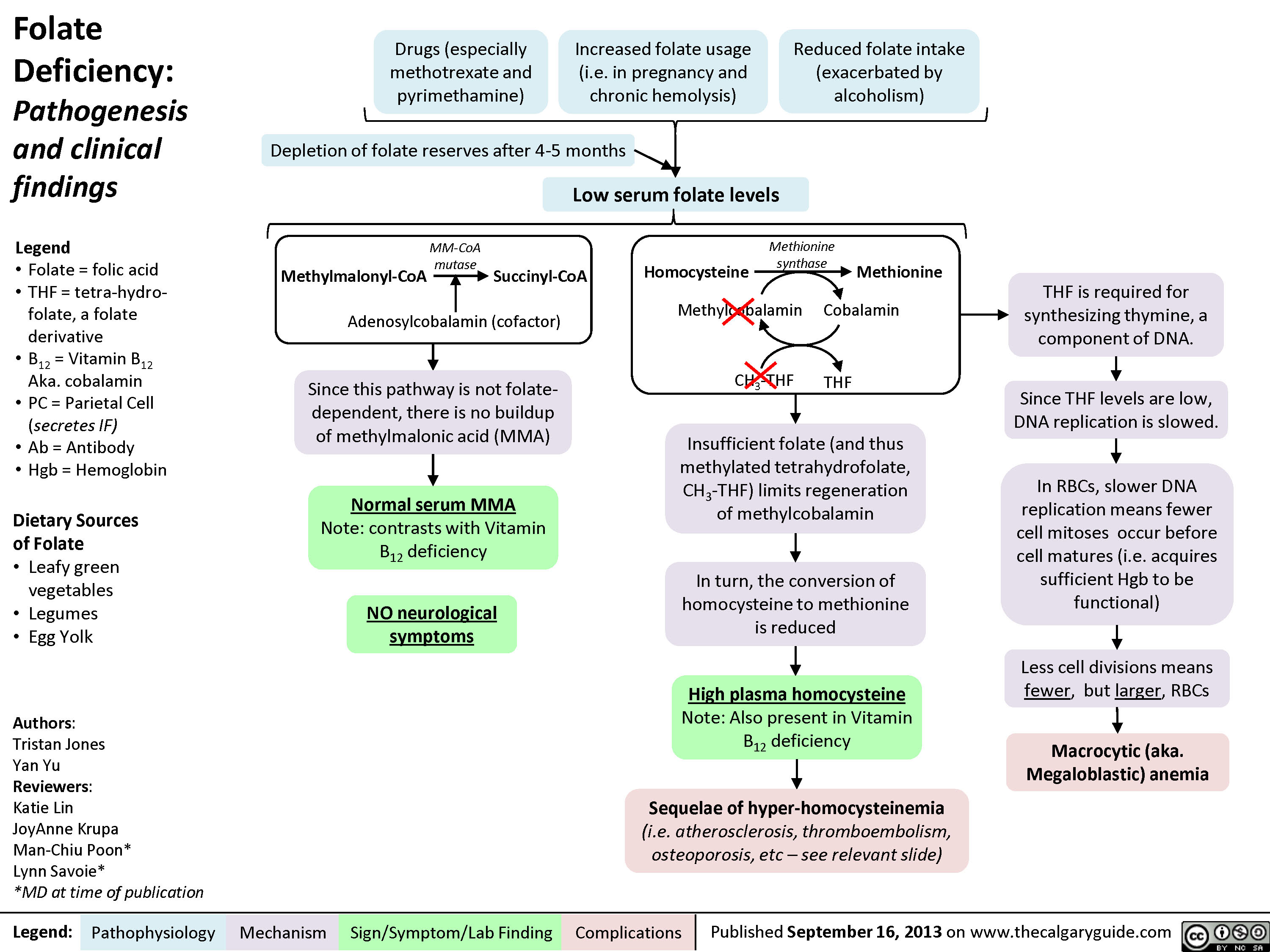
Folate Deficiency
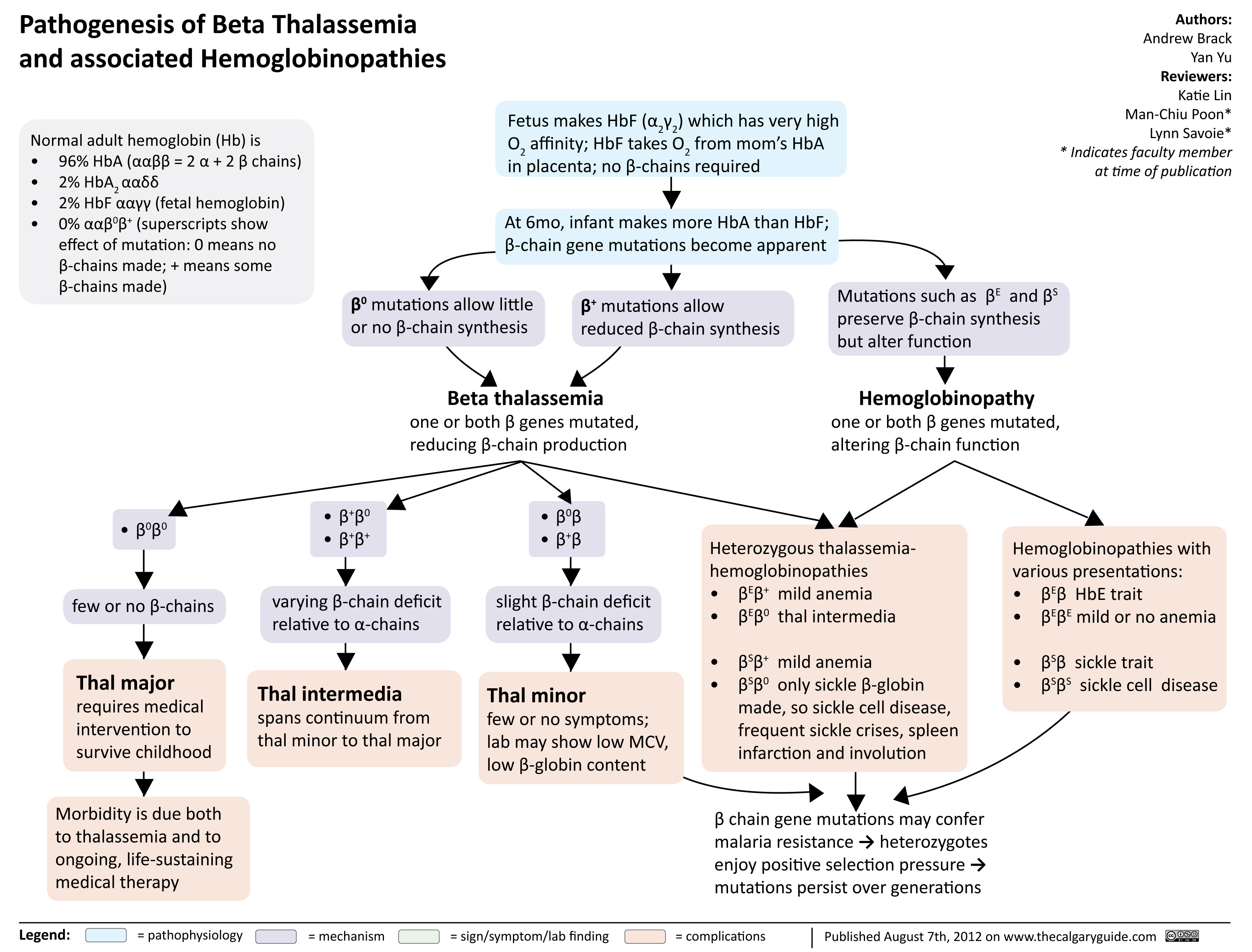
Pathogenesis of Beta Thalassemia
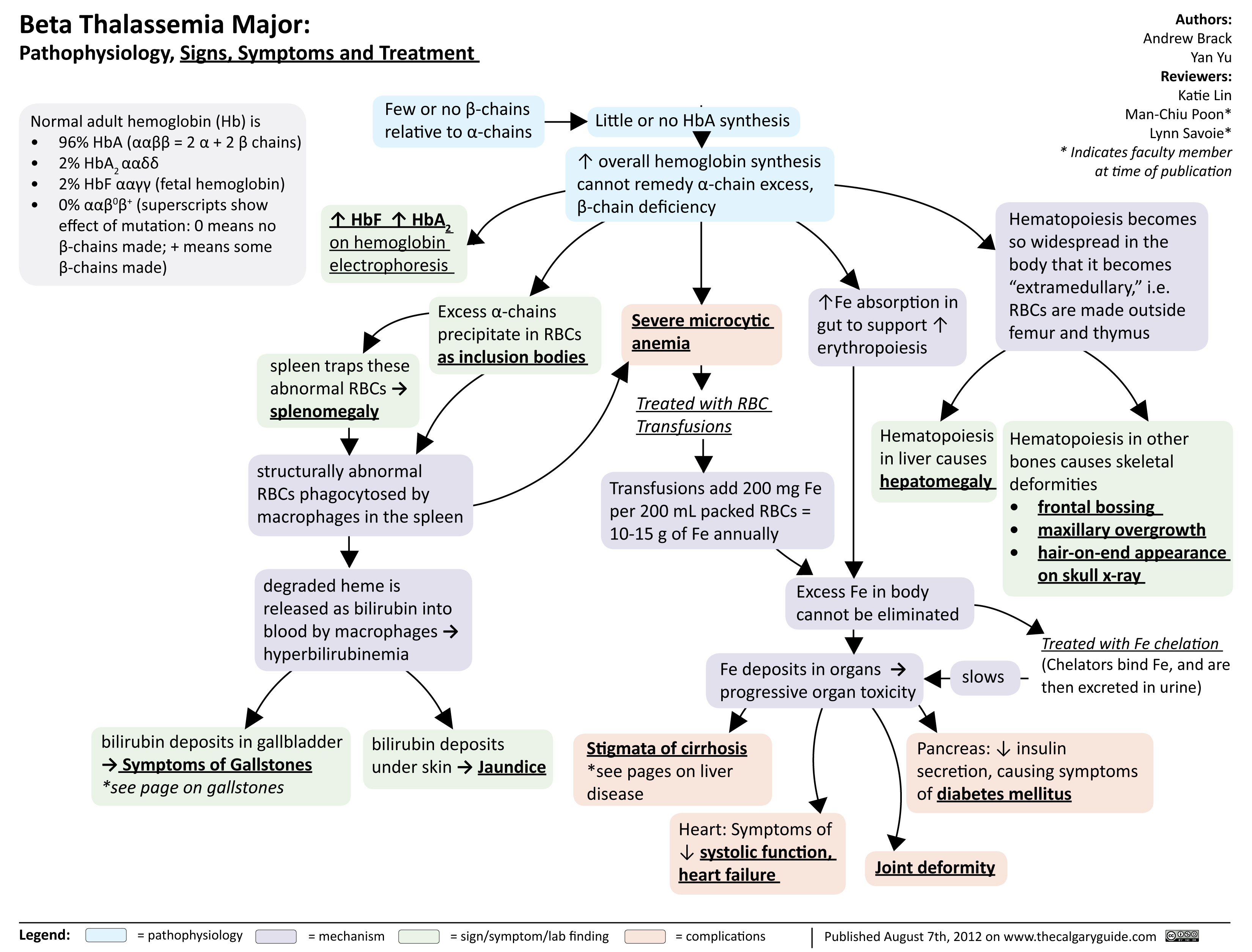
Beta Thalassemia Signs Symptoms Treatment
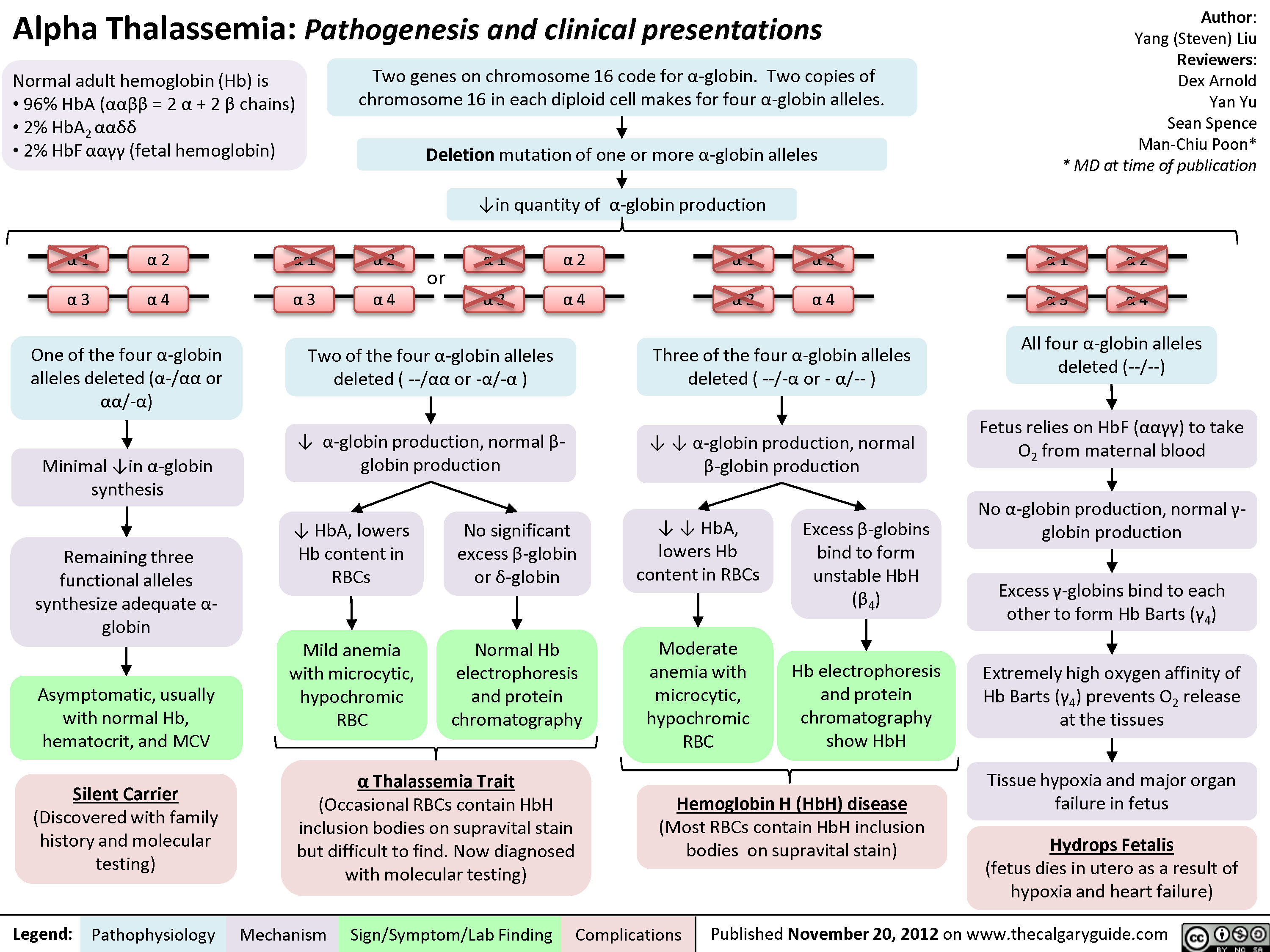
Alpha Thalassemia Pathogenesis
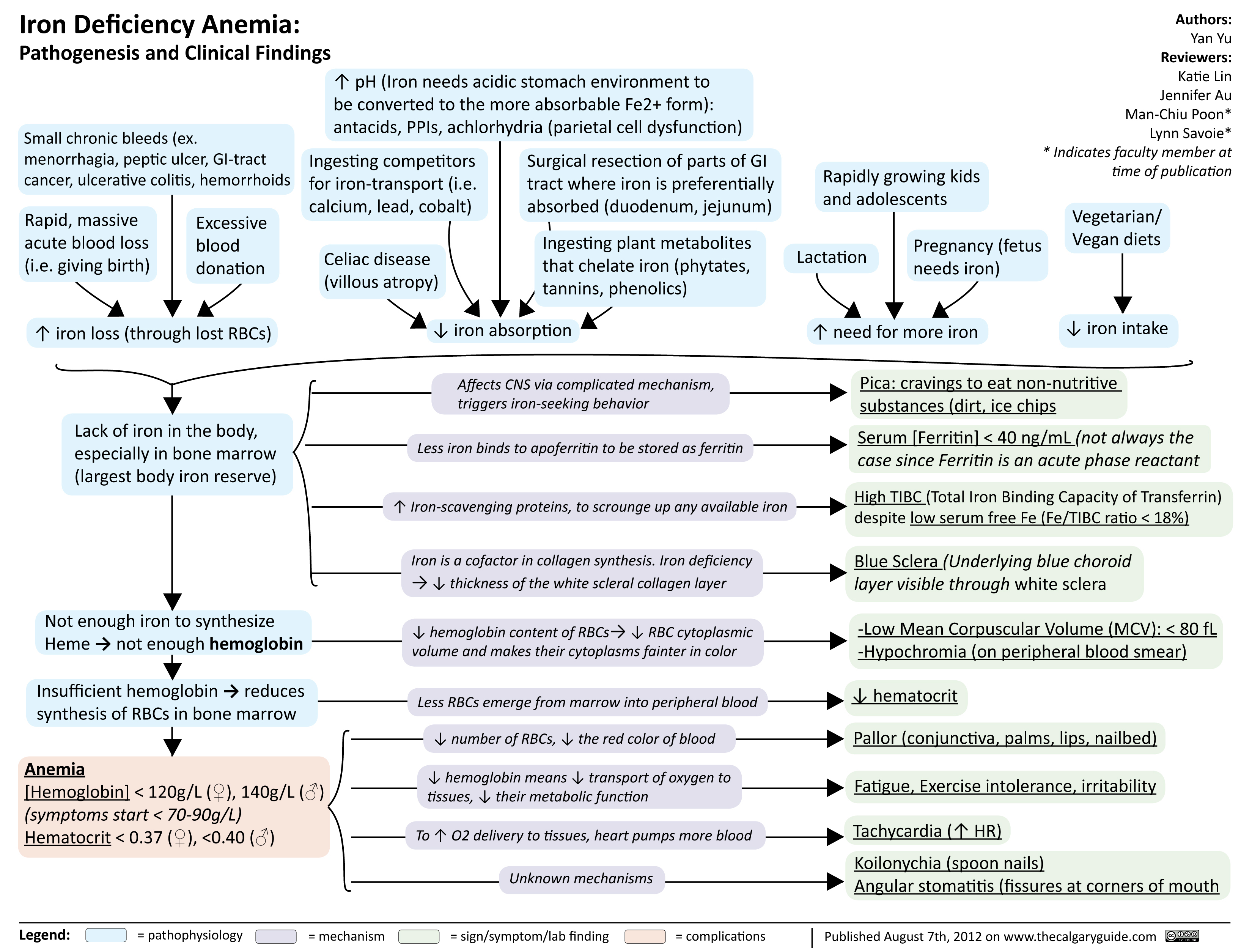
Iron Deficiency Anemia
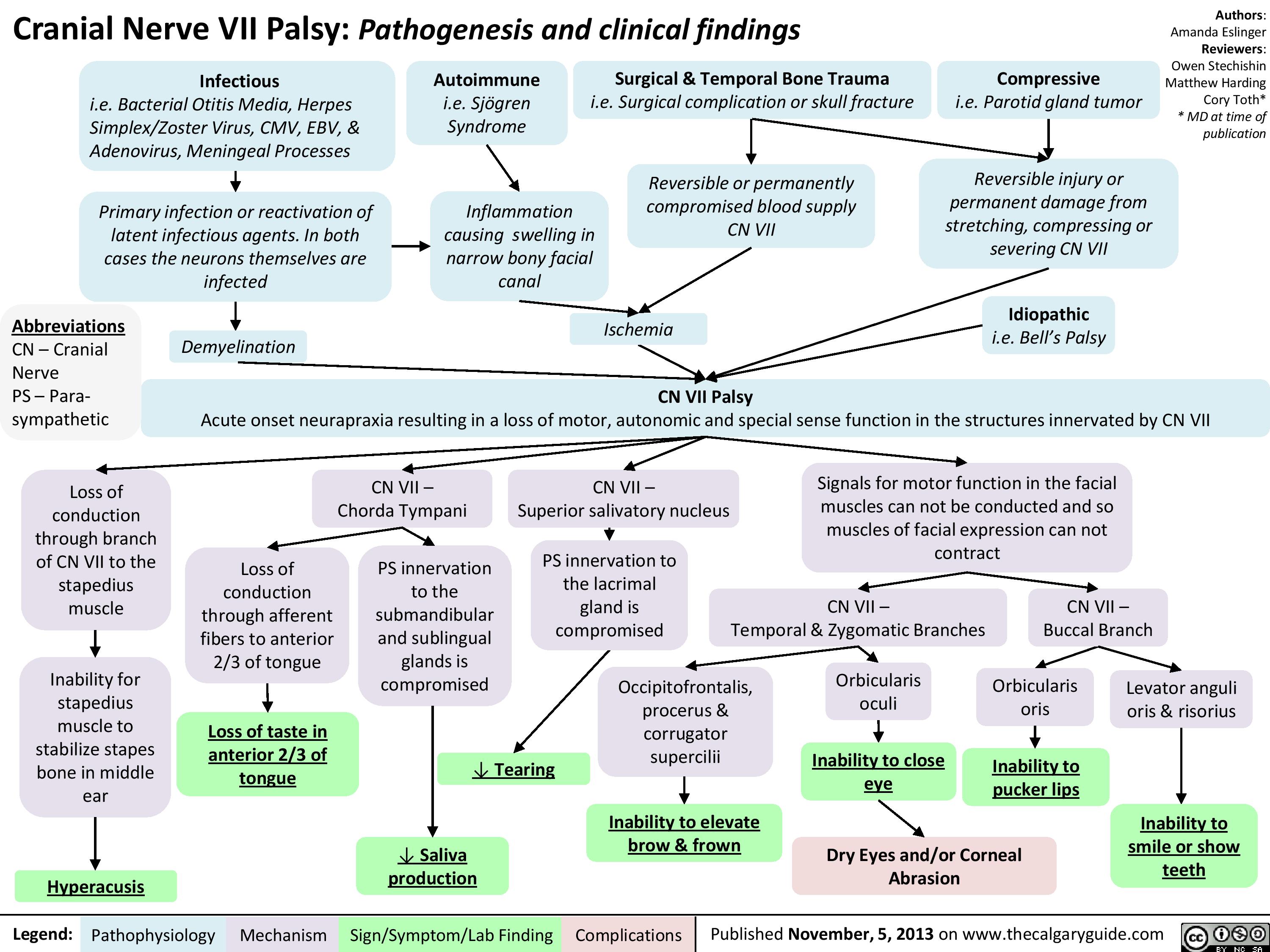
CNVII_Bells Palsy
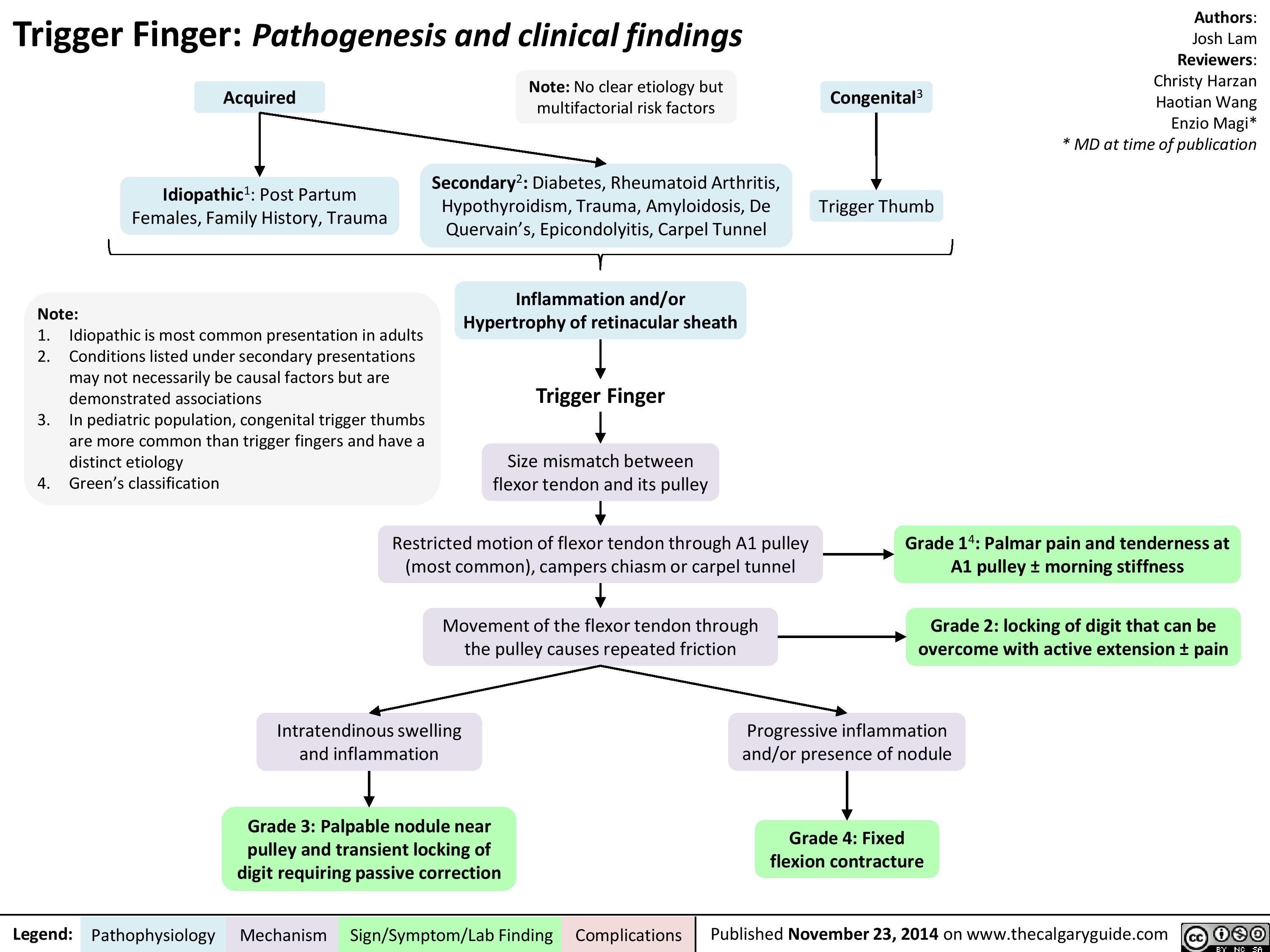
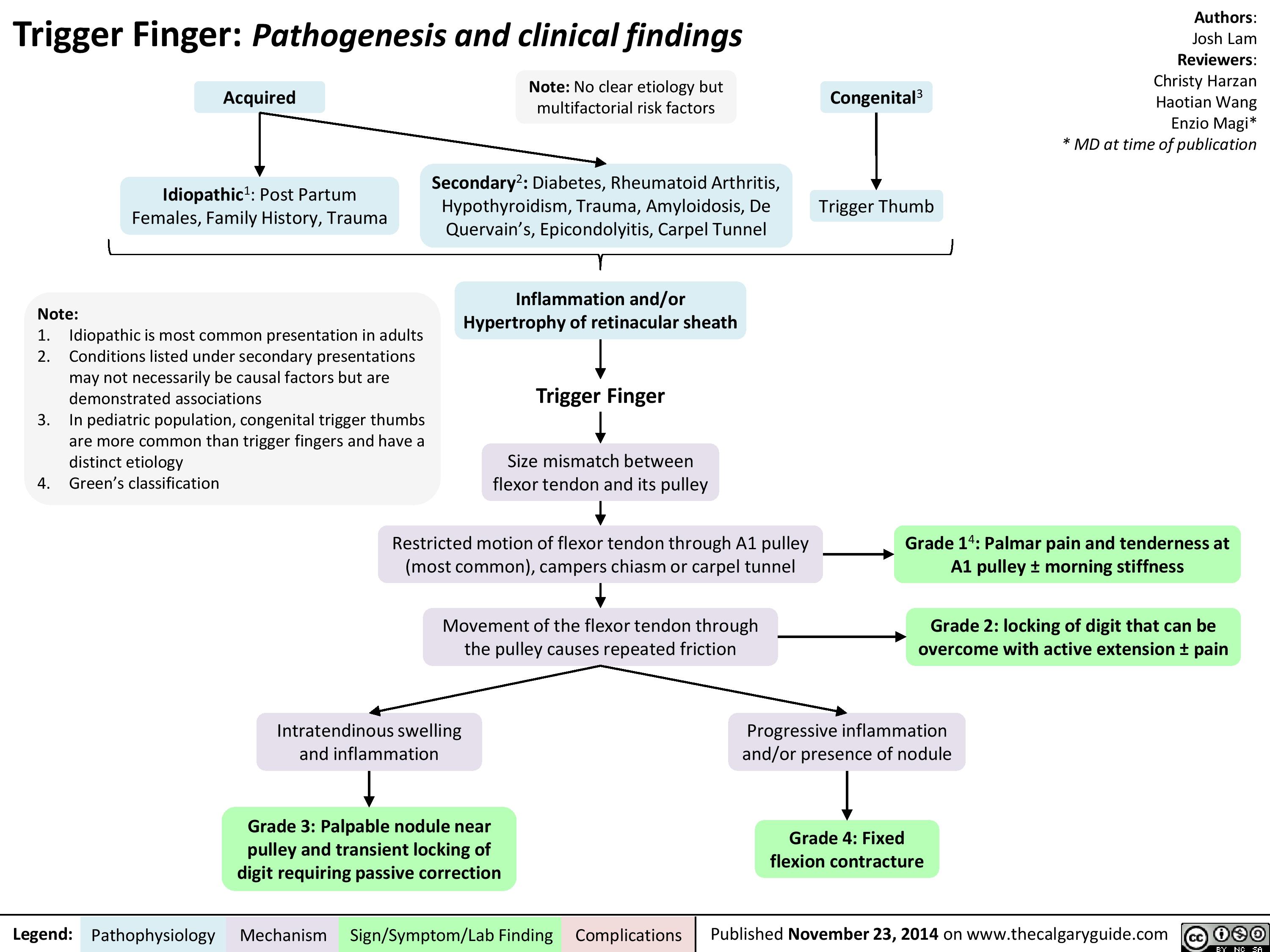
Trigger Finger
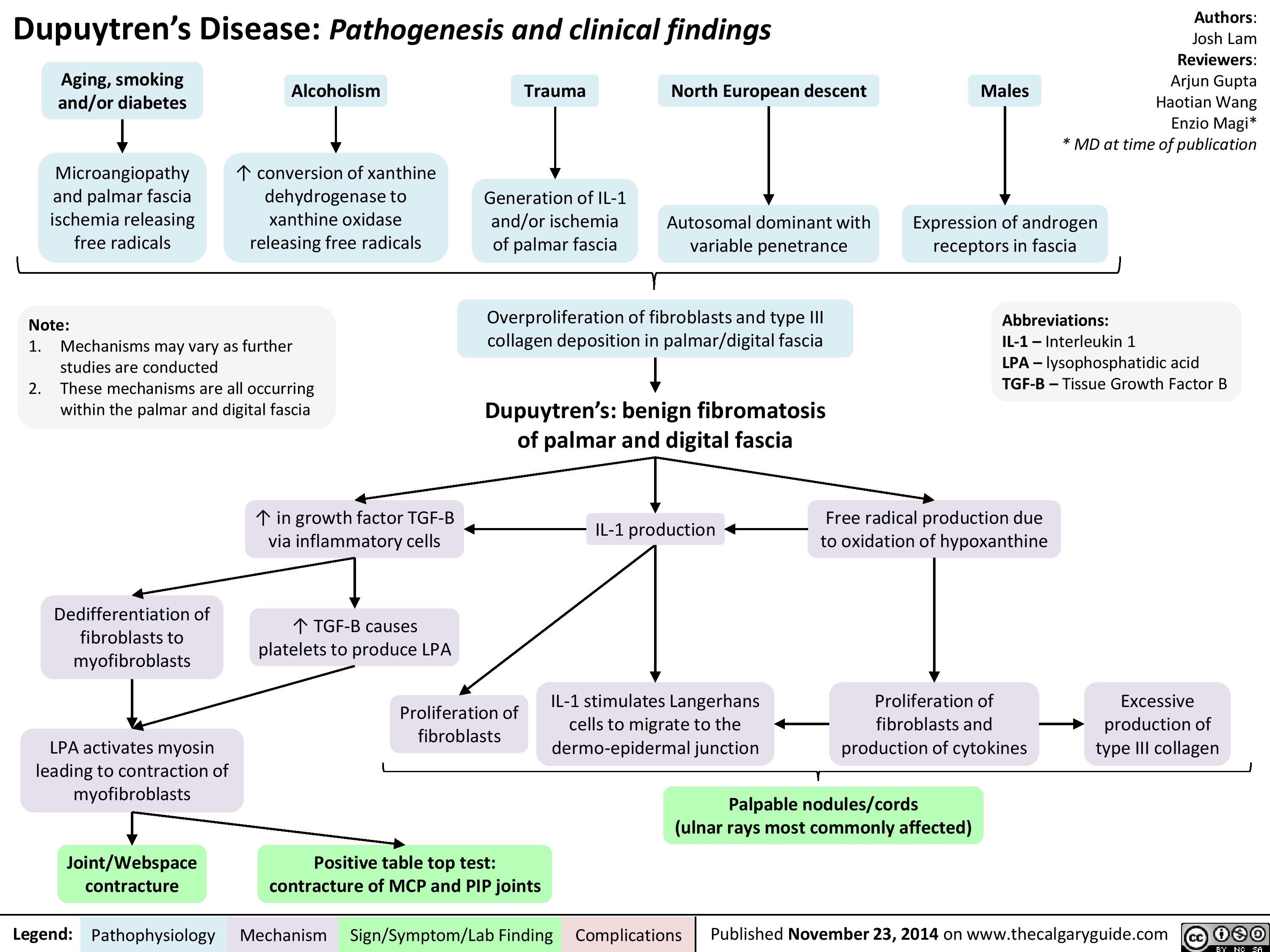
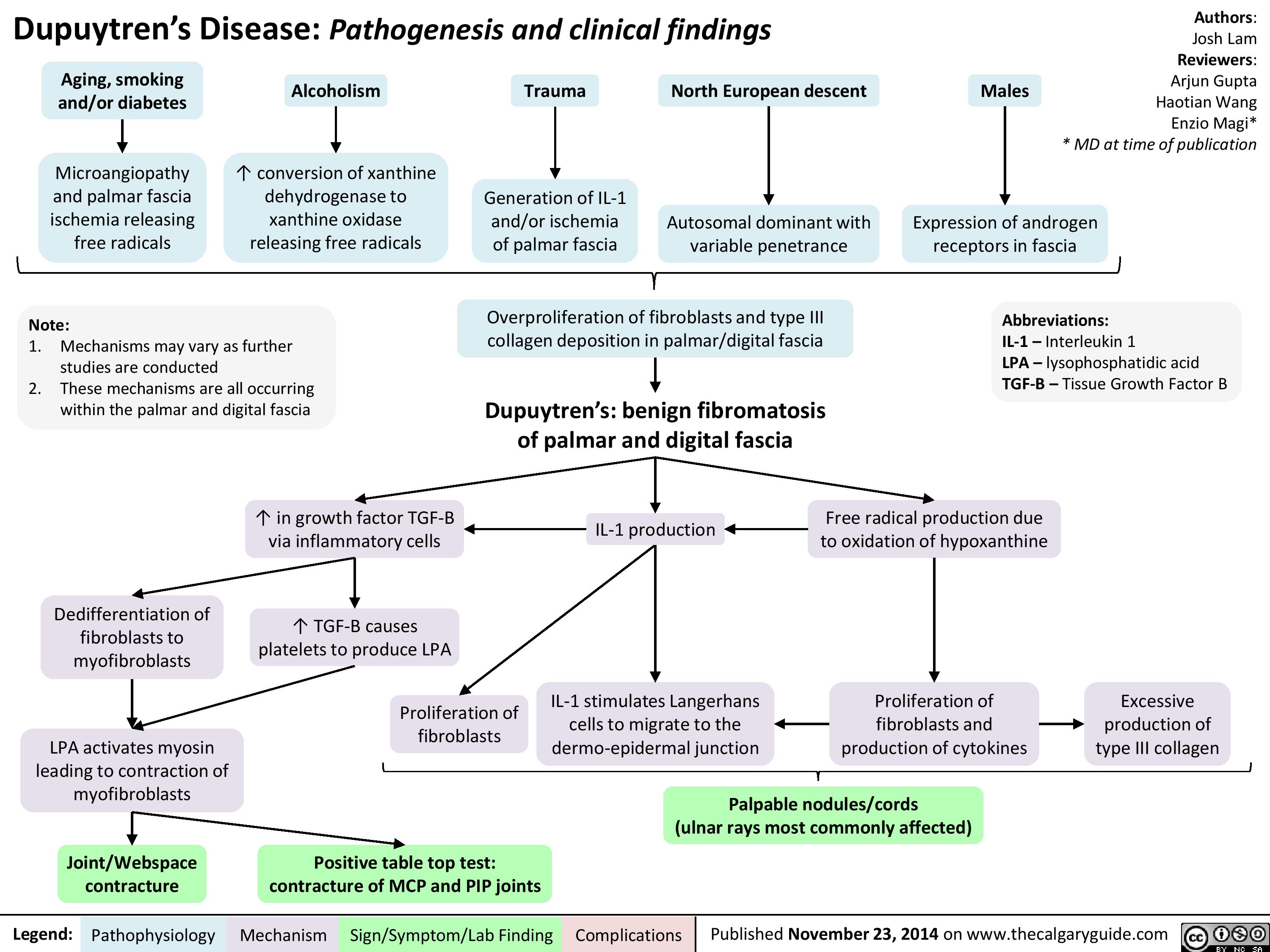
Dupuytren
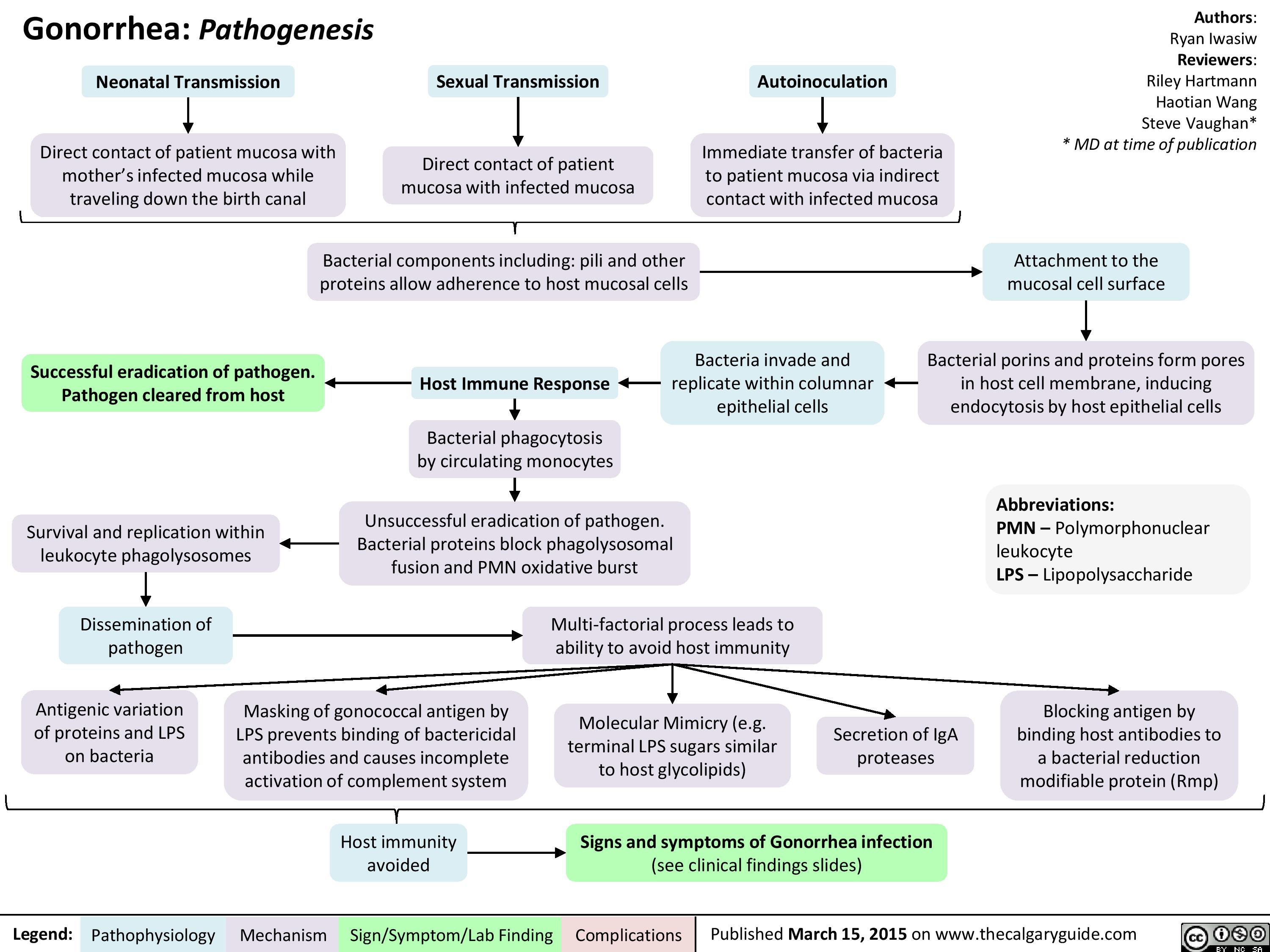
Gonorrhea Pathogenesis
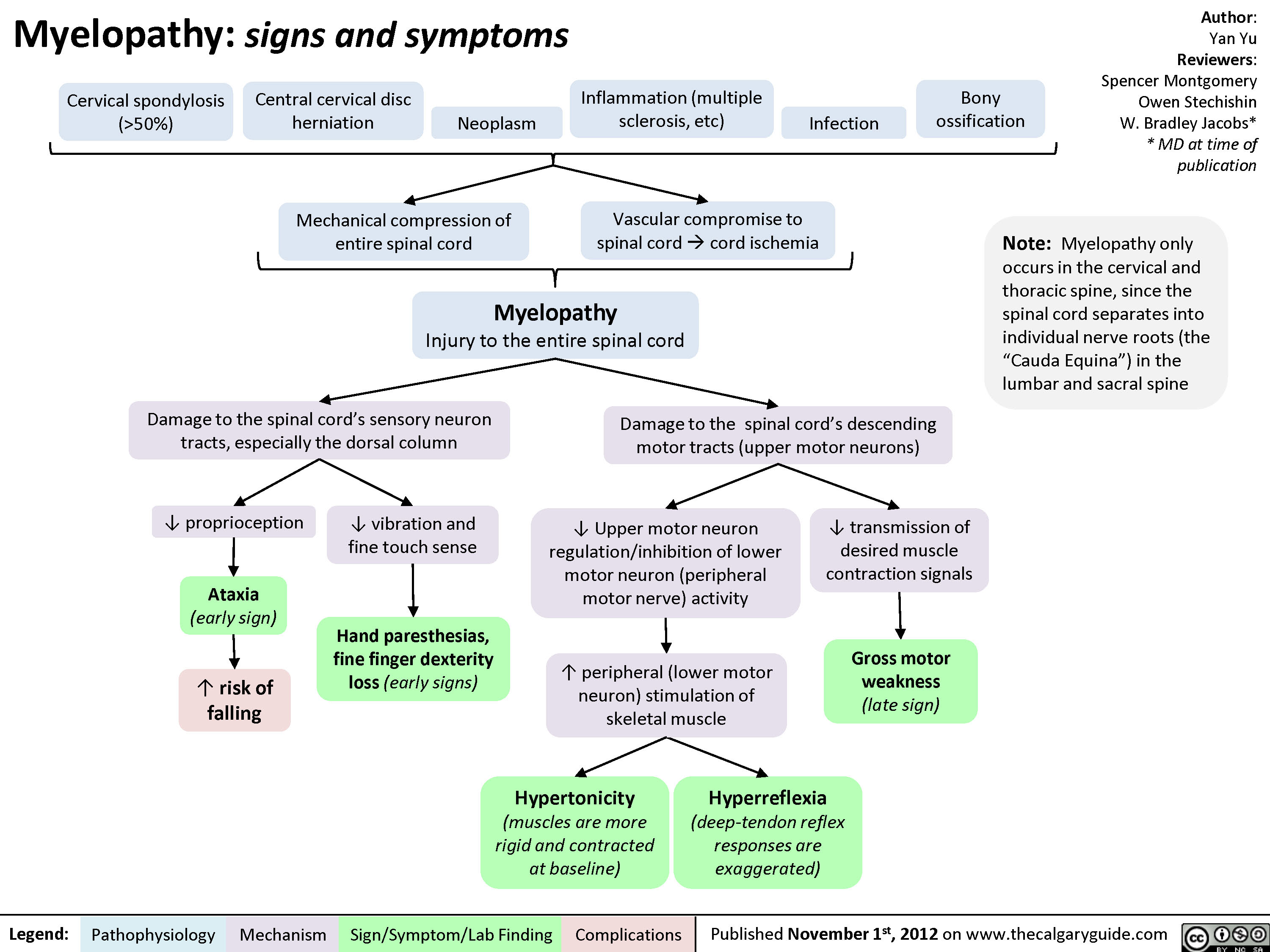
Myelopathy
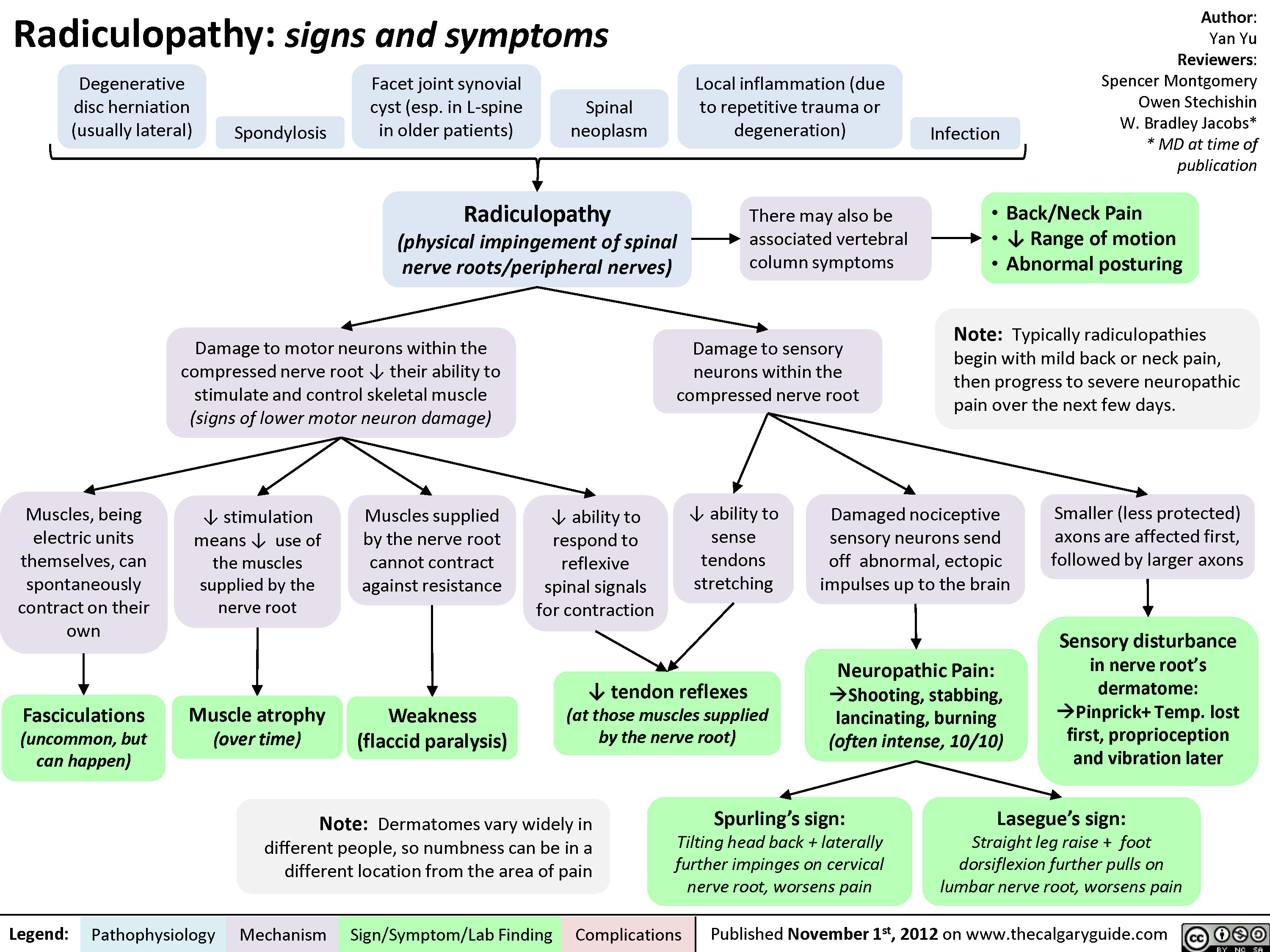
Radiculopathy
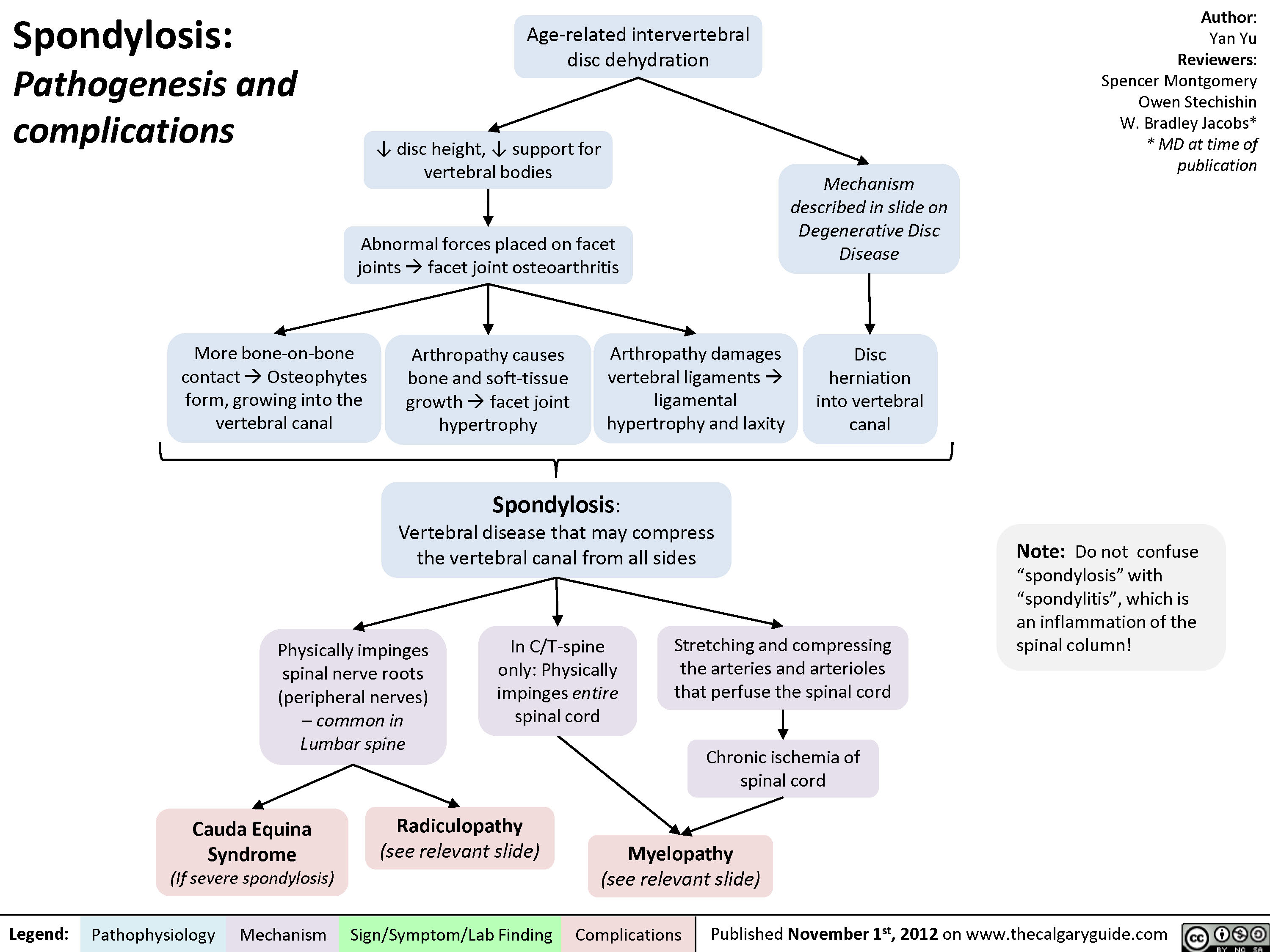
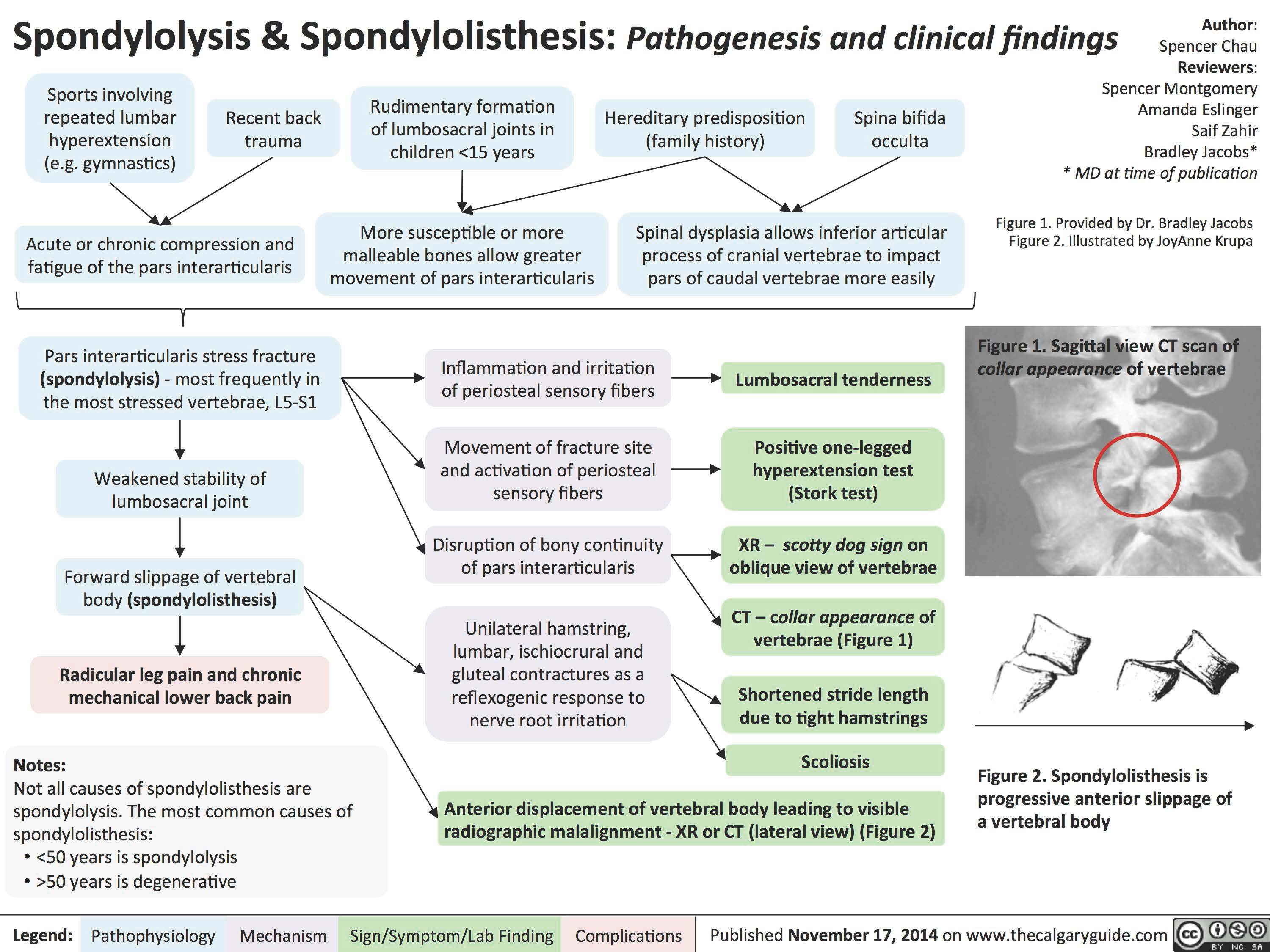
Spondylosis
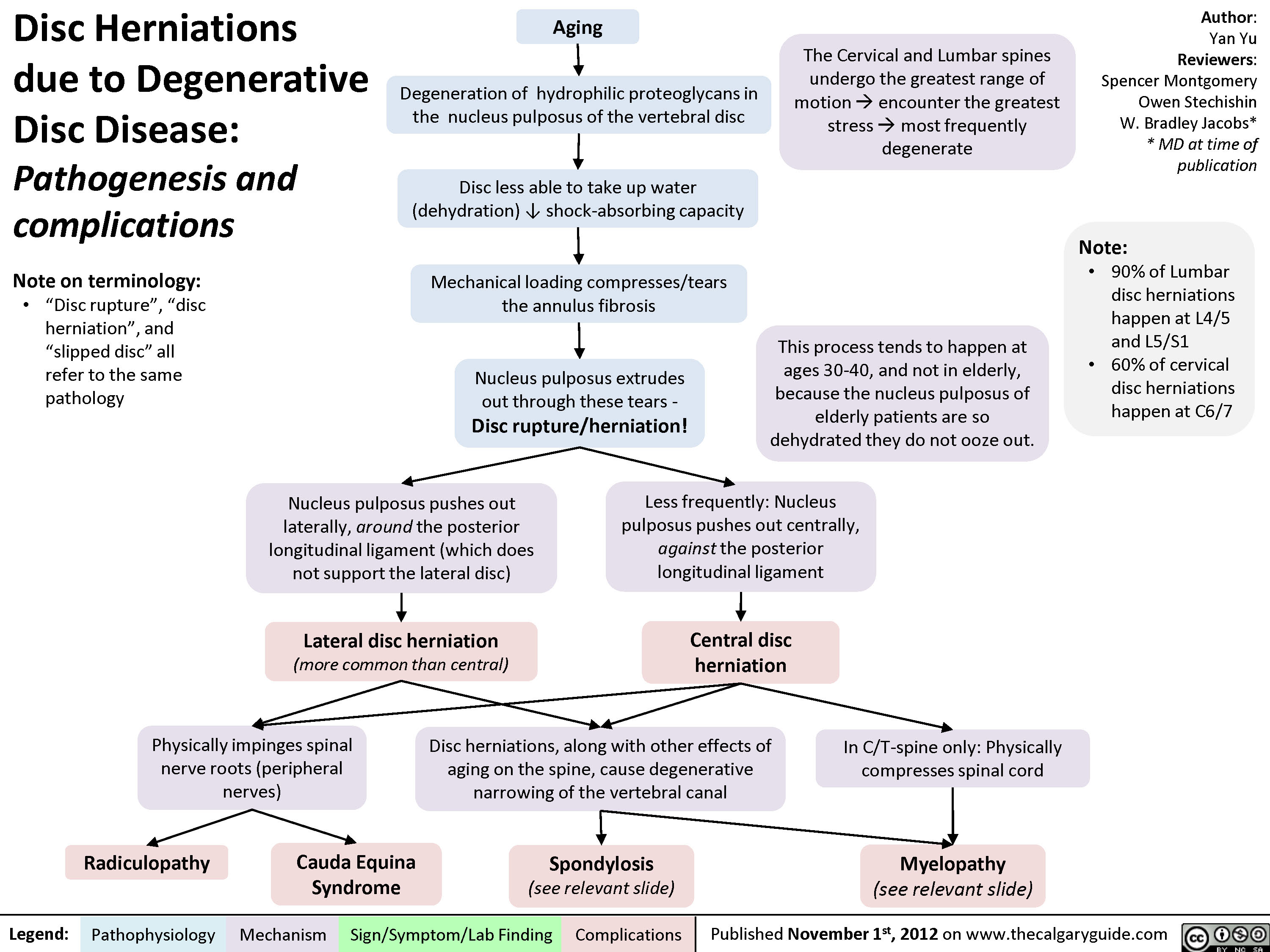
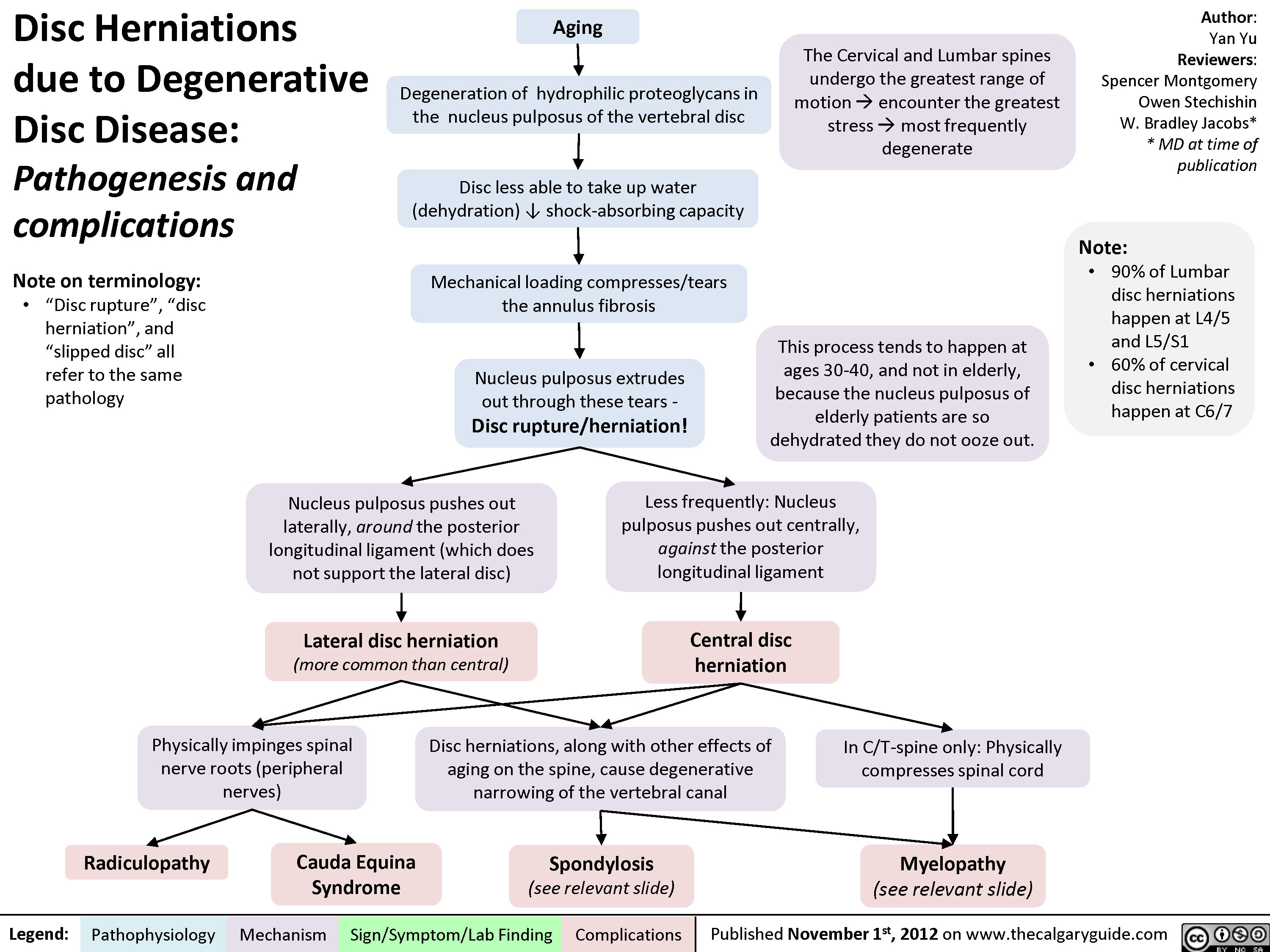
Disc Herniations
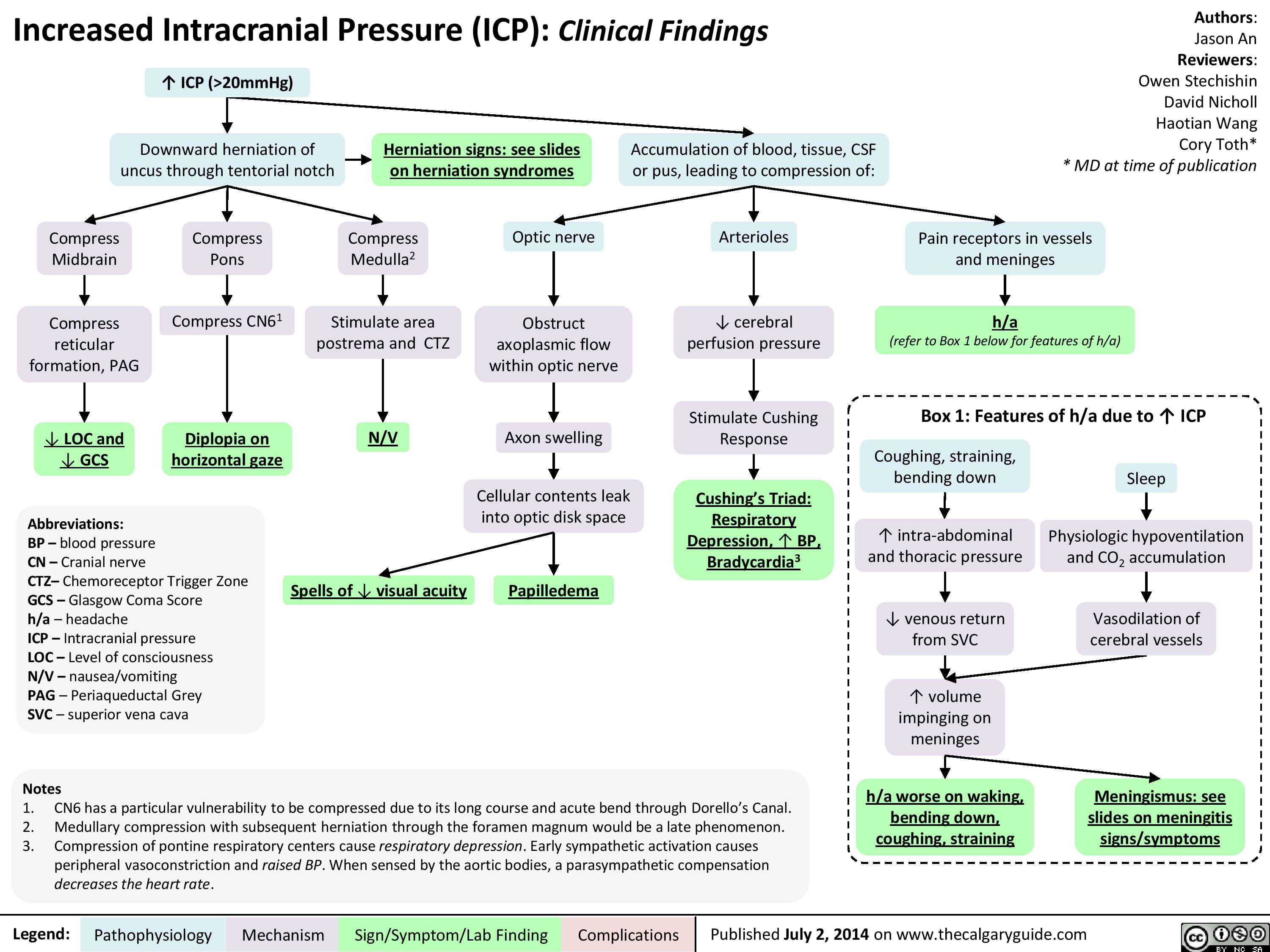
Presentation of increased ICP
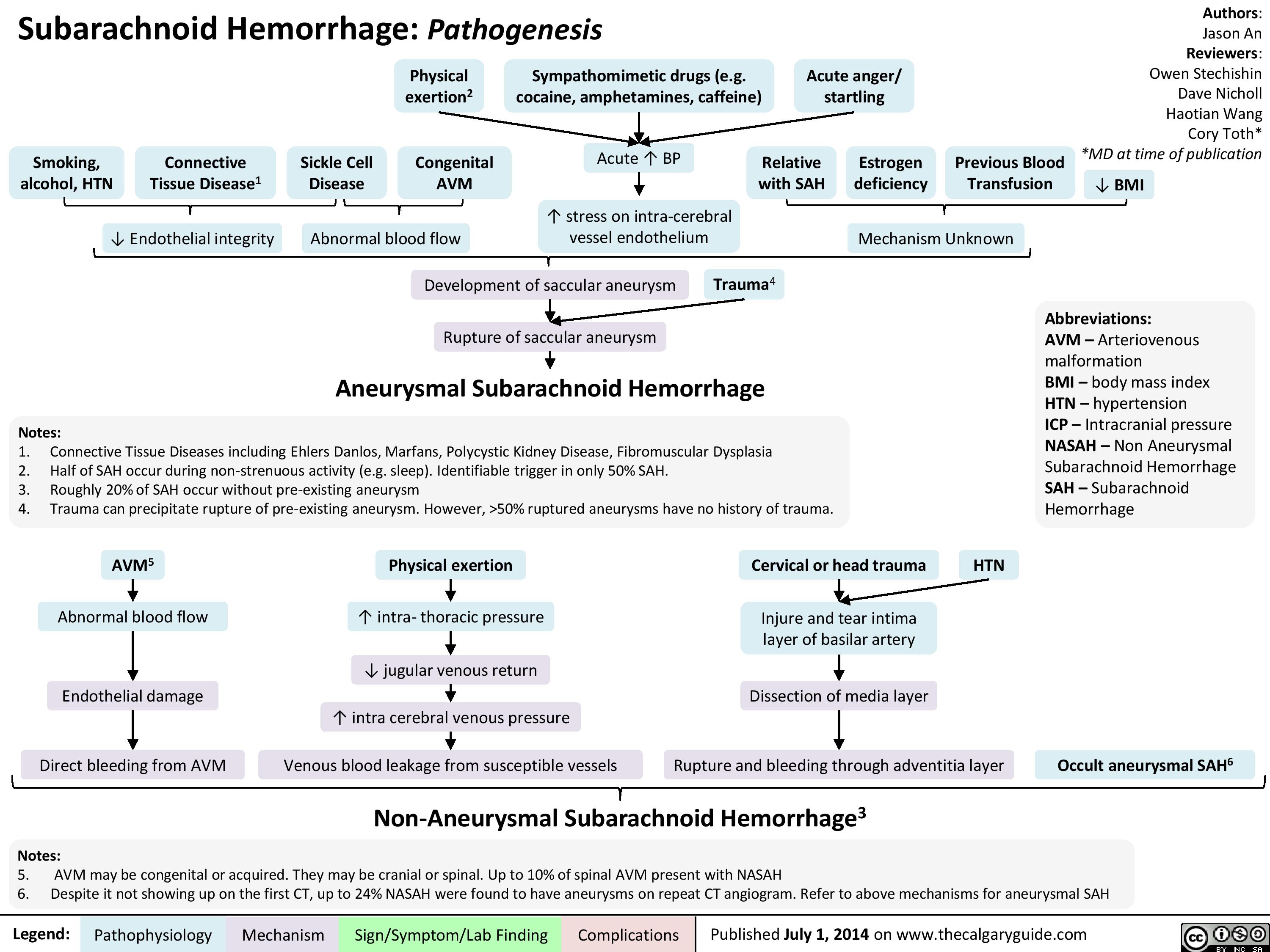
Pathogenesis of SAH
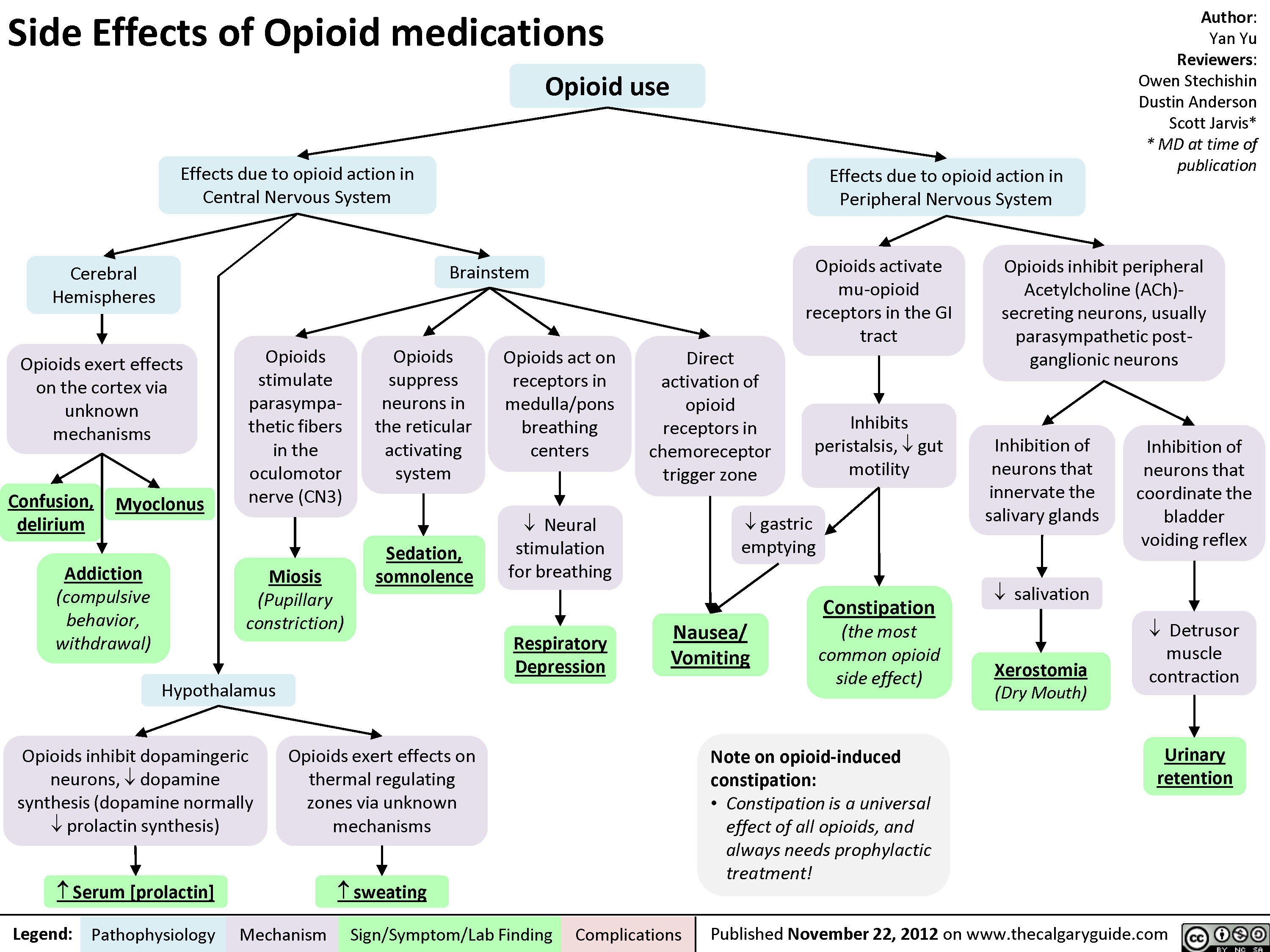
Side Effects of Opioid Medications
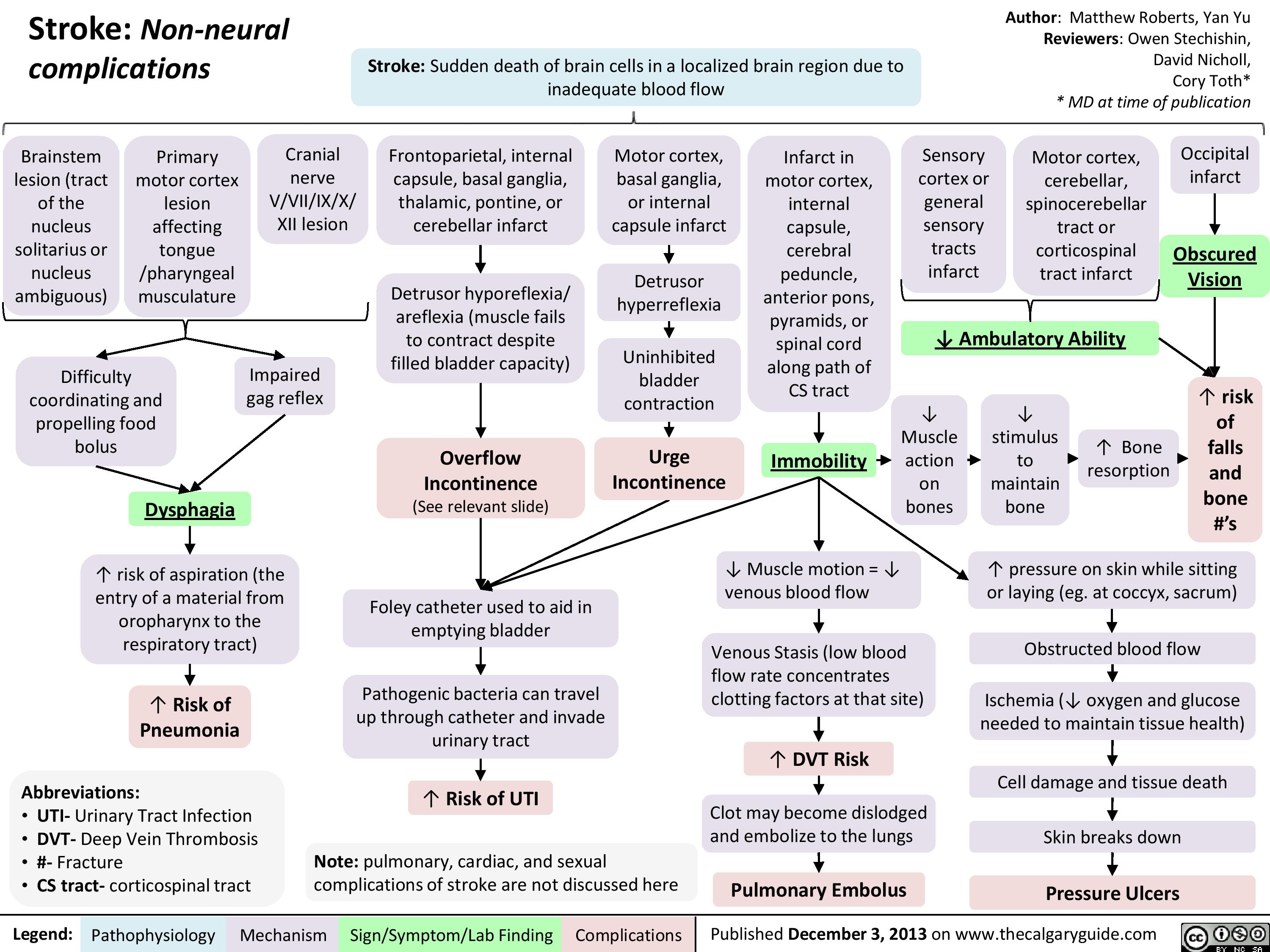
Non Neural Complications of Stroke
Giant Cell (Temporal) Arteritis - Pathogenesis and investigations
Giant Cell (Temporal) Arteritis - Clinical findings and Complications
Pain Pathways in the Head

Bacterial Meningitis Complications
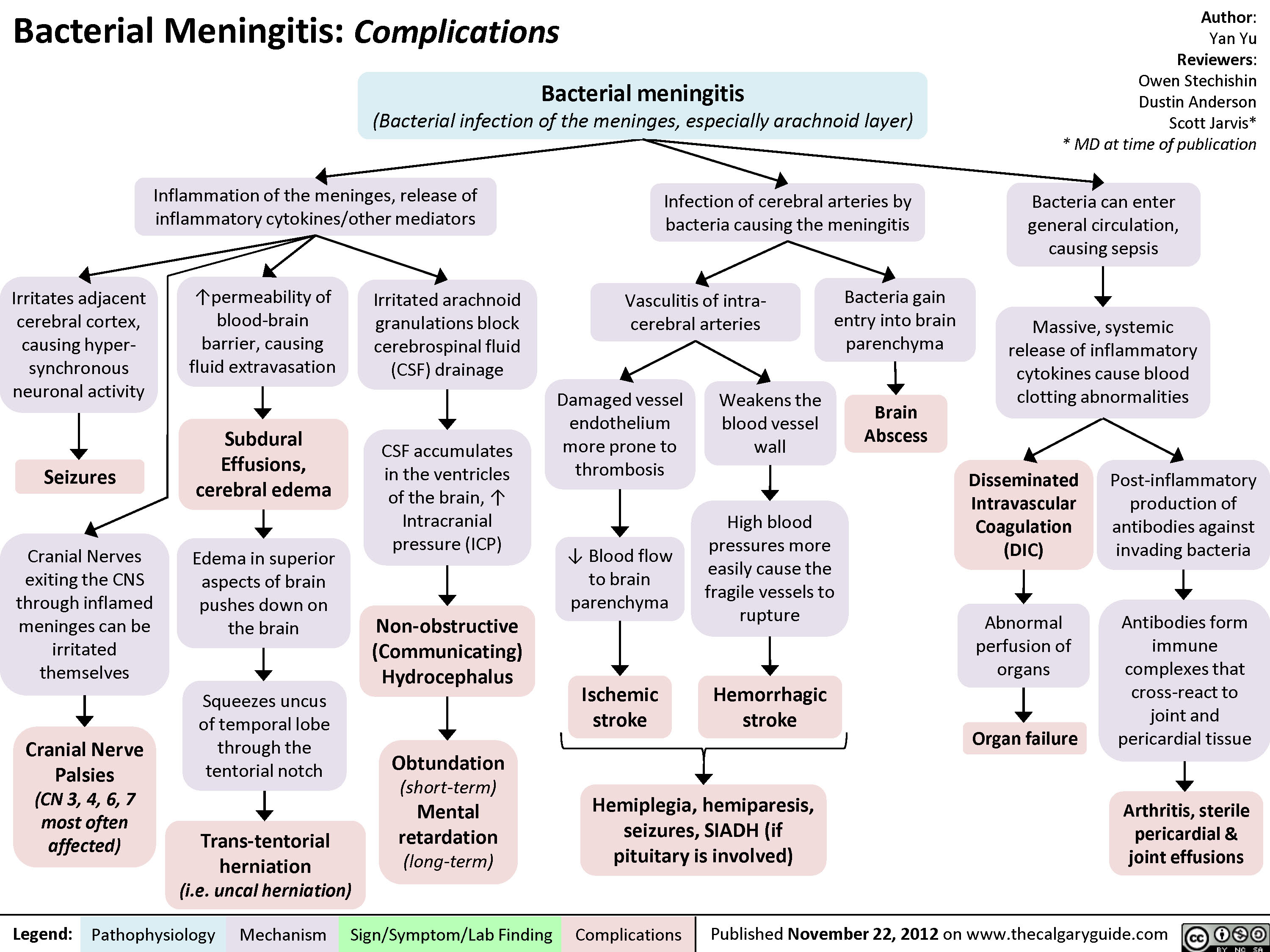
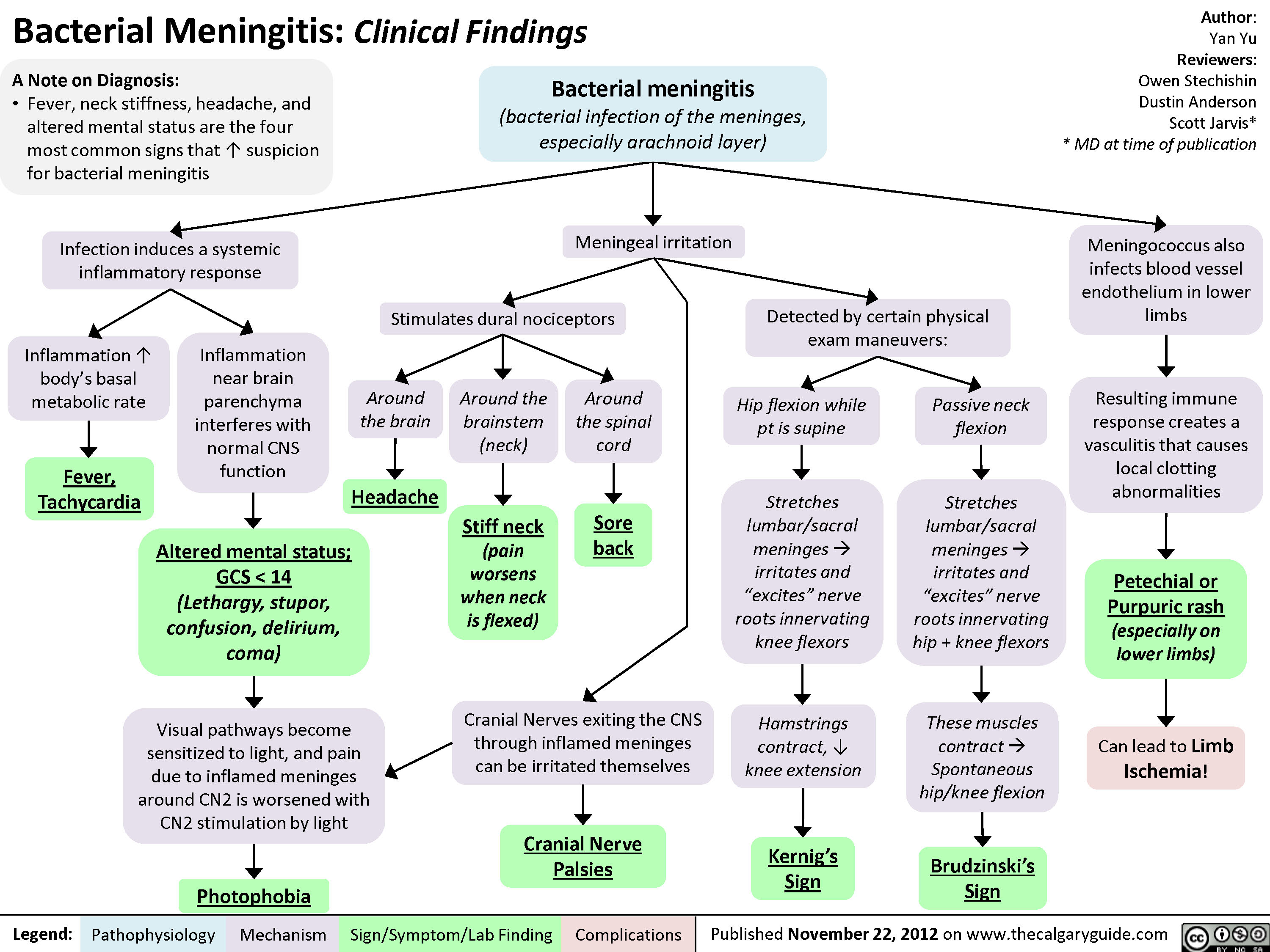
Bacterial Meningitis Clinical Findings
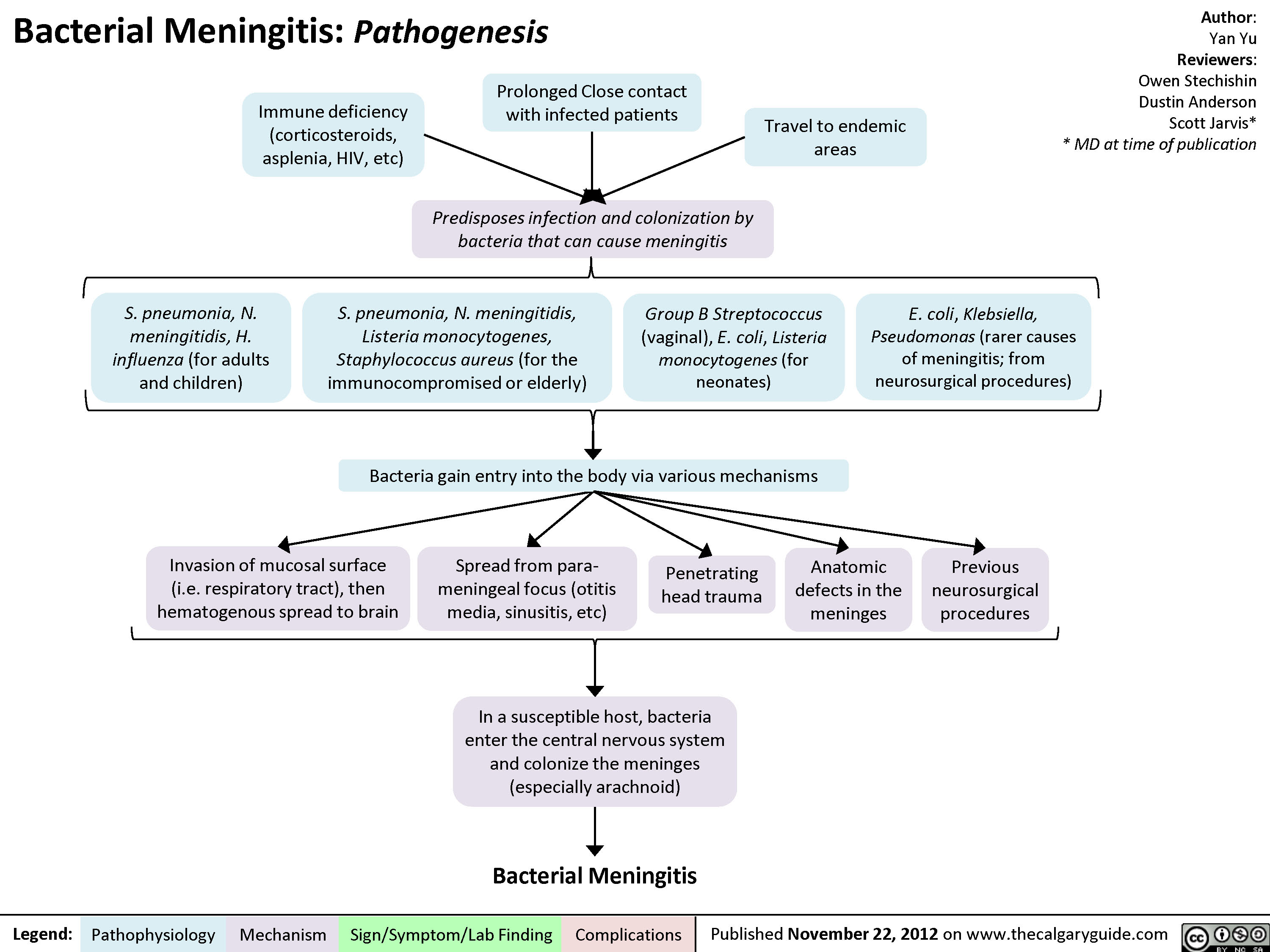
Bacterial Meningitis Pathogenesis
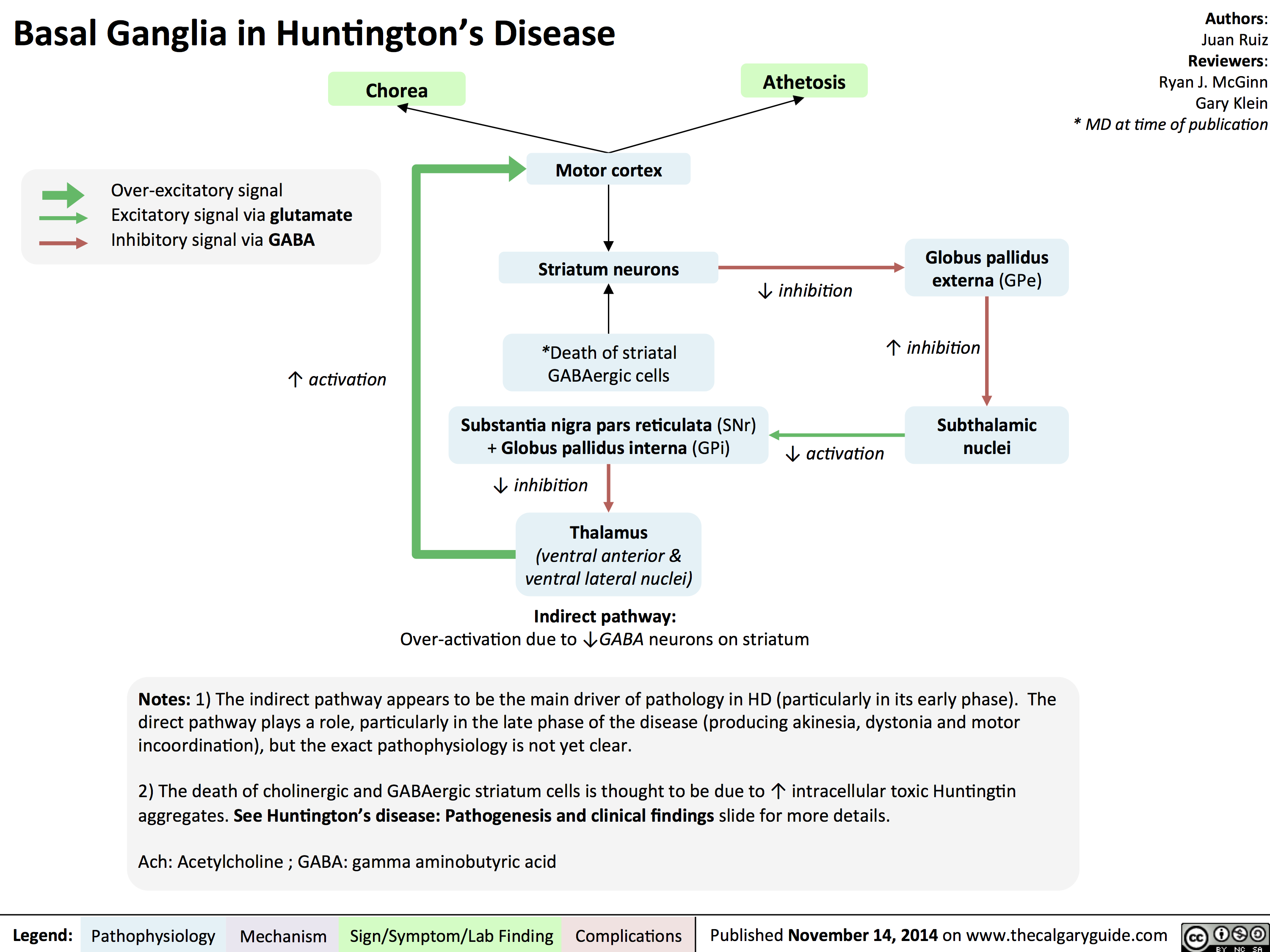
Basal Ganglia in Huntingtons Disease
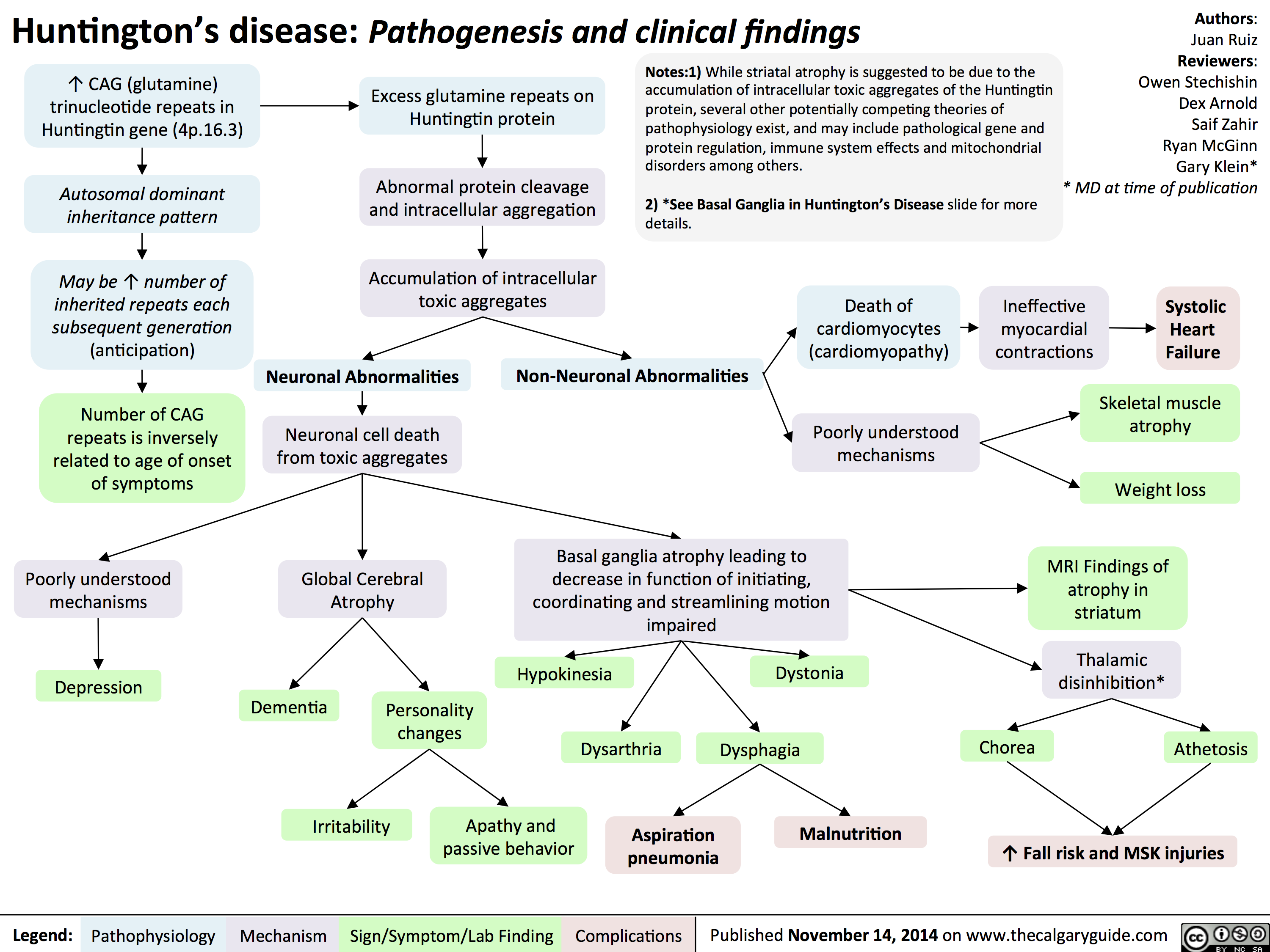
Huntingtons Disease Pathogenesis and Clinical Findings
Parkinsons Disease
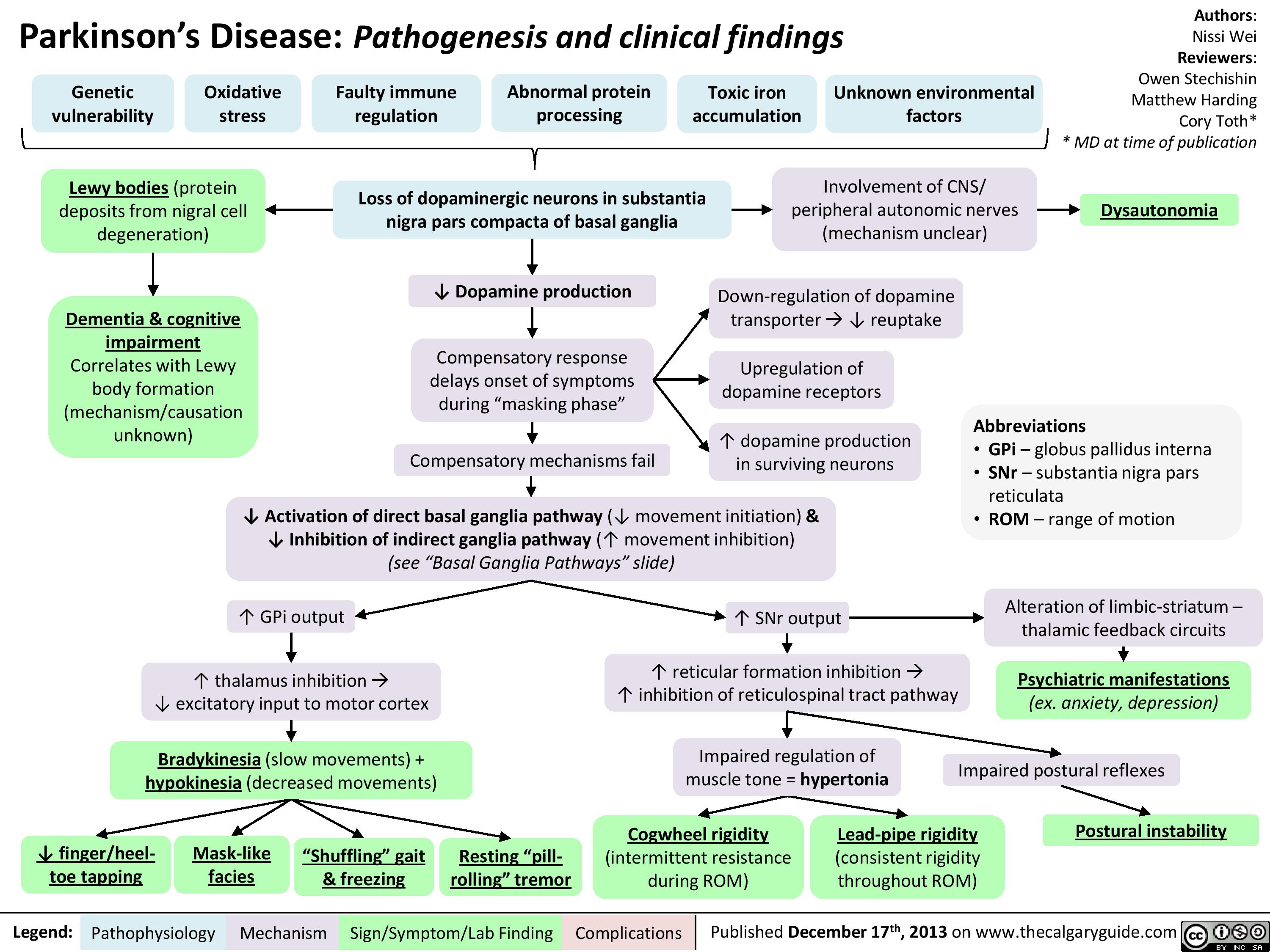
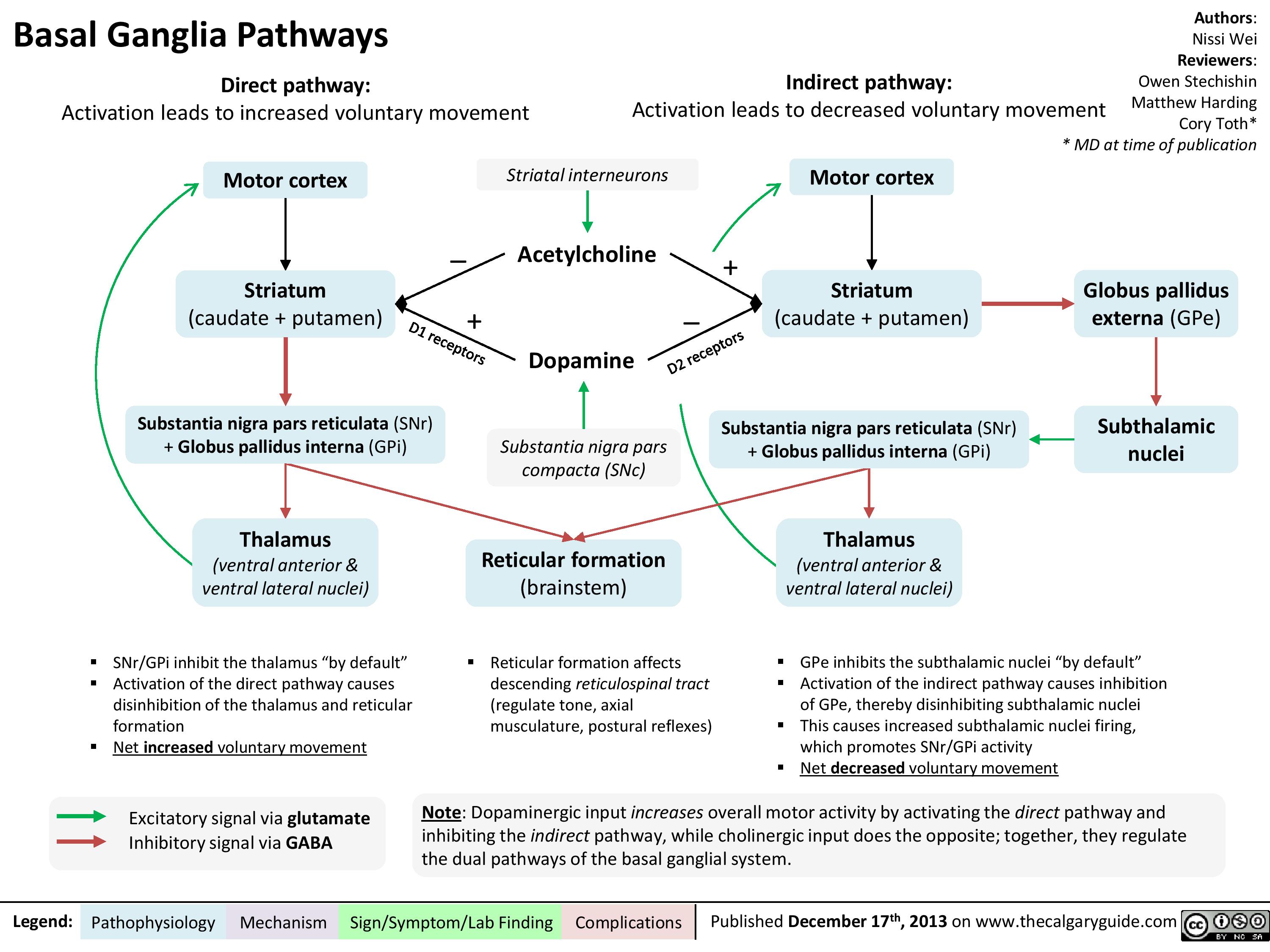
Basal ganglia pathways
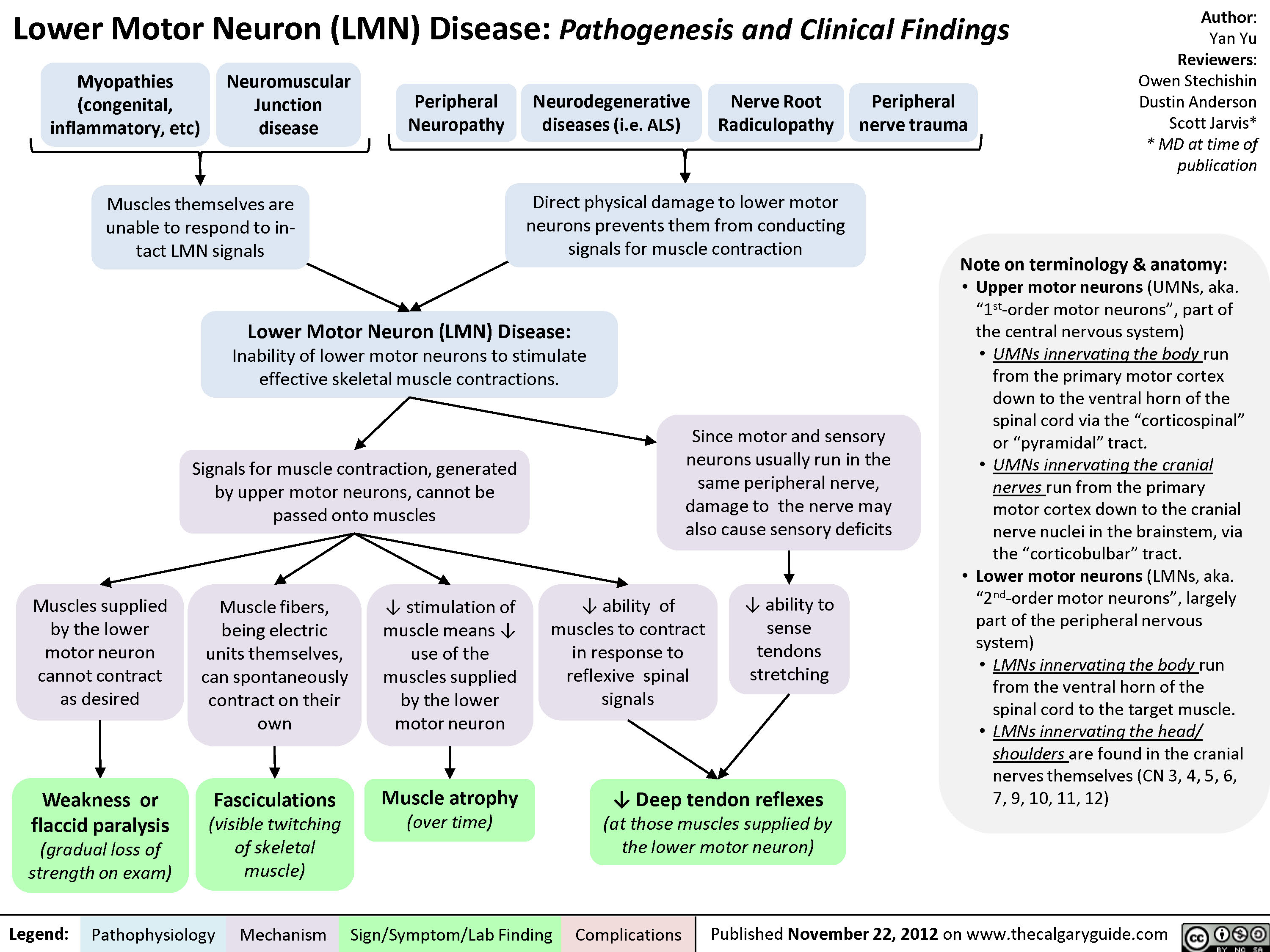
Lower Motor Neuron (UMN) Disease
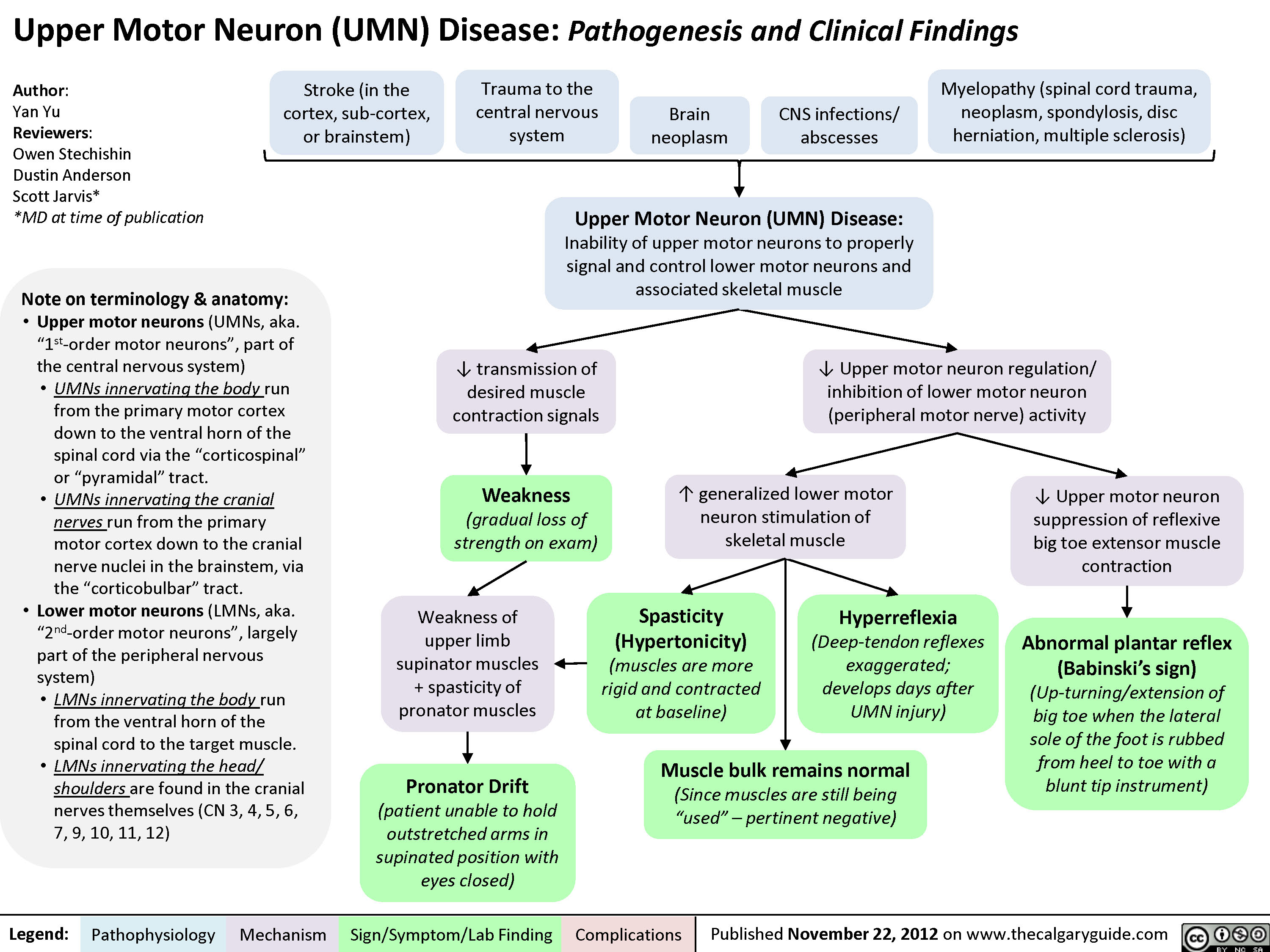
Upper Motor Neuron (UMN) Disease
Dupuytren
Trigger Finger
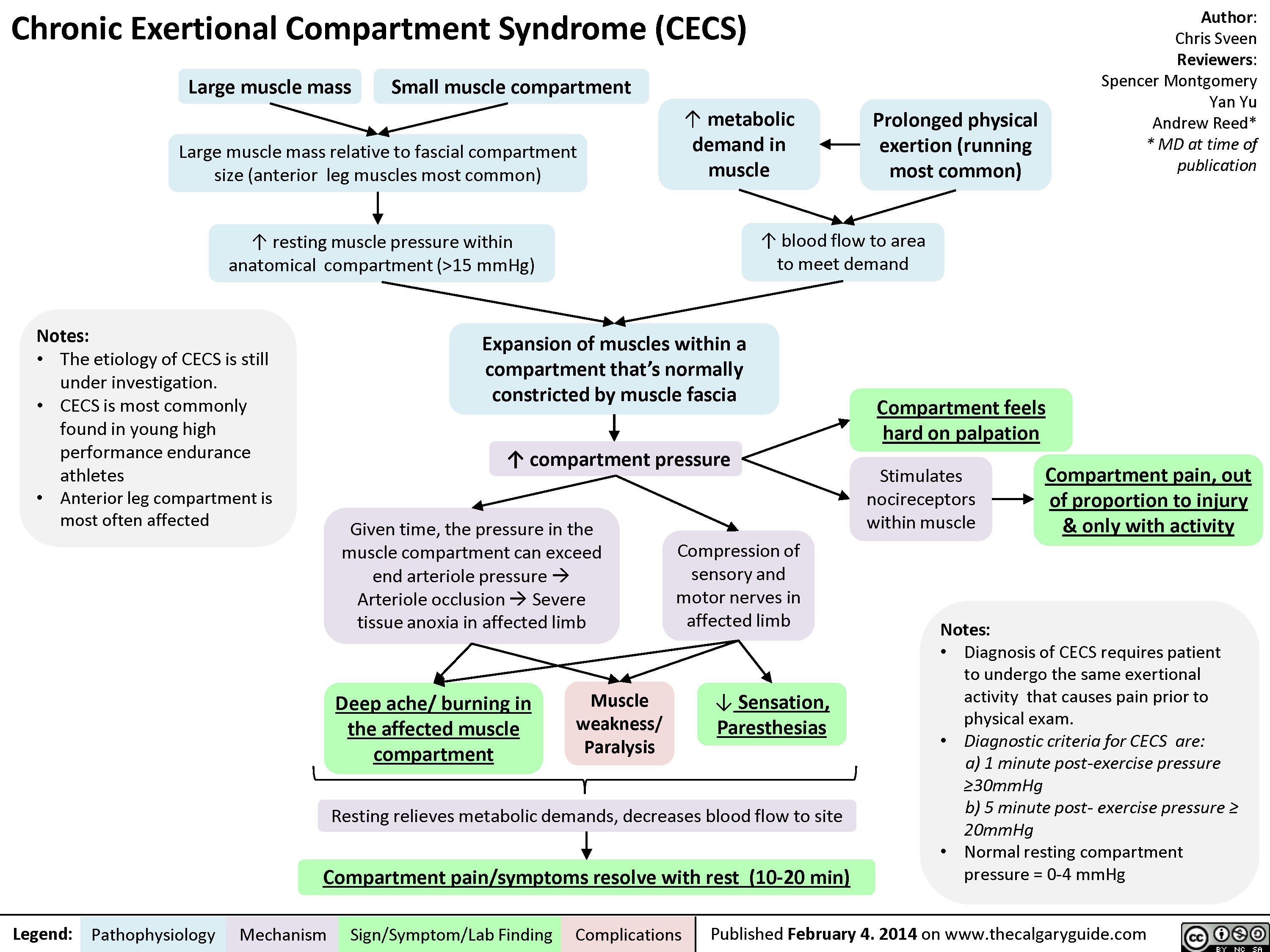
Chronic Exertional Compartment Syndrome
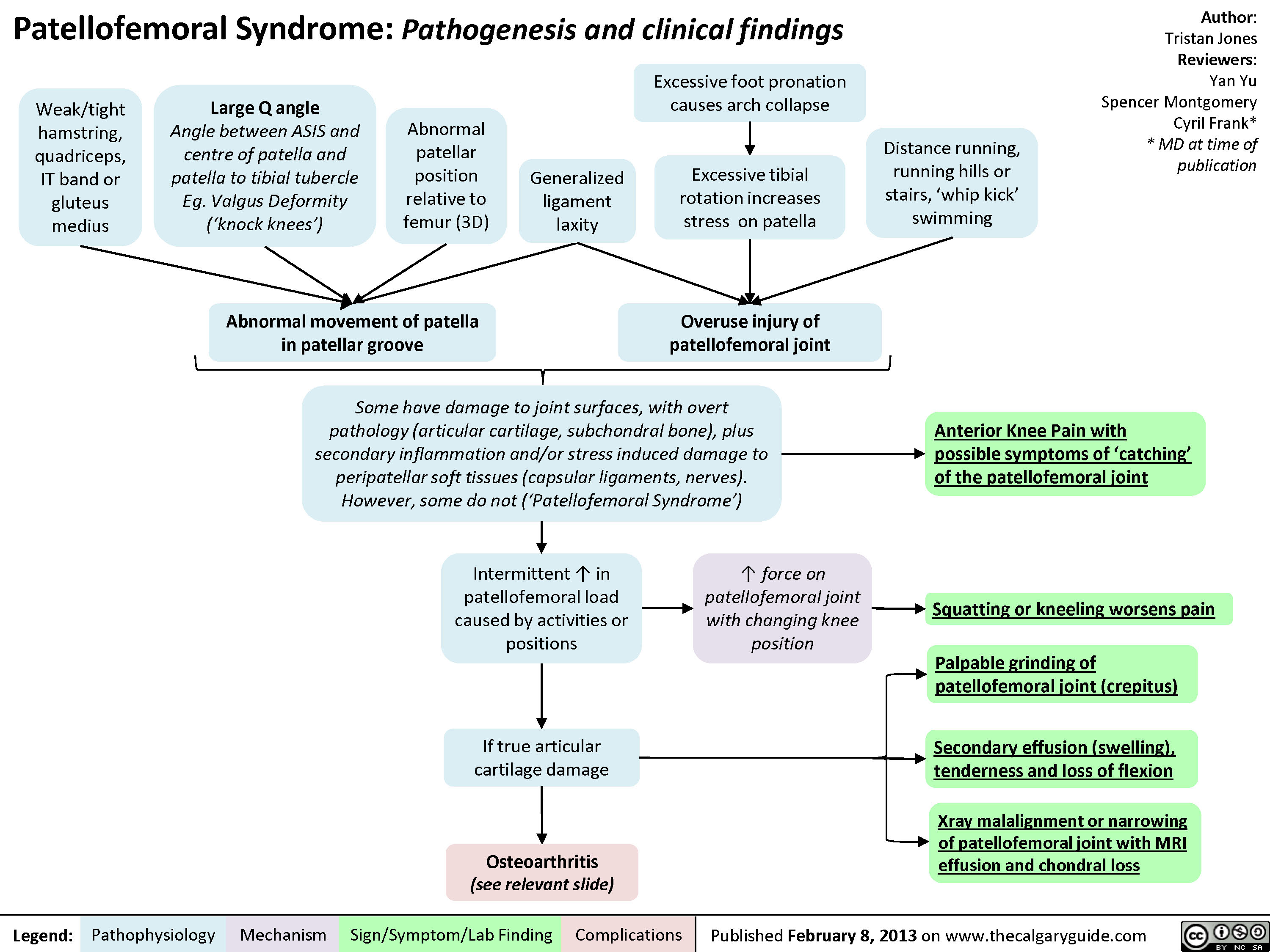
Patellofemoral Syndrome
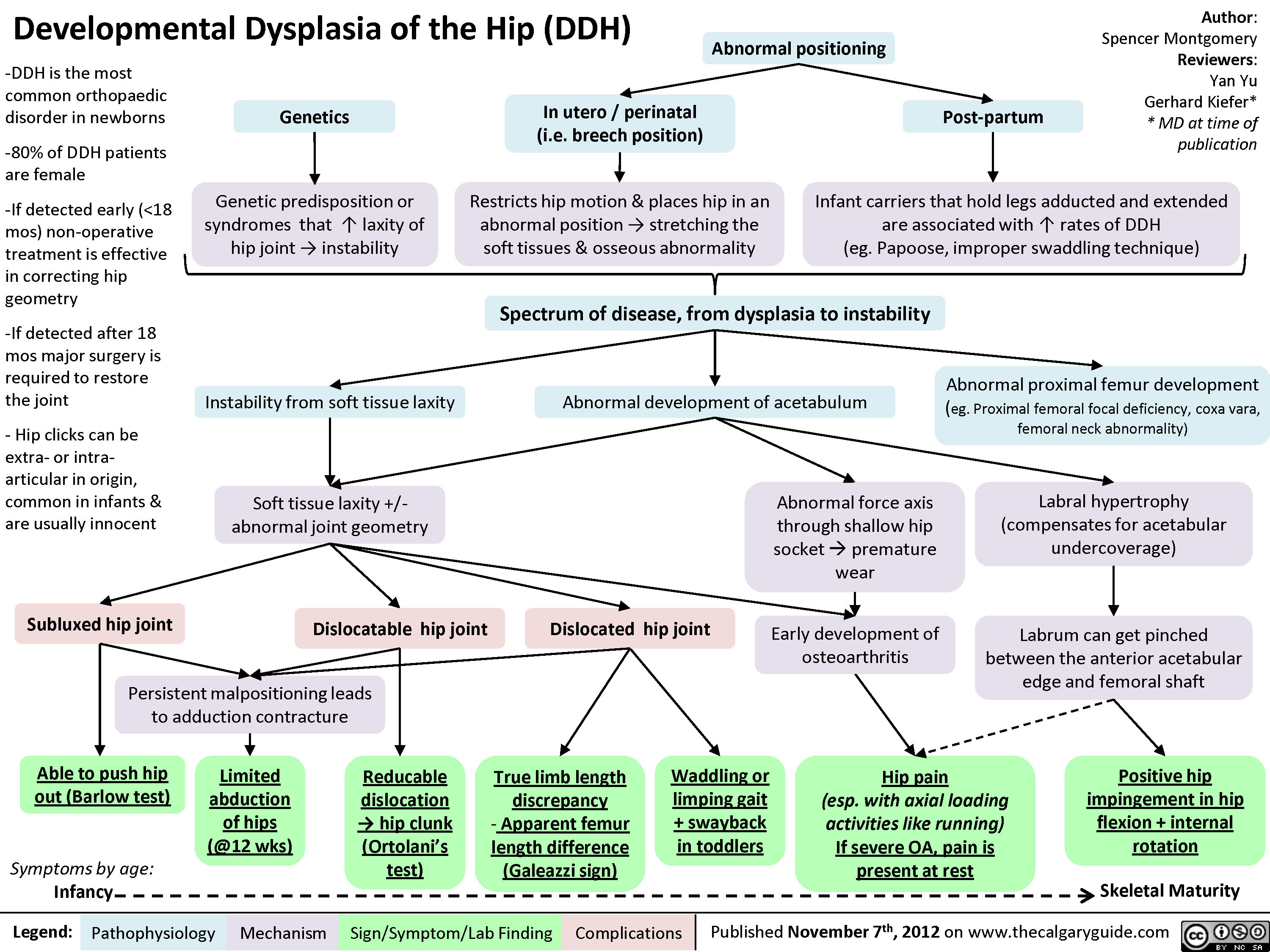
Developmental Dysplasia of the Hip (DDH)
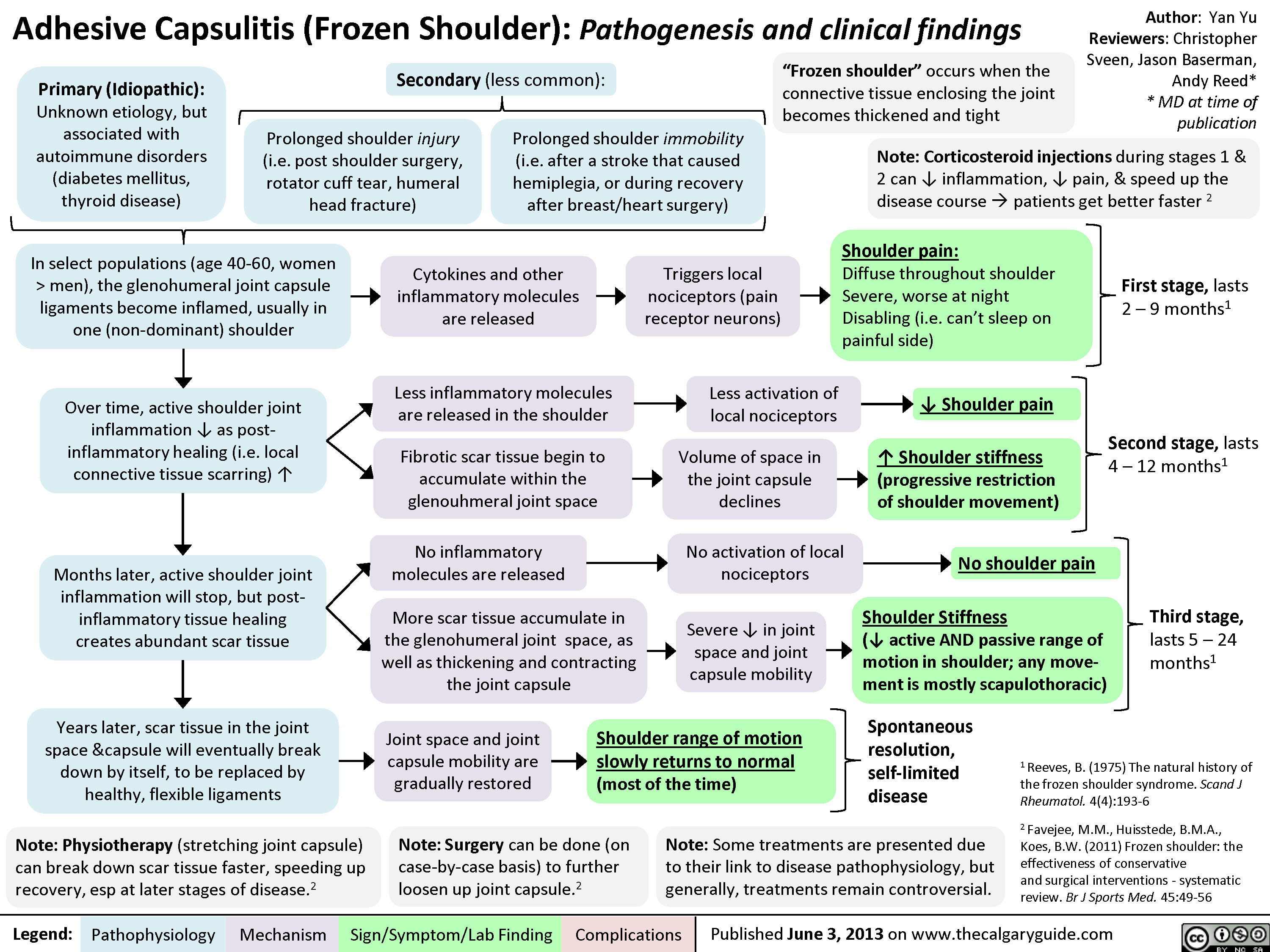
Adhesive Capsulitis
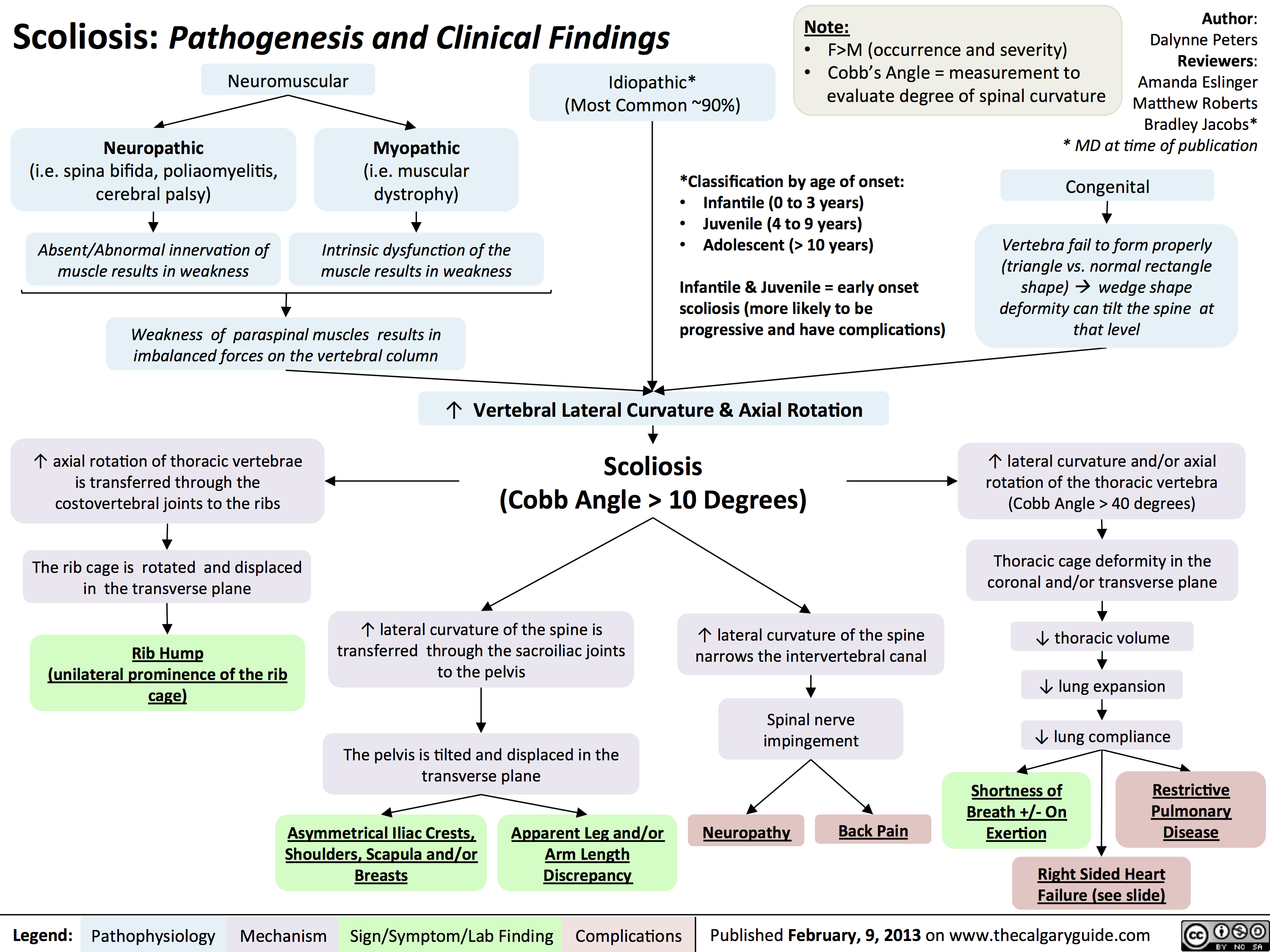
Scoliosis -Pathogenesis and Clinical Findings
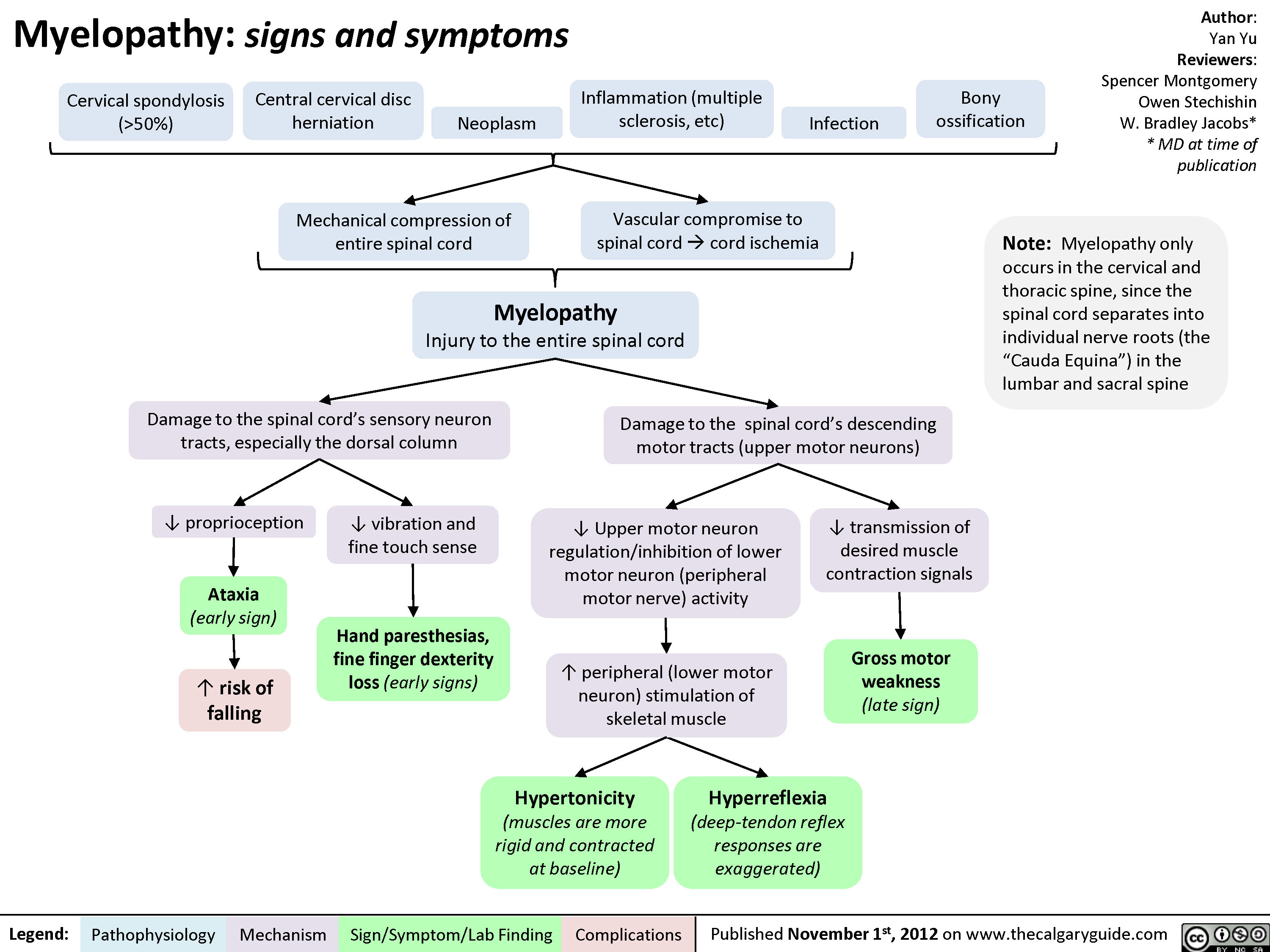
Myelopathy
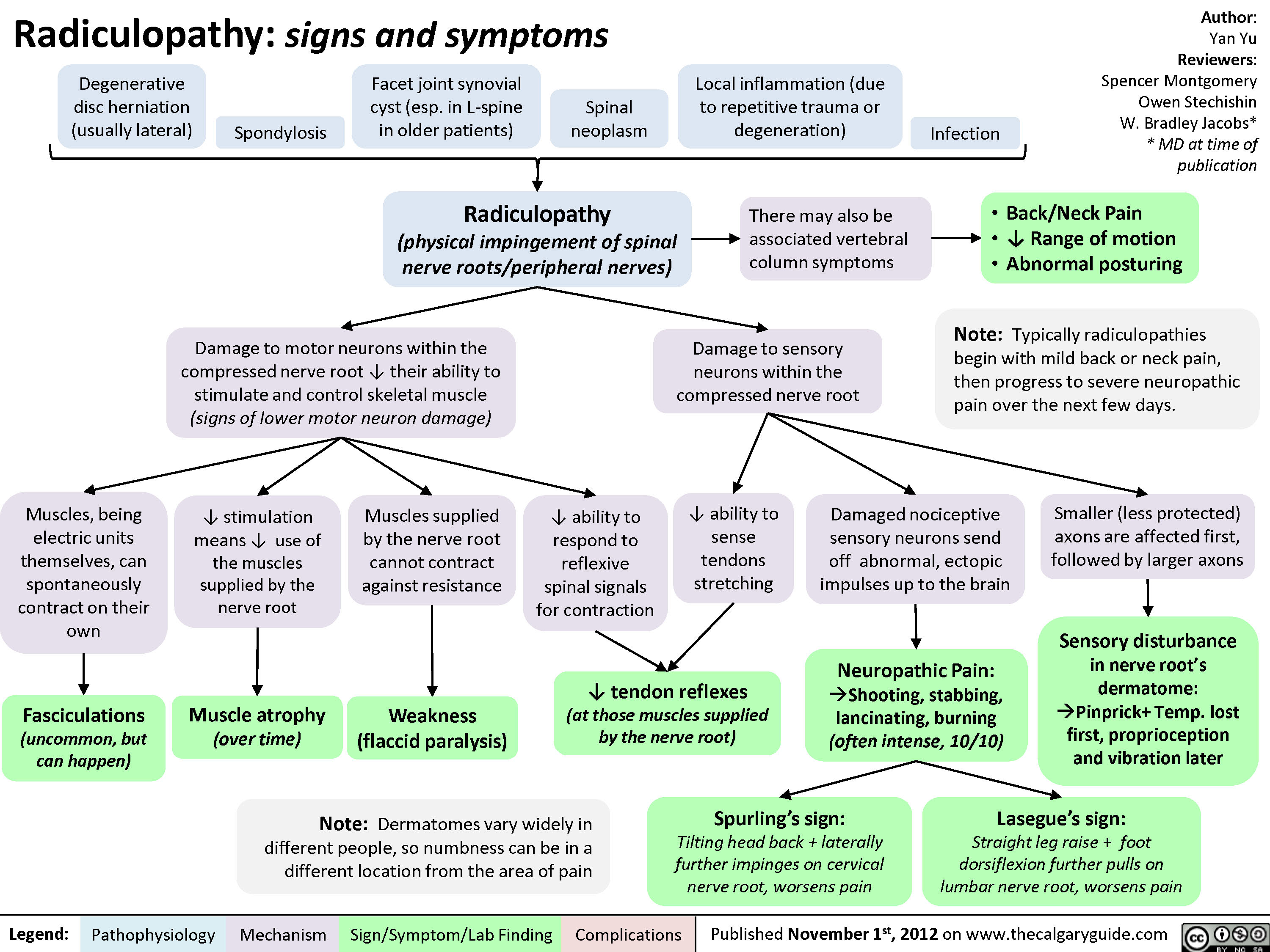
Radiculopathy
Disc Herniations
Spondylolysis _and_Spondylolisthesis Pathogenesis and Clinical Findings
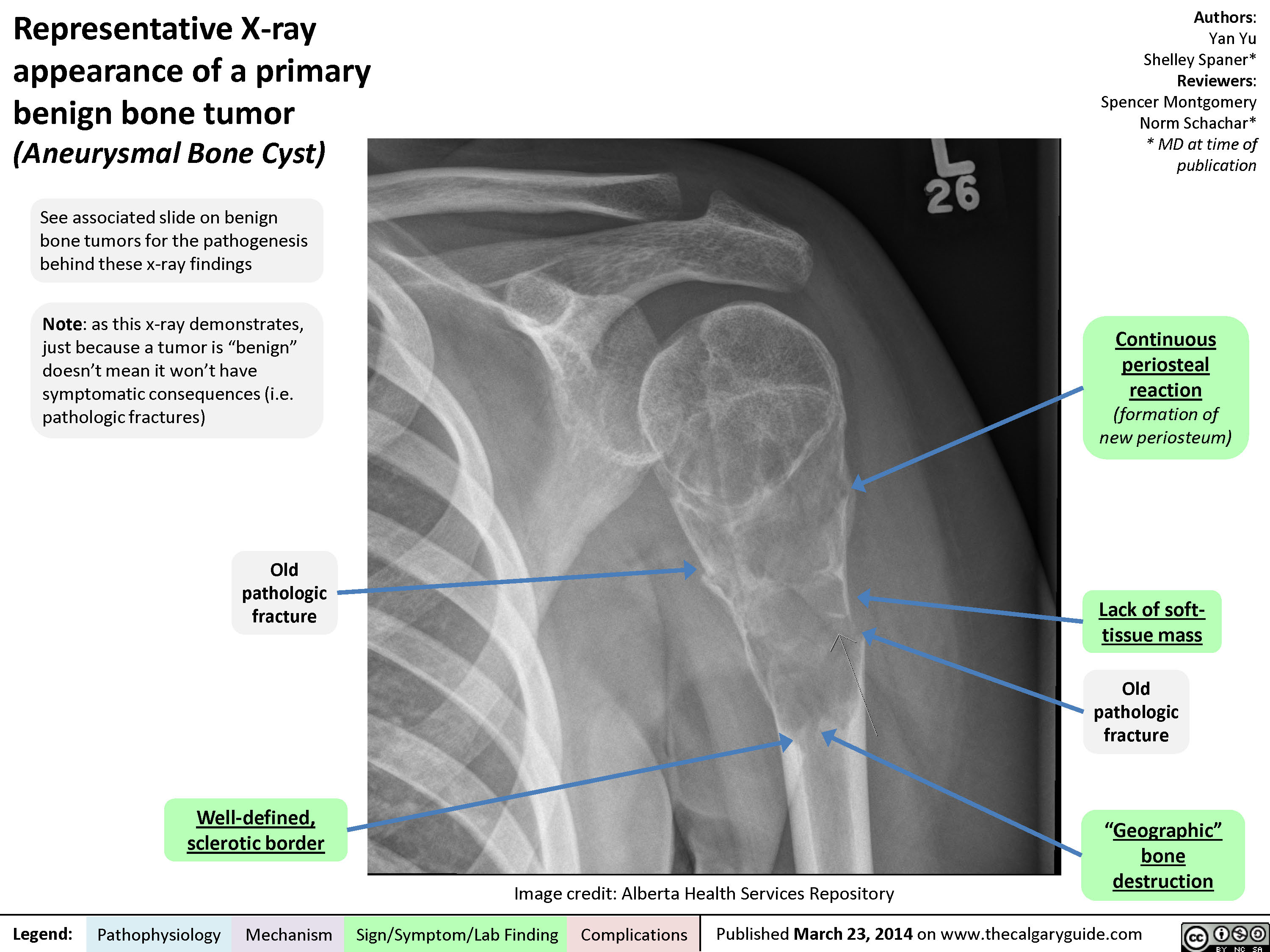
Representative X-ray appearance of a primary benign bone tumor
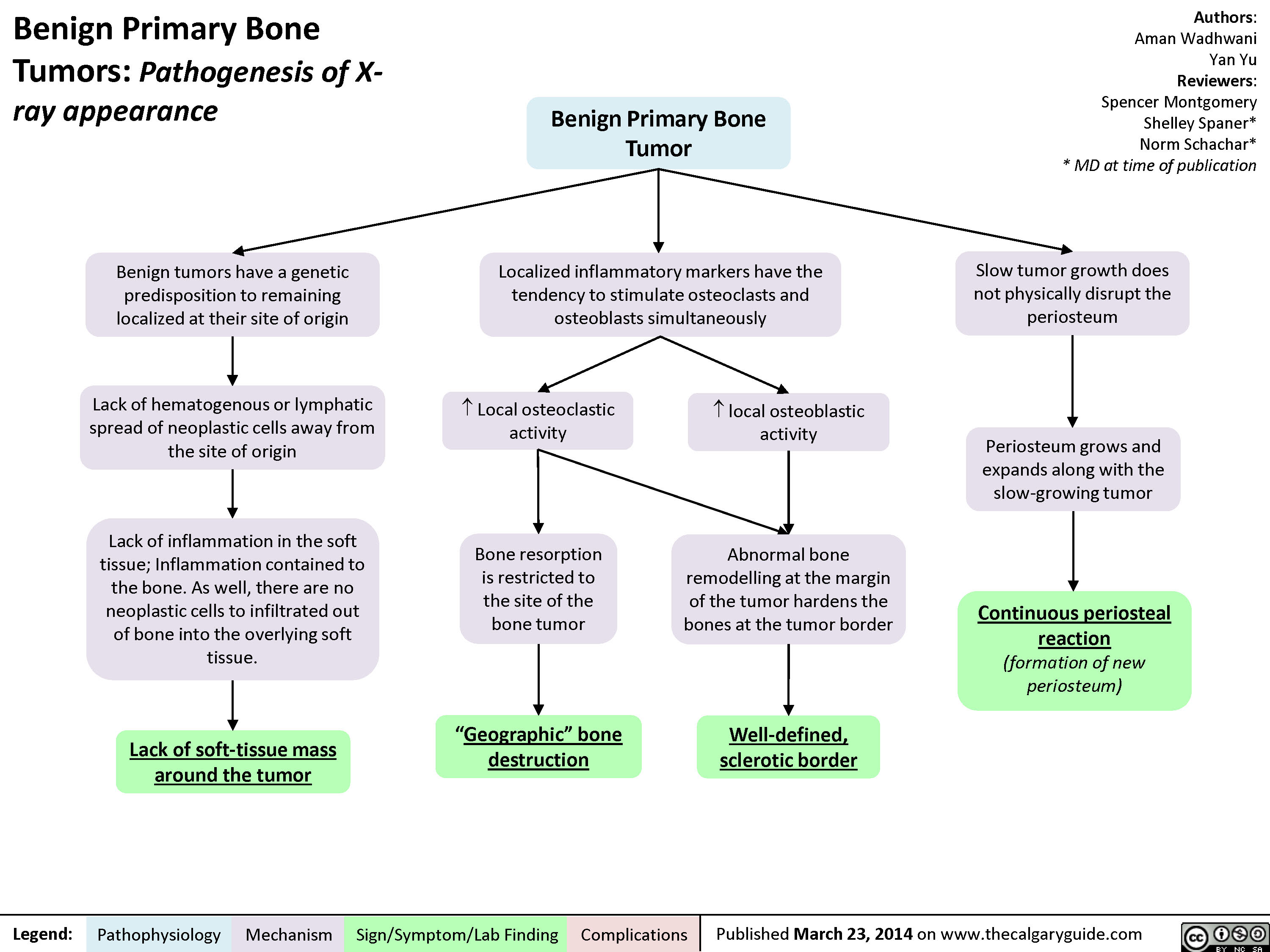
Benign Primary Bone Tumors - Pathogenesis of X-ray appearance
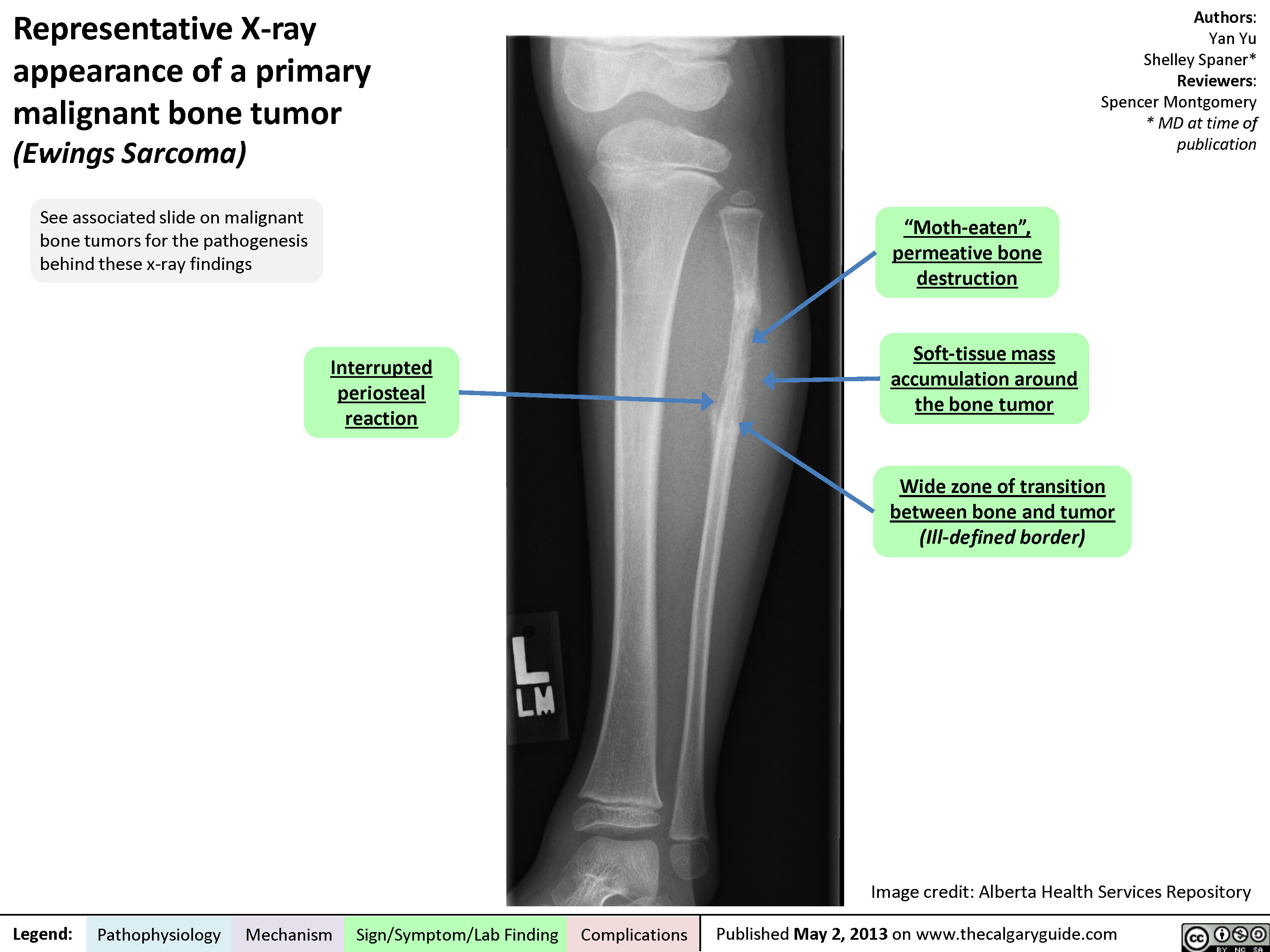
Representative X-ray appearance of a primary malignant bone tumor
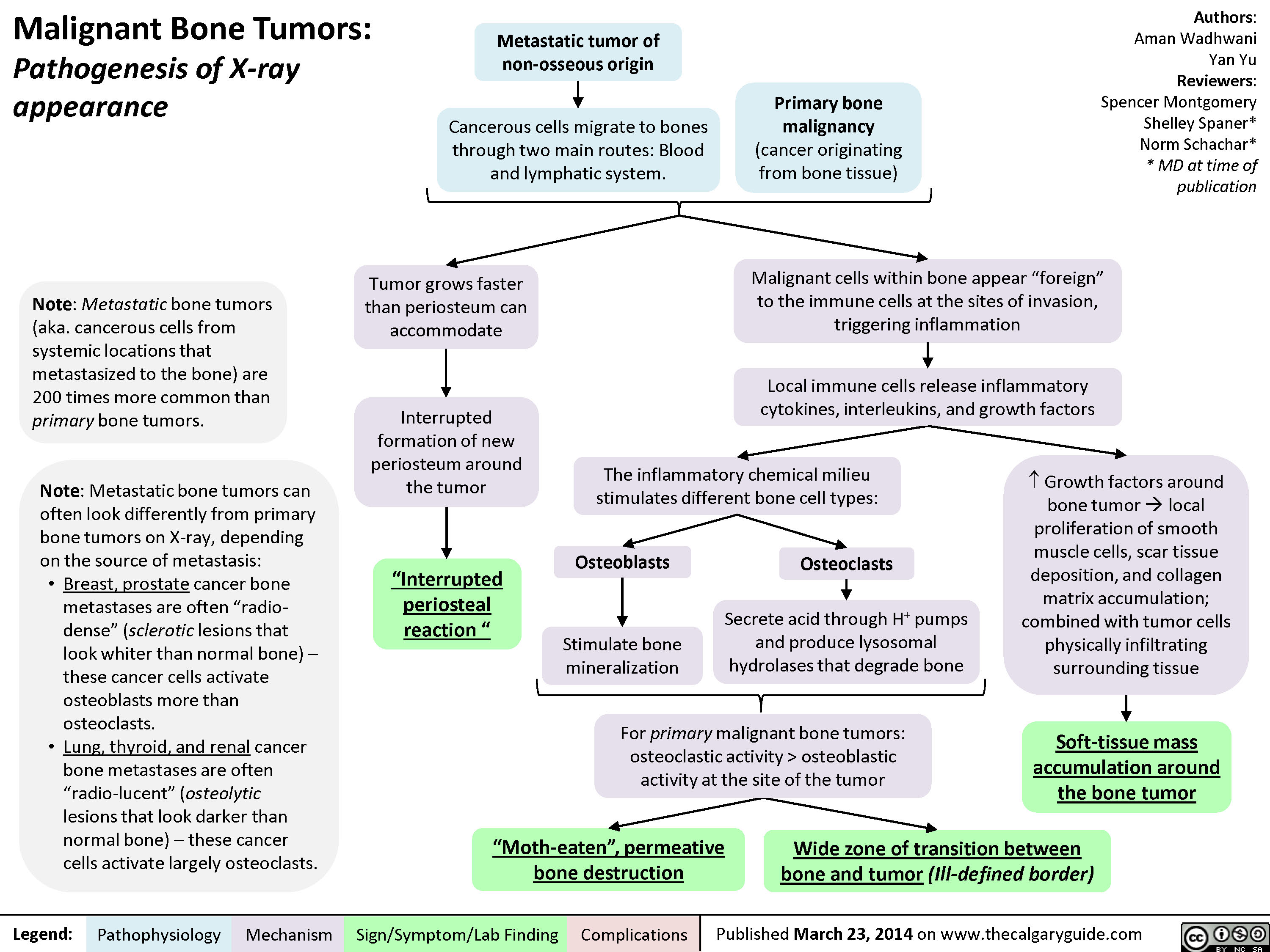
Malignant Bone Tumors - Pathogenesis of X-ray appearance
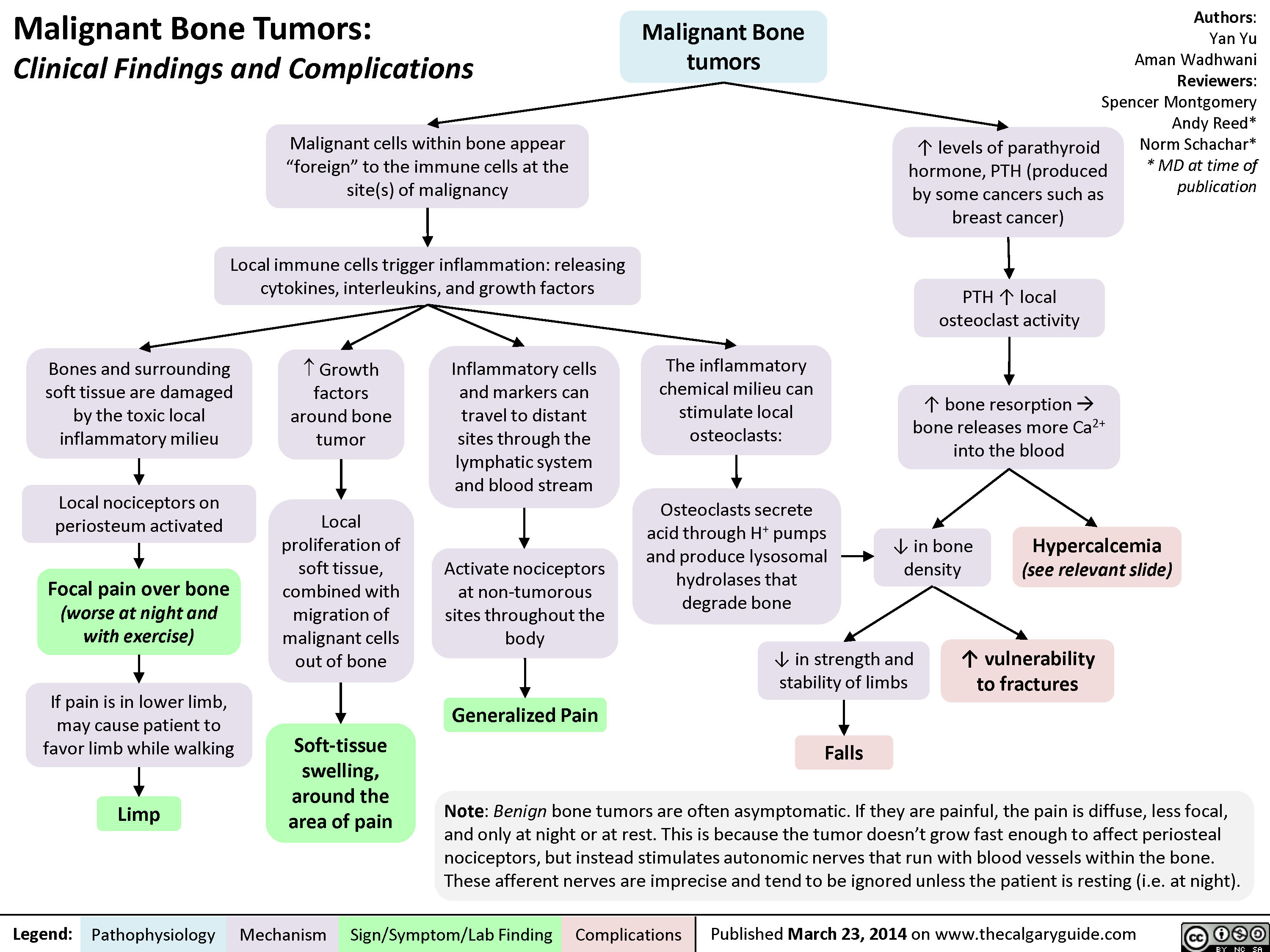
MSK tumors complications
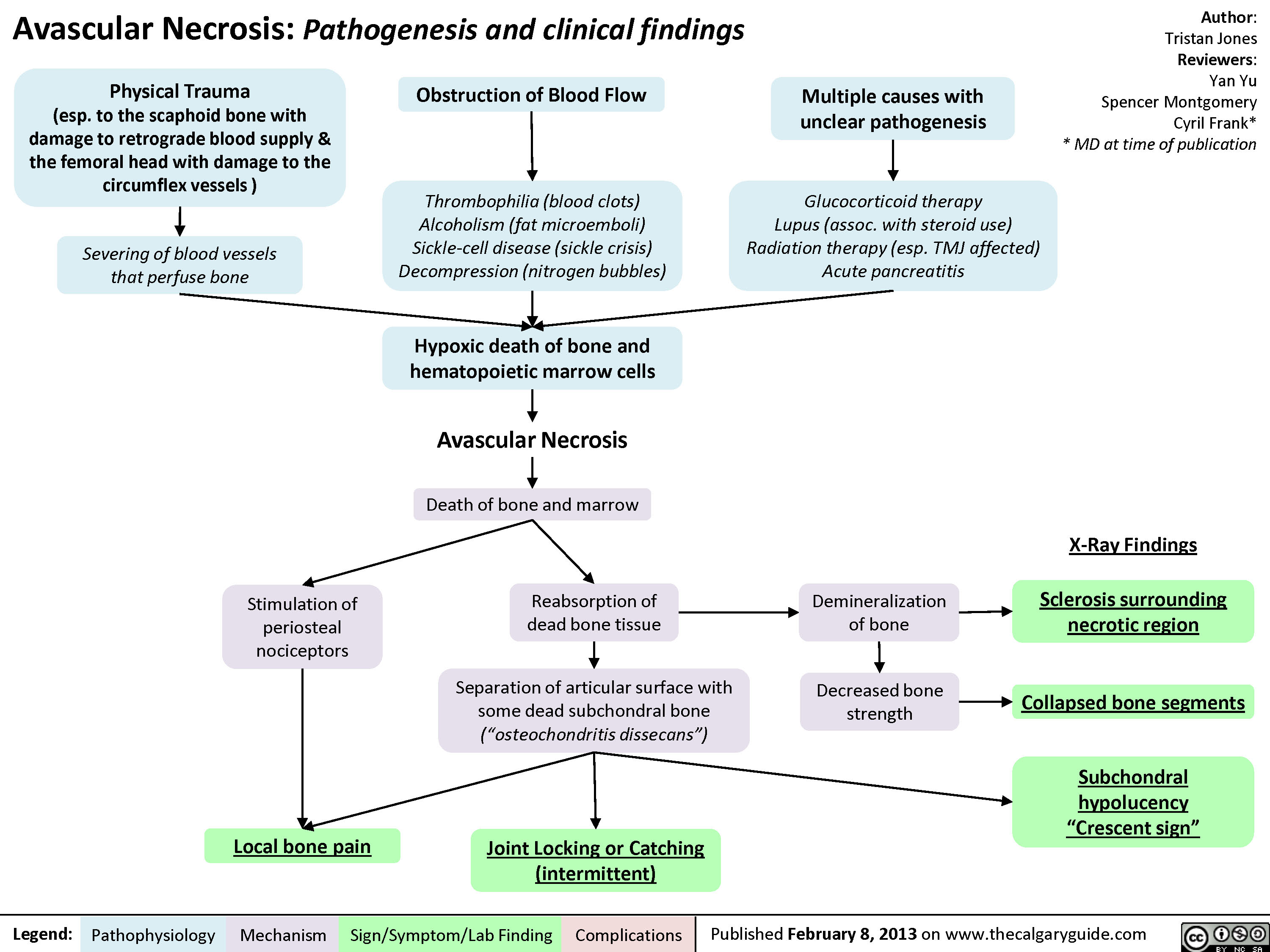
Avascular Necrosis - Pathogenesis and Clinical Findings
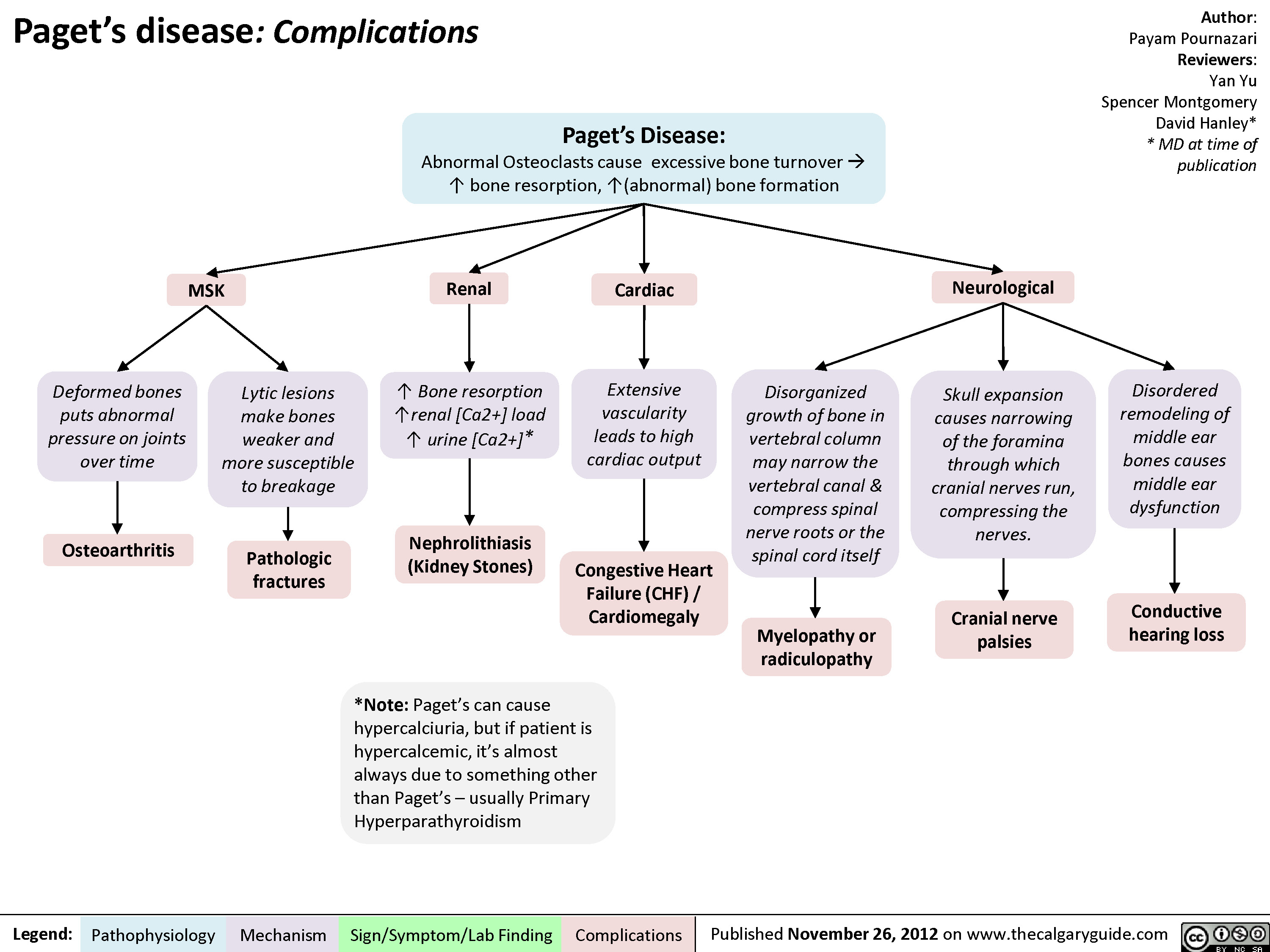
Pagets Disease Complications
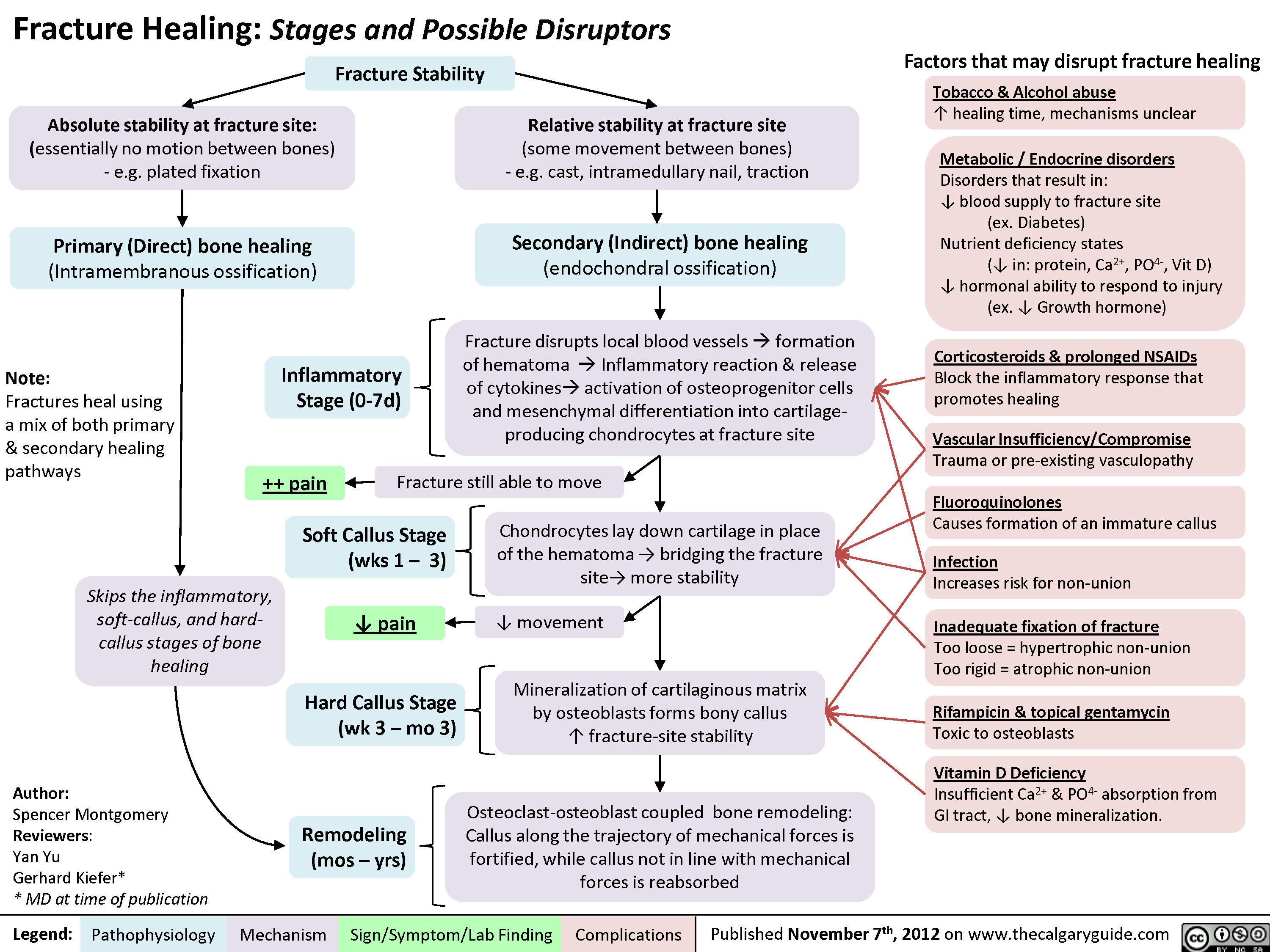
Fracture Healing (and disruptors of this process)
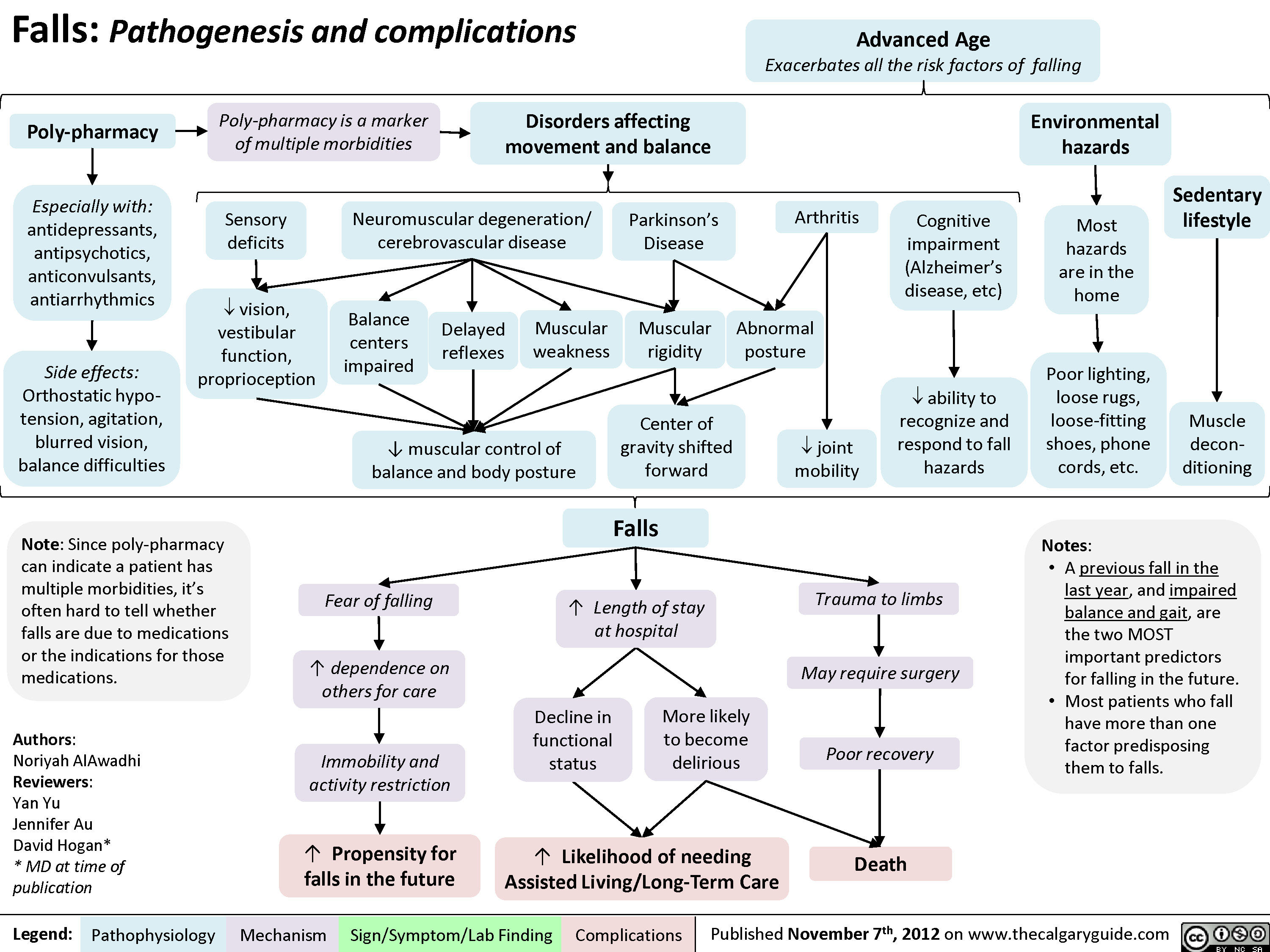
Falls
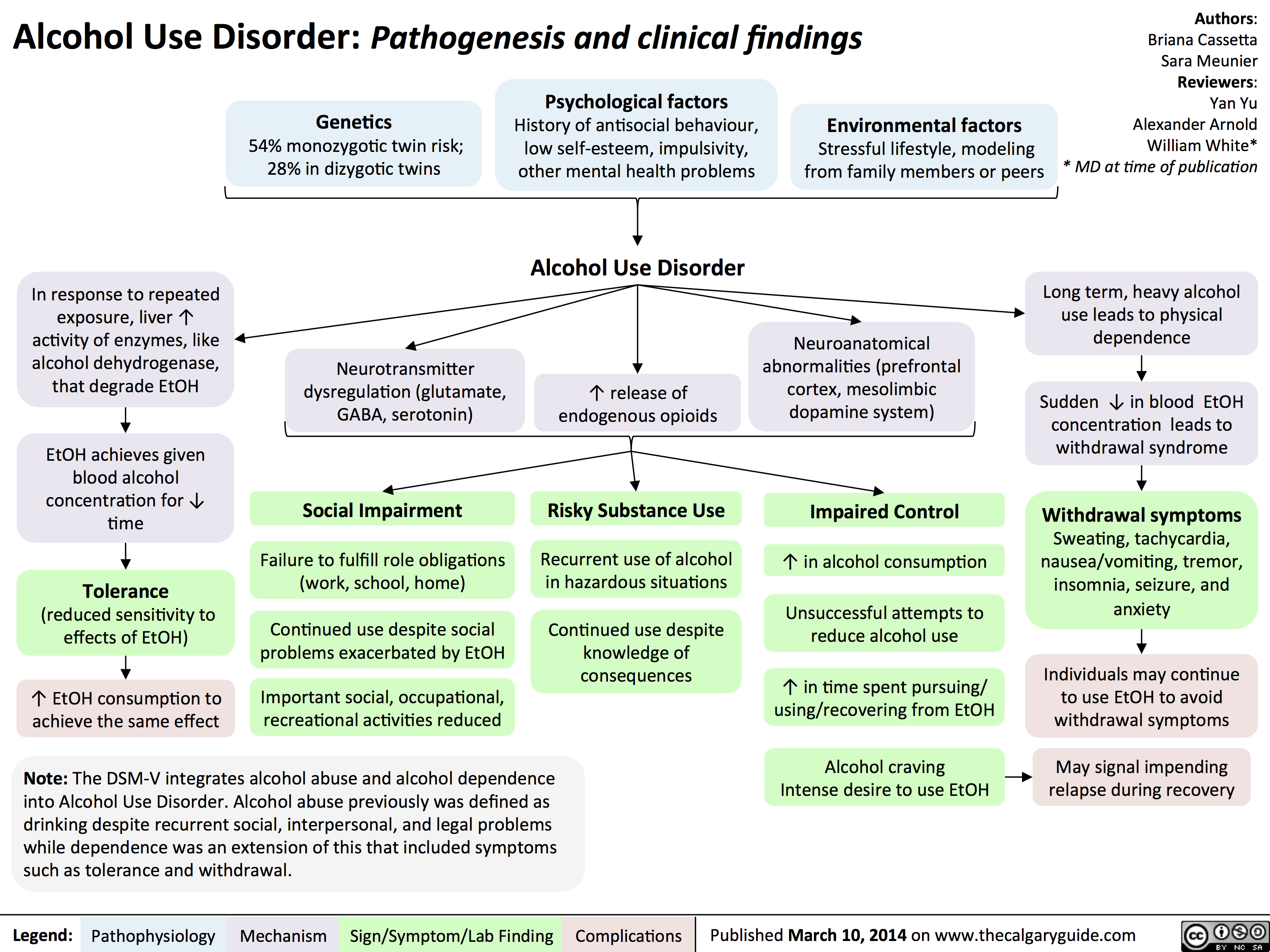
Alcohol Use Disorder
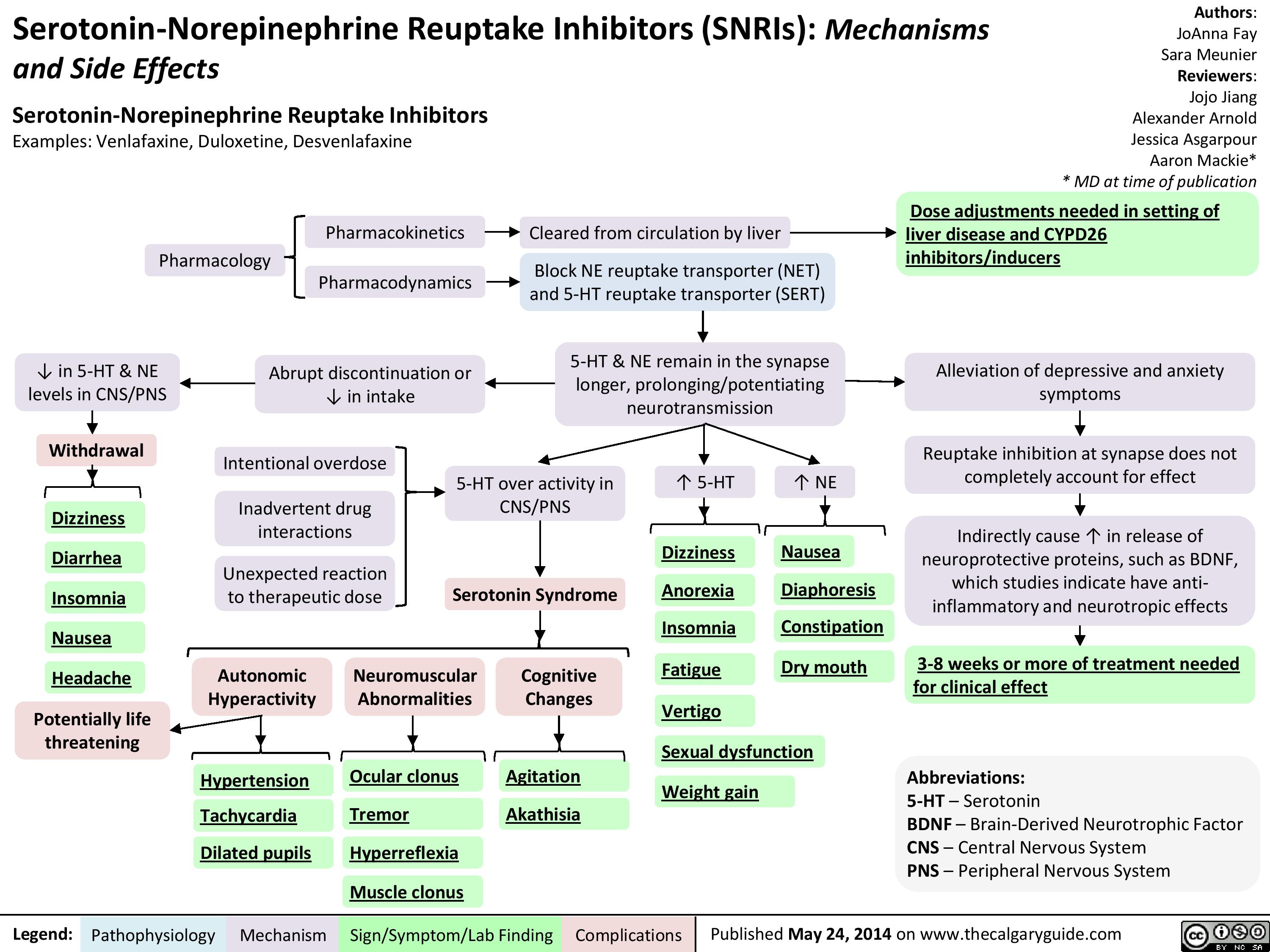
SNRIs
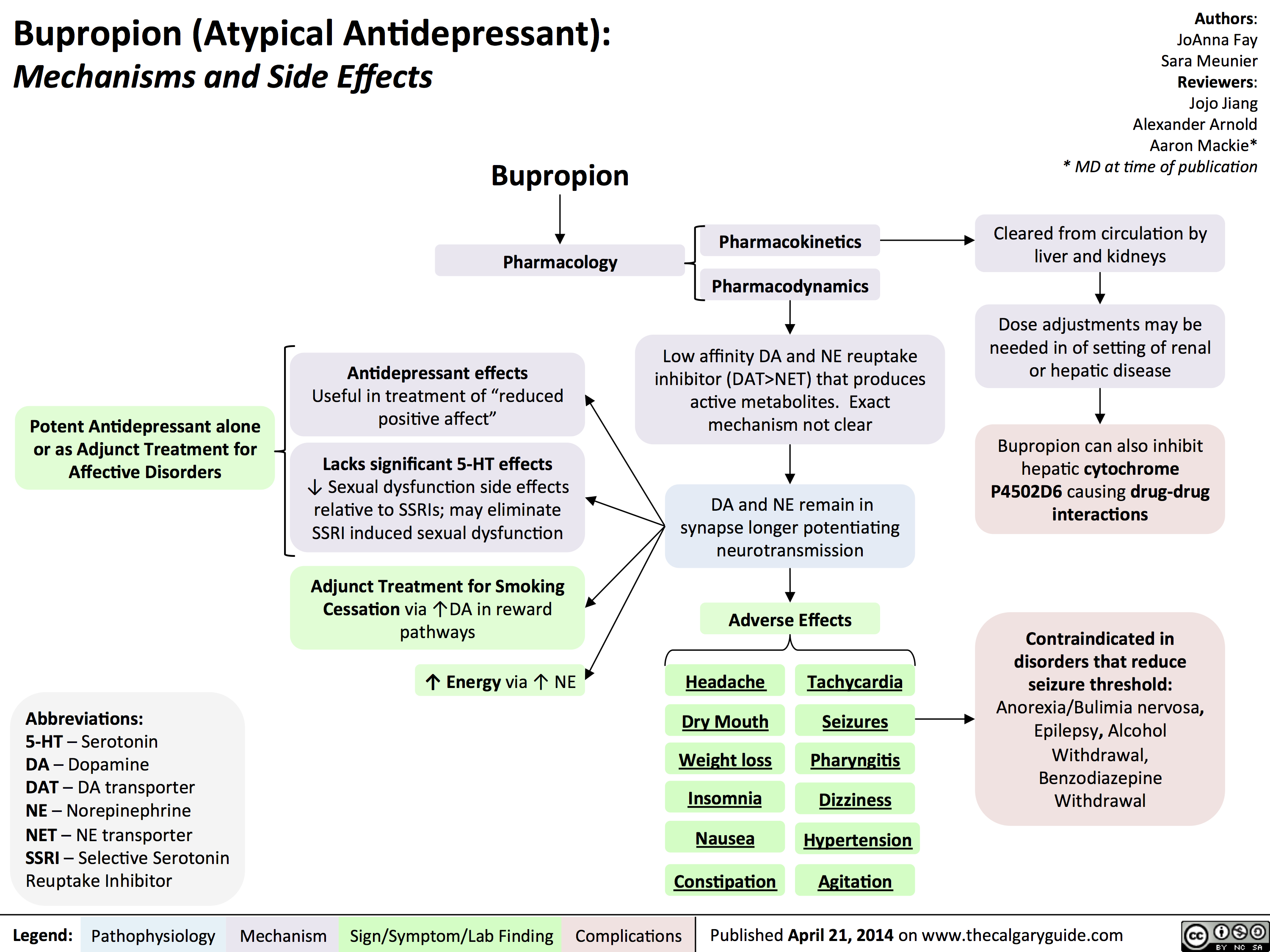
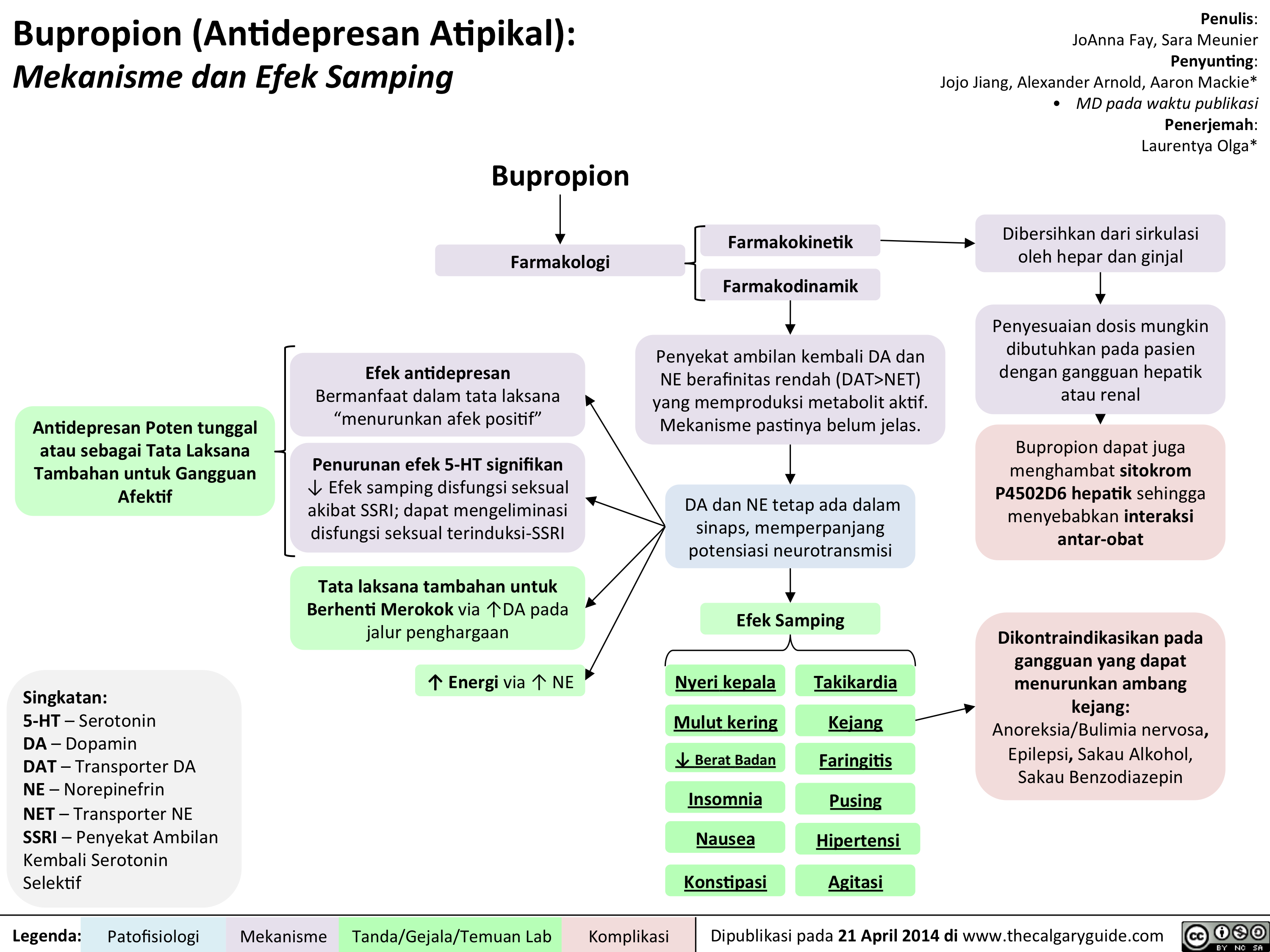
Bupropion
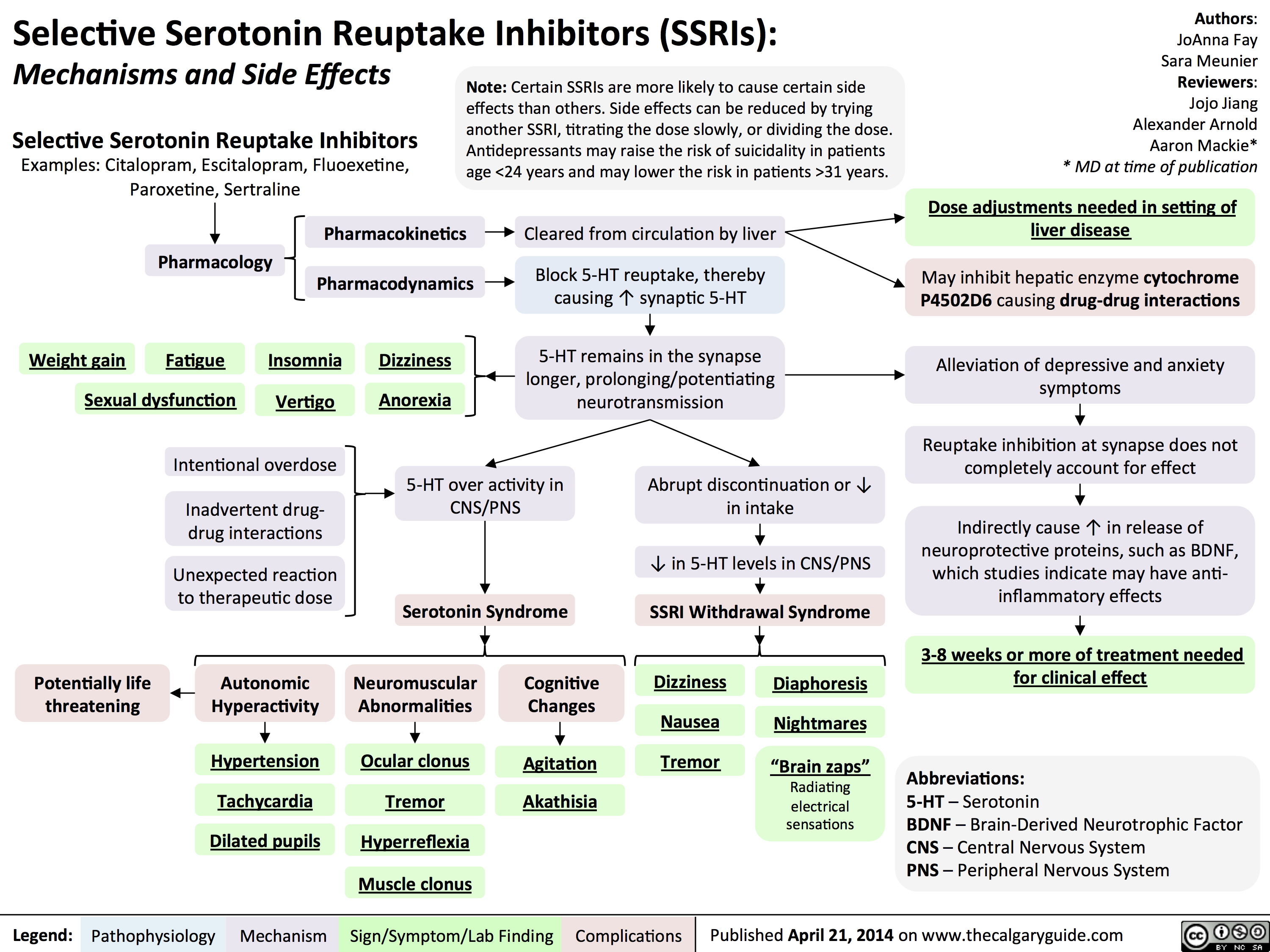
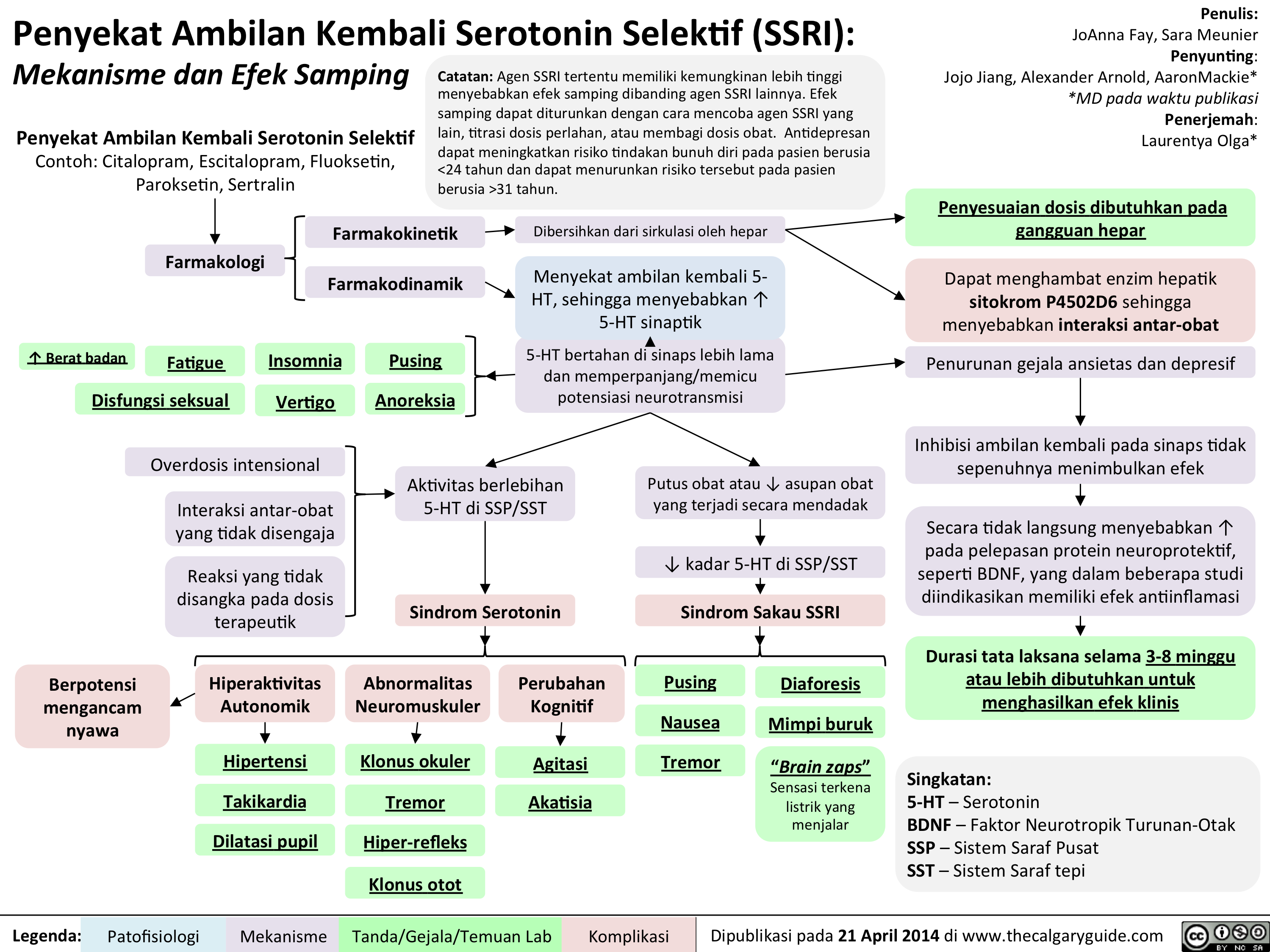
SSRIs
BipolarDisorder

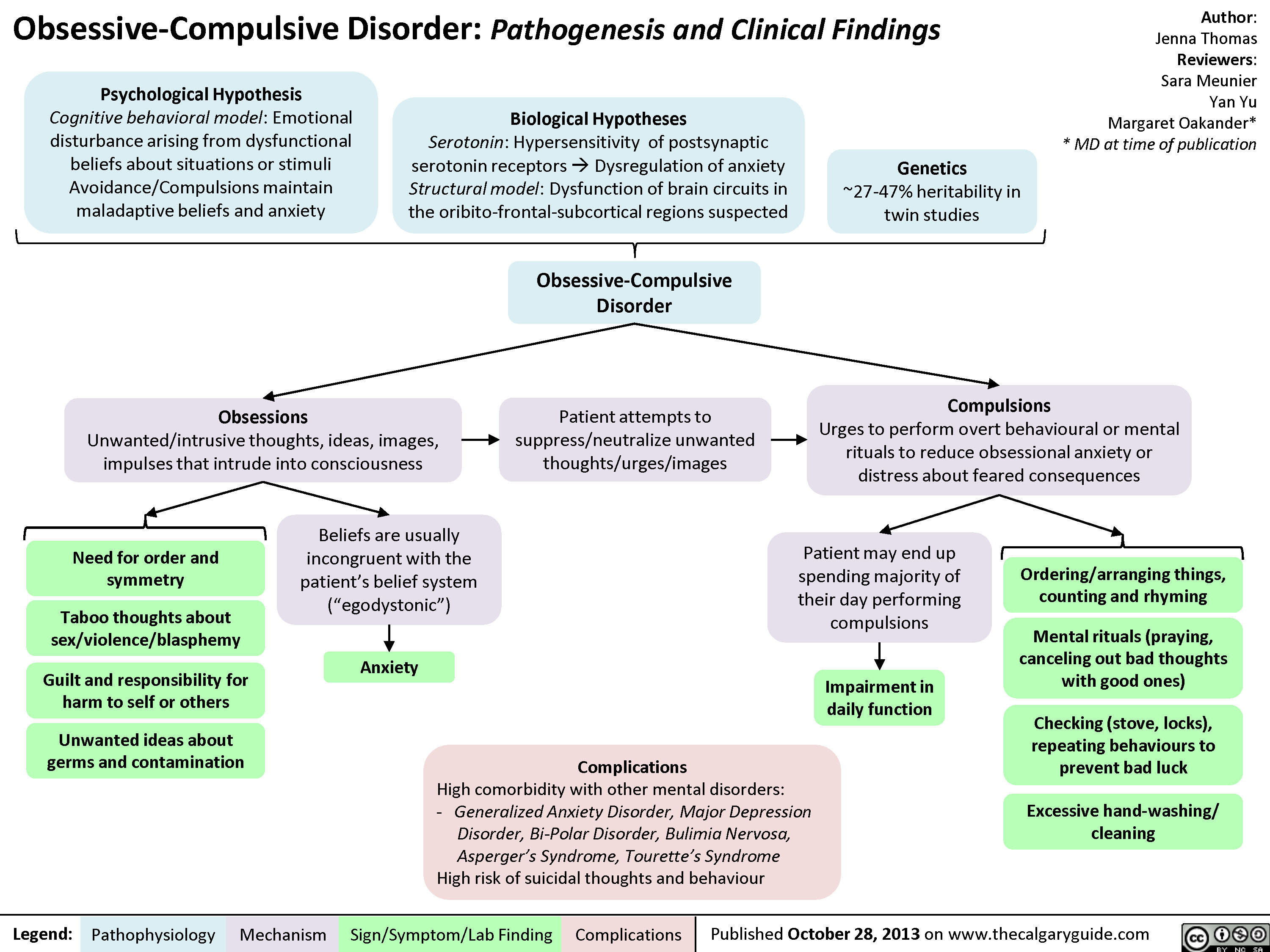
Obsessive Compulsive Disorder OCD
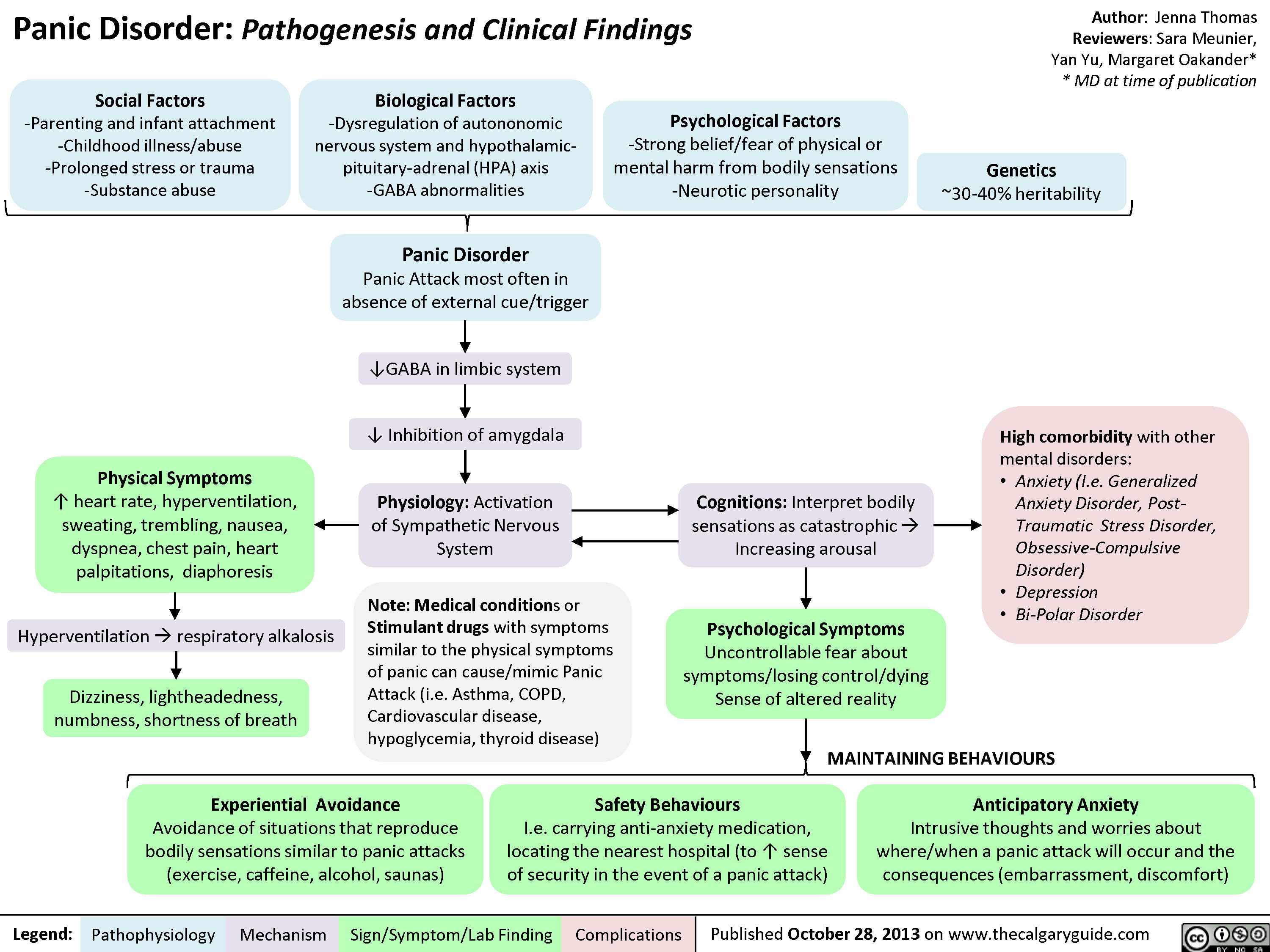
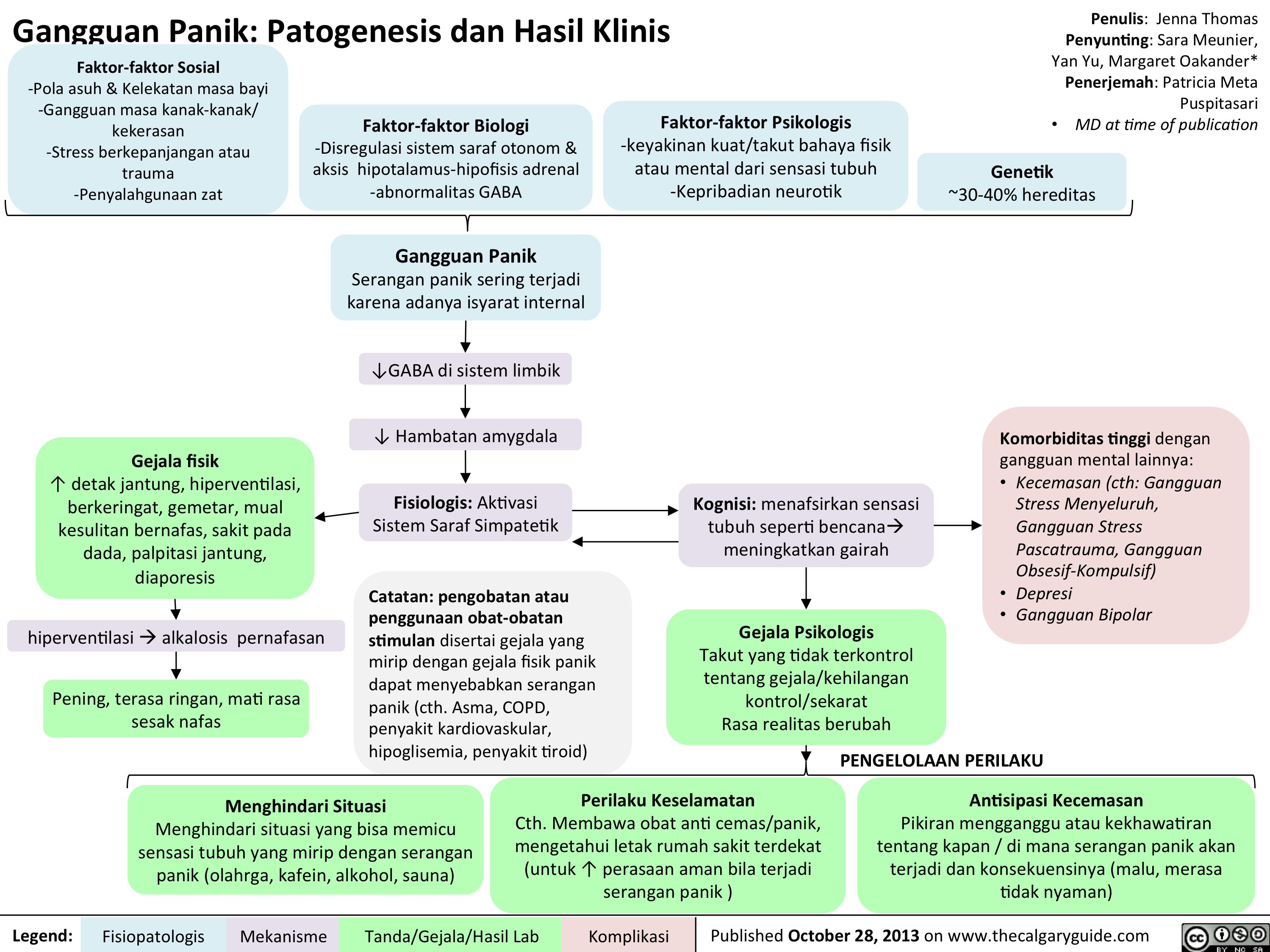
Panic Disorder
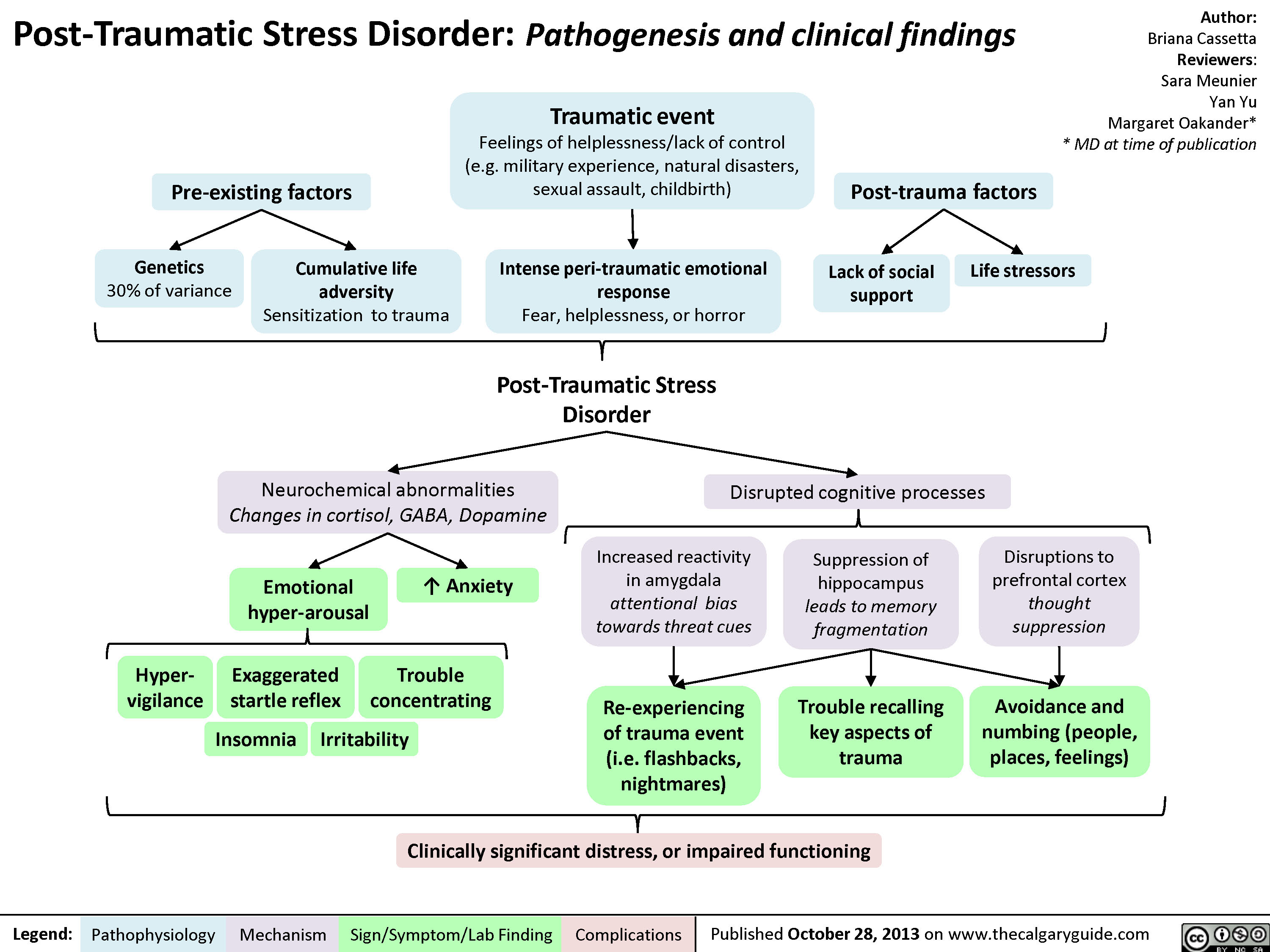
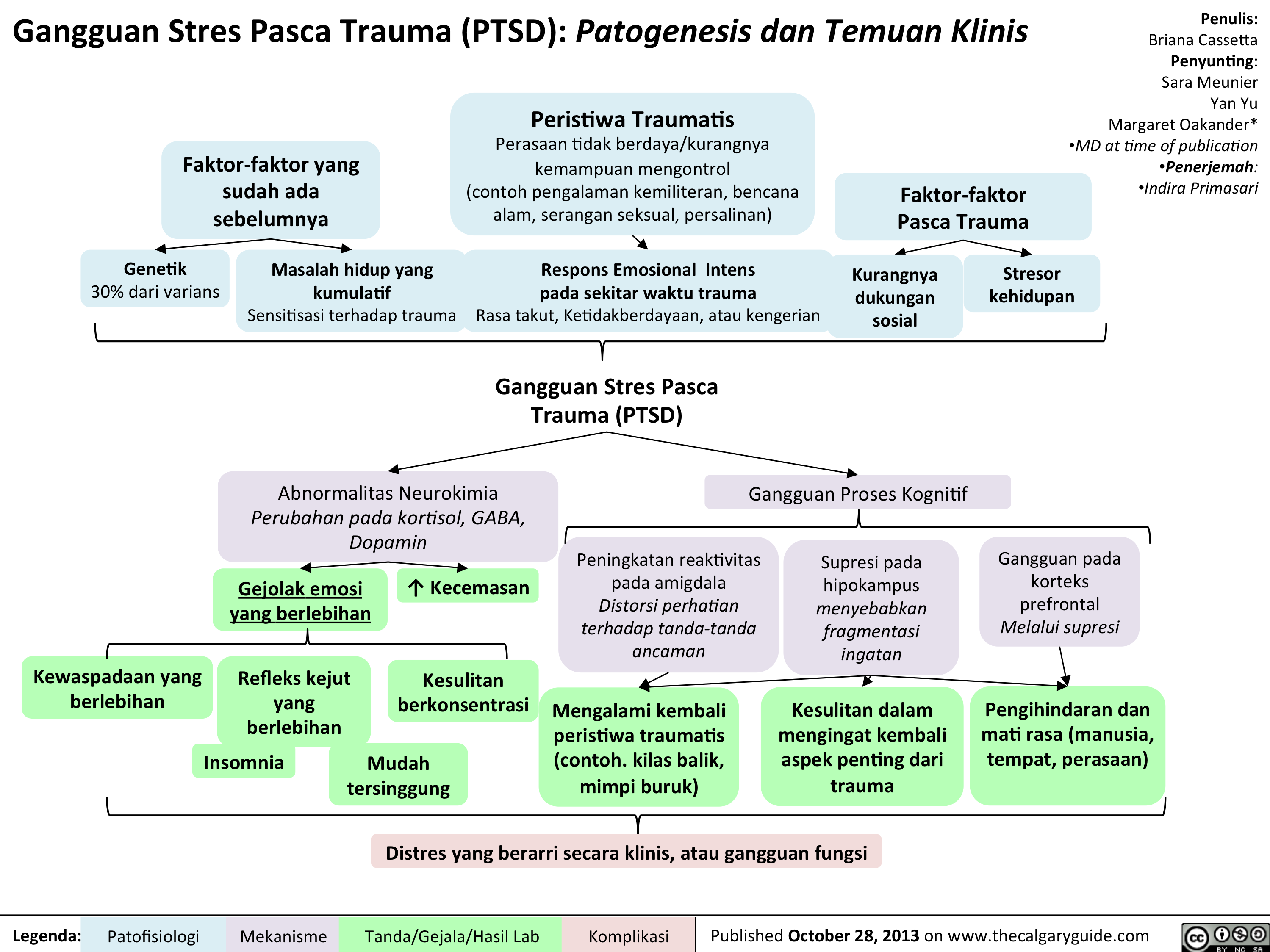
PTSD
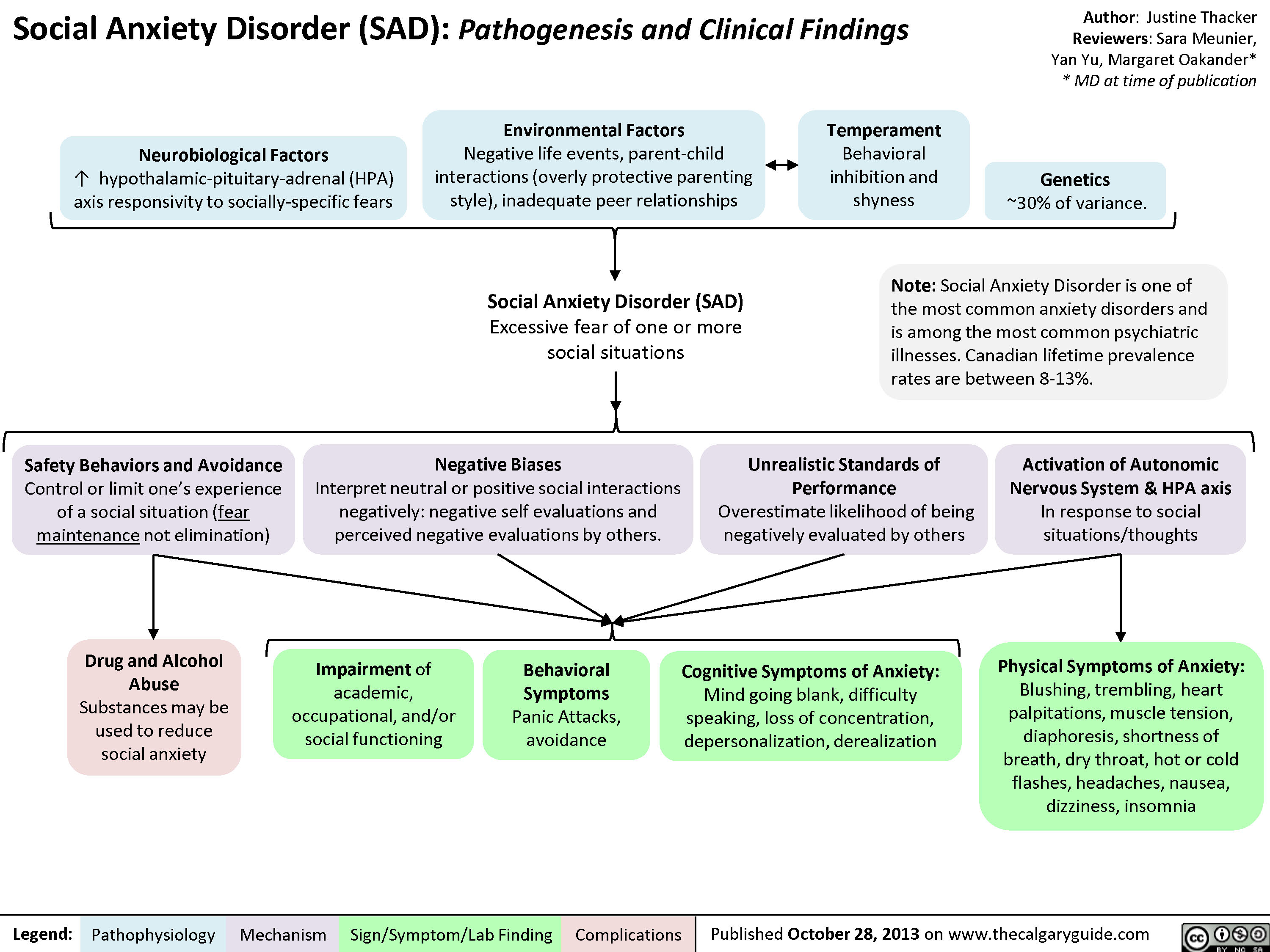
Social Anxiety
Pathogenesis of Anxiety Disorders
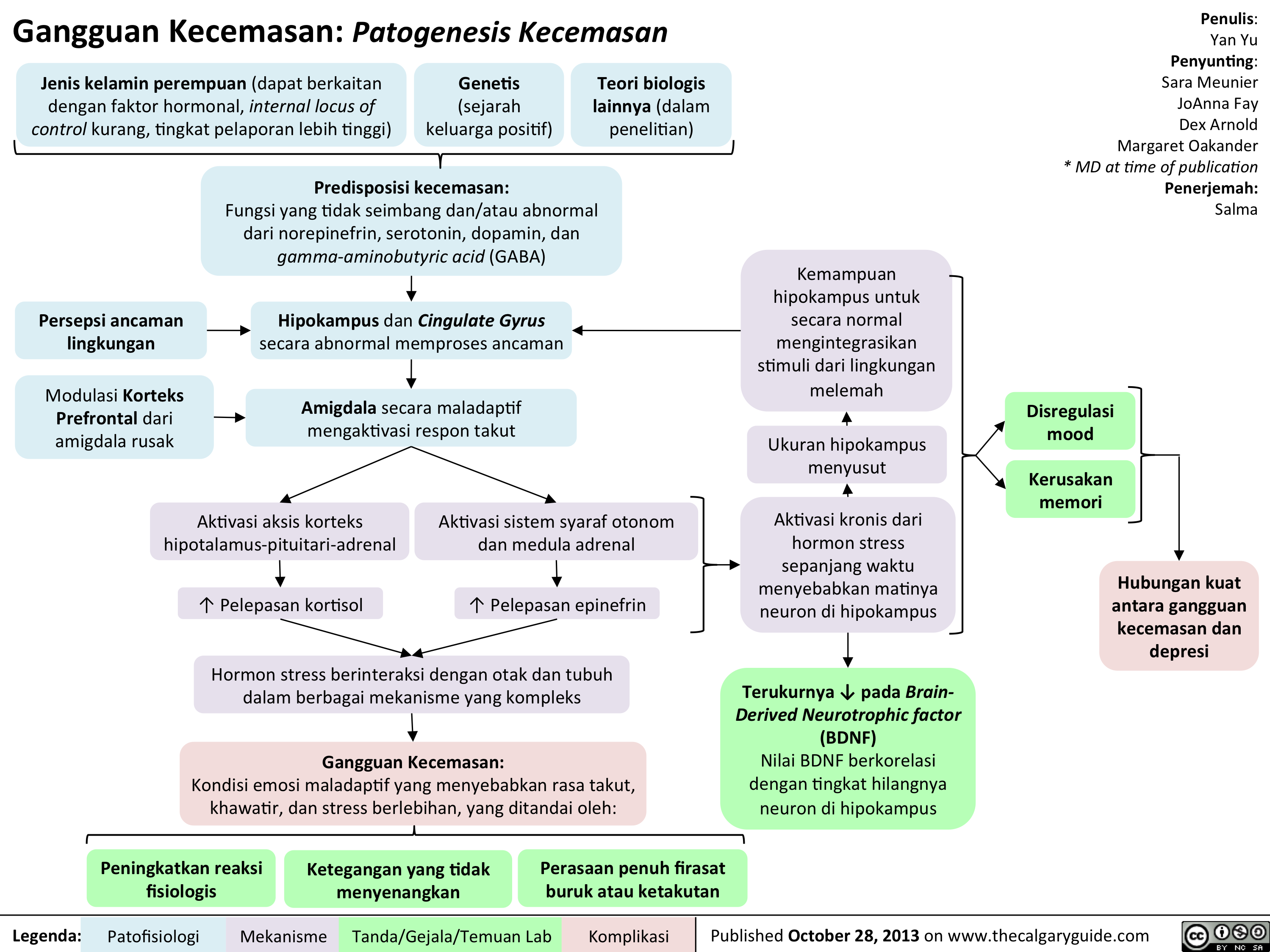
Sense of foreboding or apprehensionActivation of hypothalamus-pituitary-adrenal cortex axisPredisposition to anxiety: Imbalance and/or abnormal functioning of norepinephrine, serotonin, dopamine, and gamma-aminobutyric acid (GABA) Female gender (may be related to hormonal factors, less internal locus of control, greater reporting rates)Other biological theories (under investigation)Prefrontal Cortex modulation of amygdala impairedUnpleasant tensionAmygdala maladaptively activates fear response? Cortisol releaseChronic activation of stress hormones over time causes death of neurons in the hippocampusAnxiety Disorders:A maladaptive emotional state causing fear, worry, and excessive stress, characterized by:Perceived environmental threatHippocampus and Cingulate Gyrus abnormally process threatActivation of autonomic nervous system and adrenal medulla? Epinephrine releaseHippocampus shrinks in sizeAbility of hippocampus to normally integrate environmental stimuli is further compromisedMood dysregulationMemory impairmentStrong association between anxiety disorders and depressionMeasurable ? in Brain-Derived Neurotrophic factor (BDNF)BDNF value correlates with the degree of neuronal loss in the hippocampus
97 kB / 173 words")
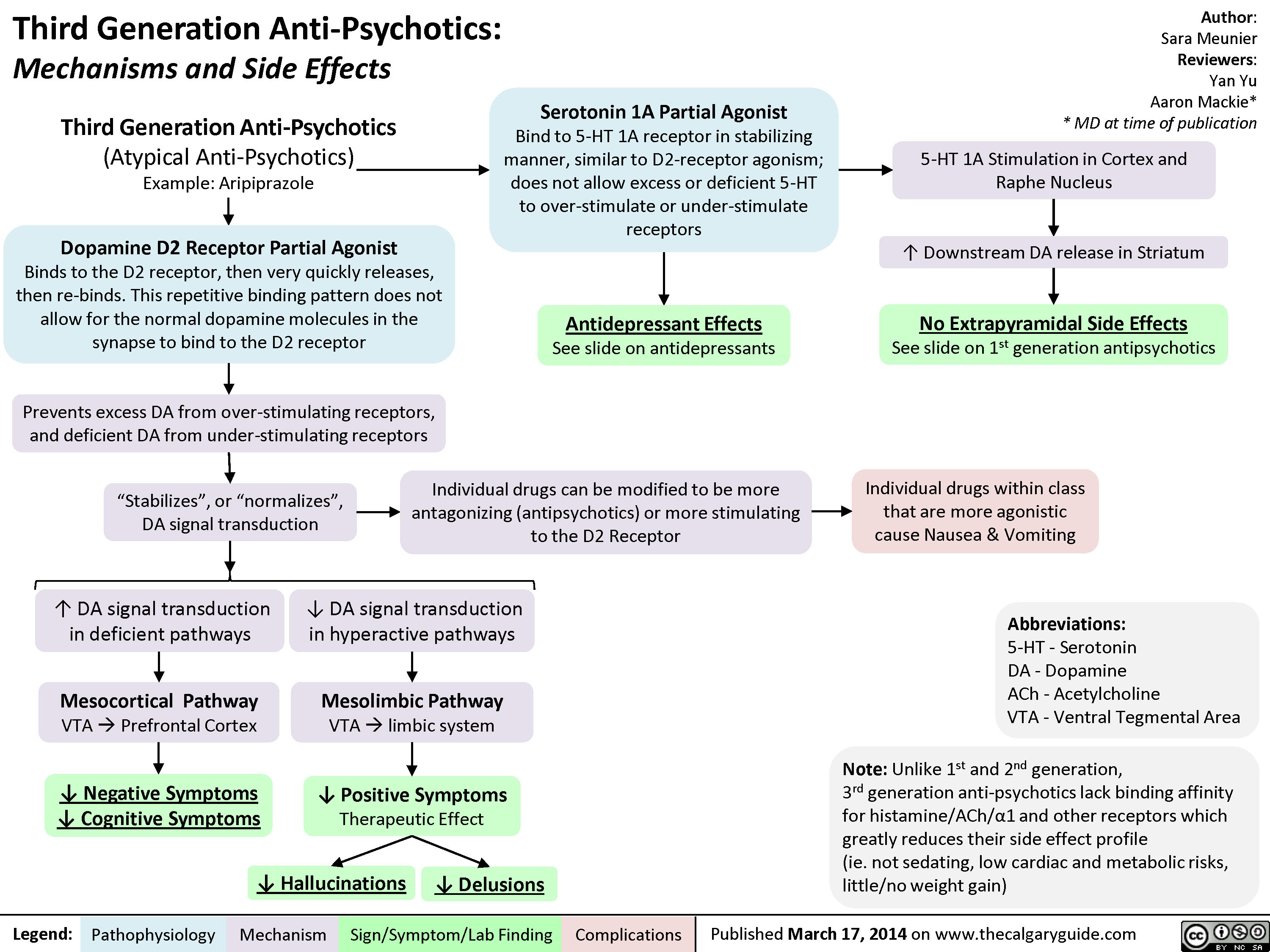
3rd gen anti-psychotics
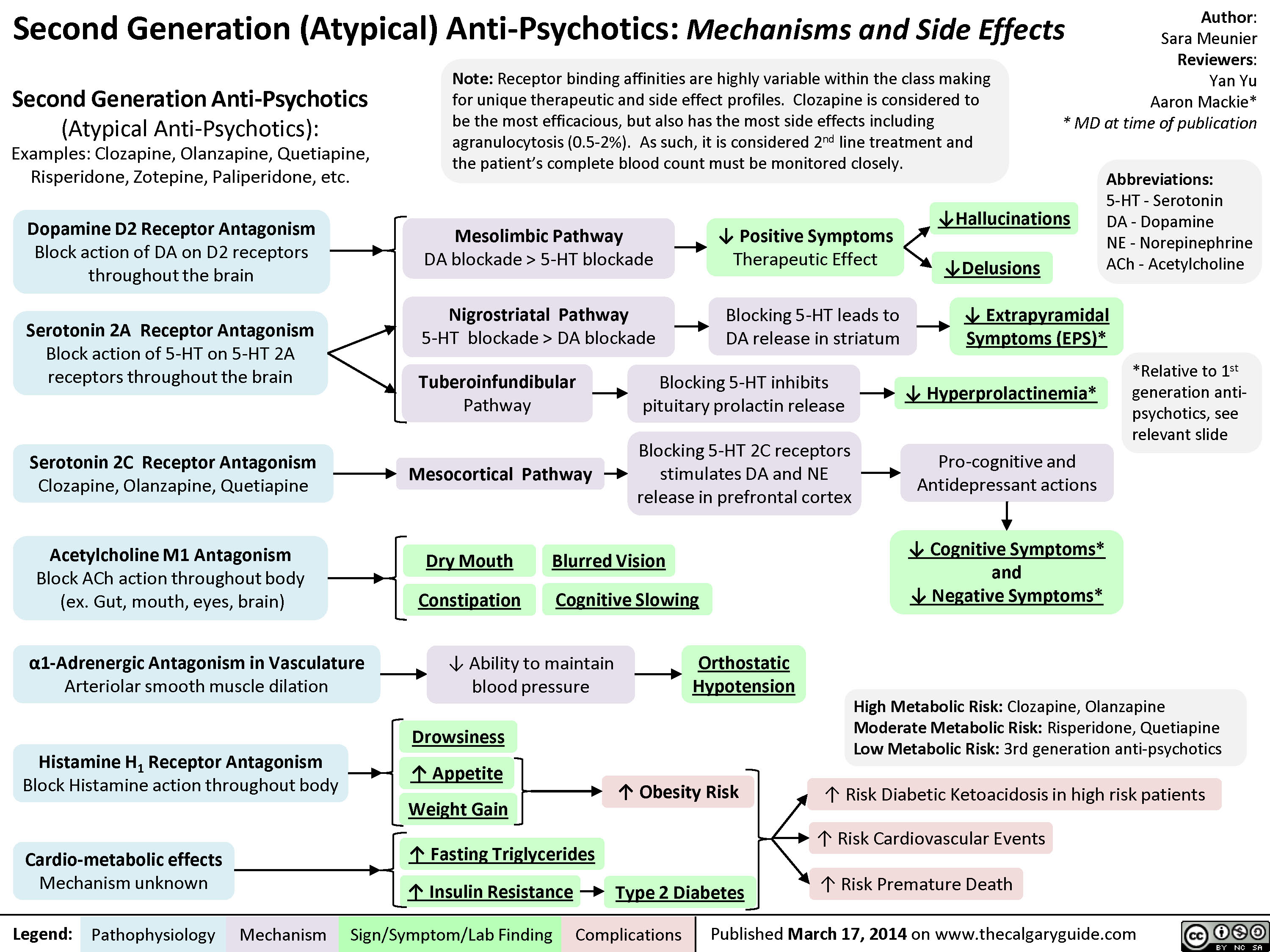
2nd generation antipsychotics
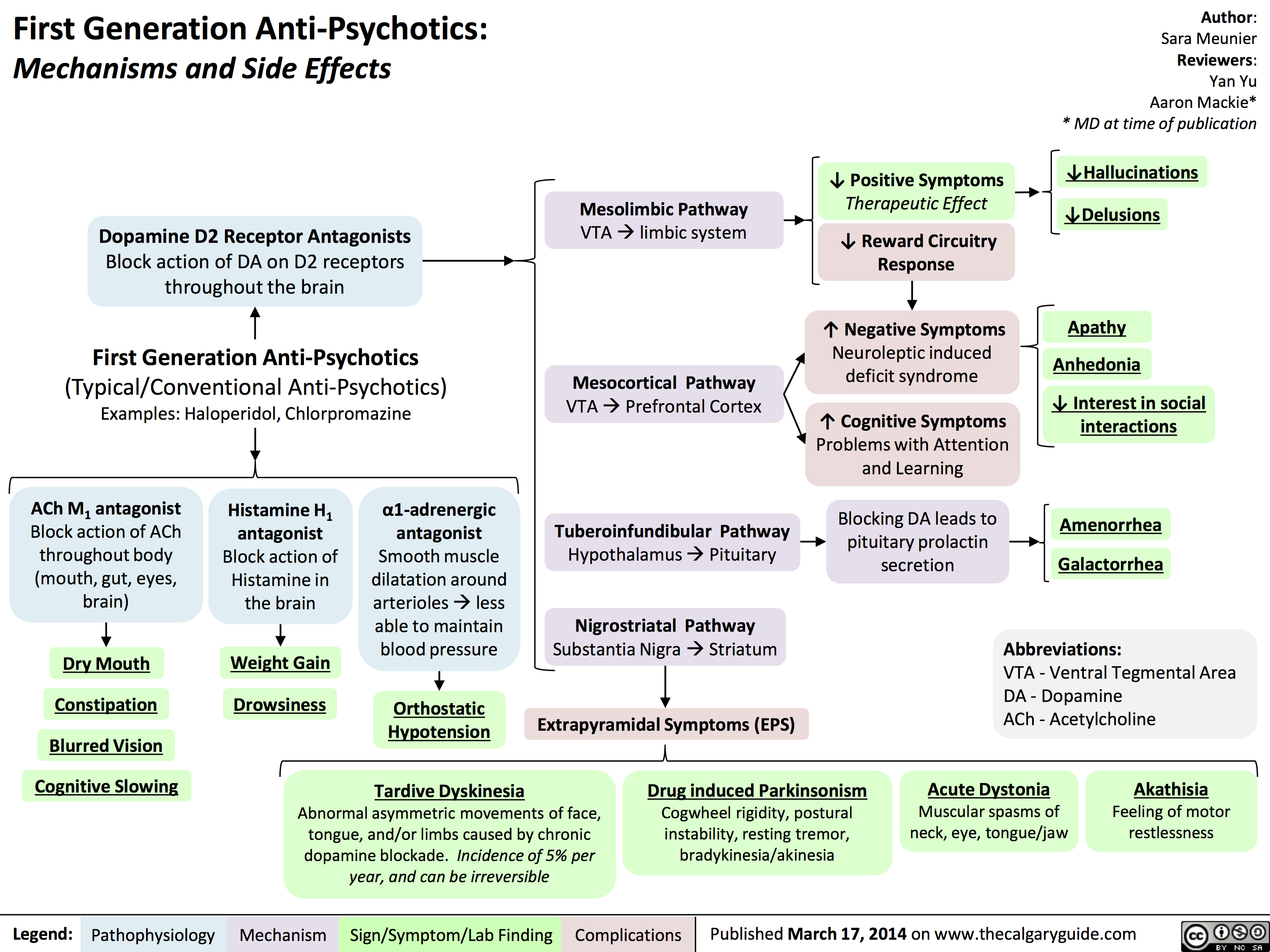
1st gen antipsychotics
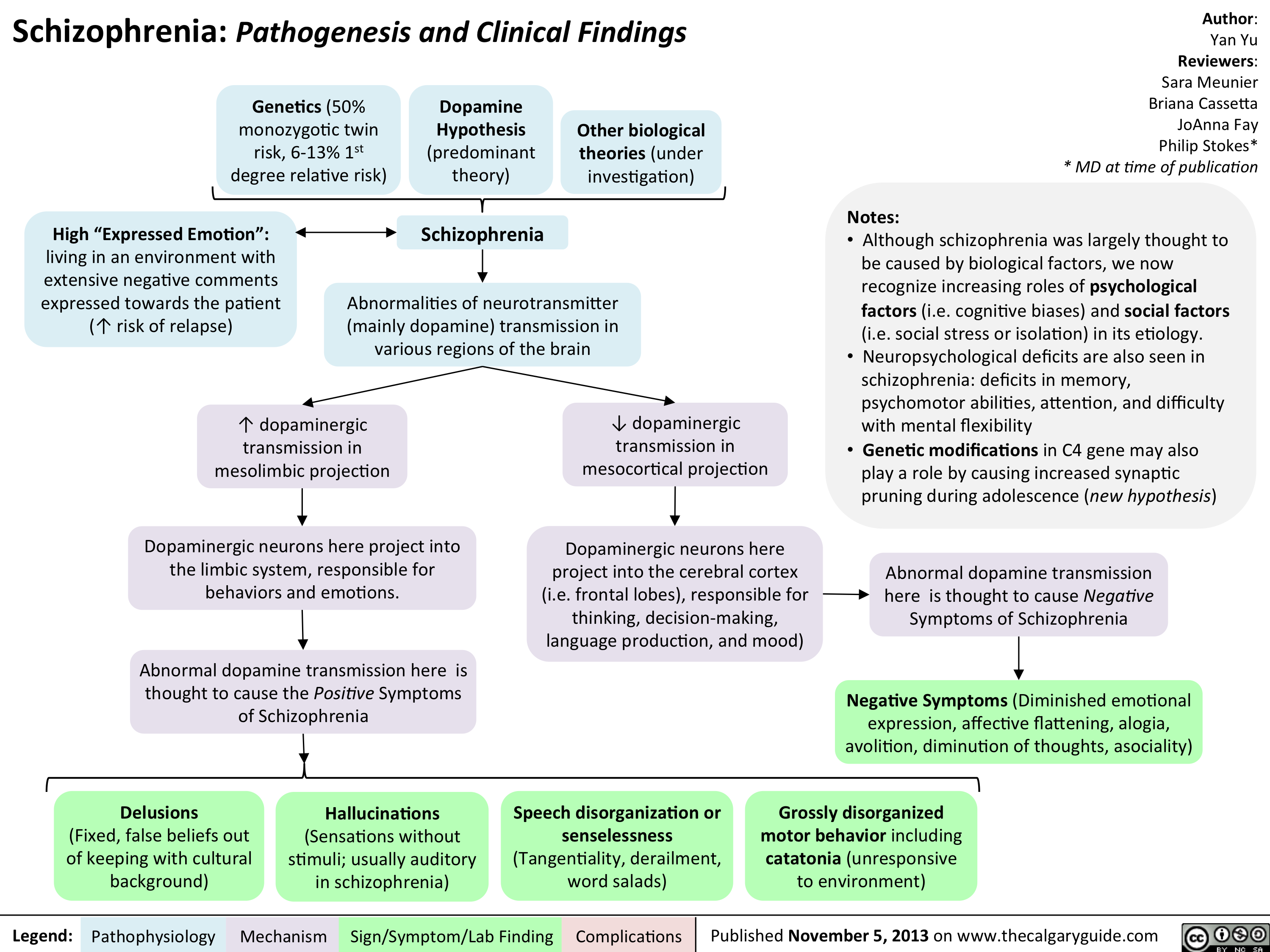
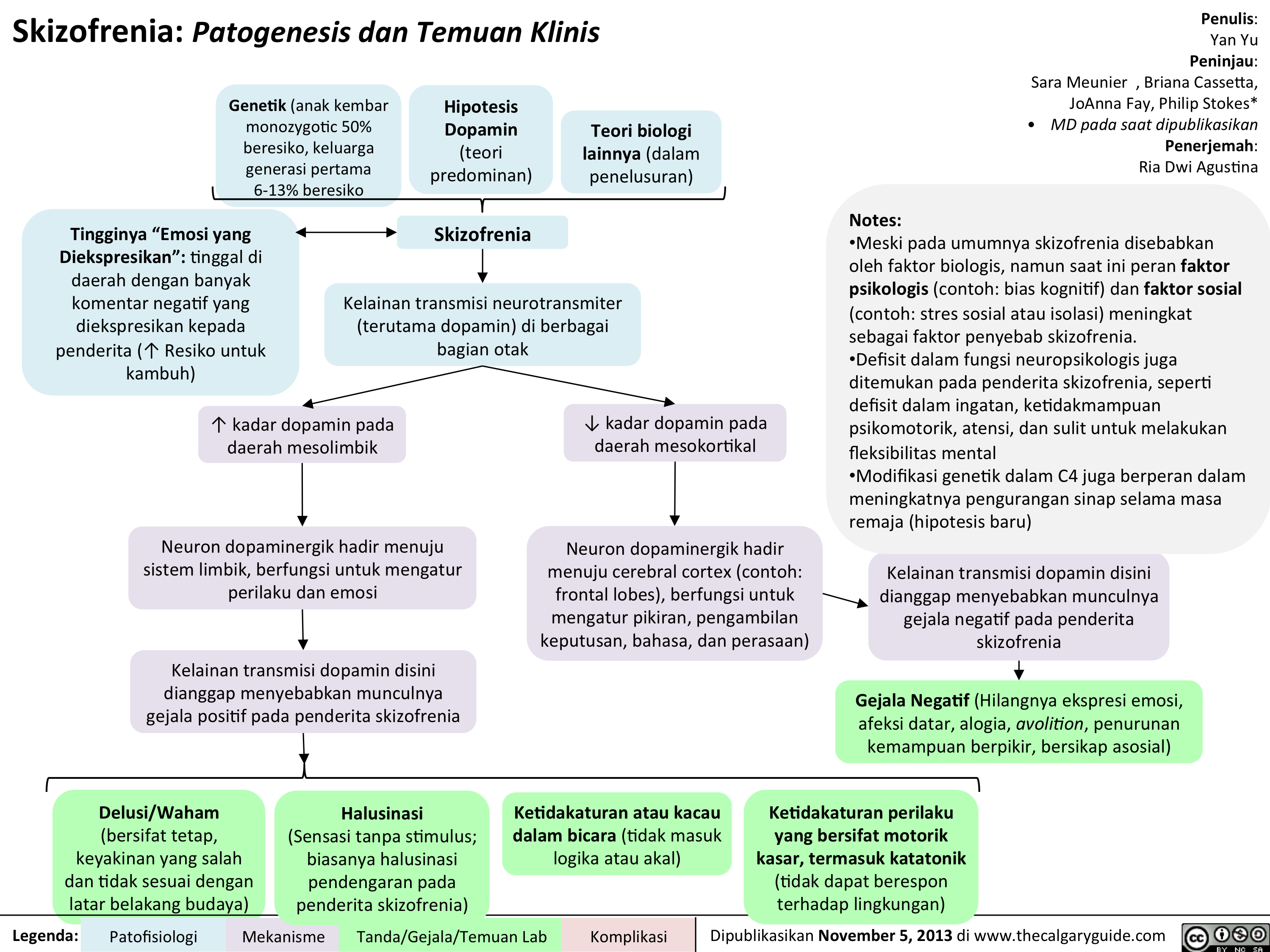
Schizophrenia Pathogenesis and Clinical Findings
? dopaminergic transmission in mesocortical projection? dopaminergic transmission in mesolimbic projection Author: Yan YuReviewers:Sara Meunier Briana CassettaJoAnna FayPhilip Stokes** MD at time of publicationLegend:Published November 5, 2013 on www.thecalgaryguide.comMechanismPathophysiologySign/Symptom/Lab FindingComplicationsGenetics (50% monozygotic twin risk, 6-13% 1st degree relative risk)Speech disorganization or senselessness (Tangentiality, derailment, word salads)Dopaminergic neurons here project into the limbic system, responsible for behaviors and emotions.SchizophreniaDopamine Hypothesis(predominant theory)Other biological theories (under investigation)High")
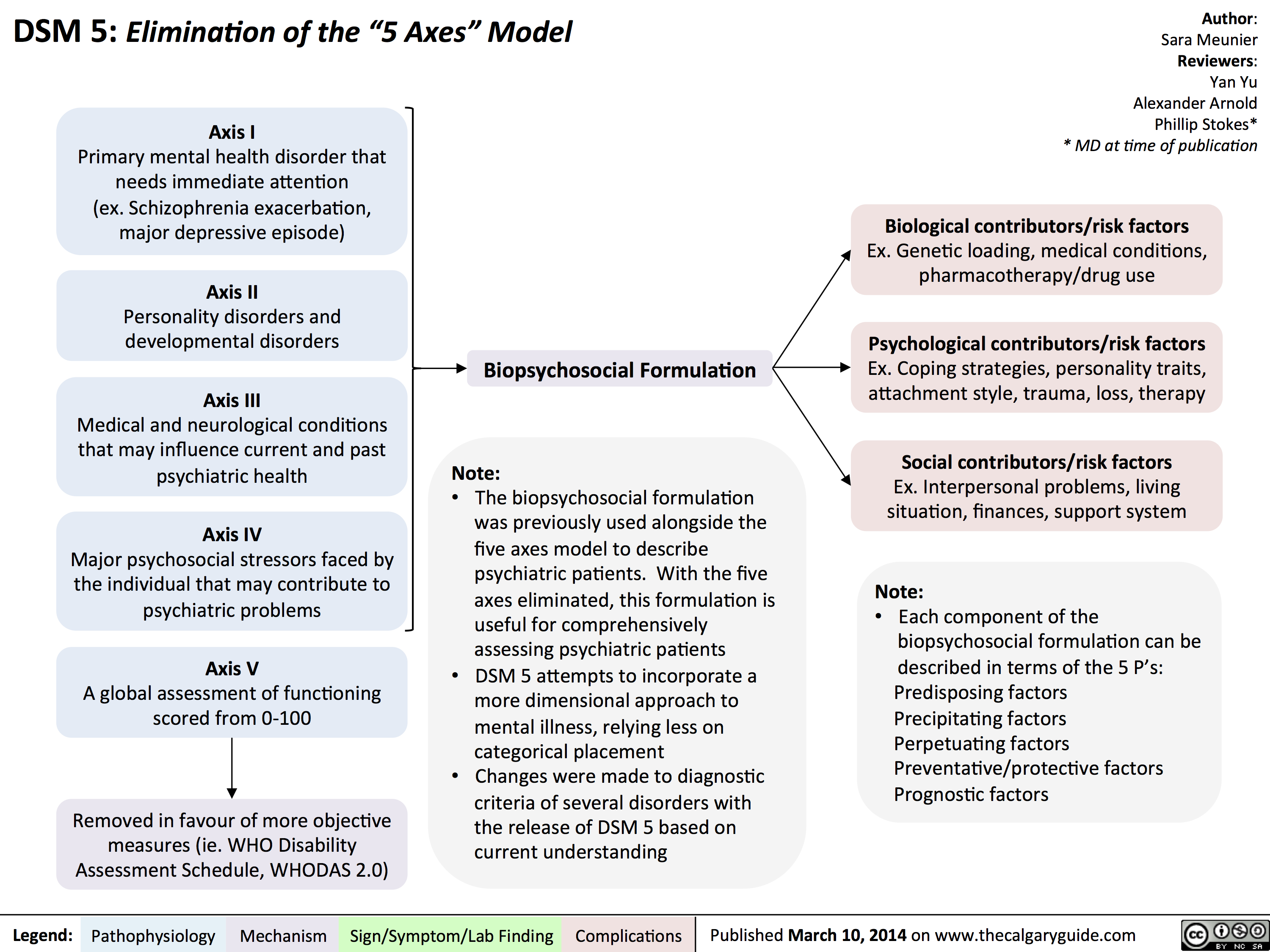
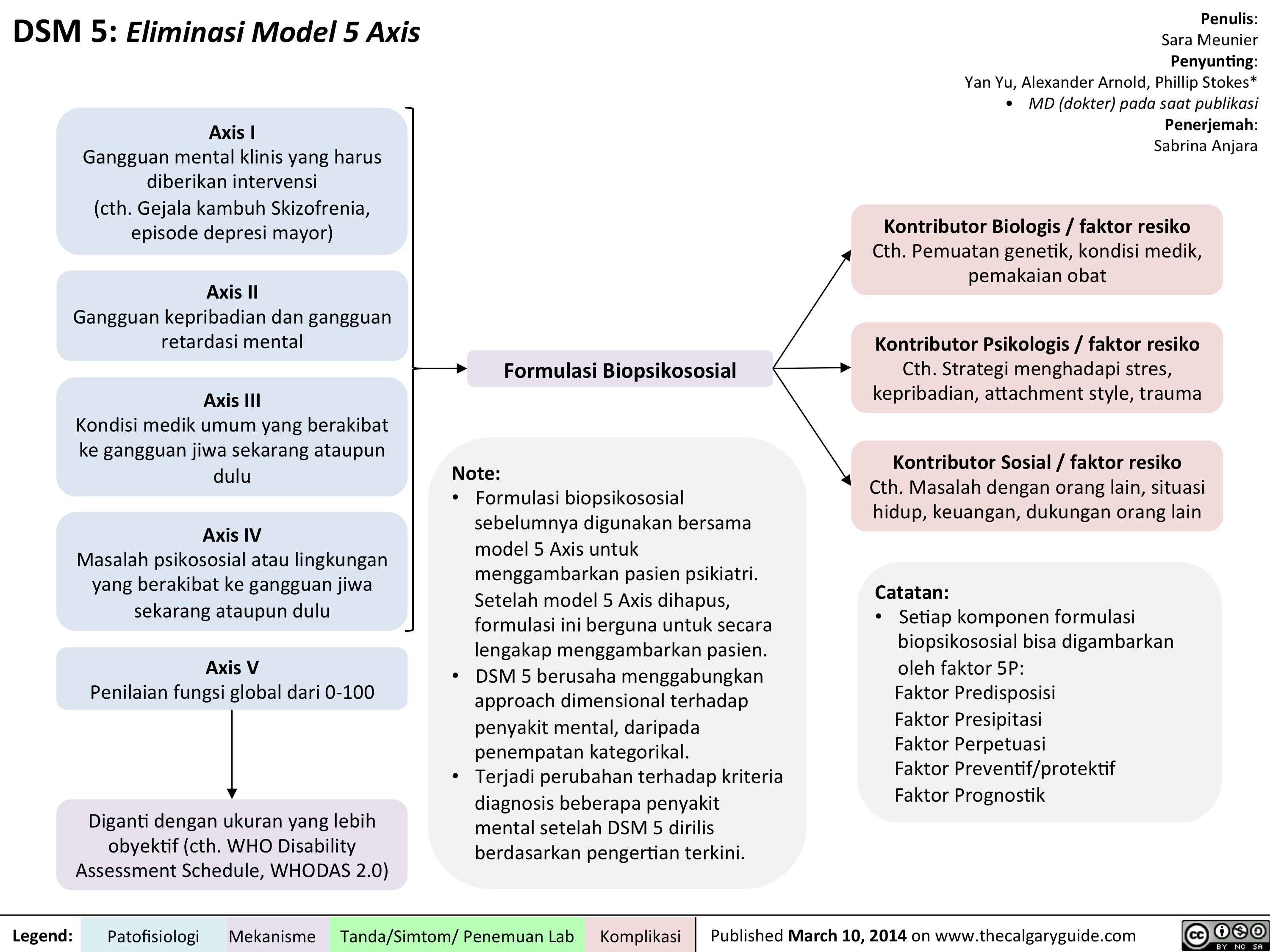
DSM - Axis to Formulation
Complications of Measles Pathogenesis and Clinical Findings
Kawasaki Disease

Physiologic Neonatal Jaundice
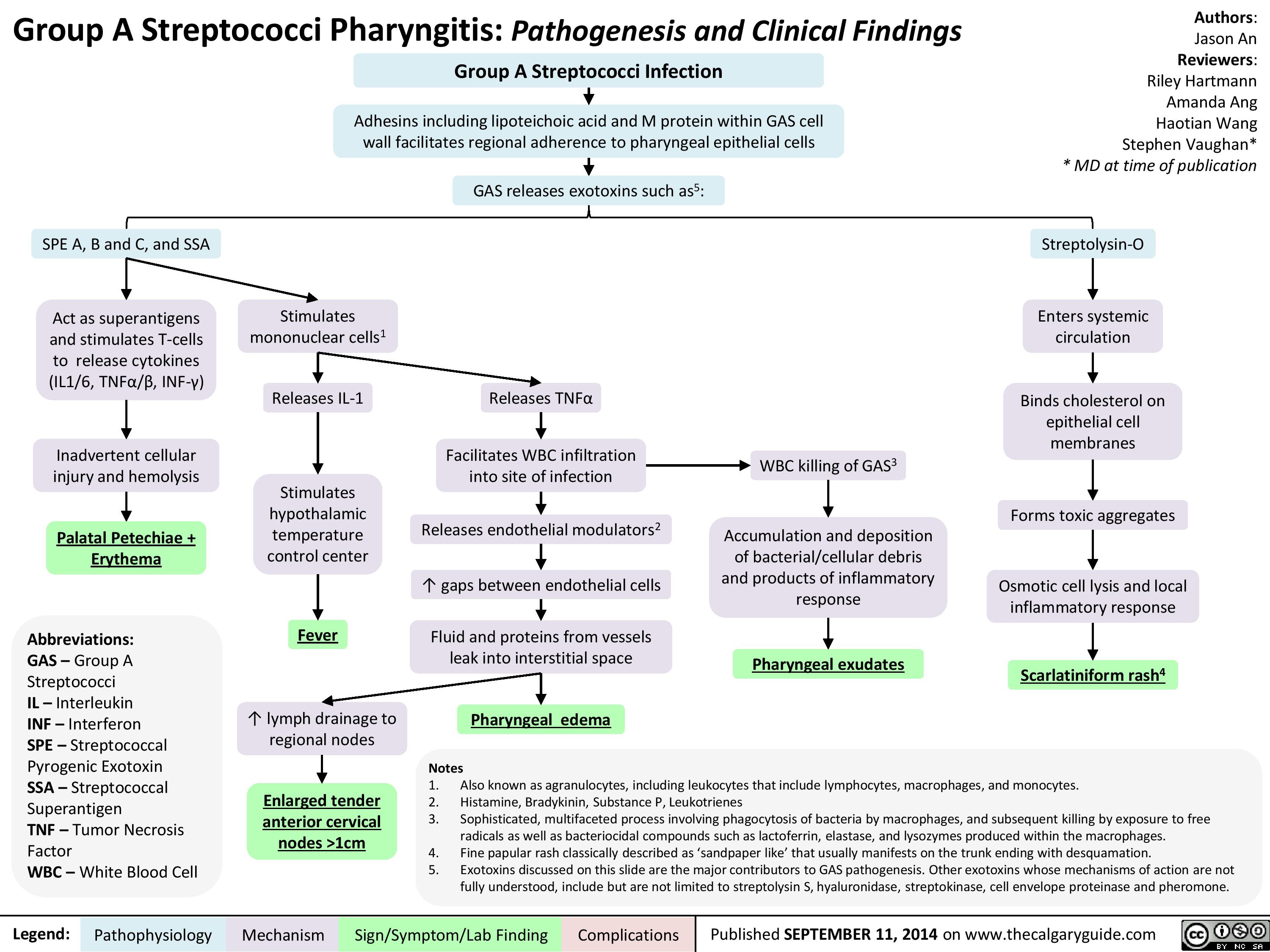
Group A Streptococci Pharyngitis Pathogenesis and Clinical Findings
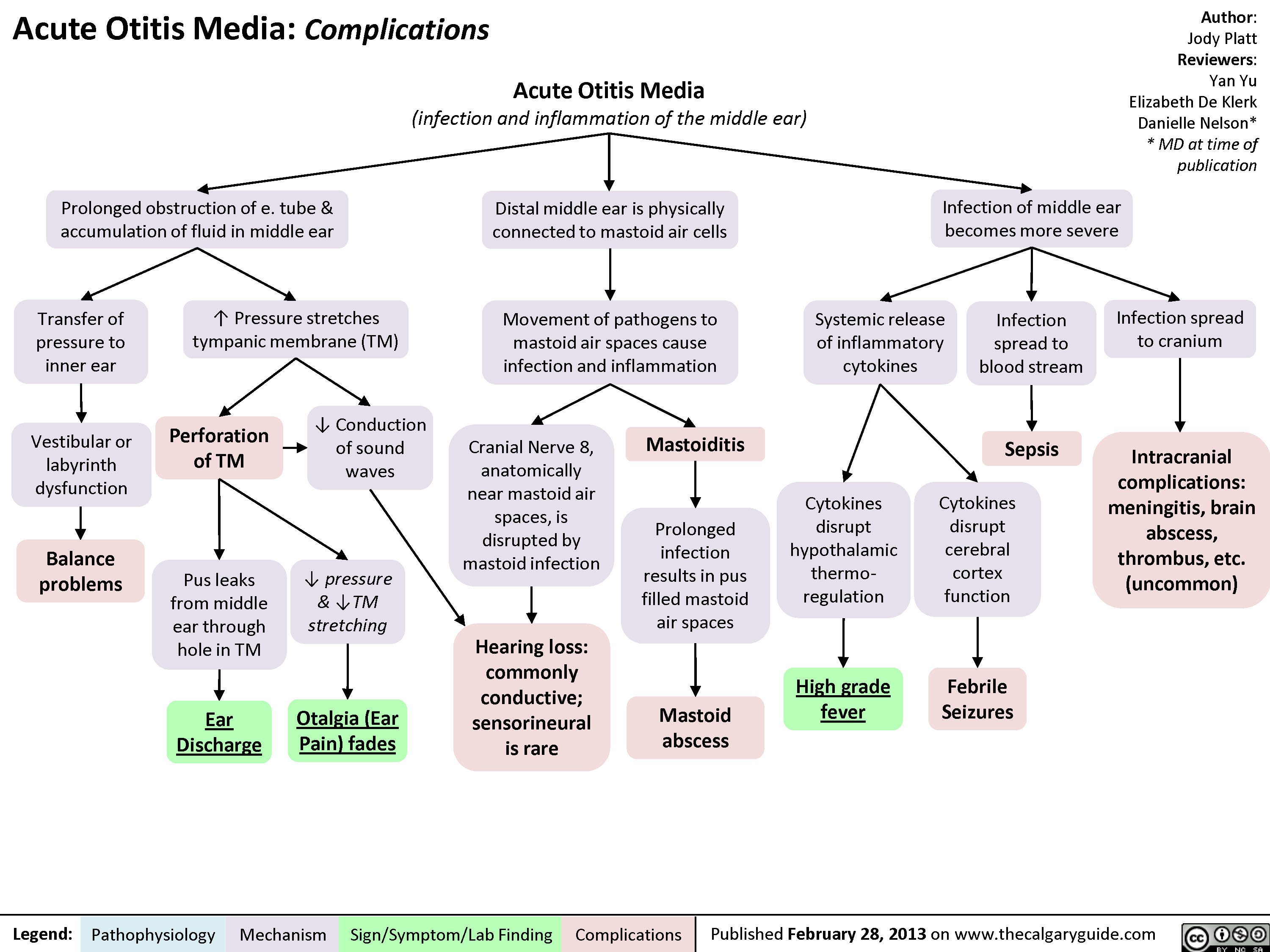
AcuteOtitisComplications
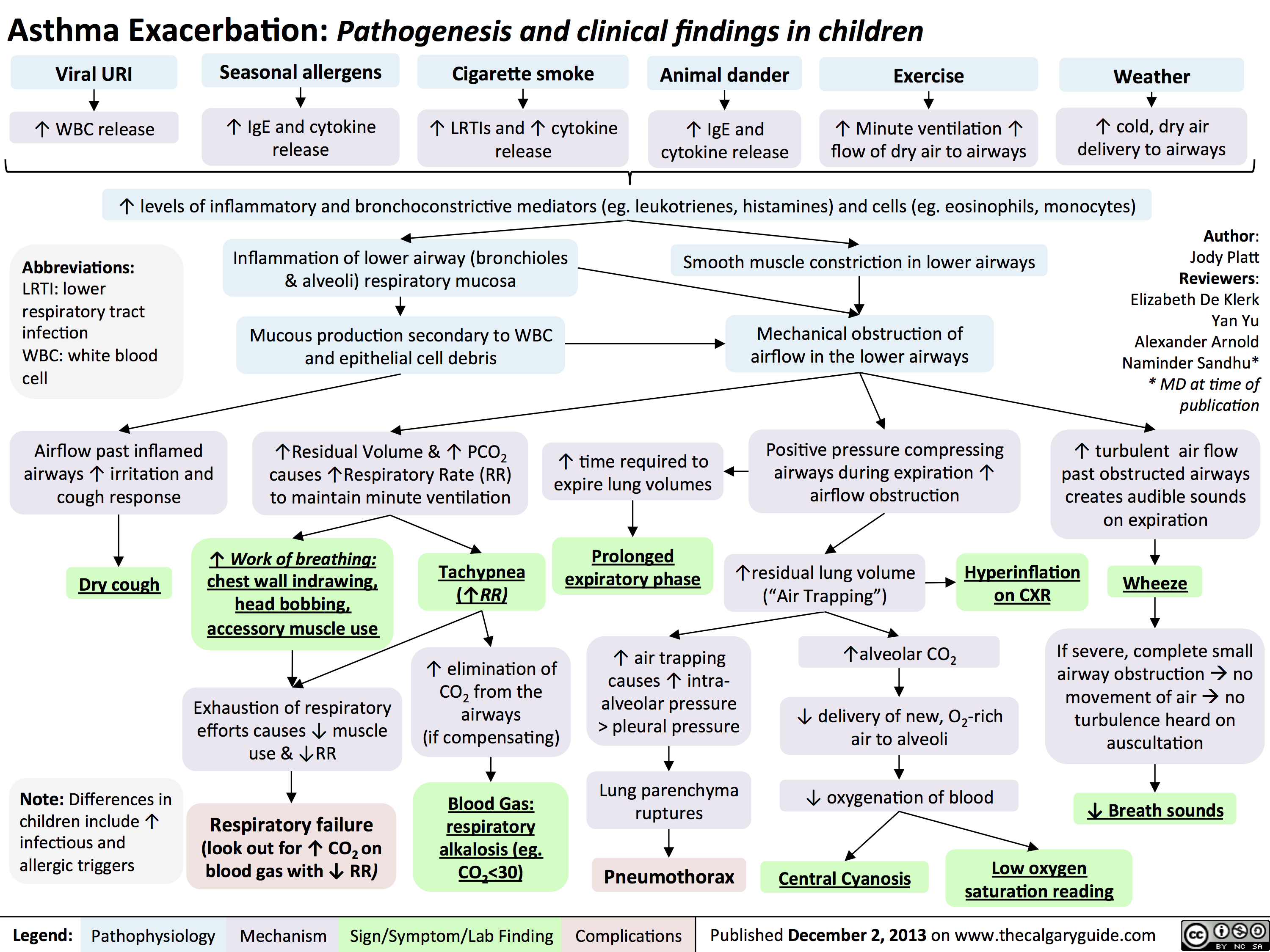
Asthma Exacerbation - Pathogenesis and Clinical Findings in Children
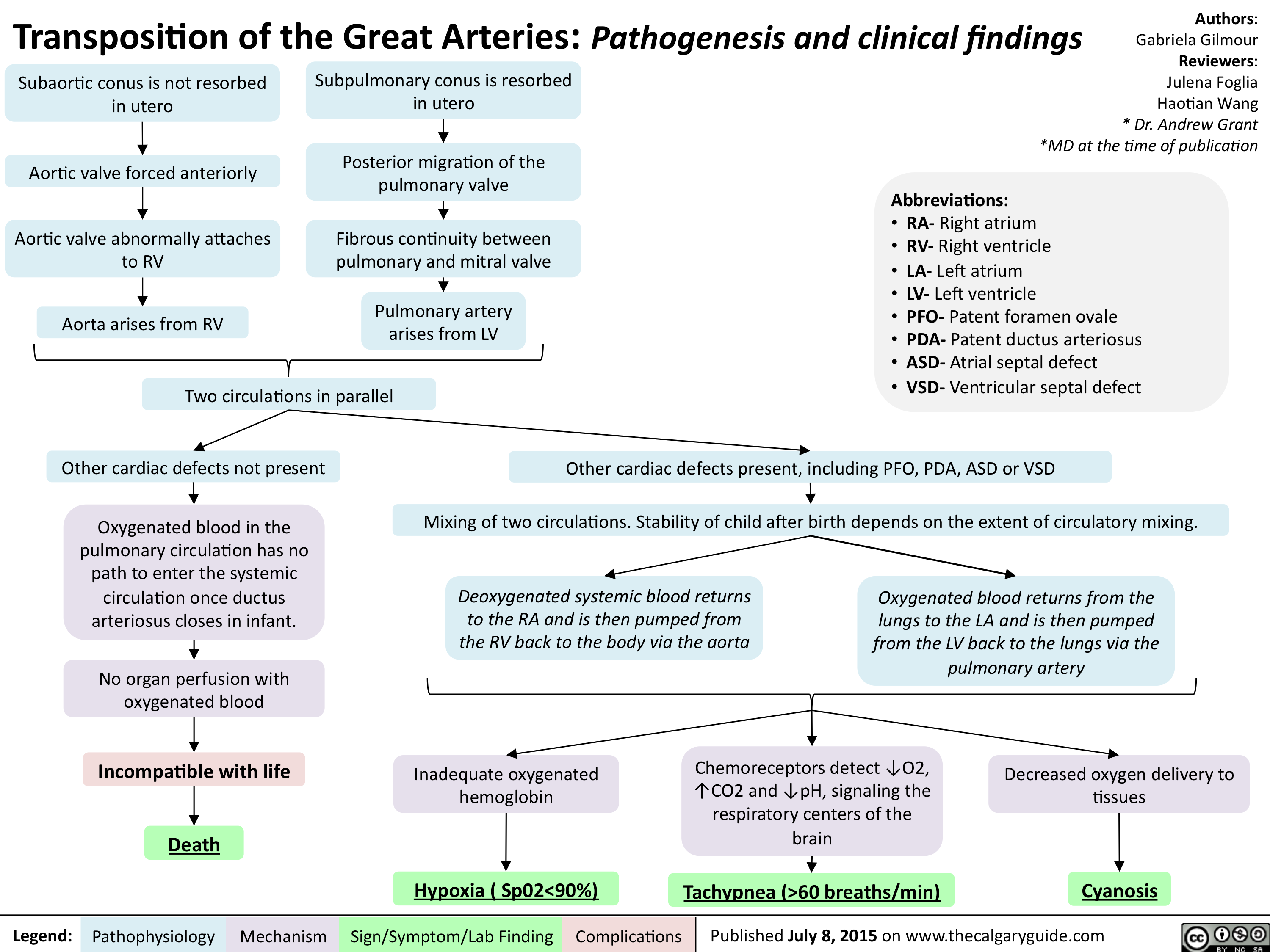
Transposition of the Great Arteries-Pathogenesis & clinical findings
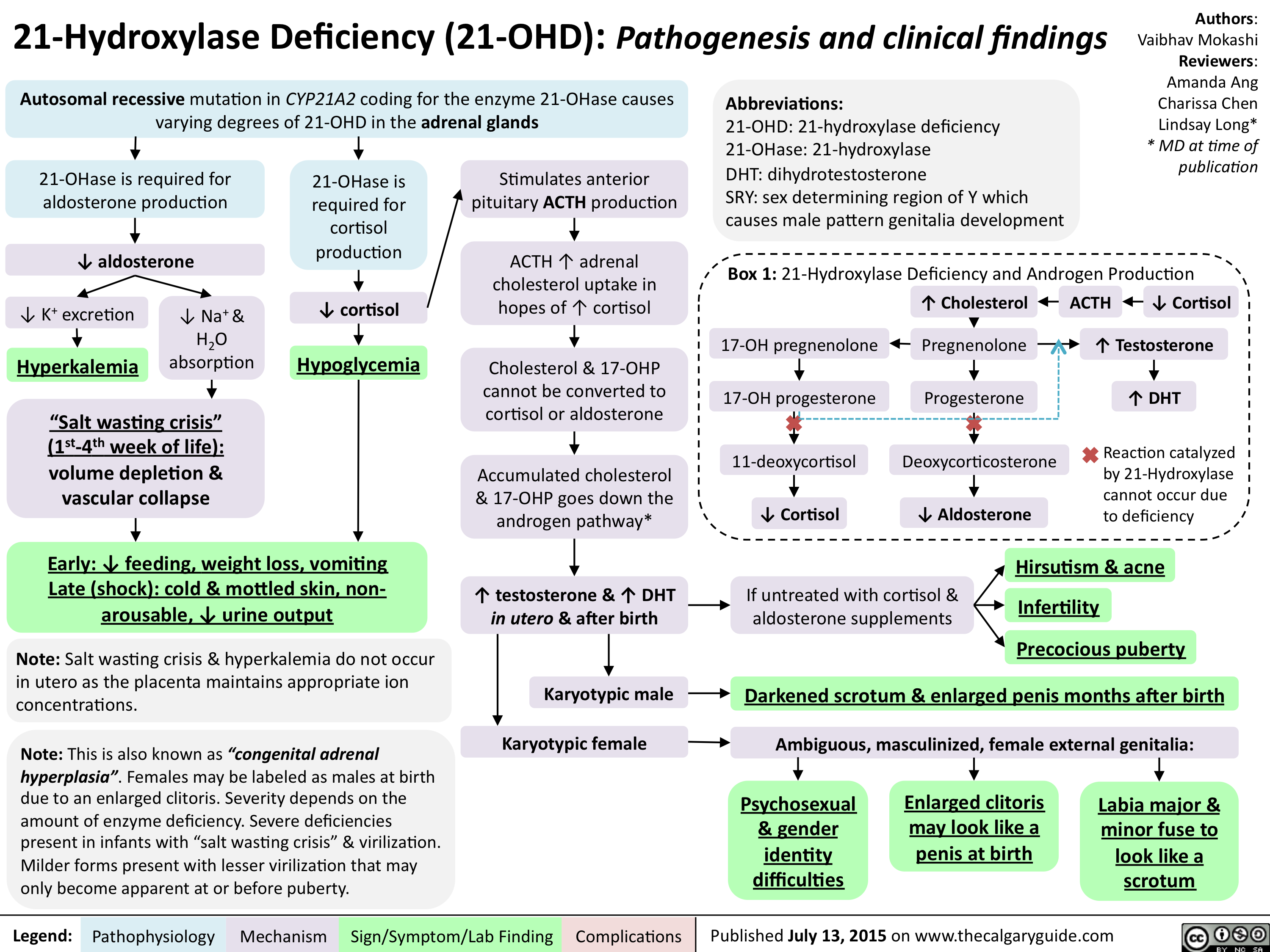
21-Hydroxylase Deficiency-Pathogenesis and clinical findings
Hallux Valgus pathogenesis and clinical findings - August 15 2015

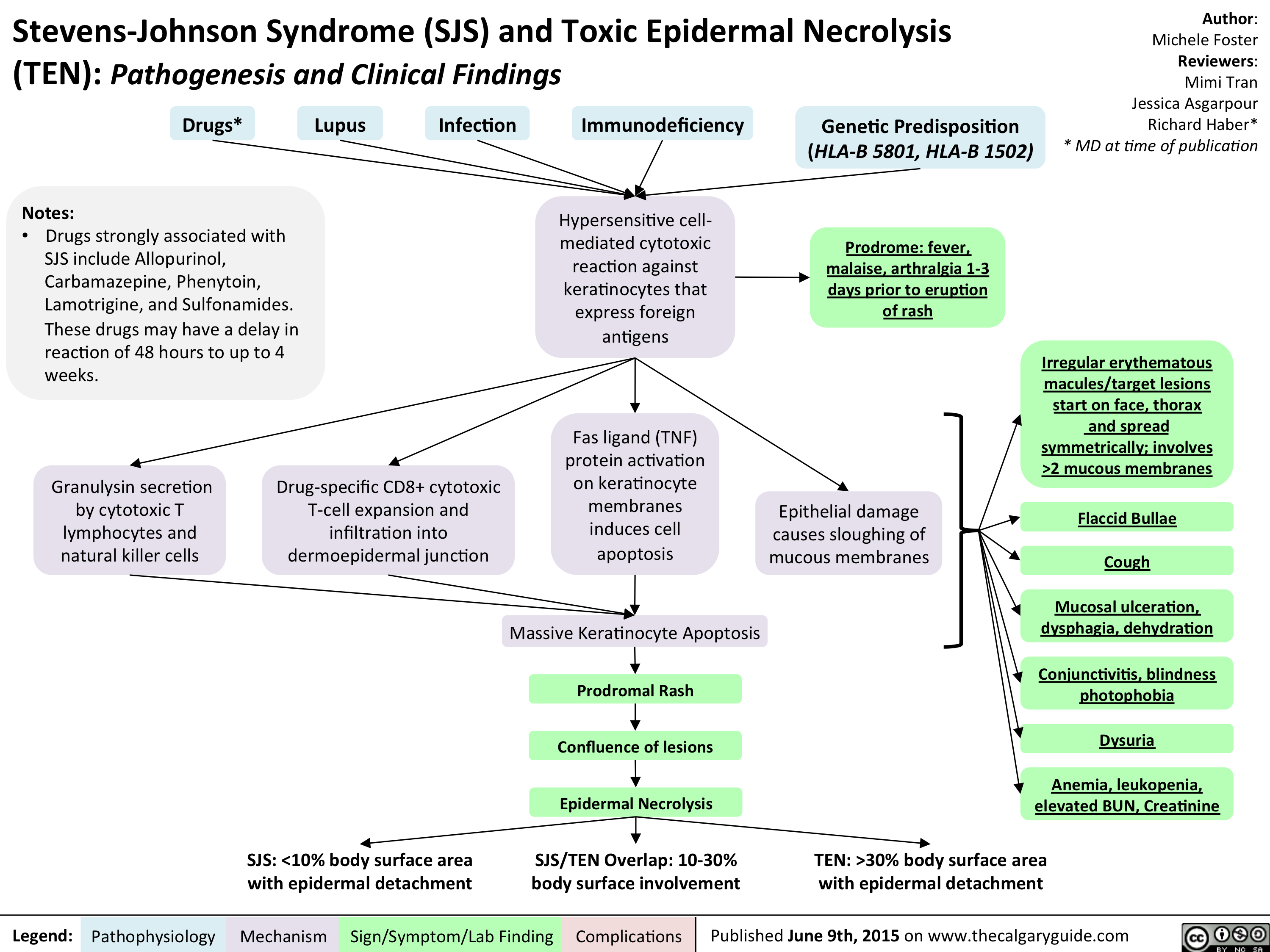
Stevens-Johnson Syndrome (SJS) and Toxic Epidermal Necrolysis (TEN) - Pathogenesis and Clinical Findings
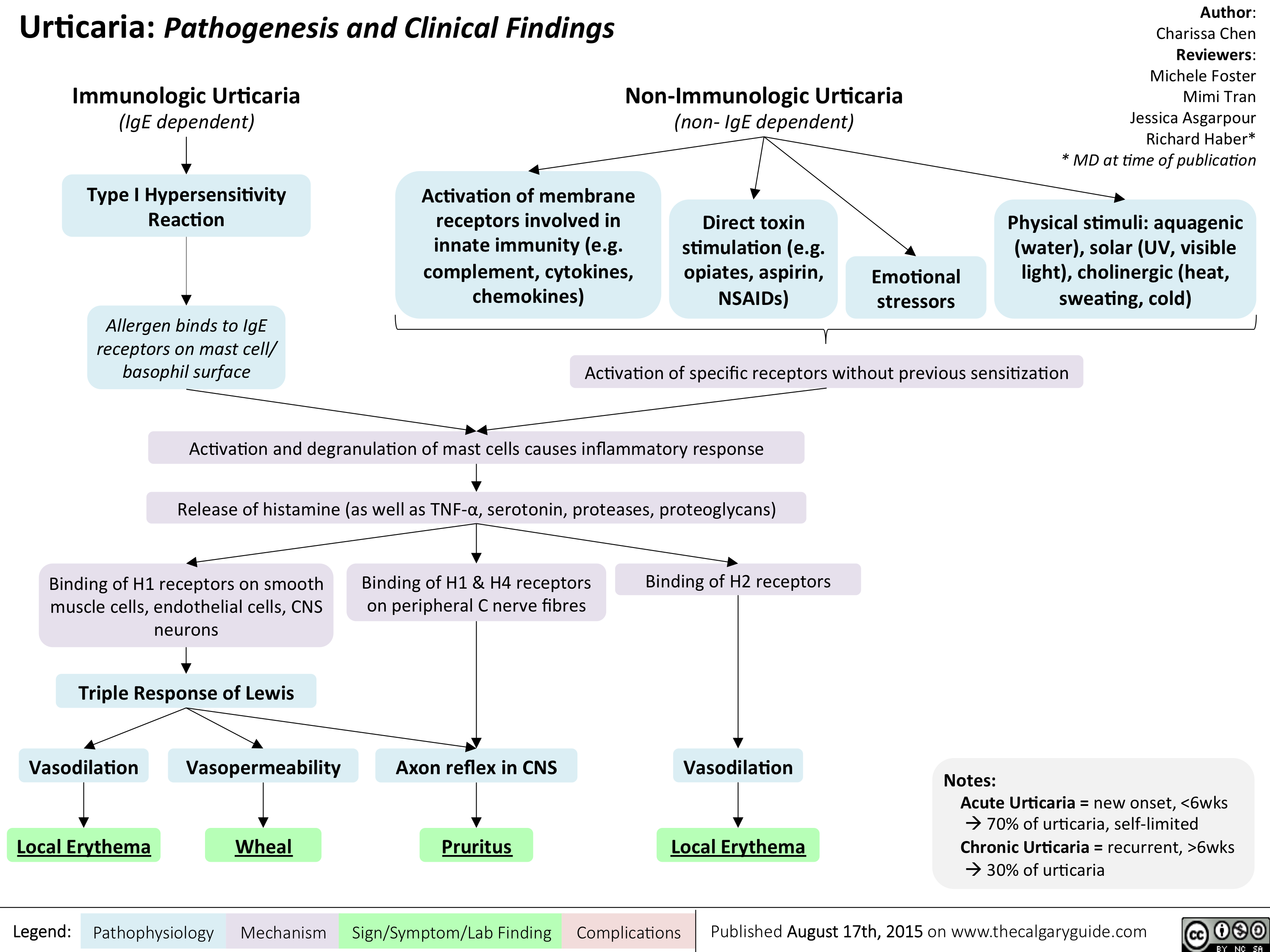
Urticaria- Pathogenesis and Clinical Findings
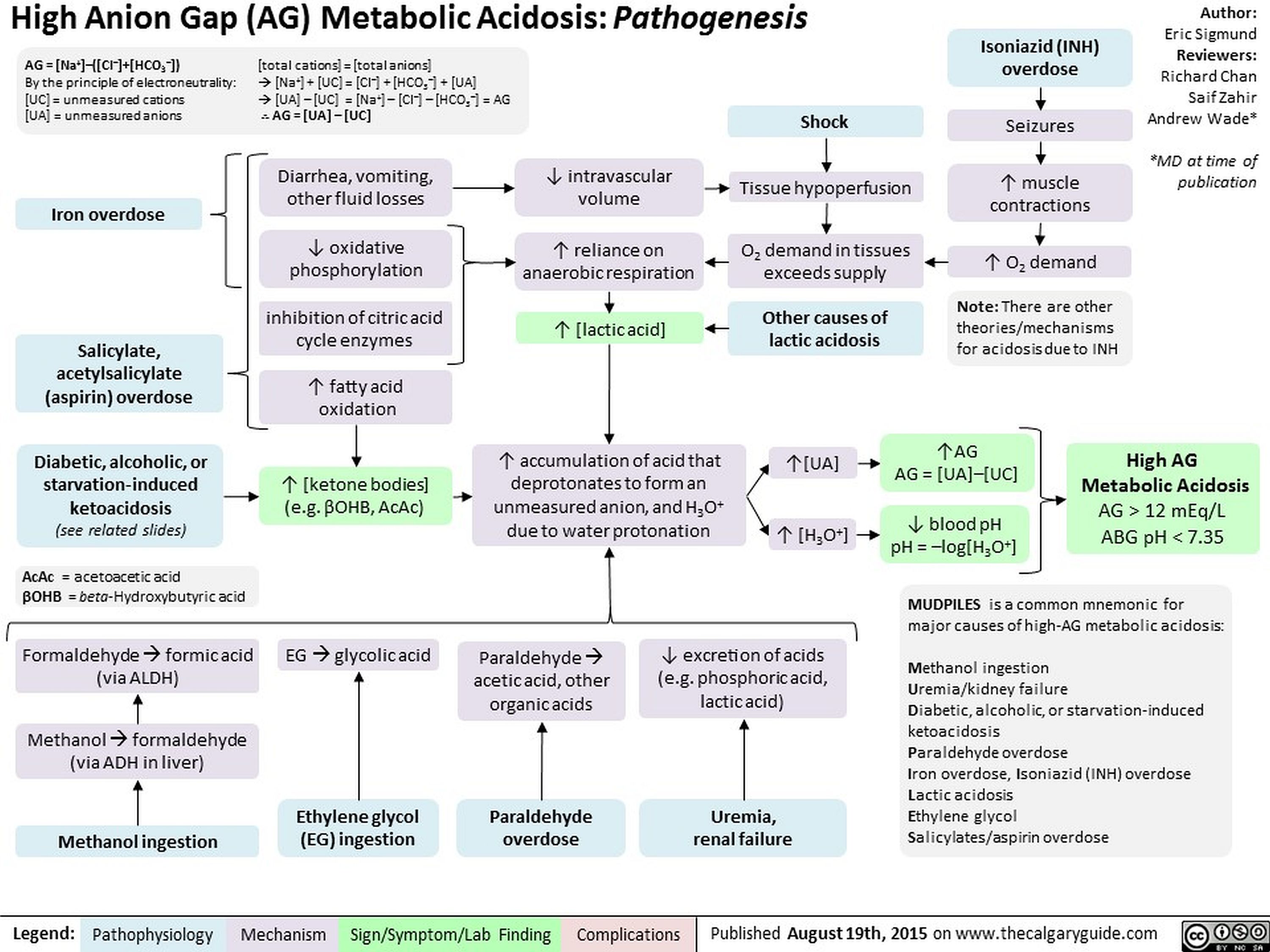
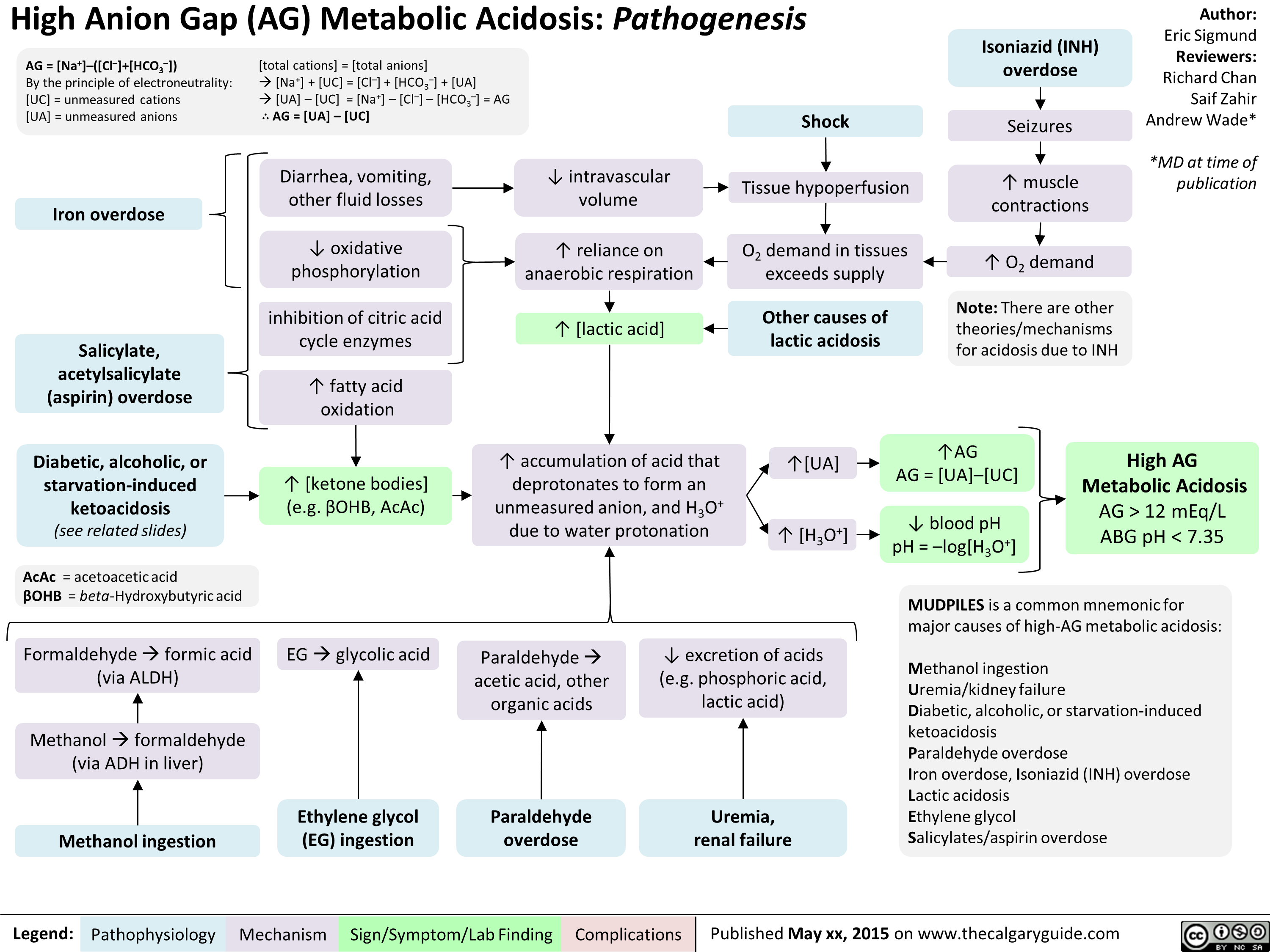
High Anion Gap Metabolic Acidosis Pathogenesis
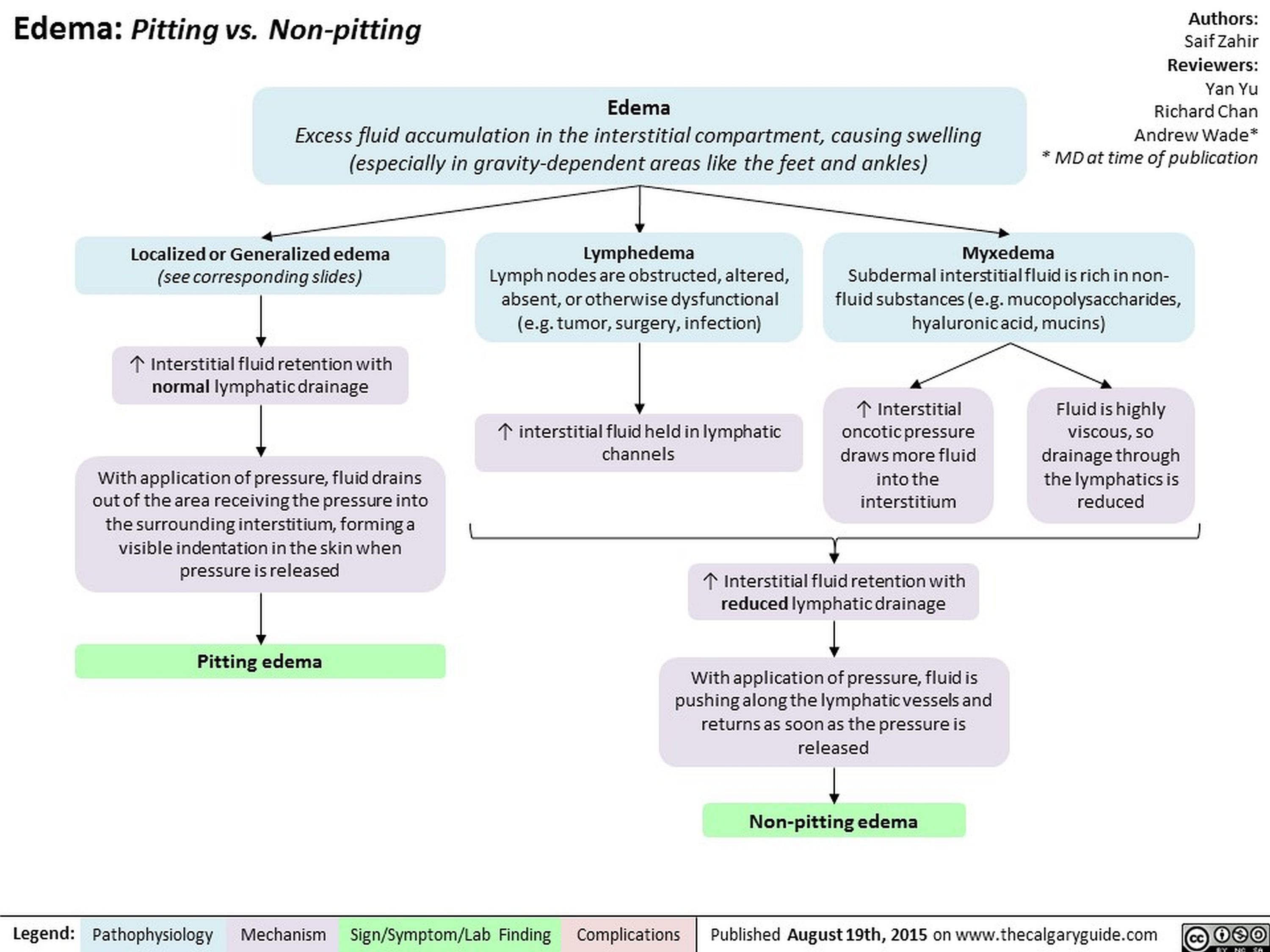
Edema Pitting vs Non-pitting
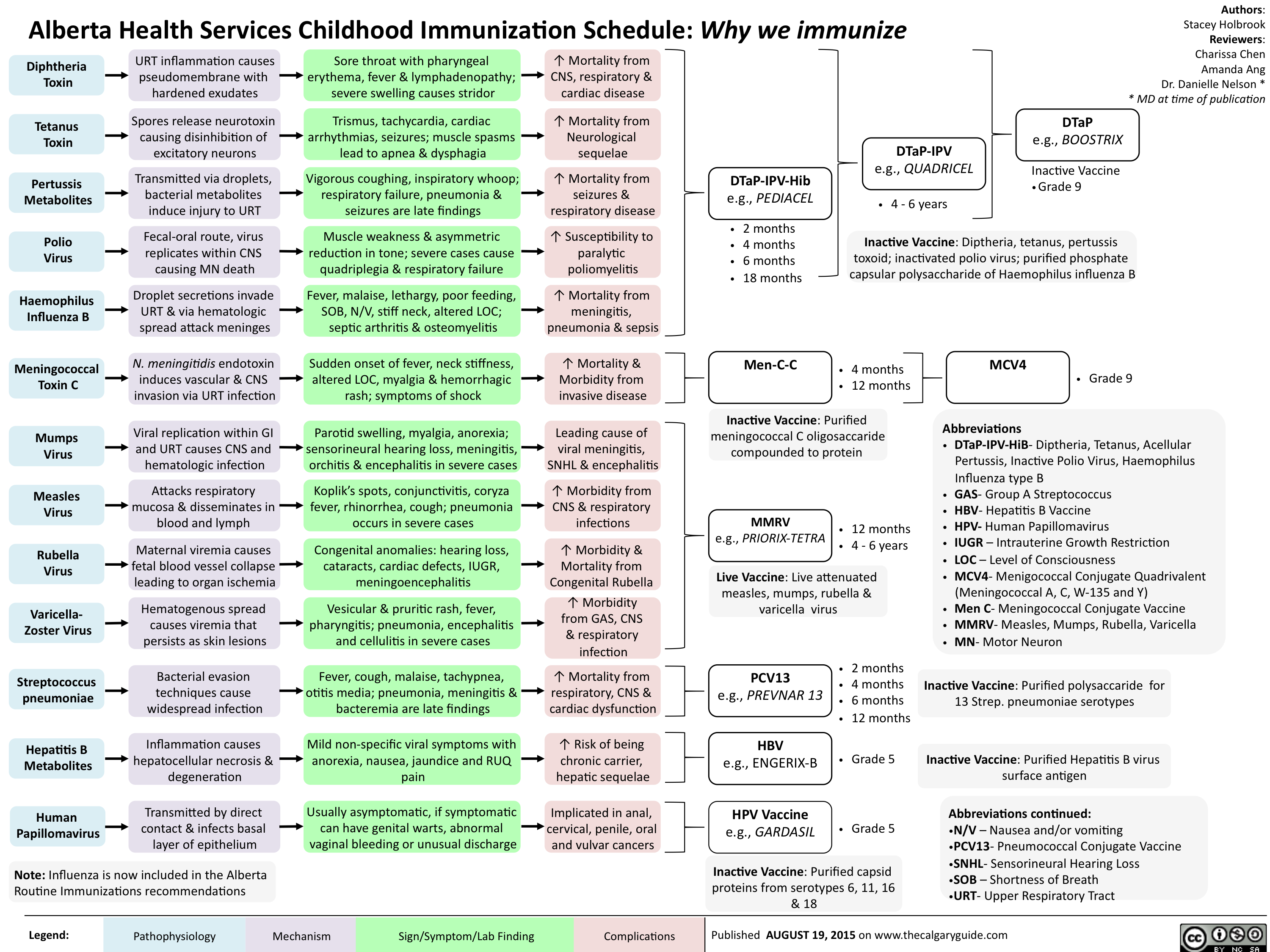
Childhood Immunization Schedule-Why we immunize
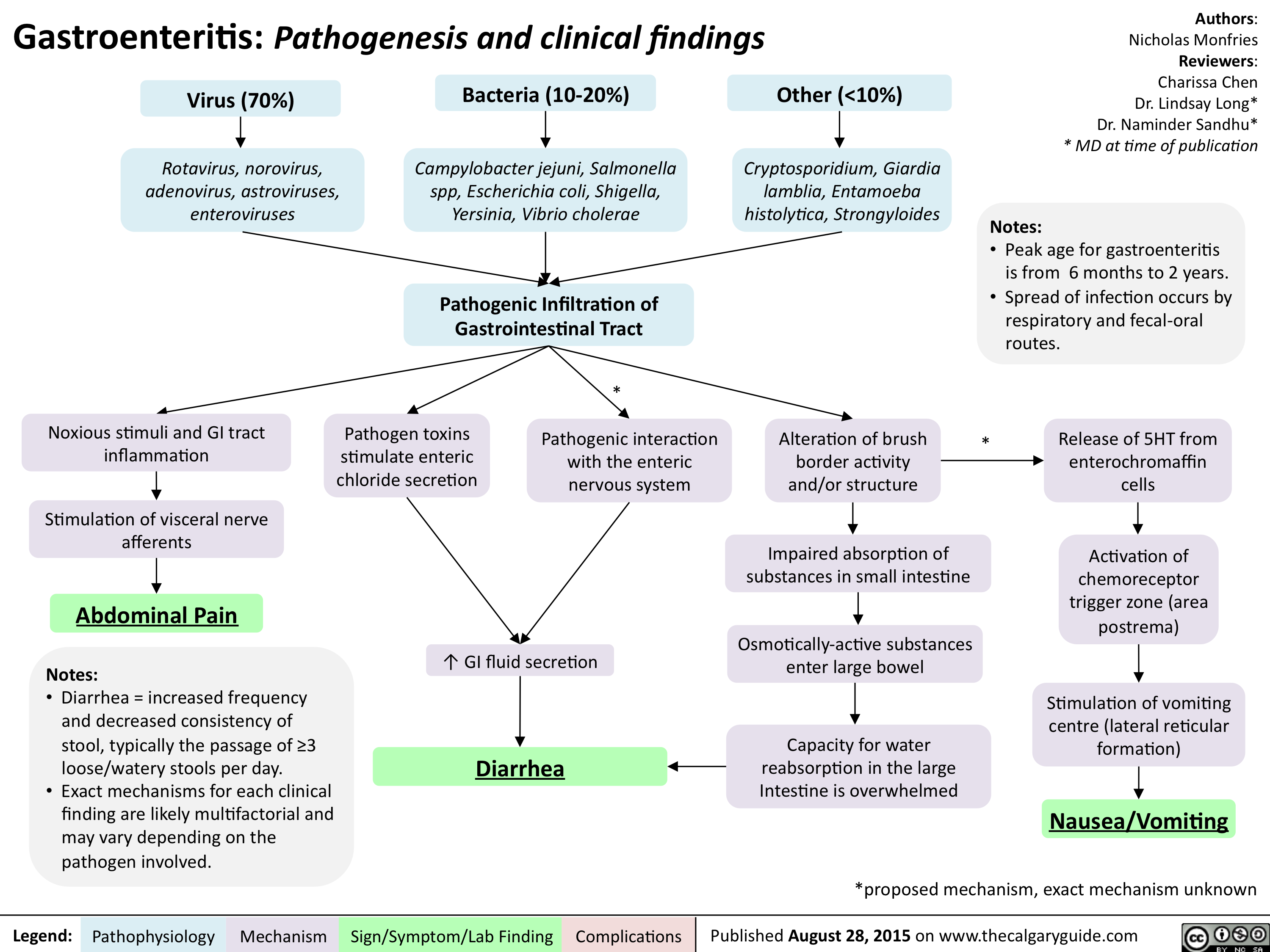
Gastroenteritis-Pathogenesis and clinical findings
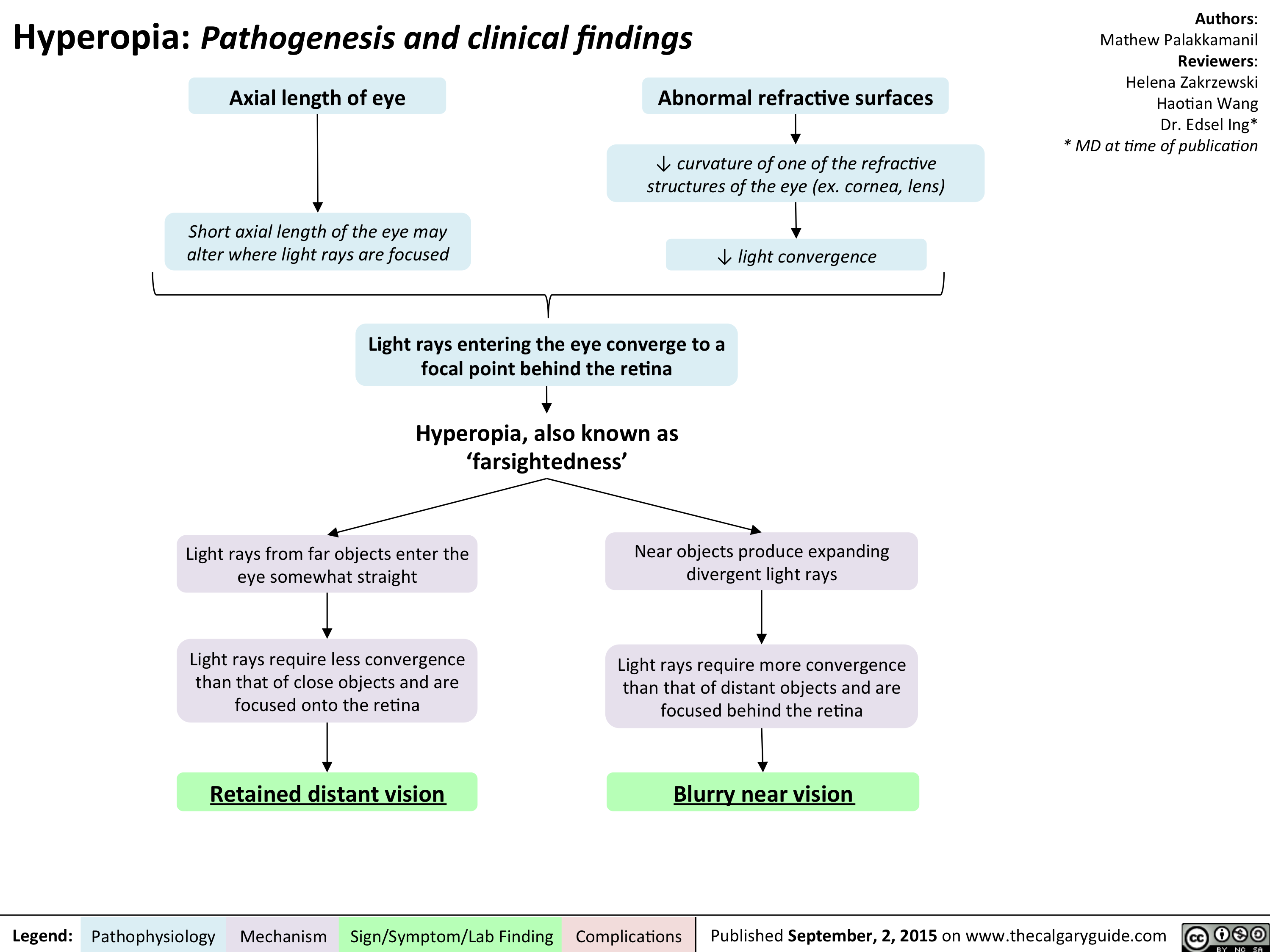
Hyperopia - Pathogenesis and Clinical Findings
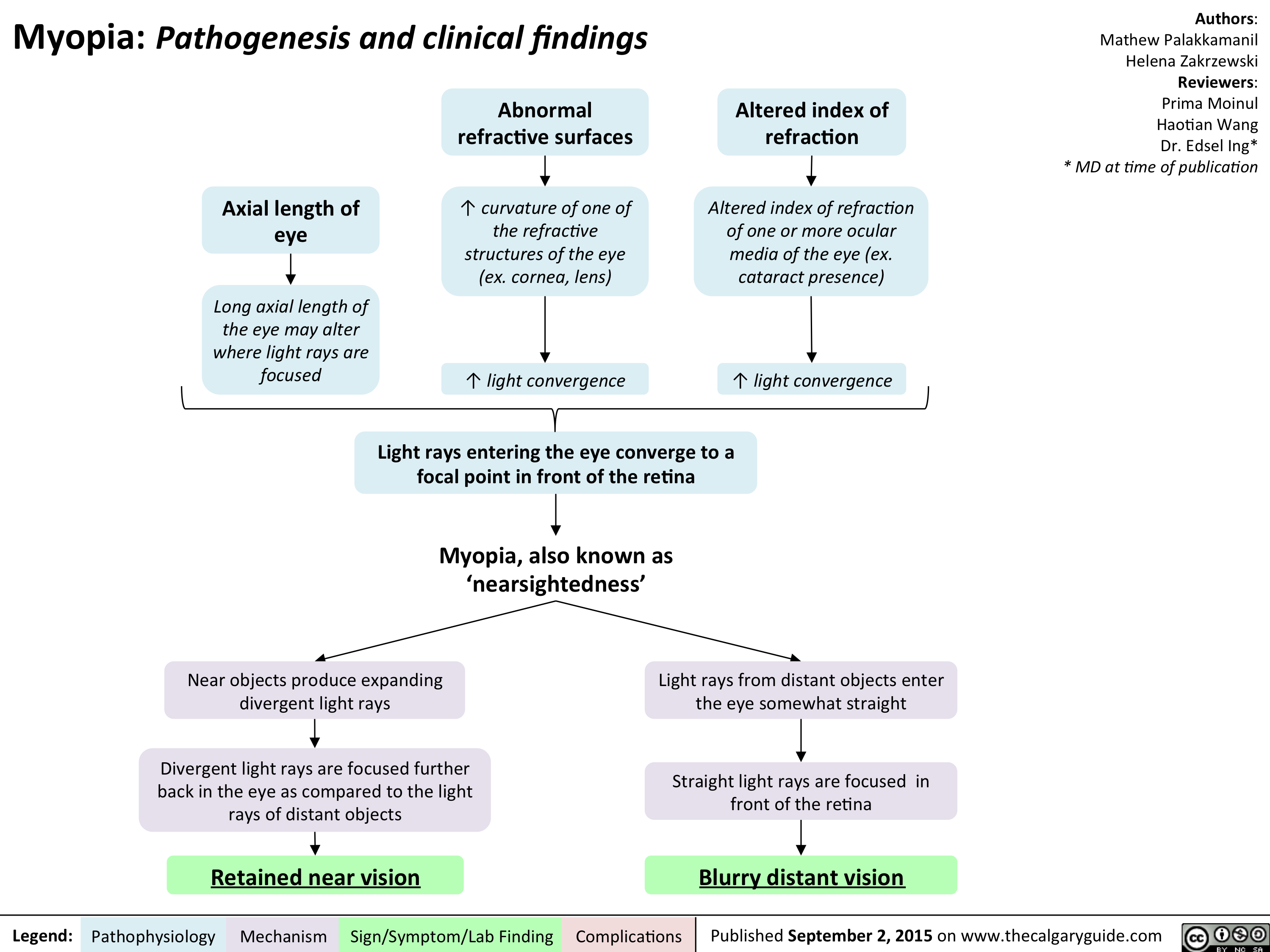
Myopia - Pathogenesis and clinical findings
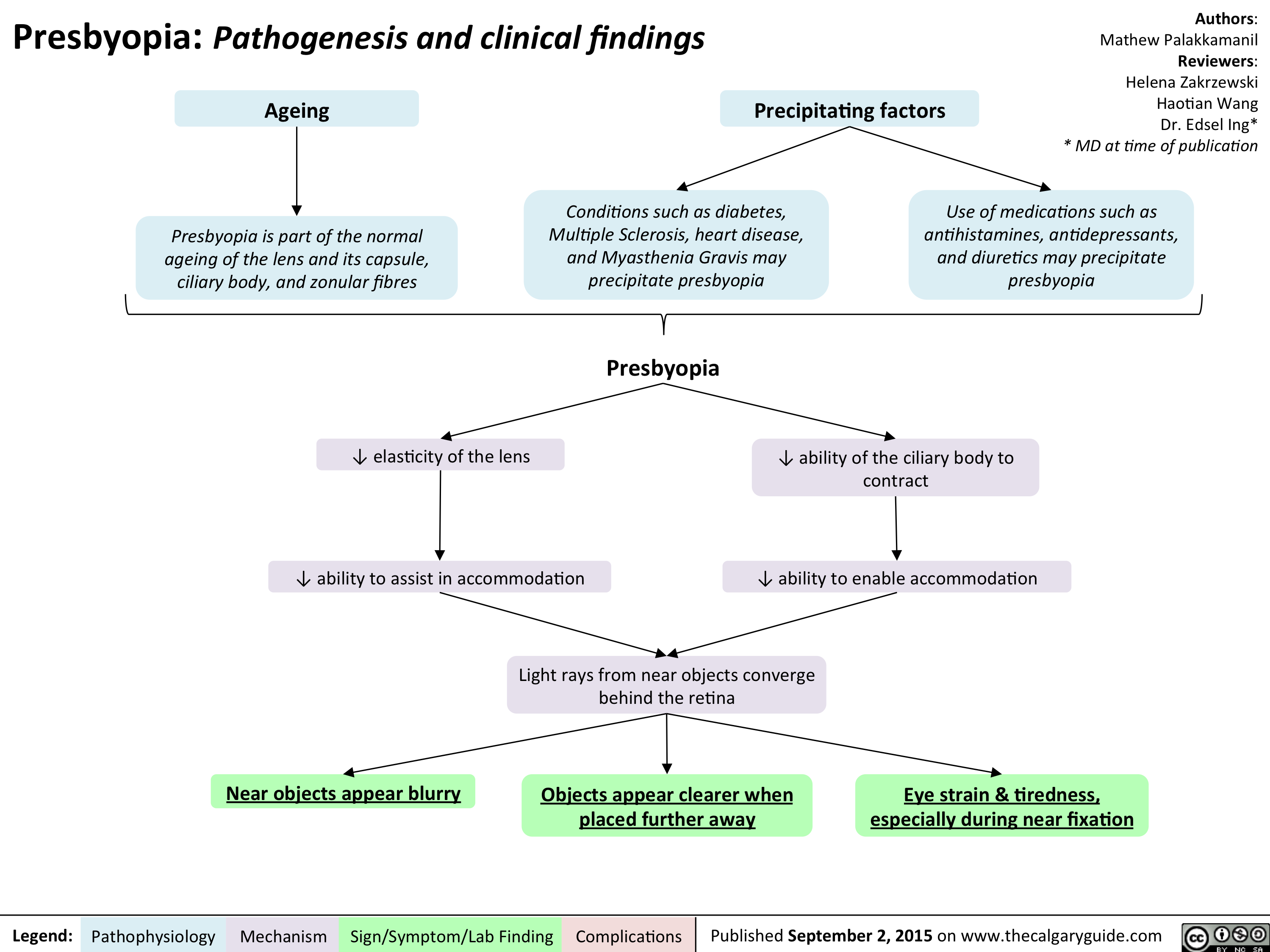
Presbyopia - Pathogenesis and clinical findings
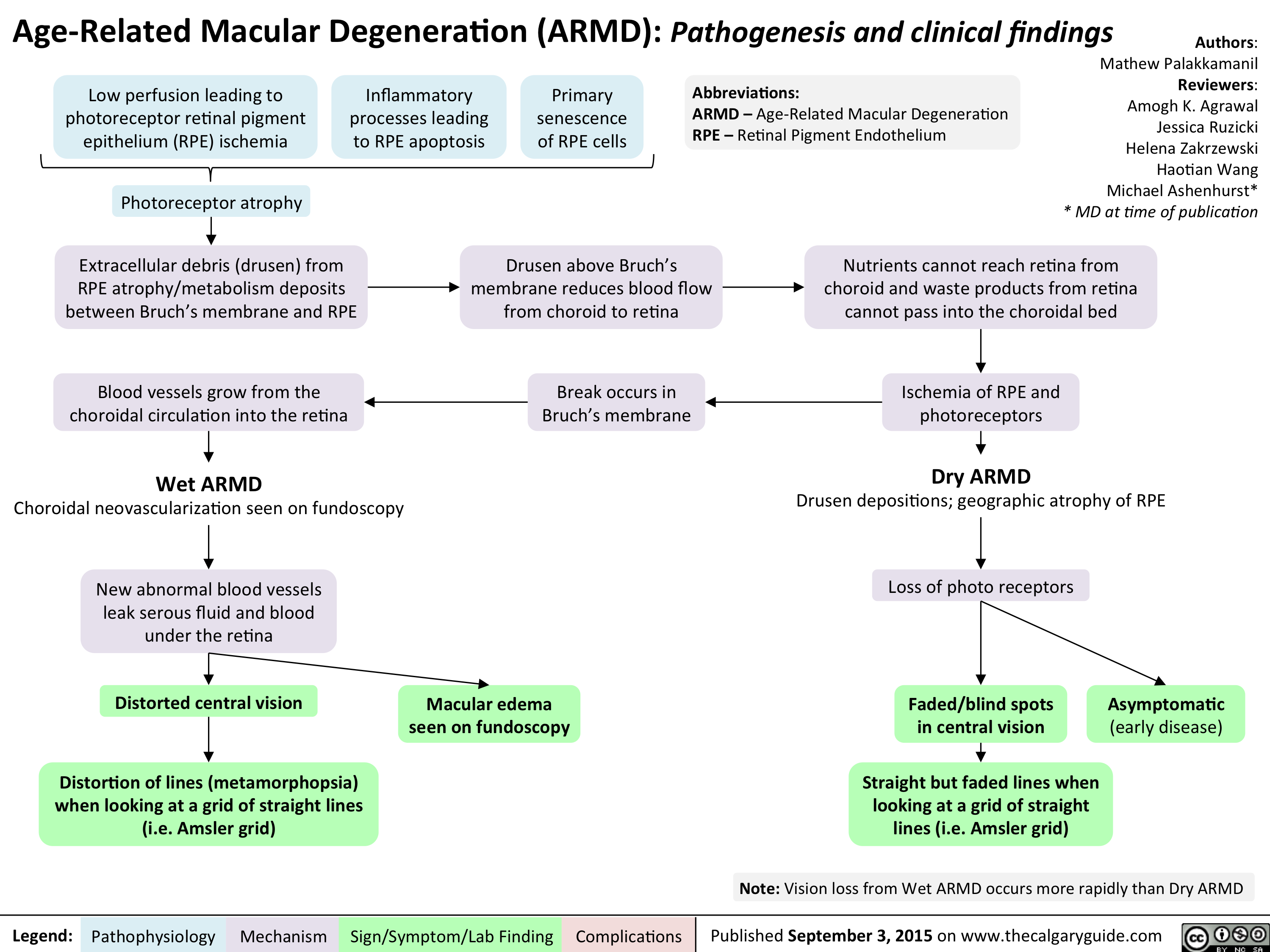
Age Related Macular Degeneration - Pathogenesis and clinical findings
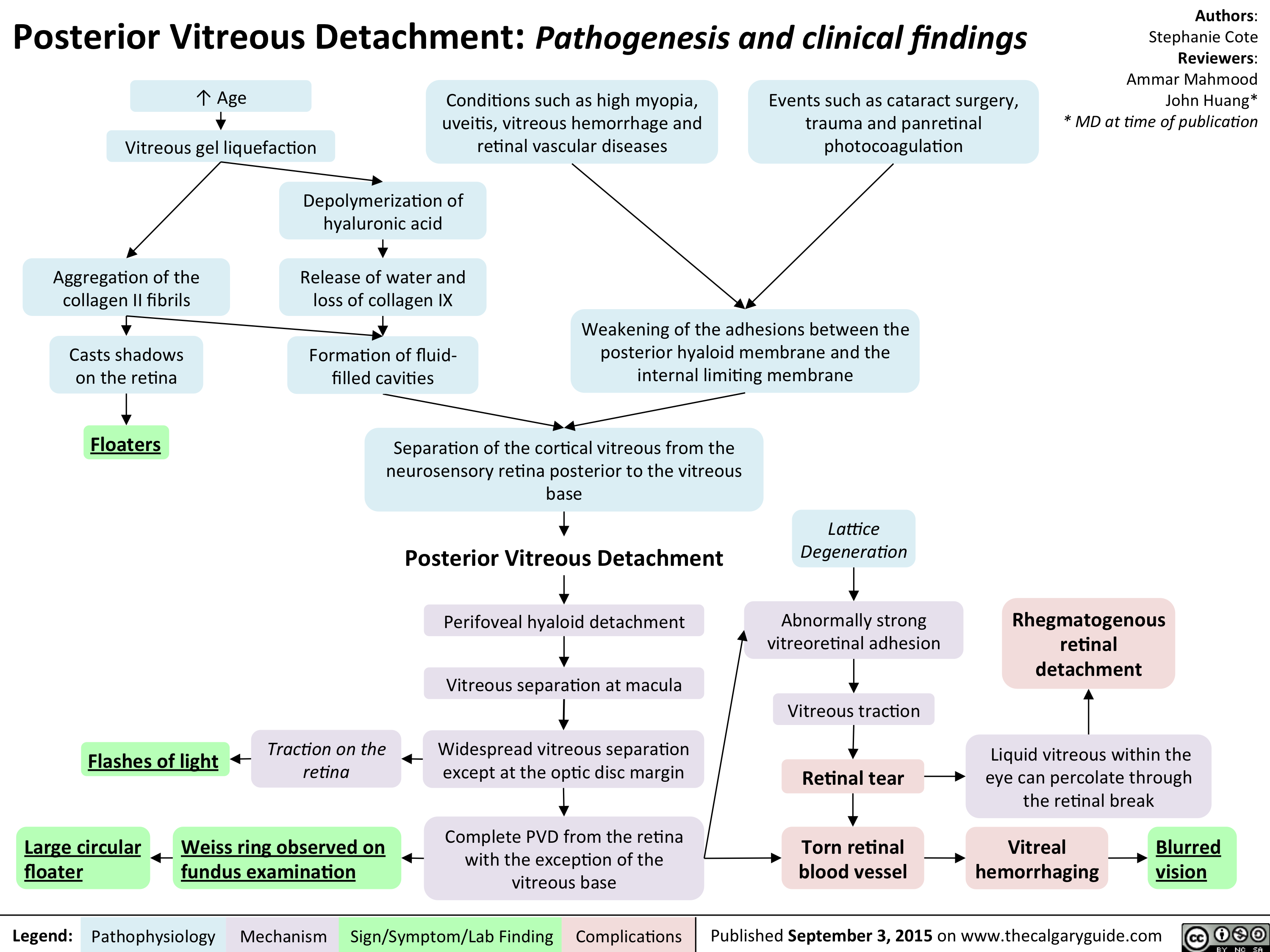
Posterior Vitreous Detachment - Pathogenesis and clinical findings
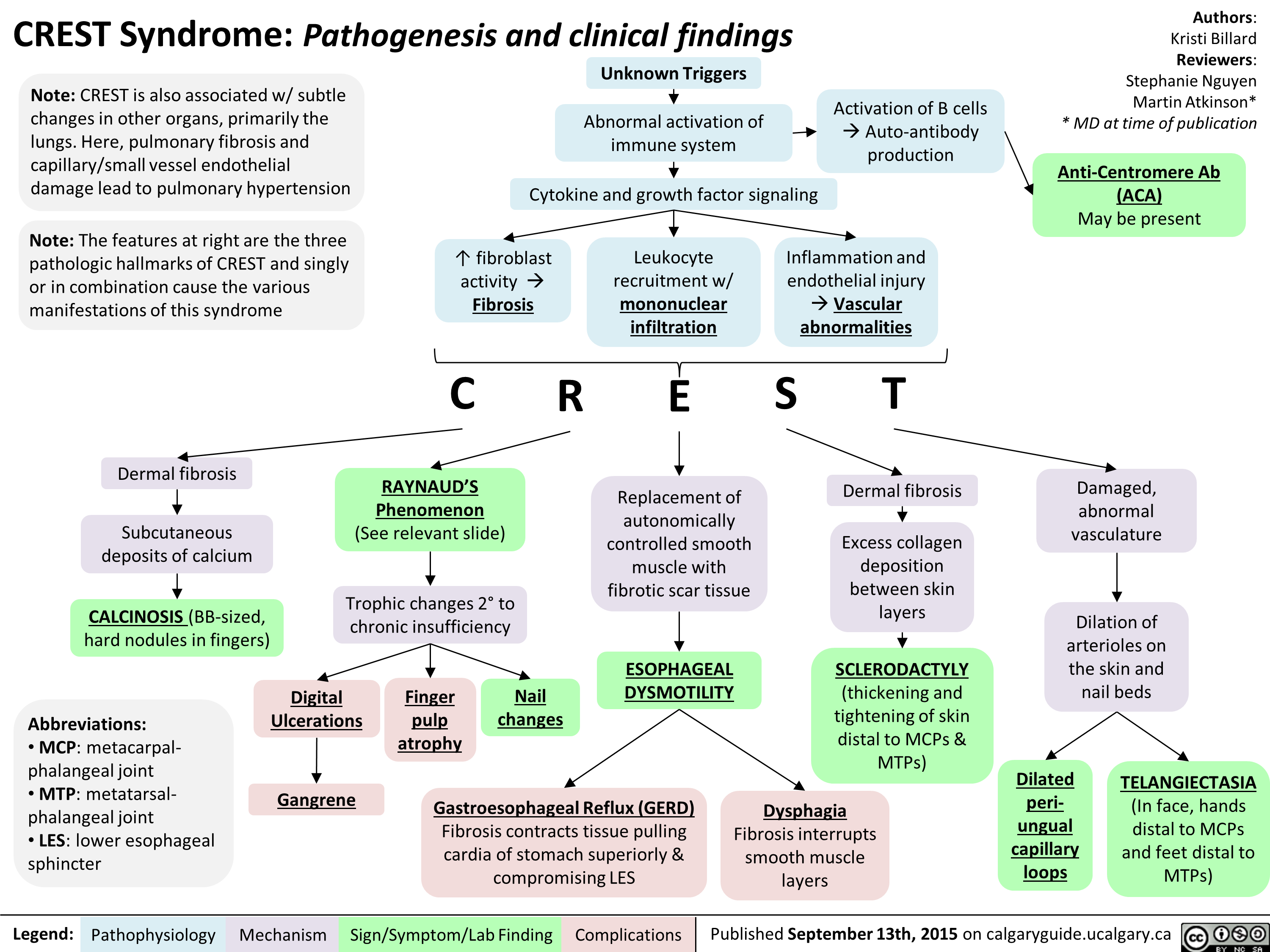
FINAL - CREST Syndrome Pathogenesis and clinical findings
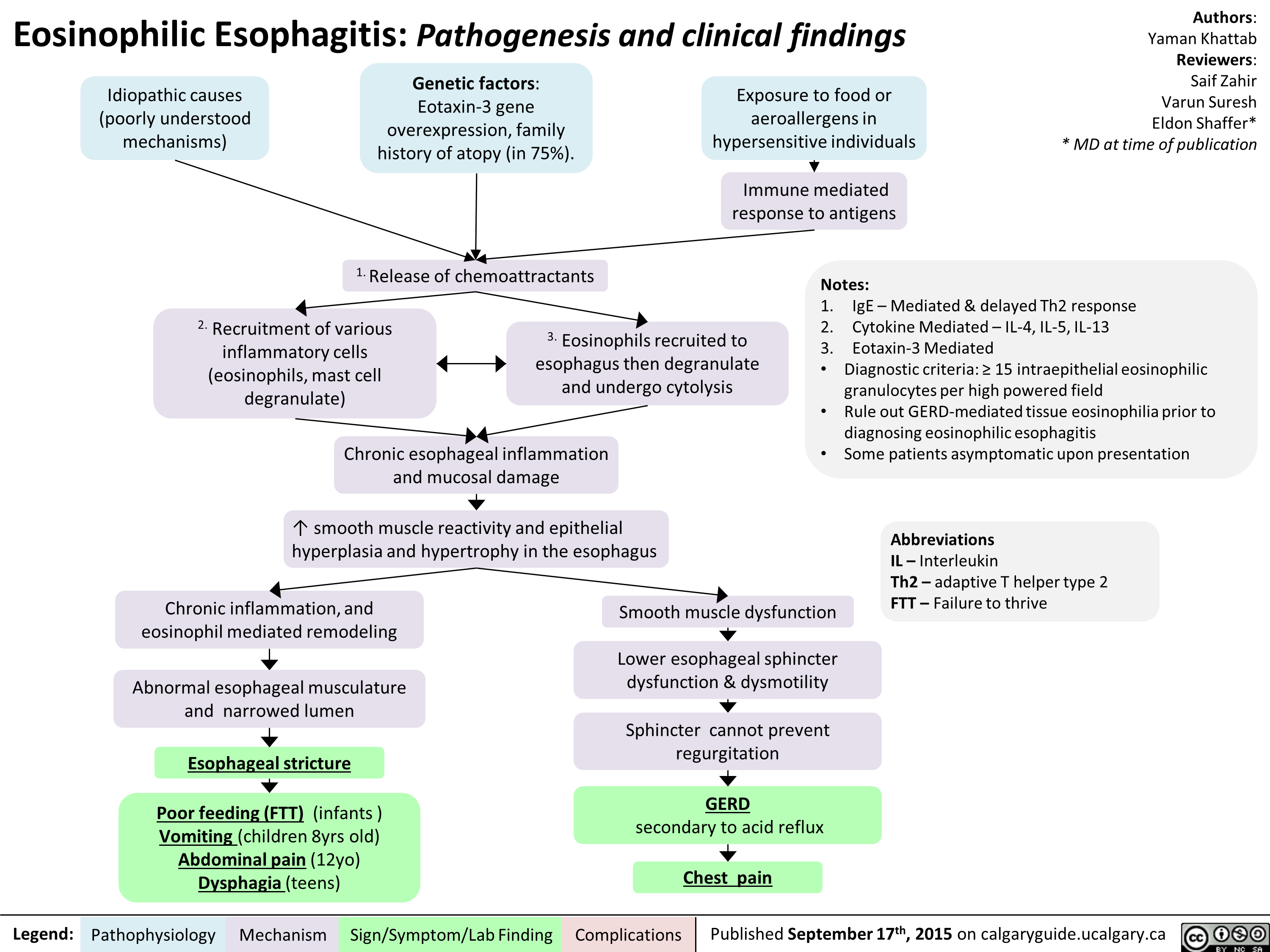
Eosinophillic Esophagitis -Kattab Yaman - Final For Publication
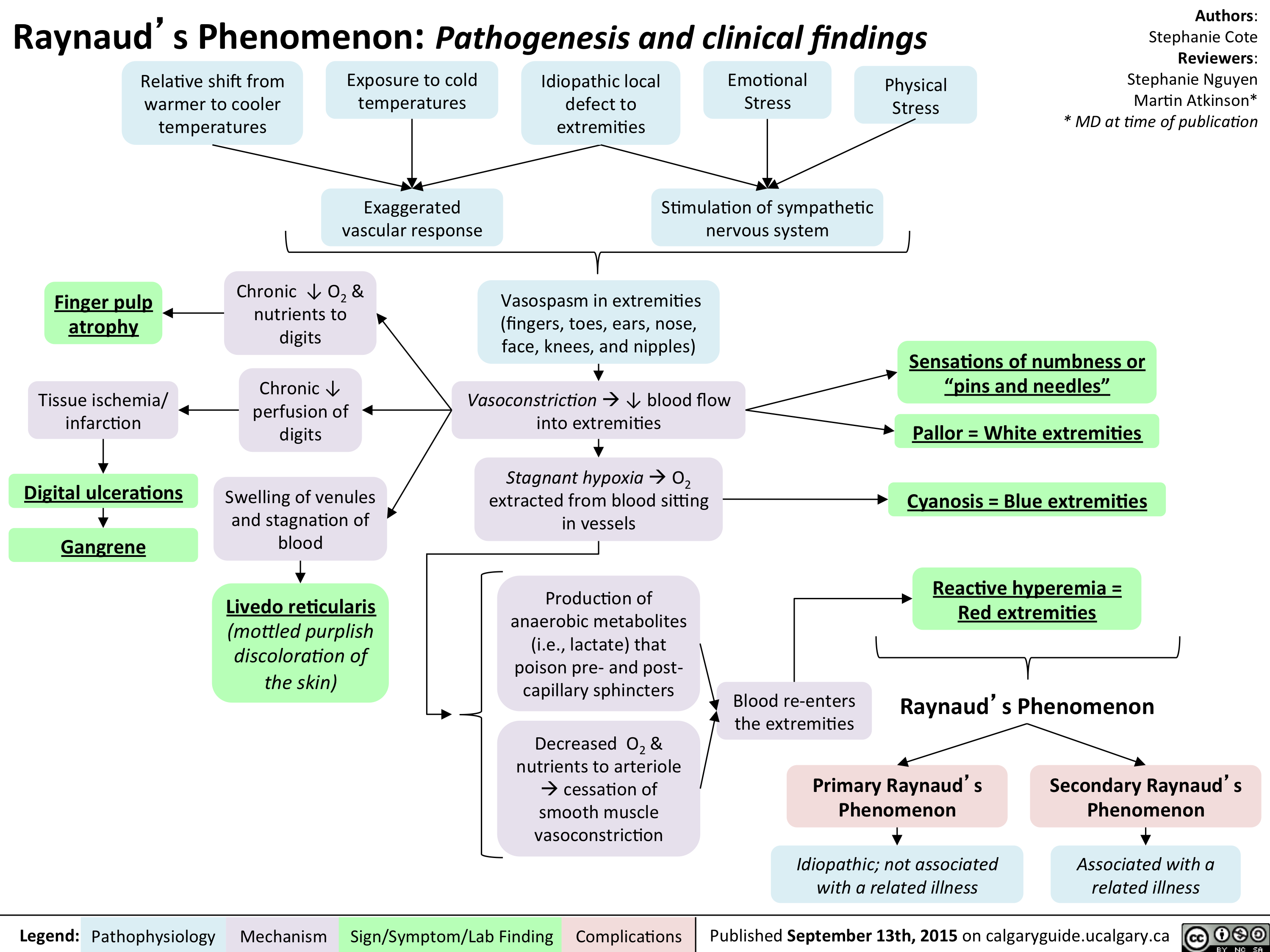
Raynaud Phenomenon Pathogenesis and Clinical Findings
Pre-Renal Acute Kidney Injury Pathogenesis

High-AG Metabolic Acidosis Pathogenesis
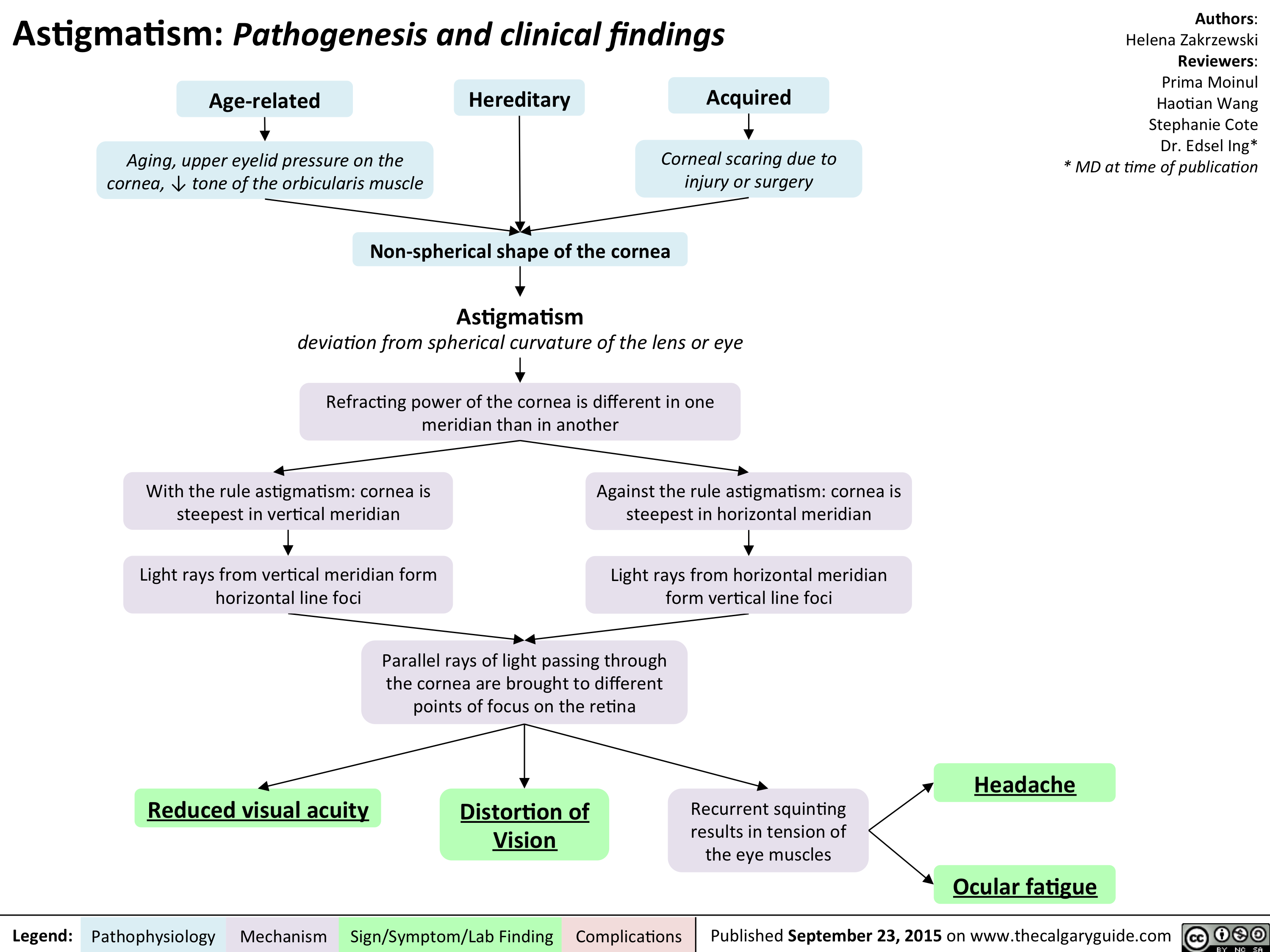
Astigmatism Pathogenesis and Clinical Findings
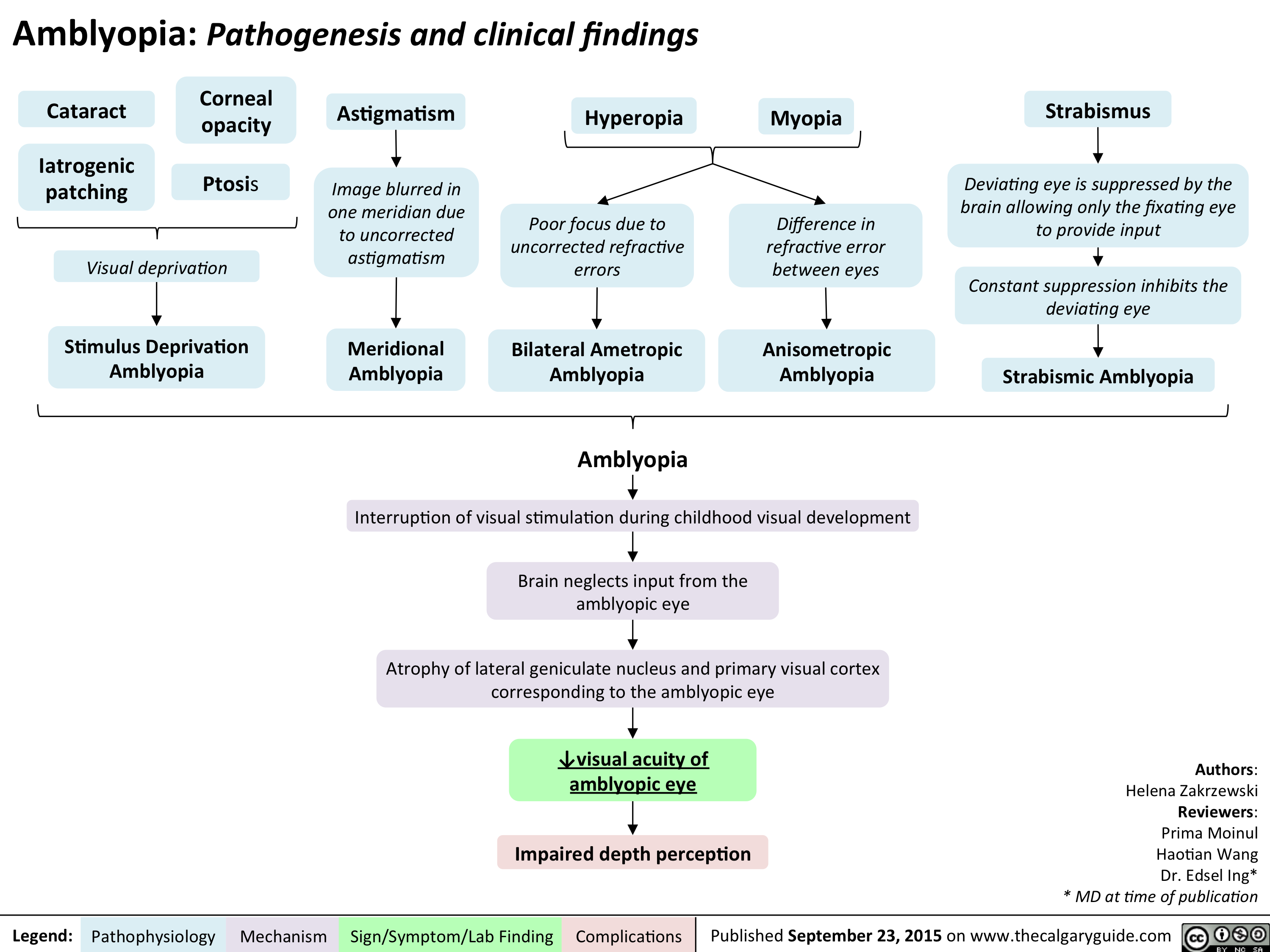
Amblyopia Pathogenesis and clinical findings
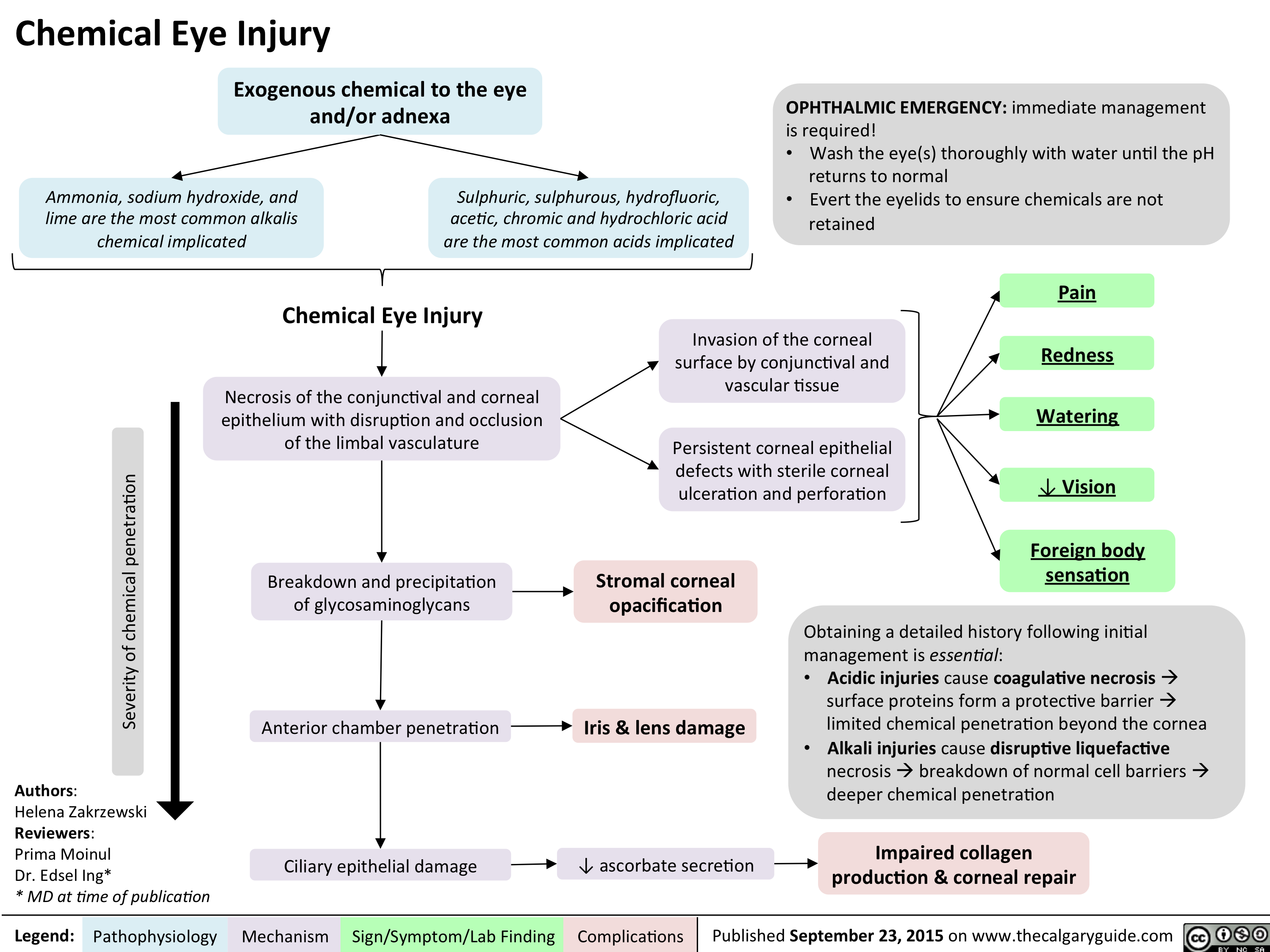
Chemical Eye Injury Pathogenesis and clinical findings
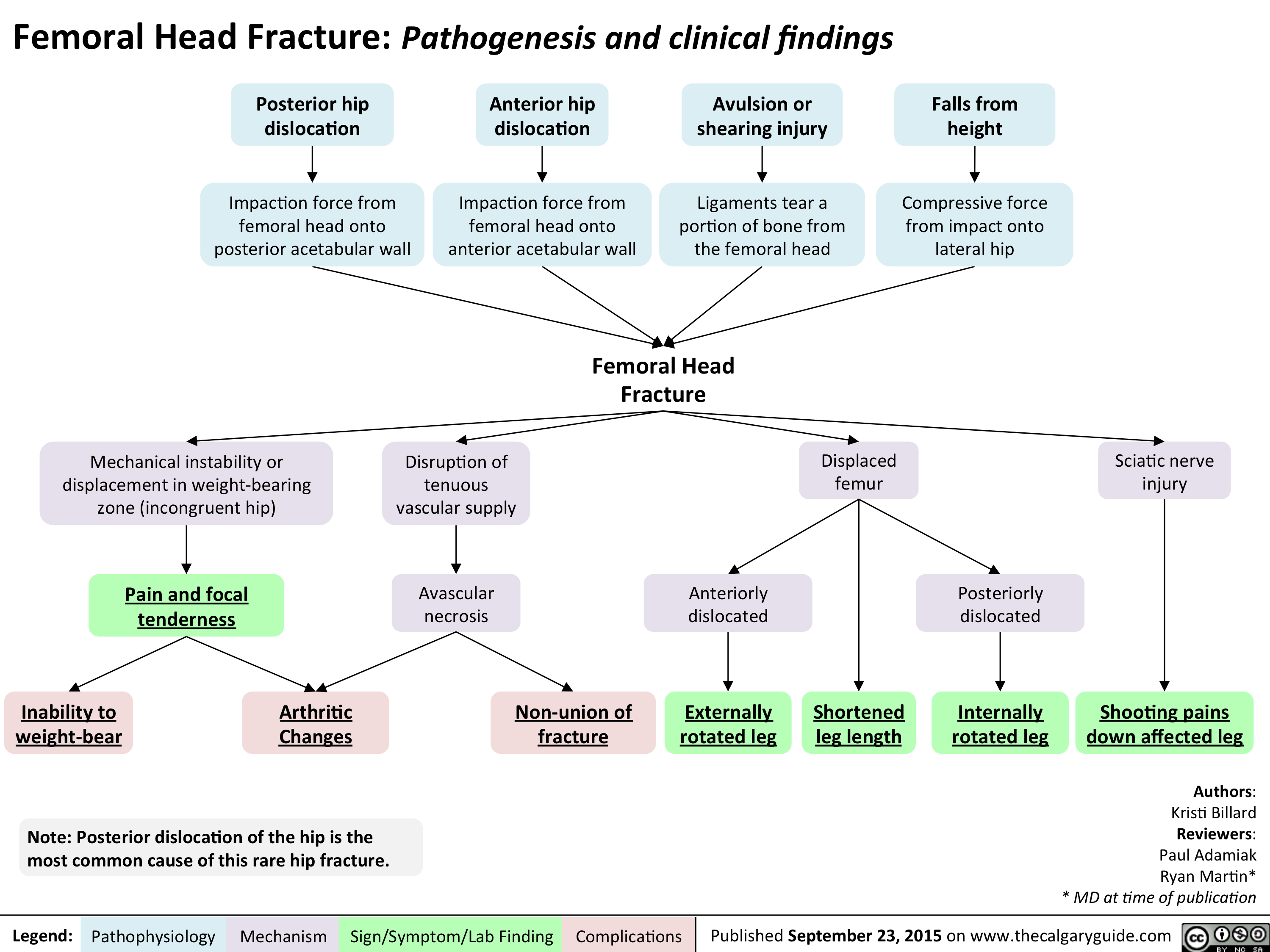
Femoral Head Fracture Pathogenesis and clinical findings
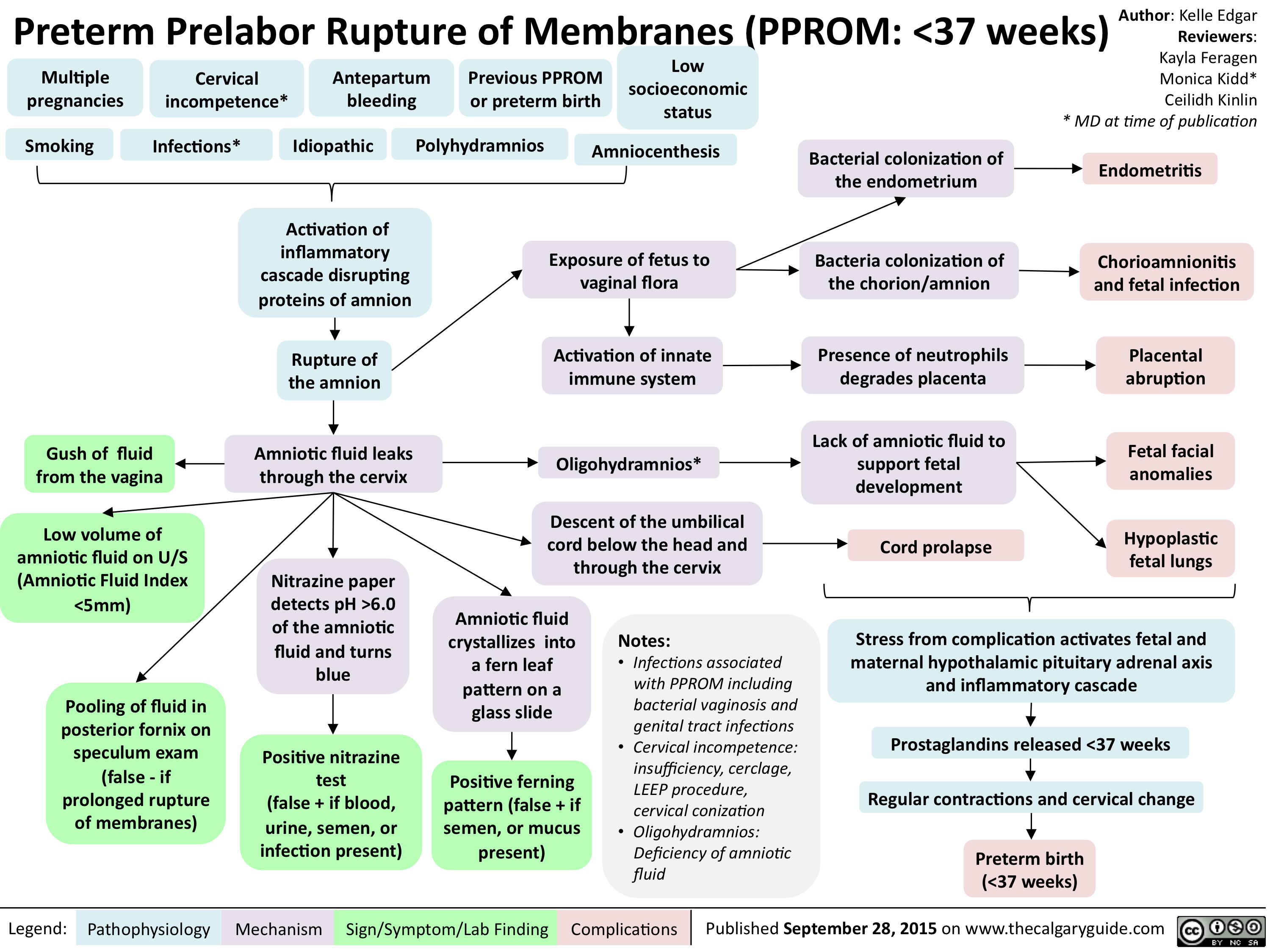
PPROM - Pathogenesis and clinical findings
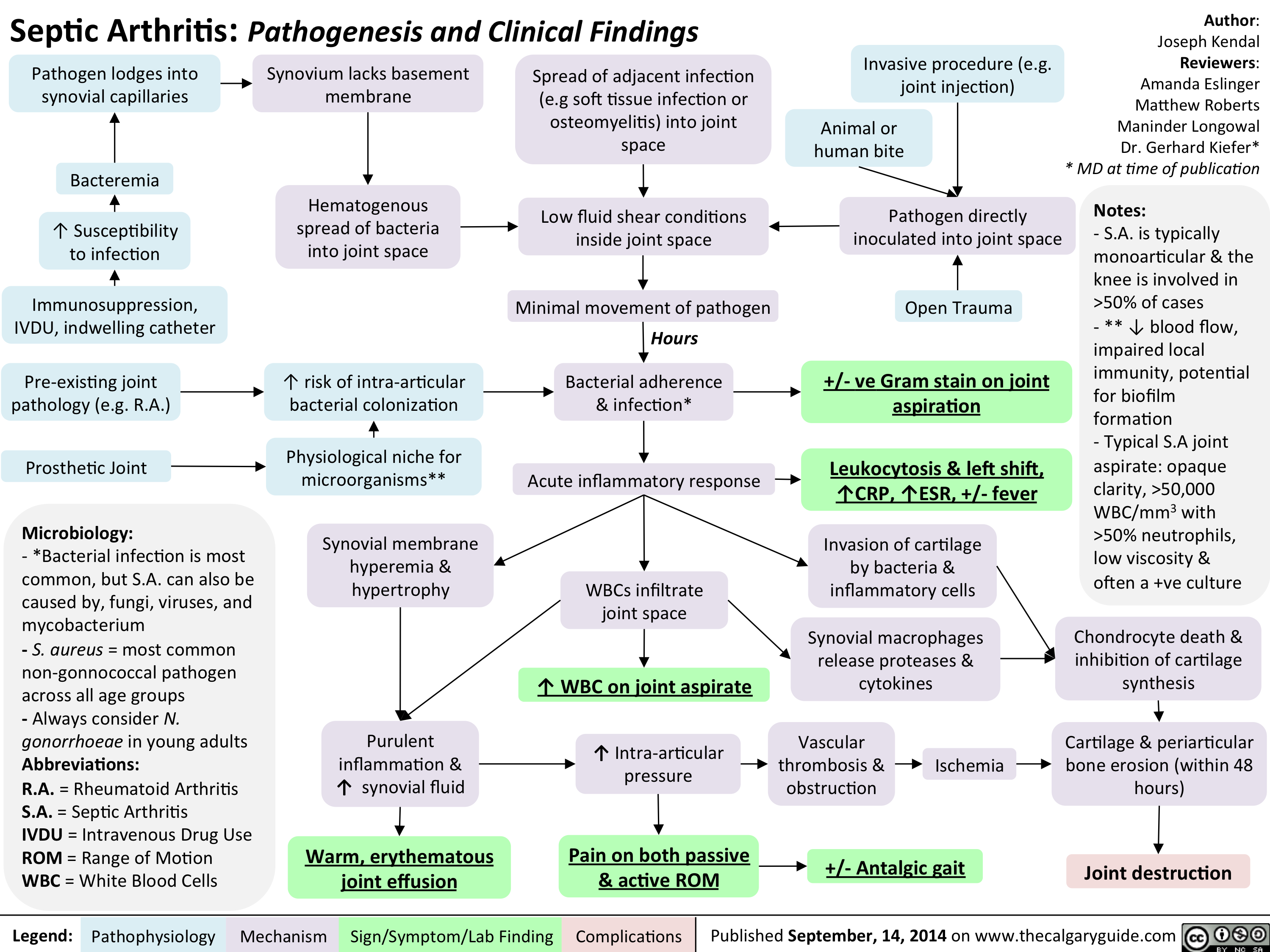
Septic arthitis pathogenesis and clinical findings
Family Med Slides Vitamin D Deficiency

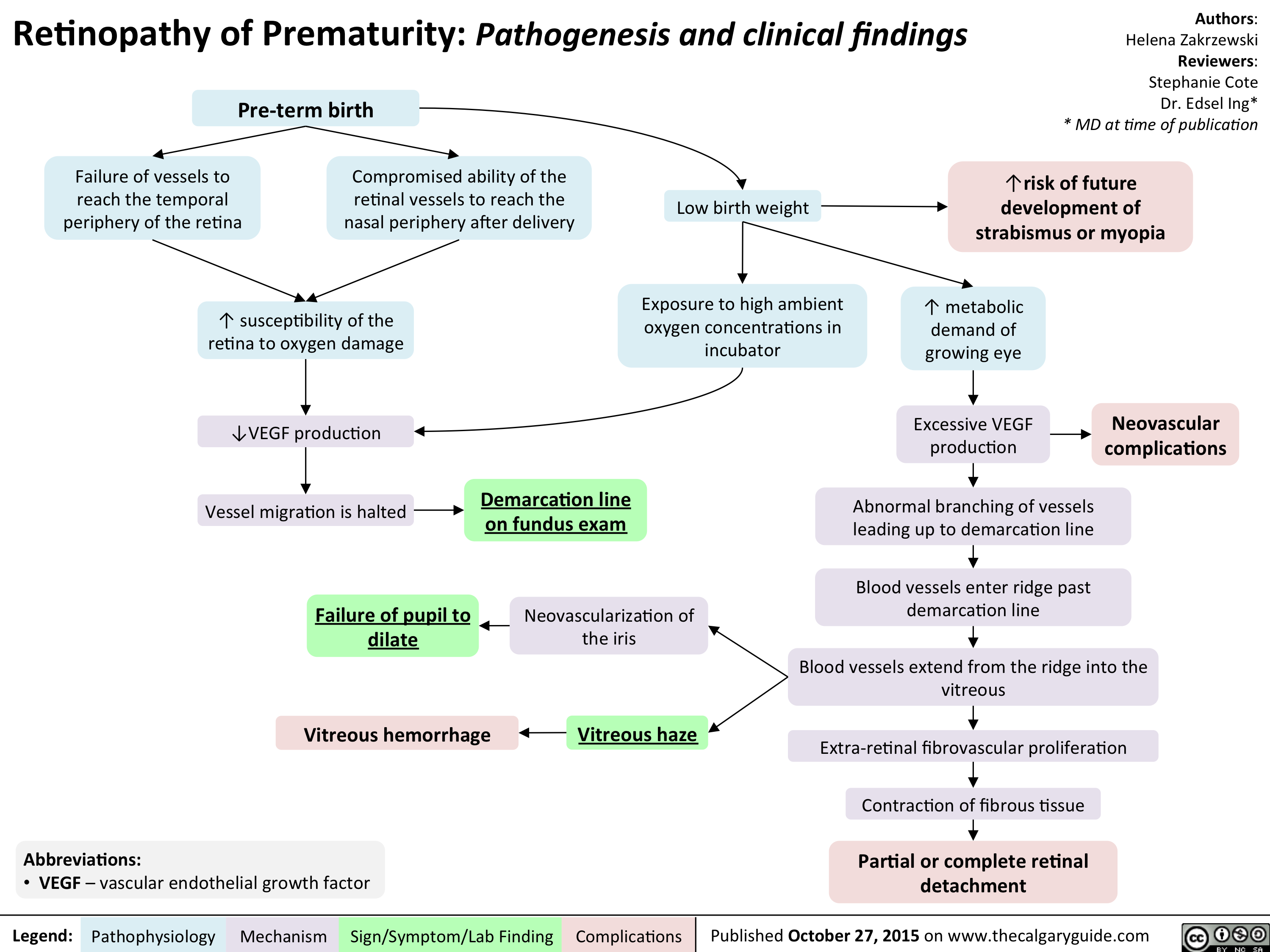
Retinopathy of Prematurity - Pathogenesis and clinical findings
Orthostatic Hypotension-Pathogenesis and clinical findings

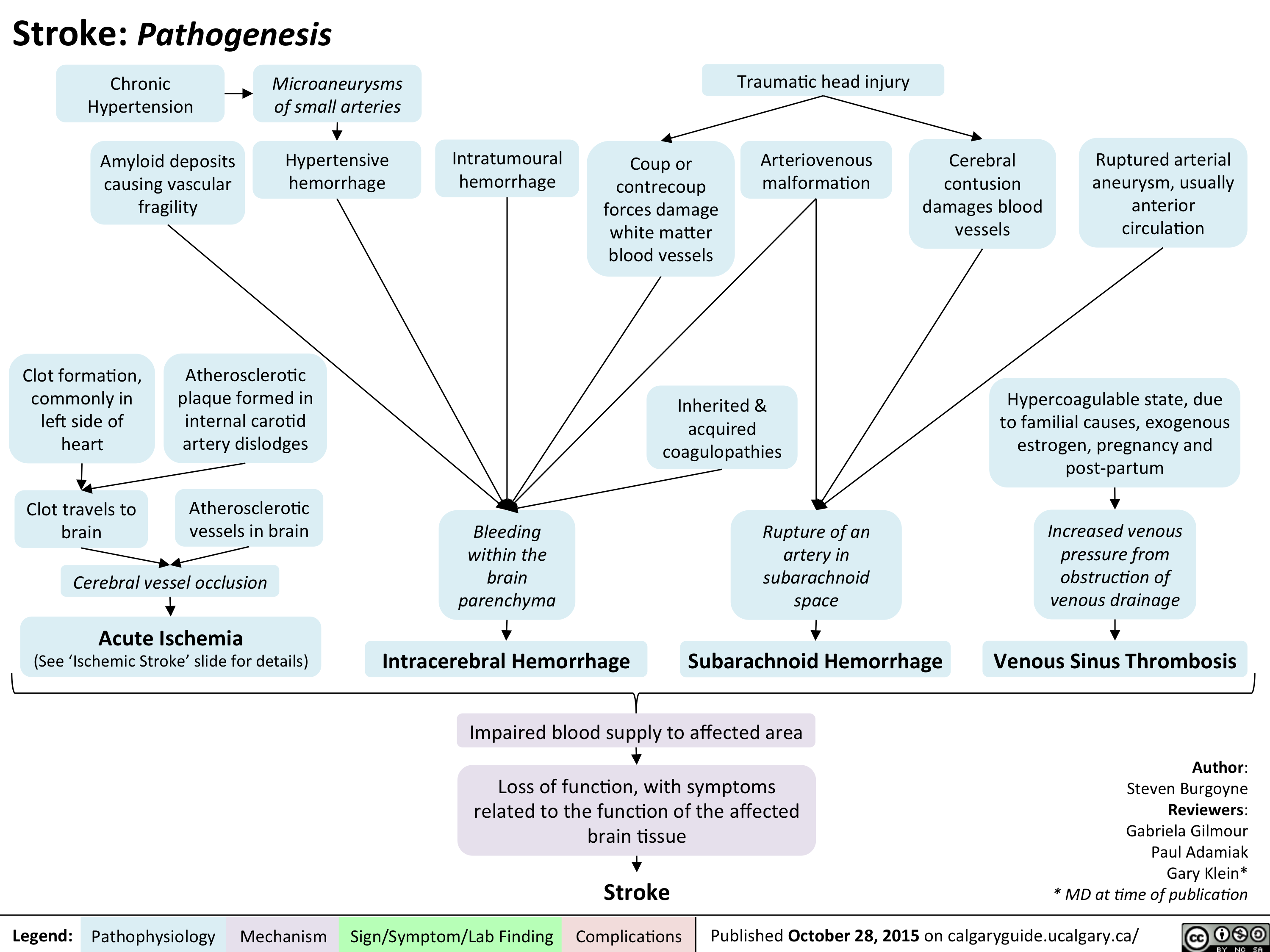
Stroke - Pathogenesis
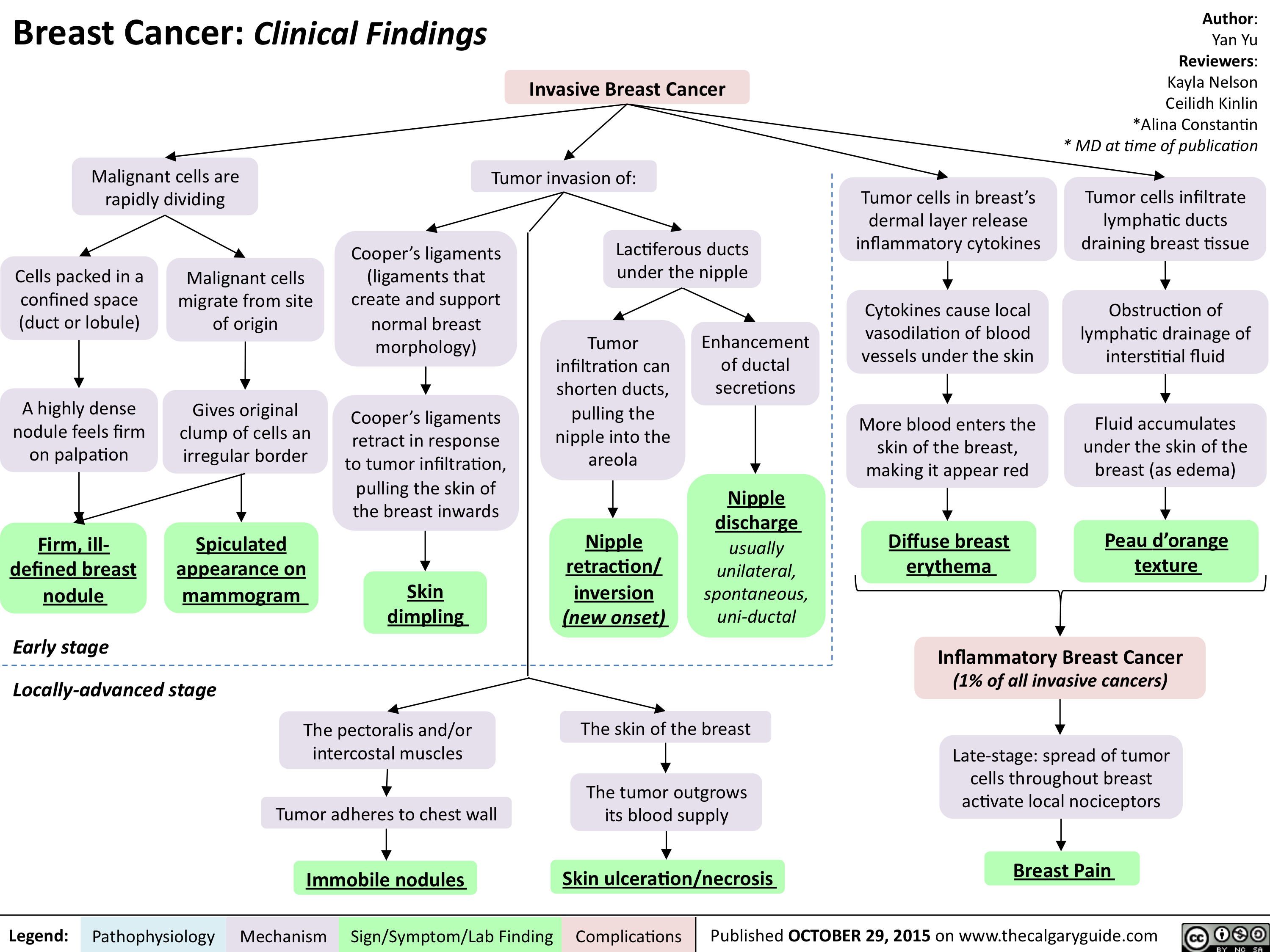
Breast Cancer - Clinical findings
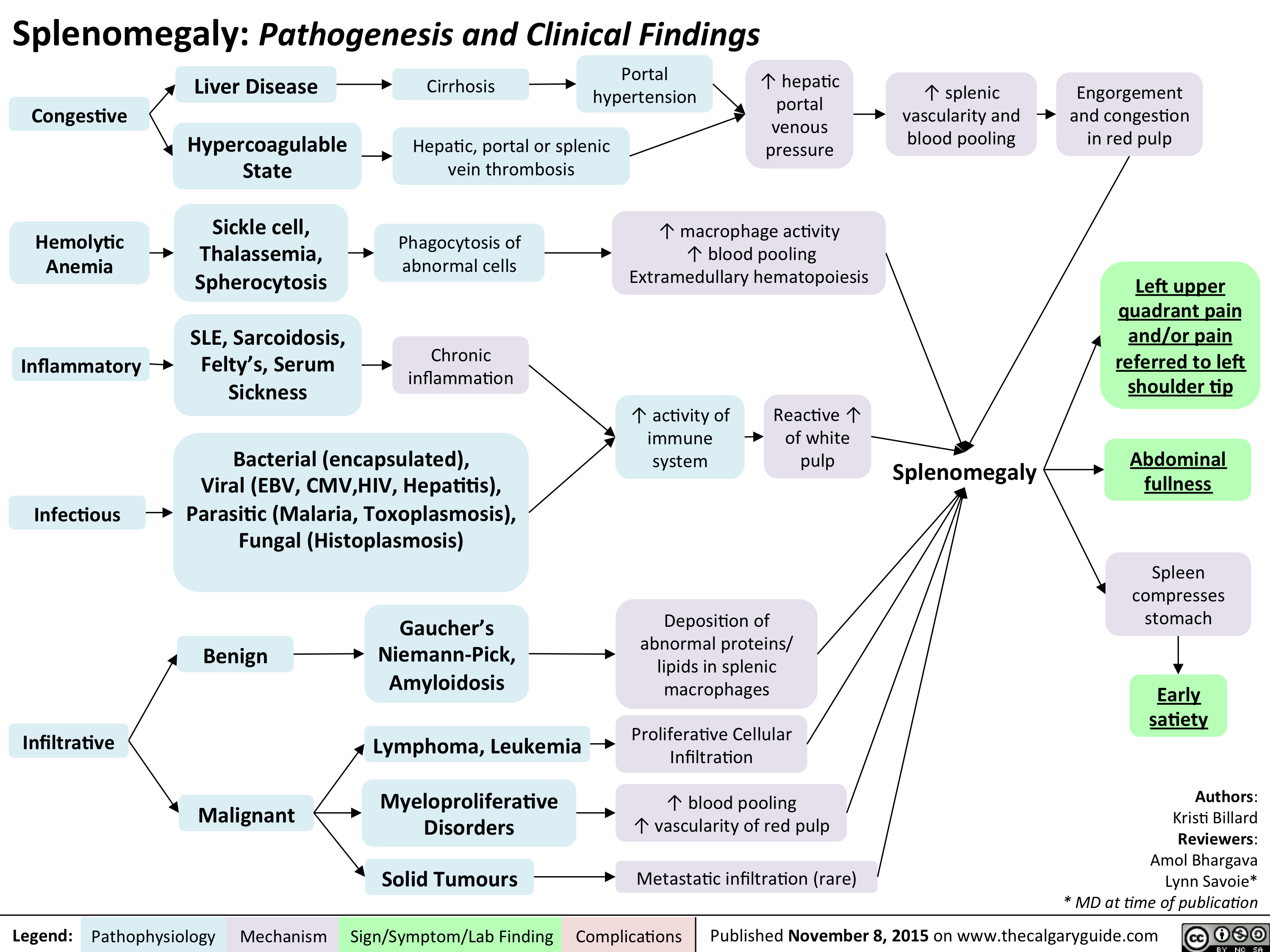
Splenomegaly - Pathogenesis and clinical findings
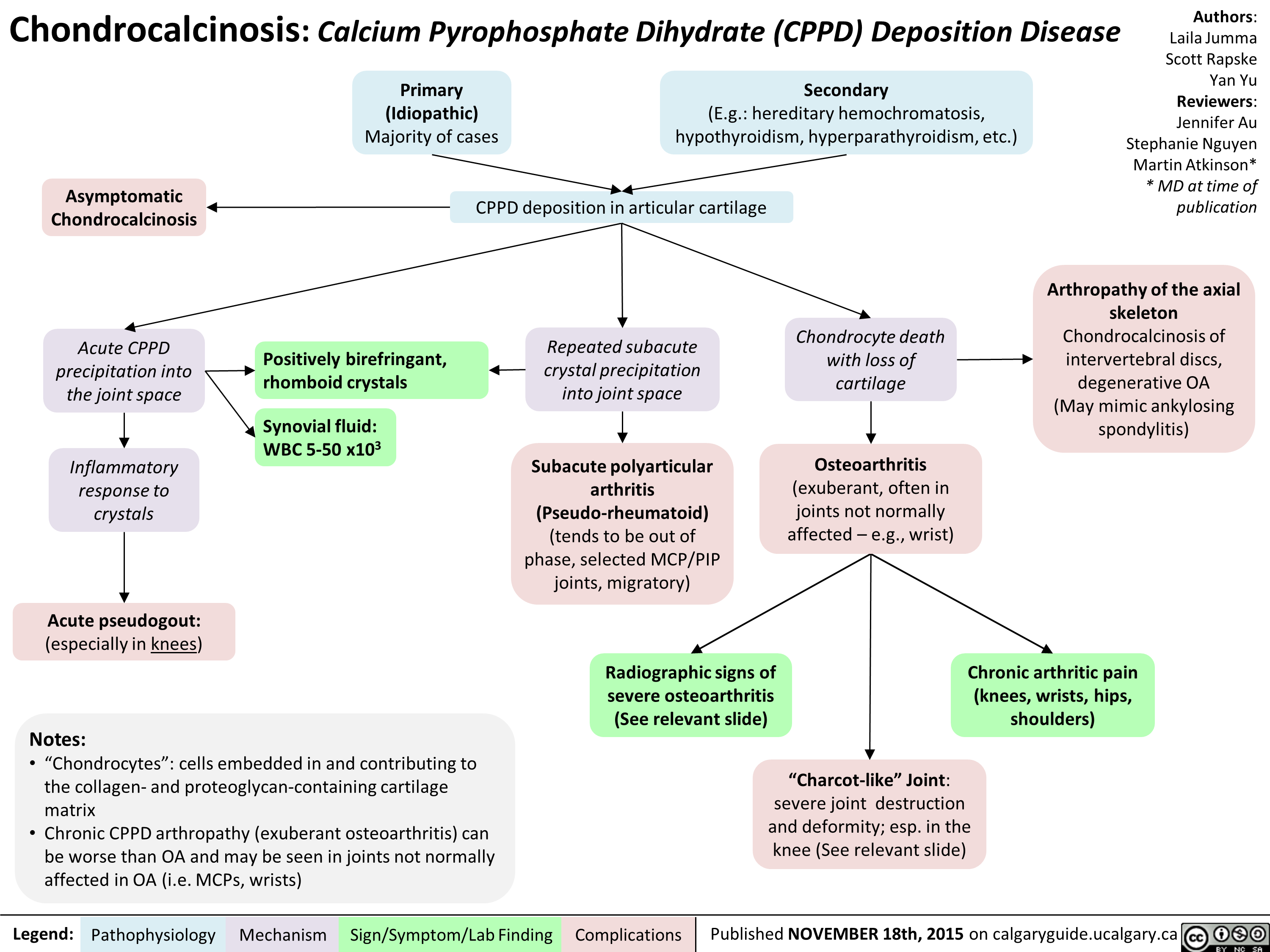
Chondrocalcinosis Calcium Pyrophosphate Dihydrate Deposition Disease
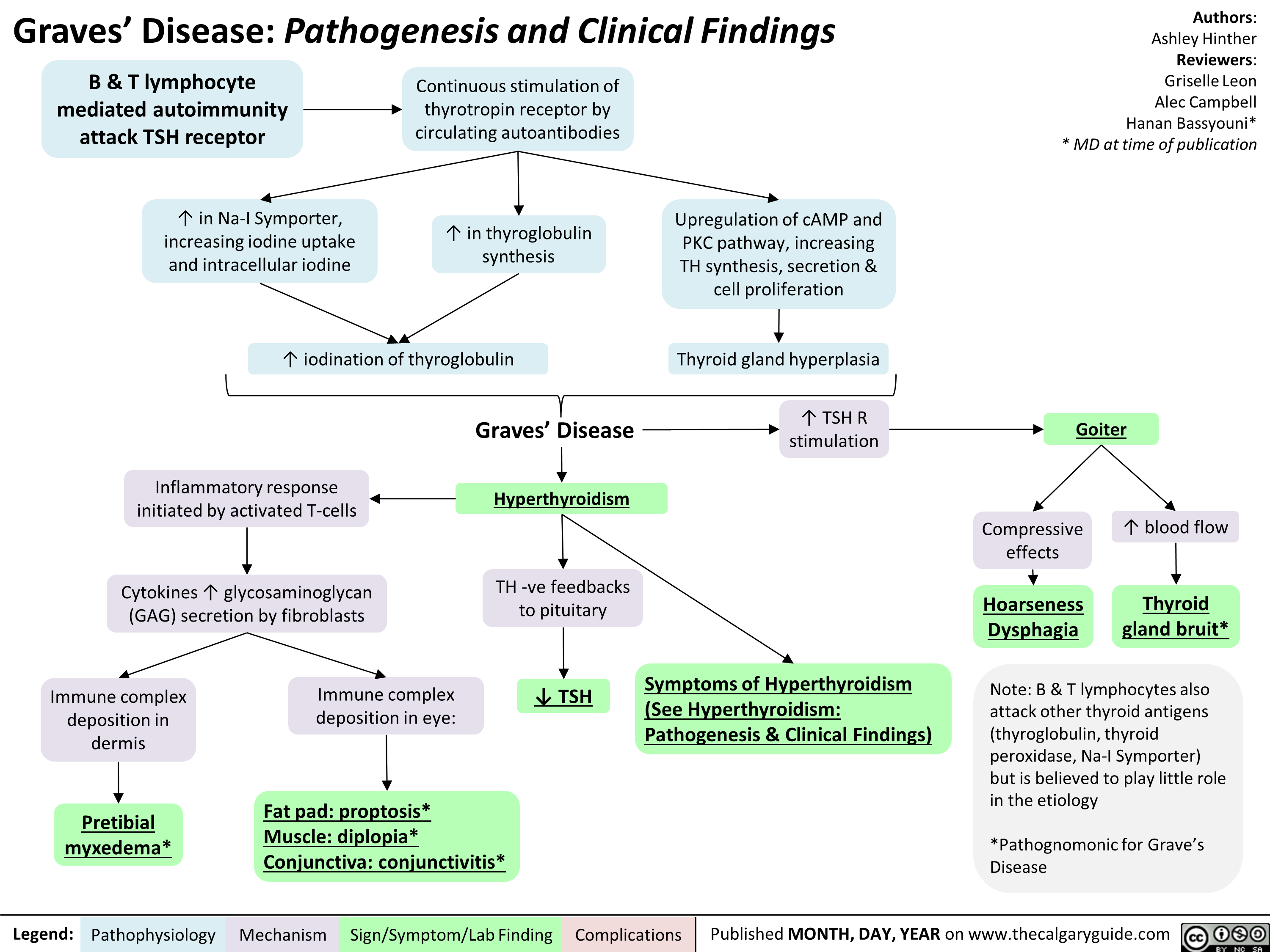
Graves' Disease Pathogenesis & Clinical Findings
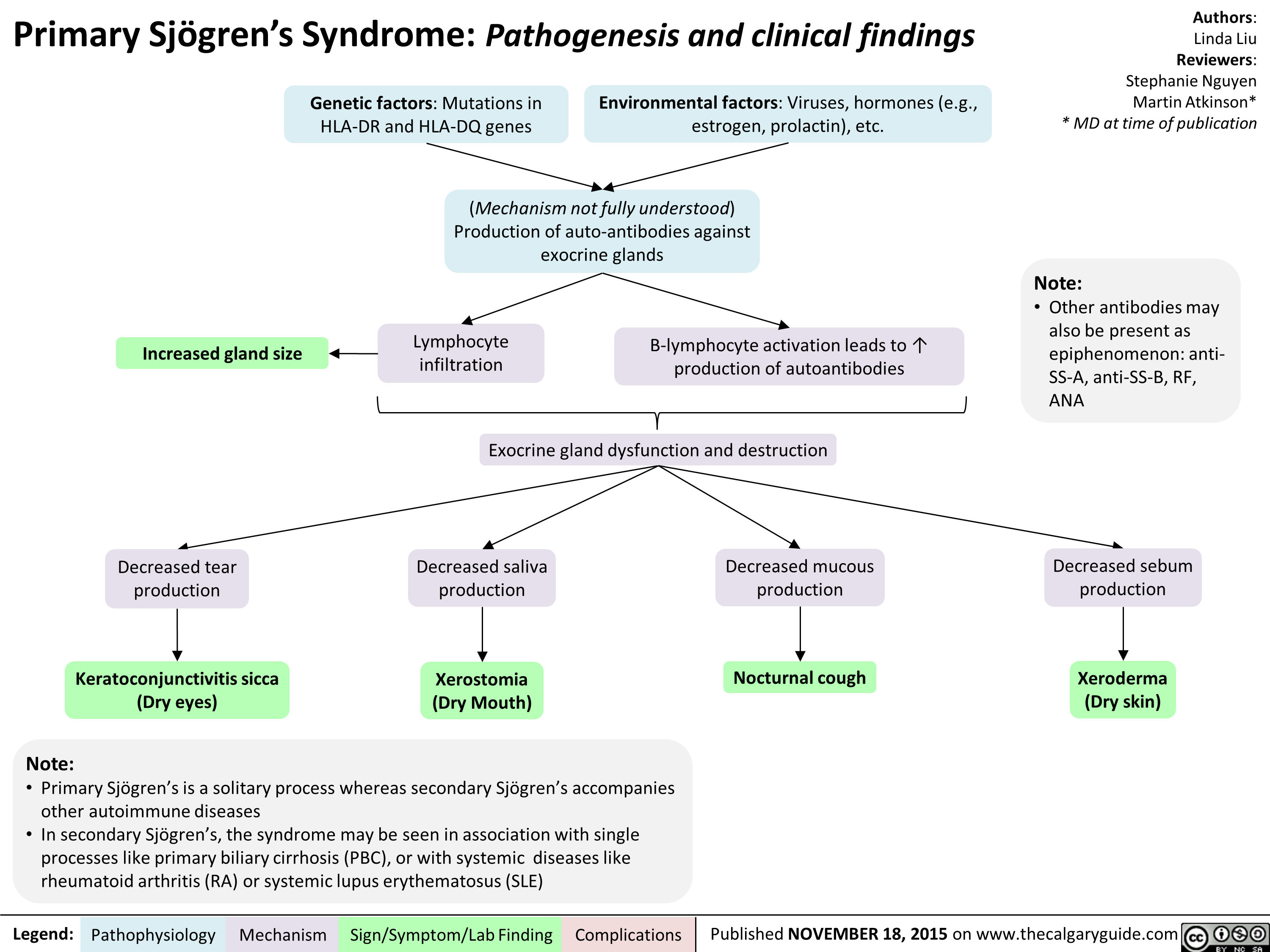
sjogrensyndrome
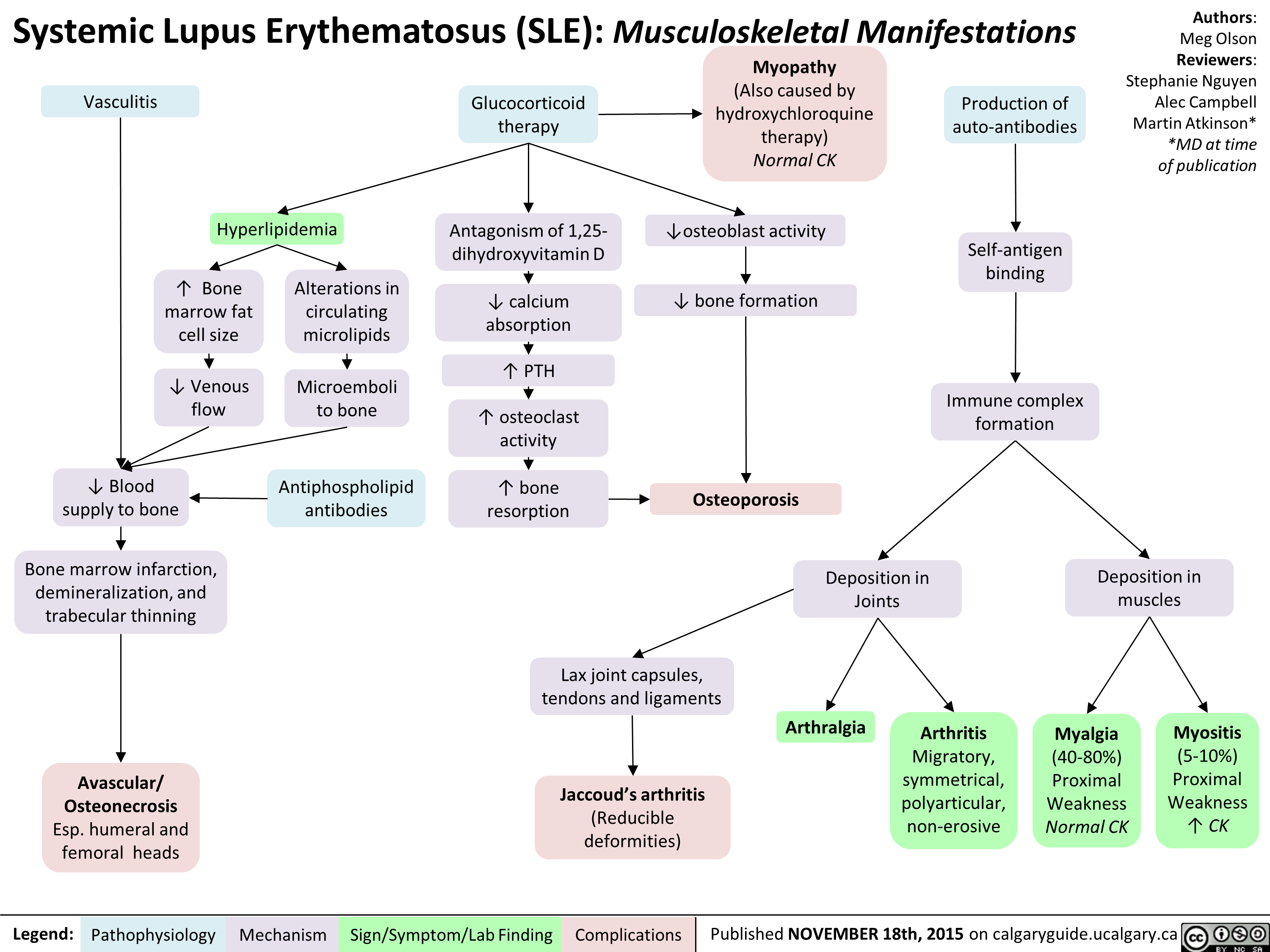
Systemic Lupus Erythematosis SLE Musculoskeletal Manifestations
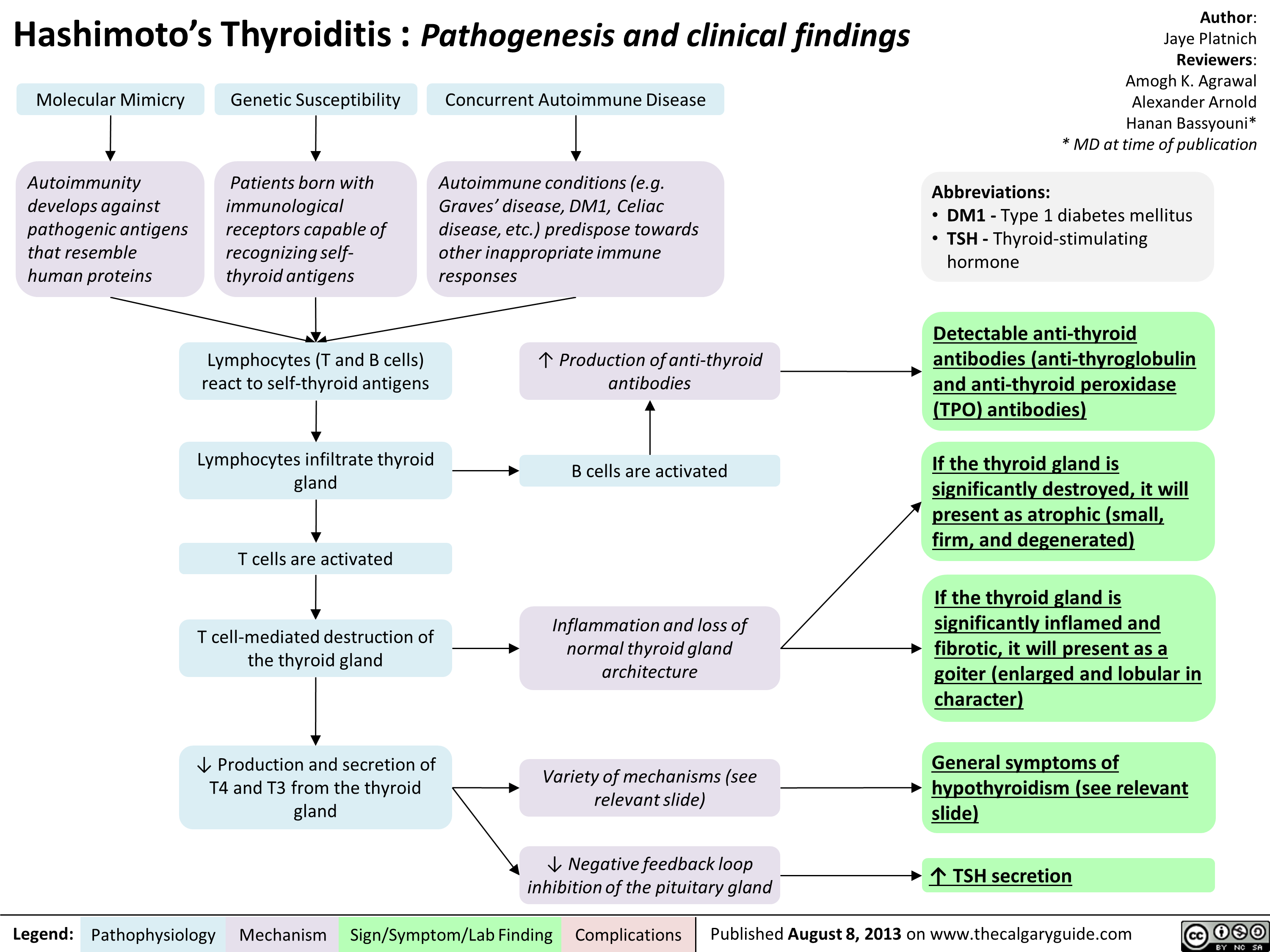
Hashimoto's Thyroiditis Natural History and Clinical Findings

Hashimoto's Thyroiditis Pathogenesis and Clinical Findings
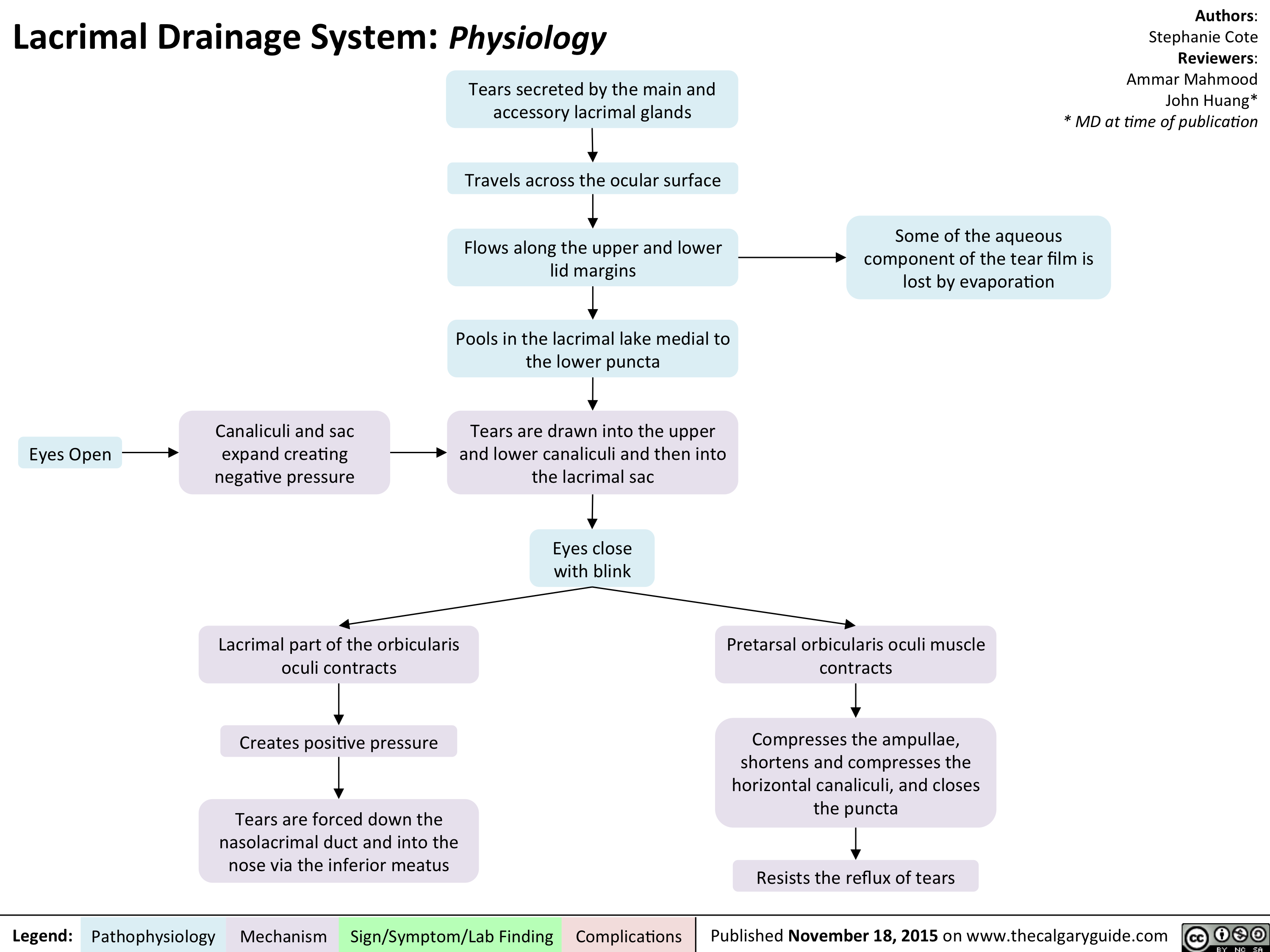
Lacrimal Drainage System - Physiology
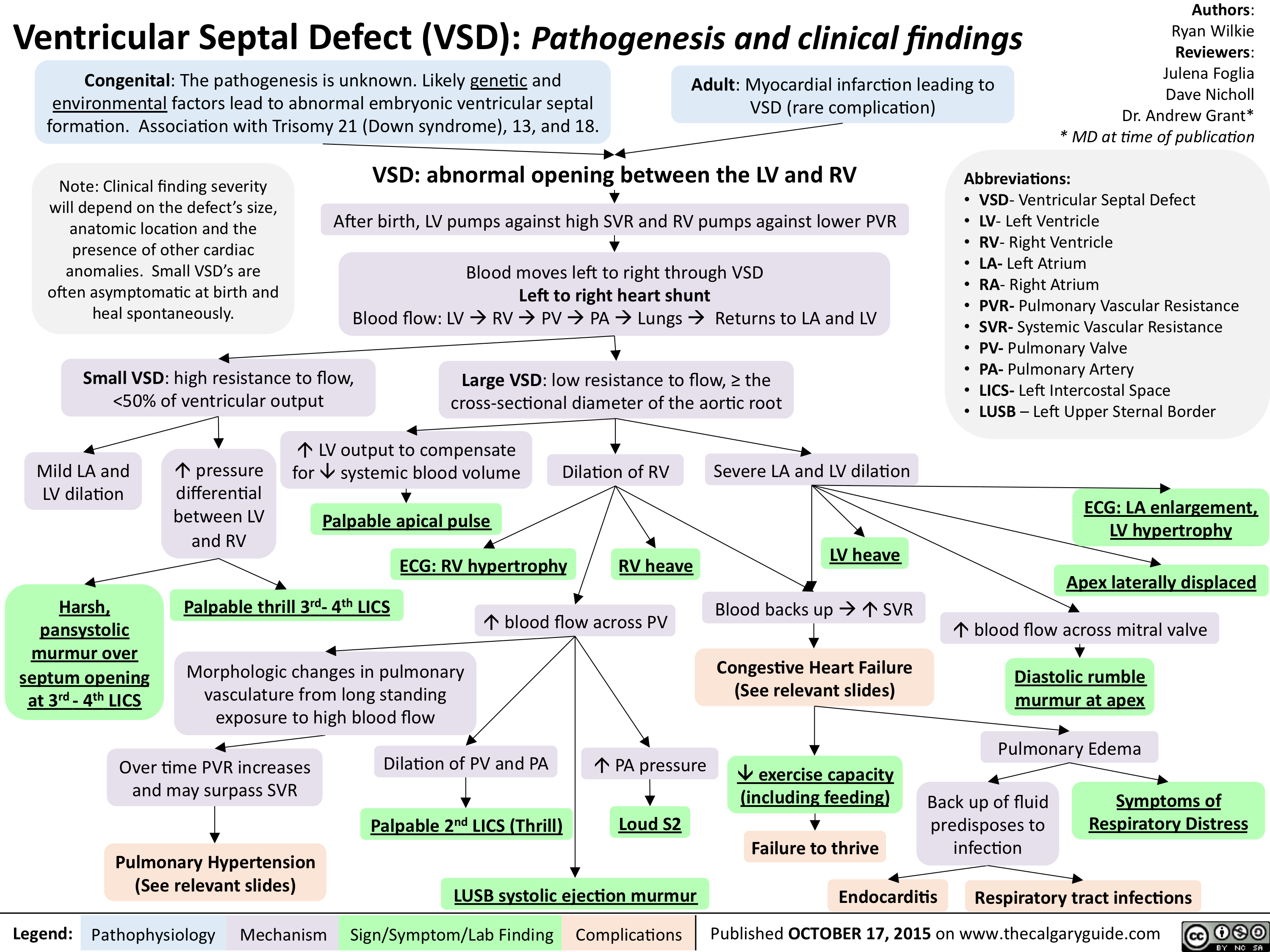
Ventricular Septal Defect (VSD)-Pathogenesis and clinical findings
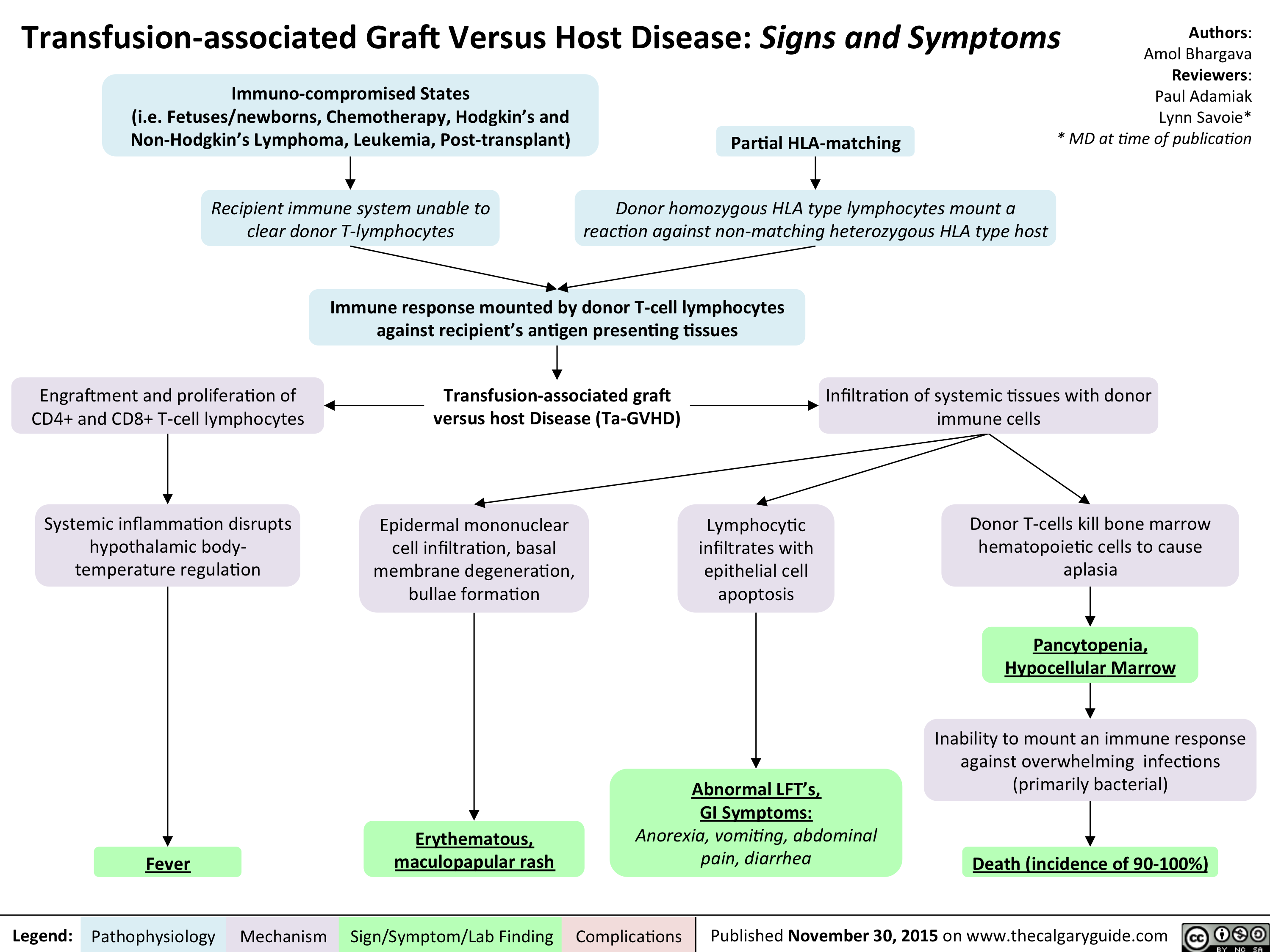
Transfusion-associated Graft Versus Host Disease - Signs and Symptoms
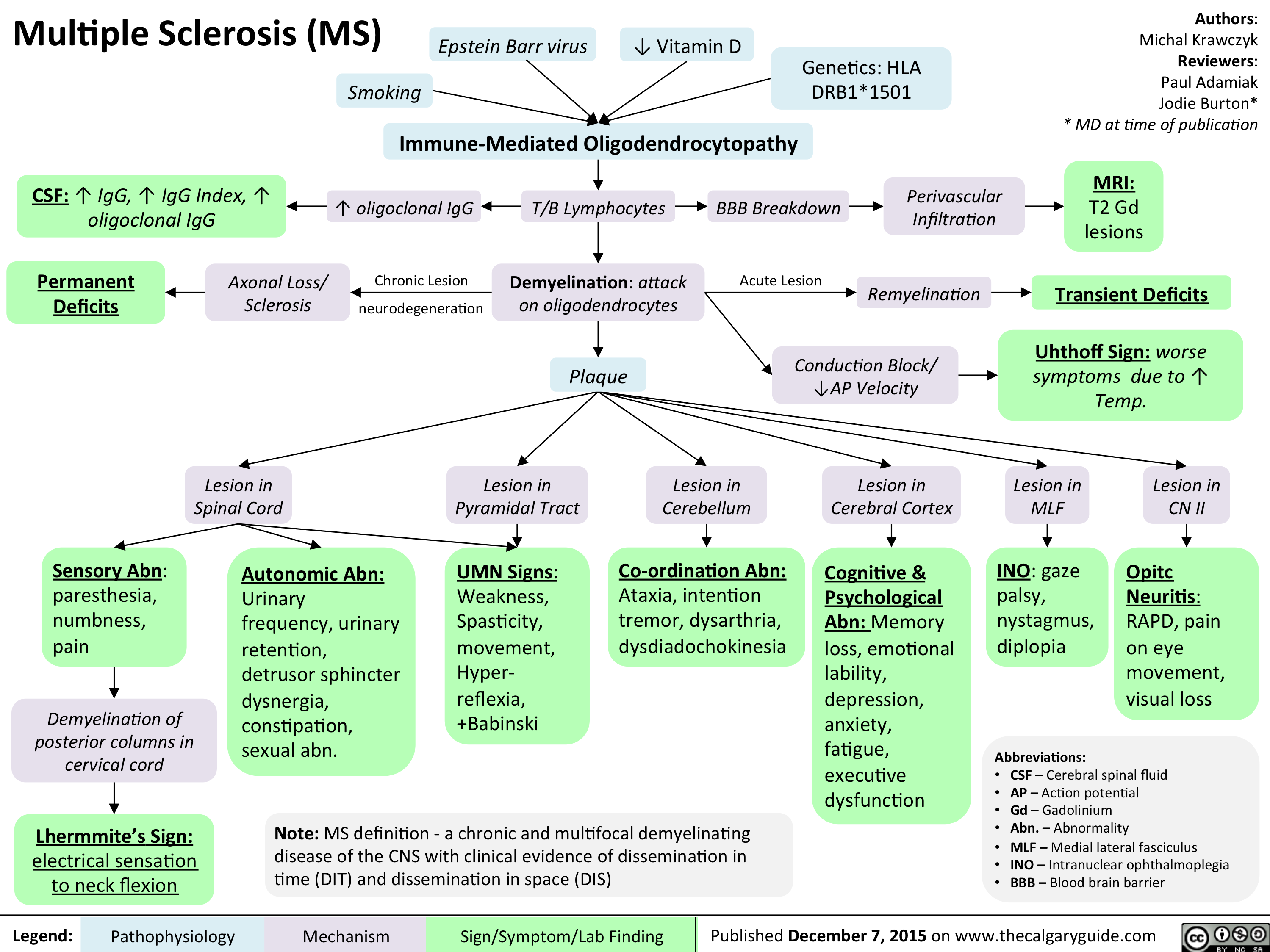
Multiple sclerosis (MS)
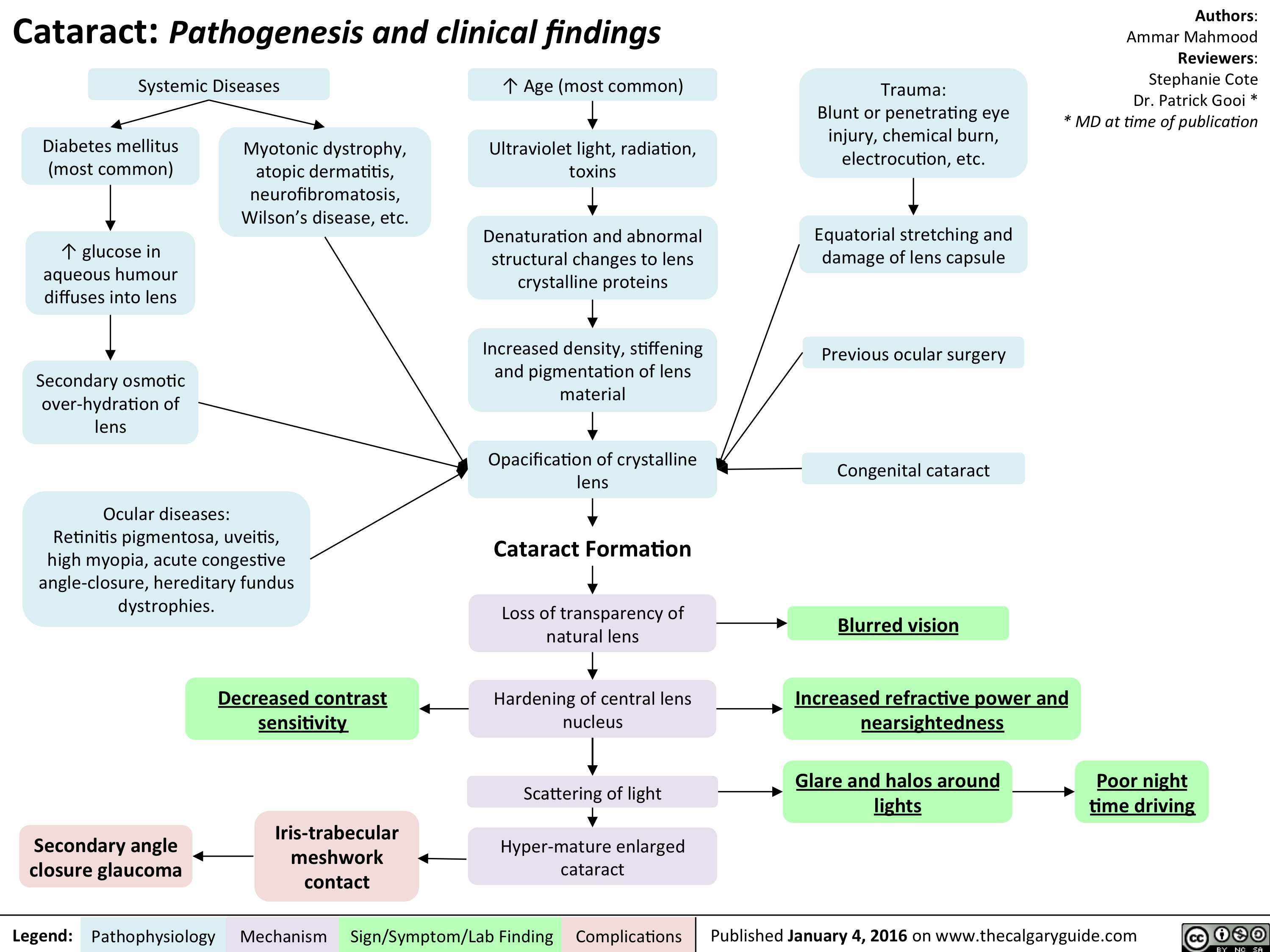
Cataracts - pathogenesis and clinical findings
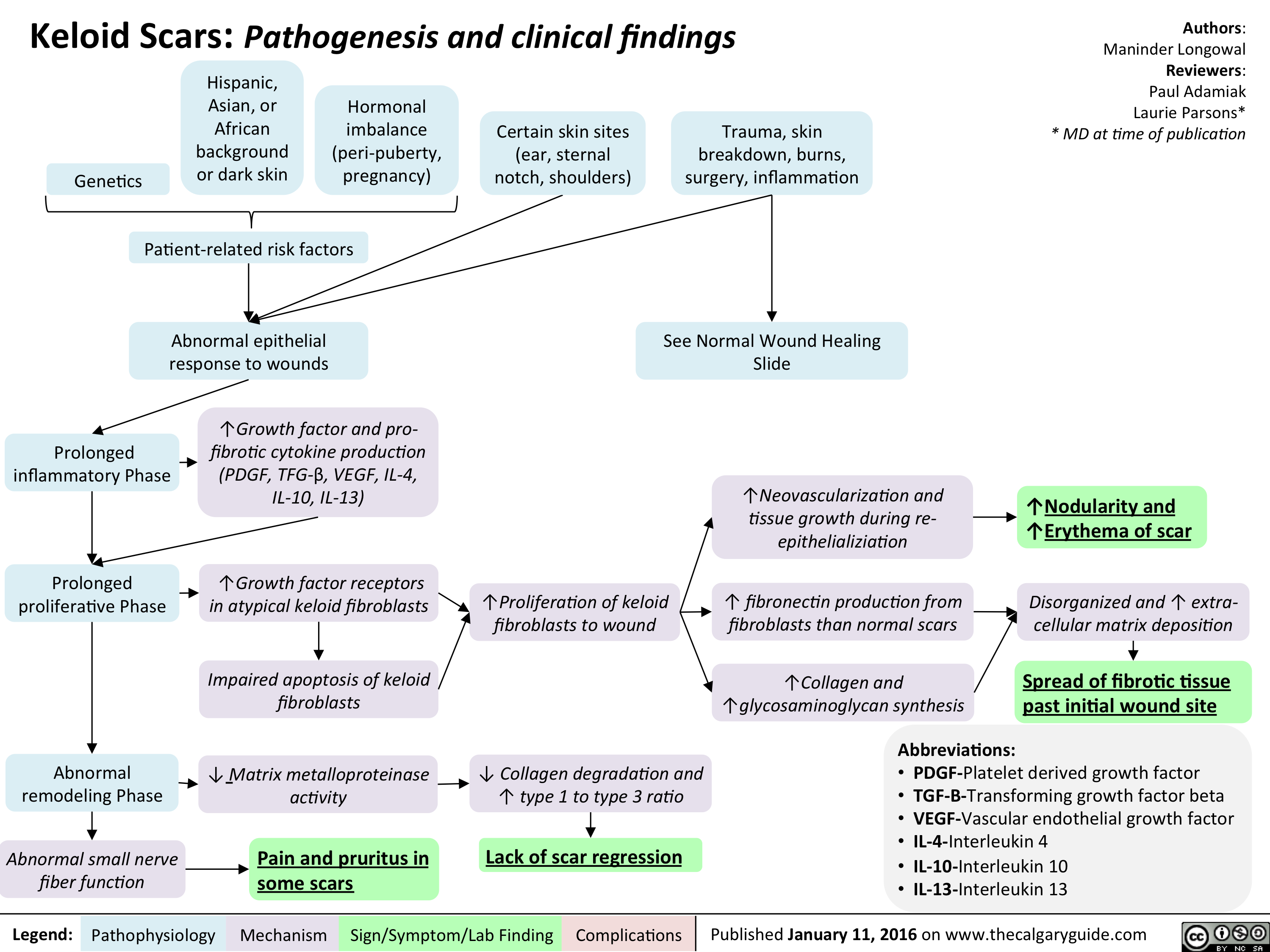
Keloid scar - pathogenesis and clinical findings
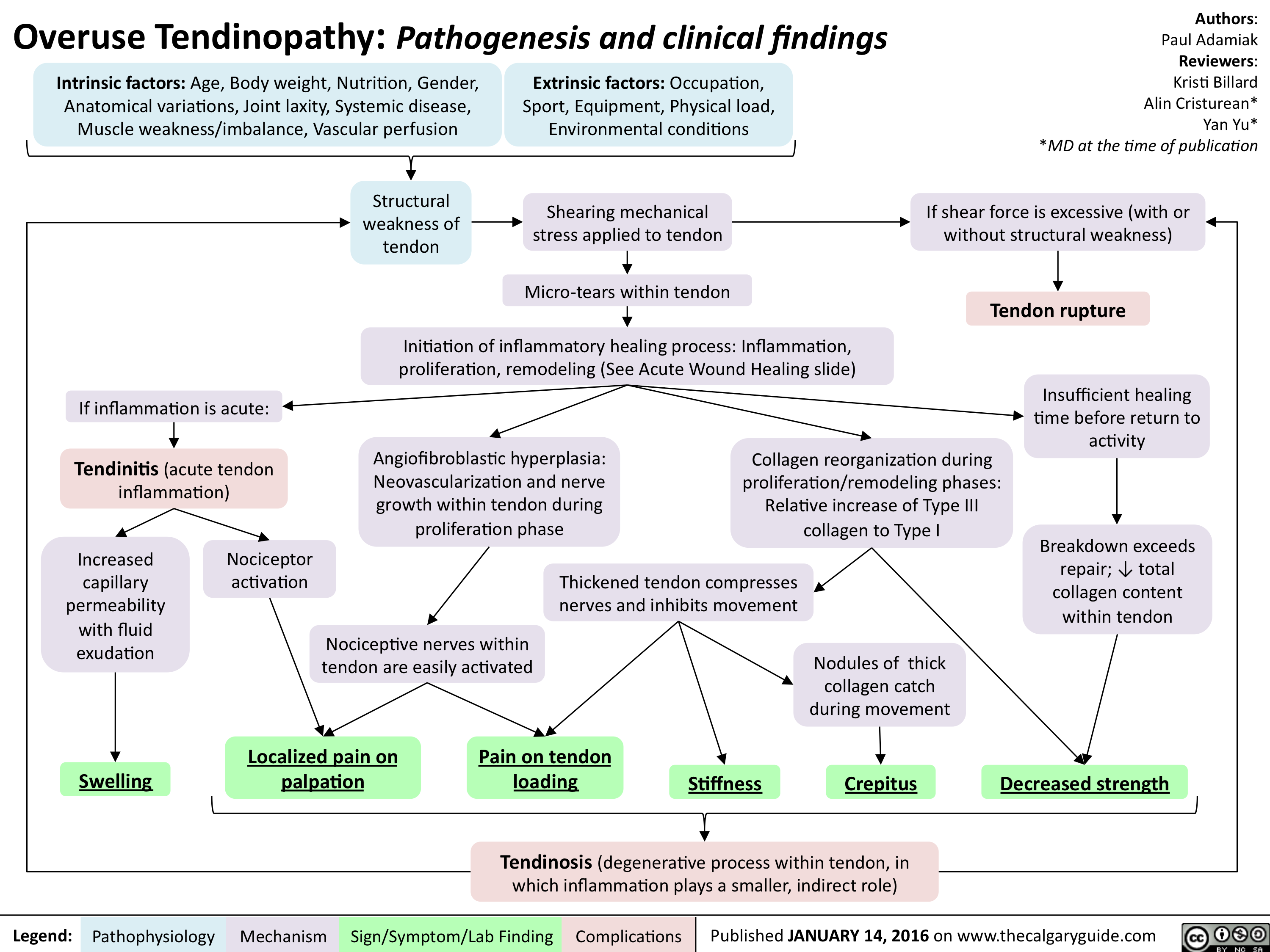
Overuse Tendinopathy -Pathogenesis and clinical findings
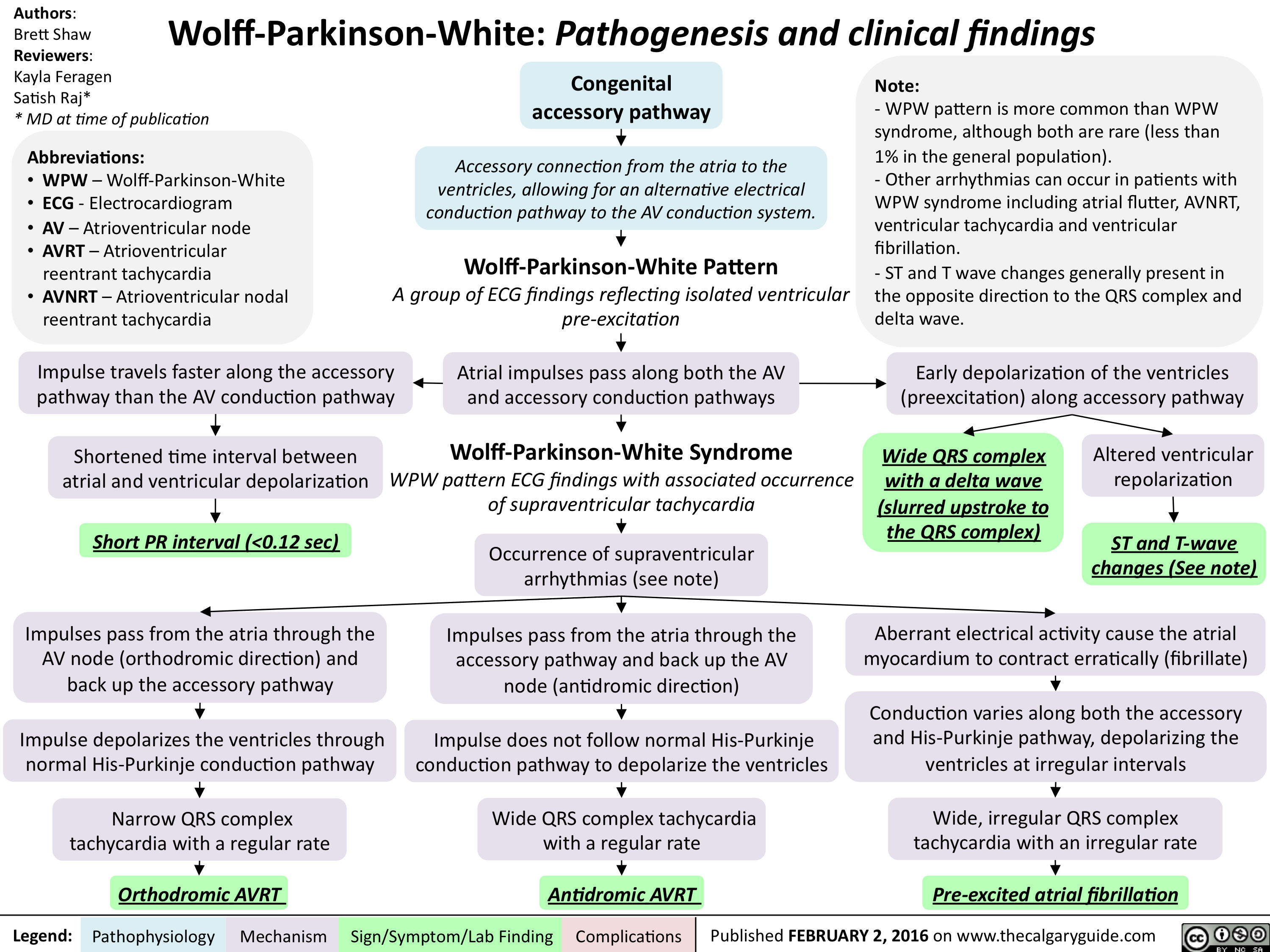
Wolff Parkinson White - Pathogenesis and clinical findings
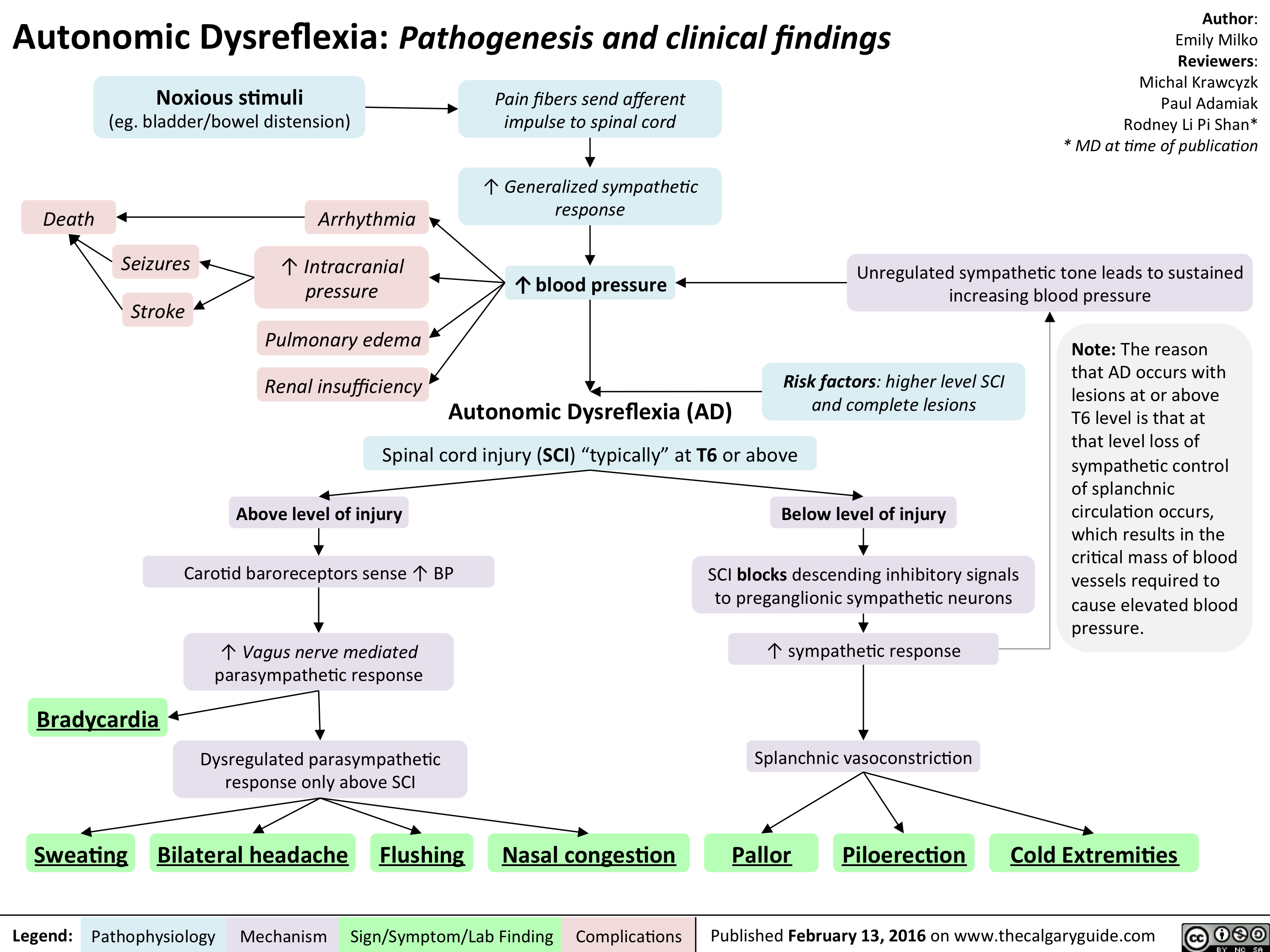
Autonomic Dysreflexia - Pathogenesis and clinical findings
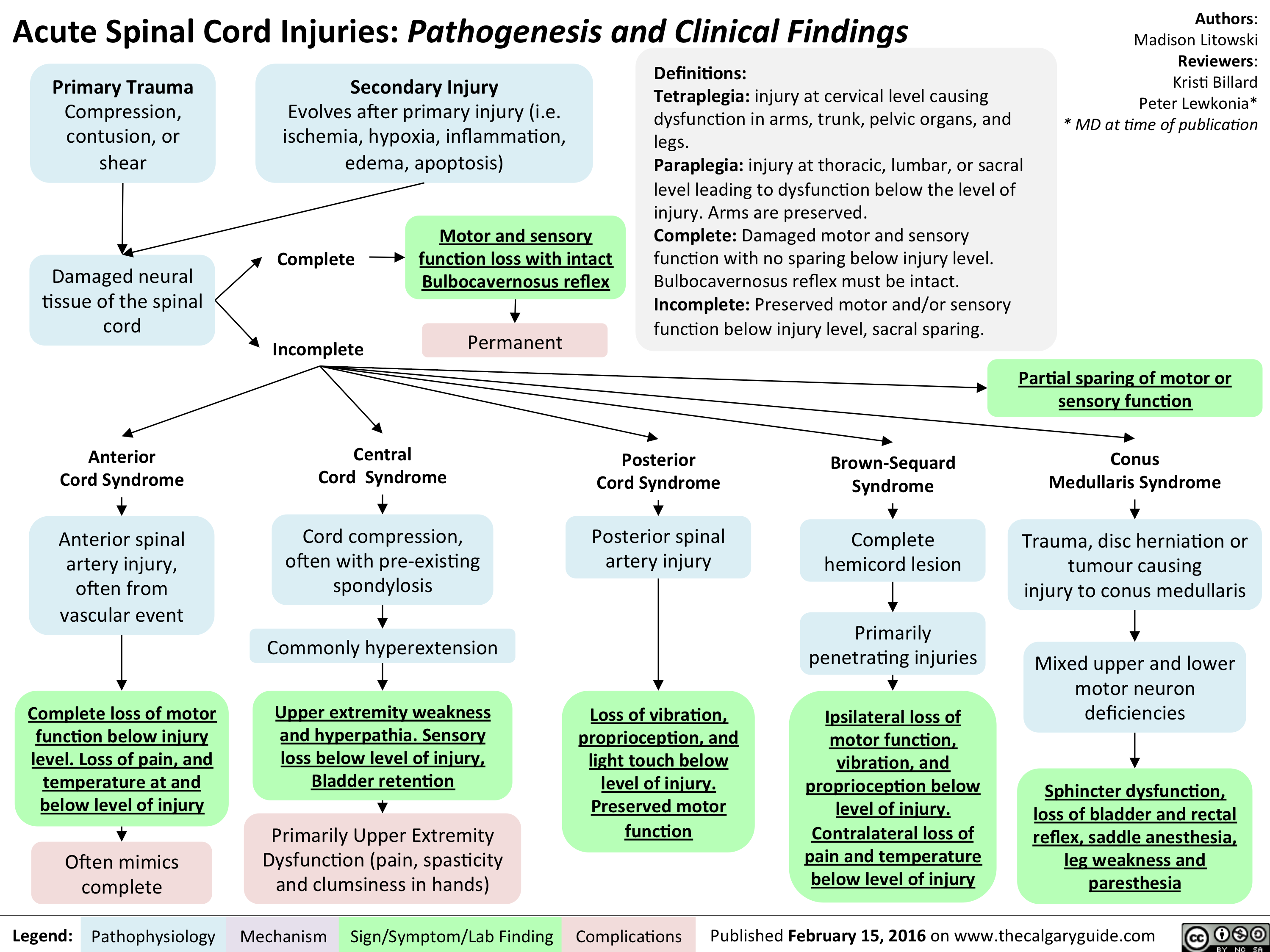
Acute Spinal Cord Injuries - Pathogenesis and clinical findings
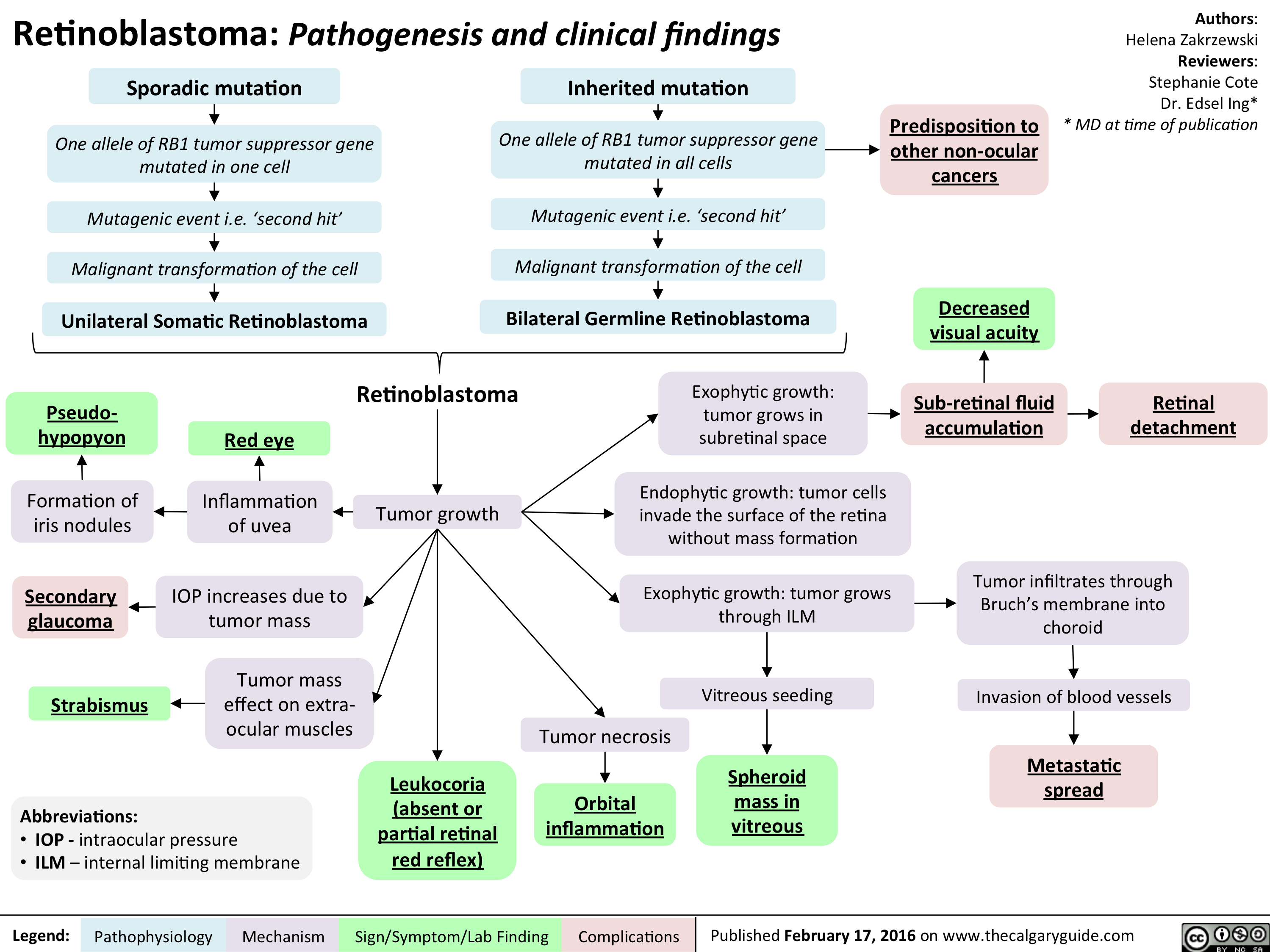
Retinoblastoma - Pathogenesis and clinical findings
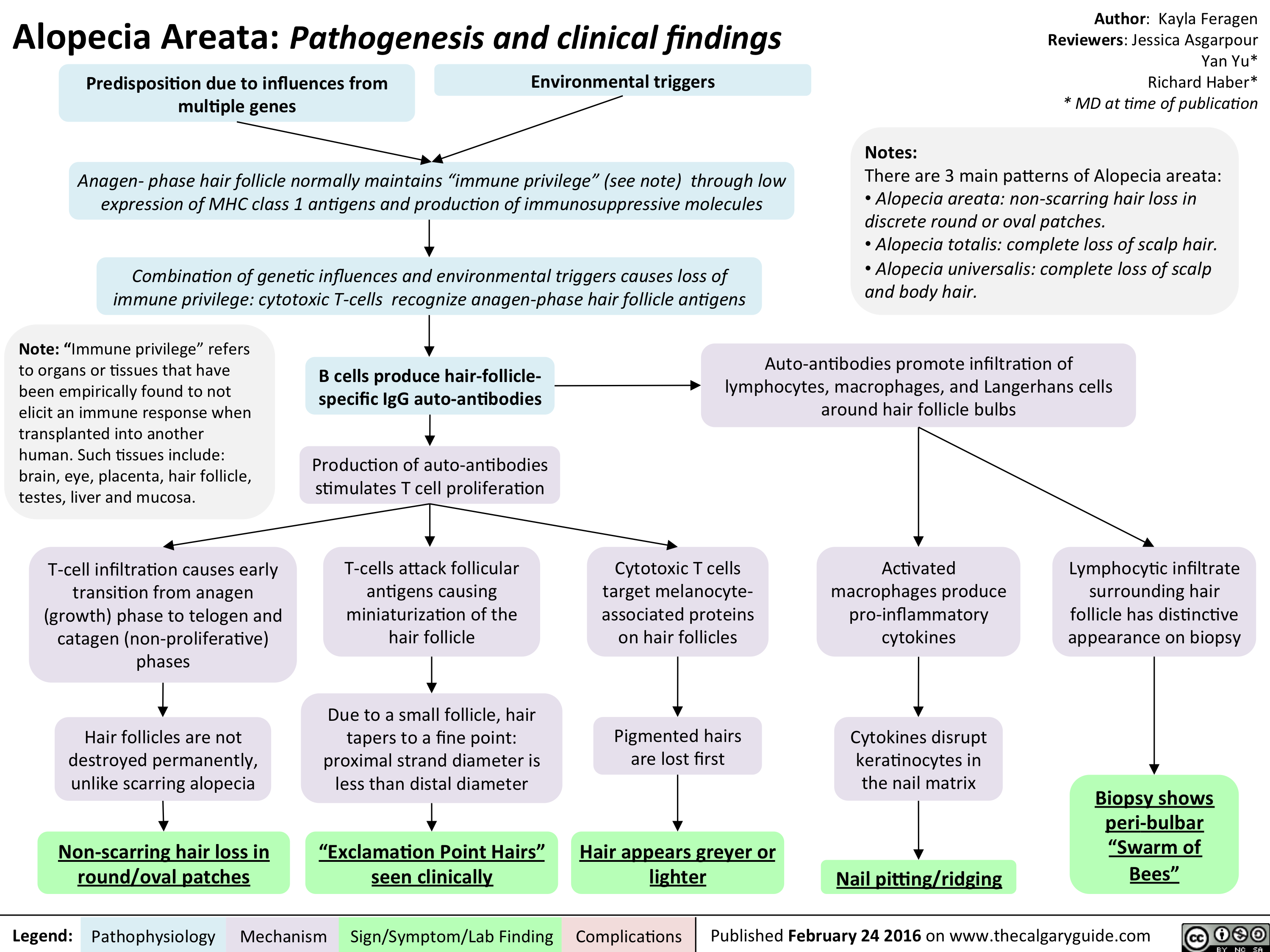
Alopecia Areata - Pathogenesis and clinical findings
Seasonal Affective Disorder - Pathogenesis and clinical findings v2

Yu, Y - Schizophrenia Pathogenesis and Clinical Findings - March 26 2016
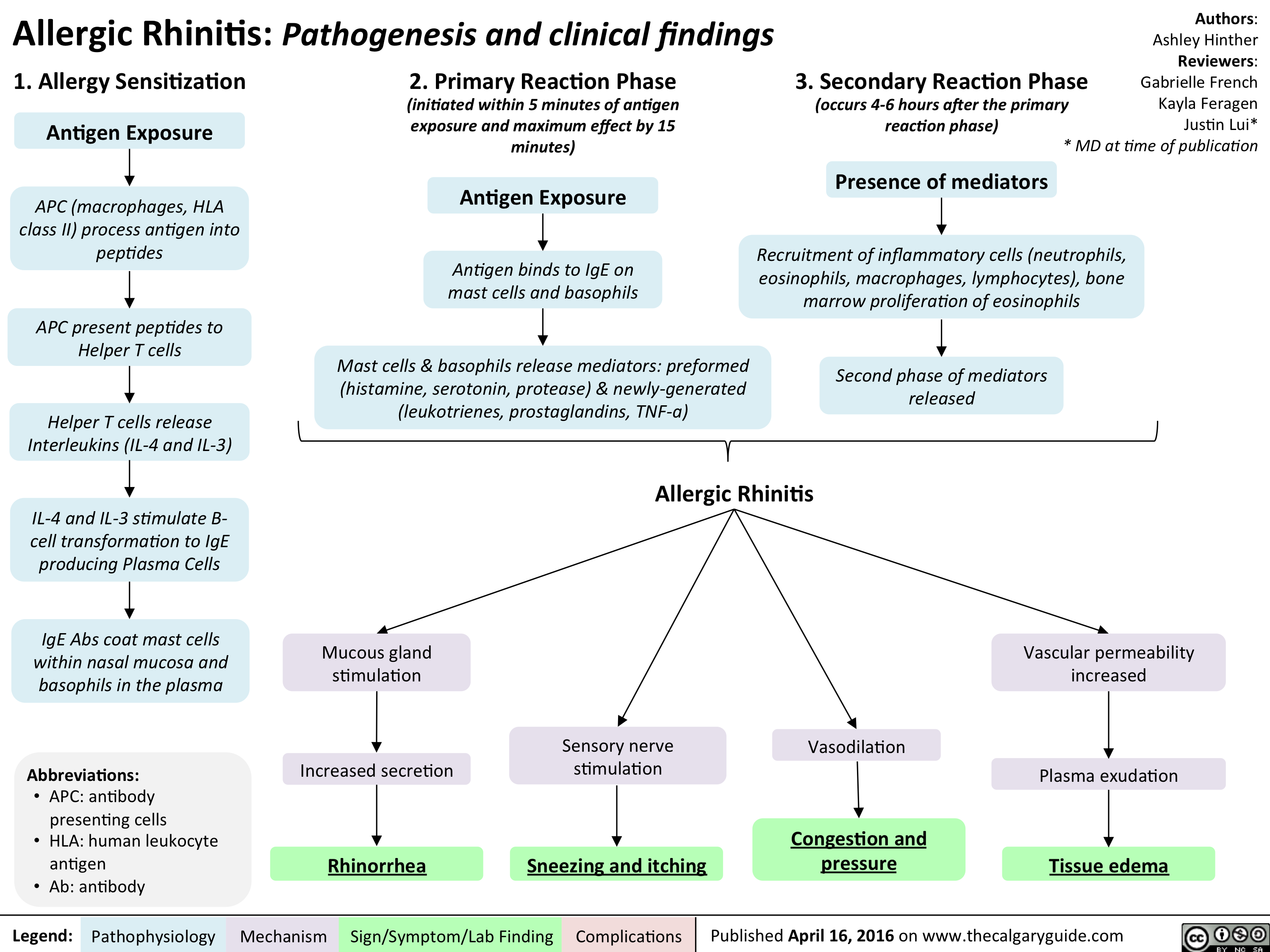
Allergic Rhinitis - Pathogenesis and clinical findings
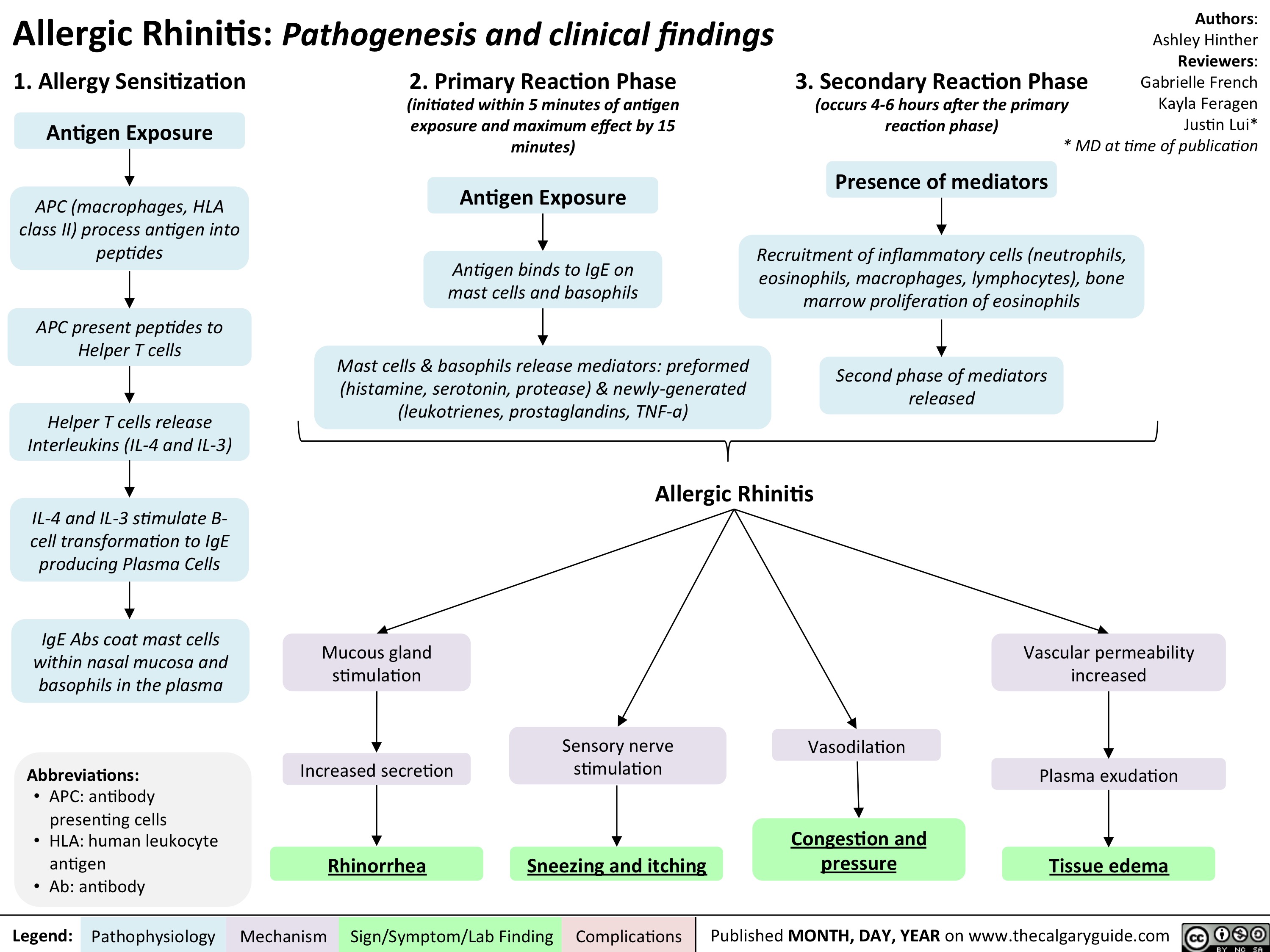
Allergic Rhinitis - Pathogenesis and clinical findings
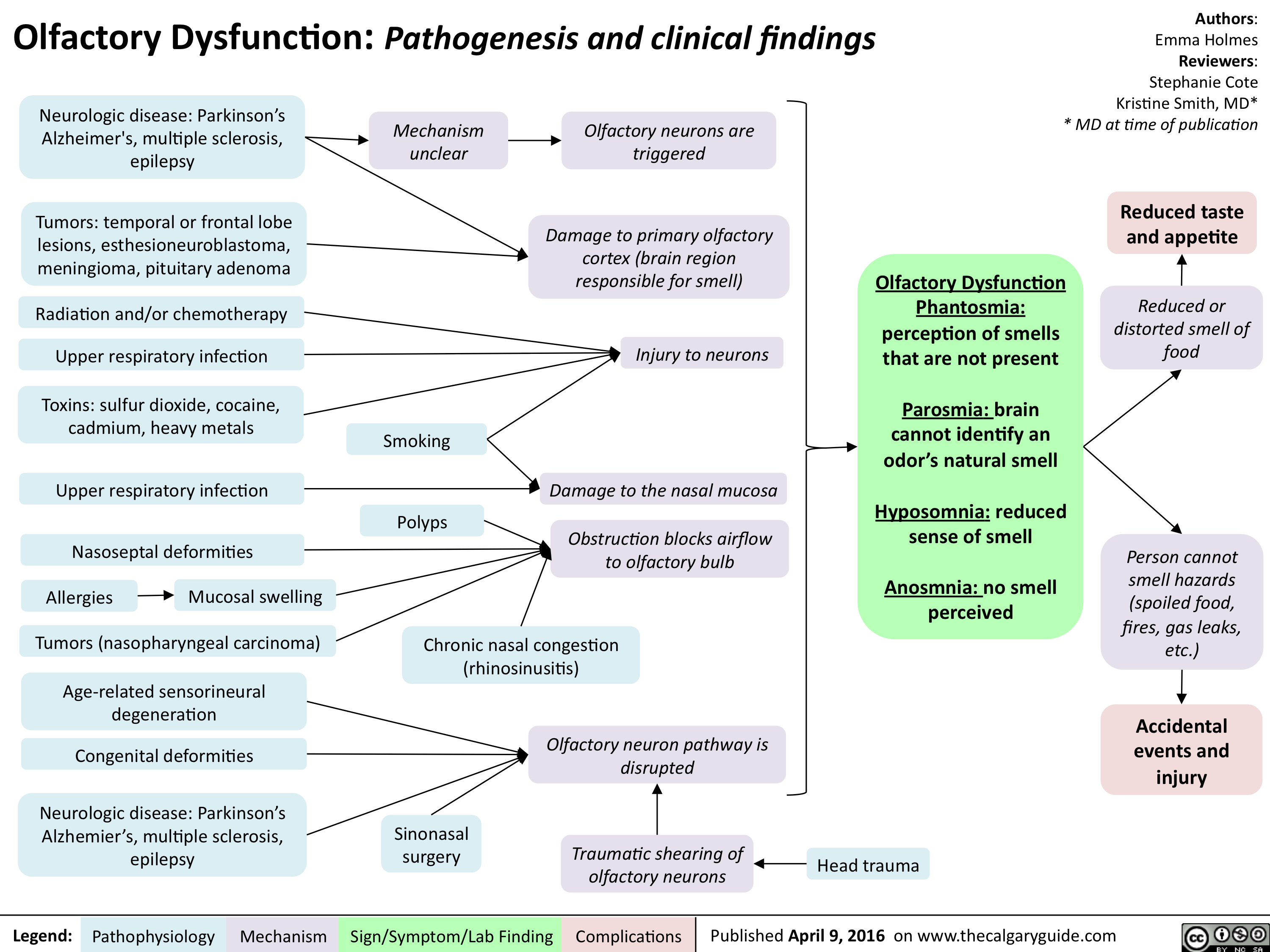
Olfactory Dysfunction - Pathogenesis and clinical findings
Tinnitus

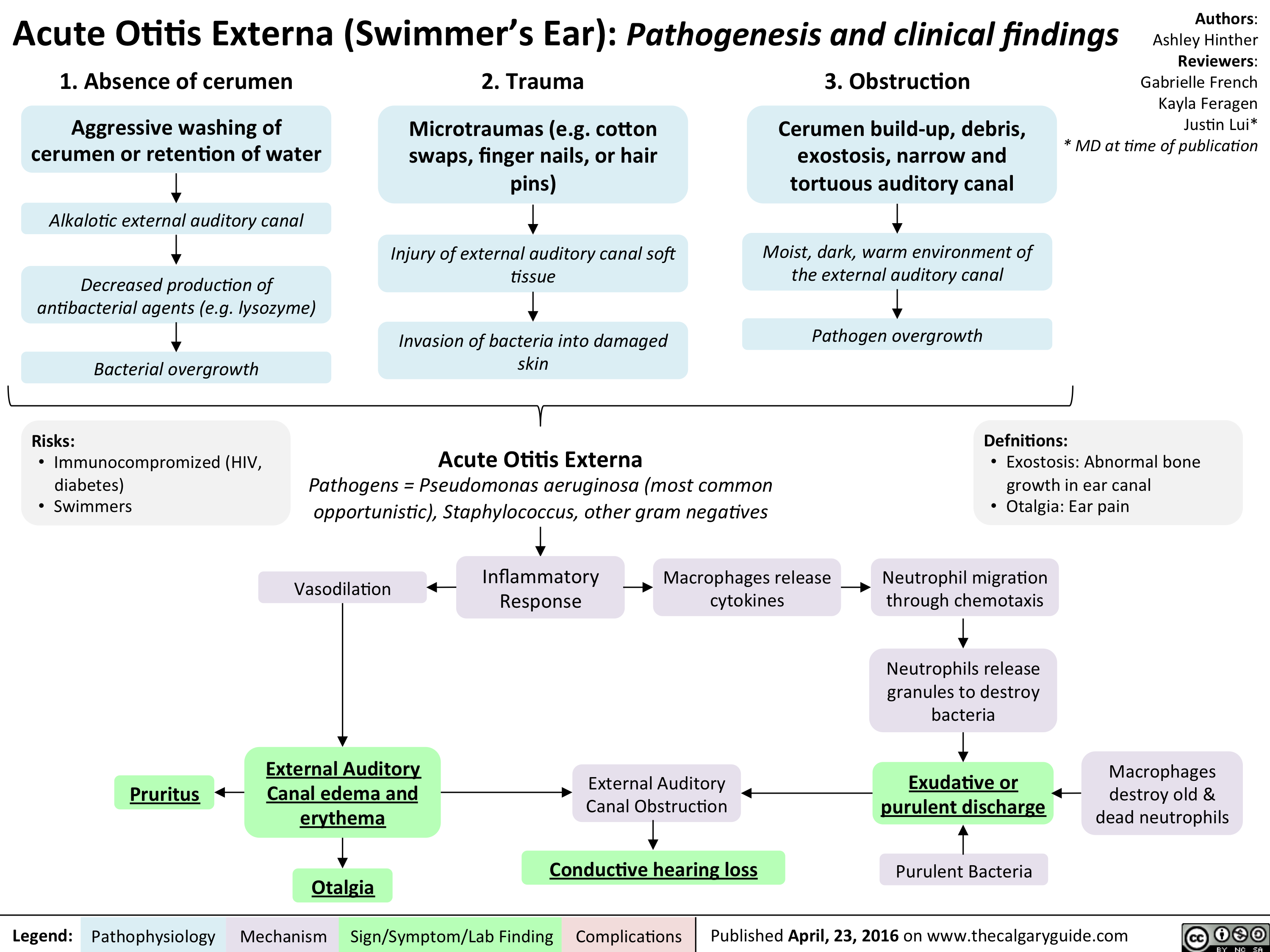
Acute Otitis Externa
Psoriasis: Pathogenesis and clinical findings

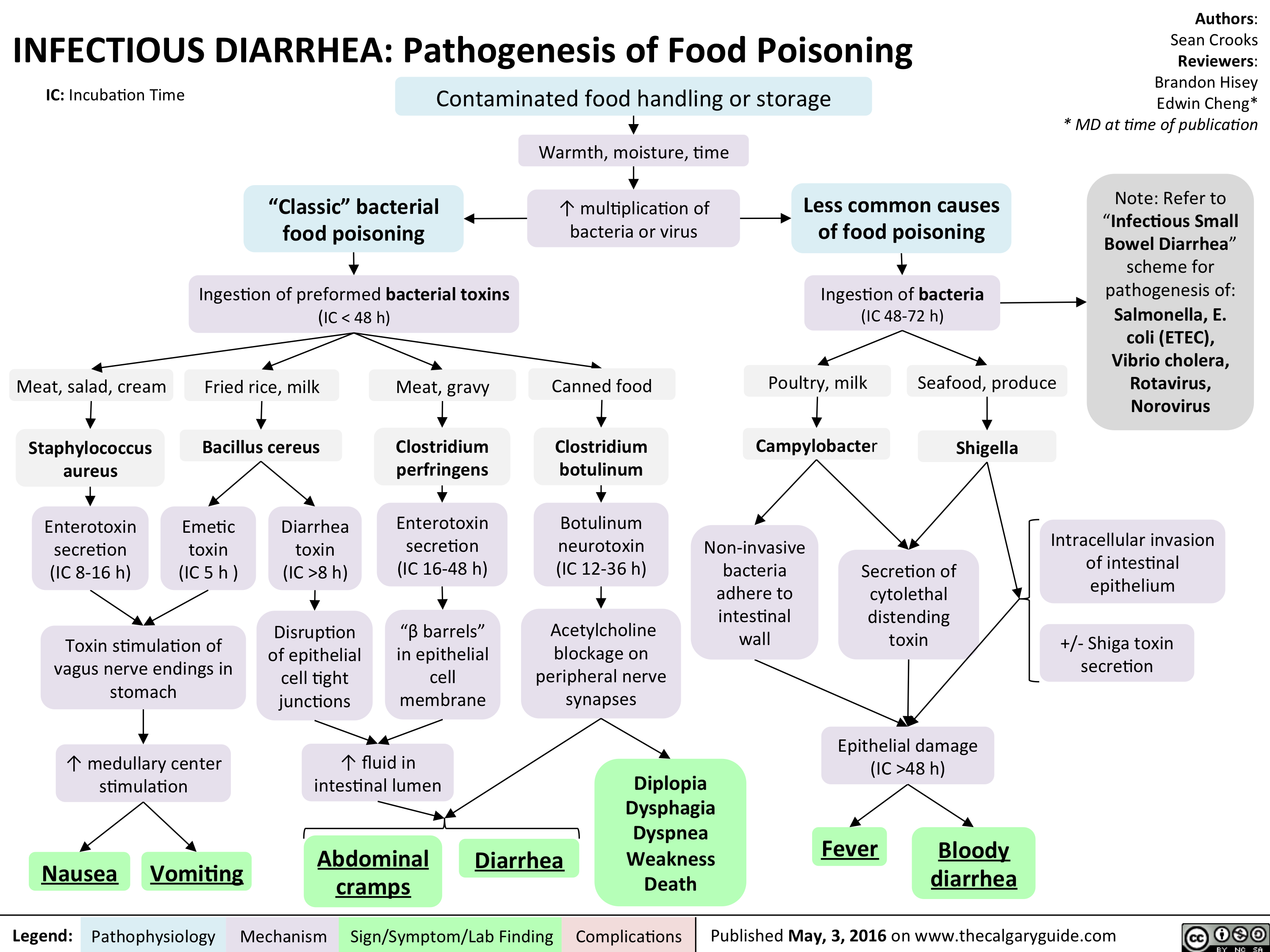
InfectiousFoodPoisoning
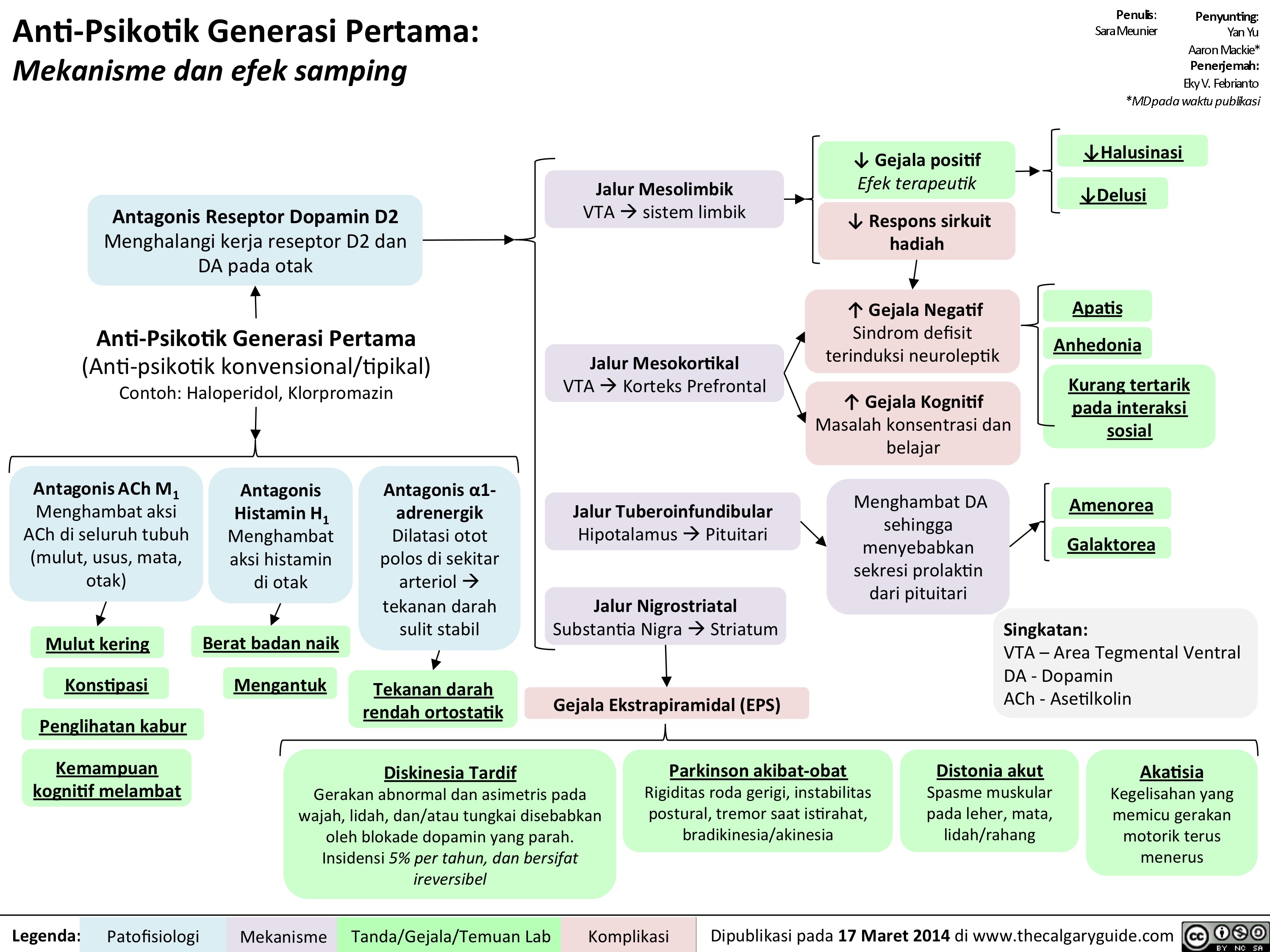
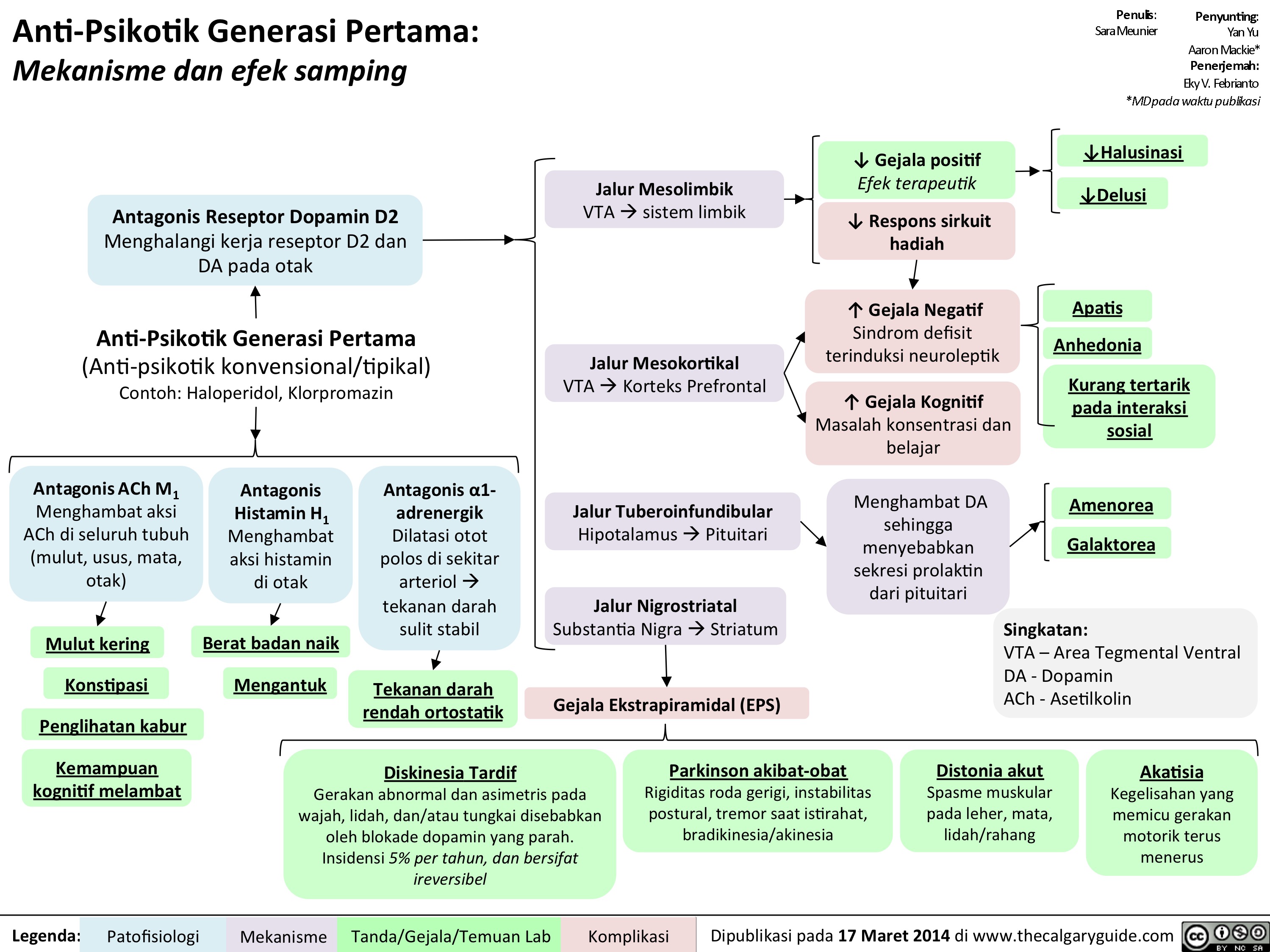
1st gen antipsychotics Translated

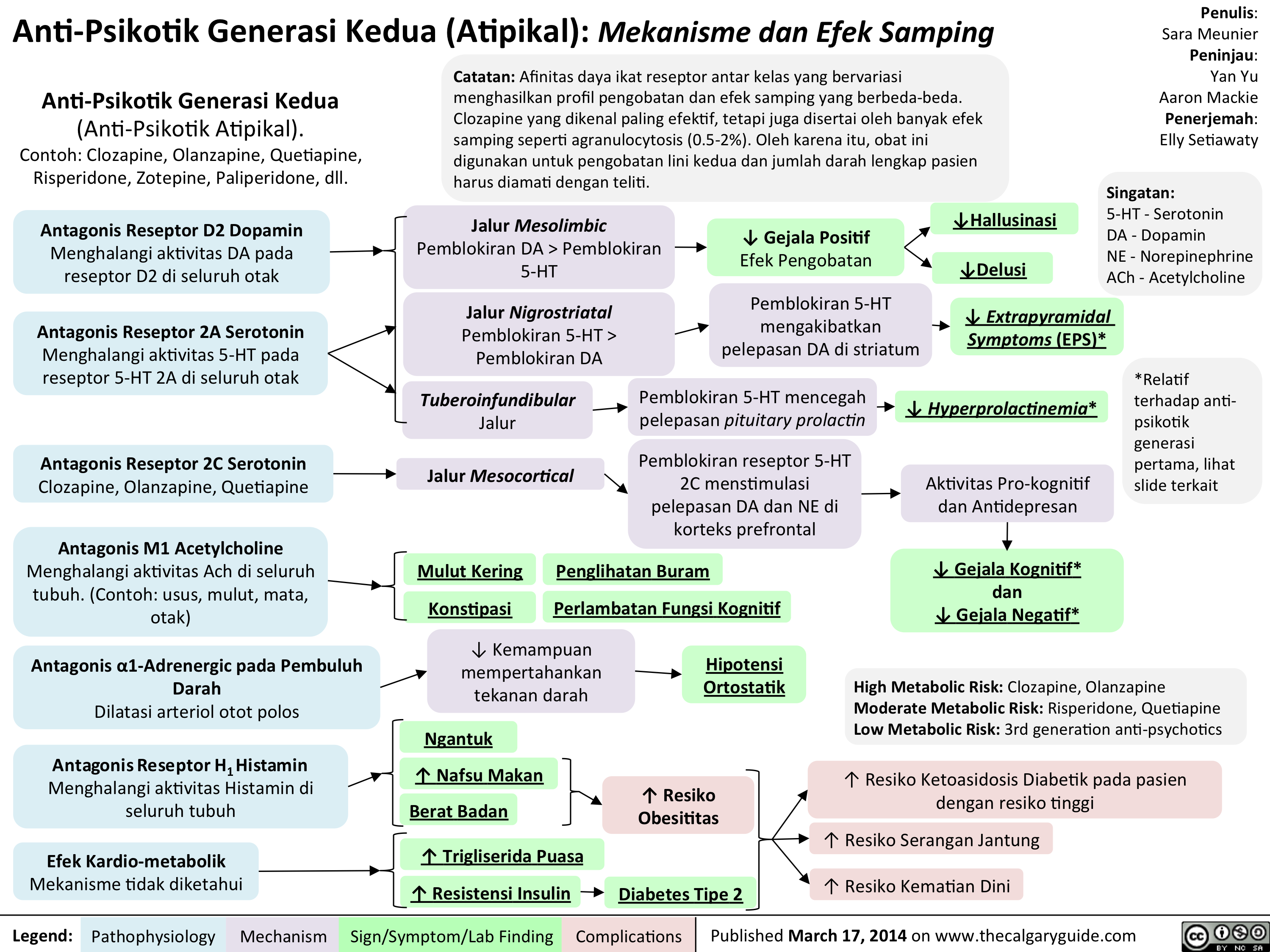
Anti-Psikotik Generasi Kedua: Mekanisme dan Efek Samping
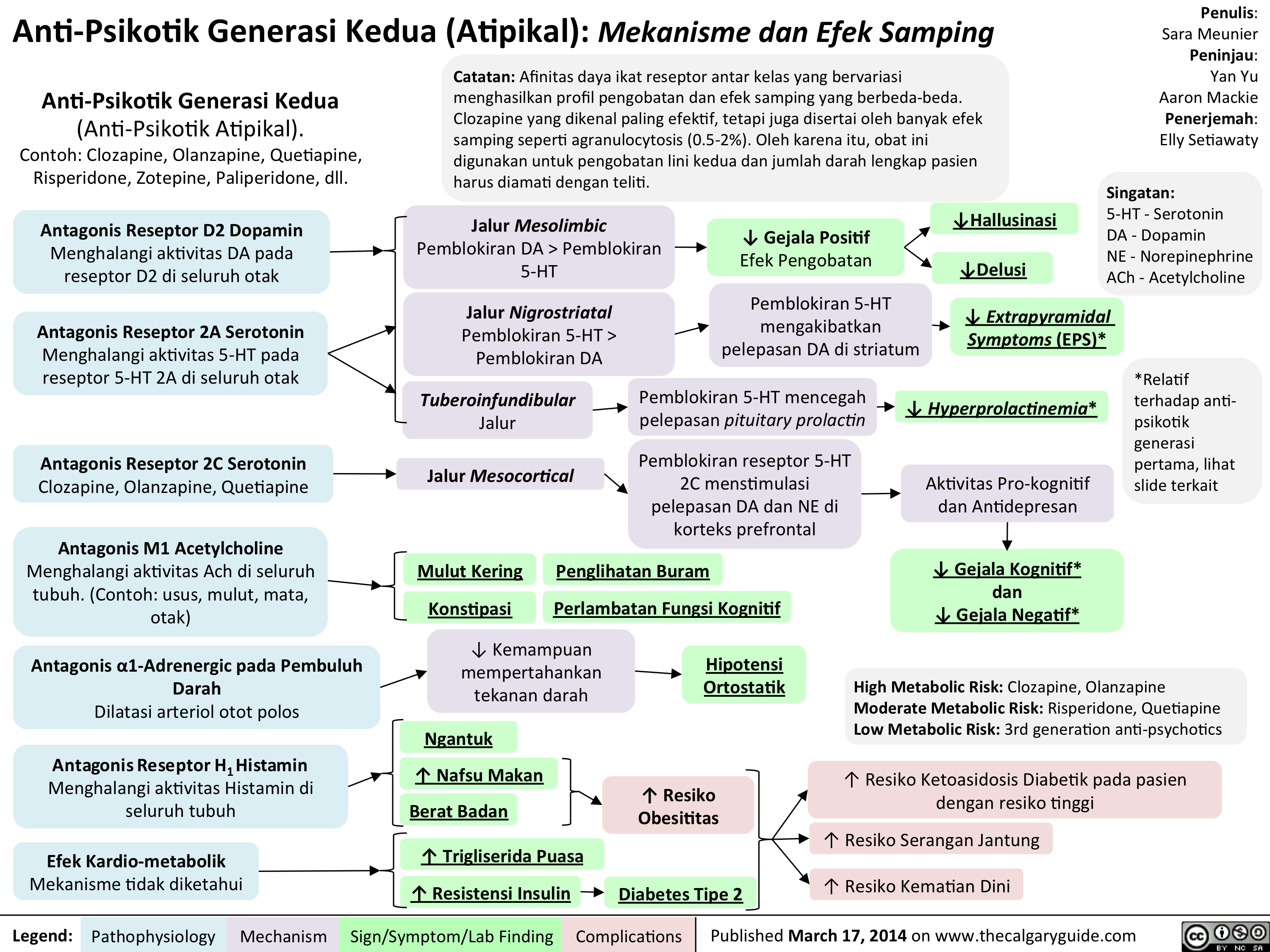
2nd generation antipsychotics Translated
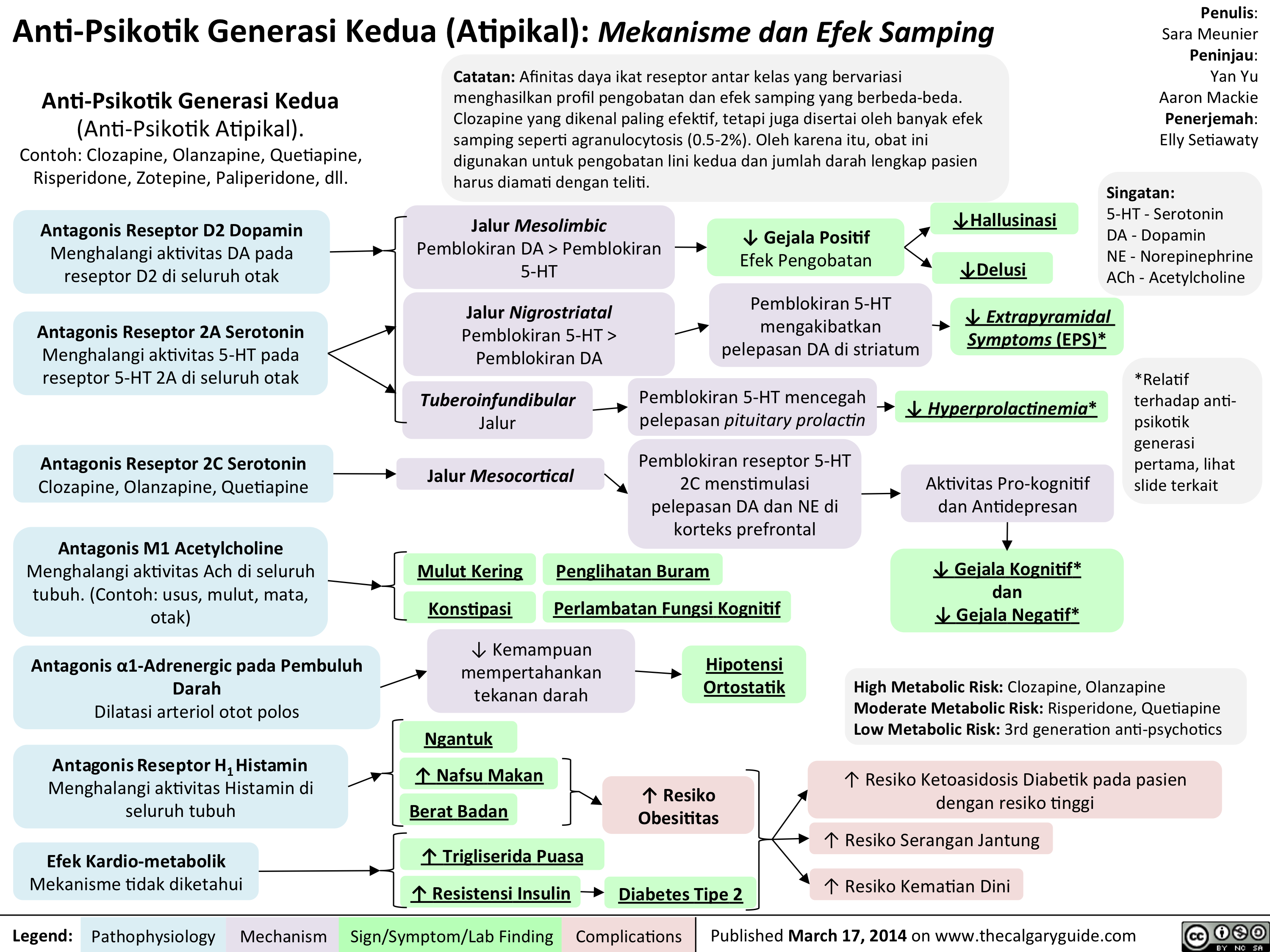
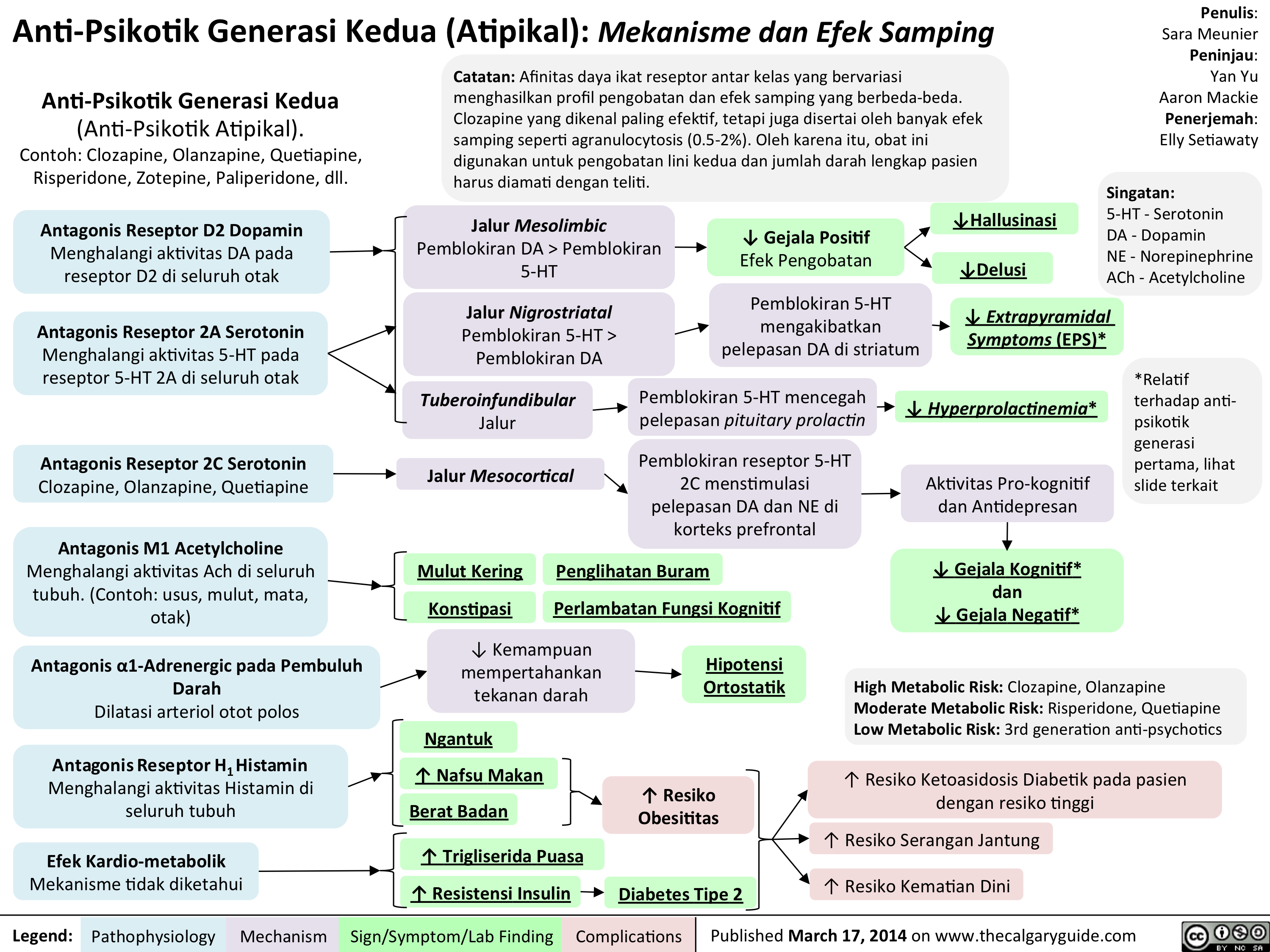
Anti-Psikotik Generasi Kedua (Atipikal): Mekanisme dan Efek Samping
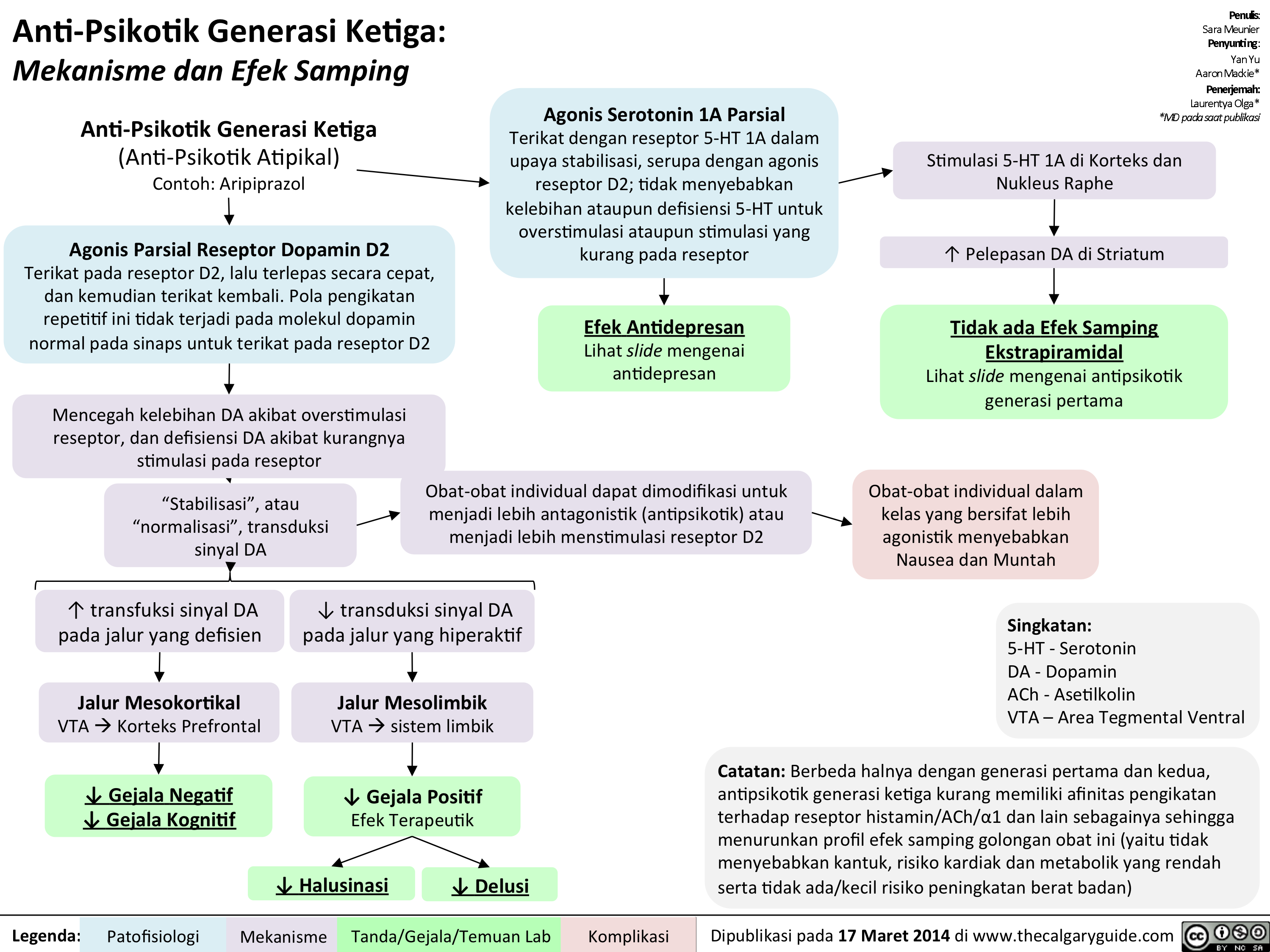
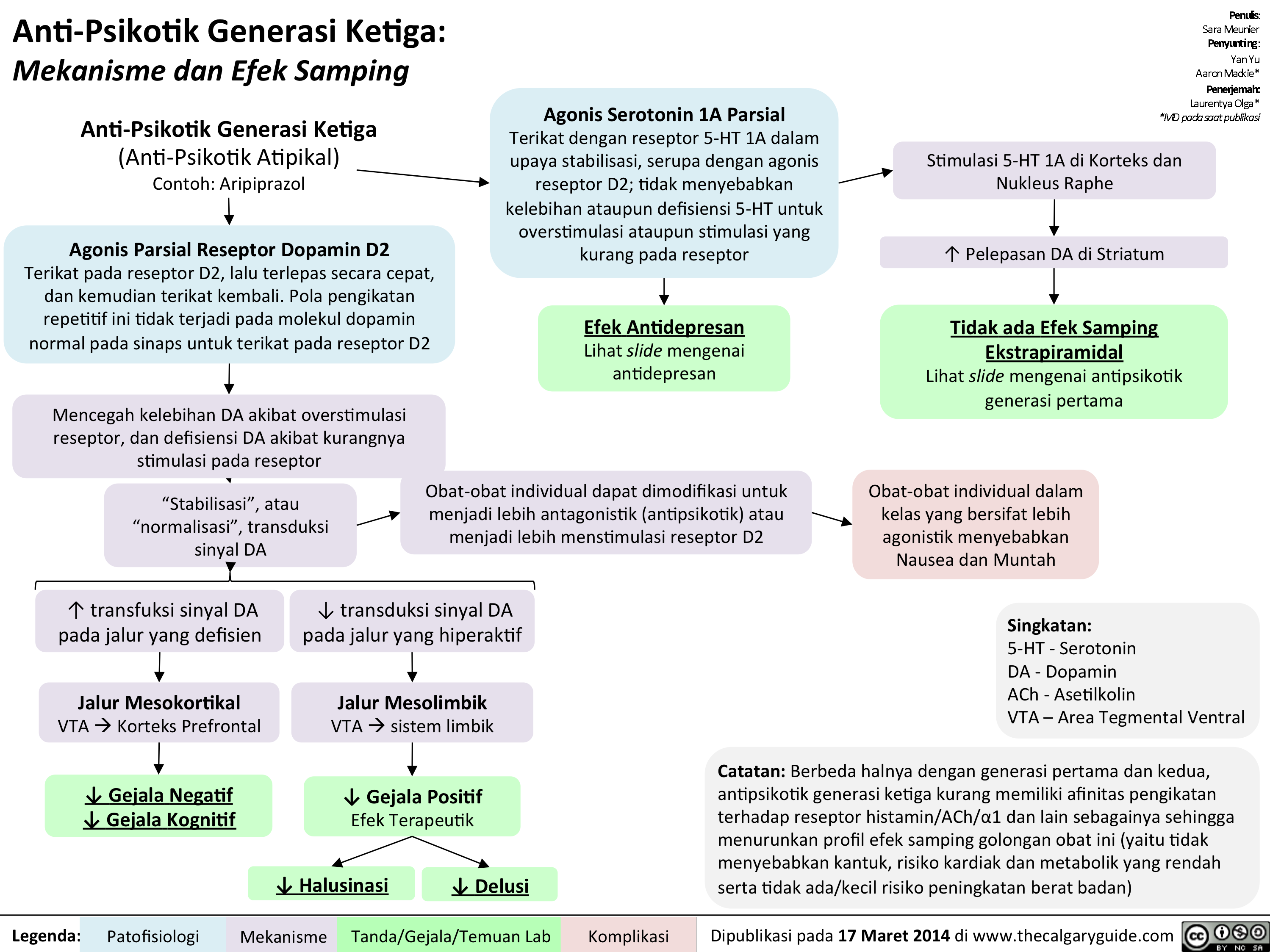
Anti-Psikotik Generasi Ketiga: Mekanisme dan Efek Samping
1st gen antipsychotics Translated
2nd generation antipsychotics Translated
1st gen antipsychotics Translated
3rd gen anti-psychotics Translated
Schizophrenia Pathogenesis Translated
Pathogenesis of Anxiety Disorders Translated
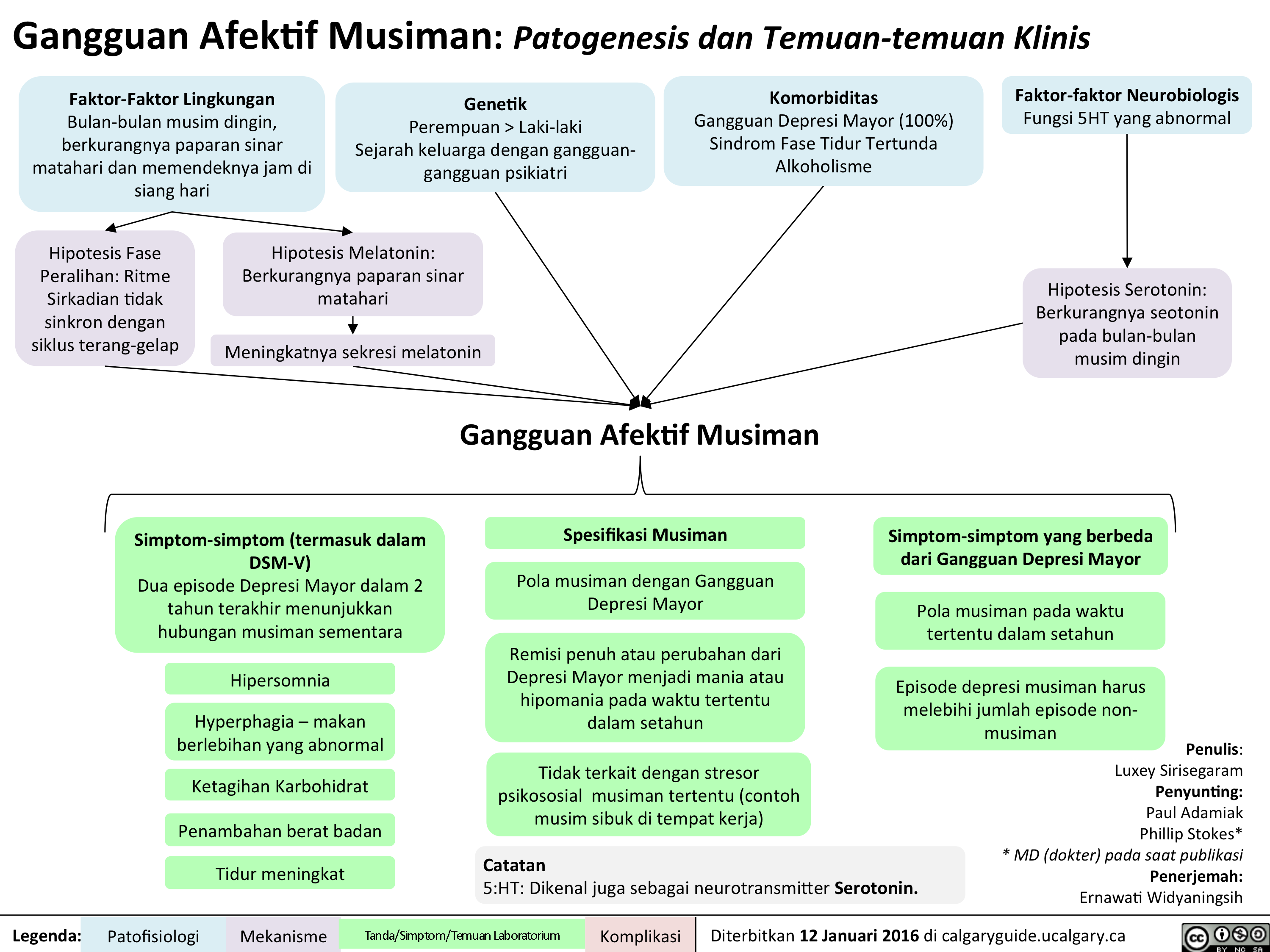
SAD Gangguan Afektif Musiman Translated
PTSD Translated
Panic Disorder Translated
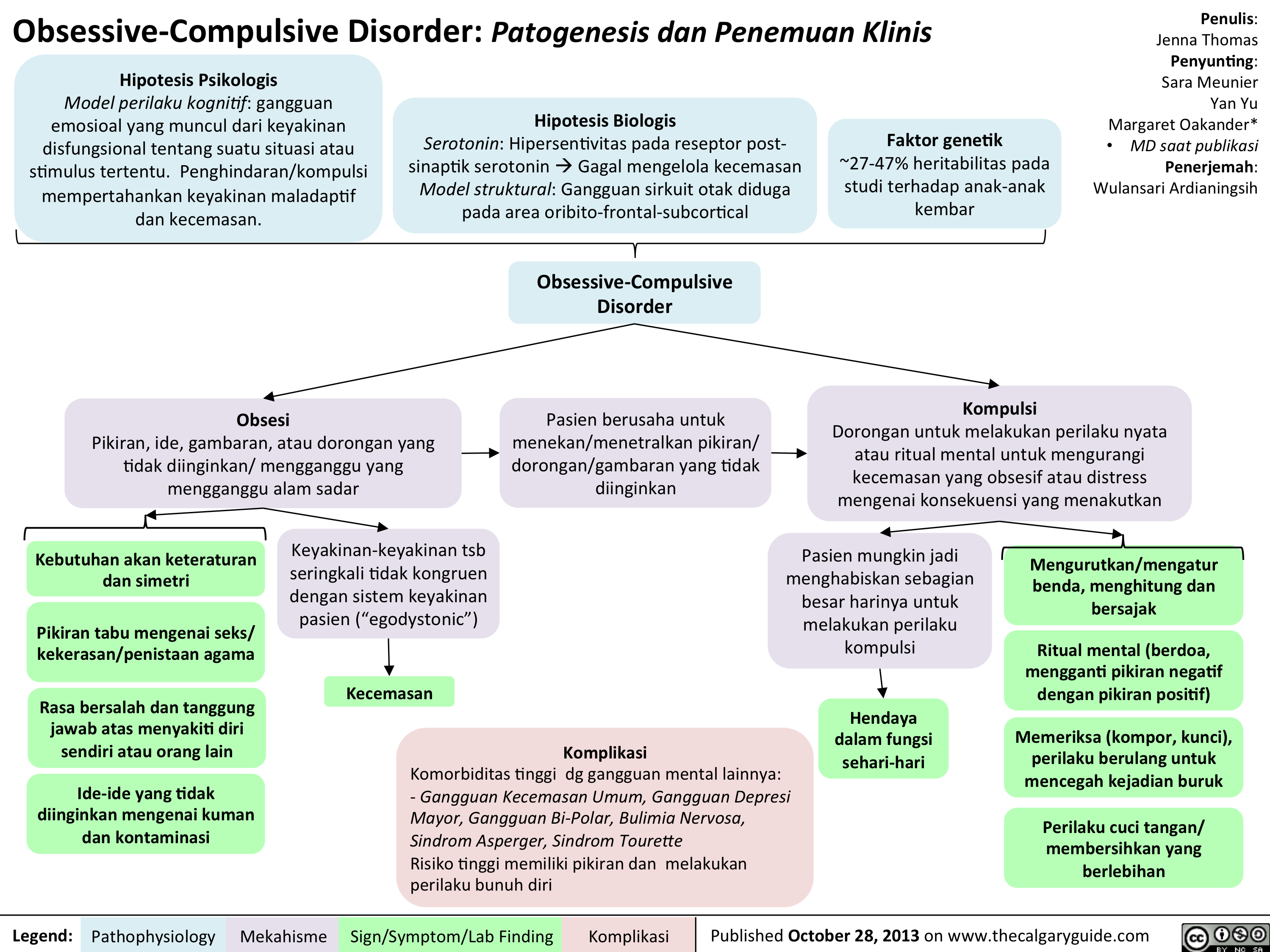
OCD Translated
Bipolar Disorder Translated

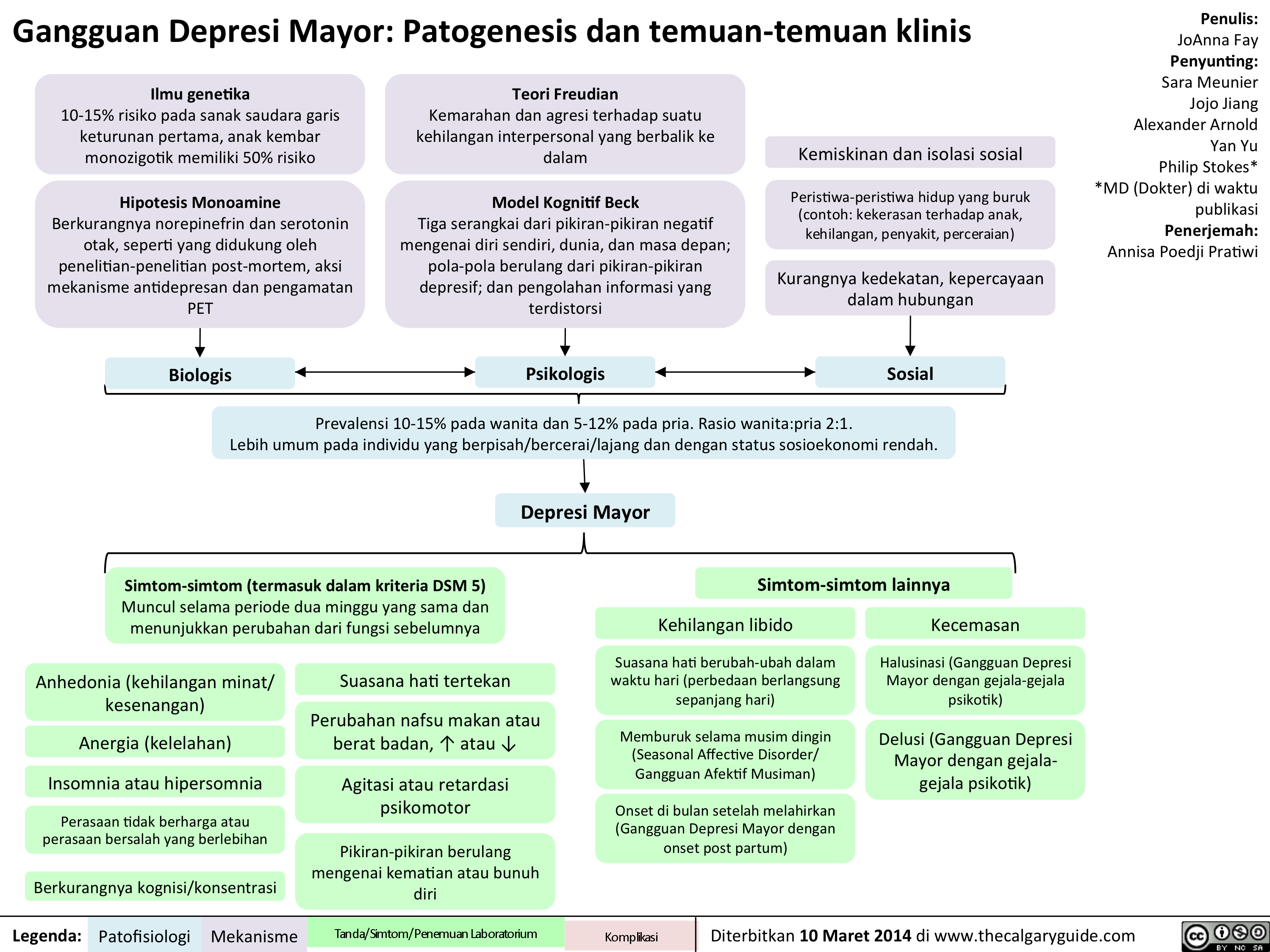
MDD Translated
SSRIs Translated
Bupropion Translated
SNRIs Translated
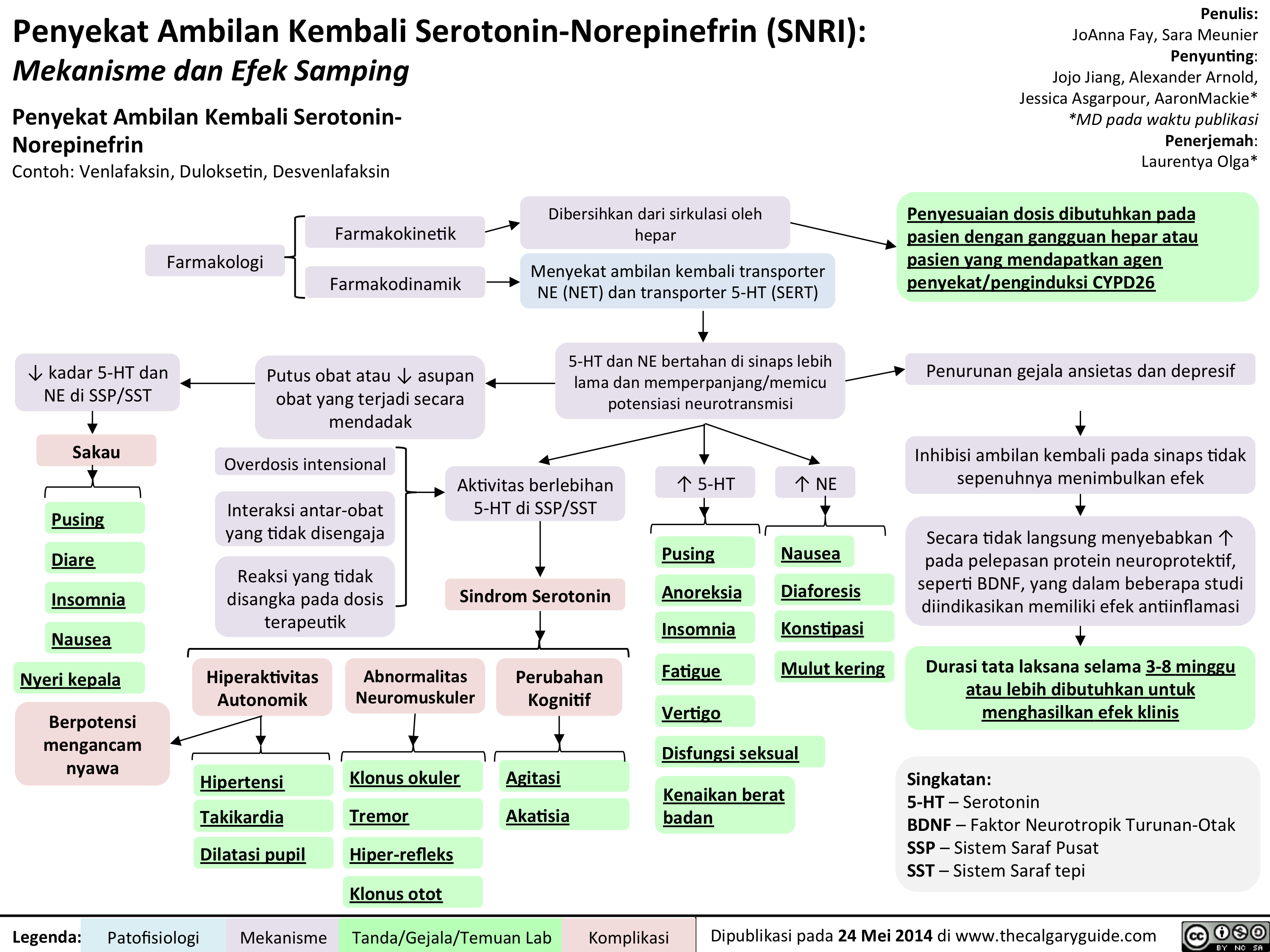
Thacker, J - Social Anxiety Translated
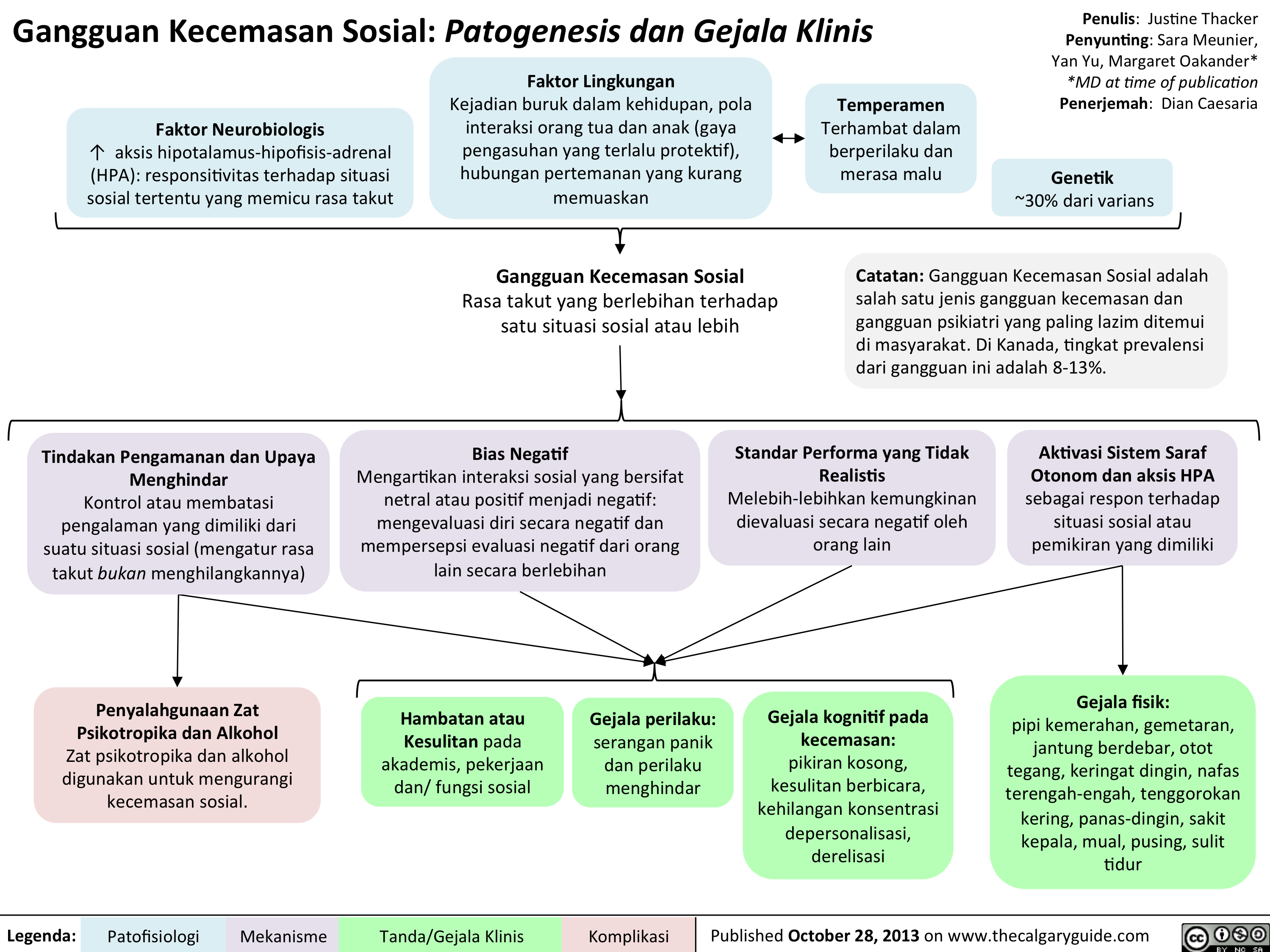
DSM - Axis to Formulation Translated
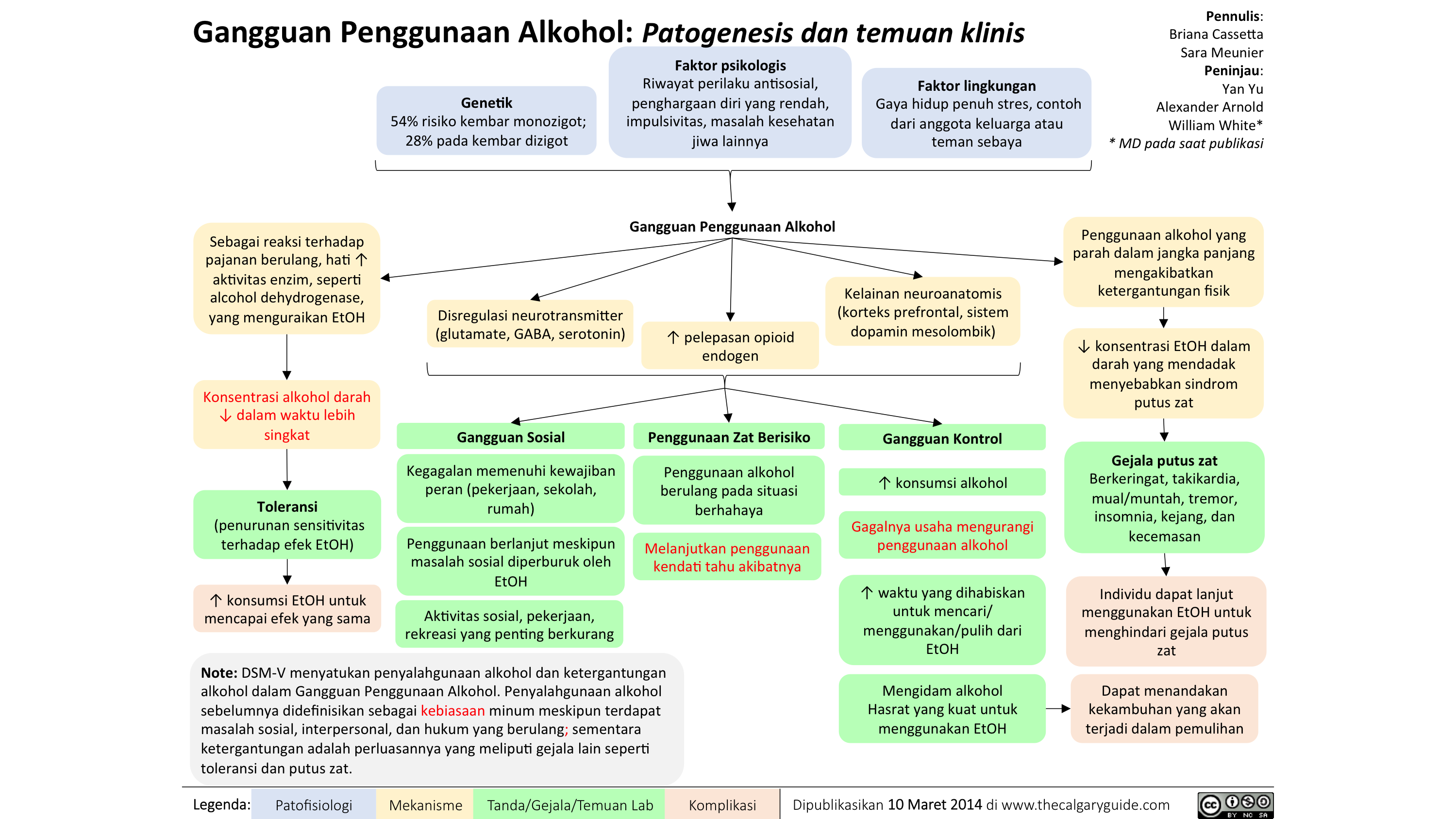
Alcohol Use Translated
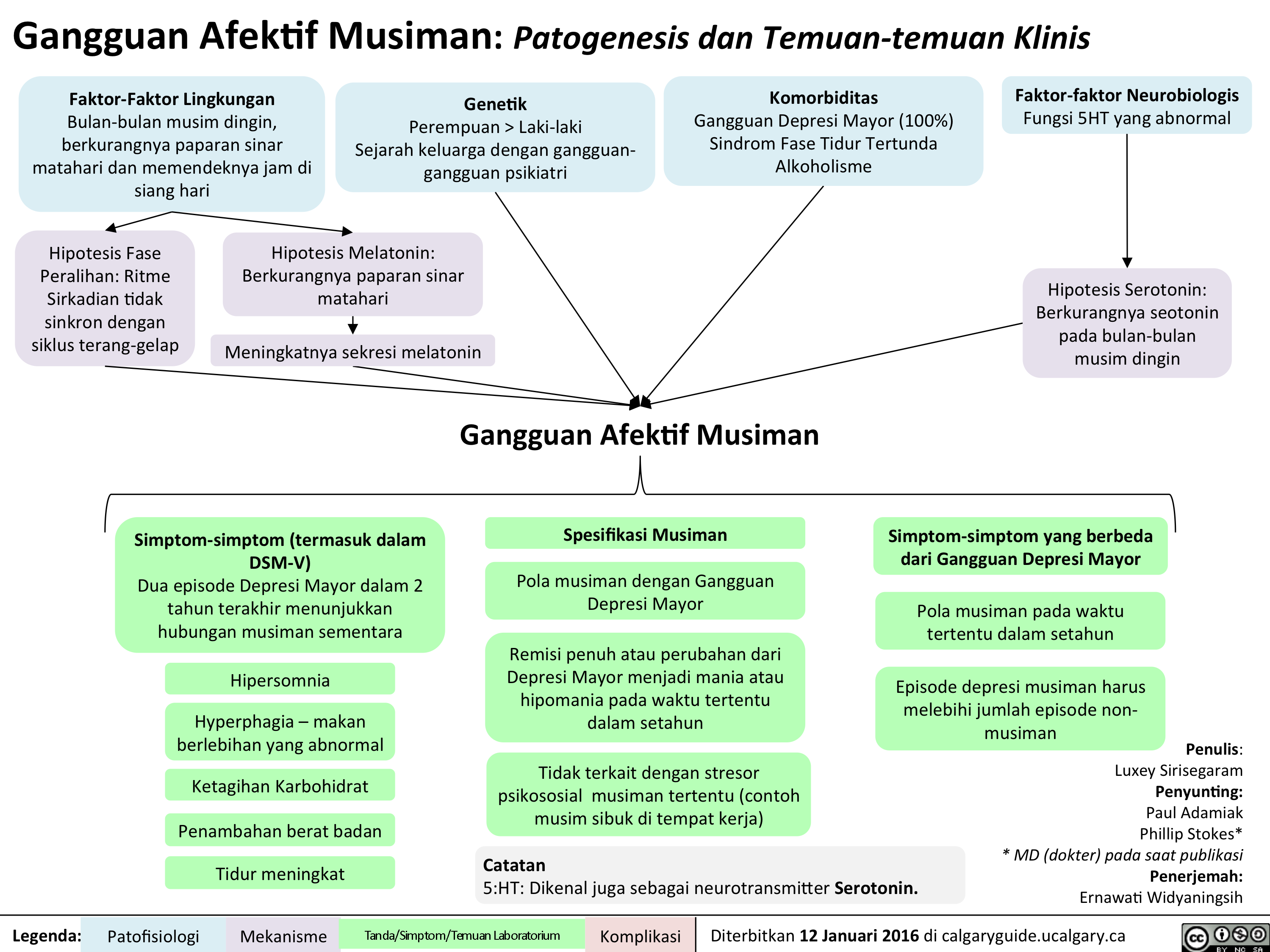
Seasonal Affective Disorder: Pathogenesis and clinical findings
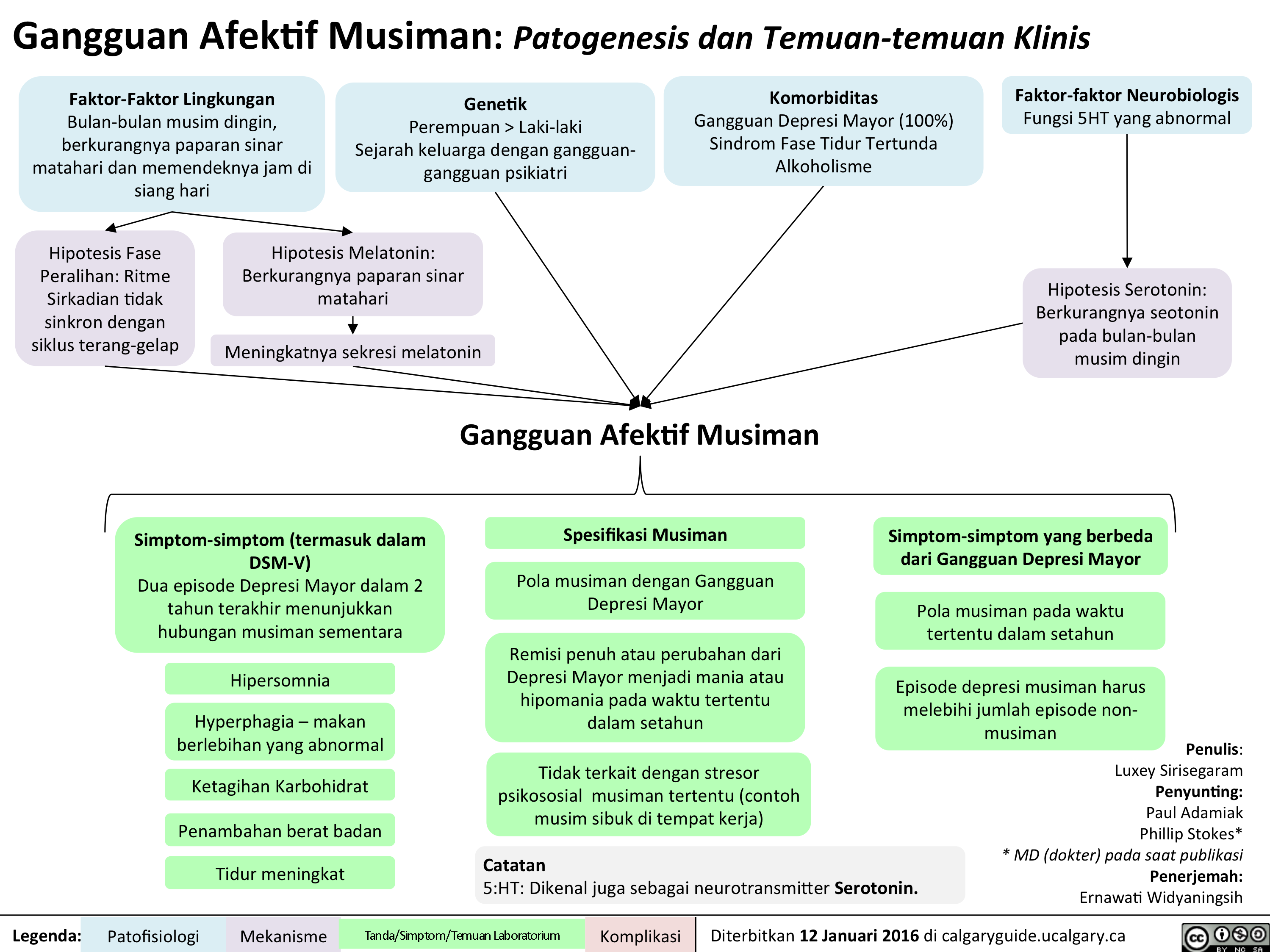
Seasonal Affective Disorder: Pathogenesis and clinical findings
Slide1
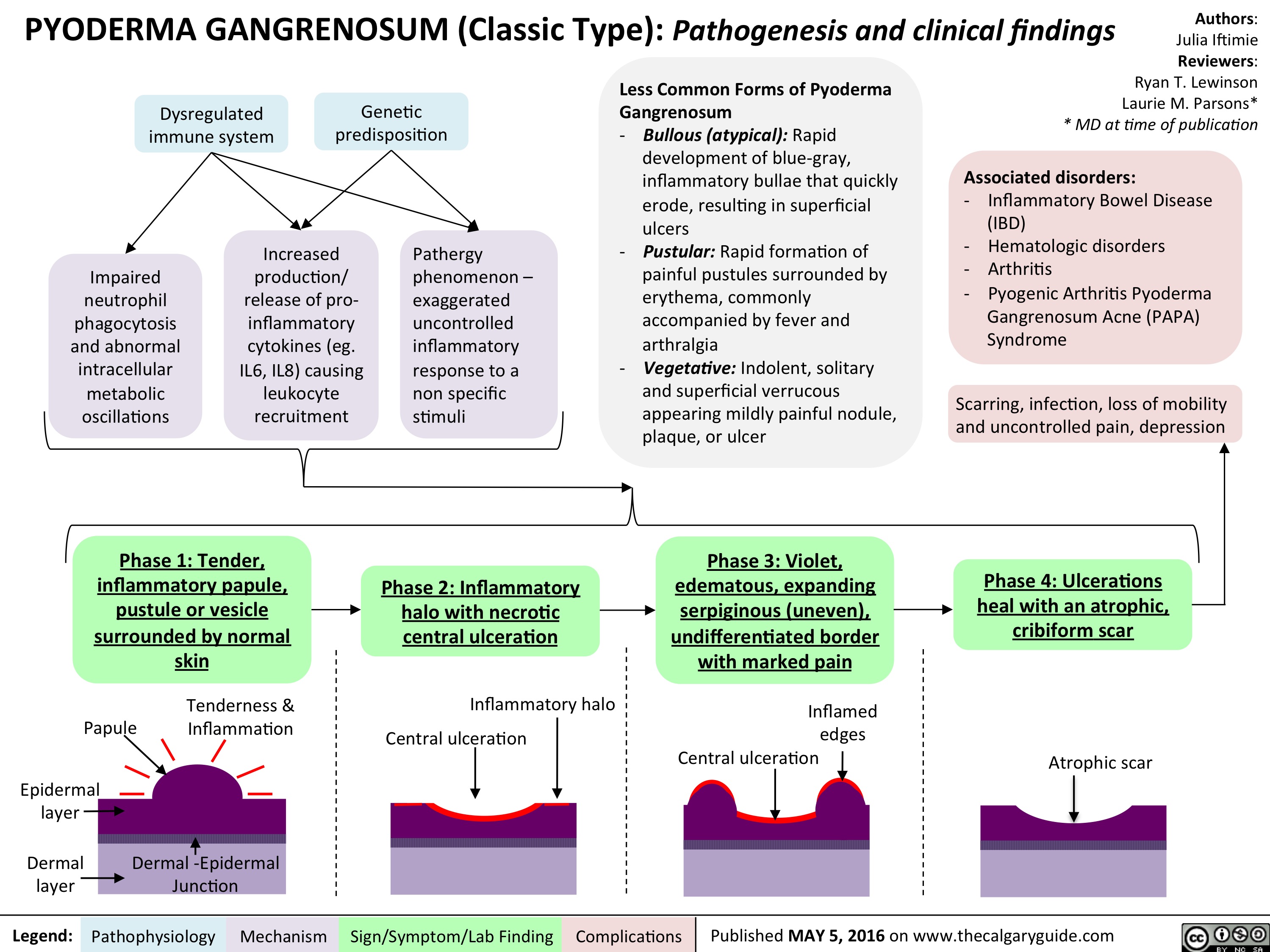
Slide1

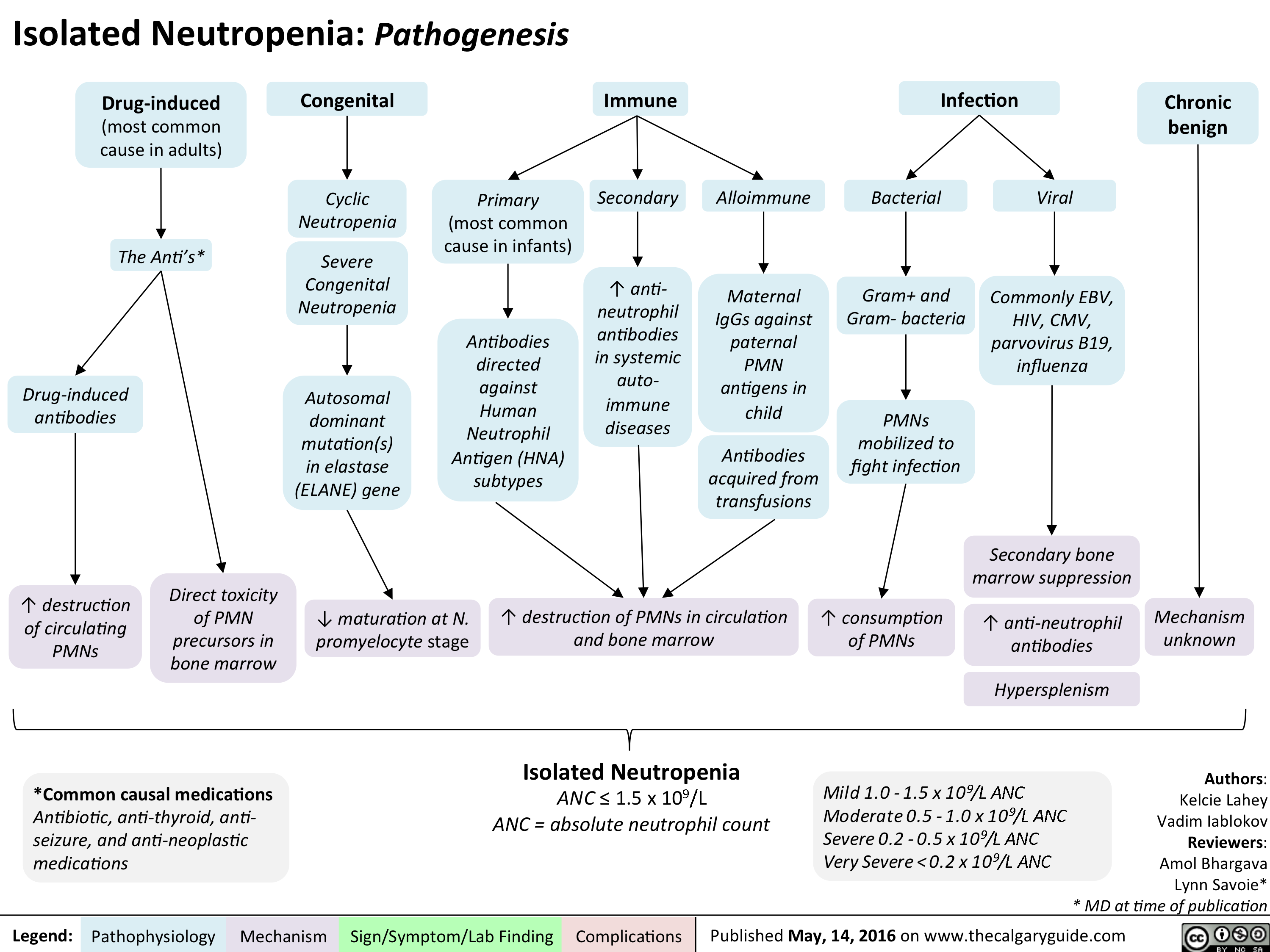
Isolated Neutropenia
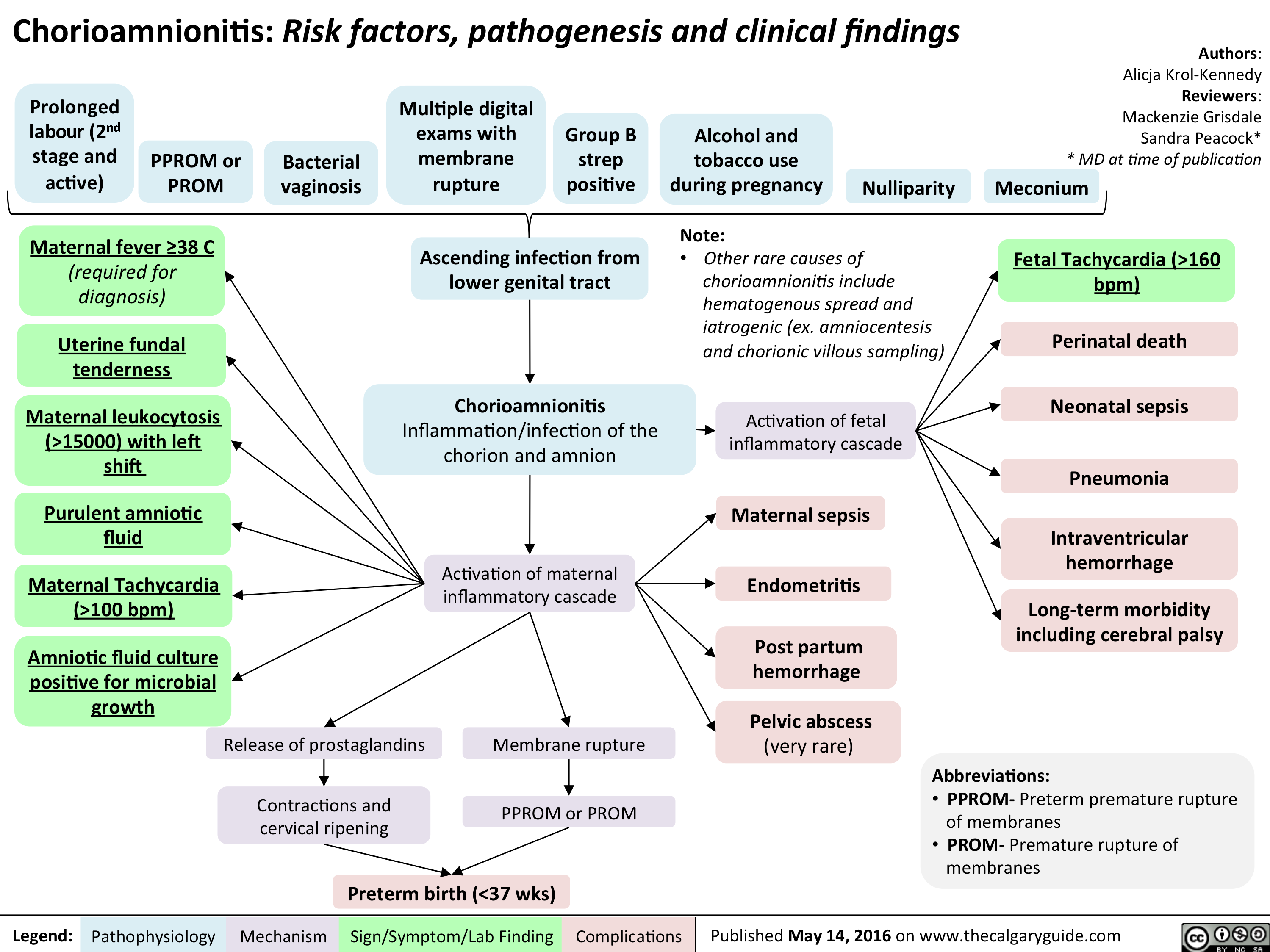
Chorioamnionitis: Risk factors, pathogenesis and clinical findings
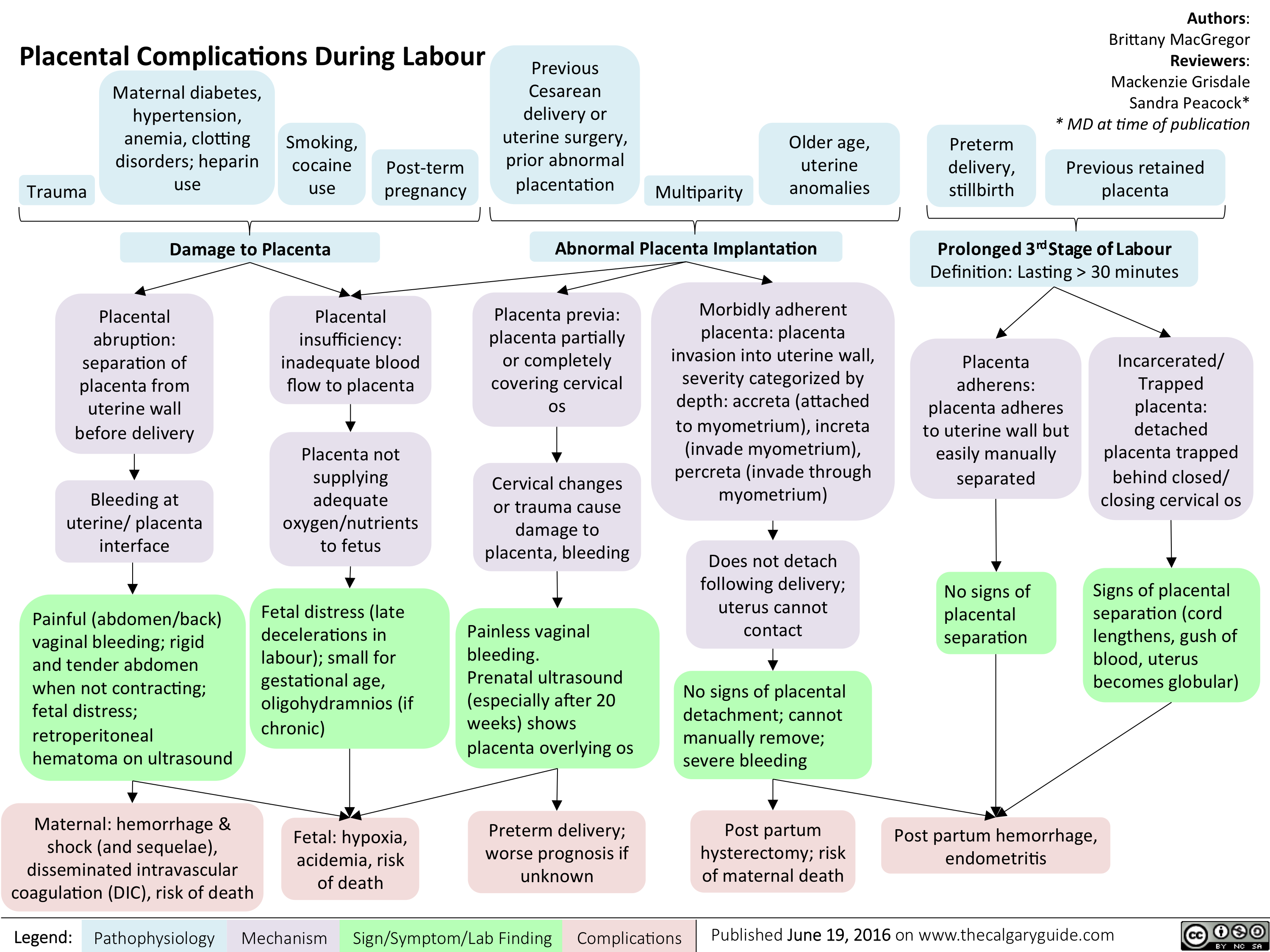
Placental Complications During Labour
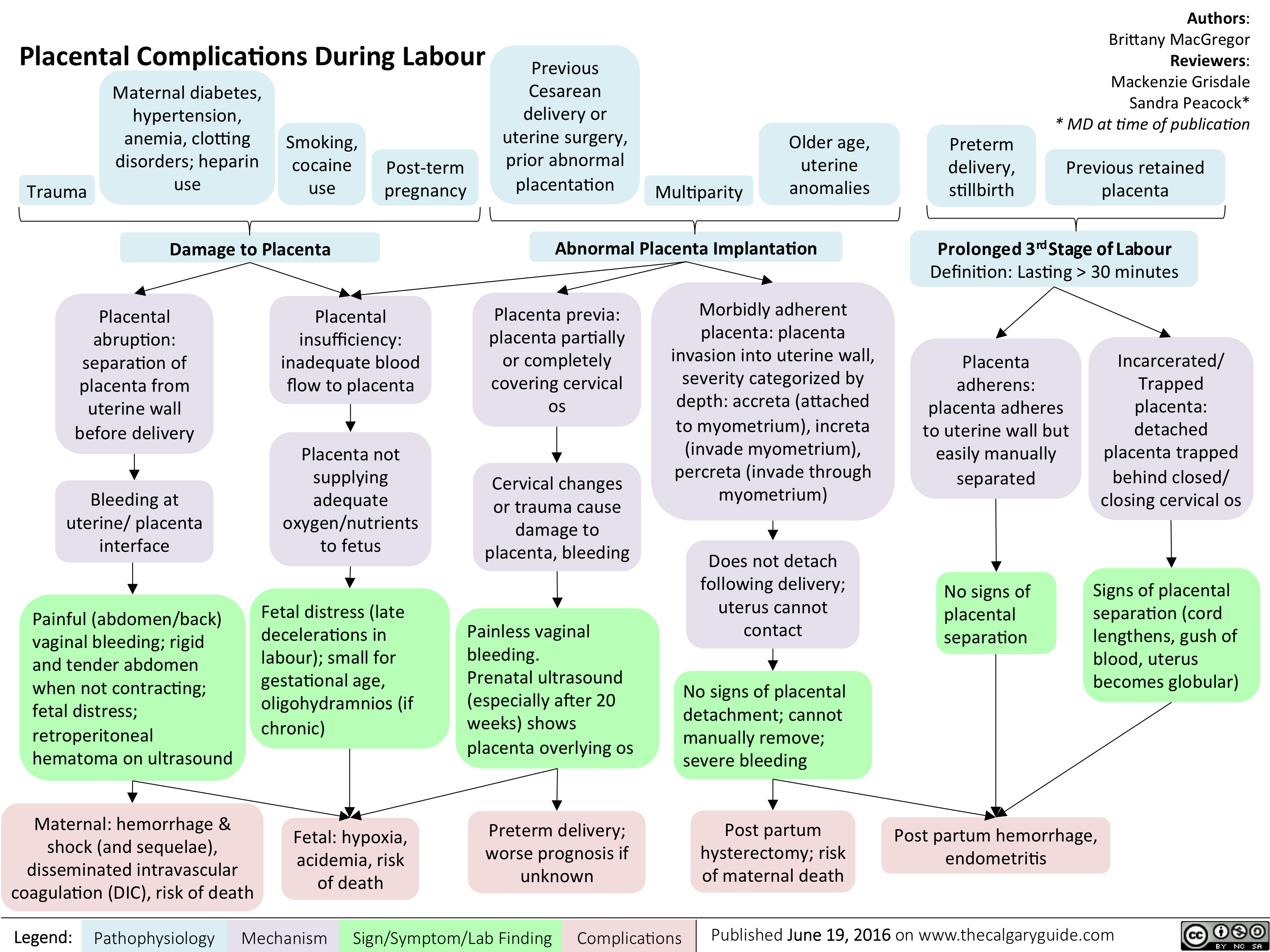
Placental Complications During Labour
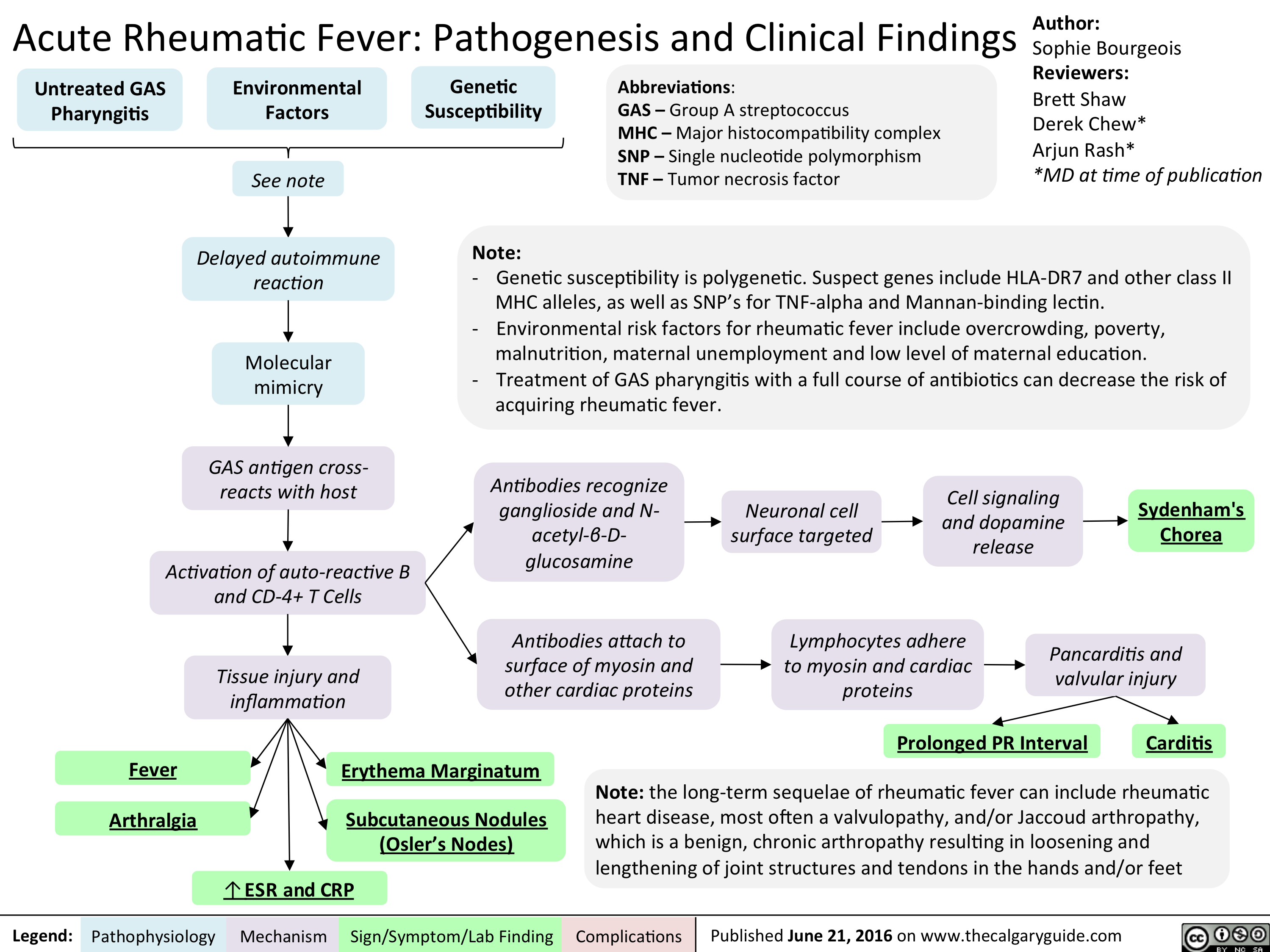
Acute Rheumatic Fever- Pathogenesis and Clinical Findings
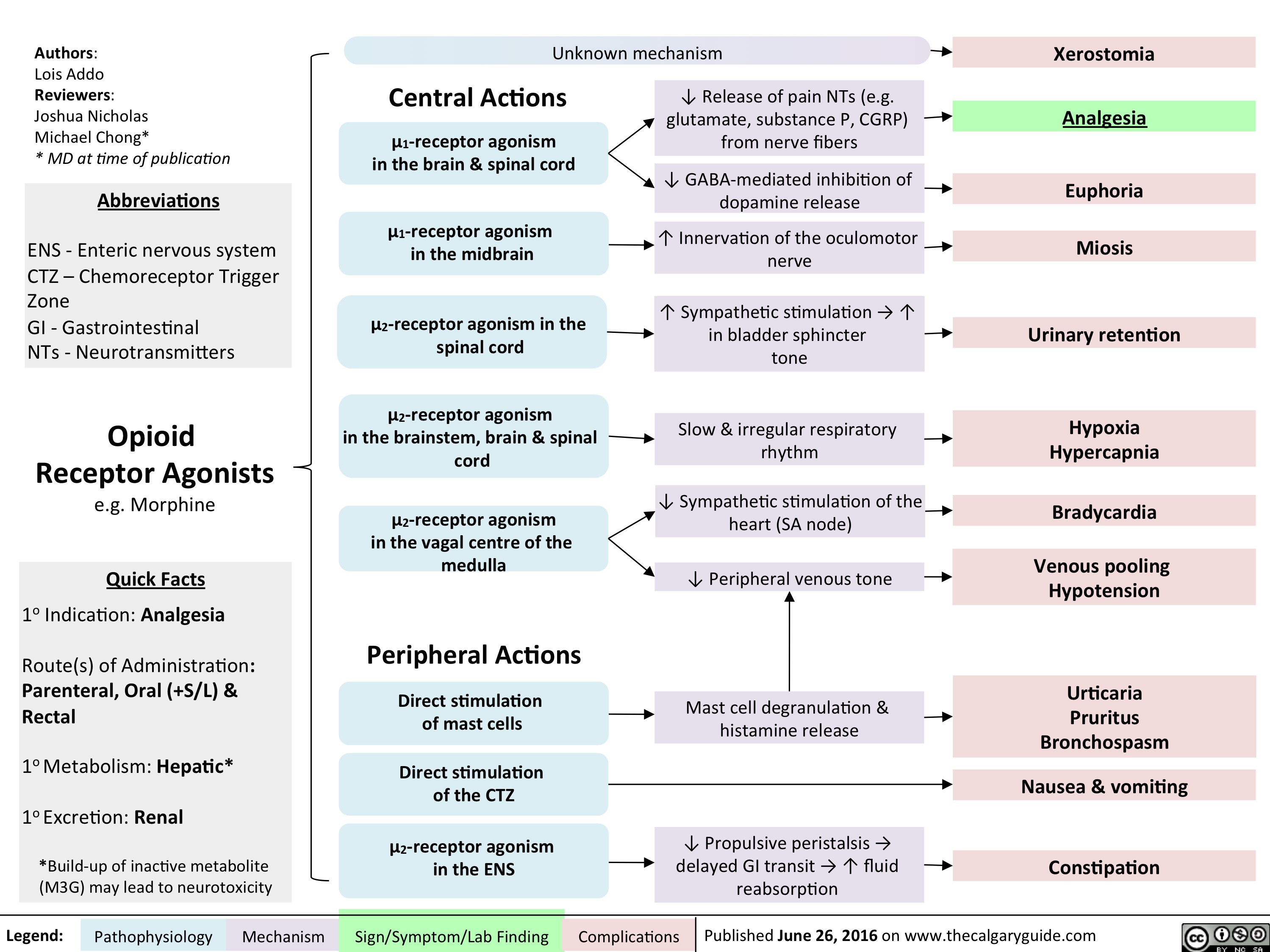
Opioid Receptor Agonists
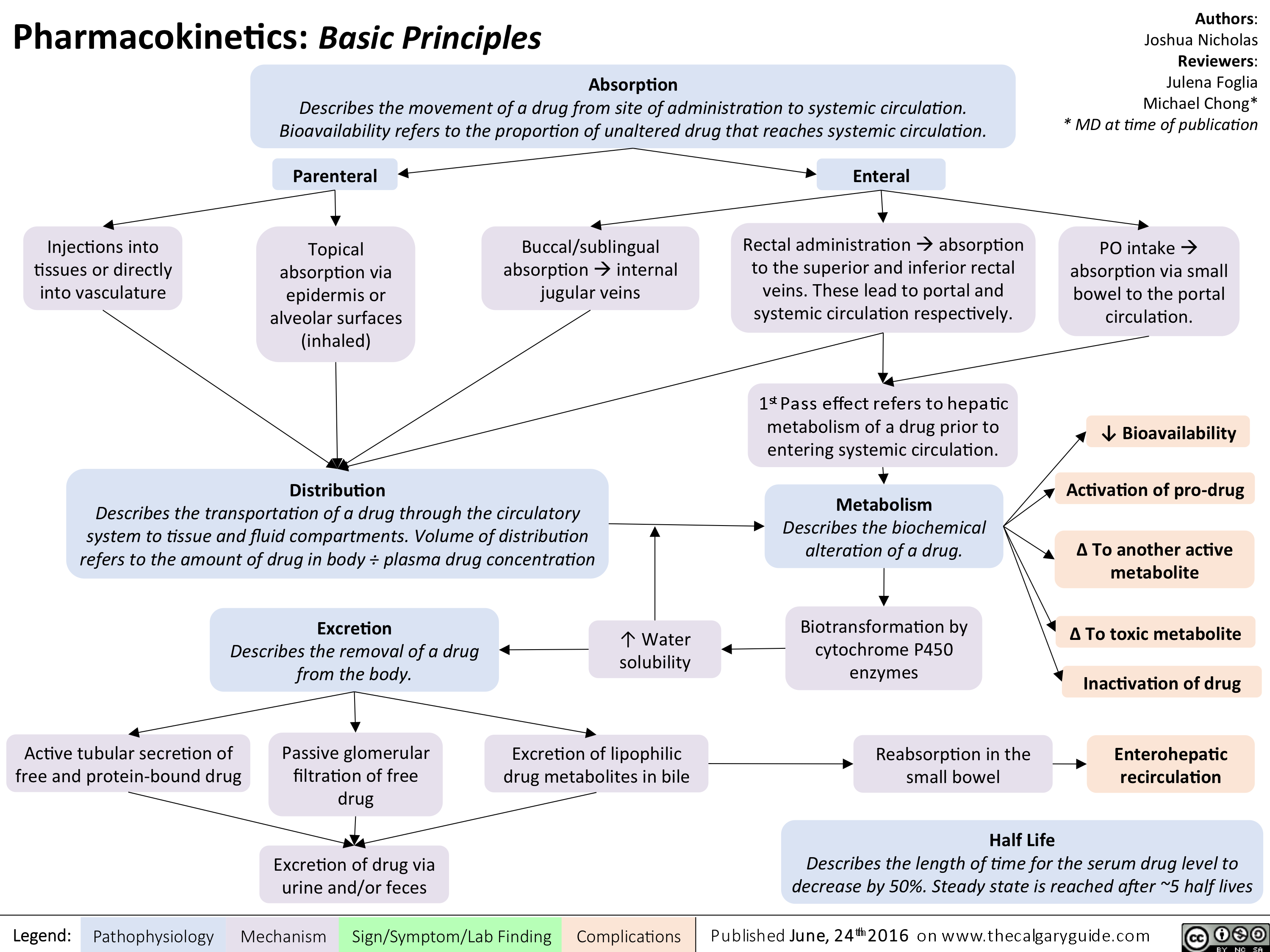
Pharmacokinetics Basic Principles
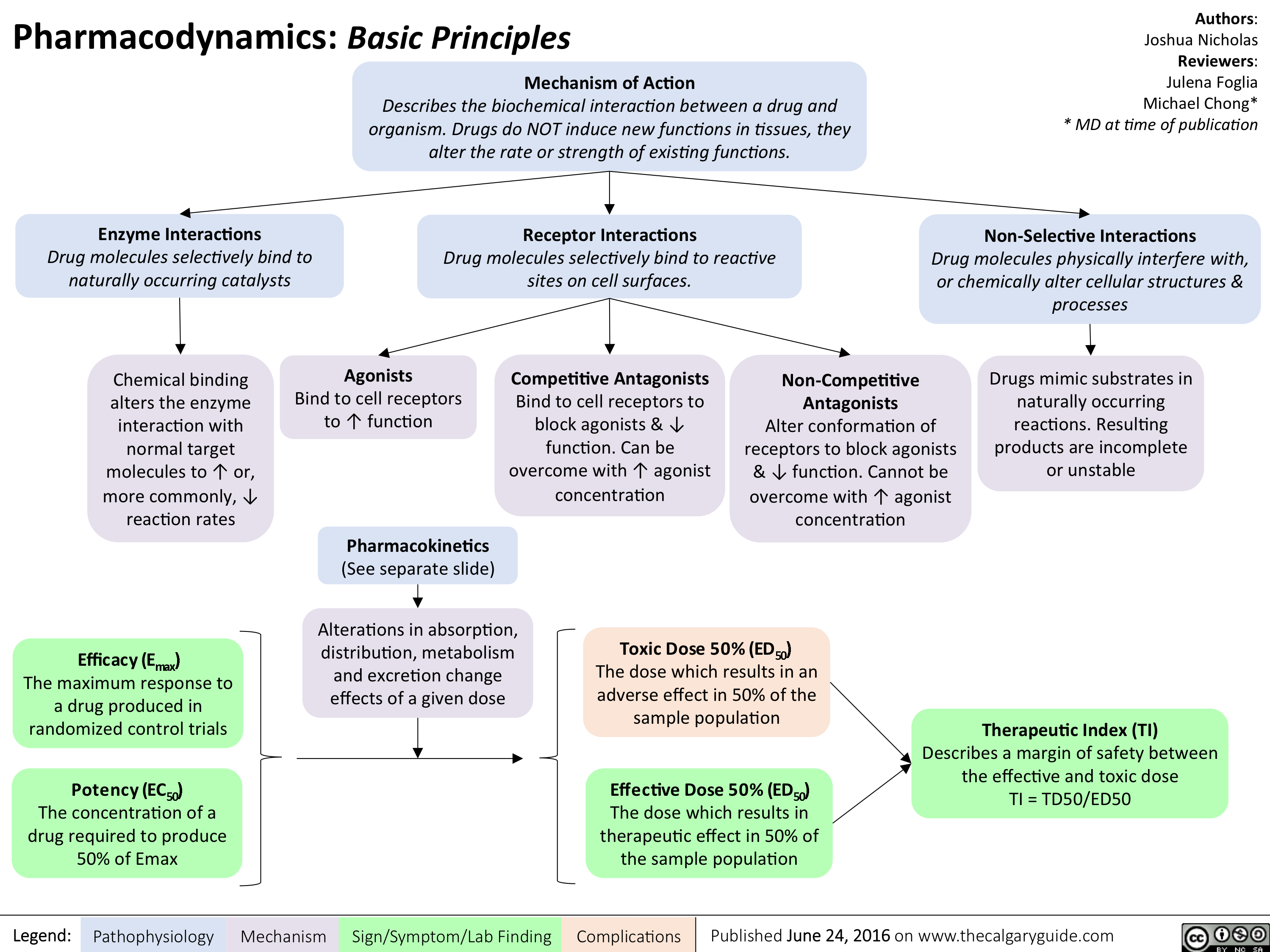
Pharmacodynamics Basic Principles
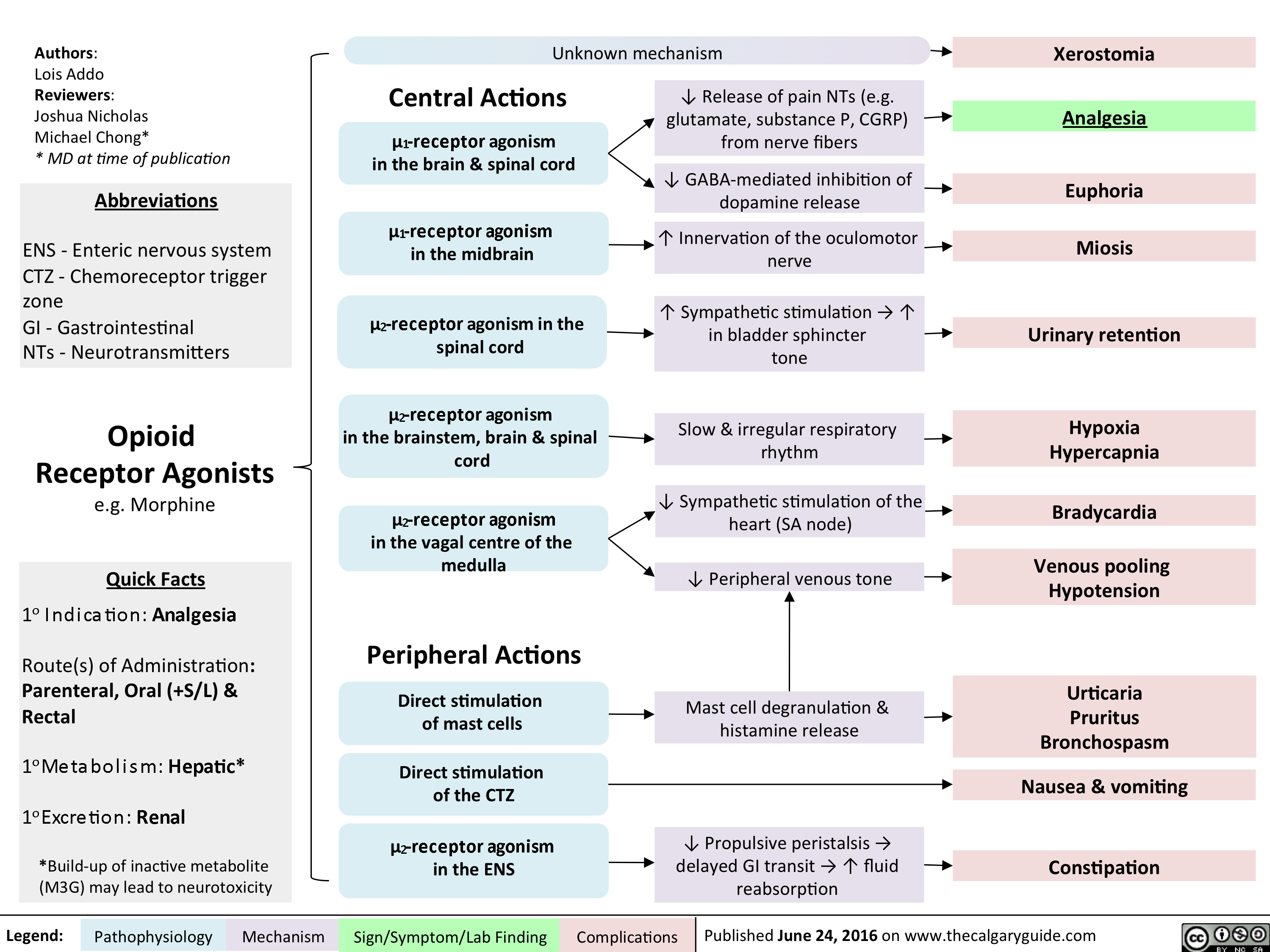
Opioid Receptor Agonists
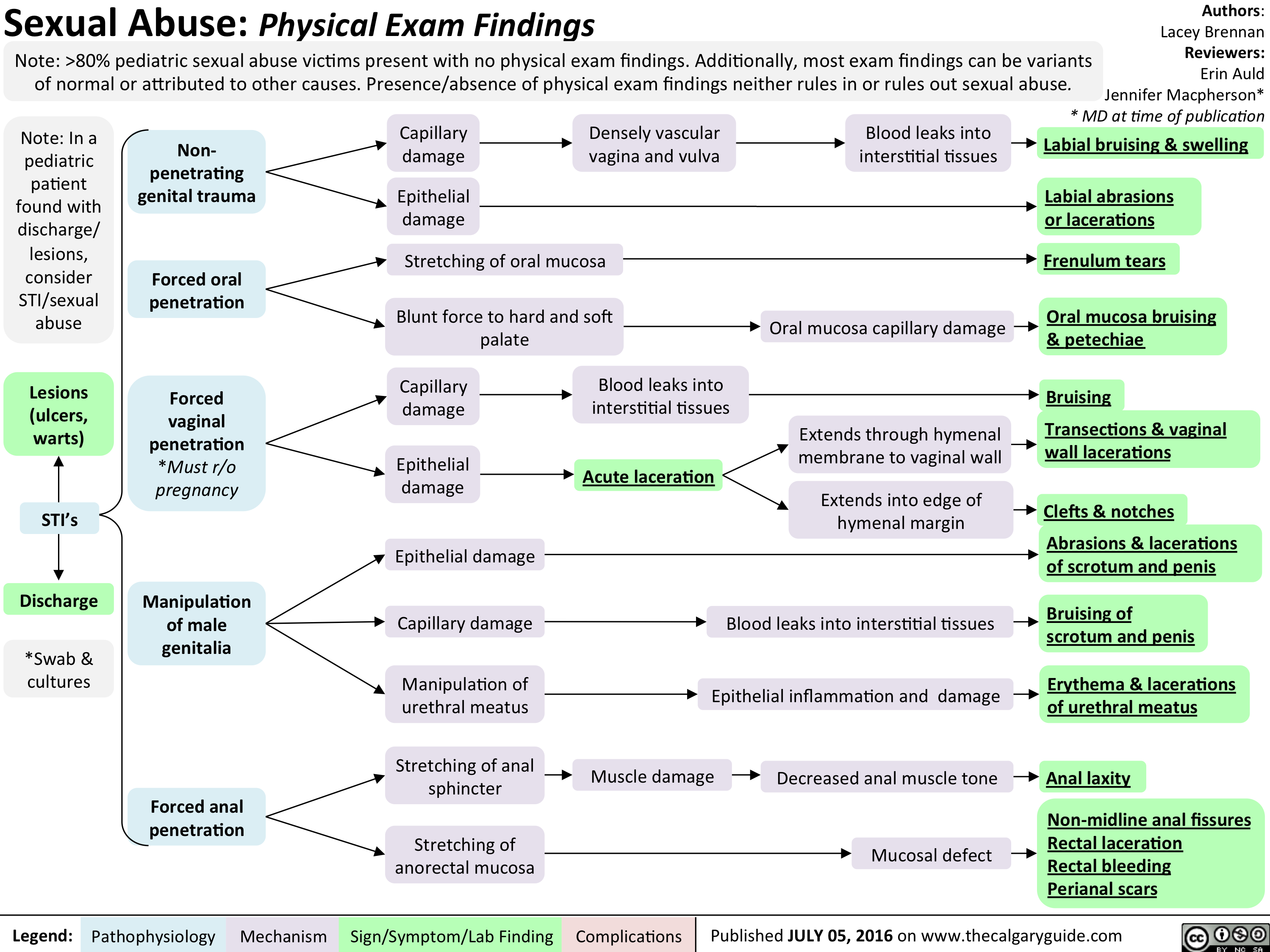
Peds Sexual Abuse
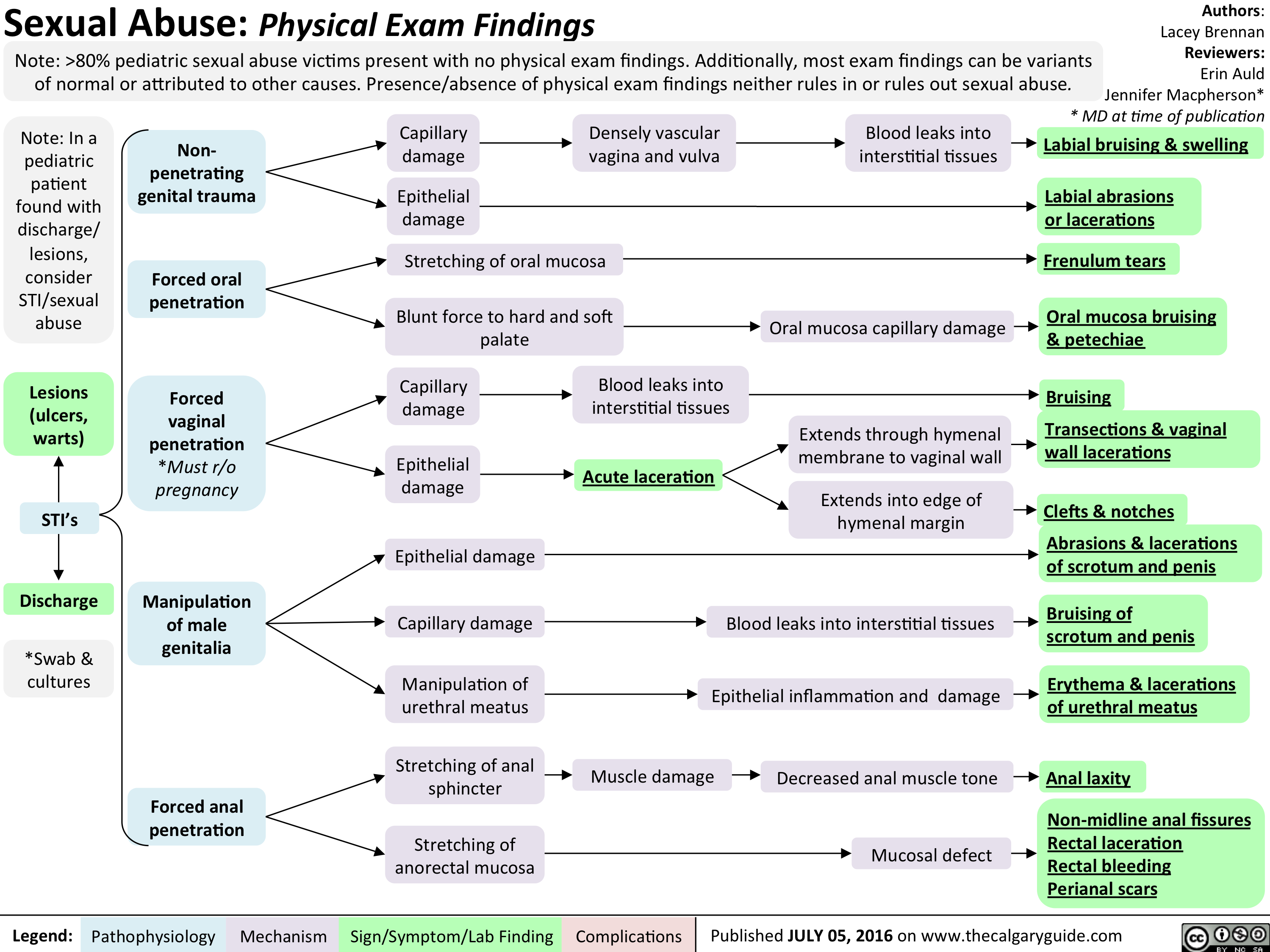
Peds Sexual Abuse
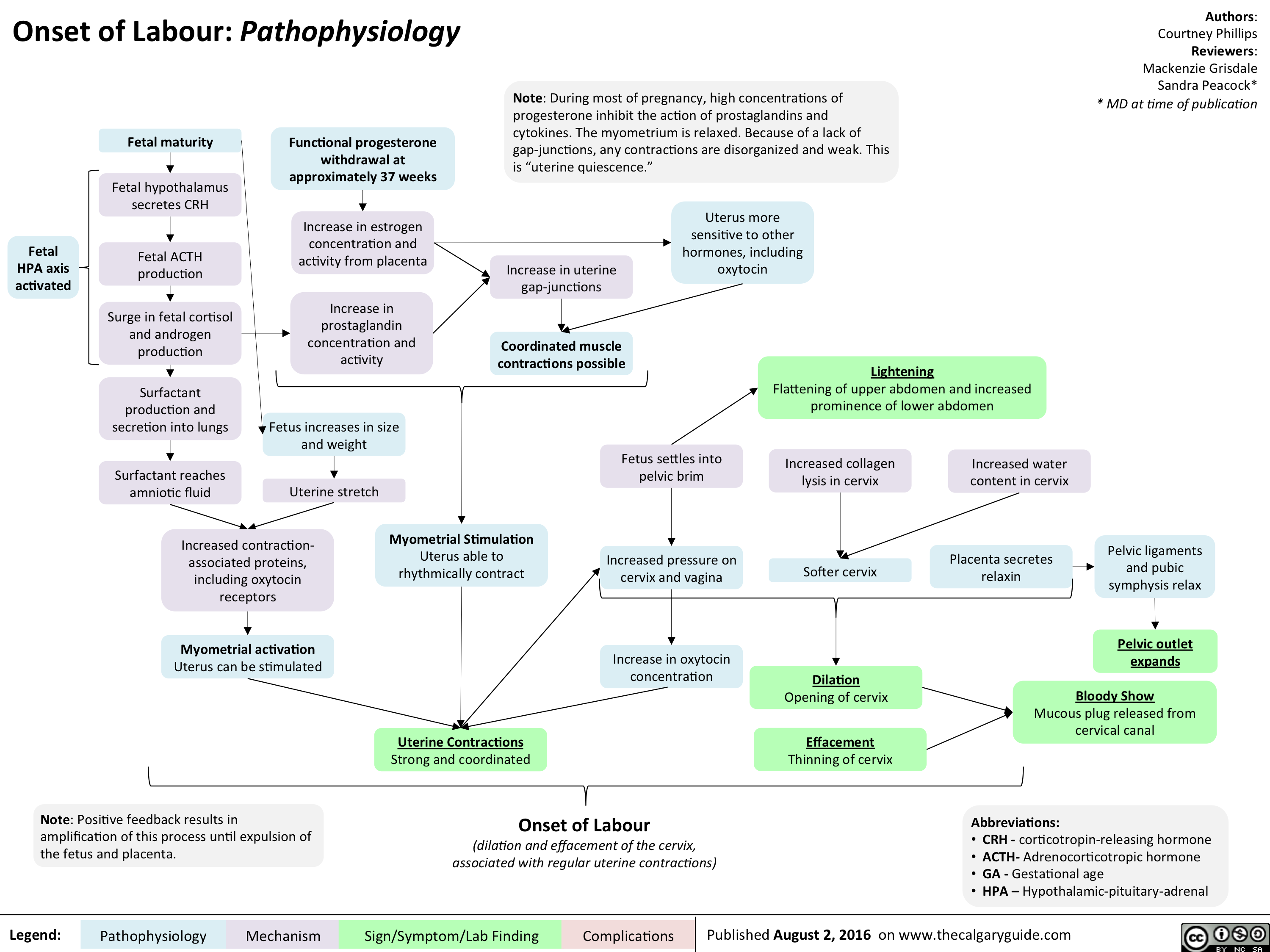
Onset of Labour- Pathophysiology
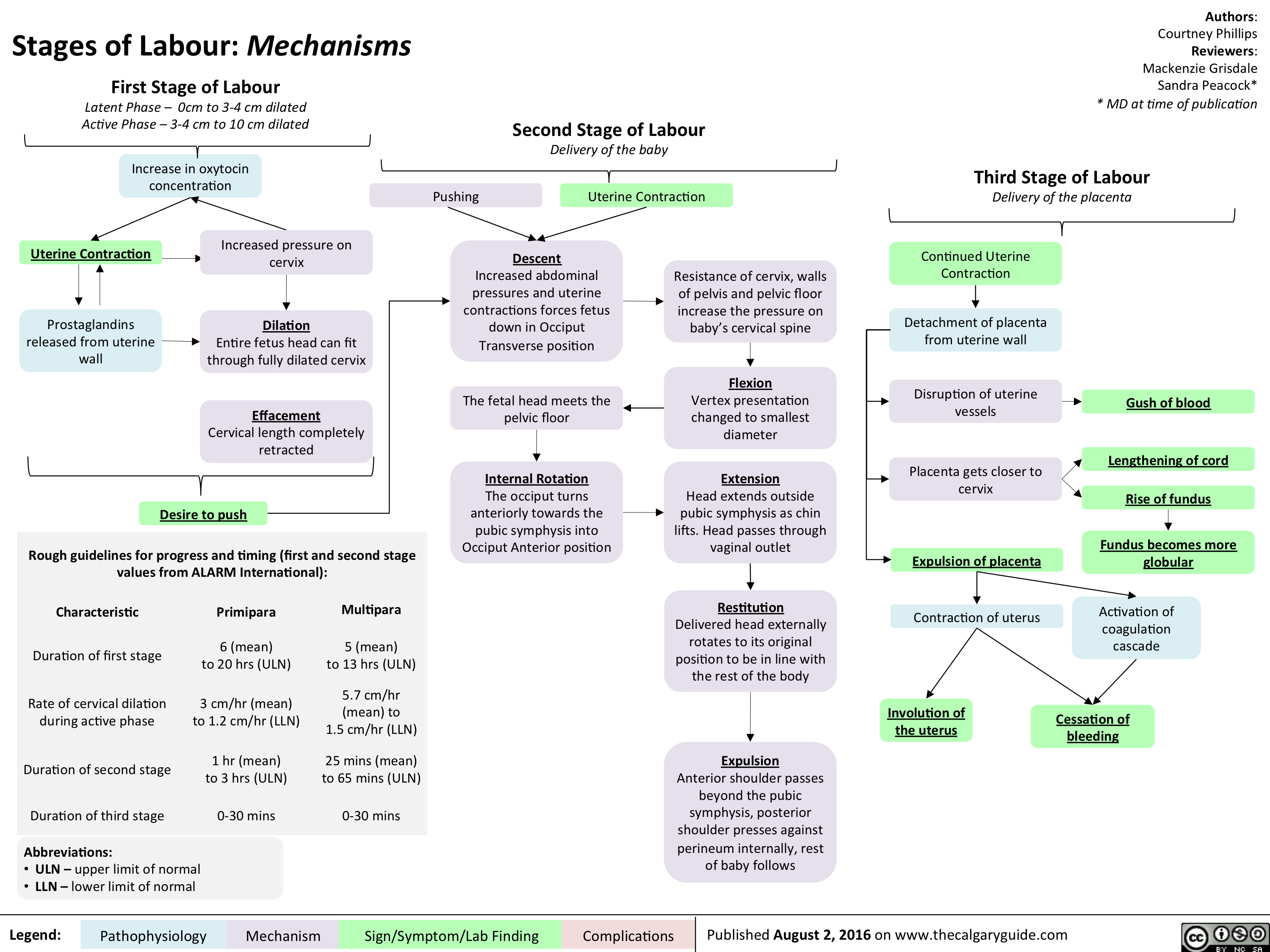
Stages of Labour- Mechanisms
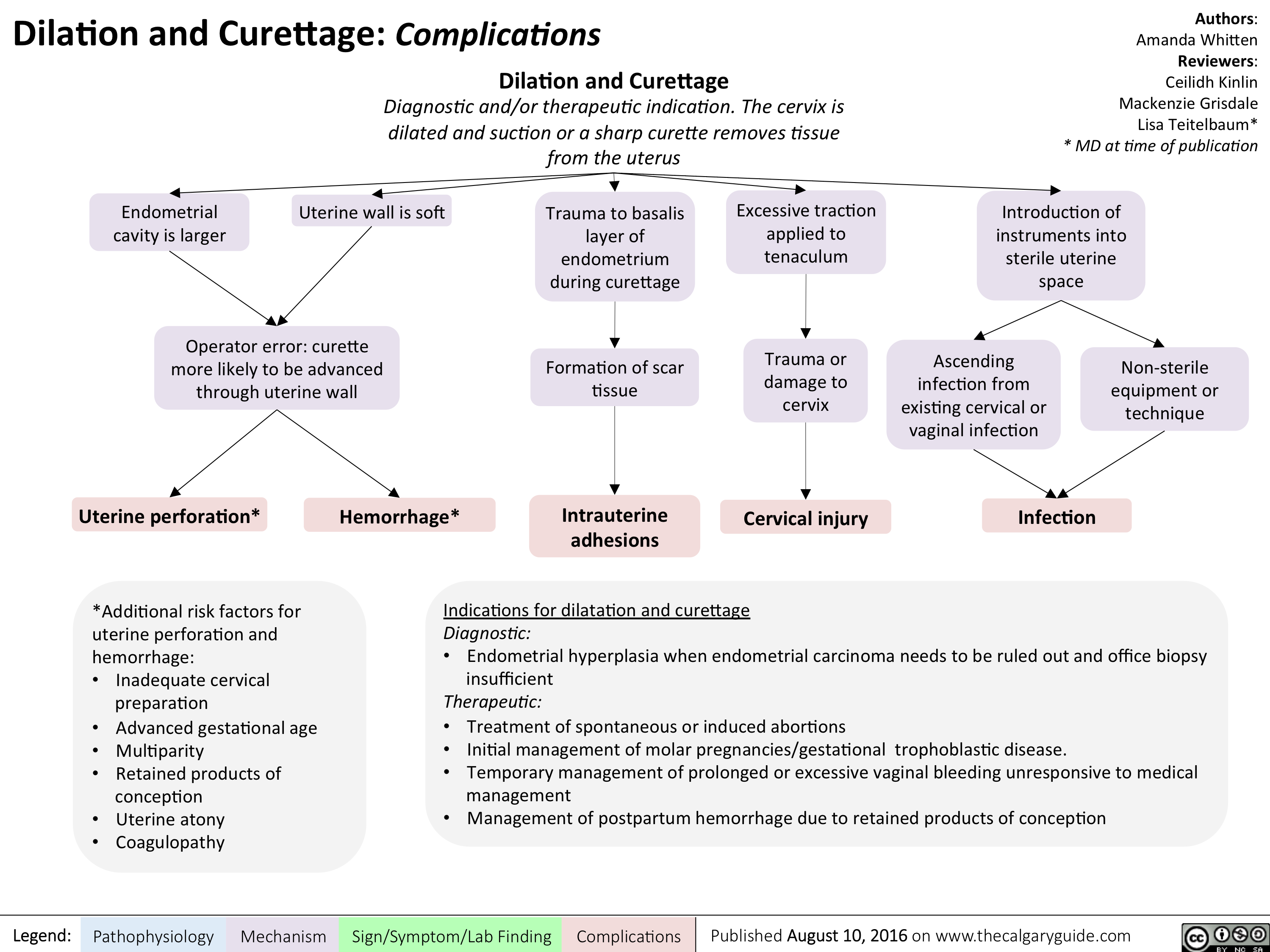
Dilatation and Curettage - Complications
myasthenia-gravis-final

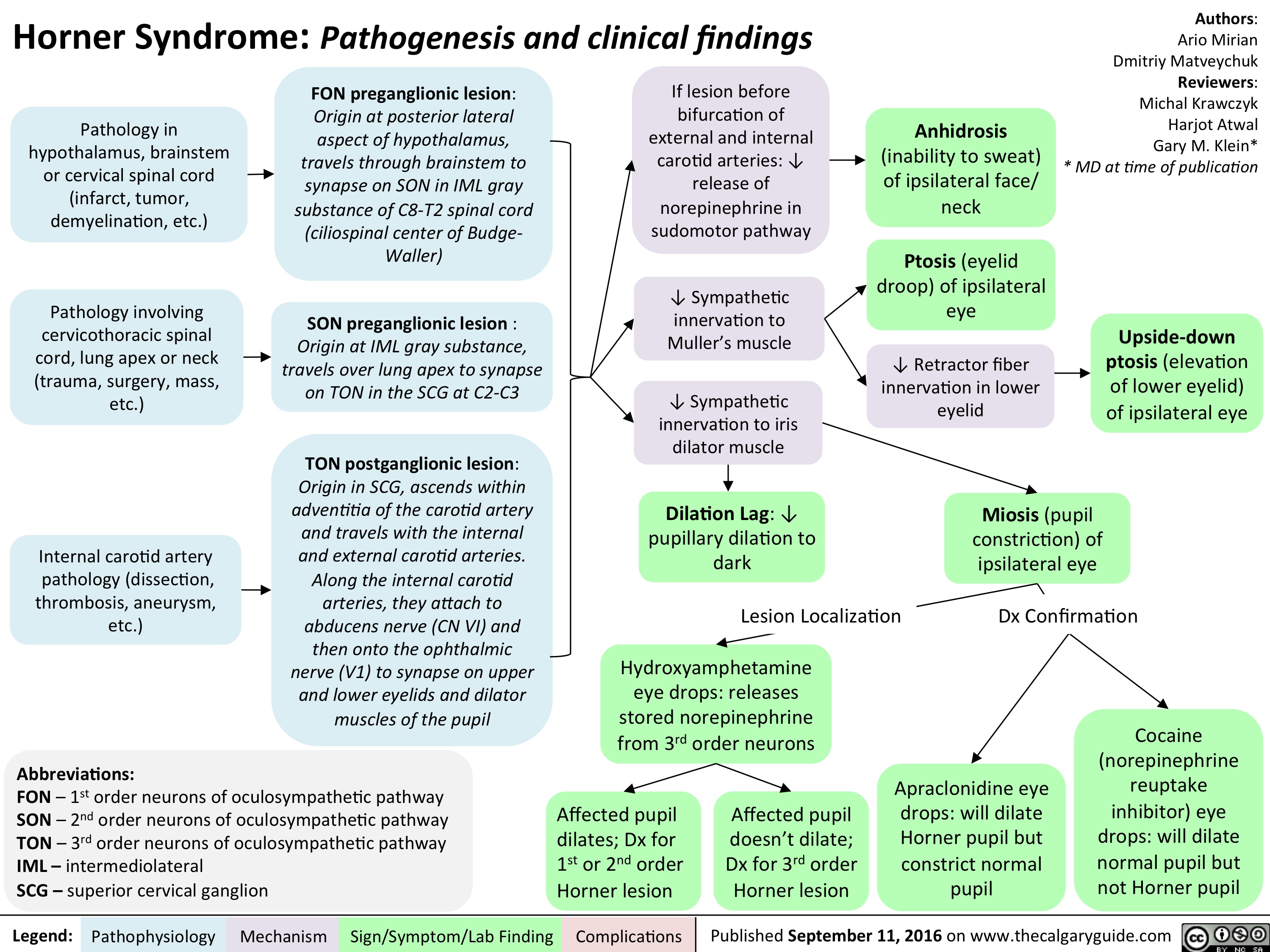
horner-syndrome
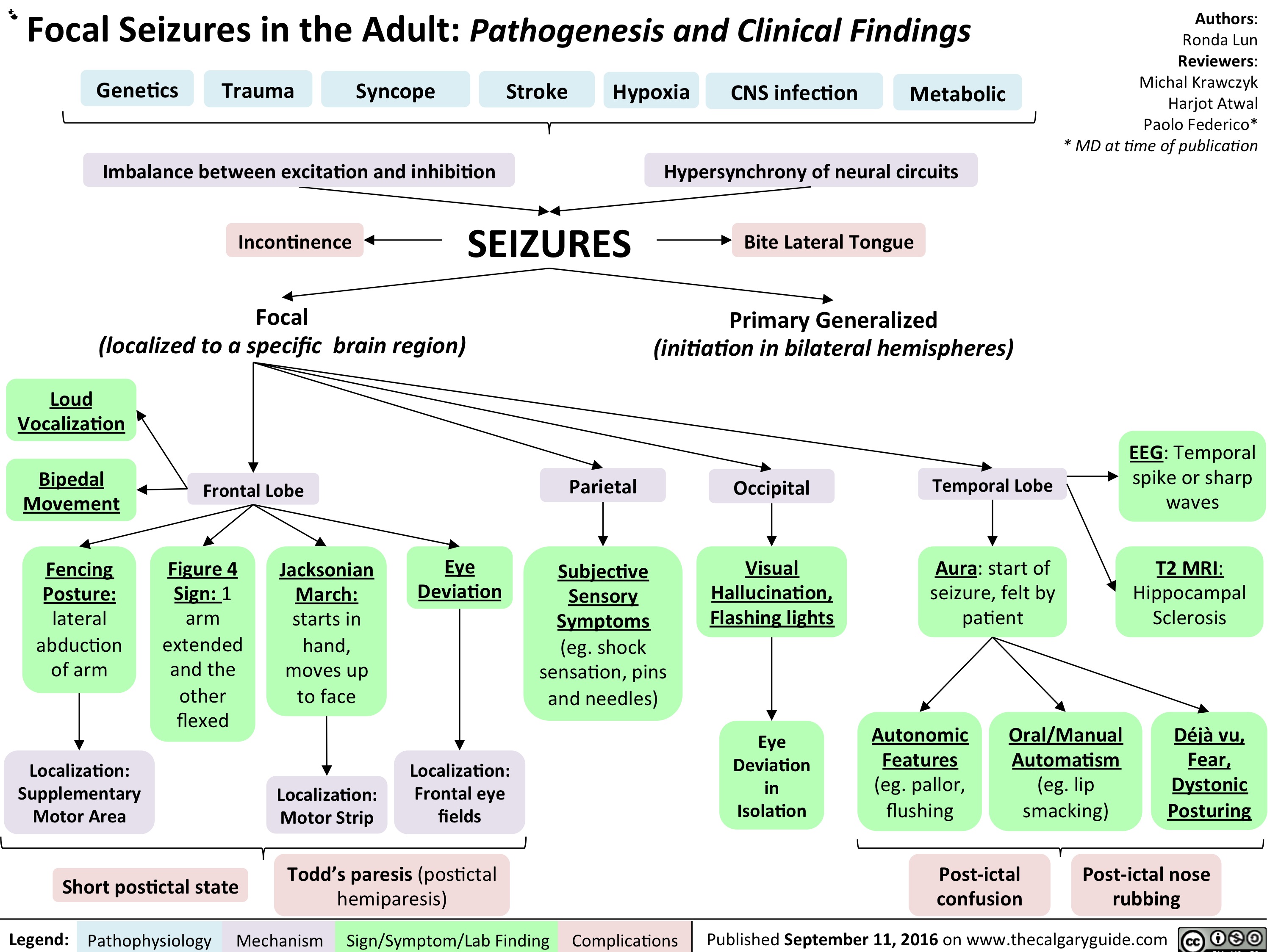
Focal Seizures in the adult: Pathogenesis and Clinical Findings
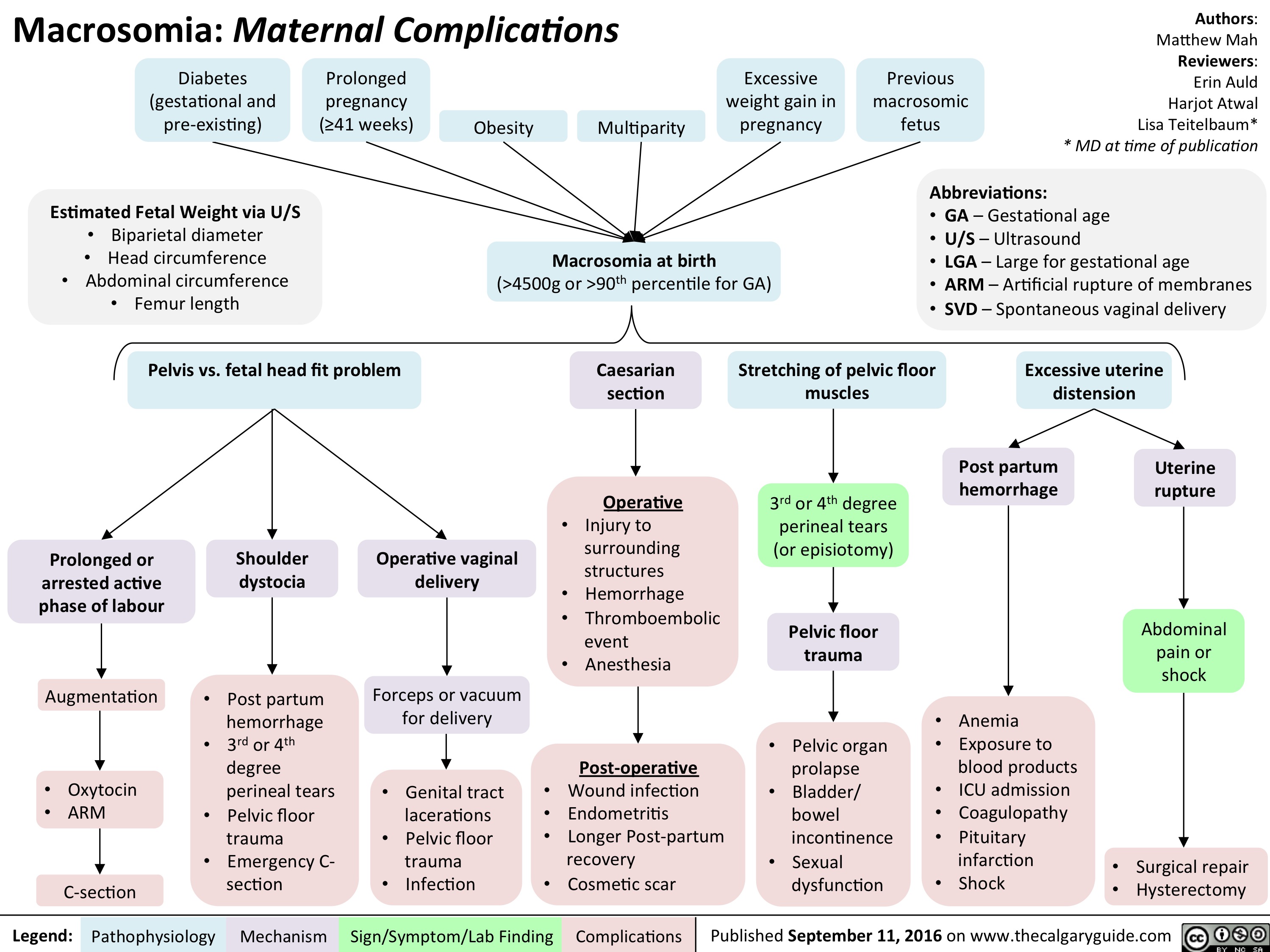
Macrosomia: Maternal Complications
Pediatric Uncompensated Shock: pathogenesis and clinical findings

Type 1 Respiratory Distress Syndrome

acute-closed-angle-glaucoma

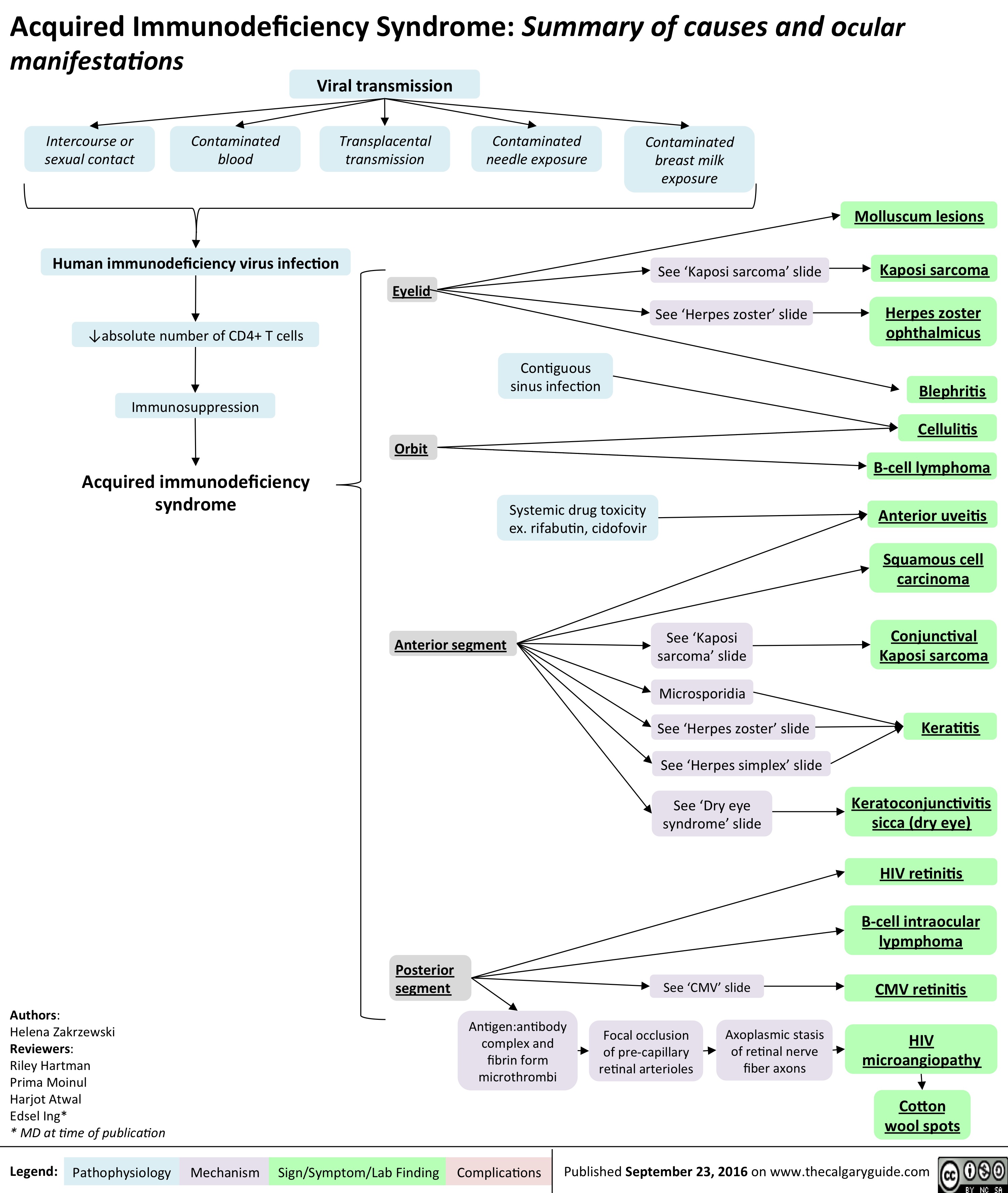
aids-and-cmv-retinitis
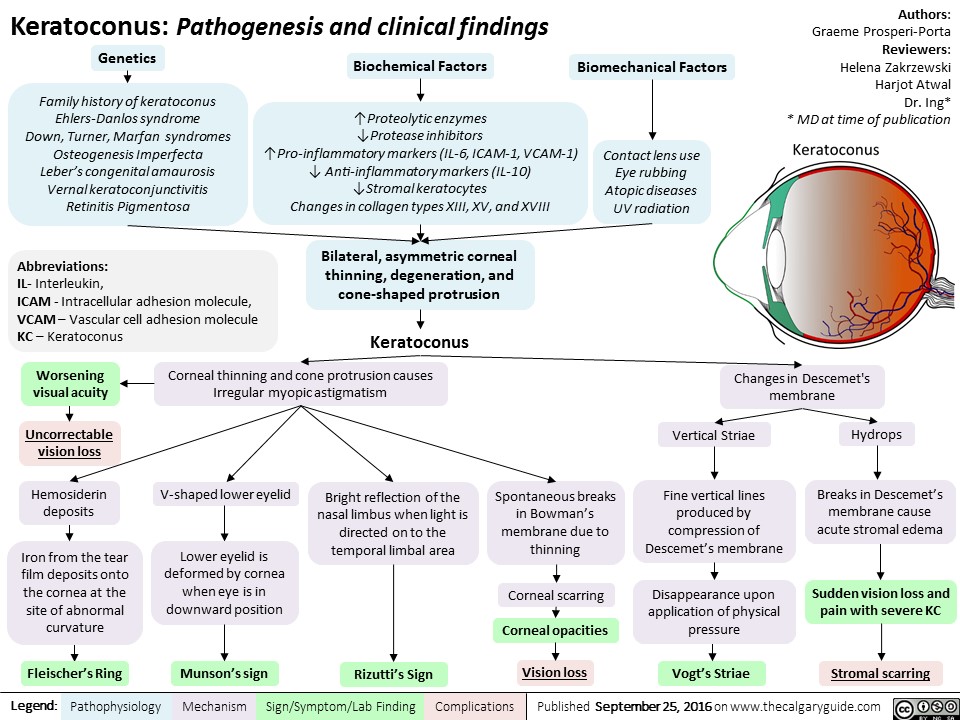
keratoconus
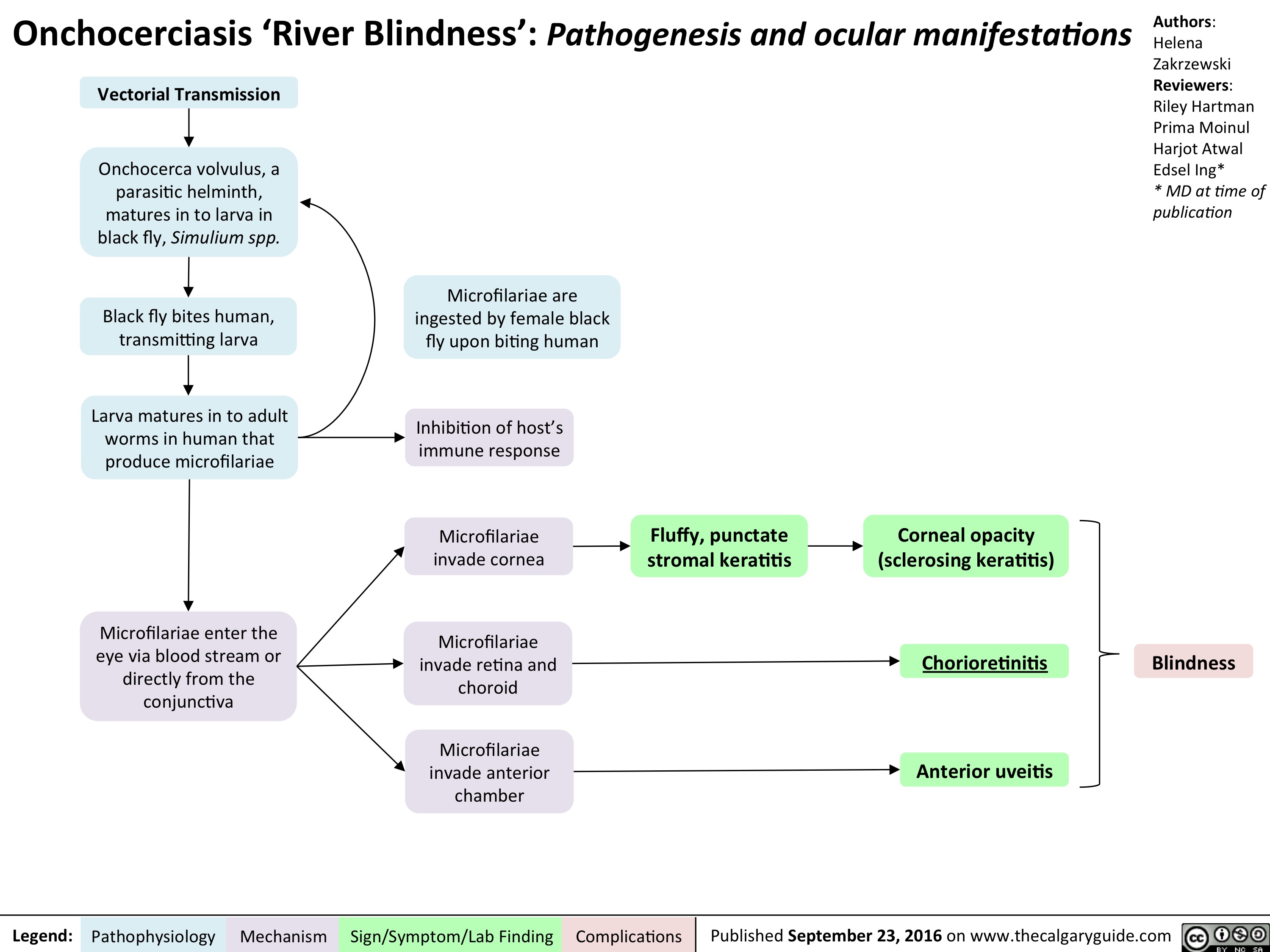
onchocerciasis
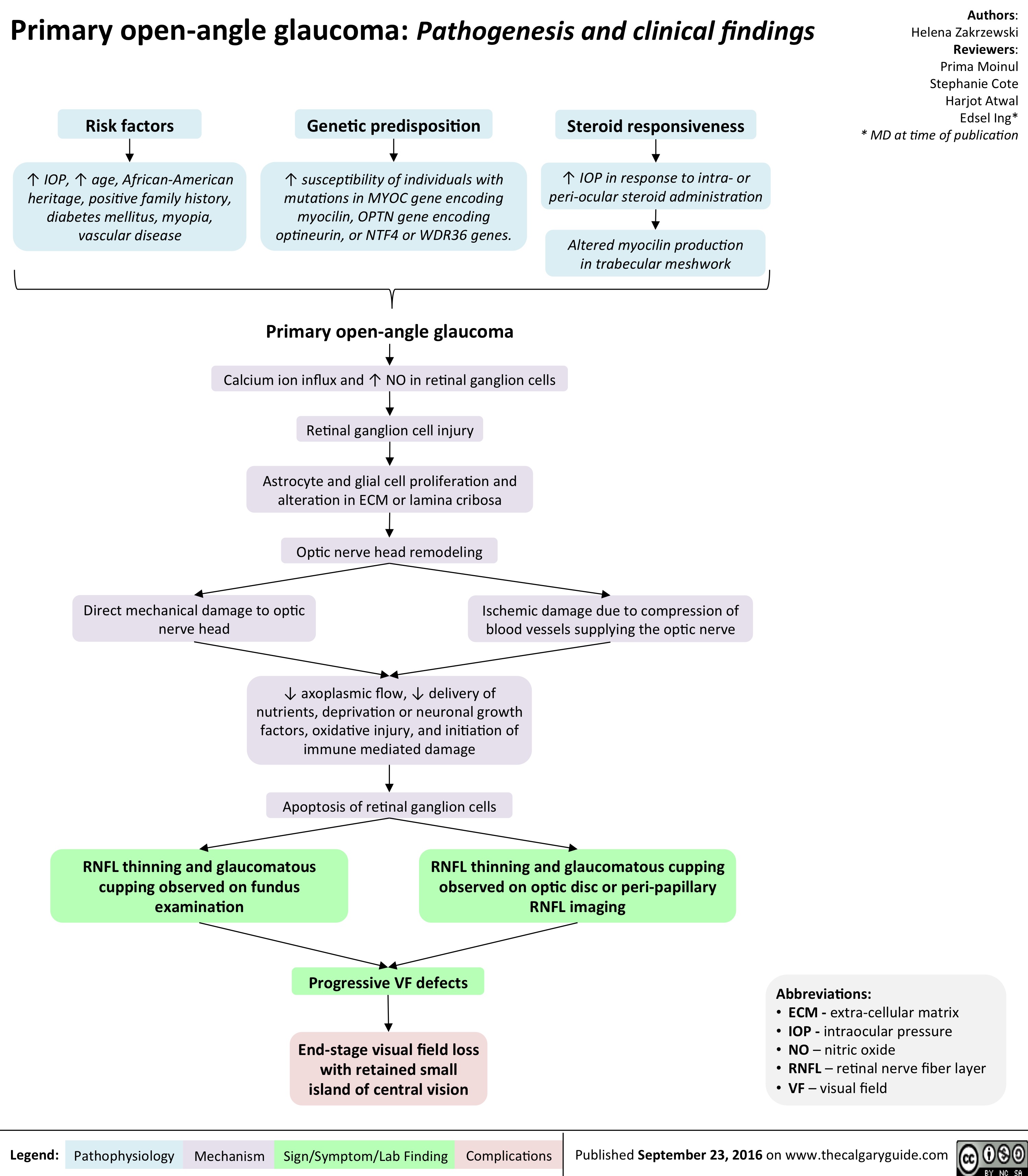
primary-open-angle-glaucoma
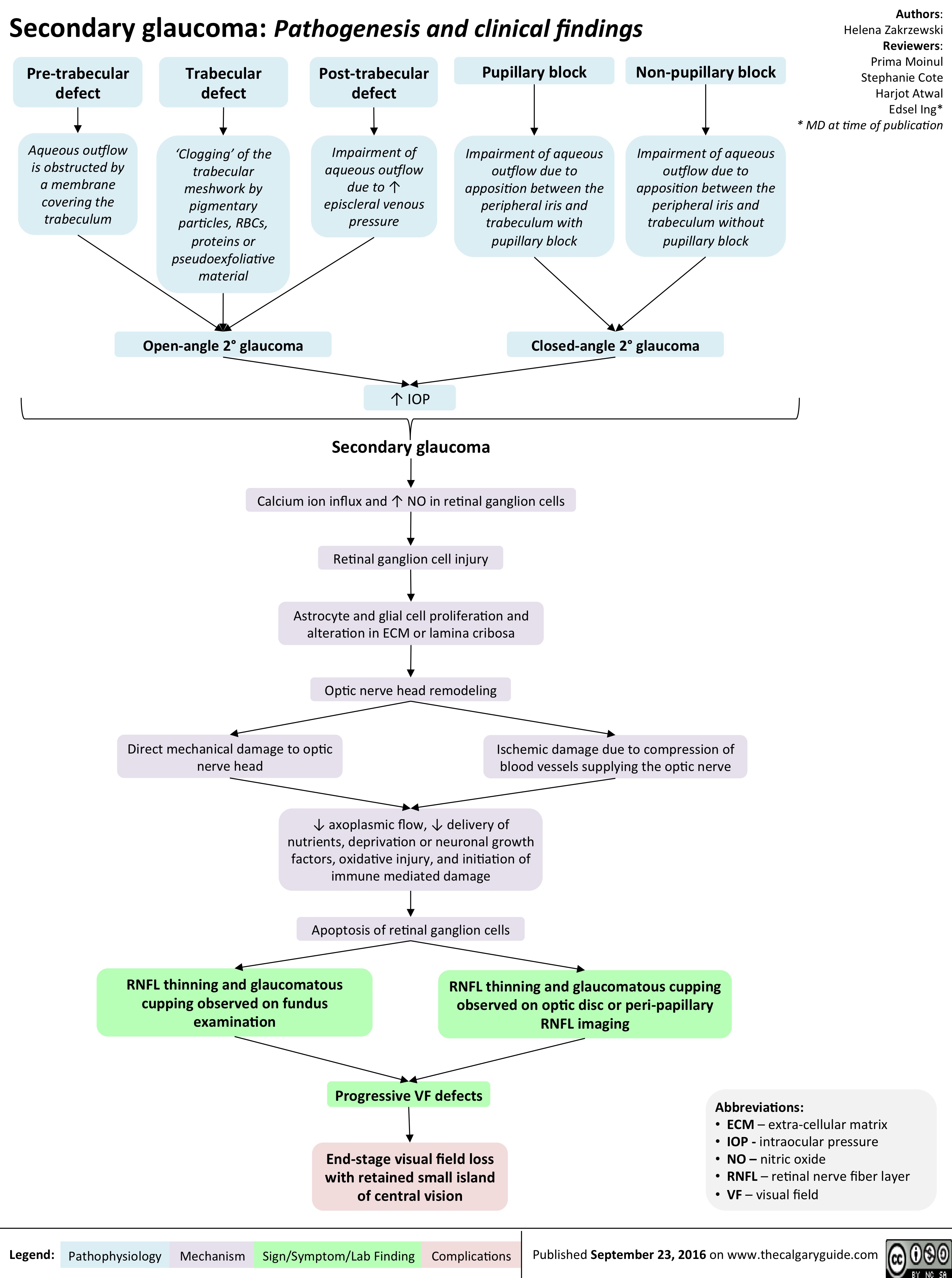
secondary-glaucoma
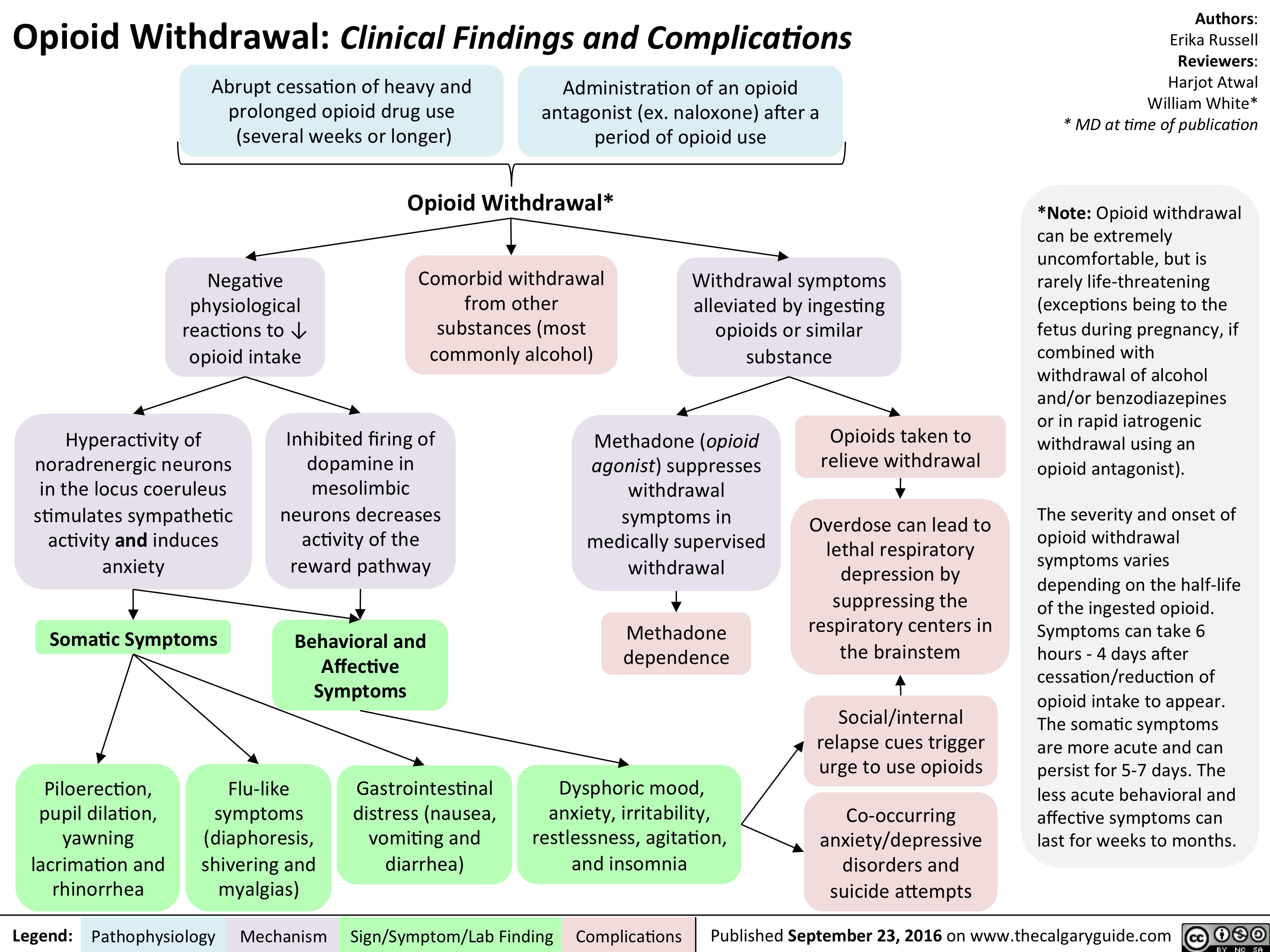
opioid-withdrawal
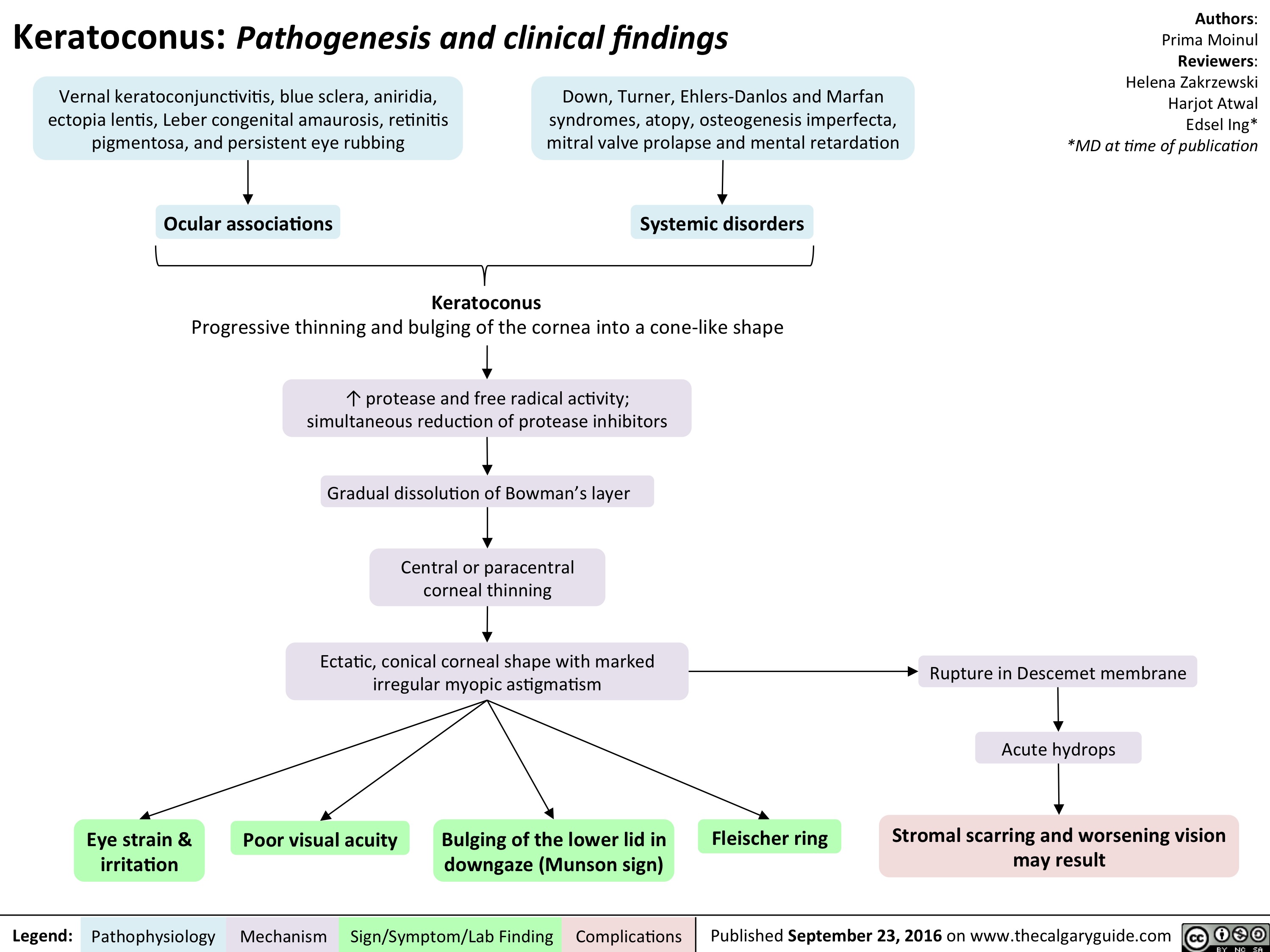
keratoconus
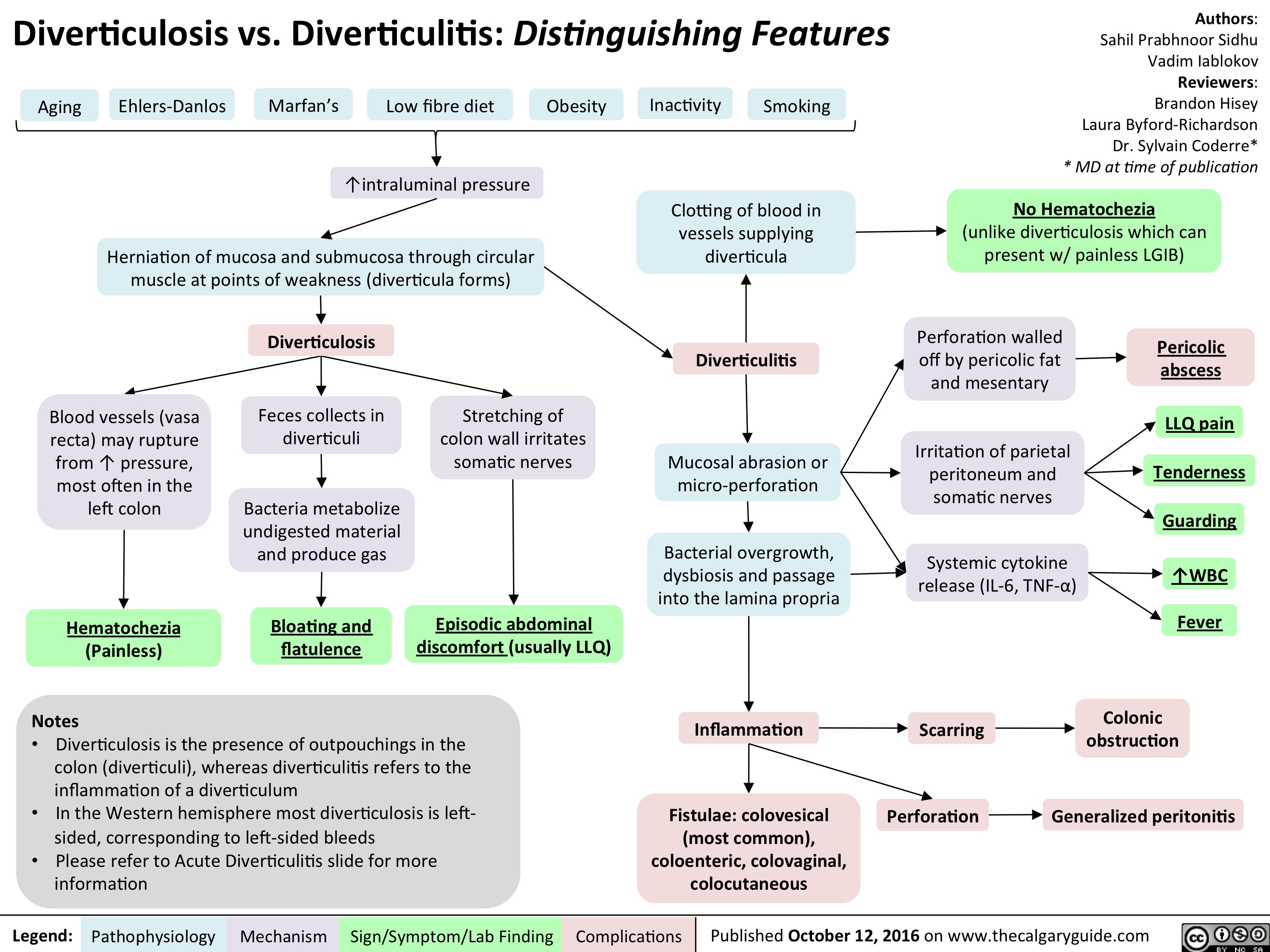
slide1
slide1
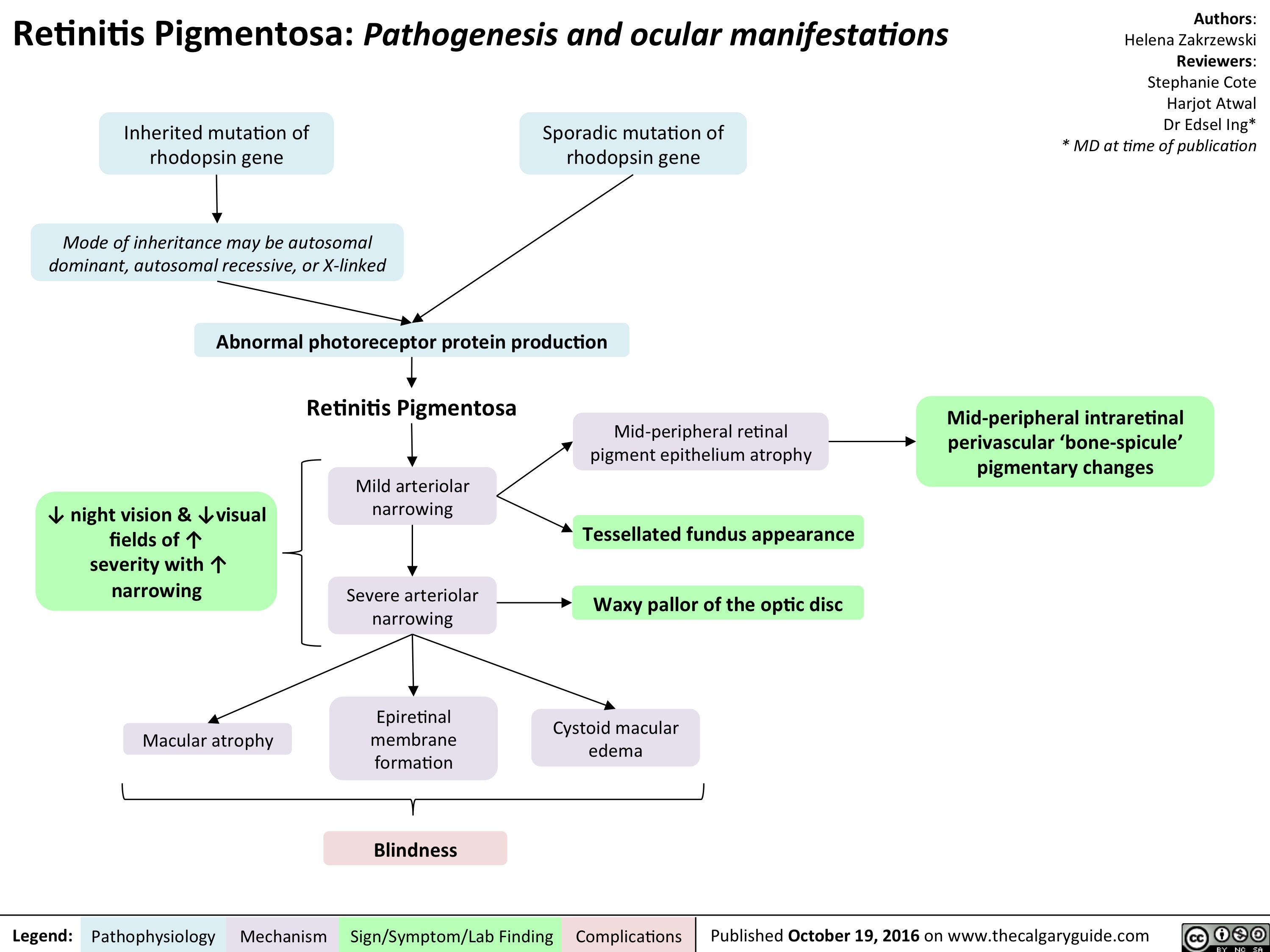
retinitis-pigmentosa-final
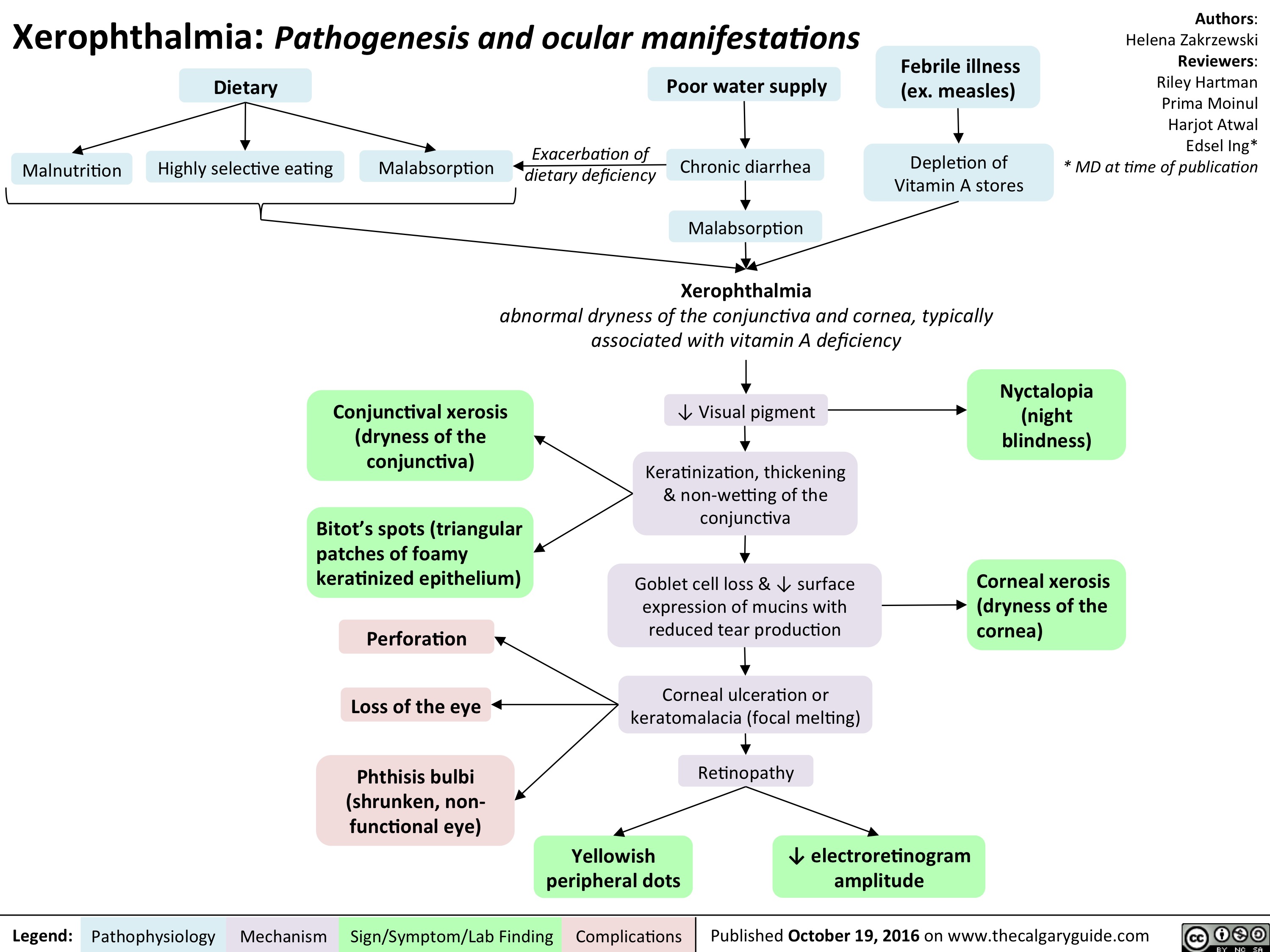
xerophthalmia-final
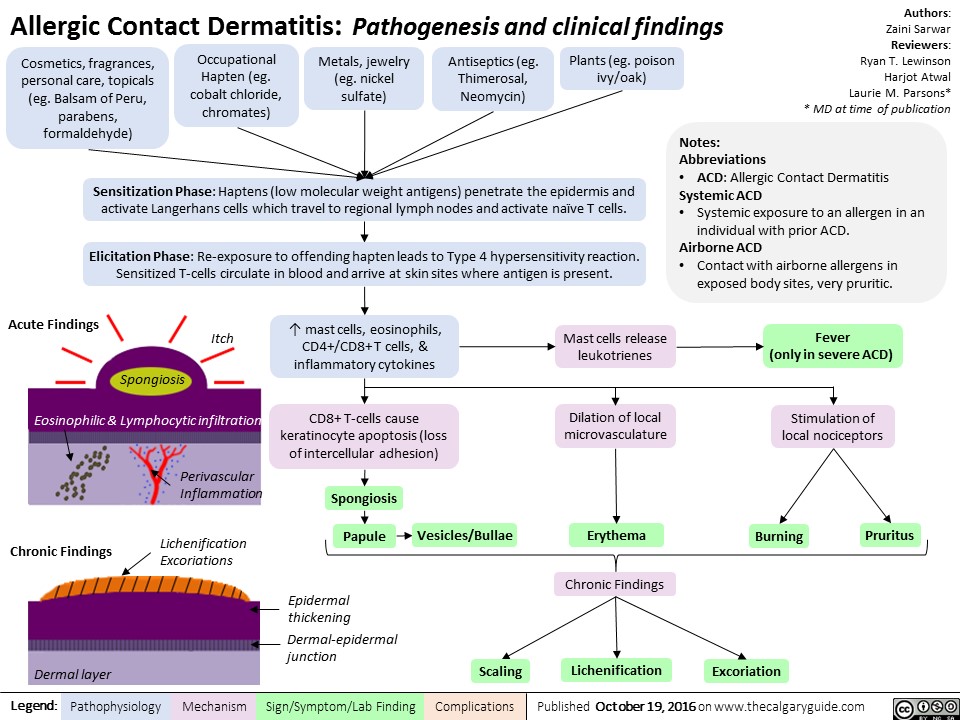
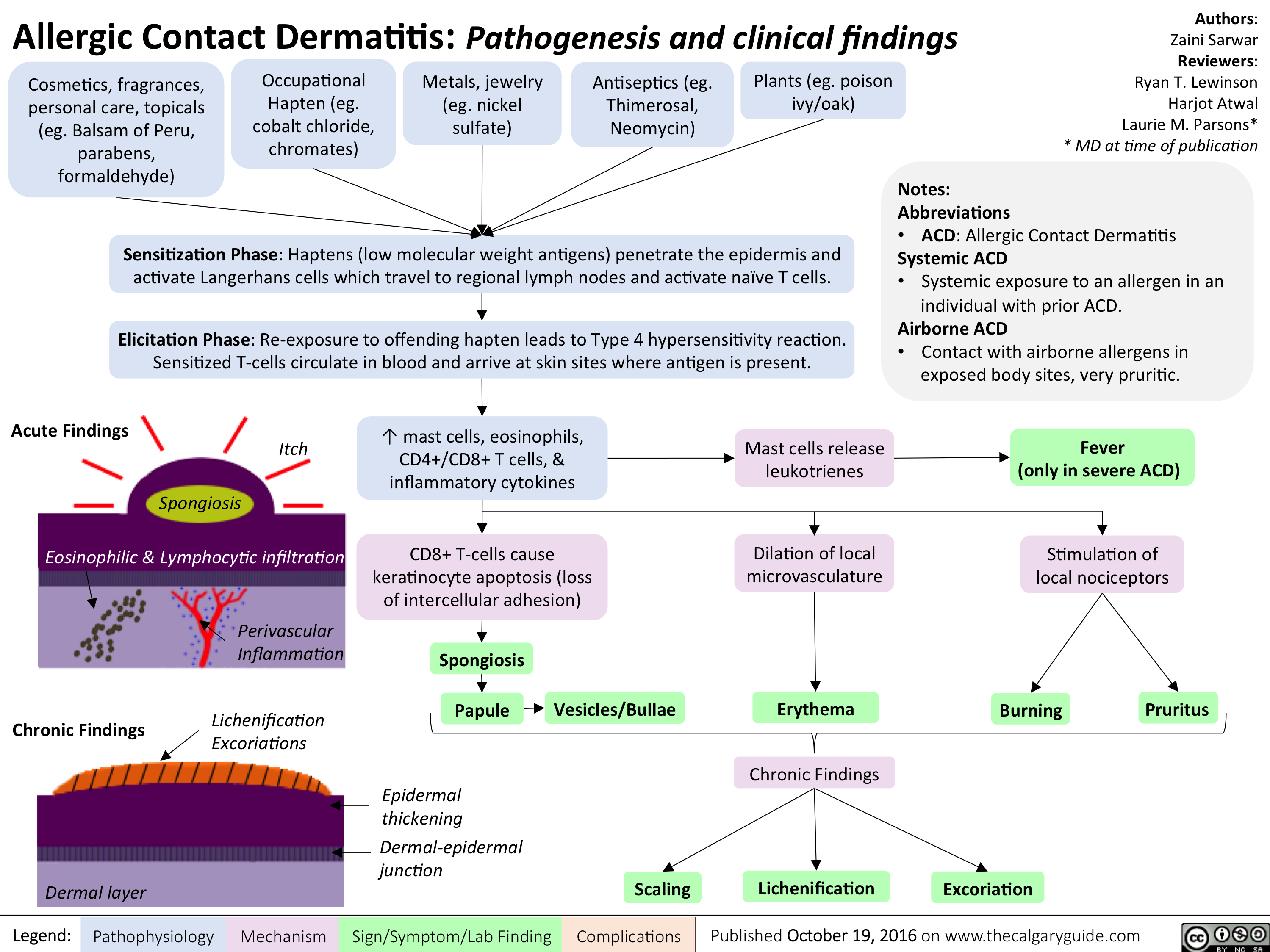
allergic-contact-dermatitis
cardio_lbbb_oct-15
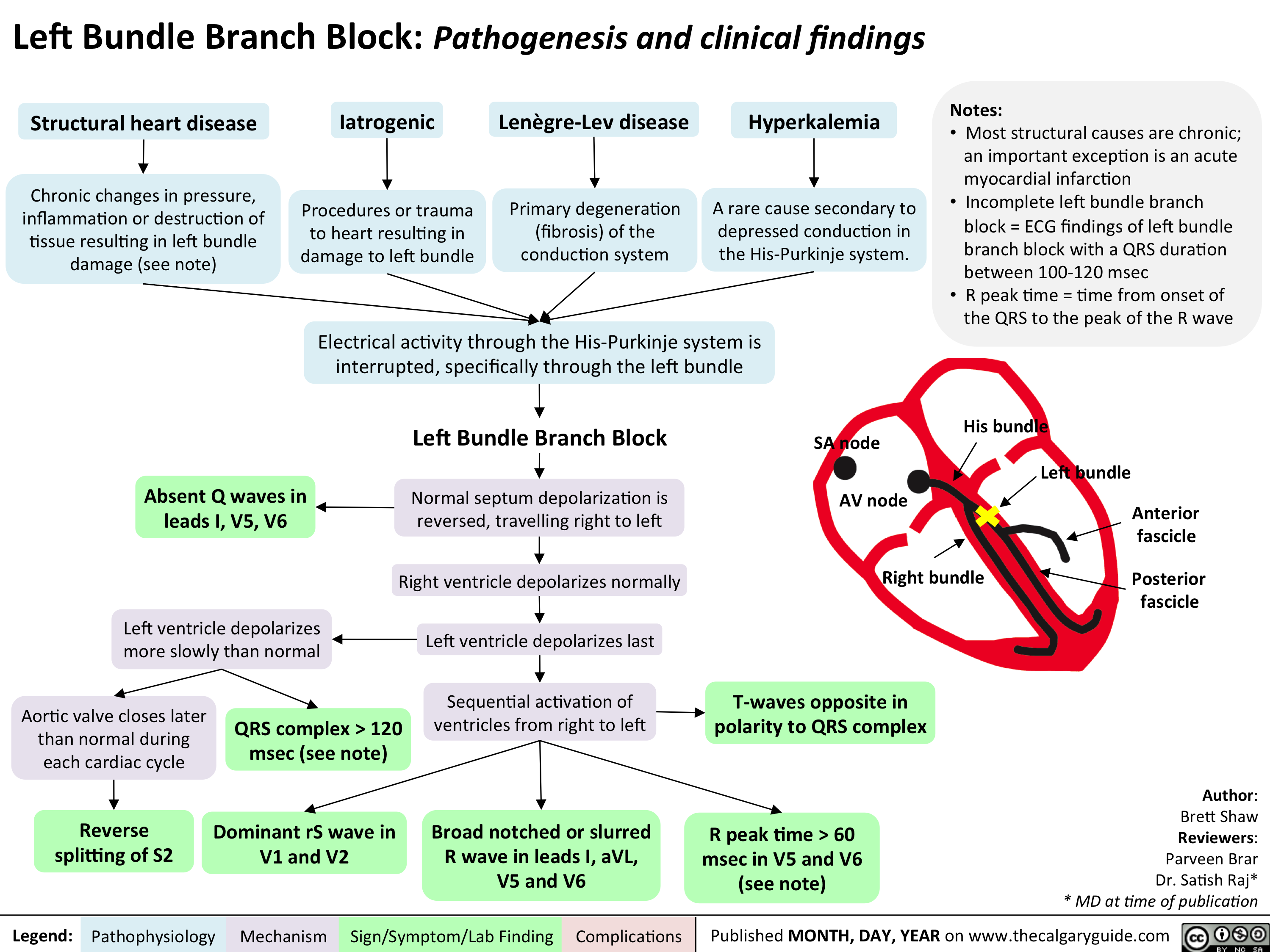
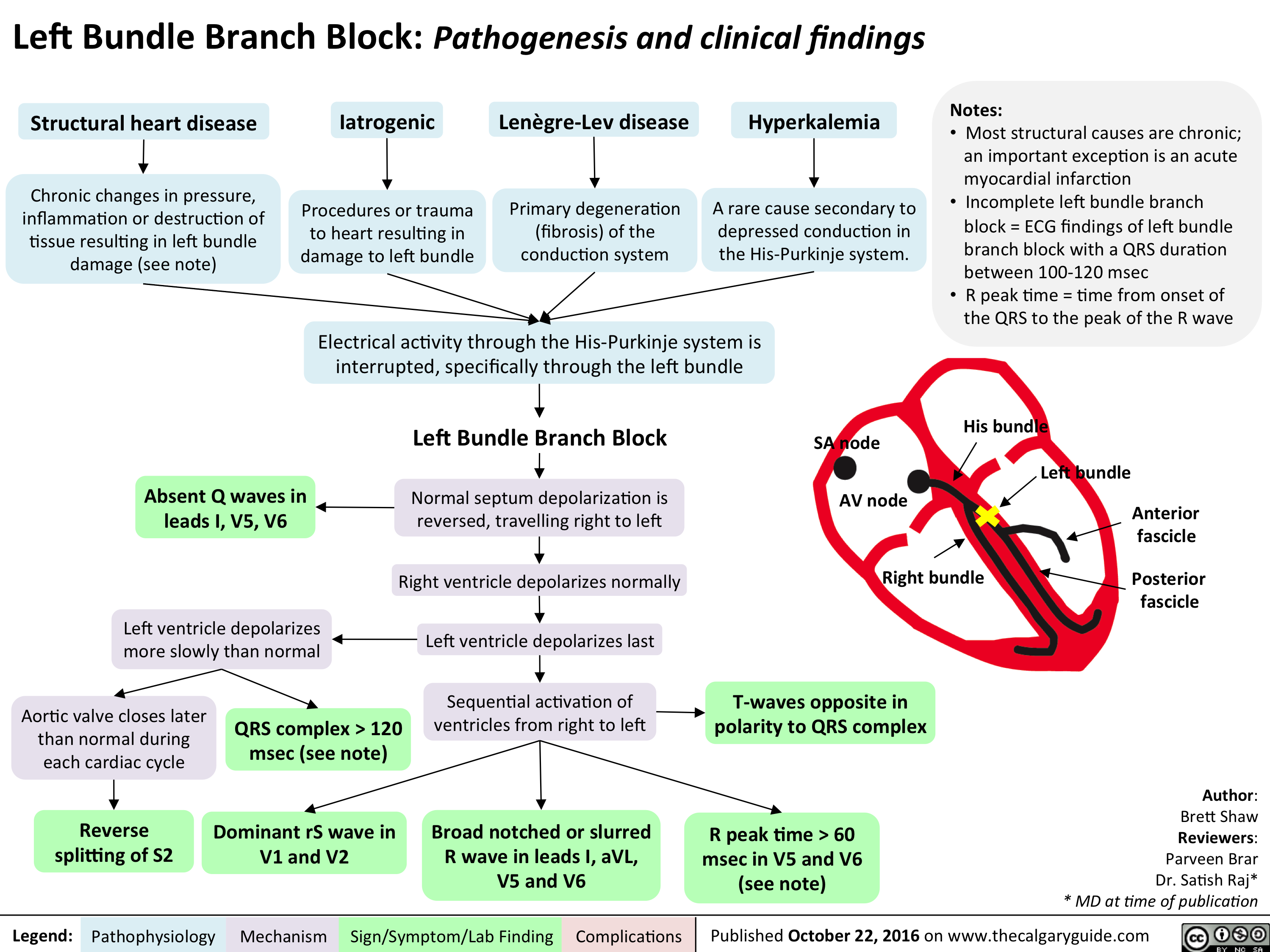
cardio_lbbb_oct-15
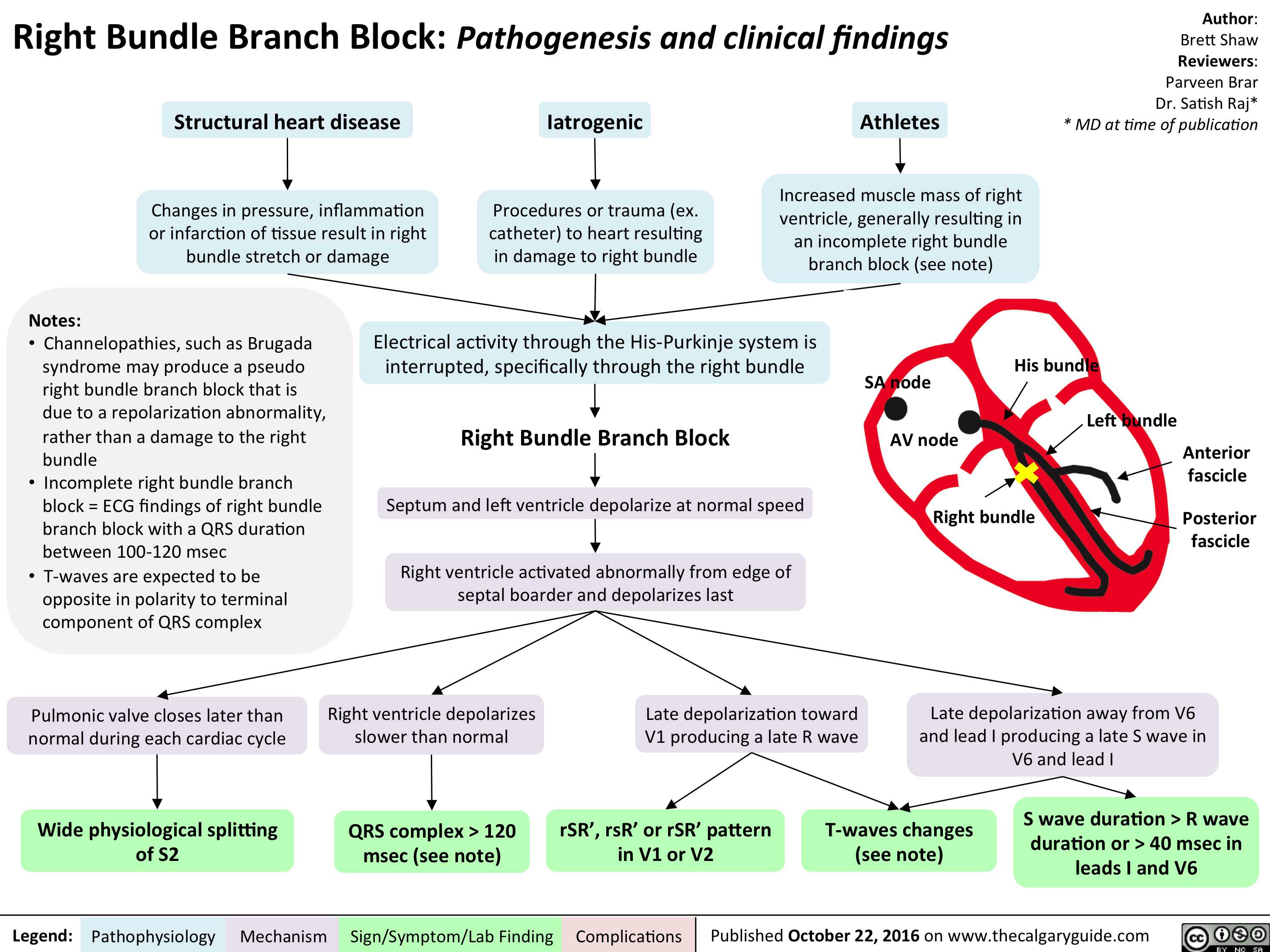
cardio_rbbb_oct-15
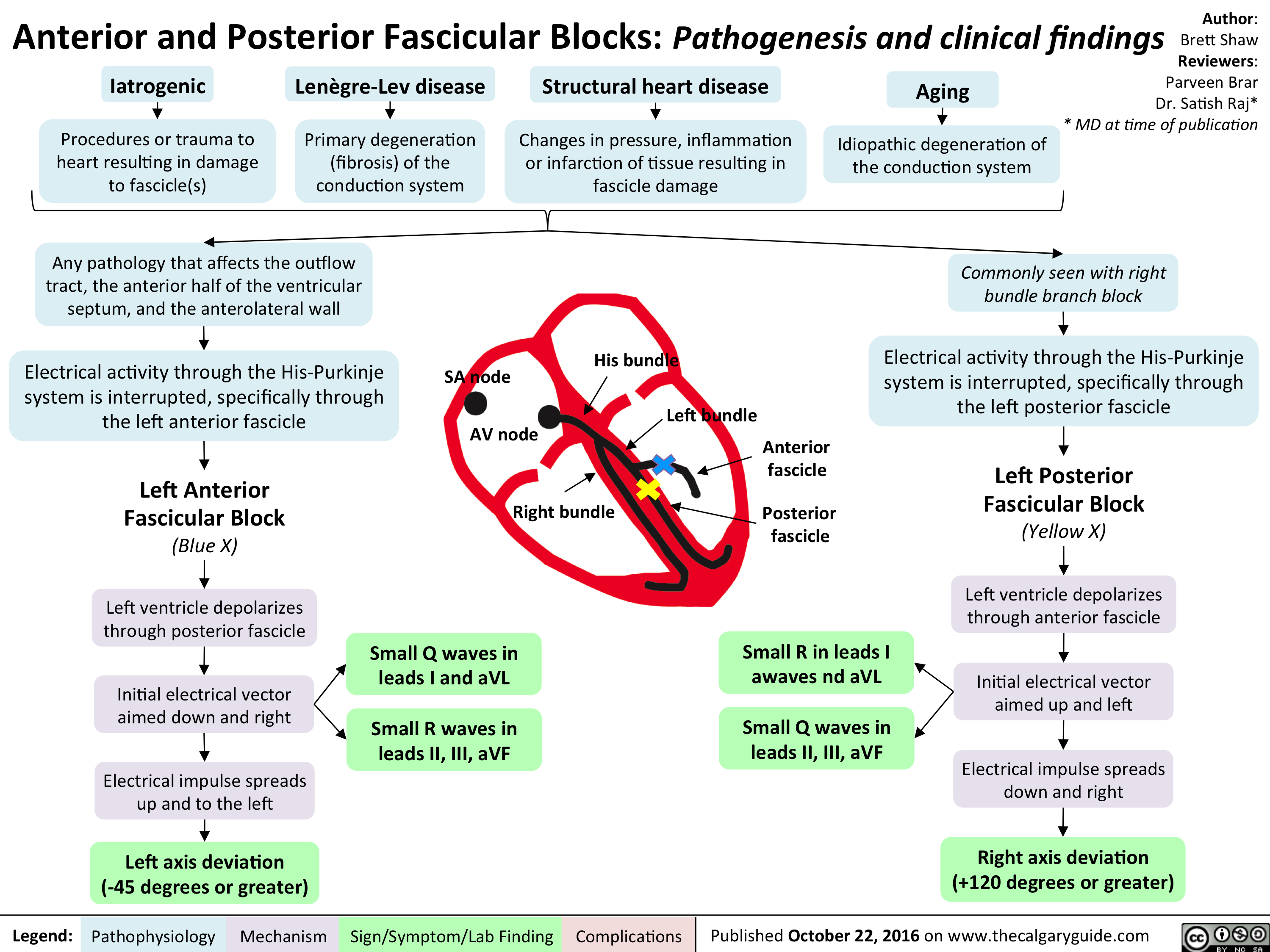
cardio_ant-and-post-fascicular-blocks_oct-15
allergic-contact-dermatitis
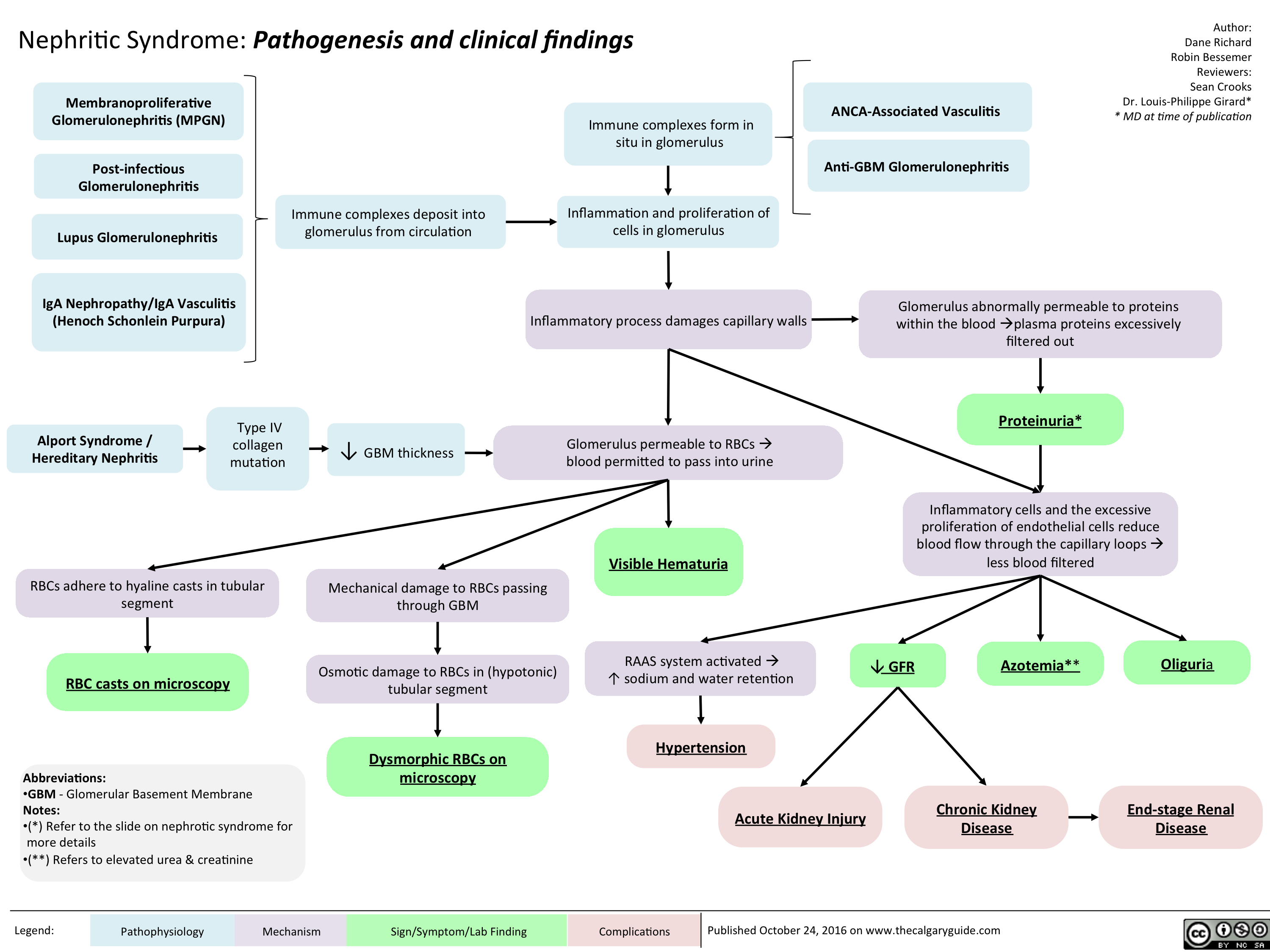
Nephritic Syndrome: Pathogenesis and clinical findings
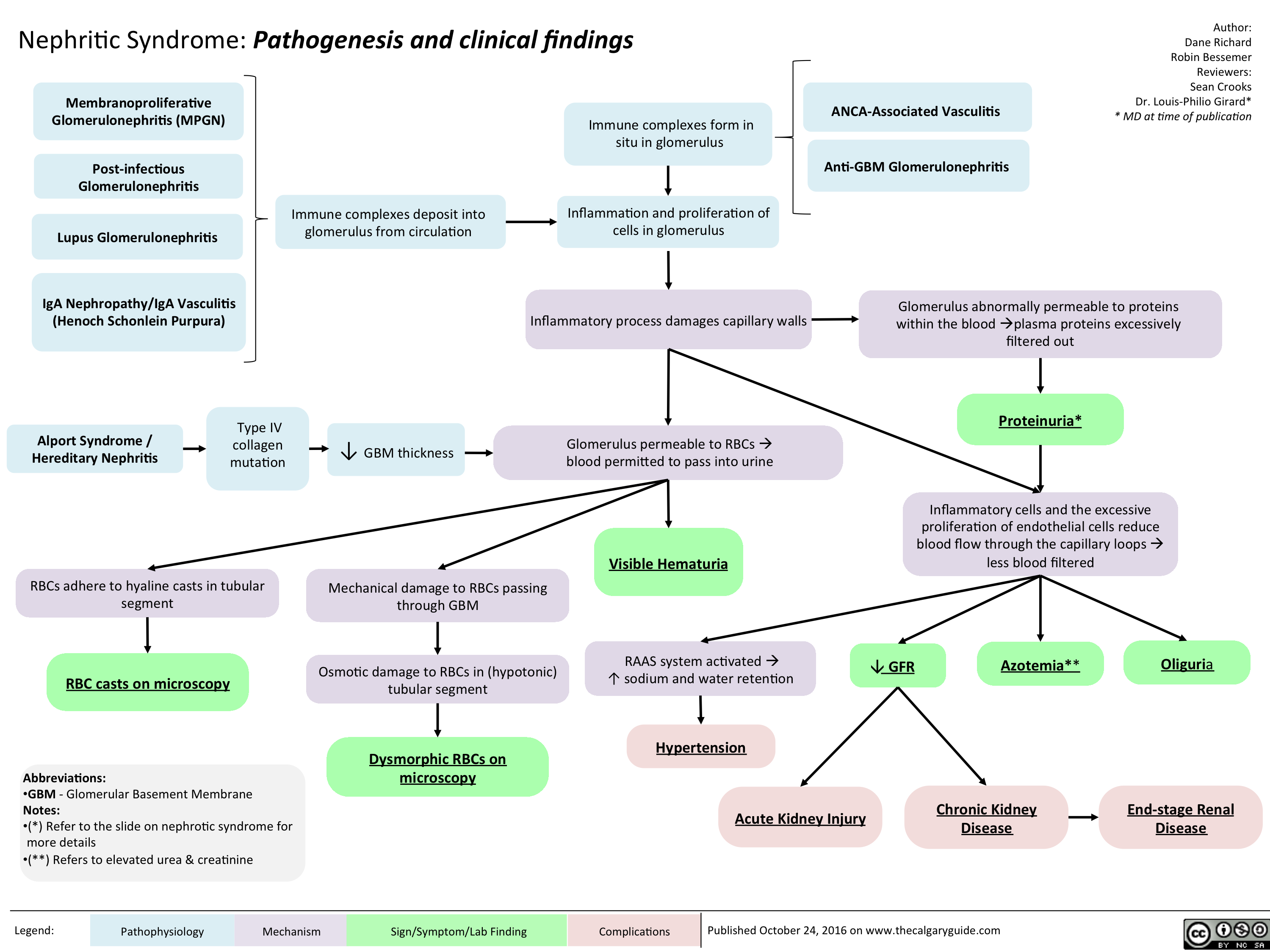
nephritic-syndrome
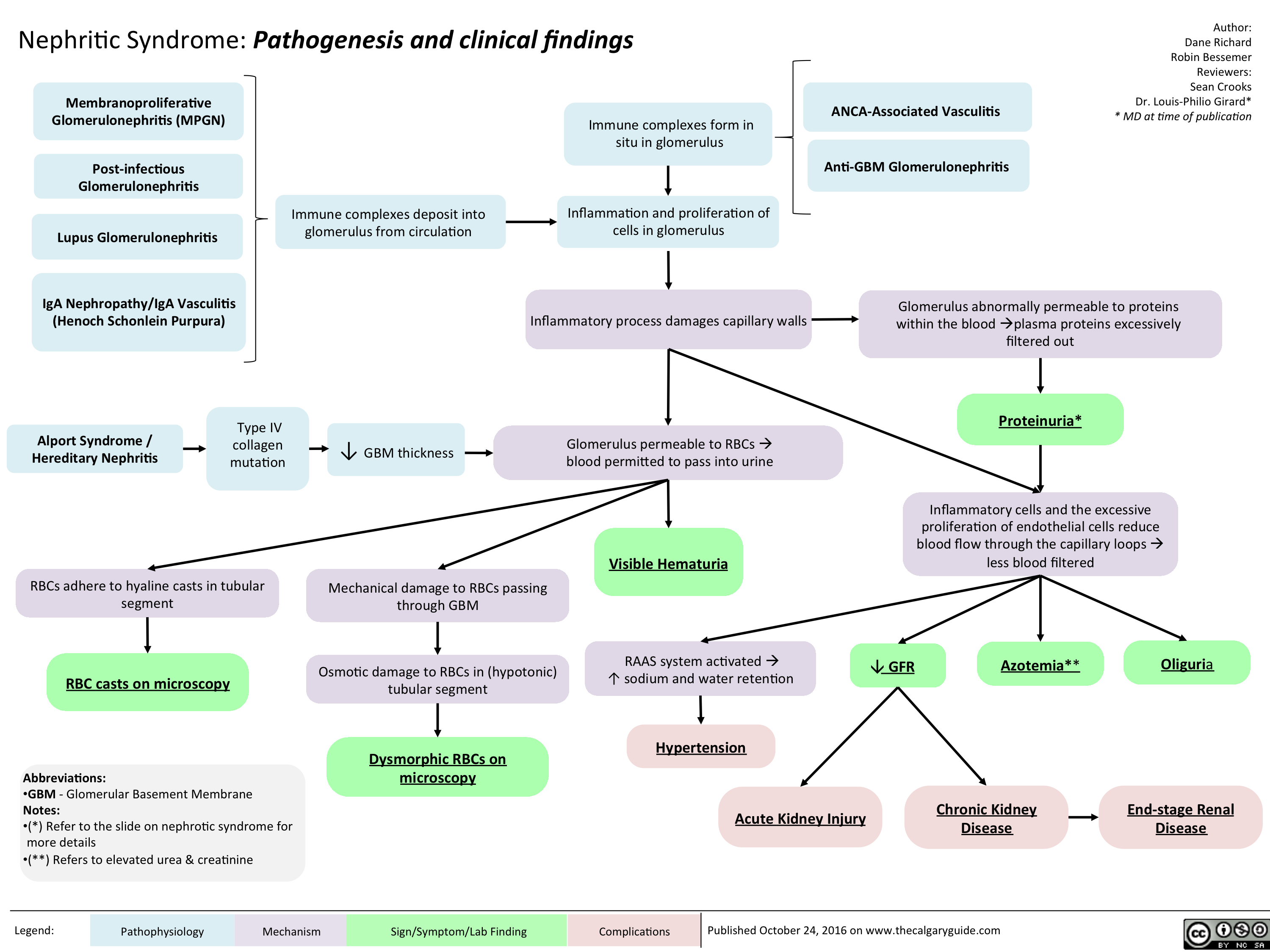
nephritic
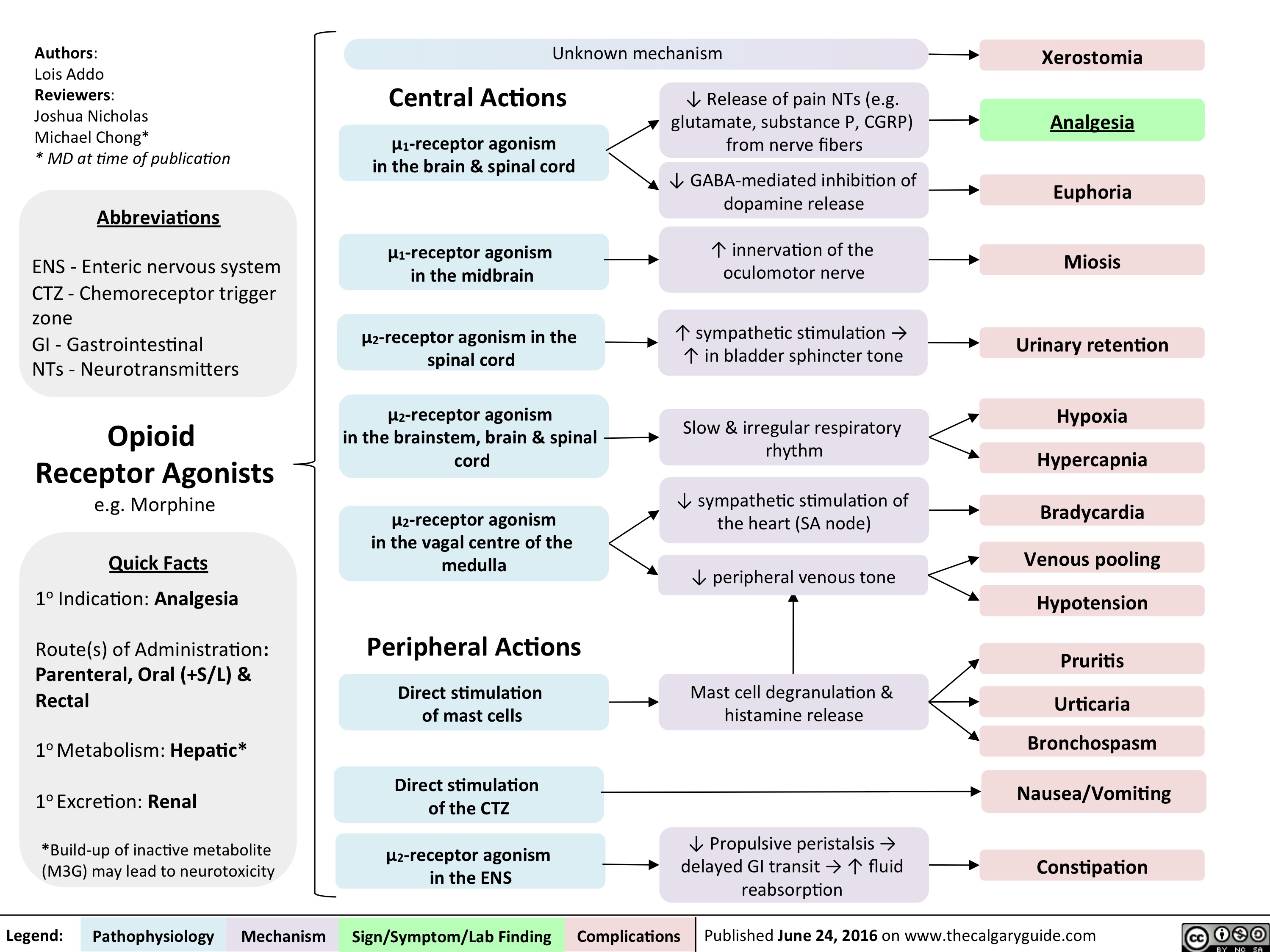
Opioid Receptor Agonists
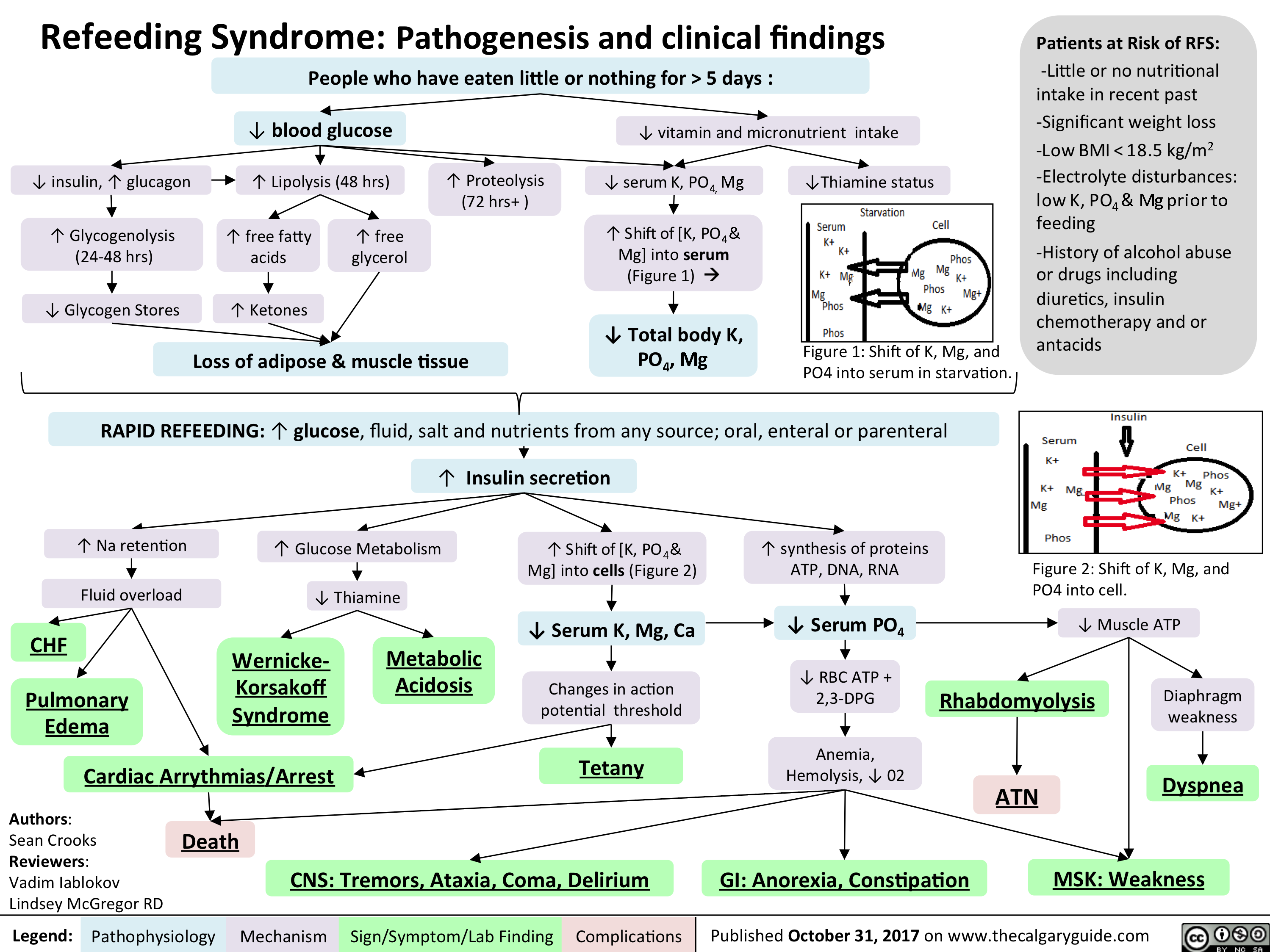
refeeding syndrome
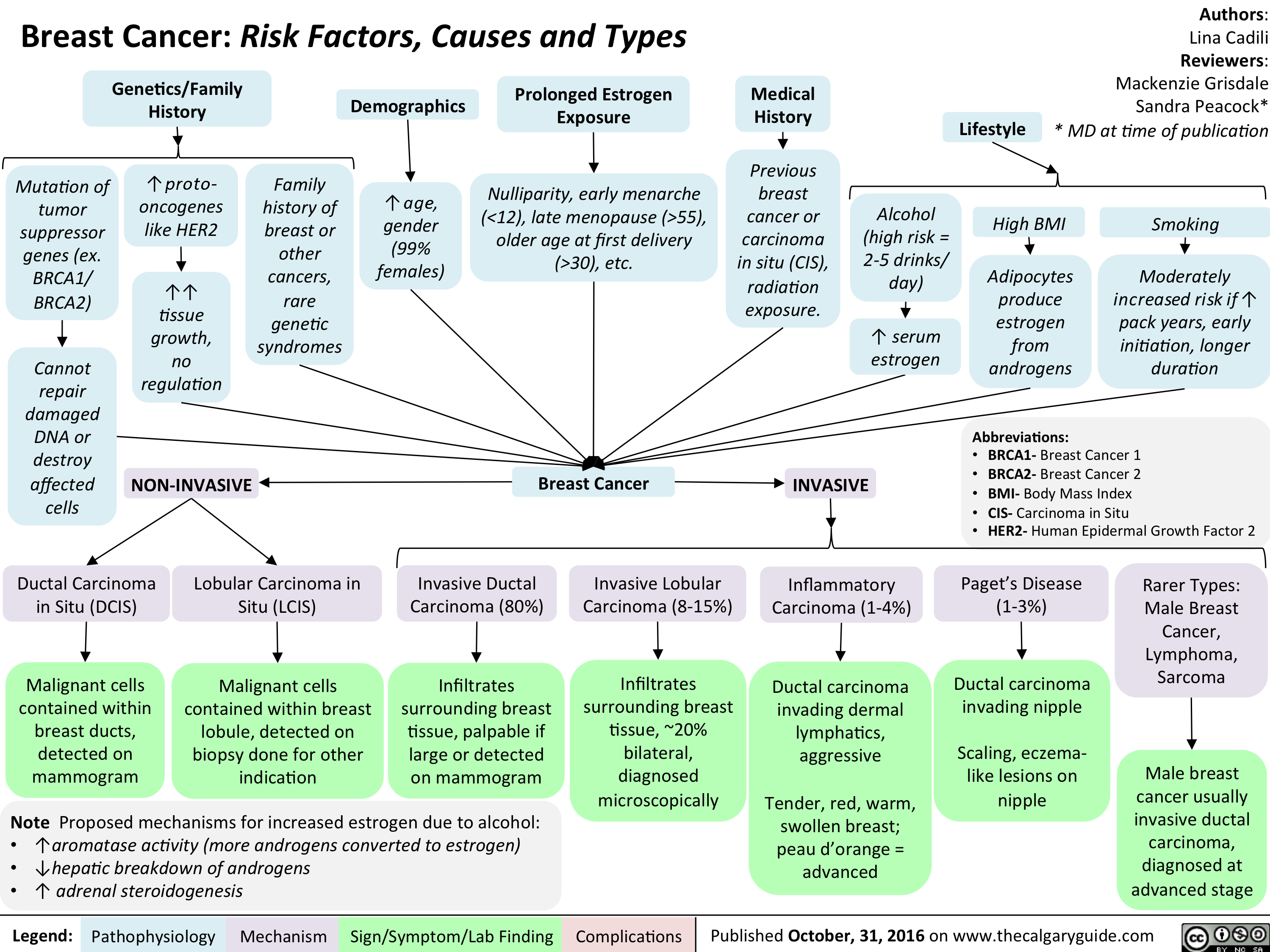
Breast Cancer Risk Factors Causes and Types
Hyperosmolar Hyperglycemic State
![Hyperosmolar Hyperglycemic State (HHS)
Note: HHS is only seen in Type II DM patients!
Note: In patients with either DKA or HHS, always look for an underlying cause (i.e. an infection)
Author: Yan Yu Reviewers:
Peter Vetere
Gill Goobie
Hanan Bassyouni* * MD at time of publication
Alters total body water & ion osmosis
Inadequate insulin production, insulin resistance, non- adherence to insulin Tx
Relative Insulin deficit
Stresses that ↑ Insulin demand: infections, pneumonia, MI, pancreatitis, etc)
Hyperglycemia
(Very high blood [glucose], higher than in DKA)
When blood [glucose] > 12mmol/L, glucose filtration > reabsorption, ↑ urine [glucose]
Glucosuria
Glucose in filtrate promotes osmotic diuresis: large- volume urine output
Polyuria
Dehydration
(↓ JVP, orthostasis: postural hypotension/ postural tachycardia, ↑ resting HR)
Some insulin still present, but not enoughsome glucose is utilized by muscle/fat cells, some remain in the blood
Cells not “starved”, but still need more energy
↑ release of Catabolic hormones: Glucagon, Epinephrine, Cortisol, GH
Body tries to ↑ blood [glucose], to hopefully ↑ cell glucose absorption
Hypothalamic cells sense low intra-cellular glucose, triggering feelings of hunger
Polyphagia
Note: the presence of some insulin directly inhibits lipolysis; thus, in HHS there is no ketone body production, and no subsequent metabolic acidosis and ketouria (unlike in DKA). If ketones are detected in an HHS patient it’s likely secondary to starvation or other mechanisms.
↓ ECF volume, ↑ ECF osmolarity (i.e. hypernatremia)
↑ Gluconeogenesis ↑ Glycogenolysis (in liver)
↓ Protein synthesis, ↑ proteolysis
(in muscle)
↑ Gluconeogenic substrates for liver If the patient doesn’t drink enough
water to replenish lost blood volume If pt is alert and
Electrolyte imbalance
water is accessible
Water osmotically leaves neurons, shrinking them
Neural damage: delirium, lethargy, seizure, stupor, coma
↓ renal perfusion, ↓ GFR
Renal Failure
(pre-renal cause; see relevant slides)
Polydipsia Note: in HHS, body K+ is lost via osmotic diuresis. But diffusion of K+ out of cells
may cause serum [K+] to be falsely normal/elevated. To prevent hypokalemia, give IV KCl along with IV insulin as soon as serum K+ <5.0mmol/L. But ensure patient has good renal function/urine output first, to avoid iatrogenic hyperkalemia!
Note: Electrolyte imbalances (i.e. hyperkalemia, hypernatremia) are worsened by the acute renal failure commonly coexisting with DKA/HHS
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published November 3, 2016 on www.thecalgaryguide.com](http://calgaryguide.ucalgary.ca/wp-content/uploads/2015/05/Hyperosmolar-Hyperglycemic-State-HHS.jpg "Hyperosmolar Hyperglycemic State (HHS)
Note: HHS is only seen in Type II DM patients!
Note: In patients with either DKA or HHS, always look for an underlying cause (i.e. an infection)
Author: Yan Yu Reviewers:
Peter Vetere
Gill Goobie
Hanan Bassyouni* * MD at time of publication
Alters total body water & ion osmosis
Inadequate insulin production, insulin resistance, non- adherence to insulin Tx
Relative Insulin deficit
Stresses that ↑ Insulin demand: infections, pneumonia, MI, pancreatitis, etc)
Hyperglycemia
(Very high blood [glucose], higher than in DKA)
When blood [glucose] > 12mmol/L, glucose filtration > reabsorption, ↑ urine [glucose]
Glucosuria
Glucose in filtrate promotes osmotic diuresis: large- volume urine output
Polyuria
Dehydration
(↓ JVP, orthostasis: postural hypotension/ postural tachycardia, ↑ resting HR)
Some insulin still present, but not enoughsome glucose is utilized by muscle/fat cells, some remain in the blood
Cells not “starved”, but still need more energy
↑ release of Catabolic hormones: Glucagon, Epinephrine, Cortisol, GH
Body tries to ↑ blood [glucose], to hopefully ↑ cell glucose absorption
Hypothalamic cells sense low intra-cellular glucose, triggering feelings of hunger
Polyphagia
Note: the presence of some insulin directly inhibits lipolysis; thus, in HHS there is no ketone body production, and no subsequent metabolic acidosis and ketouria (unlike in DKA). If ketones are detected in an HHS patient it’s likely secondary to starvation or other mechanisms.
↓ ECF volume, ↑ ECF osmolarity (i.e. hypernatremia)
↑ Gluconeogenesis ↑ Glycogenolysis (in liver)
↓ Protein synthesis, ↑ proteolysis
(in muscle)
↑ Gluconeogenic substrates for liver If the patient doesn’t drink enough
water to replenish lost blood volume If pt is alert and
Electrolyte imbalance
water is accessible
Water osmotically leaves neurons, shrinking them
Neural damage: delirium, lethargy, seizure, stupor, coma
↓ renal perfusion, ↓ GFR
Renal Failure
(pre-renal cause; see relevant slides)
Polydipsia Note: in HHS, body K+ is lost via osmotic diuresis. But diffusion of K+ out of cells
may cause serum [K+] to be falsely normal/elevated. To prevent hypokalemia, give IV KCl along with IV insulin as soon as serum K+ <5.0mmol/L. But ensure patient has good renal function/urine output first, to avoid iatrogenic hyperkalemia!
Note: Electrolyte imbalances (i.e. hyperkalemia, hypernatremia) are worsened by the acute renal failure commonly coexisting with DKA/HHS
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published November 3, 2016 on www.thecalgaryguide.com")
Neurotransmitters and Pharmacology behind Nausea and Vomiting
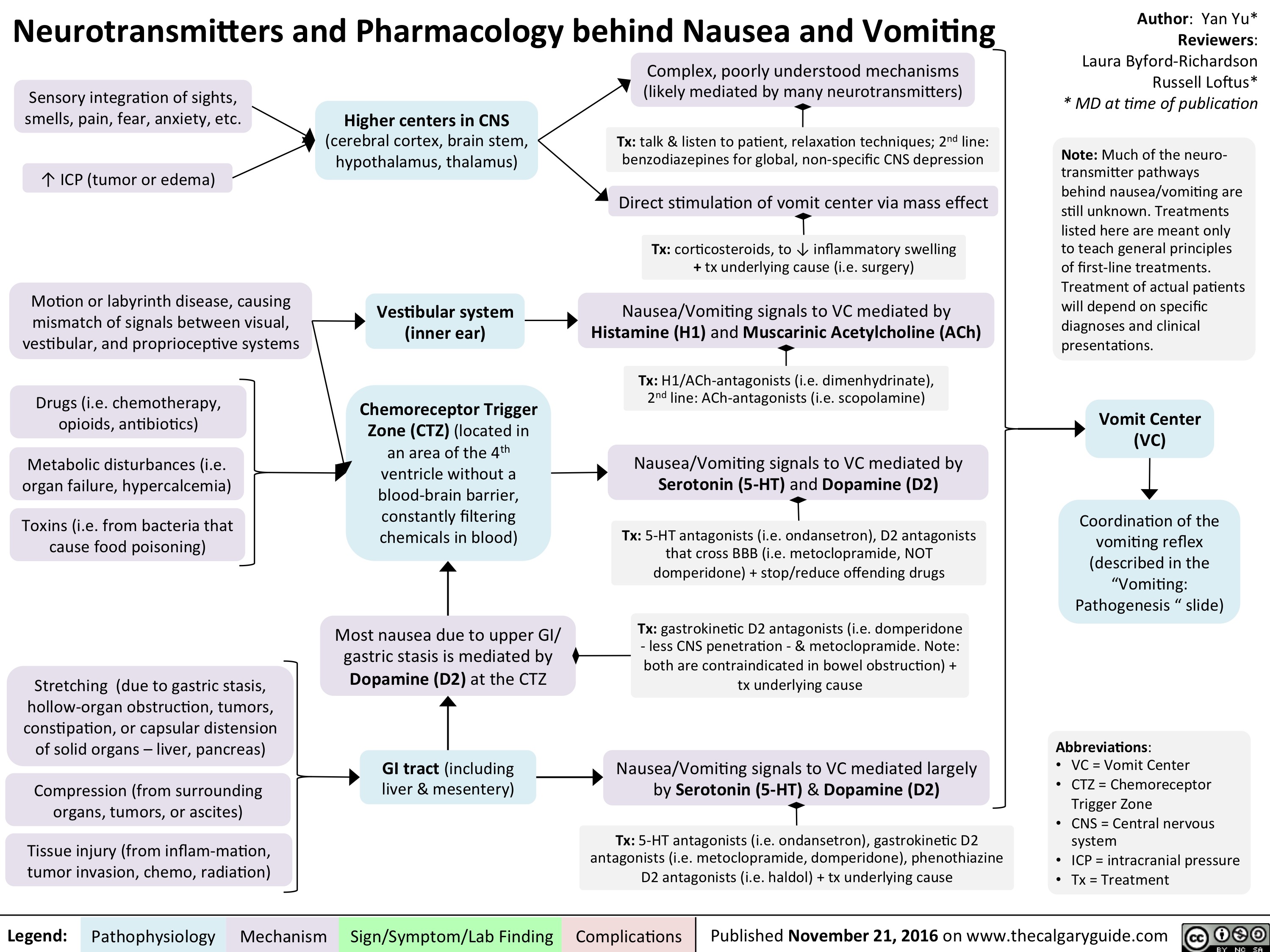
Neurotransmitters and Pharmacology behind Nausea and Vomiting
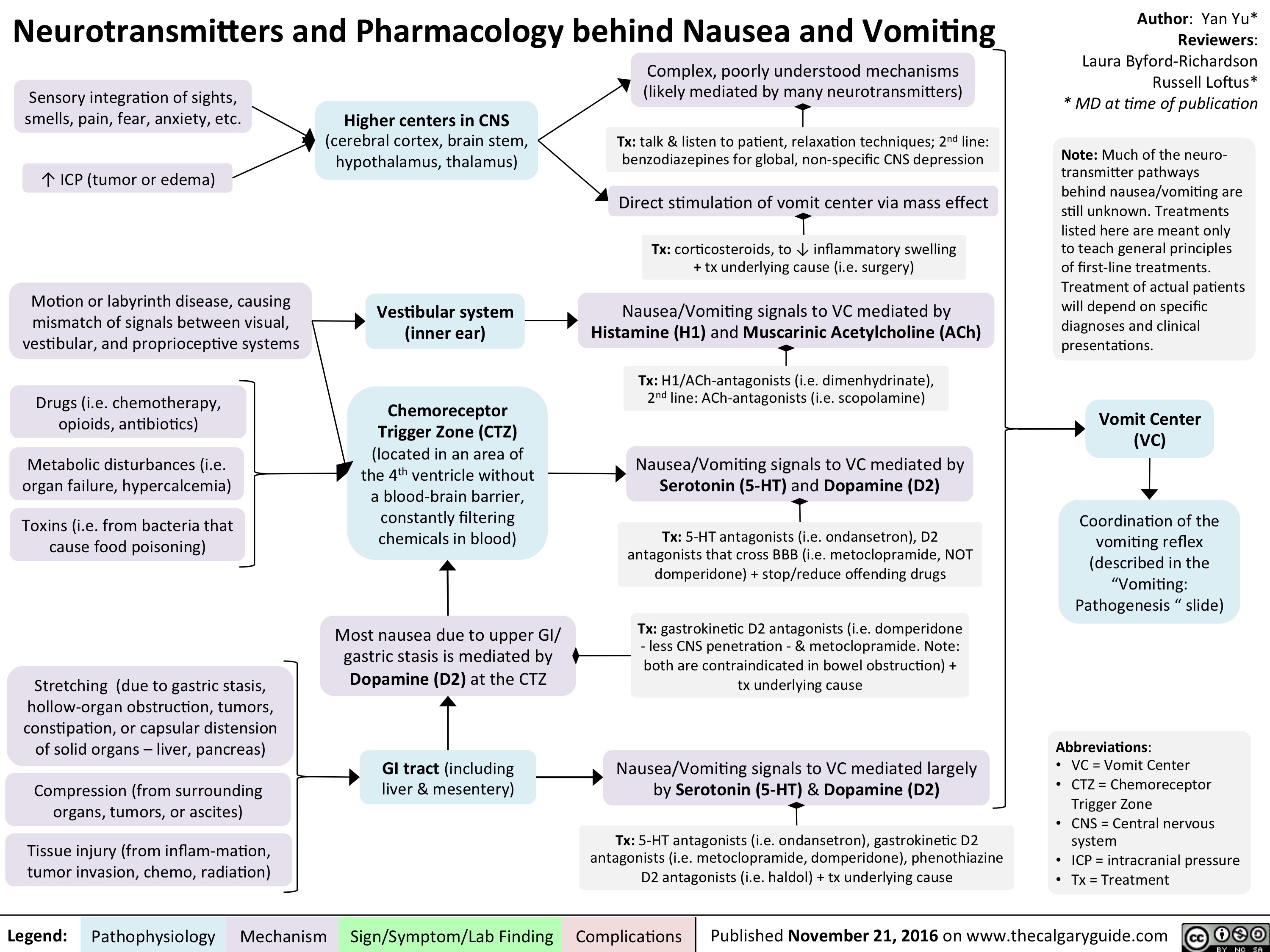
Neurotransmitters and Pharmacology behind Nausea and Vomiting
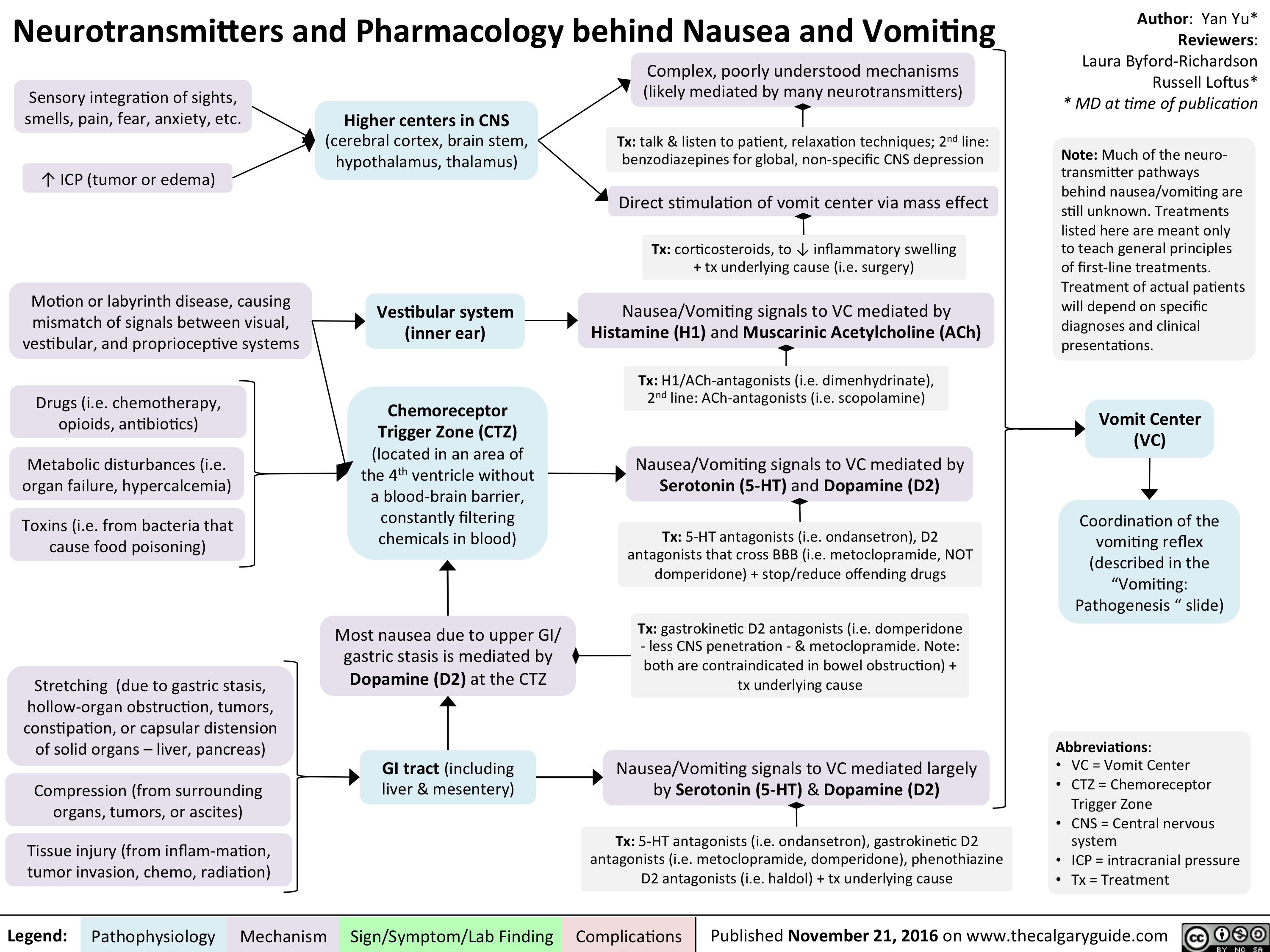
pathogenesis-of-neuropathic-pain
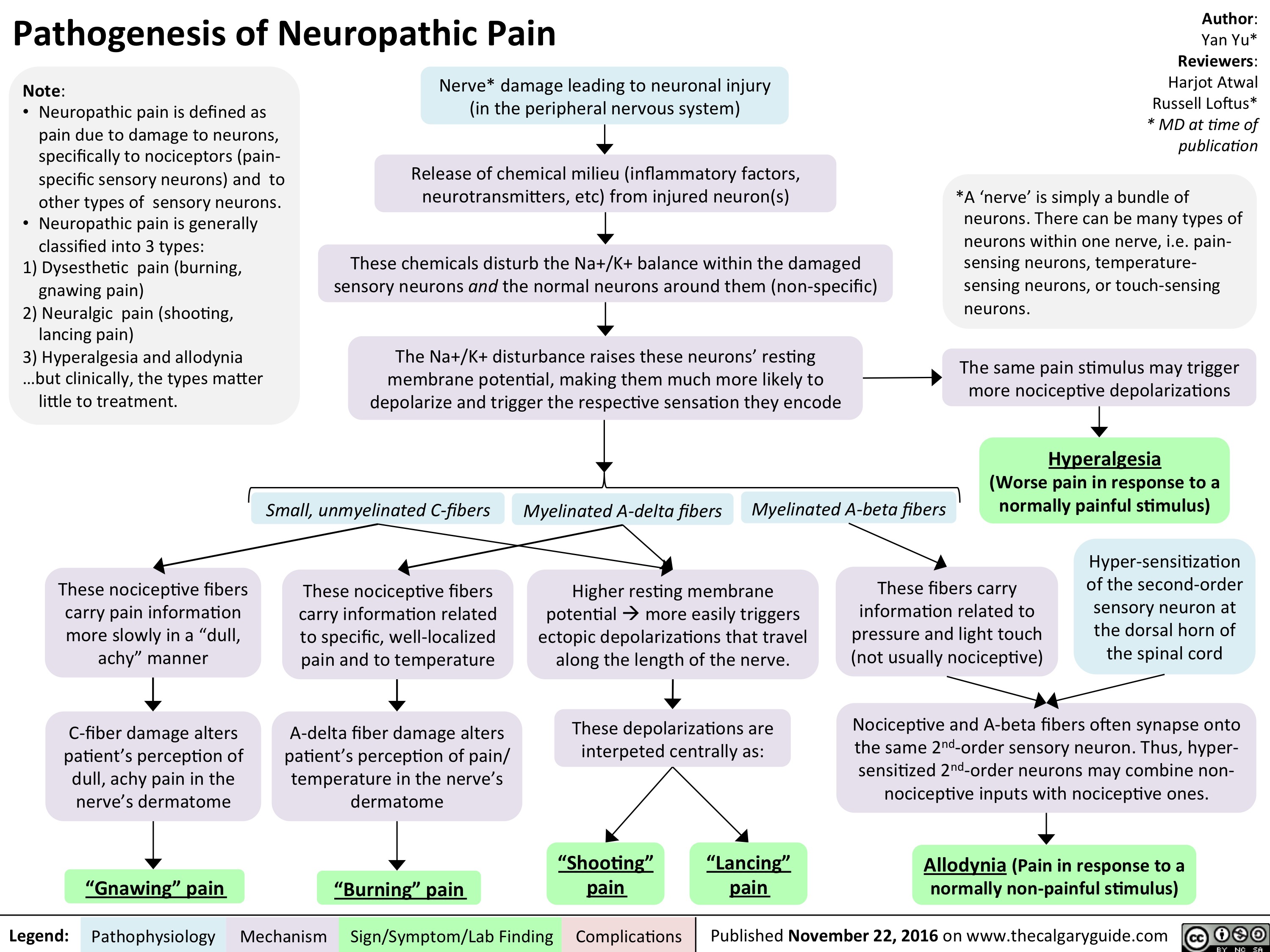
bn-pathogenesis
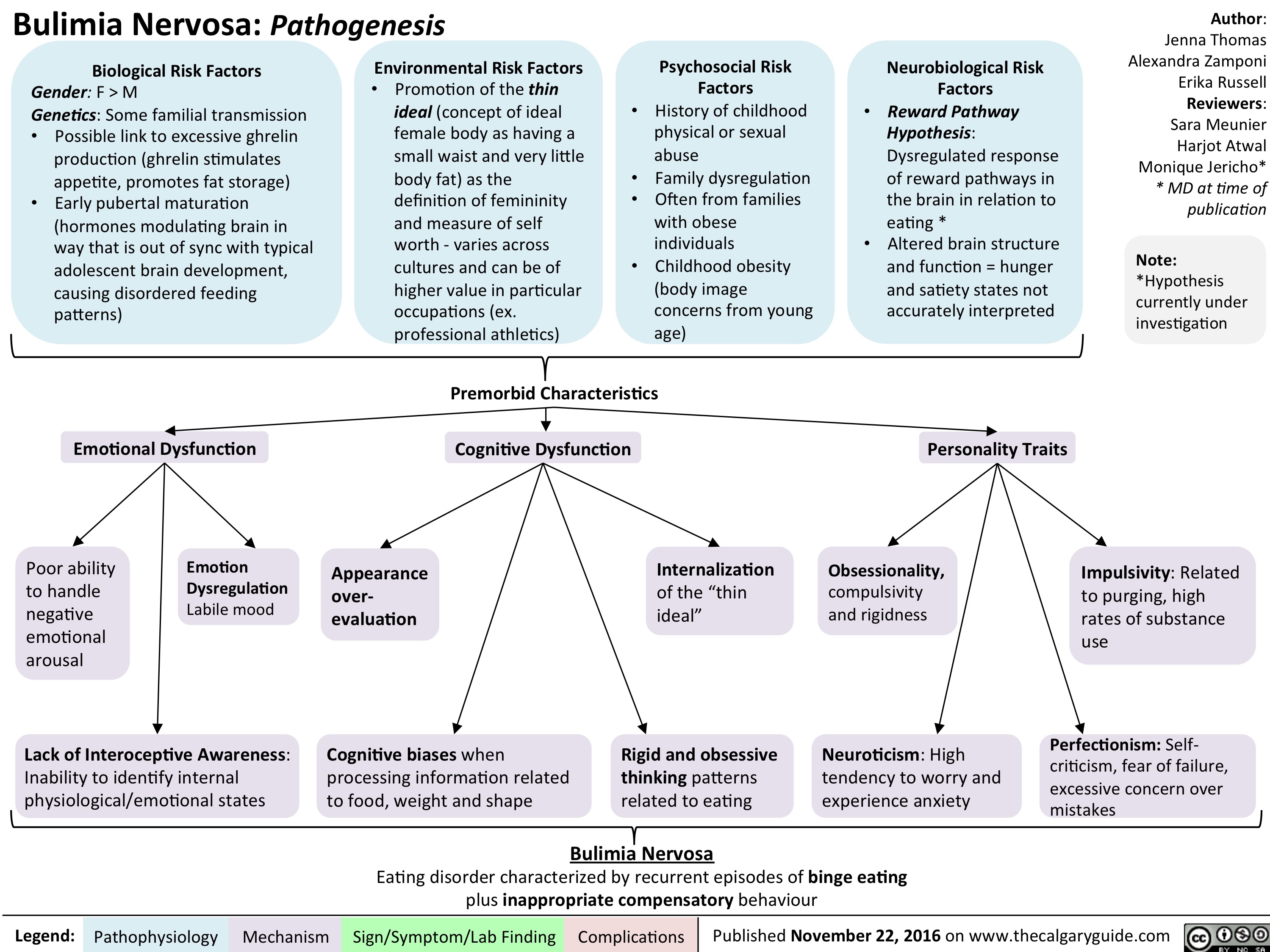
bn-signs-and-symptoms
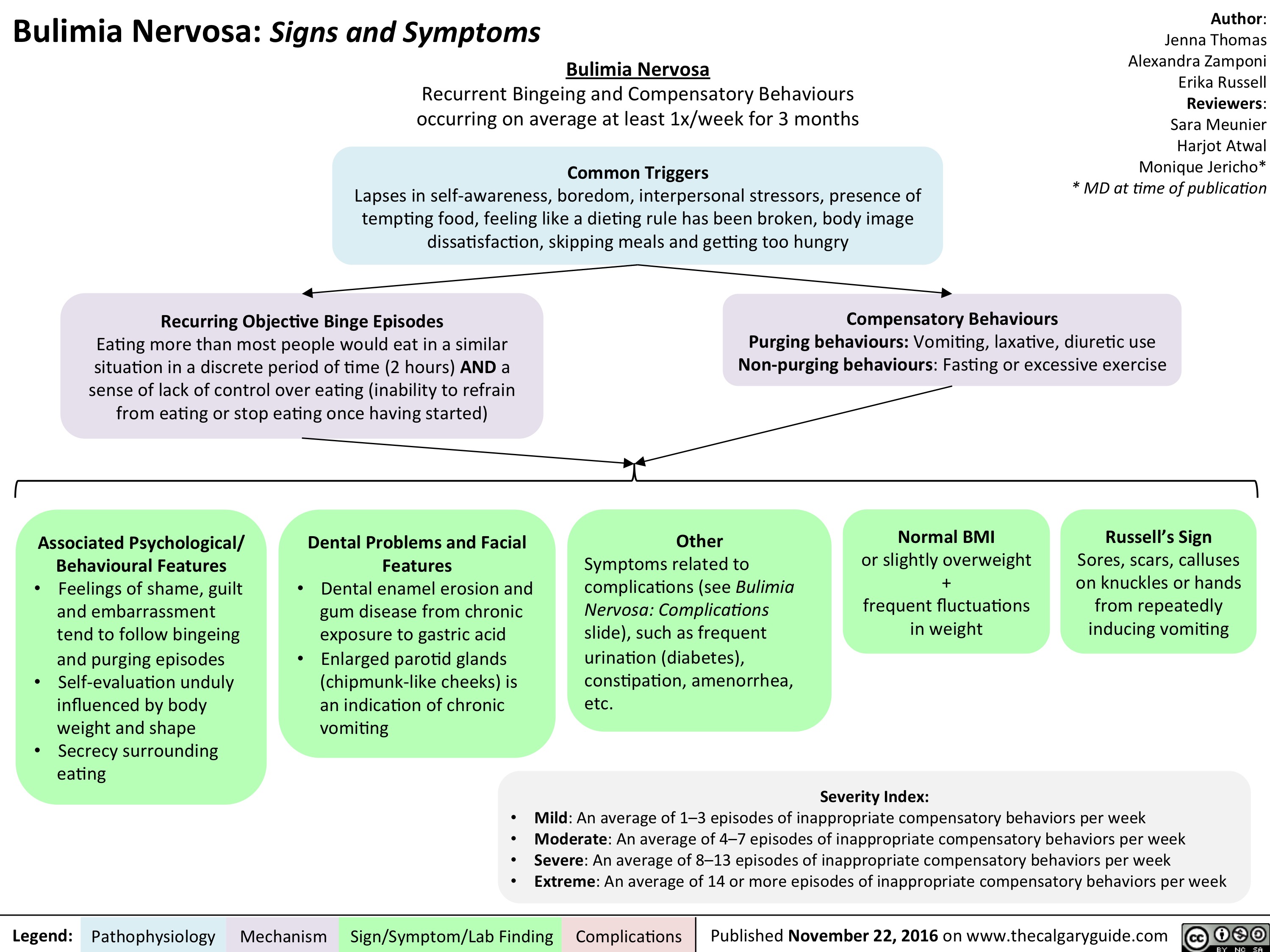
bn-complications
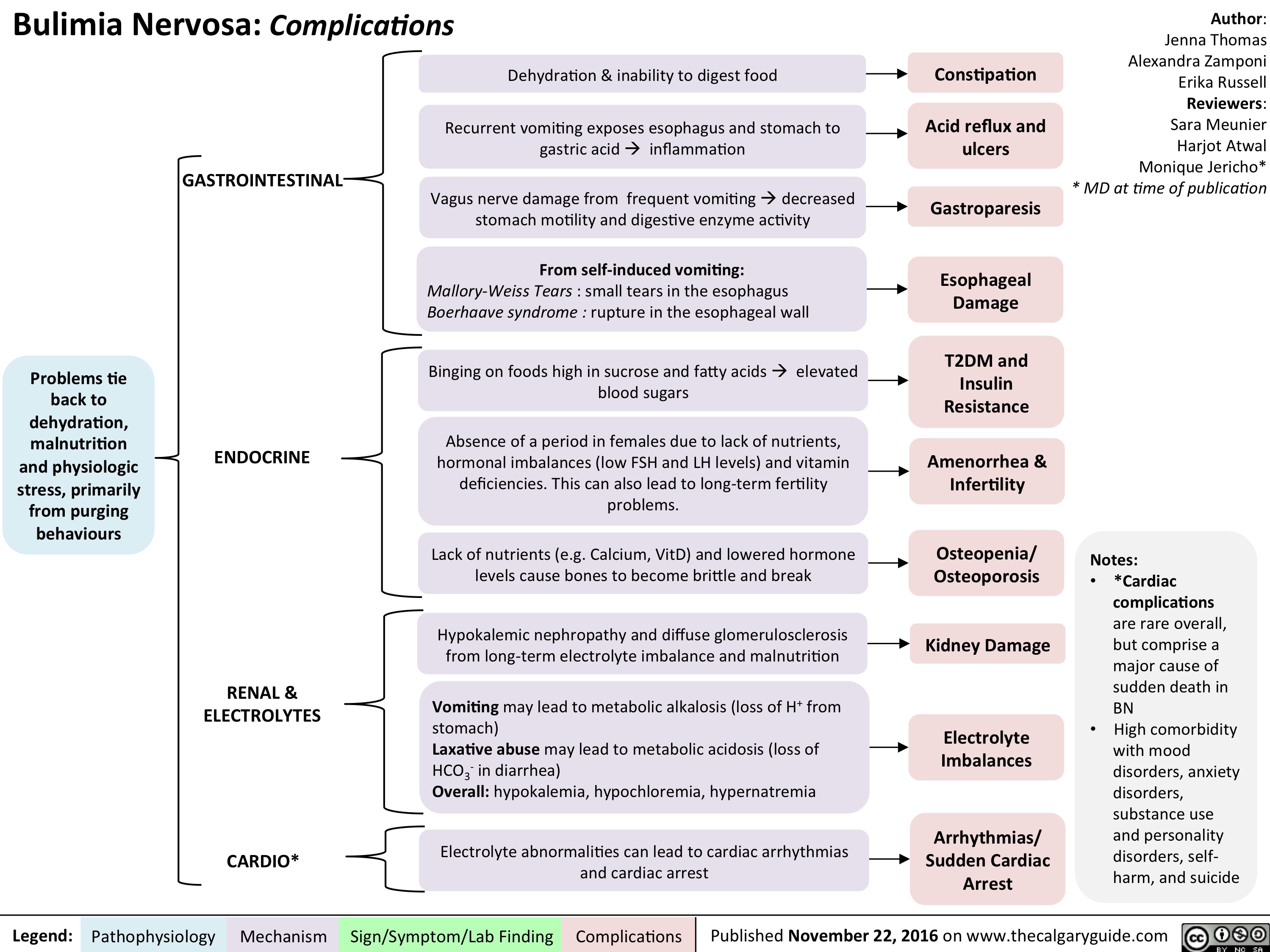
Small Bowel Bacterial Overgrowth: Pathogenesis and clinical findings
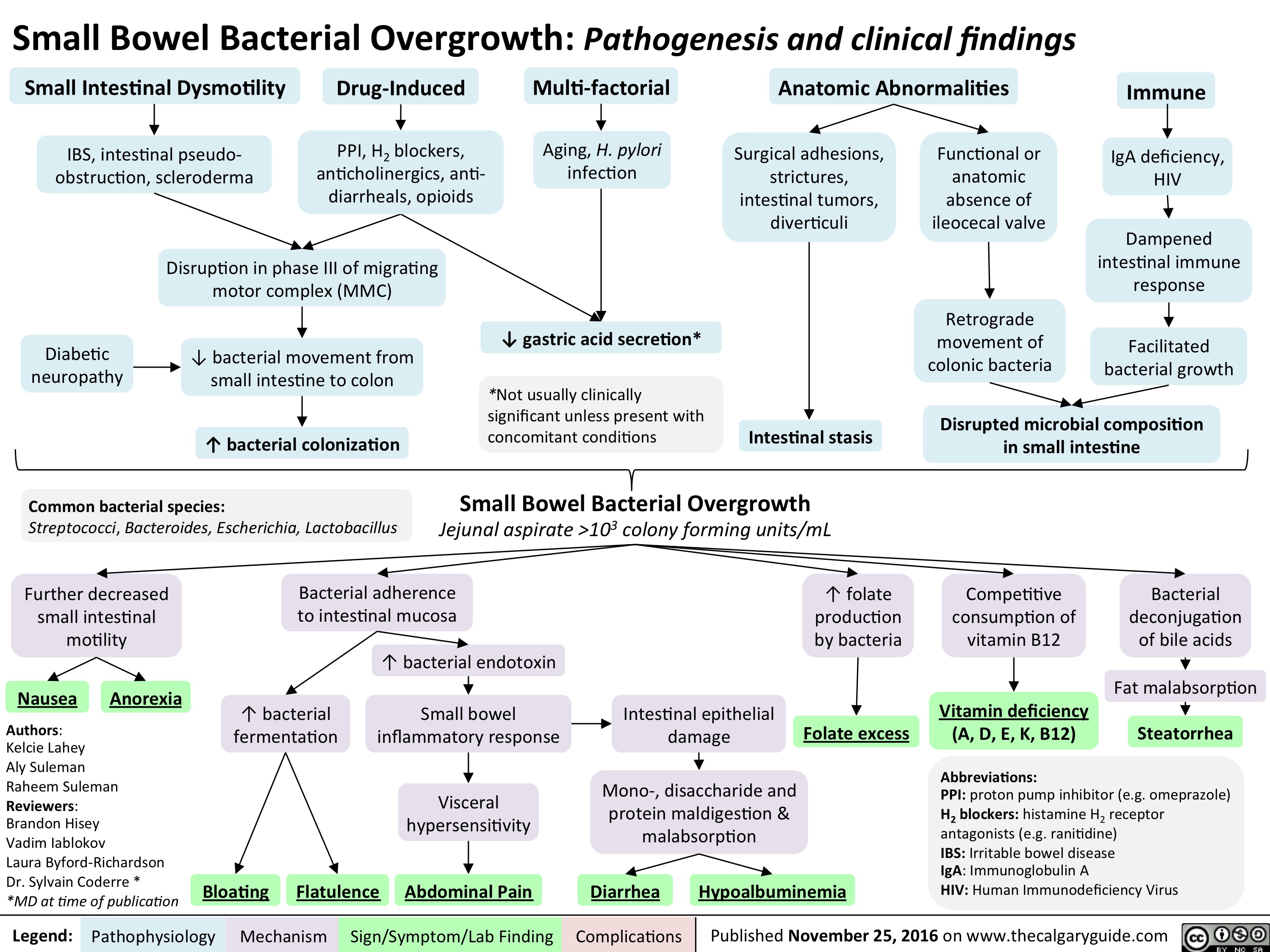
slide1
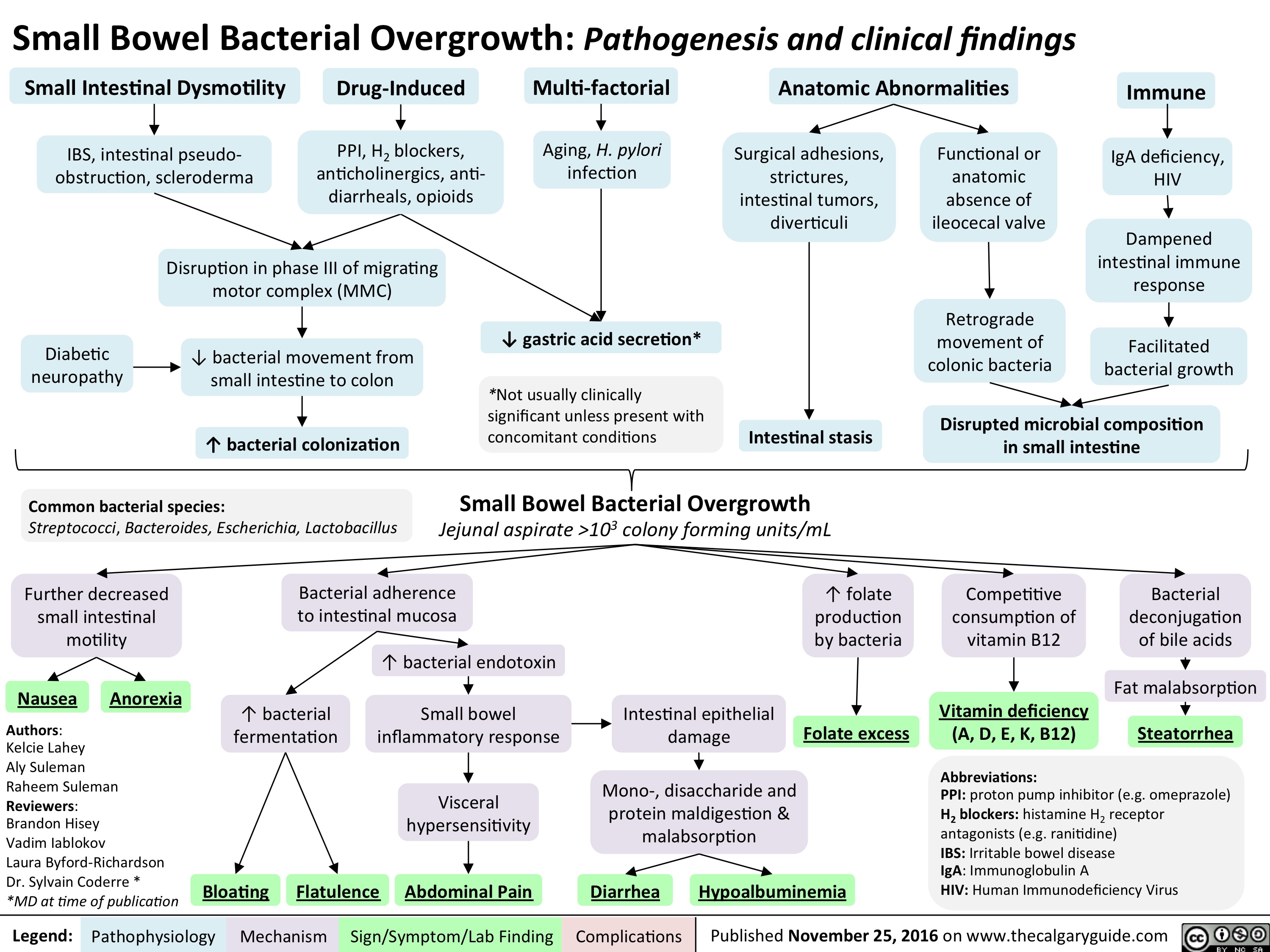
menstrual-cycle-physiology-the-hypothalamic-pituitary-ovarian-axis
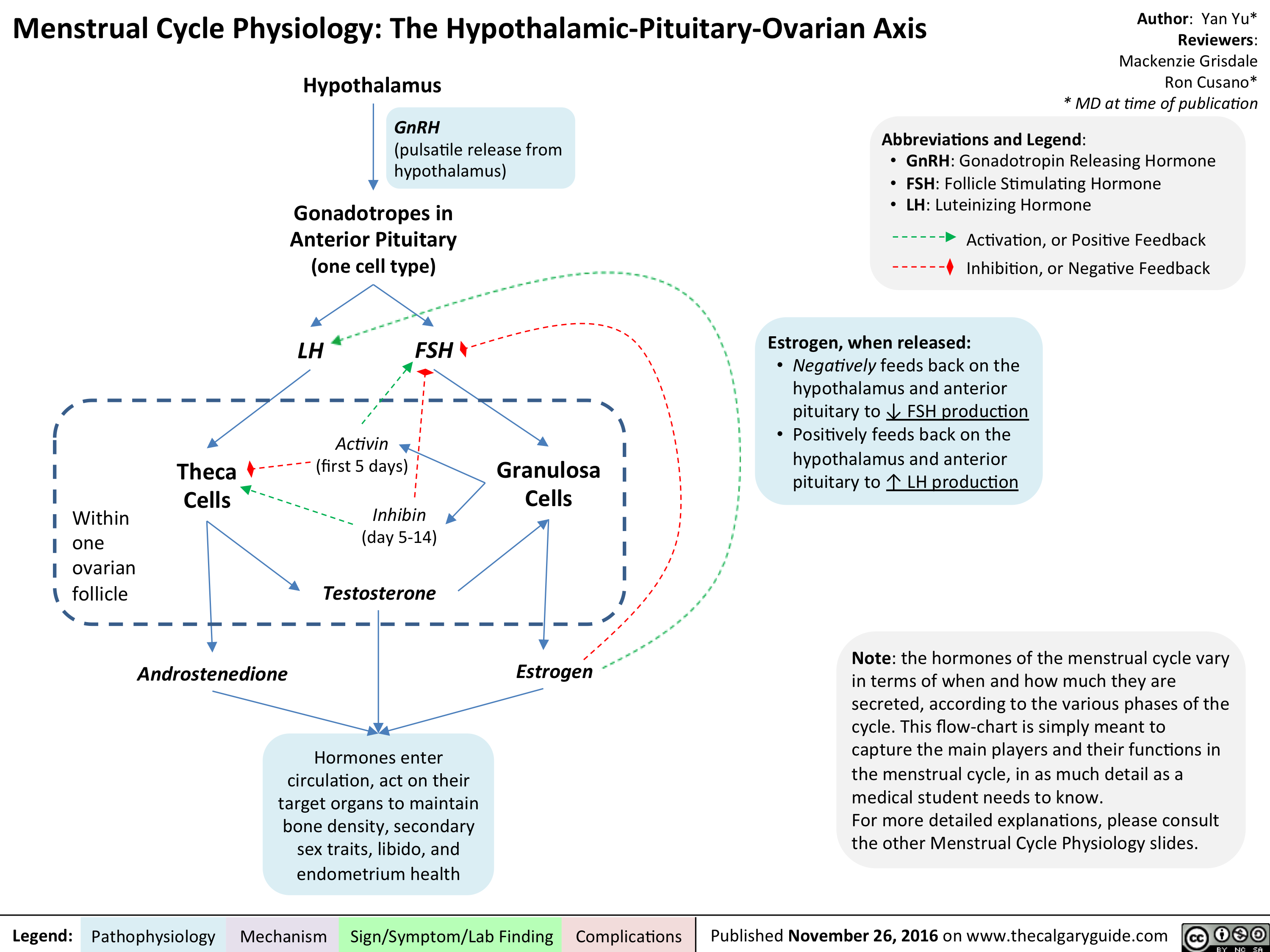
menstrual-cycle-physiology-correlating-the-ovarian-and-uterine-cycles
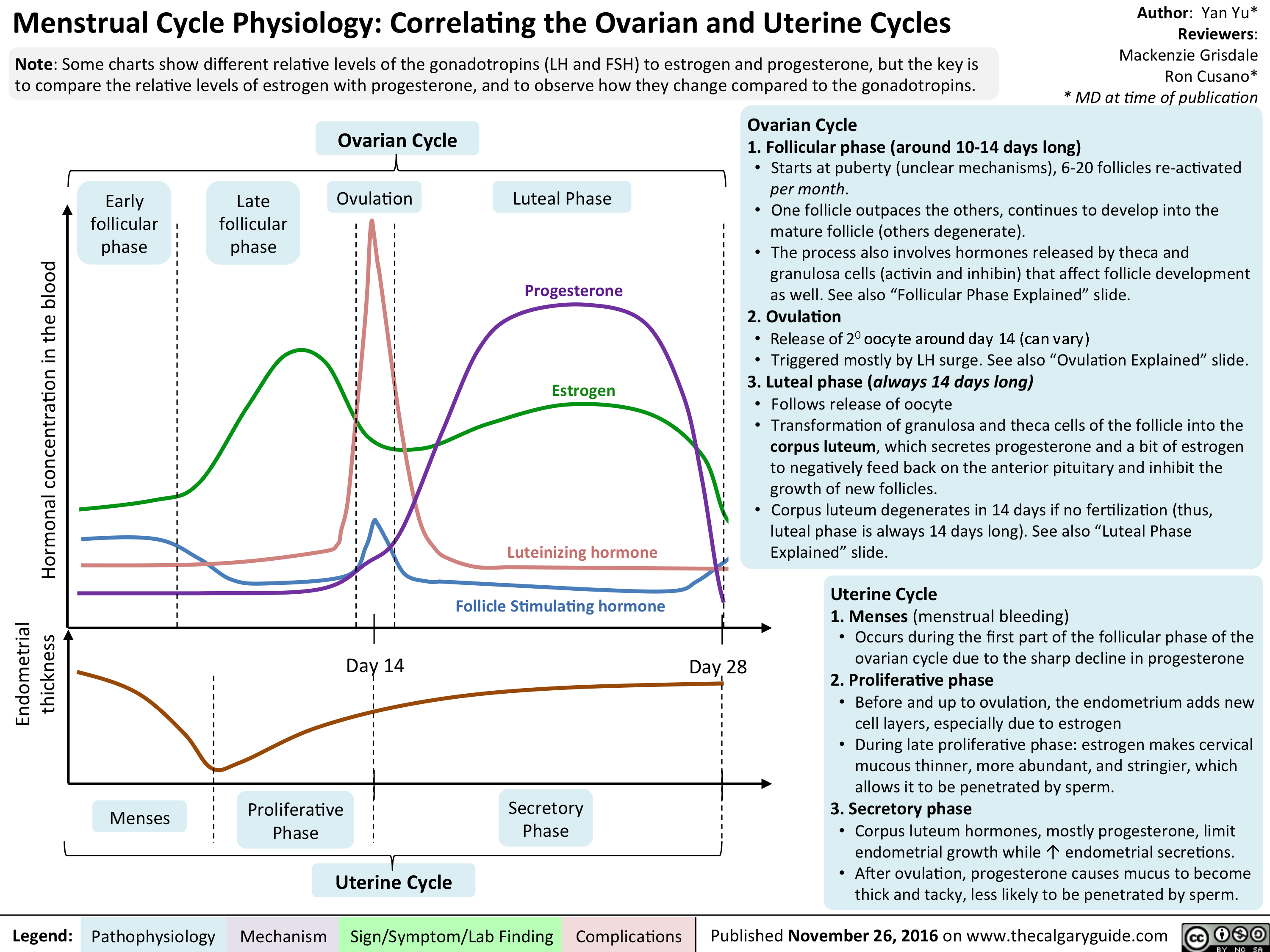
menstrual-cycle-physiology-ovarian-cycle-follicular-phase-explained
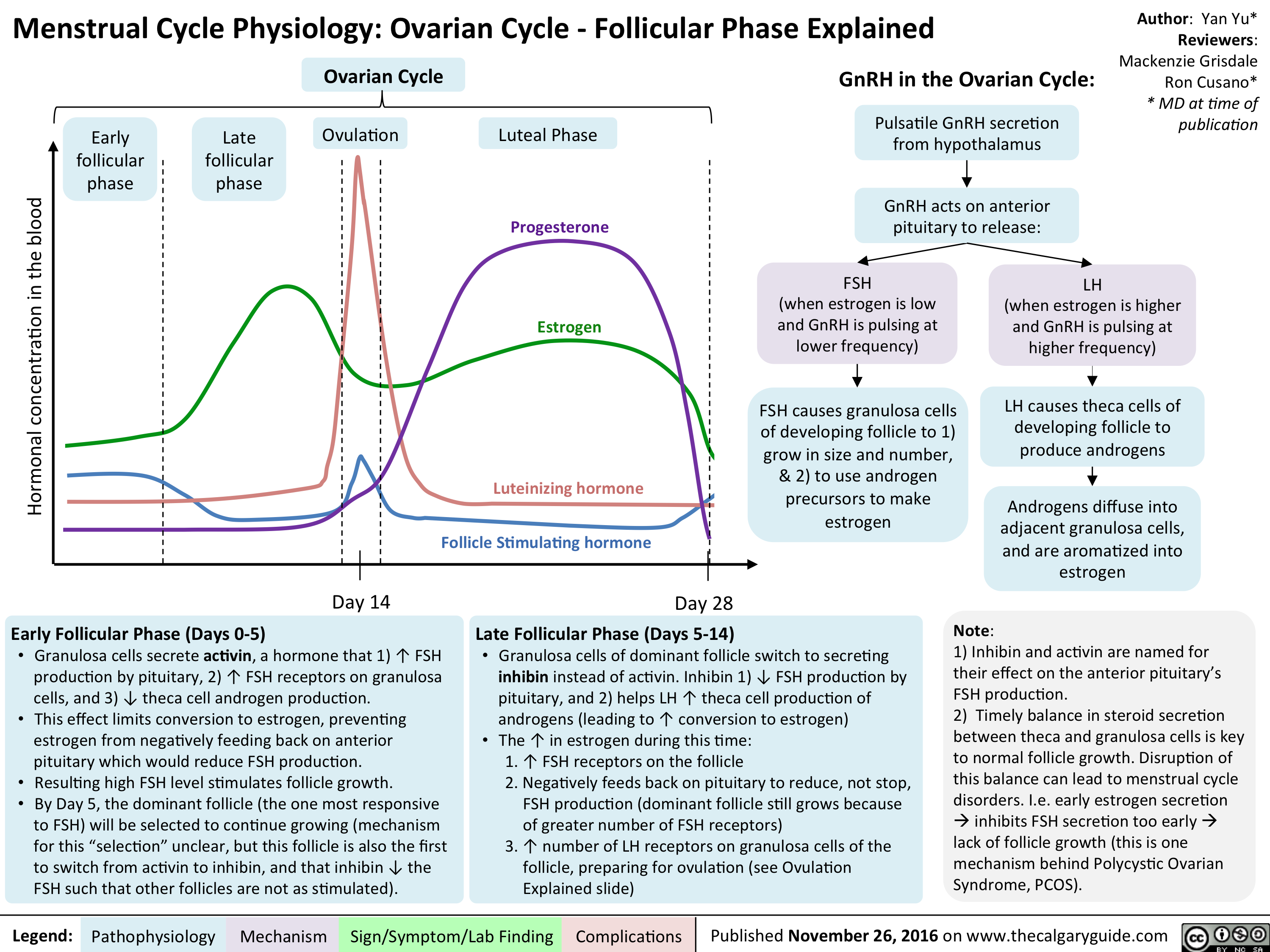
menstrual-cycle-physiology-ovarian-cycle-ovulation-explained

menstrual-cycle-physiology-ovarian-cycle-luteal-phase-explained
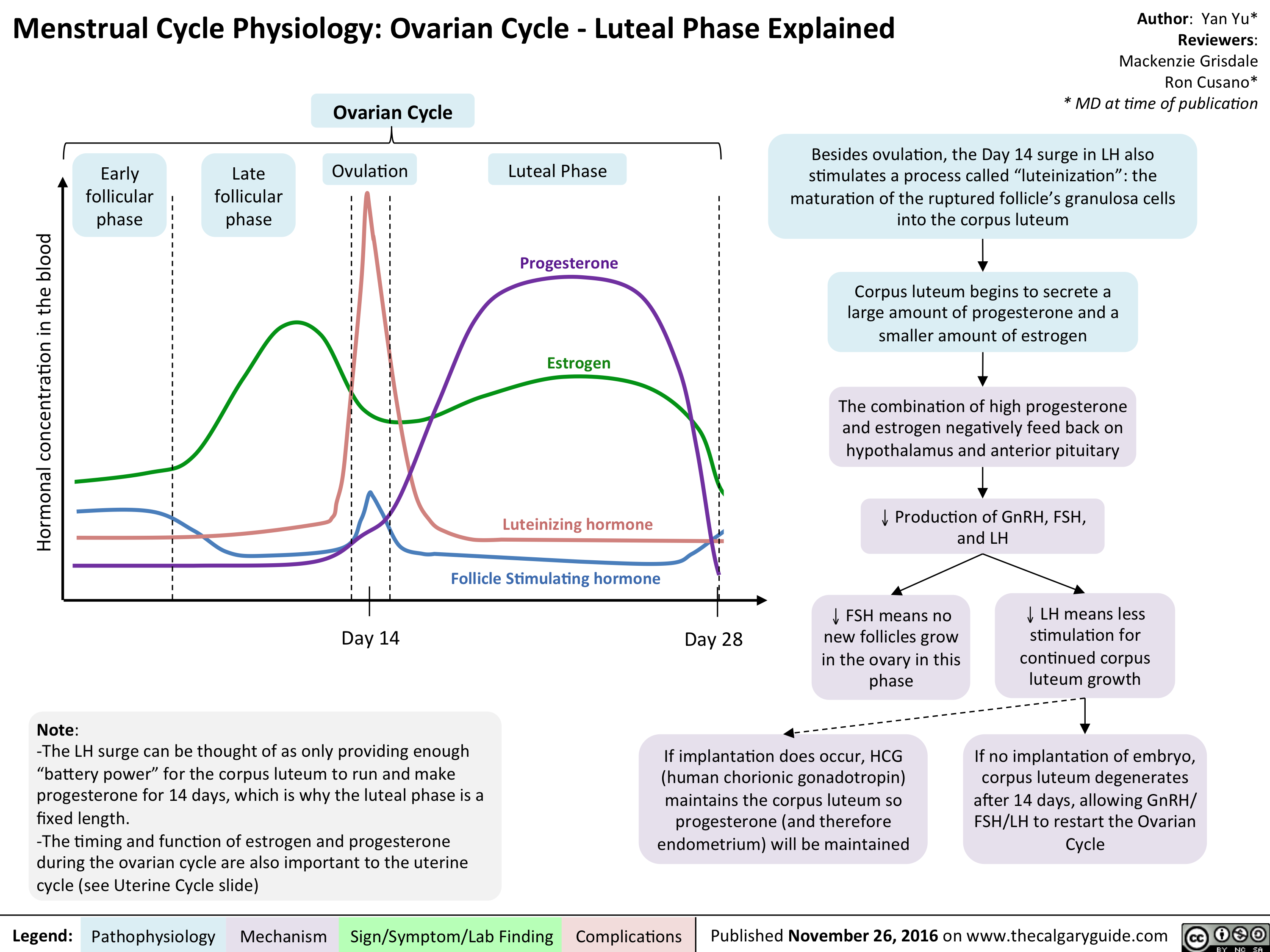
menstrual-cycle-physiology-the-uterine-cycle
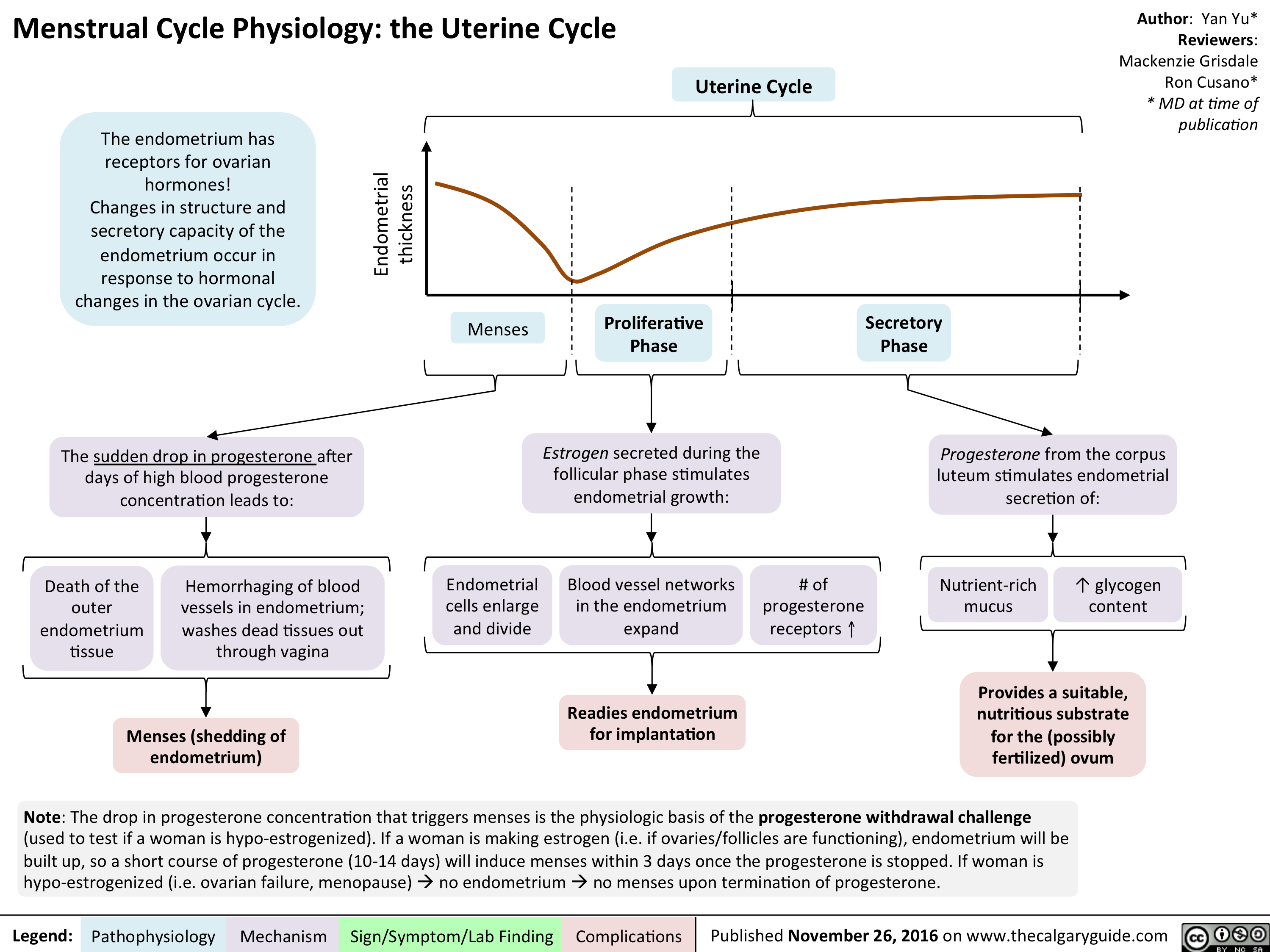
Shoulder Dystocia: Pathogenesis and Clinical Findings
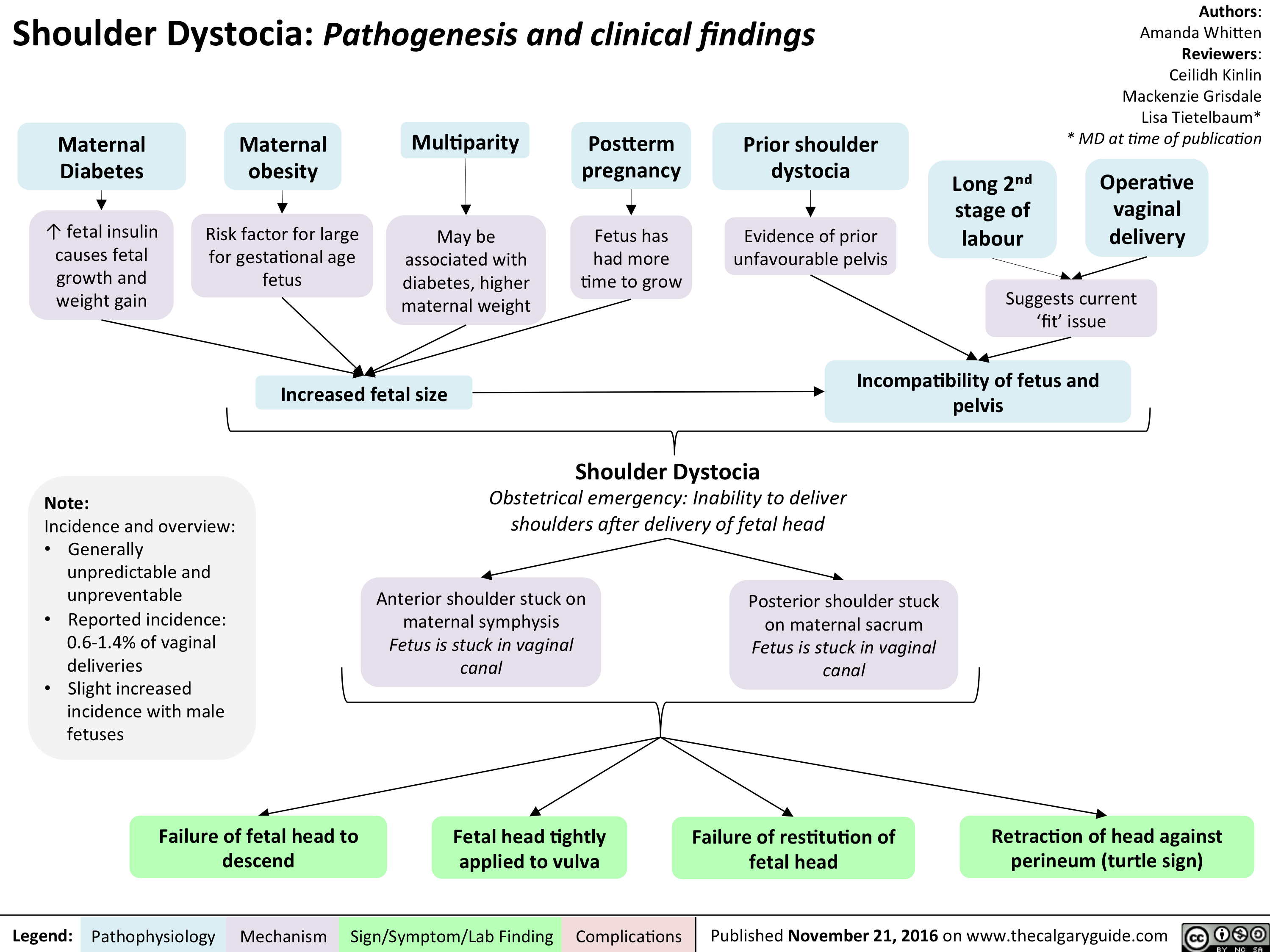
lvh-final
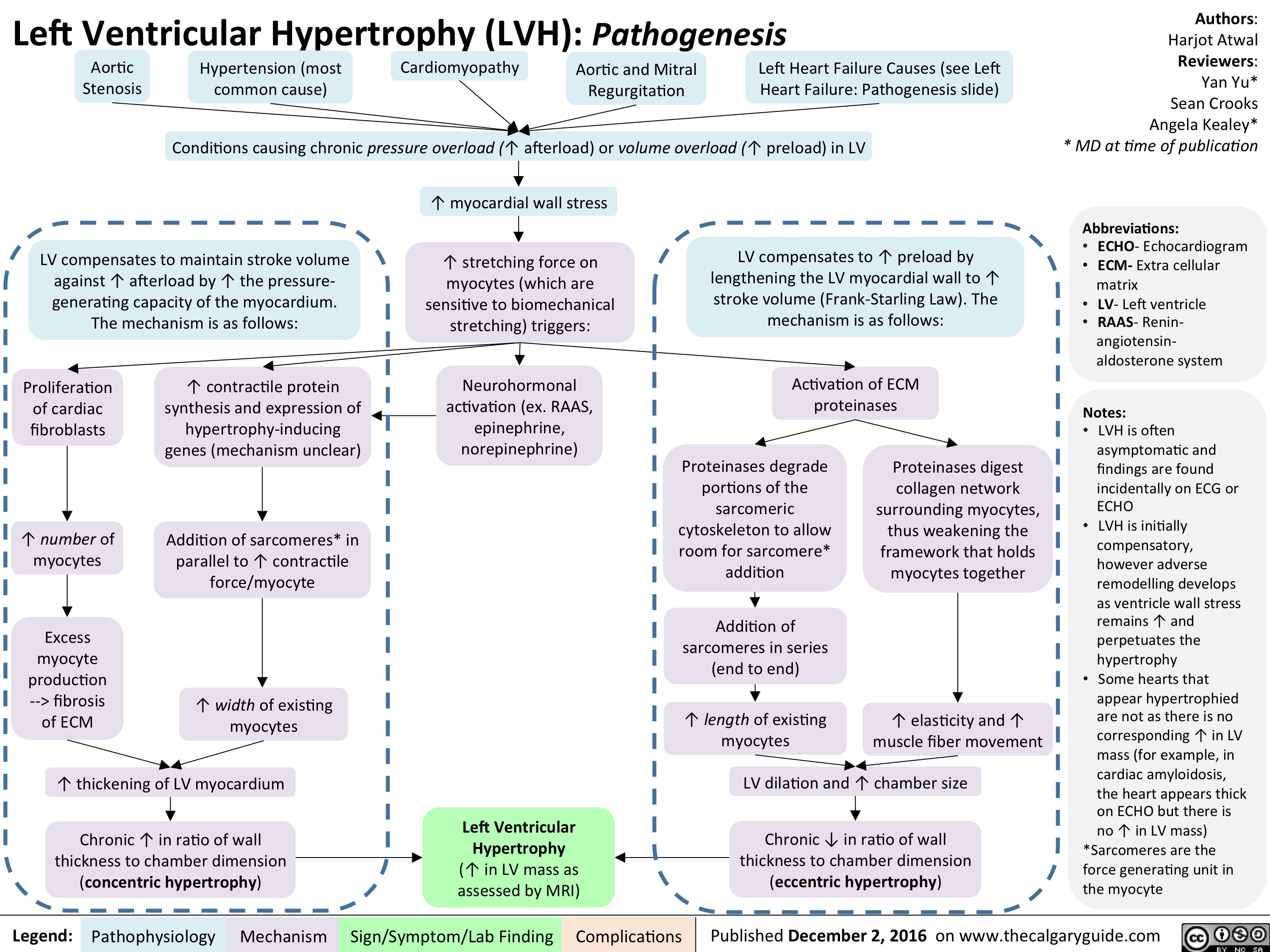
anorexia-nervosa
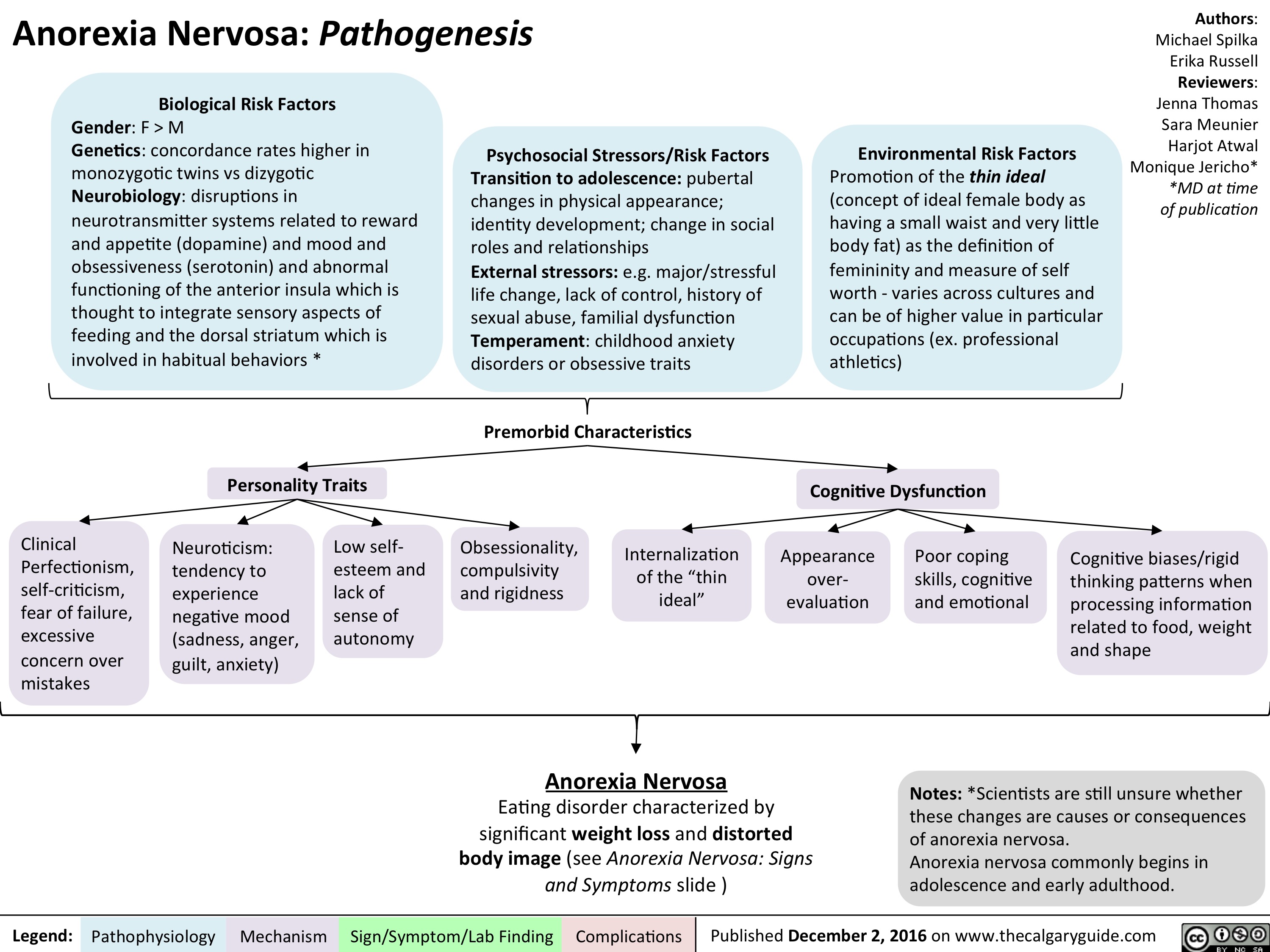
an-signs-and-symptoms
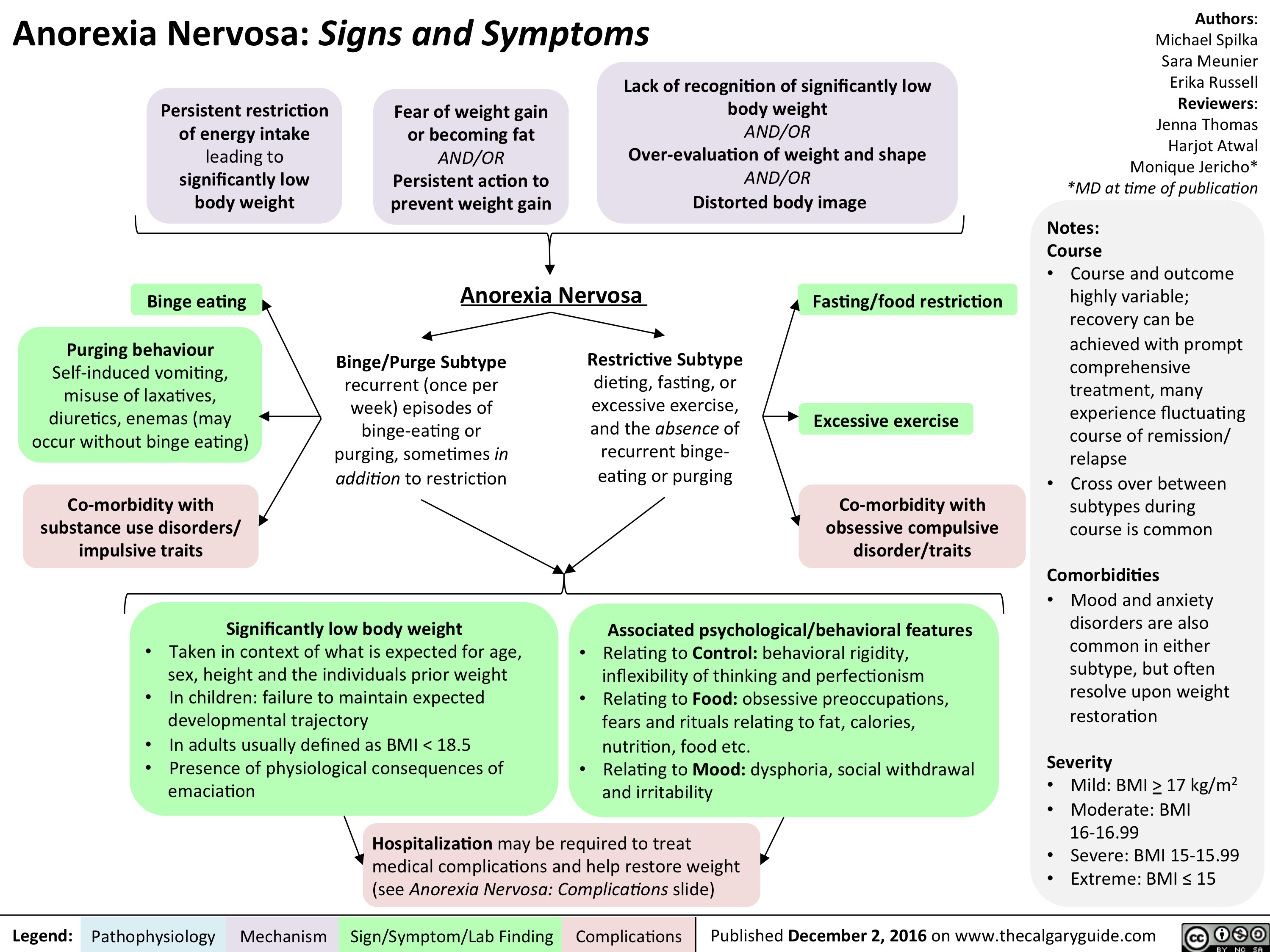
an-complications-final
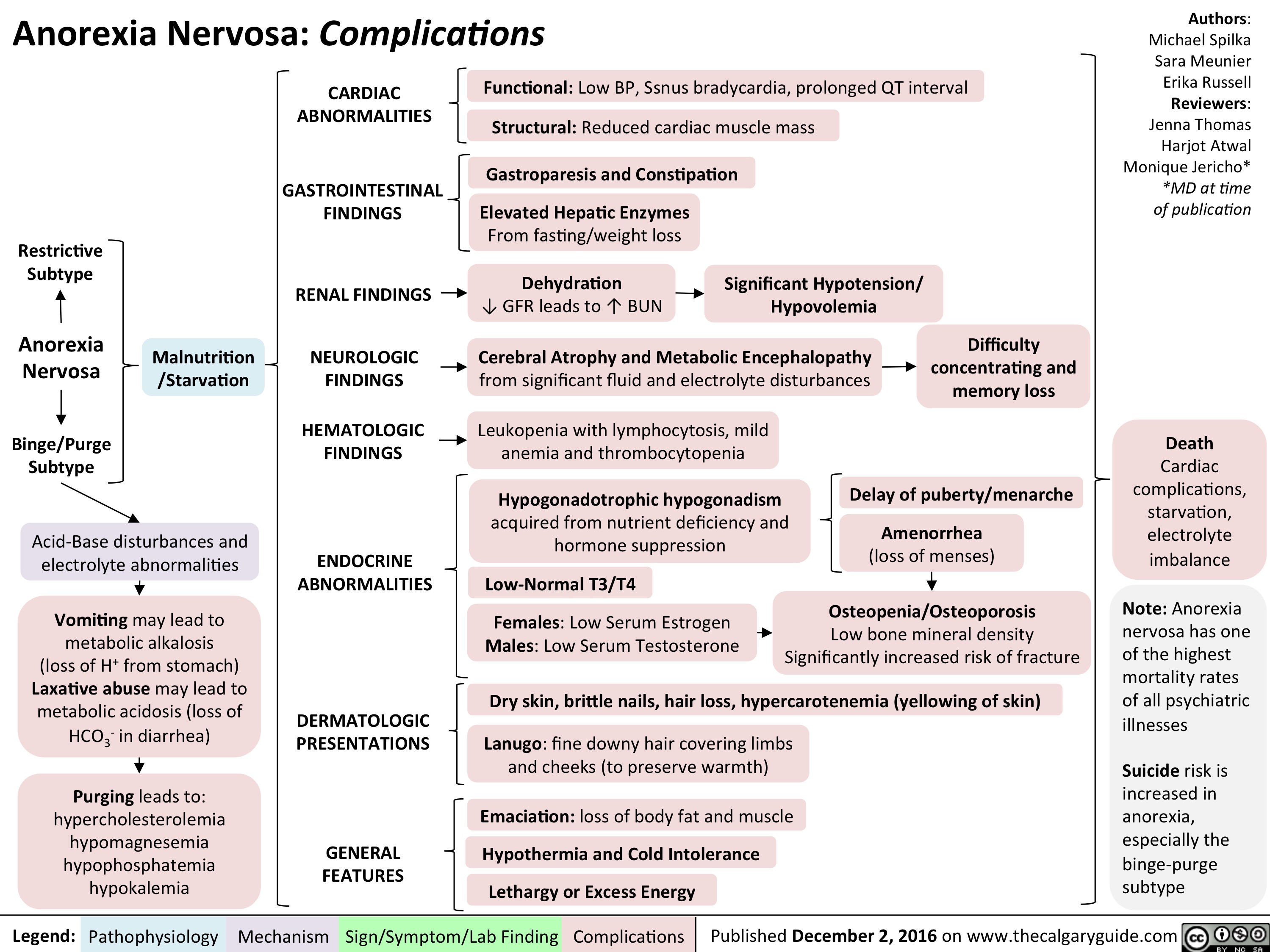
als-final
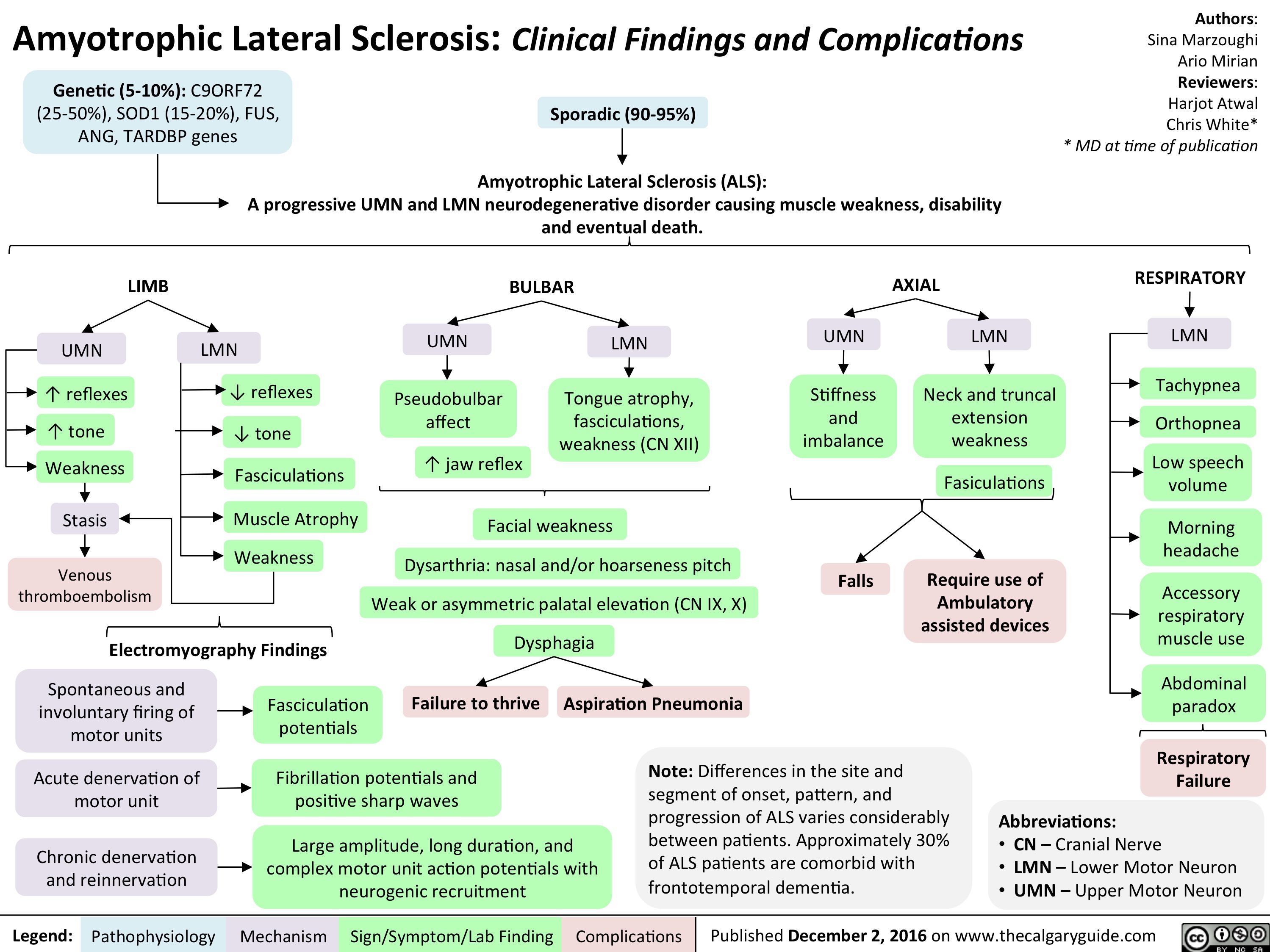
alzheimers-disease-final
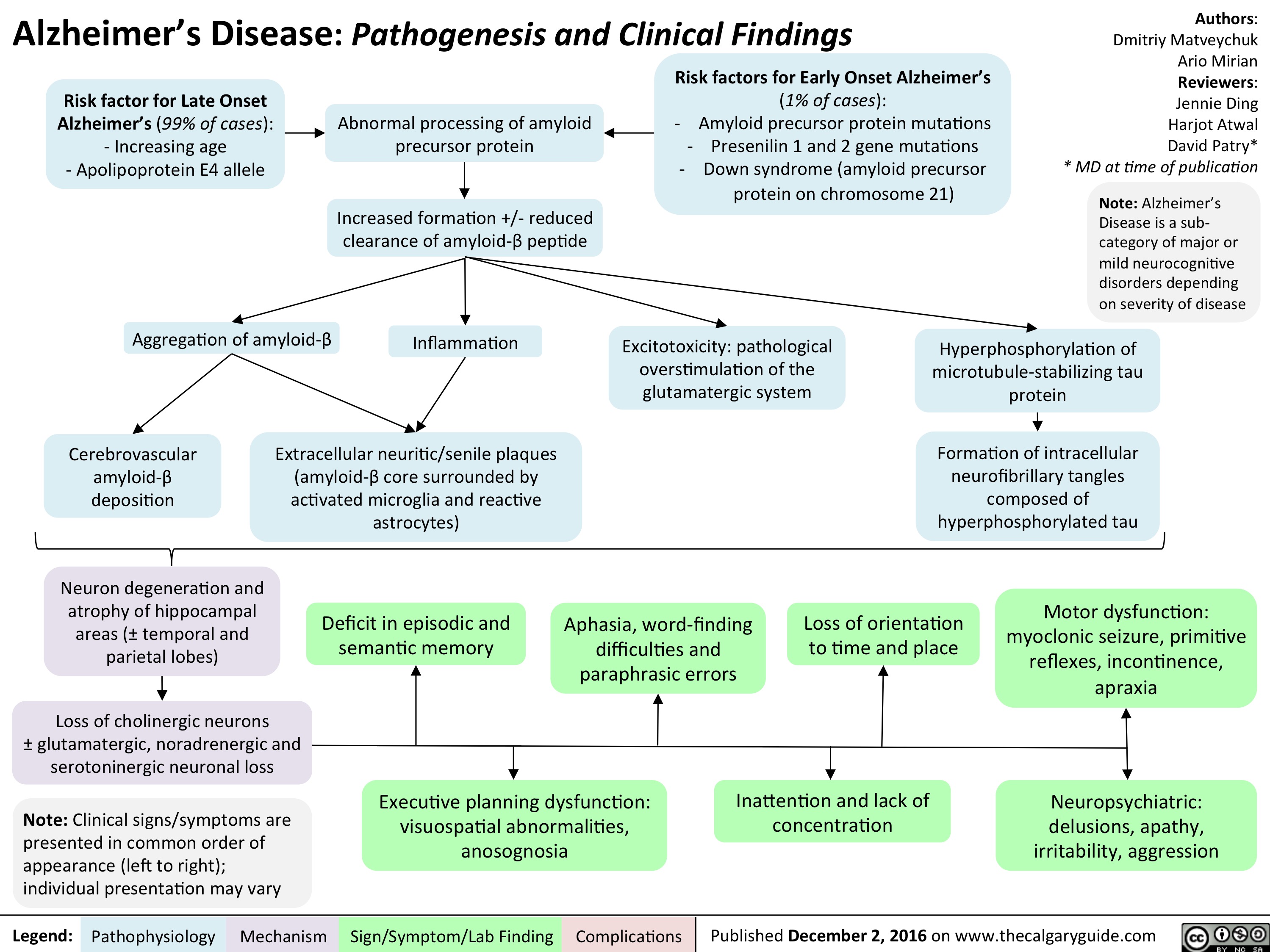
lewy-body-dementia-final
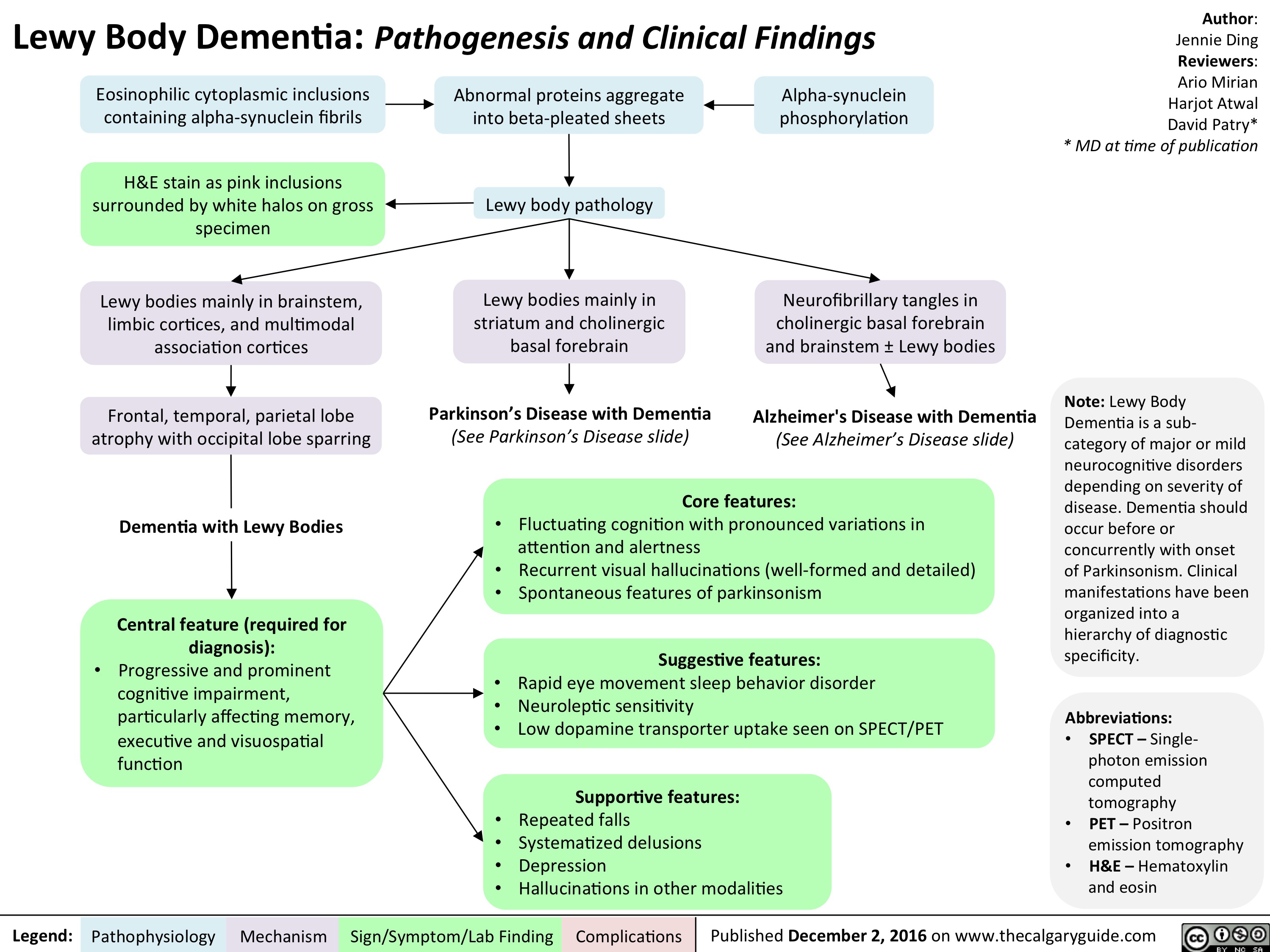
als-final
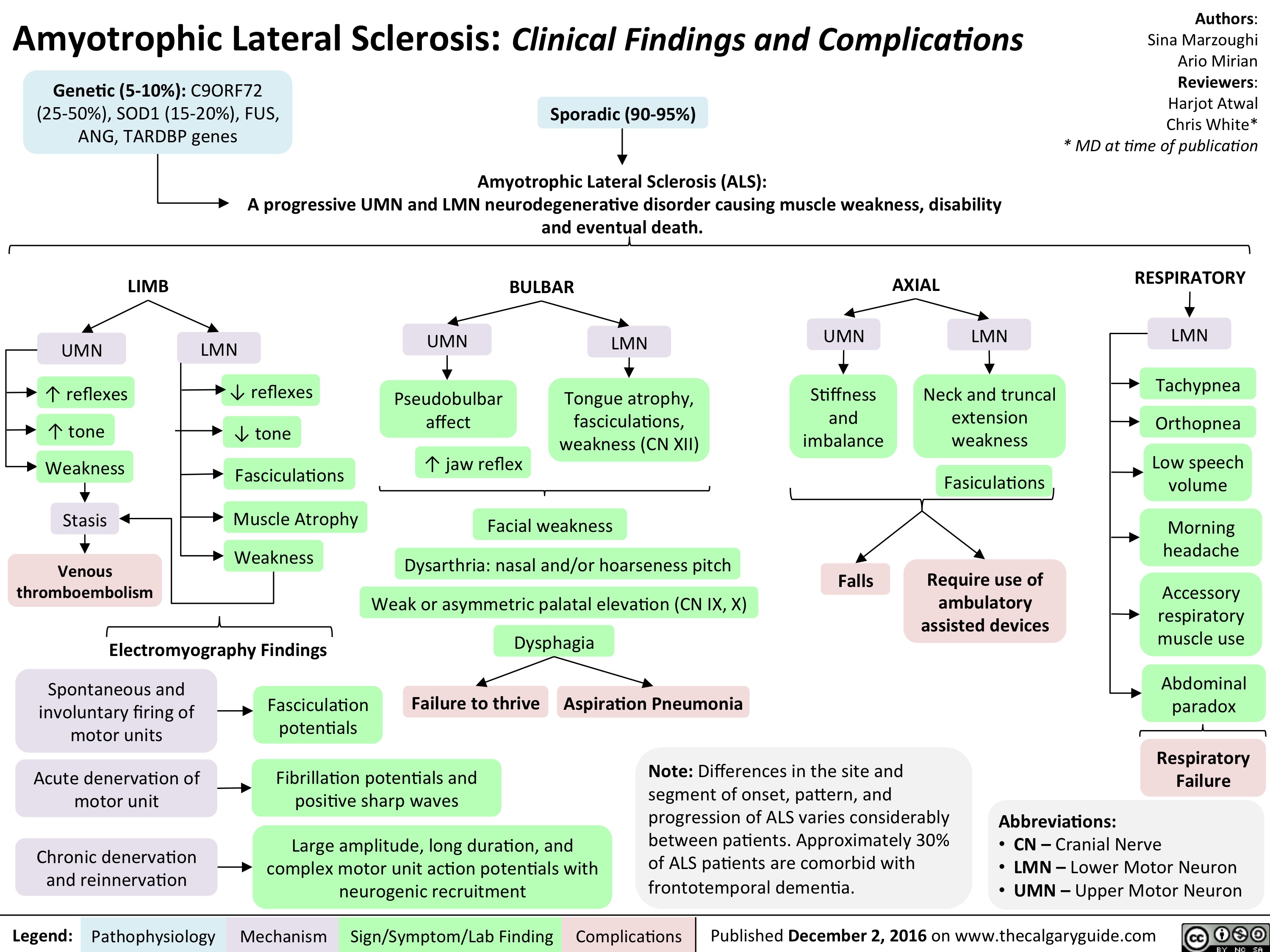
als-final
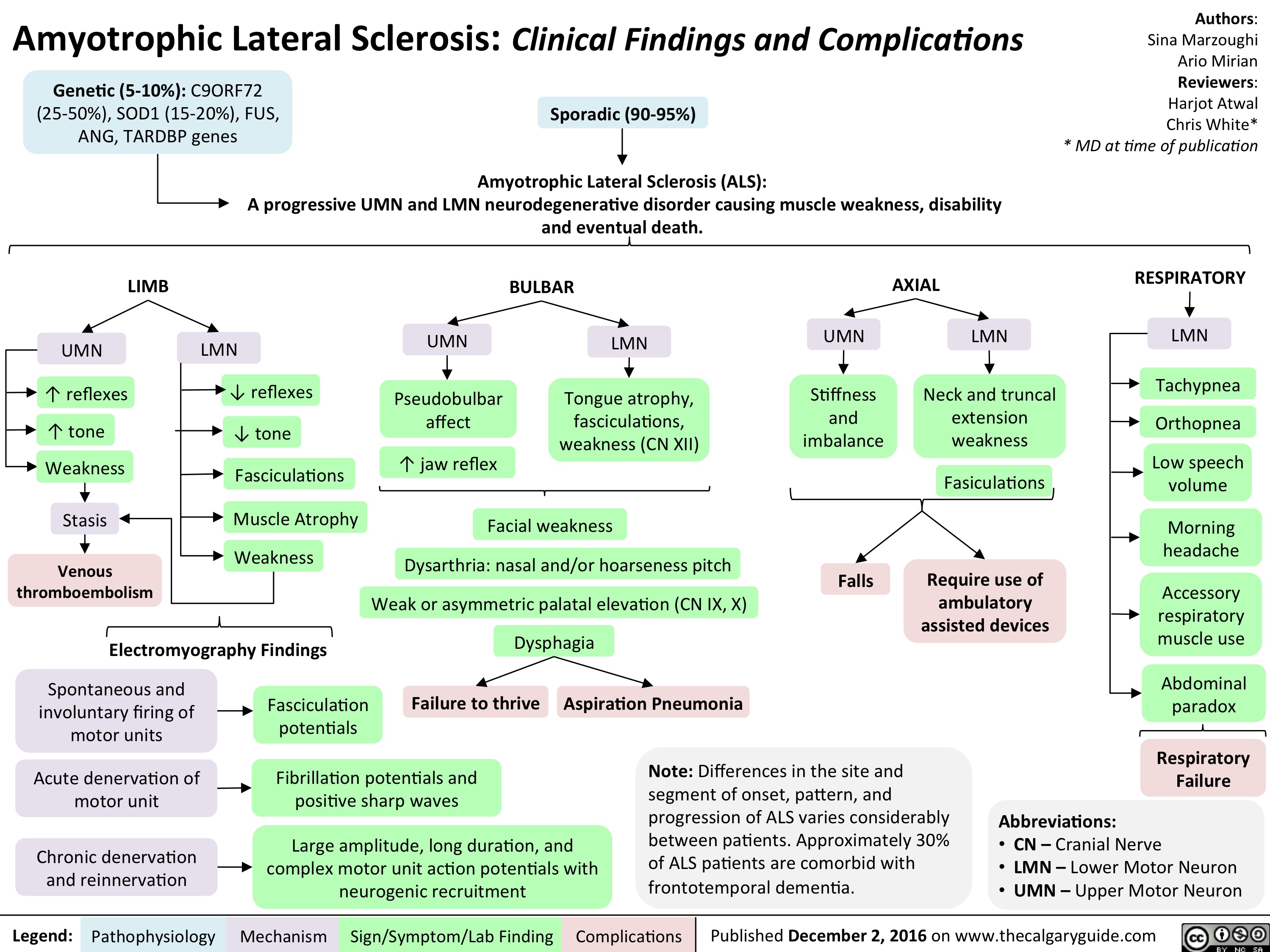
NSAIDs Final
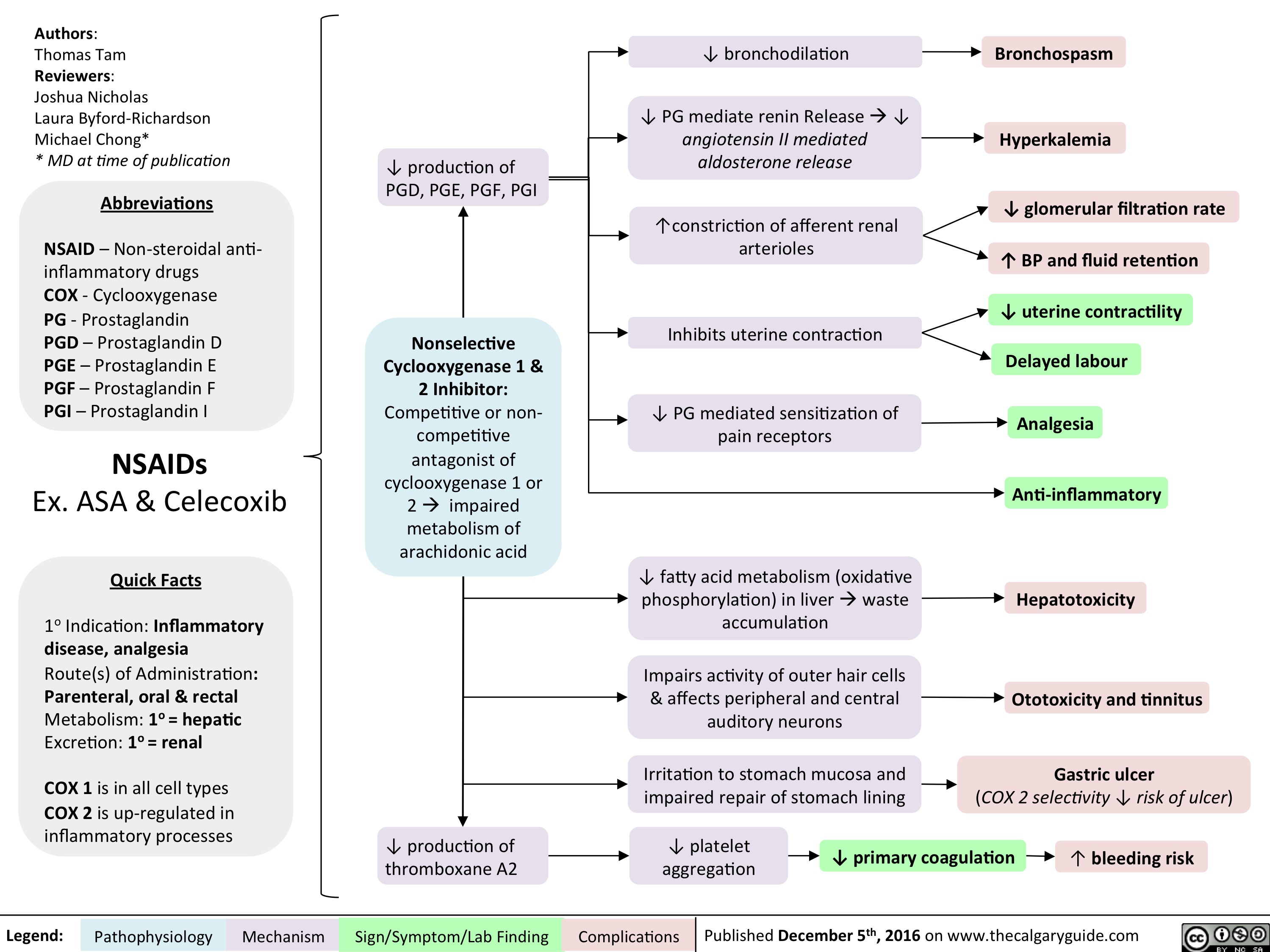
NSAIDs Final
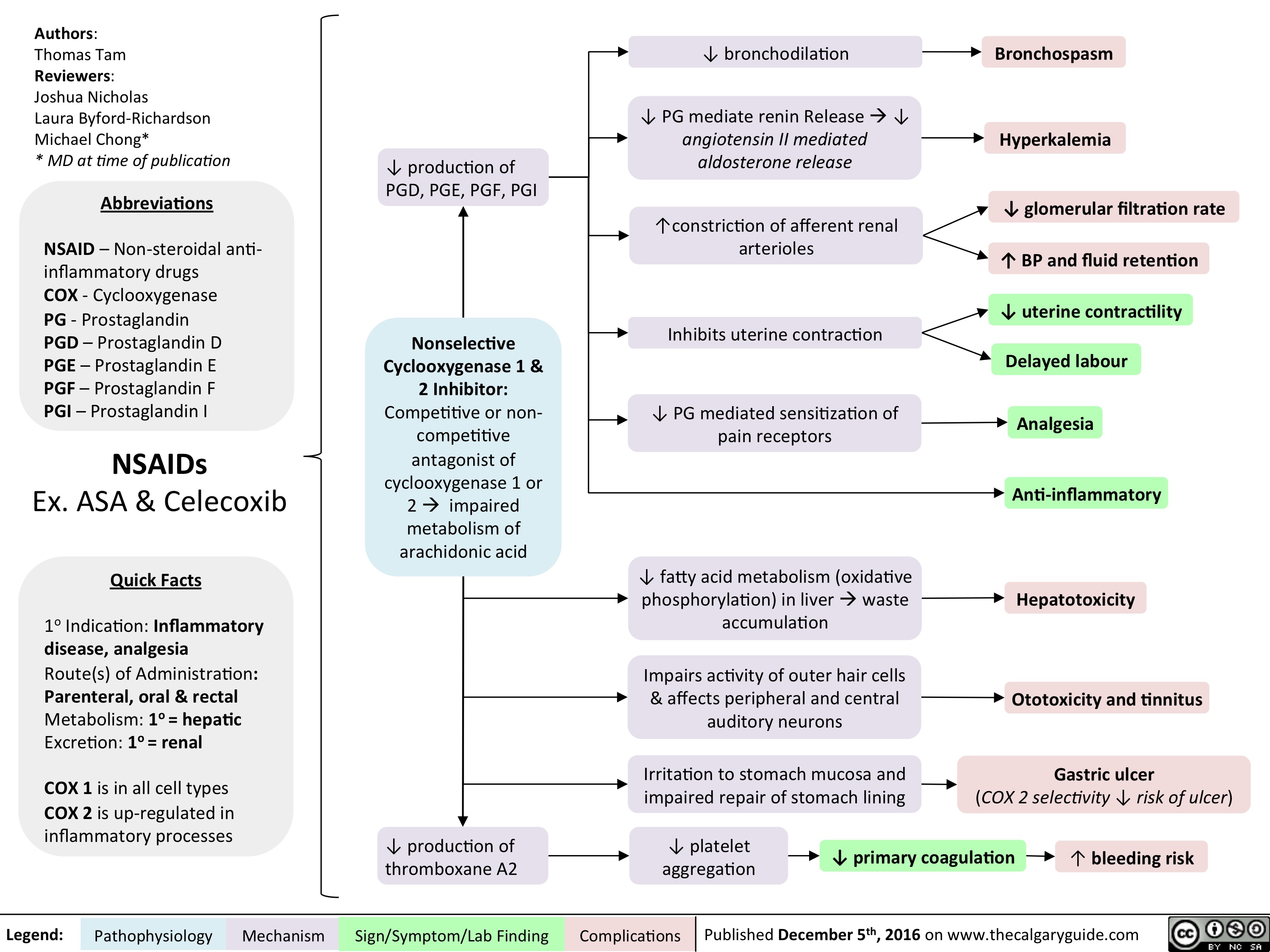
induction-of-labour-indications-and-contraindications
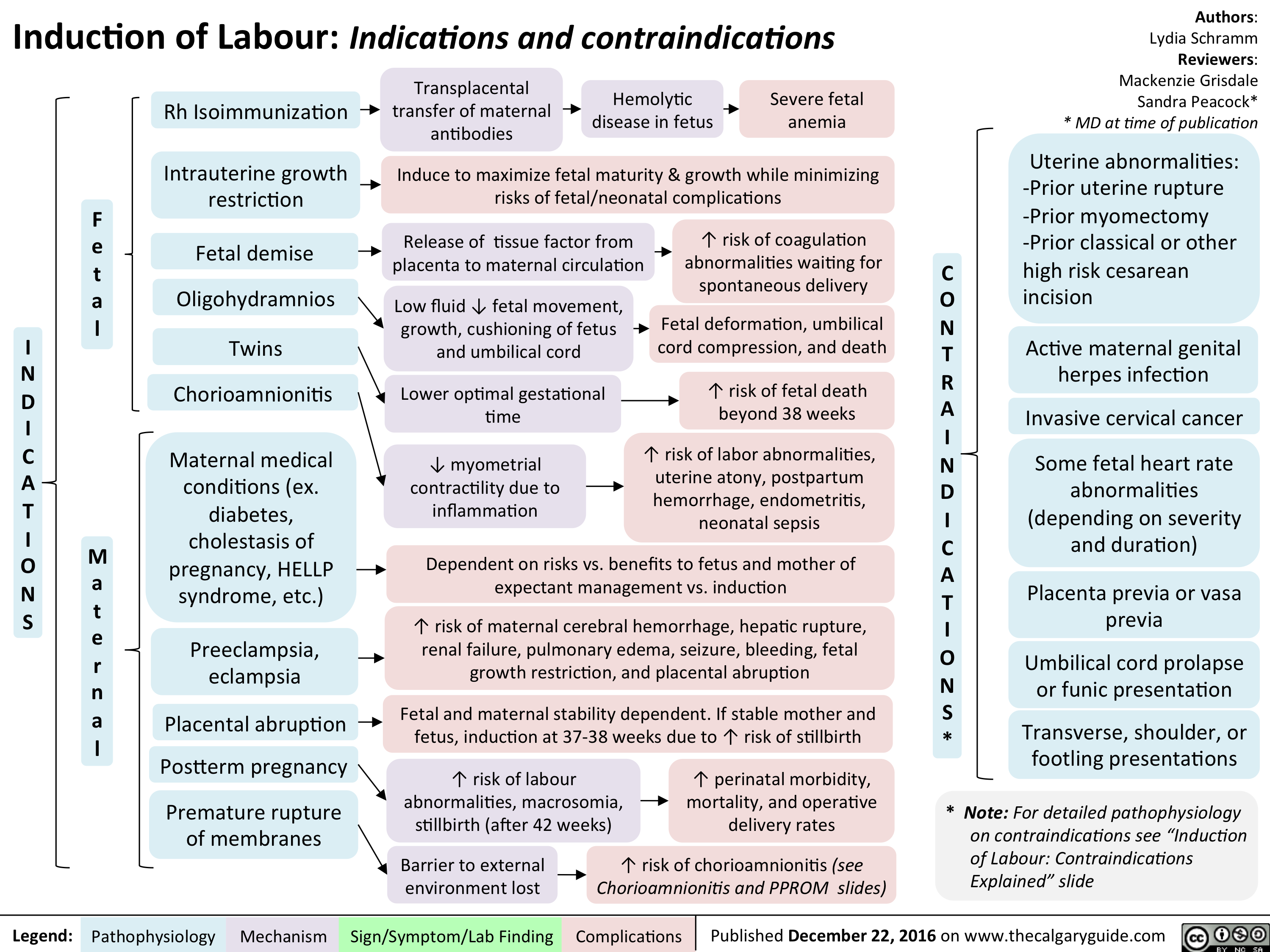
Stress Fracture v.5
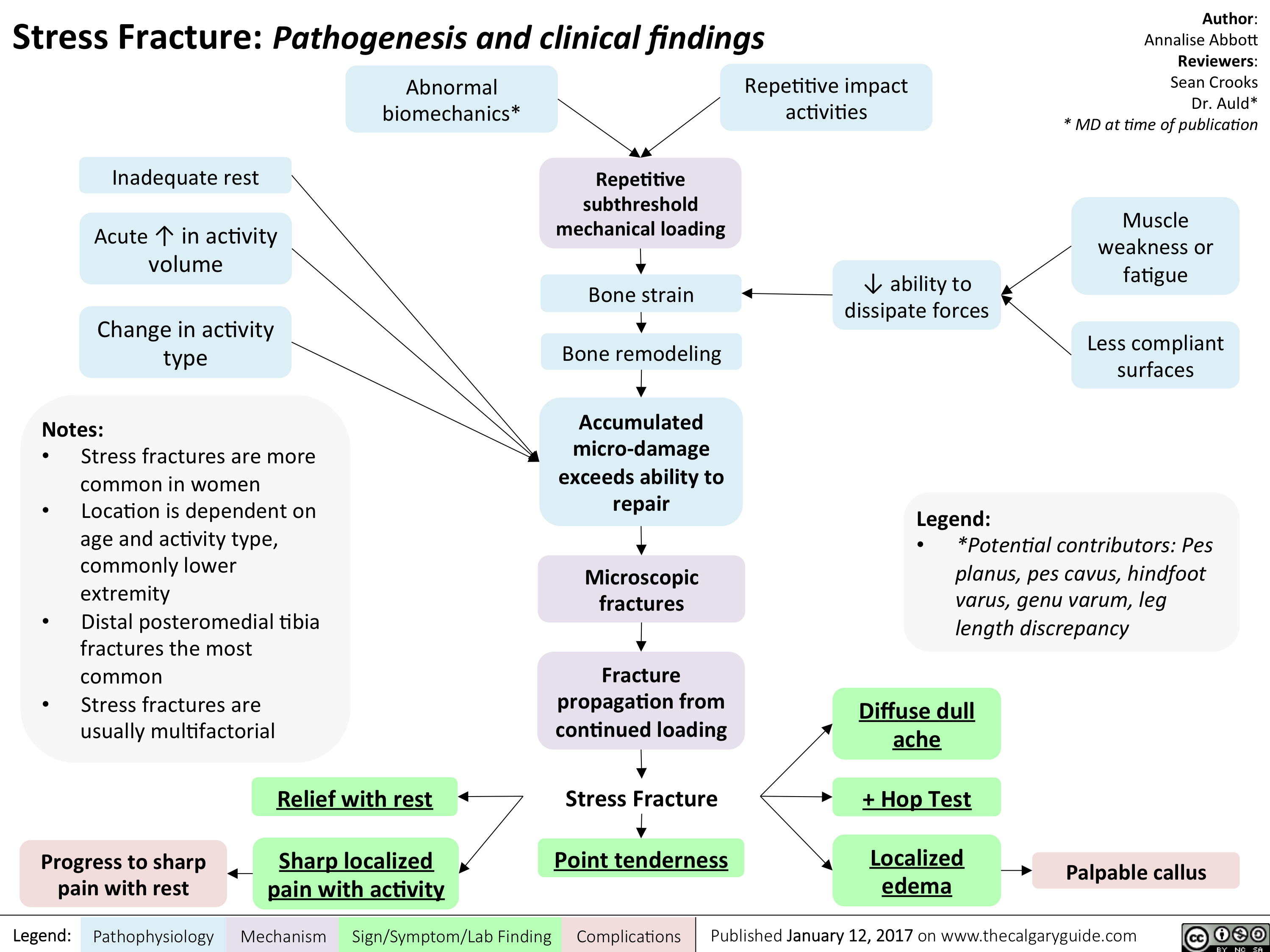
Placenta Accreta
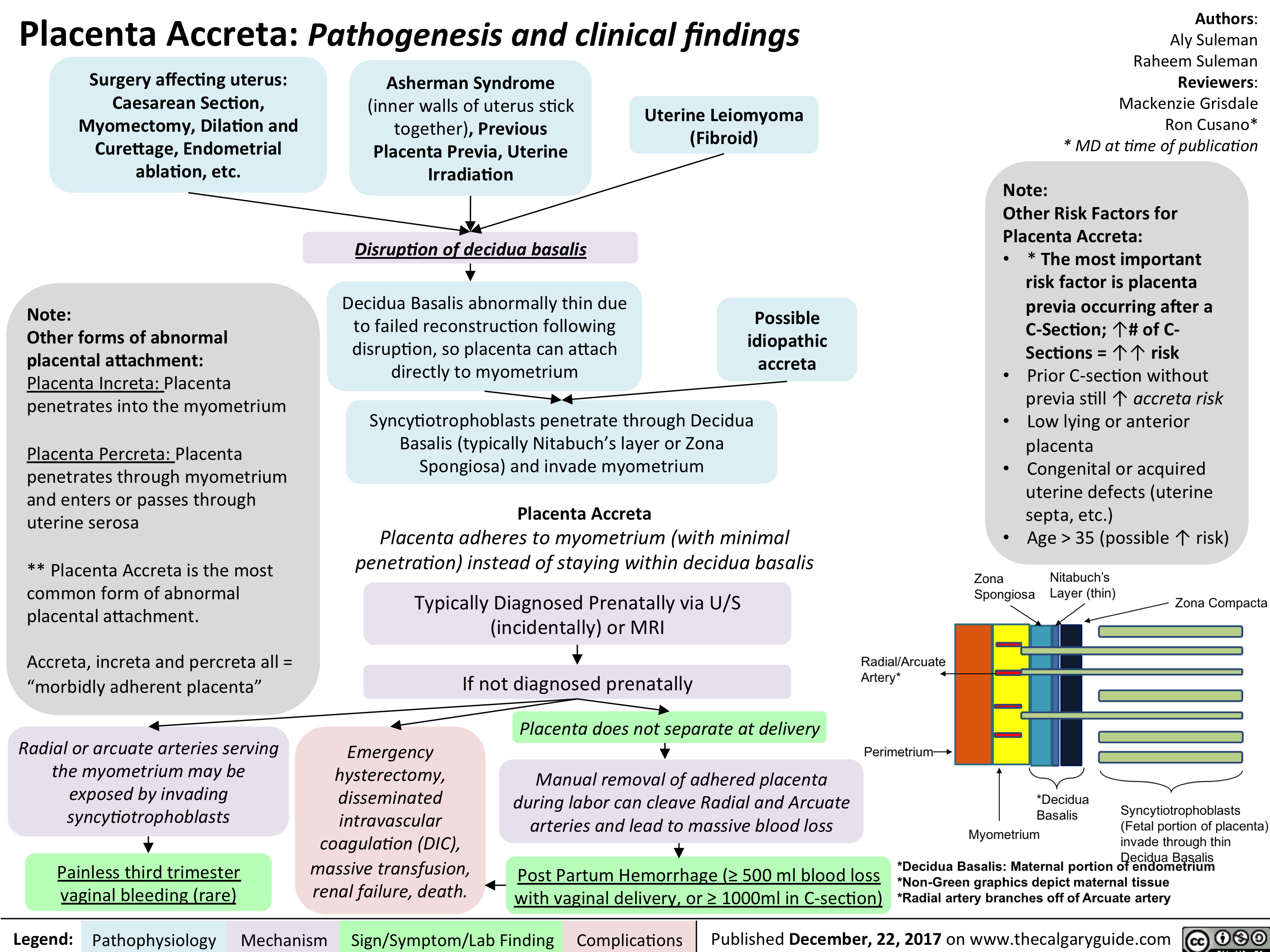
GDM: Risk factors and pathogenesis
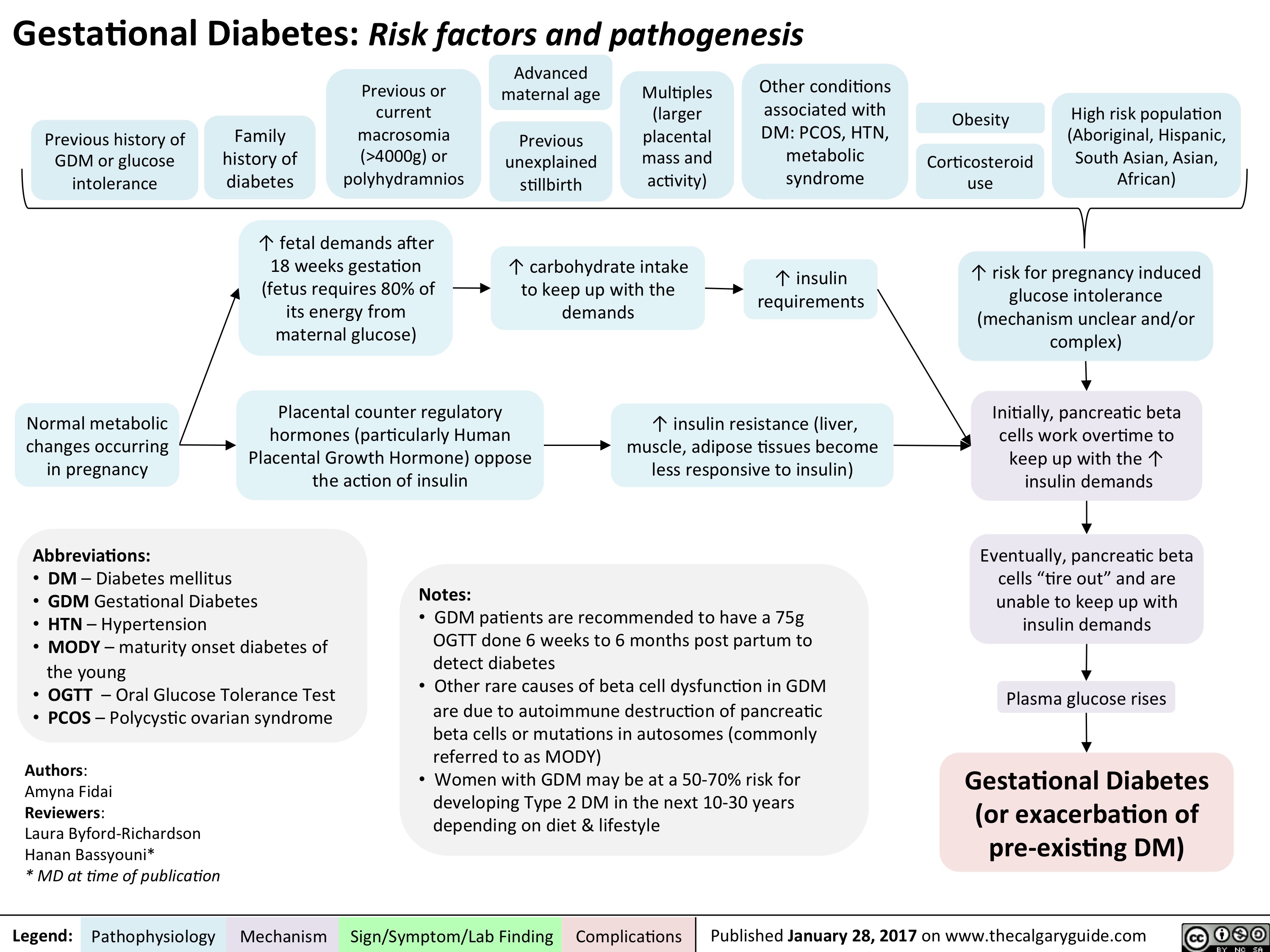
BPH Final
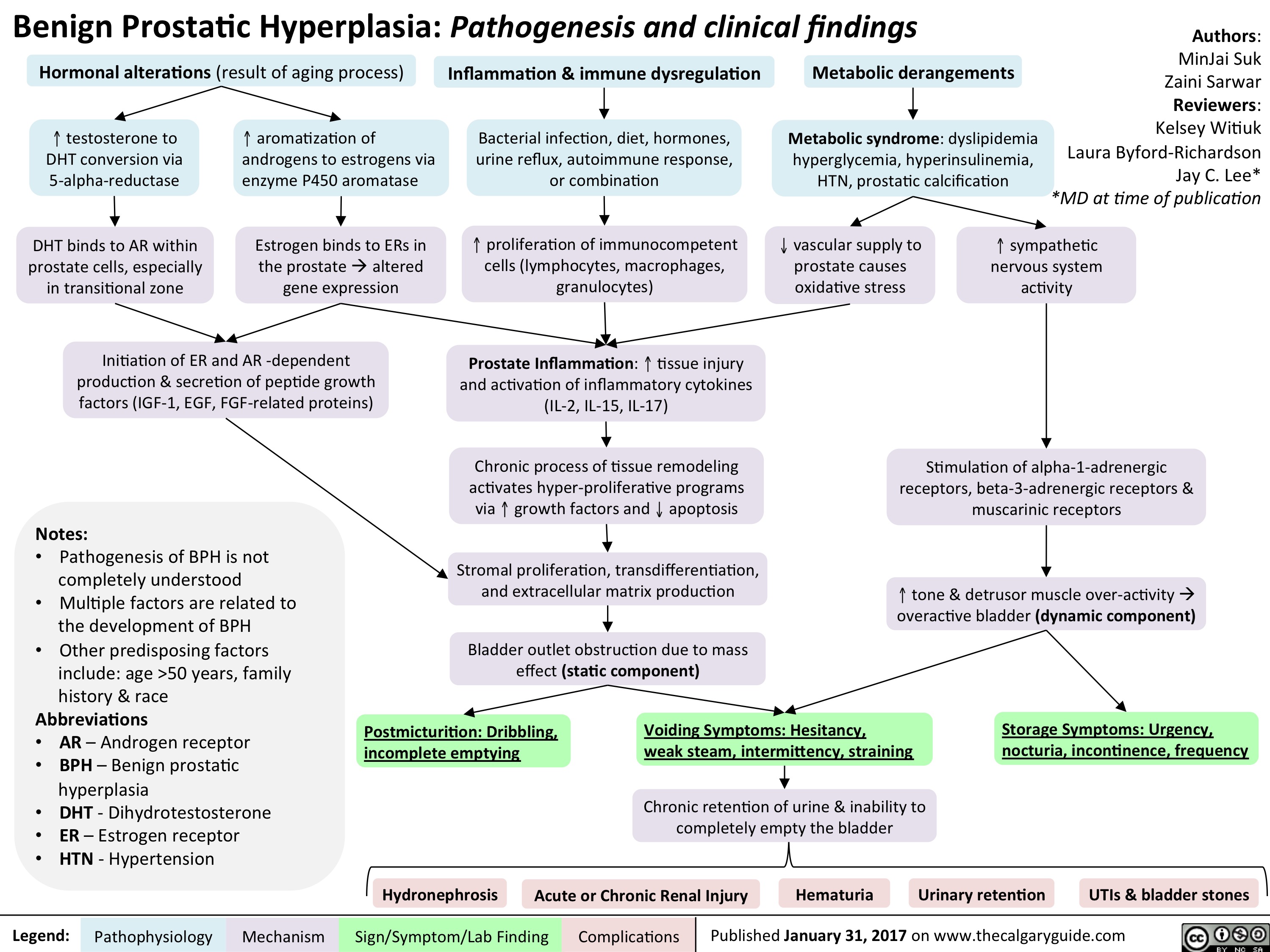
BPH Final
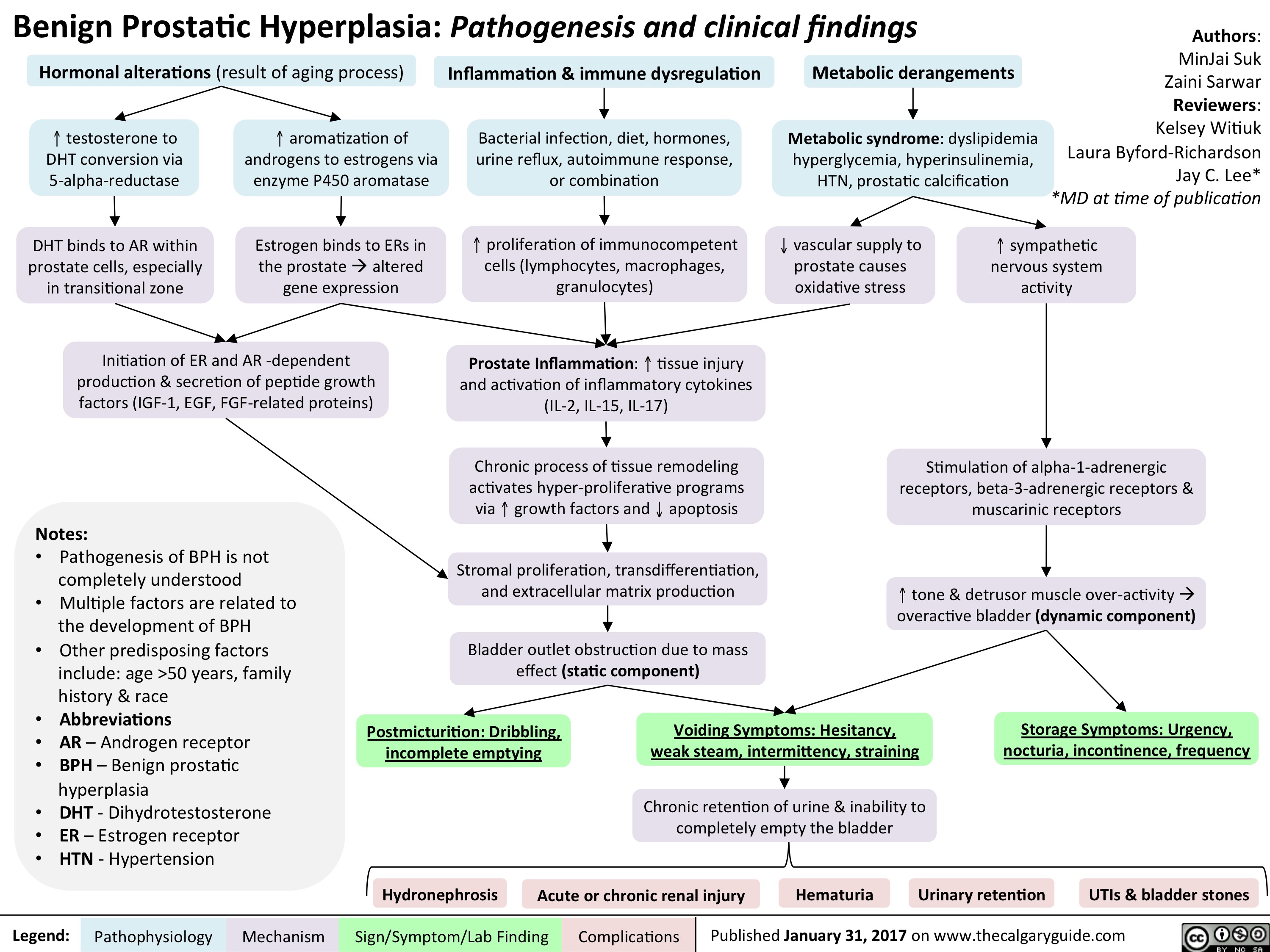
ASPD FINAL
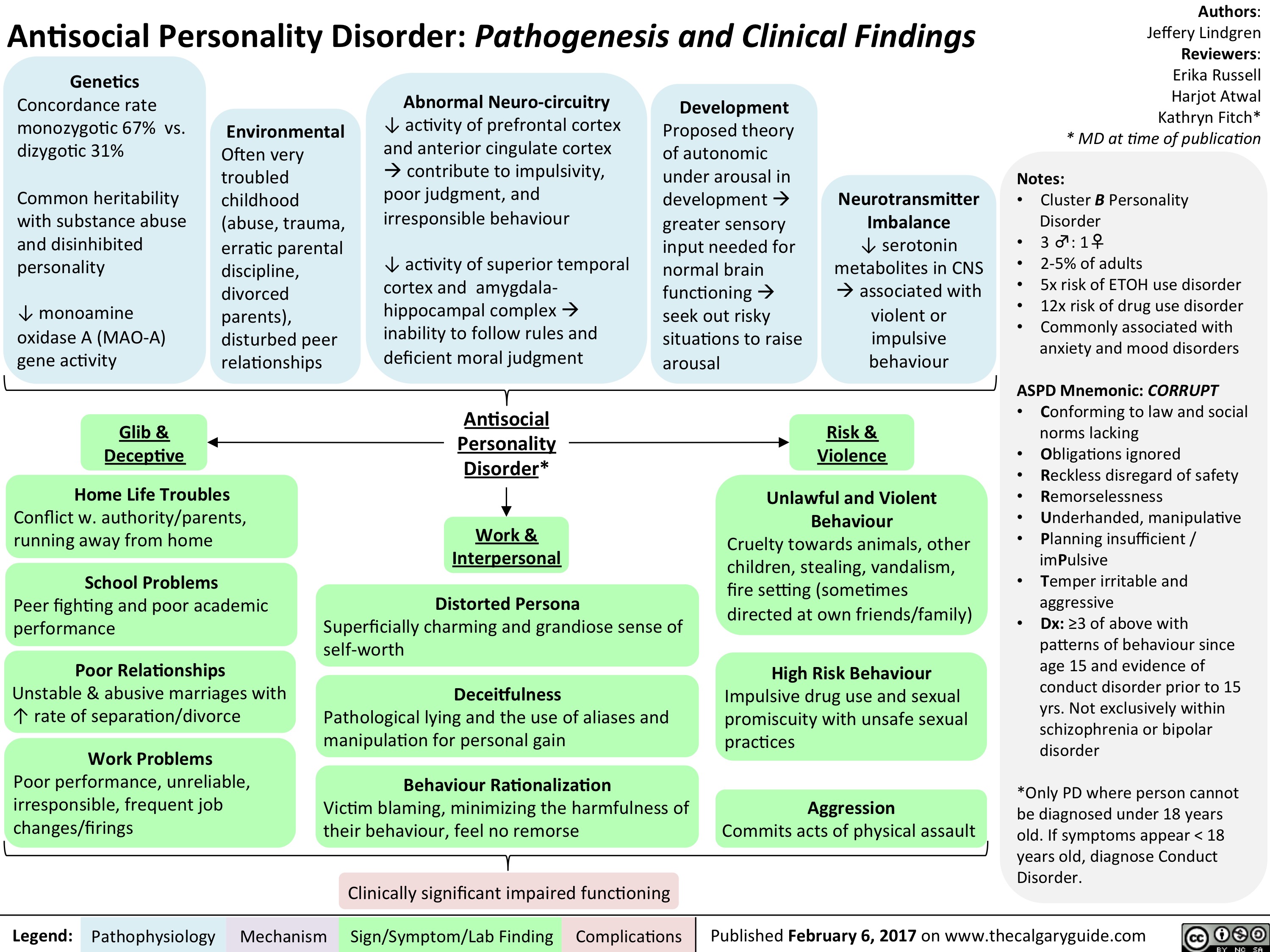
BPD FINAL
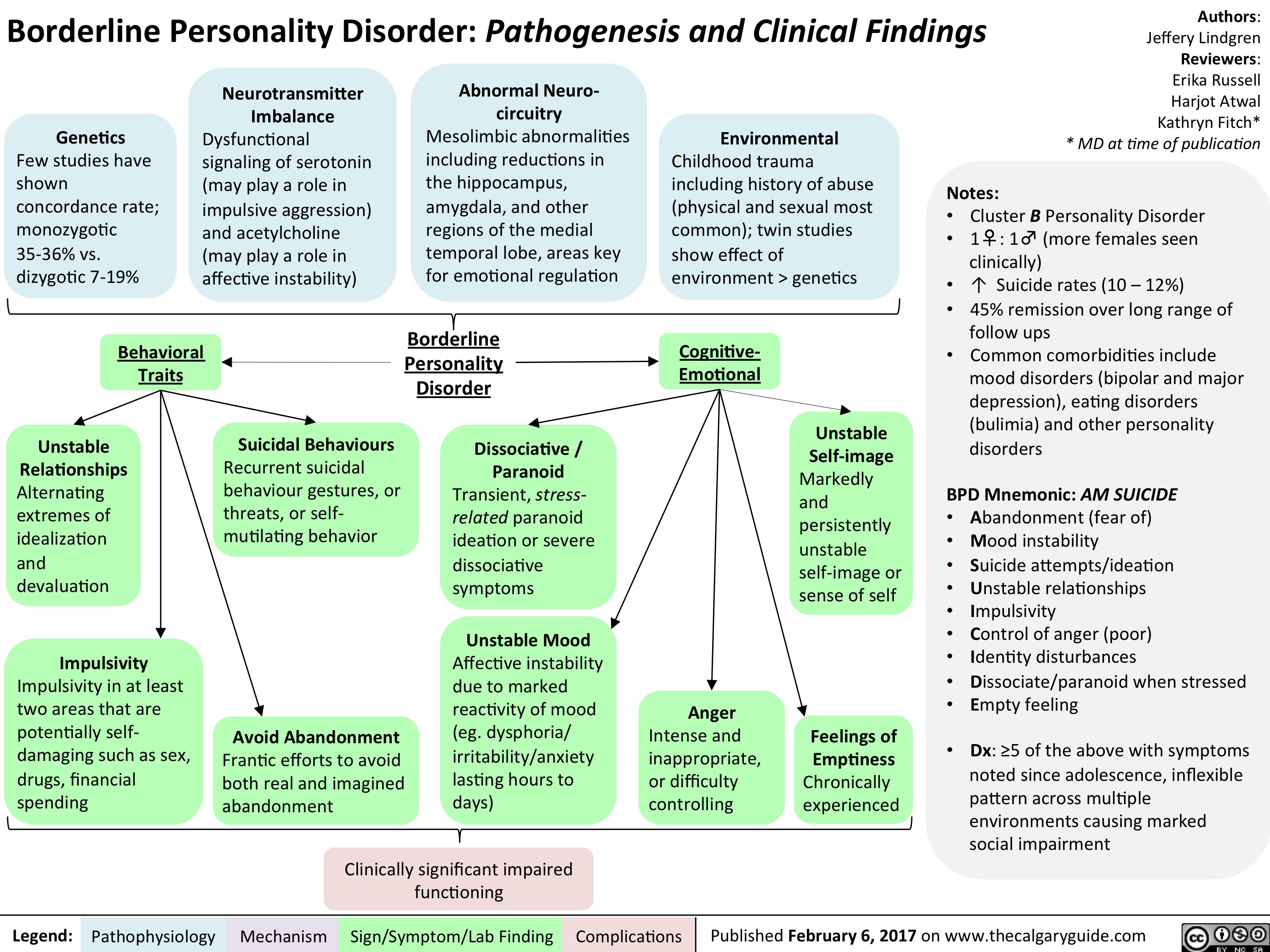
NPD FINAL
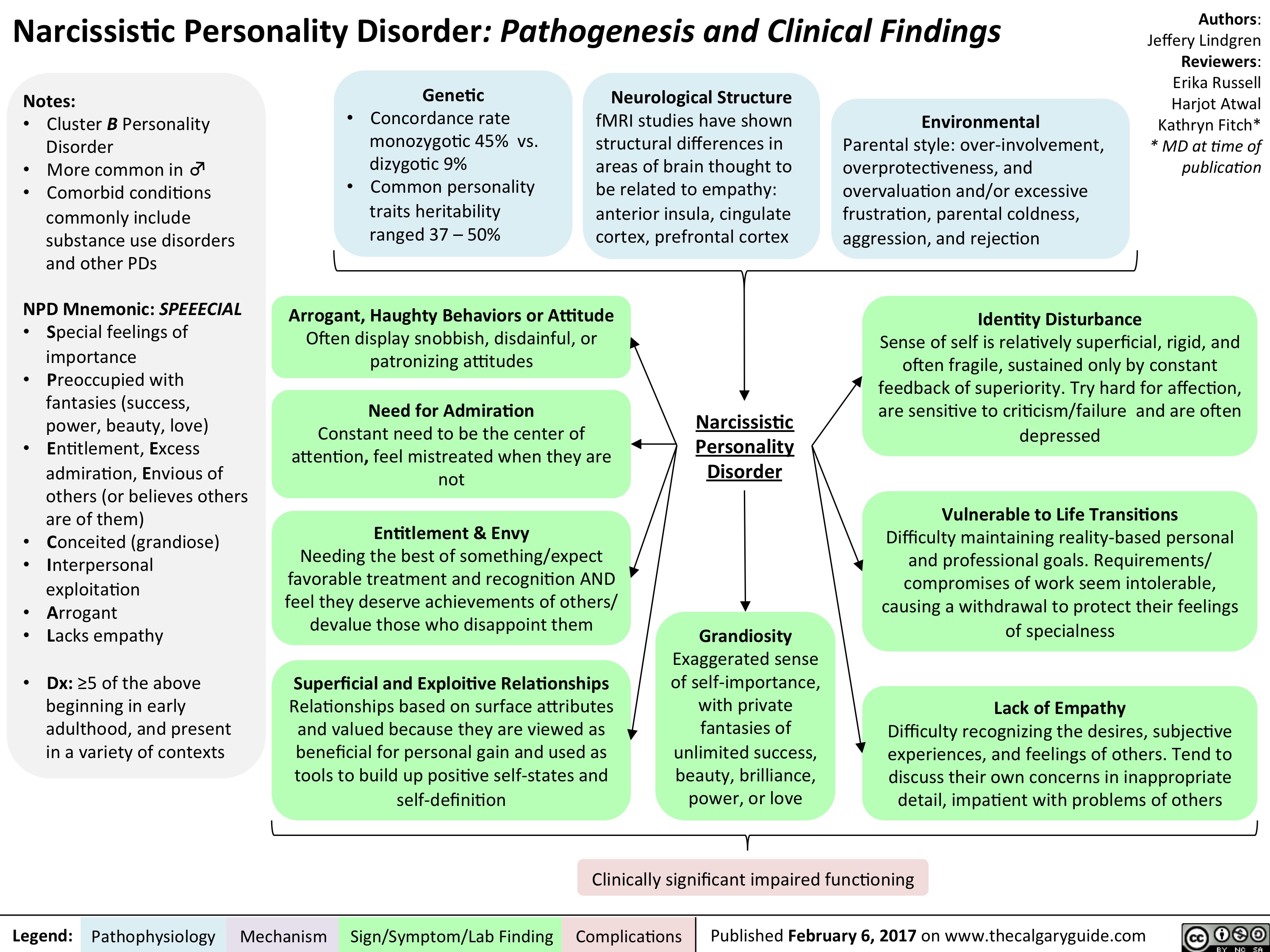
Lithium FINAL
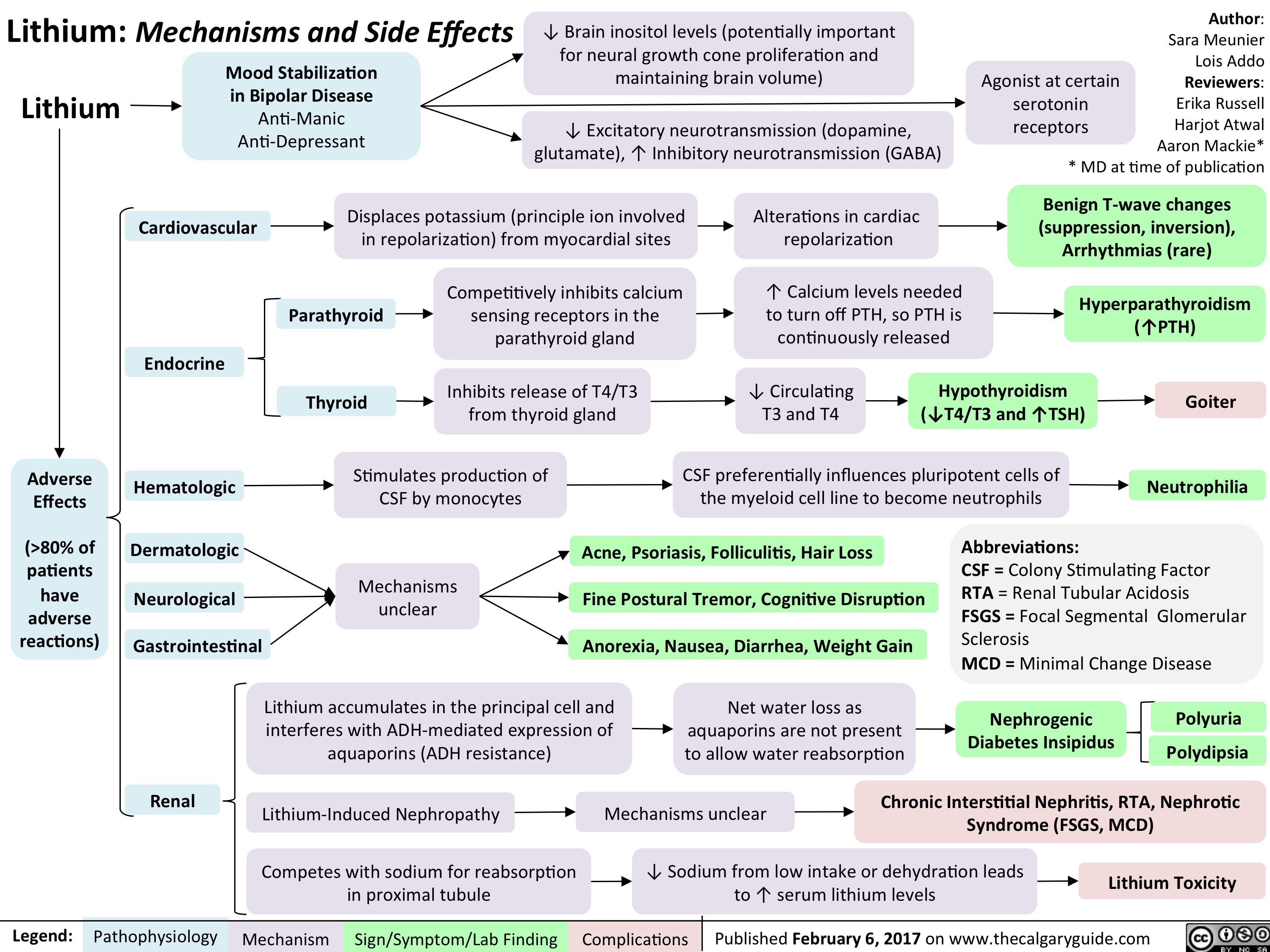
AF FINAL
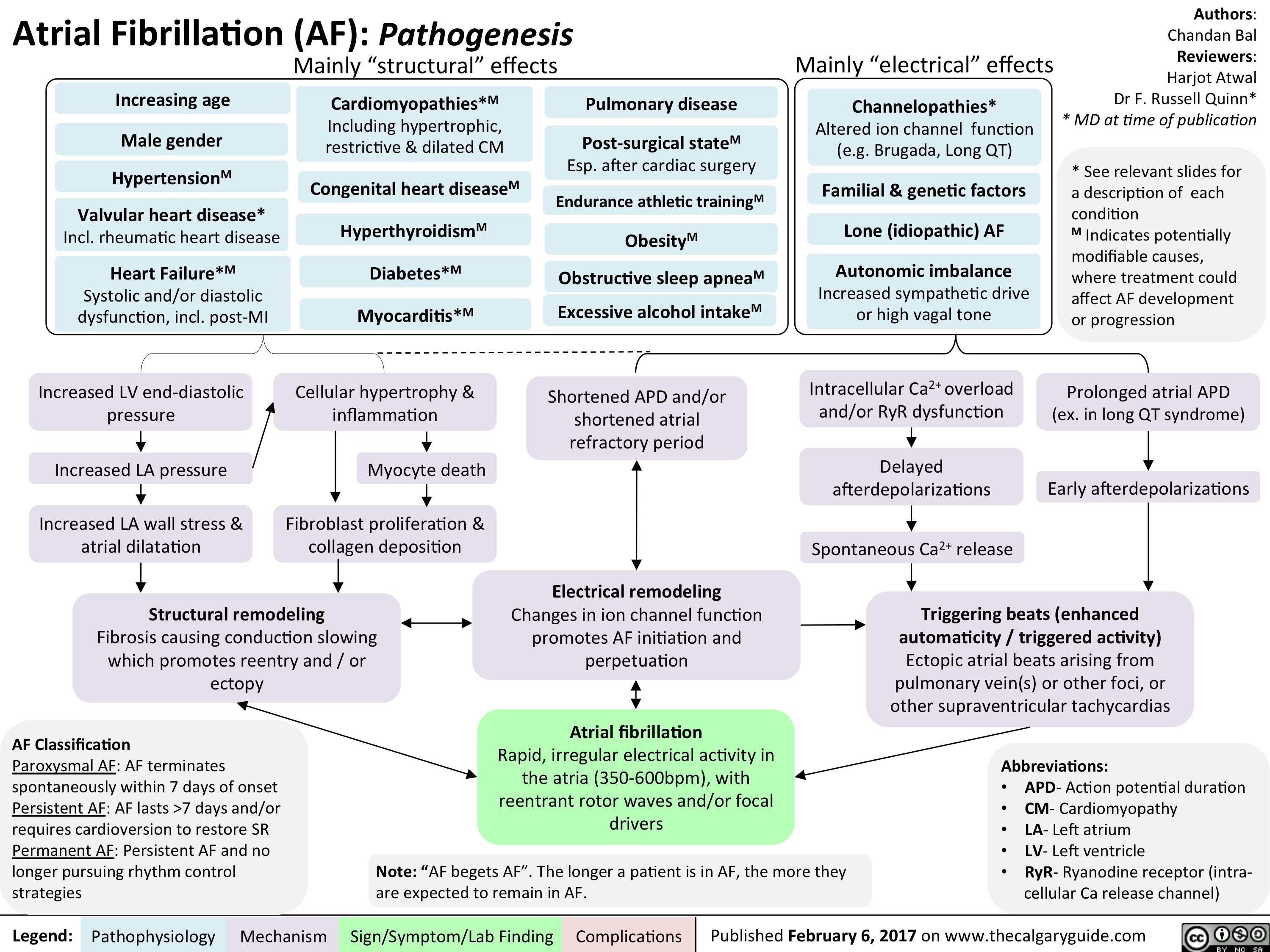
generalized-seizures-definitions

Lateral Medullary Syndrome FINAL
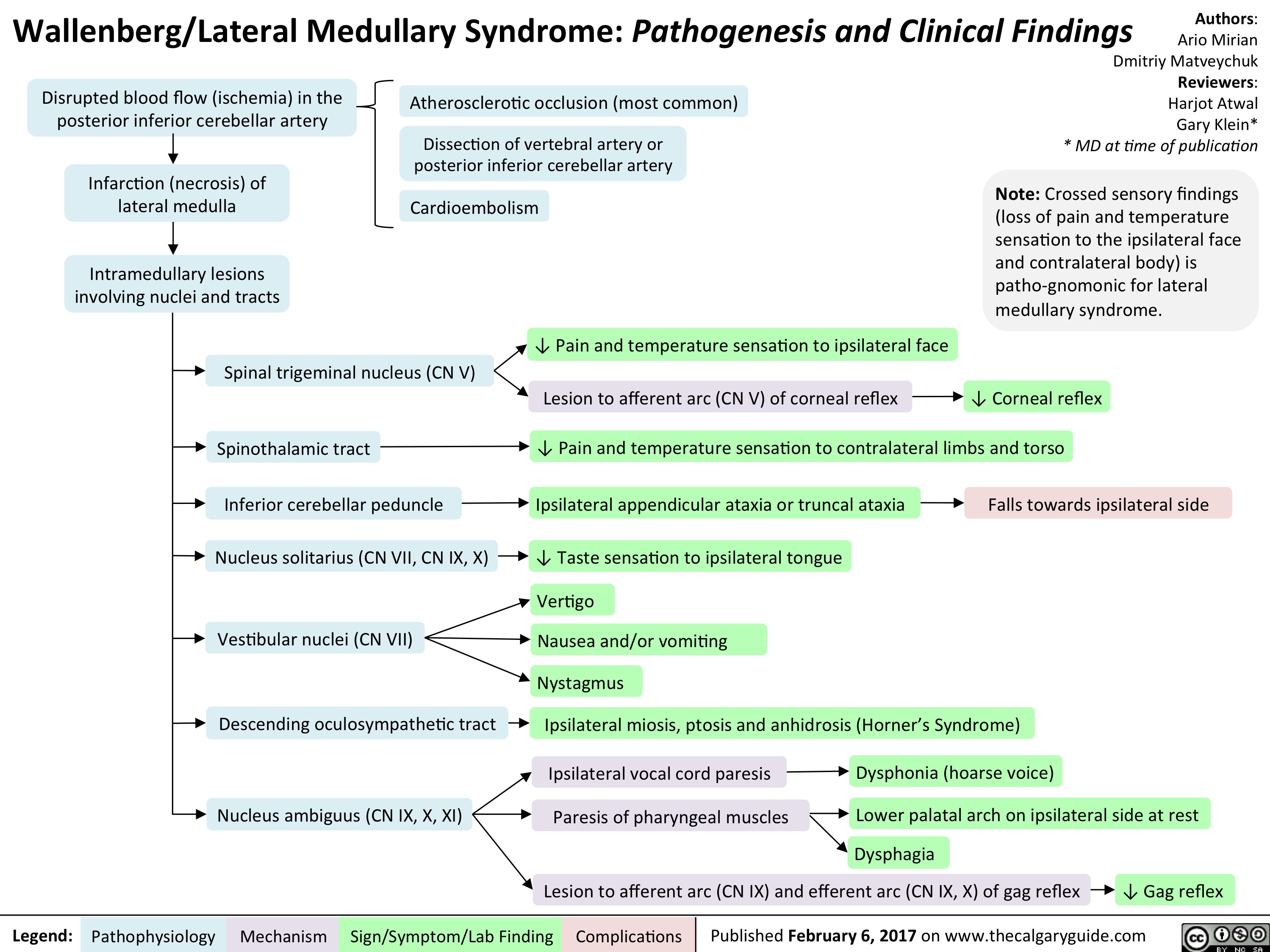
Subdural hematoma overview
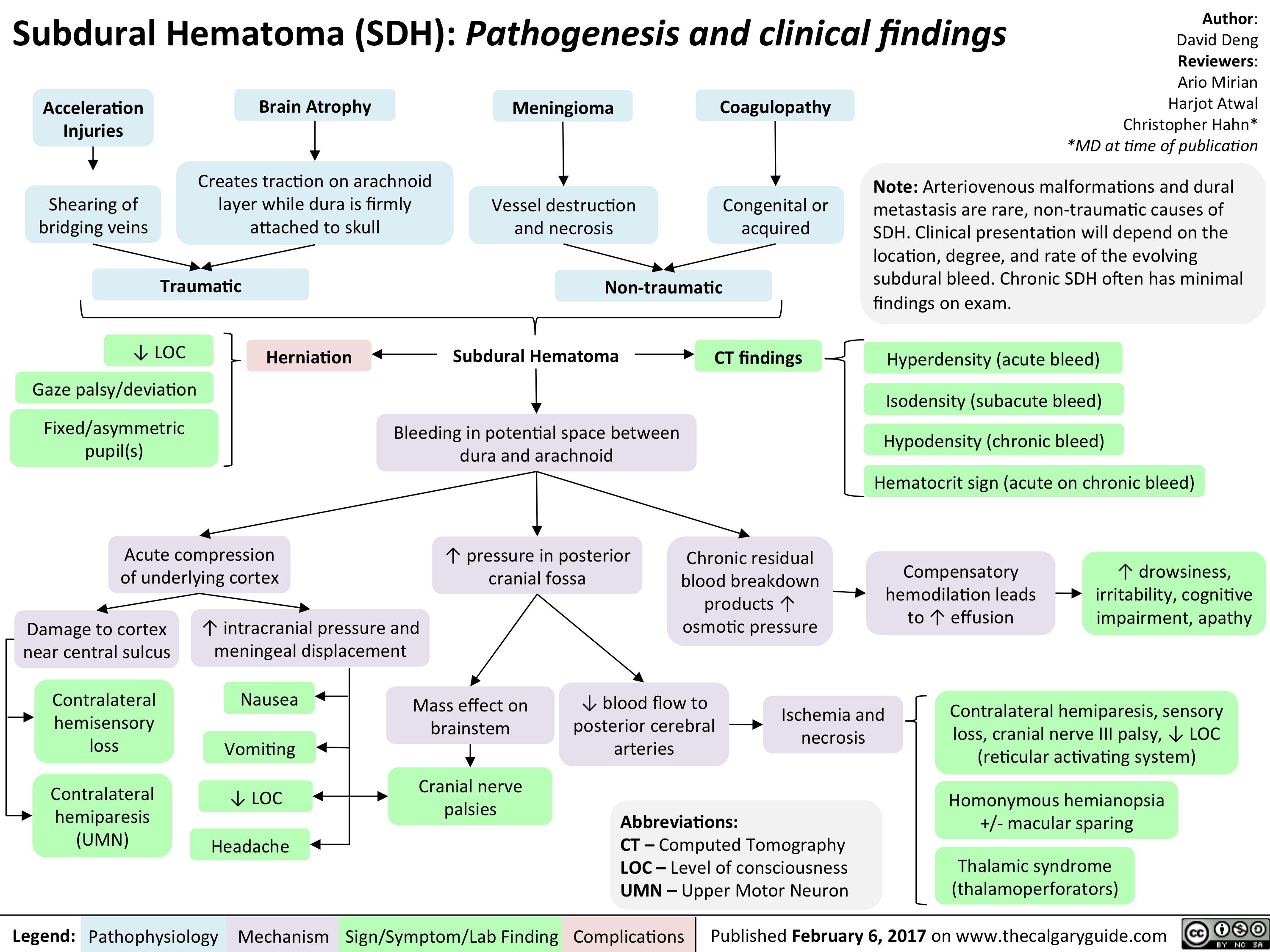
Staphylococcus Scalded Skin Syndrome FINAL
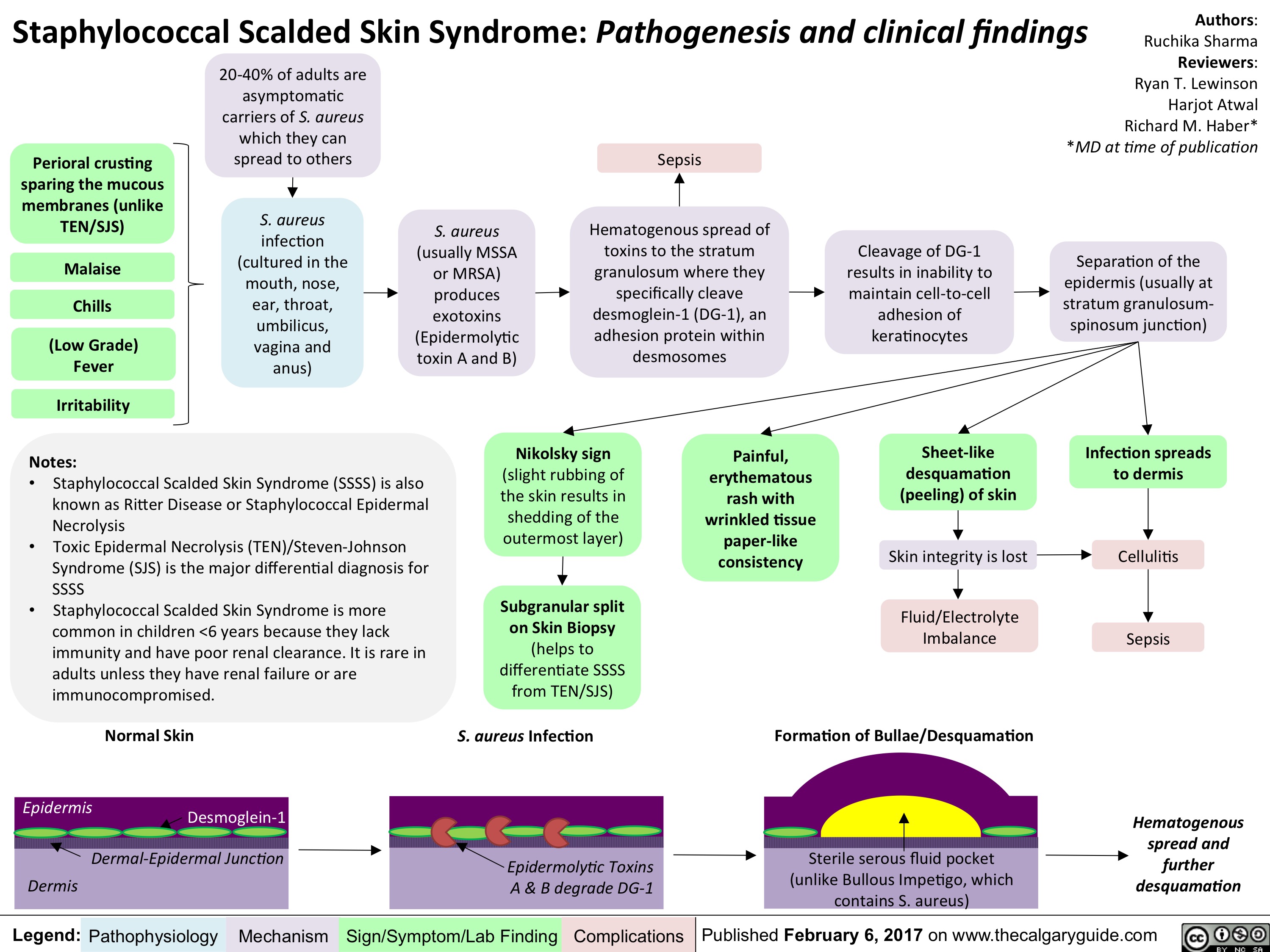
Manion contraception MOAs
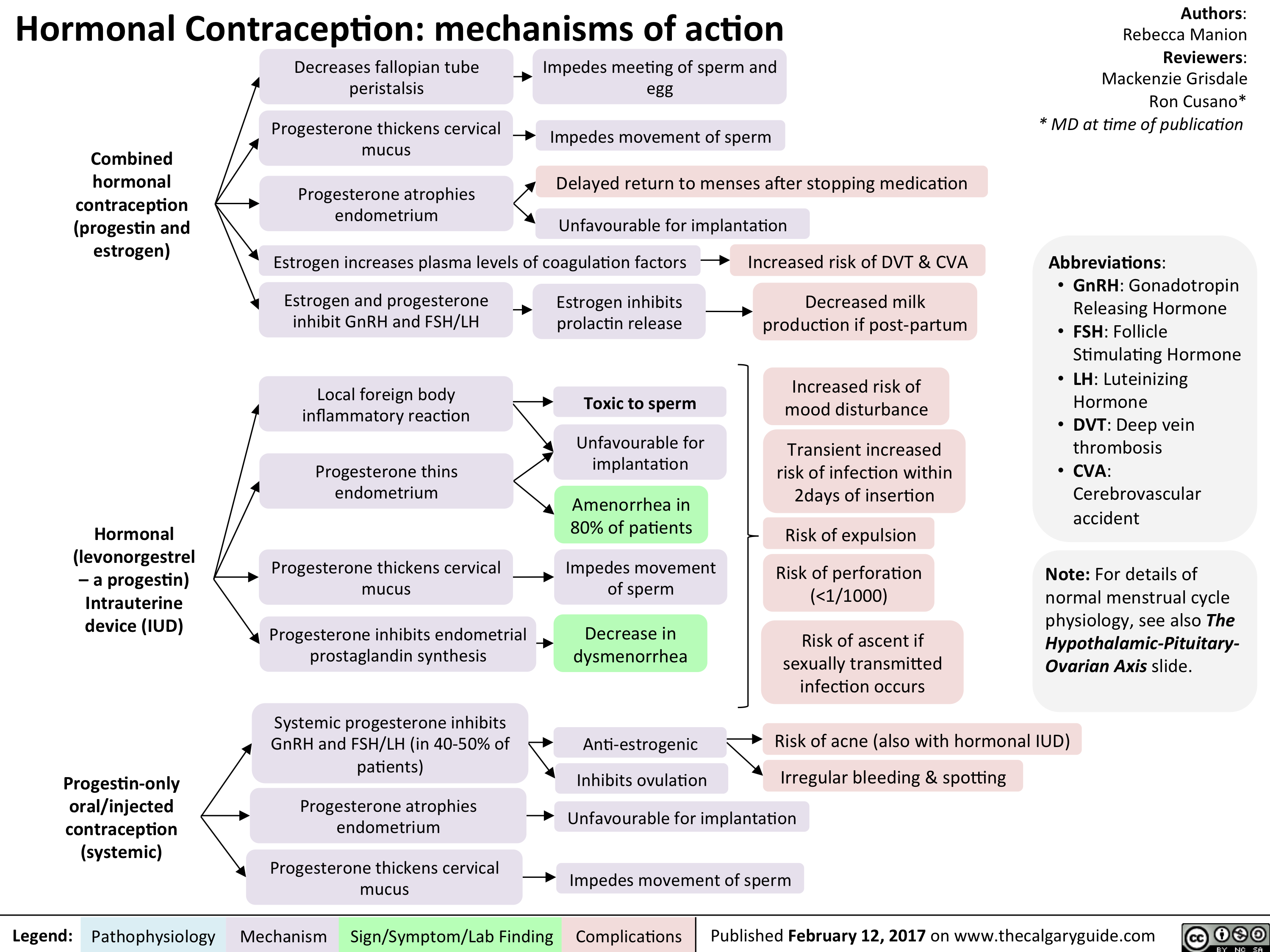
Manion contraception MOAs2
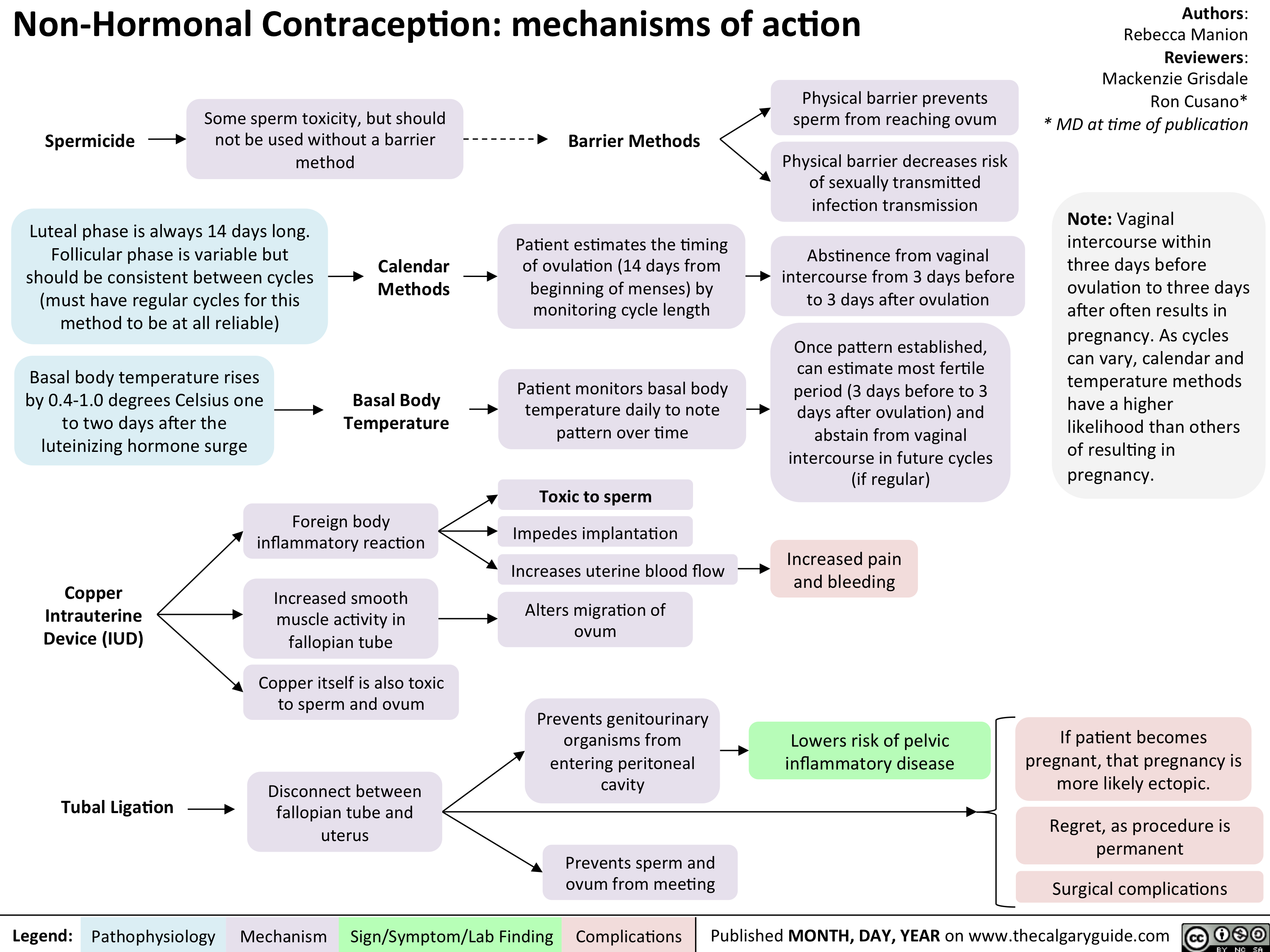
Common meds contra in Preg
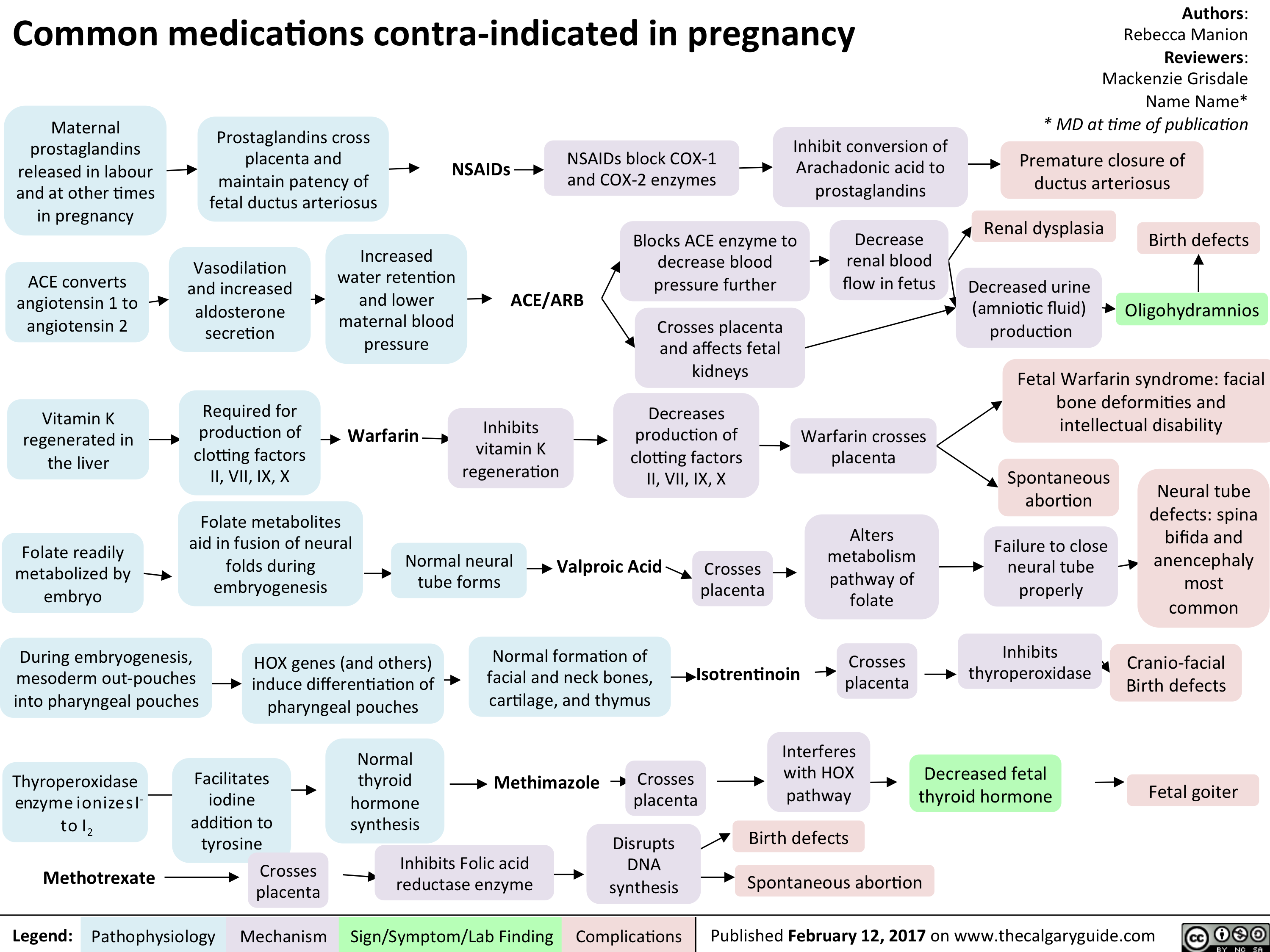
Non hormonal
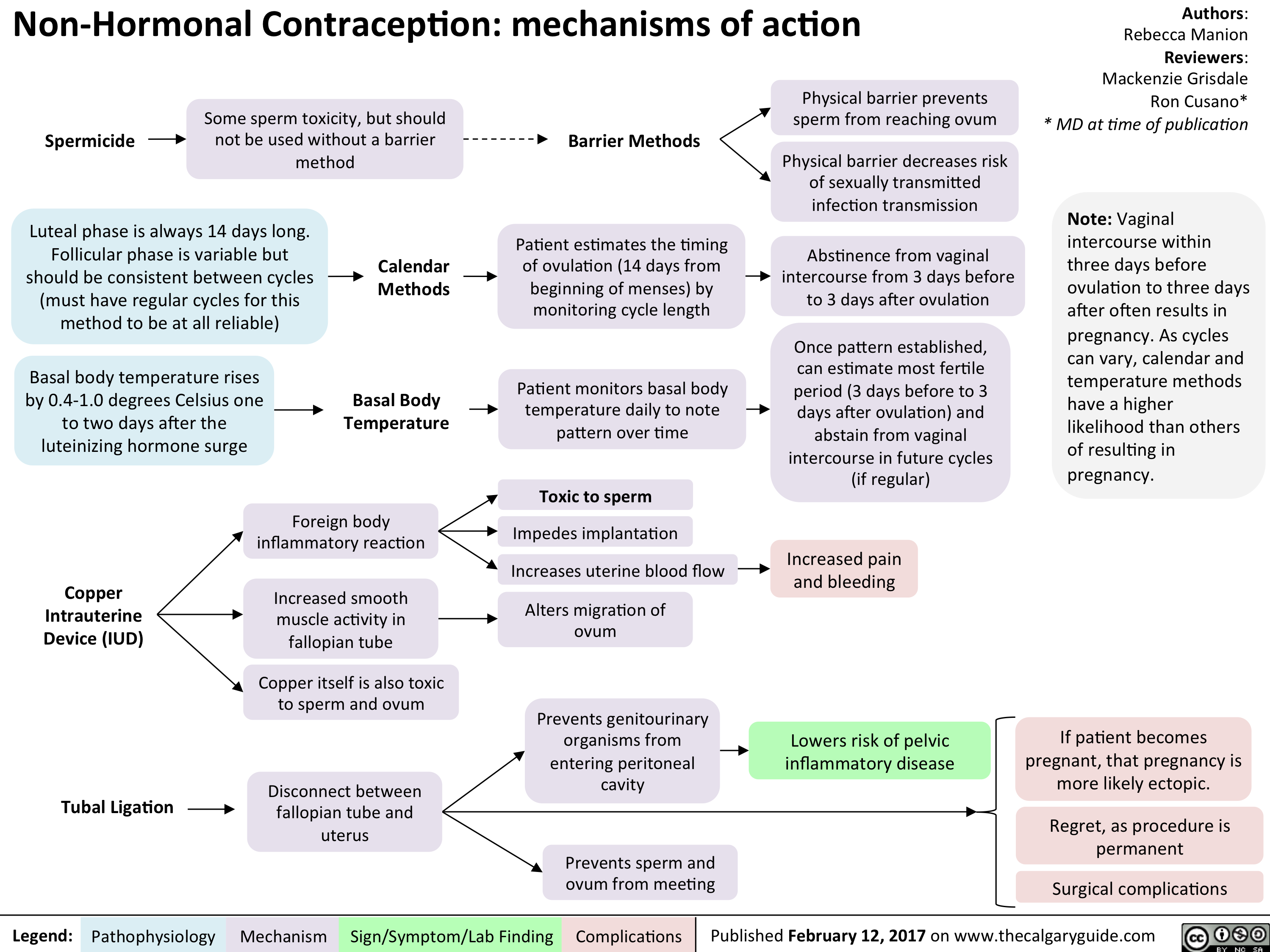
Vasa Previa
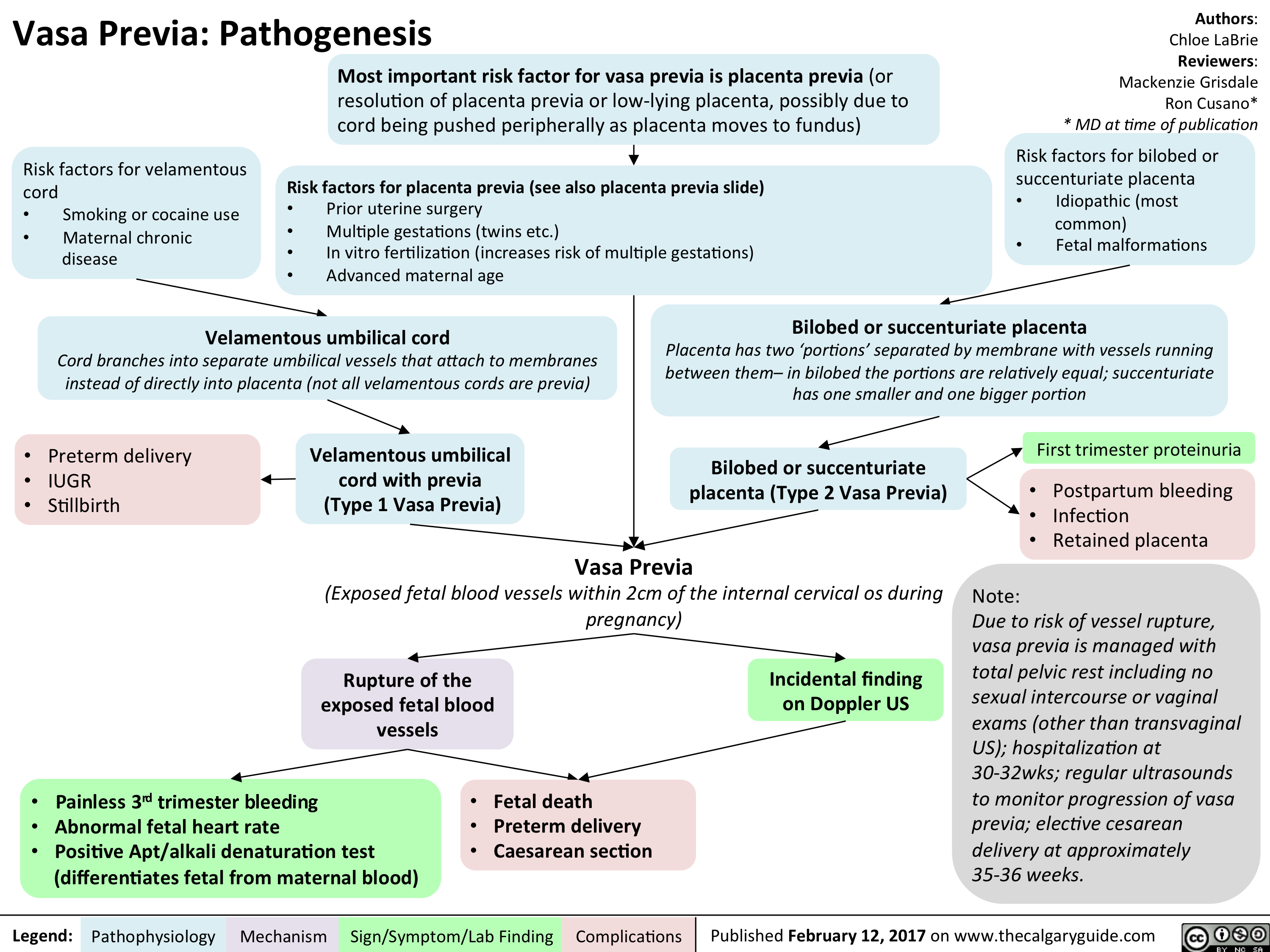
ASPD FINAL
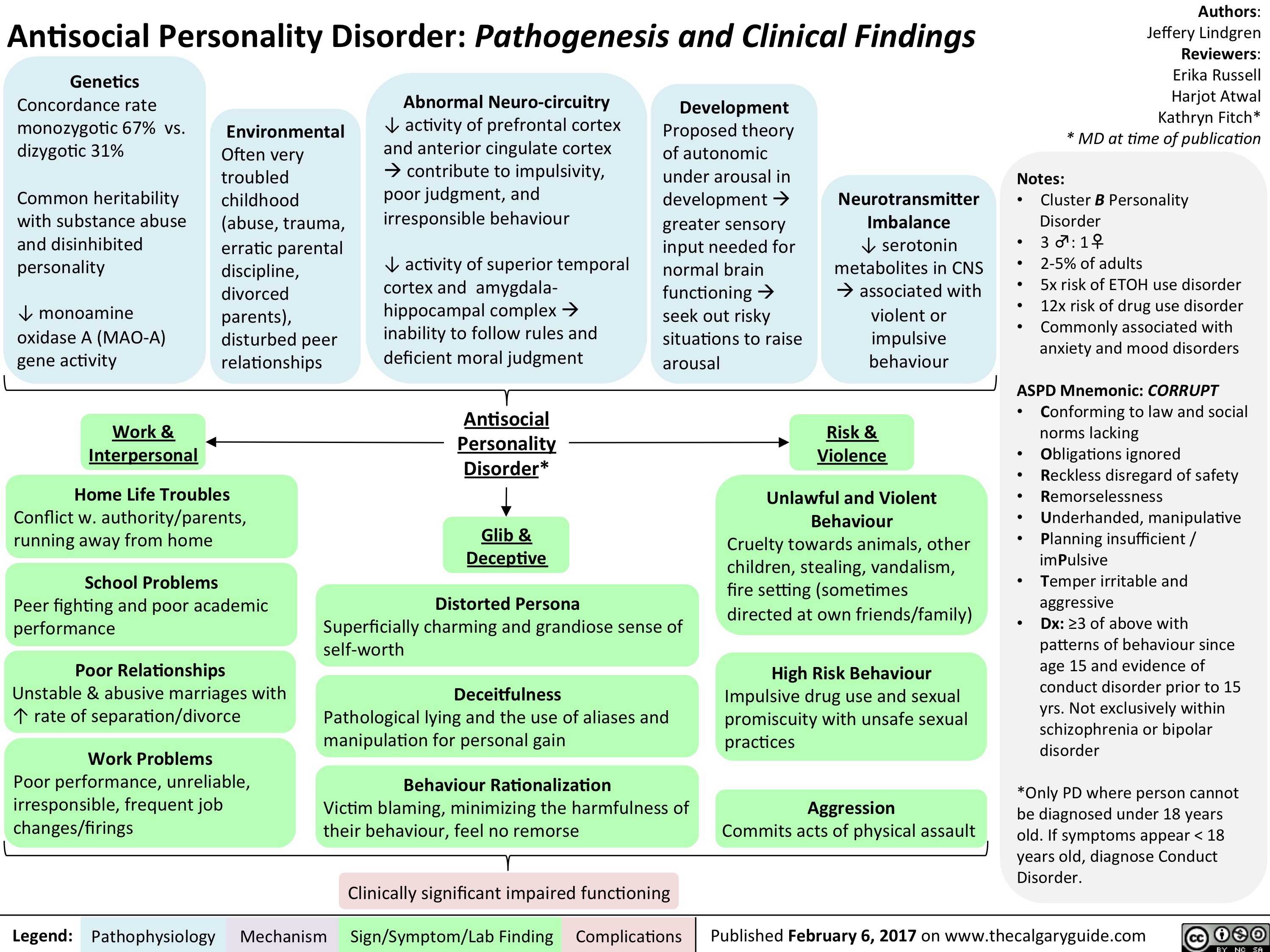
hypertension-in-pregnancy-overview-of-definitions
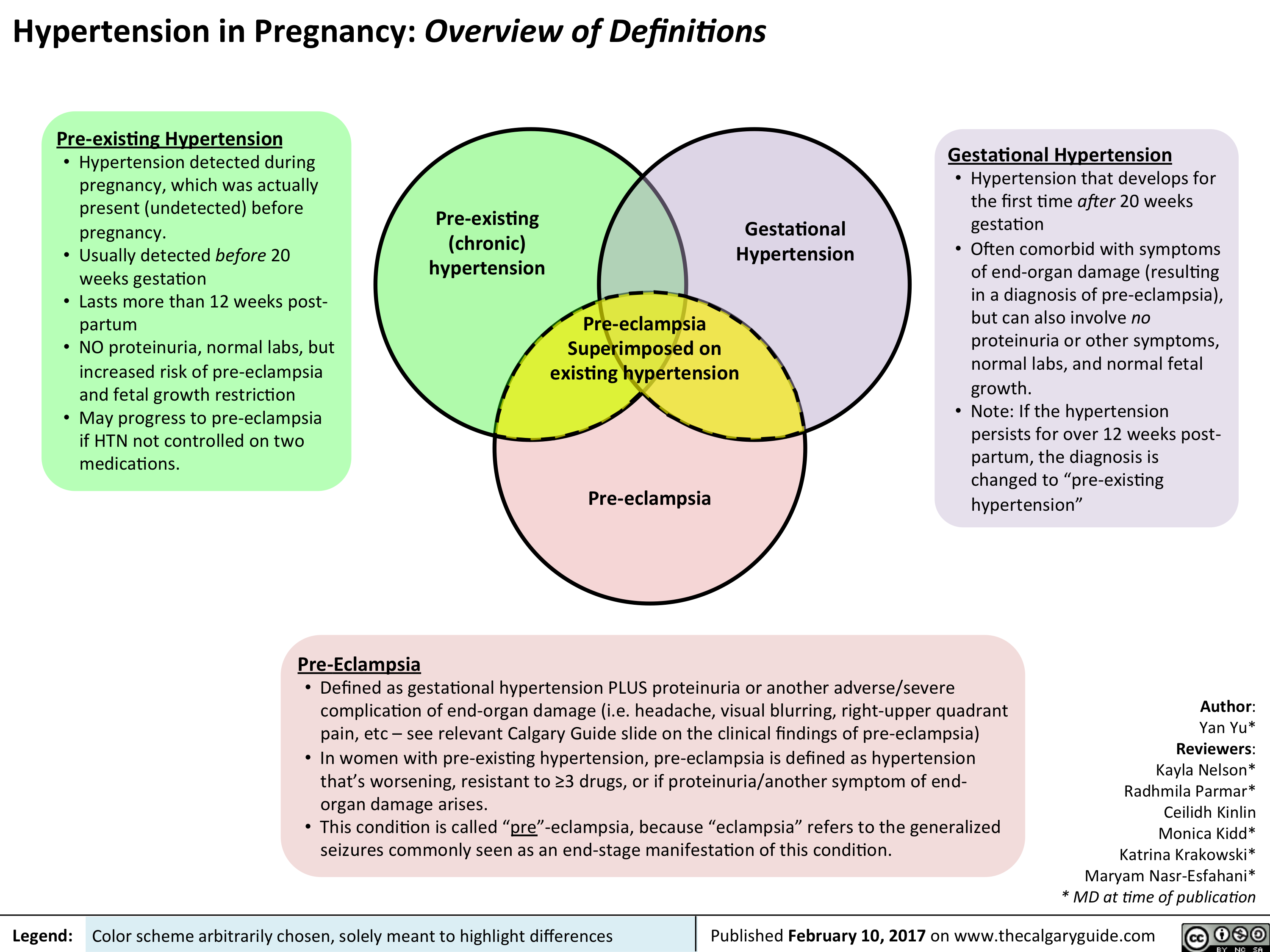
Pre-eclampsia Maternal Complications
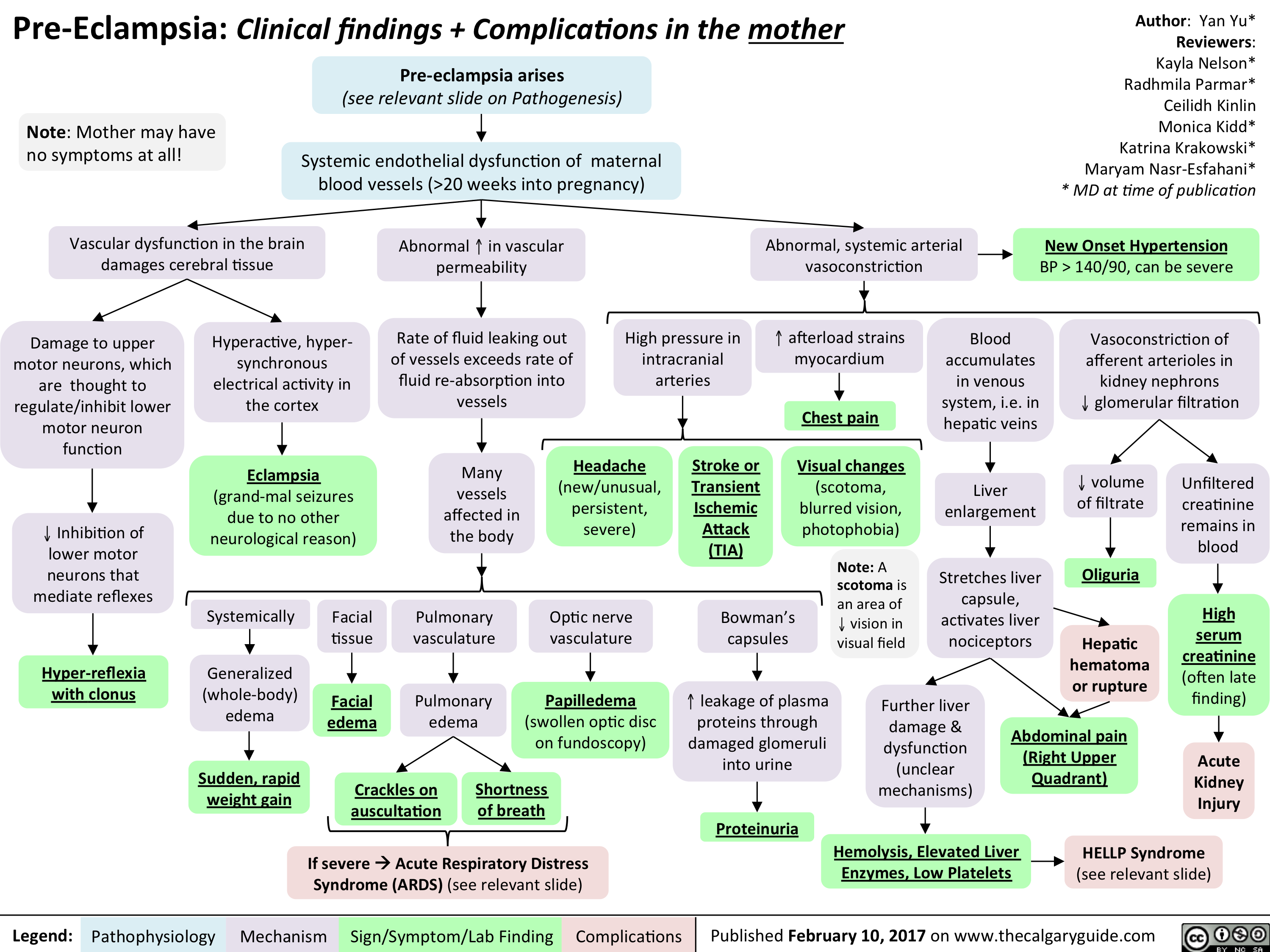
Pre-eclampsia Fetal Complications
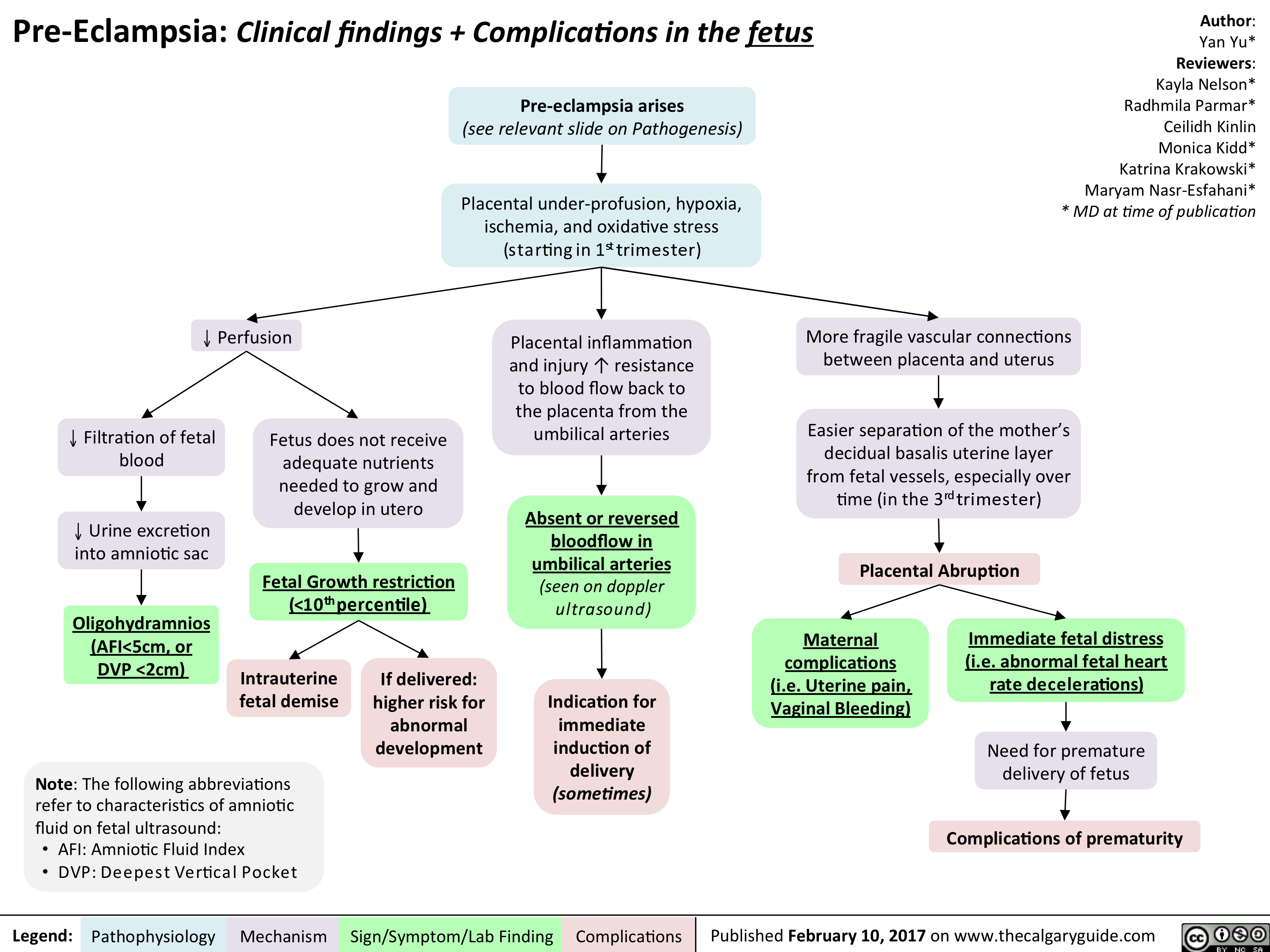
Altered Metabolism in the ICU
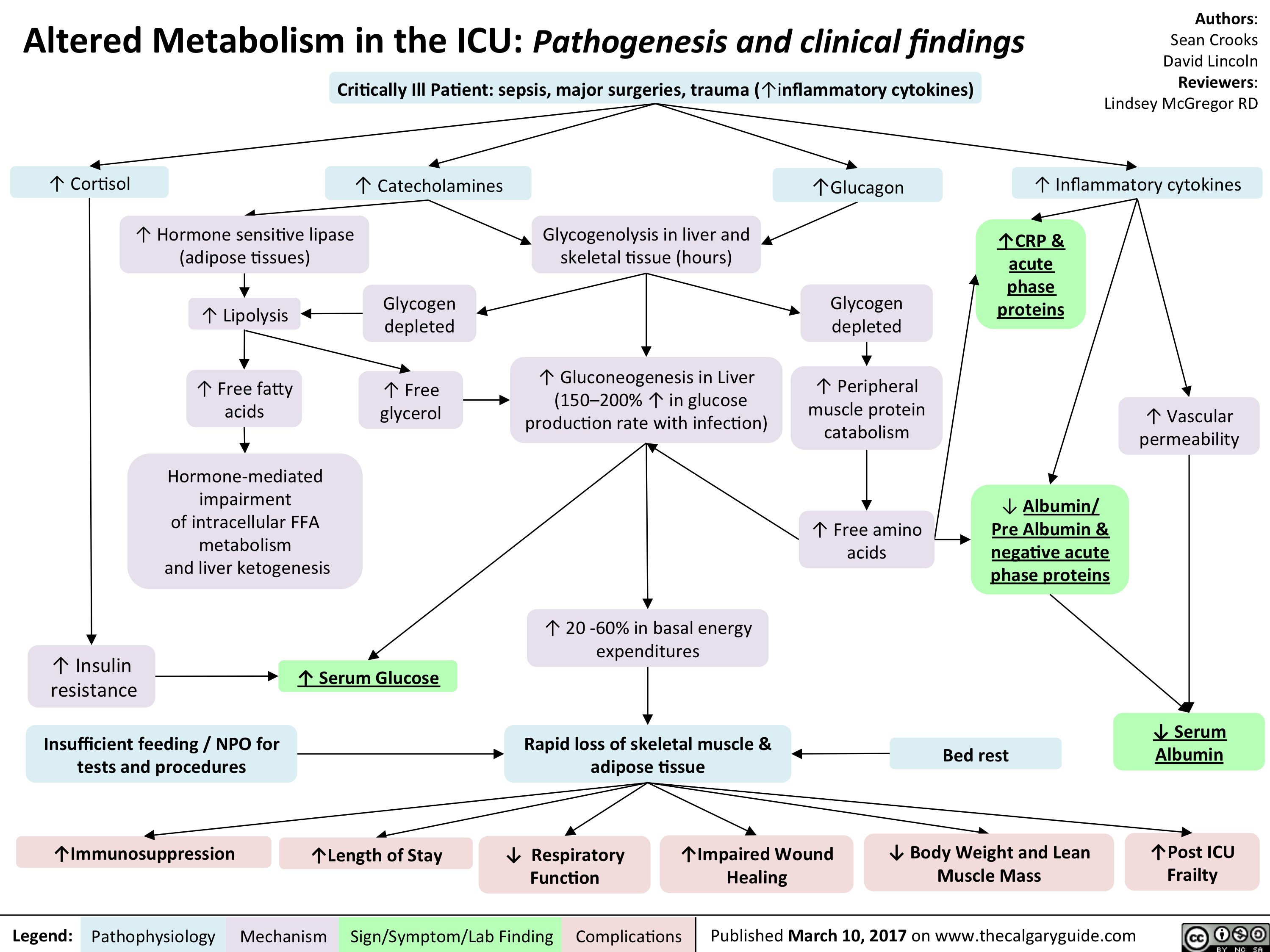
Altered Metabolism in the ICU- Pathogenesis and clinical findings
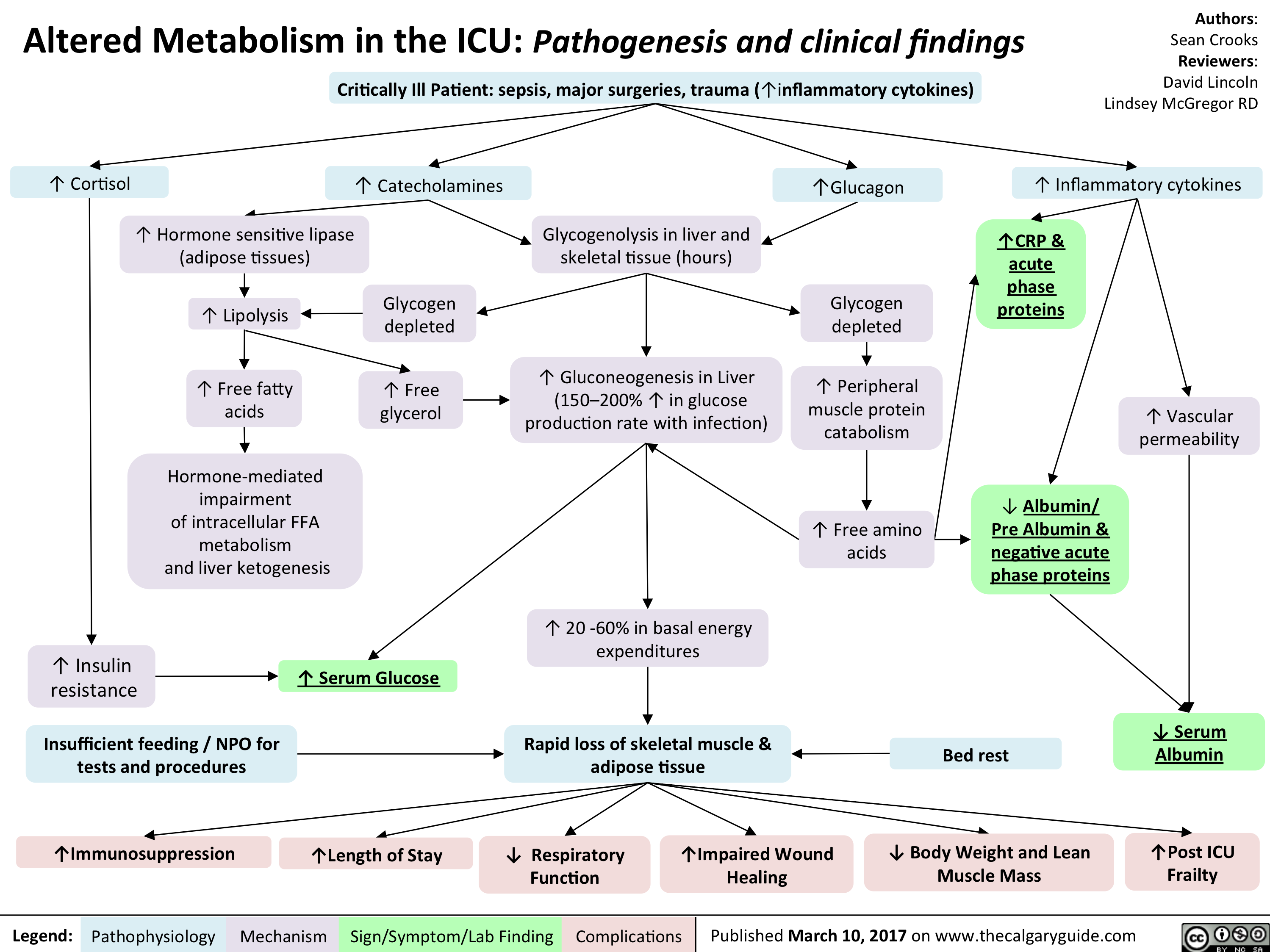
Mental Status Exam

Addiction Long-term Consequences

Addiction Pathogenesis
Non-accidental Burns
SLE-GI Manifestations
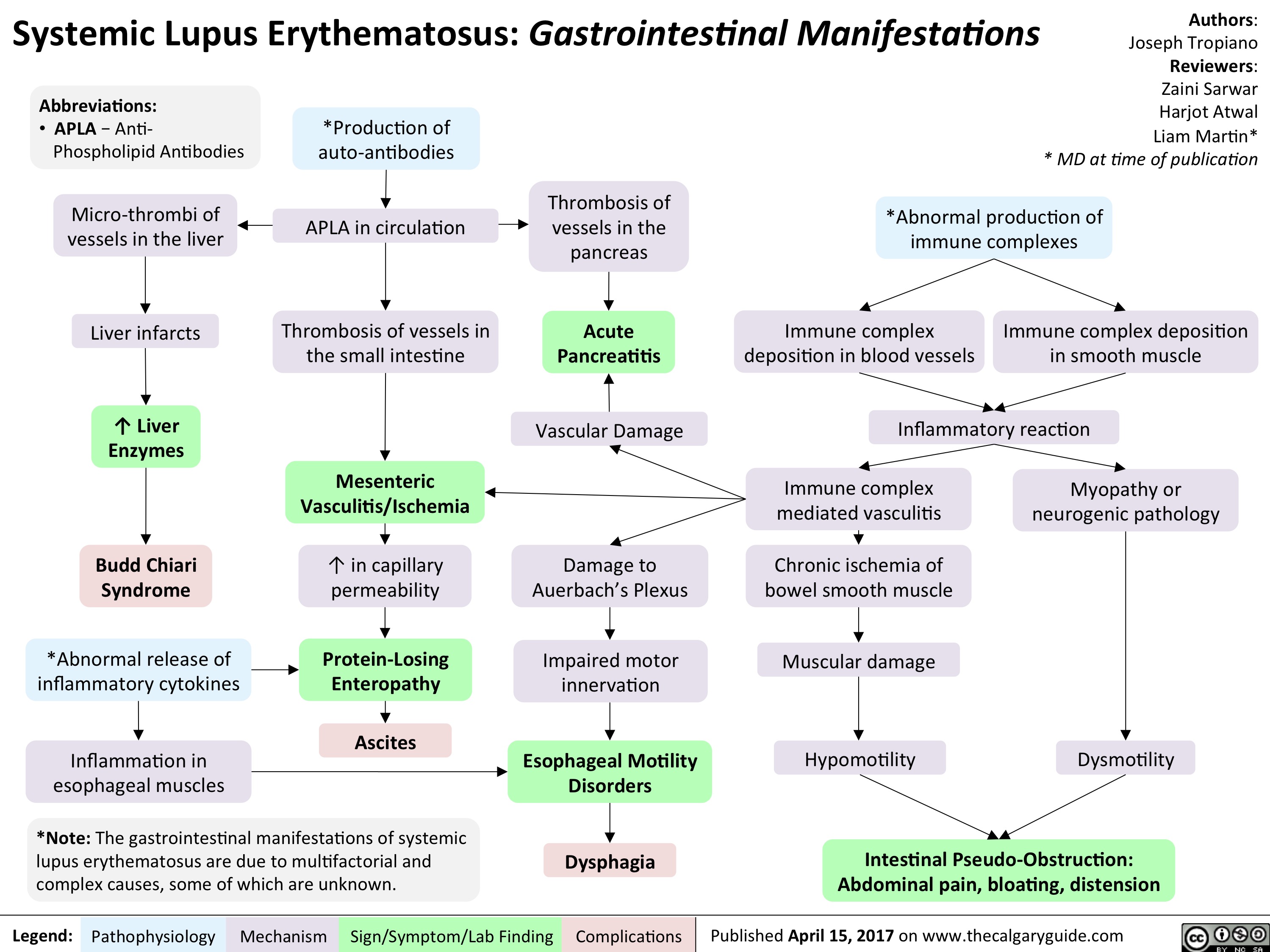
Testicular Torsion
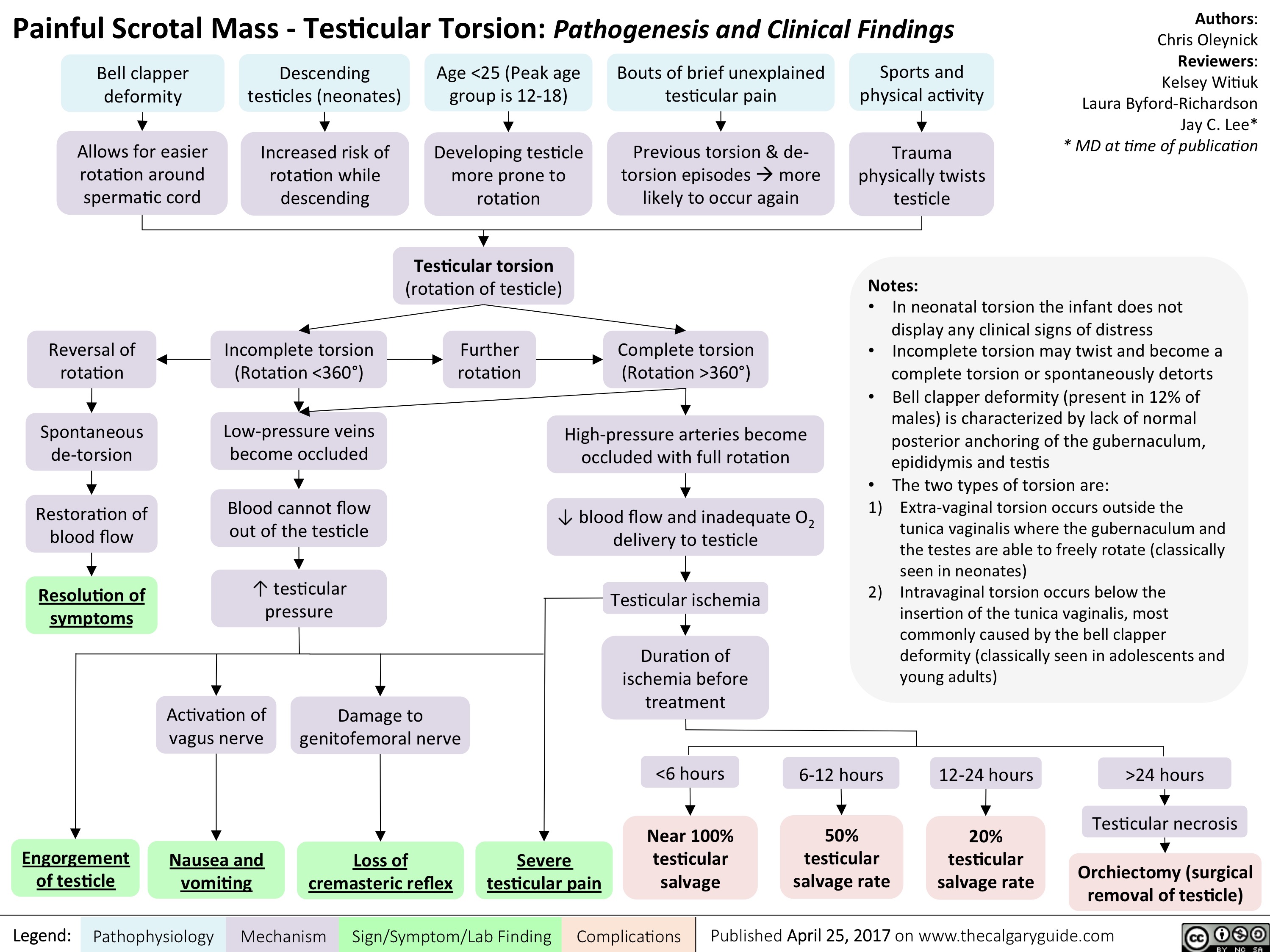
Acquired Hydrocephalus
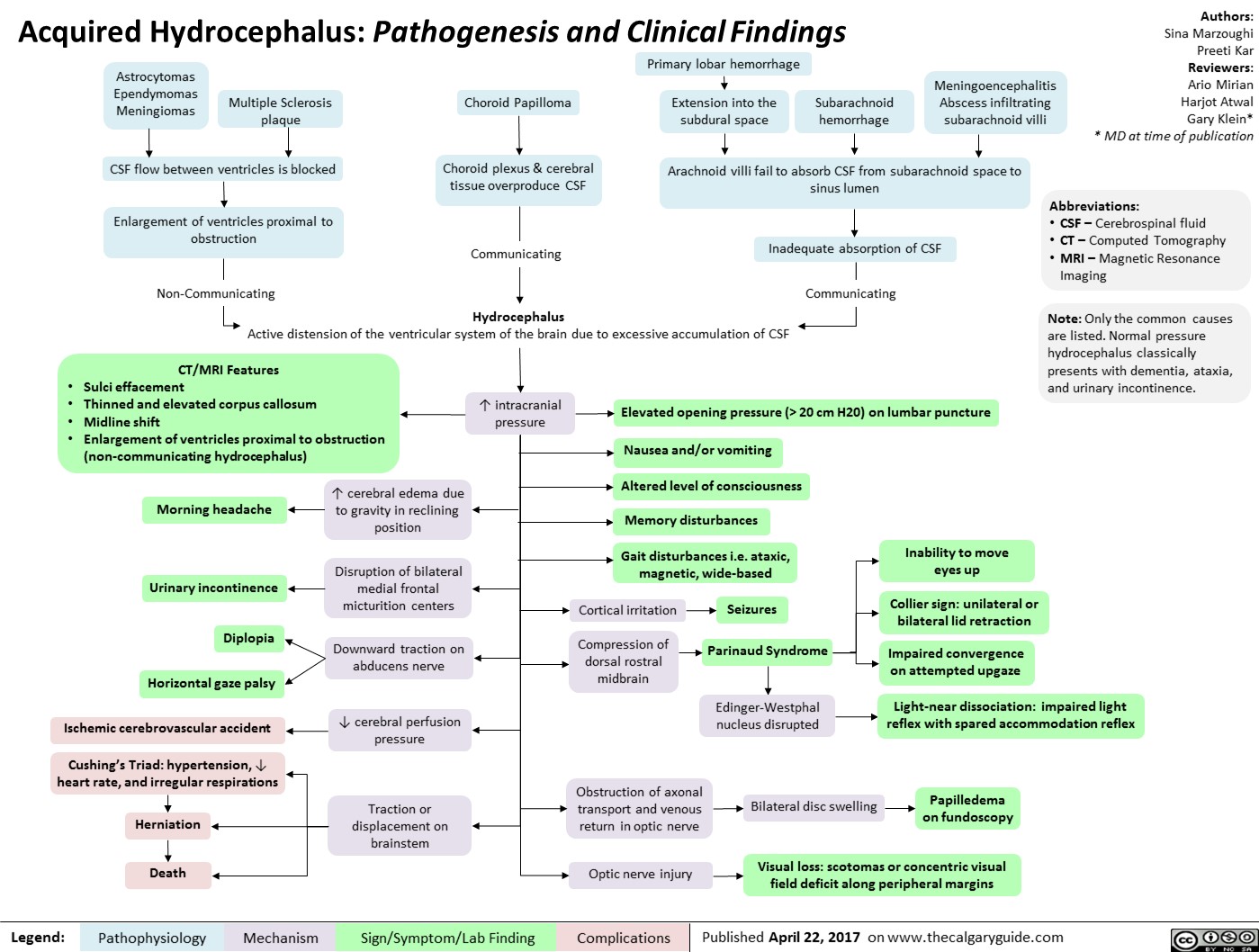
Kawasaki Disease
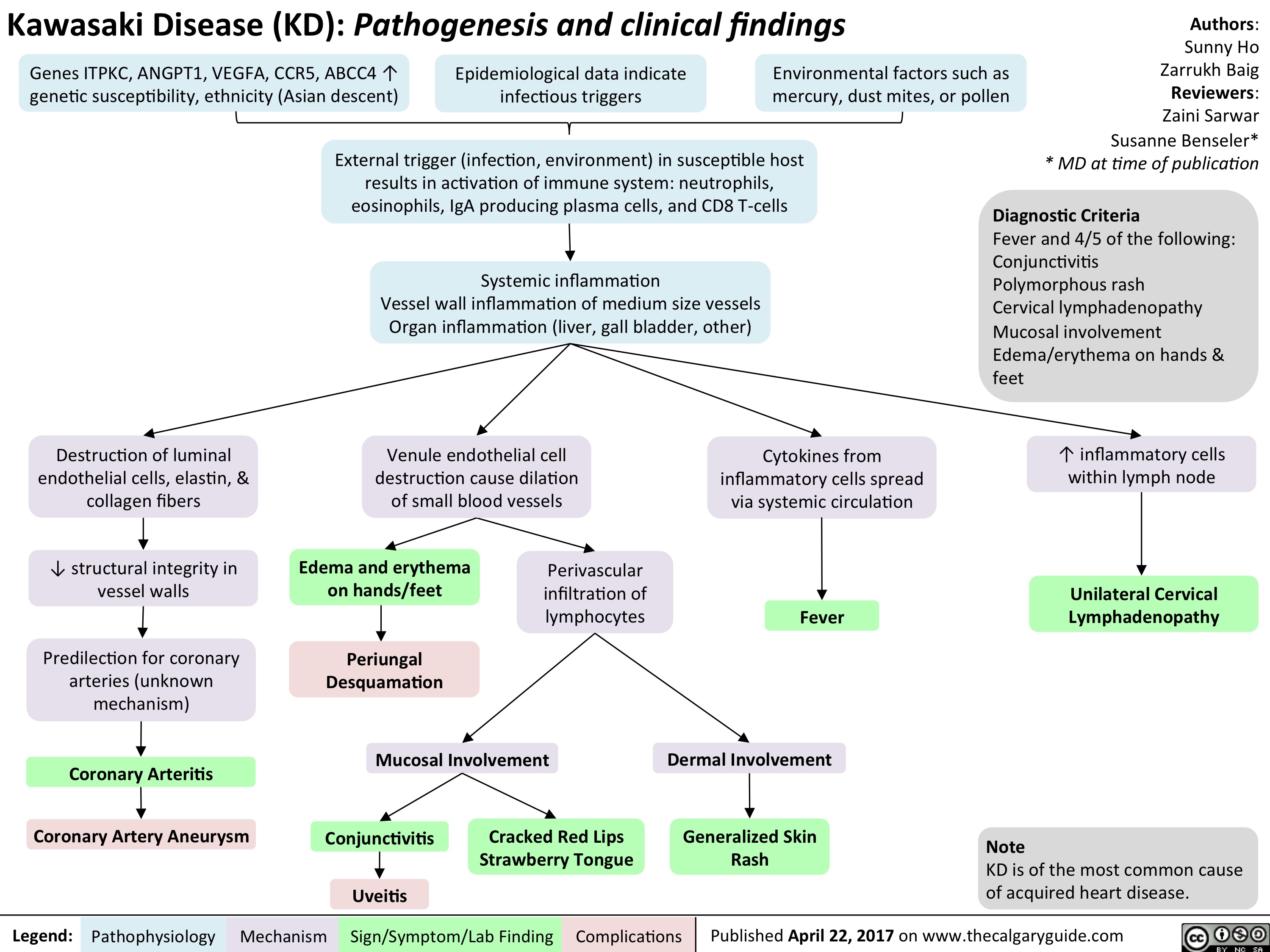
Secondary Hemostasis: Coagulation Cascade

systemic-lupus-erythematosus-gastrointestinal-manifestations

Critical Care Malnutrition
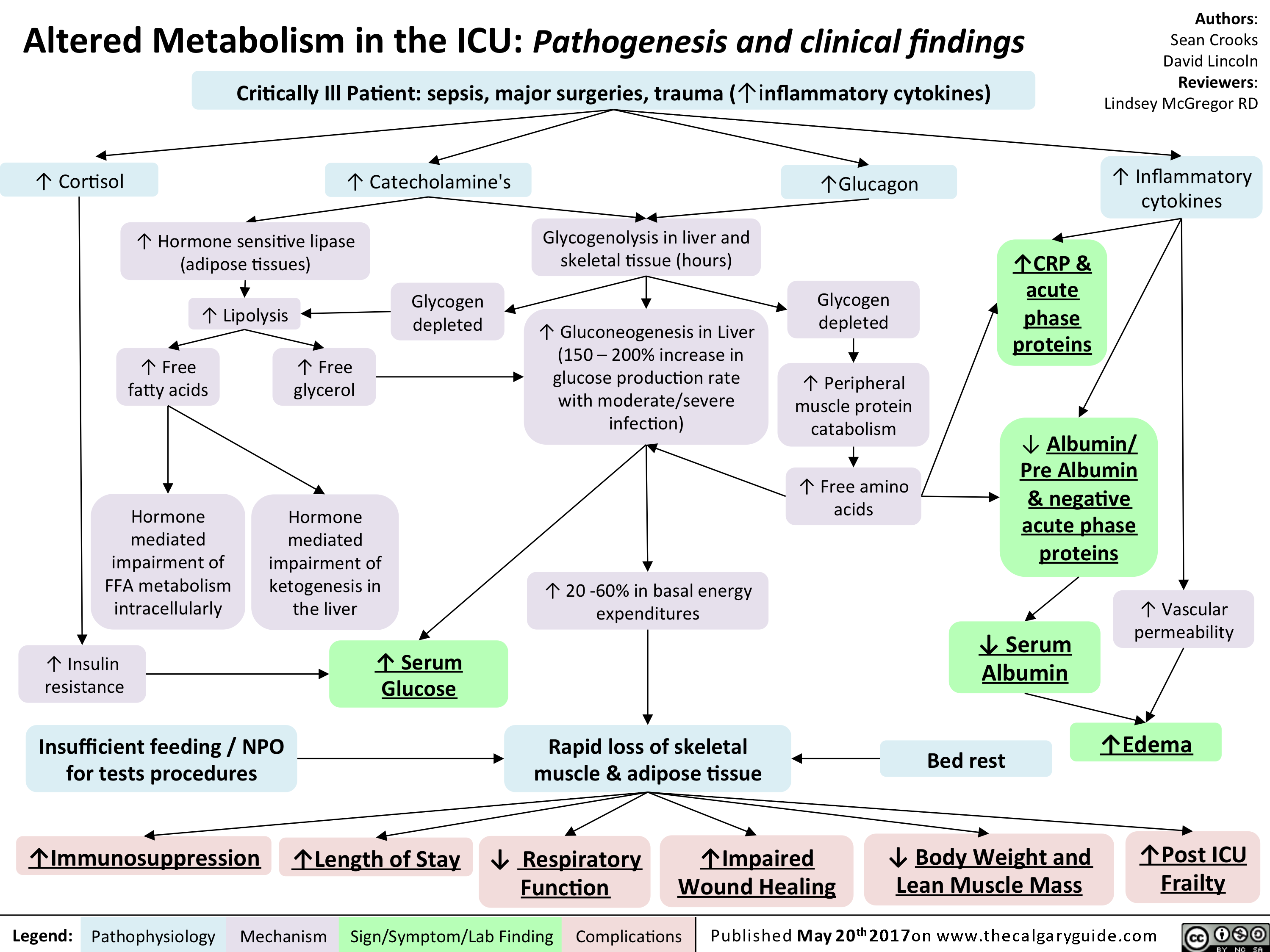
esophageal-gastric-varices

Legend:
Pathophysiology Mechanism
Sign/Symptom/Lab Finding
Complications
• Venous drainage of spleen backed up into gastric anastomoses
Tachycardia and hypotension
Anemia Death Melena Coffee ground emesis Hematemesis Bright red blood per rectum")
localized-pitting-edema

Authors: Sunny Fong Reviewers: Joseph Tropiano Adam Bass* * MD at time of publication
Central vein thrombosis
Deep vein thrombosis
T in venous pressure is transmitted to the capillaries
1` in capillary hydrostatic pressure
T in fluid extravasation from plasma into the interstitial space distal to site of obstruction or insufficiency
Localized Pitting Edema: Edema fixed at a specific anatomical site
Venous obstruction causes'` blood pooling distal to site of obstruction
Starling's Equation:
Net filtration gradient = LpS x ((Pap — Pint) Olcap
LpS = Porosity or permeability of the endothelial layer Pup = Capillary hydrostatic pressure Pint = Interstitial hydrostatic pressure ncap = Capillary oncotic pressure flint = Interstitial oncotic pressure
Note: An increase in net filtration gradient (eg. Increased capillary hydrostatic pressure or decreased capillary oncotic pressure) can lead to the formation of edema")
Neurotransmitters-and-Pharmacology-behind-Nausea-and-Vomiting - IN

2 Pusat lebih tinggi di SSP (korteks serebri, batang otak, hipotalamus, talamus)
Penyakit gerak atau labirin, menyebabkan ketidakcocokan sinyal antara sistem visual, vestibular, and proprioseptif
Obat-obatan (cth. kemoterapi, opiat, antibiotik)
Gangguan metabolik (cth. Gagal organ, hiperkalsemia)
toksin (cth. Dari bakteri penyebab keracunan makanan)
Peregangan (akibat stasis gaster, obstruksi organ berongga, tumor, konstipasi, atau distensi kapsuler organ padat — liver, pankreas)
Kompresi (dari organ sekitar, tumor, atau asites)
Jejas jaringan (dari peradanga, invasi tumor, kemoterapi, radiasi)
Legenda:
Sistem vestibular (telinga dalam)
Zona Pemicu Kemoreseptor (CTZ) (terletak di daerah ventrikel 4 tanpa sawar darah otak, secara konstan menyaring bahan kimia di darah)
Mekanisme kompleks dan belum dimengerti (mungkin dimediasi oleh banyak NT)
Tx: bicara dan dengarkan pasien, teknik relaksasi; lini-2: benzodiazepin untuk depresi SSP global, non-spesifik
Stimulasi langsung pusat muntah via efek massal
Tx: kortikosteroid, to 4, peradangan + tx penyebab (cth. pembedahan)
Sinyal mual/muntah ke pusat muntah dimediasi oleh Histamin (H1) dan Asetilkolin Muskarinik (ACh)
T
Tx: antagonis H1/ACh (cth. dimenhidrinat), Lini-2: antagonis ACh (cth. scopolamin)
Sinyal mual/muntah ke pusat muntah dimediasi Serotonin (5-HT) dan Dopamin (D2)
Tx: antagonis 5-HT (cth. ondansetron), antagonis D2 yang menembus sawar darah otak (cth. metoklopramid, bukan domperidon) + berhenti/kurangi obat penyebab
Kebanyakan m ua I a kibat stasis Tx: antagonis D2 gastrokinetik(cth. domperidon — saluran makan atas /lambung penetrasi SSP kurang - & metoklopramid. catatan: keduanya dikontraindikasikan pada obstruksi GI) + dimediasi Dopamin (D2) di CTZ tx penyebab
Patofisiologi Mekanisme
Saluran GI (termasuk liver & mesenterium)
Tanda/Gejala/Penunjang
Sinyal mual/muntah ke pusat muntah dimediasi terutama oleh Serotonin (5-HT) dan Dopamin (D2)
Tx: antagonis 5-HT (cth. ondansetron), antagonis D2 gastrokinetik (cth. metoklopramid, domperidon), fenotiazin antagonis D2 (i.e. haldol) + tx penyebab
Komplikasi
Penulis: Yan Yu* Penyunting: Laura Byford-Richardson Russell Loftus* Penerjemah: M Harmen Reza S* * MD (dokter) pada saat publikasi
Catatan: Kebanyakan jalur neurotransmiter pada mual/muntah masih belum diketahui. Tatalaksana yang tercantum di sini hanya bertujuan untuk menjelaskan prinsip umum terapi lini pertama. Terapi terapan pada pasien akan bergantung kepada diagnosis spesifik dan presentasi klinis.
Pusat Muntah
Koordinasi refleks muntah (dijelaskan di slide")
chronic-hypertensive-retinopathy-pathogenesis-and-clinical-findings
 • 1° HTN
Retinal Detachment
Vitreous Hemorrhage
Central/Branch Retinal Artery/Vein Occlusions
Risk Factors for 2° HTN (ex. Hyperaldosterone, Cushing's, Acromegaly, Chronic Kidney Disease, Obstructive Sleep Apnea, Diabetes Mellitus, Hypo/Hyper-thyroid, Adrenal Hyperplasia, Renal Artery Stenosis)
2° HTN
Ophthalmic Artery Hypertension ,17
Stage 1: Mild/vasoconstrictive
Stage 2: Moderate/sclerotic
Stage 3: Severe/exudative
Stage 4: Malignant
Abbreviations: • HTN — Hypertension • BRB — Blood-retinal barrier • RPE — Retinal pigment epithelium
Legend:
Pathophysiology
Mechanism
Acute and chronic vasospasm
Authors: Graeme Prosperi-Porta Reviewers: Stephanie Cote Usama Malik Johnathan Wong* * MD at time of publication
Diffuse and focal arterial narrowing and vascular tortuosity
Atherosclerosis and hyalinization causes arteriolar wall thickening resulting in a diffuse light reflex appearing red-brown coloured
Thickening of the arteriolar wall and/or sclerotic thickening at the arteriole/venule crossing compresses the underlying venule
BRB breakdown causes dot/blot hemorrhages in the inner retina and flame hemorrhages in the nerve fiber layer
Serum proteins and lipids leakage from damaged BRB appears as white or yellow areas with sharp margins
Occlusion of the terminal retinal arterioles causes fluffy white ischemic lesions in the inner retinal nerve fiber layer
Hyper-pigmented patches surrounded by a hypo-pigmented ring due to RPE clumping around atrophic areas in the choroid
Sign/Symptom/Lab Finding
lschemia of optic disc arterioles causes optic nerve swelling and blurred disc margins. Leakage of optic disc arterioles causes hemorrhage and disc edema.
Complications
Copper Wiring
AV nicking
Retinal Hemorrhages
Yellow Hard Exudates
Cotton-wool Spots
Elschnig's Spots
Papilledema")
central-retinal-artery-occlusion-pathogenesis-and-clinical-findings
 (i.e. Valvular, arrhythmias, congenital defects) (i.e. OCP, Protein C&S deficiency, ATIII deficiency) (i.e. leukemia/lymphoma, sickle cell, polycythemia) Endothelial cell damage Abnormal blood flow 1` coagulation and/or 1 blood viscosity and creates hypercoagulable state causing localized stasis 4, anti-coagulation inflammation
Abbreviations: • GCA — Giant Cell Arteritis • SLE — Systemic Lupus Erythematosus • GPA — granulomatosis with polyangitis • OCP — Oral contraceptive pill • ATIII — Anti-thrombin Ill
Thrombus formation
Blockage of central retinal artery
Central Retinal Artery Occlusion (CRAO)
Authors: Graeme Prosperi-Porta Reviewers: Stephanie Cote Usama Malik Johnathan Wong* * MD at time of publication Carotid Artery Atherosclerosis
Atherosclerotic plaque dislodges from carotid artery
The retina becomes pale 4, perfusion of retinal Slow retinal artery blood Acute retinal edema Ganglion cells and axons from NI, perfusion arterioles due to upstream flow allows for caused by ischemia results death due to ischemia CRAO segmentation of the blood column in a blurred appearance of the retina results in disc pallor seen months after CRAO The choroidal vessels supplying the macula via the posterior ciliary artery become more prominent within a background of retinal pallor")
Fragile X Syndrome
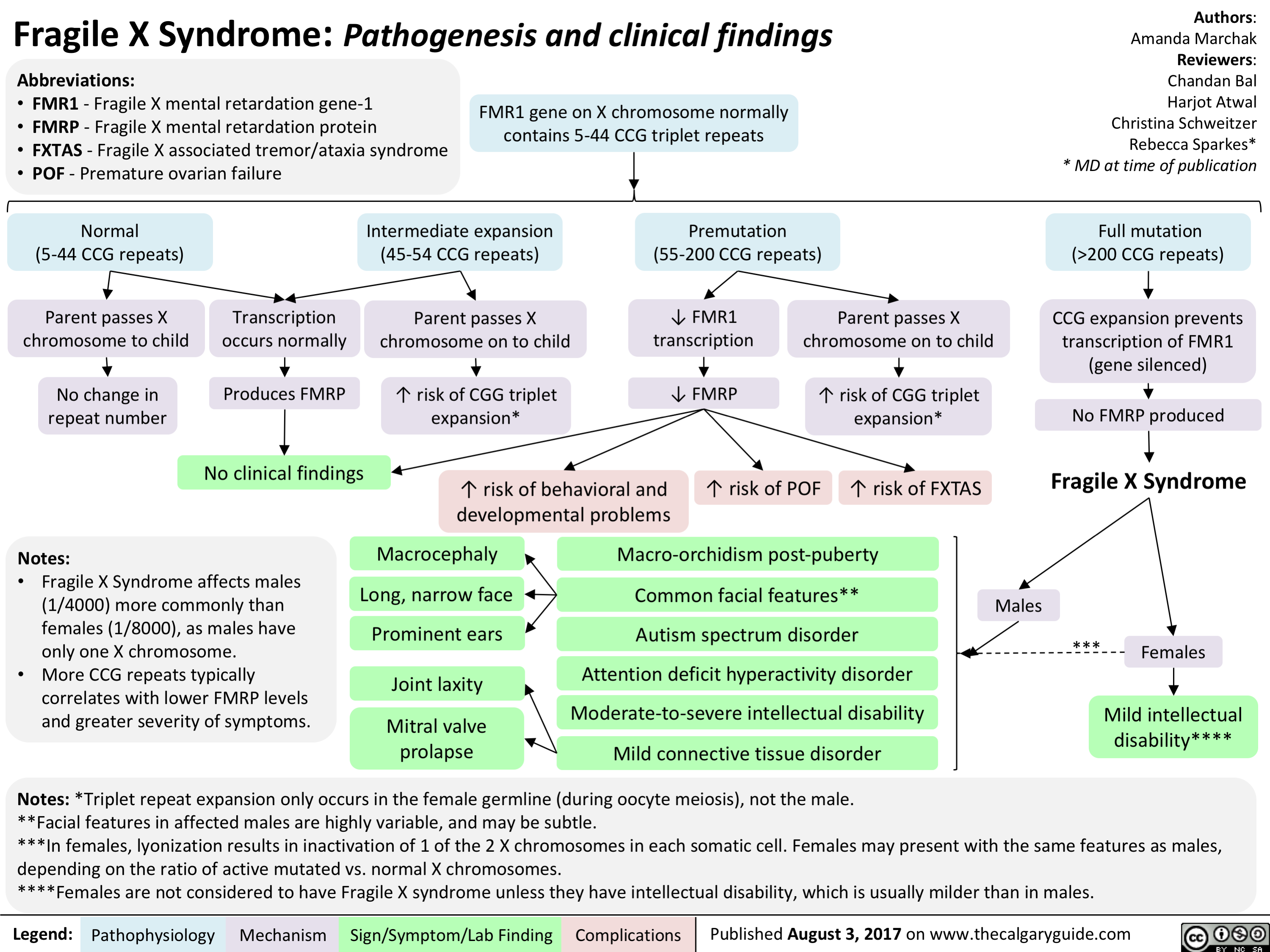
Attention Deficit Hyperactivity Disorder (ADHD)

Oppositional Defiant Disorder (ODD)
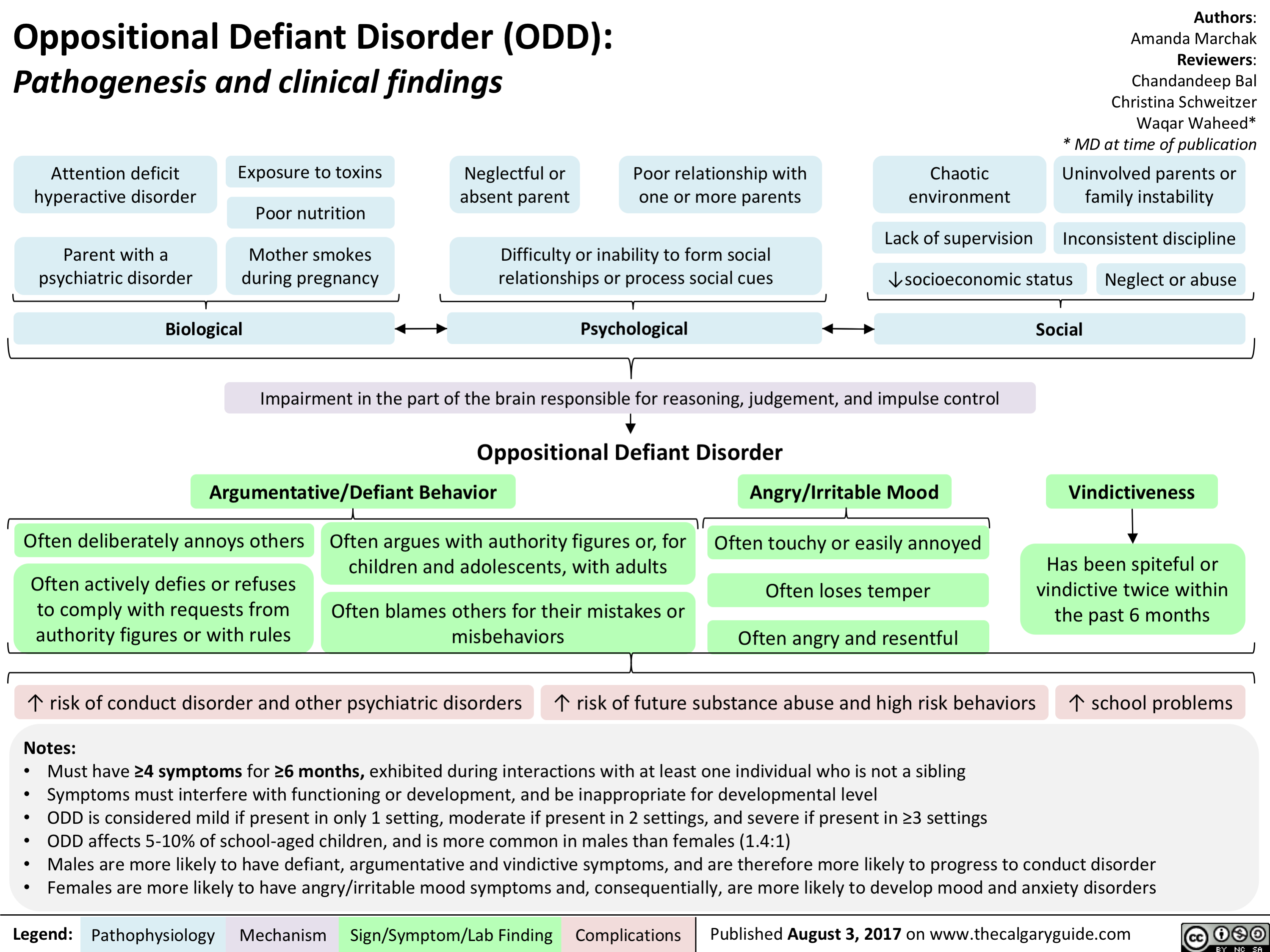
Conduct Disorder (CD)
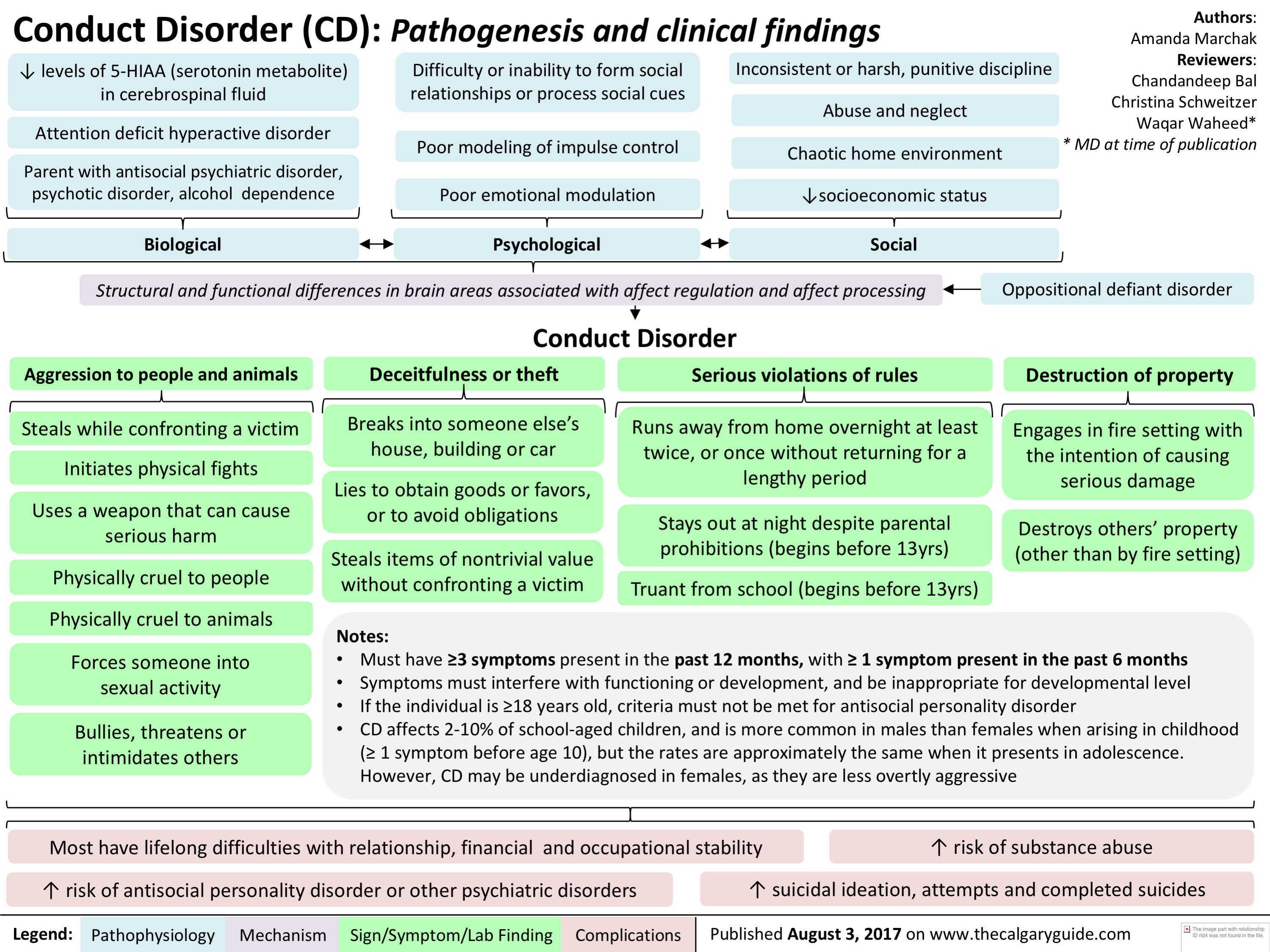
Down Syndrome

infectious-esophagitis-pathogenesis-and-clinical-findings

Herpes Simplex Infection of squamous cells and macrophages
Colonization facilitated by use of antacid therapy
Nuclear Large Superficial Squamous Macrophage inclusion bodies esophageal ulceration ulcers cell inclusion bodies aggregation Legend: Pathophysiology Mechanism Sign/Symptom/Lab Finding Complications
Spores and pseudohyphae seen on biopsy
• Invas.on of underlying blood vessels
• White plaques over erythematous base
'1' neutrophils due to inflammatory response")
The Neuroanatomy and Physiology of Emotion
 Processes higher order emotions that 1— require recall, reason, and decision making (e.g., embarrassment) Integrates sensory information from the viscera in response to the inciting •
environmental object or event with information from the amygdala
Emotional feeling (i.e., conscious experience of emotion)
Thalamus
Emotionally salient environmental stimulus
Sensory input (e.g., vision, touch, smell, hearing, taste)
Hypothalamus ► (major output of the limbic system)
Olfaction*
Primary and Association Cortices •
Amygdala Processes lower order emotions that have fast, automatic, and conditioned responses (e.g., fear) Participates in formation and retrieval of emotionally relevant memories from neocortical areas, attaching emotional significance to sensory input
Hippocampus (key for memory consolidation and retrieval)
► Motor, Endocrine and Visceral systems
Notes: • The structures involved in emotions are defined as the limbic system • The thalamus, basal ganglia and PFC together form the corticostriatalthalmocortical loop which comprises the major emotional processing pathway in the brain • The Prefrontal Cortex (PFC) includes the orbitofrontal cortex (OFC), dorsolateral prefrontal cortex and anterior cingulate cortex • *Olfaction is the only sense that bypasses the thalamus and connects directly to the primary olfactory cortex
Legend: Pathophysiology Mechanism
Sign/Symptom/Lab Finding
Authors: Andrea Moir Reviewers: Erika Russell Usama Malik Brienne McLane* * MD at time of publication
Autonomic Nervous System (ANS)
The ANS automatically and unconsciously responds to the emotional stimulus (e.g., 11` HR, blushing)
Physical response to emotional stimulus Visceral sensation
Sensory receptors are stimulated by the ANS response and transmit this information to the somatosensory and insular cortices")
Serotonin Syndrome Pathogenesis and Clinical Findings
, opioids, antiemetic agents, triptans, weight loss agents, drugs of abuse (e.g. cocaine, amphetamines)
Therapeutic drug use
• Drug interactions (esp. combo of serotonergic agents) Serotonin Syndrome Variable combination of mental status changes, autonomic instability, and neuromuscular hyperactivity ranging from mild to life-threatening with an abrupt onset (within minutes to hours) after medication ingestion and most cases resolving within 24 hours of cessation of offending medication
Intentional self-poisoning
Excessive serotonergic activity at 5-HT receptors centrally (brainstem) and peripherally
serotonin synthesis and release
serotonin reuptake and metabolism
IN receptor agonism and sensitivity
4,
Drug-induced changes in the relative ratio of non-serotonergic neurotransmitters (e.g. increase in noradrenaline)
Altered Mental Status
Anxiety, confusion, agitation, hypervigilance, pressured speech, delirium, coma
Autonomic Instability
Shivering, diaphoresis, fever, diarrhea, tachycardia, mydriasis, hypertension
Authors: Preeti Kar Reviewers: Erika Russell Usama Malik Aaron Mackie* * MD at time of publication
Notes: The Hunter Serotonin Toxicity Criteria is used to make a clinical diagnosis • History of serotonergic agent taken within past 5 weeks + any of the following clinical features: • Spontaneous clonus • Inducible clonus and either agitation or diaphoresis • Ocular clonus and either agitation or diaphoresis • Tremor and hyperreflexia • Hypertonia, temperature > 38 °C, and either ocular clonus or inducible clonus
Neuromuscular Hyperactivity
Hyperreflexia, muscle rigidity (esp. lower extremities), myoclonus, tremor, incoordination, trismus*, opisthotonus*, ocular clonus*, seizures
*Notes: • Trismus or lockjaw, is the reduced opening of the jaw • Opisthotonus is an abnormal body position where the person is usually rigid and arches their back, with their head thrown backwards • Ocular Clonus is rhythmic or equal movements of both eyes; should be distinguished from nystagmus which has a fast and slow component
Legend: Pathophysiology Mechanism
Sign/Symptom/Lab Finding
Abbreviations: • 5-HT = serotonin • SSRI = selective serotonin reuptake inhibitors • SNRI = selective noradrenaline reuptake inhibitors • MOAI = monoamine oxidase inhibitors • TCA = tricyclic antidepressants")
Side Effects of ACEi/ARBs During Pregnancy

Side Effects of Methimazole During Pregnancy
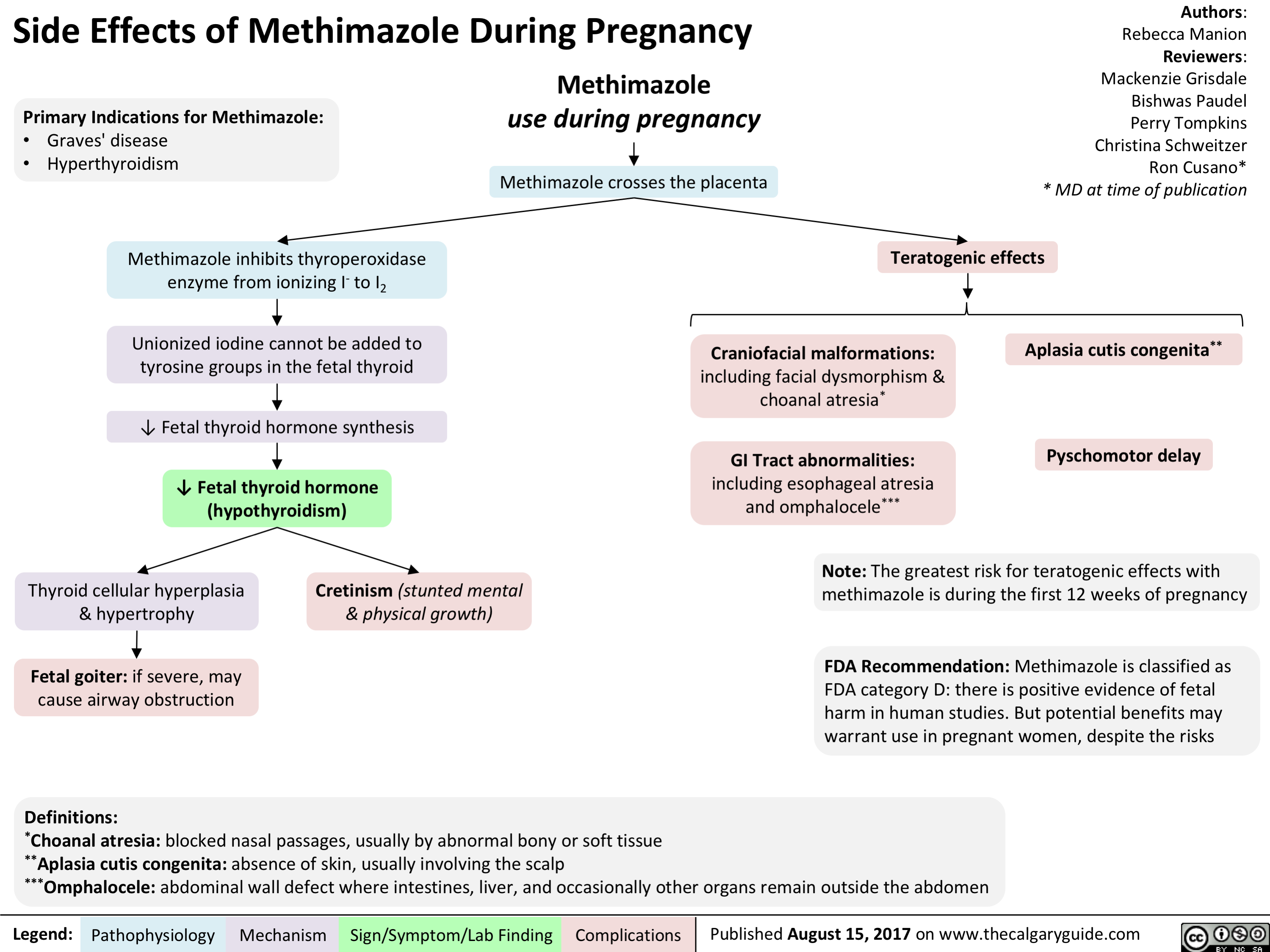
Side Effects of NSAIDs During Pregnancy

Side Effects of Valproic Acid During Pregnancy
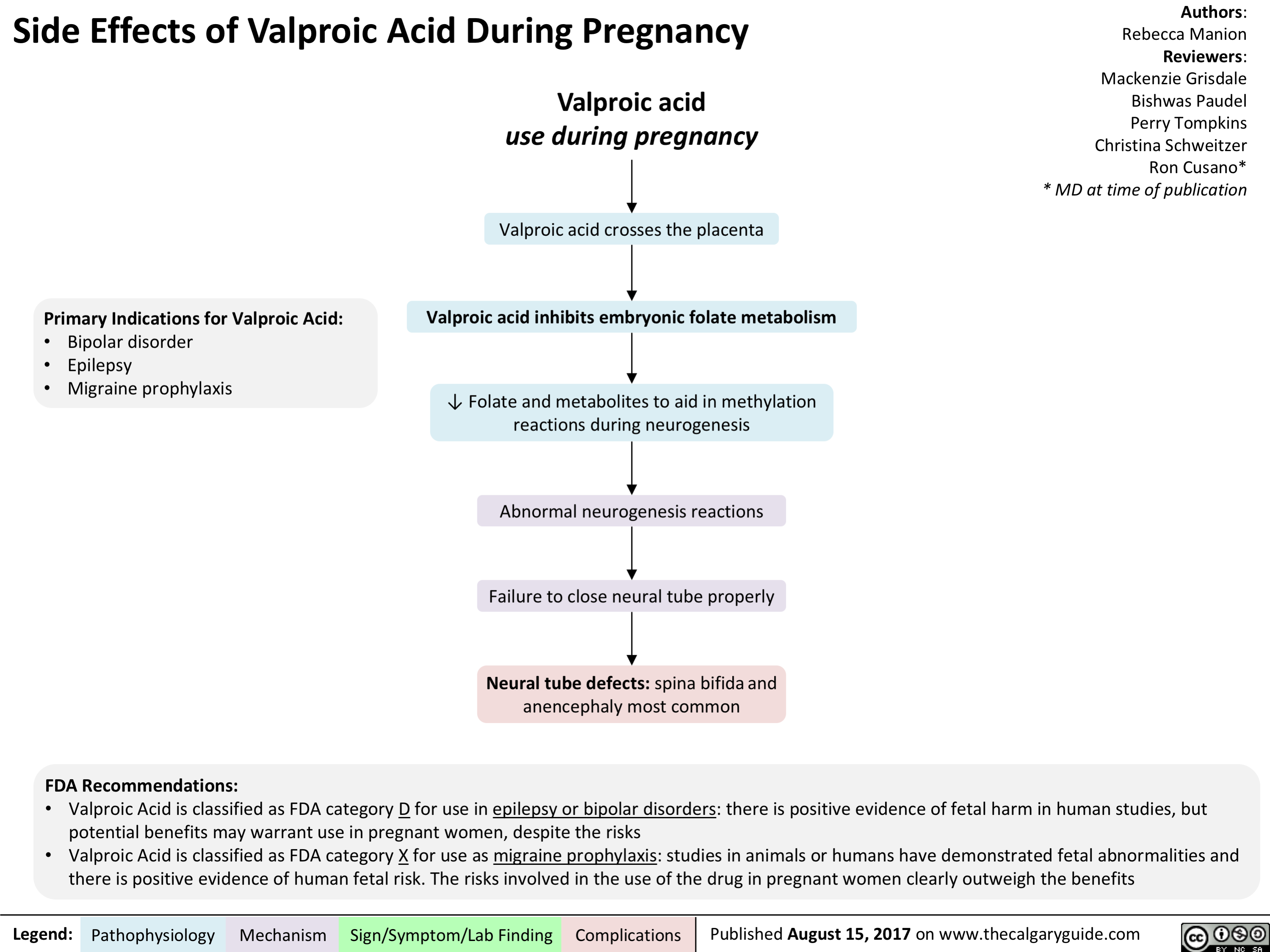
Side Effects of Warfarin During Pregnancy
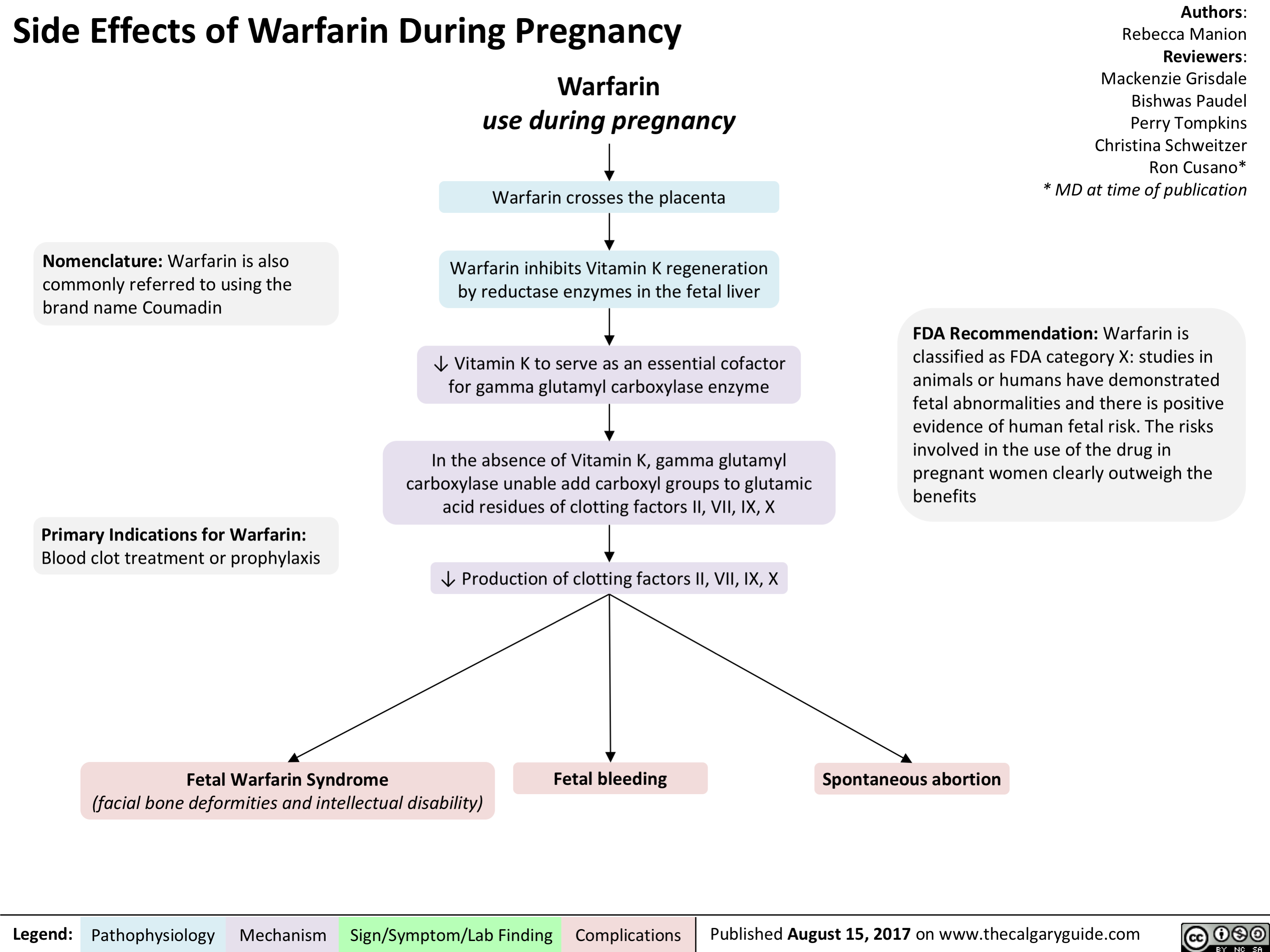
Pediatric Parasomnias and Nightmares: Pathogenesis and clinical findings

Hemolytic Disease of the Fetus and Newborn
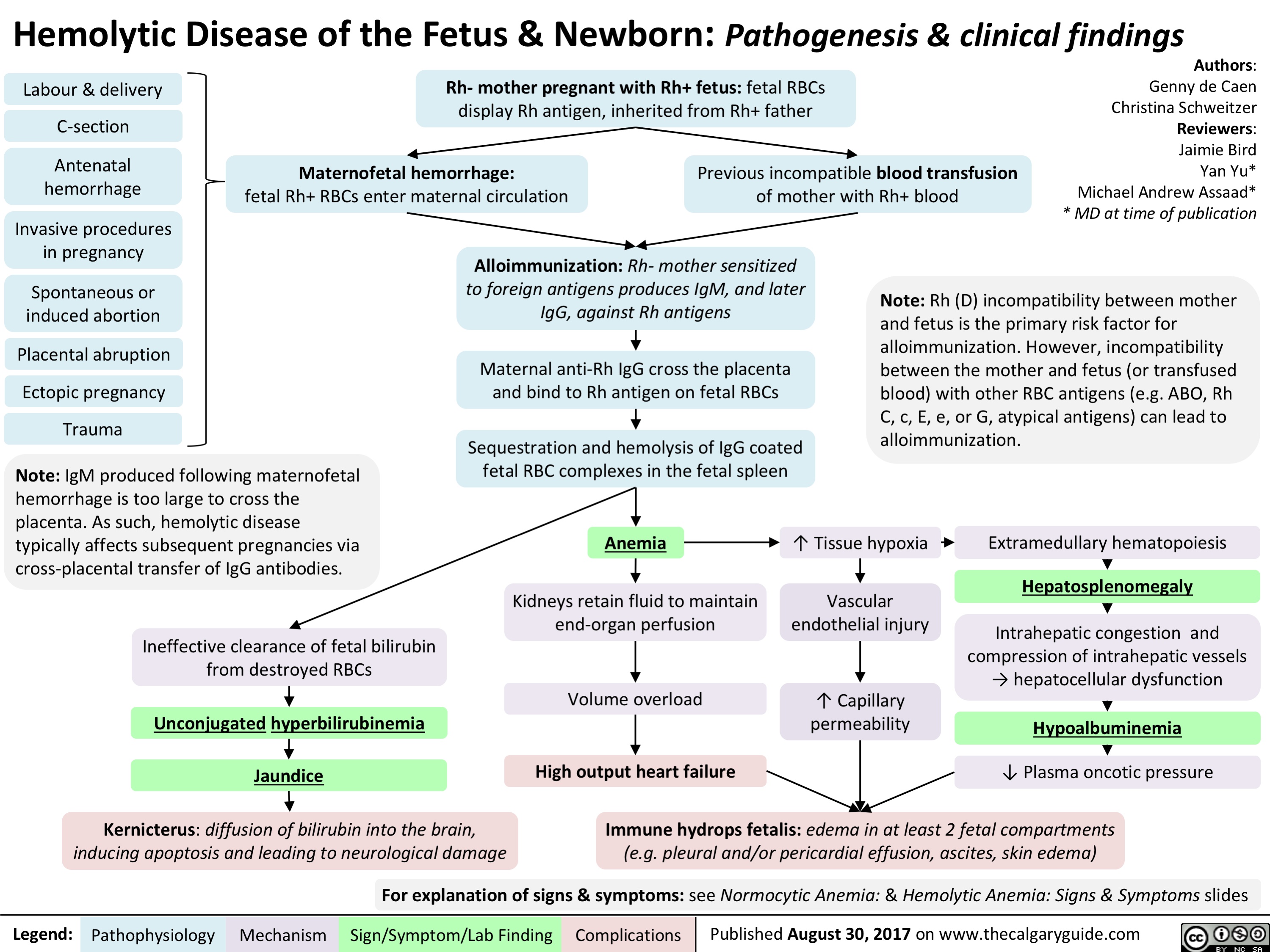
Trigeminal Neuralgia
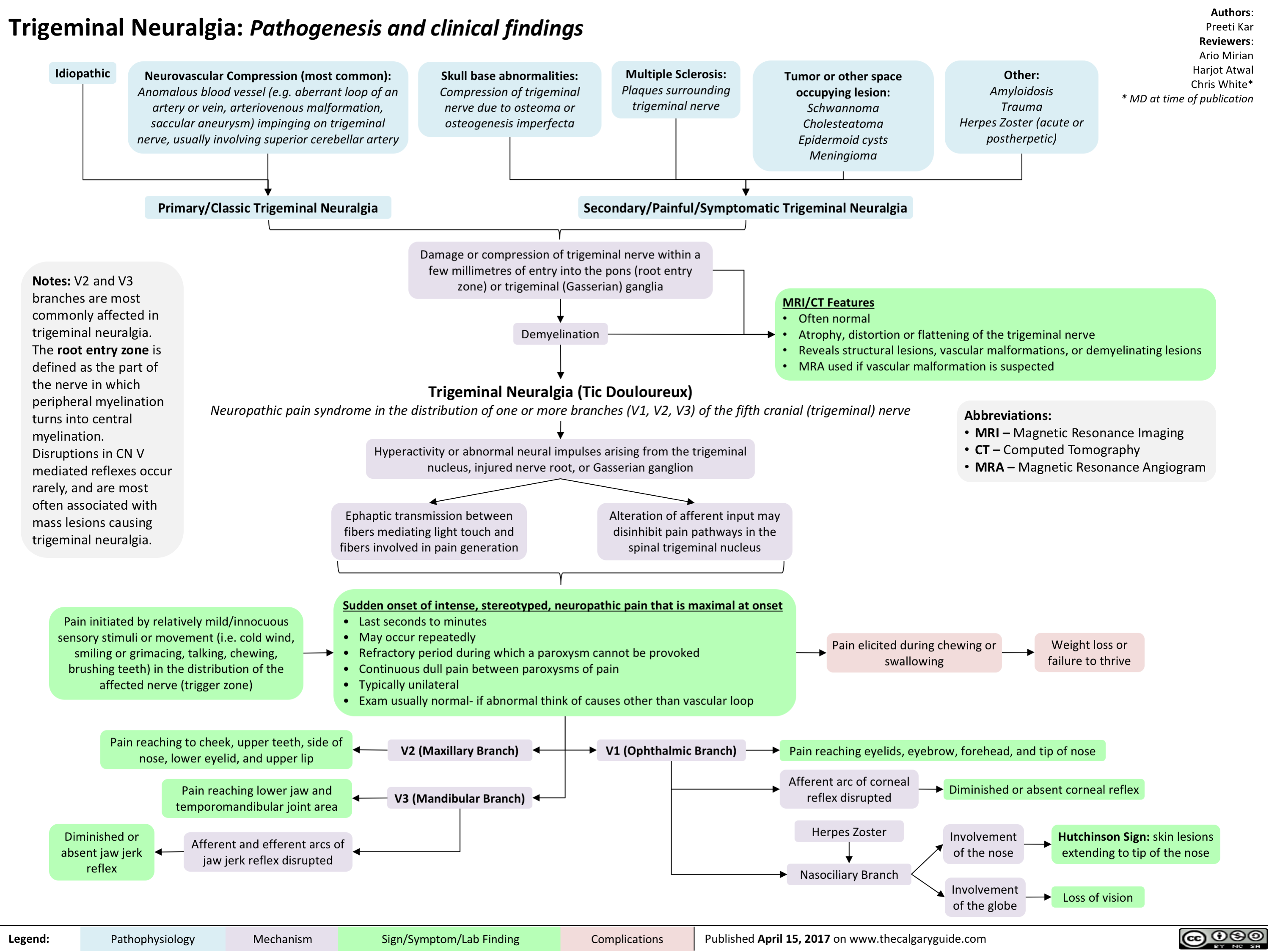
Mallory-Weiss Tear
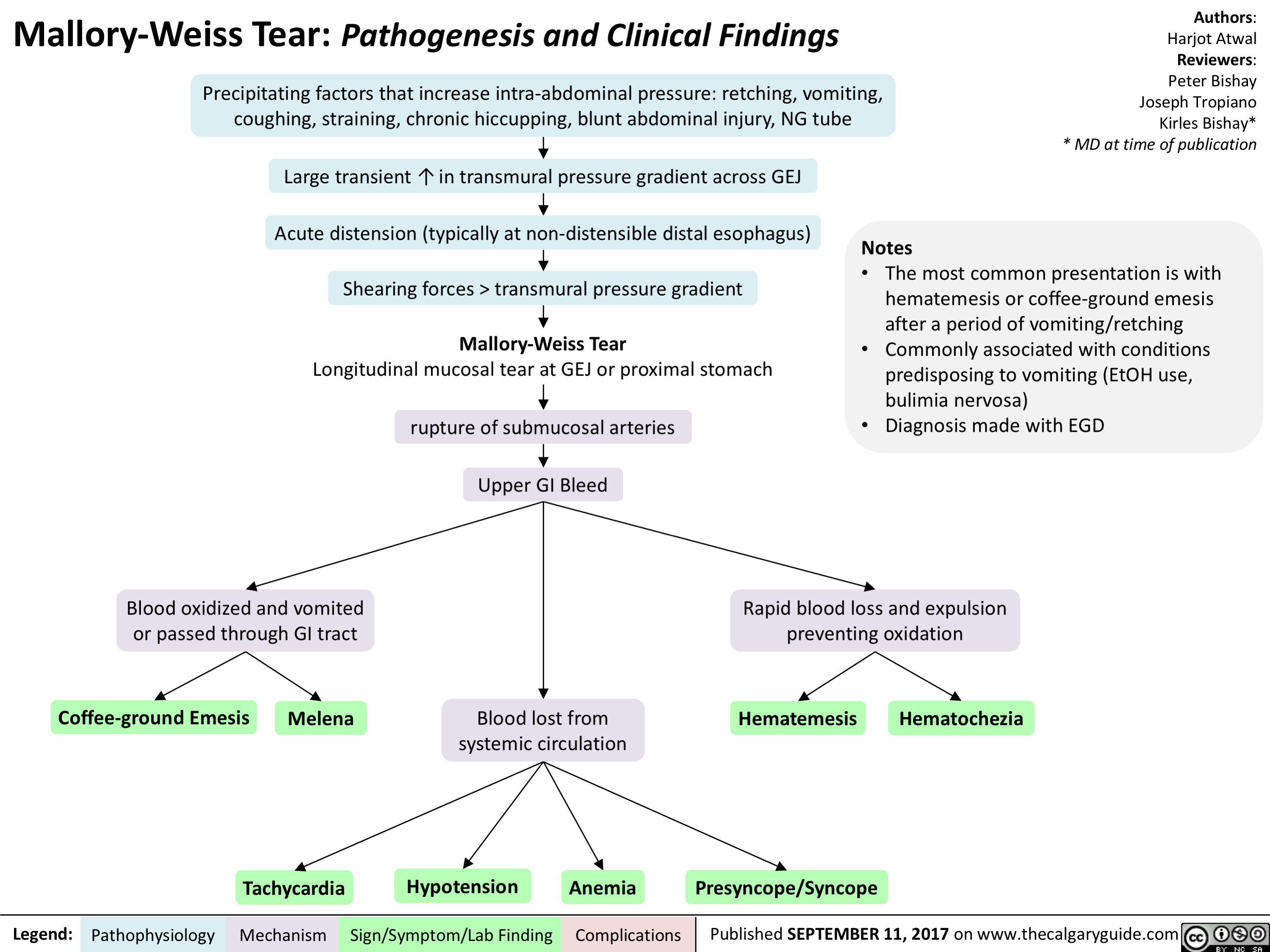
Unconjugated Neonatal Hyperbilirubinemia - Complications

pathogenesis-of-select-causes-of-constipation-in-adults-and-in-elderly
 Hypercalcemia) Hypothyroid-ism) Sclerosis) Scleroderma) Analgesics)
Mechanical I`Ca2+ = 4, Na+ Thyroid Demyelination Collagen Puborectalis Disturbance in obstruction in permeability in hormone of CNS neurons deposits into muscle and the gut-brain the bowel neurons deficiency colonic mucosa, leading to external anal sphincter fail interaction fibrosis of the gut wall to relax Interrupted 4, Excitability Possible Dysfunction of Narrowed Mechanism flow of bowel contents and tone of bowel smooth mechanisms: hormone autonomic nerves that anorectal angle and unknown, many Atrophy of the muscle receptor supply smooth muscle '`pressure of pathways changes, involuntary wall of the colon anal canal neuromuscular disorders, myopathy from bodily functions 1, Peristalsis of infiltration of 4, Ability of the Evacuation Visceral the bowel colon to contract of feces is hypersensitivity Abbreviations: the intestinal wall 4, Digestion less effective and 4, colonic and colonic motility motor • IBS-C: Irritable Bowel Syndrome with predominant constipation • CNS: Central Nervous System 4, Peristalsis of response after a meal the bowel
Opioids bind to Lt-opioid receptors on gut wall
Inhibition of excitatory neural pathways within the enteric nervous system
1, Peristaltic contractions
1
l• Colonic transit time")
1st gen antipsychotics (Slovenian translation) - FINAL VERSION
 primera: haloperidol, klorpromazin
antagonisti ACh M1 receptorjev zavirajo delovanje ACh po celem telesu (v ustih, prebavilih, o6eh, mo2ganih)
zaprtje
suha usta
zamegljen vid
kognitivna upolasnjenost
Legenda:
antagonisti histaminskih H1 receptorjev zavirajo delo-vanje histami-na v mo2ganih zaspanost
porast tel. teie
antagonisti adrenoceptorjev dilatacija gladkih miSic v stenah arteriol - te2ave pri vzdrievanju krvnega tlaka
ortostatska hipotenzija
mezolimbiZna pot VTA 4 limbicni sistem
mezokortikalna pot VTA prefrontalna skorja
tuberoinfundibularna pot hipotalamus 4 hipofiza
nigrostriatna pot substanca nigra 4 striatum
1
ekstrapiramidni simptomi (EPS)
avtorica: Sara Meunier pregledala: Yan Yu, Aaron Mackie*
* dr. med. ob objavi
4, pozitivnih simptomov terapevtski ueinek
4, odziv nagrajevalne poti
negativnih simptomov 11• kognitivnih simptomov teiave s pozornostjo in u6enjem
blokada DA vodi v izlaanje prolaktina iz adenohipofize
prevedel in priredil: Jan Kejiar, dr. med., specializant psihiatrije pregledala: doc. dr. Brigita Novak Sarotar, dr. med., spec. psih.
4, halucinacij
4, blodenj
otopelost
anhedonija
4. zanimanja za socialne stike
amenoreja
galaktoreja
okrajgave: VTA - ventral no tegmental no podraje DA - dopamin ACh - acetilholin
tardivna diskinezija Nenormalni asimetri6ni gibi obraznih mrgic, jezika in/ali udov, povzro6eni s kroni6no dopaminsko blokado. Letna incidenca pribliino 5 %, potencialno ireverzibilna!
patofiziologija mehanizem
iatrogeni parkinsonizem pojav zobatega kolesa, nestabilnost v drii, tremor v mirovanju, bradikinezija/akinezija
znak/simptom/laboratorijska najdba
akutna distonija kra migic vratu, okrog ob, jezikageljusti
akatizra obL'utek motorie'nega nemira
zaplet Objavljeno 15. junija 2017 na www.thecalgaryguide.com.")
2nd gen antipsychotics (Slovenian translation) - FINAL VERSION
: primeri: klozapin, olanzapin, kvetiapin, risperidon, paliperidon itd.
antagonisti dopaminskih D2 receptorjev zavirajo delovanje DA na D2 receptorjih po celotnih mo2ganih
antagonisti serotoninskih 2A receptorjev zavirajo delovanje 5-HT na 5-HT2A receptorjih po celotnih mo2ganih
antagonisti serotoninskih 2C receptorjev klozapin, olanzapin, kvetiapin
antagonisti ACh M1 receptorjev zavirajo delovanje ACh po telesu (v ustih, prebavilih, o6eh, mo2ganih)
antagonisti aradrenoceptorjev v oiilju dilatacija gladkih migic v stenah arteriol
antagonisti histaminskih H1 receptorjev zavirajo delovanje histamina po telesu
sano-presnovni kink' mehanizem neznan
Legenda:
patofiziologija mehanizem
Pomni: posledica velikih razlik v afiniteti za receptorska vezavna mesta so svojevrstni terapevtski in varnostni profili atipicnih a nti psi hoti kov. Kloza pi n med vsemi velja za najud nkovitejSega, a i ma tudi najve6 neZelenih uC'inkov, vkljUuja agranulocitozo (0,5-2 %). Tako predstavlja terapijo drugega izbora, potrebno je red no spremljanje bolnikove krvne slike.
mezolimbiEna pot DA blokada > 5-HT blokado
nigrostriatna pot 5-HT blokada > DA blokado
4, pozitivnih simptomov terapevtski ueinek
blokada 5-HT vodi v sprokanje DA v striatumu
tubero- blokada 5-HT zavre sprokanje infundibularna pot prolaktina v adenohipofizi
mezokortikalna pot
blokada 5-HT2c receptorjev stimulira sprokanje DA in NA v prefrontalni skorji
zamegljen vid
kognitivna upolasnjenost
sposobnost vzdrievanja krvnega tlaka
omotica
apetit
T tel. tee
ortostatska hipotenzija
tveganje za debelost
trigliceridi na tee
inzulinska odpornost —■ diabetes tipa 2
znak/simptom/laboratorijska najdba
4, halucinacii
blodeni
avtorica: Sara Meunier pregledala: Yan Yu, Aaron Mackie*
* dr. med. ob objavi
prevedel in priredil: Jan KejZar, dr. med., specializant psihiatrije pregledala: doc. dr. Brigita Novak Sarotar, dr. med., spec. psih.
4, ekstrapiramidnih simptomov (EPS)*
4, hiperprolaktinemije*
prokognitivni in antidepresivni ainki
4, kognitivnih simptomov* 4, negativnih simptomov*
* Glede na anti-psihotike prve generacije, glej ustreznogradivo!
okrajgave: 5-HT - serotonin DA - dopamin NA - noradrenalin ACh - acetilholin
visoko presnovno tveganje: klozapin, olanzapin zmerno presnovno tveganje: risperidon, kvetiapin nizko presnovnotveganje:antipsihotikitretjegeneracije
1 tveganje za diabeti6no ketoacidozo pri bolnikih z visokim tveganjem tveganje za srbo-iilne dogodke tveganje za prezgodnjo smrt
zaplet Objavljeno 15. junija 2017 na www.thecalgaryguide.com.")
3rd gen antipsychotics (Slovenian translation) - FINAL VERSION
 primer: aripiprazol
delni agonist dopaminskih D2 receptorjev Veie se na D2 receptor, se hitro sprosti in znova veie. Ta ponavljajoc vzorec receptorske vezave molekulam DA v sinapsah ne pusti, da bi se vezale na D2 receptorje.
4,
prepre6uje vikk DA zaradi preve6 stimuliranih receptorjev in primanjkljaj DA zaradi premalo stimuliranih receptorjev")
Alcohol Use Disorder (Slovenian translation) - FINAL VERSION
 sprokanje endogenih opioidov skorji in mezolimbibem dopaminskem sistemu)
etanol dose2e dano koncentracijo v krvi hitreje (tj. v 6asa)
toleranca (zmanjgana ob'6utljivost na ainke etanola)
/1` vnos etanola za dosego enakih u6inkov
socialna okrnjenost
nezmoZnost izpolnjevanja obvez (na delu, v Soli, doma)
nadaljevanje (zlo)rabe kljub druZbenim problemom, povzro6enih z alkoholom
kljuthih druthenih, poklicnih in rekreativnih aktivnosti
•
tvegana uporaba substance
ponavljajo6e se raba alkohola v 'gkodIjivih situacijah
nadaljevanje rabe navkljub poznavanju posledic
Pomni: v DSM-5 sta prej lo6eni entiteti zloraba alkohola in odvisnost od alkohola zdruZeni v motnjo rabe alkohola (an. alcohol use disorder). Zloraba alkohola je bila prej definirana kot pitje navkljub ponavljajo6im se druZbenim in medosebnim teZavam ter teZavam z zakonom, odvisnost pa je predstavljala „podaljSek")
BMR (Slovenian translation) - FINAL VERSION
 )
Pomni: • Bipolar motnja tipa 1: maniba epizoda in/ali depresivna epizoda: priblrino 10 epizod manije in depresije v 2ivljenju bolnika v primeru nezdravljene bolezni • Bipolar motnja tipa 2: hipomaniba epizoda in depresivna epizoda • hipomaniba epizoda predstavlja bla2jo obliko manije, tj. brez funkcionalne okrnjenosti, brez psihotibih simptomov, brez hospitalizacije in v trajanju vsaj 4 zaporednih dni
znak/simptom/laboratorijska najdba
avtorici: Briana Cassetta, Sara Meunier pregledali: Yan Yu, Alexander Arnold, Philip Stokes*
* dr. med. ob objavi
prevedel in priredil: Jan Kejiar, dr. med., specializant psihiatrije pregledala: doc. dr. Brigita Novak Sarotar, dr. med., spec. psih.
depresivna epizoda najmanj 2 tedna
depresivno razpoloienje
socialnega urriika
obEutkov krivde, tesnobe
brezupa
samomorilnih misli in vedenja
anhedonija
teiave s pozornostjo
A v spanju, utrujenost
A tel. teie, apetita
psihomotorna agitira nost ali upolasnjenost
zaplet Objavljeno 30. junija 2017 na www.thecalgaryguide.com.")
Bupropion (Slovenian translation) - FINAL VERSION
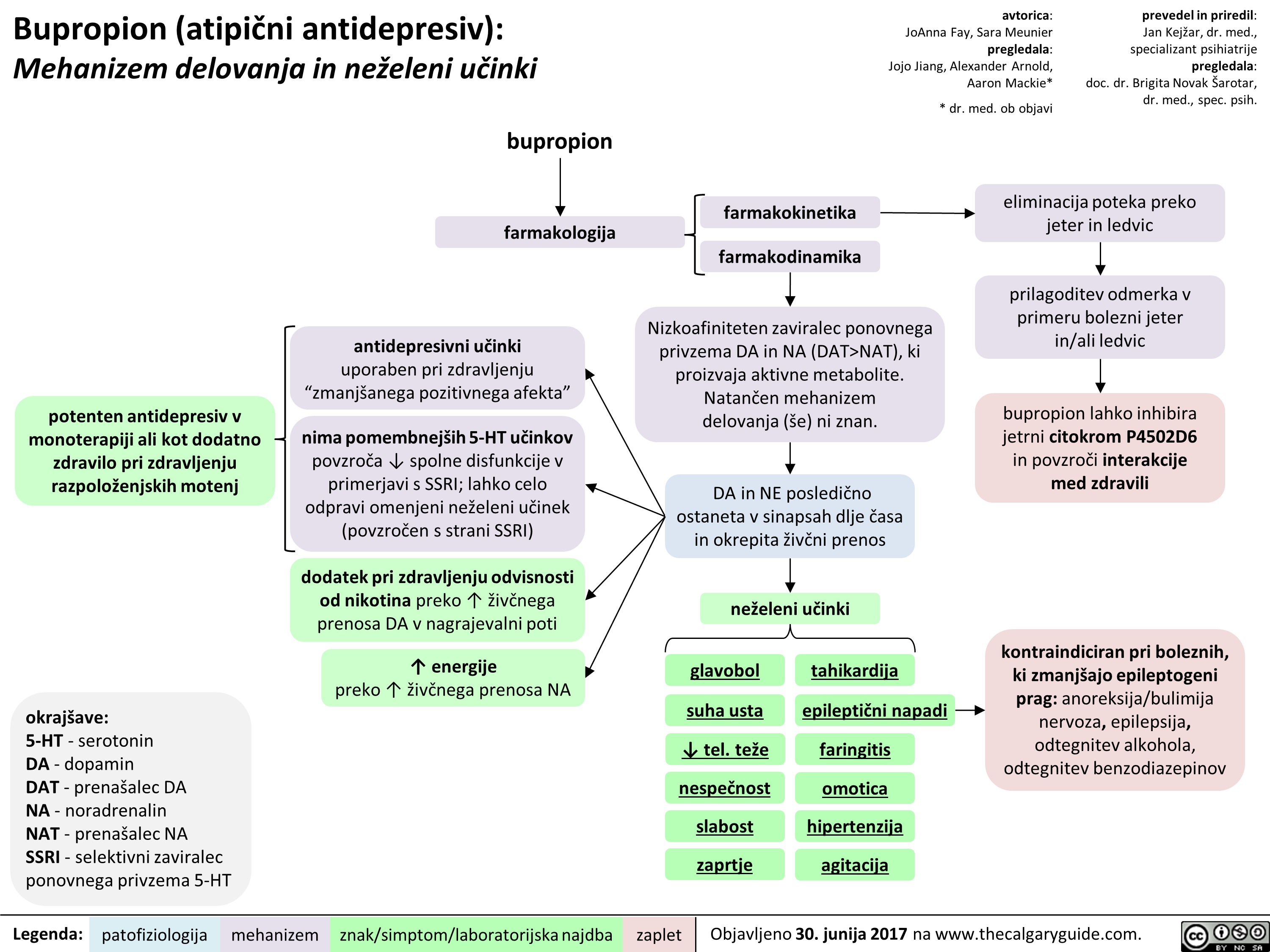 NAT), ki proizvaja aktivne metabolite. Natan'6en mehanizem delovanja (se) ni znan.
znak/simptom/laboratorijska najdba
DA in NE posledi6no ostaneta v sinapsah dlje Casa in okrepita 2iv6ni prenos
neieleni udnki
glavobol
suha usta
4, tel. tee nespeEnost slabost zaprtie
tahikardija
epileptiEni napadi
faringitis
omotica hipertenziia agitacija
prevedel in priredil: Jan Kejiar, dr. med., specializant psihiatrije pregledala: doc. dr. Brigita Novak Sarotar, dr. med., spec. psih.
eliminacija poteka preko jeter in ledvic
prilagoditev odmerka v primeru bolezni jeter in/ali ledvic
bupropion lahko inhibira jetrni citokrom P4502D6 in povzrod interakcije med zdravili
kontraindiciran pri boleznih, ki zmanIgajo epileptogeni prag: anoreksija/bulimija nervoza, epilepsija, odtegnitev alkohola, odtegnitev benzodiazepinov
zaplet Objavljeno 30. junija 2017 na www.thecalgaryguide.com.
" title="Bupropion (atipiEni antidepresiv): Mehanizem delovanja in neieleni utinki
potenten antidepresiv v monoterapiji all kot dodatno zdravilo pri zdravljenju razpoloienjskih motenj
okrajgave: 5-HT - serotonin DA - dopamin DAT - prenagalec DA NA - noradrenalin NAT - prenagalec NA SSRI - selektivni zaviralec ponovnega privzema 5-HT
Legenda:
bupropion
farmakologija
antidepresivni udnki uporaben pri zdravljenju "zmaniganega pozitivnega afekta"
nima pomembnelgih 5-HT udnkov povzraa spolne disfunkcije v primerjavi s SSRI; lahko celo odpravi omenjeni neieleni udnek (povzraen s strani SSRI)
dodatek pri zdravljenju odvisnosti od nikotina preko T iive'nega prenosa DA v nagrajevalni poti
energije preko T 2ivbega prenosa NA
patofiziologija mehanizem
farmakokinetika
farmakodinamika
avtorica: JoAnna Fay, Sara Meunier pregledala: Jojo Jiang, Alexander Arnold, Aaron Mackie*
* dr. med. ob objavi
Nizkoafiniteten zaviralec ponovnega privzema DA in NA (DAT>NAT), ki proizvaja aktivne metabolite. Natan'6en mehanizem delovanja (se) ni znan.
znak/simptom/laboratorijska najdba
DA in NE posledi6no ostaneta v sinapsah dlje Casa in okrepita 2iv6ni prenos
neieleni udnki
glavobol
suha usta
4, tel. tee nespeEnost slabost zaprtie
tahikardija
epileptiEni napadi
faringitis
omotica hipertenziia agitacija
prevedel in priredil: Jan Kejiar, dr. med., specializant psihiatrije pregledala: doc. dr. Brigita Novak Sarotar, dr. med., spec. psih.
eliminacija poteka preko jeter in ledvic
prilagoditev odmerka v primeru bolezni jeter in/ali ledvic
bupropion lahko inhibira jetrni citokrom P4502D6 in povzrod interakcije med zdravili
kontraindiciran pri boleznih, ki zmanIgajo epileptogeni prag: anoreksija/bulimija nervoza, epilepsija, odtegnitev alkohola, odtegnitev benzodiazepinov
zaplet Objavljeno 30. junija 2017 na www.thecalgaryguide.com.
" />
NAT), ki proizvaja aktivne metabolite. Natan'6en mehanizem delovanja (se) ni znan.
znak/simptom/laboratorijska najdba
DA in NE posledi6no ostaneta v sinapsah dlje Casa in okrepita 2iv6ni prenos
neieleni udnki
glavobol
suha usta
4, tel. tee nespeEnost slabost zaprtie
tahikardija
epileptiEni napadi
faringitis
omotica hipertenziia agitacija
prevedel in priredil: Jan Kejiar, dr. med., specializant psihiatrije pregledala: doc. dr. Brigita Novak Sarotar, dr. med., spec. psih.
eliminacija poteka preko jeter in ledvic
prilagoditev odmerka v primeru bolezni jeter in/ali ledvic
bupropion lahko inhibira jetrni citokrom P4502D6 in povzrod interakcije med zdravili
kontraindiciran pri boleznih, ki zmanIgajo epileptogeni prag: anoreksija/bulimija nervoza, epilepsija, odtegnitev alkohola, odtegnitev benzodiazepinov
zaplet Objavljeno 30. junija 2017 na www.thecalgaryguide.com.
" title="Bupropion (atipiEni antidepresiv): Mehanizem delovanja in neieleni utinki
potenten antidepresiv v monoterapiji all kot dodatno zdravilo pri zdravljenju razpoloienjskih motenj
okrajgave: 5-HT - serotonin DA - dopamin DAT - prenagalec DA NA - noradrenalin NAT - prenagalec NA SSRI - selektivni zaviralec ponovnega privzema 5-HT
Legenda:
bupropion
farmakologija
antidepresivni udnki uporaben pri zdravljenju "zmaniganega pozitivnega afekta"
nima pomembnelgih 5-HT udnkov povzraa spolne disfunkcije v primerjavi s SSRI; lahko celo odpravi omenjeni neieleni udnek (povzraen s strani SSRI)
dodatek pri zdravljenju odvisnosti od nikotina preko T iive'nega prenosa DA v nagrajevalni poti
energije preko T 2ivbega prenosa NA
patofiziologija mehanizem
farmakokinetika
farmakodinamika
avtorica: JoAnna Fay, Sara Meunier pregledala: Jojo Jiang, Alexander Arnold, Aaron Mackie*
* dr. med. ob objavi
Nizkoafiniteten zaviralec ponovnega privzema DA in NA (DAT>NAT), ki proizvaja aktivne metabolite. Natan'6en mehanizem delovanja (se) ni znan.
znak/simptom/laboratorijska najdba
DA in NE posledi6no ostaneta v sinapsah dlje Casa in okrepita 2iv6ni prenos
neieleni udnki
glavobol
suha usta
4, tel. tee nespeEnost slabost zaprtie
tahikardija
epileptiEni napadi
faringitis
omotica hipertenziia agitacija
prevedel in priredil: Jan Kejiar, dr. med., specializant psihiatrije pregledala: doc. dr. Brigita Novak Sarotar, dr. med., spec. psih.
eliminacija poteka preko jeter in ledvic
prilagoditev odmerka v primeru bolezni jeter in/ali ledvic
bupropion lahko inhibira jetrni citokrom P4502D6 in povzrod interakcije med zdravili
kontraindiciran pri boleznih, ki zmanIgajo epileptogeni prag: anoreksija/bulimija nervoza, epilepsija, odtegnitev alkohola, odtegnitev benzodiazepinov
zaplet Objavljeno 30. junija 2017 na www.thecalgaryguide.com.
" />
PTSD (Slovenian translation) - FINAL VERSION
: patogeneza in kliniene najdbe
predbolezenski dejavniki
genetika 30-% varianca*
skupen vpliv preteklih iivljenjskih stisk veda oUutijivost za dukvne travme
**Op. prey.: nepristranskost pozornosti (an. attentional bias) se nanak na podyrknost oz. nagnjenost Elovekovih zaz-nav k vplivu njegovih trenutnih misli. VeE v skupni bibliografiji psihologov A. Tverskyja in D. Kahnemana.
Legenda:
travmatski dogodek obaitki nemod in/ali izgube nadzora (npr. v okolikinah vojne, naravnih katastrof, spolnega napada, poroda)
intenziven Zustveni odziv ob du§evni travmi strah, nema ali groza
posttravmatska stresna motnja (PTSM)
nevrokemijske nepravilnosti spremembe v prenosu kortizola, GABA-e, dopamina
prekomerna Zustvena vzburjenost
anksioznost
posttravmatski dejavniki
pomanjkanje druibene podpore
stresni dejavniki v iivljenju
avtorica: Briana Cassetta pregledali: Sara Meunier, Yan Yu, Margaret Oakander*
* dr. med. ob objavi
prevedel in priredil: Jan Kejiar, dr. med., specializant psihiatrije pregledala: doc. dr. Brigita Novak Sarotar, dr. med., spec. psih.
*Op. prey.: z narakanjem variance se ve6a fenotipska raznolikost v populaciji.
moteni spoznavni procesi
pove6ana odzivnost amigdale nepristranskostpozornosti** zoper sign ale ogroknosti
inhibicija hipokampusa vodi v drobljenje spomina
pove'Cana opreznost pretirana bojazijivost teiave s pozornostjo podoiivljanje duKevnih travm, npr. v obliki noZnih mor ali „iivega")
Lipid Physiology Slide
 Lipid Supply
Small Intestine: Absorption of dietary triglycerides and cholesterol
Lymph, then Plasma: Chylomicrons (ApoC2 , ApoB48 ) Transport of dietary Triglycerides and Cholesterol
Peripheral Tissues: Triglyceride uptake and Chylomicron metabolism via Lipoprotein Lipase receptors (in presence of ApoC2)
Plasma: Chylomicron Remnants (ApoE)
Liver: Chylomicron Remnant uptake via surface LDL receptors and LRP1 (in presence of ApoE)
Note: Key component carrier lipoproteins are indicated in bold parentheses. i.e. (ApoC2). _DL are Density Lipoproteins which can be High (HDL), Intermediate (IDL), Low (LDL), and Very Low (VLDL) Deficiencies in these lipoproteins often result in inability to shuttle lipids and ensuing hyperlipidemias.
Endogenous Triglyceride Supply
Liver: Synthesis of VLDL from FFAs, ApoB100, and Cholesterol
Plasma: VLDL (ApoB100, ApoC2, ApoE) Transport of endogenous triglycerides and Cholesterol
Peripheral Tissues: Triglyceride uptake and VLDL cleavage via Lipoprotein Lipase receptors (in presence of ApoC2)
Endogenous Cholesterol Supply
Plasma: IDL (ApoE ) Transport of Cholesterol and Triglycerides to Liver
Liver: IDL uptake via surface LDL receptors (in presence of ApoE)
IDL metabolism to LDL via Hepatic Lipase
Plasma: LDL (ApoB100, Apo E) Transport of Cholesterol
Peripheral Tissues: Cholesterol uptake via surface LDL receptors (in presence of ApoB100)
Author: David Lincoln Reviewers: Harjot Atwal Usama Malik Dr. Alexander A-C Leung* * MD at time of publication
Reverse Cholesterol Transport
Plasma: Immature HDL (ApoAl) Nascent in plasma
•
Peripheral Tissues: Cholesterol export via surface ABCA1/ABCG1 transporters
Abbreviations: • ABCA1 - ATP-Binding Cassette Transporter Al • ABCG1 - ATP-Binding Cassette Transporter G1 • FFAs - Free Fatty Acids • LCAT - Lecithin Cholesterol Acetyl Transferase • LRP1 - LDL Receptor-related protein • SRB1 - Scavenger Receptor B1
Plasma: Mature HDL (ApoAl) Free cholesterol is esterified via LCAT.
Some cholesterol remains in plasma via CTEP-mediated transfer of Cholesterol to lower density lipoproteins (i.e. VLDL, IDL)
Liver: Cholesterol ester uptake into liver via SRB1
Legend: Pathophysiology Mechanism Sign/Symptom/Lab Finding Complications Published September, 24,2017 on www.thecalgaryguide.com")
Ovarian Torsion
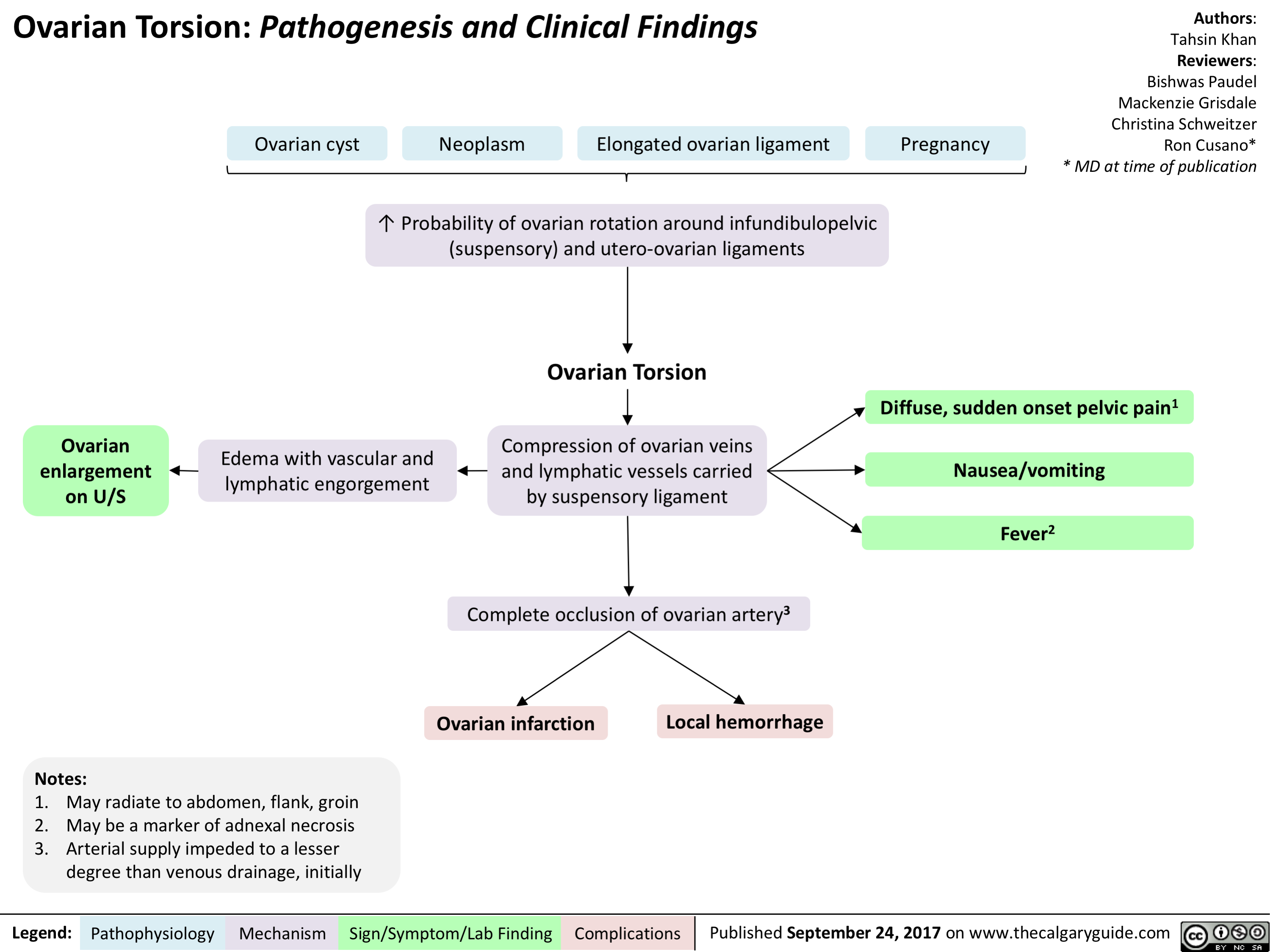
Non-Hodgkin Lymphoma
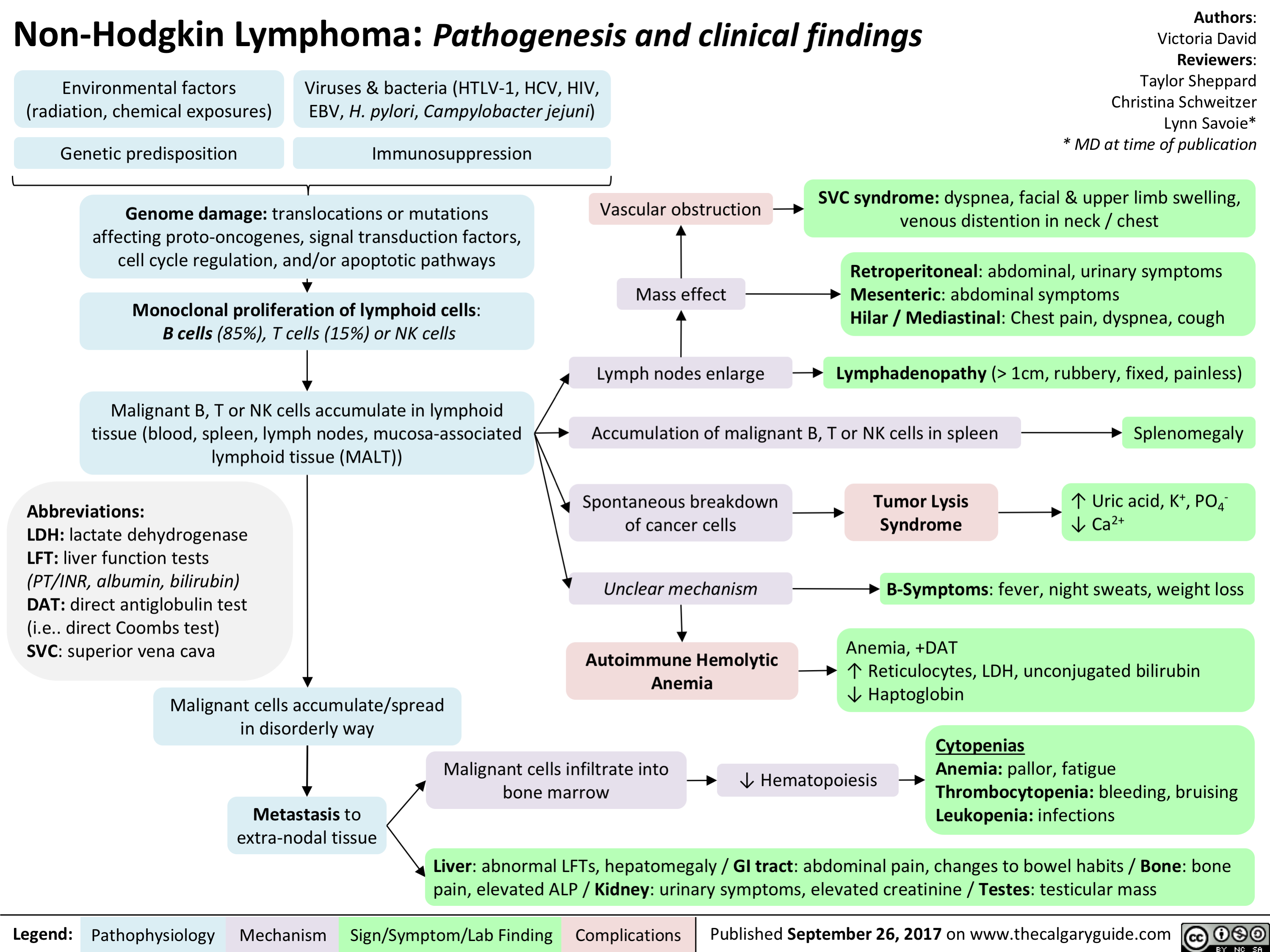
Pelvic Inflammatory Disease
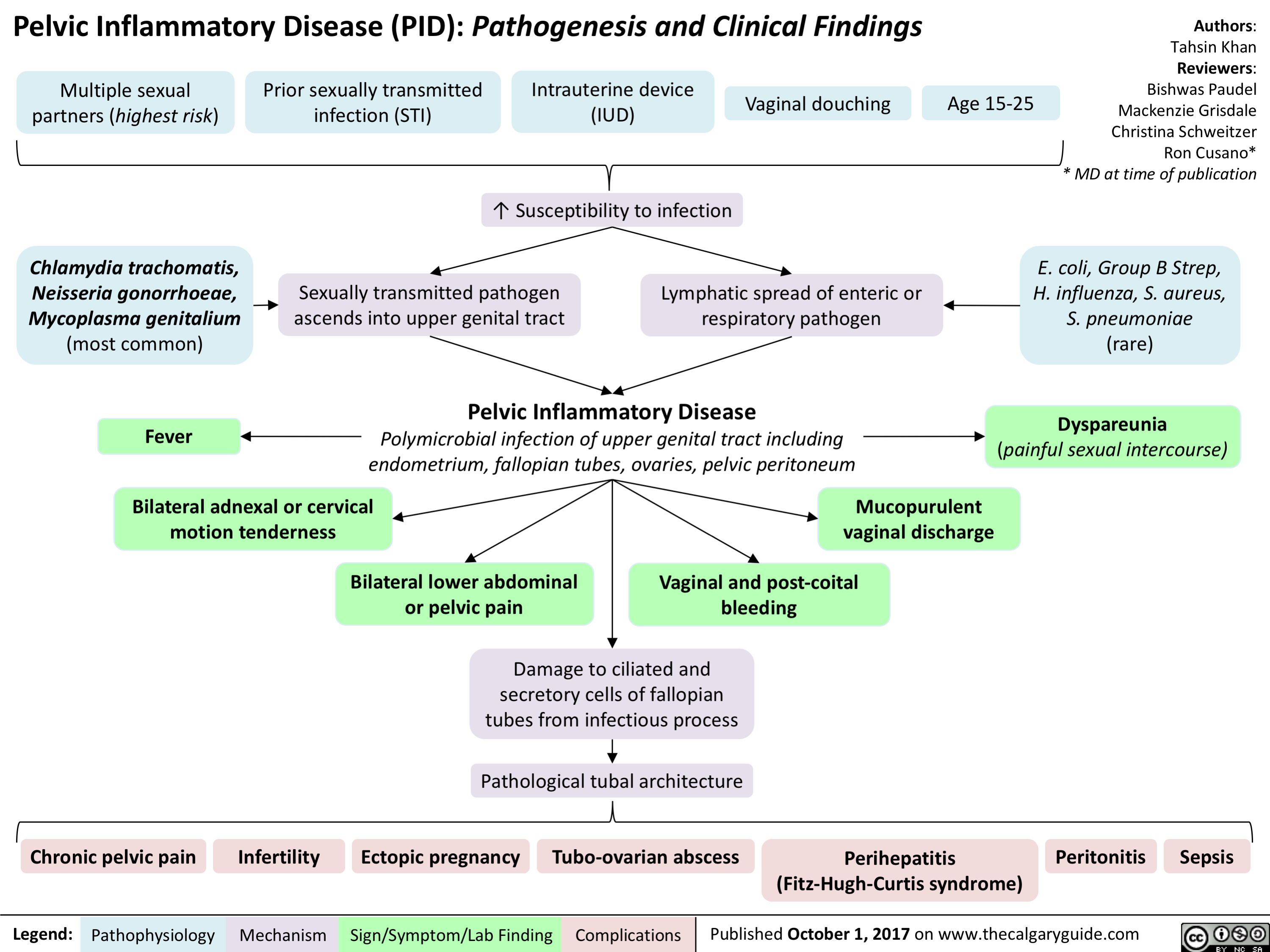
Pituitary Mass Effects
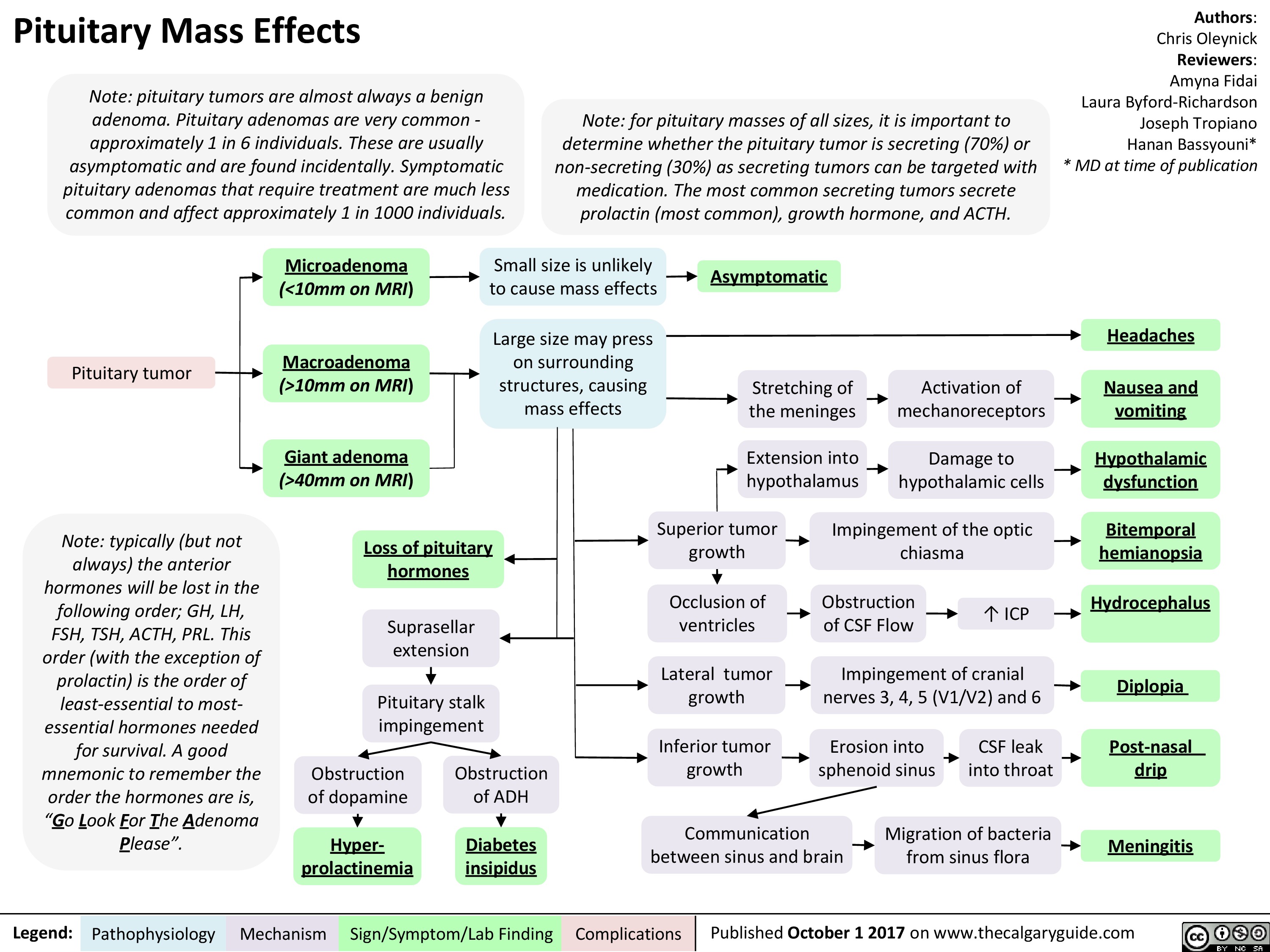 10mm on MRI) vomiting Giant adenoma Extension into hypothalamus —1■• Damage to hypothalamic cells Hypothalamic (>40mm on MRI) dysfunction Obstruction of dopamine Superior tumor growth Impingement of the optic chiasma Bitemporal Loss of pituitary hemianopsia hormones ICP Suprasellar extension Occlusion of ventricles Obstruction of CSF Flow Hydrocephalus Lateral tumor growth Impingement of cranial nerves 3, 4, 5 (V1/V2) and 6 4 Pituitary stalk impingement Diplopia Inferior tumor growth Erosion into sphenoid sinus CSF leak into throat Post-nasal Obstruction of ADH drip Communication between sinus and brain Migration of bacteria from sinus flora Hyper-Diabetes Meningitis prolactinemia insipidus
Pathophysiology Mechanism
Sign/Symptom/Lab Finding
Complications
Published October 1 2017 on www.thecalgaryguide.com
" title="Pituitary Mass Effects
Note: pituitary tumors are almost always a benign adenoma. Pituitary adenomas are very common -approximately 1 in 6 individuals. These are usually asymptomatic and are found incidentally. Symptomatic pituitary adenomas that require treatment are much less common and affect approximately 1 in 1000 individuals.
Pituitary tumor
Note: typically (but not always) the anterior hormones will be lost in the following order; GH, LH, FSH, TSH, ACTH, PRL. This order (with the exception of prolactin) is the order of least-essential to most-essential hormones needed for survival. A good mnemonic to remember the order the hormones are is, "Go Look For The Adenoma Please".
Legend:
Note: for pituitary masses of all sizes, it is important to determine whether the pituitary tumor is secreting (70%) or non-secreting (30%) as secreting tumors can be targeted with medication. The most common secreting tumors secrete prolactin (most common), growth hormone, and ACTH.
Authors: Chris Oleynick Reviewers: Amyna Fidai Laura Byford-Richardson Joseph Tropiano Hanan Bassyouni* * MD at time of publication
Microadenoma Small size is unlikely to cause mass effects (<10mm on MRI) Asymptomatic Macroadenoma Large size may press on surrounding structures, causing mass effects Headaches Stretching of the meninges Activation of mechanoreceptors Nausea and (>10mm on MRI) vomiting Giant adenoma Extension into hypothalamus —1■• Damage to hypothalamic cells Hypothalamic (>40mm on MRI) dysfunction Obstruction of dopamine Superior tumor growth Impingement of the optic chiasma Bitemporal Loss of pituitary hemianopsia hormones ICP Suprasellar extension Occlusion of ventricles Obstruction of CSF Flow Hydrocephalus Lateral tumor growth Impingement of cranial nerves 3, 4, 5 (V1/V2) and 6 4 Pituitary stalk impingement Diplopia Inferior tumor growth Erosion into sphenoid sinus CSF leak into throat Post-nasal Obstruction of ADH drip Communication between sinus and brain Migration of bacteria from sinus flora Hyper-Diabetes Meningitis prolactinemia insipidus
Pathophysiology Mechanism
Sign/Symptom/Lab Finding
Complications
Published October 1 2017 on www.thecalgaryguide.com
" />
10mm on MRI) vomiting Giant adenoma Extension into hypothalamus —1■• Damage to hypothalamic cells Hypothalamic (>40mm on MRI) dysfunction Obstruction of dopamine Superior tumor growth Impingement of the optic chiasma Bitemporal Loss of pituitary hemianopsia hormones ICP Suprasellar extension Occlusion of ventricles Obstruction of CSF Flow Hydrocephalus Lateral tumor growth Impingement of cranial nerves 3, 4, 5 (V1/V2) and 6 4 Pituitary stalk impingement Diplopia Inferior tumor growth Erosion into sphenoid sinus CSF leak into throat Post-nasal Obstruction of ADH drip Communication between sinus and brain Migration of bacteria from sinus flora Hyper-Diabetes Meningitis prolactinemia insipidus
Pathophysiology Mechanism
Sign/Symptom/Lab Finding
Complications
Published October 1 2017 on www.thecalgaryguide.com
" title="Pituitary Mass Effects
Note: pituitary tumors are almost always a benign adenoma. Pituitary adenomas are very common -approximately 1 in 6 individuals. These are usually asymptomatic and are found incidentally. Symptomatic pituitary adenomas that require treatment are much less common and affect approximately 1 in 1000 individuals.
Pituitary tumor
Note: typically (but not always) the anterior hormones will be lost in the following order; GH, LH, FSH, TSH, ACTH, PRL. This order (with the exception of prolactin) is the order of least-essential to most-essential hormones needed for survival. A good mnemonic to remember the order the hormones are is, "Go Look For The Adenoma Please".
Legend:
Note: for pituitary masses of all sizes, it is important to determine whether the pituitary tumor is secreting (70%) or non-secreting (30%) as secreting tumors can be targeted with medication. The most common secreting tumors secrete prolactin (most common), growth hormone, and ACTH.
Authors: Chris Oleynick Reviewers: Amyna Fidai Laura Byford-Richardson Joseph Tropiano Hanan Bassyouni* * MD at time of publication
Microadenoma Small size is unlikely to cause mass effects (<10mm on MRI) Asymptomatic Macroadenoma Large size may press on surrounding structures, causing mass effects Headaches Stretching of the meninges Activation of mechanoreceptors Nausea and (>10mm on MRI) vomiting Giant adenoma Extension into hypothalamus —1■• Damage to hypothalamic cells Hypothalamic (>40mm on MRI) dysfunction Obstruction of dopamine Superior tumor growth Impingement of the optic chiasma Bitemporal Loss of pituitary hemianopsia hormones ICP Suprasellar extension Occlusion of ventricles Obstruction of CSF Flow Hydrocephalus Lateral tumor growth Impingement of cranial nerves 3, 4, 5 (V1/V2) and 6 4 Pituitary stalk impingement Diplopia Inferior tumor growth Erosion into sphenoid sinus CSF leak into throat Post-nasal Obstruction of ADH drip Communication between sinus and brain Migration of bacteria from sinus flora Hyper-Diabetes Meningitis prolactinemia insipidus
Pathophysiology Mechanism
Sign/Symptom/Lab Finding
Complications
Published October 1 2017 on www.thecalgaryguide.com
" />
Acute Chest Syndrome (Sickle Cell Disease)
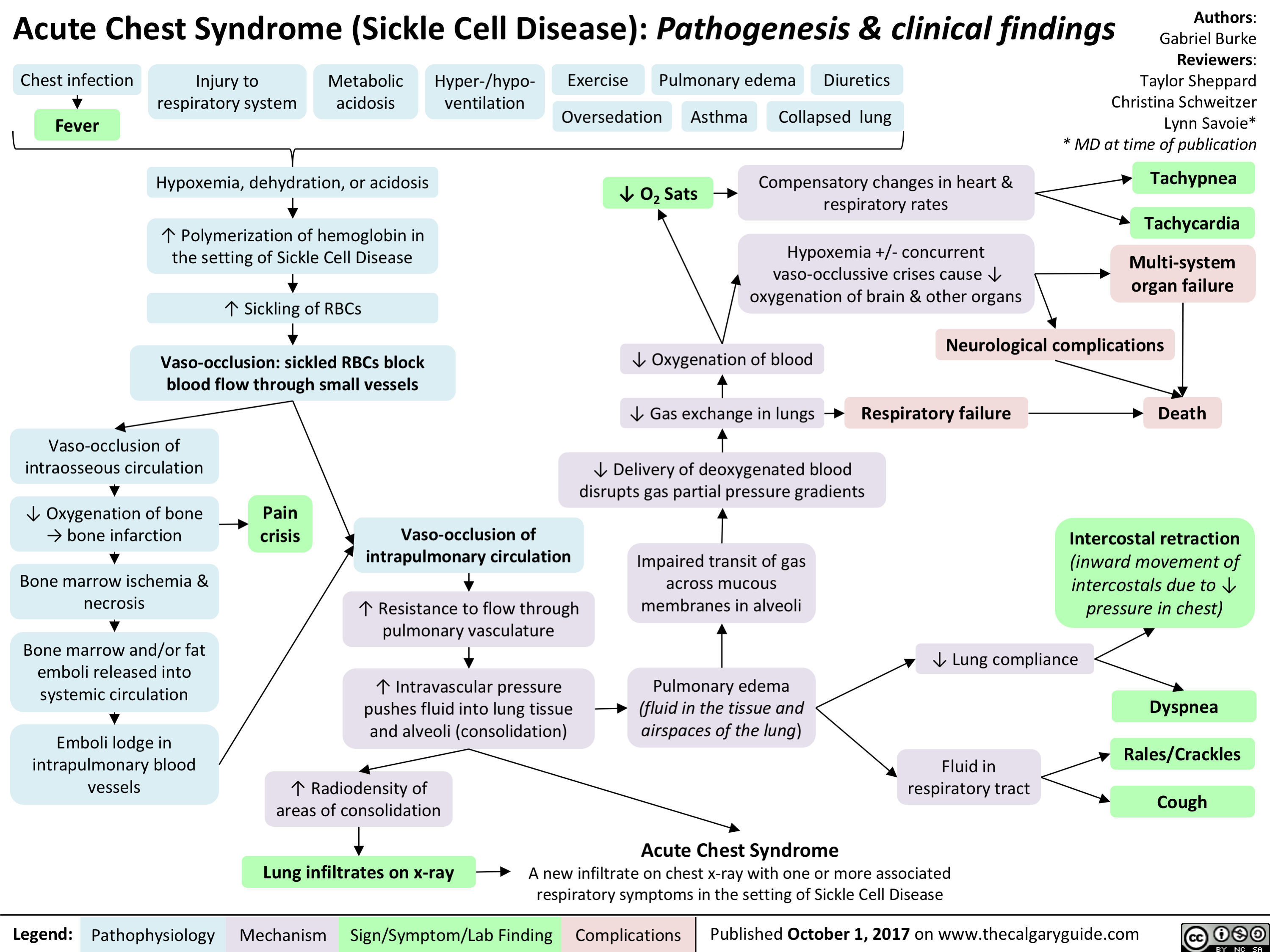
tetanus

Anaphylaxis - Pathogenesis
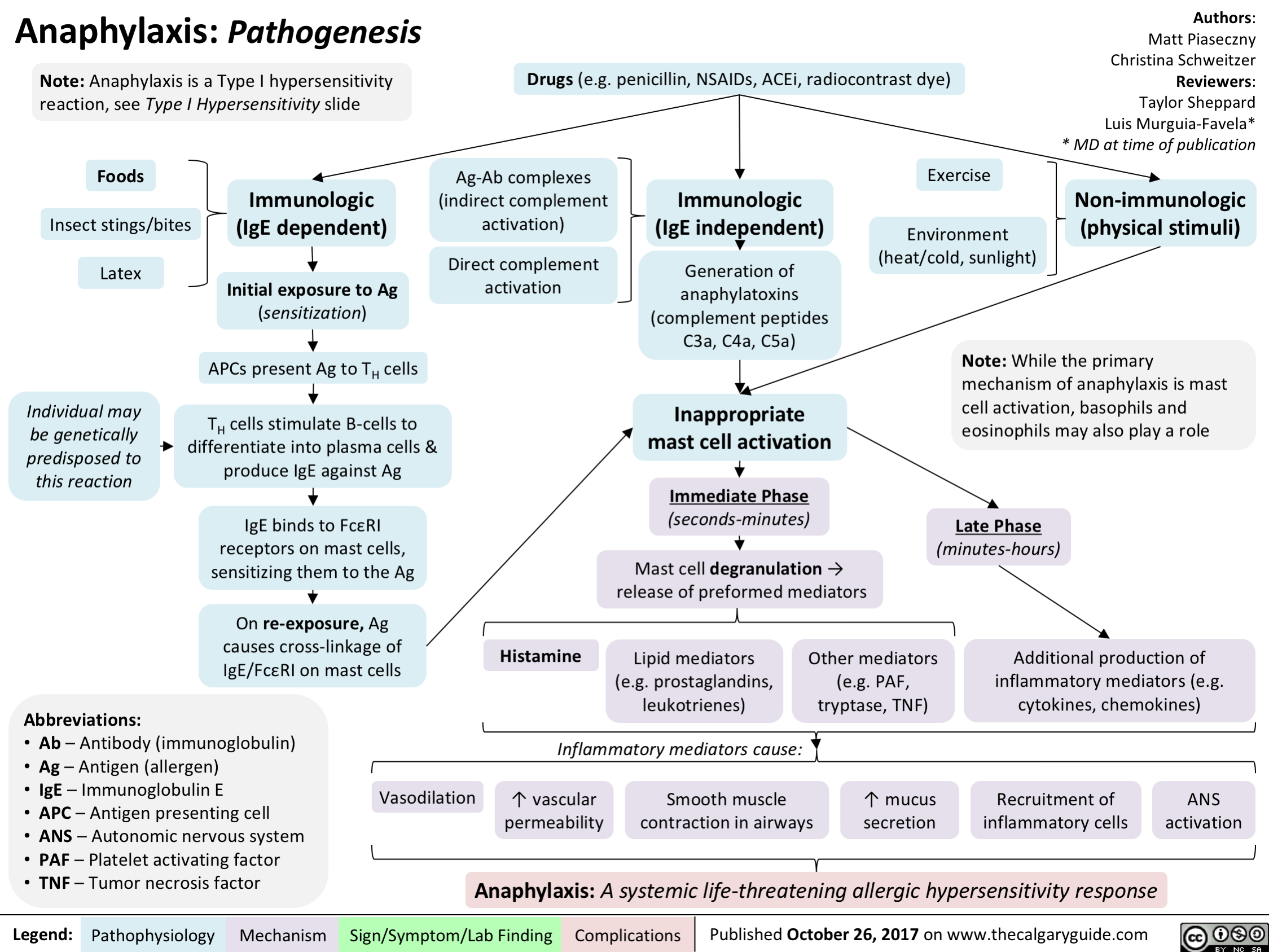
Ischemia: Pathogenesis of Cellular Injury and Death

Hepatitis C (HCV) Infections: Explaining Serology Patterns
 Infections: Explaining Serology Patterns
Seroconversion occurs on average 8-9 weeks after exposure to antigen
H CV RNA Negative
Anti-HCV Antibody Positive2
HCV RNA Positive4
1 HCV Screen
Anti-HCV Antibody Negative
Suspected acute HCV3
HCV RNA will be positive in blood within 1-3 weeks after exposure
No risk factors; likely no HCV exposure
HCV RNA Negative
No HCV exposure
HCV cleared spontaneously or with treatment or false positive antibody test6
Acute HCV (15%) 5Chronic HCV (85%)
HCV RNA negative 12 or 24 weeks after stopping therapy (SVR12 or SVR24)
Abbreviations: SVR12: sustained virologic response after 12 weeks SVR24: sustained virologic response after 24 weeks
Hepatocellular Carcinoma
Cirrhosis
Decompensation (ascites, variceal bleeding, encephalopathy)
7 Liver Transplant
Death
Authors: Emma Boyce Sarah Lacny Reviewers: Peter B i s h ay Joesph Tropiano Yin Chan* * MD at time of publication
Notes: 1Indications for HCV screen: born between 1945-1965, ↑ALT/AST, IVDU, received blood or organ transplant before 1992, received clotting factors before 1987, HIV infected or multiple sexual partners, tattoos and piercings (especially if done in prison), dialysis patients, Egyptian background 2There is no HCV vaccine; an anti-HCV positive test result indicates exposure to the virus 3Seve re l y immunocompromised, hemodialysis, possible exposure, clinical manifestations 4Assess genotype and viral load (HCVRNA), symptoms, and potential exposures to diagnose chronic versus acute HCV 5Acute HCV infection is defined as the first 6 months following exposure 6The anti-HCV antibody does not protect against future infections 7Liver transplant recipients have an 80% chance of developing a recurrent HCV infection
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published NOVEMBER 12, 2017 on www.thecalgaryguide.com")
Hepatitis C (HCV) Infection: Explaining Serology Patterns
 Infections: Explaining Serology Patterns
Seroconversion occurs on average 8-9 weeks after exposure to antigen
H CV RNA Negative
Anti-HCV Antibody Positive2
HCV RNA Positive4
1 HCV Screen
Anti-HCV Antibody Negative
Suspected acute HCV3
HCV RNA will be positive in blood within 1-3 weeks after exposure
No risk factors; likely no HCV exposure
HCV RNA Negative
No HCV exposure
HCV cleared spontaneously or with treatment or false positive antibody test6
Acute HCV (15%) 5Chronic HCV (85%)
HCV RNA negative 12 or 24 weeks after stopping therapy (SVR12 or SVR24)
Abbreviations: SVR12: sustained virologic response after 12 weeks SVR24: sustained virologic response after 24 weeks
Hepatocellular Carcinoma
Cirrhosis
Decompensation (ascites, variceal bleeding, encephalopathy)
7 Liver Transplant
Death
Authors: Emma Boyce Sarah Lacny Reviewers: Peter B i s h ay Joesph Tropiano Yin Chan* * MD at time of publication
Notes: 1Indications for HCV screen: born between 1945-1965, ↑ALT/AST, IVDU, received blood or organ transplant before 1992, received clotting factors before 1987, HIV infected or multiple sexual partners, tattoos and piercings (especially if done in prison), dialysis patients, Egyptian background 2There is no HCV vaccine; an anti-HCV positive test result indicates exposure to the virus 3Seve re l y immunocompromised, hemodialysis, possible exposure, clinical manifestations 4Assess genotype and viral load (HCVRNA), symptoms, and potential exposures to diagnose chronic versus acute HCV 5Acute HCV infection is defined as the first 6 months following exposure 6The anti-HCV antibody does not protect against future infections 7Liver transplant recipients have an 80% chance of developing a recurrent HCV infection
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published NOVEMBER 12, 2017 on www.thecalgaryguide.com")
Medical Conditions Causing Mania or Mania-Like Episodes: Pathogenesis
 pathway
4, myelin synthesis and neurotransmitter regulation
Altered Thyroid Activity Corticosteroid Use 7),
4, thyroid activity
4, central serotonin activity
4, postsynaptic beta-adrenergic receptor activity 4, in catecholamine (including dopamine) transmission
Adrenocortical hormones t Dopamine D2 receptor numbers (hypothesis)
Note: Mania is defined as at least 1 week of abnormally and persistently elevated, expansive, or irritable mood, and abnormally and persistently increased goal-directed activity or energy
By definition, a manic episode is not caused by drugs/medication or a medical illness, therefore those with a non-psychiatric underlying cause are mania-like episodes. Note as well that the conditions listed are not an exhaustive list
Legend:
Pathophysiology Mechanism
Manic episodes precipitated by Dopamine D2 receptor overactivity and 4, serotonergic regulation (hypothesis)
Additional environmental precipitating factors including lack of sleep, social stress, or change Mania or Mania-like Episode
Sign/Symptom/Lab Finding
Complications
Authors: Emily Ower Reviewers: Alexa Scarcello Usama Malik Dr. Lauren Zanussi* * MD at time of publication")
Left Heart Failure: Pathophysiology (Neurohormonal Activation)

Frank Starling Mechanism • The Frank Starling mechanism of the heart represents the relationship between preload (EDV) and SV • As preload (EDV) increases, SV increases, because higher volumes of blood in the ventricles stretch the cardiac fibers and increases cardiac contraction during systole. However, volume overload causes reduced SV.
Myocardial Dysfunction: • Left ventricular Compensatory 4, SV 4A, CO 4 Mechanisms
4, BP
Important equations: • BP = CO x SVR • CO = SV x HR Cardiac hemodynamics: • Stroke volume is affected by three factors 1) Preload (end-diastolic volume (EDV)) 2) Afterload (resistance to LV ejection) 3) Contractility (inherent strength of contraction of LV myocytes) Definition of heart failure: • Myocardial dysfunction (systolic or diastolic) results in decreased CO, such that the heart cannot meet the body's metabolic demands or can only do so at elevated filling pressures
Anti-diuretic hormone (ADH) activation: Arginine 4, BP 4 carotid sinus Vasopressin --• and aortic arch (V2) receptor baroceptors activation activation 4 I` ADH release
RAAS System: 4, BP 4 t release of renin from the juxtaglomerular kidney cells due to renal hypoperfusion
SNS System: In response to CO, 4 SNS t release of (catecholamines) norepinephrine and epinephrine
Adrenal Glands: Aldosterone release
Angiotensin II Type 1 receptor activation
al receptor activation
13, receptor activation
—•
Renal Collecting Ducts: t H2O retention
Renal Distal —• Tubules: t Na+ & H2O retention
Heart: Activation of fibroblasts 4 collagen synthesis and hypertrophy
Blood Vessels: Peripheral vasoconstriction 4 SVR
Heart: Chronic p, receptor activation 4 Ca2+ overload myocyte apoptosis
Heart: Increase HR to maintain normal CO
Maladaptive Response: t preload (EDV), —• volume overload
Abbreviations: • SV — Stroke volume • CO — Cardiac output • SVR — Systemic vascular resistance • BP — Blood pressure • RAAS — Renin-Angiotensin-Aldosterone System • SNS — Sympathetic nervous system
tin systemic and pulmonary congestion via the Frank-Starling Mechanism
Maladaptive 1` resistance Response: t BP, —• against LV afterload ejection 4 4, SV
Maladaptive Response: Adverse LV remodelling
Maladaptive Response: t myocardial oxygen demand and 4, diastolic time
4, contractility —• of the heart
4, coronary blood flow 4 myocardial ischemia
Physical signs and symptoms of congestive heart failure (see relevant slide)
Authors: Sunny Fong Reviewers: —• I Jack Fu Usama Malik Dr. Jason Waechter* *MD at time of publication")
Primary Spontaneous Pneumothorax: Pathogenesis and clinical findings

Malnutrition Smoking
Structurally compromised lung parenchyma
Notes: • PSPs usually occur at rest • Respiratory symptoms vary in severity • Suspect thoracic endometriosis in young women with recurrent PSPs that coincide with menstruation • *Pathophysiology of tension pneumothorax is described in a separate slide
Air leaks into the subcutaneous tissue
Subcutaneous emphysema
Authors: Lauren Hampton Reviewers: Kening (Midas) Kang Natalie Morgunov Sadie Kutz Usama Malik Leila Barss* * MD at time of publication
Thoracic Ischemia endometriosis
Mechanical forces of respiration create blebs Inflammation disrupts mesotheial and/or bullae ~ cell layer of the visceral pleura
47
Spontaneous rupture of blebs or bullae
47
Sudden onset pleuritic chest pain
71r
Primary spontaneous pneumothorax: Presence or introduction of air in the pleural space in a patient WITHOUT diagnosed or clinically apparent lung disease Tachycardia Abbreviations: Communication occurs between the alveoli and pleural space • FLCN- Folliculin gene • HCY- Homocystinuria • MFS- Marfan syndrome Alveolar pressure > pleural pressure • CTD- Connective tissue disease • PSP- Primary spontaneous Air from the lungs enters the pleural space pneumothorax • V/Q- ventilation/perfusion Air separates the chest from /1` intrapleural pressure • Sp02- oxygen saturation the lung parenchyma Small areas of lung collapse Blood flow to areas of On affected side: under un-opposed intrinsic atelectasis is maintained -• Si, chest wall expansion elastic recoil while ventilation 4, t resonance to percussion Si, or absent tactile fremitus Si, or absent breath sounds 4, lung compliance Shunting and V/Q mismatch Pleural line on chest x-ray 1` work of breathing Tension pneumothorax* Accessory muscle use, 4• Sp02, Sudden onset dyspnea, Tachycardia Hypotension, Juglar venous Tachypnea distension, Pulsus paradoxus")
Anksiozne motnje: patogeneza tesnobe

Depresivna epizoda/motnja: patogeneza in klinične najdbe

pomanjkanje tesnih, zaupnih odnosov
druibeni dejavniki
Prevalenca 10-15 % pri 2enskah in 5-12 % pri mogkih. Razmerje med 2enskami in mc&imi 2:1. Pogosteja pri razvezanih, laenih in samskih ter tistih z nizkim druThenoekonomskim stanjem.
depresivna epizoda/motnja simptomi (po DSM 5 merilih) prisotni tekom istega dvotedenskega obdobja in pomenijo odklon od premorbidne ravni funkcioniranja drugi simptomi pomanjkanje libida tesnoba razpolo2enje '6ez dan niha (ang. diurnal variation) huje pozimi (sezonska depresivna motnja) halucinacije (psihotiCna depresija) blodnje (psihotiena depresija) anhedonija (izguba zanimanj/u2itkov) depresivno razpolo2enje nejeknost ali povaan apetit s pridrdienimi spremembami telesne te2e (4, ali 1`) anergija (utrujenost) nespanost ali preva spanja okrajgave: 5-HT - serotonin NA - noradrenalin nastop v mesecu po porodu (poporodna depresija) obC'utki niC'vrednosti ali pretirane krivde psihomotorit'na upaasnjenost ali agitiranost upad spoznavnih sposobnosti (zlasti motnje pozornosti) ponavljajae se misli na smrt/samomor")
Obsesivno-kompulzivna motnja: patogeneza in klinične najdbe
 z bolnikovim vrednostnim sistemom (")
Panična motnja: patogeneza in klinične najdbe
.
izogibajoEe vedenje izogibanje okolikinam, ki poustvarjajo telesne ob6utke, podobne panithemu napadu (telesna vadba, u2ivanje kofeina ali alkohola, obisk savne)
Legenda:
patofiziologija mehanizem
kognitivno: bolnik si telesne obEutke razlaga kot katastrofibe 4 povet'anje vzburjenja
avtorica: Jenna Thomas pregledali: Saara Meunier, Yan Yu, Margaret Oakander*
* dr. med. ob objavi
prevedel in priredil: Jan Kejiar, dr. med., specializant psihiatrije pregledala: doc. dr. Brigita Novak Sarotar, dr. med., spec. psih.
-1110
psiholoKki simptomi nenadzorovan strah pred simptomi/izgubo nadzora/smrtjo obt'utek izgubljanja stika z realnostjo
vzdrievalno vedenje
varovalno vedenje npr. posedovanje pomirjeval, iskanje najbli2je bolnignice (z namenom oUutka varnosti v primeru panibega napada)
znak/simptom/laboratorijska najdba
pogosto pridruiene druge dugevne bolezni: • anksiozne motnje (npr. generalizirana anksiozna motnja, posttravmatska stresna motnja, obsesivno-kompulzivna motnja) • ponavljajoea se depresivna motnja • bipolama motnja razpoloienja
prilakovanje vnoviZnega napada vsiljive misli in skrbi o tern, kdaj/kje se bo zgodil naslednji panibi napad in kakgne bodo posledice (srannota, nev'nosti)")
Sezonska depresivna motnja: patogeneza in klinične najdbe
, sindrom kasne zaspanosti, odvisnost od alkohola
nevrologki dejavniki nenormalen serotoninergibi prenos
serotoninska hipoteza: zni2ana konc. serotonina v zimskih mesecih
sezonska depresivna motnja
avtorica: Luxey Sirisegaram pregledala: Paul Adamiak, Phillip Stokes* * dr. med. ob objavi
prevedel in priredil: Jan Kejiar, dr. med., specializant psihiatrije pregledala: doc. dr. Brigita Novak Sarotar, dr. med., spec. psih.
I
simptomi (po DSM 5 merilih) 2 depresivni epizodi v zadnjih 2 letih z nakazanim casovnim vzorcem pojavljanja
hipersomnija
hiperfagija
sla po u2ivanju ogljikovih hidratov
porast telesne te2e
patofiziologija mehanizem
sezonske posebnosti
sezonski vzorec pojavljanja pri depresivni epizodi/motnji
popolne remisije all obrati faze v dolaenem delu leta
ni povezave s specifithim sezonskim psihosocialnim stresorjem (npr. pove6anim obsegom dela v slu2bi)
znak/simptom/laboratorijska najdba
simptomi, po katerih se loEi od depresivne epizode/motnje
sezonski vzorec pojavljanja (torej v specifibem delu leta)
sezonske depresivne epizode morajo po 'gtevilu presegati ne-sezonske depresivne epizode")
Shizofrenija: patogeneza in klinične najdbe
 (prevladujaa teorija) predmet raziskav)
visoka stopnja izraianja Zustev • (ang. expressed emotion): iivljenje v okolju s pogostimi negativnimi komentarji na raC'un bolnika (I` tveganje za relaps)
shizofrenija
nepravilnosti v 2ivthem prenosu nevrotransmitorjev (predvsem dopamina) v razlithih predelih mo2ganov
dopaminergibega dopaminergiC'nega
prenosa v mezolimbibi poti
dopaminergibi nevroni te6ejo do Iimbicnih struktur in so odgovorni za nadzor nad razpolo2enjem in 6ustvi
moten prenos dopamina v tej poti naj bi bil kriv za pozitivne simptome shizofrenije
prenosa v mezolimbiC'ni poti
avtorica: Yan Yu pregledali: Sara Meunier, Briana Cassetta, JoAnna Fay, Philip Stokes*
* dr. med. ob objavi
prevedel in priredil: Jan Kejiar, dr. med., specializant psihiatrije pregledala: doc. dr. Brigita Novak Sarotar, dr. med., spec. psih.
Pomni: • Ceravno smo vzroke za shizofrenijo sprva iskali v biologkih dejavnikih, zdaj vse bolj prepozna-vamo vlogo tako psihologkih (npr. kognitivnih stereotipov) kot druibenih dejavnikov (npr. stresa ali osame) v njeni etiologiji. • pri shizofreniji lahko odkrijemo tudi nevropsihologke primanjkljaje: motnje spomina, psihomotorike in pozornosti ter te2ave s psihi6no prilagodljivostjo. • Genetske modifikacije C4 gena naj bi ravno tako igrale va2no vlogo v etiologiji bolezni, saj v 6asu adolescence povzro6ajo pove6ano zmanjgevanje gtevila sinaps (nova hipoteza).
dopaminergibi nevroni te6ejo do razlibih predelov mo2g. skorje (npr. moten prenos dopamina v tej frontalnega re2nja) in so odgovorni poti naj bi bil kriv za negativne za migljenje, odlaanje, tvorbo simptome shizofrenije jezika in razpolo2enje
negativni simptomi (okrnjeno izraianje 6ustev, 6ustvena otopelost, osiroma§en govor, brezvoljnost, ‘iY miselna zavrtost, nedruiabno vedenje)
blodnje (nespremenljiva, zmotna prepriEanja, odstopajoEa od bolnikovega kulturnega ozadja)
Legenda: patofiziologija mehanizem
ha lucinacije (zaznave brez dra2ljajev; obiC'ajno pri shizofreniji a kustiC'n e)
dezorganiziran/nesmisein govor (tangencialni odgovori, ohlapne asociacije, besedna solata)
znak/simptom/laboratorijska najdba
hudo dezorganizirano vedenje, vkljauja katatonijo (neodzivnost na zunanje dra2ljaje)")
Serotoninski sindrom: patogeneza in klinične najdbe
, opioidi, antiemetiki, triptani, zdravila za izgubo tel. te2e, ilegalne psihoaktivne snovi (npr. kokain, amfetamini)
terapevtska raba
1
interakcije (se posebej pri kombinacijah serotoninergikov) namerna samozastrupitev
serotoninski sindrom raznolika kombinacija sprememb v psihiZnem stanju, vegetativne nestabilnosti in iivZnomigiZne hiperaktivnosti, ki sega od blage do iivljenje ogroiajoL'e z nenadnim nastopom (v nekaj minutah do urah) po administraciji zdravil(a), v vecini primerov pa se razregi v <24 urah po prekinitvi jemanja odgovorne snovi
prekomerna serotoninergitha aktivnost 5-HT receptorjev centralno (v moiganskem deblu) in periferno
sinteza in privzem in T receptorski agonizem sprogEanje 5-HT presnova 5-HT in ok'utljivost
z zdravili povzraene spremembe v relativnem razmerju ne-serotoninergithih nevrotransmitorjev (npr. pove6anje konc. noradrenalina)
spremenjeno psihreno stanje
tesnoba, zmedenost, agitacija, hipervigilnost, dolgoveznost, delirij, koma
vegetativna nestabilnost
mrzlica/drgetanje, 6ezmerno potenje, vraina, driska, tahikardija, ra8irjene zenice, hipertenzija
avtorica: Preeti Kar pregledali: Erika Russell, Usama Malik, Aaron Mackie*
* dr. med. ob objavi
prevedel in priredil: Jan Kejiar, dr. med., specializant psihiatrije pregledala: doc. dr. Brigita Novak Sarotar, dr. med., spec. psih.
Pomni: za klinicno postavitev diagnoze uporabljamo Hunterjeva merila serotoninske toksiZnosti: - zgodovina jemanja serotoninergithih snovi v preteklih 5 tednih + kateri koli od naslednjih pogojev: • spontani klonus • inducirani klonus in bodisi agitacija bodisi L'ezmerno potenje • aesni klonus in bodisi agitacija bodisi L'ezmerno potenje • tremor in hiperrefleksija • hipertonija, tel. temperatura >38 °C in bodisi aesni klonus bodisi inducirani klonus
ihgnomigiZna hiperaktivnost
hiperrefleksija, mR")
Zaviralci privzema serotonina in noradrenalina (SNRI): mehanizem delovanja in neželeni učinki
: mehanizem delovanja in neieleni utinki
zaviralci privzema serotonina in noradrenalina predstavnika: venlafaksin, duloksetin
konc. 5-HT in NA v 02S/P2S
farmakologija
farmakokinetika farmakodinamika
nenadna prekinitev jemanja zdravila ali vnos
avtorici: JoAnna Fay, Sara Meunier pregledali: Jojo Jiang, Alexander Arnold, Jessica Asgarpour, Aaron Mackie*
iz krvnega obtoka jih odstranijo jetra
zavirajo prenagalec za privzem NA (NET) in prenaalec za privzem 5-HT (SERT)
5-HT in NA ostaneta v sinapsah dlje in podaVata oz. okrepita iiv6ni prenos
odtegnitev namerno predoziranje nenamerno povzrocene interakcije nepri6akovan odziv na terapevtski odmerek prekometna v aktivnost 02S/P2S sindrom 1` 5-HT omotica 5-HT serotoninski omotica driska nejegZnost nespeEnost nespanost slabost utrujenost glavobol vegetativna * iivEnomiKiEne kognitivne hiperaktivnost nepravilnosti spremembe vrtoglavica potencialno iivljenje ogroiajoE spolne hipertenzija olesni klonus agitacija motn'e porast tahikardija tremor akatizra tel. tee raigir-ene hiperrefleksija zenice migiEni klonus
Legenda: patofiziologija
mehanizem
znak/simptom/laboratorijska najdba
1` NA
slabost
Zezmerno potenie
* dr. med. ob objavi
prevedel in priredil: Jan Kejiar, dr. med., specializant psihiatrije pregledala: doc. dr. Brigita Novak Sarotar, dr. med., spec. psih.
v primeru ietrnih bolezni in/ali saasne rabe zaviralcev/sproncev CYPD26 ie potrebna prilagoditev odmerka
izboljganje depresivne in anksiozne simptomatike 1 blokada ponovnega privzema 5-HT in NA v sinapsah pa u6inka SNRI ne pojasni v celoti
posredno SNRI povzro6ijo sproManja nevrozaMitnih proteinov, denimo BDNF, ki bi naj imel po podatkih §tudij protivnetno delovanje
za dosego kliniEnega uZinka ie potrebnih 3-8 tednov all veZ zdravlienia
okralgave in opombe: 5-HT— serotonin NA— noradrenalin BDNF — mo2ganski nevrotrofiEni faktor 025 — osrednji 2ivEni sistem P2S — periferni 2ivEni sistem *vegetativen — nanagajoE se na avtonomni 2ivEni sistem")
Socialna anksiozna motnja: patogeneza in klinične najdbe

Selektivni zaviralci privzema serotonina (SSRI): mehanizem delovanja in neželeni učinki
: mehanizem delovanja in neieleni utinki
selektivni zaviralci privzema serotonina predstavniki: citalopram, escitalopram, fluoksetin, paroksetin, sertralin
farmakologija
Pomni: dolo6eni SSRI z ve6jo verjetnostjo povzro6ajo nekatere od na'gtetih neielenih ainkov. Zmanigamo jih lahko z zamenjavo z drugim SSRI, postopnim titriranjem odmerka ali razdelitvijo dnevnega odmer-ka. Antidepresivi lahko zvgajo tveganje za samomor pri mlajsih od 24 let, pri starelgih od 31 let pa ga lahko znilajo.
farmakokinetika
farmakodinamika
iz krvnega obtoka jih odstranijo jetra
zavirajo privzem 5-HT in povzraijo sinaptibe konc. 5-HT
avtorici: JoAnna Fay, Sara Meunier pregledali: Jojo Jiang, Alexander Arnold, Aaron Mackie*
* dr. med. ob objavi
prevedel in priredil: Jan Kejiar, dr. med., specializant psihiatrije pregledala: doc. dr. Brigita Novak Sarotar, dr. med., spec. psih.
v primeru jetrnih bolezni je potrebna prilagoditev odmerka
lahko zavrejo jetrne encime citokroma P4502D6 in povzraijo interakcije med zdravili
tel. teie omotica 4 5-HT ostane v sinapsah dlje in podaVa oz. okrepi 2iv6ni prenos izboVanje depresivne in anksiozne simptomatike spolne motnje vrtoglavica nejeknost namerno predoziranje nenadna prekinitev jemanja zdravila ali I, vnos blokada ponovnega privzema 5-HT v sinapsah pa u6inka SSRI ne pojasni v celoti prekometna aktivnost 5-HT v 02S/P2S nenamerno povzro'6ene interakcije konc. 5-HT v O2S/P2S posredno SSRI povzraijo sprokanja nevrozakitnih proteinov, denimo BDNF, ki bi naj imel po podatkih gtudij protivnetno delovanje nepriakovan odziv na terapevtski odmerek serotoninski sindrom odtegnitveni sindrom
potencialno iivljenje ogroiajoE 1 vegetativna * iivEnomiKiEne nepravilnosti kognitivne spremembe omotica tremor za dosego kliniEnega uEinka je potrebnih hiperaktivnost slabost none more 3-8 tednov ali veE zdravljenja okrajgave in opombe: 5-HT— serotonin BDNF — nnoiganski nevrotrofi6ni faktor 025 — osrednji 2ivcni sistem P2S — periferni iivcni sistem *vegetativen— nanagajo6 se na avtonomni iivcni sistem hipertenzija olesni klonus agitacija Eezmerno")
Nevroanatomija in fiziologija čustev
![Nevroanatomija in fiziologija tustev
bazalni gangliji
prefrontalna skorja obdeluje 6ustva vgje-ga reda, ki zahtevajo priklic, razum(sko) presojo in sposobnost odloCanja (npr. sramota) senzorne informacije iz notranjih organov kot odziv na spralne okoljske objekte ali dogodke integrira z informacijami iz amigdale
Zustveni obEutki (npr. zavestne izkugnje Zustev)
1—
talamus
Zustveno izstopaja okoljski drailjaj
zaznave (npr. vid, sluh, tip, oku'ganje, vonjanje)
avtorica: Andrea Moir pregledali: Erika Russell, Usama Malik, Brienne McLane*
* dr. med. ob objavi
hipotalamus (glavna izhodna [ang. output] struktura limbibega sistema)
voh*
prim. in asociacijski predeli mo2g. skorje •
amigdala obdeluje 6ustva niijega reda, torej tista s hitrimi, samodejnimi in pogojevanimi odzivi (npr. strah) sodeluje pri tvorbi in priklicu C'ustveno pomembnih spominov iz neokortikalnih podrodj, torej zaznavi pripi§e neko Custveno vrednost
hipokampus (kljaen za utrditev in priklic spominov)
motorl6ni, endokrini in visceralni sistemi
prevedel in priredil: Jan Kejiar, dr. med., specializant psihiatrije pregledala: doc. dr. Brigita Novak Sarotar, dr. med., spec. psih.
avtonomni zivcni sistem (A2S) A2S se samodejno in podzavestno odziva na okoljske dra2ljaje (npr. s '(` frekvence sr6nega utripa ali zardevanjem)
telesni odziv na Zustveni drailja(
Pomni: • Strukture, ki sodelujejo pri C'ustvovanju, se imenujejo limbiEni sistem. • Talamus, bazalni gangliji in prefrontalna skoraj skupaj tvorijo kortiko-striato-talamo-kortikalno zanko, ki predstavlja glavno pot za obdelavo C'ustev v moiganih. • Prefrontalna skorja vkljue'uje orbitofrontalno skorjo, dorzolateralno prefrontalno skorjo in sprednjo cingulatno skorjo. • *Voh je edini cut, cigar vlakna zaobidejo talamus in se stekajo neposredno v primarno olfaktorno skorjo.](http://calgaryguide.ucalgary.ca/wp-content/uploads/2017/11/The-Neuroanatomy-and-Physiology-of-Emotion-Slovenian-translation-FINAL-VERSION.png "Nevroanatomija in fiziologija tustev
bazalni gangliji
prefrontalna skorja obdeluje 6ustva vgje-ga reda, ki zahtevajo priklic, razum(sko) presojo in sposobnost odloCanja (npr. sramota) senzorne informacije iz notranjih organov kot odziv na spralne okoljske objekte ali dogodke integrira z informacijami iz amigdale
Zustveni obEutki (npr. zavestne izkugnje Zustev)
1—
talamus
Zustveno izstopaja okoljski drailjaj
zaznave (npr. vid, sluh, tip, oku'ganje, vonjanje)
avtorica: Andrea Moir pregledali: Erika Russell, Usama Malik, Brienne McLane*
* dr. med. ob objavi
hipotalamus (glavna izhodna [ang. output] struktura limbibega sistema)
voh*
prim. in asociacijski predeli mo2g. skorje •
amigdala obdeluje 6ustva niijega reda, torej tista s hitrimi, samodejnimi in pogojevanimi odzivi (npr. strah) sodeluje pri tvorbi in priklicu C'ustveno pomembnih spominov iz neokortikalnih podrodj, torej zaznavi pripi§e neko Custveno vrednost
hipokampus (kljaen za utrditev in priklic spominov)
motorl6ni, endokrini in visceralni sistemi
prevedel in priredil: Jan Kejiar, dr. med., specializant psihiatrije pregledala: doc. dr. Brigita Novak Sarotar, dr. med., spec. psih.
avtonomni zivcni sistem (A2S) A2S se samodejno in podzavestno odziva na okoljske dra2ljaje (npr. s '(` frekvence sr6nega utripa ali zardevanjem)
telesni odziv na Zustveni drailja(
Pomni: • Strukture, ki sodelujejo pri C'ustvovanju, se imenujejo limbiEni sistem. • Talamus, bazalni gangliji in prefrontalna skoraj skupaj tvorijo kortiko-striato-talamo-kortikalno zanko, ki predstavlja glavno pot za obdelavo C'ustev v moiganih. • Prefrontalna skorja vkljue'uje orbitofrontalno skorjo, dorzolateralno prefrontalno skorjo in sprednjo cingulatno skorjo. • *Voh je edini cut, cigar vlakna zaobidejo talamus in se stekajo neposredno v primarno olfaktorno skorjo.")
Diabetic Foot: Pathogenesis and clinical findings
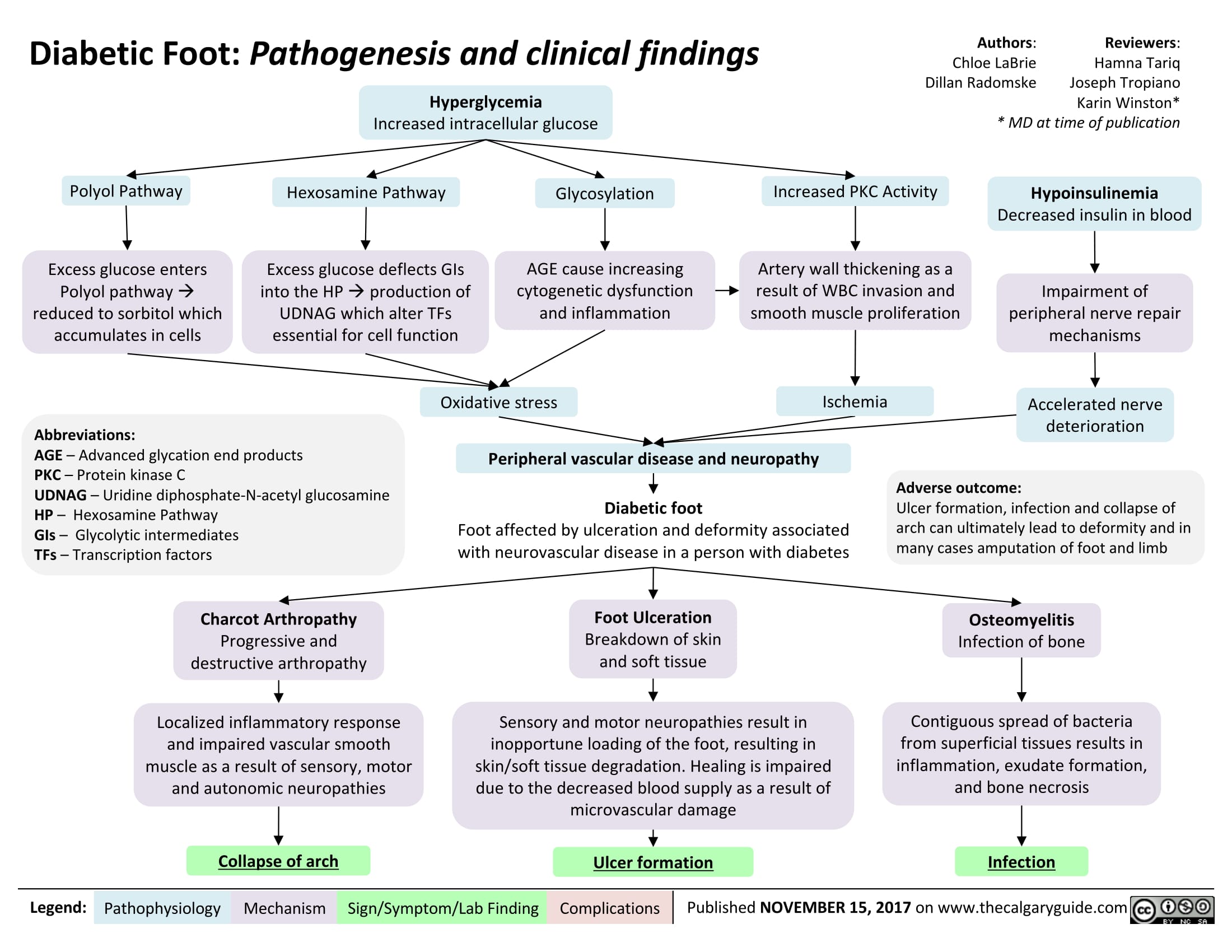
Bronchogenic Carcinoma - Pancoast Tumors Pathogenesis and clinical findings

Endothoracic fascia
1— Parietal pleura •
1—
Upper ribs
Large Cell
Carcinoma
Adenocarcinoma
Squamous Cell
Carcinoma
Primary Bronchogenic
Carcinoma
Pancoast tumor:
Local/metastatic growth in
ipsilateral lung apex
Disruption of structures
adjacent to superior
pulmonary sulcus
NSCLC (more common)
Invasion of airways
► causing obstruction
(later stages)
Author:
Bradley Stebner
Daniel Meyers
Midas (Kening) Kang
Reviewers:
Natalie Morgunov
Sadie Kutz
Usama Malik
Kerri Johannson*
*MD at time of publication
Notes:
• Pancoast Tumor: Malignant
lesion occupying the superior
pulmonary sulcus (lung apex)
Bronchogenic carcinoma:
primary malignant neoplasm
arising from epithelium of
bronchus or bronchiole
Pancoast tumors can be caused
by primary or metastatic
pulmonary neoplasms
(described here) as well as
infectious foci
Hemoptysis
Compression of C8
and T1 nerves
•
Disruption of paravertebral
sympathetic chain
Shoulder • Weakness in intrinsic Horner's • 4, sympathetic to to iris muscle Syndrome 4, sympathetic radial to eccrine sweat gland
Pain (ulnar hand muscles
nerve) 4, sympathetic innervation STM
Paresthesia in
4th /5th digits and
arm/forearm medial
Ptosis Mi o sis Anhidrosis Legend: Pathophysiology Mechanism
Sign/Symptom/Lab Finding
Compression of SVC
SVC Syndrome
•
4, venous return to
RA
4, Cardiac output to
lungs
Dyspnea
Disruption of RLN
1
Hoarse voice
4, venous drainage
from upper thoracic
cavity
Retention of fluid in
upper limb
•)r
Facial and limb swelling")
Hyperthyroidism

Hyperthyroidism
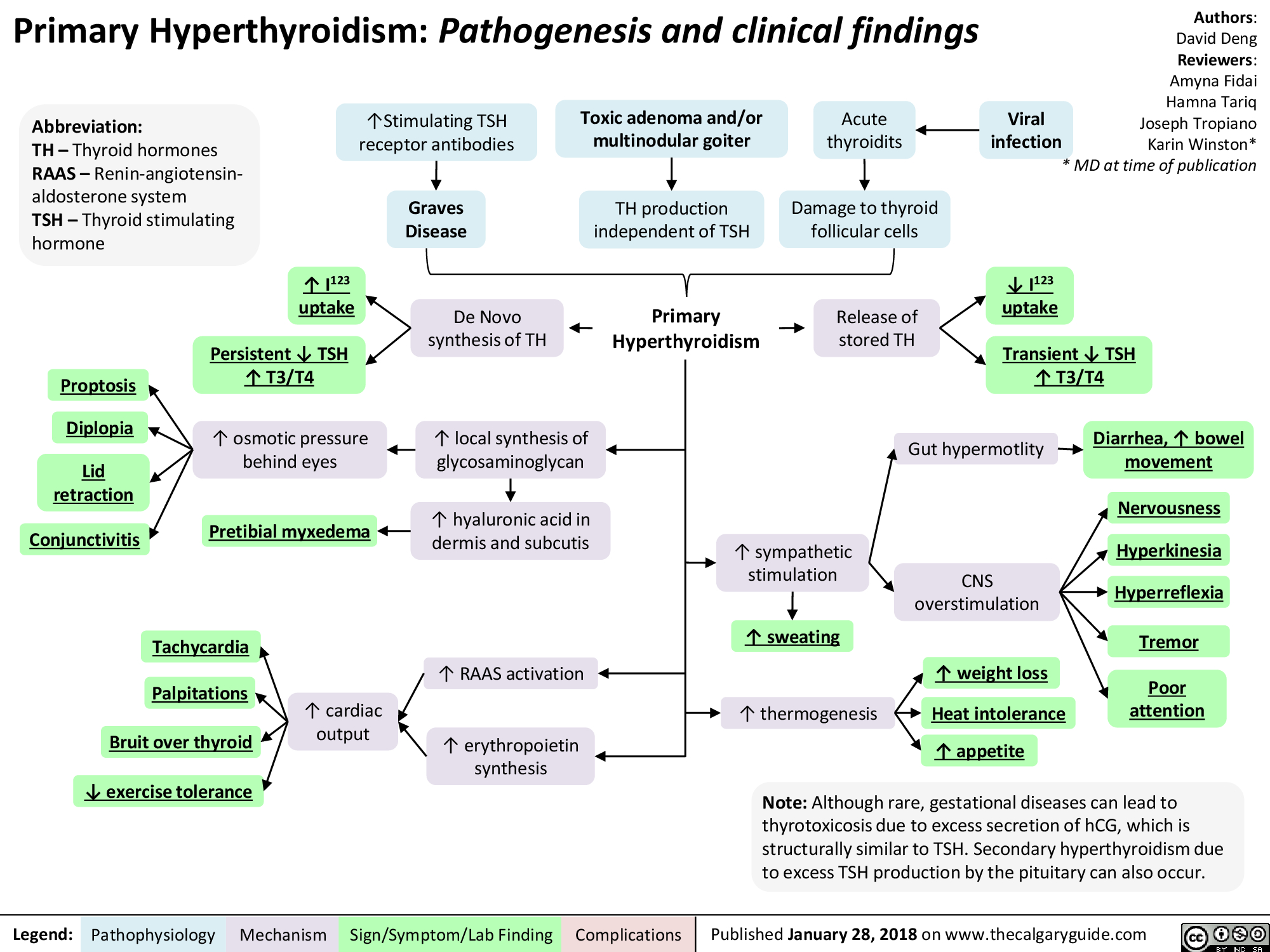
Growth Hormone Excess

Somatotroph adenoma in anterior pituitary
Microduplications in chromosome Xq26.3
4— Plain film
CT/M RI
GH secretion 1\ frequency and amplitude
► GH excess
Imaging 4— Acromegaly/Gigantism
Stimulation/ repression of other hormones
Authors: David Deng Reviewers: Amyna Fidai Hamna Tariq Joseph Tropiano Karin Winston * MD at time of publication
intracranial Pressure
Headache 4—
4, visual confrontation
4, visual acuity
Bitemporal hemianopsia
Abbreviations: • GH: Growth hormone • GHRH: Growth hormone releasing hormone • FSH: Follicle stimulating hormone • LH: Luteinizing hormone • TSH: Thyroid stimulating hormone
Legend:
4--
Local IGF-1 overproduction
Median nerve edema
Connective tissue Carpal tunnel overgrowth syndrome
Acne 4
Coarse features
Hyperhydriosis
Pathophysiology Mechanism
Hypertrichiosis
Macrognathia 1—
Enlarged hands and feet
Sign/Symptom/Lab Finding
Excess insulin secretion
\I, insulin receptor numbers and affinity
Acanthosis Nigricans
Impaired glucose tolerance
Cardiovascular disease
Weight gain
Cardiac arrhythmia
Valvular damage
ejection fraction
► HTN
Notes: • Acromegaly and gigantism share the same pathophysiology but differ mainly in terms of time of onset of GH excess. GH excess prior to growth plate fusion 4 gigantism. GH excess after growth plate fusion 4 acromegaly. • Other rare causes of acromegaly include GHRH hypothalamic tumors and ectopic secretion of GHRH by neuroendocrine tumors.
Complications
Published January 28, 2018 on www.thecalgaryguide.com")
Asthma Acute Exacerbation: Pathogenesis and Treatment
: An episode of increased symptoms due to decreases in airflow
Abbreviations • PCO2: Partial pressure of CO, in arterial blood • PEF: Peak expiratory flow • SABA: Short-acting beta-2 agonists • Sp02 : Blood oxygen saturation level
Mild to moderate exacerbation: PEF 50% of predicted
Titrate O2 toSpO2, 92%, give SABA & steroids ■
Good response: symptoms resolved, PEF > 80%
[Treat at home with SABA as needed and steroids
Dyspnea
Bronchoconstriction
1` Residual volume and 1` PCO2
Respiratory failure
1` Air trapping causes '1' intra-alveolar pressure
Severe exacerbation: PEF 50% of predicted Educate patient regarding medications, Loss of Pulsus inhaler technique & [consciousness paradoxus follow up with primary care provider I
Legend: Pathophysiology Mechanism
Titrate O2 to402 93%, give SABA, steroids & magnesium sulfate
Sign/Symptom/Lab Finding
{Worsening symptoms and/or respiratory failure: Do not delay intubation, send to ICU, give SABA, steroids & magnesium sulfate
Authors: Luke Gagnon Reviewers: Midas (Kening) Kang Usama Malik Lian Szabo* * MD at time of publication
4, Delivery of oxygen rich air to alveoli 4, Oxygenation of blood
Drowsy and confused
Central cyanosis
• Tachycardia
Pneumothorax
[Depending on 1 severity: Observation or place chest tube")
Bronchiectasis Pathogenesis and clinical findings
, MAC complex infection, COPD, allergic bronchopulmonary aspergillosis, chronic infections
Irreversibly dilated bronchi
Chronic bronchial infection and inflammation
1
Easily collapsible airways
I Bronchiectasis (persistent and progressive damage to lungs)
Chronic cough (mucopurulent)
Defect in immunity and/or mucus clearance
Persistent bacteria in airway (commonly Pseudomonas/Staph aureus)
Inflammatory response
Rhinosinusitis
Abbreviations: • A1AT — Alpha-1-antitrypsin • COPD — Chronic Obstructive Pulmonary Disease • HIV — Human Immunodeficiency Virus • MAC — Membrane Attack Complex • VQ— Ventilation/Perfusion ratio
Legend:
Pathophysiology Mechanism
Fever
Sign/Symptom/Lab Finding
Failure to thrive (children)
Authors: Rebecca (Becky) Phillips Reviewers: Midas (Kening) Kang Usama Malik Eric Leung* * MD at time of publication
Notes: • Can be focal (single lobe/segment) or diffuse (both lungs) • Mainly in elderly • 1% prevalence in children
Tissue damage
Epithelial destruction of airways
Further impairment of bacterial clearance
Persistent inspiratory adventitious sounds (crackles > wheezing)
Complications
Structural damage to bronchial walls
Obstructive pulmonary function tests
Hemoptysis
Chest pain
VQ mismatch and 4, gas exchange
4, oxygenation
Digital clubbing (rare)
Fatigue Dyspnea
Cyanosis (uncommon)")
Chronic Thromboembolic Pulmonary Hypertension (CTEPH) Pathogenesis
: Pathogenesis
Acute Pulmonary Thromboembolic Event
Mechanical breakdown, Fibrinolysis
—5%: Incomplete thrombus resolution after 2 years (Etiology unknown)
*
Clot resolution (>90%)
Hypothesis 1: Increased hypercoagulability Hypothesis 2: Impaired clot lysis Hypothesis 3: Impaired angiogenesis Hypothesis 4: Inflammatory thrombosis • Associated with 1` levels of • Fibrin more resistant to • Impaired VEG-F function (unknown if cause or effect) plasma Factor VIII plasmin-mediated lysis • Ventriculoatrial shunts • Infected pacemaker wires • Splenectomy • Inflammatory bowel disease
Unresolved thromboemboli incorporates into blood vessel wall by fibrosis leading to fibrothrombotic organization
Authors: Dinusha T. Senaratne Reviewers: Midas (Kening) Kang Usama Malik Natalie Morgunov* Lian Szabo* * MD at time of publication
Legend:
Webs, bands and slow blood flow In-situ thrombosis expanding the fibrotic thrombus and branch occlusion Abbreviations: BP: Blood pressure PE: Pulmonary embolism PH: Pulmonary hypertension RAD: Right atrial dilatation RHF: Right heart failure RVHD: Right Ventricular hypertrophy/dilatation RVP: Right ventricular pressure VEGF: Vascular endothelial growth factor Proliferation of vascular and inflammatory cells proximal to lesion sites Adaptive vascular remodeling BP in patent vessels of the pulmonary vasculature For signs and symptoms refer to PH slide ■ Progressive PH
CTEPH (Chronic Thromboembolic pulmonary hypertension): Chronic occlusion of the pulmonary arteries due to intraluminal fibrosis of thromboembolic material from unresolved PE clots
Pathophysiology Mechanism
Progressive'(` RVP causes RAD and RVH/D
Sign/Symptom/Lab Finding
Progressive RHF
Complications
For signs and symptoms refer to RHF slide")
Lung cancer clinical findings and paraneoplastic syndromes

Obstruction of proximal airway
Inability to clear inhaled pathogens Postobstructive pneumonia
Cough, fever, dyspnea
Local tumor growth
Spread of tumor to pleural surface
Chest Pleural discomfort effusion
• Obstruction or compression at local site
Uncontrolled abnormal cell growth in one or both lungs 4 Lung Cancer
Airway invasion
Hemoptysis
Lambert-Eaton syndrome (Production of auto-antibodies against Calcium channels)
Muscle weakness
I` effort to Compression at the Compression Superior vena ventilate the laryngeal nerve of brachial cava lungs nerve plexus compression Impaired innervation to the vocal cords Dyspnea Shortness of Arm/shoulder/ Face/arm breath Voice hoarseness neck pain edema
Legend: Pathophysiology Mechanism
Sign/Symptom/Lab Finding
Authors: Yoyo Chan Reviewers: Midas (Kening) Kang Usama Malik Leila Barss* * MD at time of publication
Tumor secretes biologically active substances
Paraneoplastic Syndromes 4 Associated symptoms with malignant diseases
TGF131 extracellular matrix protein
Fingers clubbing
PTHrP T calcium release from bones
Hypercalcemia Serum calcium >2.6 mmol/L
ADH 1 SIADH T water reabsorption 1
Hyponatremia Serum sodium <135mEq/L
Abbreviations: • ACTH: Adrenocorticotropic hormone • ADH: Anti-diuretic hormone • PTHrP: Parathyroid hormone-related protein • SIADH: Syndrome of inappropriate antidiuretic hormone production • TGFI31: Transforming growth factor beta 1
1` ACTH
cortisol release and production
Cushing's syndrome (symptoms and signs caused by prolonged cortisol exposure)
Muscle weakness, hyperglycemia, severe hypokalemia")
Impetigo Pathogenesis and clinical findings

Pressure Ulcers Pathogenesis and clinical findings

Must remove slough/eschar to determine stage
-110.
Kennedy terminal ulcer (often precedes death)
—1111.
Pressure Ulcers: Pathogenesis and clinical findings
Bed, wheelchair, stretcher, car seat
External physical compression
Involuntary muscle movement, passive repositioning of torso Shear forces (dermis/epidermis fixed through contact with a surface while deeper tissues are moved; vessels angulate and thrombose, creating undermining of ulcer)
Inability to move well, aging skin (loss of elasticity, blood flow, and subcutaneous fat) Friction (person dragged across surface, damaging stratum corneum)
Bowel/bladder incontinence, diaphoresis, wound drainage Moisture (skin maceration)
4, in movement (coma, neuro injury, post-surgery, etc.) Limited mobility
Unrelieved pressure greater than arterial capillary pressure (>32 mmHg, with more rapid ulcer formation at higher pressures; normal range 12-32 mmHg) Disrupts blood supply and deprives tissues of oxygen and nutrients Pressure Ulcer (local injury to skin and/or underlying tissues, often over bony prominence)
Authors: Rebecca (Becky) Phillips Reviewers: Gurleen Chahal Usama Malik Laurie M. Parsons* * MD at time of publication
Notes: • More common in ages 65+ • Risk factors: diabetes, peripheral arterial disease, immunodeficiency, steroid therapy, smoking, dementia, poor nutrition, sensory deficit, circulatory disturbance, prolonged immobility • Grading system from National Pressure Ulcer Advisory Panel
Pear- or butterfly-shaped sacral ulcer
Stage I (non-blanchable erythema of intact skin; heralds impending ulcer)
May be warmer, painful, edematous, indurated, or discolored compared to surrounding tissue
Legend:
Stage II (partial thickness skin loss)
Erosion, serum-filled blister, or shallow ulcer with red-pink wound bed
Pathophysiology Mechanism
Stage III (full thickness skin loss; damage to subcutaneous tissue but not underlying fascia)
*
Exposed subcutaneous fat. May have slough, undermining, or tunneling (nose bridge, ear, occiput, and malleolar ulcers will appear shallow due to absence of subcutaneous tissue)
Sign/Symptom/Lab Finding
Stage IV (full thickness tissue loss)
Bone, tendon, or muscle exposed. Slough or eschar may be present. Often have undermining or tunneling
Complications
Bacterial invasion via contiguous spread (commonly S. Aureus and coagulase-negative staphylococci)
Osteomyelitis")
Benign Prostatic Hyperplasia: Pathogenesis and medications

Note: MoA not fully established
PDE-5 inhibitors (e.g. tadalafil)
Bladder and prostate smooth-muscle a-1 receptor antagonism
—110.
Relaxation of bladder outlet and prostate smooth-muscle
Authors: Michael Korostensky Reviewers: Alex Tang Usama Malik Dr. Jay Lee* * MD at time of publication
Acronyms: 5-ARI = 5-a reductase inhibitors COX = cyclooxygenase DHT = dihydrotestosterone GnRH = gonadotropin-releasing hormone LUTS = lower urinary tract symptoms PDE-5-mediated cGMP degradation in prostate smooth-Improved urinary outflow muscle and associated vascular supply Relaxation of prostate smooth-muscle MoA = mechanism of action NSAID = nonsteroidal anti-inflammatory drugs PDE-5 = phosphodiesterase-5 -NO
5-ARIs (e.g. dutasteride)
LHRH receptor antagonists (e.g. cetrorel ix)
P3-adrenergic agonists (e.g. mirabegron)
anticholinergics (e.g. oxybutynin)
NSAIDs
1` Bladder pressures
Pathophysiology Mechanism
5-a-reductase activity
1, Conversion of testosterone into DHT
4, Progression of LUTS
1, Testosterone secretion from testicular Leydig cells 1, LH secretion from pituitary GnRH antagonism DHT production Relaxation of detrusor Bladder muscle 1` capacity Improved LUTS
Acetylcholine antagonism at muscarinic receptors Relaxation of bladder outlet smooth-muscle 1` volume to first detrusor contraction 4, Prostaglandin release Analgesia and 4, Prostatic ,f, COX activity ► inflammation —110. Bladder smooth-muscle hyperplasia (detrusor thickening) /1` Sensitivity (i.e. overactive detrusor) -1110. 1, Volume to first detrusor contraction LUTS")
Erectile Dysfunction: Pathogenesis

1. Assess CVD Disease risk* a. I% Blood pressure b. I% Fasting glucose or HbA1c c. TG's & cholesterol 1. Penile duplex sonography 2. Cavernosometry
Legend:
Endocrinologic Erectile Dysfunction
Hypogonadism, hyperprolactinemia, hyperthyroidism, alcoholism, iatrogenic
.J, circulating free testosterone
•
1. 4, 7 AM free testosterone* 2. l• Thyroid Stimulating Hormone 3. l• Prolactin 4. l• Follicle Stimulating Hormone 5. l• Luteinizing Hormone
4, release of NO and cGMP levels within corpora cavernosa and smooth muscle relaxation
Pathophysiology Mechanism
Neurogenic Erectile Dysfunction
Neurologic disease, trauma, iatrogenic, diabetes mellitus
Central (cerebral or spinal cord); peripheral (afferent/sensory neuropathy) or efferent (autonomic neuropathy)
4, parasympathetic nerve firing
4, NO release
Psychogenic Erectile Dysfunction
•
Sudden onset, sporadic (circumstantial), younger, nocturnal/AM erection present
•
Anxiety, depression, strained relationship, lack of sexual arousal, psychological disorder
Possible mechanisms include an imbalance of central neurotransmitters, over inhibition of spinal erection center by the brain, and sympathetic overactivity
1. Abnormal Nocturnal penile 1. Normal Nocturnal penile tumescence and rigidity* tumescence and rigidity*
Erectile Dysfunction -• (persistent or recurrent inability to achieve an erection sufficient to achieve desired sexual performance)
Sign/Symptoni/Lab Finding
Complications
Authors: Braden Milian Reviewers: Alex Tang Usama Malik Jay C. Lee* * MD at time of publication")
Mixed Urinary Incontinence Pathogenesis and clinical findings
 4, Urinary leakage preceded by a sudden, strong urge to void
Overflow Incontinence vir Overfilling of the bladder from obstruction; BOO (tumour, stone, BPH, urethral or bladder neck stricture)
Detrusor Overactivity Ilr OAB (idiopathic), CNS lesion (neurogenic), inflammation/ infection (cystitis, UTI), diabetes mellitus
4. Bladder Wall Compliance
Progressive t in intravesicle pressure during bladder filling pushing urine from the bladder
Authors: Braden Millan Reviewers: Alex Tang Usama Malik Jay C. Lee* * MD at time of publication
Stress Urinary Incontinence (SUI) + Episodic involuntary urinary leakage with sudden l• in intra-abdominal pressure
4.
Urethral hypermobility, intrinsic sphincter deficiency, or a poorly coapting urethra
4,
4, Pelvic floor muscle and ligament strength causing 4. tone of vesicoureteral sphincter unit; 4, urethral strength and associated striated and smooth muscle; iatrogenic
Legend:
Failure to Void Weak Stream (+ dribbling), Intermittent, Straining, '1` PVR if a complication of urinary retention; obstruction visible on cystoscopy
Failure to Store Frequency, Urgency, Nocturia, Dysuria if SUI or UUI not caused by obstruction
Pathophysiology Mechanism
Urodynamic Studies SUI — 4, urethral closure pressure with 11` IAP/Bladder Volume and urinary leakage UUI — involuntary detrusor contraction and/or detrusor sphincter dyssynergia
Incontinence, 4, Quality of Life, UTI's")
Penyembuhan Luka Akut: Patogenesis dan Temuan Klinis

•
0 — 7 hari
Proliferasi (kolagen, matriks ekstrasel & pembuluh darah)
4 - 14 hari
Remodeling ► (.j, pemb. darah & kolagen tersusun)
Lapisan Epidermis Batas Dermis-Epidermis Lapisan Dermis
Vasokonstriksi transien
Pembentukan sumbat platelet
Jejas endotel dan subendotel
mengaktivasijalur koagulasi
Sel mast melepaskan histamin merespon iritan
Penulis: Amanda Eslinger Penyunting: Heena Singh Yan Yu Laurie Parsons* Penedemah: M Harmen Reza S* * MD (dokter) pada saat publikasi
Inflamasi I Proliferasi I Remodeling
Perdarahan berkurang atau berhenti karena sumbat hemostatik (hemostasis)
Permeabilitas pemb. kecil terhadap neutrofil & makrofag
Aktivasi komplemen memicu sel endotel sekitar untuk melepaskan prostaglandins
TGF-8*, dibentukselama fase inflamasi, menarikfibroblas ke daerah luka
Klot Menyatukan Ujung luka Perdarahan
Leukosit proliferasi dan
menyapu kotoran dan bakteri
1 Suplai vaskuler dan vasodilatasi di daerah luka
Makrofag melepaskan transforming growth factor beta (TGF-8) *
-110.
1' Tekanan hidrostatik mendorong cairan dari pembuluh jaringan sekitar
Fibroblas& makrofag menstimulasi pertumbuhanjaringan dan angiogenesis yang menggantikan sumbat hemostatik. Akhirnya, reepiteliasisasi terjadi
8 — 365+ hari
Patofisiologi Mekanisme
Kolagen tipe 1 yang mengalami tautan silang secara luas menggantikan kolagen yang berserakan yang tersusun pada fase proliferasi
Tanda/Gejala/Hasil Lab
Komplikasi
Luka Sembuh
kadar —* protein dalam kolagen
Eritema
Edema
Keropeng
Penyembuhan
Luka Parut (Scar)")
Penyembuhan Fraktur: Tahapan dan Faktor Pengganggu
 (osifikasi intramembran)
Penyembuhan tulang tahap inflamasi, kalus halus, and kalus keras
Penulis: Spencer Montgomery Penyunting: Yan Yu Dr. Gerhard Kiefer* Penerjemah: M Harmen Reza S* * MD (dokter) pada saat publikasi
Legenda:
Fraktur
Catatan: Penyembuhan fraktur melibatkan campuran antara jalur penyembuhan primer dan sekunder
Tahap Inflamasi (0-7Hari)
Stabilitas relatif pada lokasi fraktur: (pergerakan pada ujung tulang) - e.g. bidai, paku intramedular, traksi
Penyembuhan tulang sekunder (indirek) (osifikasi endokondral)
Kerusakan pembuluh darah lokal 4 hematoma 4 4, perfusi/02 ke tulang 4 osteonekrosis pada garis fraktur 4 terbentuk inflamasi lokal
Pergerakan pada lokasi fraktur
++ nyeri
Tahap Kalus Halus (mgg 1— 3)
Tahap Kalus Keras (mgg 3 — bin 3)
Remodeling (bin — thn)
Patofisiologi Mekanisme
• 1Kondrosit menyusun tulang rawan di lokasi hematoma, menjembatani kedua ujung tulang 4 nyeri berkurang
1
Osteoblas mengendapkan Ca3(PO4)2 ke matriks tulang rawan, membentuk kalus,1` stabilitas lokasi fraktur
Faktor yang dapat mengganggu penyembuhan fraktur
Tembakau Memperlama waktu penyembuhan, mekanisme belum jelas. Tiga hipotesis: 1) Nikotin: 4, aliran darah, dapat bersifat toksik pada osteoblas 2) Karbon Monoksida: 4, 02 ke lokasi fraktur 3) Hidrogen Sianida: menginhibisi metabolisme oksidatif pada tingkat sel
Penyalahgunaan alkohol Memperlama waktu penyembuhan, mekanisme tidak diketahui
Kortikosteroid & AINS jangka panjang Menghalangi respon inflamasi yang membatu penyembuhan
Remodeling tulang oleh pasangan Osteoklas-osteoblas : mengikir kalus agar tulang dapat mencapai bentuk efisien, sepanjang jalur gaya mekanisnya
Tanda/Gejala/Penunjang
Komplikasi
Kuinolon Menyebabkan pembentukan kalus imatur
Defisiensi Vitamin C Mengurangi pembentukan kolagen (Vit C adalah kofaktor kunci sintesis kolagen)
Diabetes Produksi kalus lemah (studi hewan)
Rifampicin & gentamycin topikal Toksik terhadap osteoblas
Hipotiroidisme Menginhibisi osifikasi endokondral (studi hewan)
Defisiensi Vitamin D Kurangnya absorpsi Ca2+ & Fosfat dari saluran cerna, 4, mineralisasi tulang.")
RICE: Mekanisme Aksi

Ice (diaplikasikan pada lokasi cedera)
Compression (pembalutan lokasi cedera)
Elevation (tungkai dinaikkan lebih tinggi dari jantung)
Mengurangi aliran darah ke jaringan
Mekanisme belum dipahami
Tekanan mekanis pada lokasi cedera
_110,
pengiriman PMN dan makrofag ke lokasi luka
Mencegah jejas lanjutan terhadap jaringan yang terkena beban mekanis produksi sitokin (bahan-bahan proinflamasi) seperti TNF-a, PDGF, (3- FGF, EGF, dan TG F-(3 Inflamasi Penulis: Matthew Roberts Penyunting: Alexander Arnold Amanda Eslinger Bradley Jacobs* Penerjemah: M Harmen Reza S* * MD (dokter) pada saat publikasi • ►
Cairan berlebih terdorong kembali ke kapiler dan limfe
Gravitasi pengembalian darah vena menuju sirkulasi sistemik
Abbreviations: • PMN- Netrofil Polimorfonuklear • TNF-a- Tissue Necrosis Factor-Alpha • PDGF- Platelet Derived Growth Factor • 13-FGF- Basic Fibroblast Growth Factor • EGF- Epidermal Growth Factor • TGF-13- Transforming Growth Factor Beta
Legenda: Patofisiologi Mekanisme
4, Edema (akumulasi cairan di ruang interstisial)
*Note: Penyembuhan cedera merupakan keseimbangan antara mengontrol nyeri dan inflamasi yang cukup untuk memulai aktivitas. Tujuan utama RICE adalah untuk menurunkan inflamasi. Aktivitas itu sendiri menimbulkan nyeri dan inflamasi, namun merupakan faktor penting dalam proses rehabilitasi.
Tanda/Gejala/Penunjang
► 4, Nyeri
1% Rentang gerak (Range of motion), dan juga fungsi
1
Inisiasi olahraga rehabilitatif spesifik dini untuk memperbaiki rentang gerak, kekuatan dan propriosepsi
Tekanan pada lokasi cedera akan menyebabkan robekan mikro pada jaringan lalu menginduksi inflamasi dan perbaikan* Otot, tendon, tulang, atau ligamen yang cedera menjadi lebih kuat
Penyembuhan dini")
Hemostasis Sekunder: Kaskade Koagulasi
:
F8a
Ca 2+
F5a
Protrombin (F2*)
Uji Laboratorium Rutin: • PT — Lama terbentuknya sumbatan setelah aktivasi jalur ekstrinsik • INR — PT yang dinormalisasi • PTT — Lama terbentuknya sumbatan setelah aktivasi jalur intrinsik
Jalur ekstrinsik: Jejas pada jaringan
Kerusakan sel endotel vaskuler akan memaparkan sub-endothelial Tissue Factor (Faktor jaringan)
F10* F10a*
Ca2+
Ca2+
• Faktor jaringan (Tromboplastin)
F7a*
Kompleks faktor jaringan — F7a*
► Trombin (F2a*)
Fibrinogen (F1)
Defisiensi jalur intrinsik
Ca2+
► Fibrin (Fla) Koagulasi Pembentukan sumbatan (clot) Fibrin
Defisiensi jalur Defisiensi jalur ekstrinsik bersama
Penulis: Christina Schweitzer Penyunting: David Lincoln Yan Yu* Lynn Savoie* Penerjemah: M Harmen Reza S* * MD (dokter) pada saat publikasi
Abbreviations: • PT — Prothrombin Time • INR - International Normalized Ratio • PTT — Partial Thromboplastin Time • F — Coagulation Factor (Faktor koagulasi) • a —Activated coagulation factor (Faktor koagulasi teraktivasi) • N— Normal • HMWK — High molecular weight kininogen • * — bergantung Vitamin K (untuk info lebih lanjut, lihat slide Vitamin K Deficiency)
Jembatan keledai (tidak diterjemahkan): • PT = Ekstrinsik: Play Tennis outside • PTT = Intrinsik: Play Table Tennis Inside • Faktor intrinsik — TENET: Twelve, Eleven, Nine, Eight, Ten • Faktor jalur bersama — 10/5=2, 2/2=1 (F10, 5, 2, 1) • Faktor bergantung Vitamin K— 1972: F10, 9,
7, 2
Jalur intrinsik, ekstrinsik & bersama normal
N PT, l• PTT 1` PT, N PTT t PT, t PTT N PT, N PTT
Perdarahan Memanjang")
Fisiologi sistem Renin-Angiotensin-Aldosteron (RAAS)

Hipoperfusi Ginjal
Regangan baroreseptor pada dinding arteriol aferen
Hipotensi/ Hipovolemia
9 Pengiriman NaCI ke Makula Densa
9 Tekanan dirasakan oleh Baroreseptor Jantung & Arteri
Katekolamin di sirkulasi
Aktivitas saraf simpatis pada arteriol aferen
Stimulasi reseptorn-adrenergik pada arteriol aferen
Keseimbangan Glomerulotubuler (Mekanisme intrinsik ginjal diluar RAAS) *Hanya sebagian dari mekanisme tersebut
Angiotensin II 4 konstriksi arteriol eferen
01% Tekanan hidrostatik glomerulus
Jumlah cairan yang disaring
+ Konsentrasi sisa protein darah di glomerulus
Darah kental bergerak dari glomerulus menuju kapiler peritubuler
it pada kapiler peritubuler
9 Tekanan hidrostatik peritubular
Legenda:
Patofisiologi Mekanisme
Pelepasan Renin oleh sel-sel Jukstaglomerular pada arteriol aferen 4 berujung pada T Angiotensin II
Sekresi aldosterone dari korteks adrenal
1
Insersi KNaE pada Sel Prinsipal di DK
■
Aktivitas transporter NHE3 di dalam PCT
Reabsorpsi Na ke dalam darah, menarik H2O ke dalam darah melalui osmosis
Reabsorpsi H2O langsung ke dalam sirkulasi darah (via aliran gradien tekanan onkotik dan hidrostatik H2O)
Tanda/Gejala/Penunjang
Komplikasi
Author: David Waldner Reviewers: Yan Yu Sean Spence Sophia Chou* Penerjemah: M Harmen Reza S* * MD (dokter) pada saat publikasi
Hati secara normal mensintesis angiotensinogen dengan laju basal, melepaskannya ke dalam sirkulasi darah:
Vasokonstri ksi arteri sistemik
Tekanan darah
Daftar singkatan: • ACE: Angiotensin Converting Enzyme (disintesis oleh Ginjal dan Paru) • DK: Duktus Kolektivus • PCT: Tubulus Konturtus Proksimal • NHE3: Sodium Hydrogen Exchanger (Antiport) 3 • KNaE: Kanal Natrium (Sodium) Epitel • n:Tekanan onkotik")
Gradien pO2 Alveolus-arteri: Mengapa ada, dan mengapa penting
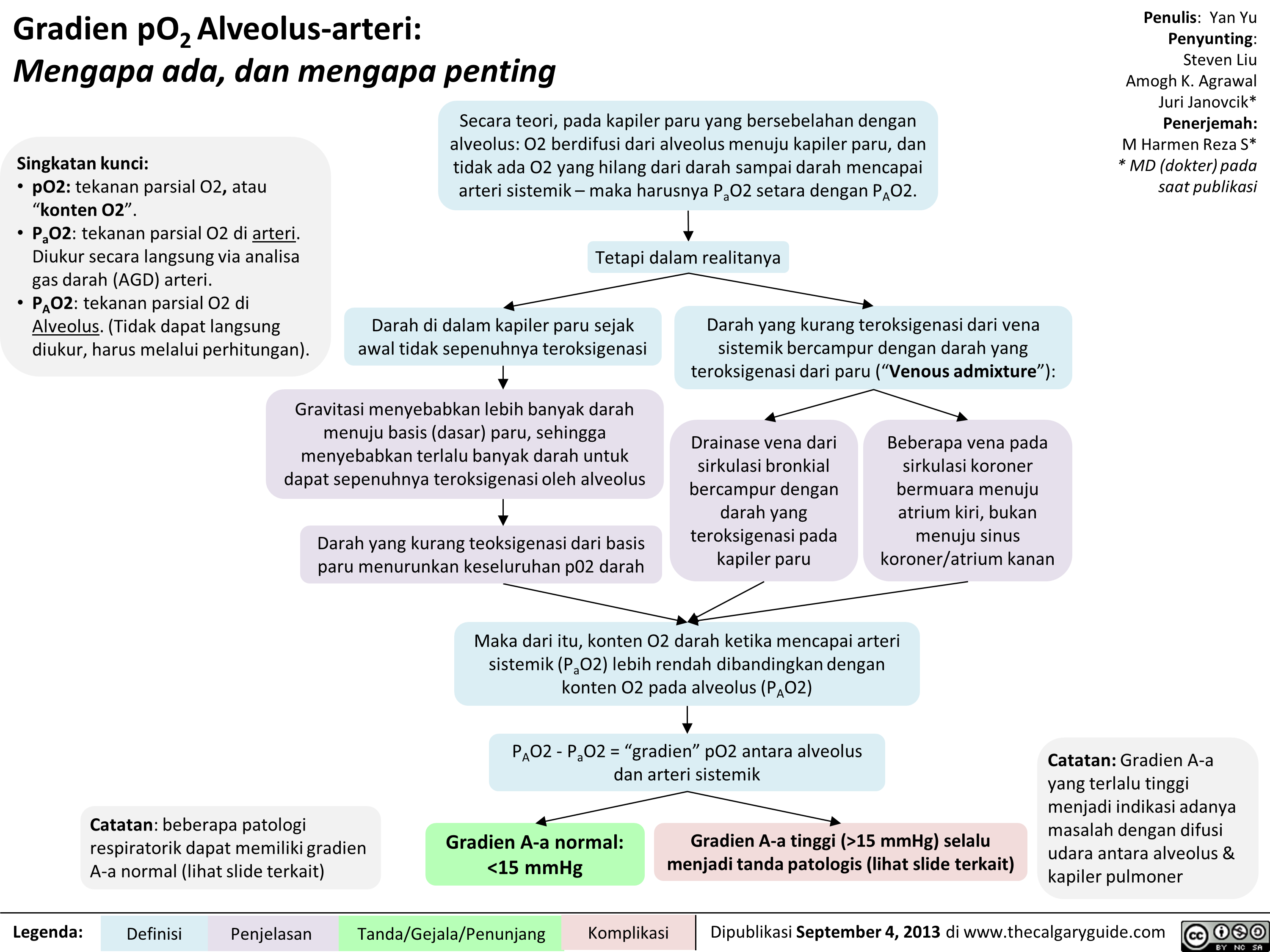 15 mmHg) selalu menjadi tanda patologis (lihat slide terkait)
Penulis: Yan Yu Penyunting: Steven Liu Amogh K. Agrawal Juri Janovcik* Penerjemah: M Harmen Reza S* * MD (dokter) pada saat publikasi
Catatan: Gradien A-a yang terlalu tinggi menjadi indikasi adanya masalah dengan difusi udara antara alveolus & kapiler pulmoner " title="Gradien pO2Alveolus-arteri: Mengapa ada, dan mengapa penting
Singkatan kunci: • p02: tekanan parsial 02, atau "konten 02". • Pa02: tekanan parsial 02 di arteri. Diukur secara langsung via analisa gas darah (AGD) arteri. • PA02: tekanan parsial 02 di Alveolus. (Tidak dapat langsung diukur, harus melalui perhitungan).
Secara teori, pada kapiler paru yang bersebelahan dengan alveolus: 02 berdifusi dari alveolus menuju kapiler paru, dan tidak ada 02 yang hilang dari darah sampai darah mencapai arteri sistemik— maka harusnya Pa02 setara dengan PA02.
1
Tetapi dalam realitanya
Darah di dalam kapiler paru sejak awal tidak sepenuhnya teroksigenasi
Gravitasi menyebabkan lebih banyak darah menuju basis (dasar) paru, sehingga menyebabkan terlalu banyak darah untuk dapat sepenuhnya teroksigenasi oleh alveolus
Darah yang kurang teoksigenasi dari basis paru menurunkan keseluruhan p02 darah
Catatan: beberapa patologi respiratorik dapat memiliki gradien A-a normal (lihat slide terkait)
Legenda: Definisi
Penjelasan
Darah yang kurang teroksigenasi dari vena sistemik bercampur dengan darah yang teroksigenasi dari paru ("Venous admixture"):
Drainase vena dari sirkulasi bronkial bercampur dengan darah yang teroksigenasi pada kapiler paru
Beberapa vena pada sirkulasi koroner bermuara menuju atrium kiri, bukan menuju sinus koroner/atrium kanan
.111••••■•
Maka dari itu, konten 02 darah ketika mencapai arteri sistemik (Pa02) lebih rendah dibandingkan dengan konten 02 pada alveolus (PA02)
PA02 - Pa02 = "gradien" p02 antara alveolus dan arteri sistemik
Gradien A-a normal: <15 mmHg
Tanda/Gejala/Penunjang
Gradien A-a tinggi (>15 mmHg) selalu menjadi tanda patologis (lihat slide terkait)
Penulis: Yan Yu Penyunting: Steven Liu Amogh K. Agrawal Juri Janovcik* Penerjemah: M Harmen Reza S* * MD (dokter) pada saat publikasi
Catatan: Gradien A-a yang terlalu tinggi menjadi indikasi adanya masalah dengan difusi udara antara alveolus & kapiler pulmoner " />
15 mmHg) selalu menjadi tanda patologis (lihat slide terkait)
Penulis: Yan Yu Penyunting: Steven Liu Amogh K. Agrawal Juri Janovcik* Penerjemah: M Harmen Reza S* * MD (dokter) pada saat publikasi
Catatan: Gradien A-a yang terlalu tinggi menjadi indikasi adanya masalah dengan difusi udara antara alveolus & kapiler pulmoner " title="Gradien pO2Alveolus-arteri: Mengapa ada, dan mengapa penting
Singkatan kunci: • p02: tekanan parsial 02, atau "konten 02". • Pa02: tekanan parsial 02 di arteri. Diukur secara langsung via analisa gas darah (AGD) arteri. • PA02: tekanan parsial 02 di Alveolus. (Tidak dapat langsung diukur, harus melalui perhitungan).
Secara teori, pada kapiler paru yang bersebelahan dengan alveolus: 02 berdifusi dari alveolus menuju kapiler paru, dan tidak ada 02 yang hilang dari darah sampai darah mencapai arteri sistemik— maka harusnya Pa02 setara dengan PA02.
1
Tetapi dalam realitanya
Darah di dalam kapiler paru sejak awal tidak sepenuhnya teroksigenasi
Gravitasi menyebabkan lebih banyak darah menuju basis (dasar) paru, sehingga menyebabkan terlalu banyak darah untuk dapat sepenuhnya teroksigenasi oleh alveolus
Darah yang kurang teoksigenasi dari basis paru menurunkan keseluruhan p02 darah
Catatan: beberapa patologi respiratorik dapat memiliki gradien A-a normal (lihat slide terkait)
Legenda: Definisi
Penjelasan
Darah yang kurang teroksigenasi dari vena sistemik bercampur dengan darah yang teroksigenasi dari paru ("Venous admixture"):
Drainase vena dari sirkulasi bronkial bercampur dengan darah yang teroksigenasi pada kapiler paru
Beberapa vena pada sirkulasi koroner bermuara menuju atrium kiri, bukan menuju sinus koroner/atrium kanan
.111••••■•
Maka dari itu, konten 02 darah ketika mencapai arteri sistemik (Pa02) lebih rendah dibandingkan dengan konten 02 pada alveolus (PA02)
PA02 - Pa02 = "gradien" p02 antara alveolus dan arteri sistemik
Gradien A-a normal: <15 mmHg
Tanda/Gejala/Penunjang
Gradien A-a tinggi (>15 mmHg) selalu menjadi tanda patologis (lihat slide terkait)
Penulis: Yan Yu Penyunting: Steven Liu Amogh K. Agrawal Juri Janovcik* Penerjemah: M Harmen Reza S* * MD (dokter) pada saat publikasi
Catatan: Gradien A-a yang terlalu tinggi menjadi indikasi adanya masalah dengan difusi udara antara alveolus & kapiler pulmoner " />
Gradien pO2 Alveolus-arteri: Penjelasan rumus (Penjelasan ringkas)

Untuk penjelasan fisiologis mengenai mengapa terdapat gradien A-a, silahkan lihat:")
Gradien pO2 Alveolus-arteri: Penjelasan rumus (Penjelasan secara ilmiah)

Untuk penjelasan fisiologis mengenai mengapa terdapat gradien A-a, silahkan lihat:")
Anticholinergics

Anticholinergics E.g. Atropine, Glycopyrrolate, Scopolamine
Abbreviations
ACh — Acetylcholine SA — Sino-atrial
Quick Facts
Primary indication in the OR = Combined with acetylcholinesterase inhibitors to prevent over-activation of the parasympathetic nervous system
Route of Administration = IV
Muscarinic (parasympathetic) effects
Competitive antagonism at muscarinic receptors in various organ systems
4, Binding sites for ACh at muscarinic receptors
Blockage of muscarinic ► receptors in SA node (esp. atropine)
► Smooth muscle relaxation in the bronchial airways
4, Respiratory tract mucosal ► secretions (esp. glycopyrrolate, scopola mine)
► Pupillary dilation
Tachycardia
Bronchorelaxation
Respiratory tract clearing
Mydriasis
► 4, Secretion of salivary glands —■ Dry mouth
► 4, Intestinal motility and peristalsis
► 4, Ureter and bladder tone
Constipation
Urinary retention
Legend: Pathophysiology Mechanism Sign/Symptom/Lab Finding Complications I Published March 3, 2018 on www.thecalgaryguide.com
0 0 4SI 0 I' it En")
non-depolarizing-neuromuscular-blocks-ndnmbs
 Eg. pancuronium, rocuronium, atracurium, vecuronium
Abbreviations NDNMBs — Non-Depolarizing Neuromuscular Blockers ACh — Acetylcholine
Quick Facts 1° Indication = Skeletal muscle paralysis to facilitate tracheal intubation, and used during indicated surgeries or mechanical ventilation
Route of Administration = IV
Metabolism & Excretion = Redistribution, hepatic clearance/renal excretion (extent varies greatly by drug). NOT degraded by acetylcholinesterase or pseudocholinesterase
See Acetylcholinesterase Inhibitors slide for reversal of NDN M Bs
Competitive antagonism at post-synaptic nicotinic receptors on muscles
Competitive antagonism at the pre-synaptic nicotinic receptors on neurons
Vagolytic effect (esp. pancuronium)
Anaphylactic/ anaphylactoid reactions
4, Binding sites for ACh at post-synaptic nicotinic receptors on muscles
4, Binding sites for ACh at pre-synaptic nicotinic receptors on neurons
Blockage of vagal muscarinic receptors in sinoatrial nodes
IgE antibodies attach to ammonium ion components of NDNMBs Non-immunologic mast cell degranulation (esp. atracurium)
4, Muscle cell depolarization
4, Positive feedback for continued ACh ► release in response to high frequency stimulation
Skeletal muscle paralysis
Tetanic fade, Train-of-Four fade
4, —• Parasympathetic Tachycardia effects on heart
Release of histamine from mast cells and basophils
Bronchospasm
Hypotension
Legend: Pathophysiology Mechanism Sign/Symptom/Lab Finding Complications I Published March 3, 2018 on www.thecalgaryguide.com
0€3,0 BY NC SA")
Propofol

4, Cerebral metabolic rate CNS —÷ 4, Cerebral oxygen consumption 4, Intra-cranial pressure
4, Systemic vascular resistance Hypotension ► Cardiovascular --■ —■ (with no change 4, Preload in heart rate) 4, Contractility
Respiratory /
♦ Hypercapnia 4, Hypoxic and --■ hypercapnic Hypoxia respiratory drive ♦ Apnea
4, Upper airway reflexes
Complications I Published MARCH 3, 2018 on www.thecalgaryguide.com
Lac.).T2")
Succinylcholine

Abbreviations ACh — Acetylcholine SA — Sino-atrial
Quick Facts 1° Indication = Skeletal muscle paralysis to allow tracheal intubation with the advantage of faster onset (30s-60s) and shorter duration (<10min) than non-depolarizing neuromuscular blocking agents
Route of Administration = IV
Metabolism & Excretion = redistribution and metabolism by pseudocholinesterase in blood plasma and liver
No reversal of succinylcholine available
Contraindicated in patients with traumatized, denervated, or immobilized muscles due to risk of cardiac arrest from hyperkalemia.
Agonist at nicotinic ACh receptors in muscles Generates action potential Not affected by synaptic acetylcholinesterase (unlike ACh)
Continuous end-plate depolarization
Inactivation of sodium channels
Prevention of repolarization and additional action potentials
Skeletal muscle paralysis
Succinylcholine mimics ACh in structure
♦ Fasciculation
♦ Myalgia
1` Serum lc (esp. in patients with muscle trauma/denervation/ immobilization)
1
Hyperkalemia
Malignant Hyperthermia (See Slide)
Agonist at nicotinic receptors in parasympathetic ganglia, sympathetic ganglia, and muscarinic receptors in SA node of heart
Parasympathetic effect (low dose succinylcholine) 4, Heart rate 4, Contractility
Cardiac arrest
1
Sympathetic effects (high dose succinylcholine)
t Heart rate t Contractility Catecholamine release
Legend: Pathophysiology Mechanism Sign/Symptom/Lab Finding Complications I Published March 3, 2018 on www.thecalgaryguide.com
0€3,0 BY NC SA")
Volatile Gases

Respiratory 4, Hypoxic and hypercapnic respiratory drive 4, Tidal volume 4, Alveolar ventilation Hypercapnia
1 t Respiratory rate
Musculoskeletal
Neuromuscular blockade and potentiation of NDNMBs
Muscle Relaxation
Malignant Hyperthermia (See Slide)
Complications I Published March 3, 2018 on www.thecalgaryguide.com
0 0 IS 0 riL - H")
non-depolarizing-neuromuscular-blocks-ndnmbs
 Eg. pancuronium, rocuronium, atracurium, vecuronium
Abbreviations NDNMBs — Non-Depolarizing Neuromuscular Blockers ACh — Acetylcholine
Quick Facts 1° Indication = Skeletal muscle paralysis to facilitate tracheal intubation, and used during indicated surgeries or mechanical ventilation
Route of Administration = IV
Metabolism & Excretion = Redistribution, hepatic clearance/renal excretion (extent varies greatly by drug). NOT degraded by acetylcholinesterase or pseudocholinesterase
See Acetylcholinesterase Inhibitors slide for reversal of NDN M Bs
Competitive antagonism at post-synaptic nicotinic receptors on muscles
Competitive antagonism at the pre-synaptic nicotinic receptors on neurons
Vagolytic effect (esp. pancuronium)
Anaphylactic/ anaphylactoid reactions
4, Binding sites for ACh at post-synaptic nicotinic receptors on muscles
4, Binding sites for ACh at pre-synaptic nicotinic receptors on neurons
Blockage of vagal muscarinic receptors in sinoatrial nodes
IgE antibodies attach to ammonium ion components of NDNMBs Non-immunologic mast cell degranulation (esp. atracurium)
4, Muscle cell depolarization
4, Positive feedback for continued ACh ► release in response to high frequency stimulation
Skeletal muscle paralysis
Tetanic fade, Train-of-Four fade
4, —• Parasympathetic Tachycardia effects on heart
Release of histamine from mast cells and basophils
Bronchospasm
Hypotension
Legend: Pathophysiology Mechanism Sign/Symptom/Lab Finding Complications I Published March 3, 2018 on www.thecalgaryguide.com
0€3,0 BY NC SA")
Succinylcholine
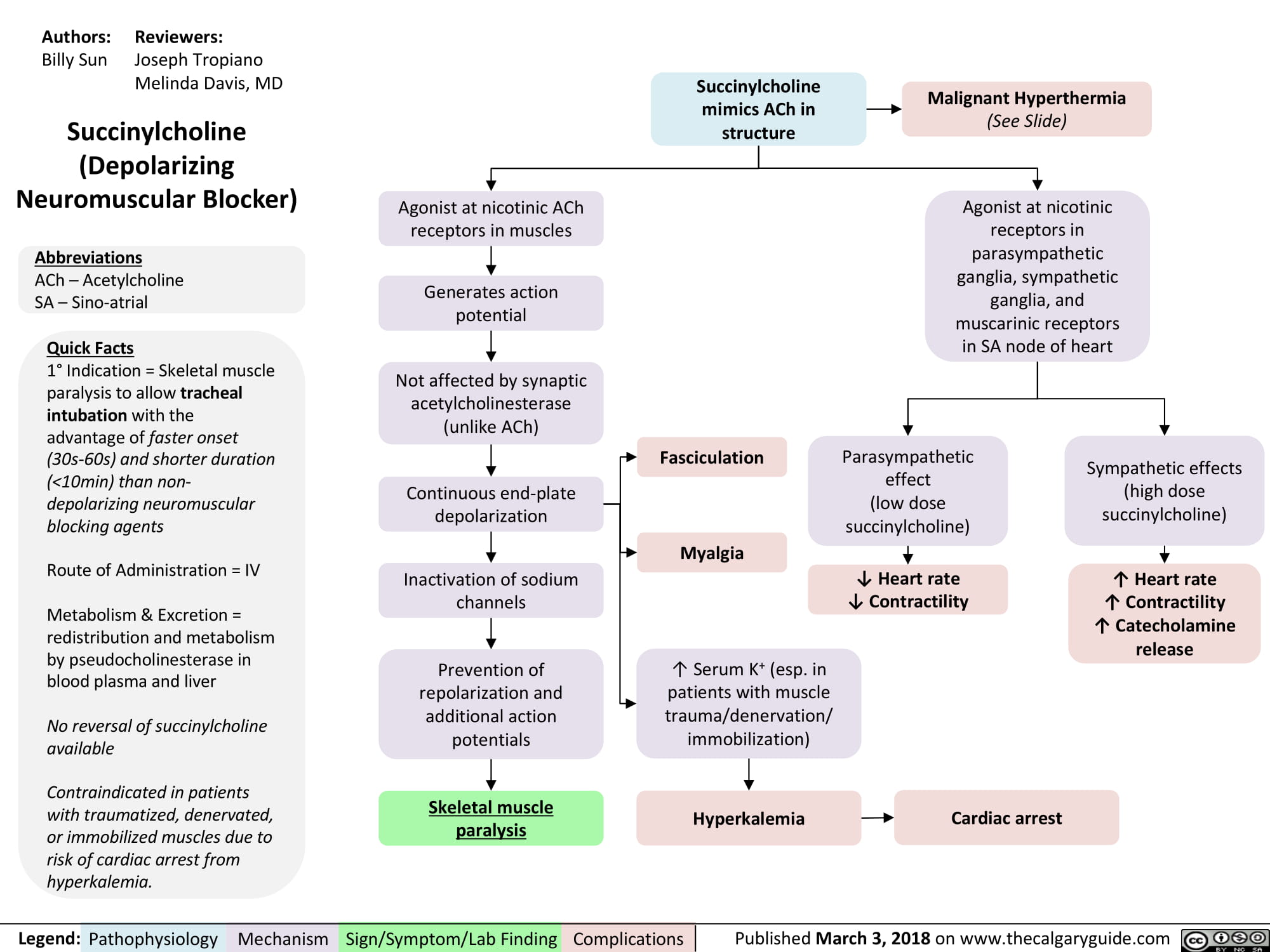
Clavicular Fracture: Pathogenesis and clinical findings
 (e.g. rowing) (87%) hand (6%) an object (7%) • • Acute fracture Stress fracture
Proximal third Group III (2.8%)
Undisplaced fracture
•
Shape of clavicle undergoes minimal change
Legend:
Middle third (mid-shaft) Group I (69%)
Clavicular Fracture
Displaced medial and lateral segments
Sternocleidomastoid muscle pulls the medial segment up
•••-•••11
Anterior Shoulder Pain
Pathophysiology Mechanism
Distal third Group II (29%)
Fracture is medial or lateral to the coracoclavicular ligaments
Pectoralis muscle and weight of arm pulls the lateral segment down and towards midline
Sign/Symptom/Lab Finding
Complications
Lateral Shoulder Pain
Authors: Jack Fu Reviewers: Reza Ojaghi Usama Malik Aaron J. Bois* * MD at time of publication
Notes: • There is no correlation between the mechanism of injury and the site of fracture (i.e., which")
Radiological findings of child abuse
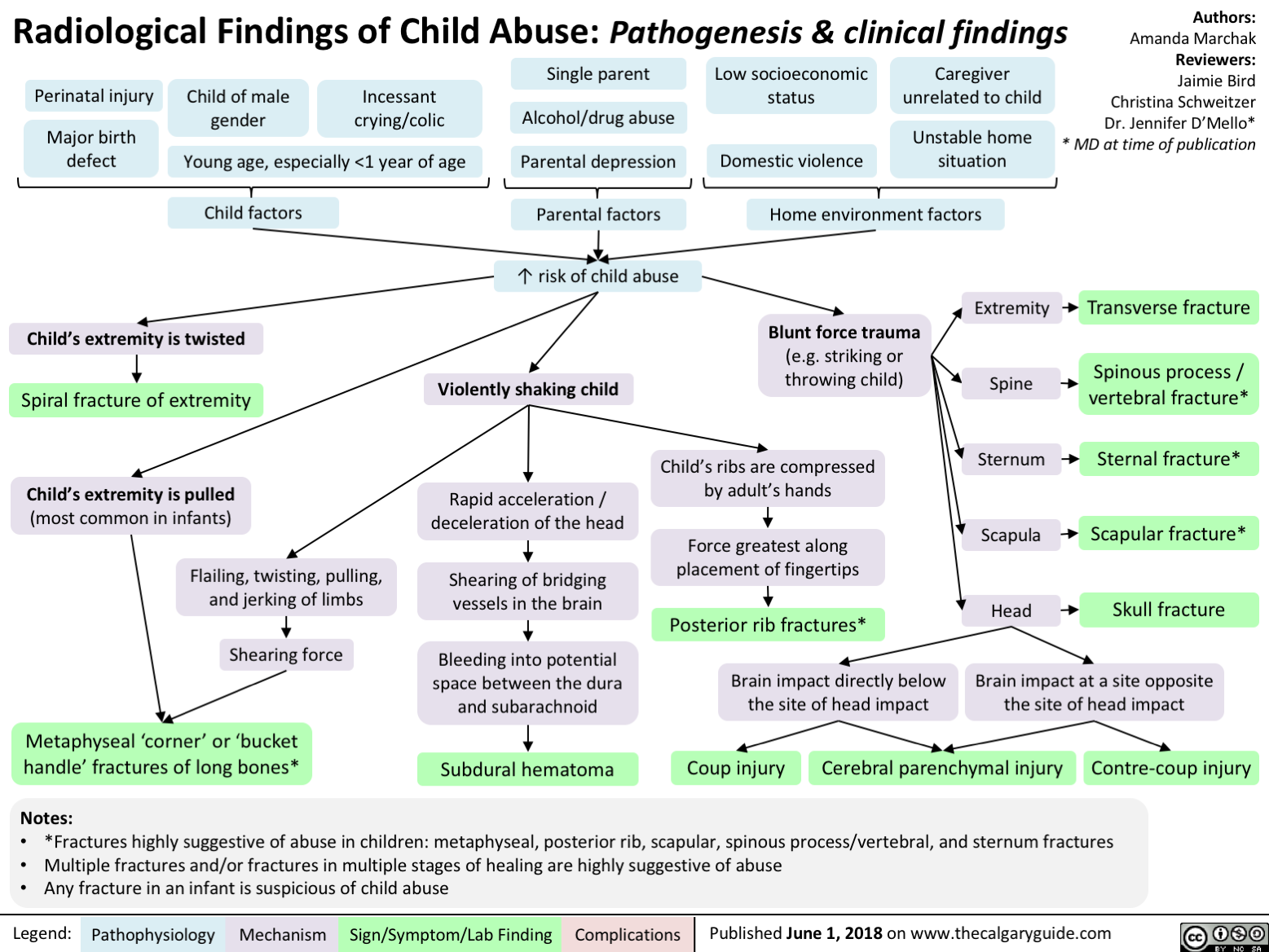
Non-Accidental Head Trauma
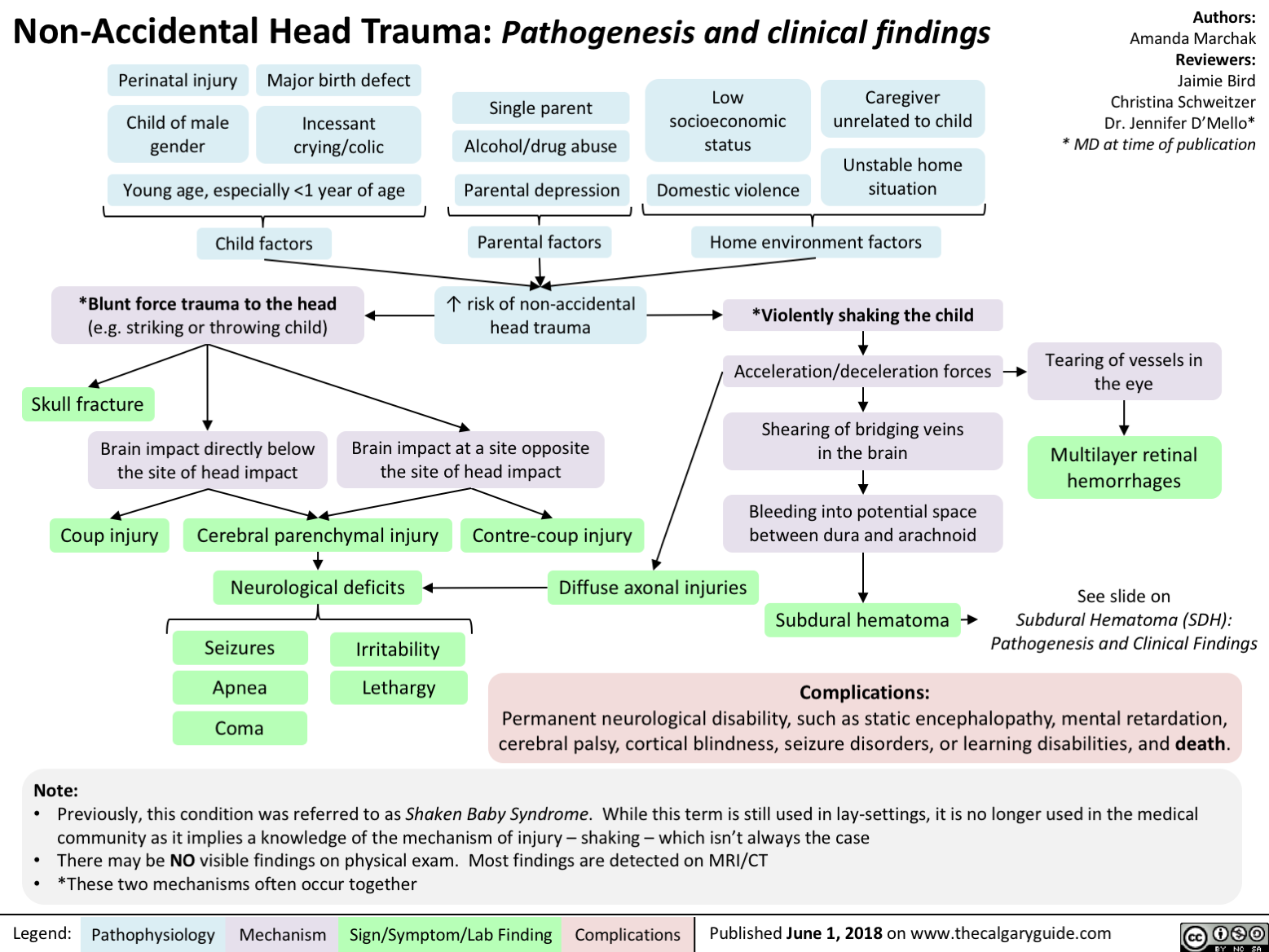
Duchenne Muscular Dystrophy
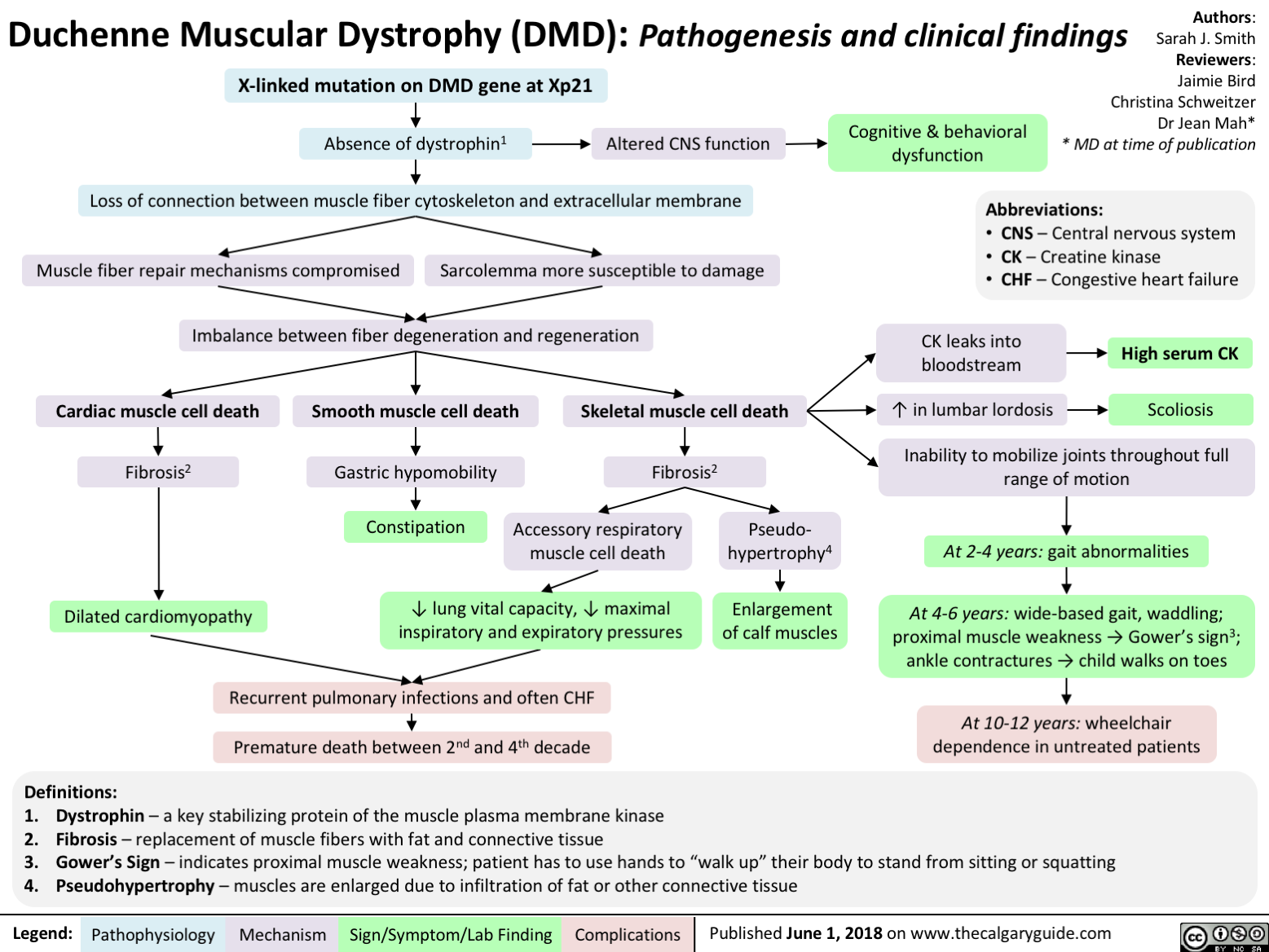
Celiac Disease: Pathogenesis and clinical findings

Genetic predisposition -■ (Northern European, Down's syndrome, Associated with HLA DQ 2,8)
Note: *The anti-TTG antibody is an IgA anti-body, therefore if the patient is IgA-deficient, absence of anti-TTG does not rule out celiac
Anti-TTG in serum*
Anti-TTG reacts with TTG in skin
Deposition of anti-TTG in renal glomeruli
Dermatitis herpetiformis
Chronic Kidney Disease
Small Bowel Biopsy: Crypts of bowel become enlarged (hyperplasia) with architectural change, villous shortening
Legend: Pathophysiology
Mechanism
Exposure to prolamins (proteins found in wheat, rye, oats, barley)
TTG alters prolamin Altered protein fits more easily into HLA
HLA activates adaptive immunity
IgA generated against prolamin-TTG
Wheat prolamin (gliandin) interacts with and activates zonulin signalling
Gut epithelium becomes more porous
Large dietary proteins in epithelium disrupt tight junctions
Author: Matthew Harding Yan Yu Peter Bishay Reviewers: Dean Percy Jason Baserman Usama Malik Kerri Novak* * MD at time of publication
Inflammation of Intestinal epithelium Inflammation disrupts structure of bowel mucosa Mechanism Unknown Lymphocytes migrate to site of inflammation Extraintestinal Complications: Arthropathy Ataxia (gluten associated) Infertility Mild Hepatitis Villi of intestine atrophy Risk of Microscopic Colitis (50x) Malabsorption Extraintestinal manifestations: Chronic watery Fatigue Failure to thrive, weight loss Anemia (Fe, B12, folate) Peripheral neuropathy (B12, Ca) Ataxia (Ca) Dysrhythmia (Ca, K) Osteoporosis (Vit D, Ca) diarrhea + Intestinal manifestations steatorrhea: Pale, foul-smelling Abdominal bloating Steatorrhea (fat in stool) Diarrhea")
Celiac Disease: Complications
 Carbohydrate Protein Fat Secretory maldigestion maldigestion malabsorption diarrhea
Legend:
Fermentation by gut bacteria 1 Gas production
Bloating
Fat retained in stool
Steatorrhea
Abdominal pain
Pathophysiology Mechanism
Sign/Symptom/Lab Finding
Growth Retardation
Authors: Yoyo Chan Reviewers: Peter Bishay Usama Malik Sylvain Coderre* * MD at time of publication
IgA response
Autoimmune IgA deposits Lymphocyte response in sub-epidermal skin layer against enamel
Dermatitis Herpetiformis (Chronic pruritic blisters)
Nutritional deficiency
Dental enamel hypoplasia
Vitamin D and calcium deficiency
Zinc, selenium Folate Iron Osteoporosis deficiency deficiency deficiency Anemia t Risk of miscarriages")
Orofacial Clefts cleft lip cleft palate
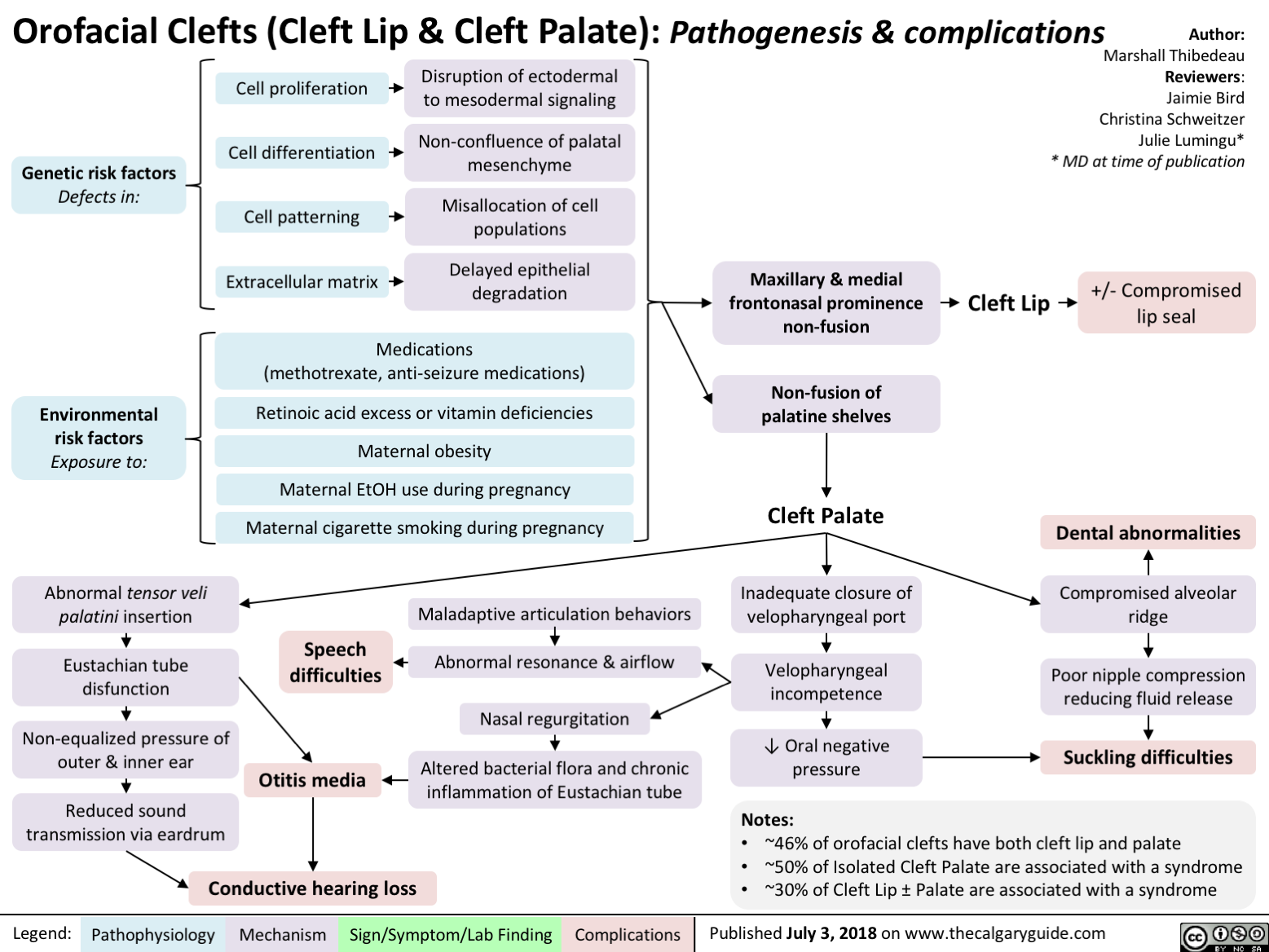
Cerebral Palsy clinical findings
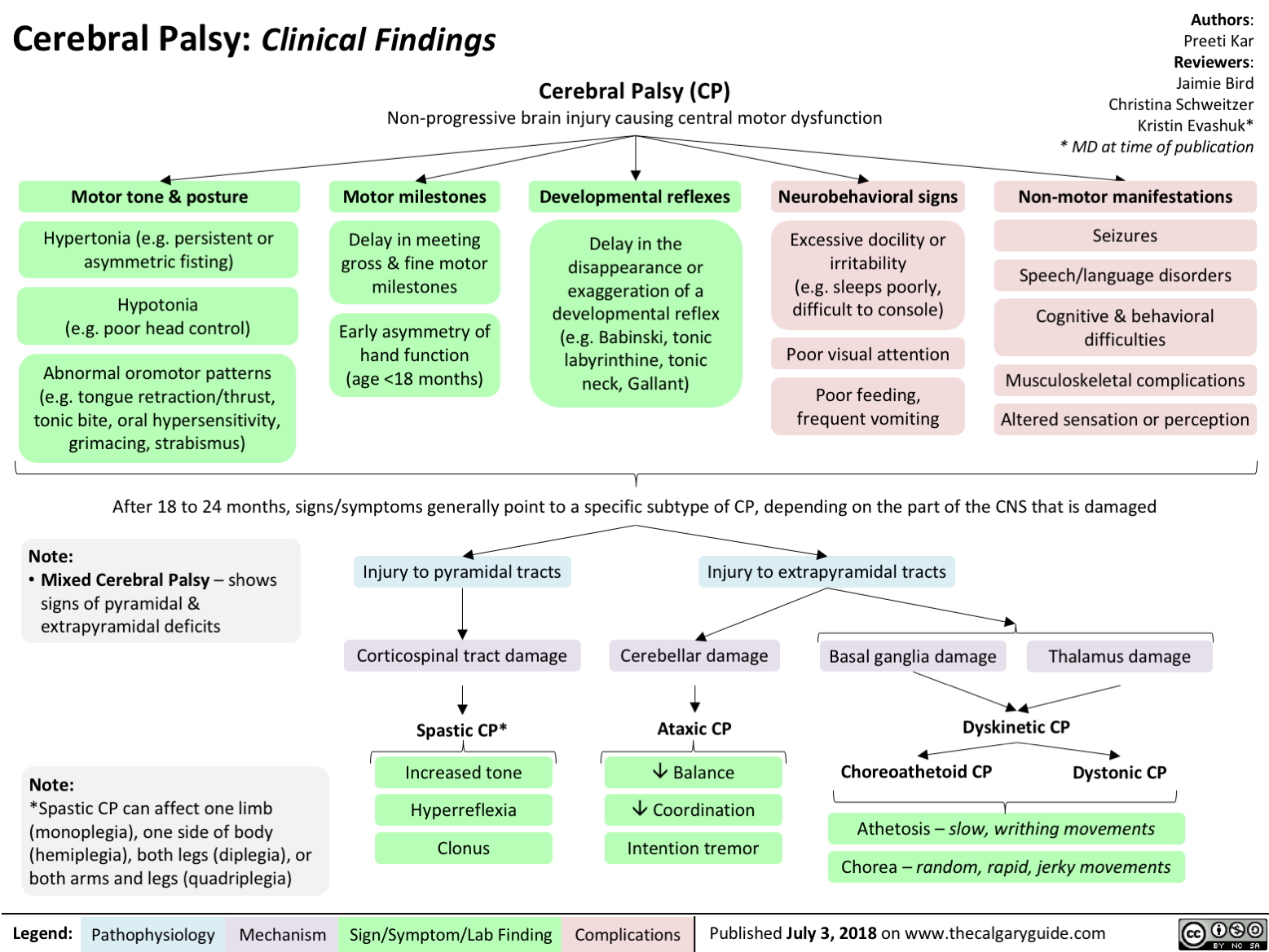
Developmental Coordination Disorder
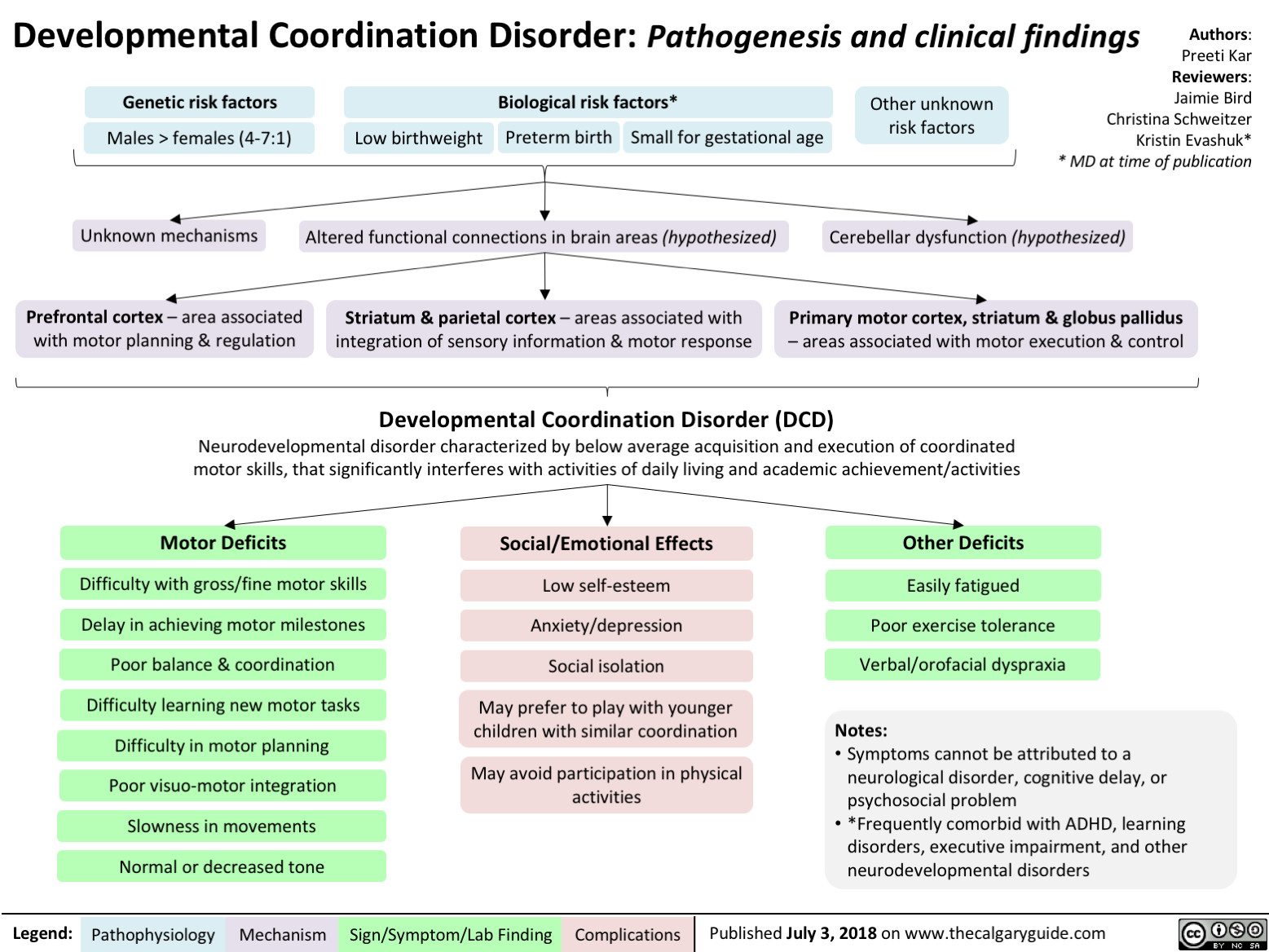
Sudden Infant Death Syndrome SIDS Triple Risk Model

Aplastic Anemia: Pathogenesis and Clinical Findings
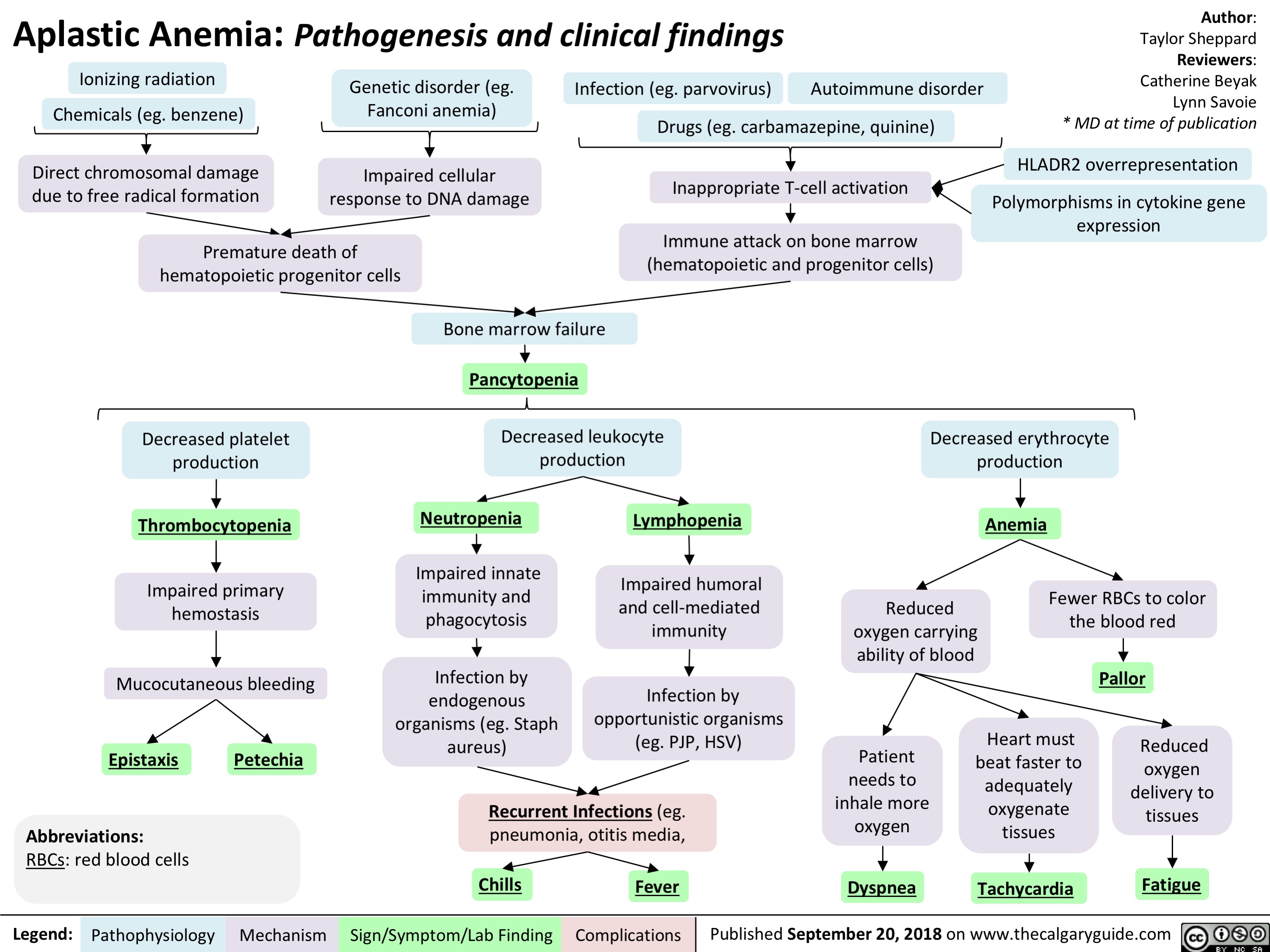
Feedback Loops Growth Hormone
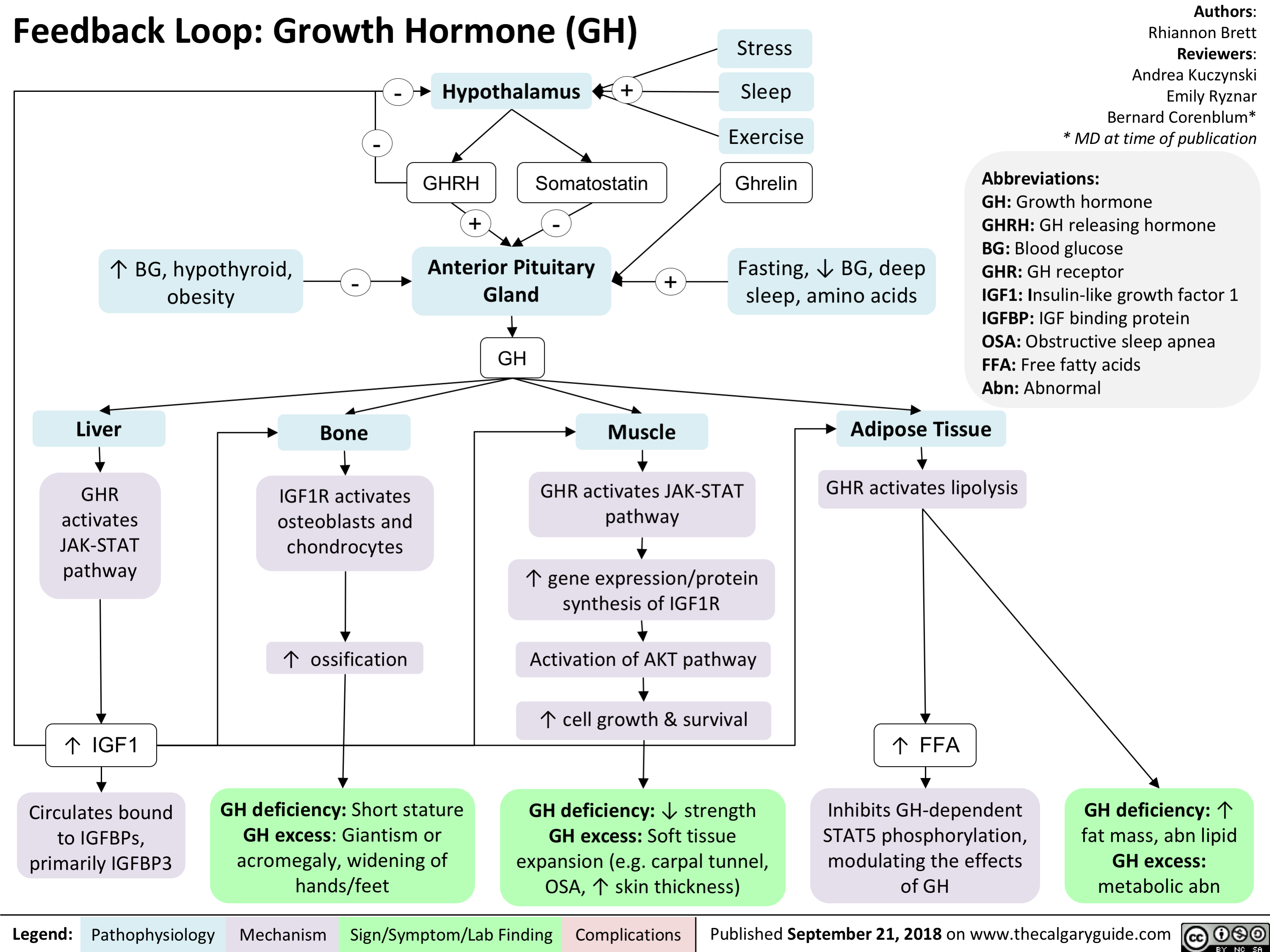
Kallmann Syndrome and Normosmotic Idiopathic Hypogonadotropism: Pathogenesis and Clinical Findings
 and Normosmic Idiopathic Hypogonadotropic Hypogonadism (nIHH): Pathogenesis and clinical findings
Authors: Danielle Lynch Reviewers: Nicola Adderley Josephine Ho* * MD at time of publication
Abbreviations:
GnRH – gonadotropin- releasing hormone
LH – luteinizing hormone FSH - follicle stimulating hormone
Notes:
• 4 male : 1 female
• KS, nIHH, and isolated
anosmia exist on a
spectrum
• KS has X-linked recessive,
autosomal dominant, autosomal recessive,
oligogenic, and idiopathic
forms
• KS and nIHH are also
associated with gene- specific features such as
cleft lip/palate, oculomotor synkinesis, hearing loss, and unilateral renal aplasia
• Diagnosis typically occurs around pubertal age, but may be earlier in males if micropenis and cryptorchidism are present at birth
Mutations in KAL1, NELF, and PROKR2
Abnormal olfactory bulb development
Mutations in FGFR1, FGF8, PROK2, PROKR2, HS6ST1, CHD7, WRD11, and SEMA3A or idiopathic
Mutations in
GNRH1, KISS1, KISS1R, TAC3, TACR3
Impaired migration of olfactory neurons from olfactory bulb to hypothalamus
Impaired migration of GnRH neurons from olfactory bulb to hypothalamus
Impaired GnRH neuron activity
Kallmann syndrome
(idiopathic hypogonadotropic hypogonadism with anosmia)
Normosmic idiopathic hypogonadotropic hypogonadism
Anosmia
Olfactory bulb aplasia
Abnormal /absent olfactory bulb on MRI
↓ GnRH ↓LH and ↓FSH
Delayed or absent puberty
Infertility
↓estrogen
In males: ↓testosterone
Cryptorchidism
Delayed epiphyseal plate closure
Micropenis (5-10%)
Uninhibited long bone growth
↑ length hands, arms, legs
Delayed bone age (hand/wrist x-ray)
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published September 29, 2018 on www.thecalgaryguide.com")
Feedback Loop: Adrenocorticotropic Hormone (ACTH)

Authors: Rhiannon Brett Reviewers: Andrea Kuczynski Bernard Corenblum* * MD at time of publication
Posterior Pituitary Gland
ADH
-
-
-
Hypothalamus
CRH
+
Anterior Pituitary Gland
ACTH, MSH, other hormones produced from POMC
ACTH
+
Adrenal Cortex
Activate ACTHR
Activate cAMP Activate PKA Activate Zona Fasciculata
Cortisol
Abbreviations:
CRH: Corticotropin Releasing Hormone
ADH: Anti-diuretic Hormone
ACTH: Adrenocorticotropic Hormone
MSH: Melanocyte Stimulating Hormone
POMC: Pro-Opiomelanocortin
ACTHR: ACTH Receptor
cAMP: Cyclic Adenosine Monophosphate
PKA: Protein Kinase A
DHEA(S): Dehydroepiandrosterone (sulfonated form) F: Female
M: Male
Excess: hirsutism, acne, oily skin, oligo- or amenorrhea, virilization in F
Deficiency: symptoms not typically seen due to gonadal production
Note:
Activate Zona Reticularis Activate Zona Glomerulosa
(minor effect)
Excess: central obesity, hirsutism, violaceous striae, weakness
Deficiency: weight loss, fatigue, weakness, nausea/vomiting, diarrhea
DHEA(S) Aldosterone
Specific to primary adrenal insufficiency: ↑ pigmentation, hyperkalemia
• Virilization is a red flag for an androgen- secreting tumor
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published October 2, 2018 on www.thecalgaryguide.com")
Feedback Loop- Thyroid Stimulating Hormone (TSH)

Authors: Josh Kariath Reviewers: Andrea Kuczynski Bernard Corenblum* * MD at time of publication
Stress
Excess: heat intolerance, tremor, proximal muscle weakness, palpitations, systolic HTN, brisk reflexes, weight loss, ↑ appetite, ↑ bowel movements
Deficiency: cold intolerance, firm gland of any size, fatigue, depression, weight gain, constipation, delayed relaxation of DTRs, diastolic HTN
Cortisol
-
+ +
Cold
Hypothalamus
TRH
TRH travels down
hypophyseal stalk through portal vessels
Anterior Pituitary Gland -
TSH
TSH travels through the blood
Thyroid Gland (Follicle) +
T3 T4
Converted into T3 in all target tissues
↑ BMR
-
Abbreviations:
TRH: Thyrotropin Releasing Hormone HTN: Hypertension
DTRs: Deep Tendon Reflexes
TSH: Thyroid Stimulating Hormone I2: iodine
CO: Cardiac Output
T3: Triiodothyronine
T4: Thyroxine
CNS: Central Nervous System
SNS: Sympathetic Nervous System BMR: Basal Metabolic Rate
-
Diet
I2
Excess: no real disorders
Deficiency: goiter
↑ CO, ↑ contractility, ↓ resistance
Hormone metabolism, bilirubin metabolism
Heart Liver
Bone growth Bone
↑ SNS activation, temperature homeostasis
CNS
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published October 2, 2018 on www.thecalgaryguide.com")
Anaphylaxis: Signs and Symptoms

Giant Cell Arteritis: Pathogenesis and Clinical Findings
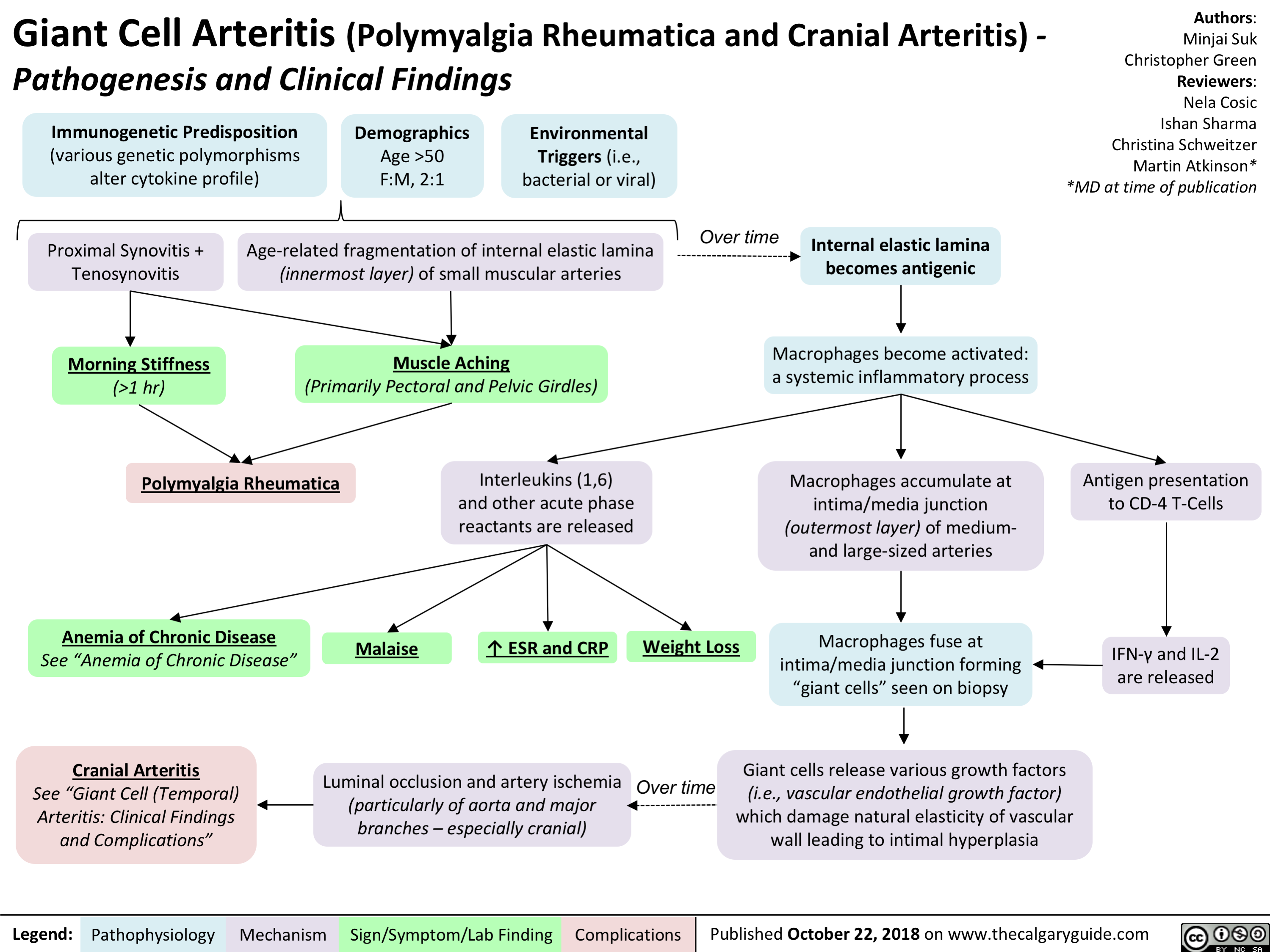
Acute Hemolytic Transfusion Reaction: Signs and Symptoms
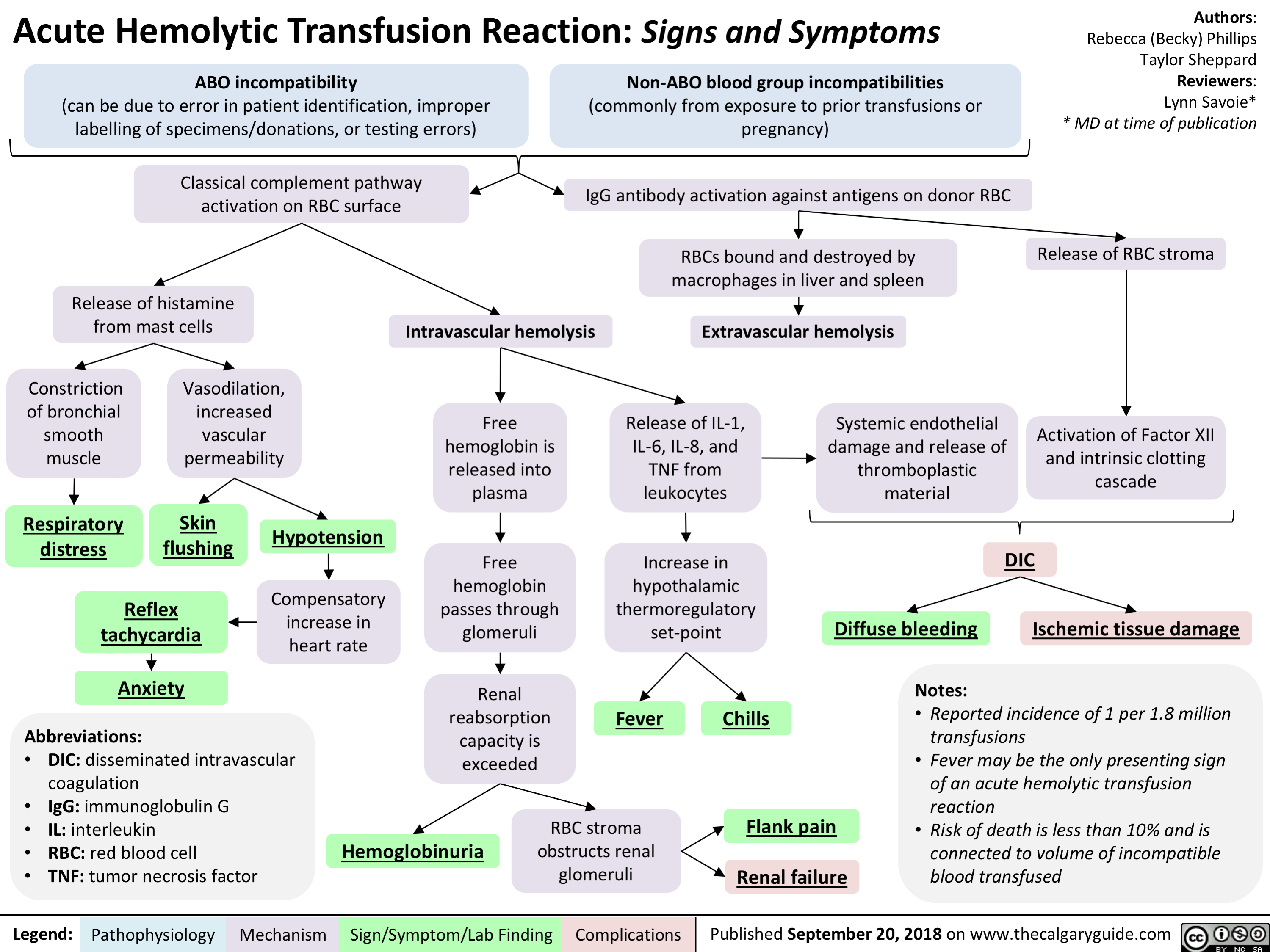
Anaphylaxis: Treatments
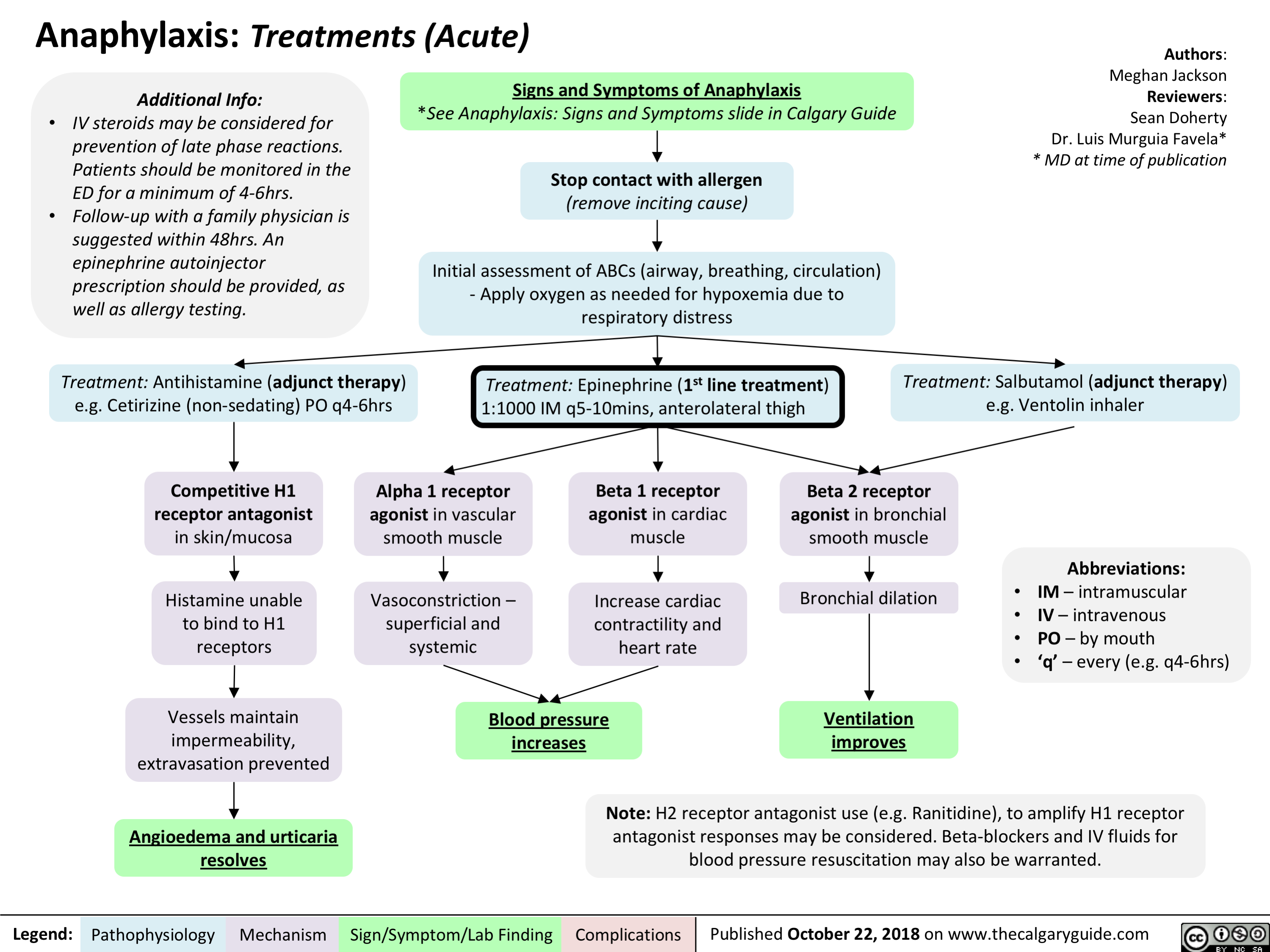
Glucocorticoid Induced Osteoporosis: Pathogenesis and Clinical Findings

intrauterine-growth-restriction-iugr-pathogenesis
: Pathogenesis
Authors: Ricki Hagen Reviewers: Jaimie Bird Sarah McQuillan* * MD at time of publication
Abbreviations:
• DM: diabetes mellitus
• HTN: hypertension • IUGR: intrauterine growth restriction
• SGA: small for gestational age
• SLE: systemic lupus erythematosus
• TORCH: Toxoplasmosis, Others, Rubella, CMV, HSV
Maternal Factors
Maternal-Fetal Factors Placental malformations
(Ex. previa, accreta, infarction, abnormal implantation, ischemia)
Gestational HTN/ Preeclampsia
Multiple gestation Gestational DM
Fetal Factors Structural anomalies
(often comorbid with cytogenetic disorders)
Congenital infections
(Ex. TORCH)
Inborn errors of metabolism
Chromosomal disorders/ genetic syndromes
Multiple unclear intrinsic fetal mechanisms
Note:
Teratogenic medications (Ex. Warafin, Valproic Acid, Folic Acid Antagonists)
High altitude living
Smoking, ETOH and/or drug use
Malnutrition/ Low pre-gestational weight
Multiple unclear extrinsic fetal mechanisms
Medical conditions
(Ex: chronic HTN, cyanotic heart disease, severe chronic anemia, kidney disease)
Autoimmune conditions (Ex. Type 1 DM, SLE)
Decreased uteroplacental blood flow
Nutrient supply to fetus compromised
Reduction of total body mass, bone
mineral content, and muscle mass
Blood flow redirected away from vital organs to brain, placenta, heart and adrenal glands
Reduction of overall fetal size to increase survival
IUGR
Failure to reach genetically determined growth potential
• IUGR is not synonymous with SGA • Constitutional SGA is due to
paternal and maternal factors such as height, weight, ethnicity, and parity; it is not associated with increased risk for infant mortality or morbidity
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published October, 30, 2018 on www.thecalgaryguide.com")
fetal-alcohol-spectrum-disorder-pathogenesis-and-clinical-findings

Maternal alcohol consumption during pregnancy (greatest risk with binge drinking and daily/chronic intake)
Prenatal alcohol exposure to fetus via placenta-umbilical transport
Notes:
Fetal Alcohol Spectrum Disorder with Sentinel Facial Features (confirmed or unknown PAE)
Fetal Alcohol Spectrum Disorder without Sentinel Facial Features
• There is no known safe amount of prenatal alcohol exposure
(PAE). Although PAE below the threshold of >7 drinks/week or > 2 binge episodes (one binge episode is >4 drinks in one sitting) has not been associated with neurodevelopmental effects, there is insufficient objective evidence to deem this level of alcohol exposure safe.
Sentinel Facial Features
(confirmed PAE) Alcohol exposure between Alcohol acts directly as a teratogen
Vasoconstriction of placental-umbilical unit ̄ blood flow and ̄ oxygen delivery to fetus
Notes (continued):
• A multidisciplinary team is necessary for an accurate and comprehensive diagnosis and subsequent recommendations. Canadian Guidelines for diagnosis, 4-digit code, the APP Toolkit, University of Washington Lip- Philtrum Guide are different methods of assessing and diagnosing patients.
• While not specific to FASD, congenital abnormalities (e.g. heart defects, renal problems, auditory/visual impairments, skeletal defects) and growth deficits (prenatal and/or postnatal height or weight ≤ 10th percentile) are often associated with this disorder.
Smooth philtrum
Thin upper lip
day 15 and 22 of & indirectly via its metabolites pregnancy (most often)
Neuronal cell death & disruption of migration & proliferation Altered signaling, neurotransmitter imbalance, & neural connectivity Evidence of impairment in 3+ neurodevelopmental domains
Small palpebral fissures
(≥2 SDs below the mean for age & sex or < 3rd percentile)
Language Memory Attention Motor skills Cognition Affect regulation
Academic achievement
Executive function (including hyperactivity & impulsivity)
Neuroanatomy/ Neurophysiology (e.g. seizure disorder, abnormal brain structure as seen on brain imaging, microcephaly (head circumference ≤ 10th percentile))
Adaptive behavior, social skills, or social communication
Failure to reach age-appropriate milestones, challenges at school, aggression, delinquency, mental health challenges, substance use disorders
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published October 30, 2018 on www.thecalgaryguide.com")
Hydrocephalus: Clinical Findings

↓ or insufficient CSF absorption
(Communicating Hydrocephalus) Hydrocephalus
Enlargement of CSF pathways
Neuroimaging: ventriculomegaly
CSF accumulation
↑ ICP
Compression of blood vessels and altered cellular metabolism
Delayed neuronal migration (neonates)
Periventricular white matter tract damage
(variable) Seizures, developmental anomalies (e.g. learning disability, impaired speech)
Compression of brainstem cranial nerve nuclei and/or tracts
Distortion of meninges and blood vessels
Headache (worse in the morning), nausea/vomiting
Midbrain/brainstem dysfunction (as hydrocephalus worsens)
Lethargy/↓ LOC, neck stiffness
Irritable, feeding difficulty
Upward gaze palsy (Setting Sun sign)
Papilledema, CN 6 palsy
Infant
Older Child
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published October 30, 2018 on www.thecalgaryguide.com")
Orbital Cellulitis: Pathogenesis and clinical findings
 Periorbital cellulitis1,2
Hematogenous spread
Contiguous spread of infection
Pathogens reach orbital tissue (posterior to the orbital septum)
Spreads to periorbital tissue (anterior to the orbital septum)
Localized inflammation
Conjunctival chemosisa
Eyelid and periorbital edema
Pain on palpation
Induration
Warmth
Orbital Cellulitis Inflammation of orbital tissue Proptosis
Spreads to surrounding structures
Subperiosteal abscess Brain abscess Cavernous sinus thrombosis Meningitis Subdural empyema Orbital abscess
Notes:
Impinges on ocular muscles
Impaired extra- ocular movements
Pain with eye
movement or opthalmoplegia
Definitions:
Impinges on nerves
Afferent pupillary defect
Decreased visual acuity
Exposes cornea
Corneal drying and scarring
a. Chemosis: Edema of the bulbar conjunctiva
b. Panopthalmitis: inflammation of all coats of the eye including intraocular structures.
c. Endopthalmitis: inflammation of the interior of the eye.
1. See slide on Periorbital Cellulitis for how sinusitis can lead to the development of periorbital cellulitis
2. The micro-organism responsible for periorbital cellulitis varies depending on how the pathogen was introduced to the system.
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published November 5, 2018 on www.thecalgaryguide.com")
Periorbital Cellulitis: Pathogenesis and Clinical Findings

Note: Also referred to as preseptal cellulitis
Dacryoadenitisa Conjunctivitisb
Acute chalazionc
Dacryocystitisd Hordeolume
Streptococcus pneumoniae, Moraxella catarrhalis, non-typable Haemophilus influenza (most common organisms)
Abrasion Insect bite
Burns Trauma
Local infection
Contiguous spread of infection
Sinusitis
Otitis media Hematogenous spread
Local break in skin Micro-organisms enter
Definitions:
Note:
Eye exam should reveal normal:
- extra-ocular
movements and globe
position
- pupillary reflex and
visual acuity
If any are abnormal, the presentation is no longer considered periorbital cellulitis, as the infection has likely spread beyond the preseptal compartment/orbital septum.
If the eye cannot be assessed, the patient NEEDS a CT scan.
Pathogens reach dermis and subcutaneous periorbital tissue
Periorbital Cellulitis
a. Dacryoadenitis: infection of the lacrimal glands
b. Conjunctivitis: inflammation of the conjunctiva
c. Chalazion: a benign, painless bump or nodule inside the upper or lower eyelid which results from healed internal hordeolums that are no longer infectious.
d. Dacryocystitis: an infection of the lacrimal sac, secondary to obstruction of the nasolacrimal duct at the junction of lacrimal sac.
e. Hordeolum: localized infection or inflammation of the eyelid margin involving hair follicles of the eyelashes or meibomian glands.
Spreads beyond preseptal compartment/orbital septum
Involves the orbit Orbital cellulitis
See slide on Orbital Cellulitis: Pathogenesis and clinical findings
Localized inflammation
Pain on palpation
Induration
Warmth
Eyelid and periorbital edema
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published November 5, 2018 on www.thecalgaryguide.com")
Child Abuse: Risk Factors and Possible Indicators
, especially <1 year old
↑ Risk
Note: Special Indicators for Sexual Abuse
Definition: Child Abuse
• Any act or omission
by a parent or caregiver that results in actual or potential harm to a child
• Includes physical, sexual, & emotional abuse, or neglect
Family Factors
Unrelated caregiver Low SES Single parent Domestic violence Social isolation
Indicators on History
Indicators on Physical Exam
Injury blamed on pet or sibling
Delays in seeking treatment
Frequent visits to different healthcare providers for injuries
Self-inflicted injury not compatible with child’s development
Child described as “difficult”
Hx of injury changes with time
History not compatible with injury
Extensive physical injury with hx of minor trauma
• • • • •
Regressive behaviour
Difficulty sleeping
Sexually inappropriate behaviour Sudden change in behaviour School difficulties
Burns (esp. immersion)
Frenulum tear in infants
Bruising: Pattern, Location (esp. face), Number, Shape (esp. ligature marks, instrumentation), Age of child (“those who don’t cruise don’t bruise”)
Abusive head trauma
See “Non-accidental Head Trauma” slide
Fractures (esp. posterior rib, shearing/twisting forces)
Evidence of injury with no hx of trauma
Suspicion of child abuse
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published November 5, 2018 on www.thecalgaryguide.com")
Mastoiditis: Pathogenesis and clinical findings

Distal middle ear is physically connected to mastoid air spaces
Pathogens spread from middle ear to the mastoid air spaces
Mucosa lining the mastoid becomes inflamed
Mastoiditis 1
Infection persists
Accumulation of pus in mastoid cavities
↑ pressure Formation of abscess cavities
Dissection of pus into adjacent areas
Infection spreads
Into intracranial compartment
See slide on Acute Otitis Media (AOM): Pathogenesis and Clinical Findings in Children
Post- operation
Trauma Infection
Notes:
1. Most common suppurative complication of AOM
Tenderness, erythema, swelling and fluctuance over the mastoid process
Inflammation spreads to external auditory canal
Cranial Nerve VII anatomically near mastoid
Cranial Nerve VIII anatomically near mastoid air space
Destroys bony septae b/t air cells (visible on CT)
Mastoid abscess
Swelling of external auditory canal
Mastoid inflammation disrupts nerve
Mastoid inflammation disrupts nerve
Petrositis
Facial nerve palsy
Sensorineural hearing loss Labyrinthitis
Osteomyelitis of the calvaria
Into adjacent bones
Underneath the periosteum Subperiosteal abscess Pinna is pushed out and
of the temporal bone
Into the neck beneath the attachment of the sternocleidomastoid and digastric muscles
forward
Dural venous thrombosis Temporal lobe abscess Meningitis
Epidural abscess Subdural abscess Cerebellar abscess
Bezold abscess
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published November 5, 2018 on www.thecalgaryguide.com")
Scarlet Fever: Pathogenesis and clinical findings

0-1 days post- pharyngitis
Pastia’s lines5
1-2 days post- pharyngitis
Appears on upper trunk and axillae
3-4 days post- pharyngitis
Spreads to remainder of body, sparring face6, palms and soles
7-10 days post- pharyngitis
Fades Desquamation7
Otitis media, sinusitis, pneumonia, bacteremia, osteomyelitis, meningitis, arthritis, erythema nodosum, hepatitis, acute poststreptococcal glomerulonephritis, and acute rheumatic fever
Fine maculopapular rash
Blanchable with Non-pruritic and pressure painless
Notes:
1. While the majority of infections are cases of GAS pharyngitis, rarely, it is possible to develop scarlet fever from a GAS skin infections. 2. Scarlet fever is most common in patients of this age group although, rarely, it can occur in adults.
3. White strawberry tongue is characterized by a white coating on the tongue through which edematous lingual papillae project.
4. Red strawberry tongue is characterized by a beefy red, edematous tongue covered in edematous lingual papillae.
5. Prominent erythema and petechiae in the body folds, especially the antecubital fossae and axillary folds. They tend to appear before the rash and persist through the desquamation phase.
6. Typically, the rash does not occur on the face, although facial flushing may be noted. When this occurs, there is perioral sparring.
7. Desquamation tends to occur ~1 week after the rash fades, most severely effecting the hands and feet, and lasts 2-6 weeks. While a classical presentation, not
everyone gets it.
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published November 5, 2018 on www.thecalgaryguide.com")
Sinusitis: Pathogenesis and clinical findings

Stomach Acid Reducing Medications - Mechanisms of Action
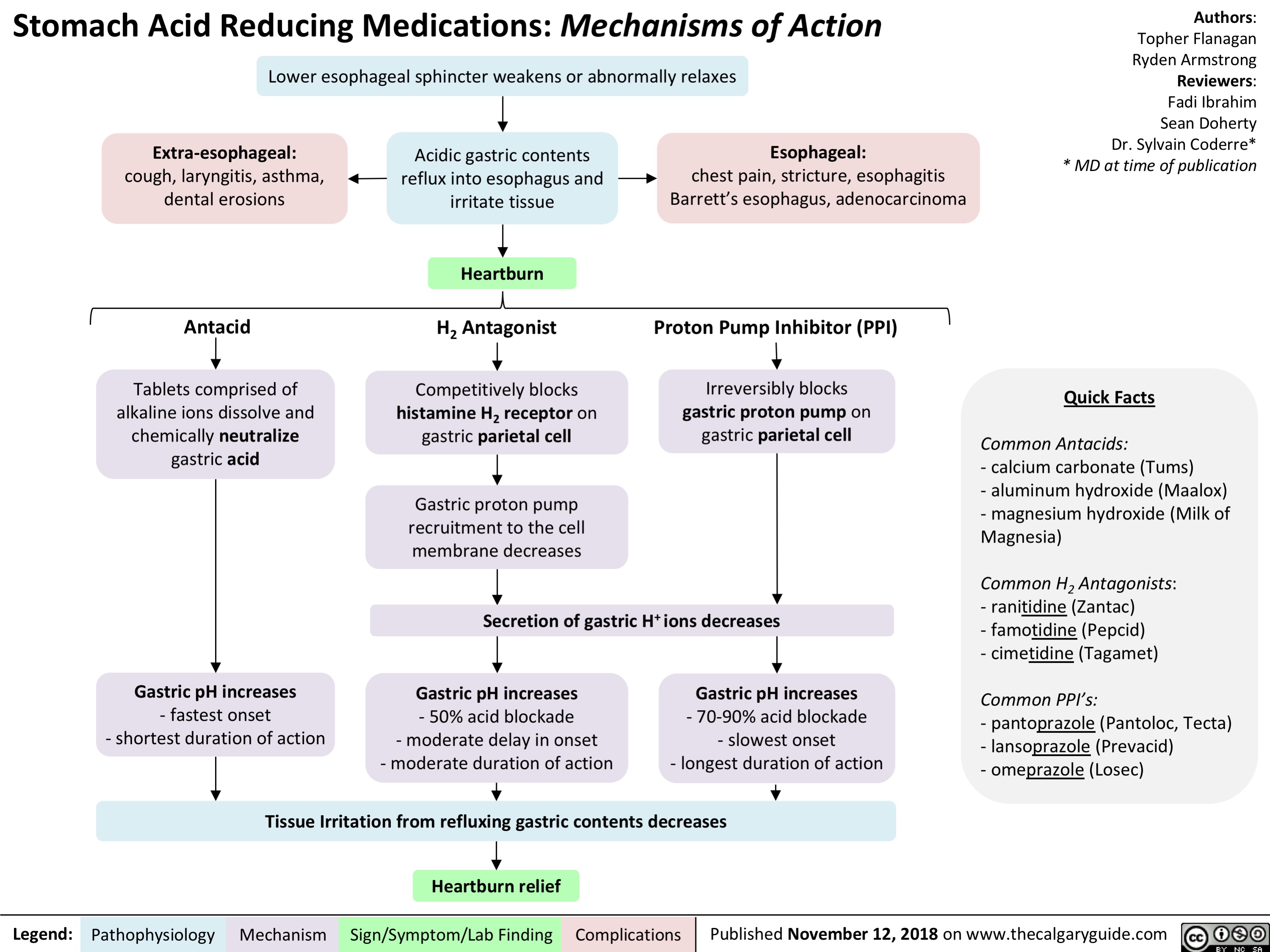
Dependent Personality Disorder Slide - Pathogenesis and clinical findings
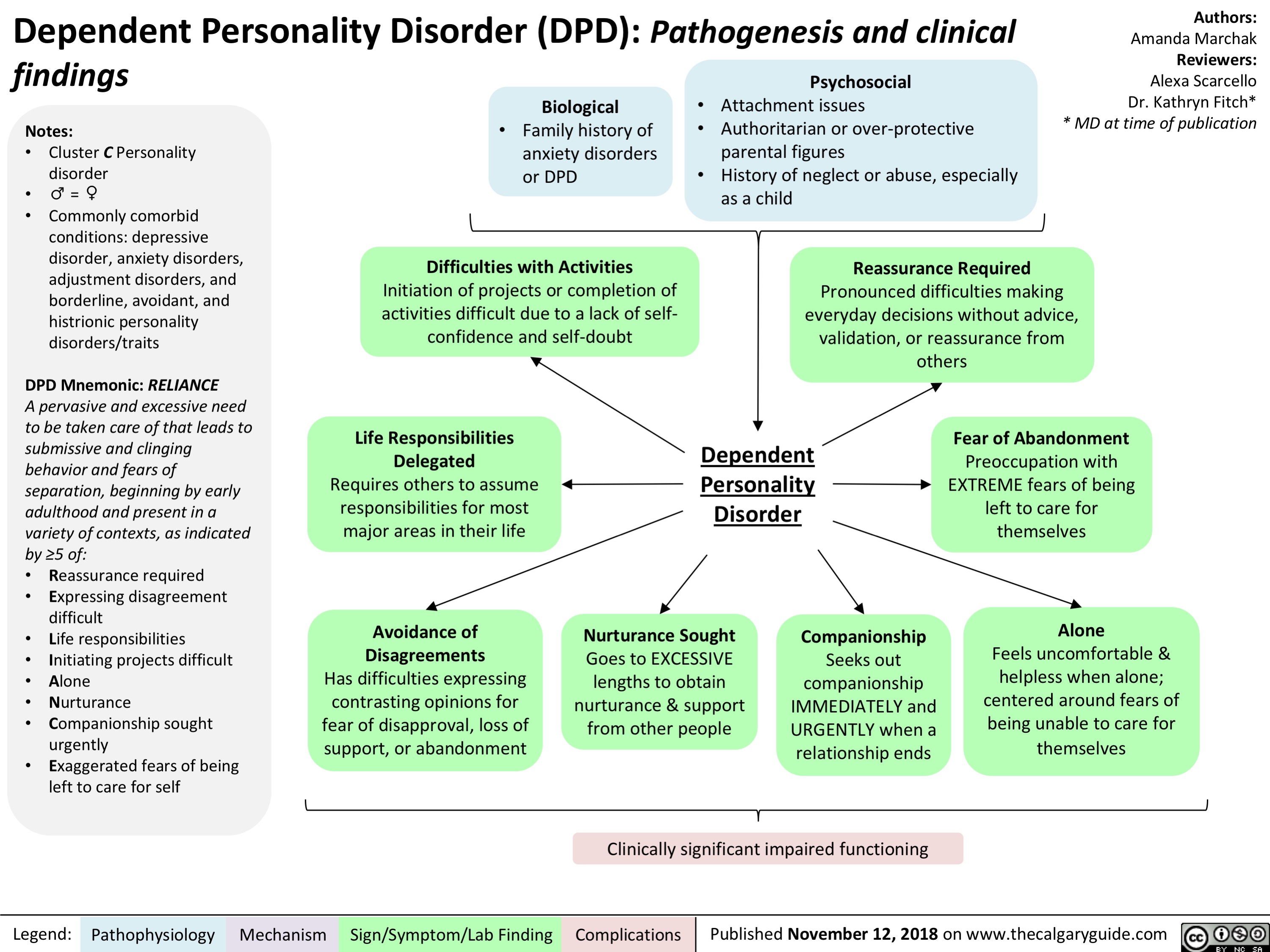
Obsessive-Compulsive Personality Disorder Slide - Pathogenesis and clinical findings

Brain Death: Pathogenesis assessment and clinical findings
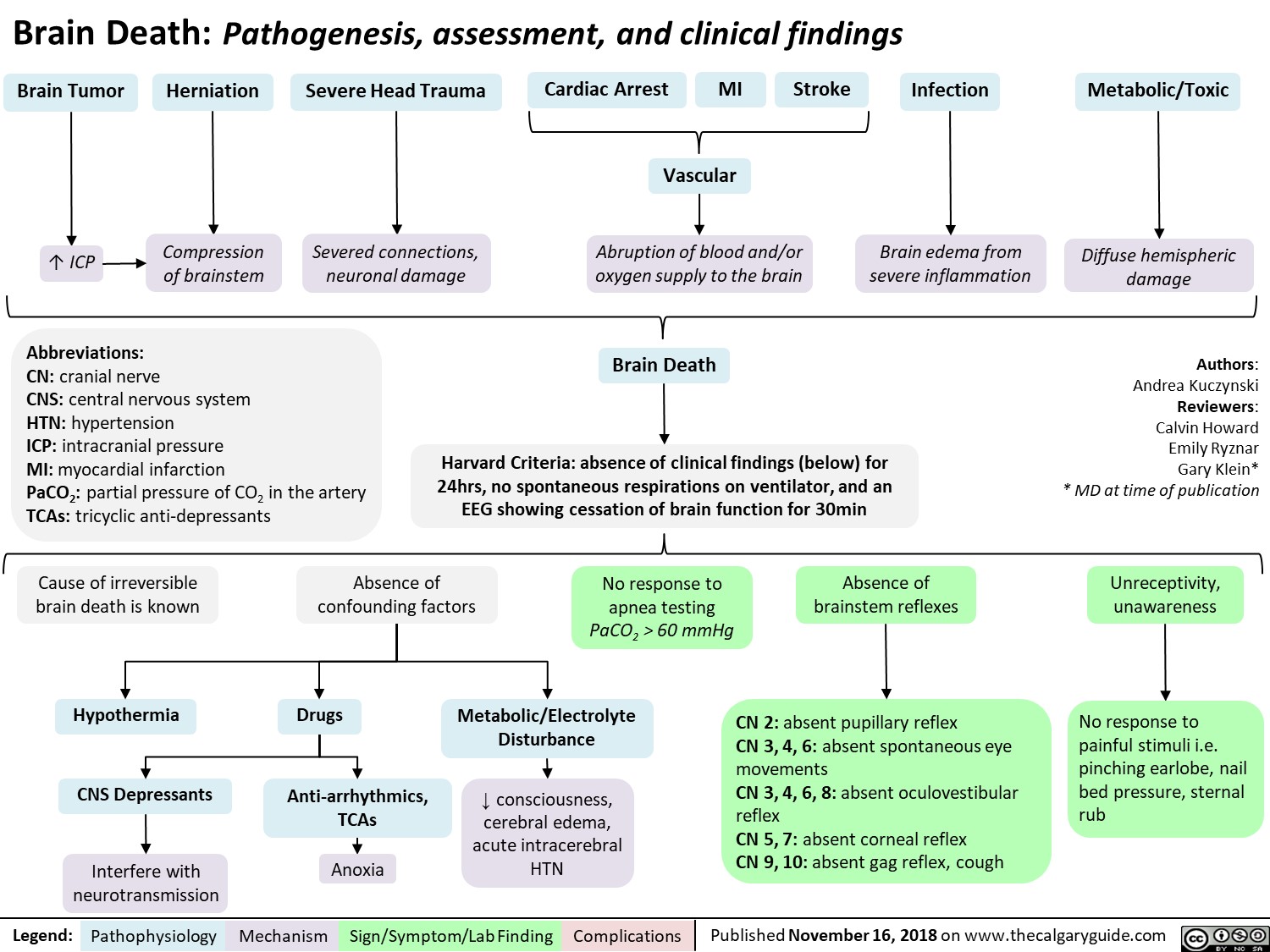
Benefits of Breast Milk: Mechanism of Action

Major Depressive Disorder: Complications

Avoidant Personality Disorder - Pathogenesis and clinical findings

Measures of Population Health
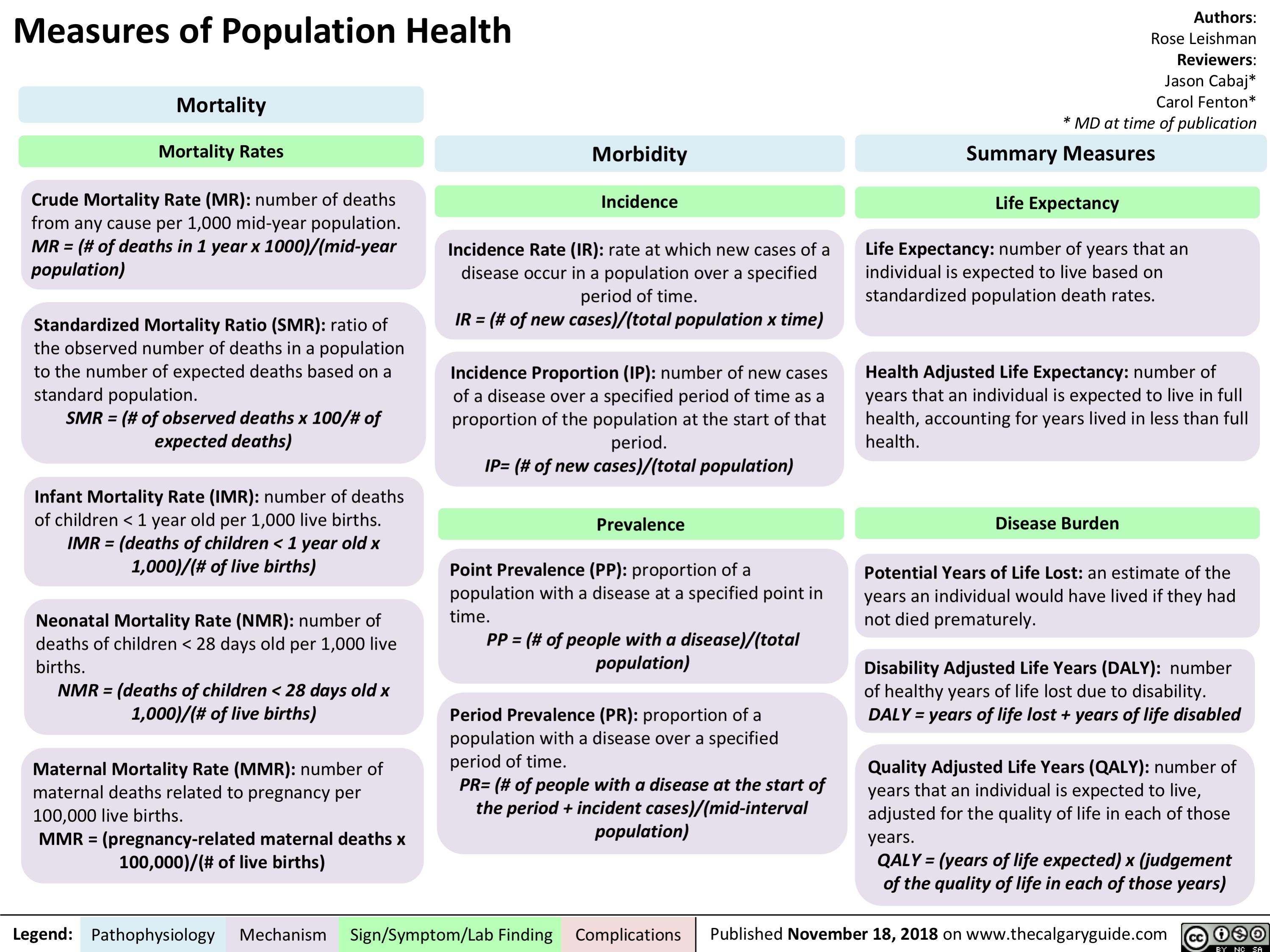
Employment as a Determinant of Health
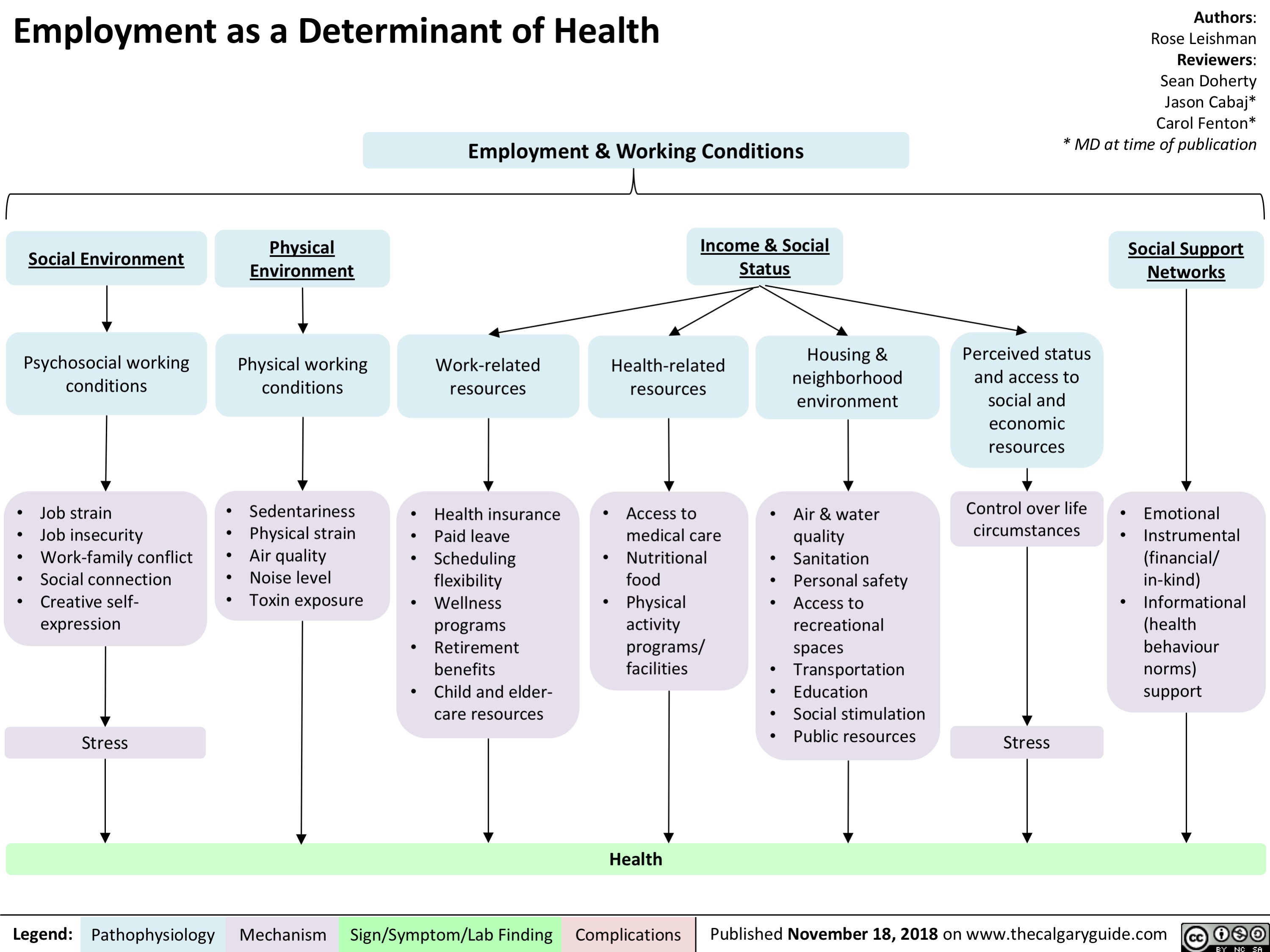
Menopause contraindications to hormone replacement therapy
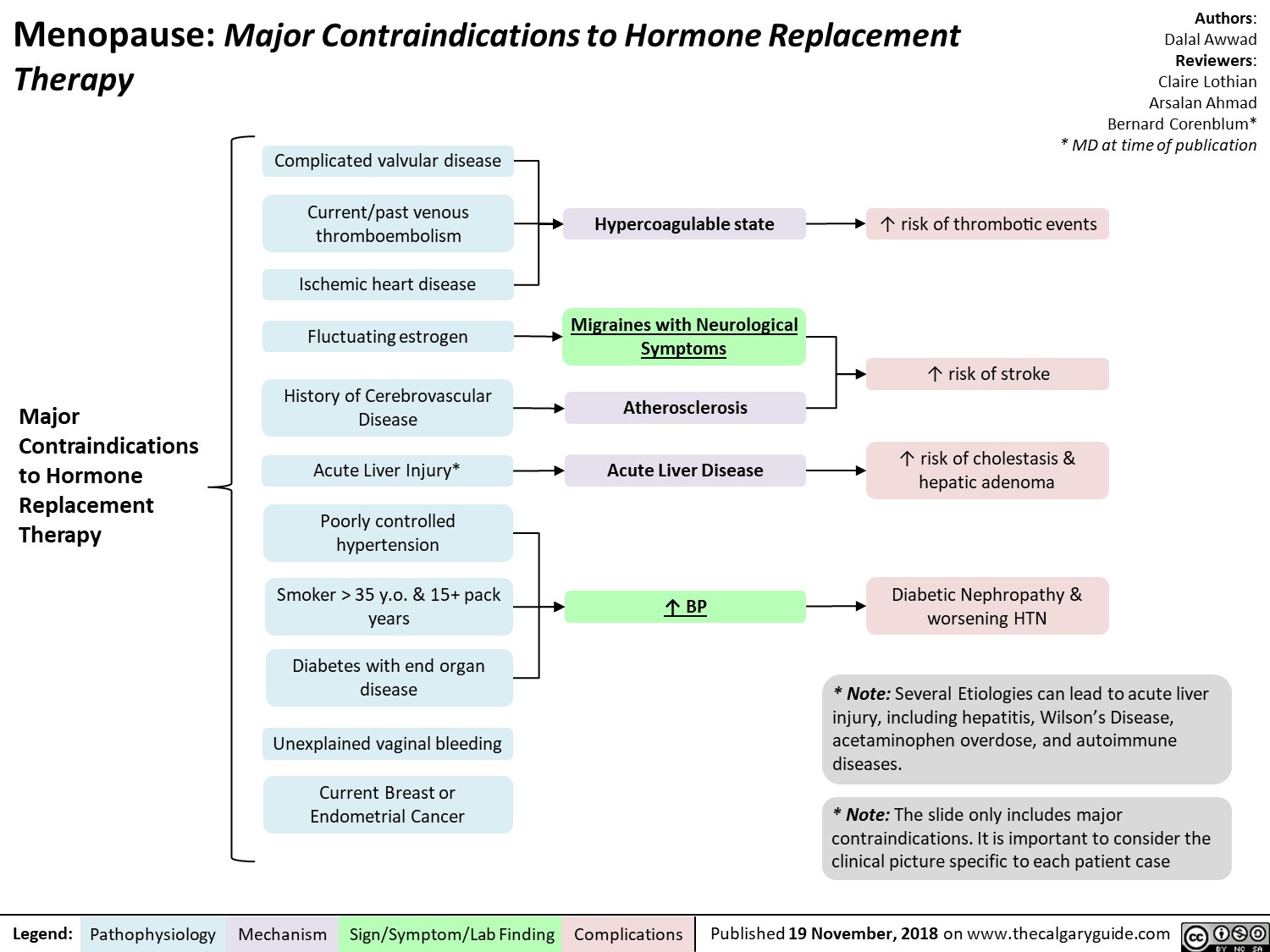
Phenylketonuria (PKU): Pathogenesis and clinical findings
![Phenylketonuria (PKU): Pathogenesis and clinical findings
Authors: Hamna Tariq Reviewers: Chandan Kaur Bal Nicola Adderley Rebecca Sparkes* * MD at time of publication
Abbreviations:
PAH – phenylalanine hydroxylase BH4 – tetrahydrobiopterin
Phe – phenylalanine
Tyr - tyrosine
LAT-1 - L-amino acid transporter 1 LNAA – large neutral amino acids NT – neurotransmitter
Biallelic mutations in PAH gene on chromosome 12q23.2
Loss of activity/deficiency of PAH
Inability to convert Phe to Tyr
Buildup of Phe and its metabolites
↑ transport of Phe via LAT-1 at the blood brain barrier
Phe outcompetes LNAA for binding sites on LAT-1
↓ LNAA transport
↓ cerebral protein synthesis and ↓ NT synthesis
White matter lesions, oxidative damage, hypomelination & demyelination
Sources of Phe
Dietary protein
Breakdown into amino acids
↓ Tyr and derivatives
↓ melanin
Fair skin and hair
Notes:
Endogenous recycling of amino acid stores
Phe oxidized to phenylacetate
Musty odor to urine, sweat and breath
• Autosomal recessive inborn error of metabolism; clinical severity depends on residual PKU activity
• This slide denotes clinical features of classical PKU in untreated patients. Symptoms develop within a few months of birth only if untreated.
• PAH, using a BH4 cofactor, converts Phe to tyrosine, which is necessary to produce epinephrine, norepinephrine, dopamine and melanin. (Rare inherited disorders of BH4 synthesis/recycling cause elevated Phe and neurologic symptoms.)
• PKU is screened for at birth in most developed nations.
• In early-diagnosed, continuously treated patients, ID,
microcephaly and neurologic features are not expected, but there is still an increased incidence of behavioral, emotional and social problems
↑ brain [Phe]
Behavioral, emotional & social problems (ADHD, mood disorders, aggression, ↓ self-esteem, ↓ social competency, ↓ autonomy)
Intellectual disability
(↓ language, memory & learning skills, executive function, IQ, school performance)
Neurological findings (tremor, shaking, seizures, poor coordination)
Microcephaly
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published November 19, 2018 on www.thecalgaryguide.com](http://calgaryguide.ucalgary.ca/wp-content/uploads/2018/11/Phenylketonuria_Final.jpg "Phenylketonuria (PKU): Pathogenesis and clinical findings
Authors: Hamna Tariq Reviewers: Chandan Kaur Bal Nicola Adderley Rebecca Sparkes* * MD at time of publication
Abbreviations:
PAH – phenylalanine hydroxylase BH4 – tetrahydrobiopterin
Phe – phenylalanine
Tyr - tyrosine
LAT-1 - L-amino acid transporter 1 LNAA – large neutral amino acids NT – neurotransmitter
Biallelic mutations in PAH gene on chromosome 12q23.2
Loss of activity/deficiency of PAH
Inability to convert Phe to Tyr
Buildup of Phe and its metabolites
↑ transport of Phe via LAT-1 at the blood brain barrier
Phe outcompetes LNAA for binding sites on LAT-1
↓ LNAA transport
↓ cerebral protein synthesis and ↓ NT synthesis
White matter lesions, oxidative damage, hypomelination & demyelination
Sources of Phe
Dietary protein
Breakdown into amino acids
↓ Tyr and derivatives
↓ melanin
Fair skin and hair
Notes:
Endogenous recycling of amino acid stores
Phe oxidized to phenylacetate
Musty odor to urine, sweat and breath
• Autosomal recessive inborn error of metabolism; clinical severity depends on residual PKU activity
• This slide denotes clinical features of classical PKU in untreated patients. Symptoms develop within a few months of birth only if untreated.
• PAH, using a BH4 cofactor, converts Phe to tyrosine, which is necessary to produce epinephrine, norepinephrine, dopamine and melanin. (Rare inherited disorders of BH4 synthesis/recycling cause elevated Phe and neurologic symptoms.)
• PKU is screened for at birth in most developed nations.
• In early-diagnosed, continuously treated patients, ID,
microcephaly and neurologic features are not expected, but there is still an increased incidence of behavioral, emotional and social problems
↑ brain [Phe]
Behavioral, emotional & social problems (ADHD, mood disorders, aggression, ↓ self-esteem, ↓ social competency, ↓ autonomy)
Intellectual disability
(↓ language, memory & learning skills, executive function, IQ, school performance)
Neurological findings (tremor, shaking, seizures, poor coordination)
Microcephaly
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published November 19, 2018 on www.thecalgaryguide.com")
Feedback Loop: Prolactin (PRL)

Authors: Nicola Adderley Reviewers: Andrea Kuczynski Bernard Corenblum* * MD at time of publication
Abbreviations:
TRH: Thyrotropin Releasing Hormone
DA: Dopamine
GnRH: Gonadotropin Releasing Hormone FSH: Follicle Stimulating Hormone
LH: Luteinizing Hormone
Note:
• PRL levels exhibit diurnal, menstrual, and age-related variation
• TRH has a stimulatory effect on PRL release; however, dopamine is the principal regulator of PRL secretion
Alveolar Cells of Breast Ducts
Milk production and release
Lactation
TRH
Suckling stimulus (via spinal afferents)
+
+
Hypothalamus + (arcuate nucleus)
DA
-
Anterior Pituitary Gland Lactotrophs
Uterus, immune cells, breast tissue, prostate
PRL
Hypothalamus
↓ GnRH secretion ↓ FSH/LH
↓ estradiol in females, ↓ testosterone in males
Limbic System
↓ libido
Immune System Organs
Promotes proliferation and maturation of immune cells
Oxytocin
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published November 19, 2018 on www.thecalgaryguide.com")
Vesicoureteric reflux (VUR): Pathogenesis and clinical findings
: Pathogenesis and clinical findings
Authors: Nicola Adderley Reviewers: Emily Ryznar *Lindsay Long * MD at time of publication
Abnormal function
Abnormal anatomy
Neurogenic bladder (e.g. cerebral palsy, constipation, spinal injury, iatrogenic)
Non-neurogenic bladder (neuropsychological)
Lower urinary tract abnormality (posterior urethral valves, meatal stenosis)
Bladder outlet obstruction
↑ pressure distorts UVJ
Upper urinary tract abnormality (ureters)
UVJ abnormality
Incomplete closure of UVJ during bladder contraction
Abbreviations
• UVJ - ureterovesicular junction • UTI – urinary tract infection
Failure of bladder sphincter to relax during bladder contraction
Vesicoureteric reflux (VUR):
Back flow of urine from the bladder into one or both ureters +/- kidneys
Migration of lower urinary tract bacteria to kidneys
Bacterial invasion of renal parenchyma
Upper UTI (pyelonephritis)
Incomplete emptying of bladder during Abnormal
↑ pressure in bladder
Bladder dilates
Dilated bladder on U/S
urination
Bacteria in bladder are not cleared during urination
voiding habits
↑ bladder capacity
Renal scarring
↓ functional renal tissue
*Chronic kidney disease (↓ GFR,
hypertension, proteinuria)
Flank tenderness
Fever, dysuria, urgency, frequency
Lower UTI (cystitis) Urinary stasis
Cloudy, foul- smelling urine
Urethral stricture
Notes
Urgency, dysuria, frequency
• First febrile UTI in an infant should trigger a work-up for VUR
• High likelihood of spontaneous resolution • *Late complication of severe VUR
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published November 19, 2018 on www.thecalgaryguide.com")
IVH Intraventricular Hemorrhage in Preterm Infants - Pathogenesis

↑ susceptibility to ischemia/reperfusion injury
GM capillary network is prone to hemorrhage
Hemorrhage in germinal matrix capillary bed, which drains into venous system
Intraventricular Hemorrhage
hemorrhage in periventricular subependymal germinal matrix
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published November 20, 2018 on www.thecalgaryguide.com")
Prader Willi syndrome: pathogenesis and clinical findings
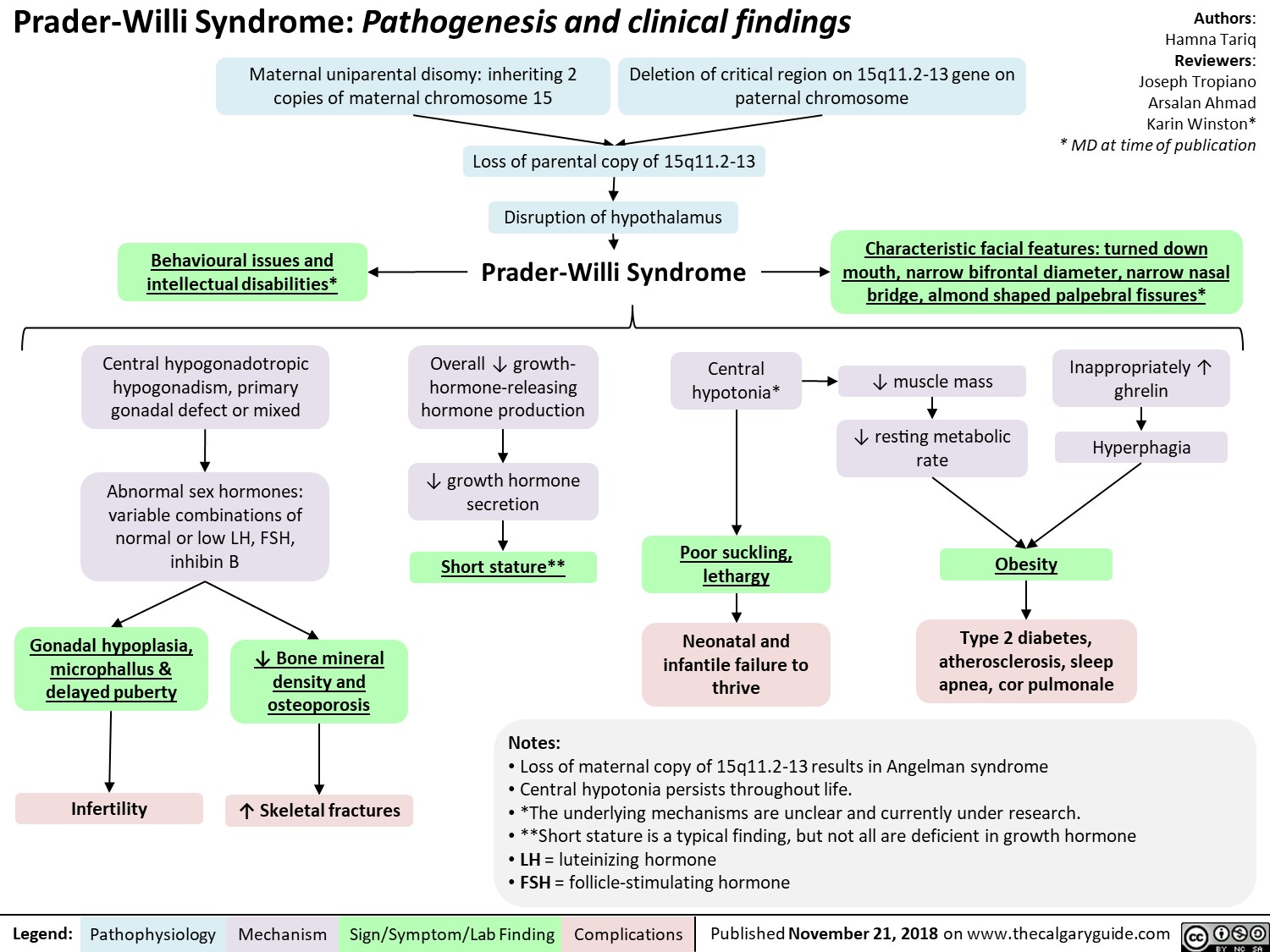
Anticonvulsants as Mood Stabilizers: Mechanism and Side-effect
, thrombocytopenia
Abbreviations: • VSSCs: Voltage-Sensitive Sodium Channels")
Benzodiazepine (BZD) withdrawal: clinical findings and complications
 withdrawal: clinical findings and complications
Abrupt cessation of chronic ingestion of BZDs
Administration of BZD antagonist (flumazenil) on patients who have developed -* tolerance/dependence to BZD
Withdrawal Seizure
Negative physiological reactions BZD intake inhibition a mygd to f, • of a la Withdrawal symptoms Benzodiazepine Withdrawal GABA receptor activity (less inhibition alleviated by ingesting BZD Tolerance GABA BZD intake Conformational changes in the GABA receptor 1, receptor's Withdrawal Insomnia Pro-excitatory 4— state of excitatory neurotransmitters) 4— to the agent activity affinity for the agent
A
Activation of ACC and OFC
Feelings of fear
Activation of PAG
Behavioural response of fight or flight
Legend: Pathophysiology Mechanism
Activation of hypothalamus '1` Cortisol CAD, T2DM, Stroke
Sign/Symptom/Lab Finding
Activation of PBN
V
t RR, SOB, Asthma, or a sense of being smothered
Activation of LC
t Sympathetic Activity
t BP, t HR variability, tremor, and diaphoresis
Authors: Usama Malik Reviewers: Sina Marzoughi Aaron Mackie* * MD at time of publication
Notes: • The onset of withdrawal can vary according to the half-life of the BZD involved. Symptoms may be delayed up to three weeks in BZDs with long half-lives, but may appear as early as 24 to 48 hours after cessation of BZDs with short half-lives.
Abbreviations: • ACC: Anterior Cingulate Cortex • BP: Blood Pressure • CAD: Coronary Artery Disease • HR: Heart Rate • LC: Locus Coeruleus • MI: Myocardial Infarction • OFC: Orbitofrontal Cortex • PAG: Periaqueductal Gray • PBN: Parabrachial Nucleus • RR: Respiratory Rate • SOB: Shortness of Breath • T2DM: Type 2 Diabetes
I` atherosclerosis, cardiac ischemia, MI, or sudden death")
Schizotypal Personality Disorder (SPD): Pathogenesis and clinical findings
: Pathogenesis and clinical findings
Genetics Environmental Neurobiological Changes Neurotransmitter /1` prevalence among first-More common among Significant atrophy of the Imbalance degree relatives of people with lower (predominantly left) lateral Dysfunction in synaptic probands with incomes and those never temporal lobe. Certain subregions dopamine degradation, Schizophrenia. Genes associated with married, divorced, separated, or widowed. of the prefrontal cortex have been shown to be enlarged. usually due to gene catechol-0- schizophrenia are also risk factors for Schizotypal PD. Volume 1, of specific temporal lobe subregions. methyltransferase (COMT).
Cognitive-perceptual •
Magical thinking Belief in paranormal or supernatural phenomena, such as superstitions, telepathy, weird fantasies
Unusual Perceptions Seeing a halo or aura, presence of unseen force, or bodily illusions
Ideas of Paranoia/ Reference Suspiciousness Believing Can range from coincidences persistent and have strong overt hostility, personal guardedness to significance pleasant and agreeable compliance
Schizotypal Personality Disorder Oddness/ disorganized
► Interpersonal
Lack Close Friends Deficit in finding social interactions gratifying, a form of social anhedonia
Constricted affect
Eccentric behavior Unusual thinking or or appearance speech Unconventional or Speech and thought idiosyncratic process can be hygiene, attire, or vague, unelaborate, social behaviors circumstantial, metaphorical, or stereotyped but not grossly incoherent or blocked
Social Anxiety Unrelenting, situationally generalized, unconditional, and does not tend to lessen with familiarity
Clinically significant impaired functioning
Authors: Usama Malik Reviewers: Sina Marzoughi Aaron Mackie* * MD at time of publication
Notes: • Cluster A Personality Disorder • 4.2CY : 3.7g - frequently diagnosed in fragile X syndrome • 3.9-4.6%of adults • More than half of these patients have at least one episode of major depression
SPD Mnemonic: ME PECULIAR • Magical thinking Experiences unusual perceptions • Paranoid / suspicious ideation • Eccentric behavior or appearance • Constricted or inappropriate affect • Unusual thinking or speech • Lacks close friends • Ideas of reference • Anxiety in social situations • Rule out psychotic or pervasive developmental disorders
• Dx: present in a variety of contexts causing marked social impairment")
Boutonniere Deformity: Pathogenesis and Complications

menstrual-cycle-physiology-ovarian-cycle-brief-overview

Microangiopathic Hemolytic Anemia: Pathogenesis and clinical findings

↓ Inhibition of complement
Immune inflammatory
↑ Pressure in
↑ Extrinsic clotting pathway activation
TTP
(See TTP-HUS Slide)
Disruption of
endothelial cell
activation perfusion/ischemia response metabolism vasculature ↑Thrombin Deficient
Placental under-
renal afferent
↑ Complement pathway activation
MAC-mediated cell lysis
Cytokine release
Endothelial injury
Shear stress on endothelium
production
↑ Fibrin clot production and deposition in small vessels
ADAMTS13 protease enzyme
↓ Cleavage and ↑ accumulation of VWF multimers
Abbreviations:
• HELLP - Hemolysis, Elevated Liver Enzymes, Low Platelets • HUS - Hemolytic-Uremic Syndrome
• DIC - Disseminated Intravascular Coagulation
• TTP - Thrombotic Thrombocytopenic Purpura
• MAC - Membrane Attack Complex
• STEC - Shiga Toxin-Producing Escherichia coli
• VWF - Von Willebrand Factor
Pro-thrombotic Environment (see Virchow’s Triad Slide)
Platelet aggregation Mechanical obstruction of the vessel lumen
Hypercoagulable state
Thrombocytopenia
↑ RBC production in bone marrow
↑ Reticulocytes
Hemoglobin released from damaged RBCs Shearing of RBCs Anemia
Binds to haptoglobin Conversion to bilirubin in liver
↓ Free haptoglobin ↑Indirect bilirubin
Shistocytes Jaundice
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published August 13, 2018 on www.thecalgaryguide.com")
Neurogenic Claudication: Pathogenesis and Clinical Findings
 and myotomal distribution (weakness, cramping) of the impaired spinal nerve when walking or standing. Relieved with rest.
Symptoms distributed along the dermatome and myotome
Neurogenic
• Position dependent
• Dermatomal distribution
• Proximal to distal progression
Vascular
• Exercise dependent
• Sclerotomal distribution
• Distal to proximal progression
Hip extension
Buckling of ligamentum flavum and overlapping of adjacent vertebral laminae and facets
↓ canal size ↑ compression
Worsening of symptoms
Myotomal:
weakness, cramping
Dermatomal:
pain, paraesthesias
Modification of symptoms with movement and position
Exercise
↑ vasodilation of
spinal arteries ↑ compression
Hip flexion
Stretching of ligamentum flavum, reduction of overlap of adjectent vertebral laminae and facets, enlarged foramina
↑ canal size
↓ compression
Slow relief of symptoms (>30 min)
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published December 7, 2018 on www.thecalgaryguide.com")
Atopic Dermatitis: Pathogenesis and Clinical Findings
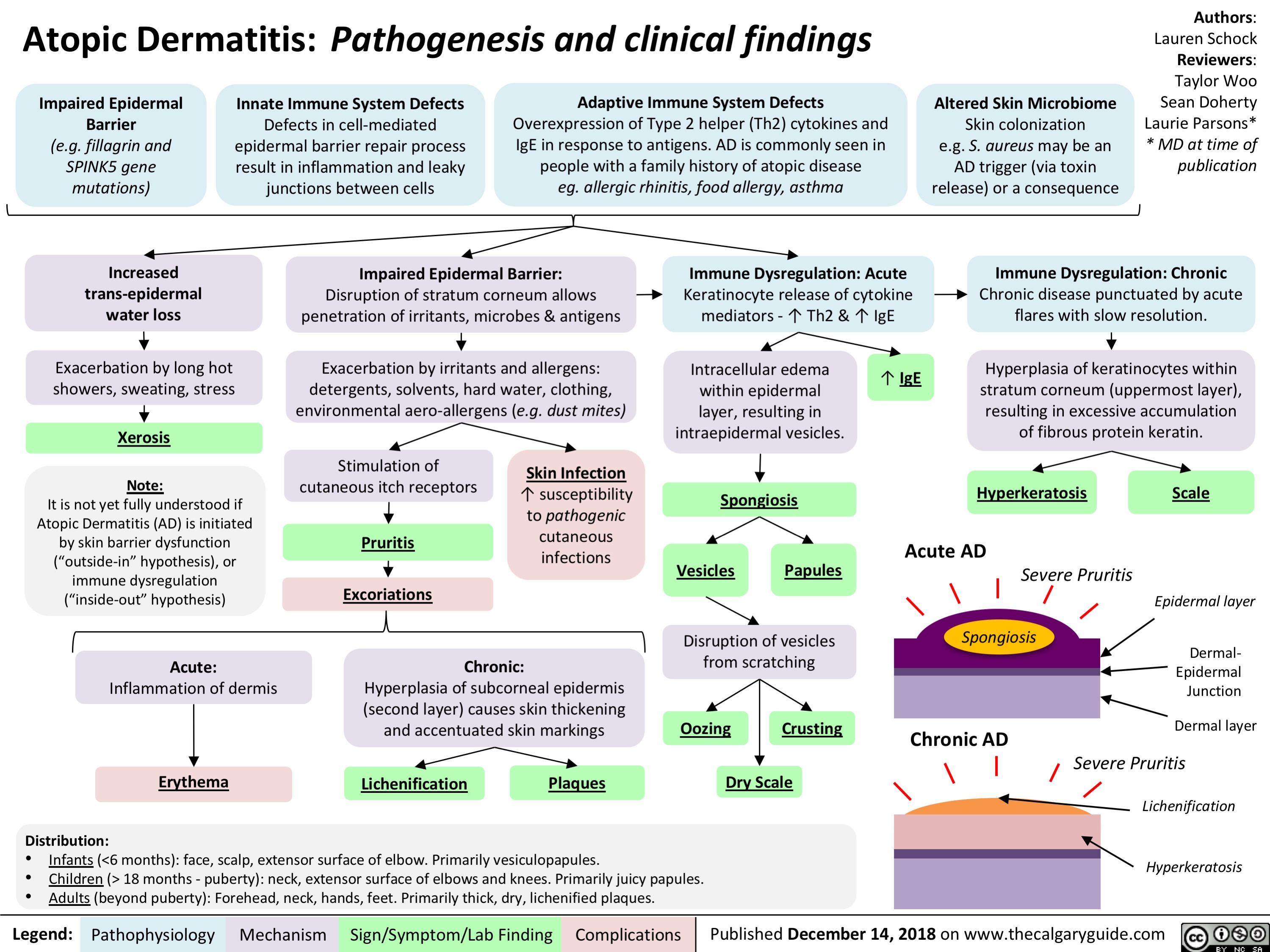
Rosacea: Pathogenesis and Clinical Findings
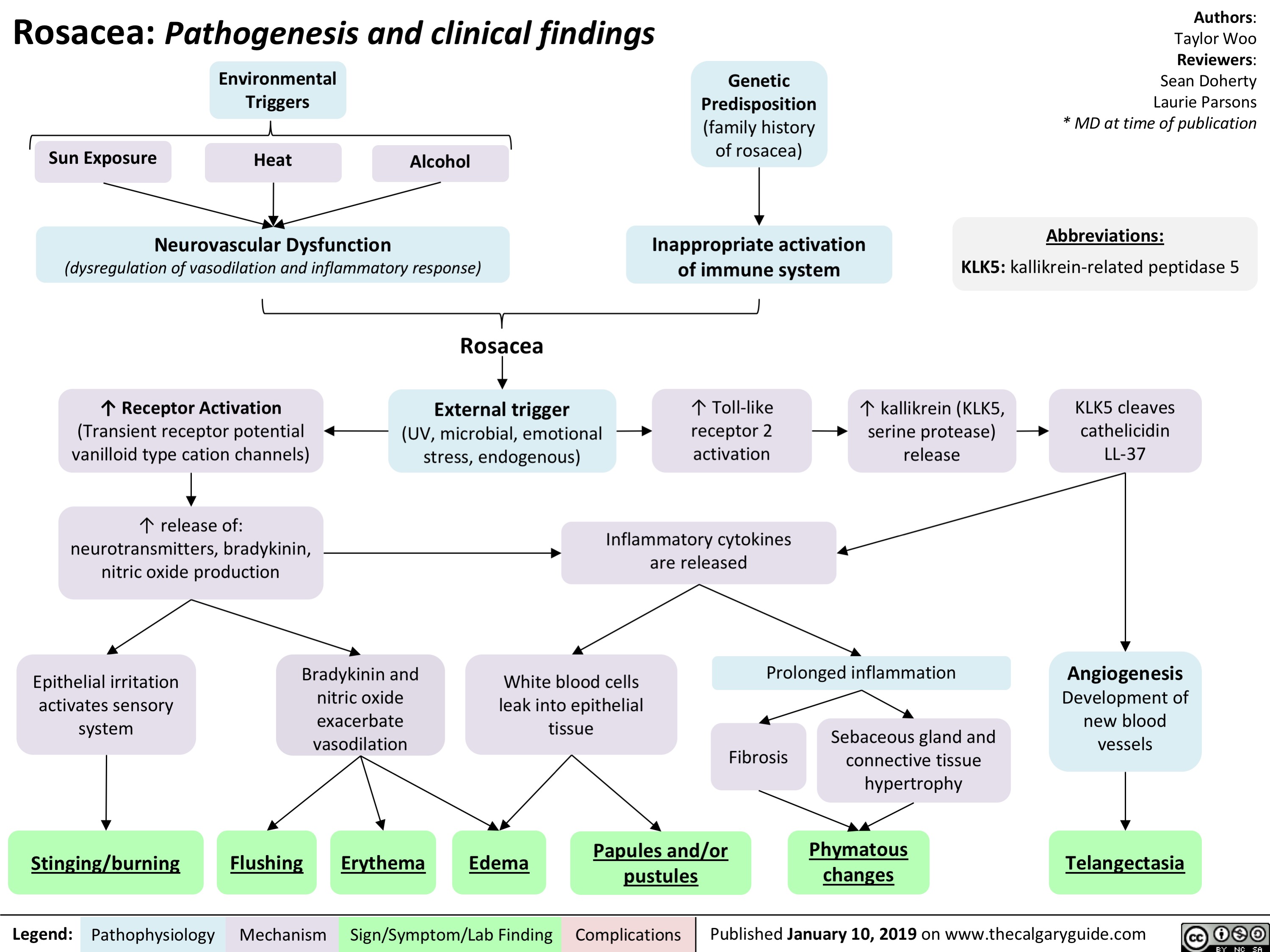
Hepatic Encephalopathy: Pathogenesis and Clinical Findings

Hypernatremia Physiology
![Hypernatremia: Physiology Unreplaced H2O loss
Hypodipsia
H2O shift into cells
Severe exercise, electroshock induced seizures
Transient ↑ cell osmolality
Na+ overload
Inappropriate IV hypertonic solution, salt poisoning
Abbreviations:
H2O: Water
GI: Gastrointestinal
DM: Diabetes Mellitus
DI: Diabetes Insipidus
Na+: Sodium ion
IV: Intravenous
ADH: Antidiuretic Hormone LOC: Level of Consciousness
Skin
Sweat, burns
GI
Vomiting, bleeding, osmotic diarrhea
Fluid [Na+] < serum [Na+]
↑ H2O loss compared to Na+ loss
Renal
DM, Mannitol, Diuretics
Absent thirst mechanism
Hypothalamic lesion impairs normal drive for H2O intake
Nephrogenic
↑ renal resistance to ADH
H2O Deprivation Test + no AVP response
↓ access to H2O
DI
Central
↓ ADH secretion
H2O Deprivation Test + AVP response
↑ [Na+] 10- 15 mEq/L within a few minutes
Weakness, irritability, seizures, coma
↑ thirst, ↓ urinary frequency and volume
Note:
Hypernatremia
Serum [Na+] > 145 mmol/L
Intracranial hemorrhage
Headache, vomiting, ↓ LOC
• Plasma [Na+] is regulated by water intake/excretion, not by changes in [Na+].
• Effects on plasma [Na+] of IV fluids or loss of bodily fluids is determined by the tonicity of the fluid, not the osmolality.
Authors: Mannat Dhillon Reviewers: Andrea Kuczynski Kevin McLaughlin* * MD at time of publication
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published January 11, 2019 on www.thecalgaryguide.com](http://calgaryguide.ucalgary.ca/wp-content/uploads/2019/01/Hypernatremia-Physiology-.jpg "Hypernatremia: Physiology Unreplaced H2O loss
Hypodipsia
H2O shift into cells
Severe exercise, electroshock induced seizures
Transient ↑ cell osmolality
Na+ overload
Inappropriate IV hypertonic solution, salt poisoning
Abbreviations:
H2O: Water
GI: Gastrointestinal
DM: Diabetes Mellitus
DI: Diabetes Insipidus
Na+: Sodium ion
IV: Intravenous
ADH: Antidiuretic Hormone LOC: Level of Consciousness
Skin
Sweat, burns
GI
Vomiting, bleeding, osmotic diarrhea
Fluid [Na+] < serum [Na+]
↑ H2O loss compared to Na+ loss
Renal
DM, Mannitol, Diuretics
Absent thirst mechanism
Hypothalamic lesion impairs normal drive for H2O intake
Nephrogenic
↑ renal resistance to ADH
H2O Deprivation Test + no AVP response
↓ access to H2O
DI
Central
↓ ADH secretion
H2O Deprivation Test + AVP response
↑ [Na+] 10- 15 mEq/L within a few minutes
Weakness, irritability, seizures, coma
↑ thirst, ↓ urinary frequency and volume
Note:
Hypernatremia
Serum [Na+] > 145 mmol/L
Intracranial hemorrhage
Headache, vomiting, ↓ LOC
• Plasma [Na+] is regulated by water intake/excretion, not by changes in [Na+].
• Effects on plasma [Na+] of IV fluids or loss of bodily fluids is determined by the tonicity of the fluid, not the osmolality.
Authors: Mannat Dhillon Reviewers: Andrea Kuczynski Kevin McLaughlin* * MD at time of publication
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published January 11, 2019 on www.thecalgaryguide.com")
Hyponatremia- Physiology
![Hyponatremia: Physiology
Authors: Mannat Dhillon Reviewers: Andrea Kuczynski Kevin McLaughlin* * MD at time of publication
Abnormal Renal H2O Handling (hypo-osmolar serum)
AKI/CKD Heart failure
↓ renal blood flow
↓ glomerular filtration
GFR < 25 mL/min, ↓ urine dilution ↑ H2O retention
Note:
• Plasma [Na+] is regulated by water intake/excretion, not by changes in [Na+].
• Artifactual hyponatremia can be differentiated by a normal or hyperosmolar serum.
Appropriate ADH secretion
↓ EABV
Hypovolemia: losses via GI, renal, skin, 3rd spacing, bleeding
Hypervolemia: heart failure, cirrhosis
↑ Na+/H2O absorption at PCT
↓ EABV, ↑ H2O retention
Urine [Na+] < 20 mmol/L
Hereditary: tubular disorders
(Bartter, Gitlemann syndromes).
Thiazide diuretics
Inappropriate: SIADH, hypothyroidism, AI
Normal EABV
Anti-diuresis
Primary polydipsia, eating disorder
↑ H2O or ↓ solute intake
↓ Osmoles
Impaired desalination
Block NCC
↑ H2O retention ↑ Na+/K+ excretion
Hyponatremia
Serum [Na+] < 135 mmol/L
Urine osmolality > 100 mmol/L
Urine osmolality < 100 mmol/L
Cerebral edema, ↑ intracranial pressure, vasoconstriction
If hypovolemic: ↓ JVP, ↓ blood pressure
Lethargy, altered mental status
Abbreviations:
AKI: Acute Kidney Injury
CKD: Chronic Kidney Disease
GFR: Glomerular Filtration Rate
H2O: Water
PCT: Proximal Convoluted Tubule
EABV: Effective Arterial Blood Volume
NCC: Na+/Cl- Co-Transporter
SIADH: Syndrome of Inappropriate ADH Secretion AI: Adrenal Insufficiency
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published January 11, 2019 on www.thecalgaryguide.com](http://calgaryguide.ucalgary.ca/wp-content/uploads/2019/01/Hyponatremia-Physiology-.jpg "Hyponatremia: Physiology
Authors: Mannat Dhillon Reviewers: Andrea Kuczynski Kevin McLaughlin* * MD at time of publication
Abnormal Renal H2O Handling (hypo-osmolar serum)
AKI/CKD Heart failure
↓ renal blood flow
↓ glomerular filtration
GFR < 25 mL/min, ↓ urine dilution ↑ H2O retention
Note:
• Plasma [Na+] is regulated by water intake/excretion, not by changes in [Na+].
• Artifactual hyponatremia can be differentiated by a normal or hyperosmolar serum.
Appropriate ADH secretion
↓ EABV
Hypovolemia: losses via GI, renal, skin, 3rd spacing, bleeding
Hypervolemia: heart failure, cirrhosis
↑ Na+/H2O absorption at PCT
↓ EABV, ↑ H2O retention
Urine [Na+] < 20 mmol/L
Hereditary: tubular disorders
(Bartter, Gitlemann syndromes).
Thiazide diuretics
Inappropriate: SIADH, hypothyroidism, AI
Normal EABV
Anti-diuresis
Primary polydipsia, eating disorder
↑ H2O or ↓ solute intake
↓ Osmoles
Impaired desalination
Block NCC
↑ H2O retention ↑ Na+/K+ excretion
Hyponatremia
Serum [Na+] < 135 mmol/L
Urine osmolality > 100 mmol/L
Urine osmolality < 100 mmol/L
Cerebral edema, ↑ intracranial pressure, vasoconstriction
If hypovolemic: ↓ JVP, ↓ blood pressure
Lethargy, altered mental status
Abbreviations:
AKI: Acute Kidney Injury
CKD: Chronic Kidney Disease
GFR: Glomerular Filtration Rate
H2O: Water
PCT: Proximal Convoluted Tubule
EABV: Effective Arterial Blood Volume
NCC: Na+/Cl- Co-Transporter
SIADH: Syndrome of Inappropriate ADH Secretion AI: Adrenal Insufficiency
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published January 11, 2019 on www.thecalgaryguide.com")
Coronary anatomy on ECG- Localizing Ischemia
 of the aortic valve left posterior left CA anterior aortic sinus right CA myocardium right coronary artery posterior descending right atrium ventricle SA AV nodes inferior MI leads II III aVF bradycardia heart blocks problems posterior MI V1 V4 leads anterolateral left main circumflex descending septum ventricle apex anteroseptal occlusion dominant T wave flattening inversion transmural reciprocal depression limb leads precordial leads")
Patent Ductus Arteriosus (PDA)- Pathogenesis and Clinical Findings
- Pathogenesis and Clinical Findings
Sun Bishay Gagnon Adderley Ryznar waechter circulating PGE2 low arterial oxygen content genetic factors char syndrome patent birth aortic pressure pulmonary artery pressure continuous flow from aorta to pulmonary artery via the PDA left to right shunt continuous flow murmur precordial activity S1 S2 accentuated pulse volume load dilation dysfunction left atrium ventricle displaced apex dynamic left ventricular impulse grade pulmonary blood flow systemic blood flow exercise intolerance wide systemic pulse pressure pulmonary artery pressure vascular changes resistance difficulty feeding infants failure to thrive heart failure respiratory distress pulmonary hypertension reversal of shunt adult complications eisenmenger syndrome clubbing cyanosis")
Arterial Insufficiency- Signs and symptoms

Venous insufficiency- Signs and symptoms

Takotsubo Cardiomyopathy- Pathogenesis and clinical findings

Meralgia paresthetica- Pathogenesis and Clinical Findings

Reactive Neutrophilia- Pathogenesis and Clinical Findings

Hypoxemia- Pathogenesis and clinical findings

Bronchopulmonary Dysplasia (BPD)- Pathogenesis and clinical findings
- Pathogenesis and clinical findings Adderley Mitchell antenatal post-natal prematurity intrauterine growth restriction genetic predisposition maternal smoking chorioamnionitis pregnancy induced hypertensive disorders post natal mechanical ventilation sepsis O2 toxicity patent ductus arteriosus insult to lungs pro inflammatory cytokines disruption pulmonary vascular alveolar development reduced pulmonary vascular resistance vascular growth and altered vasoreactivity
dysmorphic capillary beds remodelling of pulmonary arteries artery hypertension suboptimal repair abnormal remodelling interstitial fibrosis diffuse haziness interstitial thickening cxr impaired pulmonary gas exchange hypoxia signs of respiratory distress retractions wheezes crackles DLCO airway resistance obstructive lung disease FEV1 separation and alveolar hypoplasia functional alveoli surface area gas exchange")
Neuromuscular Junction (NMJ)- Physiology and pharmacology
- Physiology and pharmacology calcium ion ions voltage gated ca2+ channels acetylcholine ACh receptors nicotinic SNARE protein complex AChR receptor sodium muscle specific kinase action potential voltage gated Ca2+ channels activated release presynaptic terminal binds")
Biliary Atresia (BA)- Pathogenesis and clinical findings
- Pathogenesis and clinical findings Intrauterine environment genetic factors abnormal bile duct development toxic inflammatory response viral immunologic injury to bile duct epithelia pathophysiology poorly understood histology consistent with obstruction on liver biopsy biliary atresia progressive idiopathic fibre-obliterative disease extra-hepatic biliary tree biliary obstruction on intra-operative cholangiogram (diagnostic) partial complete bile duct obstruction delivery of bile acids to small intestine pressure in bile duct absorption of fat and soluble vitamins vitamin K+ deficiency coagulopathy INR PTT bruising petechiae acholic pale stool failure to thrive elimination of bilirubin conjugated direct bilirubin jaundice pruritus excreted urine dark urine diaper yellow pressure bile duct GGT backs up in liver cholestatic hepatitis firm enlarged liver fibrosis cirrhosis ALT AST Horwitz Adderley McKenzie")
Inflammatory-Cascade-Pathogenesis-and-Clinical-Findings
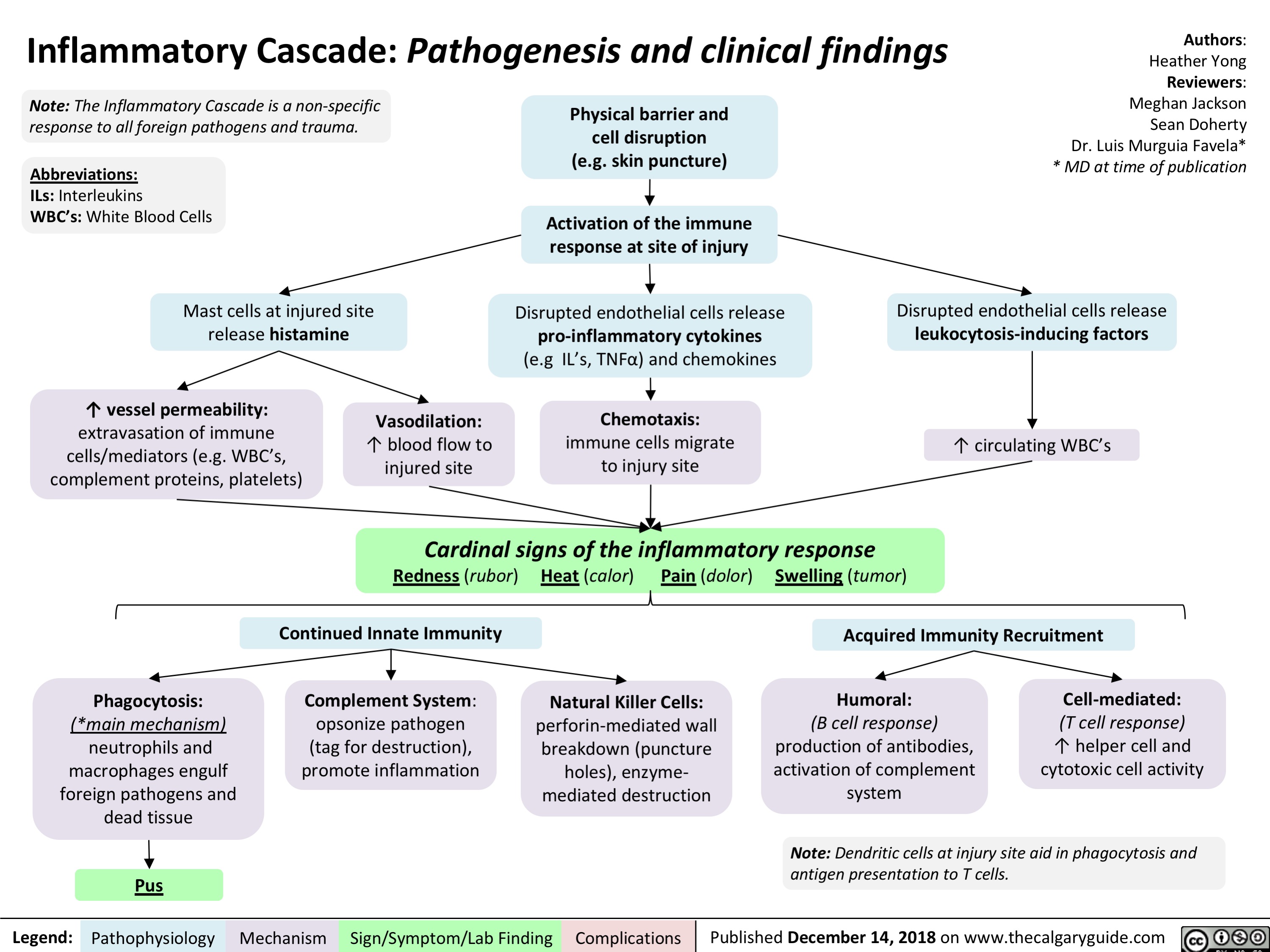
Sepsis, and Septic Shock- Pathogenesis and Clinical Findings

Comorbidities
Immunosuppression or ↑ susceptibility (e.g. splenectomy)
Pathogen virulence
Invasion and host immune avoidance
Vulnerable infection site
↑ likelihood of spread of infection & mortality
Genetics
↑ Sensitivity of innate immune response
Community acquired
Hospital acquired
Infection of host
Innate immune response
Fever, Leukocytosis/ Leukopenia, Left Shift/ Bandemia
Compensatory response
Tachypnea, Altered Level of Consciousness, Hypotension
↓ perfusion and oxygen delivery to organs
Dysregulated Host Response
Pro- and anti-inflammatory response
Life-threatening organ dysfunction caused by a dysregulated host response to infection
The Sequential Organ Failure Assessment (SOFA) or quick
SOFA (qSOFA) Scores may be used to assess mortality risk
Respiratory
↓ PaO2 / FiO2 (mmHg)
Nervous
↓ Level of consciousness
Septic Shock
Cardiovascular
↓ Mean Arterial Pressure
Organ Dysfunction
Liver
↑ Bilirubin
Kidney
↑ Creatinine, Acute oliguria
Coagulation
Thrombocytopenia, ↑ INR or aPTT
Require vasopressors to ↑ mean arterial pressure
Persistent hypotension despite adequate fluid resuscitation
↓ Mean Arterial Pressure (< 65 mmHg), ↑ Lactate (> 2 mmol/L)
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published February 12, 2019 on www.thecalgaryguide.com")
adrenergic-agonists-for-treating-hypotensionlow-blood-pressure
 β1-receptor
activation on
cardiac
myocytes
↑ contractility
α1-receptor
activation on
the smooth
muscle of blood
vessel walls
↑ intra-cellular Ca2+
in these cells, ↑
their contraction
Ephedrine
Direct
effect
Indirect
effect
↑ release of endogenous norepinephrine
from the adrenal medulla (see above)
Mimics epinephrine
(see above)
Primary
Indications:
anaphylaxis,
cardiac arrest
Primary
Indications:
Hypovolemic states
(e.g. blood loss),
Low systemic
vascular resistance
states (e.g. sepsis,
Anesthesia-induced
hypotension)
Primary
Indications:
mainly used in
Anesthesiainduced
hypotension
↑ intracellular
Ca2+ ↑ rate of
myocyte
contraction
Phenylephrine
↑ Cardiac
Output
↑ heart rate
↑ strength
of myocyte
contraction
↑ arterial
wall tone
↑
stroke
volume
Pushes more
blood to flow
back to heart
(↑ preload)
↑ systemic vascular
resistance (SVR)
More blood in
ventricles stretch
myocytes more
optimally for ↑
contractility
(Frank Starling
Law)
↑ venous
wall tone
↑ Blood
Pressure
Nonspecific activation of α and β receptors on other areas of the body
(e.g. on the autonomic nervous system) as well as on cardiac myocytes
Hypertension
Cardiac Dysrythmias: e.g.
palpitations, ventricular fibrillation
Tremors Cardiac
arrest
All four
drugs")
Hyperkalemia- Physiology
![Hyperkalemia: Physiology ↓ Renal Excretion
↑ Intake
↓ Intracellular Shift
Acute and chronic kidney disease; CHF
Principal Cell Dysfunction (TTKG < 7)
ACEi/ARB; AI; heparin
Hypovolemia (TTKG > 7)
↓ EABV
↓ distal flow of Na+ and H2O
Urine [Na+] < 20 mmol/L
Cell lysis
↑ osmolarity H2O efflux
Solvent drag
β2 inhibition α1 stimulation
Digoxin ↓ A
NAGMA ↓ insulin
↓ NHE1 activity
Diabetic nephropathy; NSAIDs
↓ A: ↓ R
K+ sparing diuretics; voltage- dependent RTA
↓ Na+/K+ ATPase activity
↓ GFR
↓ A: ↑ R ↑ A: ↑ R
↓ CCD K+ secretion
↑ K+ availability
↑ K+ release
↓ intracellular K+ influx
Chronic: Desensitize voltage- gated Na+ channels and ↓ membrane excitability
ECG: Peaked T-waves, ↑ PR interval, flat/absent P-wave, ↑ QRS, QRST “sine wave”
Hyperkalemia
Serum [K+] > 5.1 mmol/L
Acute: ↑ extracellular [K+] makes the RMP less (-)
Abbreviations:
A: Aldosterone
AI: Adrenal Insufficiency
CCD: Cortical Collecting Duct
CHF: Congestive Heart Failure
EABV: Effective Arterial Blood Volume H+: Hydrogen ion
K+: Potassium ion
Na+: Sodium ion
NAGMA: Normal Anion Gap Metabolic Acidosis
NSAIDs: Non-steroidal anti-inflammatory drugs Note:
Muscle weakness or paralysis, ↓ urinary acid excretion
R: Renin
RTA: Renal Tubular Acidosis
RMP: Resting Membrane Potential TTKG: Transtubular Potassium Gradient
• Pseudohyperkalemia should always be excluded; can be caused by thrombocytosis, leukocytosis or improper blood withdrawal technique.
Authors: Mannat Dhillon Joshua Low Reviewers: Andrea Kuczynski Kevin McLaughlin* * MD at time of publication
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published March 6, 2019 on www.thecalgaryguide.com](http://calgaryguide.ucalgary.ca/wp-content/uploads/2019/03/Hyperkalemia-Physiology-.jpg "Hyperkalemia: Physiology ↓ Renal Excretion
↑ Intake
↓ Intracellular Shift
Acute and chronic kidney disease; CHF
Principal Cell Dysfunction (TTKG < 7)
ACEi/ARB; AI; heparin
Hypovolemia (TTKG > 7)
↓ EABV
↓ distal flow of Na+ and H2O
Urine [Na+] < 20 mmol/L
Cell lysis
↑ osmolarity H2O efflux
Solvent drag
β2 inhibition α1 stimulation
Digoxin ↓ A
NAGMA ↓ insulin
↓ NHE1 activity
Diabetic nephropathy; NSAIDs
↓ A: ↓ R
K+ sparing diuretics; voltage- dependent RTA
↓ Na+/K+ ATPase activity
↓ GFR
↓ A: ↑ R ↑ A: ↑ R
↓ CCD K+ secretion
↑ K+ availability
↑ K+ release
↓ intracellular K+ influx
Chronic: Desensitize voltage- gated Na+ channels and ↓ membrane excitability
ECG: Peaked T-waves, ↑ PR interval, flat/absent P-wave, ↑ QRS, QRST “sine wave”
Hyperkalemia
Serum [K+] > 5.1 mmol/L
Acute: ↑ extracellular [K+] makes the RMP less (-)
Abbreviations:
A: Aldosterone
AI: Adrenal Insufficiency
CCD: Cortical Collecting Duct
CHF: Congestive Heart Failure
EABV: Effective Arterial Blood Volume H+: Hydrogen ion
K+: Potassium ion
Na+: Sodium ion
NAGMA: Normal Anion Gap Metabolic Acidosis
NSAIDs: Non-steroidal anti-inflammatory drugs Note:
Muscle weakness or paralysis, ↓ urinary acid excretion
R: Renin
RTA: Renal Tubular Acidosis
RMP: Resting Membrane Potential TTKG: Transtubular Potassium Gradient
• Pseudohyperkalemia should always be excluded; can be caused by thrombocytosis, leukocytosis or improper blood withdrawal technique.
Authors: Mannat Dhillon Joshua Low Reviewers: Andrea Kuczynski Kevin McLaughlin* * MD at time of publication
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published March 6, 2019 on www.thecalgaryguide.com")
Torsades de Pointes (TdP)- Pathogenesis and Clinical Findings
![Torsades de Pointes (TdP): Pathogenesis and Clinical Findings
Drugs (e.g. Class 1A [quinidine], Class III [sotalol,
amiodarone], TCAs, erythromycin, quinolones, anti-histamines)
Sinus bradycardia, AV block
Metabolic abnormality (hypo K+/Ca2+/Mg2+)
Primary heart disease: ischemic, congestive heart failure, cardiomyopathy
Acquired long QT syndrome
Congenital long QT syndrome
↓ repolarizing current/ depolarizing current in cardiomyocytes
Mutated cardiac ion channels
Author: Nicola Adderley Reviewers: Luke Gagnon Emily Ryznar *Saman Rezazadeh *George Veenhuyzen MD at time of publication*
↓ repolarizing current in cardiomyocytes
QTc interval
Prolonged ventricular action potential duration
Early after depolarization (EAD) triggering PVC
Torsades de Pointes
Illustrated changes to action potential:
Normal cardiac action potential
EAD
Abbreviations: TCAs: tricyclic antidepressants AV: atrioventricular QTc: QT interval, corrected for heart rate
PVC: premature ventricular contraction
VF: ventricular fibrillation
SR: sinus rhythm
Polymorphic ventricular tachycardia initiated by PVC in the setting of QT interval prolongation and maintained by functional re-entry
Non-sustained TdP
Asymptomatic, palpitations, syncope (length-dependent)
Illustrated changes to ECG Strip:
OR
Sinus rhythm restored
Degeneration to VF
Sudden cardiac death
Prolonged repolarization
Triggered beat (PVC)
A
BCDE
A. Prolonged QT interval B. PVC
C. PVC triggers TdP
D. Non-sustained TdP
E. Return to SR
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published March 10, 2019 on www.thecalgaryguide.com](http://calgaryguide.ucalgary.ca/wp-content/uploads/2019/03/Torsades-de-Pointes-TdP-Pathogenesis-and-Clinical-Findings-.jpg "Torsades de Pointes (TdP): Pathogenesis and Clinical Findings
Drugs (e.g. Class 1A [quinidine], Class III [sotalol,
amiodarone], TCAs, erythromycin, quinolones, anti-histamines)
Sinus bradycardia, AV block
Metabolic abnormality (hypo K+/Ca2+/Mg2+)
Primary heart disease: ischemic, congestive heart failure, cardiomyopathy
Acquired long QT syndrome
Congenital long QT syndrome
↓ repolarizing current/ depolarizing current in cardiomyocytes
Mutated cardiac ion channels
Author: Nicola Adderley Reviewers: Luke Gagnon Emily Ryznar *Saman Rezazadeh *George Veenhuyzen MD at time of publication*
↓ repolarizing current in cardiomyocytes
QTc interval
Prolonged ventricular action potential duration
Early after depolarization (EAD) triggering PVC
Torsades de Pointes
Illustrated changes to action potential:
Normal cardiac action potential
EAD
Abbreviations: TCAs: tricyclic antidepressants AV: atrioventricular QTc: QT interval, corrected for heart rate
PVC: premature ventricular contraction
VF: ventricular fibrillation
SR: sinus rhythm
Polymorphic ventricular tachycardia initiated by PVC in the setting of QT interval prolongation and maintained by functional re-entry
Non-sustained TdP
Asymptomatic, palpitations, syncope (length-dependent)
Illustrated changes to ECG Strip:
OR
Sinus rhythm restored
Degeneration to VF
Sudden cardiac death
Prolonged repolarization
Triggered beat (PVC)
A
BCDE
A. Prolonged QT interval B. PVC
C. PVC triggers TdP
D. Non-sustained TdP
E. Return to SR
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published March 10, 2019 on www.thecalgaryguide.com")
Shoulder Dystocia: Complications

Previous
shoulder
dystocia
Size discrepancy between fetal
shoulders and maternal pelvis
Multiparity
Maternal
diabetes
Inadequate
uterine tone (over
distension of
uterus, prolonged
2nd stage) and
birth canal trauma
from complicated
delivery
**Attempts to
disimpact and/or
deliver a
macrosomic fetus
Traction to head
can lead to
stretching and
tearing of
brachial plexus
nerves
Intentional or
incidental
fracture of the
fetus’:
Dysfunctional
or prolonged
labour and/or
contractions
↓ oxygenation
to fetus
Authors:
Danielle Hubbert
Risk Factors: *Up to 50% have no risk factors = Obstetrical Emergency that is challenging to predict Mark Diaz
Fetal death
Legend: Pathophysiology Mechanism Sign/Symptom/Lab Finding Complications Published March 12, 2019 on www.thecalgaryguide.com
Prolonged
2nd stage of
labour
Maternal
obesity
Operative
vaginal
delivery
Fetal anterior shoulder becomes impacted
against maternal pubis symphysis and
fails to deliver spontaneously with normal efforts
Clavicular fracture
Erbs palsy (C5-6)
Klumpke palsy (C8-T1)
(rarely permanent)
Episiotomy or
3rd-4th degree
perineal tears Postpartum
hemorrhage
Hypoxia/asphyxia (see PPH slide)
Turtle sign – fetal head
retracts tight against
perineum
Uterine
rupture
Antepartum Risks Intrapartum Risks
Cord
Compression
Weakened and
distended
musculature
Fetal Complications Maternal Complications
Clavicle, to
↓ diameter
of shoulders
Humerus, when
sweeping
posterior arm
across chest
Humeral
fracture
Reviewers:
Dalynne Peters
Angela Deane
Ingrid Kristensen*
*MD at time of publication")
Primary Myelofibrosis pathogenesis and clinical findings

within hematopoietic
stem cells
Non-driver mutations in
other myeloid genes
(e.g., LNK, CBL, TET2,
ASXL1, IDH)
Constitutive activation of
cellular proliferation
pathways
↑ cell signaling
↑ gene transcription and
expression
Cellular proliferation and
resistance to apoptosis
Proliferation of abnormal
megakaryocytes
↑ neutrophil engulfment
by megakaryocytes
↑ growth factor release
by megakaryocytes
Stimulation of
fibroblasts
Stimulation of
endothelial cells
New blood vessel formation
↑ osteoprotegerin Unbalanced osteoblast
proliferation Osteosclerosis
Fibrosis of the
bone marrow
Anemia
Bleeding and
bruising
Infections
Fatigue and pallor
Bone pain
Increased cell
turnover
Tumor lysis
syndrome
Cachexia, night sweats,
fever/chills, malaise
Expanding
marrow pushing
against bone
Extramedullary
hematopoiesis Hepatomegaly
Portal
hypertension
Splenomegaly
↑ LDH
Thrombocytosis
Leukocytosis
Secretion of
coagulation
inducing cytokines
Arterial and
venous
thromboembolism
↓ blood cell
production
&
leukoerythroblastosis
Thrombocytopenia
Leukopenia
↑ K+, PO4
2-, uric acid
↓ Ca2+
Bone pain
Periostitis
Immature granulocyte and
erythroid precursors with blasts
↑ cytokine
production
(+)
↑ sequestration of blood cells
Disseminated
intervascular
coagulation
(see MAHA slide)
Definitions:
Periostitis – Inflammation of the
membrane surrounding bone
Osteosclerosis – Abnormal hardening
and increased in density of bone")
Hemorrhoids - Pathogenesis and Clinical Findings

Increased Intra-Abdominal Pressure
I.e. pregnancy, constipation, chronic straining,
lifting, cirrhosis
Hemorrhoids: Pathogenesis and clinical findings
Dilations originate from inferior
hemorrhoidal venous plexus
Vascular cushions engorge
along anal canal
Legend: Published March 30, 2019 on www.Pathophysiology Mechanism Sign/Symptom/Lab Finding Complications thecalgaryguide.com
Authors:
Aleeza Manucot
Reviewers:
Yoyo Chan
Sean Doherty
Dr. Sylvain Coderre*
* MD at time of publication
Supporting tissues of anal cushions weaken,
disintegrate, or deteriorate
Inflammatory reaction
occurs, involving vascular
wall and connective tissue
Thrombosis
Pain
↑ mucus secretions or fecal
soiling of prolapsing
hemorrhoids
Cushion epithelium erodes via
damage from compression
Painless
rectal
bleeding
Bleeding without prolapse
Prolapse with spontaneous
reduction
Prolapse requiring manual
reduction
Irreducible
1st degree
2nd degree
3rd degree
4th degree
Infarction and thrombosis
Acute severe pain
Anal cushions prolapse (downwardly slide)
into rectum or open space
Dentate line: divides
the upper two thirds
and lower third
of the anal canal
EXTERNAL Hemorrhoids
- Found distal to the dentate line
- Somatic innervation
Somatic nerve
receptors activated
Sebaceous glands
↑ secretions around
area of hemorrhoid
Itching Perianal
irritation
Swelling
Inflammation creates
prothrombotic state
Hemorrhoids")
gastroesophageal-reflux-disease-gerd-complications
: Complications
Esophageal stricture
disease
Esophagitis
Esophageal
adenocarcinoma
Barrett’s esophagus
GERD
Reflux of gastric content into distal esophagus
Damage to squamous
esophageal epithelium
Legend: Published March 30, 2019 on www.Pathophysiology Mechanism Sign/Symptom/Lab Finding Complications thecalgaryguide.com
Authors:
Wendy Wang
Reviewers:
Yoyo Chan
Sean Doherty
Dr. Sylvain Coderre*
* MD at time of publication
Squamous esophageal
epithelium undergoes
metaplasia to become
columnar epithelium
This predisposes cells to
premalignant changes
(dysplasia)
Collagen is deposited
where ulcers heal
Asthma/Chronic Cough
Chronic Laryngitis
Laryngeal and
Tracheal Stenosis
Extra-esophageal Complications Esophageal Complications
Airway becomes
irritated
Fibroblasts proliferate
and deposit granulation
tissue in airway
Tissue deposition
leads to narrowing of
laryngeal and
tracheal space
Damage to pharyngeal
lining and airway
Esophageal tissue repeatedly
exposed to stomach acid
Pro-inflammatory cells and cytokines
are recruited to the area
Definitions:
• Metaplasia: abnormal change in the
nature of a tissue
• Pro-inflammatory cells and cytokines:
Mediators of inflammation. Examples
of cells include macrophages and T
cells, cytokines include IL-17, IL-2, IL-4
Over time, collagen fibers
contract
Bronchoconstriction
↑ vagal
tone
↑ bronchial
reactivity
Cough sensory
nerve endings are
stimulated
Vagal reflex
is activated
Activation of
cough center in
brainstem
↑ inflammation of
squamous epithelium
Ulcers form in esophagus")
autosomal-dominant-polycystic-kidney-disease-adpkd
:
Pathogenesis,
Clinical Findings,
and Complications
Author:
Yan Yu*
Reviewers:
David Waldner*
Sean Spence*
Andrew Wade*
* MD at time of
publication
Legend: Published April 14, 2019 on www.Pathophysiology Mechanism Sign/Symptom/Lab Finding Complications thecalgaryguide.com
One theory (mechanism unclear): these mutations in the polycystin
gene result in dysfunctional Ca2+ channels on epithelial cells
PKD1 mutation
(~78%)
Abnormal Ca2+ entry disrupts intracellular Ca2+ signaling
In the Kidney: all segments of the nephron develop cysts: sacs of flattened epithelium
filled with proteinaceous fluid, replacing normal parenchyma with dysfunctional tissue
PKD2 mutation
(~15%)
Expansive cell
proliferation
Low urine
Osmolality
(< 500
mmol/kg)
Abnormally expandable
basement membranes
PKD3 mutation
(rare)
↑ fluid
secretion
95% inherited, autosomal dominant mutations
5% spontaneous mutations
In adults, the same pathophysiology occurs
in epithelial tissue throughout the body
When pH of urine <5.5, uric acid is in
its protonated form & is less soluble
àprecipitates uric acid stones
Nephrolithiasis
Urine accumulates within cysts Cyst growth
Pyelonephritis
Damaged
tubules
leak
proteins
into filtrate
Proteinuria
Flank pain
Inability to
concentrate
urine
Low urine
specific
gravity
(<1.010)
¯ NH3 production,
↑ acidity of renal
tubule (¯ pH)
Activates
nociceptors Cyst hemorrhage
Urine stasis à
precipitation of
CaOxalate stones
within cysts
Multiple Renal Cysts
(bilaterally, in cortex and
medulla, on Ultrasound)
In the Brain:
expansion &
weakening of
cerebral
arterial walls
“Berry
Aneurysms” 9-
12%
(ask about this
on Family Hx!)
In many organs:
epithelial tissue
expansion &
fluid secretion
In the Heart:
abnormal valve
collagen matrix
Valve prolapse
& regurgitation
Liver (90%),
spleen/pancreas
(5-10%), thyroid
(rare) cysts
In seminal vesicles:
Cyst formation disrupts
sperm motility
Infertility
In GI tract: Cyst
formation
Herniations,
diverticuli
Dysfunctional
collecting ducts
Abdominal mass
(enlarged kidneys)
(may be palpable)
Blood leaks into
renal tubules
Hematuria (isomorphic)
Bacteria
accumulate
in the static
urine
Dysfunctional
proximal tubules
Unknown
mechanisms à
¯ urine citrate
àmore urine
Ca2+ binds to
oxalate than to
citrate à↑ Ca-
Oxalate stones
Note: PKD1 mutations have
more severe prognosis than
PKD2 mutations (earlier
disease onset, larger cysts)
Vessels
tear
more
easily
Stretches
renal
capsule
Cysts compress renal vasculature
¯ Glomerular perfusion
To ↑ perfusion à
kidneys activate RAAS
Hypertension")
Rickets and Osteomalacia: Pathogenesis and Clinical Findings

Shear forces
bend the
osteopenic
bone
Short stature
Diffuse skeletal pain
(bone tenderness)
Osteopenia
reduces bone
density and
cause fractures
with minimal
force applied
If hypophosphatemic,
production of ATP and
other high energy
molecules declines
Unequally
distributed
forces and
muscle/tendon
tension
stimulate
nociceptors
Legend: Published November 26, 2012 on www.Pathophysiology Mechanism Sign/Symptom/Lab Finding Complications thecalgaryguide.com
Calcification inhibitors
(excess exposure to Al,
Fluoride, etidronate)
Lack, or reduced
function, of
mineralization
enzymes (like ALP)
Lack of bone mineral components:
1. Phosphate: renal tubule disorders, vit D
or Phosphate deficiency, ↑FGF23
2. Calcium: severe deficiency (infants)
Rickets: Occurs before
epiphyseal closure
Epiphyseal
plates do not
fuse, impairing
bone growth
Bowed legs
Cartilage in epiphyseal
plates cannot become
ossified
Disruption in
calcium ion
homeostasis
↓ GI
absorption of
Ca2+ into
blood
↓ Kidney
reabsorption
of Ca2+ into
blood
↓ energy available
to muscle
Author:
Payam Pournazari
Reviewers:
Yan Yu
Spencer Montgomery
David Hanley*
* MD at time of
publication")
Polyarteritis Nodosa (PAN): Pathogenesis and Clinical Findings
: Pathogenesis and clinical findings
Environmental triggers
Infectious/viral agents (commonly Hepatitis B)
Medical Comorbidities Malignancies (most commonly hairy-cell leukemia)
Immunogenetic Predisposition: patient is genetically predisposed to a dysregulated immune response
Fever
↑ ESR and CRP
Postulate 1
Viral antigen-antibody complexes deposit in vasculature, causing lesions and activating cellular inflammatory response
Authors: Nela Cosic, Yan Yu* Reviewers: Sean Doherty Martin Atkinson*
* MD at time of publication
Palpable or necrotic purpura
Malignant Hypertension
Renal Insufficiency
Myocardial ischemia
Heart failure Diffuse myalgias
Postulate 2
Viral replication causes direct injury to vascular endothelial cells
↑ Anti- endothelial cell autoantibodies (AECA)
Altered cytokine profile (↑TNF-α, IL-1β, IFN-α, IL- 2)à↑ T-cell mediated immune response
Weight metabolism Loss
Autoimmune attack on various areas of the body
Malaise and/or Arthralgias (knees, ankles, elbows, wrists)
Orchitis: Testicular pain, erythema and/or swelling
Small intestine perforation GI Manifestations
Non-specific abdo pain
GI hemorrhage
Peripheral sensory changes: Distal mononeuropathy
multiplex
Polyarteritis Nodosa (PAN)
Focal segmental necrotizing leukocytoclastic vasculitis of medium or small-sized arteries
Inflammation of arteries damages the vascular endothelium of those arteries
Inflammation predisposes formation of arterial thromboses
Blockage of arteriesà tissue ischemia and possible necrosis (tissue cell death)
↑ basal
Arterial aneurysms
Inflamed subcutaneous arteries
Inflamed renal artery
àluminal narrowing and reduced blood flow to kidneys
Inflamed coronary artery à luminal narrowing, occlusion, thromboses
Segmental inflammation of muscular arteries, stimulating surrounding nociceptors. Muscle ischemia develops long-term.
Ischemia/necrosis of the testicles
Ischemia/necrosis of the small intestine
Ischemic vasculitic nerve damage: Immune complex deposition within vessel walls of arteries traveling with nerves leads to persistent vascular inflammation and ischemia of associated nerve
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published April 18, 2019 on www.thecalgaryguide.com")
dermatomyositis-dm-and-polymyositis-pm-pathogenesis-and-clinical-findings
 and Polymyositis (PM):
Authors: Merna Adly, Yan Yu* Reviewers: Nela Cosic Sean Doherty Martin Atkinson* * MD at time of publication
Pathogenesis and clinical findings
Immunogenetic and Cellular Predisposition
Genetic polymorphisms cause dysregulated immune response, cytokine profile, and protein expression in muscle cells
Demographics
F:M, 2:1
Bimodal age distribution:
- Juvenile: 7 years of age (mean) - Adult: 52 years of age (mean)
Malignancy
Tumor cells increase systemic inflammatory response, leading to increase of autoantigens associated with DM
↑ in DM-associated autoantibodies
(Ex. Anti-Mi-2/ Anti-Jo-1) Autoantibodies bind to DNA or RNA in muscles,
provoking a systemic inflammatory response
↑ chemokine and cytokine release in endothelial vasculature of muscles
Perivascular Capillary necrosis inflammation
Lack of blood supply to the myofibers causes endofascicular hypoperfusion and muscle ischemia
Muscle tissue damage:
Inflammatory infiltrates destroy cellular components of muscle (endoplasmic reticular, myofiber, and keratinocytes)
Environmental triggers
Infectious agents (ex. Picornavirus) or drugs (ex. statins) provoke immune response
Elevated Antinuclear Antibodies
Anti-Jo-1 Anti-OJ Anti-Mi2 Anti-SRP Anti-EJ Anti-PL12 Anti-PL7
These processes occur in the skin on the dorsum of the hands, forming hyperkeratotic flat red papules
These processes occur in the upper & lower eyelids, causing red-purple discoloration +/- swelling
Perifascicular atrophy
(Observed on histology)
Weaker GI tract musculature Weaker pulmonary musculature Weaker cardiac musculature
Dermatomyositis only:
Gottron Papules Heliotrope Rash
Damaged muscle cells release their internal cellular enzymes into the bloodstream
Elevation of muscle enzyme levels in serum:
Creatinine kinase (CK), lactate dehydrogenase (LD), aldolase, aspartate aminotransferase (AST), and alanine aminotransferase (ALT)
Muscle Biopsy Findings Muscle necrosis, fiber regeneration, diffuse CD8+ T lymphocytes infiltrates
Bilateral Muscle Weakness Subacute development, primarily deltoids and hip flexors affected
Dysphagia
Aspiration, respiratory compromise
Atrioventricular defects, tachyarrhythmias, dilated cardiomyopathy
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published April 18, 2019 on www.thecalgaryguide.com")
DiGeorge Syndrome: Pathogenesis and Clinical Findings
, though their role is less well understood.
Abbreviations:
• PA-VSD – pulmonary atresia
with ventricular septal defect
• TBX1 – T-box Protein 1
• VSD – ventricular septal defect
Heterozygous deletion at chromosomal region 22q11.2
The region’s main gene product, TBX1*, exhibits haploinsufficiency: even a heterozygote for this gene product, producing half the normal quantities of TBX1, is insufficient to produce a normal phenotype
Abnormal pharyngeal arch development
Hypoplastic / aplastic thymus ↓ T cells
(lymphopenia)
Craniofacial malformations (e.g. tracheomalacia, horizontal Eustachian tubes, cleft palate)
Impaired ear and sinus drainage
Abnormal conotruncus development
Heart defects
(e.g. interrupted aortic arch, tetralogy of Fallot, PA-VSD/VSD, truncus arteriosus)
Hypoplastic parathyroid glands
Hypocalcemia Seizures
(usually neonatal onset)
Memory Aid:
CATCH-22
Cyanotic congenital heart disease
Abnormal facies
Thymic hypoplasia Cognitive impairment Hypoparathyroidism, hypocalcemia
22q11.2 deletion
Abnormal T cell regulation
and development
Compromised cytotoxic T cells
Susceptibility to intracellular pathogens
Compromised helper T cells
↓ communication with memory B cells
↓immunoglobulins
(progressive hypogammaglobulinemia)
Susceptibility to extracellular pathogens
Autoimmunity and Atopy
Note: There is phenotypic variability in DiGeorge Syndrome. Other features include: thin upper lip, upslanted palpebral fissures, prominent nose, low-set ears, small mouth, hearing impairment, long tapered fingers, scoliosis, vertebral malformations, learning disabilities, and failure to thrive.
Viral Infections
Bacterial Infections
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published April 21, 2019 on www.thecalgaryguide.com")
Molluscum Contagiosum: Pathogenesis and Clinical Findings
:
• MCV I *more prevalent than other
subtypes except in
immunocompromised individuals
• MCVII
• MCV III
• MCVIV
Abbreviations:
- MCV: Molluscum contagiosum virus - NF-KB: nuclear factor kappa-light- chain-enhancer of activated B cells
(a transcription factor regulating genes responsible for innate & adaptive immune responses)
Sexually active adults: abdomen, genitals, inner thighs Children: face, trunk, limbs
Immunosuppression
Active Atopic Dermatitis Hot, humid climates Crowded living conditions
Risk factors for infection
Direct skin-to-skin contact with infected host Contact with contaminated objects
Molluscipox virus infection
of epidermal keratinocytes
Virus replicates in cytoplasm of epithelial cells
Cytoplasmic inclusion bodies form, multiply & push nucleus to edge of cell
Rupture and discharge of virus-packed inclusion bodies
MCV-released proteins inhibit NF-KB activation
Suppress host immune response to infection
Single or multiple, 2-6 mm, pearly white, discrete papules with central umbilication containing white, waxy curd-like core appearing on any cutaneous surface
Papule contains caseous plug
Squeezing the papule produces white cheesy fluid
Transmission of MCV to conjunctiva of eye
Scratching or removing lesions by curettage and cautery
Staphylococcus aureus infects lesion
Neutrophils & macrophages phagocytose bacteria
Immunosuppression
Impaired T-cell immunity fails to defend against MCV infection
Conjunctivitis
Scarring
Abscess
Widespread lesions
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published April 21, 2019 on www.thecalgaryguide.com")
acute-somatic-pain
 Thermal (eg: hot stove) Chemical (eg: inflammation)
Nociceptors activated at site of injury (1st order sensory neurons)
Nociceptive fibres (A∂ and C) carry noxious sensory information to the ipsilateral dorsal horn of the spinal cord
Excitatory neurotransmitters are released and stimulate 2nd order sensory neurons
2nd order sensory neurons immediately cross the midline of the spinal cord, and ascend up the opposite side’s anterolateral (aka spinothalamic) tracts, terminating in various locations:
Authors: Lisa Murphy Yan Yu* Reviewers: Mackenzie Gault Melinda Davis* * MD at time of publication
Nociceptors: neurons that detect noxious or painful stimuli and carry this information to the spinal cord. There are two major types:
A∂ fibres: myelinated, initial “sharp, fast” feeling
C fibres: unmyelinated, delayed, “dull, burning” feeling
To hypothalamus
2nd order neuron synapses in the hypothalamus
Hypothalamic neurons coordinate the body’s visceral response to pain
2
nd
To thalamus
order neuron terminates in thalamus
To brainstem
2nd order neuron synapses in brainstem’s reticular formation
To midbrain
2nd order neuron synapses in periaqueductal gray area (PGA) in the midbrain
In the thalamus, 2nd order sensory neurons synapse with 3rd order sensory neurons, which carry the signal to the cerebral cortex
Stimulates descending pathways to modulate the incoming pain signal
Decreased or increased perception of pain
Pain localization and sensation
Emotional and behavioural response
↑ Heart Rate
Nausea
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published April 25, 2019 on www.thecalgaryguide.com")
Small Bowel Infarction
 versus the colon’s dual blood supply (SMA and IMA)
• With decreased perfusion colonic tissue tends to suffer from ischemia rather than more serious infarction
• Colonic ischemia presents with pain, diarrhea, and rectal bleeding
Abbreviations
• SMA - Superior mesenteric
artery
• IMA - Inferior mesenteric artery
Atrial fibrillation
Blood stasis in left atria of heart more
prone to coagulation
Embolism occluding SMA
Hypertension, dyslipidemia, smoking, diabetes, + family hx
Atherosclerosis (of SMA)
Thrombosis in the superior mesenteric artery
↓ Arterial perfusion of the small intestine (↓ O2 delivery to bowel tissue)
Ischemia of bowels Infarction of bowels
Venous trauma
↓ blood flow and endothelial injury
hypercoagulable state
Mesenteric venous thrombosis, backing up arterial blood
Food in the
intestine ↑ demand for blood in gut
Death of cells under visceral peritoneum stimulates autonomic nerves
Post-prandial abdominal pain
Severe central abdominal pain
Small bowel infarction from
SMA occlusion is commonly pain progressive, and out of proportion with the patient’s physical exam findings
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Re-Published June 15, 2019 on www.thecalgaryguide.com")
Crohn's Disease

Inflammation of the GI tract lining
- Inflammation is “transmural”, spanning the entire thickness of the intestinal wall from luminal mucosa to the serosa.
- The inflammation occurs anywhere in the GI tract from the oral mucosa to the anal mucosa (from ‘gums to bum’) in skip lesion pattern.
Atrophy, scarring of the intestinal villi
Inflammatory cytokines destroy the mucosa epithelial cells of the GI tract wall, causing cell apoptosis and ulceration
↑ permeability of the blood vessels supplying the GI tract wall
Chronic inflammation impairs healing responses
Dysregulated wound healingàexcess
extracellular matrix deposition
Fibrosis leads to scar tissue and thickening of all layers of the GI tract
Strictures
Inflammation is systemic, affecting:
Joints Arthropathy Erythema
Impaired absorption of nutrients
Weight loss
Prolonged GI bleeding
Anemia
Transporter proteins responsible for Na+ reabsorption gradually disappear from the epithelium
More sodium (and thus water) is
retained in the GI tract lumen
Microperforations can penetrate through the intestinal wall
Anal fistulae (“holes” connecting the anus to the skin, bladder, peritoneum, small bowel, etc.)
Continued inflammation and/or infection can lead to:
Leakage of fluid out of capillaries into the GI tract
Luminal edema and swelling
Narrowing of GI lumenàbowel obstruction
Skin
Mouth Eyes
Liver
nodosum, pyoderma gangreno- sum
>5 canker sores
Uveitis
Iritis, scleritis
Sclerosing cholangitis
↓ fat absorption
Fatty acids (negatively charged) bind Ca2+, freeing oxalate from Ca2+
↑ oxalate absorbed into blood & filtered by kidney
Calcium oxalate kidney stones
Diarrhea
Abdominal cramping and pain
(see Bowel Obstruction page for full mechanism
Anal abscesses Inflammatory masses
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Re-Published June 15, 2019 on www.thecalgaryguide.com")
Hemophilia
 accounts for approx. 15% of cases
• Factor levels below 1% of normal constitutes severe disease, 1-5% moderate disease, and 5-40% mild disease
• The more severe the disease, the greater the odds of “spontaneous” bleeds occurring
Genetic defect on the X chromosome affecting the factor VIII or factor IX gene
Deficiency of functional factor VIII (Hemophilia A) or factor IX (Hemophilia B) in blood
Insufficient clotting action when faced with a bleeding challenge
Factor VIII/IX deficiency slows intrinsic clotting pathway
Factor VIII/IX not part of extrinsic clotting pathway
↑PTT Normal PT
Uncontrolled bleeding Occurs across entire body
Hemarthrosis (bleeding in joints) Intramuscular hematoma
Dental bleeds
Intracranial hemorrhage
GI/GU bleeds, etc.
Chronic hemophilia arthropathies
(main contributor to disability and reduced QoL)
In severe patients, repeated joint bleeds common
Recurrent bleeding into joints damages cartilage
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Re-Published June 15, 2019 on www.thecalgaryguide.com")
Signs and Symptoms of Pulmonary Embolism

Deep Vein Thrombosis – popliteal, femoral, iliac veins Clot migrates to IVCàright atrium of heartàright
ventricleàpulmonary vasculature
Large clots (saddle emboli) are lodged in pulmonary arteries
Small clots are lodged in pulmonary arterioles
Saddle embolus (pulmonary artery obstruction)
Back-up of blood into right heart
Right heart strain
↓ CO2 delivery to the lungs for exhalation
Less CO2 exhaled, CO2 builds up in the blood, triggers medullary chemoreceptors to ↑ respiratory rate
Well-ventilated (V) areas of lung do not receive adequate blood supply (Q); vice versa
V/Q mismatch
On V/Q scan
Signals brain to ↑ heart rate
Ischemic tissue becomes inflamed and adheres to pleura
Pleural friction rub
Sandpaper-like sound heard on auscultation
Pleuritic chest pain
Focal, localized chest pain that occurs with each breath
Clot ↓ pulmonary arterial/arteriolar blood flow
↓ delivery of deoxygenated blood to alveoli for oxygenation
Low O2 in blood (↓ O2 saturation) is detected by aortic/carotid chemoreceptors
Signals brain to ↑ respiratory rate
If circulation to lung periphery is cut off, sub-pleural lung tissue can become ischemic and infarct
Irritation of somatic sensory nerve endings on the parietal pleural membrane
Pain stimulates adrenergic response
Tachycardia
Dyspnea/shortness of breath (SOB)
Most sensitive indicator of PE, but not very specific
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Re-Published June 15, 2019 on www.thecalgaryguide.com")
Hemolytic Anemia - Pathophysiology
 may result in slightly macrocytic anemia (because reticulocytes have larger volumes than RBCs)
Defects in the RBC’s environment
Defects in RBC membranes
Ex. Hereditary Spherocytosis:
mutation causing deficiency of RBC structural proteins like ankyrin or spectrin
RBC membranes become weakened and form blebs that break off
↓ RBC surface area while volume remains constantà RBC becomes spherical
Spherocytes in spleen trapped and phagocytosed by splenic macrophages (extravascular hemolysis)
Defects in RBC internal contents (thalassemia, hemoglobinopathies, and metabolic defects)
Ex. Sickle Cell Disease: point mutation in hemoglobin (Hgb) structure (GluàVal)
Inappropriate Hgb polymerization in low oxygen environments due to mutationàRBC becomes rigid, forms a sickle shape
Inflexible RBCs become trapped in the spleen’s sinusoid membranes àphagocytosed by splenic macrophages (extravascular hemolysis)
Infection triggers immune system activation
Autoimmune processes
TTP/HUS (abnormal platelet aggregation blocking blood vessels)
Production of abnormal
antibodies and immune complexes targeted against RBC surface antigens
Immunoglobulin-bound RBCs are marked for
destruction by the immune system (by either the cell- mediated or complement- mediated pathways)
DIC (fibrin deposition blocking blood vessels)
Artificial heart valve
RBCs are sheared when they flow past an abnormal surface
Rate of hemolytic RBC destruction > rate of bone marrow RBC synthesis (reticulocytosis)
↓ total number of RBCs in the body (despite normal RBC production/volume)
Normocytic anemia
Authors: Yan Yu Katie Lin Man-Chiu Poon* Reviewers: Andrew Brack Julia Heighton JoyAnne Krupa Lynn Savoie* * MD at time of publication
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Re-Published June 15, 2019 on www.thecalgaryguide.com")
Wilson's Disease

Hepatic Cu accumulation, deposition in hepatocyte lysosomes
Hepatocyte injury (speculated mechanism: free radicals)
Cu leak from damaged hepatocytes
Epidemiology:
• Autosomal Recessive condition with prevalence of 1:30,000 • 60% of cases present initially with neurologic Symptoms
• Fulminant cases present with acute liver failure and massive
hemolysis, treated with liver transplant
↓ ceruloplasmin release ↓ serum ceruloplasmin
Early asymptomatic liver dysfunction
Cu movement into bloodstream
Cu deposition in vulnerable tissues
Abbreviations:
• Cu - Copper
• AST - Aspartate Aminotransferase • ALT - Alanine Aminotransferase
↑ AST, ALT, and Bilirubin
↑ Serum free Cu (total usually low due to low ceruloplasmin)
Eyes: Kayser-Fleischer rings
CNS: Neurologic disease, Psychiatric disease MSK: Arthropathies
Kidney: Fanconi syndrome, Kidney stones
Chronic hepatitis, Cirrhosis with hepatic insufficiency, Portal hypertension, Hemolysis, Acute Liver Failure
Continued hepatocyte injuryà progressive liver damage
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Re-Published June 17, 2019 on www.thecalgaryguide.com")
chronic-myeloid-leukemia
: Pathogenesis and Clinical Presentation
Translocation of a Chr 9 segment onto Chr 22, creating a Philadelphia chromosome (Chr 22) containing the BCR-ABL1 fusion gene
Mutations from ionizing radiation
Other genetic abnormalities
Authors: Yan Yu Katie Lin Reviewers: Jennifer Au Crystal Liu Danielle Chang Lynn Savoie* *Indicates faculty member at time of initial publication
These genetic abnormalities accumulate in the earliest cell of the blood cell differentiation sequence: the pluripotent hematopoietic stem cell
Hematopoietic stem cell division in the bone marrow becomes unregulated
1. Chronic Stage (85% of clinical presentation): Hematopoietic stem cell division/differentiation in the bone marrow results in ↑ production of multiple blood cell lines (detectable on CBC, but patients are usually asymptomatic at this stage)
Acquired ↑ genetic abnormalities
2. Accelerated Stage
More and more immature precursor cells (”blasts”) divide and accumulate in bone marrow (where 10-19% of blood cells are “blasts”.) Blasts start to spill over into the peripheral blood
Acquired ↑ genetic abnormalities
3. Blast crisis (transformation into AML/ALL)
Neoplastic blast cells have filled up the bone marrow (where >20% of blood cells are blasts). More blasts spill out into the peripheral blood.
Multifactorial causes, most Weight loss, malaise, fever/
with unclear mechanisms
Neoplastic division of platelet precursor cells
Neoplastic division of WBC precursor cells, especially neutrophil precursors
Dividing “blasts” limit the space and resources available for RBC synthesis
chills, night sweats Thrombocytosis
Leukocytosis:
· Neutrophilia, basophilia, & eosinophilia
· “Left shift”: ↑ neutrophil & band production
· Disorderly WBC differential: i.e. “myelocyte bulge”
Trapping of WBC’s in the spleen enlarges the spleen
Splenomegaly:
· Left upper quadrant pain · Early satiety (large spleen compresses the stomach)
· Associated hepatomegaly (if spleen is overfilled & WBCs spill over into liver)
Anemia
↓ oxygenation of blood means blood is less red & body tries to compensate
Pallor Dyspnea Tachycardia
High turnover of these cancerous cells → excess cell lysis
Release of intracellular contents (uric acid, K+, LDH) into plasma
· Hyperkalemia · High (LDH)
Hyperuricemia
Gout
Acute Kidney Injury
Expanding marrow pushing on bone
Bone marrow expands into sternum
Bone pain
Sternal tenderness
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Re-Published June 15, 2019 on www.thecalgaryguide.com")
Secondary Polycythemia

Poor lung function
Renal artery stenosis
↓ blood flow to kidney
Kidney senses ↓ O2
High affinity Hb or CO poisoning
Hb does not easily unload O2
Tissues become hypoxic
Tumors (e.g. renal, hepatic, lung)
Secretes EPO in an unregulated way, as a “paraneoplastic syndrome”
High altitude
Obstructive sleep apnea
Episodic airway obstruction during sleep
Intermittent hypoxia
Cyanotic heart disease
Shunting of blood
Venous and arterial blood mixes
Poorly oxygenated blood
↓ O2 partial pressure
↑ EPO production independent of O2 (inappropriate response)
↑ EPO production due to hypoxia (appropriate response)
Abbreviations:
• Hb- Hemoglobin
• EPO- Erythropoietin • ILD- Interstitial Lung
Disease
• COPD- Chronic
Obstructive Pulmonary Disease
“Endogenous causes” of high EPO
Secondary Polycythemia
“Exogenous causes” of high EPO
Testosterone therapy Iatrogenic EPO (results in ↑ EPO synthesis administration
within the body)
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Re-Published May 5, 2019 on www.thecalgaryguide.com")
Acute Lymphoblastic Leukemia
![Acute Lymphoblastic Leukemia (ALL): Pathogenesis and Clinical Presentation
Authors: Yan Yu, Katie Lin Reviewers: Crystal Liu, Kara Hawker, Jennifer Au, Lynn Savoie* * MD at time of initial publication
Note: ALL is much rarer than AML and is usually seen in children
Accumulation of genetic abnormalities in immature lymphoid precursor cells (B/T cell precursors)
Neoplastic lymphoid precursor cells (“blasts”) divide and accumulate in bone marrow
Abundance of blasts displaces other blood precursors from marrow, inhibiting their development/differentiati on
After neoplastic blasts fill up bone marrow, they spill out into blood
High turnover of these cancerous cells
Multifactorial causes, most with unclear mechanisms
Expanding marrow pushing on bone
Pancytopenia on CBC
20% of marrow is blasts (on bone marrow aspirate and/or biopsy)
Neoplastic blasts continue to divide and accumulate in lymph
nodes and spleen (can occur, but not that common)
Blasts detected as white blood cells on CBC
High rate of cell lysis
Weight loss, malaise, fever/chills, night sweats
Bone pain (worse than that felt in AML, especially in children)
↓ in neutrophils
↓ in RBCs
↓ in platelets, reduced blood clotting ability
Lymphadenopathy Splenomegaly
May cause leukocytosis, despite pancytopenia
Release of intracellular contents (uric acid, K+, LDH) into plasma
Greater chances of infection
Anemia
Fatigue, shortness of breath, pallor
Easy bruising and petechiae on skin
Hyperuricemia Hyperkalemia High [LDH]
Tumor lysis syndrome
Acute kidney injury
Gout
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Re-Published May 5, 2019 on www.thecalgaryguide.com](http://calgaryguide.ucalgary.ca/wp-content/uploads/2015/05/Acute-Lymphoblastic-Leukemia.jpg "Acute Lymphoblastic Leukemia (ALL): Pathogenesis and Clinical Presentation
Authors: Yan Yu, Katie Lin Reviewers: Crystal Liu, Kara Hawker, Jennifer Au, Lynn Savoie* * MD at time of initial publication
Note: ALL is much rarer than AML and is usually seen in children
Accumulation of genetic abnormalities in immature lymphoid precursor cells (B/T cell precursors)
Neoplastic lymphoid precursor cells (“blasts”) divide and accumulate in bone marrow
Abundance of blasts displaces other blood precursors from marrow, inhibiting their development/differentiati on
After neoplastic blasts fill up bone marrow, they spill out into blood
High turnover of these cancerous cells
Multifactorial causes, most with unclear mechanisms
Expanding marrow pushing on bone
Pancytopenia on CBC
20% of marrow is blasts (on bone marrow aspirate and/or biopsy)
Neoplastic blasts continue to divide and accumulate in lymph
nodes and spleen (can occur, but not that common)
Blasts detected as white blood cells on CBC
High rate of cell lysis
Weight loss, malaise, fever/chills, night sweats
Bone pain (worse than that felt in AML, especially in children)
↓ in neutrophils
↓ in RBCs
↓ in platelets, reduced blood clotting ability
Lymphadenopathy Splenomegaly
May cause leukocytosis, despite pancytopenia
Release of intracellular contents (uric acid, K+, LDH) into plasma
Greater chances of infection
Anemia
Fatigue, shortness of breath, pallor
Easy bruising and petechiae on skin
Hyperuricemia Hyperkalemia High [LDH]
Tumor lysis syndrome
Acute kidney injury
Gout
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Re-Published May 5, 2019 on www.thecalgaryguide.com")
Ulcerative Colitis

Immune response against the GI tract. (Unclear mechanism, but thought to be mediated by cytokine release and neutrophil infiltration)
Inflammation of the GI tract epithelial lining
- Starting at the rectum and moves up the colon and is continuous (does not invade the small intestine)
- Inflammation affects the mucosal and submucosal only
Diarrhea, abdo pain and cramping causing avoidance of food
Weight loss
Apoptosis of GI tract mucosa
Transporter proteins responsible for Na+ reabsorption gradually disappear from the epithelium
Ulceration, into the anus, and more severe
Prolonged Bleeding - GI and anus
Anemia, often iron deficiency
Inflammation ↑ permeability of the blood vessels supplying the GI tract wall
Fluid leak out of capillaries into GI tract wall, causes edema and swelling
Swelling narrows the GI tract lumen, causing bowel obstruction
Inflammation, ulceration, or infection at the anus (all involve the RECTUM!)
Anal irritation stimulates autonomic and somatic nerves leading up to the brain, causing the pt to want to defecate
Tenesmus, urgency, frequency (feeling or urgency to defecate, but little stool is produced)
Joints Skin
Arthroplasty/ joint pain
Erythema nodosum, pyoderma gangrenosum
Mouth >5 canker sores
More sodium (and thus water) is retained in the GI tract lumen
Bloody Diarrhea, usually bloody due to anal bleeding and ulceration bleeding
Abdominal Cramping and pain (see Bowel Obstruction page for full mechanism)
Eyes (uvea, iris, sclera)
Liver Blood
Uveitis
Iritis, scleritis
Sclerosing Cholangitis
Autoimmune hemolytic anemia
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Re-Published May 5, 2019 on www.thecalgaryguide.com")
HBV Serology

Anti-HBcAg IgM acute/recent infection
Anti-HBcAg IgG chronic/remote infection
(Due to vaccination or past exposure)
Marker of active viral disease and patient infectiousness
Detected in chronically infected patients or patients who have cleared infection
Never infected, never immunized
-
-
-
-
-
-
Chronic infection/carrier
+
+/-
+
-
+/-
+/-
Acute infection
+
+
-
-
+
-
Natural immunity (past infection)
-
-
+
+
-
+/-
Immunized
-
-
-
+
-
-
Historical Notes:
• HBeAg was previously used as a marker of viral replication
• Presence of Anti-HBeAg antibodies indicates that the antigen has been
cleared, representing seroconversion and a halt to viral replication
• HBV DNA is the current clinical marker of viral replication, and can be
used to assess viral load in the body
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Re-Published May 5, 2019 on www.thecalgaryguide.com")
Chronic Lymphocytic Leukemia

Clonal division of neoplastic B lymphocytes within lymph nodes
With continued cell division, B- cells spill over into the peripheral blood
Neoplastic B cells fail to die and continue to divide within lymph nodes over time
Neoplastic B cell precursors infiltrate the spleen and the bone marrow
A small % of CLL patients undergo Richter’s transformation, in which their disease evolves into Diffuse Large B-cell Lymphoma
Notes:
• CLL is the most common adult leukemia in developed nations.
• Prevalence of CLL ↑ with age, occurring most often in older people
Slow process of neoplastic cell division
Patients are most commonly asymptomatic at initial presentation
Multifactorial causes, most Weight loss, malaise, fever/chills, night
with unclear mechanisms
B-cell are detected as lymphocytes on a CBC
Lack of functional B cells ↓ body’s ability to produce antibodies for immune response
Neoplastic cells accumulate in lymph nodes
sweats
Lymphocytosis (B cell count >5x109/L for at least 3 months)
Hypogammaglobulinemia
Lymphadenopathy
↑ risk for infection, especially by encapsulated bacteria normally killed by antibodies
Hypercellular bone marrow (on bone narrow biopsy)
Splenomegaly
↑ sequestration of platelets and RBCs
↓ in RBCs ↓ in
platelets
Anemia
Thrombocytopenia (reduced blood clotting ability)
Fatigue, shortness of breath
Easy bleeding, easy bruising, petechiae
Abundance of B-cell precursors in marrow inhibits the development/differentiation of other cell types
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Re-Published June 30, 2019 on www.thecalgaryguide.com")
perforated-viscous
:
Author:
Yan Yu
Reviewers:
Michael Blomfield, Tony Gu, Dean Percy, Danny Guo Maitreyi Ramran* * MD at initial time of publication
Chest X-Ray (CXR)
Pathogenesis and Clinical Findings
Diverticulitis
Crohn’s disease Peptic ulcer (H. pylori
infection, NSAID use, ICU stress, etc)
Appendicitis
Malignant neoplasm
Irritates visceral peritoneum, stimulates autonomic nerves
Severe inflammation causes destruction of GI tract mucosa
Over time, Perforation of the GI tract wall
Bowel contents (air, fluids) released into peritoneal cavity
Massive peritoneal inflammation
Diagnostic investigations if a GI perforation is suspected
Dull diffuse abdominal pain
Severe, Sharp abdominal pain with peritoneal signs
Abdominal X-ray
Irritation of parietal peritoneum, stimulates somatic nerves
• Abdominal X-ray
• Intra-peritoneal air will coat the GI tract surfaces, giving them a faint white outline
under X-ray
• Chest X-ray of upright patient (Diagnostic)
• Intra-peritoneal air will rise above the peritoneal fluid when pt is upright, accumulating under the right hemi-diaphragm.
• Note: air under left hemi-diaphragm = normal gastric bubble
• CT? Most patients with suspected GI perforation will get a CT scan, but this is not the diagnostic gold standard (and access to CT can be limited, especially in rural settings)
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Re-Published June 30, 2019 on www.thecalgaryguide.com")
acanthosis-nigricans-pathogenesis-and-clinical-findings

HELLP syndrome pathogenesis and clinical findings

Abnormal maternal immune tolerance of placentation in early pregnancy
Polymorphisms of FasL and receptor gene
↑levels of placental FasL in maternal circulation
Lesions in membrane separating maternal and fetal circulation
Release of inflammatory products from placenta into maternal circulation
Systemic maternal inflammatory response (activation of coagulation and complement pathways)
Microvascular endothelial activation, dysfunction, and damage
Widespread platelet aggregation and agglutination ↓ amount of platelets measurable on complete blood count
Low platelets
HELLP Syndrome
RBCs are sheared as they flow through damaged vessels
Microangiopathic hemolytic anemia (MAHA)
Lab findings indicative of hemolysis: normocytic anemia, high LDH & bilirubin, low haptoglobin
Microthrombin in hepatic circulation
Damage to hepatocytes
Elevated liver enzymes
Hemolysis, Elevated Liver Enzymes, Low Platelets
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published July 14, 2019 on www.thecalgaryguide.com")
Lyme Disease Pathogenesis and Clinical Findings

Tick bite from Borrelia burgdorferi infected Ixodes ricinus (tick from European countries)
Tick bite from Borrelia burgdorferi infected Ixodes scapularis (Deer tick/black-legged tick in SE Canada and NE United States)
Binding of OspC (a surface protein expressed by B. Burgdorferi) to human plasminogen allowing the spirochete to spread from bite site to other host organs and tissues
B. burgdorferi spreads through skin and other tissues via bloodstream in human host.
If tick bite lasts 36-72 hours or more, this is sufficient time for ticks to transmit the infection. (<36 hours of tick attachment results in a lower rate of infection: 1.2% -1.4%)
Lyme Disease
A vector-borne, infectious multi-system disease with highest risk in late spring and summer by the spirochete Borrelia burgdorferi
Early Disease Stage (<30 days)
Macrophages and T-cells produce ↑ inflammatory (TNF- α, IFN-γ) and ↑ anti-inflammatory cytokines, causing eosinophils to concentrate adjacent to the tick bite site
Early Disseminated Disease Stage (<3 months)
B. burgdorferi attach to host cell integrins
Pro-inflammatory response with production of matrix glycosaminoglycans and extracellular matrix proteins which have an affinity to attack collagen fibrils on the heart, nerves, and joints
1. Multiple erythema migrans 2. Meningitis
3. Meningoradiculoneuritis
4. Cranial nerve palsies
5. Carditis
6. Borrelial lymphocytoma
Late Disease Stage (>3 months)
Ongoing and repeated innate and adaptive host immune response to B. burgdorferi
Chronic inflammatory state results in synovial hypertrophy, vascular
proliferation, and ↑ mononuclear cell infiltrate in large joints
Large joint arthritis (most commonly affecting the knees)
Erythema migrans (a slowly expanding red skin patch with partial central clearing resulting in a “target clearing lesion” appearance) at site of tick bite
Systemic inflammatory response
after dissemination of the spirochete to body tissues and organs
Flu-like symptoms (fever, chills, muscle aches, headache, fatigue, joint aches, swollen lymph nodes)
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published July 24, 2019 on www.thecalgaryguide.com
References
• David A. Wetter and Colin A. Ruff. CMAJ August 09, 2011 183 (11) 1281; DOI: https://doi.org/10.1503/cmaj.101533
• https://www.canada.ca/en/public-health/services/diseases/lyme-disease/causes-lyme-disease.html
• Borrelia burgdorferi Infection-Associated Surface Proteins ErpP, ErpA, and ErpC Bind Human Plasminogen. Catherine A. Brissette, Katrin Haupt, Diana Barthel, Anne E. Cooley, Amy Bowman, Christina Skerka, Reinhard Wallich, Peter F. Zipfel, Peter Kraiczy, Brian Stevenson. Infection and Immunity Dec 2008, 77 (1) 300-306; DOI: 10.1128/IAI.01133-08
• https://www.uptodate.com/contents/what-to-do-after-a-tick-bite-to-prevent-lyme-disease-beyond- the-basics
• Murray, T. S., & Shapiro, E. D. (2010). Lyme disease. Clinics in laboratory medicine, 30(1), 311–328. doi:10.1016/j.cll.2010.01.003
• Weedon, David. Weedon's Skin Pathology E-Book: Expert Consult-Online and Print. Elsevier Health Sciences, 2009.")
intraventricular-hemorrhage-in-preterm-infants-clinical-findings-and-complications
 in preterm infants:
Clinical findings and complications
Authors: Alexa Scarcello Reviewers: Nicola Adderley, Emily Ryznar, Yan Yu*, Jennifer Unrau* * MD at time of publication
Volpe Grading Grade I: germinal matrix
hemorrhage with no or minimal IVH (<10% of ventricular area)
Grade II: IVH (10-50% of ventricle) Grade III: IVH (>50% of ventricle;
usually distends lateral ventricle)
Grade IV/Intra-parenchymal echodensity (IPE): periventricular hemorrhagic infarction
Inflammation/dysfunction of arachnoid villi
↓ absorption of CSF 2° to obstruction of arachnoid villi
Communicating hydrocephalus (IVH grades II-IV)
Venous congestion
Venous infarction
Periventricular hemorrhagic necrosis
Destruction of periventricular motor tracts
Cerebral palsy
Rapid significant blood loss
↓ intravascular blood volume
Hypotension
↓ bloodflow to the brain to support brain function
Intraventricular Hemorrhage (IVH)
hemorrhage in periventricular subependymal germinal matrix
Ultrasound: blood in germinal matrix, ventricles, or cerebral parenchyma
Sudden ↓ hematocrit
Blood irritates contiguous structures
Variable neurologic findings; including altered level of consciousness, hypotonia, apnea, etc
Neuro- developmental abnormalities (varying severity)
See slide - Hydrocephalus: Clinical Findings in Pediatrics
This mechanism leads to three different possible clinical manifestations:
1. Silent Presentation (most common)
2. Stuttering/Saltatory Course: non-specific findings - hypotonia, apnea, altered level of consciousness, bradycardia, and ↓ Spontaneous movements
3. Catastrophic Deterioration (least common) Stupor or coma, decerebrate posturing, seizures, bradycardia, metabolic acidosis, bulging fontanelles, abnormal pupillary reflexes, inappropriate ADH secretion
Notes
• Incidence & severity are inversely proportional to gestational age
• 50% occur within 1st day of life, 90% by 3rd day
• As explained in the flow chart, the postnatal clinical presentations of
IVH fall into three categories (1-3)
• Symptoms of catastrophic bleeds are uncommon and usually caused
by rapid significant blood loss with subsequent neurologic findings 2° to meningeal irritation, inflammation, and potential mass effect/acute hydrocephalus; severe bleeds may also occur in the absence of clinical findings attributable to IVH
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published July 27, 2019 on www.thecalgaryguide.com")
Vitamin K Deficiency

Pancreatic insufficiency
Cholestasis
Malabsorption
Exposure to vitamin K antagonists (e.g., warfarin)
Inhibition of vitamin K epoxide reductase
↑ bleeding tendency
Gastrointestinal tract bleeds Intracranial bleeds Hemarthoses (bleeding into joints)
Easy bruising
Petechiae, purpura Heavy menstrual bleeds
Prolonged PT/INR
Note: PT/INR is more sensitive and validated for warfarin monitoring
↓ Activity of intrinsic clotting pathway
Prolonged PTT
↓ Activity of extrinsic clotting pathway
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Re-Published July 27, 2019 on www.thecalgaryguide.com")
Normocytic Anemia
![Normocytic Anemia: Causes, Signs, and Symptoms
Authors: Katie Lin Yan Yu Reviewers: Andrew Brack Jessica Tjong Man-Chiu Poon* Lynn Savoie* * MD at time of publication
Aplastic anemia: hypo-proliferation of bone marrow RBC precursors
Anemia of Chronic Disease
Splenomegaly
↑ RBC sequestration within enlarged spleen
Acute bleeding
Hemolysis (infection, autoimmune, RBC structural defects)
↓ RBC production
↑ RBC destruction/elimination
Normocytic Anemia:
[Hgb] <120g/L in females, <140g/L in males, with the RBC mean corpuscular volume (MCV) still within the normal range: 80-100 fL
RBCs that ultimately end up in the blood are still qualitatively normal/functional; there is a quantitative shortage of these RBCs in the blood relative to body needs
Spurious/False normocytic anemia:
Any fluid overload state (pregnancy, heart failure, kidney disease, etc.) can ↑ plasma volume which can dilute RBCs and cause apparent anemia, but the mean volume of each RBC is still normal
Normocytic Anemia
Heart needs to work faster to pump
sufficient oxygenated blood to tissues
↑ Heart rate
Reduced oxygen- carrying ability of blood
Patient feels oxygen- deprived, needs to inhale more oxygen as compensation
Dyspnea (shortness of breath) ↑ Respiratory rate (RR)
Not enough oxygen being delivered to body tissues, including brain
Fatigue
Reduced absolute number of RBCs means less RBCs to color the blood red
Pallor (especially conjunctival and palmar)
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Re-Published July 27, 2019 on www.thecalgaryguide.com](http://calgaryguide.ucalgary.ca/wp-content/uploads/2015/05/Normocytic-Anemia-1.jpg "Normocytic Anemia: Causes, Signs, and Symptoms
Authors: Katie Lin Yan Yu Reviewers: Andrew Brack Jessica Tjong Man-Chiu Poon* Lynn Savoie* * MD at time of publication
Aplastic anemia: hypo-proliferation of bone marrow RBC precursors
Anemia of Chronic Disease
Splenomegaly
↑ RBC sequestration within enlarged spleen
Acute bleeding
Hemolysis (infection, autoimmune, RBC structural defects)
↓ RBC production
↑ RBC destruction/elimination
Normocytic Anemia:
[Hgb] <120g/L in females, <140g/L in males, with the RBC mean corpuscular volume (MCV) still within the normal range: 80-100 fL
RBCs that ultimately end up in the blood are still qualitatively normal/functional; there is a quantitative shortage of these RBCs in the blood relative to body needs
Spurious/False normocytic anemia:
Any fluid overload state (pregnancy, heart failure, kidney disease, etc.) can ↑ plasma volume which can dilute RBCs and cause apparent anemia, but the mean volume of each RBC is still normal
Normocytic Anemia
Heart needs to work faster to pump
sufficient oxygenated blood to tissues
↑ Heart rate
Reduced oxygen- carrying ability of blood
Patient feels oxygen- deprived, needs to inhale more oxygen as compensation
Dyspnea (shortness of breath) ↑ Respiratory rate (RR)
Not enough oxygen being delivered to body tissues, including brain
Fatigue
Reduced absolute number of RBCs means less RBCs to color the blood red
Pallor (especially conjunctival and palmar)
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Re-Published July 27, 2019 on www.thecalgaryguide.com")
Appendicitis

The appendix is anatomically located in the RLQ; appendicitis may be confused with disorders of surrounding structures: Gynecological Diseases
• RuleoutpregnancywithHCG pregnancy test
• Rupturedovariancyst
• Ectopicpregnancy
• Mittelschmerz(mid-cycle
pain)
Gastro-intestinal Diseases
• Meckel’sdiverticulum (presents identically to appendicitis; surgically located 2 feet from ileocecal valve; mostly seen in children)
• Diverticulitis(presentsasleft sided appendicitis)
Non-GI Abdominal Issues
• Mesentericadenitisinkids <15: swollen mesenteric lymph nodes
• Renalcolic
Obstruction of appendiceal lumen (by fecalith, fibrosis, neoplasia, foreign bodies or lymph nodes in kids)
Appendix distension and spasms
↑ lumen pressure, ↓ blood flow to appendix
Ischemia, tissue necrosis, loss of appendix structural integrity
Bacterial invasion of the appendix wall, causing transmural inflammationandnecrosis
Stretching of visceral peritoneum, stimulation of autonomic nerves T9-T10
Progression of inflammation over several days (variable length of time)
Irritation of parietal peritoneum, stimulation of somaticnerves
If appendix not surgically removed
Perforation of colon wall, causing peritonitis, abscesses or death
Note: Symptoms hugely variable. Only 30% present with classic history. Diagnosis is mostly clinical. Further investigations:
CBC: Leukocytosis (due to inflammatory response) CT: Gold standard test. Thickened visceral membrane with enhancing (white) rim due to ↑ blood flow
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Re-Published July 27, 2019 on www.thecalgaryguide.com")
Acute GI Related Abdominal Pain

Bowel stretching, pulling, contracting
Abdominal pain type:
Diffuse, non-localized Dull, crampy, periodic Not associated with movement
Patient may writhe around, trying to get rid of the pain
Mesentery Intestinal lumen
Parietal peritoneum
(innervated by somatic nerves)
Cross-section of the GI tract
Cuts, structural damage, and inflammation in the bowel
Important Notes
• Acute abdominal pain can also result from non-
gastrointestinal causes, such as kidney stones, female reproductive tract issues, and urinary tract issues. For simplicity’s sake, only the GI-related acute abdominal pain disorders are listed here.
• The DDx of visceral abdominal pain is broad. Please consult relevant sections of the Calgary Black Book for the DDx.
• Keep in mind that visceral abdominal pain can also be caused by the “acute abdomen” diseases (if the diseases are presenting in their initial phases).
• • •
• • •
Abdominal pain type:
Sharp, well-localized
Excruciatingly painful, persistent Associated with movement of bowels
Patient often lies still to avoid abdominal vibration
Peritoneal signs
Abdominal guarding, pain with abdominal vibration (coughing, shaking, percussion, palpation)
Transition from diffuse to localized pain can indicate disease progression (e.g. from visceral to parietal peritoneal inflammation)
Note: bowel obstruction may or may not present as acute abdominal pain
Bowel Infarction
Appendicitis Diverticulitis
Acute Cholecystitis
Acute Pancreatitis
Perforated Ulcer
DDx of an “acute abdomen”:
A sudden, non-traumatic disorder of the abdomen that needs urgent diagnosis and treatment. Each topic will be further explored in their respective slides.
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Re-Published July 27, 2019 on www.thecalgaryguide.com")
Operative vaginal delivery Complications of Vacuum
 Vacuum
Authors: Alexa Scarcello Reviewers: Claire Lothian, Crystal Liu, Yan Yu*, Ron Cusano* * MD at time of publication
Maternal trauma to surrounding structures to fit operative device
Traction and torsion on flexion point of fetal head, between anterior and posterior fontanelles
Pressure of device directly on fetal scalp
Pop-offs of vacuum cup
Rapid decompression and compression forces
Separation of underlying structures
Bleeding between skull and periosteom of neonate
Cephalohematoma
↑ breakdown of RBCs
↑ release of hemoglobin
Hemoglobin breakdown à↑ release of bilirubin into the bloodstream
Potential interference with spontaneous rotation of trunk
↑ risk of shoulder dystocia
Delivery during shoulder dystocia may traumatize infant’s shoulder
Brachial plexus injury
Head injury leads to potential ocular trauma
Retinal hemorrhage
Hyperbilirubinemia
Fetal scalp lacerations
Tentorial tears
Intracranial bleeding
Perineal lacerations
1° Lacerations: skin, subcutaneous tissue, vaginal epithelium 2° Lacerations: into superficial perineal muscles
3° Lacerations: extending to rectal sphincter
4° Lacerations: extending to rectal mucosa
Urethral injury
Rupture of emissary veins (connections between dural sinus and scalp veins)
Blood accumulation between epicranial aponeurosis and periosteom
Subgaleal hemorrhage
Large space in between these tissue layers ↑ capacity for blood accumulation here
Hypovolemia Death
Factors independent of operative device
Notes:
• •
Vacuums are now used more often (compared to forceps) due to ↓ maternal trauma
Caution in use if fetus <34wks
Jaundice
Less operator experience Higher fetal station
Poor head Rotation Longer active 2nd stage
↑ complication rate
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published Aug 4, 2019 on www.thecalgaryguide.com")
Ataxia Telangiectasia Pathogenesis and Clinical Findings
; involuntary muscle contractions, hypotonia, IQ decline, and abnormal eye movement
Loss of ATM leads to mitotic defects and arrest in gamete genetic recombination process
Gonadal dysgenesis and delayed puberty
DNA damage to tumor suppressors such as p53 and BRCA1
Impaired signaling of downstream cell cycle regulators
Impaired genome stability and increased disposition to cancer
↑ Acute Lymphocytic Leukemia of T cell origin (in children) and Chronic Lymphoblastic Leukemia (in adults)
Impaired recombination of DNA in immune cells
Thymic hypoplasia; humoral & cellular immunodeficiency
↓ or absent functional immunoglobulins IgA, IgE, and IgG2 that function to prevent respiratory infections
Respiratory infections with bronchiectasis and pneumonia
Cells less able to undergo apoptosis in response to ionizing radiation
Accumulation of DNA defects in the cells of sun exposed areas such as skin, hair, and conjunctiva
Mucocutaneous telangiectasia on the bulbar conjunctiva and ears between 2-6 years of age
May progress to involve periorbital skin, trunk, extremities, body folds, and other mucosal surfaces
Sterility
DNA damage and genomic instability
Premature melanocyte stem cell differentiation
Premature graying of skin and hair
Abbreviations:
• ATM – Ataxia-telangiectasia
mutated protein
• p53 – Tumor protein 53
• BRCA 1 – Breast cancer
susceptibility protein.
• IgA, IgE, IgG2 – Immunoglobulins
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published August 4, 2019 on www.thecalgaryguide.com")
Diverticulosis and Angiodysplasia

Angiodysplasia
Autoimmune Diseases
Unclear Mechanisms
These patients tend to have “arterial-venous malformations” that rise up to the mucosa of the lower GI tract
Irritation of these malformations leads to bleeding into the GI tract lumen
Lower GI bleed
(occult, slow, asymptomatic, large- volume blood loss, usually associated with iron deficiency anemia)
Renal Failure
Older Age
Gradual expansion over
time thins the diverticular wall
Capillaries within diverticuli burst and leak blood into the colon lumen
Lower GI bleed
(usually stops by itself)
Colon wall forms little outpouchings (diverticuli)
Stretching of colonic serosa stimulates
somatic sensory nerves innervating the colon
Bloating, cramping
(But most often PAINLESS)
Trapping of feces in the colonic diverticuli
Bacteria have more time to metabolize the undigested materials, producing gas
Flautulence
Irregular defecation
Both conditions share similar features:
• Both are common, and their prevalence increases with age
• Are relatively benign and most are easy to treat (80% stop w/o intervention)
• Both present without pain in a previously-well patient
• Together, account for 50-80% of lower GI bleeds (diverticulosis > angiodysplasia)
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Re-Published August 4, 2019 on www.thecalgaryguide.com")
Erythema Nodosum pathogenesis and clinical findings
 ~28-48% of cases
Medications (Ex. Birth Control Pills, Sulfa drugs) ~3-10% of cases
Malignancy (ex. Lymphoma)
Autoimmune conditions (ex. Sarcoidosis and
Inflammatory Bowel Disease) ~11-25% of cases
Pregnancy ~1-3% of cases
Antigenic Stimuli / Bacteria / Viruses / Chemical Agents all could trigger the following process: Phase 1. Neutrophils Infiltrate the fibrous septa between fat lobules in the subcutaneous fat
Phase 2. Neutrophils release reactive oxygen species, leading to oxidative tissue damage and inflammation
Phase 3. Opening of inter-endothelial junction and the migration of more inflammatory cells into the septal venules, including macrophages, histocytes, and eosinophils
Phase 4. Macrophages secrete inflammatory cytokines, which stimulates the proliferation of more helper T cells (Th1)
Phase 5. Th1 cells secrete more cytokines, leading to the further release of Th1 cytokines and mediating the immune complexes deposition in the septal venules of the subcutaneous fat (panniculitis). The Th1 immune reaction is called Type IV Delayed Hypersensitivity Reaction
Phase 6. Activated macrophages produce hydrolytic enzymes and transform into multi- nucleated giant cells, called Miescher’s Radial Granulomas. These consist of small, well defined aggregations of small histocytes arranged radially around a small cleft of variable shapes in the septal venules of the subcutaneous fat
Phase 1-4. Lesions are red tender nodules, poorly defined, vary in size from 2-6 cm, and usually on shins ( 1st week)
Fat Lobules T lymphocytes
Macrophages
Note: we’ve done extensive research and can’t figure out why erythema nodosum happens mostly on the shins. If you have an answer, please email us!
Phase 5. Lesions become tense, hard, and painful; and they change in color into bluish or livid. (2nd week)
Phase 6. Lesions become fluctuant as in abscess, but do not ulcerate. Lesions fade to a yellowish color
Epidermal layer Dermal-Epidermal Junction
Dermal layer Subcutaneous Fat Layer
Phase 6. Miescher’s Radial Granulomas
Fat Lobules
T lymphocytes
Macrophages
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published August 25, 2019 on www.thecalgaryguide.com")
Colorectal Carcinoma pathogenesis and clinical findings

↓ appetite
Local bleeding
Abdominal Abscess
Weight loss
Acute blood loss
Iron deficiency anemia
Classification of tumours/abnormal growths:
1. Adenoma–abenigntumorfromglandular structures
2. Carcinoma–cancerarisingfromtheepithelial tissue of the skin or the lining of internal organs
3. Sarcoma–cancerarisingfromconnectivetissue
Mechanical bowel Obstruction
Ribbon (thin) stool
In rectum
Mass effect
Tumor ↓ bowel lumencaliber
Backed up contents mayberegurgitated
Cancerinvadesrectal sphincters, muscles, vessels&nerves
Compressing ureters, urine backs up into kidney
Compressing stomach
Abdominal distension/pain Invades blood vessels
Inflammatory
Bowel Disease Smoking
Abdominal radiation
Tubular adenomas
(pre- cancerous polyps)
Obesity
Local growth of tumor
Cell line mutations
Idiopathic
Uncontrolled cell division in the colon and rectum
Hereditary syndromes
Colorectal Carcinoma
(Develops over time)
Outside bowel serosa
Bowel perforation
Bowel to bowel/local organ fistulisation
Host immune cells release cytokines to combat cancer
Bowel contents leak
Metabolic abnormalities, ↑energy use
Tumor Spread Mechanisms:
1. Hematogenous 2. Lymphatic
3. Contiguous
4. Transperitoneal
Tumor cells spread distally
Friable vessels supply tumor
Metastatic Disease
Vessels Rupture
Occult bleeding &
melena (black stools) depletes stores of iron
Authors:
Karly Nikkel
Reviewers:
Michael Blomfield
Tony Gu
Yan Yu*
Edwin Cheng*
* MD at time of publication
Tumors develop in liver, lungs, brain, peritoneum and lymph nodes
Hematochezia (passage of fresh blood in stool)
Slow, chronic blood loss
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published August 25, 2019 on www.thecalgaryguide.com")
iga-vasculitis-henoch-scholein-purpura-pathogenesis-and-clinical-findings
 : Pathogenesis and clinical findings
Authors: Mia Koegler Nela Cosic Reviewers: Crystal Liu Yan Yu* Martin Atkinson* * MD at time of publication
Infectious Agents
50% have preceding upper respiratory tract infections, i.e., influenza virus or Group A Strep
Drugs
I.e., antibiotics (penicillin, erythromycin), NSAIDs and biologics (tumor necrosis factor α inhibitors)
Immunogenetic and cellular predisposition
Various genetic polymorphisms alter cell- mediated immune response, IgA levels elevated in 50% of people
↑ Circulating galactose-deficient IgA1 (GD-IgA1). Deficiency in galactosylation of IgAà↓ IgA serum clearanceàadhesion of IgA complexes, which then deposit into the endothelial lining of blood vesselsàattraction of various inflammatory cells to the area:
Formation of Secretion of Interleukin 8 (IL8) - cytokine that induces Neutrophils infiltrate Activation of complement immune complexes neutrophilic chemotaxis and macrophage phagocytosis the tissue site factors (C3, C4)
Leukocytoclastic vasculitis (histopathologic term for small vessels inflamed by neutrophilic autoimmune response)
Inflamed cutaneous vessels become enlarged in clusters
Symmetrical palpable purpura (red/purple, non- blanchable papules) distributed on lower limbs and buttocks areas
Cutaneous small vessel vasculitis (100%)
Inflamed gastric vessels - hemorrhage and edema within bowel wall
Gastrointestinal (85%)
Colicky abdominal pain (commonly in the periumbilical region), nausea, vomiting
Gastrointestinal
GI bleeding (hematemesis, melena), Intussusception
Glomerular mesangial proliferation and inflammation
↑ mast cell deposition in joints
Joints (60-85%)
Arthralgia's (common), arthritis (especially knees and ankles)
Arthralgia often transient. No permanent sequelae
Sympathetic nervous system activation
Glomerulosclerosis, tubulointerstitial and podocyte damage
Renal tissue ischemia
↑ Na sensitivity in renal tubules (↑ Na and water retention)
Renal (10-50%)
Increased renin secretion
HTN, nephrotic/nephritic syndrome, renal insufficiency
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published September 1, 2019 on www.thecalgaryguide.com")
Negative-Pressure-Pulmonary-Edema

Thrombotic Thrombocytopenic Purpura-Hemolytic Uremic Syndrome (TTP-HUS): Pathogenesis and clinical findings
: Pathogenesis and clinical findings
Thrombotic Thrombocytopenic Purpura
Hemolytic Uremic Syndrome
Shiga toxin from E. coli 0157:H7 infection
Acquired
ADAMTS13 auto-antibodies
Pregnancy/Post-partum Unclear mechanism
Failure to cleave large vWF multimers
Accumulation of large vWF multimers (Usually broken down by ADAMTS13)
Platelet aggregation and formation of platelet thrombi
Widespread microthrombi occluding blood vessels causing tissue damage/necrosis
Congenital ADAMTS13 mutation
Triggering event
ADAMTS13 deficiency
Drug dependent antibodies target platelets
Direct tissue toxicity (unclear mechanism)
ADAMTS 13 Inhibition Widespread hemolysis
Shut down of complement regulatory genes
Uninhibited membrane attack complex formation
Drugs (Quinine, Chemotherapy)
Abbreviations:
• vWF – von Willebrand Factor
• ADAMTS13 (vWF cleaving protein) – A
Disintegrin And Metalloprotease with a ThromboSpondin type 1 motif, member 13.
Formation of fibrin strands
Microangiopathic Hemolytic Anemia (schistocytosis)
Cellular damage due to fibrin deposit (intravascular, non- autoimmune hemolysis)
↑ serum indirect bilirubin
↑ serum lactate dehydrogenase ↓ serum haptoglobin
Authors:
Sean Spence Nicole Burma Reviewers: Tristan Jones Yan Yu Man-Chiu Poon* Lynn Savoie*
* MD at time of publication
Uncontrolled platelet activation and consumption
Thrombocytopenia
Reduced clotting ability
Purpura
Central Nervous System (CNS)
Confusion, severe headache, focal neurological findings
Heart
Patchy necrosis of myocardium
Arrhythmia, Sudden Cardiac Death
Kidney
Glomerular damage à↓ GFR + protein leakage into urine
Proteinuria, Acute Kidney Injury (AKI)
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published September 1, 2019 on www.thecalgaryguide.com")
virchows-triad-and-deep-vein-thrombosis-dvt
:
Authors: Dean Percy Yan Yu Reviewers: Tristan Jones Ryan Brenneis Man-Chiu Poon* Maitreyi Raman* * MD at time of publication
Pregnancy, Oral Contraceptives (OCP)
Pathogenesis and Complications
Platelet Activation
Increased clot formation
Hypercoagulable State
↑ ability for the blood to coagulate upon stimulation
Inherited Disorders
Congenital defect in coagulation (ie. Factor V Leiden, Factor II
mutation, Protein S/C deficiency) ↑ blood clotting ability
Estrogen promotes
hypercoagulability, especially in presence of other risk factors
Notes:
• Venous thrombus causes pulmonary embolism, arterial thrombus causes stroke
• Previous DVT is risk factor for current DVT
Trauma/Surgery
Malignancy
Abnormal release of coagulation-promoting cytokines
Systemic injuryà activation of coagulation cascade
Hypertension
Bacteria Artificial Valve
Physically damages blood vessel walls
Adhere/invade vessel wall
Abnormal surface
Vessel Injury
Exposes tissue factor on damaged cells and subendothelium for vWF binding
Virchow’s Triad
Venous Stasis
Low blood flow rate over site of vessel injury, concentrating blood clotting factors at that site
Fat contains more aromatase, converts more androgens to estrogen
Sedentary lifestyle, poor venous return
Obesity
Clot formation typically occurs in leg veins
Deep, large veins allow for blood pooling (stasis, hypercoagulability) Venous return from legs often against gravity (stasis)
Valves in leg veins prone to backflow (stasis)
↓ muscle motion = ↓ venous blood flow
Fracture, immobilization, bedrest, long vehicle/airplane ride
Destruction of vein valve by clot
Venous Insufficiency
Clot prevents blood from returning to heart. Blood accumulating in the leg results in unilateral leg edema and venous inflammation (redness, warmth, tenderness)
1. 2. 3.
Clot embolizes to the lungs
Thromboembolus
-*Pulmonary embolism (acute life threatening complication)
-Chronic thromboembolic pulmonary hypertension
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Re-Published September 1, 2019 on thecalgaryguide.com")
Acute-Pancreatitis
![Acute Pancreatitis: Pathogenesis and Clinical Findings
Authors: Yan Yu Reviewers: Laura Craig Noriyah AlAwadhi Ryan Brenneis Maitreyi Raman* * MD at time of publication
Associated signs due to intra- abdominal hemorrhage from an unknown mechanism (classically associated with pancreatitis, but happens in <1% of cases):
Note:
It is not enough to just diagnose “acute pancreatitis”. Full management requires determining underlying etiology with further work-up.
Alcohol
↑ Toxic metabolites within pancreas and Spincter of Oddi Spasms
Gallstones
Migration to common bile duct blocks Sphincter of Oddi
Hypertriglyceridemia
Unknown
mechanism (rare)
Idiopathic
Further investigations:
CBC: Cell counts elevated, due to sever hypovolemia
Serum [Lipase]: Gold Standard Diagnostic Test; rupture of pancreatic cells releases lipase into circulation
Pancreatic secretions back up, ↑ pressure within pancreas
Hypercalcemia (Rare; Ca2+ depositions in bile ducts block outflow of pancreatic secretions)
Since pancreas is retroperitoneal, somatic
nerves in the parietal peritoneum are directly stimulated
Inflammation triggers cytokine release
Inflamed pancreas irritates adjacent intestines, causing ileus
Inflamed, more permeable blood vessels leak fluid into pancreas
• •
Cullen’s sign (bruising in peri-umbilical region) Grey-Turner’s sign (bruises along both flanks)
Sudden, severe epigastric pain (with peritoneal signs), radiates to the center of the back
Fever, nausea/vomiting
(general signs of inflammation)
Diminished bowel sounds Profound dehydration
(flat JVP, hypotension, tachycardia, oliguria) – may happen, not always
1. Pressure compresses pancreatic blood vessels, causing tissue ischemia.
2. Activation of inactive proteases (zymogens) digesting pancreatic tissue
Necrosis (death) of pancreatic cells
Inflammation self- perpetuates
Massive systemic inflammatory response
2 main complications, usually detected on CT;
may happen, but not always
1. Pancreatic pseudocyst (enlargement of the
pancreas due to fluid accumulation)
2. Pancreatic necrosis/abscesses (death of a part of the pancreas)
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Re-published September 1, 2019 on thecalgaryguide.com](http://calgaryguide.ucalgary.ca/wp-content/uploads/2019/09/Acute-Pancreatitis.jpg "Acute Pancreatitis: Pathogenesis and Clinical Findings
Authors: Yan Yu Reviewers: Laura Craig Noriyah AlAwadhi Ryan Brenneis Maitreyi Raman* * MD at time of publication
Associated signs due to intra- abdominal hemorrhage from an unknown mechanism (classically associated with pancreatitis, but happens in <1% of cases):
Note:
It is not enough to just diagnose “acute pancreatitis”. Full management requires determining underlying etiology with further work-up.
Alcohol
↑ Toxic metabolites within pancreas and Spincter of Oddi Spasms
Gallstones
Migration to common bile duct blocks Sphincter of Oddi
Hypertriglyceridemia
Unknown
mechanism (rare)
Idiopathic
Further investigations:
CBC: Cell counts elevated, due to sever hypovolemia
Serum [Lipase]: Gold Standard Diagnostic Test; rupture of pancreatic cells releases lipase into circulation
Pancreatic secretions back up, ↑ pressure within pancreas
Hypercalcemia (Rare; Ca2+ depositions in bile ducts block outflow of pancreatic secretions)
Since pancreas is retroperitoneal, somatic
nerves in the parietal peritoneum are directly stimulated
Inflammation triggers cytokine release
Inflamed pancreas irritates adjacent intestines, causing ileus
Inflamed, more permeable blood vessels leak fluid into pancreas
• •
Cullen’s sign (bruising in peri-umbilical region) Grey-Turner’s sign (bruises along both flanks)
Sudden, severe epigastric pain (with peritoneal signs), radiates to the center of the back
Fever, nausea/vomiting
(general signs of inflammation)
Diminished bowel sounds Profound dehydration
(flat JVP, hypotension, tachycardia, oliguria) – may happen, not always
1. Pressure compresses pancreatic blood vessels, causing tissue ischemia.
2. Activation of inactive proteases (zymogens) digesting pancreatic tissue
Necrosis (death) of pancreatic cells
Inflammation self- perpetuates
Massive systemic inflammatory response
2 main complications, usually detected on CT;
may happen, but not always
1. Pancreatic pseudocyst (enlargement of the
pancreas due to fluid accumulation)
2. Pancreatic necrosis/abscesses (death of a part of the pancreas)
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Re-published September 1, 2019 on thecalgaryguide.com")
Peptic Ulcer Disease

Toxin release
Inflammatory response
Inhibition of H+ detection in gastric antrum
H+ over-secretion
NSAID use
(including ASA
+/- other anti- platelet agents)
Ischemia
Zollinger–Ellison Syndrome (↑ acid secretion due to gastrin secreting tumor)
Stress
(in ICU setting)
Crohn’s disease
Cancer
(adenocarcinoma, SCC, lymphoma)
↑ Mucosal Injurious Substances: Acid: Gastrin, Histamine, AChàpromote
acid secretion in parietal cell
Toxins: drugs (for instance, NSAIDs) directly toxic to gastric epithelial cells, drugs that reduce platelet adhesion/plug
Authors: Dean Percy Yan Yu Reviewers: Sean Spence, Jason Baserman Paige Shelemey, Tony Gu, Kerri Novak* * MD at time of publication
COX-1 inhibition
Decreased prostaglandins
↑ H+ production, ↓
gastric mucous production
Toxicity to Gastric epithelial cells
↓ Gastroprotective Factors:
• Mucous: barrier between cells and HCl
• Blood flow: removes H+, supports mucosal cells
• Epithelial cells: physical barrier, HCO3- buffer
• Prostaglandins: ↓ H+ secretion, ↑ mucous
• •
production
Erosion of Mucosa
Erosion proceeds into blood vessels
Irritation of somatic innervation (T5-T8)
Bleeding into stomach
Bleeding into esophagus
Hematemesis
(Vomitting Blood)
Blood passes through GI tract, becomes oxidized by HCl & digestion
Melena
(black, tarry, foul- smelling stool)
Blood gets oxidized by HCl but moves back into esophagus
‘Coffee-ground’ Emesis
Notes:
Dyspepsia, Epigastric Pain
Nausea
• COX-2 inhibitors not always effective for gastroprotection
• In elderly patients and patients on NSAIDs, mucosal erosion can be silent (asymptomatic)
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Re-Published September 22, 2019 on thecalgaryguide.com
Mechanism not well understood")
Rapid Sequence Induction and Intubation (RSII): Clinical Approach
: Clinical Approach
Authors:
Sandy Ly Reviewers: Wendy Yao
Melinda Davis*
* MD at time of publication
Reversible with Sugammadex (selective relaxant binding agent)
Responds to acetylcholinesterase
Reversible with acetylcholinesterase inhibitors
Not immediately reversible due to high dose
Classical RSII
Modified RSII
Induction Agent e.g. Ketamine or Propofol
(2 mg/kg)
Inhibitory effect on central nervous system
Cricoid pressure
(10 lbs pressure posteriorly)
Esophagus at the level of the cricoid obstructed
Reduced gastric regurgitation
Succinylcholine (2 mg/kg) acts similar to Ach
Depolarize end plate nicotinic receptors in skeletal muscle
Non-competitive with no antagonist
Rapid skeletal muscle paralysis
(<30 seconds) with short duration (<10 minutes)
Irreversible
Preoxygenation
with 100% O2 displaces nitrogen.
Functional residual capacity (2.5L) is filled with O2
Oxygen consumption (250 mL/min)
Extend time to desaturation (Ideal condition: 10 minutes)
Gastric distension with use of bag valve mask ventilation (positive pressure)
High dose Rocuronium (1 mg/kg) competitively antagonizes Ach
Decreased Ach binding on
nicotinic receptors in skeletal muscle
Rapid skeletal muscle paralysis (<60 seconds) with long duration (>45 minutes)
Quick Facts
Induction of anesthesia
Abbreviations
Ach – acetylcholine
See other pathways for more detailed pathophysiology
• •
•
RSII is used in patients with increased risk of gastric aspiration. Cricoid pressure is NOT THE SAME as BURP (backward, upward, rightward pressure).
Other induction agents possible (e.g. etomidate).
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published September 22, 2019 on www.thecalgaryguide.com")
Priapism
 pathways
Post-cavernosal venous drainage is mechanically obstructed e.g. sickle cell disease, dialysis etc.
Ischemic (Low-flow):
Inadequate venous function
Blood pools in corpora cavernosa
Cavernosal cell metabolism uses O2 and releases CO2 into pooled blood
Trabecular smooth muscle relaxes
Cavernosal artery smooth muscle relaxes
Lack of norepinephrine action on penile SM
Post-cavernosal venules are compressed against tunica albuginea
Trabecular smooth muscle and cavernosal artery smooth muscle do not contract
↑ pressure in corpora
↑PCO2 ↓pH
Acidosis
↓PO2
Tissue hypoxia
Tissue damage
Irreversible fibrosis leading to erectile dysfunction
Priapism
Prolonged erection lasting more than 4 hours; in absence of sexual stimulation; not relieved by ejaculation
Trauma to penis or adjacent areas
Authors:
Arsalan Ahmad
Reviewers:
Alec Mitchell
Darren Desantis*
Yan Yu*
* MD at time of publication
Cavernosal artery is damaged
Excessive, unregulated arterial blood flow into corpora cavernosa
Pain
Non-ischemic (High-flow):
Uncontrolled arterial flow
↑ volume of blood in corpora
↑ pressure in corpora
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published September 22, 2019 on www.thecalgaryguide.com")
Infantile Colic
 factors
↑ bile acid wasting ↓ bile acid production in immature in immature gut enterohepatic circulation
↓ bile acid availability
Psychosocial factors
Infant temperament Over/understimulation
Parental variables (e.g. parental stress)
Collectively, these factors influence the infant’s reactivity to adverse stimuli and the caregiver’s perception of whether crying is problematic.
Other biologic factors
Poor feeding techniques: under/ overfeeding, swallowing air, infrequent burping
Altered gut motility
GI discomfort
Infantile Colic
Dietary intolerances: cow’s milk protein, lactose
↑ gas production and gut distension
↓ mucosal barrier function
Loss of bacteriostatic effects of bile acids
Immature enteric nervous system
Intestinal microbial imbalance
↑ intestinal permeability
↑ systemic inflammation
Altered central and enteric neuronal function via microbiota-gut-brain axis
Altered perception of pain and other GI stimuli
Crying for no apparent reason that lasts > 3 hours/day and occurs ≥ 3 times/week for > 3 weeks in an otherwise healthy infant < 3 months old. There must be normal growth, development, and physical exam. Colic itself is a benign, self-limiting condition that resolves with time.
Facial flushing or grimacing Tense or distended abdomen
Drawing up of legs Clenching of fingers Stiffening of arms Arching of back
Distress expressed via behaviour
Loud, high- pitched, urgent cry
↑ risk of non- accidental trauma
GI factors directly affect infant’s behaviour
Hypothesized role of immature CNS regulation of circadian rhythm
↑ parental stress
Immature regulation of behaviour
Episodes cluster in evening and/or late afternoon
↑ risk of post- partum depression
Paroxysmal crying Inconsolable
Authors: Simonne Horwitz Nicola Adderley Reviewers: Crystal Liu Yan Yu* Danielle Nelson* * MD at time of publication
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published September 28, 2019 on www.thecalgaryguide.com")
Macrosomia-Fetal-Complications

Size discrepancy between fetal shoulders and maternal pelvic inlet
Anterior shoulder becomes impacted behind the symphysis pubis during delivery
Shoulder Dystocia
(↑risk in infants of diabetic mothers – see slide on Gestational Diabetes)
Injuries acquired as a result of the birthing process in an infant with shoulder dystocia
↑ incidence of preterm birth
Surfactant deficiency
Respiratory Distress Syndrome
Umbilical cord compression
↓ delivery of oxygenated blood to fetus
Hypoxia/Asphyxia
Infant gasping
Perinatal aspiration of stained amniotic fluid
Meconium Aspiration Syndrome
↑ incidence of cesarean deliveries
↓ duration/absence of labour
↓ release of maternal epinephrine and glucocorticoids
↓ activation of epithelial sodium channels on type II pneumocytes
Delayed resorption of fetal lung fluid
Transient Tachypnea of the Newborn
Notes
↑ oxygen demands
Fetal hypoxia
↑ production of erythropoetin
Polycythemia Neonatal Jaundice
Maternal diabetes
↑ intrauterine exposure to
excessive nutrients and glucose
2Fetal Hyperinsulinism
↑ glucose utilization and suppression of hepatic glucose production
Termination of the maternal glucose supply at delivery
Hypoglycemia
Brachial Plexus Injury
Clavicular Fracture
Humeral Fracture
1. Complications of macrosomia ↑ with birth weight. Risk of stillbirth ↑ above 5 kg 2. Most common in the setting of poorly controlled maternal diabetes; however,
hyperinsulinism may be absent if macrosomia is secondary to a different etiology (e.g. post-term fetus, genetic conditions).
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published September 22, 2019 on www.thecalgaryguide.com")
intrauterine-devices-iuds-mechanism-of-action
: Mechanisms of Action
Authors: Danielle Chang Reviewers: Mirna Matta Crystal Liu Aysah Amath* * MD at time of publication
Intrauterine Device (IUD)
Contraceptive device that sits in the fundus of the uterus
Two Types
Copper
T-shaped polyethylene with fine copper wrapped around the vertical stem and arms
Releases copper in the uterus
Progesterone Releasing
IUD made of plastic containing levorgestrel (synthetic progesterin, progesterone receptor agonist)
Releases levorgestrel in the uterus Stimulate progesterone receptors
May inhibit ovulation through negative feedback on hypothalamus (not consistent evidence)
Foreign body reaction in endometrium
IUD creates local lysosomal activation and uterine inflammatory response Mechanism not fully understood
Spermicidal
High copper concentrations toxic to sperm
Alters sperm mobility
Decreases chance of fertilization
Suppresses endometrium
Thickening of the cervical mucus
Barrier to sperm penetration
Disturbs function of endometrium
Changes uterine environment
Thins the endometrium
Decreases number of blood vessels to the endometrium
Suppresses proliferation
Atrophies endometrial glands
Sloughing of the endometrium
Inflammatory reaction against blastocyst if fertilization occurred
Hostile environment for implantation
Decreased menstrual blood loss and eventual amenorrhea
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published October 12, 2019 on www.thecalgaryguide.com")
X-linked Severe Combined Immunodeficiency (SCID)
: Pathogenesis & clinical findings
Abbreviations:
• Ig: Immunoglobulin
• IL: Interleukin
• NK: Natural Killer
• Th2: T helper 2 cell
• Treg: T regulatory cell
↓ IL-15 signaling ↓NK cell differentiation
and proliferation
↓ or absent NK cells in the blood, bone marrow and peripheral lymphoid tissues
Impaired innate immune system
Chronic Stress Response
Mutation of the IL2-RG gene encoding the interleukin receptor common gamma chain located on the X-chromosome
↓ IL-2 signaling ↓T cell
proliferation
Sequence analysis shows mutated, duplicated or deleted IL2-RG gene
↓ IL-7 signaling Global ↓ in
lymphocyte survival
Absent thymic shadow on X-ray
↑ susceptibility to fungal infections
Complete defect in cell mediated and humoral immunity
↓ antibody responses to vaccinations
↑ susceptibility to extracellular pathogens, most notably bacteria
Authors: Kyo Farrington Reviewers: Paul Adamiak Jessica Tjong Louis Girard* *MD at time of publication
↓ IL-4 signaling ↓Th2
differentiation
↓T cell help for B cell activation and class switching
Dysfunctional B cells
Genetic Predisposition
↓ Treg development
↑ risk of developing an autoimmune disease
↓ T cell response to mitogens or
anti-CD3 stimulation
↓ or absent T lymphocytes in the blood, bone marrow and peripheral lymphoid tissues
↑ susceptibility to viral infections (often gastrointestinal ones like rotavirus and/or enterovirus)
Chronic diarrhea
Hypoplastic lymphoid tissues (I.e. tonsils, adenoids, lymph nodes)
Hypermetabolic State
Failure to thrive
↓ antibody production in response to antigen exposure
↓ IgA, IgM and IgG serum concentrations
Note: IL2-RG gene encodes the interleukin receptor common gamma chain, which is a sub-unit common to the receptor complexes for IL-2, IL-4, IL-7, IL-9,
IL-15 and IL-21. The bolded/italicized cytokines contribute most to the pathophysiology of X-linked Severe Combine Immunodeficiency (SCID).
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Re-Published October 12, 2019 on www.thecalgaryguide.com")
Ischemic Colitis
 supply blood to colon
Surgical repair of aorta
Borders of SMA and IMA collaterals at the splenic flexure and rectosigmoid junction are vulnerable to ischemia (“watershed” areas)
Atherosclerosis and narrowing of mesenteric arteries
Low flow state
(e.g., CHF, hypotension, arrhythmia)
Underlying CAD/PVD
Atrial fibrillation, endocarditis
Embolic arterial occlusion of SMA and/or IMA
Trauma, infection, clotting abnormalities
Mesenteric vein thrombosis
Vascular risk factors (e.g., smoking, hypertension)
Thrombotic arterial occlusion of SMA and/or IMA
Endograft coverage of IMA
Nonocclusive hypoperfusion
Inadequate blood flow to meet the cellular metabolic needs in the colon
Ischemic Colitis
Tachypnea Tachycardia Hyperthermia Hypotension
Ischemic period
Loss of oxygen and nutrients to bowel
Reperfusion period
Influx of O2àreacts to produce more oxygen free radicals
Lipid peroxidation
Systemic inflammatory response syndrome*
Nausea and vomiting
Abdominal pain (generally left sided)
Peritonitis
Leukocytosis
Systemic shock
(inadequate perfusion to tissue)
Author: Audrey Caron Michael Blomfield Reviewers: Tony Gu Yan Yu* Edwin Cheng* * MD at time of publication
Systemic shock
(inadequate perfusion to body tissue)
Hematochezia (Bloody stool)
Gangrene (tissue death)
Hemorrhage
Tissue damage/cell death (starting from mucosa and submucosa going outwards to serosa)
Mucosal ulceration
Colonic inflammation
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published November 10, 2019 on www.thecalgaryguide.com")
Incisional-Hernia
 that ↑ risk of infection
Post-op wound Infection
Obesity
Chronic constipation
Chronic cough Pregnancy
↓ clotting factors
Vigorous cough
Severe Hypertension
Seroma
Post-op hematoma Bulging fluid separates High risk
Sutures unsuitable Poor surgical for tension technique
↑ intraabdominal pressure
Fascial Incision separates
Notes:
fascial incision
surgeries* High Risk Surgeries*
Connective tissue disorder
Suboptimal fascial closure
• • •
Emergency surgeries Midline incisions
Acute abdominal surgeries
↓ wound healing/collagen synthesis
Fascial defect at previous incision site
Incisional Hernia:
Protrusion of tissues through prior fascial incision
• Deep wound infection = most common cause of incisional hernias
• Diagnosis on physical exam +/- CT scan if patient is obese
• Treatment = surgery
Bulge at prior incision site
Palpable fascial defect
Bowel and other abdominal contents protrude through defect
Mechanical bowel obstruction (see relevant slide)
Constipation /obstipation
Contents unable to be pushed back through defect (incarceration)
Vascular supply is compromised to herniated contents
Contents become ischemic (strangulated)
Prolonged pressure on skin & bowel over time
Ulceration & ischemia
↓ blood flow to skin layers
Discoloration of skin
Bulge ↑ with coughing/straining
Ulcers extend through bowel wall
Authors: Karly Nikkel Meaghan Ryan Reviewers Michael Blomfield Tony Gu Yan Yu* Edwin Cheng* *MD at time of publication
Colo-enteric fistula
Bowel Perforation
Abdominal Pain
Abdominal Distension
Nausea/ Vomiting
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published November 13, 2019 on www.thecalgaryguide.com")
Endometritis

Uterine tenderness
Pelvic and/or abdominal pain
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published November 26, 2019 on www.thecalgaryguide.com")
Hydrocele

Transillumination
Testicular Atrophy
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published November 26, 2019 on www.thecalgaryguide.com")
Varicocele
.
• Small varicoceles can be identified by preforming the Valsalva maneuver (decreases venous return).
• Unilateral right varicoceles are uncommon and should be investigated for underlying pathology causing obstruction.
Primary
Anatomically: the left spermatic vein drains into the left renal vein
Nutcracker Effect: The left renal vein can get pinched by the abdominal aorta and superior mesenteric artery
Backup of blood in left renal vein ↑ pressure in left spermatic vein
Secondary
Renal cell carcinoma or retroperitoneal masses
Inferior vena cava thrombus
External compression of spermatic vein
Obstruction of blood flow
↑ spermatic vein pressure
Vein valve leaflet failure & retrograde bloodflow back towards testicle
Dilation of pampiniform plexus and scrotal vein plexus
Varicocele
↑ scrotal blood volume ↑ volume in a closed
space
↑ pressure and distension of scrotal layers
↑ scrotal vein plexus pressure
Compliant veins distend, becoming visible through scrotum
Blood heats up the structures it flows through
Scrotal hyperthermia
Unsuitable environment for spermatogenesis
Loss of germ cell mass
Bag of Worms Sign
Dull ache/heaviness
Decreased fertility Testicular atrophy
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published November 26, 2019 on www.thecalgaryguide.com")
Aphasia
: Pathogenesis and clinical findings
Authors: Davis Maclean Reviewers: Heather Yong Tony Gu Yan Yu* Scott Jarvis* *MD at time of publication
Ischemic stroke (common)
Local Invasion (e.g. by a tumour, Head infection, or hemorrhage) Trauma
Intracerebral Hemorrhage
Dementia (e.g. Fronto- temporal Dementia)
Episodic occurrences (e.g., migraine, epilepsy)
Damage to language-dominant cerebral hemisphere (the left hemisphere, for the majority of humans):
Damage affecting Broca’s Area in the Inferior frontal gyrus (area 44 & 45)
Damage affecting Wernicke’s Area in the posterior part of the superior temporal gyrus (area 22)
Localization: Inferior frontal gyrus, superior sylvian fissure
Blood supply: superior division M2 branch middle cerebral artery
Localization: Posterior perisylvian region, temporal lobe
Blood supply: inferior division M2 middle cerebral artery
Sensory speech
areas still intact (posterior superior temporal lobe)
Intact comprehension (intact hearing & reading)
Impaired function of Broca’s Area
↓ output or generation of speech/ text
If function of nearby motor areas is also impaired
Contralateral hemiparesis (face, arm > leg)
If function of other nearby areas is also impaired
Impaired naming and repetition
Motor speech areas still intact (inferior frontal lobe)
Fluent (but non-sensical) speech output
Impaired function of Wernicke’s Area
Impaired compre- hension (i.e. cannot understand speech or text)
Loss of sensory speech input to motor areas
Errors in word usage, tense, structure
If function of nearby sensory areas is impaired
Contralateral sensory deficits
Broca’s Aphasia
Wernicke's Aphasia
(Expressive language impairment: non-Fluent)
Notes/Definitions:
(Receptive language impairment/Fluent: the person can talk but their speech is nonsensical)
• Dysarthria ≠ Aphasia (Dysarthria: disruption to neurons controlling the muscles that produce sounds, resulting in slurred/disjointed speech. Aphasia: acquired deficit in language comprehension or generation/output usually due to disruption of neurons in the cerebral cortex.)
• “Global” aphasia affects both receptive and expressive language.
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published January 1, 2020 on www.thecalgaryguide.com")
Slide-authoring-process-FINAL-2

A1AT-Deficiency
 allele(s)
Accumulation of mutant α1AT protein as ordered polymers in endoplasmic reticulum of hepatocytes
↓ α1AT inhibition of Hepatocyte Injury tissue proteases (Mechanism unclear)
↓ release from hepatocytes
Cirrhosis, chronic hepatitis, hepatocellular carcinoma
↓ lung elasticity, ↓ ability for lung to expel air on expirationà gas trapping, hyperinflation, airway collapse over time
Role of Genetics:
Low serum α1AT
In the skin: Subcutaneous proteases > Anti- proteases
Unopposed proteolysis in subcutaneous tissues
Panniculitis (Rare, most cases associates with ZZ Genotype)
Lung Proteases > Anti-proteases àproteolytic destruction of lung parenchymaàpanacinar emphysema (accelerated by smoking)
Stigmata of chronic liver disease (ascites, jaundice, spider nevi, petechiae, etc.)
Symptoms of chronic obstructive pulmonary disease (COPD): barrel chest/High residual volume (RV), low vital capacity (VC), wheezes on auscultation, etc) – see relevant slide
Degree of α1AT deficiency dependent on genotype:
• MM gives normal α1AT levels
• MZ genotype produces levels ~ 35% of normal
• ZZ genotype produces severe deficiency ( <10% of normal)
• Null phenotype is completely deficient of α1AT
N.B. Heterozygotes almost never develop phenotypic α1AT deficiency syndromes. Even some homozygotes don’t manifest the disease.
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Re-Published January 12, 2020 on www.thecalgaryguide.com")
Viral-Hepatitis
 of hepatocytes
Infection with chronic viruses (HBV and HCV) persist over time and additional symptoms may develop
RUQ pain/tenderness
If infection is prolonged or severe, inflammation becomes systemic
Release of hepatocyte’s cellular contents into the bloodstream
Infection with acute viruses (HAV and HEV) resolve over time, and the symptoms above normalize
Notes:
• HDV can only infect people with concomitant HBV infection
• HAV and HBV vaccines are the only ones that currently exist
• Not all patients with viral hepatitis will develop each of these symptoms. The presentations vary.
Fever, nausea, vomiting ↑ serum ALT, AST
↓ Hepatic metabolic activity (e.g. reduction of gluconeogenesis) ↓Serum Glucose
↓ Synthesis of plasma proteins (albumin, clotting factors, etc) ↓ Albumin, ↑ INR
Abbreviations:
• HAV - Hepatitis A Virus
• HBV - Hepatitis B Virus
• HCV - Hepatitis C Virus
• HEV - Hepatitis E Virus
• RUQ - Right Upper Quadrant
• ALT - Alanine Aminotransferase
• AST - Aspartate Aminotransferase
• INR - International Normalized Ratio
↓ Bilirubin clearance from blood, bilirubin ends up under the skin Jaundice Portal Hypertension
Encephalopathy, Splenomegaly, Esophageal Varices, Ascites, Caput Medusae, Edema
Encephalopathy, Muscle Wasting, Metabolic Bone Disease, Terry’s Nails, Ascites, Bruising, Clubbing, Edema
Spider Nevi, Altered Hair Patterns, Testicular Atrophy, Gynecomastia, Palmar Erythema
Progressive deterioration in liver function, possibly ending up in cirrhosis. (See slide on “Cirrhosis: pathogenesis and complications” for more details on mechanisms and full explanations.)
Hepatic Insufficiency Hyperestrogenism
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Re-Published January 12, 2020 on www.thecalgaryguide.com")
Multiple-Myeloma
 immunoglobulins
Normal SPEP: no spike in the gamma (γ) region
Notes:
• MGUSismuchmorecommon than MM!
• “Plasmacell”:Blymphocytes that produce antibodies/ immunoglobulins
Stimulation by specific antigens
Genetic changes/mutations accumulate over time in one type of plasma cell
Abbreviations/Definitions: • SPEP - Serum Protein
Electrophoresis
• Ig – Immunoglobulin • Monoclonal – “of one
specific genetic strain or subtype”
One type of plasma cell starts to proliferate abnormally
Monoclonal Gammopathy of Undetermined Significance (MGUS) (Premalignant, mild monoclonal plasma cell proliferation; asymptomatic)
In 1-2% of cases, further cytogenetic changes over time stimulate further proliferation of this plasma cell line
Multiple Myeloma (MM)
(extensive monoclonal B lymphocyte proliferation, causing end organ damage)
Slightly more monoclonal plasma cells will produce slightly more monoclonal Immunoglobulins
Small spike in the gamma (γ) region of the SPEP (less prominent compared to the spike in MM)
More monoclonal plasma cells result in far greater amounts of monoclonal immunoglobulins being secreted
Large spike in gamma (γ) region of the SPEP
Ig light chains accumulate in the tubules of kidney nephrons
Light chain casts obstruct tubules
Renal Insufficiency (↓GFR)
Osteoblasts ↑ expression of RANK-ligand (RANKL, an apoptosis regulator), and ↓ expression of Osteoprotegerin (OPG, a decoy receptor for RANKL)
↑ osteoclast activity vs osteoblast activityàbone loss
Clonal plasma cells overrun normal bone marrow, crowding out production of red blood cells, ↓ red blood cell counts
Anemia
Authors: Tristan Jones, Tyler Anker, Yan Yu Reviewers: Jennifer Au, Crystal Liu, Man- Chiu Poon*, Lynn Savoie* * MD at time of publication
Damaged bones hurt, & become more brittle
Bony pain, pathologic fractures
Osteolytic bone lesions
Osteoclasts release calcium from bone and into blood
Hypercalcemia
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published January 12, 2020 on www.thecalgaryguide.com")
Pathophysiology-Behind-the-Leukemias

Chromosomal Abnormality (duplication, loss, recombination error)
Combinations of these genetic defects causes reduced tumor suppressor gene expression and/or increased oncogene expression
Initiating Mutational Event
ALL
Any combination of mutations, chromosomal
alterations, or other genetic abnormalities that creates a neoplastic cell (incapable of regulating cell growth/division).
AML
CLL
CML
Translocation between Chr 9 and Chr 22à Philadelphia chromosome ( abnormal Chr 22)
àBCR-ABL1 oncogene (along with other genetic abnormalities)
•
In White Blood Cells and their precursors: Lack of cell growth inhibition and / or apoptosis.
• Over stimulation of cell division/growth Neoplastic blood cell incapable of regulated cell
division
Neoplastic cells uncontrollably divide in a monoclonal way: one neoplastic cell originates all successive cells
Genes regulating differentiation/maturation disrupted, affected neoplastic cells are incapable of further differentiation/maturation
Genes regulating maturation remain intact (affected neoplastic cell is capable of further differentiation/maturation)
Some neoplastic cells take time to mature furtheràless rapid disease progression (more indolent disease); cells don’t die
CLL: Chronic Lymphoid Leukemia
Note:
Although it is tempting to group the leukemias together for study purposes, it is best to learn the 4 main types of leukemias independently of one another, as they have a uniquely different pathophysiology and clinical presentation
Specific mutations cause slower disease progression
CML: Chronic Myeloid Leukemia
Degeneration during CML’s ”blast crisis”
Specific mutations cause rapid division and buildup of existing neoplastic cells àAcute/rapid disease progression.
ALL: Acute Lymphoblastic Leukemia AML: Acute Myeloid Leukemia
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Re-Published January 19, 2020 on www.thecalgaryguide.com")
Aqueous-Humor-Production-and-Drainage
 through combined hydrostatic pressure + active transport
AH enters the posterior chamber
AH flows around the lens through the pupil into the anterior chamber
Temperature gradient cause aqueous humor to flow convectively: downwards at the cornea (colder, fluid denser); upwards at lens (warmer)
Abbreviations:
• AH – Aqueous Humor
• TM – Trabecular Meshwork
Provides nutrition, removes wastes, and transports neurotransmitters to the avascular lens and cornea.
Circulates inflammatory cells to avascular structures
Maintains structural integrity of ocular structures via intraocular pressure (10-21 mmHg)
AH leaves the anterior chamber via two pathways located near the limbus
Limbus
Trabecular Meshwork Pathway (~60-80 % of flow)
AH passes through the TM layers: Uveal, corneoscleral, and juxtacanalicular
Flows into Schlemm’s Canal
Flows through internal outflow channels into the external outflow channels, aqueous veins, intrascleral venous plexus, and the deep scleral venous plexus
Drains into the episcleral and conjunctival veins
Uveoscleral Pathway (~20-40 % of flow)
AH enters uveal meshwork and anterior ciliary body
Flows in suprachoroidal space
Passes outward through the sclera
Enters the scleral, and conjunctival veins
Drains back into systemic venous circulation
Inflow
Trabecular Outflow
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published January 19, 2020 on www.thecalgaryguide.com")
Innate-Immune-Response

Pathogens overcome physical barriers (e.g., epithelium, cilia)
Trauma
Damage-associated molecular patterns
Pathogen-associated molecular patterns
Examples of tissue-resident macrophages: • Alveolar macrophages – Lung
• Histiocytes – Connective tissue
• Kupffer cells – Liver
Recognition by pattern recognition receptors (e.g., toll-like receptors)
• Mesangial cells – Kidney • Microglial cells – Brain
• Osteoclasts – Bone
Microbe engulfed and exposed to oxidative burst
Microbes destroyed
Pus
Pro-inflammatory chemokines
Recruitment of circulating
granulocytes and monocytes
Pro-inflammatory cytokines (e.g., IL-1β, TNFα, IL-6)
Tissue-resident macrophage activation
Antimicrobial proteins
Unresolved infection/ inflammation
Antigen presented to T cells
Recruitment of adaptive immune response
Enhanced immune responses
Acute phase protein production by liver (i.e., C- reactive protein)
Prostaglandin production in the hypothalamus
Fever
Endothelial tight junctions on vasculature disrupted
Intravascular fluid leak into extravascular space
Edema
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published January 19, 2020 on www.thecalgaryguide.com")
vomiting-pathogenesis
 Center
Toxins circulating in bloodstream: Chemotherapy, Opioids
Offending substance travels through circulation and binds to receptors in the CTZ, outside the blood brain barrier
Abbreviations:
GERD: Gastroesophageal Reflux Disease PUD: Peptic Ulcer Disease
IBD: Inflammatory Bowel Disease
CTZ: Chemoreceptor Trigger Zone
CNX: Cranial Nerve Ten
H1: Histamine Receptor
M1: Muscarinic Receptor
Disrupted inner ear balance: Motion Sickness
Activation of H1 & M1 receptors in vestibular center traveling via Cerebellum
Stimulates Solitary Tract Nucleus (Medulla)
(Medulla)
Vagus Nerve (CNX) and enteric nervous system activation, resulting in:
Gastric relaxation, ↓ pylorus tone, retrograde duodenal peristalsis
Downward diaphragm contraction, abdominal & chest wall muscles contract: ↑ intra-gastric pressure
Vomiting
(Forceful expulsion of material from stomach and intestines)
Upper and lower esophageal sphincter relaxation and glottis closure
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Re-published February 16, 2020 on www.thecalgaryguide.com")
Pseudogout
 crystals from joints is inhibited by iron
Hypomagnesia
The relative absence of magnesium impairs pyrophosphatase activity, reduces pyrophosphate breakdown
Hypophosphatasia
Defective mineralization of calcium and phosphorous in bones
Idiopathic (vast majority of cases)
Mechanism unknown
↑ serum concentrations of Ca2+ or Pyrophosphate
Enhanced mineralization in chondrocytes (cells that make cartilage)
Abbreviations
• NTPPPH nucleoside triphosphate
pyrophosphohydrolase
• CPPD – Calcium Pyrophosphate
Dihydrate
Notes:
• There are different types of calcium pyrophosphate crystal deposition (CPPD) disease. This slide only covers “pseudogout”.
• Pyrophosphate (PPi) = 2 phosphate molecules = P2O74−
• Pyrophosphate is made from the breakdown of Adenosine triphosphate (ATP): ATP -> AMP + PPi
Once in cartilage, high levels of either calcium ions or pyrophosphate can result in them binding together, forming CPPD crystals
Aggregated CPPD crystals shed into synovial fluid
Neutrophils enter joint to phagocytose the crystals and release pyrophosphatase enzyme
Repeated crystal precipitation into joint space over time (subacute process)
CPPD crystals collect on collagen fibers in articular cartilage
Chondrocalcinosis, seen on high-resolution ultrasound and/or x-ray
CPPD crystals exhibit unique properties on polarizing microscopy
Inflammatory cascade
Positively birefringent (crystals appear blue parallel to axis of polarizer)
PAINFUL, warm, swollen joint (sudden onset)
↑ C-reactive protein (CRP); erythrocyte sedimentation rate (ESR)
Knees and wrist subjected to more trauma over a person’s lifetime
Knee > Wrist >>> any other joint affected
Wearing down of joint cartilage over time
Rapidly progressive osteoarthritis (see osteoarthritis slide)
Subchondral sclerosis & cysts, joint space narrowing, and osteophytes seen on x-ray
bone loss, hemarthrosis
Nerve damage over time
“Charcot-like” joint: severe weightbearing to areas that joint destruction/deformity,
Painless joint
Lack of sensation can cause tolerate it poorly
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published February 16, 2020 on www.thecalgaryguide.com")
C5-C9-deficiency
 causes Secondary (acquired) causes
All are autosomal recessive Biologic therapy ex. eculizumab Absence or suboptimal functioning of
Abbreviations:
• MAC: Membrane Attack
Complex
• CH50: Classic Hemolytic
Complement Test
• AH50: Alternative Hemolytic
Complement Test
• CNS: Central Nervous System • CSF: Cerebrospinal Fluid
≥1 terminal complement proteins
C5-C9 deficiency
Inability to form MAC
↑ susceptibility to systemic Neisseria infection
Commonly N. meningitidis Rarely N. monorrhoeae
Nasopharyngeal colonization of N. meningitidis, ↑ susceptibility to bacteremia
CH50 ± AH50 assay No lysis
Note:
Total complement activity assay <10% activity C5, C6, C7, C9 <50% activity C8
Varied bactericidal action via other complement proteins
• Risk of invasive meningococcal disease is 1000-10000X higher in C5-C9 deficiency than in the general population
• Reason is unknown
• C5-C9 deficient patients are not at greater
risk for contracting other gram (-) infections • Clinical meningitis in C5-C9 deficiency is less
severe and fatality is rare
Bacterial lysis
Especially gram (-)ve bacteria like Neisseria
Bacteria cross the blood-brain barrier, causing swelling and damaging brain tissue
Fatigue, fever, headache, altered mental status, etc.
Inflammation of CSF and meninges
Activation of dura and pia mater fibres
Headache, neck stiffness
Bacteria release toxins
Damage to surface blood vessels
Maculopapular rash
Exact mechanism unknown
Recurrent meningitis
CNS damage due Sepsis to recurrent
meningitis
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published February 16, 2019 on www.thecalgaryguide.com")
tinea-capitis-tinea-corpora-and-tinea-pedis
:
1. Trichophyton
2. Epidermophyton 3. Microsporum
Direct contact with infected humans, animals, or inanimate objects
Dermatophyte invades uppermost, non-living, keratinized layer of skin, the stratum corneum
Dermatophyte produces enzyme keratinase Keratinase catalyzes degradation of keratin
proteins in the skin Dermatophyte burrows deeper into skin
Keratinocytes release inflammatory cytokines in reaction to dermatophyte antigens
Dermatophyte moves outwards, away from site of infection, to new areas around the initial site
Classic pink-to-red ringed lesion, with central healing
Tinea corporis
Scaling of skin (such as between the toes: “maceration”)
Invasion of hair follicle
Hair shaft breaks
Alopecia (loss of hair) Pruritis(itchiness) Erythema (redness) Induration (swelling) Heat
Local inflammatory response in skin
Tinea capitis
Tinea pedis
Infection of the foot (aka. Athlete’s foot)
Infection of the scalp Infection of the trunk and extremities of the body
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published February 17, 2020 on www.thecalgaryguide.com")
Brain-Neoplasms

Mutagen exposure (e.g., radiation, carcinogens)
Errors of DNA replication
Acquired cell mutations leading to uncontrolled cell division in the brain
Inherited diseases (e.g., neurofibromatosis, tuberous sclerosis)
Primary Brain Tumours
(e.g., gliomas, meningiomas, pituitary adenomas)
Brain Neoplasms
Tumors in the brain arising from brain tissue itself (primary) or from non-brain tissue (metastatic)
Tumor produces vascular endothelial growth factor (VEGF) which generates new vessels (angiogenesis)
Tumor occupies intracranial space
↑Intracranial pressure
Tumor irritates grey matter
Seizures
Headaches Papilledema
Tumor outgrows and disrupts its blood supply
Cerebral ischemia and/or necrosis
Critically located tumors may damage specific neural pathways
Tumor invades, infiltrates, or replaces normal brain parenchyma
Friable blood vessels within tumor àeasy bleeding
Brain hemorrhage
Disrupted blood- brain barrier
↓ blood vessel integrity à ↑ serum leaks out
Edema
Injury to localized brain regions; symptoms vary depending on location of brain affected:
↑ penetration of substances
(e.g., drugs, toxins)
Mass pressing on surrounding structures (mass effect on brain)
Frontal lobe damage
Personality change
Cerebellar damage
Ataxia
Occipital lobe damage
Visual deficits
If adjacent to 3rd/4th ventricles,
tumor will impede flow of cerebrospinal fluid
Obstructive Hydrocephalus
Stretching of meninges; activation of mechanoreceptors affecting the chemoreceptor trigger zone
Vomiting Nausea
Brain tissue pushed down beyond the
tentorium cerebelli, squeezing on brain stem
Brain Herniation
Note: Clinical findings tend to be similar for primary brain tumors and intracranial metastases.
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published February 17, 2020 on www.thecalgaryguide.com")
Umbilical-Cord-Prolapse
 position for delivery e.g. transverse presentation
Abnormal placentation
Poly- hydramnios
If membranes are ruptured, there is a large volume of fluid that may push the umbilical cord through the cervix
Multiple gestation
(second twin)
Multiparity
Multiple fetuses distend uterus
↑ laxity of the uterus ↑ space for cord to come down
Smaller fetal size
Fetus has not shifted into optimal delivery position
↑ space for fetus to move around in once first twin delivered
Placenta implanted near or over cervical opening
Large intrauterine space ↑ risk of mal- presentation
Abbreviations:
• PROM: Premature
The fetus does not adequately block the cervical opening Umbilical cord comes out through the cervix before the fetus does
Umbilical Cord Prolapse
Obstetrical emergency: Umbilical cord descends through the cervix before or alongside the fetus
Authors: Gabrielle Wagner Reviewers: Danielle Chang, Crystal Liu, Yan Yu*, Aysah Amath* * MD at time of publication
membranes
• PPROM: Preterm premature
rupture of membranes
Visible or palpable umbilical cord
Cord compression
Presenting part of the fetus compresses the cord as the fetus descends through the birth canal
Umbilical vasospasm
Exposure of the cord to cold or touch causes arterial vasospasms within the cordà↓ blood flow to fetus
Ante/intrapartum
Fetal hypoxia Post-Partum
Neonatal hypoxic-ischemic Cerebral Neonatal encephalopathy palsy death
Complicated variable fetal Prolonged fetal heart rate decelerations bradycardia
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published February 17, 2020 on www.thecalgaryguide.com")
mechanisms-of-opioid-analgesia-in-the-peripheral-nervous-system

Exogenous
Opioids derived from external source
Semi-synthetics
Synthesized from natural sources (ex. heroin)
Synthetics
Entirely created via synthesis (ex. fentanyl)
Endogenous
Opioids produced naturally in the body
Released in response or in anticipation of to painful stimuli
Endorphins
Enkephalins
Dynorphins
Inhibition of adenyl cyclase
↓ conductance of presynaptic Ca2+ channels
↑ conductance of postsynaptic K+ channels
↓Activity and production of cAMP
Activation of G- protein coupled receptors
↓ neuro- transmitter release
Inhibition of postsynaptic neuron
Blockage of nerve transmission
Binding to opioid receptor subtypes in nervous tissues mu (μ), kappa (k) delta (δ) - Majority of clinically relevant analgesic effects relate to mu (μ) receptor
↓ Release of Substance P and Glutamate in the dorsal horn of spinal cord
↓ Peripheral nociceptive signaling to thalamus and other brain areas
Inhibition of pain transmission to brain
Inhibition of GABA- releasing neurons in Periaqueductal grey (PAG) area
Activation of descending inhibitory pathways from PAG
Other/Systemic Side Effects
(For complete list and mechanism see Calgary Guide – Side effects of Opioids
Abbreviations:
cAMP (cyclic Adenosine mono-phosphate) – Cellular signaling molecule
GABA (gamma-Aminobutyric acid) – Inhibitory neurotransmitter
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published March 4, 2020 on www.thecalgaryguide.com")
pharmaceuticals-under-investigation-by-who-for-treating-covid-19-proposed-mechanisms
:
Yan Yu*, Stephen Vaughan*
* MD at time of publication
Note: This slide is based on literature available up to 03/30/2020. Medicines shown here are those included in the WHO SOLIDARITY trial, which were selected based on in vitro work and clinical data from MERS and SARS. Mechanisms are preliminary and there is insufficient data to support or refute the use of these agents for COVID- 19. Research is ongoing.
Viral replication is terminated
Fewer new cells infected
↓ activation of inflammatory and immune responses
Improvement or halt in progression of clinical signs of infection*
*See slide on pathophysiology and clinical findings of COVID19
Proposed Mechanisms
COVID-19 Viral Replication Pathway
Virus adheres to Angiotensin Converting Enzyme 2 (ACE-2) receptor on body cells
Endocytosis of virus in clathrin coated vesicles
Vesicles mature through endolysosomal pathway
Virus membrane fuses with mature endolysosome releasing viral RNA into cytosol
Viral RNA uses host cell ribosomes to make new viral proteins like RNA-polymerase
Viral RNA-polymerase incorporates nucleotides from the host cell
New viral RNA is produced
Viral RNA and proteins packaged into new viral particles
Viral particles released from cell
Chloroquine or Hydroxychloroquine (CQ, HCQ)
Weak basicity leads to ↑ pH of endosomes and lysosomes
N-terminal glycosylation of ACE-2 in Golgi is inhibited
Abnormal ACE-2 receptor expressed on cell surface
Viral membrane cannot fuse with immature endosome
Altered virus- ACE-2 interaction impairs entry into host cell
Viral contents are not released
Interferon-β (IFNβ) (Given with LPV/RTV)
Ritonavir (RTV)
(given with LPV)
Inhibition of CYP450, a drug metabolizing enzyme
↓ degradation of Lopinavir (LPV)
↑ plasma half life and duration of action of LPV
Binding to interferon receptor
Impaired maturation of endosomes
Activation of JAK/STAT pathway
Transcription of IFN- regulated genes
↑ expression of antiviral and immunomodulatory proteins
Antiviral effects may ↑ response to LPV/RTV (mechanism uncertain)
Remdesivir (RDV)
RDV is phosphorylated to RDV-triphosphate (RDV-TP)
Lopinavir (LPV)
Inhibition of viral 3- chymotripsin-like protease
Inhibition of viral replication (multiple mechanisms)
RDV-TP competes with ATP for binding to viral RNA polymerase
Viral protein precursors are not cleaved into mature viral proteins
Incorporation of RDV-TP terminates growing RNA
Newly formed viral particles can’t infect new cells
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published March 30, 2020 on www.thecalgaryguide.com")
GI-changes-during-pregnancy
 Tract
Pregnancyàhormonal and physical changes in the body
Mechanisms poorly understood
↑ human chorionic ↑ estrogen ↑ progesterone gonadotropin (hCG)
↑ uterus size
Uterus rises into
abdominal cavity
↑ intra-gastric pressure
↑ backup of stomach contents
Nausea & vomiting
In the extreme case:
Hyperemesis gravidarum (extreme vomiting causing weight loss, dehydration, ketosis)
Liver displaced upwards
Liver edge generally not palpable on exam
↑ blood pressure in veins within the abdomen
Veins around rectum & anus stretch under pressure
↑ blood flow to the gum tissue
↑ tendency for gingival bleeding & ulceration
Gingivitis
↑ neo- vascularization in lesions on skin
Pyogenic granuloma of pregnancy (shiny red papule with a raspberry-like surface)
Mechanism poorly understood
Ptyalism (excessive salivation)
Difficulty swallowing excess saliva
↑ gallbladder stasis
Biliary
sludge given time to solidify within gallbladder
Gallstones
↓ mobilization of intracellular calcium within smooth muscle cells
Smooth muscle relaxation in tissues such as the gallbladder & GI tract
Changes in taste perception
Dysgeusia
Cultural influences and psychological factors
Change in diet and dietary cravings
↓ lower esophageal sphincter tone
Retrograde transport of gastric contents into esophagus
Gastroesophageal reflux
↓ GI motility
Delayed gastric and intestinal emptying
Stool builds up in colon, and hardens as water is resorbed
Constipation
Pooling of blood within rectal veins àvenous thrombosis
Hemorrhoids
Fragile veins, more easily torn
Rectal bleeding
Change in gut microbiome
Authors: Simonne Horwitz, Yan Yu*
Reviewers: Claire Lothian, Crystal Liu, Ronald Cusano* * MD at time of publication
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published April 29, 2020 on www.thecalgaryguide.com")
Fecal-Incontinence

↓ anal resting tone
Aging:
Degene- ration of muscle fibers
Movement disorders (e.g. arthritis, Parkinson’s); aging is a risk factorà ↓ mobility
↓ timely access to bathrooms
Inflammation
of colon (e.g., Ulcerative colitis, Radiation proctitis)
↓ capacity of
rectal smooth muscle to stretch
↓ capacity to store stool
↑ urgency of defecation
↑ reflex relaxation of internal anal sphincter
Chronic diarrhea, diarrhea- predominant irritable bowel syndrome, laxatives
Yan Yu* Erika Dempsey* * MD at time of publication
Stretch injury of Pelvic surgery
Chronic constipation
Build up of solid, immobile mass of stool in the rectum
Loose stool is able to flow around impacted stool, exiting anal canal (overflow diarrhea)
Sensory neuro- pathy (e.g. Diabetes)
Altered
mental conditions (e.g. stroke, dementia)
pudendal nerve (innervating the pelvic muscles and external anal sphincter)
Local neuronal damage
Impaired pelvic muscle and external anal sphincter motor control
Pelvic trauma
Rectal prolapse
Direct external anal sphincter impairment
↑ Stool volume
↑ Loose stools
Rectal hyposensitivity (↓ perception of rectal distension)
Patient fails to sense rectal fullness and voluntarily releases their external anal sphincter
Voluntary external anal sphincter contraction is no longer sufficient in closing the anus
Loose stool is more prone to escape through anal canal compared to solid stool
Continence mechanisms are impaired
Fecal Incontinence: The unintentional loss of solid or liquid stool
Skin Skin
Continence mechanisms are intact, but overwhelmed or ignored
infection Skin erythema
erosion
Inability to control what is widely considered a basic, fundamental bodily process
↑ caretaker burden Social stigma
↑ skin contact with acidic irritant (stool)
↑ rate of institutionalization, (e.g., admission into long-term care)
↓ confidence, sense of agency
↑ stress, anxiety
Skin inflammation
↓ social activity, work ↓ help-seeking ↓ treatment
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published May 2, 2020 on www.thecalgaryguide.com")
Placenta-Previa

• If patient presents with bleeding, a pelvic exam = risk of damaging placentaàmore bleeding
• Use transvaginal ultrasound to confirm location of placenta
Previous C/S Multiple gestation Maternal smoking
Placenta Previa
Presence of placental tissue that extends over the internal cervical os. (Pathogenesis unknown; preceding textboxes are risk factors only)
Previous placenta previa Increased maternal age Increased parity
Total placenta previa
Placenta completely covers the cervix
Partial placenta previa
Placenta covers cervix partially
Marginal placenta previa
Placenta near the edge of the cervix
Diagnosed early in pregnancy on routine abdominal ultrasound at 18-20 weeks Stretching of lower segment of uterus during 3rd trimester
OR
Alternate scenario:
One scenario:
This stretching elongates the space between the cervix and the placenta, relocating the stationary lower edge of the placenta away from the cervical os
Placenta previa resolves on its own
Reassuring: Placenta >2cm from cervical os on ultrasound
This stretching fails to move the placental away from the cervical os
Previa persists as uterus changes in preparation for labour:
Thinning of the lower segment of the uterus
Uterine contractions
Shearing forces to the placental attachment site
Painless bright red vaginal bleeding (90%)
↑ risk of clinically significant hemorrhage
Cervix becomes thinner (effaced) and opens (dilates)
Bleeding limits oxygen delivery to placenta, injuring placental tissue
Tissue injuryàActivates intracellular G-protein signalling pathways
Release of stored intracellular calcium àmyometrial contraction
Uterine contraction and bleeding (10%)
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published May 2, 2020 on www.thecalgaryguide.com")
Humoral-Immunity
 or circulate in the blood
Memory B cells proliferate and differentiate into plasma cells in response to re- exposure to antigen
↑ Rate and amplitude of secondary immune response on repeat exposure
Antigens (Ag) are produced from pathogens (bacteria, viruses, fungi, parasites) or the patient (via trauma, tumor, metabolism), & circulate in plasma, lymph, or other tissue
Clonal expansion
(proliferation of the activated B cells)
↑ WBCs (lymphocytosis) Autoimmune disease if B
cells recognize self-antigen
T cell-dependent Ag:
Ag-presenting cells (such as dendritic cells or macrophages) present Ag to CD4+ helper T cells and activate them. Activated helper-T cells then stimulate B cells
T cell-independent Ag:
Ags such as peptides, carbohydrates and lipids
may be directly recognized by B cells, triggering their activation
Complement:
Circulating serum complement proteins detect and bind Ag. Ags tagged with C3 complement fragment bind B cell co-receptor complex and enhance B cell activation.
Abbreviations:
Ab – Antibody
Ag – Antigen
Ig – Immunoglobulin WBC – White Blood Cell
Naïve B cells
Activated B cells
(in secondary lymphoid organs, such as the spleen or lymph nodes)
Plasma cells first produce IgM
Cytokines and T cells stimulate Ig class switching of B cells (changing the heavy chain constant regions of the Ig molecule)
Ig production switches from IgM to IgG, IgA, IgE, or IgD
IgG is the most common Ig in immune reactions. IgA concentrates at mucosa, IgE degranulates mast cells, IgD helps mature B cells.
Differentiation (into memory B cells or plasma cells)
Plasma cells produce antibodies, which contribute to immunity in 3 ways:
Opsonization: Abs coat pathogens, helping recognition by phagocytes
Neutralization: Abs bind to pathogen surface molecules that are needed to invade host cells, thereby neutralizing them
Activate Complement: Abs activate complement proteins via the classical pathway (see Complement Activation slide)
Clearance of pathogen by adaptive immune response
↑ Serum Ig
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published May 2, 2020 on www.thecalgaryguide.com")
Diabetic-Nephropathy

↑ Intrarenal angiotensin II
↑ Advanced glycation end products (AGE)
Shunting of glucose through non- glycolytic pathways (e.g. polyol)
Activation of Protein Kinase C (PKC) pathway
Excessive production and accumulation of glycolytic intermediates (e.g. sorbitol, hexosamine, succinate)
hyperglycemia
Succinate via GPR91
↑ Free radical production (oxidative stress)
Activation of cellular signalling, transcription factors and cytokines (e.g. TGF-β-Smad-MAPK, IGF-1, NF-κB)
↑NADPH oxidase activity
↑Blood volume ↑ Blood pressure ↑ Renal perfusion
Relative afferent arteriole dilatation, efferent arteriole constriction
Initial ↑ in glomerular filtration rate (GFR)
Podocyte loss/injury
Authors: Steven Chen Shannon Gui Yan Yu* Reviewers: Julia Heighton Ryan Brenneis Sophia Chou* * MD at time of publication
Initial glomerular hyperfiltration at time of diagnosis
↓ Production of matrix metallo- proteinases
Aberrant extracellular matrix (ECM) protein expression and accumulation
Sheer stress to glomeruli à pressure-induced damage
↑ Glomerular basement membrane permeability to proteins like albumin
↓ Extracellular matrix regulation
“Metabolic Pathway”
Mesangial matrix expansion
Kimmelstiel-
Wilson lesions (pink
hyaline nodules due to accumulation of damaged proteins)
Tubular fibrosis
Scarred glomeruli are less able to effectively filter blood
↓ in glomerular filtration rate (GFR)
Albuminuria
Usually occurs after ~5 years from time of diagnosis in T1DM; can occur at time of diagnosis in T2DM
Abbreviations
IGF: Insulin-like growth factor
MAPK: Mitogen-activated protein kinases NADPH: Nicotinamide adenine dinucleotide phosphate
NF-κB: Nuclear factor kappa-light-chain- enhancer of activated B cells
TGF-β: Transforming growth factor-β
Protein endocytosis into tubular cells causing inflammation
“Hemodynamic Pathway”
Diabetic Nephropathy
Overt diabetic nephropathy may take upwards of 15-25 years to develop
Note: The mechanisms presented here have been simplified. The cross- talk and signaling between the metabolic and hemodynamic factors do not manifest in a step-wise fashion, but rather occur in parallel.
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published May 3, 2020 on www.thecalgaryguide.com")
Infarctus du myocarde: Antécédents médicaux

Auteur: Yan Yu Traductrice: Olivia Genereux Réviseurs: Sean Spence Tristan Jones Nanette Alvarez* Marie Giroux* *MD au moment de publication
Sang du ventricule gauche reflue à l’oreille[e gauche et finalement s'accumule dans le système vasculaire pulmonaire
Haute pression du système vasculaire pulmonaire force fluide hors des capillaires et dans l'interstitium pulmonaire et les alvéoles
Inflammation myocardique locale
Médiateurs inflammatoires irritent les nerfs cardiaques (plexus cardiaque)
̄ Fonc8on systolique (myocarde nécro6que ne peut pas se contracter suffisamment)
Invasion généralisée des cytokines inflammatoires
Cytokines agissent les régulateurs de température hypothalamique
Fièvre légère
Irritation des nerfs sympathiques afférents (T1-T4)
Signal entre moelle épinière, a/n dermatomes T1-T4
Cerveau perçoit irritation des nerfs comme douleur des dermatomes T1-T4
Douleur écrasante, pression, oppression de la poitrine: Souvent rétrosternale, avec radiation à épaule, cou et l'intérieur des deux bras (D>G) (Apparition: au repos, crescendo)
IrritaNon des branches cardiaques du nerf vague
AcNvaNon réponse vasovagale
Fatigue, étourdissements, nausée, vomissement
↓ volume systolique (VS) ↓ debit cardiaque (DC)
Activité sympathique (pour essayer de maintenir DC)
Sueurs (diaphorèse)
Peau moite
VasoconstricNon généralisé
Vasoconstriction des artérioles de peaux
Peau froide
Interstitium
pulmonaire
mou ↓
compliance
pulmonaire
d
Fluide compresse les voies respiratoires, ↑ résistance au flux d’air
Abrévia8ons:
• a/n – au niveau
À Noter: Douleur myocardique ischémique peut présenter différemment entre les patients, mais les symptômes récurrents habituellement présentent les mêmes pour un patient donné.
Muscles respiratoires travaillent plus fort pour venNler les poumons
Essoufflement
(difficulté à respirer)
Légende:
Physiopathologie
Mécanisme
Signe/Symptôme/Résultats de Laboratoire
Complications
Publié Janvier 20, 2013 sur www.thecalgaryguide.com")
Marfan-Syndrome

Dural ectasia
(widening of the dural sac)
Diminished and disorganized dural elastic fibres
Abnormalities in connective tissues
Tear in the aortic intima (innermost layer of aorta)
Aortic dissection
Type A (tear in ascending aorta) > Type B (tear in descending aorta)
Back pain
Sensory and motor deficits
Ectopia lentis
(lens dislocation)
Development of lung bullae and blebs
Rupture of bullae/blebs
Pneumothorax
** Abnormal properties of lens + cornea
** Scoliosis
** Myopia
Tall stature Chest wall (pectus)
Inherited (autosomal dominant) or de novo mutation in FBN1 gene
Distortion of neural roots
Thinning of ciliary zonules of the eye
Weakness and rupture of alveolar tissue
Production of aberrant or reduced fibrillin-1
Formation of unstable microfibrils in extracellular matrix of connective tissues
**
inactivate TGF-β1
↑ production of matrix metalloproteinases
↑ cellular signaling cascades
↑ production of growth factors in the endocardium
Cell proliferation and apoptosis suppression in mitral valve leaflets
Change in valvular architecture
Mitral prolapse
Mitral regurgitation
↑ degradation of extracellular matrix
Thinning of the aortic media
Weakness of the aortic wall
Inability of fibrillin- 1 to sequester and
↑ TGF-β1 signalling
Abbreviations
• TGF-β: Transforming
growth factor beta (a cytokine)
Notes
**The underlying
mechanisms are unclear
Authors:
Tony Gu Reviewers: Amanda Nguyen Davis Maclean Yan Yu* Michelle Keir*
* MD at time of publication
Aortic root dilation
Aortic valve leaflets stretched outwards, unable to fully close
Aortic regurgitation
Aneurysmal dilation of the abdominal & thoracic aorta
Aortic rupture
Stroke
Blood enters and pressurizes a ‘false lumen’
Obstruction of aortic branches
End organ malperfusion
** deformities
** Joint hypermobility
Thumb sign: Thumb tip extends from palm of hand when thumb is folded into closed wrist
Wrist sign: thumb and fifth finger of the hand overlap when grasping opposite wrist
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published June 28, 2020 on www.thecalgaryguide.com")
GU-changes-in-pregnancy

Mechanism unclear, possibly 1) ↑ circulating anti- angiogenic factorsà ↑ permeability of glomerular basement membrane, or 2) ↑ plasma volumeà↓ oncotic pressure of plasma at the glomerulus
Proteinuria
> 300 mg/day, abnormal in pregnancy
↑ hCG level
Dilation of renal vasculature
↑ renal vascular & interstitial volume
↑ filtration surface area
Overwhelming load of glucose to proximal tubule
↑ urine output
↑ relaxin secreted by placenta
Mechanism not well understood
↓ osmotic threshold for ADH release & thirst
↑ ADH secretion & ↑ oral hydration
Incomplete reabsorption of glucose
Glucosuria
Encourage bacterial growth in the urine
Urinary tract infection (e.g. cystitis, pyelonephritis)
↑ serum progesterone
Uterine rotation as uterus enlarge due to presence of large bowel
Ureter compression (R > L)
Ureterovesical reflux (back-flow of urine into the ureters/kidneys)
Dilatation of ureters (R > L) (hydroureter) & renal pelvis (hydronephrosis)
Progesterone competes with aldosterone
↓ sodium reabsorption
↓ plasma sodium concentration
Hyponatremia of pregnancy* (pathological if < 130 mEq/L)
*Despite factors favoring sodium excretion, there is a net retention of sodium during pregnancy from adaptation of the renal tubules
↑ urinary stasis
↑ excretion of creatinine and urea in urine
↓ serum creatinine and urea
↓ ureteral toneà ↑potential for ureter dilation
Mechanism not well understood
↓ peristalsis of ureters
↑ urinary frequency (voiding > 7x/day)
↑ nocturia (voiding ≥ 2x/night)
↓ oncotic pressure of plasma intra-vascularly àwater leaves blood, enters interstitial tissue, and stays there in gravity-dependent regions of the body
Pedal +/- ankle edema
Author: Simonne Horwitz Reviewers: Claire Lothian, Crystal Liu, Ronald Cusano*, Candace O’Quinn*, Yan Yu* * MD at time of publication
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published June 28, 2020 on www.thecalgaryguide.com")
Retinal-Detachment-Pathogenesis
 e.g. macular degeneration
Formation of fibrous tissue bands in vitreous cavity
Bands contract and exert a tractional (pulling) force on the retina
Retina is pulled off the choroid layer at the back of the eye by tractional forces, without a retinal tear
Tractional Retinal Detachment (TRD):
Retina is pulled off the choroid layer at the back of the eye in the absence of retinal tears
Posterior Vitreous Detachment
For complete pathogenesis and clinical findings see: Calgary Guide – Posterior Vitreous Detachment: Pathogenesis and Clinical Findings
Note: Not every retinal tear leads to retinal detachment
Parts of the gel- like vitreous humour detach from the retina
↑ Age
Vitreous gel liquification
Vascular Damage Optic disc Anomalies Degenerative Conditions
During rotational eye movement, the vitreous humour moves within the vitreous cavity
Strong tractional forces transmitted to retina through remaining attachments
Retinal Tear: Physical defect in the retina associated with vitreous traction
Defect held open by vitreoretinal traction
Defect in retina allows vitreous fluid to gain access into sub retinal space
Tumour/ Malignancy
Idiopathic
Rhegmatogenous Retinal Detachment (RRD): Accumulation of sub retinal fluid due to a tear in the retina allowing liquid vitreous gel to get underneath the retina
Separation of the retina from choroid layer at the back of the eye
Inflammatory/Infectious Conditions
↑ Permeability of choroid or retinal blood vessels
Fluid leaks out of retinal/choroid blood vessels
Accumulation of fluid beneath retina and without retinal tear or vitreous traction
Exudative Retinal Detachment (ERD):
Fluid accumulation in subretinal space in the absence or tears or traction from the vitreous
Retinal Detachment:
Sight threatening condition, considered ocular emergency
See Calgary Guide slide: Retinal Detachment: Clinical findings
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published July 11, 2020 on www.thecalgaryguide.com")
Retinal-Detachment-Clinical-Findings

Concave elevation (in tractional retinal detachment)
Authors: Natalie Arnold, Davis Maclean Reviewers: David Sia*, Yan Yu* * MD at time of publication
Rhegmatogenous retinal detachment starts from periphery = curtain pattern of vision
Relative afferent pupillary defect: Affected pupil dilates when light moved from unaffected eye to affected eye
Lower intraocular pressure Vitreous hemorrhage
Detachment: Pathogenesis
Elevation of retina on fundus examination
Retinal Detachment: Separation of the retina from choroid layer at the back of the eye
If involving retinal tears and/or posterior vitreous detachments:
Retinal Tear: only present in Rhegmatogenous Retinal Detachments
Posterior Vitreous Detachment: usually seen in Rhegmatogenous Retinal Detachments (See Calgary Guide - Posterior vitreous detachment)
Separation of retina from the underlying choroid
As the choroid contains blood vessels feeding the retina, retinal cells are deprived of blood and become ischemic
Death of photoreceptors
Light detected by fewer melanopsin retinal ganglion cell photoreceptors
Loss of function in detached retina
Smaller percentage of action potentials travel to dorsal midbrain
Vision Loss
Retinal pigment epithelium cell proliferation and formation of contractile membrane (scar tissue) in vitreous
Proliferative vitreoretinopathy
Communication between vitreous and exposed choroid
Physical defect in the retina involves tearing of blood vessels within the retina
↑ outflow of fluid by active pumping through Retinal Pigment Epithelium (small layer of cells between the sensory retina and choroid)
Blood released into the neurosensory retina blocks light’s pathway to retina
Blood directly block light from stimulating photoreceptors on retina
Retinal pigment epithelial cells released
Shafer’s sign: (pigment in anterior vitreous seen with slit lamp)
Released cells directly block light from stimulating photoreceptors on retina
Floaters: Small circles, dots or lines in field of vision
Aggregation of Collagen II fibers and blood released from optic disc during posterior vitreous detachment
Tractional forces from vitreous movement are transmitted to areas where vitreous remains attached to the retina
Collagen fibers cast shadow on the retina
Fibers directly block light from stimulating photoreceptors on retina
Vitreoretinal traction causes physical Photopsia stimulation of photoreceptors in the retina (Flashes)
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published July 11, 2020 on www.thecalgaryguide.com")
preterm-labour-pathogenesis-maternal-complications

Authors: Skye Russell Reviewers: Danielle Chang, Crystal Liu, Yan Yu*, Nicholas Papalia*
*MD at time of publication
Digest and weaken amniotic membrane
Preterm Premature Rupture of Membranes
↑ myometrial sensitivity to oxytocin
Uterine contractions Preterm Labour
Uterine contractions and cervical change occurring <37wk gestational age ↑ Risk of future preterm labour and preterm birth
Abbreviations:
• LEEP – Loop electrosurgical
excision procedure
• ACTH – adrenocorticotropic
hormone
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published July 12, 2020 on www.thecalgaryguide.com")
Hereditary Hemorrhagic Telangiectasia (Osler-Weber-Rendu disease)
:
Pathogenesis and Clinical Findings
Inherited or de novo mutation in the ACVRL1, ENG, or Smad4 genes
Abnormal signalling within the transforming growth factor ß (TGF-ß) pathway
Unclear mechanismsàInability of vascular mural cells to stabilize and remodel newly formed blood vessels
Excessive proliferation of endothelial cells and ensuing overgrowth of blood vessels
Authors: Tony Gu Reviewers: Brian Rankin Yan Yu* Laurie Parsons* * MD at time of publication
Formation of friable telangiectasias
(small dilated vessels apparent near the surface of skin or mucous membranes)
Formation of Arteriovenous malformations (AVMs):
Direct connection between arteries and veins without intervening capillary bed
Nasal telangiectasias
Epistaxis (nosebleeds)
Gastrointestinal telangiectasias
Gastrointestinal bleeding
Mucocutaneous telangiectasias
Cerebral AVMs
Hepatic AVMs
Left to right shunting of blood
Heart works harder to perfuse tissues
Heart failure
Pulmonary AVMs
Rupture
High flow left to right shunting of blood (the steal effect)
Cerebral ischemia
No oxygenation at capillaries
Hypoxemia
↑ erythropoietin production
Secondary polycythemia
No filtering from capillaries
Hemorrhage, shock, death
Venous emboli enter arteries (paradoxical embolism)
Stroke
Venous bacteria enter arteries
Cerebral abscess
Iron deficiency anemia
↓ serum iron is associated with ↑ coagulation factor VIII levels (mechanism unclear)
Venous thromboembolism
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published July 28, 2020 on www.thecalgaryguide.com")
cg-logo

splash-header-v2

splash-header-v3

splash-header-v4

splash-header-ipad

acute-pancreatitis-complications
:
Interstitial edematous pancreatitis
Local accumulation of fluid in the pancreas
<2 weeks after onset
Acute peripancreatic fluid collection (not encapsulated)
Walled off by fibrous & granulation tissue
>2 weeks after onset
Pancreatic pseudocyst
(completely encapsulated)
Peritoneal irritation à pain
Large cyst can (very rarely) compress surrounding bowel
Acute Pancreatitis
Inflammatory cytokines are released from damaged pancreas
If recurrentàchronic pancreatitis (see relevant slide)
Inflammation damages pancreatic exocrine
cellsàInappropriate release of pancreatic enzymes into surrounding tissue & vasculature àdigesting pancreatic parenchyma
Authors: Nissi Wei, *Yan Yu Reviewers: Dean Percy, Miles Mannas, Varun Suresh, Brandon Hisey, *Kerri Novak, *Sylvain Coderre * MD at time of publication
complete resolution (most cases)
Necrotic tissue is vulnerable to
infection (esp. Gram neg GI bacteria)
inflammation & necrosis activate cytokine cascade
Severe, necrosis (15%): Necrotizing pancreatitis
Local infection
Severe pancreatic inflammation shifts body fluid into retroperitoneal spaceàintravascular volume depletion
Systemic Inflammatory Response Syndrome (SIRS) (see relevant slide)
Organ failure (may be sole feature on presentation)
Stagnant fluid can more easily become infected
Infection spreads to bloodstream
Cardiac failure Hypovolemic shock Renal failure
Local accumulation of fluid & necrosis in the pancreas
< 4 weeks after onset:
Acute necrotic collection (not encapsulated)
Walled off by fibrous & granulation tissue
> 4 weeks after onset
walled-off necrosis
(completely encapsulated)
When treated with excess fluid resuscitation:
Intra- abdominal hypertension
Respiratory failure (ARDS)
Disseminated intravascular coagulation (DIC)
Bowel obstruction Gastric outlet (see relevant slide) obstruction
Infected pancreatic necrosis
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published September 20, 2016, updated September 7, 2020 on www.thecalgaryguide.com")
Slide-authoring-process-FINAL3

Pulmonary Hypertension

↓ contractility and/or diastolic relaxation
↓ left ventricle cardiac output Backup of blood in left ventricle and atrium Backup of blood in pulmonary vasculature
↑ pulmonary capillary wedge pressure– estimate of blood pressure in left atrium
Chronic Anemia
↓ plasma hemoglobin content
↓ oxygen carrying capacity per unit blood
Compensatory ↑ in heart rate to maintain tissue oxygen supply
↑ cardiac output
Lung disease (chronic obstructive pulmonary disease)
Tissue breakdown and ↓ lung elasticity
Chronic thromboembolism
Pulmonary vessel disease (pulmonary arterial hypertension, scleroderma)
Vascular obstruction/fibrosis
↓ lungs’ ventilation ability
↓ surface areaà↓ gas exchange
Lung vasculature undergo reflexive, localized vaso- constriction, to shunt blood to better ventilated areas
Chronic hypoxemia
↓ local alveolar partial pressure of oxygen
↓ blood vessel compliance
↑ Pulmonary vascular resistance (PVR)
Impaired gas exchange across thickened vessel walls
↓ blood partial pressure of O2 and ↑ partial pressure of CO2
Insufficient O2 provision & CO2 removal from tissues
Reflexive mechanisms trigger harder & faster breathing to compensate
Vascular fibrosis due to chronically increased pressures
↓ circulation of blood to left heart and ↓ filling of left ventricle
↓ left ventricle cardiac output
Elevated blood pressure in the lung arteries Pulmonary Hypertension
↑ right-ventricle afterload (pressure against which the heart contracts to eject blood)
↓ right ventricle cardiac output
↑ residual volume in right heart after cardiac contraction
Backup of blood in systemic circulation ↑ blood volume in venous system
Myocardial hypertrophy develops over time (eccentric & concentric)
↑ tissue volume
↑ myocardial oxygen demand
Myocardial ischemia (supply/demand mismatch)
↑ risk of chest pain in times of ↑ oxygen demand
Peripheral edema
Formation of aberrant conduction pathways and ectopic electrical foci
Dysrhythmias
Palpitations
↓ tissue perfusion
Fatigue
Dyspnea
↓ brain perfusion
Syncope
Authors: Grant E. MacKinnon Davis Maclean Hannah Yaphe Reviewers: Yan Yu* Jason Weatherald* * MD at time of publication
↑ volume and blood pressure in capillaries Fluid pushed from vessels into interstitial space of tissues
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published September 8, 2020 on www.thecalgaryguide.com")
insuffisance-du-coeur-gauche-la-pathogenese
 précharge cardiaque (volume télédiastolique)
• 2) postcharge cardiaque (résistance à l’éjection du ventricule gauche)
• 3) contractilité (la force de contraction des myocytes du ventricule gauche)
• Fraction d’éjection= volume d’éjection systolique/volume télédiastolique
• Le % du volume télédiastolique du ventricule gauche éjecté lors de la systole • Fraction d’éjection normale = 55-75%
Dégénération des myocytes et presence de fibrose interstitielle
Surcharge volumique chronique à causant une dilatation et hypertrophie du ventricule gauche
Myocytes nécrotiques et non fonctionnels
̄ de la contractilité du ventricule gauche
Dilatation, hypertrophie et fibrose du ventricule gauche
Dysfonction systolique:
Le ventricule gauche ne peut se comprimer/ou se vider, aussi ̄ de la fraction d’éjection
Insuffisance cardiaque
gauche
̄ Volume d’éjection systolique, reduction du débit cardiaque: le sang n'est donc pas pompé suffisamment au niveau de la circulation systémique pour répondre aux besoins métaboliques corporels. Le sang est donc reflué au niveau des poumons, ce qui provoque une congestion pulmonaire. Il en résulte alors des symptômes et des signes de diminution du débit cardiaque (insuffisance cardiaque antérograde) et de congestion (insuffisance cardiaque rétrograde).
(voir les diapositives correspondantes)
↑ résistance à l'éjection du ventricule gauche (↑ la postcharge)
̄ relaxation du ventricule gauche et ̄ de l’entrée du sang lors de la diastole
↑ de la rigidité de la paroi ventriculaire gauche
̄ volume télédiastolique du ventricule gauche
Dysfonction diastolique:
Le ventricule gauche ne peut alors pas se dilater/se remplir adéquatement même si la fraction d'éjection est préservée
Surcharge chronique de la pression du ventricule gauche
Note: L'insuffisance cardiaque gauche est la manifestation la plus importante (stade ultime) de nombreuses pathologies cardiaques.
Emma Hofland-Burry Jean-François Lemay*
Auteurs:
Yan Yu Editeurs: Sean Spence Jason Baserman Nanette Alvarez* Jean-François Lemay* * MD au moment de la publication
Traductrice/Traducteur:
Voir les diapositives correspondantes pour une description plus détaillée de chacun de ces diagnostics
Légende:
Pathophysiologie
Mécanisme
Signe/Symptôme/Trouver laboratoire
Complications
Publié le 10 Janvier 2013 à www.thecalgaryguide.com")
insuffisance-cardiaque-gauche-resultats-de-lanamnese

↑ de la pression hydrostatique veineuses forçant un écoulement liquidien dans les tissus interstitiels, surtout au niveau des zones dépendantes de la gravité
Oedème de la cheville
Insuffisance du ventricule gauche
Débit cardiaque réduit (“insuffisance cardique antérograde”):
le sang n'est pas suffisamment pompé au niveau de la circulation systémique pour répondre aux besoins métaboliques corporels
Congestion pulmonaire (“insuffisance cardiaque rétrograde”):
Le sang reflue du ventricule gauche vers l'oreillette gauche causant ainsi une accumulation dans le système pulmonaire vasculaire
Activation sympathique (pour essayer d' ↑ le débit cardiaque)
Tachycardie, palpitations et diaphorèse
Perfusion tissulaire limitée (i.e. au niveau musculaire)
Faiblesse et fatigue
Une pression artérielle pulmonaire veineuse élevée force l’evacuation des fluides des capillaires vers l'interstitium et les alvéoles pulmonaires
La congestion pulmonaire reflue le sang vers le cœur droit, et ultimement vers la circulation veineuse systémique
Ce fluide est dépendant de la gravité ; il s'installe dans les lobes pulmonaires inférieurs.
Progressivement, les vaisseaux sanguins des lobes pulmonaires supérieurs (qui sont alors mieux ventilés) se dilatent par réflexe, ce qui ↑ les échanges gazeux (")
insuffisance-cardiaque-gauche-les-resultats-de-lexamen-physique

Dysfonction systolique
̄ Perfusion tissulaire (au niveau cerebral, renal etc.)
Vasoconstriction périphérique àdetournement limité du volume d’ejection systolique vers les organes principaux
Extrémités froides, cyanose périphérique
̄ Du débit urinaire
̄ De l’état de conscience
Hypoxie tissulaire la respiration anaérobique, production d’acide lactique (acidose)
Les centres respiratoires tentent de compenser
Expulsion des fluides des capillaires vers les alvéolesà oedème pulmonaire transudatif
La congestion vasculaire et les alvéoles oedématiées compriment les voies respiratoires, provoquant une turbulence du flux d'air
Un flux d'air turbulent est entendu lors de l'auscultation
Respiration sifflante (wheezing)
Flux turbulent dans le ventricule gauche distendu au début de la diastole
B3
Choc
de la pointe diffuse
̄ Du flux sanguin artériel pendant la systole
̄ Tension artérielle en systole
De la
tension artérielle lors de la diastole
̄ Pouls cardiaque
Fermeture des valves pulmonaires avec une force supérieure à la normale
B2 pulmonaire
̄ De l’oxygenation sanguine
Le transsudat obstrue les petites voies aériennes et alvéolaires
Si le ventricule gauche est en surcharge de pression ou si hypertrophié (i.e. sténose aortique, hypertension sévère)
Choc de la pointe soutenue
De l'activité du système sympathique (pour tenter de rétablir le débit cardiaque)
Pendant l'inspiration, de petites zones de compartiments aériennes s'ouvrent
Crépitements pulmonaires
(ils sont généralement bilatéraux et surtout au niveau de la base pulmonaire)
Auteur: Yan Yu
Editeurs: Sean Spence, Jason Baserman, Nanette Alvarez*
Traductrice/Traducteur:
Emma Hofland-Burry, Jean-François Lemay* *MD au moment de la publication
Activité des glandes sudoripares
Diaphorèse
Activité cardiaque
Tachycardie
Fréquence respiratoire
Tachypnée
Remarque: l'insuffisance cardiaque gauche étant la cause principale de l'insuffisance cardiaque droite, elle peut également se présenter par des signes d'insuffisance cardiaque droite. Les autres caractéristiques de l'insuffisance cardiaque gauche dépendent de la cause sous-jacente.
Si la pression vasculaire artérielle pulmonaire est chroniquement élevéeàporvequera un surmenage prolongé de la function du ventricule droit:
Insuffisance cardiaque droite: Le patient présente des conséquences directes de sa congestion veineuse systémique: œdème
périphérique (i.e.cheville), épanchements pleuraux, congestion hépatique/ascites (s.v.p. voir la diapositive correspondante)
Publié le 10 Janvier 2013 à www.thecalgaryguide.com")
Essential Tremor
: Pathogenesis and clinical findings Most common tremor in adults
Authors: Davis Maclean Evan Allarie Reviewers: Hannah Yaphe Gary Klein* Yan Yu* * MD at time of publication
↑ Age
Unknown environmental factors
Oscillatory network hypothesis: diffuse
oscillating neuronal activity in thalamic, inferior olivary and cerebellar networks
↑ activity in the cerebellothalamocortical circuit
Alternating contractions of antagonistic muscles
Involuntary rhythmic/oscillatory
movements with generally constant frequency and variable amplitude
Indeterminate cerebellar and locus coeruleus (pons) pathology
3 non-mutually exclusive mechanisms proposed
Polygenic genetic predisposition (up to 70% have family history)
Approximately 50% of cases are associated with an autosomal dominant inheritance pattern – specific gene has yet to be identified
GABAergic hypothesis: ↓ GABA (Gamma amino
butyric acid – Inhibitory neurotransmitter) activity in cerebellar and locus coeruleus circuits
Neurodegeneration hypothesis: Possible cerebellar degeneration (this hypothesis is currently controversial and requires more evidence)
Potential neuro- degenerative process
May lead to difficulty with gait
and mild cognitive impairment seen in a minority of ET patients
Alcohol binds to GABA receptors in the cerebellum
↑ neuronal inhibition
Bilateral and symmetric tremor
↓ activity in overactive (cerebellar) tremor circuits
Alcohol consumption ↓ tremor in majority of patients
Issues with writing, cooking, eating, occupational duties or other instrumental activities of daily living
Pathologic changes in the brain affect both sides of the motor system
Unknown mechanism
Typically involves the hands and arms but may present or progress to involvement of head, voice, legs, face, jaw and trunk
Embarrassment or other social consequences Slowly progressive tremor of 6-12Hz frequency
Tremor is persistent and visible
Postural tremor (evident during sustained anti-gravity postures), and/or a kinetic tremor (evident during voluntary movement)
Tremor becomes apparent when holding outstretched arms against gravity
Tremor typically increases at the end of goal directed movement
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published September 19, 2020 on www.thecalgaryguide.com")
Cellulitis
 and entry of pathogen
Risk Factors: Immunocompromised Host: -Diabetes mellitus+ -Lymphedema -Malnourishment
-Older patient+
-Obesity+
-Peripheral vascular disease General Infection Risk: -History of cellulitis+ +highest risk factors
Risk Factors for MRSA Cellulitis: Increased exposure to MRSA: -Contact sports
-Crowded living conditions -Health care workers -Indigenous descent
-Sharing towels, equipment
Increased susceptibility:
-Immunodeficiency -Young age
Direct inoculation (e.g. trauma) Organism virulence overwhelms host defense mechanisms (related to risk factors)
Cellulitis: A bacterial infection in which pathogens penetrate deep dermis and/or subcutaneous fat
Cytokines activate immune response
Accumulation of pus (bacteria, white blood cells, dead skin)
Abscess formation
Infection spreads to nearby lymph nodes
Lymphadenitis
Infection spreads through lymph vessels
Ascending lymphangitis
Local inflammatory response in skin
Pain Warmth Edema Erythema (redness)
with indistinct margins
Vesicles and bullae
Organisms penetrate blood vessels
Bacteremia (presence of bacteria in blood)
Systemic inflammation
Distant spread to bone
Osteomyelitis
Distant spread to endocardium (inner lining of heart chambers and valves)
Endocarditis
Fever Malaise
Chills
Sepsis
(rarely)
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published September 27, 2020 on www.thecalgaryguide.com")
pertussis-pathogenesis-clinical-findings-and-complications
 which damage mucosal cells
Pertussis toxin produces cyclic AMP (cAMP) and disrupts normal intracellular signalling, impairing the immune response initially
Pertussis (“Whooping Cough”) Respiratory syndrome consisting of severe fits of paroxysmal coughing and stridor
Nasopharyngeal swab produces positive culture and/or positive PCR result (either is diagnostic)
Initial immune dampening allows the bacteria to take hold and begin replicating. During this “incubation period”, the bacteria has not yet replicated to the point of causing symptoms.
1. Catarrhal Stage (5-10 days) After a few days, continued
damage to nasopharynx epithelial cells stimulates the immune system to ↑ its response once again
2. Paroxysmal Stage (1-2 weeks)
Tracheal cytotoxin released by B. pertussis impairs normal cilia function and ciliary beating in the trachea
3. Convalescent stage (2 weeks - months) Immune defenses successfully
eliminate the majority of B. pertussis from the respiratory tract
↑ mucus production from goblet cells of the respiratory epithelium
Mucus blocks airway, prevents air entry
Collapsed lung
Rarely, areas of chest or abdominal wall are weakened, allowing contents to bulge out
Hernia
↑ proinflammatory cytokine production
Mild fever
Cold-like symptoms
Mild dry cough, runny nose, sneezing, nasal congestion
↓ fluid clearance from the respiratory tract
Fluid in the trachea narrows tracheal diameter
“Whooping” cough
Severe, rapid and sequential coughing fits, followed by characteristic “whooping” sound on inspiration due to a stridor from a narrower trachea
Fluid build up in the lungs
Environment more susceptible to co-infection
Other bacteria colonize the lungs
Pneumonia
Paroxysmal coughing fits ↓ in frequency and number
Cough may sound louder (mechanism unknown)
but overall symptoms ↓
Some B. pertussis still remain
Residual cough flares
This stage may be prolonged in unvaccinated individuals who eliminate the bacterium more slowly
Intense cough can break ribsàsharp rib ends puncture lungàair leaks out
↑ pressure on bladder
If weak urethral sphincters:
Urinary incontinence
Cough ↑ intra- abdominal pressure
If dripping mucus triggers gag reflex while a cough is contracting abdominal muscles:
Vomiting
Coughing fits disrupt regular inspiration and ↓ oxygenation
Hypoxia
If hypoxia is profound enough to affect brain
Seizures
Abdominal muscles tire from coughing, and coughing fits make it difficult to sleep
Extreme fatigue
Rarely, violent coughing causes trauma to head
Intracranial hemorrhage
Vertebral or carotid dissection
Cerebral ischemia Coma or death
Notes:
• B. pertussis is a Gram- negative strict aerobe
• An effective vaccine exists to prevent infection by B. pertussis
• Pertussis most commonly infects children <18 months prior to completion of scheduled vaccination series, or adolescents with ↓ immunity
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published October 4, 2020 on www.thecalgaryguide.com
Include a sentence!
When sending in the final draft of a slide, please include in the email a sentence that describes the slide that you've produced in detail. This will help us boost the relevance of the slide with search engines.
E.g., Disease X presents with symptom Y due to pathophysiology Z
Pertussis presents with fits of severe paroxysmal coughing due to impaired mucociliary clearance.")
Creutzfeldt-Jakob-Disease
: Autosomal dominant inheritance of pathologic prion protein (PRNP) genes
Sporadic (85%)
Pathologic prion protein (PrPSc) form spontaneously from normal prion protein isoforms (PrPc)
Acquired (<1%)
Variant: consuming bovine spongiform encephalopathy (BSE) infected beef (mad cow disease)
Iatrogenic: related to medical intervention (e.g instruments contaminated with PrPSc)
Note: The normal prion protein isoform (PrPc) is expressed predominantly in neurons. It is not intrinsically pathological.
Presence of PrPSc kickstarts an “autocatalytic”
process by which existing PrPSc converts more normal prion protein (PrPc) into pathological protein.
Pathological prion proteins (PrPSc) enter host
Astrogliosis: ↑ astrocyte cells infiltrate and occupy the space of lost neurons
Vacuolation or spongiform changes: clusters of small vacuoles in synaptically dense cortical areas
Pathologic prion proteins (PrPSc) are insoluble and aggregate within neurons Mechanism unknown
Neuronal loss
Creutzfeldt-Jakob Disease
Fatal infectious brain disorder causing neuronal death
Diffuse brain atrophy
Exact mechanism unknown
Frontal lobe atrophy
The frontal lobes are responsible for executive function and personality
Occipital lobe atrophy
The occipital lobes are responsible for interpreting vision
Basal ganglia atrophy
The basal
ganglia is responsible for sequencing voluntary movements
Loss of inhibitory cortical neurons
Hyper-excitable cortical neurons
Myoclonus: quick involuntary muscle jerks
Cerebellar atrophy
The cerebellum is responsible for coordination and fine correction of movements
Gait Slurred
Extensive neuronal loss in the brainstem reticular activating system
Coma
Impaired control of respiratory muscles
Pneumonia and respiratory failure
~ 75% mortality within 1 year
Rapidly progressive dementia
Anxiety, depression
Visual hallucinations
Double vision
Bradykinesia:
Tremor
Rigidity: ↑ muscle tone
ataxia
speech Incoordination
Personality changes
Irritability
slow movements
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published October 4, 2020 on www.thecalgaryguide.com")
Epidural-Hematoma-Findings-on-CT
 occurs beneath
the pterion (location where frontal, sphenoidal, temporal and parietal bone meet)
The expansion of the hematoma (collection of blood) is limited by cranial sutures (firm attachments of the dura)
Multiple bony connections
decrease stability in this region
Pterion region of the skull is
the thinnest portion of the skull
Hematoma has limited range to expand in anterior posterior direction
High rate of fractures in this area from skull trauma
The middle meningeal artery traverses under the temporal bone and pterion, and is susceptible to injury from fractures in this area
Hematoma near or under temporal bone
Hematoma must expand medially, or
inwards, towards brain parenchyma
Biconvex hyperintensity
Can lead to brain herniation (See Calgary Guide: Brain Herniation Types and Clinical Findings)
Estimating Size of Hematoma (A x B x C)/ 2
• A= Maximum hemorrhage diameter at largest point of hematoma
• B= Measurement of diameter 900 to Measurement A
• C= The number of CT slices where hematoma is visible
multiplied by thickness of CT slices
CT or MRI?
• CT is rapidly available in most North American emergency departments and is the best imaging modality for detecting acute intracranial hemorrhage.
Increased pressure can cause shift of brain structures
Midline structures shifted away from hematoma (Midline shift)
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published October 14, 2020 on www.thecalgaryguide.com")
Pathogenesis-of-Female-Infertility
 from hypothalamus
↓ release of Luteinizing hormone (LH) & Follicle stimulating hormone (FSH) by pituitary
↓ release of estrogen by ovaries
Anovulation (oocyte is not released)
Fewer follicles available to ovulate
* Causes of Hyperprolactinemia include: prolactinoma (prolactin-producing tumor), hypothalamic infiltrate or mass, chest wall irritation, hypothyroidism, renal or liver disease (↓ prolactin clearance), dopamine antagonists that ↑ prolactin secretion (antipsychotics, anti- depressants, anti-emetics)
Polycystic ovary syndrome (see PCOS: Pathogenesis and Clinical findings)
↑ androgen production & ↑ estrogen earlier in the menstrual cycle
↓ FSHà↓ follicle growth
↑ rate of follicle depletion
Oocyte not available every month for fertilization
Premature ovarian insufficiency due to unexplained causes, chemotherapy, radiation, autoimmune ovarian destruction, Turner’s & Fragile X Syndromes
Damage in germ cells that accumulates over a woman’s lifetime
Age-related changes in quality of granulosa cells surrounding oocyte
Genetic damage accumulates, such as ↑ rates of meiotic nondisjunction (failure of chromosomes to separate during gamete cell division)
Tubal occlusion or ↓ transport of oocyte tubal cilia dysfunction through fallopian tube
↓ quality of oocytes
Normal transport of oocyte & sperm through fallopian tube is impaired
↓ facilitation of sperm transportation
Inhibits normal zygote implantation
Chlamydial or gonorrhoeal pathogens
Previous tubal surgery or ectopic pregnancy surgery tissue removal ↓ transport of oocyte through fallopian tube
Female Infertility
Previous abdominal infection or surgery Endometriosis
Congenital malformations or trauma / surgery to cervix
Uterine leiomyomata (benign smooth muscle monoclonal tumor) or polyp
Intrauterine procedures
Pelvic adhesions (scar-like tissue that tether together abdominal organs) may distort the shape and normal anatomy of the fallopian tube
Ectopic endometrial cells implant & Local inflammatory response grow along pathway of egg/sperm further ↓ egg/sperm mobility
Inability of cervix to produce normal mucus, and/or sperm physically unable to enter the cervix
Submucosal or intracavitary component disrupts uterine lining
Trauma to basalis layer of endometrium
Intrauterine scarring or synechiae (adhesions)
↓ vascularization & endometrial regrowth
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published October 25, 2020 on www.thecalgaryguide.com")
Anesthetic-Considerations-Aortic-Stenosis
 for Patients with Aortic Stenosis Undergoing Non-Cardiac Surgery
CRRAP Goals:
Contractility, Rate, Rhythm, Afterload, Preload
Pathophysiology Driving Anesthetic Management Hemodynamic Anesthetic Intervention (CRRAP) Goals
Author:
Ryan Brenneis Reviewers: Stephen Chrusch Hannah Yaphe Yan Yu*
Karl Darcus*
* MD at time of publication
Aortic stenosis = Narrow aortic valve opening
Notes:
-Cardiac Output = Heart Rate x Stroke Volume
-Stroke volume has 3 determinates:
1. Contractility 2. Afterload
3. Preload
If the patient’s heart cannot ↑ contractility to maintain cardiac output...
↑ resistance to forward blood flow àheart must ↑ its contractility (↑ the forcefulness of its contractions) to overcome this resistance
Applying ↑ force over time causes left ventricle to undergo concentric hypertrophy
Contractility deteriorates over time
Heart rate must compensate for maintaining cardiac output
Coronary Perfusion Pressure = Diastolic BP (DBP) – Left Ventricular End Diastolic Pressure (LVEDP)
↑ cardiac muscle massà↑ myocardial metabolism and oxygen demand
↑ left ventricular wall stiffnessà↓ LV filling while relaxed (diastolic dysfunction)
Intraoperative ↓ in contractility compromises cardiac output
Bradycardia ↓ cardiac output
Tachycardia ↓ filling time of left ventricle (↓ preload)
Coronary perfusion occurs during diastole
Coronaries require a high DBP to maintain perfusion
Tachycardia ↓ perfusion time
Hypotension ↓ coronary perfusion pressure
Possible myocardial ischemiaà ↓ blood pumped into vessels
40% of LV preload supplied from atrial kick
Loss of atrial kick with arrhythmias à↓ cardiac output
Note: Aortic stenosis severity (see slide on aortic stenosis) and the type/risk of surgery guide the hemodynamic consequences and need for intervention
Adequate intravascular volume required to passively fill stiff ventricle
Contractility
↓ use of negative inotropic Maintain drugs, e.g. calcium channel
contractility blockers (“Inotrope”: drug that alters heart’s contractility)
Rate
Keep heart rate above 60 bpm
Keep heart rate below 80 bpm
Consider transcutaneous pacing, anticholinergics, & sympathetic agonists
↑ anesthetic depth, consider beta blocker (e.g. Esmolol)
Afterload
Maintain a Mean Arterial Pressure >70mmHg
Consider sympathomimetic drugs to treat hypotension
Monitor blood pressure closely via arterial line
Consider increasing anesthetic depth for severe hypertension
Rhythm
Maintain Sinus Rhythm
Consider presurgical placement of defibrillator pads & crash cart
Amiodarone ready & available during operation, to terminate any arrhythmias
Preload
Maintain Euvolemia
Possible use of transesophageal echo to monitor preload
Ensure adequate venous access- consider central venous catheter and large bore IV’s
Legend:
Pathophysiology
Mechanism
Goal
Anesthetic Intervention
Published October 25, 2020 on www.thecalgaryguide.com")
Tumour-Lysis-Syndrome

Though rare, aggressive tumours can spontaneously lyse without treatment
Intracellular potassium released into bloodstream
Intracellular phosphate released
Hyperphosphatemia
↑ serum phosphate
Intracellular lactate dehydrogenase (LDH) released
↑ serum LDH
Intracellular nucleic acids released
Nucleic acids metabolized to uric acid
Hyperuricemia
↑ serum uric acid
Hyperkalemia
↑ serum potassium
(see Hyperkalemia: clinical findings)
Uric acid (a crystallizing substance) ↑ precipitation of calcium phosphate
↑ filtration of poorly soluble uric acid into acidic environment of renal tubules
Serum phosphate binds serum calcium, forming solid calcium phosphate precipitate crystals
High levels of calcium phosphate ↑ uric acid precipitation
Uric acid precipitates as crystals and deposits in kidney tubules and collecting ducts
Tubular injury and/or intraluminal obstruction
Endothelial dysfunction in renal vasculature
Renal inflammation, vasoconstriction, and
impaired renal vascular autoregulation
Decreased renal filtration
Acute Kidney Injury
(see Acute Kidney Injury Overview)
Crystal deposition in the heart
Depletion of soluble calcium
Hypocalcemia
Calcium phosphate crystals deposit in kidneys
Main mechanism of Acute Kidney Injury
Cardiac Arrythmias
↓ serum calcium
(see Hypocalcemia: clinical findings)
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published October 25, 2020 on www.thecalgaryguide.com")
Small-Bowel-Obstruction-findings-on-X-Ray
 proximal to the obstruction
Gas rises above the fluid
If exclusively or mostly accumulation of fluid (and not gas) occurs
Any small amounts of gas/air present (not enough to create an
air-fluid level) will rise and become trapped in valvulae conniventes (small bowel folds)
Small bowel loops are anatomically
central compared to large bowel
Bowel contents physically push on the bowel walls, dilating them
Dilated bowel loops are “central” in location on the x-ray
Dilated bowel loops (>3cm)
Valvulae Conniventes Visible: Anatomical folds of the small bowel that becomes more
apparent when small bowel is distended & allows differentiation from large bowel
Air-fluid level (on erect/upright study)
- Dark area above level = Gas/Air
- Bright/white area below level = Fluid
‘Gasless’ abdomen (not seen here): Refers to the lack of gas/air (dark on X-ray) in the bowel loops - only fluid (bright/white on X- ray) is seen in bowel loops
String of pearls sign (not seen here):
Small gas bubbles seen arranged in a “string of pearls” pattern instead of a large air fluid level
Image Credit: Alberta Health Services Repository
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published October 25, 2020 on www.thecalgaryguide.com")
Calcium-Oxalate-Kidney-Stones
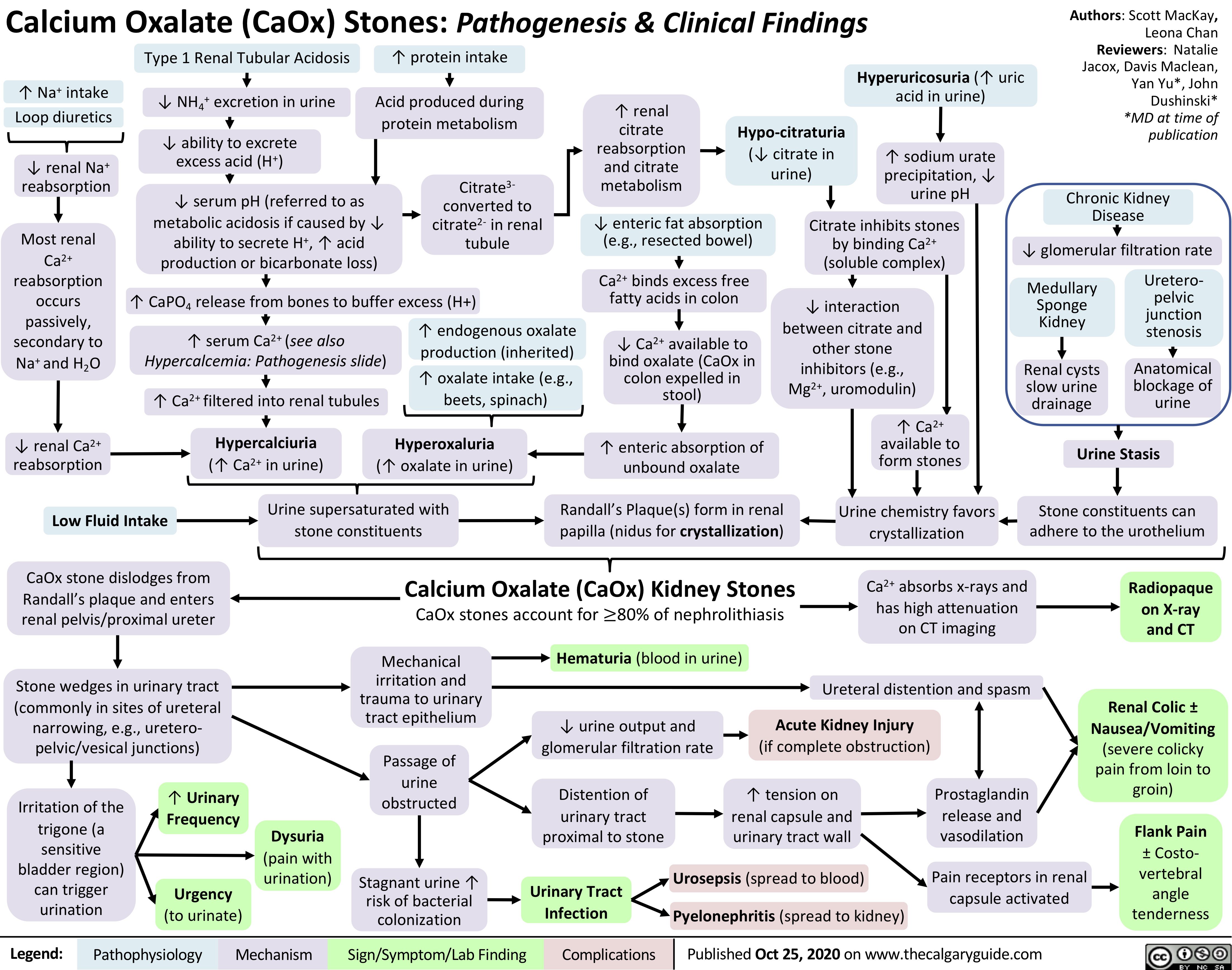
Multiple-Sclerosis-on-Brain-MRI
 on Brain MRI
Authors: Evan Allarie Davis Maclean Viesha Ciura* Reviewers: Yan Yu* * MD at time of publication
infiltrates in the infratentorial region
Note: variation in findings exist. The findings shown here are not exhaustive but are some of the most common areas implicated on brain MRI in MS. Most common sites that lesions are observed are: juxtacortical regions, periventricular, infratentorial, spinal cord, and the optic nerve. However lesions can occur anywhere there is myelin in the CNS.
Active inflammation and destruction allows for gadolinium contrast to cross the blood- brain barrier, which can be visualized as a marker of active inflammation in MS
Classic optic nerve (CN II) lesion seen in optic neuritis. Gadolinium enhancement (↑ signal
intensity of lesion after gadolinium injection) in this T1 image demonstrates active inflammation
Image Credits: Dr. Viesha Ciura
For further pathogenesis, see the Calgary Guide slide: Multiple Sclerosis (MS): Pathogenesis and Clinical Findings
Inflammation within the central nervous system (CNS) T-cell, B-cell, and macrophages infiltrate CNS
Infiltration occurs through veinsàlocal inflammation Perivenular infiltrates around the medullary veins
(perpendicular to ventricles)
Inflammation and blood-brain barrier destruction ↑ extravascular inflammatory fluid around lesions, which appears hyperintense/bright
Loss of neuronal/axonal density in affected region, replaced by gliosis over time
Lesions extend out around veins, creating characteristic ovoid lesions
Classic hyperintense T2/FLAIR perpendicular periventricular plaques following the medullary veins – ‘Dawson’s Fingers’
Middle cerebellar peduncle T2/FLAIR hyperintense lesion in classic location for MS
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published October 25, 2020 on www.thecalgaryguide.com")
Uterine-Fibroids
: Pathogenesis and clinical findings
Early Menarche (onset of period)
↑ estrogen exposure in
Obesity
↑ adipose tissue
↑ conversion of androgens into estrogen
Ethnicity (African)
↑ amount of aromatase enzymes
Family History
Complex chromosomal rearrangements
Myometrial Injury
Hypoxia of myometrial cells
Low Parity
Lack of protective pregnancy-induced remodeling to myometrium
Note:
Approximately three-quarters of women have fibroids. Of these, only about one-quarter become symptomatic. Fibroids generally decrease in size after menopause and symptoms improve.
Intracavitary
Fibroid projects into uterine cavity
↑ Endometrial Surface Area ↑ endometrium to proliferate and
lose during menstruation
Age 40-50
lifetime Estrogen stimulates proliferation of uterine smooth muscle cells
Benign proliferation of monoclonal myometrial (uterine wall/muscle) cells into discrete masses
Uterine Fibroids (Leiomyomas)
Benign tumours originating in and consisting of uterine muscle tissue
Fibroids can be located in different areas of the uterus, including the following locations
Authors: Emilee Anderson Reviewers: Danielle Chang Crystal Liu Yan Yu* Aysah Amath* * MD at time of publication
Subserosal
Fibroid grows adjacent to perimetrium into uterine muscle
Enlarged uterus or pelvic mass on bimanual exam
Irregularities in uterine cavity
Transformation of normal myocytes into abnormal myocytes
Submucosal
Fibroid grows adjacent to endometrium into uterine muscle
Intramural
Fibroid is within the thickness of the myometrium
Pedunculated
Fibroid extends into pelvic cavity or uterine cavity on a stalk
Spherical mass on ultrasound
Fibroid ↑ intra-abdominal pressure and puts pressure on adjacent organs
Enlarged Uterine/Pelvic Mass
Repeat shedding over time
Iron Deficiency Anemia
Pelvic Pain
Compression of stomach
Fibroid takes up space
Enlarged Abdomen
Fibroid puts pressure on the cervix
Dyspareunia
Fibroid compress the bladder
Urinary incontinence
Fibroid compress the bladder outlet
Difficulty with voiding
Fibroid compress rectum
Constipation
Embryo cannot implant
Infertility and/or recurrent pregnancy loss
Menorrhagia Dysmenorrhea
Sensation of abdominal fullness
Early Satiety
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published November 5, 2020 on www.thecalgaryguide.com")
Bronchiolitis-updated
 - but can be others such as rhinovirus,
adenovirus, parainfluenza, influenza, and coronaviruses - initially colonizes the nasopharyngeal mucosa Virus travels via the epithelium to the lower airways to the terminal bronchioles (small airways)
Authors: Nick Baldwin Rebecca Lindsay Reviewers: Kayla Nelson, Yan Yu Timothy Fu, Danielle Nelson* *MD at time of publication
Upper airway mucosal inflammation
Bronchiolitis
(bronchiole inflammation)
Apnea
(cessation of breathing; via unknown mechanism, potentially apnea reflex)
RSV-fusion protein facilitates fusion of the virus to the host cell and directs viral penetration as well as facilitates fusion of the infected cell with its healthy neighbors
Forms syncytia (multinucleated cells)
Cytokines are released into circulation
↑ thermo- regulatory set- point at the hypothalamus
Mild Fever
Copious coryza
(nasal discharge)
Protein and fluid leak into nasopharyngeal interstitium
↑ Capillary permeability
Protein and fluid leak into the bronchiole interstitium, accumulating around airway walls
Airway wall becomes thickened, more readily apparent on x-ray
Peribronchial cuffing
(X-ray finding: bronchi appear like thickened ‘cuffs’ when viewed head-on)
Inflammation stimulates the upregulation of mucous secreting goblet cells
↑ mucous production
Mucous within alveoli ↑ intra- alveolar surface tensionà collapsing alveolar walls
During inspiration, air enters the collapsed alveoli if airway is not yet occluded
↑ intra-alveolar pressure causes the alveoli to suddenly pop open
Inspiratory crackles on auscultation
Syncytia slough off the bronchial epithelium into airways
Airways become narrower and occlude
Disruption of the ciliated epithelial cells (which transport mucous out of the airways and into the pharynx, to be swallowed or evacuated)
↓ mucous clearance from airways
Excess airway mucous triggers cough reflex
Cough
Interstitial edema
Nasal Congestion
Air is absorbed distal to occlusion (gas trapping)
When all air is absorbed, alveoli collapse (resorptive atelectasis)
↓ gas exchange between blood and air in remaining alveoli
↓ O2 saturation & ↑ CO2 content of blood
Narrower airways, especially during expiration, causes audible turbulent airflow
Wheeze on auscultation
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Re-Published November 29, 2020 on www.thecalgaryguide.com")
mrna-vaccines-against-coronavirus-disease-2019-covid-19-production-and-mechanism-of-action
:
Production and Mechanism of Action
Vaccine Production
The “spike protein” is known to be a major viral surface
Authors: Ryan Brenneis, Yan Yu* Reviewers: Davis MacLean, Hannah Yaphe, Timothy Fu, Stephen Vaughan* * MD at time of publication
References
1. ACS Nano 2020, 14, 10, 12522–12537, Publication Date: October 9, 2020, https://doi.org/10.1021/acsnano.0c07197
2. NEJM 2020, Publication Date: December 10, 2020, DOI: 10.1056/NEJMoa2034577
3. Expert Review of Vaccines 2017, 16, 9, 871-881, Publication Date: 2017, DOI: 10.1080/14760584.2017.1355245
4. NEJM 2020, 383, 2439-2450, Publication Date: December 17, 2020, DOI: 10.1056/NEJMoa2027906
5. NEJM 2020, 383, 2427-2438, Publication Date: December 17, 2020, DOI: 10.1056/NEJMoa2028436
6. BMJ 2000, 321, 7271, 1237-1238, Publication Date: November 18, 2000, DOI: 10.1136.bmj.321.7271.1237
Notes:
SARS-CoV-2 (RNA virus causing COVID-19) collected from an infected patient (ie. with a nasopharyngeal swab)
Various reagents are added to the sample containing both human cells and viral constituents
Reagents cause cell/viral membrane lysisàspilling cell contents, viral particles, and viral RNA
Fats, proteins, and carbohydrates removed through various washing reagents, leaving nucleic acids (like RNA)
Reverse transcriptase polymerase chain reaction produces complementary DNA (cDNA) from viral RNA
cDNA library allows for SARS-CoV-2 genome to be mapped through whole-genome sequencing technology
SARS-CoV-2’s spike protein DNA sequence is identified, and is used as a template to create synthetic viral spike protein mRNA
Extra RNA bases are added to this mRNA strand to promote its stabilityàresulting RNA strand is now called “nucleoside-modified RNA” (“modRNA”)
antigen (substance that elicits an immune response) from studies of other coronaviruses (e.g. SARS- CoV-1 and MERS-CoV)
Pfizer/BNT162b2 vaccine contents:
Moderna mRNA-1273 vaccine:
Note: Lipid nano- particles are spherical hollow “balls” made of an outer lipid membrane plus other emulsifiers and membrane stabilizers.
Lipid nanoparticles are capable of engulfing smaller molecules (like RNA) and merging with normal cell membranes
Spike protein modRNA is then isolated (using a series of precipitation, extraction, and chromatography methods)
Final modRNA lipid nanoparticle vaccine is now created and ready for intramuscular injection
The modRNA vaccine is injected intramuscularly into a healthy person 2nd dose after 3-4 weeks needed to strengthen the immune response
(to a level exceeding the immune response in patients recovered from Covid-19), boosting vaccine efficacy especially in older individuals4,5
Lipid nanoparticle fuses with human cells’ phospholipid membranes via endocytosis, releasing modRNA into the cell’s cytosol
modRNA is translated by human ribosomes naturally found in the cell’s cytosol, producing viral spike protein components
•
•
Foreign substance can cause local tissue inflammation
The spike proteins encoded by the modRNA of each of the two vaccines are similar
It is the proprietary lipid nanoparticle formulation (unknown to the public) that is unique to each vaccine
Pain, redness, swelling at injection site (Transient)
Proprietary Pfizer/ BioNTech lipid nanoparticle
The modRNA encodes a
full-length spike protein modified with two proline amino acids (for stability and immunogenicity)2
The modRNA encodes a full-length spike protein modified with two proline amino acids (for stability)1
Proprietary Moderna lipid nanoparticle
Encapsulating this modRNA within Pfizer/ BioNTech’s lipid nanoparticle creates the 162b2 vaccine
This specific formulation requires colder storage temperatures (-700C)
Encapsulating this modRNA within Moderna’s lipid nanoparticle creates the mRNA-1273 vaccine
This specific formulation can be stored at slightly warmer temperatures (-200C)
Muscles are preferred injection sites as they have greater blood supply than other body tissues
Vaccine Action
Able to bring in immune cells faster to process foreign antigens6
Able to drain away foreign vaccine material fasterà minimizing local reactions6
Cell-mediated Immunity
Spike protein degraded by intracellular enzymes into fragments
Humoral Immunity
Natural cellular processes release spike protein components from the cell into the bloodstream
Spike protein components are engulfed by antigen presenting cells (dendritic cells, B cells, macrophages), fragmented, & bound to unique MHC Class II proteins
MHC Class II proteins bring spike protein fragments to the antigen presenting cell’s surface, to present them to circulating naïve CD4+ (helper) T cells
Some naïve helper T cells are able to successfully bind to the spike protein-MHC Class II protein complexes
Binding activates these spike-protein specific helper T cells
Spike protein fragments bound by MHC Class I proteins
MHC Class I proteins bring spike protein fragments to the human cell surface
MHC Class I proteins present spike protein fragments to naïve CD8+ T cell
Naïve CD8+ T cells that able to bind to the spike protein-MHC Class I protein complex become activated, and travel to the lymphatic system to mature
MHC = Major Histocompatability Complex; cell surface proteins key to immune function
CD = Cluster of Differentiation; glycoproteins on T cell surfaces that are co-receptors and facilitate T cell binding to antigens/MHC complexes. They also distinguish the types of T cells.
Some of these T-Cells mature into cytotoxic T cells that now recognize the viral spike protein
Cytotoxic T-cells bind to human cells infected with SARS-CoV-2 expressing spike protein or spike protein fragments
Cytotoxic T cell releases enzymes perforating infected cell, causing cell death to occur
Immune system identifies and destroys human cells infected with SARS-CoV-2, slowing viral spread
Other T cell’s can mature into memory T cells (stimulated by cytokines released by helper T cells)
Memory T cells travel to lymphatic tissue, awaiting activation from future exposure to spike protein
More rapid cell-mediated immune response to future SARS-CoV-2 infection (immunity)
Activated helper T cells specific to the viral spike protein secrete cytokines to stimulate immune activity
Systemic cytokine releaseàsystemic reactions like fever, chills, fatigue, myalgias (Transient)
Some B cells mature into plasma cells that produce IgG antibodies against the viral spike protein
Antibodies to spike protein mark SARS-CoV-2, allowing immune system to destroy virus
Eradication of SARS-CoV-2 in extracellular compartments
Activated helper T cell interacts with
naïve B cells in lymphatic tissue
Some B cells mature into memory B cells specific to SARS-CoV- 2 spike protein
Future exposure to spike protein re-activates memory B cell in lymphatic tissue & creates plasma cells, producing antibodies more rapidly
Rapid humoral immune response to future SARS-CoV-2 infection (immunity)
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Clinical Finding
End Result
Published December 19, 2020 on www.thecalgaryguide.com")
generalized-absence-seizures-petit-mal
 Seizure Hyperventilation: A common trigger of absence seizures
in pediatric patients with existing absence seizures ↑ respirationà↑ CO2 expelled from body & blood
↓ acidic CO2 levels in bloodà↑ blood pH (↓ blood acidity)
Predisposes neurons to fire spontaneously and asynchronouslyà↓ seizure threshold
Pathogenesis of absence seizure is complex and not yet fully elucidated, but evidence supports the cortical focus theory:
Hyperexcitable focal neurons on cerebral cortex send activation signals down to thalamocortical neuron network
Activated neurons in thalamus interact with cortical neurons to produce rhythmic oscillatory neuronal firing (brain waves) between these two regions of the brain
Abnormal rhythmic and bilaterally synchronous activation of the cerebral cortex during wakefulness
Between seizures (inter-ictal)
Inter-ictal changes in neuronal firing patterns and connectivity in sensorimotor cortices (mechanism unclear)
First degree relative with absence epilepsy
Genetic predisposition/idiopathic (>90%)
No single identified cause such as a structural lesion or single genetic mutation
Multiple gene mutations that predispose to epilepsy when occurring together
Authors: Alyssa Federico, Davis Maclean, Erika Russell, Harjot Atwal Reviewers: Ario Mirian, Shaily Singh*, Kim Smyth*, Yan Yu* * MD at time of publication
Monogenetic mutation (<10%)
Single gene mutation predisposing to epilepsy
Mutations involve genes encoding voltage-gated calcium channels and gamma aminobutyric acid (GABA) receptors, which are important in regulating thalamocortical activity
Absence Seizure:
Brief lapse of consciousness with a vacant stare lasting 3-10 seconds, without convulsions or loss of motor tone. May occur up to 100 times per day.
Seizure features (ictal phase)
Absence seizures generally occur in the context of an epilepsy syndrome and present in childhood
Childhood absence epilepsy: Most common form of pediatric epilepsy, characterized by absence seizures
Juvenile absence epilepsy:
Characterized by absence seizures +/- generalized tonic-clonic seizures
Juvenile myoclonic epilepsy:
Characterized by myoclonic seizures +/- absence seizures
Post-seizure (post-ictal)
Mechanisms unclear
Consciousness regained immediately after seizure
No post-ictal symptoms
No memory of event
Neuropsychiatric symptoms (poor attention, memory, mood, cognition) seen in 60% of children
Smooth/rapid transition to seizure
No aura (warning sign) prior to seizures
Seizure activity directly or indirectly impairs communication between neural networksà alters activity of brain structures involved in maintaining awareness
Some simple response to internal or external stimuli
may remain intact (mechanisms unclear)
Automatisms: Eye movements (fluttering), oral (lip smacking, swallowing, chewing), manual (finger tapping, scratching)
Brain wave oscillations generated in the thalamus
EEG findings: Generalized 3 Hz
spike-wave activity with maximum amplitude in both frontal lobes
Impaired awareness
Inhibition of
response to external stimuli
Impaired school performance and social interactions
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published January 2, 2021 on www.thecalgaryguide.com")
Primary-Aldosteronism

↑ Serum aldosterone level
↑ Circulating aldosterone activates mineralocorticoid receptors on cardiac myocytes
↑ Transcription of proinflammatory and profibrotic genes in cardiac myocytes
Cardiac fibrosis and hypertrophy
In the rest of the body, K+ moves from intracellular to extracellular environment (down it's concentration gradient) to compensate for decreased serum K+
Loss of positively charged K+ànegative intracellular chargeàcells take up H+ to remain electrostatically neutral
↑ Expression of epithelial sodium channels in
principal cells of cortical collecting duct
↑ Na+ reabsorption from cortical collecting duct into blood vessels
+ Water follows Na
into the blood vessels to balance the osmotic pressure between the blood and the renal tubules
↑ blood volume within the volume-
constrained space of blood vessels
↑ Activation of Na+/K+ ATPase in principle cells of cortical collecting duct
Removal of positively charged Na+ from tubular lumenà lumen becomes electronegative versus the interstitial space & inside tubular epithelial cells
Positively charged K+ follows the electrical gradient and is secreted into tubular lumen
↓ Serum K+ concentration
↑ Expression of H+ ATPase in the apical (luminal)
membrane of intercalated cell of cortical collecting duct
H+ is secreted into cortical collecting duct → renal loss of H+ from the body
Metabolic Alkalosis
(blood becomes more basic; ↑ serum pH)
Hypertension
Muscle weakness, fatigue, polyuria
↓ pH in intercalated cell
↓ pH in cells of proximal convoluted tubule
↑ loss of H+ through kidneys
H+ secretion into cortical collecting duct
↑ H+ ATPase activity in intercalated cell
Activation of glutaminase in proximal convoluted tubule
HCO3- pumped into blood
↑ production of HCO3- (product of glutamine breakdown)
↑ breakdown of glutamine
Hypokalemia
(See Hypokalemia: Clinical Findings slide)
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published January 16, 2020 on www.thecalgaryguide.com")
Maternal-complications-after-labor-and-vaginal-delivery

Scar tissue forms at site of placental detachment
After complete hemostasis (cessation of bleeding) and vessel healing, scar tissue is shed from uterus
Lochia
(vaginal discharge/ bleeding) & eschar (scar tissue) shedding
Placental tissue may be retained in the uterus
Uterus fails to contract fully to seal off uterine blood vessels
Post-partum hemorrhage (See relevant slide)
Foreign substances trigger systemic inflammatory response in mother
Disseminated Intravascular Coagulation, DIC (see relevant slide)
Rarely, torn blood vessels let amniotic
fluid (with fetal cells & meconium) enter maternal circulation
Amniotic fluid embolism (clumps of foreign fetal cells and meconium in maternal circulation)
Viscous amniotic fluid can block maternal blood vessels
Obstructing blood flow out of lungs
Blood backs up before lungsàless preload for heart
Damaged tissue & blood in the uterus
Ample nutrients for bacteria to infect uterus
Endometritis
Uterine pain, radiating throughout the abdomen
Pulmonary edema
Hypotension
Perineal tears
(1st-4th degree);
Hemorrhoids
Perineal Pain
Unilateral leg pain & swelling
Dyspnea, Cough
If profound
Cardiac arrest
Tissue damage activates blood coagulation factors
Coagulation in areas of hemostasis (e.g. veins)
Deep Vein Thrombosis
Post-partum fever
(see relevant slide)
Cardiovascular collapse
Lack of perfusion to heart
Note: Post-partum depression is commonly seen in at least 10% of newly-delivered mothers.
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Initially published September 5, 2013 on www.thecalgaryguide.com Updated and re-published January 23, 2021")
Beta-Blockers-Mechanism-of-Action-and-Side-Effects
 to these receptors, ↓ their normal adrenergic tone
Beta-2 receptor antagonism Beta-1 receptor antagonism
Lungs Eyes Central nervous system Heart Kidneys ↓ cAMP (intracellular messenger) productionàcomplex, tissue-specific intracellular mechanisms resulting in a variety of effects in different tissues:
Throughout body tissue
Epinephrine (via cAMP) indirectly ↑ the activity of the Na+/K+ pump on cell membranes (a pump that moves 3 Na+ out of cells per 2 K+ moved into cells)
Blocking epinephrine from binding
the beta-2 receptor and producing cAMPà↓ activity of Na+/K+ pump à↓K+ moved into cells
↑ proportion of K+ now resides in extracellular fluid, detectable in serum (total body K+ remains the same)
Hyperkalemia (see Calgary Guide: Hyperkalemia – Clinical findings)
Blocking sympathetic hormonesà↓ relaxation of smooth muscle circumferentially wrapped around airways
↑ resting airway muscle toneà bronchoconstriction
↑ resistance to airflow
Wheezing, dyspnea, chest tightness
Exacerbation of underlying airway disease (e.g. asthma)
↓ ciliary epithelium’s production of aqueous humor (fluid that fills anterior chamber of the eye)
Reduced intraocular pressure
Blocking adrenergic response mediated by epinephrine and norepinephrine (e.g. the physiologic “fight- or-flight” response to stress)
↓ tremor, irritability, anxiety
↓ ability to produce adrenergic symptoms in response to hypoglycemia
Hypoglycemia unawareness
Coronary perfusion pressure = diastolic blood pressure in aorta – LV end diastolic pressure
↓ inotropy (contractility of cardiac muscle)
↓ chronotropy (heart rate and conduction velocity)
↓ renin releaseà↓ creation of angiotensin II & aldosterone
+ ↓ reabsorption of Na
and H2O in nephron
↑ urinary Na+ & H2O loss
↓ total blood volume
Decompensation of acute heart failure
Dizziness and fatigue Hypotension (Blood pressure = cardiac
output x systemic vascular resistance)
↓ O2 demand of myocardial tissue
Bradycardia
Inability to ↑ heart rate in response to stress (e.g. shock, sepsis)
↓ stroke volume
↓ cardiac output
Beta blockers ↓ diastolic blood pressure, & thus may ↓ coronary perfusion pressure
Before giving beta blockers, ensure blood pressure isn’t too low
Otherwise, may worsen acute myocardial ischemia
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published Jan 14, 2021, updated Feb 7, 2021 on www.thecalgaryguide.com")
Adenovirus-Vector-Vaccines-Against-COVID19-Production-and-Mechanism-of-Action
, a mild human adenovirus, is isolated
Previous exposure to Ad5 or Ad26 may have sensitized immune system to the adenovirus vector1
Potential for human adenovirus vaccine to fail due to previous exposureàimmunity not built against spike protein
CanSino
Adenovirus type 5 (Ad5), a mild human adenovirus, is isolated
Oxford/AstraZeneca
Chimpanzee adenovirus AZD1222 (ChAdOx1), previously shown to be safe & to elicit an immune response in humans2, is isolated
Vaccine Production
SARS-CoV-2 (virus causing COVID-19) synthetic DNA library sequenced from viral RNA using reverse transcriptase polymerase chain reaction and whole genome sequencing technology
Spike protein DNA sequence isolated from SARS-CoV-2 genome
A promoter sequence is added to the spike protein DNA sequence, allowing human RNA polymerase to recognize and transcribe the spike protein DNA when introduced into human cells
Recombinant genetic technology inserts the modified spike protein DNA into a plasmid: a circular piece of DNA that acts as a shuttle allowing for the insertion of new genes (such as the spike protein gene) into host genomes (like the adenovirus vector DNA genome)
Adenoviral DNA isolated using various lytic & washing reagents (chemicals that break open cell membranes and remove non- nucleic acid cellular materials)
Adenoviral DNA sequenced using whole genome sequencing, then modified as follows:
Chimpanzee virus negates possibility of previous immunity to the viral vector1
Chimpanzee viral vector more likely to successfully
generate immune response to the spike protein
E1 region of adenoviral genome E3 region of adenoviral genome deleted to create deleted to block viral replication3 room for insertion of SARS-CoV-2 spike protein DNA3
Adenovirus used in the final vaccine cannot replicate
within human cells and cannot cause human disease
References
1. ACS Nano 2020, 14, 10, 12522–12537, Publication Date: October 9, 2020, https://doi.org/10.1021/acsnano.0c07197
Modified adenoviral DNA genome is reinserted into The viral vector & the spike protein plasmid are mixed together, and DNA recombination
the adenovirus particle, creating the “viral vector”
technology inserts the spike protein gene from the plasmid into the adenovirus DNA2
Adenovirus containing transcribable SARS-CoV-2 spike protein DNA is introduced into a special cellular culture, allowing the virus to replicate despite its modified DNA2, 3
Authors: Ryan Brenneis, Yan Yu* Reviewers: Davis MacLean, Hannah Yaphe Stephen Vaughan* * MD at time of publication
2. Nature 2020, 586, 578–582, Publication Date: October 20, 2020, https://doi.org/10.1038/s41586- 020-2608-y
3. Frontiers in Immunology 2018, 9, 1963, Publication Date: September 19, 2018, doi: 10.3389/fimmu.2018.01963
4. NPJ Vaccines 2020, 5, 69, Publication Date: July 27, 2020, doi: 10.1038/s41541-020-00221-3
5. The Lancet 2020, Publication Date: Dec. 8, 2020, https://doi.org/10.1016/S0140-6736(20)32623-4
6. BMJ 2000, 321, 7271, 1237-1238, Publication Date: November 18, 2000,
DOI: 10.1136.bmj.321.7271.1237
7. NEJM 2021, Publication Date: Jan. 13, 2021, DOI: 10.1056/NEJMoa2034201
Adenovirus containing transcribable SARS-CoV-2 spike protein DNA is isolated and concentrated to a high enough level for administration as a vaccine
Adenoviruses have an outer protein layer (called a capsid) to protect its DNA
DNA is more stable than mRNA due to deoxyribose sugar backbone and intermolecular bonds between strands
Enhanced stability compared to mRNA lipid nanoparticle vaccines
Can be stored at 2-8°C for up to 3-6 months
Muscles are preferred injection sites as they have greater blood supply than other body tissues
Immune cells arrive faster to The viral vector vaccine is injected intramuscularly into a healthy person process foreign antigens6
Foreign substance can cause local tissue inflammation
Pain, redness, swelling at injection site (Transient)
Note: The Johnson and Johnson vaccine may be 90% effective after a single dose7
Foreign vaccine material drains away fasterà minimizing local reactions6
2nd dose after 28 days recommended to strengthen the immune response (to a level exceeding the immune response in patients recovered from Covid-19), boosting vaccine efficacy especially in older individuals5
Vaccine Action
Cell-mediated Immunity
Spike protein degraded by intracellular enzymes into fragments
Adenovirus surface antigens interact with human cellular receptors, allowing viral entry into human cell via endocytosis3 Adenovirus vector travels to cell nucleus, merges with nuclear envelope and injects its DNA (including the spike protein DNA) into the nucleus
RNA polymerases in the nucleus transcribe the viral DNA, making messenger RNA (mRNA) for SARS-CoV-2 spike protein
mRNA is transported back into the cytosol & translated by ribosomes naturally found there, producing full length SARS-CoV-2 spike protein
Humoral Immunity
Natural cellular processes release spike protein components from the cell into the bloodstream
Spike protein components are engulfed by antigen presenting cells (dendritic cells, B cells, macrophages), fragmented, & bound to unique MHC Class II proteins
MHC Class II proteins bring spike protein fragments to the antigen presenting cell’s surface, to present them to circulating naïve CD4+ (helper) T cells
Some naïve helper T cells are able to successfully bind to the spike protein-MHC Class II protein complexes
Binding activates these spike-protein specific helper T cells
Spike protein fragments are bound by MHC Class I proteins
MHC Class I proteins bring spike protein fragments to the human cell surface MHC Class I proteins present spike protein fragments to naïve CD8+ T cell
Naïve CD8+ T cells that able to bind to the spike protein-MHC Class I protein complex become activated, and travel to the lymphatic system to mature3
MHC = Major Histocompatability Complex; cell surface proteins key to immune function
CD = Cluster of Differentiation; glycoproteins on T cell surfaces that are co-receptors and facilitate T cell binding to antigens/MHC complexes. They also distinguish the types of T cells.
Some of these T cells mature into cytotoxic T cells that now recognize the SARS-CoV-2 spike spike protein
Cytotoxic T cells bind to human cells expressing spike protein or spike protein fragments (e.g. future COVID-19 infection)
Cytotoxic T cells release enzymes perforating infected human cells, causing cell death to occur
Immune system can now more quickly identify & destroy human cells showing signs of COVID-19 infection
Some T cell’s can mature into memory T cells (stimulated by cytokines released by helper T cells)
Memory T cells travel to lymphatic tissue, awaiting activation from exposure to spike protein in the future
More rapid cell-mediated immune response to
future SARS-CoV-2 infection (immunity)
Activated helper T cells specific to the viral spike protein secrete cytokines to stimulate immune activity
Systemic cytokine releaseàsystemic reactions like fever, chills, fatigue, myalgias (Transient)
Note: Duration of cellular/ humoral immunity is unknown
Some B cells mature into plasma cells that produce IgG antibodies against the viral spike protein
Antibodies to spike protein mark SARS-CoV-2, allowing immune system to destroy virus
Eradication of SARS-CoV-2 in extracellular compartments
Activated helper T cell interacts with naïve B cells in lymphatic tissue
Some B cells mature into memory B cells specific to SARS-CoV- 2 spike protein
Future exposure to spike protein re-activates memory B cell in lymphatic tissue & creates plasma cells, producing antibodies more rapidly
Rapid humoral immune response to future SARS-CoV-2 infection (immunity)3
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Clinical Finding
End result
Published February 11, 2021 on www.thecalgaryguide.com")
Potassium-Sparing-Diuretics-Mechanism-of-Action-and-Side-Effects
 Inhibit Na+ channels (involved in Na+ reabsorption) in the luminal membrane of the principal cells in the cortical collecting duct
Aldosterone Antagonists (Spironolactone and Eplerenone) Competitively blocks mineralocorticoid receptors and ↓ aldosterone effect in the renal tubules and throughout the body
↓ aldosterone effectà↓ expression of basolateral Na+/K+ pumps & luminal epithelium Na+ channels on the principal cells of cortical collecting duct
Spironolactone’s molecular structure is similar to that of steroid
hormonesàspironolactone can also block androgen receptors
Anti-androgenic effects
(due to ↓ androgen function in reproductive organs, skin, and on brain centers)
Triamterene can form triamterene crystals
and granular casts (unclear mechanism)
↓ Na+ reabsorption by principal cells
↓ in serum Na+ concentration: Hyponatremia
↓ K+ pumped out into the tubule by principal cells
Crystals & casts obstruct tubular lumen → inflammation (unclear mechanism)
Triamterene crystals are
directly cytotoxic to tubular cells
Only ~2-5% of Na+ filtered by the glomerulus is normally reabsorbed in the
cortical collecting ductàthe ↑ in Na+ retained in cortical collecting duct is mild
Water follows Na+ to maintain a balanced osmotic pressureà↑ in water in cortical collecting duct available for excretion
↑ positive charges in lumen relative to surroundings generates an electropositive tubular lumen
Unfavorable electrical gradient ↓ secretion of positive charged ions into electropositive lumen
↓ libido
Menstrual abnormalities (in women)
↓ acne Gynecomastia
(in men)
↓ secretion of K+
↑ in serum K+ concentration:
Hyperkalemia
Cardiac Arrythmias (See relevant slide on Hyperkalemia: Clinical Findings)
Interstitial inflammation and tubular injury
Drug-induced Nephrotoxicity
Mild Diuresis
↓ blood volume
↓ Blood pressure/ Hypotension
↓ secretion of H+
↑ in serum H+ concentration:
Metabolic Acidosis
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published February 15, 2021 on www.thecalgaryguide.com")
Avascular-necrosis-of-the-femoral-head
 of the Femoral Head:
Findings on X-Ray
Blood supply to the subchondral bone is disrupted (for full pathogenesis, see Calgary Guide slide Avascular Necrosis: Pathogenesis and clinical findings)
Hypoxia at boneàOsteocyte (primary bone cell) apoptosis
Authors: Daniel Cusano, Evan Allarie, Reviewers: Yan Yu* Davis Maclean, Shelley Spaner* *MD at time of publication
↓ local osteoprotegerin (OPG) production
OPG acts to 1) inhibit osteoclast formation, & 2) bind and sequester RANKL, a chemical that stimulates osteoclast activation & proliferation
↓ OPGà↑ osteoclast activation & proliferationà↑ bone resorption
Areas that absorb ↑ X-ray radiation appear brighter on X-ray
RANKL - Receptor activator of nuclear factor kappa-Β ligand
Basic X-Ray Physiology
Bone absorbs more X-ray radiation than other tissues in the body (e.g fat and muscle)
Areas of ↓ bone density are darker: described as (radio)lucency on X-ray
Areas of ↑ bone density are whiter: described as sclerotic on X-ray
Image Credit: Alberta Health Services Repository
↓ Production of vascular endothelial growth factor (VEGF), which normally maintains & builds healthy bone & vasculature (via a multifactorial process)
Osteopenia: generalized ↓ in bone density, visualized as ↑ radiolucency (darkness) of bone
Bone demineralization and collagen matrix destruction
Scattered Cysts: radiolucent areas of resorbed bone
The femoral head’s trabecular bone is normally more porous (less dense) than the overlying cortical boneàosteopenia affects trabecular bone first, structurally weakening the femoral head
Osteoclast activity, and a shift in the balance of multiple cell signaling factorsàcompensatory activation of osteoblasts (bone forming cells)
Altered femur structure changes associated joint alignments (e.g. in hip, knee)
Repetitive microtrauma and abnormal frictional forces in affected joints
Degenerative arthritis:
joint space narrowing, osteophytes, acetabular degeneration (late, chronic findings not pictured here) For examples, see Calgary Guide slide Osteoarthritis (OA): X-Ray Findings
Fracturing of trabecular bone underneath the cortical bone of the femoral head
Fracture creates a curved space between subchondral trabecular bone and overlying subchondral cortical bone
Continued net resorption of bone & subcortical fracturingà overlying cortical bone collapses
Flattening of femoral head
(late, chronic finding; not pictured here)
Deposition of collagen matrix and bone re-mineralization
Patchy Sclerosis
Dense (white) areas of bone deposition
Crescent Sign
Subcortical lucency indicative of fracture there (atypical outside of instances of avascular necrosis)
• X-ray provides a fast and inexpensive imaging modality compared to MRI, however X-ray sensitivity varies from 41- 71% and is best suited to detect more advanced disease
• MRI sensitivity is higher at 71-100% across different studies, with the added benefit of being able to detect internal bone changes earlier than X-ray
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published February 26, 2021 on www.thecalgaryguide.com")
AAA-Pathogenesis
: Pathogenesis
Different parts of the aorta have different embryologic origins
Atherosclerosis
Hypertension
Age > 65
Progressive deterioration of aorta structural integrity over life span
Connective Tissue Disease
Structurally abnormal protein or protein organization in aorta
Autoimmunity
Infection (e.g Chlamydia, Mycoplasma pneumoniae, Helicobacter pylori, human cytomegalovirus, herpes simplex virus)
Antigens (substance that causes immune response) on virus or bacteria resemble local proteins in abdominal aorta
Antibodies produced in response to infection inappropriately target host cells in the aorta
Antibodies tag cells in the abdominal aorta for destruction by T-lymphocytes
Immune-mediated destruction of aorta
Smoking
Genetics
Unclear mechanisms
Subacute (not clinically detectable) inflammation of aortic tissue
Inflammatory cytokines are released and immune cells are recruited
↑ pressure on aorta and other vessel walls
Infiltration of vessel wall by lymphocytes and macrophages
Production of enzymes that break down elastin & collagen proteins (which provide most tensile strength to aorta)
Aorta susceptible to damage
Degradation of aortic connective tissue
Biomechanical stress on vessels
Authors: Olivia Genereux Davis Maclean Reviewers: Jason Waechter* Amy Bromley* Yan Yu* *MD at time of publication
The exact mechanisms are complex, debatable, and an area of intensive research – the 3 mechanisms and associated pathophysiology presented here are generally thought to be the main causes of abdominal aortic aneurysms
Infrarenal aorta has poorly developed vaso vasorum (dedicated blood supply to vessel wall)
Infrarenal aorta relies solely on nutrient diffusion from aortic blood that crosses abdominal aorta
Infrarenal aortic wall has fewer “lamellar” units (fibromuscular units) than other regions of the aorta
Infrarenal aorta is less elastic & less able to distribute stress
Loss of smooth muscle cells & thinning of tunica media
Destruction of elastin in tunica media
Normal layers of the aortic wall
↓ aortic tensile strength (ability to withstand stretching) Aorta expands and dilates due to internal pressure
Tunica Intima (inner-most tissue layer of aorta)
Tunica Media (layers of elastic
tissue (elastin) and muscle fibers)
Adventitia (thin outermost collagenous layer)
(longitudinal section)
Aortic aneurysms are usually infrarenal (85%)
Abdominal Aortic Aneurysm
Infrarenal aorta more prone to ischemia and has impaired repair potential
Abnormal, irreversible dilation of a focal area of abdominal aorta (area of aorta between diaphragm & aortic bifurcation) to twice the diameter of adjacent normal artery segment
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Re-Published February 27, 2021 on www.thecalgaryguide.com")
AAA-Clinical-Findings-and-Complications
: Clinical Findings and Complications AAA = Abnormal, irreversible dilation of a focal area of abdominal aorta (area of aorta between
diaphragm & aortic bifurcation) to twice the diameter of adjacent normal artery segments
Asymptomatic, non-ruptured aneurysm:
Most AAAs are asymptomatic until rupture or days before impending rupture
Asymptomatic AAAs are only detectable on imaging or by palpation
Given their structural weakness, AAAs are at risk of rupture (risk ↑ with ↑ size of aneurysm)
Ruptured AAA: a medical emergency
Aorta lies in between the peritoneal and retroperitoneal space
Symptomatic, non-ruptured aneurysm: rarely, an unruptured AAA can cause symptoms or complications (0.1%-1% of AAAs)
Authors: Olivia Genereux, Davis Maclean, Yan Yu* Reviewers: Jason Waechter*, Amy Bromley*, Sandeep Aggarwal*, Gregory Samis* *MD at time of publication
Posterior aortic wall rupture
Retroperitoneal hemorrhage
Sudden and severe abdominal and/or back pain
Anterior aortic wall rupture
Peritoneal hemorrhage
Peritoneal space is larger and holds larger volumes of blood
Peritoneal hemorrhages are large (entire blood volume can pool in the peritoneal space in minutes)
Prior to rupture, the adventitia (thin outermost collagenous layer of the aorta) may dilate significantly
Adventitia is the only layer of the aorta that contains sensory innervation à nociceptors there can be activated by adventitia dilation
Very rarely (0.1%), areas of stagnant blood flow within the AAA allow for blood & clotting factors to accumulate
Thrombi (blood clots) develop in these aneurysms
Thrombi may dislodge and travel (embolize) to distal vasculature, cutting off blood- flow to these areas
This process (referred to as tamponade) is crucial in preventing catastrophic blood loss, allowing for a window of opportunity for treatment
Compensated Hypovolemic Shock: Low blood pressure &
poor organ perfusion, but blood loss has temporarily stopped. This state of shock is compatible with life if patient is otherwise healthy (e.g. no coronary artery disease)
↓ space for blood to accumulate in the retroperitoneal space (compared to the peritoneal space)
Rapid pressure ↑ in retroperitoneal space overcomes the pressure in aorta
Since blood will only travel from areas of ↑ pressure to areas of ↓ pressure, this pressure gradient prevents further blood loss from ruptured aorta
Massive hemorrhage due to high blood flow volume through aorta
Hypotension and rapid progression to hypovolemic shock
Abdominal and/or àorgan ischemia
back pain
The most common symptom of a symptomatic non- ruptured aneurysm, and may be the first sign of an impending aneurysm rupture
Death within minutes
(rare)
If low blood pressure is now (inappropriately) treated with fluid resuscitation, the blood pressure will ↑
Differential pressure gradient is reversed (aortic blood pressure > pressure in retroperitoneal space)
Bleeding into retroperitoneal space resumes
Decompensation of hypovolemic shock Possible death
Kidney ischemia
Lower limb ischemia
Time exists to transfer patient to tertiary care center for surgical treatment
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Re-Published February 28, 2021 on www.thecalgaryguide.com")
Cubital-Tunnel-Syndrome-Ulnar-Neuropathy
: Pathogenesis and clinical findings
Osteoarthritis
Rheumatoid arthritis
Diabetes mellitus (see Calgary Guide slide on Diabetic Neuropathy)
Degenerative bone & joint changes
Autoimmune damage to joints
Abnormal bone structure/lesions
Idiopathic
Hyperglycemia causes nerve damage (complex mechanisms)
Elbow trauma
Repetitive elbow flexion
Authors: Chris Oleynick Alexandros Mouratidis Yan Yu* Reviewers: Annalise Abbott Sean Crooks Davis Maclean Hannah Koury Jeremy LaMothe* Ian Auld* * MD at time of publication
Inflammation or edema in cubital tunnel (space between medial epicondyle of humerus and olecranon of ulna) where ulnar nerve is found
↓ size of the cubital tunnelà↑ pressure on internal contents (e.g. ulnar nerve)
Axonal conduction is interrupted
Myelin sheath is damaged
↓ blood supply to nerve
Weakness of adductor pollicus longus (works to adduct thumb)
↑ compensatory activity of flexor
pollicis longus with pinching
↓ activity of hypothenar muscles (which move the 5th digit)
↓ activity of 5th digit palmar interosseus muscles (which adduct the 5th digit)
Cubital Tunnel syndrome: compression neuropathy
Paresthesia
Abnormal sensations of skin (“pins & needles”, tingling, burning, and/or numbness) in ulnar nerve sensory distribution
ulnar nerve
↓ activity of muscles innervated by ulnar nerve (medial forearm flexors, hypothenar muscles, interossei muscles, adductor pollicis longus)
+ Tinel sign
over cubital tunnel (tapping at medial elbow causes discomfort and/or paresthesia)
Weak pinch & ↓ grip strength
+ Froment’s sign
(thumb hyperflexion compensates for weak pinch)
Hypothenar atrophy
+ Wartenburg’s sign
(involuntary, excess abduction of 5th digit)
of the
Altered sensation in areas innervated by ulnar nerve (medial forearm and wrist, 5th digit, and medial half of 4th digit)
+ elbow flexion test
(flexing elbow & extending wrist further ↓ volume of the cubital tunnelà paresthesia in ulnar nerve sensory distribution
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
First published January 12, 2017, re-published February 28, 2021 on www.thecalgaryguide.com")
acute-mca-territory-ischemic-stroke-findings-on-non-contrast-ct
 strokes are generally caused by emboli (blood clot that travelled from elsewhere in the body) and less commonly caused by thrombus (blood clot that has developed locally in the MCA)
Embolism (or thrombus) occludes flow through the MCA
Acute embolism
(or thrombus) is more dense than surrounding tissue
Acute embolism (or thrombus) material attenuates (absorbs more X-rays) more than surrounding tissue (results in area of brightness)
Hyperdense MCA sign
↓ blood supply to MCA vascular territory (e.g. basal ganglia or insula)
↓ oxygen for aerobic metabolism (produces large amounts of ATP)
↓ ATP production in area of ischemia
↓ energy for ATP-dependent Na+/K+ pumps (that move Na+ out of cells) in affected neurons (grey matter)
Neurons have a higher metabolic rate than other nervous system cells (e.g oligodendrocytes that make myelin) and thus are more vulnerable to hypoperfusion and ischemia
As grey matter contains more neurons than white matter, grey matter is more vulnerable to hypoperfusion and ischemia
Grey matter shows changes on CT earlier than does white matter
Normal grey versus white matter on CT
On CT, structures that are more dense (e.g. bone, tissues) absorb more X-raysà brighter. Less dense regions (e.g. water, fluids, fat) absorb less X-raysàdarker.
Authors:
Evan Allarie Davis Maclean Viesha Ciura* Yan Yu* Reviewers:
Katie Lin* Aravind Ganesh* Gary Klein*
*MD at time
of publication
↓ extra-cellular Na+ concentration
Na+ in extracellular space is replenished via capillaries
Water follows the Na+ out of the capillaries
↑ intra-cellular Na+ concentration
Change in osmotic gradientàwater moves from extracellular space into cells
↑ water content inside & around affected neurons (grey matter)
Affected grey matter looks darker on CT (more similar to white matter)
White matter contains more fatty myelin (lower density than grey matter)à appears darker on CT
Normal insular ribbon (difficult to see here): grey matter usually seen lateral to red line shown here
Grey matter, with more neurons, has a higher densityà appears brighter on CT
Normal basal ganglia: Lentiform nucleus (putamen + globus pallidus) outlined here
Loss of grey-white differentiation throughout MCA vascular territory Difficult to see on this window (adjustable brightness of image) but is present on this scan and easily seen at different window levels
Areas with the least collateral circulation
(e.g. basal ganglia or insula) show findings first
Insular ribbon sign
(loss of insular ribbon)
Disappearing basal ganglia sign (loss of visible basal ganglia)
Comparison to normal anatomy helps outline pathologic findings
Comparison to normal anatomy helps outline pathologic findings
Images presented here are multiple CT images from the same patient with a large MCA territory ischemic stroke (Image credit: Alberta Health Services Repository)
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published March 7, 2021 on www.thecalgaryguide.com")
Achilles-Tendon-Rupture

Impaired ability to heal from microtrauma
Mechanical factors
Abnormal biomechanics:
bowleg, flatfoot, excessive supination, obesity, change in terrain
Participation in vigorous exercise
Excessive heat generation
(hyperthermia) in Achilles tendon induces changes in the extracellular matrix
Intra-tendinous collagen degeneration, disorientation, and thinningàcompromised Achilles tendon integrity
Sudden shear stress and high tension in an already weakened Achilles tendon, stretching it beyond its capacity
Haglund’s deformity: enlargement of the calcaneal postero- superior tuberosity
Bony enlargement rubs on soft tissue and causes inflammation of Achilles tendon
↓ tendon strength from
changes in extracellular matrix
Episodic participation or sudden ↑ in exercise
Irregular use renders Achilles tendon weaker
Microtrauma to the
and less flexible Achilles tendon on Achilles Tendon
Abnormal loading
Sudden, forced dorsiflexion of foot
Eccentric contraction of the gastrocnemius-soleus complex
The gastrocnemius muscle normally attaches to Achilles tendon to control plantar flexion of the foot
With the Achilles tendon ruptured, contraction of the gastrocnemius muscle cannot induce plantar flexion
Achilles tendon rupture
Achilles tendon innervated by the sural nerve and tibial nerve
Pain over rupture site (e.g. “like being kicked in the back of the leg”)
Antalgic gate: abnormal gait developed to avoid pain while walking
High tension at the origin and insertion points of the tendon causes the ends of the tear to separate
The Achilles tendon contains blood vessels. Rupturing the tendon also ruptures the blood vessels withinàblood leaks out into tendon rupture site
Bruising and swelling over rupture site
↓ resting ankle plantar flexion (Observed in prone position with knees flexed at 90 degrees)
Passive contraction (e.g. squeezing) of the gastrocnemius does not cause plantar flexion
+ Thompson (calf squeeze) test
Weak ankle plantar flexion
Lack of push-off in gait
Audible “pop” at time of injury
Palpable gap between rupture ends
Difficulty ambulating
↓ muscle use over time
Calf atrophy
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Re-Published March 21, 2021 on www.thecalgaryguide.com")
epidural-hematoma-pathogenesis-and-clinical-findings

Authors: Krusang Patel Yan Yu* Reviewers: Davis Maclean Austin Laing Gary Klein* * MD at time of publication
General force of trauma transiently disturbs structures involved in consciousness (e.g., axonal injury from shear force)
Initial Loss of Consciousness (LoC)
Patient awakens as initial insult to the
brain has subsided, not enough to permanently affect consciousness, and normal action potentials resume
Briefly regain consciousness (Lucid Interval), until:
Pressure stimulates emetic (vomiting) centers in the medulla
Nausea
Pressure interrupts area in parietal cortex responsible for up-down spatial orientation
Dizziness
Pressure compresses brain tissue
Headache
Most (75%) occurs beneath the pterion (location where frontal, sphenoidal, temporal and parietal bone meet)
Multiple bony connections ↓ stability in this region
Pterion region of the skull is the thinnest portion of the skull
Middle Meningeal Artery located under temporal bone & pterion
This area of skull susceptible to traumatic fractures Fracture lacerates the Middle Meningeal Artery
Bleeding into the epidural space, where Middle Meningeal Artery runs
Blood accumulates in the epidural space, but not yet to the extent that consciousness is affected
Blood continues to build up in the epidural space
Suture lines (attaches dura to the overlying bone) limit hematoma expansion anteriorly and posteriorly - Hematoma can only expand medially (inwards) towards brain parenchyma
More blood accumulatesà ↑ intracranial pressure (ICP)
EPIDURAL HEMATOMA:
Collection of blood in the epidural space between the inner layer of the skull and outer layer of the dura mater
For more information on imaging findings of Epidural hematoma, see Calgary Guide slide - Acute Epidural Hematoma: Findings on CT
Cerebellar tonsil (located on the underside of the cerebellum) herniates through foramen magnum, an opening in the base of the skull
Cerebellar tonsil compresses brainstem
Brainstem (controls functions like breathing, swallowing, blood pressure, and consciousness) loses function
Coma or Death
Uncus (processes emotions) on baso-medial surface of temporal lobe herniates through tentorial notch, an opening between supra and infratentorial spaces
Uncus compresses oculomotor nerve (CN III), needed for ocular movement
Muscles for ocular movement (except lateral
rectus and superior oblique) and parasympathetic innervation to the pupil are unable to function
Eye pulled down and out and pupillary dilation
Intracranial pressure becomes greater than mean arterial pressure
ICP compresses arterioles in brain
↓ cerebral blood flow
Activates α1 adrenergic receptors (part of
sympathetic nervous system)
Arteries in the body constrict to divert blood flow to the brain
Hypertension Cushing’s Triad
Baroreceptors in aortic arch
detect ↑ blood pressure
Stimulates vagus nerve (part of parasympathetic nervous system)
Vagus nerve releases acetylcholine, binding M2 muscarinic receptors on SA node membrane, inhibiting sympathetic activity and slowing SA node activity
Bradycardia
↑ ICP compresses respiratory centers in the medulla
↓ Respiration
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published April 11, 2021 on www.thecalgaryguide.com")
COVID-Public-Health-Control-Measures

R0 = Average # of people that one contagious person will infect when all are susceptible to infection. NOTE: R0 for COVID-19 ↑↑ in indoor, poorly ventilated, crowded settings.
People infected by SARS-CoV-2 are more likely to pass the virus onto others than people infected by other viruses (e.g. flu)
↑ Production of ventilators
↑ Available hospital beds & staff (e.g. cancel elective surgeries, discharge inpatients earlier)
↑ Supply of personal protective equipment (higher- grade masks like N95 respirators, goggles, gowns, gloves)
Hospital administration response
Epidemic curve with no intervention
Smaller respiratory droplets (aerosols) containing the virus can remain airborne
Wear a high-quality, well- fitting face mask (with minimal gaps between mask & face) in public, especially indoors
Some infected individuals can rapidly infect large numbers of other people
↑ Sick/critically ill patients
Acute care (e.g. hospital) system risks becoming overwhelmed by patient volume
Risk of insufficient medical & protective equipmentàrisk of nosocomial viral spread
# of cases
Larger respiratory droplets containing the virus can travel ~2 meters
Maintain physical distance of 2 meters from other people
Unmet health needs
Capacity of health system
Time
Once settled on a surface, the virus may remain viable for up to 72 hours, but can be destroyed by soap or ethanol
Frequently wash hands and surfaces (soap + water or ethanol-based hand rub)
Infection can
occur if virus contacts mucous membranes
Avoid touching mouth, nose, eyes & facemask; get vaccinated against the virus
After host is infected, virus has a incubation period up to 14 days. Host is infectious for ~10 days after symptom onset, as long as all symptoms other than cough have resolved
Individual-level prevention measures
Protection
measures needed
Remove source of infection (e.g. mandatory mask policies; closing borders to travel)
14 day quarantine for returning travelers & close contacts of COVID patients; minimum 10 day isolation for symptomatic patients
↓ Crowding & time spent indoors (e.g. closing schools, limiting hospital/long term care visitation, cancelling indoor mass gatherings)
↑ mortality among elderly and those with co-morbidities
Improving indoor ventilation (e.g. opening windows), ↑ air filtration
Break chain of infection
↑ Surveillance & testing for COVID-19
Contact tracing, isolating close contacts & those exposed
Assessment of population to identify at-risk groups (e.g. ↑ protection of elderly) and to continuously inform public measures (e.g. assess whether interventions are working)
Disaster and emergency management
(e.g. coordinate with emergency experts and first responders)
Research/Evaluation: developing and appraising the evidence to optimize the response (e.g. best use of masks and supplies, develop treatments & vaccines)
Public health control measures
Community (e.g. out-of-hospital) measures to reduce the number of people infected and hospitalized. Goals include 1) removing the infection sources, 2) breaking the chain of transmission, and 3) protecting unexposed individuals
↑ Resources for urgent and critical patient care
Limit viral spread within communities
Cases spread over longer time
# of cases
↑ capacity of health system
End goal: Minimize serious illness and overall death in the population
Time
Legend:
Pathophysiology
Public Health Intervention
Result
Complications
First published April 2, 2020; updated May 1, 2021 on www.thecalgaryguide.com")
Bacterial-Osteomyelitis

Trauma:
Penetrating injury or open fracture introduces bacteria directly to bone
Hematogenous:
Blood carries bacteria from distant site of infection to bone
Contiguous:
Neighboring site of infection seeds bacteria directly to bone
Bacterial Osteomyelitis
Innate Immune cells and local mast cells release vasoactive cytokines
Capillaries dilate to ↑blood flow to affected area and become more permeable to allow for exit of immune cells and plasma contents
Localized Immune response
Poorly understood mechanismsàinnate immune systems unable to clear the bacteria in some individualsàchronic infection
Immune cells produce pyrogenic intermediates (i.e. PGE2, IL-6, and IL-1)
Pyrogenic intermediates travel through the
bloodstream to the hypothalamus and alters the body’s thermal setpoint
Fever
Immune cells produce inflammatory cytokines (i.e. TNFα and IL6)
Systemic inflammatory cytokines result in the production of Non-specific acute- phase reactants
High Serum CRP and ESR
Warmth Erythema
Progressive blood vessel occlusion by immune cellular and bacterial debris causing infarct and necrosis of bone
More permeable capillaries allow plasma to leak into surrounding periosteal tissue
More permeable capillaries allow macrophages and neutrophils to exit the capillaries at the site of infection
Immune cells phagocytize bacteria, producing an of opaque off-white fluid
Pus
Immune cells sequester necrotic bone tissue into a regional abscess within medullary bone
Sequestrum seen as localized opacity on CT or late X-ray imaging
↑ Osteoclast activity to remove damaged medullary bone and ↑ Osteoblast activity to reform new bone
Osteoclasts and osteoblasts remodel and deposit new bone in area surrounding necrotic tissue
Swelling
↑ Periosteal fluid applies ↑ pressure to surrounding nerve endings
Bone Pain manifesting as refusal to use limb
New layer of bone formed
Positive Bone Scan in area of infection
Abbreviations:
• IL – Interleukin
• PGE2 – Prostaglandin E2
• TNFα – Tumor necrosis factor alpha
Involucrum / Periosteal Reaction: seen as regional luminance surrounding sequestrum on CT or late X-ray imaging
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published May 21, 2021 on www.thecalgaryguide.com")
Thyroïdite

Symptômes généraux de l’hyperthyroïdie
Post-natal
Médicaments/ Radiation
Thyroïdite bactérienne
Subaigüe (post-virale)
Abbréviations:
• TSH- Hormone stimulant de la
thyroïde
Inflammation de la glande thyroïde
Plusieurs méchanismes
Brianna Ghali Sylvain Coderre* Phillippe Couillard*
Translator:
Infiltration de la glande thyroïde avec des globules blancs
Destruction immunitaire de la glande thyroïde
↑ libération de T4 et T3 stockés par glande thyroïde endommagée
durant 2-6 semaines (phase hyperthyroïde)
↓ sécrétion de T4 et T3 de la glande thyroïde endommagée
durant des semaines-mois (phase hypothyroïde)
Souvent une résolution à un état euthyroïde (normal), mais peut aussi rester dans un état hypothyroïde
↑ T4/T3 inhibe la
libération de la TSH par la ↓ sécrétion TSH
Réaction inflammatoire altère la fonction folliculaire normale (production T4/T3 et importation d’iode)
glande pituitaire
↓ Absorption de l'iode par la glande thyroïde
↓ Capture de l'iode radioactif à la scintigraphie de médecine nucléaire
Plusieurs méchanismes Symptômes généraux de l’hypothyroïdie
↓ T4/T3 stimule la
liberation de la TSH par la ↑ sécrétion TSH
Dommage altère la fonction folliculaire normale, (production T4/T3 et importation d’iode)
glande pituitaire
↓ Absorption de l'iode par la glande thyroïde
↓ Capture de l'iode radioactif à la scintigraphie de médecine nucléaire
Légende:
Pathophysiologie
Méchanisme
Signes/Symptômes/Résultats labo
Complications
Publié June 19, 2013 on www.thecalgaryguide.com")
VITT
: Pathogenesis and Clinical Findings
Current leading theory: COVID-19 viral vector vaccines (Johnson and Johnson and AstraZeneca) contain an anionic molecule (currently unspecified), which binds to the cationic platelet factor 4 (PF4) molecule in the bloodstream, forming vaccine/PF4 complexes
Authors: Brooke Fallis Reviewers: Yan Yu* Katie Lin* * MD at time of publication
Spleen macrophages remove antibody/platelet complexes from circulation
Fewer unbound platelets in circulation detected on complete blood count (CBC)
Thrombocytopenia: Platelets <150x!")
Lambert-Eaton-Myasthenic-Syndrome-Pathogenesis-and-Clinical-Findings
 in >50% of patients
Tumour membrane expresses voltage-gated calcium channels (VGCCs), which normally exist on neurons and function in neurotransmission
Note:
A paraneoplastic syndrome is a condition that arises due to cancer elsewhere in the body; possibly an immune response against tumour cells
Positive anti-VGCC IgG on serology
↓ Stimulation of salivary glands à↓ production of saliva
↓ Nitric oxide & prostaglandin production by cavernosal endothelial cellsàImpaired vasodilation of penile arteries
↓ Acetylcholine-induced gastric motility
↓ Acetylcholine available to mediate muscle reflexes
↑ Variability in action potential initiation along muscle fibers
Immune response to foreign cancer cells triggers production of antibodies against VGCCs on the cell surfaces of presynaptic neurons
Antibodies bind VGCCs, blocking Ca2+ Antibodies bind, cross-link, and
from entering presynaptic neurons
↓ Ca2+ influx into the presynaptic neuron during its depolarization
internalize VGCCsà↓ VGCC on neuron surface
Xerostomia (dry mouth)
Erectile dysfunction
Constipation
↓ Deep tendon reflexes
Unstable motor unit action potentials on electromyography
↓ Baseline compound muscle action potentials (CMAPs:
summated action potentials of all motor endplates in one muscle) on nerve conduction studies
Since intracellular Ca2+ mediates neurotransmitter vesicle fusion with the presynaptic membrane, ↓ Ca2+ influx ↓release of neurotransmitters like acetylcholine into the synaptic cleft
However, with repeated stimulation of the presynaptic neuron (e.g. exercise), there is ↑ Ca2+ accumulation within the axon terminal, allowing for more neurotransmitter vesicle fusion with the presynaptic membrane
↑ Acetylcholine available to mediate muscle reflexes
With high frequency repetitive nerve stimulation, ↑ number of compound CMAPS can be generated
↓ Acetylcholine release into synapses leading to autonomic nerves
↓ Acetylcholine release into neuromuscular junction
Autonomic dysfunction
↑ Deep tendon reflex amplitude
Temporary improvement in muscle strength
↓ Number of
muscle fibers activated by each action potential
Post-activation facilitation: Repeated stimulation improves symptoms
Larger proximal muscles involved in movement (i.e. walking) do not recruit sufficient number of muscle fibers for proper function
Can affect any muscle group, but muscles involved in speech, swallowing, and periocular muscles are often afflicted
Gait disturbance
Symmetric skeletal muscle weakness
Dysarthria (difficulty speaking)
Dysphagia (difficulty swallowing)
Ptosis (drooping of upper eyelid)
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Re-Published July 18, 2021 on www.thecalgaryguide.com")
Corneal-Abrasion

Trichiasis
(Misdirected eyelashes directly abrading the ocular surface)
↓ tear production (See Calgary Guide - Dry eye pathogenesis)
↓ quantity or quality of tear film
Any condition causing incomplete or inadequate eyelid closureà↑ exposure of eye surface to atmosphere (Bell’s palsy, proptosis/ exophthalmos)
↑ Tear evaporation
↓ lubrication of eye surface
Dryness and desiccative damage to ocular surface structures
↓ protective barriers against mechanical or foreign body damage
Infection of epithelial defect by pathogens
Damage deepens to inner layers of the cornea
Immune response causes white blood cells to accumulate
Pathogen and immune cells obstruct blood flow, causing tissue necrosis
Infectious corneal ulcer
Any condition affecting the trigeminal nerve and/or peripheral corneal nerves (e.g. Herpes zoster/simplex infection, diabetes, medications, surgery)
Tight lens
↓ O2 reaching cornea
Corneal epithelium hypoxiaà cellular damage
Contact Lens use
Extended use Lens dehydrates
Corneal epithelium adheres to lens and is removed with the lens
Neurotrophic keratopathy
(corneal damage secondary to loss of innervation)
↓ stimulation of the cornea by neurotransmitters (complex and multifactorial)
↓ Blinking (if bilateral)
Area of defect stained bright green under cobalt-blue filtered light
Impaired sensory innervation of the
corneaàImpaired Reflex Tearing
↓ adhesion between corneal epithelium and Bowman’s layer
Trauma from insertion or removal of contact lens
Previous traumatic abrasion with damage to Bowman’s layer or inadequate epithelial adherence (e.g. corneal dystrophy)
Spontaneous erosion (occurs without antecedent injury or foreign body)
↓ structural integrity of corneal epithelium
Fluorescein dyes the exposed Bowman’s layer (inner corneal layer between epithelium and stroma)
Corneal Epithelial Damage (the transparent portion of the eye that covers the anterior portion of the eye and covers the pupil iris and anterior chamber)
Corneal Abrasion: Focal area of epithelial loss - outermost layer of cornea (damage may extend to the bowman's layer below as well)
Abrasions that overlay the pupil
Epithelium (outermost portion on cornea)
Bowman’s layer
Stroma
Descemet’s membrane
Endothelium
Cornea (cross section)
Layers of the cornea
Concurrent damage to other anterior
segment structures in trauma
Can induce traumatic uveitis
Irritation and spasms of the iris/ciliary body muscle complex
Light stimulus induces further movement of
irritated/inflamed structures
Damaged tissues & ensuing inflammatory responseà Inflammatory mediator release
Conjunctival injection (vasodilation of vessels in the conjunctiva)
Red eye
Damage to corneal nerves stimulates corneal nociceptors
Pain/ Foreign body sensation
prevent light entry through the pupil
Acute vision loss
Hyperalgesia (lowered peripheral nerve
threshold for firing while damaged tissues are healing, during which normally non- noxious stimuli like light, wind or temperature change - can induce pain)
Nociceptors stimulate afferent neurons in trigeminal nerve, which then activates efferent neurons in the facial nerve
Stimulation of the lacrimal gland
Tearing
Anterior chamber of the eye
Photophobia (Light sensitivity or light-induced pain/ discomfort) – Pathophysiology is complex and multifactorial
Authors: Yejun Hong, Davis Maclean Reviewers: Mehul Gupta, Adam Muzychuk*, Victor Penner*, Yan Yu* *MD at time of publication
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published July 19, 2021 on www.thecalgaryguide.com")
Fat-Embolism-Syndrome

Non-trauma related (rare)
Long bone fracture
Pelvic fracture
Orthopedic Trauma
Intraosseous access
Soft tissue injuries
Chest compressions
Bone marrow transplant
Pancreatitis
Diabetes mellitus
Fat from bone marrow is disrupted and leaks into bloodstream via damaged blood vessels
Fat globules obstruct dermal capillaries
Capillaries rupture
Blood leaks into the skin
Petechial rash
Non-Orthopedic Trauma (less common)
Fat from injured adipose tissue is released from adipocytes into bloodstream
Metabolic disturbance mobilizes stored fat and moves it into circulation
Fat Embolism Syndrome
the presence of fat globules in circulation
Fat globules damage blood vessel walls
Platelets stick to damaged areas Platelet aggregation
↑ circulating free fatty acids
↑ inflammatory cytokines (TNF, IL1, IL6)
↑ serum C Reactive Protein (an acute phase reactant)
C reactive protein binds to lipid vesicles in circulation
↑ formation of lipid complexes in the blood
Obstruction of cerebral vasculature
↓ blood flow and oxygen delivery to brain tissue
Neurological findings: ranging from ↓ level of consciousness to seizures
Notes:
Large quantities of fat globules can obstruct pulmonary vasculature
Blood clots form throughout the body
Disseminated intravascular coagulopathy
Back up of blood into right heart àRight ventricular dysfunction
↓ pulmonary arterial blood flow à↓ gas exchange in the lungs
Higher CO2 & lower O2 levels in blood àdetected by chemoreceptors
Chemoreceptors stimulate respiratory centre in the brain to ↑ rate of respiration
Dyspnea / Tachypnea
Authors: Tabitha Hawes Reviewers: Hannah Koury, Alyssa Federico, Davis MacLean, Mehul Gupta, Yan Yu*, Jeremy Lamothe* * MD at time of publication
• Underlined findings indicate classic triad of symptoms (petechial rash, neurologic findings, dyspnea/tachypnea)
• Clinical presentation of fat embolism syndrome is variable and may present with any or all of these findings
↓ pumping of blood into systemic circulation
Hypotension Obstructive shock
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published July 19, 2021 on www.thecalgaryguide.com")
Complications-Accouchement-Vaginale
 et la cicatrisation des vaisseaux, le tissu cicatriciel est éliminé de l'utérus.
Lochies (pertes/ saignements vaginaux) et perte d'escarres (tissu cicatriciel)
Le tissu placentaire peut être retenu dans l'utérus.
L'utérus ne se
contracte pas complètement pour fermer les vaisseaux sanguins utérins.
Hémorragie post- partum (Voir la diapositive correspondante)
Les substances étrangères déclenchent une réaction inflammatoire systémique chez la mère
Coagulation intravasculaire disséminée, CIVD (voir diapositive correspondante)
Dans de rares cas, des vaisseaux sanguins déchirés laissent le liquide amniotique (contenant des cellules fœtales et du méconium) pénétrer dans la circulation maternelle.
Embolie de liquide amniotique (amas de cellules fœtales et de méconium dans la circulation maternelle)
Le liquide amniotique visqueux peut bloquer les vaisseaux sanguins maternels
Obstruction de la circulation du sang dans les poumons
Bas debit cardiaque avec reduction de la précharge.
Tissus endommagés et sang dans l'utérus
Des nutriments pour que les bactéries infectent l'utérus.
Endométrite
Douleur
utérine, irradiant dans tout l'abdomen
Œdème pulmonaire
Hypotension
Manque de perfusion au cœur Arrêt cardiaque
Le passage du fœtus distend les muscles pubo- vésiculaires et pubo-rectaux.
L'urètre/ rectum ne sont plus suffisamment courbés pour empêcher les fortes pressions intra- abdominales de forcer l'urine ou les selles.
Incontinence de stresse
(de la vessie et des intestins ; généralement temporaire)
Remarque: la dépression post-partum est couramment observée chez au moins 10 % des mères qui viennent d'accoucher.
Déchirures
périnéales (1er-4e degré) ; Hémorroïdes
Douleur périnéale
Douleur et gonflement
unilatéral de la jambe
Dyspnée, Toux
Les lésions tissulaires activent les facteurs de coagulation du sang.
Coagulation
dans les zones d'hémostase (par exemple, les veines)
Thrombose veineuse profonde
La fièvre du post- partum
(voir la diapositive correspondante)
L'insertion d'un tube étranger dans la vessie facilite la colonisation de la vessie et des voies urinaires par les bactéries.
Infections des voies urinaires
Si grave
Effondrement cardio-vasculaire
Légende:
Pathophysiologie
Méchanisme
Signe/Symptôme/Résultat Laboratoire
Complications
Publié 19 juin 2013 à www.thecalgaryguide.com")
Hypertriglycéridémie primaire

La LPL présente dans le cœur,
les muscles squelettiques, les tissus adipeux et d'autres tissus est incapable d'éliminer les TG des CM et des VLDL.
Hyperlipidémie combinée familiale
Surproduction de TG-riches VLDL, LDL et/ou ApoB100
Dysbétalipoprotéinémie familiale
Mutation de ApoE (l'apoprotéine responsable de l'élimination des CMs et VLDLs de la circulation)
VLDLs & CMs riches en TG s’accumulent dans le sang
Hypertriglycéridémie
Primaire versus Secondaire:
- L'hypertriglycéridémie primaire est due à une cause génétique. -L'hypertriglycéridémie secondaire est due à une cause médicale ou à un mode de vie acquis.
Mécanismes inconnus du risque ↑ CV, distincts du risque d'athérosclérose associé à l'hypercholestérolémie.
MC Prématurée
(âge d'apparition avant 50 ans chez les hommes et 55 ans chez les femmes)
Remarque : les signes les plus pathognomoniques de l'hypertriglycéridémie sont les xanthomes éruptifs et la lipémie rétinienne.
La diffusion de la lumière par les TG sanguins fait paraître les vaisseaux de la rétine blanc au fond d’oeil
Lipemia Retinalis
La lipase pancréatique libère les FFA des CM et des VLDL.
La concentration plasmatique des FFA dépasse la capacité de fixation de l'albumine.
Les AGF s'accumulent dans le pancréas, provoquant une inflammationàla lyse des cellules pancréatiques et la libération d'enzymes digestives pancréatiques.
Pancréatite
(le risque de pancréatite augmente considérablement avec un taux de TG >10mmol/L)
Des niveaux excessifs de CM et de TG traversent les vaisseaux sanguins pour atteindre le derme ou la cornée.
TGs engloutis par les macrophages dermiques or cornéens (histiocytes)
Xanthomes éruptifs
(papules jaunes de 1 à 3 mm généralement observées sur les tendons et surfaces d'extension comme les coudes, les genoux, les fesses, ainsi que sur le dos, la poitrine et les extrémités proximales).
Légende:
Pathophysiologie
Méchanisme
Signe/Symptôme/Résultat Laboratoire
Complications
Publié 19 juin 2013 à www.thecalgaryguide.com")
Nodule-thyroïdien
 thyroïdien(s) hyperfonctionnel(s): Pathogénie et résultats cliniques
Traduction:
Radiation Tabagisme
↑ Âge Génétiques
Brianna Ghali Philippe Couillard* Autheur: Jaye Platnich Rédacteurs: Amogh Agrawal Alexander Arnold Hanan Bassyouni* *MD au moment de publication
ADN endommagée et mutée
La mutation du récepteur de la TSH dans le tissu thyroïdien entraîne une activation constitutive
(une activation indépendante de la TSH)
↑ Signalisation de la protéine G (spécifiquement Gs alpha)
Le tissu thyroïdien affecté devient hyperfonctionnel et
indépendant des mécanismes normaux de la régulation.
↑ Production et sécrétion de T4 et T3 à partir du nodule hyperfonctionnel.
↑ T4/T3 supprime la production de TSH par la glande pituitaire
Abbréviations:
• TSH - Hormone de stimulation de la thyroïde
Note: Les nodules thyroïdiens hyperfonctionnels sont très rarement malins : plus de 98 % de ces nodules sont bénins. La capacité à capter l'iode et à produire de la T4 et de la T3 suggère un niveau de différenciation cellulaire rarement atteint par les cellules malignes.
Prolifération localisée des cellules folliculaires de la thyroïde.
↑ Capture de l'iode radioactif et du technitium-99 par le tissu thyroïdien hyperfonctionnel.
Variety of mechanisms (see hyperthyroidism slide)
↓ Stimulation du tissu thyroïdien normal par la TSH
Nodule(s) palpable(s) sur la glande thyroïde
Captation de l'iode radioactif montre une ↑ quantitative de l'activité thyroïdienne.
La scintigraphie thyroïdienne au technitium 99 permet de visualiser qualitativement le ou les nodules hyperactifs (")
Physiology-of-the-Normal-ECG-Waveform-Lead-II

ECG Waveform (Lead II)
A group of pacemaker cardiomyocytes in the right atrium (the Sino-Atrial (SA) node) rely on slow Ca2+ channels on their surface to spontaneously reach depolarization threshold
Depolarization wave travels through the atria, contracting atrial myocytes (thus causing atrial contraction)
AV node relays conduction to Bundle of His, which delays the depolarization
Depolarization conducts down the ventricular septum via the bundle branches, towards the apex
At the apex, depolarization conducts up the Purkinje fibers within the ventricular walls, towards the base of the heartàventricular contraction
During ventricular contraction, repolarization starts in the atria àatrial relaxation
Re-polarization proceeds to ventricles, and occurs from the epicardium to the endocardium, with a net direction from the apex towards the base of the heart
Depolarizations occur at regular intervals of 60-100 beats per minute
Normal heart rate Regular cardiac rhythm
Authors:
Davis MacLean, Mehul Gupta, Amanda Nguyen, Yan Yu*
Reviewers:
Luke Gagnon, Paige Shelemey, Katie Lin*
* MD at time of publication
Regular intervals of P- waves followed by QRS complexes (60- 100 P-waves/min, 60- 100 QRS/min) – ”normal sinus rhythm”
R
Depolarization moves towards Lead II, thus causing upward deflection on ECG. Atria have less myocytesàsmaller depolarizationà smaller upward deflection on ECG Lead II
Normal P Wave
Flat segment between P-wave and QRS
Depolarization directed inferior and left (compared to patients' vertical axis), in the direction of Lead IIà upward deflection on ECG Lead II
Depolarization is now directed away from Lead IIàdownward deflection on ECG Lead II
Electrical signal from atrial repolarization is too weak to stand out and is “buried” in the QRS
Ventricular myocytes are contractedàno net movement of electrical charge
Normal R wave
Normal S wave
Q
Normal QRS Complex
S
Note: for simplicity, we will only use Lead II to explain the direction of ECG deflections. However, the same principle applies to all leads:
Depolarization moving towards the leadàupward deflection on ECG Depolarization moving away from the leadàdownward deflection on ECG
Absence of any ECG components representing atrial relaxation
• •
PR Interval
Represents time between start of atrial depolarization to start of ventricular depolarization
SA node to atrial and AV node depolarization takes 120-200 milliseconds (ms)
No ECG deflection occurs
Isoelectric ST segment
(Interval between end of ventricular depolarization and beginning of repolarization)
QRS Width
Represents time required for ventricular depolarization
Depolarization spreads along fast conduction pathways of bundle branches and Purkinje fibers
Rapid conduction results in ventricular depolarization in 60- 110 ms
“Narrow” QRS <110 ms
QT Interval
Represents time required for ventricular depolarization and repolarization
Without the influence of factors including genetics, electrolyte abnormalities, or drugs, the QT segment is of a normal length
Normal adult QT interval: <440ms in males and <460ms in females
The opposite charge (re-polarization) is now moving away from the positive electrode of Lead II
An upward deflection in Lead II is produced, but is slower and smaller in amplitude compared to the QRS, which reflects the slower repolarization process
Normal T Wave
End of waveform
Lead number and orientation
Base
Apex
Note:
• Q Waves: Small Q-waves can be seen in lead II and are non-
pathologic as they represent the left to right depolarization of the ventricular septum. Larger Q waves are pathologic and are usually indicative of previous infarct/ischemia.
• Axis: For an explanation of how to tell the Axis from an ECG, please consult the relevant Calgary Guide slide on the subject.
Note: Normally, the apex of the heart points down and to the left of the body, in the direction of leads I, II, and aVF. Since QRS deflections on ECG reflect the direction of ventricular depolarization, an upward QRS deflection in these leads means the heart’s apex is pointed in a normal direction (normal “axis”).
Action potential conduction velocity is slowed at AV node
PR interval is 120-200 ms (normally)
Normally, each action potential travels through the AV node to the ventricular conducting system to cause ventricular depolarization
If abnormal conduction through AV node or down the ventricular septum:
Heart Block
Legend:
Pathophysiology
Mechanism
Normal Findings
ECG Components
Published July 31, 2021 on www.thecalgaryguide.com")
Dry-Eye-Syndrome-Pathogenesis
: Pathogenesis
The Pathophysiology of Dry eye disease is complex and an area of active investigation – The mechanism and causes presented here represented the highest yield causes and mechanism for students
Post laser eye surgery
Disruption of corneal nerves
↓ corneal sensitivity
Damage to trigeminal nerve, the sensory innervation of the eye (due to: Herpes Zoster, tumor, trauma, etc)
Blepharitis (eyelid inflammation)
Many medications can cause dry eye via
multiple mechanisms presented here (e.g. ↓ corneal sensitivity, Meibomian gland dysfunction, lacrimal gland atrophy)
Sex Hormones (e.g. androgens & estrogens) play a complex and poorly understood role in mediating dry eye disease (net effect is that women are more often affected by dry eye)
Obstructed meibomian glands
Eyelid damage Gland atrophy
These items represent general causes of meibomian gland dysfunction – exact causes are numerous, their pathophysiology is beyond the scope of this slide
Contact lens (long term use)
Corneal nerve adaptation to chronic mechanical stimulation
Autoimmune disease (e.g. Sjögren's syndrome)
Chronic inflammatory infiltration of the lacrimal gland (and salivary gland)
Autoimmune Lymphocytic infiltration
Inflammatory cytokine release
Autoantibody production
Cell death and apoptosis
Lacrimal gland degeneration
Meibomian gland dysfunction (Located along the eyelid margins, these glands produce meibum, an oily substance that prevents evaporation of the tear film)
↓ meibum secretion Loss of lipid layer
covering the eye, ↓ the barrier that blocks evaporation of tear film
Tear Film instability
Lifestyle
Extended reading or
TV or electronic device uses
Exposure Keratopathy (any condition causing dryness due to incomplete or inadequate eyelid closure, e.g. Bell’s Palsy)
↓ activity of the afferent portion of
corneal reflex arc (responsible for reflex tearing: tearing in response to irritation of the eye)
Mechanical damage to goblet cells
secrete mucins – a substance that lubricates the eye and preserves tear film
↓ blink rate
↑ time and area
↓ normal reflex tearing
for evaporation
Dry climate Wind exposure
Infiltrative diseases
(e.g. sarcoidosis)
Lacrimal gland infiltration
↓ Lacrimal gland secretion of the the watery aqueous layer of the tear film (Aqueous deficient dry eye)
Deficient or unstable tear film (Evaporative dry eye)
↑ tear evaporation
Direct damage to lacrimal gland (e.g. infection or trauma of the eye)
Authors: Davis Maclean, Yan Yu*, Michael Penny, O.D.
Reviewers: Natalie Arnold, Saleel Jivraj, O.D., Adam Muzychuk*, Victor Penner* *MD at time of publication
Hyperosmolar Tear Film (hyperosmolarity = ↑ solutes and ↓ solvent)
(Further) Tear Film instability
Corneal and conjunctival epithelial
cells dry out, including goblet cells (which secrete mucins – a substance that lubricates the eye)
Inflammatory immune response àRecruitment and activation of CD4+ (Helper) T-Cells, further produce cytokines
Further irritation and damage to ocular surface structures (cornea, conjunctiva and Meibomian glands) and lacrimal glands
See Calgary Guide: “Dry Eye Syndrome
(Kerato- conjunctivitis sicca):Clinical Findings” for signs and symptoms
Dry Eye Syndrome (Keratoconjunctivitis sicca): A multifactorial disease of the ocular surface and tears characterized by loss of tear film homeostasis, tear film hyperosmolality and inflammation
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published August 7, 2021 on www.thecalgaryguide.com")
Dry-Eye-Syndrome-Clinical-Findings
:
Clinical Findings
If dry eye is chronic or severe (e.g Sjögren’s syndrome) corneal ulcerations or scarring may occur
Impaired visual acuity/ Blindness (does not improve with lubrication)
See Calgary Guide: “Dry Eye Syndrome (Keratoconjunctivitis sicca): Pathogenesis” for explanation of etiology and pathogenesis
Dry Eye Syndrome (Keratoconjunctivitis sicca): Disease of the ocular surface and tears characterized by loss of tear film homeostasis, tear film hyperosmolality and inflammation
Tear film instability & hyperosmolarity
Ocular surface inflammation: Immune cell (specifically T-Cell) destruction of corneal and conjunctival epithelium
↓ quantity and/or quality of tear film
Inflammatory factors vasodilate conjunctival vessels, making the eye look redder in appearance
Conjunctival injection
Red eye
Corneal surface irregularities
Light rays pass through
disrupted tear film and ocular surface structures as they enter the eye
Degraded image quality as light passes through the eye
Impaired visual acuity
(improves with lubrication)
↓ lubrication over the eye surface
↑ friction on the eye during blinking or when opening eyes after sleep
May cause corneal abrasion (See Calgary Guide: Corneal Abrasion)
↑ stimulation of corneal and conjunctival nerve endings
Irritation and damage to corneal and conjunctival cells
↑ stimulation of nerve endings
Chronic inflammation surrounding corneal nerves in conjunctiva and epithelium can lead to decreased neuronal firing thresholds (Neurosensory dysfunction / hyperalgesia)
↑ activity of the afferent portion of corneal reflex arc (responsible for reflex tearing: tearing in response to irritation of the eye)
Foreign body sensation
(scratching / feeling as if a foreign body, like a grain of sand, is in the eye)
Minor (typically non-painful stimuli) may cause pain due to hypersensitive corneal nerves
Corneal neuropathic pain (pain not fully explained by ocular surface changes, often resistant to standard treatments, sometimes still present following ocular surface anaesthesia if central pathways affected)
Pain with temperature change and wind exposure
Exact mechanism unknown
Photophobia
Burning sensation Paradoxical reflex tearing (↑ tearing, which does
not improve dry eye due to altered tear composition, poor surface coverage or overflow of temporary tearing out of the eye
Authors: Michael Penny, O.D. Davis Maclean Yan Yu*
Reviewers: Natalie Arnold Saleel Jivraj, O.D. Adam Muzychuk* Victor Penner* *MD at time of publication
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published August 6. 2021 on www.thecalgaryguide.com")
medial-collateral-ligament-mcl-injury
 Injury
Previous MCL injury
Ligament healing requires
adequate blood supply to bring oxygen and nutrients to the site of injury
Incomplete healing of MCL due to poor blood supply
Authors: Alyssa Federico Reviewers: Hannah Koury, Mehul Gupta, Yan Yu*, Alexander Rezansoff* * MD at time of publication
Sports involving significant twisting or torsion of a planted lower extremity (soccer, basketball, tennis, skiing)
Tibial rotation
External rotation of tibia while foot is planted on the ground
High-energy trauma
Contact sports (wresting, hockey, football, rugby, soccer)
Contact trauma
External (valgus) force on flexed knee
↓ stability and resistance to contact trauma and tibial rotation
Grade 1
Mild injury
Grade 2
Moderate injury
Grade 3
Severe injury
Similar forces affect surrounding structures in the knee
MCL injuries commonly co-occur with anterior cruciate ligament and medial meniscus injuries
Medial knee innervated by the saphenous nerve (branch of the femoral nerve)
MCL Injury
Classified into three grades, each with characteristic findings
MCL is located outside of the knee joint capsule
Microscopic tear (<10% of collagen fibers torn)
Incomplete tear (>10% of collagen fibers)
Complete tear of MCL
Normal valgus laxity (0-5 mm) at 30° knee flexion with perceived endpoint
↑ valgus laxity (5-10 mm) at 30° knee flexion with a perceived endpoint
Significantly ↑ valgus laxity (>10 mm) at 0° and 30° knee flexion with no endpoint
↓ stability and resistance to valgus stress
+ Valgus stress test
with 0° and/or 30° of knee flexion
Excessive valgus laxity, medial joint opening and/or medial knee pain
MCL stretched beyond capacityàtears collagen fibres in the ligament
Medial knee pain
Medial knee tenderness on palpation
MCL bleedingà blood accumulates below the skin
Bruising on medial aspect of knee
Localized, extra- articular swelling at site of injury
Soft tissue swelling on medial aspect of knee
Audible “pop” at
time of injury
Knee instability from extreme laxity
Favouring the injured side to avoid pain while walking
Antalgic gate
Stiffness and inability to bend or straighten the knee
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published August 14, 2021 on www.thecalgaryguide.com")
lateral-collateral-ligament-lcl-injury
 Injury
Previous LCL injury
Ligament healing requires
adequate blood supply to bring oxygen and nutrients to the site of injury
Incomplete healing of LCL due to poor blood supply
Authors: Alyssa Federico Reviewers: Mehul Gupta Yan Yu* Alexander Rezansoff* * MD at time of publication
Sports involving high velocity pivoting or jumping (tennis, gymnastics, soccer, basketball, skiing, football, hockey)
High-energy trauma
Contact sports (wresting, hockey, football, rugby, soccer)
Hyperextension
Knee joint extended beyond normal range
Similar forces affect other structures in the knee
LCL injuries commonly co- occurs with anterior cruciate ligament, posterior cruciate ligament, and posterolateral corner injuries
Lateral knee innervated by the posterior articular branches of tibial nerve
↓ stability and resistance to varus force and hyperextension
Varus force
Extreme force placed on medial knee
Contact trauma
Direct blow causing varus force and hyperextension
LCL stretched beyond physiologic capacityàtears collagen fibres in the ligament
Grade 1
Mild injury
Grade 2
Moderate injury
Grade 3
Severe injury
Microscopic tear (<10% of collagen fibers torn)
Incomplete tear (>10% of collagen fibers)
Complete tear
Knee instability from extreme laxity
Normal varus laxity (0-5 mm) with a perceived endpoint
↑ varus laxity (5-10 mm) with a perceived endpoint
Significantly ↑ varus laxity (>10 mm) with no endpoint
↓ stability and resistance to varus stress
+ Varus stress test
with 0° and 30° of knee flexion
Excessive varus laxity, lateral joint opening and/or lateral knee pain
LCL Injury
Classified into three grades, each with characteristic
findings
LCL is located outside of the knee joint capsule
Lateral knee pain
Lateral knee tenderness on palpation
LCL bleedingà blood accumulates below the skin
Bruising on lateral aspect of knee
Localized, extra- articular swelling at site of injury
Soft tissue swelling on lateral aspect of knee
Audible “pop” at time of injury
Favouring the injured side to avoid pain while walking
Antalgic gate
Varus thrust: visible lateral movement of the knee upon weight bearing on ambulation (lateral shift of the head of the fibula and upper tibia)
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published August 14, 2021 on www.thecalgaryguide.com")
Congenital-Thrombophilia
 Sharma, Man-Chiu Poon* *MD at time of publication
Group I
Hereditary deficiencies of coagulation inhibitors
Group II
Hereditary disorders with ↑ levels or function of coagulation factors
Antithrombin (AT) deficiency
AT inhibits thrombin & activated coagulation e.g. factor X (FXa)
Autosomal dominant mutation of the
SERPINC1 gene causes ↓ production of AT
↓ AT concentrations allows thrombin & FXa to promote secondary hemostasis
Protein C (PC) deficiency
Protein S (PS) deficiency
Factor V Leiden
Autosomal dominant point mutation of the Factor V gene
Single nucleotide substitution (G1691A) results in mutated form of FVa protein
FVa becomes resistant to aPC & PS inactivation
FVa is broken down at ↓ rate
Prothrombin mutation
Autosomal dominant point mutation of the Prothrombin gene
Single nucleotide substitution (G20210A) of the gene’s 3’ untranslated region
Accumulation of prothrombin mRNA & protein copies of prothrombin
↑ concentration of prothrombinà ↑ conversion to thrombin
↑ clotting factor quantity or function due to mutations intrinsic to these clotting factors
Activated Protein C (aPC) & PS combine to inhibit
activated clotting Factors V & VIII (FVa & FVIIIa)
Autosomal dominant mutation of the PROC & or PROS1 gene ↓ production of PC and/or PS
↓ aPC & PS concentrationsà less breakdown of FVa & FVIIIa, ↑ blood’s clotting ability
↑ clotting factor function due to ↓ inhibition or breakdown of clotting factors
Congenital Thrombophilia
An inherited abnormality / imbalance of blood coagulation factors that increases the risk of thrombus formation
↑ generation of thrombin
↑ clotting tendency, especially in veins where blood flow is slower/prone to stasis (see Calgary Guide slide on Virchow’s Triad)
Venous Thromboembolism
Deep Vein Thrombosis Pulmonary Embolism
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published August 15, 2021 on www.thecalgaryguide.com")
epithelial-ovarian-cancer-pathogenesis-and-clinical-findings

lesions of the fallopian tube fimbria develop overtime
STIC cells may break off from the fallopian tube & become trapped during inclusion cyst formation
As part of the ovarian cycle, the coelomic epithelium (CE) of the ovary is ruptured & repaired after ovulation
Incomplete repair of the CE results in invagination of the
rupture site, forming a benign cortical inclusion cyst
CE cells trapped during cyst formation undergoes metaplasia to tubal or other types of epithelium
Endometriosis
Endometriosis causes endometrial tissue to start growing on ovarian CE
Endometrial cells trapped during cyst formation progresses to endometrioma or “chocolate cysts”
Immune cell infiltration & cytokine release inside the
ovary results in dysregulated inflammation
Cancer cells proliferate in distant organs, invading & destroying native cells
Organ failure
Metastatic spread to liver, lung, brain & lymph nodes
Lymphadenopathy
Systemic immune activation
↑ metabolic consumption
↑ capillary surface area & permeability
Fatigue & weight loss ↑ fluid entry into Ascites
peritoneal cavity
Further somatic mutation accumulation leads to malignant transformation of epithelium
Tumor growth stimulates new blood vessel formation to supply itself with nutrients & O2
Omental / peritoneal seeding of cancer cells
Early-stage disease
Asymptomatic
Epithelial Ovarian Cancer
In late-stage disease, tumor may secrete a glycoprotein called mucin 16, also known as CA-125 (sensitive but non-specific biomarker for ovarian cancer)
↑ serum CA-125 levels
Cancer cells proliferate inward & eventually ruptures the ovary
Cancer cells are released into the peritoneal cavity
Tumor growth in local organs (bladder/uterus) progresses to symptomatic cancer
↑ tumor volume & local tumor spread directly disrupts neighboring structures
Pelvic/abdominal pain
Palpable pelvic/abdominal mass Altered urinary frequency
Compression of
colon/bladder Change in bowel habits
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published August 15, 2021 on www.thecalgaryguide.com")
covid-19-pathophysiology-and-clinical-findings
: Pathophysiology and Clinical Findings
Authors: Ryan Brenneis, Yan Yu* Reviewers: Ciara Hanly, Yonglin Mai ()*, Stephen Vaughan* * MD at time of publication
-Respiratory failure -Septic shock -Multiple organ dysfunction
Critical
Respiratory droplet production (cough, sneeze, talking, breathing) by human host or animal vector infected with SARS-CoV-2
Note: Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2) is the name of the betacoronavirus (a positive sense, single stranded RNA virus). COVID-19 is the name of the disease caused by this virus.
rate, ↓ oxygen saturation -Bilateral interstitial infiltrates on Chest X-ray, progressively worsening
Small droplets (<5μm in diameter) are aerosolized and become airborne
Inhalation of aerosolized particles that are suspended in air
Patient exposed to the virus (SARS-CoV-2)
Droplet contacts the mucus membranes (eyes, nose, mouth) of recipient
Live viral particles can adhere to inanimate objects called fomites (e.g. doorknobs)
Symptoms are on a continuum -Worsening dyspnea: ↑ respiratory
-Fever
-Cough
-Myalgia, fatigue -Nausea/vomiting -Diarrhea
-Loss of taste/smell
Recipient touching infected fomite subsequently touches any of their mucus membranes
Mild
Moderate
Severe
Virus spreads in the body via 1) mucus membrane spread to surrounding cells and 2) entering the blood
Virus adheres to angiotensin-converting enzyme 2 (ACE-2) receptor on body cells, mimics ACE-2, & gains access into cell
COVID-19
Symptomatic infection with SARS-CoV-2
Viral proliferation in cells of tissues with more ACE-2 receptors: lungs (type II pneumocytes); vasculature (endothelial cells), kidneys (proximal tubular epithelium), heart (myocardium), GI tract (enterocytes)
Cell death and ↑ in inflammatory cytokines triggers immune response
Neutrophils move to lungs, release reactive oxygen species and cytokines
Alveolar/capillary damage
Fluid accumulates in interstitium and alveoli
↑ Distance for O2 to diffuse from alveoli to capillaryà ↓ blood O2 saturation
Cytokines induce the hypothalamus to release prostaglandins
↑ body temperature set point to fight infection
Virus disassembles, releasing viral RNA, which uses host cell’s ribosomes to make new viral proteins like RNA-polymerases
Newly made viral RNA-polymerases use cell’s own nucleotides to synthesize new viral RNA
Bilateral ground-glass opacities (CT lung) & interstitial infiltrates on Chest X-ray
Dyspnea
Airway irritation
Cough
Myocardial cell damage
↑ Troponin
↑ skeletal muscle cell damage
Viral RNA & proteins packaged into new viral particles
New virus assembled and released from cell, killing the cell and contributing to disease symptoms
Average incubation period (time from initial infection to symptom onset) is 4-5 days; can be up to 14 days
Heart tries to compensate for
Fever Myalgia
hypoxemiaà↑ cardiac output Arrhythmogenic
and strain on myocardium
state
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published March 22, 2020, updated Aug 18, 2021 on www.thecalgaryguide.com")
cystic-fibrosis-findings-on-chest-x-ray-and-lung-window-ct-scan
, Mark Montgomery* * MD at time of publication
Collapsed alveoli appears white on x-ray
Peribronchial Cuffing
secretions become more viscous. (See relevant slide for CF pathogenesis.)
As early as at birth, secretions collect in bronchial lumen, delaying mucociliary clearance
Mucus plugs and obstructs bronchial lumen
Air trapped distal to obstruction and cannot leave lungs
Lung Hyperinflation
Diaphragm domes below 10th posterior rib and 6th anterior rib on PA CXR
Flattened hemidiaphragm, enlarged retrosternal space on lateral CXR
With time, pulmonary capillaries gradually absorb gases in alveoli distal to obstruction àalveolar collapse (Atelectasis)
Collapsed alveoli more solid and radiodense than airà↓X-ray penetration
Adjacent structures may shift towards atelectasis on CXR
In first few years of life, retained bronchial secretions serve as nidus for recurrent bacterial colonization and infection
Late findings at 10- 30 years oldà secretion accumulation blocks inhaled air to affected lung segments àchronic hypoxia
Inflammatory response (cytokines, nitric oxide and radical oxygen species) leads to bronchial and peribronchial destruction
Hypoxia induces pulmonary capillary vasoconstriction
Some airways are blocked more than others
Leakage of fluid into bronchial walls & peri- bronchial regions.
Inflammatory cytokines destroy elastic components of bronchi
Accumulation of inflammatory exudate in bronchi
Pulmonary artery hypertension à Blood backs up in pulmonary arteries
Some segments of lung underventilated
Fluid around bronchial walls is more radiodense than air à↓X-ray penetration àappears white
Bronchiectasis: dilated, thickened, untapered bronchi on CXR/CT
Fluid/mucus in bronchi is more radiodense than air, thus appearing white on imaging
Edematous bronchi viewed end on are thickened and appear ring-like on CT/CXR
Tramlines (AKA Tramtracking)
Bronchus wider than corresponding blood vessel on CXR/CT
Advanced bronchiectasis is saccular in appearance
Mucus-filled bronchi form tubular opacities giving appearance of white fingers on CXR/CT
Signet Ring Sign
Bronchiectatic Cavities
Mucoid Impaction (AKA Finger in glove sign)
↑ Blood dilates pulmonary arteries
Blood backs up in right ventricle, dilating it
Pulmonary arteries look wider on CXR.
Main pulmonary artery >30 mm diameter on CT
Right Ventricle Enlargement on CXR/CT
Underventilated lung segments appear as lower intensity (darker) than normal ventilated segments which appear as normal intensity (brighter)
Mosaic Changes on CT(non- contrast
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published June 13, 2013, updated Aug 18, 2021 on www.thecalgaryguide.com")
asthma-pathogenesis
 Naushad Hirani* * MD at time of publication
Genetic factors
(i.e. HLA gene mutations, defects in bronchial airway epithelium)
Environmental factors
(i.e. excess hygiene, fewer siblings, antibiotics within the first two years)
Asthma:
Defined as airway hyper-responsiveness causing variable and reversible airflow obstruction
Atopy:
predisposition to allergic hyper-sensitivity in airways
First exposure to triggers*
sensitizes helper T cells
Stimulation of B-cells to produce IgE, which binds to mast cell surfaces
Activated Helper-T cells & IgE-sensitized mast cells now line the airways
Triggers of airway hyper- responsiveness include:
Upper respiratory tract infections (URTIs)
Allergens (pollen, animal dander, dust, mold, etc)
Air pollution, cigarette smoke, other chemicals
Drugs (aspirin, NSAIDs, Beta- blockers)
Cold air
Exercise
Early response (0-2 hrs)
Delayed response (3-4 hrs)
Allergens cross-link IgEs on mast cells
Activated mast cells & helper T cells release cytokines
Mast cells release histamines, leukotrienes, and other inflammatory mediators
Induce maturation of granular WBCs like eosinophils
Eosinophils migrate into:
Vascular permeabilityà edema of airway mucosa
Goblet cell hyperplasia à mucus secretion
Bronchial smooth muscle contraction
Airway obstruction
Second exposure to triggers
Asthma Airways Bronchiole constriction
Eyes Conjunctivitis Nose Rhinitis
Note: Delayed response presents within 3-4 hrs, peaks within 6-8 hrs, and resolves within 24 hrs
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published Dec 17, 2012, updated Aug 19, 2021 on www.thecalgaryguide.com")
disseminated-intravascular-coagulation
: Pathogenesis and clinical findings
Authors: Emily Wildman Mehul Gupta Sean Spence Yan Yu* Reviewers: Kiera Pajunen Wendy Yao Tristan Jones Man-Chiu Poon* Lynn Savoie* * MD at time of publication
Sepsis
Microorganisms express pathogen- associated molecular patterns (PAMPs)
Severe Trauma
Damaged endothelial cells release damage- associated molecular patterns (DAMPs)
Solid and Hematologic Malignancies
Cancer cells express tissue factor and release procoagulants
Immune cells recognize DAMPs and PAMPs and begin expressing tissue factor and releasing procoagulants
Systemic activation of coagulation cascade
Activation of coagulation cascade results in the factor X mediated conversion of large quantities of prothrombin (factor II) to its active form, thrombin (factor IIa)
Thrombin cleaves fibrinogen (factor I) circulating in blood to an activate form known as fibrin (factor 1a)
↓ Serum fibrinogen
Diffuse coagulation causes imbalances between coagulation and anticoagulation pathways
Consumption of coagulation factors (including platelets, fibrinogen, prothrombin, factor V, factor VIII) exceeds rate of production
Fibrin, in conjunction with platelets, form widespread thrombi within vasculature
Fibrin thrombi accumulate in the microvasculature and shear transiting red blood cells
Microangiopathic hemolytic anemia (MAHA)
↑ thrombi formation ↑ fibrinolysis, a parallel process that naturally degrades fibrin thrombi
Relative deficiency of coagulation factors leads to a bleeding diathesis (tendency to bleed) despite widespread clots
↑ Prothrombin time (PT)
↑ Partial thromboplastin time (PTT)
↑ D-dimer Fibrin thrombi
↑ Fibrin degradation products
Platelets are used up forming thrombià less free platelets in circulation
↓ Platelets on Complete Blood Count
accumulate in microvasculature of major organs
Deposition of thrombi occlude blood flow resulting in ischemia of organ parenchyma
Lack of coagulation factors can predispose spontaneous hemorrhage in various organs
Multiple organ dysfunction syndrome (MODS) - (renal failure, hepatic dysfunction, stroke, pulmonary disease)
Clinical manifestations of bleeding, including petechiae (pinpoint red spots on skin), ecchymoses (bruising), weeping wound sites, bleeding mucous membranes
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
First published Aug 7, 2012, updated July 27, 2019 & Aug 29, 2021 on www.thecalgaryguide.com")
convulsions-febrile

Brianna Ghali Eddy Lang*
Infection (souvent HHV6 ou la grippe) chez l'enfant (généralement âgé de 6 mois à 6 ans)
Température cérébrale élevée de manière exogène (par example un bain chaud)
Mutation de la sous-unité du transporteur sodium
Synthèse de l'IL-1β dans l'hippocampe
Fièvre ≥380C
Mutation du récepteur GABAA
Conditions physiologiques
Susceptibilité génétique familiale ou sporadique aux températures élevées
La température élevée a un impact sur les canaux ioniques
↑ glutamate et/ou ↓ GABA
↑ excitabilité et synchronisation de l'activité
Crise fébrile simple
Convulsions fébrile complexes
L'étiologie sous-jacente diffère de celle des crises fébriles simples.
Signes focaux pendant la crise (déviation des yeux, rotation de la tête, etc.)
Prolongée >15 min État épileptique fébrile
Une ponction lombaire doit être envisagée pour exclure une infection grave, c'est-à- dire une méningite.
Crise tonico-clonique généralisée unique sans signes focaux d'une durée de <15 minutes
EEG et IRM normaux (l'IRM n'est généralement pas indiquée)
Légende:
Physiopathologie
Mécanisme
Signe/Symptôme/ Résultat Laboratoire
Complications
Publié 21 janvier 2019 à www.thecalgaryguide.com")
Localisation-des-AVC
 dans l'ACA
↓ perfusion (ischémie) dans l'ACM
M1-ACM
M2-ACM
↓ perfusion (ischémie) dans l’ACP
↓ perfusion (ischémie) dans l’ABV
↓ perfusion (ischémie) dans l’AB
Faiblesse et perte sensorielle controlatérale dans la membre inférieure
Abréviations:
ACA : artère cérébrale antérieure AB : artère basilaire
ACM : artère cérébrale moyenne ACP : artère cérébrale postérieure UE : membre supérieur
Lésion de l’hémisphère gauche
Aphasie
Déficits de la perception visuelle et Lésion de l’hémisphère droite négligence (agnosie du côté gauche)
ABV : artère basilaire vertébrale Aucune hémianopsie homonyme
Hémiparésie et déficits sensoriels controlatéraux, déficits du champ visuel, aphasie, agnosie, apraxie, agraphie.
Affect seulement la member supérieur et le visage
Aphasie expressive de Broca (moteure)
Hémianopsie homonyme controlatérale Aphasie réceptive de Wernicke (sensorielle)
Perte sensorielle, perte de mémoire, hémianopsie homonyme controlatérale, alexie.
Problèmes des nerfs crâniens : dysarthrie (IX, X), diplopie, paresthésie du visage (VII), syndrome de Foville.
Déficits moteurs : Syndrome de Millard-Gubler, syndrome de Raymond, syndrome de Wallenburg, ataxie.
Perte sensorielle unilatérale ou bilatérale (↓ sensation de la position et des vibrations).
Problèmes des nerfs crâniens : regard dysconjugué (III, IV, VI), hypoalgésie faciale ipsilatérale (V), paralysie faciale motoneurone inférieure unilatérale (VII), vertiges, dysarthrie (IX, X).
Déficits moteurs : hémiparésie controlatérale, quadriplégie. Hypoalgésie du membre controlatéral
Définitions :
Agnosie : incapacité à traiter les informations sensorielles. Agraphie : incapacité à communiquer par écrit
Alexie : difficulté de lecture
Aphasie : trouble du langage
Apraxie : déficit de la planification motrice
Ataxie : anomalie de la démarche
Aphasie de Broca : incapacité de produire du langage
Diplopie : vision double
Dysarthrie : troubles de l'élocution.
Syndrome de Foville : ataxie cérébelleuse ipsilatérale, syndrome de Horner, parésie du regard conjugué et hémiparésie controlatérale, paralysie faciale, douleur et hypoesthésie thermique.
Hémianopsie : perte du champ visuel
Hypoalgésie : diminution de la sensibilité à la douleur
Syndrome de Millard-Gubler : lésion de la protubérance dorsale.
Aphasie de Wernicke : discours fluide, compréhension affectée Syndrome de Wallenburg : déficits sensoriels du membre controlatéral, du visage ipsilatéral.
Légende:
Pathophysiologie
Méchanisme
Signe/Symptôme/Résultat Laboratoire
Complications
Publié 3 février 2018 à www.thecalgaryguide.com")
Pneumoconioses

Inhalation of carbonaceous dust
(Carboconiosis)
Inhalation of metal dust
(Metaloconiosis)
Inhalation of silica dust
(Silicosis)
“-Coniosis”: disease which comes from inhaling dust particles
Internalized asbestos fibers disrupt cellular processes through a complex series of theorized mechanisms
Inhaled dust particles (1-5μm in size) are trapped and deposited in the alveolar airspaces
Alveolar macrophages ingest dust particles, which activates the macrophages and sometimes causes apoptosis
Activated macrophages create an inflammatory microenvironment by releasing pro-inflammatory cytokines, chemokines and reactive oxygen species
Inflammatory products (e.g. reactive oxygen species) damage alveolar epithelial cells, causing activation and release of inflammatory cytokines into the alveolar space
Inhaled dust particles, inflammatory products & other pro- tussive mediators activate airway vagal afferent receptors
Activated receptors stimulate the cough center located in the medulla oblongata
Cough
Alveolar capillaries vasoconstrict in response to hypoxia à↑ Pulmonary vascular resistance
Pulmonary Hypertension
Right heart must pump blood into lungs against higher pressure àcardiomyocyte growth (via sarcomeres formed in parallel within myofibrils)àconcentric hypertrophy of right heart
Cor Pulmonale (right heart failure due to pulmonary hypertension)
Asbestos fibers accumulate in the airspace and translocate to the pleural surface
Macrophage apoptosis: ↓ alveolar macrophages
Fibroblasts are recruited to the alveolar wall and are activated
Activated fibroblasts produce and deposit collagen in the
extracellular space between alveoli
Thickening of tissue between alveoli and capillaries ↑ the diffusion distance of atmospheric and blood gasses
↓ Diffusion of CO2 from blood to alveoli and ↓ diffusion of O2 from alveoli to blood
↑ Respiratory rate to maintain minute ventilation due to ↓ lung volumes and diffusion limitations
Dyspnea and Exertional Hypoxemia
↓ Innate immune response in the lungs
Chest X-Ray: Nodular and reticulonodular opacities are seen in varying lung regions depending on the underlying inhaled dust
Excessive collagen
deposition ↓ lung compliance and ↓ lung expansion
↓ Diffusing capacity for carbon monoxide (DLCO) on pulmonary function test
↓ Arterial oxygen contentà ↑ deoxyhemoglobin and ↓ oxyhemoglobin
↑ Deoxyhemoglobin within the vasculature causes the skin and mucous membranes to appear blue
Cyanosis
↑ Risk of respiratory infections. Mycobacterial infections (e.g. tuberculosis) associated primarily with silicosis
↓ Inhalation volume
↓ Expiratory volume
Hypoxemia
↓Total lung capacity (TLC) and ↓ residual volume (RV) on pulmonary function test
↓Forced vital capacity (FVC) and ↓ forced expiratory volume in 1 second (FEV1) on pulmonary function test
Note:
Forced Vital Capacity: the volume of air that can forcibly be blown out after full inspiration
Forced Expiratory Volume in 1 second: the volume of air that can forcibly be blown out in first 1 second, after full inspiration
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published September 12, 2021 on www.thecalgaryguide.com")
COVID-19 (Corona Virus Disease 2019): Pathophysiologie und Klinische Befund
: Pathophysiologie und Klinische Befund
Autoren:: Ryan Brenneis, Yan Yu* Rezensenten: Ciara Hanly, Yonglin Mai (麦泳琳)*, Stephen Vaughan* Übersetzerin: Sarah Schwarz Übersetzungsprüfer: Gesche Tallen* * MD zum Zeitpunkt der Veröffentlichung
Produktion von respiratorischen Tröpfchen (husten, sprechen, niessen) durch Mensch oder Tier, welche mit SARS-CoV-2 infiziert sind
Beachte: Schweres Akutes Respiratorisches Syndrom Coronavirus 2 (SARS- CoV-2) ist der Name des Betacoronavirus (mit positiver Einzelstrang RNA ). COVID-19 ist der Name der Krankheit die das Virus verursacht
Kleine Tröpfchen (Durchmesser <5μm) werden aerosoliert & verteilen sich in der Luft
Inhalation der Aerosole
Exposition des Patienten gegenüber dem Virus (SARS-CoV-2)
Kontakt der Tröpfchen mit Schleimhäuten (Augen, Nase, Mund) des Empfängers
Kontaminierung eines unbelebten Gegenstandes (z.B. einer Türklinke) mit aktiven Viruspartikeln
-Fieber
-Husten -Gliederschmerzen -Schwäche -Übelkeit/Erbrechen -Durchfall -Geruchs-und Geschmacksverlust
Mild
Symptome nach Schweregrad
-Dyspnoe: Atemfrequenz↑, Sauerstoffsättigung ↓, -Bilaterale interstitielle Infiltrationen im Röntgen -progrediente Verschlechterung
-Respiratorische Insuffizienz -Septischer Schock -Multiorganversagen
Kritisch
Zelltot & vermehrte Zytokinausschüttung aktiviren die Immunantwort
Zytokine stimulieren die hypothalamische Prostaglandinaussc hüttung
Erhöhung der Körpertemperatur zur Bekämpfung der Infektion
Empfänger berührt den infizierten Gegenstand & anschließend einen Schleimhautbereich
Moderat
Schwer
Virusausbreitung im Körper 1) über die Schleimhäute in die Umgebung & 2) die Blutzirkulation
Das Virus imitiert das Angiotensin-Converting-Enzyme 2 (ACE2), haftet an dessen Rezeptoren & kann so in verschiedene Körperzellen eindringen
COVID-19
Symptomatische Infektion mit SARS-CoV-2
Virale Proliferation in Geweben mit vermehrter ACE2-Rezeptor Expression: in Lunge (Typ II Pneumozyten), Gefäßen (Endothelzellen), Niere (Zellen des Proximalen Tubulus), Herz (Myokardzellen), GIT (Enterozyten)
Virusanteile setzen virale RNA frei, die die Ribosomen der infizierten Zelle nutzen, um Proteine wie die RNA- Polymerase zu synthetisieren
Die neue Polymerase benutzt die Nukleotide der Zelle um neue Virus - RNA zu produzieren
Bilaterale Milchglastrübung (CT Lunge) & interstitielle Infiltrate im Röntgen
Luftnot
Neutrophile setzen reaktive Sauerstoffspezies & Zytokine in der Lunge frei
Alveolärer und kapillärer Schaden
Flüssigkeitsansammlungen in Alveolaren und Interstitium
Diffusionsstrecke für O2 zw. Alveole und Kapillare ↑, Sauerstoffsättigung↓
Irritation der Atemwege
Husten
Myokardschaden
Troponin ↑
Aus viraler RNA und Proteinhülle entstehen neue Viruspartikel
Die neuen Viren werden freigesetzt, die Zelle geht dabei unter, was zu den Symptomen der Krankheit beiträgt
Die durchschnittliche Inkubationszeit (von Infektion bis zu ersten Symptomen) beträgt 4-5 Tage, kann aber bis zu 14 Tagen andauern
Herz versucht, Hypoxie zu kompensieren, HZV ↑, erhöhte Herzbelastung
Herzrythmusstörung
Skelettmuskels chaden
Fieber Myalgie
Legende:
Pathophysiologie
Mechanismen
Symptome/Klinische Befunde
Komplikationen
Veröffentlicht: 22. März, 2020, aktualisiert Aug 18, 2021 on www.thecalgaryguide.com")
Pathogenese des Diabetes Mellitus (DM), Typ II
, Typ II
Genetische Prädisposition:
Poly- oder monogenetische Faktoren (z.B. „Maturity- onset diabetes of the young“ (MODY)) können eine Insulinresistenz prädispositionieren
Altern: Betazellen verlieren im Rahmen des Alterungsprozesses an Masse, wodurch sich, bei bereits bestehender Pädisposition, im Alter ein DM Typ II entwickeln kann .
Medikamente: z.B. Cortison, Psychopharmaka, hochaktive antiretrovirale Therapie(HAART), Minipille (nur auf Progesteron basierende Kontrazeptiva)
Blutzucker bleibt im Normbereich
Zuckervergiftung: Hyperglykämien wirken toxisch auf Betazellen
Hyperglykämie
Beachte: Es besteht eine beträchtliche genetische Komponente: bei familiärer Vorbelastung ist die Erkrankungswahrscheinlichkeit sehr hoch ( bei eineiigen Zwillingen bis zu 90%). Bei Verwandtschaftsgrad ersten Grades ist das Risiko einer Erkrankung 5-10 Mal höher
Ungesunder Lebensstil (z.B. Übergewicht, Bewegungsmangel)
Intraabdominell akkumuliert „Viszeralfett“ (Bauchfett) welches als endokrines Organ funktioniert:
Beachte: „Adipokine“ sind von Fettgewebe produzierte Entzündungsmediatoren (z.B. TNFalpha). Je mehr Fettgewebe ein Mensch besitzt, desto mehr Adipokine werden produziert.
Entzündungsm ediatoren
Freie Fettsäuren
Adipokine
Komplexe, teils unklare Interaktionen mit dem Gewebe
Insulinresistenz
(Die Wirkung des Insulins auf Leber, Muskel und Fettgewebe ist vermindert, Glucose kann deshalb nicht als Energiequelle genutzt werden )
Zunächst kompensieren Betazellen des Pankreas die verminderte Insulinwirkung durch vermehrte Produktion
Fettvergiftung:
Längerfristig nimmt die Kompensationsfähigkeit der Betazellen ab, Insulinproduktion ↓ (relativer Insulinmangel)
Nach Jahren nimmt die Leistungsfähigkeit der Betazellen soweit ab, dass kein Insulin mehr produziert wird (absoluter Insulinmangel)
Diabetes Mellitus Typ II
Freie Fettsäuren blockieren den GLUT2 der Betazellen, Glucoseimport ↓
Betazellen erkennen den erhöhten Blutzucker nicht, Insulinproduktion sinkt
Da der Körper keine Glukose nutzen kann, werden Triglyceride zu freien Fettsäuren umgewandelt damit diese als Energiequelle dienen können
Autor: Yan Yu Rezensenten: Peter Vetere Gillian Goobie Doreen Rabi* Übersetzerin: Sarah Schwarz Übersetzungsprüfer: Gesche Tallen* * MD zum Zeitpunkt der Veröffentlichung
Legende:
Pathophysiologie
Mechanismen
Symptome/Klinische Befunde
Komplikationen
Veröffentlicht 11. Juli 2013 auf www.thecalgaryguide.com")
Pathogenese des Diabetes Mellitus (DM), Typ I
, Typ I
Genetische Prädisposition
IDDM1 (HLA) Mutation IDDM2 (Insulingene) Mutationen
Diet (Kuhmilch, Nitrosamine)
Viren (Röteln, Coxsackie-Virus, Masern)
Drogen/Toxine
(Pyrinuron,Alloxan, Streptozocin, Pentamidine)
Direkter Schaden an Betazellen,
sodass deren Antigene dem
Immunsystem ausgesetzt werden
Stress (häufige Infektionen, Operationen, Pupertät)
Externe Risikofaktoren
Reifende T-Zellen im Thymus sind nicht in der Lage, die Fähigkeit der Erkennung von Insulingenen zu entwickeln T-Zellen greifen Insulin produzierende Betazellen an
Mutationen des HLA (MCH) Gens können die Bindung von T-Zellen an Betazellen fördern aber auch verhindern
Körperfremde Antigene imitieren Betazellantigene (Molekulare Mimiky), sodass sich die Immunantwort gegen diese Antigene auch gegen Betazellen richtet
Beeinträchtigung der Toleranz des Immunsystem gegen körpereigene pankreatische Betazellen
Autoimmunreaktion gegen Betazellen
(sowohl durch angeborenes, als auch durch erworbenes Immunsystem: Infiltration der Langerhans-Inseln durch Monozyten und Zerstörung durch T-Zellen, auch als Insulitis bezeichnet )
Atrophie der Langerhans-Inseln auf die Hälfte ihrer Ausgangsmasse
Längerfristig sind nur noch <10% der Betazellen funktionstüchtig
Beachte: Die Anzahl an Autoantikörpern im Körper korrelieren mit der Wahrscheinlichkeit einen DM Typ I zu entwickeln
Autor: Yan Yu
Rezensenten:
Peter Vetere
Gillian Goobie
*Doreen Rabi
Übersetzerin: Sarah Schwarz Übersetzungsprüfer: Gesche Tallen*
* MD zum Zeitpunkt der Veröffentlichung
Produktion von Autoantikörper gegen Betazellen
“Prä-diabetes”
Diabetes Mellitus Typ I Absoluter Insulinmangel
Anti-Insulin & Anti-GAD65 nachweisbar im Serum
Noch asymptomatisch Hoher postprandialer Blutzucker
Abgeschwächte Insulinreaktion nach i.v. Glucosegabe
Legende:
Pathophysiologie
Mechanismen
Symptome/Klinische Befunde
Komplikationen
Veröffentlicht: 11. Juli 2013 auf www.thecalgaryguide.com")
Primary-Adrenal-Insufficiency

Note: “Primary” refers to pathology in the gland that produces the functional hormone (in this case, the adrenal gland), as opposed to “secondary” or “tertiary” which refers to pathology in glands that indirectly control the primary gland.
Bilateral adrenal Autoimmune damage (cancer,
↓ Androgen levels in women (in whom adrenal glands are the primary source)
↓ Libido
↓ Axillary and pubic hair
Muscle/joint pain
Anorexia, weight loss
Abdominal pain, nausea, vomiting
Diseases bleeding, etc)
↓ Androgen production from the zona reticularis
↓ Cortisol production from the zona fasciculata
↓ Serum cortisol
Patients could present with many severe symptoms of
adrenal insufficiency, e.g. low blood pressure refractory to fluid resuscitation
Significant bilateral damage to the adrenal glands
Entire adrenal cortex function impaired
↓ Serum levels of adrenal steroid hormones exert positive feedback effect on pituitary gland, ↑ pituitary gland’s production of adrenocorticotropic hormone (ACTH)
↑ Serum levels of ACTH
The same gene that encodes ACTH also encodes melanocyte stimulating hormone (MSH)
Coincidental ↑ in MSH production by the pituitary
↓ Serum DHEAS, testosterone and androstenedione
Unknown mechanism(s)
Malaise
Adrenal Crisis
↓ Cortisol mediated gluconeogenesis and glycogenolysis
↑ Cortisol Releasing Hormone (CRH) from the hypothalamus
Lack of cortisol-driven vasoconstriction of blood vesselsà↑ Vasodilation
↓ Blood pressure, postural dizziness
Water follows sodiumà ↑ excretion of water
↓ Sodium resorbed from kidney tubulesà↑ Sodium excretion
↓ Potassium pumped into kidney tubules à↑ potassium retained in serum
Hypoglycemia (↓ blood glucose)
↑↑ Antidiuretic hormone (ADH) release
Kidneys reabsorb water à↑ total body water
Hyponatremia (↓ serum sodium)
↑ Salt craving
Hyperkalemia (high serum potassium level)
↓ Aldosterone production from the zona glomerulosa
↓ Serum aldosterone
MSH triggers ↑ production of melanin by the melanocytes in skin
↓ Excretion of hydrogen ions from kidneys (See type IV RTA slide for full mechanism)
Metabolic acidosis (low pH, low bicarbonate)
↓ Sodium channel and Na/K ATPase
expression on principle cells of the kidney
Hyperpigmentation of the skin and mucosal surfaces
Authors: Jaye Platnich, Brooke Fallis, Yan Yu* Reviewers: Mark Elliott, Alexander Arnold, Hanan Bassyouni*, David Campbell* * MD at time of publication
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published November 13, 2013, updated October 1, 2021 on www.thecalgaryguide.com")
A-a Gradient: why it exists and why we care
, Zesheng Ye (叶泽生), Yonglin Mai (麦泳琳)*, Juri Janovcik* * MD at time of publication
Why it exists, and why we care
Key Abbreviations:
• pO2: partial pressure of O2,
aka. “O2 content”.
• PaO2: partial pressure of O2 in
the arteries. Measured directly
via Arterial Blood Gas (ABGs). • PAO2: partial pressure of O2 in
the Alveoli. (Can’t be measured directly, must be calculated).
Theoretically, in lung capillaries adjacent to alveoli: O2
should diffuse from alveoli into lung capillaries, and no O2 should be lost from the blood until the blood reaches the systemic arteries – so the PaO2 should equal PAO2.
But in reality...
Blood in the lung capillaries are not fully oxygenated to begin with:
Since gravity causes more blood to settle to the lung bases, there is too much blood there for it all to be fully oxygenated by the alveoli.
Less well-oxygenated blood from lung bases reduces the blood’s overall pO2
Less oxygenated blood from systemic veins are mixed into oxygenated blood from the lungs (“Venous admixture”):
Venous drainage from the bronchial artery directly mixes with the pulmonary veins (oxygenated blood)
Some veins in the coronary circulation drain into the left ventricle, instead of into the coronary sinus/R atrium
Thus, O2 content of blood when it finally reaches the systemic arteries (PaO2) is less than the O2 content in the alveoli (PAO2)
PAO2 - PaO2 = the pO2 “gradient” between the alveoli and the systemic arteries
Note: Abnormally high A- a gradients indicate issues with gas diffusion between alveoli & the pulmonary capillaries.
Note: some respiratory pathologies can present with a normal A-a gradient as well (see relevant slides)
Normal A-a Gradient: <15 mmHg
High A-a Gradient (>15 mmHg) is always a sign of pathology (see relevant slides)
Legend:
Definitions
Explanations
Sign/Symptom/Lab Finding
Complications
Published September 4, 2013, updated October 5, 2021 on www.thecalgaryguide.com
The Alveolar-arterial pO2 gradient: Explainingtheformula A-aGradient=PO –PO
(The simplified explanation)
A 2
a
2
For an explanation of the physiology behind
why an A-a gradient exists, please consult: “The Alveolar-arterial pO2 gradient: Why it exists, and why we care”
Fraction of inhaled air made up of O2 (21%, if atmospheric)
Partial Pressure of O2 within the alveoli:
Partial Pressure of O2 in systemic arteries Measured directly via Arterial Blood Gas (ABG)
PAO2 =
(Total pressure of O2 inhaled into alveoli)
(Air pressure generated by CO2 – molecules that are produced by
the consumption of O2 molecules
PAO2 = FiO2(PB – PH2O) – (PaCO2/0.8)
Barometric Pressure Total air pressure inhaled into lungs (760mmHg at sea level, and less at higher altitudes)
Partial pressure of water (47 mmHg): Inspired air is humidified once inside airways. Water vapor adds to total inhaled air pressure, and must be accounted for
Partial pressure of dissolved CO2 in arteries, directly measured by an ABG
Respiratory quotient: conversion ratio reflecting how, for every 5 molecules of O2 the blood absorbs or consumes, it releases approximately 4 CO2 molecules into the alveoli
Partial pressure of gaseous CO2 in alveoli (PACO2)
CO2 produced by body metabolism, exhaled into alveoli
Author: Yan Yu Reviewers: Steven Liu, Amogh K. Agrawal, Ciara Hanly, Xiumei Deng (邓秀梅), Zesheng Ye (叶泽生), Yonglin Mai (麦泳琳)*, Juri Janovcik* * MD at time of publication
Legend:
Definitions
Explanations
Sign/Symptom/Lab Finding
Complications
Published September 4, 2013, updated October 5, 2021 on www.thecalgaryguide.com
The Alveolar-arterial pO2 gradient:
Explaining the formula
(The scientifically-correct explanation)
For an explanation of the physiology behind
why an A-a gradient exists, please consult: “The Alveolar-arterial pO2 gradient: Why it exists, and why we care”
A-a Gradient = PAO2 – PaO2 Partial Pressure of O2 within the alveoli:
Partial Pressure of O2 in systemic arteries
Measured directly via Arterial Blood Gas (ABG)
Rationale: While O2 is being absorbed from the alveoli into the capillary, CO2 is being released from the capillary into the alveoli. Normal body metabolism is such that the amount of CO2 released is directly proportional to the amount of O2 absorbed.
PAO2 =
(Total pO2 entering alveoli)
Can be calculated easily
– (total pO2 leaving alveoli and absorbed into blood)
Cannot be directly measured; need to estimate it using the level of CO2 in the alveoli, which we can measure
PAO2 = FiO2(PB – PH2O) – (PaCO2/0.8)
Fraction of inhaled air made up of O2 (21%, if atmospheric)
Barometric Pressure Total air pressure inhaled into lungs (760mmHg at sea level, and less at higher altitudes, ie: 660mmHg in Calgary)
47 mmHg: Inspired air is humidified once inside airways. Water vapor adds to total inhaled air pressure, and must be accounted for
Partial pressure of dissolved CO2 in arteries, directly measured by an ABG
Respiratory quotient (RQ): the ratio of the amount of CO2
released from the arterial blood to
the amount of O2 absorbed or
consumed by the blood. The value
of the RQ can vary depending
upon the diet consumed
Note: The respiratory quotient (carbohydrates, fats, and proteins)
(RQ) is always <1 because
and the metabolic state. RQ is
around 0.82 for the average human diet.
Author: Yan Yu Reviewers: Steven Liu, Amogh K. Agrawal, Ciara Hanly, Xiumei Deng (邓秀梅), Zesheng Ye (叶泽生), Yonglin Mai (麦泳琳)*, Juri Janovcik* * MD at time of publication
Legend:
Definitions
Explanations
Sign/Symptom/Lab Finding
Complications
Published September 4, 2013, updated October 5, 2021 on www.thecalgaryguide.com")
A-a Gradient: Explaining the formula simplified
, Zesheng Ye (叶泽生), Yonglin Mai (麦泳琳)*, Juri Janovcik* * MD at time of publication
Why it exists, and why we care
Key Abbreviations:
• pO2: partial pressure of O2,
aka. “O2 content”.
• PaO2: partial pressure of O2 in
the arteries. Measured directly
via Arterial Blood Gas (ABGs). • PAO2: partial pressure of O2 in
the Alveoli. (Can’t be measured directly, must be calculated).
Theoretically, in lung capillaries adjacent to alveoli: O2
should diffuse from alveoli into lung capillaries, and no O2 should be lost from the blood until the blood reaches the systemic arteries – so the PaO2 should equal PAO2.
But in reality...
Blood in the lung capillaries are not fully oxygenated to begin with:
Since gravity causes more blood to settle to the lung bases, there is too much blood there for it all to be fully oxygenated by the alveoli.
Less well-oxygenated blood from lung bases reduces the blood’s overall pO2
Less oxygenated blood from systemic veins are mixed into oxygenated blood from the lungs (“Venous admixture”):
Venous drainage from the bronchial artery directly mixes with the pulmonary veins (oxygenated blood)
Some veins in the coronary circulation drain into the left ventricle, instead of into the coronary sinus/R atrium
Thus, O2 content of blood when it finally reaches the systemic arteries (PaO2) is less than the O2 content in the alveoli (PAO2)
PAO2 - PaO2 = the pO2 “gradient” between the alveoli and the systemic arteries
Note: Abnormally high A- a gradients indicate issues with gas diffusion between alveoli & the pulmonary capillaries.
Note: some respiratory pathologies can present with a normal A-a gradient as well (see relevant slides)
Normal A-a Gradient: <15 mmHg
High A-a Gradient (>15 mmHg) is always a sign of pathology (see relevant slides)
Legend:
Definitions
Explanations
Sign/Symptom/Lab Finding
Complications
Published September 4, 2013, updated October 5, 2021 on www.thecalgaryguide.com
The Alveolar-arterial pO2 gradient: Explainingtheformula A-aGradient=PO –PO
(The simplified explanation)
A 2
a
2
For an explanation of the physiology behind
why an A-a gradient exists, please consult: “The Alveolar-arterial pO2 gradient: Why it exists, and why we care”
Fraction of inhaled air made up of O2 (21%, if atmospheric)
Partial Pressure of O2 within the alveoli:
Partial Pressure of O2 in systemic arteries Measured directly via Arterial Blood Gas (ABG)
PAO2 =
(Total pressure of O2 inhaled into alveoli)
(Air pressure generated by CO2 – molecules that are produced by
the consumption of O2 molecules
PAO2 = FiO2(PB – PH2O) – (PaCO2/0.8)
Barometric Pressure Total air pressure inhaled into lungs (760mmHg at sea level, and less at higher altitudes)
Partial pressure of water (47 mmHg): Inspired air is humidified once inside airways. Water vapor adds to total inhaled air pressure, and must be accounted for
Partial pressure of dissolved CO2 in arteries, directly measured by an ABG
Respiratory quotient: conversion ratio reflecting how, for every 5 molecules of O2 the blood absorbs or consumes, it releases approximately 4 CO2 molecules into the alveoli
Partial pressure of gaseous CO2 in alveoli (PACO2)
CO2 produced by body metabolism, exhaled into alveoli
Author: Yan Yu Reviewers: Steven Liu, Amogh K. Agrawal, Ciara Hanly, Xiumei Deng (邓秀梅), Zesheng Ye (叶泽生), Yonglin Mai (麦泳琳)*, Juri Janovcik* * MD at time of publication
Legend:
Definitions
Explanations
Sign/Symptom/Lab Finding
Complications
Published September 4, 2013, updated October 5, 2021 on www.thecalgaryguide.com
The Alveolar-arterial pO2 gradient:
Explaining the formula
(The scientifically-correct explanation)
For an explanation of the physiology behind
why an A-a gradient exists, please consult: “The Alveolar-arterial pO2 gradient: Why it exists, and why we care”
A-a Gradient = PAO2 – PaO2 Partial Pressure of O2 within the alveoli:
Partial Pressure of O2 in systemic arteries
Measured directly via Arterial Blood Gas (ABG)
Rationale: While O2 is being absorbed from the alveoli into the capillary, CO2 is being released from the capillary into the alveoli. Normal body metabolism is such that the amount of CO2 released is directly proportional to the amount of O2 absorbed.
PAO2 =
(Total pO2 entering alveoli)
Can be calculated easily
– (total pO2 leaving alveoli and absorbed into blood)
Cannot be directly measured; need to estimate it using the level of CO2 in the alveoli, which we can measure
PAO2 = FiO2(PB – PH2O) – (PaCO2/0.8)
Fraction of inhaled air made up of O2 (21%, if atmospheric)
Barometric Pressure Total air pressure inhaled into lungs (760mmHg at sea level, and less at higher altitudes, ie: 660mmHg in Calgary)
47 mmHg: Inspired air is humidified once inside airways. Water vapor adds to total inhaled air pressure, and must be accounted for
Partial pressure of dissolved CO2 in arteries, directly measured by an ABG
Respiratory quotient (RQ): the ratio of the amount of CO2
released from the arterial blood to
the amount of O2 absorbed or
consumed by the blood. The value
of the RQ can vary depending
upon the diet consumed
Note: The respiratory quotient (carbohydrates, fats, and proteins)
(RQ) is always <1 because
and the metabolic state. RQ is
around 0.82 for the average human diet.
Author: Yan Yu Reviewers: Steven Liu, Amogh K. Agrawal, Ciara Hanly, Xiumei Deng (邓秀梅), Zesheng Ye (叶泽生), Yonglin Mai (麦泳琳)*, Juri Janovcik* * MD at time of publication
Legend:
Definitions
Explanations
Sign/Symptom/Lab Finding
Complications
Published September 4, 2013, updated October 5, 2021 on www.thecalgaryguide.com")
A-a Gradient: Explaining the formula scientific
, Zesheng Ye (叶泽生), Yonglin Mai (麦泳琳)*, Juri Janovcik* * MD at time of publication
Why it exists, and why we care
Key Abbreviations:
• pO2: partial pressure of O2,
aka. “O2 content”.
• PaO2: partial pressure of O2 in
the arteries. Measured directly
via Arterial Blood Gas (ABGs). • PAO2: partial pressure of O2 in
the Alveoli. (Can’t be measured directly, must be calculated).
Theoretically, in lung capillaries adjacent to alveoli: O2
should diffuse from alveoli into lung capillaries, and no O2 should be lost from the blood until the blood reaches the systemic arteries – so the PaO2 should equal PAO2.
But in reality...
Blood in the lung capillaries are not fully oxygenated to begin with:
Since gravity causes more blood to settle to the lung bases, there is too much blood there for it all to be fully oxygenated by the alveoli.
Less well-oxygenated blood from lung bases reduces the blood’s overall pO2
Less oxygenated blood from systemic veins are mixed into oxygenated blood from the lungs (“Venous admixture”):
Venous drainage from the bronchial artery directly mixes with the pulmonary veins (oxygenated blood)
Some veins in the coronary circulation drain into the left ventricle, instead of into the coronary sinus/R atrium
Thus, O2 content of blood when it finally reaches the systemic arteries (PaO2) is less than the O2 content in the alveoli (PAO2)
PAO2 - PaO2 = the pO2 “gradient” between the alveoli and the systemic arteries
Note: Abnormally high A- a gradients indicate issues with gas diffusion between alveoli & the pulmonary capillaries.
Note: some respiratory pathologies can present with a normal A-a gradient as well (see relevant slides)
Normal A-a Gradient: <15 mmHg
High A-a Gradient (>15 mmHg) is always a sign of pathology (see relevant slides)
Legend:
Definitions
Explanations
Sign/Symptom/Lab Finding
Complications
Published September 4, 2013, updated October 5, 2021 on www.thecalgaryguide.com
The Alveolar-arterial pO2 gradient: Explainingtheformula A-aGradient=PO –PO
(The simplified explanation)
A 2
a
2
For an explanation of the physiology behind
why an A-a gradient exists, please consult: “The Alveolar-arterial pO2 gradient: Why it exists, and why we care”
Fraction of inhaled air made up of O2 (21%, if atmospheric)
Partial Pressure of O2 within the alveoli:
Partial Pressure of O2 in systemic arteries Measured directly via Arterial Blood Gas (ABG)
PAO2 =
(Total pressure of O2 inhaled into alveoli)
(Air pressure generated by CO2 – molecules that are produced by
the consumption of O2 molecules
PAO2 = FiO2(PB – PH2O) – (PaCO2/0.8)
Barometric Pressure Total air pressure inhaled into lungs (760mmHg at sea level, and less at higher altitudes)
Partial pressure of water (47 mmHg): Inspired air is humidified once inside airways. Water vapor adds to total inhaled air pressure, and must be accounted for
Partial pressure of dissolved CO2 in arteries, directly measured by an ABG
Respiratory quotient: conversion ratio reflecting how, for every 5 molecules of O2 the blood absorbs or consumes, it releases approximately 4 CO2 molecules into the alveoli
Partial pressure of gaseous CO2 in alveoli (PACO2)
CO2 produced by body metabolism, exhaled into alveoli
Author: Yan Yu Reviewers: Steven Liu, Amogh K. Agrawal, Ciara Hanly, Xiumei Deng (邓秀梅), Zesheng Ye (叶泽生), Yonglin Mai (麦泳琳)*, Juri Janovcik* * MD at time of publication
Legend:
Definitions
Explanations
Sign/Symptom/Lab Finding
Complications
Published September 4, 2013, updated October 5, 2021 on www.thecalgaryguide.com
The Alveolar-arterial pO2 gradient:
Explaining the formula
(The scientifically-correct explanation)
For an explanation of the physiology behind
why an A-a gradient exists, please consult: “The Alveolar-arterial pO2 gradient: Why it exists, and why we care”
A-a Gradient = PAO2 – PaO2 Partial Pressure of O2 within the alveoli:
Partial Pressure of O2 in systemic arteries
Measured directly via Arterial Blood Gas (ABG)
Rationale: While O2 is being absorbed from the alveoli into the capillary, CO2 is being released from the capillary into the alveoli. Normal body metabolism is such that the amount of CO2 released is directly proportional to the amount of O2 absorbed.
PAO2 =
(Total pO2 entering alveoli)
Can be calculated easily
– (total pO2 leaving alveoli and absorbed into blood)
Cannot be directly measured; need to estimate it using the level of CO2 in the alveoli, which we can measure
PAO2 = FiO2(PB – PH2O) – (PaCO2/0.8)
Fraction of inhaled air made up of O2 (21%, if atmospheric)
Barometric Pressure Total air pressure inhaled into lungs (760mmHg at sea level, and less at higher altitudes, ie: 660mmHg in Calgary)
47 mmHg: Inspired air is humidified once inside airways. Water vapor adds to total inhaled air pressure, and must be accounted for
Partial pressure of dissolved CO2 in arteries, directly measured by an ABG
Respiratory quotient (RQ): the ratio of the amount of CO2
released from the arterial blood to
the amount of O2 absorbed or
consumed by the blood. The value
of the RQ can vary depending
upon the diet consumed
Note: The respiratory quotient (carbohydrates, fats, and proteins)
(RQ) is always <1 because
and the metabolic state. RQ is
around 0.82 for the average human diet.
Author: Yan Yu Reviewers: Steven Liu, Amogh K. Agrawal, Ciara Hanly, Xiumei Deng (邓秀梅), Zesheng Ye (叶泽生), Yonglin Mai (麦泳琳)*, Juri Janovcik* * MD at time of publication
Legend:
Definitions
Explanations
Sign/Symptom/Lab Finding
Complications
Published September 4, 2013, updated October 5, 2021 on www.thecalgaryguide.com")
copd-overview-and-definitions
”
Author: Yan Yu Reviewers: Jason Baserman, Jennifer Au, Ciara Hanly, Zesheng Ye (叶泽生), Yonglin Mai (麦泳琳)*, Naushad Hirani*, Juri Janovcik* * MD at time of publication
Cystic Fibrosis
Multisystem disease due to CFTR gene mutation, that presents in the lungs as bronchiectasis
Bronchiectasis, Cystic Fibrosis, etc
COPD
Systemic disease, largely manifesting as an airflow-obstructing respiratory disorder; can manifest in the form of any of the following disorders:
Emphysema
Lung tissue destruction & abnormal, permanent enlargement of lung acini: airspaces distal to terminal bronchioles
Chronic Bronchitis
Chronic, productive cough for a total duration of 3 months per year, over 2 continuous years
Asthma
Asthma that does not
remit completely with treatment (thus, chronic airflow obstruction) is defined as asthma-COPD overlap syndrome (ACOS)
Emphysema
Bronchiectasis
Destruction and widening of large airways, resulting in mucus hyper-secretion and recurrent infections
Chronic Bronchitis
Most common COPD manifestations
(most patients suffer from a combination of emphysema and chronic bronchitis)
Clinically, COPD is seen as:
• Progressive, partially reversible airflow obstruction and lung hyperinflation (causing respiratory symptoms like cough, sputum production, and dyspnea)
• Post-bronchodilator spirometry results: FEV1/FVC ratio <0.7 (FEV1 is not a defining feature of COPD, but a marker of severity)
• ↑ frequency & severity of acute exacerbations
• Systemic manifestations such as
deconditioning and muscle weakness
Chronic Obstructive Pulmonary Disease (COPD)
Asthma
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published January 7, 2013, updated October 5, 2021 on www.thecalgaryguide.com")
Hypersensitivity-Pneumonitis
*, Kerri Johannson* * MD at time of publication
Farming and Compost
Farmer’s Lung (Common)
Bird and Animal Proteins
Bird Fancier’s Lung (Common)
Water Contamination and Ventilation
Organic antigen identified by dendritic cells
Manufacturing and Chemical Workers
Grain and Flour Processors
Notes:
Immune complex - Production of IgG antibodies (Type III hypersensitivity)
Cell mediated - Sensitization of helper T cells (Type IV hypersensitivity)
• Thereare3typesofhypersensitivity pneumonitis: acute, subacute, chronic
• InacuteHP,removalofincitingantigen results in resolution of symptoms within days.
*Lymphoplasmocytic interstitial infiltrate
with bronchiolocentric distribution on pathology
Epithelial injury (exact mechanism unknown)
*Organizing pneumonia
Dyspnea, tachypnea, and crackles
Abbreviations:
• HP – Hypersensitivity Pneumonitis
• PFT – Pulmonary Function Test
↑ Neutrophils, mast cells, macrophages, CD8+ T cells, & inflammatory cytokines
• *TriadofmainfindingsforsubacuteHP Systemic release of
cytokines disrupts hypothalamic regulation
Fever
Macrophages ingest antigens
*Poorly formed granulomas on pathology
Tissue breakdown from neutrophils activates fibroblasts, which deposit collagen
CT: Normal or diffuse ground- glass opacity (acute)
Chronic deposition of collagen replaces normal lung parenchyma by scar tissues (Chronic Findings)
Neutrophil elastase breaks down lung elastic fibers
Tissue destruction of alveolar walls creates larger air spaces
CT: Emphysema
CT: Honeycombing
(End stage Iung disease)
Pathology: Advanced interstitial fibrosis
PFT: Restrictive pattern ↓FEV1, ↓FVC, and ↓ DLCO
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published September 1, 2016, updated October 5, 2021 on www.thecalgaryguide.com")
Tension Pneumothorax
, Yonglin Mai (麦泳琳)*, Naushad Hirani*, Yan Yu* * MD at time of publication
Contralateral Trachea Shift
Contralateral Mediastinal Shift
Absence of lung markings
Collapsed lung (visceral pleural line)
Rib splaying
Depressed Hemidiaphragm
Tachycardia Hypotension
↑ Jugular venous pressure
↓ blood oxygenation (partial pressure of O2)
Tachypnea (↑ respiratory rate)
Secondary Spontaneous Pneumothorax (underlying lung disease)
Procedures
(E.g. thoracentesis, central line insertion, lung nodule biopsy)
Positive Pressure Ventilation
Barotrauma
Primary Spontaneous Pneumothorax (no underlying lung disease)
Traumatic Pneumothorax
E.g. penetrating wound, rib fracture
Iatrogenic Pneumothorax
Disruption of parietal or visceral pleura or the tracheobronchial tree
Injured tissue forms a one- way valve into pleural space
Air enters and fills pleural space (due to physiologic negative pleural pressure), but cannot leave
↑ Pressure in ipsilateral hemithorax ↑ Pressure collapses ipsilateral lung
Irritation of highly sensitive parietal pleura
Acute onset severe, stabbing, pleuritic chest pain (can radiate to shoulder)
Contralateral cardiac silhouette shift
Air between visceral & parietal pleura separates the two layers, allowing visualization of a pleural “line”
Visible visceral pleural line
Ipsilateral rib splaying (↑ space between ribs)
Depressed hemidiaphragm
Contralateral cardiac silhouette
and tracheal Deviation
Compresses superior vena cava and/or inferior vena cava
↑ sound resonance through air compared to lung tissue
Tympany/ hyper-resonant (on manual percussion over affected area)
Sound vibrations unable to travel from the larynx through lung tissue to chest wall as normal
↓ vocal fremitus (palpable vibration of chest wall during speech)
No air entry in to affected lung
↓ chest wall expansion
Lack of lung markings beyond the pleural line
↓ breath sounds
Dyspnea
Mediastinal shift away from the affected side
↓ cardiac output
Blood backs up into venous system
Potential contralateral lung impingement
Impaired gas exchange in the affected alveoli
↓ blood return to right atrium
Risk of ipsilateral lung collapse
Ventilation/Perfusion mismatch and blood shunt
Hypoxia
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
X-Ray Findings
Initially published December 11, 2013, reviewed & updated October 24, 2020 and October 5, 2021 on www.thecalgaryguide.com")
COPD Acute Exacerbations
, Zesheng Ye (叶泽生), Yonglin Mai (麦泳琳)*, Kerri Johannson* * MD at time of publication
Notes:
• The triggers for acute exacerbations of COPD are unknown in approximately 1/3 of cases
• Changes in pulmonary function (e.g. FEV1) are poorly sensitive in the individual diagnosis of acute exacerbations of COPD due to individual variability
Acute Bacterial Infection
Activation of proinflammatory cytokines and recruitment of neutrophils
Acute Viral Infection
Epithelial cell secretion of cytokines and ↑ airway lymphocythemia
Acute ↑ in airway inflammation
(accumulation of inflammatory cells and release of harmful mediators, such as reactive oxygen species and proteases, into the lung tissue/airways)
Air pollution
(including cigarette smoke)
↑ reactive oxygen species in lungs
Airway inflammation ↑ secretions that accumulate in the airway lumen
Airway wall edema
Limits outflow of air from lungs on expiration
Airway bronchoconstriction in response to inflammation
Constricted airways create audible turbulent airflow on expiration
Systemic spread of inflammatory markers via the bloodstream cause inflammation throughout the body
↑ Cardiac Morbidity
↑ CRP
Worsening of respiratory symptoms
Irritation of cough reflexes in airways
Cough
↑ Sputum production and sputum purulence
↑ Wheeze Unexpired air becomes trapped in
the lungsàDynamic Hyperinflation
Bloodflow to lungs continue to perfuse under- ventilated regions of lungs àV/Q Mismatch
Lungs are larger àmore blood
remains in lungs à↓ preload
Acute Respiratory Failure
Tachypnea
↑ Dyspnea
Accessory Muscle Use
Attempts to restore normal arterial CO2 and O2 levels
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published April 21, 2019, updated October 6, 2021 on www.thecalgaryguide.com")
Hypercortisolemia
: Clinical Findings
Cortisol is a net catabolic hormone affecting many body systems, serving to release energy into the blood in response to stress. Excess cortisol also impacts circulation and impairs immune function. Excess Serum Cortisol affects:
Authors: Samin Dolatabadi, Yan Yu* Reviewers: Meena Assad, Amanda Henderson, Brooke Fallis, David Campbell* * MD at time of publication
Bone and Calcium Metabolism
Kidney and vasculature
Excess cortisol in the renal tubule saturates the enzyme 11β- HSD2, which converts cortisol to cortisone
Capacity of body to convert cortisol to cortisone is exceeded
Excess cortisol can mimic aldosterone
and bind to mineralocorticoid receptors (cortisone can’t bind to these receptors)
↑ Aldosterone effect → ↑ Na+ reabsorption from the cortical collecting duct into blood vessels
Liver and Peripheral Tissue
Cortisol ↑ gluco- neogenesis in liver, and ↑ insulin resistance by body tissue (unclear mechanisms)
Hyper- glycemia
Reproductive System
Cortisol exerts negative feedback on hypothalamus
à↓ gonadotropin releasing hormone (GnRH) secretion
↓ GnRH → ↓ LH/FSH → ↓ estrogen and testosterone production (especially important in females)
Infertility, ↓ Libido, Irregular Menses
Adipose Tissue
Cortisol ↑ fat breakdown (lipolysis)
Selective expression of cortisol receptor on different adipose tissuesàcentral, facial, dorsal fat is less broken down than in other areas (mechanism unclear)
Combined with cortisol ↑ appetite:
Skin & Connective Tissue
↑ Serum cortisol à↓ Fibroblast proliferation → ↓ Collagen synthesis
Skin atrophy with loss of connective tissue
Muscle
↑ Proteolysis & ↓ Protein synthesisà↓ muscle growth and function
Immune System
Normal serum cortisol protects against damaging effects of uncontrolled inflammatory and immune responses
↑ Serum cortisolà over-suppression of inflammation and impaired cell- mediated immunity
↑ Serum cortisol leads to ↓ Intestinal Ca2+ absorption and ↓ renal Ca2+ reabsorption
↓ Serum Ca2+ ↑ PTH secretion
↑ Serum cortisolà↑ RANKL:OPG ratio
↓ Osteoblast activity & ↑ Osteoclast activity
Cardiac muscle
Cardio- myopathy, Heart Failure
Skeletal
muscle, especially upper arms & thighs (for unclear reasons)
Proximal Muscle Weakness
Easy Bruising
↑ abdominal size stretches the fragile skin to become thinneràvenous blood of the underlying dermis becomes visible
Purple Striae
If hypertension is chronic
Ca2+ resorption from bone into the blood
Osteoporosis
Supraclavicular & Dorsal Fat Pads
Central Obesity
Poor Wound Healing
Susceptibility to infection
Round Face (Moon Face)
Abbreviations:
• RANKL – Receptor activator of nuclear factor kappa-Β ligand • OPG – Osteoprotegerin
• 11β-HSD2 – 11β-hydroxysteroid dehydrogenase type 2
Water follows Na+ into blood vessels to balance the osmotic pressure between the blood and renal tubules
Water reabsorption → Expansion of blood volume
Hypertension
Both primary Cushing’s (e.g. adenomas that extend into zona reticularis of the adrenal cortex) and central/secondary Cushing’s (e.g. ↑ ACTH stimulation of zona reticularis) are associated with ↑ adrenal androgen secretion
Removal of positively charged Na+ from tubular lumen creates a negative luminal environment
K+ follows the electrical gradient and is secreted into tubular lumen
↓ Serum K+ concentration
Hypokalemia
Arrhythmia, Paralysis, Cramps
(see Hypokalemia: Clinical Findings slide)
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published October 17, 2021 on www.thecalgaryguide.com
Hirsutism, Acne")
Twins Mechanisms and Complications

Allows multiple follicles to mature
IVF (multiple embryo transfer)
Ovulation induction/ superovulation fertility medications
Embryo splits during days 0-3
Embryo splits during days 4-8, after trophoblast cells (outer layer of blastocyst) have already invaded the endometrium and formed part of the placenta
Each embryo forms its own amniotic sac, both fed by 1 placenta
Monochorionic (1 placenta) Diamniotic (2 amniotic sacs) Monozygotic Twins
Embryo splits during days 9-12, when both placental and amniotic sac development have started
The embryos must now develop within a shared amniotic sac & placenta
Monochorionic (1 placenta) Monoamniotic (1 amniotic sac) Monozygotic Twins
Two ova ovulatedàEach ovum fertilized by a separate sperm
Cells still undifferentiated at this stage, allows each embryo to develop it’s own placenta and amniotic sac
Dichorionic (2 placentas) Diamniotic (2 amniotic sacs) Monozygotic Twins
Two embryos develop that implant separately into the endometrium and develop their own placenta and amniotic sac
Dizygotic Twins (Dichorionic, Diamniotic by default)
Complications of all types of twins:
Overdistention of the uterus
Weakened uterine muscles leading to uterine atony
Postpartum Hemorrhage (see relevant slide)
Larger placental mass without ↑ placental blood flow
Placental hypoperfusion (plus other complex mechanisms)
Gestational Hypertension and Pre- Eclampsia
↑ plasma volumeà diluting red blood cells, plus ↑ fetal iron consumption from mother’s stores
Anemia
High β- HCG
Mechanism unknown
Hyperemesis Gravidarum
Operative Delivery
Preterm Birth
Insufficient maternal nutritional supplies
Abnormal placental vascular connections
Fetal Death
Interplacental vascular connections lead to uneven distribution of nutrients and blood flow
Twin-Twin Transfusion Syndrome (growth discordance between twins, see relevant slide)
Note: all complications are more likely in monozygotic twins
Umbilical cords become entangled within shared amniotic sac
Cord Entanglement
Authors: Jemimah Raffé- Devine, Yan Yu* Reviewers: Brianna Ghali Jadine Paw* *MD at the time of publication
Uterine crowding
↑ risk of fetal malpresentation
Less space in uterus for continued growth
Congenital Anomalies
Intrauterine Growth Restriction
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published October 18, 2021 on www.thecalgaryguide.com")
Ectopic Pregnancy

Endometriosis
Tubal surgery or disorders
Age >35
Risk factor accumulation over time
Smoking
Impairment in tubal motility; impaired immunity (risk factor for PID)
Tubal scarring leading to adhesions, obstruction, and alteration of tubal function
Ectopic Pregnancy:
Implantation of developing blastocyst outside the uterine cavity, most commonly in fallopian tube (other locations: interstitial > cornual > cervical > ovarian > abdominal)
Embryo releases human chorionic gonadotropin (β-hCG), which supports corpus luteum to continue producing progesterone
On transvaginal ultrasound: Extrauterine gestational sac with a yolk sac or embryo
Embryo & trophoblast deathàloss of hormone support for the decidua (modified endometrial lining)
Progesterone maintains the endometrial lining, preventing it from shedding
Missed period
Penetration of ovum into muscular wall of fallopian tube
Tubal distention àTubal rupture
Intra-abdominal hemorrhage
Pregnancy cannot survive without the uterine endometrium
Maternal blood extrudes through fimbriae of fallopian tubes and into peritoneal cavity
Lower abdominal pain (including peritonitis in cases of hemoperitoneum)
Hemoperitoneum
(blood in the peritoneal cavity)
Sloughing of decidua out of the uterus through the vagina
Vaginal bleeding (usually in first trimester)
Cessation of human chorionic gonadotropin (β-hCG) release from embryo
β-hCG plateaus or decreases
Authors: Jemimah Raffé-Devine Tahsin Khan Yan Yu* Reviewers: Brianna Ghali Bishwas Paudel Mackenzie Grisdale Christina Schweitzer Ron Cusano* Jadine Paw* * MD at time of publication
Syncope
↓ Level of consciousness
Positive β-hCG, but rising <35% over 2 days
Discriminatory zone: β-hCG >2000 + absence of intrauterine pregnancy
Hypotension
Shock
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published Oct 1, 2017, updated Oct 19, 2021 on www.thecalgaryguide.com")
Acne Vulgaris Complications
, leukotrienes (LTC4, LTD4), and thromboxane A2 are released in response to inflammation
Atrophic Acne Scars
Collagen degradation and disordered deposition during healing
Formation of round-
rectangular depressions with defined edges
Box Car Scar
True scars with textural change, very slow autoresolution over time
The pigmentary changes and true scar formation resulting from acne may be more psychologically distressing than the original acne lesions
Healing processes favoring deposition of Type 1 & Type 3 collagen
Proliferation beyond the borders of original acne lesion
Keloid Formation
Healing processes favoring deposition of Type 4 collagen
Growth remains within the margins of acne lesion
Hypertrophic Scar Formation
Dermal inflammation: Due to disruption of basal cell layer, melanin released and trapped by macrophages in the dermis.
Epidermal inflammation: ↑ melanin production and transfer of melanin to keratinocytes
Microvascular dilatation and ↑presence of erythrocytes
Erythematous macules appear where acne was present
Post- inflammatory Erythema (PIE). More common in Fitzpatrick Skin Types I-III
Formation of V- shaped tracts with sharp margins
Ice Pick Scar
Formation of a
shallow edged depression layer anchored to dermal layer and subcutis
Rolling Scar
Pigmented
macules or patches appear where acne was present
Post-inflammatory Hyperpigmentation (PIH). More common in
Fitzpatrick Skin Types III-VI
No textural change, slow
autoresolution over time
True scars with textural change, do not resolve over time
Psychosocial Concerns (Depression, Anxiety, Social Isolation)
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published October 19, 2021 on www.thecalgaryguide.com")
Drug Reaction with Eosinophilia and Systemic Symptoms (DRESS)
:
Authors: Lauren D. Lee, Harry C. Liu* Reviewers: Mehul Gupta, Brian Rankin, Julia Chai, Stephen Williams Yan Yu* Laurie Parsons* *MD at time of publication
Pathogenesis and clinical findings
Genetic susceptibility
Certain HLAs (e.g., HLA-B*58:01, HLA-B*57:01, and HLA-A*31:01)
HLA alleles encode for MHC structure and may influence how specific drugs/drug metabolites interact with T cell receptors and MHC proteins on antigen-presenting cells
Exposure to offending drug
E.g., Aromatic anticonvulsants (lamotrigine, carbamazepine, and phenytoin), allopurinol, and sulfonamides
Drug-specific CD4+ and CD8+ T cells produce tumor necrosis factor-alpha and interferon gamma
↑ activated T-cells 2-6 weeks after drug exposure
Drug Reaction with Eosinophilia and Systemic Symptoms (DRESS)
Latent Viral infection
Latent viruses (HHV-6, HHV- 7, CMV, and EBV) concealed in regulatory T-cells
Reactivation of latent viruses may be contributory or secondary to T cell activation by drugs
Eosinophilia +/- atypical lymphocytes
T helper type 2 cells recruit and activate eosinophils by releasing cytokines
Activated leukocytes create a humoral immune and allergic response
Dysfunction of regulatory T cells, resulting in failure to control unwanted immune responses against “self”
Autoimmune processes with single- or multi-organ involvement
CMV- Cytomegalovirus EBV- Epstein-Barr Virus HHV- Human Herpesvirus HLA- Human Leukocyte Antigens
MHC – Major Histo- compatibility Complex
Endocrine system
mechanism of dysfunction unclear but may be linked to autoreactive T cells
Autoimmune Type 1 thyroiditis diabetes
Lymphadenopathy
Fever
Facial edema
Liver
lobular inflammation, dispersed foci of necrotic hepatocytes, granulomatous infiltrates consisting of eosinophils
Hepatitis
Kidney
Interstitial edema and infiltrates of lymphocytes, histiocytes, eosinophils, and plasma cells
Acute interstitial nephritis
Lung
increased pulmonary infiltrate and edema
Morbilliform Skin Eruption
Spongiosis
Epidermal layer
Dermal- Epidermal Junction Dermal layer
Interface dermatitis
Skin eruption
(morbilliform to diffuse erythema with follicular accentuation)
Interstitial pneumonitis
Pleural effusion
Eosinophilic infiltration
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published October 19, 2021 on www.thecalgaryguide.com")
Vestibular Neuritis
 ganglion
Inflammatory cell infiltration leads to degeneration and atrophy of the vestibular nerve
Superior Vestibular Neuritis
(40-48%) Inflammatory cells traverse through only one long bony canal, easier for inflammatory cell infiltration
Loss of afferent innervation from the superior and horizontal semicircular canal (SCC), utricle, and parts of the saccule
Combined Superior Vestibular Neuritis and Inferior Vestibular Neuritis (34-56%)
Inferior Vestibular Neuritis (1.3-18%) Inflammatory cells must pass through two separate bony canals, making inflammatory cell infiltration more difficult
Loss of afferent innervation from the posterior semicircular canal (SCC) and saccule
Utricular degeneration
Displacement of otoliths/ otoconia (commonly into the posterior SCC)
BPPV
(can occur several months after onset of neuritis)
Loss of horizontal SCC afferent neuron signaling to the brain
Unilateral Loss or ↓ of normal nystagmus response to Caloric Testing (insertion of cold and warm water/air into the ear canal while supine)
Loss of utricular afferent neuron signaling to the brain
↓ or absent Ocular Vestibular- Evoked Myogenic Potentials (VEMPs)
and Normal Cervical VEMPs
↓ unilateral vestibular input to the brain leads to acute phase symptoms (over time, brain can compensate which allows for some symptom improvement)
Loss of posterior SCC and saccular afferent neuron signaling to the brain
↓ or absent
Cervical
VEMPs, but Ocular VEMPs are normal
↓ or absent input to the ipsilateral vestibular nuclei elicits a vestibular nucleus response similar to contralateral SCC excitation
Acute-Phase Spontaneous Nystagmus Fast phase beats away from the affected side (3- 10 days)
And
Loss of ocular fixation on Head Impulse Test
↓ or absent saccular input to the lateral vestibular nuclei (e.g., lateral vestibulospinal tract) results in the loss of lower limb postural adjustability
Postural Instability
(e.g., abnormal Romberg/sharpened Romberg, Fukuda step test)
Bilateral mismatch of vestibular
information to the brain
Peripheral Vertigo (lasts several hours to days; rapid onset, severe, constant)
Nausea and Vomiting
Only the vestibular
part of the vestibulo- cochlear nerve is affected, not the cochlear nerve
No Hearing
Loss or Tinnitus
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published October 19, 2021 on www.thecalgaryguide.com")
Summary of Cyanotic Congenital Heart Diseases

Authors: Winnie Nagesh, Gaya Narendran, Deborah Fruitman* Reviewers: Austin Laing, Yan Yu* * MD at time of publication
Right heart normally carries deoxygenated blood to the pulmonary circulation while the left heart carries oxygenated blood to the systemic circulation.
Cyanosis can be due to varied pathophysiology, most involve a R-L shunt (described below)
Typically presents in the newborn period but depends on the severity and the type of lesion
Tetralogy Of Fallot (TOF)
Main features: 1.VSD, 2. Overriding aorta, 3. RVH, 4. Pulmonary Valve stenosis
1. VSD causes equal systolic pressure in R and L ventricles
2. Pulmonary Valve stenosisàRight Ventricle Outflow
Tract Obstructionà↓ pulmonary blood flow (degree
depends on severity of obstruction)
3. RVàLV flow across VSD
Presents in the 1st days to weeks, depending on severity of pulmonary valve stenosis
On exam: LUSB SEM; loud S2; hypoxemia (degree depends on severity of Right Ventricle Outflow Tract Obstruction)
CXR: “boot-shaped” heart, decreased pulmonary vasculature
Presents at birth
On exam: Often no murmur, no respiratory distress, severe cyanosis/hypoxemia
CXR: “egg on a string”, normal pulmonary vasculature
Presents in the 1st days to weeks as CHF symptoms develop On exam: Systolic ejection click, Single S2 , SEM, mild hypoxemia due to mixing of blood, +/- tachypnea, hepatomegaly (CHF symptoms)
CXR: normal or ↑ pulmonary vasculature
Presentation varies based on the degree of the outflow tract obstruction
On exam: +/- Holosystolic murmur at LLSB (from VSD), or SEM if outflow tract obstruction, +/- severe cyanosis dependent on severity of pulmonary stenosis
CXR: normal or ↓ pulmonary vasculature
Presents in early infancy (earlier and more severe presentation if obstructed)
On exam: +/-split S2, SEM LUSB, tachypnea, mild cyanosis CXR: cardiomegaly with ↑ pulmonary vasculature
Note: see relevant Calgary Guide slides for each heart condition for full explanation of their pathophysiology. Figures are hand-drawn by the authors.
Transposition of the Great Arteries (TGA)
Aorta is the outflow for RV and pulmonary artery is the outflow for the LV (parallel circulation)
Truncus Arteriosus
1. Single vessel (truncus) fails to divide into pulmonary artery and aorta, 2. Single outlet overriding VSD
Tricuspid Atresia
Tricuspid valve fails to develop normally with a
hypoplastic right heart and VSD
1. Deoxygenated blood pumped from RVàaortaà systemic circulation, bypassing the lungs.
2. Oxygenated blood pumped from LVàPA
1. RV + LV pumped through VSDàoverriding truncal artery 2. Mixed deoxygenated and oxygenated blood enters
systemic, pulmonary and coronary circulations
1. Blood cannot enter RV due to lack of tricuspid valve
2. Deoxygenated blood pumped from RAàASDàLA and
mixes with oxygenated blood
3. Mixed blood enters systemic circulation
Total Anomalous Pulmonary Venous Return (TAPVR)
Pulmonary veins return blood to systemic venous circulation (most common type of supracardiac congential heart disease)
1. Oxygenated blood flows through pulmonary veinsàenters Superior or Inferior Vena CavaàRight atrium (all systemic & pulmonary venous blood returns to the RA, resulting in mixing of blood)
2. Blood follows pressure gradient, flows from RAàLA via ASD
3. Flow from LAàLVàAortaàSystemic Circulation
Abbreviations: VSD: Ventral Septal Defect; RVH: Right Ventricular Hypertrophy; RV: Right Ventricle, LLSB: Left Lower Sternal Border, LUSB: Left Upper Sternal Border; SEM: Systolic ejection Murmur; RVOTO: Right Ventricle Outflow Tract Obstruction
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published October 21, 2021 on www.thecalgaryguide.com")
Summary of Acyanotic Congenital Heart Diseases

Authors: Gaya Narendran, Winnie Nagesh Reviewers: Jack Fu, Usama Malik, Yan Yu*, Deborah Fruitman* * MD at time of publication
Asymptomatic M, ↑ respiratory tract infections, rarely: failure to thrive
Left to Right Shunt
↑ flow from left to right heart
Dilation of chambers exposed to ↑ flow
Atrial Septal Defect (ASD)
Presents later in childhood – often asymptomatic
Note: These conditions tend to be acyanotic in presentation. Clinical severity will depend on the defect’s size, anatomic location and the presence of other cardiac anomalies. Please see relevant Calgary Guide slides for each heart condition for full explanation of their pathophysiology.
Figures are hand-drawn by the authors.
L to R physical communication between atria
1. Pressure in LA > pressure in RA à blood shunts from LA to RA
2. Dilation of RAàdilation of RV 3. ↑ pulmonary blood flow
On exam: Systolic Ejection Murmur at LUSB, fixed split S2, RV heave, tachypnea
CXR: +/- ↑ pulmonary vasculature
Ventricular Septal Defect (VSD) 1. Pressure in LV > pressure in RV (after 4-6wks old)
On exam: harsh pansystolic M +/- diastolic rumble at LLSB, +/- hepatomegaly, WOB, tachypnea, +/- ↓ perfusion signs e.g. pallor CXR: ↑ vascular markings, cardiomegaly, pulmonary edema
Feeding difficulties, failure to thrive, congestive heart failure (CHF)
L to R physical communication between ventricles
2. This causes blood in LV to flow to RV in systole
3. ↑ flow to RVà↑ flow to pulmonary arteriesà
↑ pulmonary blood flow
4. ↑ blood returning to LA & LVàLA & LV dilation
Presents usually at 4-6 weeks as PVR falls to normal (after birth) Patent Ductus Arteriosus (PDA)
1. Pressure in aorta > pressure in pulmonary arteries (PA) à continuous flow from aorta to PA
2. ↑ bloodflow load in the PA
3. ↑ flow in PA/lung vasculature à ↑ return to left
Note: Adult presentation or unrepaired large VSD may cause pulmonary HTN, leading to Eisenmenger’s Syndrome (see relevant slide)
Vessel linking descending aorta & pulmonary arteries remains after birth
On exam: Continuous machine-like murmur in sub-clavicular region, wide pulse pressure, tachypnea
CXR: ↑ pulmonary vasculature, LV enlargement, cardiomegaly, prominent PA
Asymptomatic M , less commonly: failure to thrive, congestive heart failure
heart à Dilation of the LA and LV
Typically presents later in infancy – dependent on shunt size. Presentation and management may differ in preterm infants.
Atrioventricular septal defects (AVSD)
Defect in the crux/ center of heart involving both atria and ventricles, with AV abnormalities on a spectrum
1. Pressure in left heart > pressures in right heart
2. Thus blood shunts left à right at atrial and ventricular levels 3. As PVR falls (as part of normal newborn development) à ↑
pulmonary blood flow, +/- AV regurgitation
4. ↑ pulmonary flow à ↑ return to left heart à Cardiomegaly
On exam: Systolic Ejection
Murmur at LUSB, hepatomegaly,
mild O2 desaturation in children
CXR: ↑ pulmonary vasculature,
cardiomegaly defect
Similar to VSD – may present earlier, dependent on severity of defect – associated with Trisomy 21
Abbreviations: AV: Atrioventricular valve; CHF: Congestive Heart Failure; CXR: Chest X-ray; LA: Left Atrium; LV: Left Ventricle; LUSB: Left Upper Sternal Border; LLSB: Left Lower Sternal Border; M : Murmur; PA: Pulmonary Artery; PVR: Pulmonary Vascular Resistance; RA: Right Atrium; RV: Right Ventricle; WOB: Work of Breathing
Same as VSD; dependent on severity of
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published Nov 15, 2017, updated Oct 21, 2021 on www.thecalgaryguide.com")
2-DM-II-pathogenesis-1
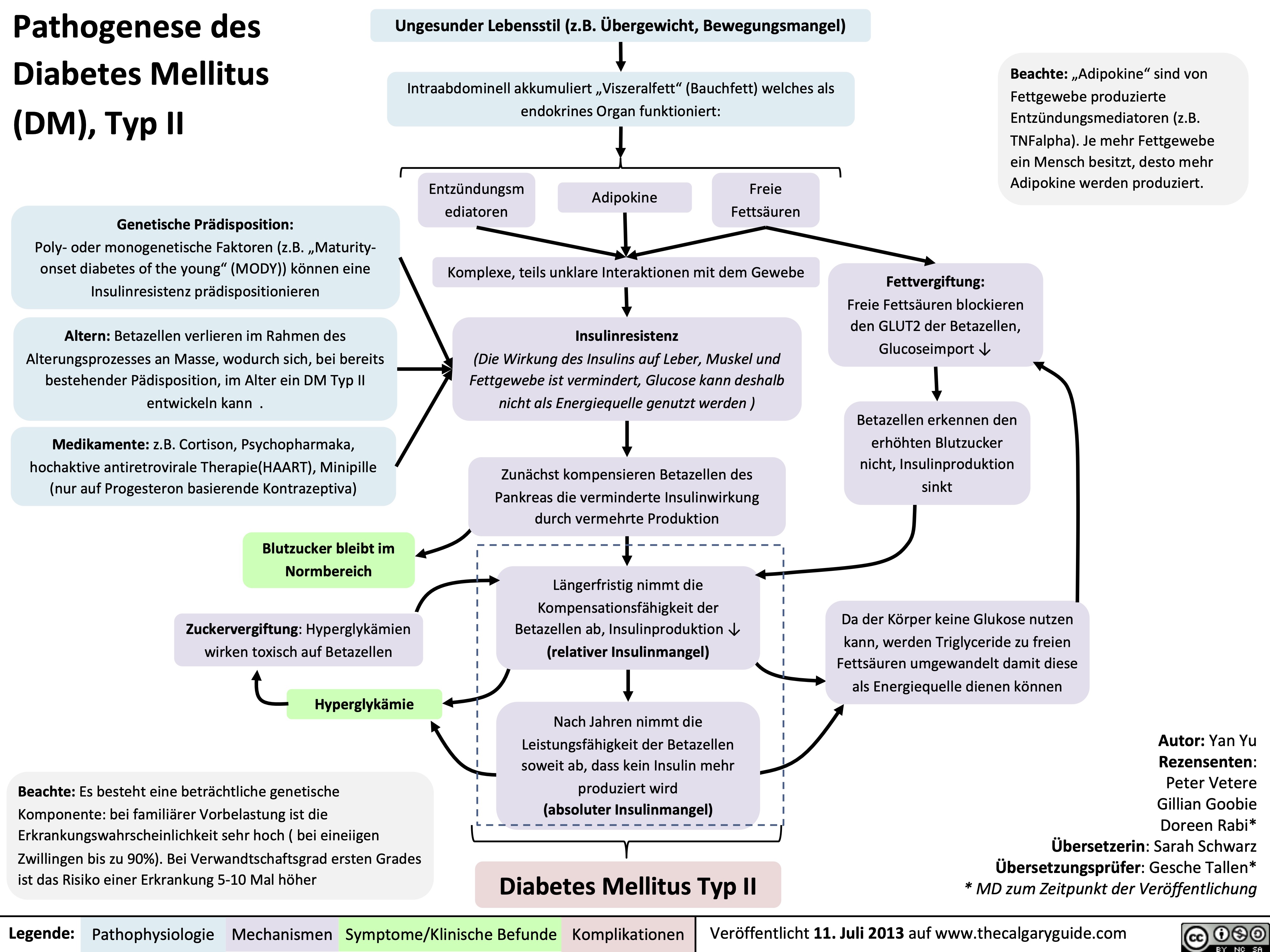
EPOC-Descripcion-general-y-definiciones
: Definiciones
Autor: Yan Yu Revisores: Jason Baserman, Jennifer Au, Ciara Hanly, Zesheng Ye (叶泽生), Yonglin Mai (麦泳琳)*, Naushad Hirani*, Juri Janovcik* * MD at time of publication Traductor: Margith Izaguirre María Rosario Talavera*
Fibrosis quística
Enfermedad multisistémica debida a la mutación del gen CFTR, que se presenta en los pulmones como bronquiectasia
Bronquiectasia, Fibrosis quística, etc
EPOC
Enfermedad sistémica, manifestado en gran parte como un trastorno en la obstrucción del flujo del aire, se puede manifestar como alguna de las siguientes enfermedades
Ensifema
Destrucción tisular anormal de pulmón, agrandamiento de los acinos pulmonares: espacios aéreos distales de los bronquios terminales
Bronquitis crónica
Tos productiva crónica por una duración total de 3 meses al año, por 2 años continuos.
Asma
Asma que no mejora completamente con tratamiento(por lo tanto, obstrucción aerea crónica) Definido como síndrome de solapamiento asma-EPOC
Ensifema
Bronquiectasia
Destrucción y ensanchamiento de las vías aéreas mayores, resultando hipersecreción de moco e infecciones recurrentes
Bronquitis crónica
Manifestaciones más communes del EPOC
(la mayoría de los pacientes sufren una combinación de enfisema y bronquitis crónica)
Clínicamente, el EPOC es visto como:
• Progresivo,reduccióndelflujoaéreo parcialmente reversible, e hiperinflación del pulmón (causando síntomas respiratorios como tos, producción de esputo y disnea)
• Resultadodeespinometríapost broncodilatador: FEV1/FVC razón <0.7 (FEV1 no es una característica definida de EPOC, pero es un marcador de severidad)
• Aumentalafrecuenciaeintensidadde exacerbaciones agudas
• Manifestacionessistémicascomofaltade energía y debilidad muscular
Enfermedad pulmonar obstructiva crónica (EPOC)
Asma
Legend:
Fisiopatología
Mecanismo
Signos/Sintomas/Hallazgo de laboratorio
Complicaciones
Publicado 7 Enero, 2013, Actualizado 5 Octubre, 2021 en www.thecalgaryguide.com")
Hypersensitivity-Definitions
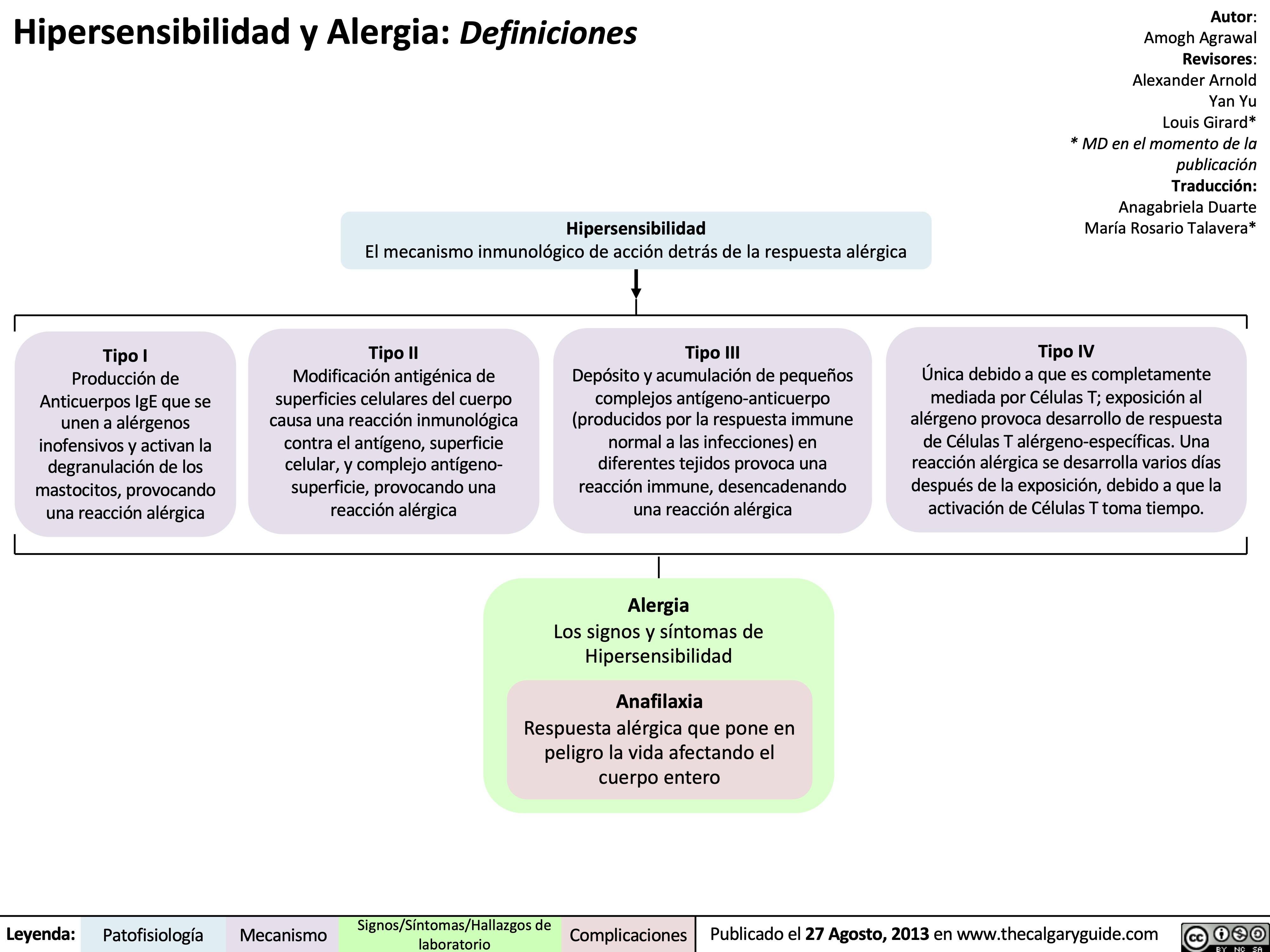
Type-1-HS
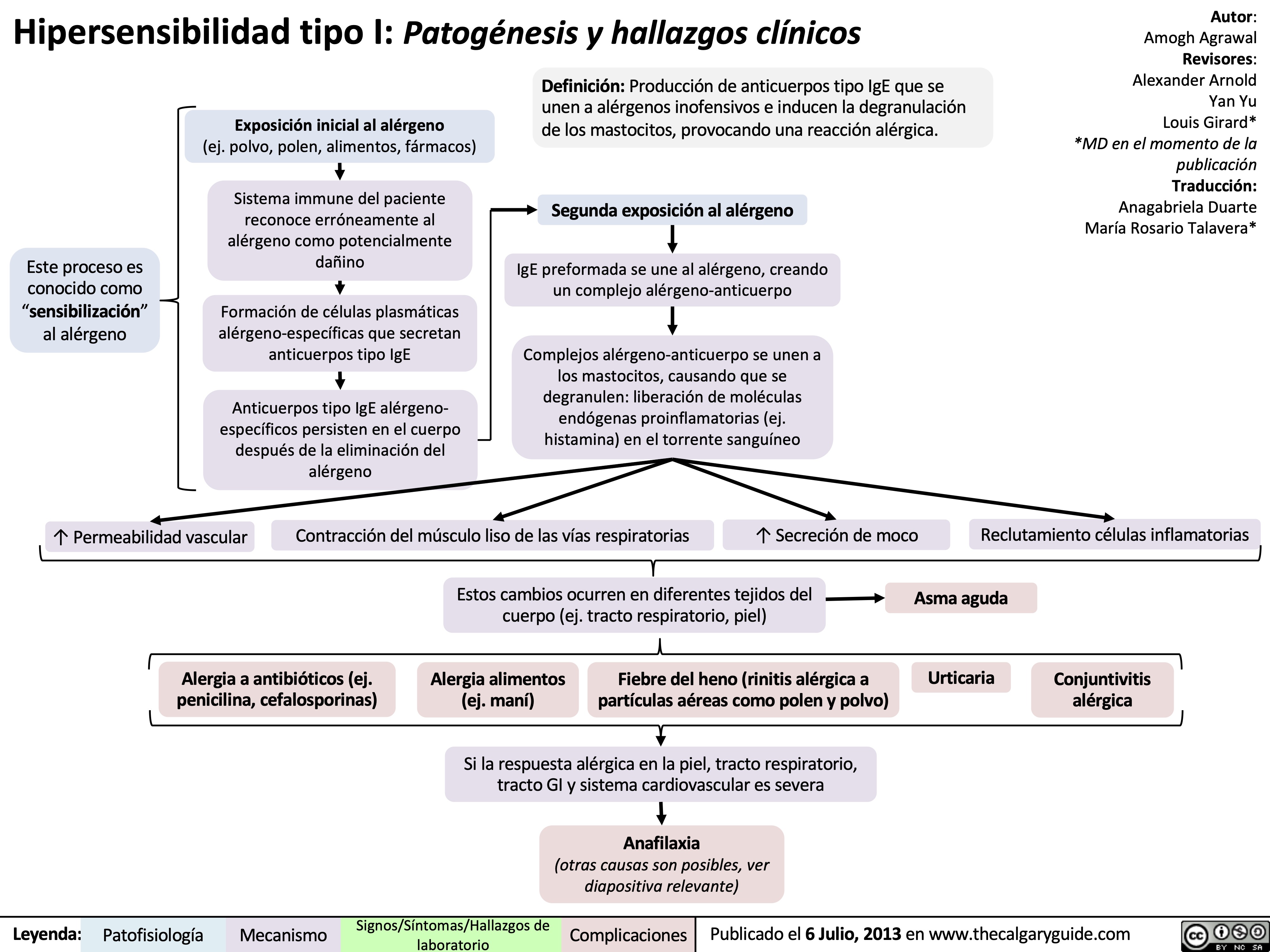
Type-2-HS
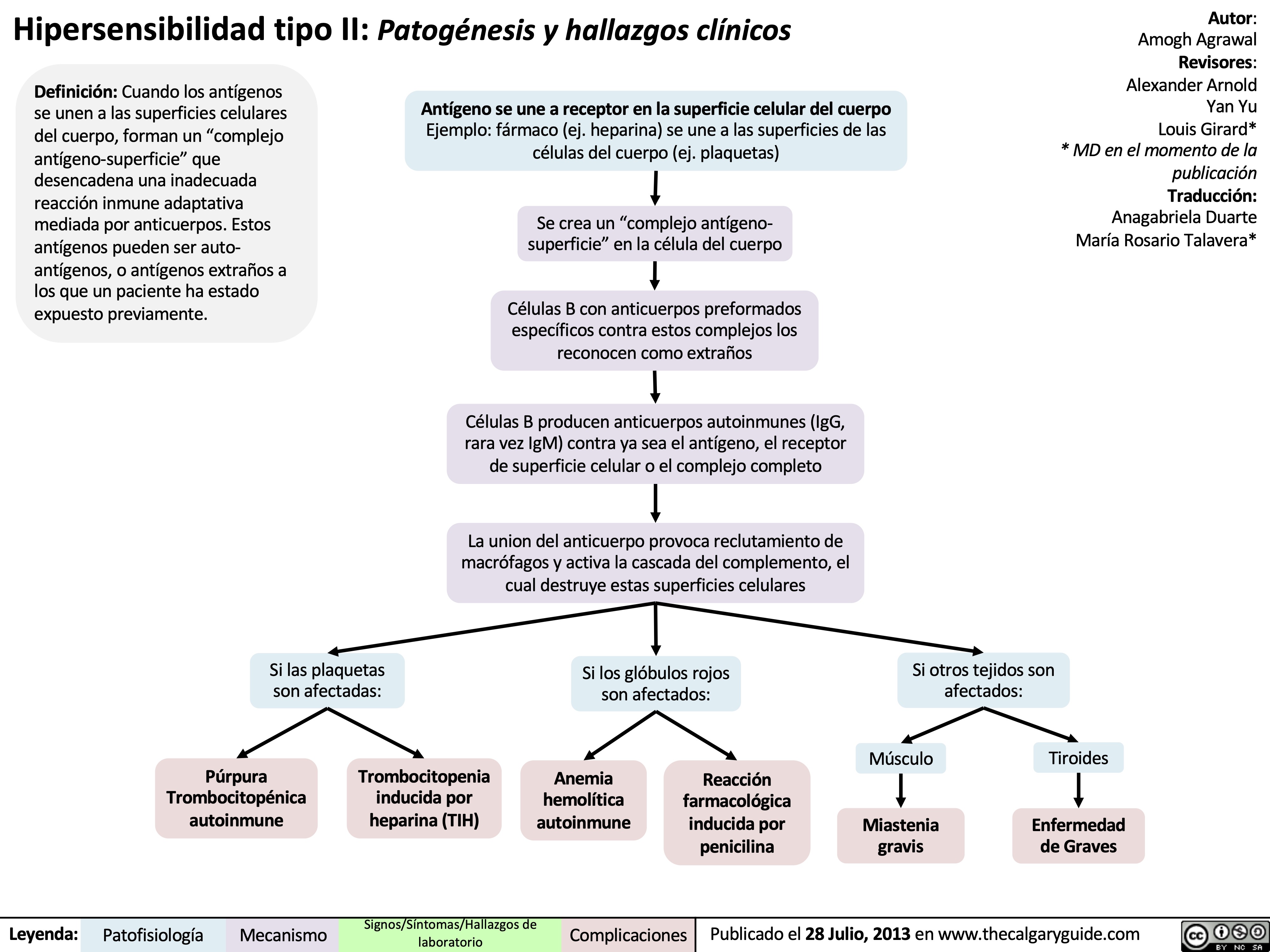
Type-3-HS
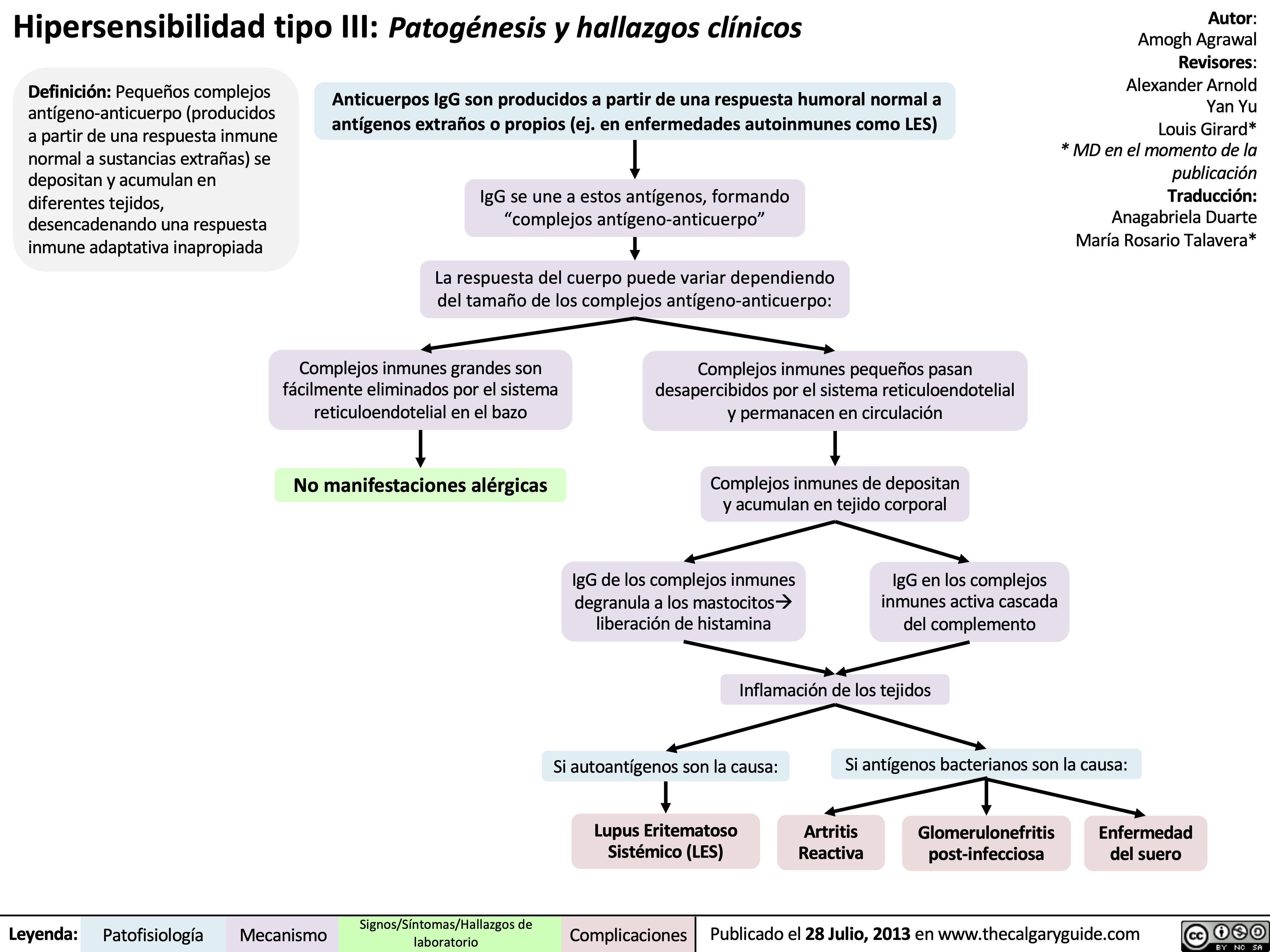
Type-4-HS
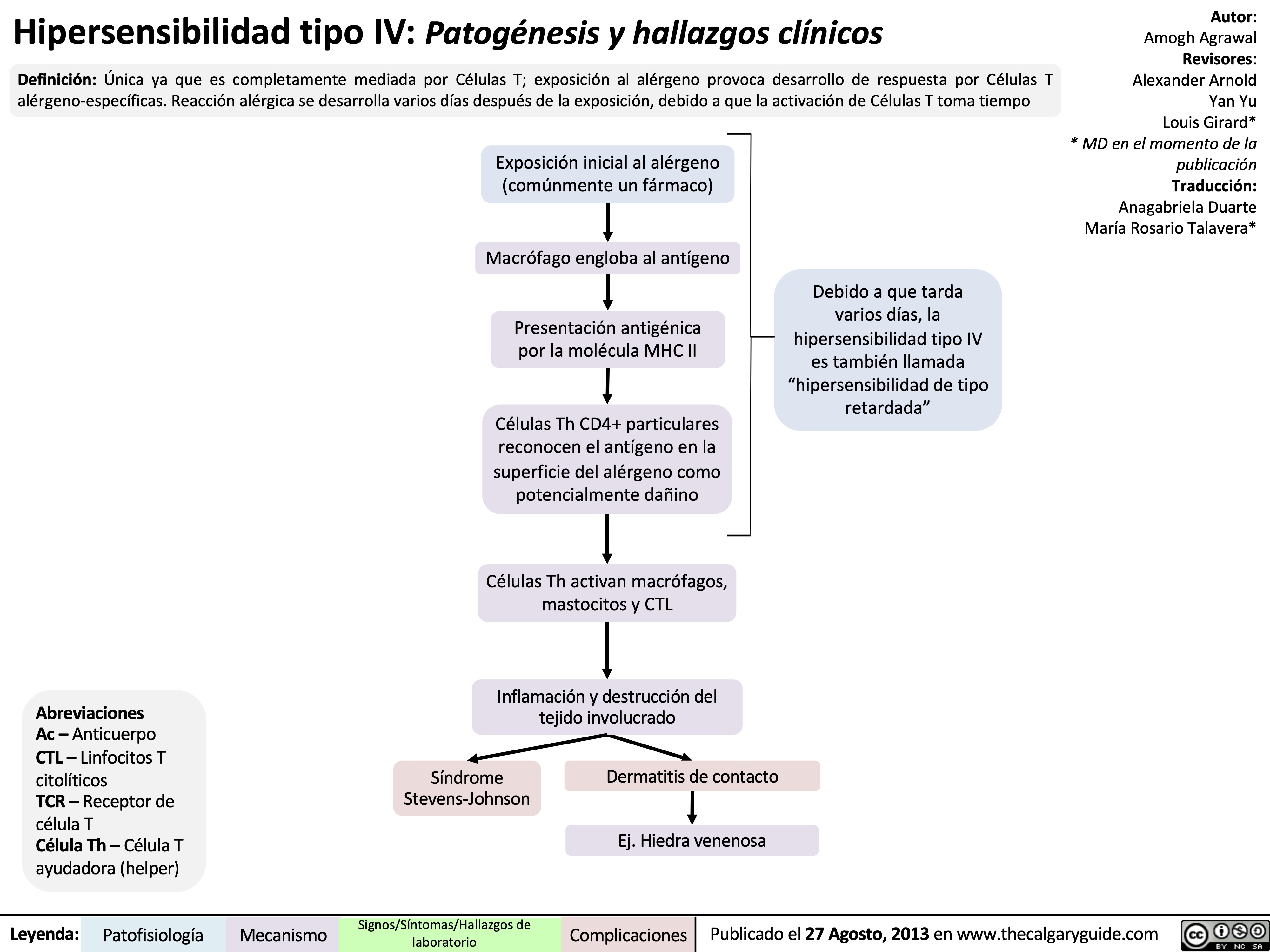
Femoroacetabular Impingement (FAI)
: Pathogenesis and clinical findings
Hip dysplasia
Overcorrection of deformity during surgery
Developmental abnormalities
Malunion of femoral neck fracture
Altered femoral head/neck shape
Repetitive Leisure or Occupational Activity
Repetitive movement of the legs beyond normal range of motion
Altered hip joint structure Excess bone or abnormal orientation between the femoral head and acetabulum:
Residual pediatric hip conditions (slipped capital femoral epiphysis, Perthe’s disease)
Structurally abnormal hip joint prior to skeletal maturity
Hip joint remodeling
Femoroacetabular impingement (FAI)
Pincer lesion: extra bone extending over rim of acetabulum
Cam lesion: extra bone growth on femoral head
Combined lesion: both pincer and cam lesions
Decreased femoral anteversion: femoral neck rotated backwards in relation to the femoral shaft
Abnormal contact between the femoral head and acetabulum with specific movements (sitting, leaning forward, walking, climbing stairs, squatting, pivoting)
Repetitive bumping of bone against articular cartilage and the joint labrum
Bones rubbing together activates nociceptors within the hip joint
Bone on bone contact restrict movement of the hip joint, especially the movements that further narrow the joint space
Cartilage degeneration tear
Secondary hip osteoarthritis
Authors: Alyssa Federico, Yan Yu* Reviewers: Mehul Gupta, Kelly Johnston* * MD at time of publication
Labral
Sensation of clicking, catching, or locking in the hip
Hip and groin pain
Patient restricts use of surrounding muscles to prevent further pain
Weakness of hip flexor and abductor muscles
Favouring the non- injured leg to avoid pain while walking
Antalgic gait
+ FADIR test: pain with flexion, adduction, and internal rotation of the hip joint
Restricted flexion and internal rotation
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published November 1, 2021 on www.thecalgaryguide.com")
Sekundäre Hämostase: Koagulationskaskade
PTT – Aktivierte Partielle
Thromboplastinzeit
• F – Koagulationsfaktor
• a – Aktivierter Koagulationsfaktor
• N – Normal
• HMWK – Hochmolekulares Kininogen • * – Vitamin K abhängig (für weitere
Erläuterung siehe Vitamin K-Mangel Folie)
Eselsbrücken:
• Faktoren der Endstrecke: 10/5=2, 2/2=1 (F10, 5, 2, 1)
• Vitamin K abhängige Faktoren– 1972 F10, 9, 7, 2
Normales extrinsisches und, intrinsisches System, Normale Endstrecke
N TPZ, N (a)PTT
Intrinsischer Weg:
Extrinsischer Weg: Gewebeschaden
Durch vaskulären Endothelschaden wird der subendotheliale Gewebefaktor freigesetzt
F12
F12a
F9*
Aktivierung durch Kontakt mit negativ geladenen Oberflächen (z.B. HMWK Kollagen, Krallikrein)
Gewebefaktor (Thromboplastin)
F7a*
F11
F11a
F10*
F10a*
F9a*
F8a
Ca2+
Ca2+
Gewebefaktor– F7a* Komplex
Gemeinsame Endstrecke:
F5a Prothrombin (F2*)
Ca2+
Thrombin (F2a*)
Ca2+
Gängige Gerinnungstest:
• TPZ, Quick-Wert, PT –
Zeit bis zur Bildung eines Thrombus bei Aktivierung des extrinsischen Weges
• INR – Standardisierung der TPZ
• (a)PTT – Zeit bis zur Bildung eines Thrombus bei Aktivierung des intrinsischen Weges
Koagulation
Bildung eines Fibrinthrombus
Fibrinogen (F1)
Störung des intrinsischen Systems
N TPZ, ↑ (a)PTT
Fibrin (F1a)
Störung des extrinsischen Systems
↑TPZ, N (a)PTT Verlängerte Blutungszeit
Störung der gemeinsamen Endstrecke
↑TPZ, ↑(a)PTT
Legende:
Pathophysiologie
Mechanismen
Symptome/Klinische Befunde
Komplikationen
Veröffentlicht: 12. Mai 2017 auf www.thecalgaryguide.com")
Pneumonie: Pathogenese und klinische Befunde
 und kann unterteilt werden in: Ambulant erworbene, nosokomial (im Krankenhaus) erworbene Pneumonie
Die Immunantwort variiert je nach eingedrungenem Erreger (z.B. Pneumokokken verursachen ein lobär betontes,
H. influenzae ein interstitiell betontes Entzündungsbild)
Rauchen unterdrückt die Funktionsfähigkeit der neutrophilen Granulozyten und schädigt das Lungenepithel
Chronische Lungenerkrankungen z.B. COPD, Asthma oder Lungenkrebs zerstören das Lungengewebe und bieten Krankheitserregern mehr Angriffsfläche für Infektionen
Durch Immunsuppression z.B. bei HIV, Sepsis, Glucocorticoid- oder Chemotherapie wird die Immunantwort eingeschränkt
Systemisch kommt es zu einer inflammatorischen Immunantwort
Durch systemische Zytokinfreisetzung wird die
hypothalamische Thermoregulation beeinträchtigt
Erregersexposition durch Inhalation, Aspiration, Kontakt- oder hämatologische Übertragung
Anfällige Person und/oder virulenter Erreger
Proliferation des Erregers in unteren Atemwegen und Alveolen
Lokal reagiert das Alveolarepithel mit einer Chemokinausschüttung um neutrophile Granulozyten an den Entzündungsort zu mobilisieren
LOBÄR betont: Lokal begrenzte Akkumulation von neutrophilen Granulozyten und Plasmaexudat in Alveolen
INTERSTITIELL betont: Zelluläre Infiltrate (Immunzellen und Immunzellfragmente) in den Alveolarwänden (zwischen Alveolen und Kapillaren)
Fieber
Anmerkungen:
Schüttelfrost
Irritation der Atemwege mit Hustenreiz
Flüssige Infiltrate in Alveolen führen zur Schleimbildung
Produktiver Husten
Das Exudat vermindert die
Röntgenstrahlendur chlässigkeit, entzündete Areale erscheinen im Röntgenbild heller/weiß.
Verschattung im Röntgen
Alveolen sind durch Flüssigkeitsansamml ungen blockiert
Verdickung der Alveolarwände, Diffusionsstrecke ↑
Irritation der Alveolarwände mit Hustenreiz
Bei ausschließlich interstitieller Infiltration -> Husten ohne Schleimproduktion
Trockener Husten
• Andere Symptome der Pneumonie sind: Brustschmerzen, Nutzung der Atemhilfsmuskulatur, auskultatorisch Rasselgeräusche, Müdigkeit
• Diese Symptome sind jedoch unspezifisch
O2 und CO2- Austausch vermindert
Hypoxie
Periphere & zentrale Chemorezeptoren werden aktiviert, Atemfrequenz ↑
Luftnot
Legende:
Pathophysiologie
Mechanismen
Symptome/Klinische Befunde
Komplikationen
Veröffentlicht: 26. September 2016 auf www.thecalgaryguide.com")
Diabetische Ketoazidose (DKA)

Betrifft hauptsächlich Patienten mit Diabetes Mellitus Typ 1: Bestimmte Situationen (z.B. Infektionen) erhöhen den Insulinbedarf, welcher durch fehlende Produktion/unzureichende Substitution nicht gedeckt werden kann
Autor: Yan Yu Rezensenten: Peter Vetere, Gill Goobie, Sean Spence, Hanan Bassyouni* Übersetzung: Sarah Schwarz Übersetzungsprüfung: Dr. Gesche Tallen * MD zum Zeitpunkt der Veröffentlichung
Hyperglykämie
(erhöhter Blutzucker)
Bei >12mmol/L, Glukosefiltration > - resorption, Glukose verbleibt im Urin
Glukosurie
Glukose ist osmotisch wirksam und zieht große Mengen Wasser mit sich (Osmotische Diurese)
Polyurie
Schwere Dehydrierung(bis zu 4-5L)
(ZVD↓, Orthostase: posturale Hypotension/ posturale Tachykardie, Ruhepuls ↑ )
Insulinmangel enthemmt die Lipolyse, sodass der Körper Energie aus Triglyceriden produziert
Freisetzung von freien Fettsäuren aus Fettgewebe
Absoluter
Insulinmangel
Hypothalamische Zellen induzieren bei geringem intrazellulären Glukosespiegel ein starkes Hungergefühl
Glukose bleibt im Blut & kann nicht durch Muskel- /Fettgewebe verstoffwechselt werden
“Ausgehungerte” Zellen triggern die Freisetzung kataboler Hormone: Glucagon, Katecholamine, Cortisol, Somatotropin
Damit intrazellulärer Glukosespiegel ↑, erhöht der Körper den Blutzucker
Hydrolyse von freien Fettsäure in
der Leber (Ketogenese) Polyphagie
↓ Proteinsynthese, ↑ Proteolyse (in Muskelgewebe)
↑ Gluconeogenese, ↑ Glykogenolyse (in der Leber)
Acetyl Co-A
Energiequelle für “ausgehungerte” Zellen
Beachte: Neben Glukose sind Ketonkörper die einzigen Energieträger die von Nervenzellen metabolisiert werden können. Deshalb werden sie vom Körper bei geringem Glukoseangebot produziert.
Ketonkörper
( β-Hydroxybutyrat, Acetoacetat, Aceton, akkumulieren im Blut)
Ketonurie
Metabolische Azidose
(↑Anionenlücke: Ketonkörper verbrauchen den HCO3--Puffer)
Substrate der Gluconeogenese↑
Durch ↓ Extrazellulärflüssigkeit, werden Ketonkörper konzentriert → Azidose
Stört das enterische Nervensystem, Magenentleerung ↓, Ileus
Bauchschmerzen, Übelkeit, Erbrechen
(Dehydration↑)
Abatmen von CO2 um Azidose auszugleichen
Kussmaul- Atmung (Tiefe, schnelle Züge)
Abatmen von Keton- körpern
Azeton- geruch der Atemluft
Beeinträchtigung elektrischer Signale zum Gehirn, Rücken- mark und Nervenzellen
Perfusion des Gehirns, Rückenmark, Nervenzellen↓
Verschiebung des Wasser- und Elektrolythaushalts
Falls Trinkwasser vorhanden
Polydipsie Beachte: Während der DKA verliert der Körper K+ durch
Elektrolytstörung
Schwäche, Delirium +/- Koma
osmotische Diurese und Erbrechen. Da gleichzeitig allerdings K+ aus den Zellen diffundiert, kann das Serumkalium unauffällig/erhöht sein. Um Hypokaliämien zu verhindern sollte bei K+ <5.0mmol/L KCl zusammen mit Insulin i.v. verabreicht werden. Cave: gute Nierenfunktion des Patienten
Therapie: 1) +++ Flüssigkeit. 2) Insulin + KCl. 3) Auffüllen der Anionenlücke. 4) Auslösende Faktoren identifizieren. 5) Wenig PO4 (typischerweise wenige Stunden bis einen Tag nach der Ketoazidose durch ↑ ATP-Produktion.)
Legende:
Pathophysiologie
Mechanismen
Symptome/Klinische Befunde
Komplikationen
Publikationsdatum: 17 Juni 2013 auf www.thecalgaryguide.com
Hyperosmolares/ Hyperglykämisches Koma (HHS)
Beachte: betrifft nur Patienten mit Diabetes mellitus Typ II
Beachte: Es sollte stets nach auslösenden Faktoren z.B. einer Infektion gesucht werden
Autor: Yan Yu Rezensenten:
Peter Vetere
Gill Goobie
Hanan Bassyouni* Übersetzung:
Sarah Schwarz Übersetzungsprüfung: Dr. Gesche Tallen
* MD zum Zeitpunkt der Veröffentlichung
Verschiebung des Wasser- und Elektrolythaushalts
Elektrolytstörung
Unzureichende Insulin- produktion, Insulinresistenz, Nichtansprechen auf Insulintherapie
Relativer Insulinmangel
Situationen mit ↑Insulinbedarf: Infekt, Pneumonie, Herzinfarkt, Pancreatitis, etc.)
Hyperglykämie
(Stark erhöhter Blutzucker, höher als bei der DKA)
Bei >12mmol/L, Glukosefiltration > - resorption, Glukose verbleibt im Urin
Glukosurie
Glukose ist osmotisch wirksam und zieht große Mengen Wasser mit sich (Osmotische Diurese)
Polyurie Dehydration
(ZVD↓, Orthostase: posturale Hypotension/posturale Tachykardie, Ruhepuls ↑ )
Durch das wenige noch vorhandene Insulin, wird ein Teil der Glukose von Muskel- /Fettgewebe verstoffwechselt, ein Teil verbleibt im Blut
Körperzellen brauchen eine weitere Energiequelle
Freisetzung kataboler Hormone: Glucagon, Katecholamine, Cortisol, Somatrotropin
Damit intrazellulärer Glukosespiegel ↑, erhöht der Körper den Blutzucker
Hypothalamische Zellen induzieren bei geringem intrazellulären Glukosespiegel ein starkes Hungergefühl
Polyphagie
Beachte: Durch das wenige noch
vorhandene Insulin wird die Lipolyse gehemmt und werden keine Ketonkörper produziert. Es kommt nicht zur Azidose und Ketonurie (Gegensatz zur DKA). Sollte es doch zur Ketourie kommen, ist das meist Folge von Hungerzuständen oder anderen Mechanismen
Extrazellulärflüssigkeit ↓ mit erhöhter Osmolarität (z.B. Hypernatriämie)
↑ Gluconeogenese, ↑ Glykogenolyse (in der Leber)
↓ Proteinsynthese, ↑ Proteolyse (in Muskelgewebe)
Substrate der Gluconeogenese ↑
Sollte der Patient nicht ausreichend trinken um das Volumendefizit auszugleichen
Falls Trinkwasser vorhanden
Polydipsie
Wasser verlässt Zellen entlang des osmotischen Gradienten, Neuronen schrumpfen
Neuronaler Schaden: Delirium, Krampfanfall, Benommenheit, Koma
Renale Perfusion↓, GFR ↓
Nierenversagen
(prä-renal, siehe entsprechende Folie)
Beachte: Während der DKA verliert der Körper K+ durch osmotische Diurese. Da gleichzeitig allerdings K+ aus den Zellen diffundiert, kann das Serumkalium unauffällig/erhöht sein. Um Hypokaliämien zu verhindern sollte bei K+ <5.0mmol/L KCl zusammen mit Insulin i.v. verabreicht werden.
Cave: gute Nierenfunktion des Patienten
Beachte: Elektrolytstörungen (z.B. Hyperkaliämien, Hypernatriämien) verschlechtern sich bei akutem Nierenversagen, welches bei der DKA &HK häufig auftritt
Legende:
Pathophysiologie
Mechanismen
Symptome/Klinische Befunde
Komplikationen
Veröffentlicht: 3. November 2016 auf www.thecalgaryguide.com")
Hyperosmolares/ Hyperglykämisches Koma (HHS)

Betrifft hauptsächlich Patienten mit Diabetes Mellitus Typ 1: Bestimmte Situationen (z.B. Infektionen) erhöhen den Insulinbedarf, welcher durch fehlende Produktion/unzureichende Substitution nicht gedeckt werden kann
Autor: Yan Yu Rezensenten: Peter Vetere, Gill Goobie, Sean Spence, Hanan Bassyouni* Übersetzung: Sarah Schwarz Übersetzungsprüfung: Dr. Gesche Tallen * MD zum Zeitpunkt der Veröffentlichung
Hyperglykämie
(erhöhter Blutzucker)
Bei >12mmol/L, Glukosefiltration > - resorption, Glukose verbleibt im Urin
Glukosurie
Glukose ist osmotisch wirksam und zieht große Mengen Wasser mit sich (Osmotische Diurese)
Polyurie
Schwere Dehydrierung(bis zu 4-5L)
(ZVD↓, Orthostase: posturale Hypotension/ posturale Tachykardie, Ruhepuls ↑ )
Insulinmangel enthemmt die Lipolyse, sodass der Körper Energie aus Triglyceriden produziert
Freisetzung von freien Fettsäuren aus Fettgewebe
Absoluter
Insulinmangel
Hypothalamische Zellen induzieren bei geringem intrazellulären Glukosespiegel ein starkes Hungergefühl
Glukose bleibt im Blut & kann nicht durch Muskel- /Fettgewebe verstoffwechselt werden
“Ausgehungerte” Zellen triggern die Freisetzung kataboler Hormone: Glucagon, Katecholamine, Cortisol, Somatotropin
Damit intrazellulärer Glukosespiegel ↑, erhöht der Körper den Blutzucker
Hydrolyse von freien Fettsäure in
der Leber (Ketogenese) Polyphagie
↓ Proteinsynthese, ↑ Proteolyse (in Muskelgewebe)
↑ Gluconeogenese, ↑ Glykogenolyse (in der Leber)
Acetyl Co-A
Energiequelle für “ausgehungerte” Zellen
Beachte: Neben Glukose sind Ketonkörper die einzigen Energieträger die von Nervenzellen metabolisiert werden können. Deshalb werden sie vom Körper bei geringem Glukoseangebot produziert.
Ketonkörper
( β-Hydroxybutyrat, Acetoacetat, Aceton, akkumulieren im Blut)
Ketonurie
Metabolische Azidose
(↑Anionenlücke: Ketonkörper verbrauchen den HCO3--Puffer)
Substrate der Gluconeogenese↑
Durch ↓ Extrazellulärflüssigkeit, werden Ketonkörper konzentriert → Azidose
Stört das enterische Nervensystem, Magenentleerung ↓, Ileus
Bauchschmerzen, Übelkeit, Erbrechen
(Dehydration↑)
Abatmen von CO2 um Azidose auszugleichen
Kussmaul- Atmung (Tiefe, schnelle Züge)
Abatmen von Keton- körpern
Azeton- geruch der Atemluft
Beeinträchtigung elektrischer Signale zum Gehirn, Rücken- mark und Nervenzellen
Perfusion des Gehirns, Rückenmark, Nervenzellen↓
Verschiebung des Wasser- und Elektrolythaushalts
Falls Trinkwasser vorhanden
Polydipsie Beachte: Während der DKA verliert der Körper K+ durch
Elektrolytstörung
Schwäche, Delirium +/- Koma
osmotische Diurese und Erbrechen. Da gleichzeitig allerdings K+ aus den Zellen diffundiert, kann das Serumkalium unauffällig/erhöht sein. Um Hypokaliämien zu verhindern sollte bei K+ <5.0mmol/L KCl zusammen mit Insulin i.v. verabreicht werden. Cave: gute Nierenfunktion des Patienten
Therapie: 1) +++ Flüssigkeit. 2) Insulin + KCl. 3) Auffüllen der Anionenlücke. 4) Auslösende Faktoren identifizieren. 5) Wenig PO4 (typischerweise wenige Stunden bis einen Tag nach der Ketoazidose durch ↑ ATP-Produktion.)
Legende:
Pathophysiologie
Mechanismen
Symptome/Klinische Befunde
Komplikationen
Publikationsdatum: 17 Juni 2013 auf www.thecalgaryguide.com
Hyperosmolares/ Hyperglykämisches Koma (HHS)
Beachte: betrifft nur Patienten mit Diabetes mellitus Typ II
Beachte: Es sollte stets nach auslösenden Faktoren z.B. einer Infektion gesucht werden
Autor: Yan Yu Rezensenten:
Peter Vetere
Gill Goobie
Hanan Bassyouni* Übersetzung:
Sarah Schwarz Übersetzungsprüfung: Dr. Gesche Tallen
* MD zum Zeitpunkt der Veröffentlichung
Verschiebung des Wasser- und Elektrolythaushalts
Elektrolytstörung
Unzureichende Insulin- produktion, Insulinresistenz, Nichtansprechen auf Insulintherapie
Relativer Insulinmangel
Situationen mit ↑Insulinbedarf: Infekt, Pneumonie, Herzinfarkt, Pancreatitis, etc.)
Hyperglykämie
(Stark erhöhter Blutzucker, höher als bei der DKA)
Bei >12mmol/L, Glukosefiltration > - resorption, Glukose verbleibt im Urin
Glukosurie
Glukose ist osmotisch wirksam und zieht große Mengen Wasser mit sich (Osmotische Diurese)
Polyurie Dehydration
(ZVD↓, Orthostase: posturale Hypotension/posturale Tachykardie, Ruhepuls ↑ )
Durch das wenige noch vorhandene Insulin, wird ein Teil der Glukose von Muskel- /Fettgewebe verstoffwechselt, ein Teil verbleibt im Blut
Körperzellen brauchen eine weitere Energiequelle
Freisetzung kataboler Hormone: Glucagon, Katecholamine, Cortisol, Somatrotropin
Damit intrazellulärer Glukosespiegel ↑, erhöht der Körper den Blutzucker
Hypothalamische Zellen induzieren bei geringem intrazellulären Glukosespiegel ein starkes Hungergefühl
Polyphagie
Beachte: Durch das wenige noch
vorhandene Insulin wird die Lipolyse gehemmt und werden keine Ketonkörper produziert. Es kommt nicht zur Azidose und Ketonurie (Gegensatz zur DKA). Sollte es doch zur Ketourie kommen, ist das meist Folge von Hungerzuständen oder anderen Mechanismen
Extrazellulärflüssigkeit ↓ mit erhöhter Osmolarität (z.B. Hypernatriämie)
↑ Gluconeogenese, ↑ Glykogenolyse (in der Leber)
↓ Proteinsynthese, ↑ Proteolyse (in Muskelgewebe)
Substrate der Gluconeogenese ↑
Sollte der Patient nicht ausreichend trinken um das Volumendefizit auszugleichen
Falls Trinkwasser vorhanden
Polydipsie
Wasser verlässt Zellen entlang des osmotischen Gradienten, Neuronen schrumpfen
Neuronaler Schaden: Delirium, Krampfanfall, Benommenheit, Koma
Renale Perfusion↓, GFR ↓
Nierenversagen
(prä-renal, siehe entsprechende Folie)
Beachte: Während der DKA verliert der Körper K+ durch osmotische Diurese. Da gleichzeitig allerdings K+ aus den Zellen diffundiert, kann das Serumkalium unauffällig/erhöht sein. Um Hypokaliämien zu verhindern sollte bei K+ <5.0mmol/L KCl zusammen mit Insulin i.v. verabreicht werden.
Cave: gute Nierenfunktion des Patienten
Beachte: Elektrolytstörungen (z.B. Hyperkaliämien, Hypernatriämien) verschlechtern sich bei akutem Nierenversagen, welches bei der DKA &HK häufig auftritt
Legende:
Pathophysiologie
Mechanismen
Symptome/Klinische Befunde
Komplikationen
Veröffentlicht: 3. November 2016 auf www.thecalgaryguide.com")
Asthma: Pathogenese
 Naushad Hirani* Übersetzung: Sarah Schwarz Übersetzungsprüfung: Dr. Gesche Tallen * MD zum Zeitpunkt der Veröffentlichung
Genetische Faktoren
(z.B. Mutationen im HLA- Gen, Defekte im bronchialen Endothel)
Umwelteinflüsse
(z.B. übermäßige Hygiene, wenige Geschwister, Antibiotikaeinnahme in den ersten 2 Lj.)
Asthma:
Eine chronisch-entzündliche Erkrankung welche durch hyperreagible Atemwege zu variablen, reversiblen Obstruktionen führt
Trigger für Hyperreagibilität der Atemwege:
Atopie:
Neigung zu allergischen Überempfindlichkeitsreaktionen der Atemwege
Erstexposition gegenüber des Triggers
Sensibilisierung der T-Helferzellen
Stimulation der B-Zellen zur IgE Produktion, welche an Mastzellen binden und diese aktivieren
Aktivierte T-Helferzellen und Mastzellen säumen die Atemwege
Infektionen des oberen Respirationstrakts
Allergene (Pollen, Haus- staub, Tierhaar etc.)
Luftverschmutzung, Zigarettenrauch, Chemikalien
Medikamente (ASS, NSAR, Beta-Blocker)
kalte Luft
Bewegung
Mastzellen setzen Histamine, Leukotriene
und andere Entzündungsmediatoren frei
Induzieren Zellreifung eosinophiler Granulozyten
Eosinophile migrieren in:
Gefäßpermeabilität ↑, Schleimhautödem in Bronchien
Becherzellhyperplasie, Schleimsekretion ↑
Kontraktion der Bronchialmuskulatur
Zweitkontak t mit dem Trigger
Frühe Reaktion (0-2 h)
Verspätet e Reaktion (3-4 h)
Allergene binden an IgEs auf Mastzellen
Aktivierte T- Helferzellen & Mastzellen setzen Zytokine frei
Obstruktion der Atemwege
Asthma
Bronchokonstriktion Konjunktivitis
Beachte: Verspätete Reaktionen beginnen nach 3-4h, erreichen ihren Höhepunkt nach 6-8h und klingen nach 24h ab
Atemwege Augen
Nase
Rhinitis
Legende:
Pathophysiologie
Mechanismen
Symptome/Klinische Befunde
Komplikationen
Veröffentlicht: 17. Dez. 2012, aktualisiert: 19. Aug. 2021 auf www.thecalgaryguide.com")
Nephrotisches Syndrom: Pathogenese und klinische Befunde

Lupus-Nephritis
Postinfektiöse Glomeruloneprhitis
IgA Nephropathie
Membranöse Glomerulonephritis
Antikörper greifen Podozyten an, Verdickung der glomerulären Basalmembran
Fokal-segmentale Glomerulosklerose (FSSGN)
Schädigung des glomerulären Endo- und Epithels
Minimal Change Glomerulo- nephritis (MCNG)
Diabetes mellitus
Autor: Yan Yu Rezensenten: Alexander Arnold David Waldner
Sean Spence
Stefan Mustata* Übersetzung:
Sarah Schwarz Übersetzungsprüfung: Gesche Tallen*
* MD zum Zeitpunkt der Veröffentlichung
Podozyten-Schädigung auf der epithelialen Seite des Glomerulums (Abflachung der Podozytenfortsätze)
Glomeruläre Immunkomplex- ablagerungen
Chronische Hyperglykämien schädigen das Glomerulum
Geschädigter Proteinfilter (v.a. für geladene Proteine)
Ablagerungen von Immunglobulin-Leichtketten im Glomerulum
Amyloidose
Geschädigte Glomeruli --> gestörte Filterbarriere v.a. für Proteine --> übermäßige Filtration von Plasmaproteinen
Vermehrte renale Ausscheidung von Immunglobulin-Leichtketten (Filtration>Resorption)
Hypoalbuminämie*
Übermäßiger Verlust an Albumin über den Urin
Proteinurie >3.5g/Tag*
Verlust an Antikoagulationsproteinen (Antithrombin, Plasminogen, Protein C & S) über den Urin
Koagulations- /Gerinnungsfaktoren (z.B. 1,7,8, 10) sind in Überzahl
Proteinurie >3.5g/Tag*
Thrombophilie
Anasarka
(Generalisiertes Ödem)
Lidödem
(klassisches Frühzeichen)
Lipidurie
(zeigt unter
gekreuztem polarisiertem Licht eine Malteserkreuz- form)
50% des Serumkalziums sind an Albumin gebunden, sodass Serumkalzium- spiegel ↓
Serum- Ca2+ repräsentiert nicht mehr das Gesamt-Ca2+
Beachte:
• DerklassischeSymptomkomplex(*)desnephrotischen Syndroms besteht aus: Proteinurie (>3,5g/Tag), Ödemen, Hypoalbuminämie,Hyperlipidämie
• Fürjede10g/LmitAlbumin<40:
➔ Addiere 2.5 zur errechneten Anionenlücke um
dessen “wahren” Wert zu bekommen
➔ Addiere 0,2mmol/L zum Gesamt-Ca um den
Wert des ionisierten Kalziums zu errechnen
Blut neigt zur Bildung von Thromben
Ödeme*
↑ Renale Filtrationder Lipide und Ausscheidung über den Urin
Die Anionenlücke ergibt sich hauptsächlich aus negativ geladenem Serumalbumin
Anionenlücke↓
Kolloid- osmotischer Druck ↓
Flüssigkeit kann nicht mehr in den Blutgefäßen gehalten werden und diffundiert ins Gewebe
Signalisiert der Leber die Albuminproduktion zuerhöhen,Albumin wird aber weiterhin über die Nieren verloren
Synthese- arbeit der Leber↑, auch ↑ Lipoprotein- synthese
Hyperlipidämie*:
(Serum-LDL, -VLDL, - TG ↑ )
Legende:
Pathophysiologie
Mechanismen
Symptome/Klinische Befunde
Komplikationen
Veröffentlicht: 19. August 2013 auf www.thecalgaryguide.com")
COPD-发病机制
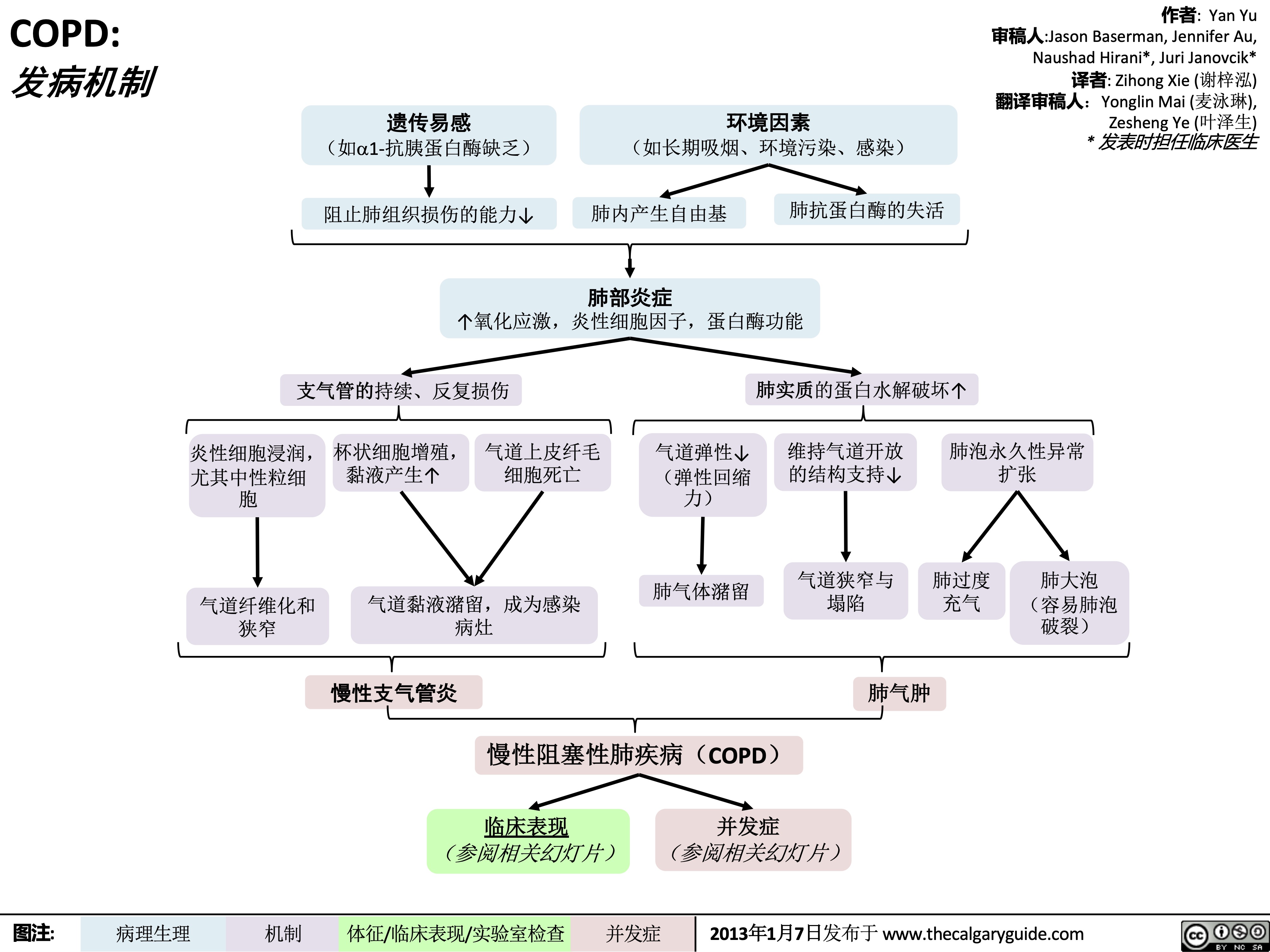 45
(on ABGs)
Ventilation- perfusion mismatch
High A-a gradient
(calculated from ABGs)
Low, flat diaphragm, >10 posterior ribs
(on frontal CXR)
High TLC and VC
(on spirometry)
• •
PaO2: partial pressure of O2 in arterial blood PaCO2: partial pressure of CO2 in arterial blood
• In the setting of fever and productive cough, especially if lung field opacifications are seen on CXR: consider sputum gram stain and culture to rule out pneumonia.
Air does not block X-ray beams, will appear black on X-ray film
Chronic hypercapnia makes breathing centers less sensitive to the high PaCO2 stimulus for breathing, & more reliant on the low PaO2 stimulus
(“CO2 retention”)
Give O2 carefully to these patients (high PaO2 may suppress patients’ hypoxic respiratory drive, ↓ their breathing, & ↑↑↑ PaCO2)
↑ retrosternal air space
(on lateral CXR)
Hyper-lucent
(darker) lung fields, ↓ lung markings (on frontal CXR)
• Arterial Blood Gasses (ABGs)
• Chest X-Ray (CXR): frontal and
lateral
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published January 7, 2013 on www.thecalgaryguide.com
COPD: !"#$ 气流阻塞
肺泡通气↓ 呼气时,胸膜腔正压挤压气 道à 阻塞↑
作者: Yan Yu 审稿人: Jason Baserman, Jennifer Au, Naushad Hirani*, Juri Janovcik* 译者:Zihong Xie (谢梓泓) 翻译审稿人: Yonglin Mai (麦泳琳), Zesheng Ye (叶泽生) * 发表时担任临床医生
慢性阻塞性肺疾病 (COPD)
肺组织损伤
没有弹性回缩力将
气体排出肺
肺实质与血管分布减少导 致气体交换面积↓
弥散功能↓ (肺功能检查)
更多的CO2残留 并扩散到血液中
高碳酸血症: PaCO2 > 45
(动脉血气)
血流灌注通气不良的肺泡
时无法获得足够的氧气
总呼气时长较正常长
FEV1/FEV < 0.7
(肺功能检查)
肺无法完全排空
更多空气潴留在肺部
(肺过度充气)
低氧血症: PaO2 < 70mmHg
(动脉血气)
通气-灌注不匹配
肺泡-动脉氧分压差↑ (可通过动脉血气分析计算得出)
横膈低平, 下移至第10肋后端 及以下部位 (胸部正位片)
TLC与VC增大 (肺功能检查)
缩写: • • FEV1: 1秒用 •
VC:肺活量
PaO2: 动脉血 力呼气量 氧分压
空气不会阻挡X射线, 在X光片上呈现为黑色
慢性高碳酸血症使呼吸中枢对PaCO2 刺激呼吸的敏感性下降 & 更依赖于低PaO2的刺激 (“二氧化碳潴留”)
给患者吸氧时需注意(高PaO2
可能会抑制患者低氧时对呼吸的 刺激,使呼吸驱动↓ & PaCO2↑↑↑ )
• FVC: 用力肺 • 活量
• TLC:肺总量 慢阻肺相关检查 :
PaCO2: 动脉 血二氧化碳 分压
胸骨后间隙↑
(胸部侧位片) 肺纹理↓
• 肺功能检查
• 动脉血气分析(Arterial Blood Gasses, ABGs)
• 胸部正侧位片
• 当患者发热和湿咳,特别是胸片上见肺野不清晰时:
肺透亮度↑, (胸部正位片)
考虑进行痰革兰氏染色及痰培养以排除肺炎可能
图注:
病理生理
机制
体征/临床表现/实验室检查
并发症
2013年1月7日发布于 www.thecalgaryguide.com
COPD: Complications Lung inflammation
Chronic Obstructive Pulmonary Disease (COPD)
Airway obstruction ↓ inhaled air in alveoli and terminal bronchioles
Rupture of emphasematous bullae on surface of lung
Inhaled air leaks into pleural cavity and is trapped there
Pneumothorax
Feeling a loss of control over one’s life, and hopelessness for the future
Goblet cell proliferation, ↑ mucus production
Death of airway
epithelium ciliated cells
↓ oxygenation of the blood passing through the lungs
Chronic hypoxemia
Kidneys compensate by ↑ erythropoietin (EPO) production
↑ Hemoglobin and red blood cell synthesis
Polycythemia (secondary)
Hypoxic alveoli cause the pulmonary arterioles perfusing them to reflexively vasoconstrict
Since most alveoli in the lungs are hypoxic, hypoxic vasoconstriction occurs across entire lung
Vasoconstriction ↑ blood pressure within lung vasculature
Pulmonary hypertension
↑ workload of the right ventricle (to pump against higher pressures)
To compensate, the right ventricle progressively hypertrophies and dilates, but over time its output ↓
Cor pulmonale
(Right heart failure in isolation, not due to Left heart failure)
Mucus trapped in airways, serve as nidus for infection
Acute exacerbation of COPD (AECOPD)
Pneumonia
The chronic, systemic inflammation in COPD is a hyper-metabolic state that consumes calories
Macro-nutrient deficiency
Trouble with respiration lead to inactivity and deconditioning
Wasting, muscle atrophy
More inactivity and deconditioning perpetuates the cycle
Depression
Author: Yan Yu Reviewers: Jason Baserman Naushad Hirani* Juri Janovcik* * MD at time of publication
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published January 7, 2013 on www.thecalgaryguide.com
COPD: !"# 肺部炎症
杯状细胞增殖, 气道上皮纤毛 粘液产生↑ 细胞死亡
黏液潴留呼吸道,成为感
染的病灶
慢性阻塞性肺疾病 (COPD) 气道阻塞à 吸入肺泡和终末细
肺大疱破裂
吸入的空气渗入
并潴留于胸腔
气胸
感觉生活失控,对未
来感到绝望
抑郁
作者: Yan Yu 审稿人: Jason Baserman, Naushad Hirani*, Juri Janovcik* 译者: Zihong Xie (谢梓泓) 翻译审稿人: Yonglin Mai (麦泳琳), Zesheng Ye (叶泽生) * 发表时担任临床医生
支气管的空气 ↓
流经肺的血液进行气 缺氧的肺泡à灌注肺泡的肺小动
慢性阻塞性肺疾 病急性加重期 (AECOPD)
肺炎
体交换↓ 慢性低氧血症
肾脏合成促红细胞 生成素进行代偿↑
血红蛋白与红 细胞合成↑
红细胞增多症 (继发性)
脉发生反射性血管收缩
肺大部分肺泡缺氧à整个肺 都出现缺氧性血管收缩
肺血管收缩 à 肺血管压力↑ 肺动脉高压
↑ 右心室负荷(泵血时对抗高压) 为了代偿,右心室逐渐肥大和扩张,
但随着病程进展,右心室输出量 ↓
肺心病 (单独出现右心衰竭,非左心衰)
COPD所致的慢性全身 呼吸困难导致活 性炎症会使机体处于高 动量减少和活动
代谢状态,消耗能量 耐量降低
宏量营养 素缺乏症
消瘦,肌肉萎缩
运动量下降和活动耐量
的降低造成恶性循环
图注:
病理生理
机制
体征/临床表现/实验室检查
并发症
2013年1月7日发布于 www.thecalgaryguide.com
" title="COPD: 发病机制
作者: Yan Yu 审稿人:Jason Baserman, Jennifer Au, Naushad Hirani*, Juri Janovcik* 译者: Zihong Xie (谢梓泓) 翻译审稿人:Yonglin Mai (麦泳琳), Zesheng Ye (叶泽生) * 发表时担任临床医生
/012
(如a1-抗胰蛋白酶缺乏) 阻止肺组织损伤的能力↓
+,-.
(如长期吸烟、环境污染、感染)
肺内产生自由基
34*5
肺抗蛋白酶的失活
↑氧化应激,炎性细胞因子,蛋白酶功能
支气管的持续、反复损伤
炎性细胞浸润, 杯状细胞增殖, 气道上皮纤毛 尤其中性粒细 黏液产生↑ 细胞死亡
气道弹性↓ (弹性回缩
肺实质的蛋白水解破坏↑ 维持气道开放 肺泡永久性异常
的结构支持↓ 扩张
胞 力)
肺气体潴留 气道狭窄与 肺过度 肺大泡
气道黏液潴留,成为感染 狭窄 病灶
塌陷 充气
肺气肿
(容易肺泡 破裂)
气道纤维化和
%&'()*
慢性阻塞性肺疾病(COPD)
临床表现 并发症 (参阅相关幻灯片) (参阅相关幻灯片)
图注:
病理生理
机制
体征/临床表现/实验室检查
并发症
2013年1月7日发布于 www.thecalgaryguide.com
COPD: Clinical Findings Lung tissue
Chronic Obstructive Pulmonary Disease (COPD)
damage
↓ elastic recoil to push air out of lungs on expiration
Lungs don’t fully empty, air is trapped in alveoli (lung hyperinflation)
↑ lung volume means diaphragm is tonically contracted (flatter)
If occurring around airways
Airflow obstruction
↑ mucus production
↓ number of epithelial ciliated cells to clear away the mucus (the cells have been killed by airway inflammation)
Chronic cough with sputum
Author: Yan Yu Reviewers: Jason Baserman Jennifer Au Naushad Hirani* Juri Janovcik* * MD at time of publication
During expiration, positive pleural pressure squeezes on airwaysà↑ obstruction
↓ ventilation of alveoli
↓ oxygenation of blood (hypoxemia)
↓ perfusion of body tissues (i.e. brain, muscle)
Fatigue; ↓ exercise tolerance
Total expiration time takes longer than normal
Prolonged expiration
More effort needed to ventilate larger lungs
Respiratory muscles must work harder to breathe
Turbulent airflow in narrower airways is heard on auscultation
Expiratory Wheeze
Diaphragm can’t flatten much further to generate deep breaths
To breathe, chest wall must expand out more
Dyspnea
Shortness of breath, especially on exertion
Breathes are rapid & shallow
If end-stage:
Chronic fatigue causes deconditioning
Muscle weakness & wasting
Barrel chest
If end-stage: diaphragm will be “flat”. Continued
Patient tries to expire against higher mouth air pressure, forcing airways to open wider
Pursed-lip breathing
Patient breathes with accessory muscles as well as diaphragm to try to improve airflow
inspiratory effort further contracts diaphragmà pull the lower chest wall inwards
Hoover’s sign
(paradoxical shrinking of lower chest during inspiration)
Tripod sitting position (activates pectoral muscles)
Neck (SCM, scalene) muscles contracted
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published January 7, 2013 on www.thecalgaryguide.com
COPD: !"#$
慢性阻塞性肺疾病 (COPD) 如果出现在气道周围 气流阻塞
肺不能完全排空
气体,气体潴留
于肺泡(肺过度
充气)
总呼气时长大于 正常时长
呼气相延长
肺组织损伤
呼气时,将空气排出肺外 的弹性回缩力↓
肺不能完全排空气体,
气体潴留于肺泡内
(肺过度充气)
肺容积↑,膈肌紧张 性收缩(膈肌平坦)
呼气时,胸膜腔正压挤压气道 à 气道阻塞↑
肺泡通气↓ 血液氧合↓ (低
氧血症)
身体组织灌注 量↓ (比如脑、 肌肉)
疲劳; 运动耐量↓
黏液生成↑ 清除黏液的上皮纤
毛细胞数量↓ (受 气道炎症损伤)
慢性咳嗽伴咳 痰
作者: Yan Yu
审稿人: Jason Baserman, Jennifer Au, Naushad Hirani*, Juri Janovcik* 译者: Zihong Xie (谢梓泓) 翻译审稿人: Yonglin Mai (麦泳琳),
Zesheng Ye (叶泽生)
* 发表时担任临床医生
容积较大 的肺需要
更加努力 才能通气
呼吸肌必须
更用力才能 呼吸
听诊闻及狭窄气
道中的湍流气流
呼气喘鸣音
呼吸困难 气促,尤其是劳累
膈肌无法进一步收缩以
产生深呼吸
呼吸浅快
为了呼吸,
胸壁必须延
展得更大
桶状胸
晚期病人:
患者试图在较高的口 慢性疲劳导致 患者动用辅助呼吸肌和膈肌呼吸,
腔内气压下进行呼气, 活动耐量下降 从而使气道更开放
以改善气流
晚期病人:膈肌 “平坦” ,持续吸气进一步压 缩膈肌à 向内拉季肋部胸壁
胡佛征 (吸气时,胸廓下侧季肋部内收)
缩唇呼吸
肌肉无力 & 消瘦
端坐呼吸 (调动胸肌)
颈部肌肉收
缩(胸锁乳
突肌、斜角
肌)
图注:
病理生理
机制
体征/临床表现/实验室检查
并发症
2013年1月7日发布于 www.thecalgaryguide.com
COPD: Findings on Investigations
Chronic Obstructive Pulmonary Disease (COPD)
Author: Yan Yu Reviewers: Jason Baserman Jennifer Au Naushad Hirani* Juri Janovcik* * MD at time of publication
Airflow obstruction
Lung tissue damage
↓ ventilation of alveoli
Blood perfusing ill- ventilated alveoli does not receive normal amounts of oxygen
During expiration, positive pleural pressure squeezes on airwaysà↑ obstruction)
No elastic recoil to push air out of lungs
Loss of lung parenchyma and vasculature ↓ surface area for gas exchange
↓ diffusion capacity
(on spirometry)
Hypoxemia: PaO2 < 70mmHg (on ABGs)
Abbreviations:
• FEV1: Forced expiratory volume in 1 second
• FVC: Forced vital capacity
• TLC: Total lung capacity
• VC: Vital Capacity
Investigations for COPD :
• Spirometry (Pulmonary function test)
Total expiration time takes longer than normal
FEV1/FEV < 0.7
(on spirometry)
Lungs don’t fully empty
More air trapped within lungs (hyperinflation)
More CO2 remains and diffuses into the blood
Hypercapnia: PaCO2 > 45
(on ABGs)
Ventilation- perfusion mismatch
High A-a gradient
(calculated from ABGs)
Low, flat diaphragm, >10 posterior ribs
(on frontal CXR)
High TLC and VC
(on spirometry)
• •
PaO2: partial pressure of O2 in arterial blood PaCO2: partial pressure of CO2 in arterial blood
• In the setting of fever and productive cough, especially if lung field opacifications are seen on CXR: consider sputum gram stain and culture to rule out pneumonia.
Air does not block X-ray beams, will appear black on X-ray film
Chronic hypercapnia makes breathing centers less sensitive to the high PaCO2 stimulus for breathing, & more reliant on the low PaO2 stimulus
(“CO2 retention”)
Give O2 carefully to these patients (high PaO2 may suppress patients’ hypoxic respiratory drive, ↓ their breathing, & ↑↑↑ PaCO2)
↑ retrosternal air space
(on lateral CXR)
Hyper-lucent
(darker) lung fields, ↓ lung markings (on frontal CXR)
• Arterial Blood Gasses (ABGs)
• Chest X-Ray (CXR): frontal and
lateral
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published January 7, 2013 on www.thecalgaryguide.com
COPD: !"#$ 气流阻塞
肺泡通气↓ 呼气时,胸膜腔正压挤压气 道à 阻塞↑
作者: Yan Yu 审稿人: Jason Baserman, Jennifer Au, Naushad Hirani*, Juri Janovcik* 译者:Zihong Xie (谢梓泓) 翻译审稿人: Yonglin Mai (麦泳琳), Zesheng Ye (叶泽生) * 发表时担任临床医生
慢性阻塞性肺疾病 (COPD)
肺组织损伤
没有弹性回缩力将
气体排出肺
肺实质与血管分布减少导 致气体交换面积↓
弥散功能↓ (肺功能检查)
更多的CO2残留 并扩散到血液中
高碳酸血症: PaCO2 > 45
(动脉血气)
血流灌注通气不良的肺泡
时无法获得足够的氧气
总呼气时长较正常长
FEV1/FEV < 0.7
(肺功能检查)
肺无法完全排空
更多空气潴留在肺部
(肺过度充气)
低氧血症: PaO2 < 70mmHg
(动脉血气)
通气-灌注不匹配
肺泡-动脉氧分压差↑ (可通过动脉血气分析计算得出)
横膈低平, 下移至第10肋后端 及以下部位 (胸部正位片)
TLC与VC增大 (肺功能检查)
缩写: • • FEV1: 1秒用 •
VC:肺活量
PaO2: 动脉血 力呼气量 氧分压
空气不会阻挡X射线, 在X光片上呈现为黑色
慢性高碳酸血症使呼吸中枢对PaCO2 刺激呼吸的敏感性下降 & 更依赖于低PaO2的刺激 (“二氧化碳潴留”)
给患者吸氧时需注意(高PaO2
可能会抑制患者低氧时对呼吸的 刺激,使呼吸驱动↓ & PaCO2↑↑↑ )
• FVC: 用力肺 • 活量
• TLC:肺总量 慢阻肺相关检查 :
PaCO2: 动脉 血二氧化碳 分压
胸骨后间隙↑
(胸部侧位片) 肺纹理↓
• 肺功能检查
• 动脉血气分析(Arterial Blood Gasses, ABGs)
• 胸部正侧位片
• 当患者发热和湿咳,特别是胸片上见肺野不清晰时:
肺透亮度↑, (胸部正位片)
考虑进行痰革兰氏染色及痰培养以排除肺炎可能
图注:
病理生理
机制
体征/临床表现/实验室检查
并发症
2013年1月7日发布于 www.thecalgaryguide.com
COPD: Complications Lung inflammation
Chronic Obstructive Pulmonary Disease (COPD)
Airway obstruction ↓ inhaled air in alveoli and terminal bronchioles
Rupture of emphasematous bullae on surface of lung
Inhaled air leaks into pleural cavity and is trapped there
Pneumothorax
Feeling a loss of control over one’s life, and hopelessness for the future
Goblet cell proliferation, ↑ mucus production
Death of airway
epithelium ciliated cells
↓ oxygenation of the blood passing through the lungs
Chronic hypoxemia
Kidneys compensate by ↑ erythropoietin (EPO) production
↑ Hemoglobin and red blood cell synthesis
Polycythemia (secondary)
Hypoxic alveoli cause the pulmonary arterioles perfusing them to reflexively vasoconstrict
Since most alveoli in the lungs are hypoxic, hypoxic vasoconstriction occurs across entire lung
Vasoconstriction ↑ blood pressure within lung vasculature
Pulmonary hypertension
↑ workload of the right ventricle (to pump against higher pressures)
To compensate, the right ventricle progressively hypertrophies and dilates, but over time its output ↓
Cor pulmonale
(Right heart failure in isolation, not due to Left heart failure)
Mucus trapped in airways, serve as nidus for infection
Acute exacerbation of COPD (AECOPD)
Pneumonia
The chronic, systemic inflammation in COPD is a hyper-metabolic state that consumes calories
Macro-nutrient deficiency
Trouble with respiration lead to inactivity and deconditioning
Wasting, muscle atrophy
More inactivity and deconditioning perpetuates the cycle
Depression
Author: Yan Yu Reviewers: Jason Baserman Naushad Hirani* Juri Janovcik* * MD at time of publication
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published January 7, 2013 on www.thecalgaryguide.com
COPD: !"# 肺部炎症
杯状细胞增殖, 气道上皮纤毛 粘液产生↑ 细胞死亡
黏液潴留呼吸道,成为感
染的病灶
慢性阻塞性肺疾病 (COPD) 气道阻塞à 吸入肺泡和终末细
肺大疱破裂
吸入的空气渗入
并潴留于胸腔
气胸
感觉生活失控,对未
来感到绝望
抑郁
作者: Yan Yu 审稿人: Jason Baserman, Naushad Hirani*, Juri Janovcik* 译者: Zihong Xie (谢梓泓) 翻译审稿人: Yonglin Mai (麦泳琳), Zesheng Ye (叶泽生) * 发表时担任临床医生
支气管的空气 ↓
流经肺的血液进行气 缺氧的肺泡à灌注肺泡的肺小动
慢性阻塞性肺疾 病急性加重期 (AECOPD)
肺炎
体交换↓ 慢性低氧血症
肾脏合成促红细胞 生成素进行代偿↑
血红蛋白与红 细胞合成↑
红细胞增多症 (继发性)
脉发生反射性血管收缩
肺大部分肺泡缺氧à整个肺 都出现缺氧性血管收缩
肺血管收缩 à 肺血管压力↑ 肺动脉高压
↑ 右心室负荷(泵血时对抗高压) 为了代偿,右心室逐渐肥大和扩张,
但随着病程进展,右心室输出量 ↓
肺心病 (单独出现右心衰竭,非左心衰)
COPD所致的慢性全身 呼吸困难导致活 性炎症会使机体处于高 动量减少和活动
代谢状态,消耗能量 耐量降低
宏量营养 素缺乏症
消瘦,肌肉萎缩
运动量下降和活动耐量
的降低造成恶性循环
图注:
病理生理
机制
体征/临床表现/实验室检查
并发症
2013年1月7日发布于 www.thecalgaryguide.com
" />
45
(on ABGs)
Ventilation- perfusion mismatch
High A-a gradient
(calculated from ABGs)
Low, flat diaphragm, >10 posterior ribs
(on frontal CXR)
High TLC and VC
(on spirometry)
• •
PaO2: partial pressure of O2 in arterial blood PaCO2: partial pressure of CO2 in arterial blood
• In the setting of fever and productive cough, especially if lung field opacifications are seen on CXR: consider sputum gram stain and culture to rule out pneumonia.
Air does not block X-ray beams, will appear black on X-ray film
Chronic hypercapnia makes breathing centers less sensitive to the high PaCO2 stimulus for breathing, & more reliant on the low PaO2 stimulus
(“CO2 retention”)
Give O2 carefully to these patients (high PaO2 may suppress patients’ hypoxic respiratory drive, ↓ their breathing, & ↑↑↑ PaCO2)
↑ retrosternal air space
(on lateral CXR)
Hyper-lucent
(darker) lung fields, ↓ lung markings (on frontal CXR)
• Arterial Blood Gasses (ABGs)
• Chest X-Ray (CXR): frontal and
lateral
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published January 7, 2013 on www.thecalgaryguide.com
COPD: !"#$ 气流阻塞
肺泡通气↓ 呼气时,胸膜腔正压挤压气 道à 阻塞↑
作者: Yan Yu 审稿人: Jason Baserman, Jennifer Au, Naushad Hirani*, Juri Janovcik* 译者:Zihong Xie (谢梓泓) 翻译审稿人: Yonglin Mai (麦泳琳), Zesheng Ye (叶泽生) * 发表时担任临床医生
慢性阻塞性肺疾病 (COPD)
肺组织损伤
没有弹性回缩力将
气体排出肺
肺实质与血管分布减少导 致气体交换面积↓
弥散功能↓ (肺功能检查)
更多的CO2残留 并扩散到血液中
高碳酸血症: PaCO2 > 45
(动脉血气)
血流灌注通气不良的肺泡
时无法获得足够的氧气
总呼气时长较正常长
FEV1/FEV < 0.7
(肺功能检查)
肺无法完全排空
更多空气潴留在肺部
(肺过度充气)
低氧血症: PaO2 < 70mmHg
(动脉血气)
通气-灌注不匹配
肺泡-动脉氧分压差↑ (可通过动脉血气分析计算得出)
横膈低平, 下移至第10肋后端 及以下部位 (胸部正位片)
TLC与VC增大 (肺功能检查)
缩写: • • FEV1: 1秒用 •
VC:肺活量
PaO2: 动脉血 力呼气量 氧分压
空气不会阻挡X射线, 在X光片上呈现为黑色
慢性高碳酸血症使呼吸中枢对PaCO2 刺激呼吸的敏感性下降 & 更依赖于低PaO2的刺激 (“二氧化碳潴留”)
给患者吸氧时需注意(高PaO2
可能会抑制患者低氧时对呼吸的 刺激,使呼吸驱动↓ & PaCO2↑↑↑ )
• FVC: 用力肺 • 活量
• TLC:肺总量 慢阻肺相关检查 :
PaCO2: 动脉 血二氧化碳 分压
胸骨后间隙↑
(胸部侧位片) 肺纹理↓
• 肺功能检查
• 动脉血气分析(Arterial Blood Gasses, ABGs)
• 胸部正侧位片
• 当患者发热和湿咳,特别是胸片上见肺野不清晰时:
肺透亮度↑, (胸部正位片)
考虑进行痰革兰氏染色及痰培养以排除肺炎可能
图注:
病理生理
机制
体征/临床表现/实验室检查
并发症
2013年1月7日发布于 www.thecalgaryguide.com
COPD: Complications Lung inflammation
Chronic Obstructive Pulmonary Disease (COPD)
Airway obstruction ↓ inhaled air in alveoli and terminal bronchioles
Rupture of emphasematous bullae on surface of lung
Inhaled air leaks into pleural cavity and is trapped there
Pneumothorax
Feeling a loss of control over one’s life, and hopelessness for the future
Goblet cell proliferation, ↑ mucus production
Death of airway
epithelium ciliated cells
↓ oxygenation of the blood passing through the lungs
Chronic hypoxemia
Kidneys compensate by ↑ erythropoietin (EPO) production
↑ Hemoglobin and red blood cell synthesis
Polycythemia (secondary)
Hypoxic alveoli cause the pulmonary arterioles perfusing them to reflexively vasoconstrict
Since most alveoli in the lungs are hypoxic, hypoxic vasoconstriction occurs across entire lung
Vasoconstriction ↑ blood pressure within lung vasculature
Pulmonary hypertension
↑ workload of the right ventricle (to pump against higher pressures)
To compensate, the right ventricle progressively hypertrophies and dilates, but over time its output ↓
Cor pulmonale
(Right heart failure in isolation, not due to Left heart failure)
Mucus trapped in airways, serve as nidus for infection
Acute exacerbation of COPD (AECOPD)
Pneumonia
The chronic, systemic inflammation in COPD is a hyper-metabolic state that consumes calories
Macro-nutrient deficiency
Trouble with respiration lead to inactivity and deconditioning
Wasting, muscle atrophy
More inactivity and deconditioning perpetuates the cycle
Depression
Author: Yan Yu Reviewers: Jason Baserman Naushad Hirani* Juri Janovcik* * MD at time of publication
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published January 7, 2013 on www.thecalgaryguide.com
COPD: !"# 肺部炎症
杯状细胞增殖, 气道上皮纤毛 粘液产生↑ 细胞死亡
黏液潴留呼吸道,成为感
染的病灶
慢性阻塞性肺疾病 (COPD) 气道阻塞à 吸入肺泡和终末细
肺大疱破裂
吸入的空气渗入
并潴留于胸腔
气胸
感觉生活失控,对未
来感到绝望
抑郁
作者: Yan Yu 审稿人: Jason Baserman, Naushad Hirani*, Juri Janovcik* 译者: Zihong Xie (谢梓泓) 翻译审稿人: Yonglin Mai (麦泳琳), Zesheng Ye (叶泽生) * 发表时担任临床医生
支气管的空气 ↓
流经肺的血液进行气 缺氧的肺泡à灌注肺泡的肺小动
慢性阻塞性肺疾 病急性加重期 (AECOPD)
肺炎
体交换↓ 慢性低氧血症
肾脏合成促红细胞 生成素进行代偿↑
血红蛋白与红 细胞合成↑
红细胞增多症 (继发性)
脉发生反射性血管收缩
肺大部分肺泡缺氧à整个肺 都出现缺氧性血管收缩
肺血管收缩 à 肺血管压力↑ 肺动脉高压
↑ 右心室负荷(泵血时对抗高压) 为了代偿,右心室逐渐肥大和扩张,
但随着病程进展,右心室输出量 ↓
肺心病 (单独出现右心衰竭,非左心衰)
COPD所致的慢性全身 呼吸困难导致活 性炎症会使机体处于高 动量减少和活动
代谢状态,消耗能量 耐量降低
宏量营养 素缺乏症
消瘦,肌肉萎缩
运动量下降和活动耐量
的降低造成恶性循环
图注:
病理生理
机制
体征/临床表现/实验室检查
并发症
2013年1月7日发布于 www.thecalgaryguide.com
" title="COPD: 发病机制
作者: Yan Yu 审稿人:Jason Baserman, Jennifer Au, Naushad Hirani*, Juri Janovcik* 译者: Zihong Xie (谢梓泓) 翻译审稿人:Yonglin Mai (麦泳琳), Zesheng Ye (叶泽生) * 发表时担任临床医生
/012
(如a1-抗胰蛋白酶缺乏) 阻止肺组织损伤的能力↓
+,-.
(如长期吸烟、环境污染、感染)
肺内产生自由基
34*5
肺抗蛋白酶的失活
↑氧化应激,炎性细胞因子,蛋白酶功能
支气管的持续、反复损伤
炎性细胞浸润, 杯状细胞增殖, 气道上皮纤毛 尤其中性粒细 黏液产生↑ 细胞死亡
气道弹性↓ (弹性回缩
肺实质的蛋白水解破坏↑ 维持气道开放 肺泡永久性异常
的结构支持↓ 扩张
胞 力)
肺气体潴留 气道狭窄与 肺过度 肺大泡
气道黏液潴留,成为感染 狭窄 病灶
塌陷 充气
肺气肿
(容易肺泡 破裂)
气道纤维化和
%&'()*
慢性阻塞性肺疾病(COPD)
临床表现 并发症 (参阅相关幻灯片) (参阅相关幻灯片)
图注:
病理生理
机制
体征/临床表现/实验室检查
并发症
2013年1月7日发布于 www.thecalgaryguide.com
COPD: Clinical Findings Lung tissue
Chronic Obstructive Pulmonary Disease (COPD)
damage
↓ elastic recoil to push air out of lungs on expiration
Lungs don’t fully empty, air is trapped in alveoli (lung hyperinflation)
↑ lung volume means diaphragm is tonically contracted (flatter)
If occurring around airways
Airflow obstruction
↑ mucus production
↓ number of epithelial ciliated cells to clear away the mucus (the cells have been killed by airway inflammation)
Chronic cough with sputum
Author: Yan Yu Reviewers: Jason Baserman Jennifer Au Naushad Hirani* Juri Janovcik* * MD at time of publication
During expiration, positive pleural pressure squeezes on airwaysà↑ obstruction
↓ ventilation of alveoli
↓ oxygenation of blood (hypoxemia)
↓ perfusion of body tissues (i.e. brain, muscle)
Fatigue; ↓ exercise tolerance
Total expiration time takes longer than normal
Prolonged expiration
More effort needed to ventilate larger lungs
Respiratory muscles must work harder to breathe
Turbulent airflow in narrower airways is heard on auscultation
Expiratory Wheeze
Diaphragm can’t flatten much further to generate deep breaths
To breathe, chest wall must expand out more
Dyspnea
Shortness of breath, especially on exertion
Breathes are rapid & shallow
If end-stage:
Chronic fatigue causes deconditioning
Muscle weakness & wasting
Barrel chest
If end-stage: diaphragm will be “flat”. Continued
Patient tries to expire against higher mouth air pressure, forcing airways to open wider
Pursed-lip breathing
Patient breathes with accessory muscles as well as diaphragm to try to improve airflow
inspiratory effort further contracts diaphragmà pull the lower chest wall inwards
Hoover’s sign
(paradoxical shrinking of lower chest during inspiration)
Tripod sitting position (activates pectoral muscles)
Neck (SCM, scalene) muscles contracted
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published January 7, 2013 on www.thecalgaryguide.com
COPD: !"#$
慢性阻塞性肺疾病 (COPD) 如果出现在气道周围 气流阻塞
肺不能完全排空
气体,气体潴留
于肺泡(肺过度
充气)
总呼气时长大于 正常时长
呼气相延长
肺组织损伤
呼气时,将空气排出肺外 的弹性回缩力↓
肺不能完全排空气体,
气体潴留于肺泡内
(肺过度充气)
肺容积↑,膈肌紧张 性收缩(膈肌平坦)
呼气时,胸膜腔正压挤压气道 à 气道阻塞↑
肺泡通气↓ 血液氧合↓ (低
氧血症)
身体组织灌注 量↓ (比如脑、 肌肉)
疲劳; 运动耐量↓
黏液生成↑ 清除黏液的上皮纤
毛细胞数量↓ (受 气道炎症损伤)
慢性咳嗽伴咳 痰
作者: Yan Yu
审稿人: Jason Baserman, Jennifer Au, Naushad Hirani*, Juri Janovcik* 译者: Zihong Xie (谢梓泓) 翻译审稿人: Yonglin Mai (麦泳琳),
Zesheng Ye (叶泽生)
* 发表时担任临床医生
容积较大 的肺需要
更加努力 才能通气
呼吸肌必须
更用力才能 呼吸
听诊闻及狭窄气
道中的湍流气流
呼气喘鸣音
呼吸困难 气促,尤其是劳累
膈肌无法进一步收缩以
产生深呼吸
呼吸浅快
为了呼吸,
胸壁必须延
展得更大
桶状胸
晚期病人:
患者试图在较高的口 慢性疲劳导致 患者动用辅助呼吸肌和膈肌呼吸,
腔内气压下进行呼气, 活动耐量下降 从而使气道更开放
以改善气流
晚期病人:膈肌 “平坦” ,持续吸气进一步压 缩膈肌à 向内拉季肋部胸壁
胡佛征 (吸气时,胸廓下侧季肋部内收)
缩唇呼吸
肌肉无力 & 消瘦
端坐呼吸 (调动胸肌)
颈部肌肉收
缩(胸锁乳
突肌、斜角
肌)
图注:
病理生理
机制
体征/临床表现/实验室检查
并发症
2013年1月7日发布于 www.thecalgaryguide.com
COPD: Findings on Investigations
Chronic Obstructive Pulmonary Disease (COPD)
Author: Yan Yu Reviewers: Jason Baserman Jennifer Au Naushad Hirani* Juri Janovcik* * MD at time of publication
Airflow obstruction
Lung tissue damage
↓ ventilation of alveoli
Blood perfusing ill- ventilated alveoli does not receive normal amounts of oxygen
During expiration, positive pleural pressure squeezes on airwaysà↑ obstruction)
No elastic recoil to push air out of lungs
Loss of lung parenchyma and vasculature ↓ surface area for gas exchange
↓ diffusion capacity
(on spirometry)
Hypoxemia: PaO2 < 70mmHg (on ABGs)
Abbreviations:
• FEV1: Forced expiratory volume in 1 second
• FVC: Forced vital capacity
• TLC: Total lung capacity
• VC: Vital Capacity
Investigations for COPD :
• Spirometry (Pulmonary function test)
Total expiration time takes longer than normal
FEV1/FEV < 0.7
(on spirometry)
Lungs don’t fully empty
More air trapped within lungs (hyperinflation)
More CO2 remains and diffuses into the blood
Hypercapnia: PaCO2 > 45
(on ABGs)
Ventilation- perfusion mismatch
High A-a gradient
(calculated from ABGs)
Low, flat diaphragm, >10 posterior ribs
(on frontal CXR)
High TLC and VC
(on spirometry)
• •
PaO2: partial pressure of O2 in arterial blood PaCO2: partial pressure of CO2 in arterial blood
• In the setting of fever and productive cough, especially if lung field opacifications are seen on CXR: consider sputum gram stain and culture to rule out pneumonia.
Air does not block X-ray beams, will appear black on X-ray film
Chronic hypercapnia makes breathing centers less sensitive to the high PaCO2 stimulus for breathing, & more reliant on the low PaO2 stimulus
(“CO2 retention”)
Give O2 carefully to these patients (high PaO2 may suppress patients’ hypoxic respiratory drive, ↓ their breathing, & ↑↑↑ PaCO2)
↑ retrosternal air space
(on lateral CXR)
Hyper-lucent
(darker) lung fields, ↓ lung markings (on frontal CXR)
• Arterial Blood Gasses (ABGs)
• Chest X-Ray (CXR): frontal and
lateral
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published January 7, 2013 on www.thecalgaryguide.com
COPD: !"#$ 气流阻塞
肺泡通气↓ 呼气时,胸膜腔正压挤压气 道à 阻塞↑
作者: Yan Yu 审稿人: Jason Baserman, Jennifer Au, Naushad Hirani*, Juri Janovcik* 译者:Zihong Xie (谢梓泓) 翻译审稿人: Yonglin Mai (麦泳琳), Zesheng Ye (叶泽生) * 发表时担任临床医生
慢性阻塞性肺疾病 (COPD)
肺组织损伤
没有弹性回缩力将
气体排出肺
肺实质与血管分布减少导 致气体交换面积↓
弥散功能↓ (肺功能检查)
更多的CO2残留 并扩散到血液中
高碳酸血症: PaCO2 > 45
(动脉血气)
血流灌注通气不良的肺泡
时无法获得足够的氧气
总呼气时长较正常长
FEV1/FEV < 0.7
(肺功能检查)
肺无法完全排空
更多空气潴留在肺部
(肺过度充气)
低氧血症: PaO2 < 70mmHg
(动脉血气)
通气-灌注不匹配
肺泡-动脉氧分压差↑ (可通过动脉血气分析计算得出)
横膈低平, 下移至第10肋后端 及以下部位 (胸部正位片)
TLC与VC增大 (肺功能检查)
缩写: • • FEV1: 1秒用 •
VC:肺活量
PaO2: 动脉血 力呼气量 氧分压
空气不会阻挡X射线, 在X光片上呈现为黑色
慢性高碳酸血症使呼吸中枢对PaCO2 刺激呼吸的敏感性下降 & 更依赖于低PaO2的刺激 (“二氧化碳潴留”)
给患者吸氧时需注意(高PaO2
可能会抑制患者低氧时对呼吸的 刺激,使呼吸驱动↓ & PaCO2↑↑↑ )
• FVC: 用力肺 • 活量
• TLC:肺总量 慢阻肺相关检查 :
PaCO2: 动脉 血二氧化碳 分压
胸骨后间隙↑
(胸部侧位片) 肺纹理↓
• 肺功能检查
• 动脉血气分析(Arterial Blood Gasses, ABGs)
• 胸部正侧位片
• 当患者发热和湿咳,特别是胸片上见肺野不清晰时:
肺透亮度↑, (胸部正位片)
考虑进行痰革兰氏染色及痰培养以排除肺炎可能
图注:
病理生理
机制
体征/临床表现/实验室检查
并发症
2013年1月7日发布于 www.thecalgaryguide.com
COPD: Complications Lung inflammation
Chronic Obstructive Pulmonary Disease (COPD)
Airway obstruction ↓ inhaled air in alveoli and terminal bronchioles
Rupture of emphasematous bullae on surface of lung
Inhaled air leaks into pleural cavity and is trapped there
Pneumothorax
Feeling a loss of control over one’s life, and hopelessness for the future
Goblet cell proliferation, ↑ mucus production
Death of airway
epithelium ciliated cells
↓ oxygenation of the blood passing through the lungs
Chronic hypoxemia
Kidneys compensate by ↑ erythropoietin (EPO) production
↑ Hemoglobin and red blood cell synthesis
Polycythemia (secondary)
Hypoxic alveoli cause the pulmonary arterioles perfusing them to reflexively vasoconstrict
Since most alveoli in the lungs are hypoxic, hypoxic vasoconstriction occurs across entire lung
Vasoconstriction ↑ blood pressure within lung vasculature
Pulmonary hypertension
↑ workload of the right ventricle (to pump against higher pressures)
To compensate, the right ventricle progressively hypertrophies and dilates, but over time its output ↓
Cor pulmonale
(Right heart failure in isolation, not due to Left heart failure)
Mucus trapped in airways, serve as nidus for infection
Acute exacerbation of COPD (AECOPD)
Pneumonia
The chronic, systemic inflammation in COPD is a hyper-metabolic state that consumes calories
Macro-nutrient deficiency
Trouble with respiration lead to inactivity and deconditioning
Wasting, muscle atrophy
More inactivity and deconditioning perpetuates the cycle
Depression
Author: Yan Yu Reviewers: Jason Baserman Naushad Hirani* Juri Janovcik* * MD at time of publication
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published January 7, 2013 on www.thecalgaryguide.com
COPD: !"# 肺部炎症
杯状细胞增殖, 气道上皮纤毛 粘液产生↑ 细胞死亡
黏液潴留呼吸道,成为感
染的病灶
慢性阻塞性肺疾病 (COPD) 气道阻塞à 吸入肺泡和终末细
肺大疱破裂
吸入的空气渗入
并潴留于胸腔
气胸
感觉生活失控,对未
来感到绝望
抑郁
作者: Yan Yu 审稿人: Jason Baserman, Naushad Hirani*, Juri Janovcik* 译者: Zihong Xie (谢梓泓) 翻译审稿人: Yonglin Mai (麦泳琳), Zesheng Ye (叶泽生) * 发表时担任临床医生
支气管的空气 ↓
流经肺的血液进行气 缺氧的肺泡à灌注肺泡的肺小动
慢性阻塞性肺疾 病急性加重期 (AECOPD)
肺炎
体交换↓ 慢性低氧血症
肾脏合成促红细胞 生成素进行代偿↑
血红蛋白与红 细胞合成↑
红细胞增多症 (继发性)
脉发生反射性血管收缩
肺大部分肺泡缺氧à整个肺 都出现缺氧性血管收缩
肺血管收缩 à 肺血管压力↑ 肺动脉高压
↑ 右心室负荷(泵血时对抗高压) 为了代偿,右心室逐渐肥大和扩张,
但随着病程进展,右心室输出量 ↓
肺心病 (单独出现右心衰竭,非左心衰)
COPD所致的慢性全身 呼吸困难导致活 性炎症会使机体处于高 动量减少和活动
代谢状态,消耗能量 耐量降低
宏量营养 素缺乏症
消瘦,肌肉萎缩
运动量下降和活动耐量
的降低造成恶性循环
图注:
病理生理
机制
体征/临床表现/实验室检查
并发症
2013年1月7日发布于 www.thecalgaryguide.com
" />
COPD-临床表现
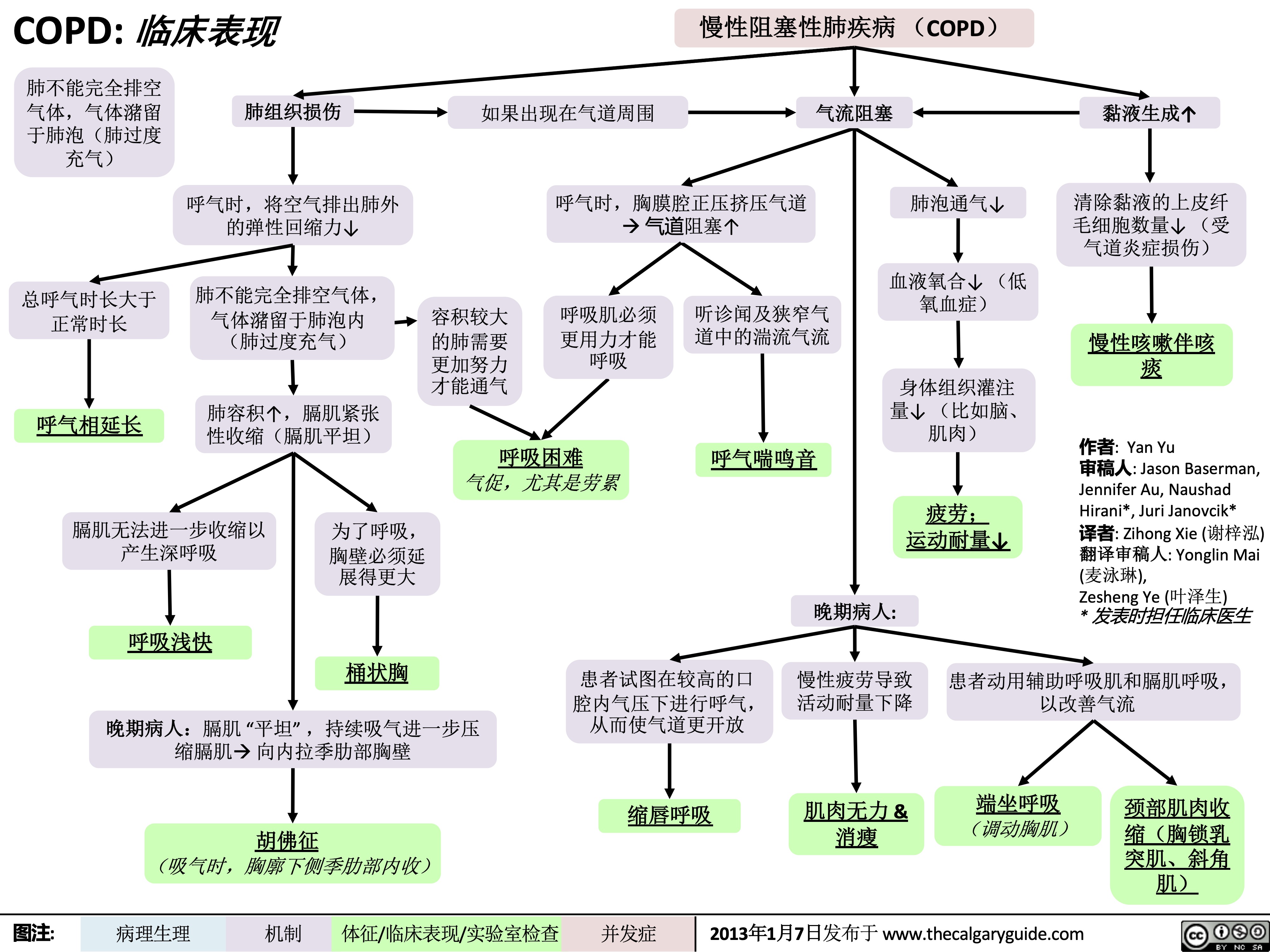 45
(on ABGs)
Ventilation- perfusion mismatch
High A-a gradient
(calculated from ABGs)
Low, flat diaphragm, >10 posterior ribs
(on frontal CXR)
High TLC and VC
(on spirometry)
• •
PaO2: partial pressure of O2 in arterial blood PaCO2: partial pressure of CO2 in arterial blood
• In the setting of fever and productive cough, especially if lung field opacifications are seen on CXR: consider sputum gram stain and culture to rule out pneumonia.
Air does not block X-ray beams, will appear black on X-ray film
Chronic hypercapnia makes breathing centers less sensitive to the high PaCO2 stimulus for breathing, & more reliant on the low PaO2 stimulus
(“CO2 retention”)
Give O2 carefully to these patients (high PaO2 may suppress patients’ hypoxic respiratory drive, ↓ their breathing, & ↑↑↑ PaCO2)
↑ retrosternal air space
(on lateral CXR)
Hyper-lucent
(darker) lung fields, ↓ lung markings (on frontal CXR)
• Arterial Blood Gasses (ABGs)
• Chest X-Ray (CXR): frontal and
lateral
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published January 7, 2013 on www.thecalgaryguide.com
COPD: !"#$ 气流阻塞
肺泡通气↓ 呼气时,胸膜腔正压挤压气 道à 阻塞↑
作者: Yan Yu 审稿人: Jason Baserman, Jennifer Au, Naushad Hirani*, Juri Janovcik* 译者:Zihong Xie (谢梓泓) 翻译审稿人: Yonglin Mai (麦泳琳), Zesheng Ye (叶泽生) * 发表时担任临床医生
慢性阻塞性肺疾病 (COPD)
肺组织损伤
没有弹性回缩力将
气体排出肺
肺实质与血管分布减少导 致气体交换面积↓
弥散功能↓ (肺功能检查)
更多的CO2残留 并扩散到血液中
高碳酸血症: PaCO2 > 45
(动脉血气)
血流灌注通气不良的肺泡
时无法获得足够的氧气
总呼气时长较正常长
FEV1/FEV < 0.7
(肺功能检查)
肺无法完全排空
更多空气潴留在肺部
(肺过度充气)
低氧血症: PaO2 < 70mmHg
(动脉血气)
通气-灌注不匹配
肺泡-动脉氧分压差↑ (可通过动脉血气分析计算得出)
横膈低平, 下移至第10肋后端 及以下部位 (胸部正位片)
TLC与VC增大 (肺功能检查)
缩写: • • FEV1: 1秒用 •
VC:肺活量
PaO2: 动脉血 力呼气量 氧分压
空气不会阻挡X射线, 在X光片上呈现为黑色
慢性高碳酸血症使呼吸中枢对PaCO2 刺激呼吸的敏感性下降 & 更依赖于低PaO2的刺激 (“二氧化碳潴留”)
给患者吸氧时需注意(高PaO2
可能会抑制患者低氧时对呼吸的 刺激,使呼吸驱动↓ & PaCO2↑↑↑ )
• FVC: 用力肺 • 活量
• TLC:肺总量 慢阻肺相关检查 :
PaCO2: 动脉 血二氧化碳 分压
胸骨后间隙↑
(胸部侧位片) 肺纹理↓
• 肺功能检查
• 动脉血气分析(Arterial Blood Gasses, ABGs)
• 胸部正侧位片
• 当患者发热和湿咳,特别是胸片上见肺野不清晰时:
肺透亮度↑, (胸部正位片)
考虑进行痰革兰氏染色及痰培养以排除肺炎可能
图注:
病理生理
机制
体征/临床表现/实验室检查
并发症
2013年1月7日发布于 www.thecalgaryguide.com
COPD: Complications Lung inflammation
Chronic Obstructive Pulmonary Disease (COPD)
Airway obstruction ↓ inhaled air in alveoli and terminal bronchioles
Rupture of emphasematous bullae on surface of lung
Inhaled air leaks into pleural cavity and is trapped there
Pneumothorax
Feeling a loss of control over one’s life, and hopelessness for the future
Goblet cell proliferation, ↑ mucus production
Death of airway
epithelium ciliated cells
↓ oxygenation of the blood passing through the lungs
Chronic hypoxemia
Kidneys compensate by ↑ erythropoietin (EPO) production
↑ Hemoglobin and red blood cell synthesis
Polycythemia (secondary)
Hypoxic alveoli cause the pulmonary arterioles perfusing them to reflexively vasoconstrict
Since most alveoli in the lungs are hypoxic, hypoxic vasoconstriction occurs across entire lung
Vasoconstriction ↑ blood pressure within lung vasculature
Pulmonary hypertension
↑ workload of the right ventricle (to pump against higher pressures)
To compensate, the right ventricle progressively hypertrophies and dilates, but over time its output ↓
Cor pulmonale
(Right heart failure in isolation, not due to Left heart failure)
Mucus trapped in airways, serve as nidus for infection
Acute exacerbation of COPD (AECOPD)
Pneumonia
The chronic, systemic inflammation in COPD is a hyper-metabolic state that consumes calories
Macro-nutrient deficiency
Trouble with respiration lead to inactivity and deconditioning
Wasting, muscle atrophy
More inactivity and deconditioning perpetuates the cycle
Depression
Author: Yan Yu Reviewers: Jason Baserman Naushad Hirani* Juri Janovcik* * MD at time of publication
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published January 7, 2013 on www.thecalgaryguide.com
COPD: !"# 肺部炎症
杯状细胞增殖, 气道上皮纤毛 粘液产生↑ 细胞死亡
黏液潴留呼吸道,成为感
染的病灶
慢性阻塞性肺疾病 (COPD) 气道阻塞à 吸入肺泡和终末细
肺大疱破裂
吸入的空气渗入
并潴留于胸腔
气胸
感觉生活失控,对未
来感到绝望
抑郁
作者: Yan Yu 审稿人: Jason Baserman, Naushad Hirani*, Juri Janovcik* 译者: Zihong Xie (谢梓泓) 翻译审稿人: Yonglin Mai (麦泳琳), Zesheng Ye (叶泽生) * 发表时担任临床医生
支气管的空气 ↓
流经肺的血液进行气 缺氧的肺泡à灌注肺泡的肺小动
慢性阻塞性肺疾 病急性加重期 (AECOPD)
肺炎
体交换↓ 慢性低氧血症
肾脏合成促红细胞 生成素进行代偿↑
血红蛋白与红 细胞合成↑
红细胞增多症 (继发性)
脉发生反射性血管收缩
肺大部分肺泡缺氧à整个肺 都出现缺氧性血管收缩
肺血管收缩 à 肺血管压力↑ 肺动脉高压
↑ 右心室负荷(泵血时对抗高压) 为了代偿,右心室逐渐肥大和扩张,
但随着病程进展,右心室输出量 ↓
肺心病 (单独出现右心衰竭,非左心衰)
COPD所致的慢性全身 呼吸困难导致活 性炎症会使机体处于高 动量减少和活动
代谢状态,消耗能量 耐量降低
宏量营养 素缺乏症
消瘦,肌肉萎缩
运动量下降和活动耐量
的降低造成恶性循环
图注:
病理生理
机制
体征/临床表现/实验室检查
并发症
2013年1月7日发布于 www.thecalgaryguide.com
" title="COPD: 临床表现
作者: Yan Yu 审稿人:Jason Baserman, Jennifer Au, Naushad Hirani*, Juri Janovcik* 译者: Zihong Xie (谢梓泓) 翻译审稿人:Yonglin Mai (麦泳琳), Zesheng Ye (叶泽生) * 发表时担任临床医生
/012
(如a1-抗胰蛋白酶缺乏) 阻止肺组织损伤的能力↓
+,-.
(如长期吸烟、环境污染、感染)
肺内产生自由基
34*5
肺抗蛋白酶的失活
↑氧化应激,炎性细胞因子,蛋白酶功能
支气管的持续、反复损伤
炎性细胞浸润, 杯状细胞增殖, 气道上皮纤毛 尤其中性粒细 黏液产生↑ 细胞死亡
气道弹性↓ (弹性回缩
肺实质的蛋白水解破坏↑ 维持气道开放 肺泡永久性异常
的结构支持↓ 扩张
胞 力)
肺气体潴留 气道狭窄与 肺过度 肺大泡
气道黏液潴留,成为感染 狭窄 病灶
塌陷 充气
肺气肿
(容易肺泡 破裂)
气道纤维化和
%&'()*
慢性阻塞性肺疾病(COPD)
临床表现 并发症 (参阅相关幻灯片) (参阅相关幻灯片)
图注:
病理生理
机制
体征/临床表现/实验室检查
并发症
2013年1月7日发布于 www.thecalgaryguide.com
COPD: Clinical Findings Lung tissue
Chronic Obstructive Pulmonary Disease (COPD)
damage
↓ elastic recoil to push air out of lungs on expiration
Lungs don’t fully empty, air is trapped in alveoli (lung hyperinflation)
↑ lung volume means diaphragm is tonically contracted (flatter)
If occurring around airways
Airflow obstruction
↑ mucus production
↓ number of epithelial ciliated cells to clear away the mucus (the cells have been killed by airway inflammation)
Chronic cough with sputum
Author: Yan Yu Reviewers: Jason Baserman Jennifer Au Naushad Hirani* Juri Janovcik* * MD at time of publication
During expiration, positive pleural pressure squeezes on airwaysà↑ obstruction
↓ ventilation of alveoli
↓ oxygenation of blood (hypoxemia)
↓ perfusion of body tissues (i.e. brain, muscle)
Fatigue; ↓ exercise tolerance
Total expiration time takes longer than normal
Prolonged expiration
More effort needed to ventilate larger lungs
Respiratory muscles must work harder to breathe
Turbulent airflow in narrower airways is heard on auscultation
Expiratory Wheeze
Diaphragm can’t flatten much further to generate deep breaths
To breathe, chest wall must expand out more
Dyspnea
Shortness of breath, especially on exertion
Breathes are rapid & shallow
If end-stage:
Chronic fatigue causes deconditioning
Muscle weakness & wasting
Barrel chest
If end-stage: diaphragm will be “flat”. Continued
Patient tries to expire against higher mouth air pressure, forcing airways to open wider
Pursed-lip breathing
Patient breathes with accessory muscles as well as diaphragm to try to improve airflow
inspiratory effort further contracts diaphragmà pull the lower chest wall inwards
Hoover’s sign
(paradoxical shrinking of lower chest during inspiration)
Tripod sitting position (activates pectoral muscles)
Neck (SCM, scalene) muscles contracted
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published January 7, 2013 on www.thecalgaryguide.com
COPD: !"#$
慢性阻塞性肺疾病 (COPD) 如果出现在气道周围 气流阻塞
肺不能完全排空
气体,气体潴留
于肺泡(肺过度
充气)
总呼气时长大于 正常时长
呼气相延长
肺组织损伤
呼气时,将空气排出肺外 的弹性回缩力↓
肺不能完全排空气体,
气体潴留于肺泡内
(肺过度充气)
肺容积↑,膈肌紧张 性收缩(膈肌平坦)
呼气时,胸膜腔正压挤压气道 à 气道阻塞↑
肺泡通气↓ 血液氧合↓ (低
氧血症)
身体组织灌注 量↓ (比如脑、 肌肉)
疲劳; 运动耐量↓
黏液生成↑ 清除黏液的上皮纤
毛细胞数量↓ (受 气道炎症损伤)
慢性咳嗽伴咳 痰
作者: Yan Yu
审稿人: Jason Baserman, Jennifer Au, Naushad Hirani*, Juri Janovcik* 译者: Zihong Xie (谢梓泓) 翻译审稿人: Yonglin Mai (麦泳琳),
Zesheng Ye (叶泽生)
* 发表时担任临床医生
容积较大 的肺需要
更加努力 才能通气
呼吸肌必须
更用力才能 呼吸
听诊闻及狭窄气
道中的湍流气流
呼气喘鸣音
呼吸困难 气促,尤其是劳累
膈肌无法进一步收缩以
产生深呼吸
呼吸浅快
为了呼吸,
胸壁必须延
展得更大
桶状胸
晚期病人:
患者试图在较高的口 慢性疲劳导致 患者动用辅助呼吸肌和膈肌呼吸,
腔内气压下进行呼气, 活动耐量下降 从而使气道更开放
以改善气流
晚期病人:膈肌 “平坦” ,持续吸气进一步压 缩膈肌à 向内拉季肋部胸壁
胡佛征 (吸气时,胸廓下侧季肋部内收)
缩唇呼吸
肌肉无力 & 消瘦
端坐呼吸 (调动胸肌)
颈部肌肉收
缩(胸锁乳
突肌、斜角
肌)
图注:
病理生理
机制
体征/临床表现/实验室检查
并发症
2013年1月7日发布于 www.thecalgaryguide.com
COPD: Findings on Investigations
Chronic Obstructive Pulmonary Disease (COPD)
Author: Yan Yu Reviewers: Jason Baserman Jennifer Au Naushad Hirani* Juri Janovcik* * MD at time of publication
Airflow obstruction
Lung tissue damage
↓ ventilation of alveoli
Blood perfusing ill- ventilated alveoli does not receive normal amounts of oxygen
During expiration, positive pleural pressure squeezes on airwaysà↑ obstruction)
No elastic recoil to push air out of lungs
Loss of lung parenchyma and vasculature ↓ surface area for gas exchange
↓ diffusion capacity
(on spirometry)
Hypoxemia: PaO2 < 70mmHg (on ABGs)
Abbreviations:
• FEV1: Forced expiratory volume in 1 second
• FVC: Forced vital capacity
• TLC: Total lung capacity
• VC: Vital Capacity
Investigations for COPD :
• Spirometry (Pulmonary function test)
Total expiration time takes longer than normal
FEV1/FEV < 0.7
(on spirometry)
Lungs don’t fully empty
More air trapped within lungs (hyperinflation)
More CO2 remains and diffuses into the blood
Hypercapnia: PaCO2 > 45
(on ABGs)
Ventilation- perfusion mismatch
High A-a gradient
(calculated from ABGs)
Low, flat diaphragm, >10 posterior ribs
(on frontal CXR)
High TLC and VC
(on spirometry)
• •
PaO2: partial pressure of O2 in arterial blood PaCO2: partial pressure of CO2 in arterial blood
• In the setting of fever and productive cough, especially if lung field opacifications are seen on CXR: consider sputum gram stain and culture to rule out pneumonia.
Air does not block X-ray beams, will appear black on X-ray film
Chronic hypercapnia makes breathing centers less sensitive to the high PaCO2 stimulus for breathing, & more reliant on the low PaO2 stimulus
(“CO2 retention”)
Give O2 carefully to these patients (high PaO2 may suppress patients’ hypoxic respiratory drive, ↓ their breathing, & ↑↑↑ PaCO2)
↑ retrosternal air space
(on lateral CXR)
Hyper-lucent
(darker) lung fields, ↓ lung markings (on frontal CXR)
• Arterial Blood Gasses (ABGs)
• Chest X-Ray (CXR): frontal and
lateral
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published January 7, 2013 on www.thecalgaryguide.com
COPD: !"#$ 气流阻塞
肺泡通气↓ 呼气时,胸膜腔正压挤压气 道à 阻塞↑
作者: Yan Yu 审稿人: Jason Baserman, Jennifer Au, Naushad Hirani*, Juri Janovcik* 译者:Zihong Xie (谢梓泓) 翻译审稿人: Yonglin Mai (麦泳琳), Zesheng Ye (叶泽生) * 发表时担任临床医生
慢性阻塞性肺疾病 (COPD)
肺组织损伤
没有弹性回缩力将
气体排出肺
肺实质与血管分布减少导 致气体交换面积↓
弥散功能↓ (肺功能检查)
更多的CO2残留 并扩散到血液中
高碳酸血症: PaCO2 > 45
(动脉血气)
血流灌注通气不良的肺泡
时无法获得足够的氧气
总呼气时长较正常长
FEV1/FEV < 0.7
(肺功能检查)
肺无法完全排空
更多空气潴留在肺部
(肺过度充气)
低氧血症: PaO2 < 70mmHg
(动脉血气)
通气-灌注不匹配
肺泡-动脉氧分压差↑ (可通过动脉血气分析计算得出)
横膈低平, 下移至第10肋后端 及以下部位 (胸部正位片)
TLC与VC增大 (肺功能检查)
缩写: • • FEV1: 1秒用 •
VC:肺活量
PaO2: 动脉血 力呼气量 氧分压
空气不会阻挡X射线, 在X光片上呈现为黑色
慢性高碳酸血症使呼吸中枢对PaCO2 刺激呼吸的敏感性下降 & 更依赖于低PaO2的刺激 (“二氧化碳潴留”)
给患者吸氧时需注意(高PaO2
可能会抑制患者低氧时对呼吸的 刺激,使呼吸驱动↓ & PaCO2↑↑↑ )
• FVC: 用力肺 • 活量
• TLC:肺总量 慢阻肺相关检查 :
PaCO2: 动脉 血二氧化碳 分压
胸骨后间隙↑
(胸部侧位片) 肺纹理↓
• 肺功能检查
• 动脉血气分析(Arterial Blood Gasses, ABGs)
• 胸部正侧位片
• 当患者发热和湿咳,特别是胸片上见肺野不清晰时:
肺透亮度↑, (胸部正位片)
考虑进行痰革兰氏染色及痰培养以排除肺炎可能
图注:
病理生理
机制
体征/临床表现/实验室检查
并发症
2013年1月7日发布于 www.thecalgaryguide.com
COPD: Complications Lung inflammation
Chronic Obstructive Pulmonary Disease (COPD)
Airway obstruction ↓ inhaled air in alveoli and terminal bronchioles
Rupture of emphasematous bullae on surface of lung
Inhaled air leaks into pleural cavity and is trapped there
Pneumothorax
Feeling a loss of control over one’s life, and hopelessness for the future
Goblet cell proliferation, ↑ mucus production
Death of airway
epithelium ciliated cells
↓ oxygenation of the blood passing through the lungs
Chronic hypoxemia
Kidneys compensate by ↑ erythropoietin (EPO) production
↑ Hemoglobin and red blood cell synthesis
Polycythemia (secondary)
Hypoxic alveoli cause the pulmonary arterioles perfusing them to reflexively vasoconstrict
Since most alveoli in the lungs are hypoxic, hypoxic vasoconstriction occurs across entire lung
Vasoconstriction ↑ blood pressure within lung vasculature
Pulmonary hypertension
↑ workload of the right ventricle (to pump against higher pressures)
To compensate, the right ventricle progressively hypertrophies and dilates, but over time its output ↓
Cor pulmonale
(Right heart failure in isolation, not due to Left heart failure)
Mucus trapped in airways, serve as nidus for infection
Acute exacerbation of COPD (AECOPD)
Pneumonia
The chronic, systemic inflammation in COPD is a hyper-metabolic state that consumes calories
Macro-nutrient deficiency
Trouble with respiration lead to inactivity and deconditioning
Wasting, muscle atrophy
More inactivity and deconditioning perpetuates the cycle
Depression
Author: Yan Yu Reviewers: Jason Baserman Naushad Hirani* Juri Janovcik* * MD at time of publication
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published January 7, 2013 on www.thecalgaryguide.com
COPD: !"# 肺部炎症
杯状细胞增殖, 气道上皮纤毛 粘液产生↑ 细胞死亡
黏液潴留呼吸道,成为感
染的病灶
慢性阻塞性肺疾病 (COPD) 气道阻塞à 吸入肺泡和终末细
肺大疱破裂
吸入的空气渗入
并潴留于胸腔
气胸
感觉生活失控,对未
来感到绝望
抑郁
作者: Yan Yu 审稿人: Jason Baserman, Naushad Hirani*, Juri Janovcik* 译者: Zihong Xie (谢梓泓) 翻译审稿人: Yonglin Mai (麦泳琳), Zesheng Ye (叶泽生) * 发表时担任临床医生
支气管的空气 ↓
流经肺的血液进行气 缺氧的肺泡à灌注肺泡的肺小动
慢性阻塞性肺疾 病急性加重期 (AECOPD)
肺炎
体交换↓ 慢性低氧血症
肾脏合成促红细胞 生成素进行代偿↑
血红蛋白与红 细胞合成↑
红细胞增多症 (继发性)
脉发生反射性血管收缩
肺大部分肺泡缺氧à整个肺 都出现缺氧性血管收缩
肺血管收缩 à 肺血管压力↑ 肺动脉高压
↑ 右心室负荷(泵血时对抗高压) 为了代偿,右心室逐渐肥大和扩张,
但随着病程进展,右心室输出量 ↓
肺心病 (单独出现右心衰竭,非左心衰)
COPD所致的慢性全身 呼吸困难导致活 性炎症会使机体处于高 动量减少和活动
代谢状态,消耗能量 耐量降低
宏量营养 素缺乏症
消瘦,肌肉萎缩
运动量下降和活动耐量
的降低造成恶性循环
图注:
病理生理
机制
体征/临床表现/实验室检查
并发症
2013年1月7日发布于 www.thecalgaryguide.com
" />
45
(on ABGs)
Ventilation- perfusion mismatch
High A-a gradient
(calculated from ABGs)
Low, flat diaphragm, >10 posterior ribs
(on frontal CXR)
High TLC and VC
(on spirometry)
• •
PaO2: partial pressure of O2 in arterial blood PaCO2: partial pressure of CO2 in arterial blood
• In the setting of fever and productive cough, especially if lung field opacifications are seen on CXR: consider sputum gram stain and culture to rule out pneumonia.
Air does not block X-ray beams, will appear black on X-ray film
Chronic hypercapnia makes breathing centers less sensitive to the high PaCO2 stimulus for breathing, & more reliant on the low PaO2 stimulus
(“CO2 retention”)
Give O2 carefully to these patients (high PaO2 may suppress patients’ hypoxic respiratory drive, ↓ their breathing, & ↑↑↑ PaCO2)
↑ retrosternal air space
(on lateral CXR)
Hyper-lucent
(darker) lung fields, ↓ lung markings (on frontal CXR)
• Arterial Blood Gasses (ABGs)
• Chest X-Ray (CXR): frontal and
lateral
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published January 7, 2013 on www.thecalgaryguide.com
COPD: !"#$ 气流阻塞
肺泡通气↓ 呼气时,胸膜腔正压挤压气 道à 阻塞↑
作者: Yan Yu 审稿人: Jason Baserman, Jennifer Au, Naushad Hirani*, Juri Janovcik* 译者:Zihong Xie (谢梓泓) 翻译审稿人: Yonglin Mai (麦泳琳), Zesheng Ye (叶泽生) * 发表时担任临床医生
慢性阻塞性肺疾病 (COPD)
肺组织损伤
没有弹性回缩力将
气体排出肺
肺实质与血管分布减少导 致气体交换面积↓
弥散功能↓ (肺功能检查)
更多的CO2残留 并扩散到血液中
高碳酸血症: PaCO2 > 45
(动脉血气)
血流灌注通气不良的肺泡
时无法获得足够的氧气
总呼气时长较正常长
FEV1/FEV < 0.7
(肺功能检查)
肺无法完全排空
更多空气潴留在肺部
(肺过度充气)
低氧血症: PaO2 < 70mmHg
(动脉血气)
通气-灌注不匹配
肺泡-动脉氧分压差↑ (可通过动脉血气分析计算得出)
横膈低平, 下移至第10肋后端 及以下部位 (胸部正位片)
TLC与VC增大 (肺功能检查)
缩写: • • FEV1: 1秒用 •
VC:肺活量
PaO2: 动脉血 力呼气量 氧分压
空气不会阻挡X射线, 在X光片上呈现为黑色
慢性高碳酸血症使呼吸中枢对PaCO2 刺激呼吸的敏感性下降 & 更依赖于低PaO2的刺激 (“二氧化碳潴留”)
给患者吸氧时需注意(高PaO2
可能会抑制患者低氧时对呼吸的 刺激,使呼吸驱动↓ & PaCO2↑↑↑ )
• FVC: 用力肺 • 活量
• TLC:肺总量 慢阻肺相关检查 :
PaCO2: 动脉 血二氧化碳 分压
胸骨后间隙↑
(胸部侧位片) 肺纹理↓
• 肺功能检查
• 动脉血气分析(Arterial Blood Gasses, ABGs)
• 胸部正侧位片
• 当患者发热和湿咳,特别是胸片上见肺野不清晰时:
肺透亮度↑, (胸部正位片)
考虑进行痰革兰氏染色及痰培养以排除肺炎可能
图注:
病理生理
机制
体征/临床表现/实验室检查
并发症
2013年1月7日发布于 www.thecalgaryguide.com
COPD: Complications Lung inflammation
Chronic Obstructive Pulmonary Disease (COPD)
Airway obstruction ↓ inhaled air in alveoli and terminal bronchioles
Rupture of emphasematous bullae on surface of lung
Inhaled air leaks into pleural cavity and is trapped there
Pneumothorax
Feeling a loss of control over one’s life, and hopelessness for the future
Goblet cell proliferation, ↑ mucus production
Death of airway
epithelium ciliated cells
↓ oxygenation of the blood passing through the lungs
Chronic hypoxemia
Kidneys compensate by ↑ erythropoietin (EPO) production
↑ Hemoglobin and red blood cell synthesis
Polycythemia (secondary)
Hypoxic alveoli cause the pulmonary arterioles perfusing them to reflexively vasoconstrict
Since most alveoli in the lungs are hypoxic, hypoxic vasoconstriction occurs across entire lung
Vasoconstriction ↑ blood pressure within lung vasculature
Pulmonary hypertension
↑ workload of the right ventricle (to pump against higher pressures)
To compensate, the right ventricle progressively hypertrophies and dilates, but over time its output ↓
Cor pulmonale
(Right heart failure in isolation, not due to Left heart failure)
Mucus trapped in airways, serve as nidus for infection
Acute exacerbation of COPD (AECOPD)
Pneumonia
The chronic, systemic inflammation in COPD is a hyper-metabolic state that consumes calories
Macro-nutrient deficiency
Trouble with respiration lead to inactivity and deconditioning
Wasting, muscle atrophy
More inactivity and deconditioning perpetuates the cycle
Depression
Author: Yan Yu Reviewers: Jason Baserman Naushad Hirani* Juri Janovcik* * MD at time of publication
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published January 7, 2013 on www.thecalgaryguide.com
COPD: !"# 肺部炎症
杯状细胞增殖, 气道上皮纤毛 粘液产生↑ 细胞死亡
黏液潴留呼吸道,成为感
染的病灶
慢性阻塞性肺疾病 (COPD) 气道阻塞à 吸入肺泡和终末细
肺大疱破裂
吸入的空气渗入
并潴留于胸腔
气胸
感觉生活失控,对未
来感到绝望
抑郁
作者: Yan Yu 审稿人: Jason Baserman, Naushad Hirani*, Juri Janovcik* 译者: Zihong Xie (谢梓泓) 翻译审稿人: Yonglin Mai (麦泳琳), Zesheng Ye (叶泽生) * 发表时担任临床医生
支气管的空气 ↓
流经肺的血液进行气 缺氧的肺泡à灌注肺泡的肺小动
慢性阻塞性肺疾 病急性加重期 (AECOPD)
肺炎
体交换↓ 慢性低氧血症
肾脏合成促红细胞 生成素进行代偿↑
血红蛋白与红 细胞合成↑
红细胞增多症 (继发性)
脉发生反射性血管收缩
肺大部分肺泡缺氧à整个肺 都出现缺氧性血管收缩
肺血管收缩 à 肺血管压力↑ 肺动脉高压
↑ 右心室负荷(泵血时对抗高压) 为了代偿,右心室逐渐肥大和扩张,
但随着病程进展,右心室输出量 ↓
肺心病 (单独出现右心衰竭,非左心衰)
COPD所致的慢性全身 呼吸困难导致活 性炎症会使机体处于高 动量减少和活动
代谢状态,消耗能量 耐量降低
宏量营养 素缺乏症
消瘦,肌肉萎缩
运动量下降和活动耐量
的降低造成恶性循环
图注:
病理生理
机制
体征/临床表现/实验室检查
并发症
2013年1月7日发布于 www.thecalgaryguide.com
" title="COPD: 临床表现
作者: Yan Yu 审稿人:Jason Baserman, Jennifer Au, Naushad Hirani*, Juri Janovcik* 译者: Zihong Xie (谢梓泓) 翻译审稿人:Yonglin Mai (麦泳琳), Zesheng Ye (叶泽生) * 发表时担任临床医生
/012
(如a1-抗胰蛋白酶缺乏) 阻止肺组织损伤的能力↓
+,-.
(如长期吸烟、环境污染、感染)
肺内产生自由基
34*5
肺抗蛋白酶的失活
↑氧化应激,炎性细胞因子,蛋白酶功能
支气管的持续、反复损伤
炎性细胞浸润, 杯状细胞增殖, 气道上皮纤毛 尤其中性粒细 黏液产生↑ 细胞死亡
气道弹性↓ (弹性回缩
肺实质的蛋白水解破坏↑ 维持气道开放 肺泡永久性异常
的结构支持↓ 扩张
胞 力)
肺气体潴留 气道狭窄与 肺过度 肺大泡
气道黏液潴留,成为感染 狭窄 病灶
塌陷 充气
肺气肿
(容易肺泡 破裂)
气道纤维化和
%&'()*
慢性阻塞性肺疾病(COPD)
临床表现 并发症 (参阅相关幻灯片) (参阅相关幻灯片)
图注:
病理生理
机制
体征/临床表现/实验室检查
并发症
2013年1月7日发布于 www.thecalgaryguide.com
COPD: Clinical Findings Lung tissue
Chronic Obstructive Pulmonary Disease (COPD)
damage
↓ elastic recoil to push air out of lungs on expiration
Lungs don’t fully empty, air is trapped in alveoli (lung hyperinflation)
↑ lung volume means diaphragm is tonically contracted (flatter)
If occurring around airways
Airflow obstruction
↑ mucus production
↓ number of epithelial ciliated cells to clear away the mucus (the cells have been killed by airway inflammation)
Chronic cough with sputum
Author: Yan Yu Reviewers: Jason Baserman Jennifer Au Naushad Hirani* Juri Janovcik* * MD at time of publication
During expiration, positive pleural pressure squeezes on airwaysà↑ obstruction
↓ ventilation of alveoli
↓ oxygenation of blood (hypoxemia)
↓ perfusion of body tissues (i.e. brain, muscle)
Fatigue; ↓ exercise tolerance
Total expiration time takes longer than normal
Prolonged expiration
More effort needed to ventilate larger lungs
Respiratory muscles must work harder to breathe
Turbulent airflow in narrower airways is heard on auscultation
Expiratory Wheeze
Diaphragm can’t flatten much further to generate deep breaths
To breathe, chest wall must expand out more
Dyspnea
Shortness of breath, especially on exertion
Breathes are rapid & shallow
If end-stage:
Chronic fatigue causes deconditioning
Muscle weakness & wasting
Barrel chest
If end-stage: diaphragm will be “flat”. Continued
Patient tries to expire against higher mouth air pressure, forcing airways to open wider
Pursed-lip breathing
Patient breathes with accessory muscles as well as diaphragm to try to improve airflow
inspiratory effort further contracts diaphragmà pull the lower chest wall inwards
Hoover’s sign
(paradoxical shrinking of lower chest during inspiration)
Tripod sitting position (activates pectoral muscles)
Neck (SCM, scalene) muscles contracted
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published January 7, 2013 on www.thecalgaryguide.com
COPD: !"#$
慢性阻塞性肺疾病 (COPD) 如果出现在气道周围 气流阻塞
肺不能完全排空
气体,气体潴留
于肺泡(肺过度
充气)
总呼气时长大于 正常时长
呼气相延长
肺组织损伤
呼气时,将空气排出肺外 的弹性回缩力↓
肺不能完全排空气体,
气体潴留于肺泡内
(肺过度充气)
肺容积↑,膈肌紧张 性收缩(膈肌平坦)
呼气时,胸膜腔正压挤压气道 à 气道阻塞↑
肺泡通气↓ 血液氧合↓ (低
氧血症)
身体组织灌注 量↓ (比如脑、 肌肉)
疲劳; 运动耐量↓
黏液生成↑ 清除黏液的上皮纤
毛细胞数量↓ (受 气道炎症损伤)
慢性咳嗽伴咳 痰
作者: Yan Yu
审稿人: Jason Baserman, Jennifer Au, Naushad Hirani*, Juri Janovcik* 译者: Zihong Xie (谢梓泓) 翻译审稿人: Yonglin Mai (麦泳琳),
Zesheng Ye (叶泽生)
* 发表时担任临床医生
容积较大 的肺需要
更加努力 才能通气
呼吸肌必须
更用力才能 呼吸
听诊闻及狭窄气
道中的湍流气流
呼气喘鸣音
呼吸困难 气促,尤其是劳累
膈肌无法进一步收缩以
产生深呼吸
呼吸浅快
为了呼吸,
胸壁必须延
展得更大
桶状胸
晚期病人:
患者试图在较高的口 慢性疲劳导致 患者动用辅助呼吸肌和膈肌呼吸,
腔内气压下进行呼气, 活动耐量下降 从而使气道更开放
以改善气流
晚期病人:膈肌 “平坦” ,持续吸气进一步压 缩膈肌à 向内拉季肋部胸壁
胡佛征 (吸气时,胸廓下侧季肋部内收)
缩唇呼吸
肌肉无力 & 消瘦
端坐呼吸 (调动胸肌)
颈部肌肉收
缩(胸锁乳
突肌、斜角
肌)
图注:
病理生理
机制
体征/临床表现/实验室检查
并发症
2013年1月7日发布于 www.thecalgaryguide.com
COPD: Findings on Investigations
Chronic Obstructive Pulmonary Disease (COPD)
Author: Yan Yu Reviewers: Jason Baserman Jennifer Au Naushad Hirani* Juri Janovcik* * MD at time of publication
Airflow obstruction
Lung tissue damage
↓ ventilation of alveoli
Blood perfusing ill- ventilated alveoli does not receive normal amounts of oxygen
During expiration, positive pleural pressure squeezes on airwaysà↑ obstruction)
No elastic recoil to push air out of lungs
Loss of lung parenchyma and vasculature ↓ surface area for gas exchange
↓ diffusion capacity
(on spirometry)
Hypoxemia: PaO2 < 70mmHg (on ABGs)
Abbreviations:
• FEV1: Forced expiratory volume in 1 second
• FVC: Forced vital capacity
• TLC: Total lung capacity
• VC: Vital Capacity
Investigations for COPD :
• Spirometry (Pulmonary function test)
Total expiration time takes longer than normal
FEV1/FEV < 0.7
(on spirometry)
Lungs don’t fully empty
More air trapped within lungs (hyperinflation)
More CO2 remains and diffuses into the blood
Hypercapnia: PaCO2 > 45
(on ABGs)
Ventilation- perfusion mismatch
High A-a gradient
(calculated from ABGs)
Low, flat diaphragm, >10 posterior ribs
(on frontal CXR)
High TLC and VC
(on spirometry)
• •
PaO2: partial pressure of O2 in arterial blood PaCO2: partial pressure of CO2 in arterial blood
• In the setting of fever and productive cough, especially if lung field opacifications are seen on CXR: consider sputum gram stain and culture to rule out pneumonia.
Air does not block X-ray beams, will appear black on X-ray film
Chronic hypercapnia makes breathing centers less sensitive to the high PaCO2 stimulus for breathing, & more reliant on the low PaO2 stimulus
(“CO2 retention”)
Give O2 carefully to these patients (high PaO2 may suppress patients’ hypoxic respiratory drive, ↓ their breathing, & ↑↑↑ PaCO2)
↑ retrosternal air space
(on lateral CXR)
Hyper-lucent
(darker) lung fields, ↓ lung markings (on frontal CXR)
• Arterial Blood Gasses (ABGs)
• Chest X-Ray (CXR): frontal and
lateral
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published January 7, 2013 on www.thecalgaryguide.com
COPD: !"#$ 气流阻塞
肺泡通气↓ 呼气时,胸膜腔正压挤压气 道à 阻塞↑
作者: Yan Yu 审稿人: Jason Baserman, Jennifer Au, Naushad Hirani*, Juri Janovcik* 译者:Zihong Xie (谢梓泓) 翻译审稿人: Yonglin Mai (麦泳琳), Zesheng Ye (叶泽生) * 发表时担任临床医生
慢性阻塞性肺疾病 (COPD)
肺组织损伤
没有弹性回缩力将
气体排出肺
肺实质与血管分布减少导 致气体交换面积↓
弥散功能↓ (肺功能检查)
更多的CO2残留 并扩散到血液中
高碳酸血症: PaCO2 > 45
(动脉血气)
血流灌注通气不良的肺泡
时无法获得足够的氧气
总呼气时长较正常长
FEV1/FEV < 0.7
(肺功能检查)
肺无法完全排空
更多空气潴留在肺部
(肺过度充气)
低氧血症: PaO2 < 70mmHg
(动脉血气)
通气-灌注不匹配
肺泡-动脉氧分压差↑ (可通过动脉血气分析计算得出)
横膈低平, 下移至第10肋后端 及以下部位 (胸部正位片)
TLC与VC增大 (肺功能检查)
缩写: • • FEV1: 1秒用 •
VC:肺活量
PaO2: 动脉血 力呼气量 氧分压
空气不会阻挡X射线, 在X光片上呈现为黑色
慢性高碳酸血症使呼吸中枢对PaCO2 刺激呼吸的敏感性下降 & 更依赖于低PaO2的刺激 (“二氧化碳潴留”)
给患者吸氧时需注意(高PaO2
可能会抑制患者低氧时对呼吸的 刺激,使呼吸驱动↓ & PaCO2↑↑↑ )
• FVC: 用力肺 • 活量
• TLC:肺总量 慢阻肺相关检查 :
PaCO2: 动脉 血二氧化碳 分压
胸骨后间隙↑
(胸部侧位片) 肺纹理↓
• 肺功能检查
• 动脉血气分析(Arterial Blood Gasses, ABGs)
• 胸部正侧位片
• 当患者发热和湿咳,特别是胸片上见肺野不清晰时:
肺透亮度↑, (胸部正位片)
考虑进行痰革兰氏染色及痰培养以排除肺炎可能
图注:
病理生理
机制
体征/临床表现/实验室检查
并发症
2013年1月7日发布于 www.thecalgaryguide.com
COPD: Complications Lung inflammation
Chronic Obstructive Pulmonary Disease (COPD)
Airway obstruction ↓ inhaled air in alveoli and terminal bronchioles
Rupture of emphasematous bullae on surface of lung
Inhaled air leaks into pleural cavity and is trapped there
Pneumothorax
Feeling a loss of control over one’s life, and hopelessness for the future
Goblet cell proliferation, ↑ mucus production
Death of airway
epithelium ciliated cells
↓ oxygenation of the blood passing through the lungs
Chronic hypoxemia
Kidneys compensate by ↑ erythropoietin (EPO) production
↑ Hemoglobin and red blood cell synthesis
Polycythemia (secondary)
Hypoxic alveoli cause the pulmonary arterioles perfusing them to reflexively vasoconstrict
Since most alveoli in the lungs are hypoxic, hypoxic vasoconstriction occurs across entire lung
Vasoconstriction ↑ blood pressure within lung vasculature
Pulmonary hypertension
↑ workload of the right ventricle (to pump against higher pressures)
To compensate, the right ventricle progressively hypertrophies and dilates, but over time its output ↓
Cor pulmonale
(Right heart failure in isolation, not due to Left heart failure)
Mucus trapped in airways, serve as nidus for infection
Acute exacerbation of COPD (AECOPD)
Pneumonia
The chronic, systemic inflammation in COPD is a hyper-metabolic state that consumes calories
Macro-nutrient deficiency
Trouble with respiration lead to inactivity and deconditioning
Wasting, muscle atrophy
More inactivity and deconditioning perpetuates the cycle
Depression
Author: Yan Yu Reviewers: Jason Baserman Naushad Hirani* Juri Janovcik* * MD at time of publication
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published January 7, 2013 on www.thecalgaryguide.com
COPD: !"# 肺部炎症
杯状细胞增殖, 气道上皮纤毛 粘液产生↑ 细胞死亡
黏液潴留呼吸道,成为感
染的病灶
慢性阻塞性肺疾病 (COPD) 气道阻塞à 吸入肺泡和终末细
肺大疱破裂
吸入的空气渗入
并潴留于胸腔
气胸
感觉生活失控,对未
来感到绝望
抑郁
作者: Yan Yu 审稿人: Jason Baserman, Naushad Hirani*, Juri Janovcik* 译者: Zihong Xie (谢梓泓) 翻译审稿人: Yonglin Mai (麦泳琳), Zesheng Ye (叶泽生) * 发表时担任临床医生
支气管的空气 ↓
流经肺的血液进行气 缺氧的肺泡à灌注肺泡的肺小动
慢性阻塞性肺疾 病急性加重期 (AECOPD)
肺炎
体交换↓ 慢性低氧血症
肾脏合成促红细胞 生成素进行代偿↑
血红蛋白与红 细胞合成↑
红细胞增多症 (继发性)
脉发生反射性血管收缩
肺大部分肺泡缺氧à整个肺 都出现缺氧性血管收缩
肺血管收缩 à 肺血管压力↑ 肺动脉高压
↑ 右心室负荷(泵血时对抗高压) 为了代偿,右心室逐渐肥大和扩张,
但随着病程进展,右心室输出量 ↓
肺心病 (单独出现右心衰竭,非左心衰)
COPD所致的慢性全身 呼吸困难导致活 性炎症会使机体处于高 动量减少和活动
代谢状态,消耗能量 耐量降低
宏量营养 素缺乏症
消瘦,肌肉萎缩
运动量下降和活动耐量
的降低造成恶性循环
图注:
病理生理
机制
体征/临床表现/实验室检查
并发症
2013年1月7日发布于 www.thecalgaryguide.com
" />
COPD-检查结果
 翻译审稿人: Yonglin Mai (麦泳琳), Zesheng Ye (叶泽生) * 发表时担任临床医生
慢性阻塞性肺疾病 (COPD)
肺组织损伤
没有弹性回缩力将
气体排出肺
肺实质与血管分布减少导 致气体交换面积↓
弥散功能↓ (肺功能检查)
更多的CO2残留 并扩散到血液中
高碳酸血症: PaCO2 > 45
(动脉血气)
血流灌注通气不良的肺泡
时无法获得足够的氧气
总呼气时长较正常长
FEV1/FEV < 0.7
(肺功能检查)
肺无法完全排空
更多空气潴留在肺部
(肺过度充气)
低氧血症: PaO2 < 70mmHg
(动脉血气)
通气-灌注不匹配
肺泡-动脉氧分压差↑ (可通过动脉血气分析计算得出)
横膈低平, 下移至第10肋后端 及以下部位 (胸部正位片)
TLC与VC增大 (肺功能检查)
缩写: • • FEV1: 1秒用 •
VC:肺活量
PaO2: 动脉血 力呼气量 氧分压
空气不会阻挡X射线, 在X光片上呈现为黑色
慢性高碳酸血症使呼吸中枢对PaCO2 刺激呼吸的敏感性下降 & 更依赖于低PaO2的刺激 (“二氧化碳潴留”)
给患者吸氧时需注意(高PaO2
可能会抑制患者低氧时对呼吸的 刺激,使呼吸驱动↓ & PaCO2↑↑↑ )
• FVC: 用力肺 • 活量
• TLC:肺总量 慢阻肺相关检查 :
PaCO2: 动脉 血二氧化碳 分压
胸骨后间隙↑
(胸部侧位片) 肺纹理↓
• 肺功能检查
• 动脉血气分析(Arterial Blood Gasses, ABGs)
• 胸部正侧位片
• 当患者发热和湿咳,特别是胸片上见肺野不清晰时:
肺透亮度↑, (胸部正位片)
考虑进行痰革兰氏染色及痰培养以排除肺炎可能
图注:
病理生理
机制
体征/临床表现/实验室检查
并发症
2013年1月7日发布于 www.thecalgaryguide.com")
COPD-并发症
 气道阻塞à 吸入肺泡和终末细
肺大疱破裂
吸入的空气渗入
并潴留于胸腔
气胸
感觉生活失控,对未
来感到绝望
抑郁
作者: Yan Yu 审稿人: Jason Baserman, Naushad Hirani*, Juri Janovcik* 译者: Zihong Xie (谢梓泓) 翻译审稿人: Yonglin Mai (麦泳琳), Zesheng Ye (叶泽生) * 发表时担任临床医生
支气管的空气 ↓
流经肺的血液进行气 缺氧的肺泡à灌注肺泡的肺小动
慢性阻塞性肺疾 病急性加重期 (AECOPD)
肺炎
体交换↓ 慢性低氧血症
肾脏合成促红细胞 生成素进行代偿↑
血红蛋白与红 细胞合成↑
红细胞增多症 (继发性)
脉发生反射性血管收缩
肺大部分肺泡缺氧à整个肺 都出现缺氧性血管收缩
肺血管收缩 à 肺血管压力↑ 肺动脉高压
↑ 右心室负荷(泵血时对抗高压) 为了代偿,右心室逐渐肥大和扩张,
但随着病程进展,右心室输出量 ↓
肺心病 (单独出现右心衰竭,非左心衰)
COPD所致的慢性全身 呼吸困难导致活 性炎症会使机体处于高 动量减少和活动
代谢状态,消耗能量 耐量降低
宏量营养 素缺乏症
消瘦,肌肉萎缩
运动量下降和活动耐量
的降低造成恶性循环
图注:
病理生理
机制
体征/临床表现/实验室检查
并发症")
毛细支气管炎-发病机制及临床表现
, 但也可以是其他病毒,如鼻病毒、腺病毒、
副流感病毒、流感病毒和冠状病毒,最初在鼻黏膜定植
病毒通过呼吸道上皮迁移至下呼吸道及终末细支气管(小气道)
毛细支气管炎 (细支气管炎症 )
作者: Nick Baldwin,Rebecca Lindsay 审稿人: Kayla Nelson, Yan Yu,Timothy Fu, Danielle,Nelson* 译者: Yonglin Mai(麦泳琳) 翻译审稿人 : Zesheng Ye (叶泽生) *发表时担任临床医生
呼吸道合胞病毒融合蛋白促进病毒与宿
主细胞融合,直接介导病毒侵袭并感染邻 近正常组织细胞
合胞体形成 (多核巨细胞)
合胞体从支气管上 纤毛上皮细胞破坏
上呼吸道黏膜 炎症反应
细胞因子释 蛋白质和液 放到血液循 体渗漏至鼻 环系统 咽间质
下丘脑体温 间质水肿 调节点↑
低烧 流鼻涕 鼻塞
↑毛细血 管通透性
蛋白质和液
体渗漏至细
支气管及周
围间质,并
积聚在支气
管壁周围
气管管壁增厚,X 光征象更为明显
支气管周围袖口征
(X光征象正位可见支 气管壁增厚,如增厚 “袖口”)
炎症刺激使分泌黏液的
杯状细胞增多
↑黏液分泌 肺泡中的黏液使得肺泡内表
面张力↑à肺泡壁塌陷
在吸气过程中,
如气道尚未阻塞,
空气可以进入塌
陷的肺泡当中
肺泡内压力↑使得塌陷 的肺泡骤然打开
听诊时可闻及吸气 性爆裂音(湿啰音)
呼吸暂停 (呼吸停止;具体机
制未明,可能触发呼 吸暂停反射 )
皮脱落至气道
气道变窄及闭塞
(其功能是清除粘 液排出气道,将其 送入咽部被吞咽或 者被排出)
↓ 气道粘 液清除
过多气道粘液触
发咳嗽反射
咳嗽
堵塞支气管远端 的气体被吸收 ( 气体潴留)
气体被全部吸收后,肺 泡会塌陷 (吸收性肺不 张)
在剩余肺泡中,血气屏障 处气体交换↓
↓ 血氧(O2 )饱和度 & ↑ 血二氧化碳(CO2)浓度
尤其在呼
气时,更
狭窄的气
道会产生
湍流气流
听诊可闻及呼 气相哮鸣音
图注:
病理生理
机制
体征/症状/实验室检查
并发症
2020年11月29日重新发布于 www.thecalgaryguide.com")
支气管扩张-胸部 X线和 CT扫描的表现
, Yang Xiang(向阳)
翻译审稿人:Ran Zhong(钟然), Yonglin Mai(麦泳琳), Zesheng Ye(叶泽生) * 出版时担任临床医生
支气管扩张: 支气管的 异常损伤和扩张
在一些炎症过程下,中性粒细胞、细胞因 子、一氧化氮和氧自由基积聚在肺内
支气管和支气管周围组织炎性破坏 支气管的弹性成分被破坏
弹性组织的丢失使得支气管壁的完整性受 损,使其结构更松散
当支气管向远端延伸时,支气管仍 扩张(但通常情况支气管是变细)
异常扩张的支气管比其 伴行的动脉更宽 (支气管:动脉直径比1.5)
支气管内的空气呈黑色 ,对比之下周围动脉内 的血液呈白色,形成环 状外观
CT下的印戒征
成像时显示支气 管扩张增厚轮廓
胸片下的轨道征
支气管扩张使远
端支气管在影像
学上更明显
CT可见距胸膜 1cm内的支气管
囊状支气管扩张可
能出现聚集成簇
横断面CT扫描显 示葡萄串征
图注:
病理生理
机制
体征/症状/实验室检查
并发症
2013年7月30日发布于 www.thecalgaryguide.com")
哮喘急性发作-发病机制和治疗
 Kang,Usama Malik,Lian Szabo*
译者:Yonglin Mai (麦泳琳) 翻译审核人:Zesheng Ye (叶泽生) * 发表时担任临床医生
下呼吸道炎症
免疫系统激活:气道上皮趋化因子、淋巴细胞和巨噬细胞激活, 白三烯产生↑ 炎症介质释放
哮喘轻中度急性发作: PEF ≥ 预计值50%
疗效良好: 症状缓 解, PEF ≥ 80%
患者日常在家 服用糖皮质激 素及必要时使 用SABA
支气管狭窄
残气量↑ 及PaCO2↑
气体潴留↑,肺 泡内压力 ↑
奇脉
气体通过 发炎的呼 吸道产生 的刺激↑
咳嗽和喘息
Notes
呼吸 困难
黏膜水肿会导 致气流湍急
喘息
吸O2使SpO2 ≥ 92%, 给予SABA & 糖皮质激素 治疗
重度急性发作: PEF ≤预计值 50%
呼吸困难
呼吸急促 呼吸衰竭
意识丧失
↓ 向肺泡传送 空气氧含量
血氧饱和度↓
•
•
Asthma哮喘: Airway hyper- responsiveness causing airflow obstructions气道高反应性引起气流受 限
Acute Exacerbation (Asthma)哮喘急性 发作: An episode of increased symptoms due to decreases in airflow气流减少而 引起一系列症状加重
Abbreviations
• PaCO2动脉二氧化碳分压:Partial
pressure of CO2 in arterial blood
• PEF最大呼气流量/呼气流量峰值: Peak
expiratory flow
• SABA短效beta-2受体激动剂: Short-
acting beta-2 agonists
• SpO2血氧饱和度: Blood oxygen
saturation level
昏睡
气胸
中枢性紫绀 心动过速
患者宣教: 正确服用药物,使用 药物吸入装置 &全科医生密切医学随 访
吸 O 使 SpO ≥ 2 2
症状恶化和/或呼吸衰竭 :及时气 管插管, 送往ICU, 给予SABA, 糖皮 质激素 & 硫酸镁
取决于气胸严 重程度: 密切观 察或者置入胸 腔引流管
93%, 给予 SABA, 糖皮质激素 & 硫 酸镁
图注:
病理生理
机制
体征/症状/实验室检查
并发症
2018年 2月6日发布于 www.thecalgaryguide.com")
新冠肺炎腺病毒载体疫苗-制备及其作用机制
![新冠肺炎腺病毒载体疫苗:制备及其作用机制
牛津/阿斯利康 56%&'H^89:;H^< 56%&'789:;7< 56CDD%&':EFGHHH9IJ:;KLGB9MNO
美国强生 康希诺生物
=>?@4A%&'B =>?@4A%&'B PQRS4TUVWA)XYZ[*]B2
*+4-.
利用逆转录聚合酶链反应(RT-PCR)和全基因组测序技术,将 SARS-CoV-2(导致COVID-19的病毒)的RNA合成DNA文库
从SARS-CoV-2基因组中提取出刺突蛋白的DNA序列 将启动子添加到刺突蛋白的DNA序列中,使该序列在导入细胞
后能被RNA聚合酶识别并转录
应用基因重组技术将修饰后的刺突蛋白DNA序列插入到质粒中。 质粒:一种环状DNA,能使新基因(如刺突蛋白基因)插入到
先前感染Ad5或Ad26可能导致机 腺病毒DNA可通过多种细胞裂解剂以及 黑猩猩病毒可免去先前对该病
体已对腺病毒载体产生免疫1
人腺病毒疫苗可能因曾感染Ad5或 Ad26而失效 ,因为免疫力并不是针 对刺突(Spike)蛋白而建立的
洗涤剂提取(可打破细胞膜并去除非核
酸细胞物质的化学试剂)
毒载体产生免疫的可能性1 黑猩猩病毒载体使机体更好地
使用全基因组测序对腺病毒DNA 进行测序,并进行以下修饰:
对刺突蛋白产生免疫反应 去除腺病毒基因组的E1区, 去除腺病毒基因组的E3区,为SARS-CoV-2
宿主基因组中(如腺病毒载体DNA基因组)
以阻断病毒复制3 刺突蛋白DNA的插入腾出空间3 修饰后的腺病毒DNA基因组将重新插入到腺病毒 将病毒载体与刺突蛋白质粒混合在一起,DNA重组技
颗粒中,形成“病毒载体” 术使质粒中的刺突蛋白基因插入到腺病毒DNA中2
作者: Ryan Brenneis, Yan Yu* 审稿人: Davis MacLean, Hannah Yaphe Stephen Vaughan*
译者: Zihong Xie (谢梓泓) 翻译审稿人: Yonglin Mai (麦泳琳) * 出版时担任临床医生
最终制备出的疫苗中的腺
病毒不能在人体细胞内复
制,也不会引起人类疾病
参考文献
1. ACS Nano 2020, 14, 10, 12522–12537, Publication Date: October 9, 2020, https://doi.org/10.1021/acsnano.0c07197
含有可转录SARS-CoV-2刺突蛋白DNA的腺病毒放置于 一种特殊的细胞培养环境中,该环境允许病毒DNA在修 饰后仍进行复制2, 3
2. Nature 2020, 586, 578–582, Publication Date: October 20, 2020, https://doi.org/10.1038/s41586- 020-2608-y
3. Frontiers in Immunology 2018, 9, 1963, Publication Date: September 19, 2018, doi: 10.3389/fimmu.2018.01963
4. NPJ Vaccines 2020, 5, 69, Publication Date: July 27, 2020, doi: 10.1038/s41541-020-00221-3
5. The Lancet 2020, Publication Date: Dec. 8, 2020, https://doi.org/10.1016/S0140-6736(20)32623-4
6. BMJ 2000, 321, 7271, 1237-1238, Publication Date: November 18, 2000,
DOI: 10.1136.bmj.321.7271.1237
7. NEJM 2021, Publication Date: Jan. 13, 2021, DOI: 10.1056/NEJMoa2034201
提取含有可转录SARS-CoV-2刺突蛋 白DNA的腺病毒,并将其配置到疫 苗注射的浓度
腺病毒有一个外层蛋白质层(称为衣 壳)来保护它的DNA
DNA比mRNA更稳定,因为DNA具有脱 氧核糖骨架以及双链间的分子键
与mRNA脂质纳米粒 疫苗相比,腺病毒疫 苗的稳定性更强
可在2-8°C 中保存长 达3-6个月
注射部位红、肿、痛
(一过性)
注意: 强生疫苗在单剂接种 后有效率可能为90%7
肌肉是首选的注射 部位,因为肌肉血 供比其他组织多
疫苗作用
细胞免疫
能使免疫细胞更快处 理外来抗原6
能让外来的疫苗物质更快 扩散,使局部反应最小化6
病毒载体疫苗通过肌肉注射到健康人体内 建议在28天后接种第二剂疫苗以增强免疫反应(超过COVID-19康复者
的免疫反应水平),从而提高疫苗效力,特别是对老年人5
外来异物可引起局 部炎症反应
腺病毒表面抗原与人体细胞受体相互作用,使病毒通过内吞作用进入人体细胞3 腺病毒载体进入细胞核,与核被膜融合,并将其DNA(包括刺突蛋白DNA)注入细胞核 细胞核内的RNA聚合酶转录病毒的DNA,合成SARS-CoV-2刺突蛋白的信使RNA(mRNA) mRNA被运回胞浆,由细胞自身的核糖体进行翻
体液免疫
刺突蛋白被胞内酶降解为片段 刺突蛋白片段与MHC-I类分子结合
MHC-I类分子将刺突蛋白片段转移到人体细胞表面
MHC-I类分子将刺突蛋白片段呈递给幼稚CD8+ T细胞
与刺突蛋白-MHC-I类分子复合物结合后,幼稚CD8+ T细胞活化, 并转移到淋巴系统进一步成熟3
译合成全长SARS-CoV-2刺突蛋白
MHC = 主要组织相容性复合体(Major Histocompatability Complex);细胞表面蛋 白,对免疫功能至关重要
CD = 分化簇(Cluster of Differentiation); T细胞表面的糖蛋白,可作为辅助受体,促 进T细胞与抗原/MHC复合物的结合,同时 可用来区分T细胞类型
刺突蛋白的成分从细胞内释放到血液中 刺突蛋白被抗原提呈细胞 (树突状细胞、B细胞、巨噬
细胞)吞噬、碎裂,与特异的MHC-II类分子结合 MHC-II类分子将刺突蛋白片段转移到抗原提呈细胞表面,
将它们提呈给血循环里的幼稚CD4+ (辅助)T细胞 与复合物结合后,活化刺突蛋白特异性辅助T细胞
结合成刺突蛋白特异性辅助T细胞后被激活
部分T细胞成熟后成为细胞毒性T 细胞,可识别SARS-CoV-2刺突蛋白
细胞毒性T细胞与感染SARS-CoV-2并表达刺 突蛋白的人体细胞或刺突蛋白片段结合
细胞毒性T细胞释放酶,使感染 的细胞穿孔,导致细胞死亡
免疫系统可以更快地识
别和消灭受感染的细胞
部分T细胞成熟后成为记忆 性T细胞 (在辅助性T细胞 释放的细胞因子的刺激下)
记忆T细胞转移到淋巴组 织后进入待定状态,在下 一次接触刺突蛋白时活化
以后SARS-CoV-2 感染时 可介导更快的细胞免疫 (获得免疫力)
活化的刺突蛋白 特异性辅助T细胞 分泌细胞因子, 促进免疫活性
全身性细胞因子释放 可导致全身反应,如 发烧、寒战、疲劳以 及肌痛 (一过性)
注意: 细胞/体液免疫 持续的时间尚不清楚
部分B细胞成熟为浆细胞,产 生病毒刺突蛋白的IgG抗体
刺突蛋白抗体识别标记SARS- CoV-2,使免疫系统能消灭病毒
根除细胞外SARS-CoV-2
淋巴组织中,活化 的辅助性T细胞与幼 稚B细胞相互作用
部分B细胞成熟为 SARS-CoV-2刺突蛋 白特异性记忆B细胞
以后一旦接触刺突蛋白,就能再次活 化淋巴组织里的记忆B细胞,使其变成 浆细胞,更快地合成抗体
以后SARS-CoV-2 感染时可介导更快的体液免疫 (获得免疫力)
图注:
病理生理
机制
体征/临床表现/实验室检查
最终结果
2021年2月11日发布于www.thecalgaryguide.com](https://calgaryguide.ucalgary.ca/wp-content/uploads/2021/11/Adenovirus-Vector-Vaccines-Against-COVID-19-Production-and-Mechanism-of-Action.jpg "新冠肺炎腺病毒载体疫苗:制备及其作用机制
牛津/阿斯利康 56%&'H^89:;H^< 56%&'789:;7< 56CDD%&':EFGHHH9IJ:;KLGB9MNO
美国强生 康希诺生物
=>?@4A%&'B =>?@4A%&'B PQRS4TUVWA)XYZ[*]B2
*+4-.
利用逆转录聚合酶链反应(RT-PCR)和全基因组测序技术,将 SARS-CoV-2(导致COVID-19的病毒)的RNA合成DNA文库
从SARS-CoV-2基因组中提取出刺突蛋白的DNA序列 将启动子添加到刺突蛋白的DNA序列中,使该序列在导入细胞
后能被RNA聚合酶识别并转录
应用基因重组技术将修饰后的刺突蛋白DNA序列插入到质粒中。 质粒:一种环状DNA,能使新基因(如刺突蛋白基因)插入到
先前感染Ad5或Ad26可能导致机 腺病毒DNA可通过多种细胞裂解剂以及 黑猩猩病毒可免去先前对该病
体已对腺病毒载体产生免疫1
人腺病毒疫苗可能因曾感染Ad5或 Ad26而失效 ,因为免疫力并不是针 对刺突(Spike)蛋白而建立的
洗涤剂提取(可打破细胞膜并去除非核
酸细胞物质的化学试剂)
毒载体产生免疫的可能性1 黑猩猩病毒载体使机体更好地
使用全基因组测序对腺病毒DNA 进行测序,并进行以下修饰:
对刺突蛋白产生免疫反应 去除腺病毒基因组的E1区, 去除腺病毒基因组的E3区,为SARS-CoV-2
宿主基因组中(如腺病毒载体DNA基因组)
以阻断病毒复制3 刺突蛋白DNA的插入腾出空间3 修饰后的腺病毒DNA基因组将重新插入到腺病毒 将病毒载体与刺突蛋白质粒混合在一起,DNA重组技
颗粒中,形成“病毒载体” 术使质粒中的刺突蛋白基因插入到腺病毒DNA中2
作者: Ryan Brenneis, Yan Yu* 审稿人: Davis MacLean, Hannah Yaphe Stephen Vaughan*
译者: Zihong Xie (谢梓泓) 翻译审稿人: Yonglin Mai (麦泳琳) * 出版时担任临床医生
最终制备出的疫苗中的腺
病毒不能在人体细胞内复
制,也不会引起人类疾病
参考文献
1. ACS Nano 2020, 14, 10, 12522–12537, Publication Date: October 9, 2020, https://doi.org/10.1021/acsnano.0c07197
含有可转录SARS-CoV-2刺突蛋白DNA的腺病毒放置于 一种特殊的细胞培养环境中,该环境允许病毒DNA在修 饰后仍进行复制2, 3
2. Nature 2020, 586, 578–582, Publication Date: October 20, 2020, https://doi.org/10.1038/s41586- 020-2608-y
3. Frontiers in Immunology 2018, 9, 1963, Publication Date: September 19, 2018, doi: 10.3389/fimmu.2018.01963
4. NPJ Vaccines 2020, 5, 69, Publication Date: July 27, 2020, doi: 10.1038/s41541-020-00221-3
5. The Lancet 2020, Publication Date: Dec. 8, 2020, https://doi.org/10.1016/S0140-6736(20)32623-4
6. BMJ 2000, 321, 7271, 1237-1238, Publication Date: November 18, 2000,
DOI: 10.1136.bmj.321.7271.1237
7. NEJM 2021, Publication Date: Jan. 13, 2021, DOI: 10.1056/NEJMoa2034201
提取含有可转录SARS-CoV-2刺突蛋 白DNA的腺病毒,并将其配置到疫 苗注射的浓度
腺病毒有一个外层蛋白质层(称为衣 壳)来保护它的DNA
DNA比mRNA更稳定,因为DNA具有脱 氧核糖骨架以及双链间的分子键
与mRNA脂质纳米粒 疫苗相比,腺病毒疫 苗的稳定性更强
可在2-8°C 中保存长 达3-6个月
注射部位红、肿、痛
(一过性)
注意: 强生疫苗在单剂接种 后有效率可能为90%7
肌肉是首选的注射 部位,因为肌肉血 供比其他组织多
疫苗作用
细胞免疫
能使免疫细胞更快处 理外来抗原6
能让外来的疫苗物质更快 扩散,使局部反应最小化6
病毒载体疫苗通过肌肉注射到健康人体内 建议在28天后接种第二剂疫苗以增强免疫反应(超过COVID-19康复者
的免疫反应水平),从而提高疫苗效力,特别是对老年人5
外来异物可引起局 部炎症反应
腺病毒表面抗原与人体细胞受体相互作用,使病毒通过内吞作用进入人体细胞3 腺病毒载体进入细胞核,与核被膜融合,并将其DNA(包括刺突蛋白DNA)注入细胞核 细胞核内的RNA聚合酶转录病毒的DNA,合成SARS-CoV-2刺突蛋白的信使RNA(mRNA) mRNA被运回胞浆,由细胞自身的核糖体进行翻
体液免疫
刺突蛋白被胞内酶降解为片段 刺突蛋白片段与MHC-I类分子结合
MHC-I类分子将刺突蛋白片段转移到人体细胞表面
MHC-I类分子将刺突蛋白片段呈递给幼稚CD8+ T细胞
与刺突蛋白-MHC-I类分子复合物结合后,幼稚CD8+ T细胞活化, 并转移到淋巴系统进一步成熟3
译合成全长SARS-CoV-2刺突蛋白
MHC = 主要组织相容性复合体(Major Histocompatability Complex);细胞表面蛋 白,对免疫功能至关重要
CD = 分化簇(Cluster of Differentiation); T细胞表面的糖蛋白,可作为辅助受体,促 进T细胞与抗原/MHC复合物的结合,同时 可用来区分T细胞类型
刺突蛋白的成分从细胞内释放到血液中 刺突蛋白被抗原提呈细胞 (树突状细胞、B细胞、巨噬
细胞)吞噬、碎裂,与特异的MHC-II类分子结合 MHC-II类分子将刺突蛋白片段转移到抗原提呈细胞表面,
将它们提呈给血循环里的幼稚CD4+ (辅助)T细胞 与复合物结合后,活化刺突蛋白特异性辅助T细胞
结合成刺突蛋白特异性辅助T细胞后被激活
部分T细胞成熟后成为细胞毒性T 细胞,可识别SARS-CoV-2刺突蛋白
细胞毒性T细胞与感染SARS-CoV-2并表达刺 突蛋白的人体细胞或刺突蛋白片段结合
细胞毒性T细胞释放酶,使感染 的细胞穿孔,导致细胞死亡
免疫系统可以更快地识
别和消灭受感染的细胞
部分T细胞成熟后成为记忆 性T细胞 (在辅助性T细胞 释放的细胞因子的刺激下)
记忆T细胞转移到淋巴组 织后进入待定状态,在下 一次接触刺突蛋白时活化
以后SARS-CoV-2 感染时 可介导更快的细胞免疫 (获得免疫力)
活化的刺突蛋白 特异性辅助T细胞 分泌细胞因子, 促进免疫活性
全身性细胞因子释放 可导致全身反应,如 发烧、寒战、疲劳以 及肌痛 (一过性)
注意: 细胞/体液免疫 持续的时间尚不清楚
部分B细胞成熟为浆细胞,产 生病毒刺突蛋白的IgG抗体
刺突蛋白抗体识别标记SARS- CoV-2,使免疫系统能消灭病毒
根除细胞外SARS-CoV-2
淋巴组织中,活化 的辅助性T细胞与幼 稚B细胞相互作用
部分B细胞成熟为 SARS-CoV-2刺突蛋 白特异性记忆B细胞
以后一旦接触刺突蛋白,就能再次活 化淋巴组织里的记忆B细胞,使其变成 浆细胞,更快地合成抗体
以后SARS-CoV-2 感染时可介导更快的体液免疫 (获得免疫力)
图注:
病理生理
机制
体征/临床表现/实验室检查
最终结果
2021年2月11日发布于www.thecalgaryguide.com")
急性呼吸窘迫综合征: 发病机制及临床表现
、溺水及吸入化学物质(如胃内容物或直接吸入性肺损伤)。
作者 : David Olmstead
审稿 : Midas (Kening) Kang, Usama Malik, Kevin Solverson* 译者:Xiumei Deng(邓秀梅)
翻译审稿人: Yonglin Mai (麦泳琳),Zesheng Ye(叶泽生) • 出版时担任临床医生
间接性肺损伤
病因包括非肺源性的脓毒血症、创伤、严重烧伤、输血相关
性肺损伤以及胰腺炎。
Note: 急性呼吸窘迫综合征是一种以急性肺损伤为 主要表现的临床综合征,表现为严重低氧血症及 双侧肺泡损伤,不伴左心压力增高。
肺组织炎症
渗出期: 由于炎症性刺激,中性 粒细胞迁移聚集至肺泡
Note: 虽然ARDS的三个病理阶段相 继发生,但是肺组织的每一个区 域并非同时处于相同的病理阶段, 因此三个病理阶段常重叠存在。
增生期: 机体试图修复肺损伤, 若修复失败,则进入纤维化期
以中性粒细胞为主的炎性渗出液
破坏肺泡表面活性物质的功能
中性粒细胞浸润及促炎细
胞因子导致肺水肿、肺功
能障碍及继发肺上皮损伤
缩略词表:
PaO2: Partial pressure of oxygen in arterial blood(动脉氧分压)
SpO2: Peripheral oxygen saturation(外周 血氧饱和度).
CXR: Chest radiograph(肺部影像学).
由于缺乏肺泡表面活性物
质,导致肺泡塌陷
肺上皮损伤,气体交换障碍
肺毛细血管不能
完全吸收渗出液
机体尝试修复肺损伤,肺
透明膜形成
通气/血流 比值失调
肺水肿 气体扩散障碍
↓ PaO2, ↓SpO2
呼吸急促
心动过速
呼吸困难
CXR:双肺 浸润影
↓ PaO2, ↓SpO2 ↑ PaCO2
↑ PaO2, ↓PaCO2
呼吸恢复正常 ↓需O 量
吞噬细胞清除肺泡腔里细胞碎片
肺泡上皮细胞修复
疾病迁延导致机体功能损伤
成纤维细胞的作用导
致胶原蛋白沉积于肺
泡腔及肺毛细血管内
气体交换的有效 肺泡表面积↑
肺泡上皮的修复,有
助于渗出液的重吸收
2 CXR:肺浸润影消散
抑郁, 焦虑,创 伤后应激障碍
咳嗽/呼吸困难 神经源性肌无力 杵状指
纤维化期: 肺修复能力不足导 致长期的肺损伤(少见)
肺纤维化
肺动脉高压 疲劳
慢性呼吸功能障碍
图注:
病理生理学
机制
体征/症状/实验室检查
并发症
2018年2月6日发布于 www.thecalgaryguide.com")
原发性自发性气胸-发病机制和临床发现
 营养不良
呼吸过程中产生的机械力会引 起肺小疱和/或大疱产生
肺小疱或大疱自发破裂
原发性自发性气胸: 在没有确诊肺部疾病或临床表现患者的胸膜腔中存在或进入空气
肺泡和胸膜腔相通
肺泡内压力> 胸膜腔内压力 肺部空气进入胸膜腔
肺实质结构受损
注意:
• PSPs通常在休息时发生。
• 呼吸系统症状的严重程度不
同。年轻女性在月经期间反 复出现PSP,应怀疑胸部子宫 内膜异位症
• *张力性气胸的病理生理学在 另一幻灯片中描述
空气渗入皮下组织 皮下气肿
作者: Lauren Hampton
审稿人: Kening (Midas) Kang, Natalie Morgunov, Sadie Kutz, Usama Malik, Leila Barss*
译者:Huiting Wang (王慧婷), Yang Xiang (向阳)
翻译审稿人:Ran Zhong (钟然), Yonglin Mai (麦泳琳), Zesheng Ye (叶泽生)
* 发表时担任临床医生
空气将肺实质从胸部分离
患侧肺: 胸壁活动度↓ 叩诊音↑ 或语音震颤↓ 或消失呼吸音↓ 胸片出现气胸线
张力性气胸* 低血压, 颈静脉怒张, 奇脉
胸腔内压力↑ 在非抵抗性内源性弹性回缩力
作用下,小面积肺萎陷 肺顺应性↓
呼吸做功↑ 辅助性肌肉使用,呼吸急促
图注:
病理生理
机制
体征/症状/实验室检查
并发症
2017年11月14日发布于 www.thecalgaryguide.com")
支气管肺癌-肺尖部肺癌肺上沟瘤-pancoast-瘤-发病机制和
: 发病机制和临床表现 Abbreviation:
作者: Bradley Stebner , Daniel Meyers, Midas (Kening) Kang 审稿人: Natalie Morgunov, Sadie Kutz, Usama Malik, Kerri Johannson*
译者: Ran Zhong (钟然) 翻译审稿人: Yonglin Mai (麦泳
琳), Zesheng Ye (叶泽生) *发表时担任临床医生
Note:
• Pancoast 瘤: 在肺上沟 (肺尖)的恶性病灶
• 支气管源性癌:支气管
或细支气管上皮引起的
原发性恶性肿瘤
• Pancoast肿瘤可能是由
原发性或转移性肺肿瘤
(在此描述)以及感染
灶引起的
影响RLN 声音嘶哑
↓上胸腔的静脉回流 上肢的液体潴留
• • • • • •
NSCLC- 非小细胞肺癌 Non Small Cell Lung Cancer
SCLC – 小细胞肺癌 Small Cell Lung Cancer
RLN –喉返神经 Recurrent Laryngeal Nerve
SVC –上腔静脉 Superior Vena Cava RA–右心房
Right Atrium
STM –上睑板肌 Superior Tarsal Muscle
胸痛 胸内筋膜 胸膜摩擦 胸膜壁层
肩痛 上肋骨
压迫C8和T1神经
肩痛(尺神经)
大细胞癌
肺腺癌
非小细胞肺癌 (常见)
肺鳞癌
小细胞肺癌 (少见)
原发性支气管肺癌
Pancoast 瘤:位于同侧肺尖 部的肺癌/转移癌
压迫肺上沟部邻近结构
压迫破坏椎旁交感神经链
Horner综合征
↓对瞳孔开大 肌的交感控制
侵犯气道并引起阻塞 (后期)
呼吸困难
咯血 咳嗽
手部肌肉无力
压迫SVC 上腔静脉阻塞综合征
↓ 静脉回流到 RA ↓ 心脏到肺的血流
第4/5手指或者手 臂内侧/前臂 出现感觉异常
↓ 对STM的交 感控制
↓对外分泌汗 腺的交感控制
上眼睑下垂
瞳孔缩小
无汗
呼吸困难
面部和四肢水肿
图注:
病理生理
机制
体征/症状/实验室检查
并发症
2017年11月16日发布于 www.thecalgaryguide.com")
新型冠状病毒肺炎(COVID-19)mRNA疫苗-制备及其作用机制
mRNA疫苗: 制备及其作用机制
作者: Ryan Brenneis, Yan Yu* 审稿人: Davis MacLean, Hannah Yaphe, Timothy Fu, Stephen Vaughan* 译者: Zihong Xie (谢梓泓) 翻译审稿人: Yonglin Mai (麦泳琳) * 发表时担任临床医生
参考文献
1. ACS Nano 2020, 14, 10, 12522–12537, Publication Date: October 9, 2020, https://doi.org/10.1021/acsnano.0c07197
2. NEJM 2020, Publication Date: December 10, 2020, DOI: 10.1056/NEJMoa2034577
3. Expert Review of Vaccines 2017, 16, 9, 871-881, Publication Date: 2017, DOI: 10.1080/14760584.2017.1355245
4. NEJM 2020, 383, 2439-2450, Publication Date: December 17, 2020, DOI: 10.1056/NEJMoa2027906
5. NEJM 2020, 383, 2427-2438, Publication Date: December 17, 2020, DOI: 10.1056/NEJMoa2028436
6. BMJ 2000, 321, 7271, 1237-1238, Publication Date: November 18, 2000, DOI: 10.1136.bmj.321.7271.1237
注意:
• 这两种疫苗的 modRNA所编码的 刺突蛋白是相似的
• 两种疫苗的主要区 别是各家专利的脂 质纳米粒配方(未 公开)
注射部位红、 肿、痛 (一过性)
疫苗制备
通过用鼻咽拭子等方法采集感染患者身上的SARS-CoV-2(引起COVID-19的RNA病毒) 将各种试剂加入到含有人体细胞和病毒成分的样本中 试剂使细胞/病毒膜溶解,释放出细胞内容物、病毒颗粒以及病毒RNA 用各种洗涤剂去除脂肪、蛋白质和碳水化合物,只留下核酸(比如RNA) 利用逆转录聚合酶链反应(RT-PCR)合成病毒RNA的互补DNA(cDNA) 通过其cDNA文库,利用全基因组测序技术绘制SARS-CoV-2的基因组图谱 找到SARS-CoV-2刺突蛋白的DNA序列,并作为模板构建合成病毒刺突蛋白的mRNA 将额外的RNA碱基添加到这条mRNA上以提高它的稳定性,由此产生的RNA链称为“核酸修饰的RNA”(“modRNA”)
用一系列沉淀、提取和层 析等方法分离出刺突蛋白 modRNA
最后,modRNA脂质纳米 粒疫苗研制完成,可用于 肌肉注射
modRNA疫苗通过肌肉注射到健康人体内 3-4周后需接种第二剂疫苗以增强免疫反应(超过COVID-19康复者
通过对其他冠状病 毒(如SARS-CoV-1和 MERS-CoV)的研究
可知,“刺突(Spike) 蛋白”是一种主要的
病毒表面抗原
辉瑞/BNT162b2疫苗成分:
莫德纳mRNA-1273疫苗:
注解: 脂质纳米粒 是一种空心“球”, 由外层脂膜加上其 他乳化剂和膜稳定 剂制成。 脂质纳米粒能够包 裹较小的分子(如 RNA),并与正常 细胞膜结合。
肌肉是首选的注射 部位,因为肌肉血 供比其他组织多
疫苗作用
能使免疫细胞更快处理外 来抗原6
能让外来的疫苗物质更快扩 散,使局部反应最小化6
细胞免疫
的免疫反应水平),从而提高疫苗效力,特别是对老年人
4,5
外来异物可引起局部 炎症反应
辉瑞/ BioNTech专 利脂质纳米 粒
modRNA可编码一个由 两个脯氨酸修饰的全 长刺突蛋白(为了稳 定性和免疫原性)2
modRNA可编码一个 由两个脯氨酸修饰的 全长刺突蛋白(为了 稳定性)1
莫德纳专利 脂质纳米粒
将这种modRNA封装在辉瑞/ BioNTech的脂质纳米颗粒中,得到 162b2疫苗
这种疫苗需要更低的储存温度((-700C)
将这种modRNA封装在莫德纳的脂 质纳米粒种,得到mRNA-1273疫苗
这种疫苗可以在稍微温暖的温 度下储存(-200C)
刺突蛋白被胞内酶降解为片段
刺突蛋白片段与MHC-I类分子结合
MHC = 主要组织相容性复合体(Major Histocompatability Complex);细胞表 面蛋白,对免疫功能至关重要
CD = 分化簇(Cluster of Differentiation); T细胞表面的糖蛋白,可作为辅助受体, 促进T细胞与抗原/MHC复合物的结合, 同时可用来区分T细胞类型
刺突蛋白的成分从细胞内释放到血液中 刺突蛋白被抗原提呈细胞 (树突状细胞、B细胞、巨噬细
胞)吞噬、碎裂,与特异的MHC-II类分子结合 MHC-II类分子将刺突蛋白片段转移到抗原提呈细胞表面,
将它们提呈给血循环里的幼稚CD4+ (辅助)T细胞 部分幼稚CD4+ 辅助T细胞能够与刺突蛋白-MHC-II类
分子复合物顺利结合
与复合物结合后,活化刺突蛋白特异性辅助T细胞
脂质纳米粒通过内吞作用与人体细胞的磷脂膜融合,将
modRNA释放到细胞的胞浆中 体液免疫
modRNA由人体细胞中自带的核糖体翻译,合成刺突蛋白 相关成分
MHC-I类分子将刺突蛋白片段转移到人体细胞表面
MHC-I类分子将刺突蛋白片段呈递给幼稚CD8+ T细胞
与刺突蛋白-MHC-I类分子复合物结合后,幼稚CD8+ T 细胞活化并转移到淋巴系统进一步成熟
部分T细胞成熟后成为细胞毒性T细 胞,可识别刺突蛋白
细胞毒性T细胞与感染SARS-CoV-2并表 达刺突蛋白的人体细胞或刺突蛋白片 段结合
细胞毒性T细胞释放酶,使感染的细 胞穿孔,导致细胞死亡
免疫系统可以更快地识别和消灭 SARS-CoV-2感染的人体细胞,使病 毒扩散速度减缓
部分T细胞可以成熟为记 忆性T细胞 (在辅助性T 细胞释放的细胞因子的 刺激下)
记忆T细胞转移到淋巴组 织后进入待定状态,在下 一次接触刺突蛋白时活化
以后SARS-CoV-2 感染时可 介导更快的细胞免疫 (获得免疫力)
活化的刺突蛋白 特异性辅助T细胞 分泌细胞因子, 促进免疫活性
全身性细胞因子释放 导致全身反应:如发 烧、寒战、疲劳以及 肌痛 (一过性)
部分B细胞成熟为浆细胞,产 生病毒刺突蛋白的IgG抗体
刺突蛋白抗体识别标记SARS- CoV-2,使免疫系统能消灭病 毒
根除细胞外的SARS-CoV-2
淋巴组织中,活 化的辅助性T细胞 与幼稚B细胞相 互作用
部分B细胞成熟为 SARS-CoV-2刺突蛋白 特异性记忆B细胞
以后一旦接触刺突蛋白,就能再次活化淋巴 组织里的记忆B细胞,使其变成浆细胞,更 快地合成抗体
以后SARS-CoV-2 感染时可介导更快的体 液免疫 (获得免疫力)
图注:
病理生理
机制
体征/临床表现/实验室检查
最终结果
2020年12月19日发布于 www.thecalgaryguide.com")
肺癌:临床发现和副癌综合征
 Kang Usama Malik, Leila Barss* 译者: Huiting Wang (王慧婷), Yang xiang (向阳) 翻译审稿人: Ran Zhong (钟然), Yonglin Mai (麦泳琳), Zesheng Ye (叶泽生) * 发表时担任临床医生
肿瘤分泌生物活性物质
副癌综合征à 与恶性疾病相关的症状
注意: 肺癌的大多数表现隐匿,具有非特异性的 症状和体征(即发烧、体重减轻、全身不适)
单侧或双侧肺部发生的不受控 异常细胞增生à肺癌
近端气道梗阻
无法清除吸入的 病原体
局部肿瘤生长
肿瘤向胸膜 气道侵袭 表面扩散
胸部不适 胸腔积液 咯血
肺癌梗阻性肺炎 局部阻塞 或
压迫
咳嗽、发热、呼 吸困难
肺通气做功↑ 喉神经压迫
↑ ADH
合征 水重吸收↑
低钠血症 血钠 <135mEq/L
Lambert-Eaton 综合征 (肌无力样综合征)
(产生针对钙通道的自身 抗体)
肌肉无力
上腔静脉 压迫
↑ TGFβ1
细胞外基 质蛋白↑
杵状指
↑ PTHrP
骨骼钙 ↑释放
高钙血症 血钙 >2.6 mmol/L
↑ ACTH
皮质醇的产生 分泌异常综 和释放↑
抗利尿激素
臂丛神经 压迫
库欣综合征 (长期皮质醇增 多所引起的症状 和体征)
肌肉无力、高血
糖、严重低钾血 症
呼吸困难 呼吸急促
支配声带的神经受损
声音嘶哑
手臂/肩膀/颈 面部/手臂 部疼痛 水肿
缩写:
• ACTH:促肾上腺皮质激素
• ADH: 抗利尿激素
• PTHrP: 甲状旁腺激素相关肽
• SIADH:抗利尿激素分泌异常综合征
• TGFβ1: 转化生长因子β1
图注:
病理生理
机制
体征/症状/实验室检查
并发症
2018年2月6日发布于 www.thecalgaryguide.com")
新生儿呼吸窘迫-临床表现

新生儿用鼻孔呼吸(6个月前) 通过鼻腔时气流受阻(由
于上呼吸道感染,鼻胃导
管,后鼻孔闭锁等)
鼻翼煽动 喂食困难 ( (扩大鼻孔以 鼻腔受阻时,
下气道阻
塞(如:
细支气管)
喘息(通 常为呼气 相)
喉部以上部 位阻塞(如. 咽, 鼻等,)
鼾音
↓ O2 储备, 氧
↓ 气流阻力)
由于潮气量受
限,新生儿只 能通过↑ RR(呼吸频率) 增加通气量
新生儿在呼吸疾
病的早期即可表
现出呼吸急促
辅助呼吸肌收缩,
试图抬高肋骨
由于肋骨无法上抬,
因此肌肉将头拉低
点头呼吸
肺泡更容易塌
陷à肺不张 气饱和度下降
口腔进食与
鼻腔呼吸的 协调难度↑)
作者: Yan Yu 审稿人:Elizabeth de Klerk Hailey Barker
Naminder Sandhu*
译者: Xiumei Deng (邓秀梅) 翻译审稿人: Yonglin Mai
(麦泳琳), Zesheng Ye (叶泽生)
* 发表时担任临床医生
喉、气管或支气管阻塞
喘鸣(通常为吸气性,
也可以是双相的)
为了防止肺不张 & 氧气饱和度下降, 新生儿通常不会完全呼气:肺内残留 气体可维持肺泡开放
新生儿发生呼吸窘迫时,呼吸动作异常明显
呼吸肌试图 潮气量, 但由于胸腔容积无 法进一步扩大,呼吸无效做功,长此以往 新生儿显得疲惫不堪
̄ 呼吸频率(RR); 可能出现呼吸衰竭
用力呼气冲开声门, 肺泡内气体压力 ↑, 以维持肺泡开放
鼾音
深吸气使得胸腔 内压↓,以抵抗 胸廓的顺应性
胸廓凹陷
图注:
病理生理
机制
体征/症状/实验室检查
并发症
2013年1月15日发布于 www.thecalgaryguide.com")
异物吸入: 发病机制和临床表现

作者: Nick Baldwin
审稿人: Elizabeth de Klerk, Yan Yu, Naminder Sandhu*
译者: Huiting Wang (王慧婷), Yang Xiang (向阳)
翻译审稿人: Ran Zhong (钟然), Yonglin Mai (麦泳琳), Zesheng Ye (叶泽生)
*发表时担任临床医生
气球易压缩及表面光 滑,容易通过气道
气球与气道贴合, 导致气道完全梗阻
口腔探索行为
口腔和吞咽运动 功能不协调
异物在后咽部引起不适,引起儿童猛然吸气 将异物吸入到气道中
异物(FB)吸入: 异物进入呼吸道
Note:
Note: 临床实际中 气球误吸十分罕见
死亡率最高
上呼吸道阻塞:从咽部到喉部
• 窒息是一个重要的l体征,但并不易被人 察觉
• 最常见于3岁以下儿童
• 异物误吸最常发生于右肺,由于右主支气
管较左侧管径更粗、走向较垂直
下气道阻塞: 从气管到肺泡
部分性
完全性: 肺和外界没有气体交换
氧气和二氧化碳交换量↓
缺氧:组织供氧不足
心肌供氧不足 心肌供氧不足
心脏停搏
空气通气量↓ 氧气和二氧化
碳交换量↓ 缺氧
氧气需求增多
呼吸中枢刺激的呼吸 驱动↑
呼吸急促,用力呼 吸
气道狭窄
经过狭窄 处时,湍 流气流会 产生独特 的声音
吸气时气道变宽,空气 能通过异物与气道空隙, 但在呼气时气道狭窄引 起气体潴留
单向阀让空气只进不出
湍流气流通过 狭窄的上呼吸
喉刺激
声带振动受损
空气进入 道à刺耳, 量↓,肺
高调呼吸音
部无法充 分通气
吸气性喘 焦躁不安,用 声音嘶哑 鸣音 力呼吸
呼气相喘 呼气相X线提 呼吸音 息: 听诊可 示单侧肺过度 减弱
闻及 充气
图注:
病理生理
体制
体征/症状/实验室检查
并发症
2013年5月29日发布于 www.thecalgaryguide.com")
囊性纤维化-发病机理临床表现及并发症
基因突变à
囊性纤维化跨膜传导调节( CFTR )蛋白 (一种表达在外分泌组织中的氯离子通道蛋白)功能紊乱
作者: Spencer Montgomery
审稿人: Yan Yu, Kayla Nelson, Mark Montgomery*
译者: Huiting Wang (王慧婷), Yang Xiang (向阳)
翻译审稿人:Ran zhong (钟然),
Yonglin Mai (麦泳琳), Zesheng Ye (叶泽生) *发表时担任临床医生
异常氯离子通道蛋白影响Cl-跨膜运输
注意:
• CFTR突变呈常染色体隐性遗传
• 已知有> 1700种不同CFTR基因突变类型, ∆F508突变占了高加索人群发病总
数的67%
• 囊性纤维化的诊断依据:汗液氯离子浓度↑, CFTR基因突变 & ≥1个相关内
在汗腺中, CFTR蛋白主要 功能为Cl-重吸收
重吸收↓ =汗 液中Cl- 浓度↑
汗液中Cl- 浓度↑
在母体 子宫内 输精管
中肾管、输精
管及相关结构
退行性变
男性不育
在身体其他部位的管道上皮组织中, CFTR蛋白促进 Cl- 扩散到分泌物中
外周纤毛液体层Cl- ↓ → 外周纤毛液 体层的含水量↓
黏液-纤毛系统清除分泌物↓ 分泌物异常聚集并阻塞全身各处的
分泌管腔
下呼吸道
脏有器官系统的 临床症状
儿童/成人: 远端肠梗阻综合征(DIOS)
胃肠道
胆道系统
鼻息肉
胃肠道内容
物蠕动↓ 新生儿:
胎粪性 肠梗阻
胎粪潴留↑→
肝硬化 & 门静脉高压
新生儿黄疸 胆红素重吸收↑ 时间延长
胆汁排出延迟 →肝炎症反应 上呼吸道 胰腺
淤积在胰腺内
的消化酶消化 胰腺自身
炎症 ;瘢痕增生 & 脂肪组织渗 透 ; 胰岛细胞 受损
II型糖尿病
慢性湿咳
诊断肺部阻塞标志: i.e. 肺过度充气(X线), 肺功能检测异常
鼻窦内分泌
胰腺分泌的消化酶无法进入 物潴留 → 细 胃肠道中 (胰液不足)
分泌物在气道内潴留→ 细菌增殖à 气道感染 & 炎症 持续呼吸道感染可进展为慢性支气管炎
± 支气管扩张(囊性纤维化最主要的死亡原因)
菌增殖
慢性鼻窦炎
脂质和蛋白 脂溶性维生素吸收 质吸收不良 ↓
发育不良
血清Vit. D ↓ 骨质疏松症
图注:
病理生理
机制
体征/症状/实验室检查
并发症
2013年1月21日发布于 www.thecalgaryguide.com")
哮吼-发病机制和临床表现
导致管道内流速↑以 及压力↓ (Bernoulli's principle伯努利原理)
真空的产生,
使得狭窄处远
端气道塌陷
气流通过狭窄的气管
会产生湍流,从而发
出独特的声音
肺通气做功↑
用力呼气: 吸气:
“犬吠样”咳嗽 喘鸣: 刺耳、高音调
的声音
呼吸窘迫: (动用辅助呼吸肌, 鼻翼煽动, 胸壁凹陷), 呼吸急促
作者: Nick Baldwin
审稿人: Jody Platt, Elizabeth de Klerk, Yan Yu, Naminder Sandhu* 译者:Yonglin Mai (麦泳琳)
翻译审稿人:Zesheng Ye (叶泽生)
*发表时担任临床医生
图注:
病理生理
机制
体征/症状/实验室结果
并发症
2013年5月28日发布于 www.thecalgaryguide.com")
世界卫生组织对治疗covid-19新型冠状病毒肺炎的药物研
的药物研
作者: Hannah Yaphe 审稿人: Davis Maclean, Timothy Fu, Yan Yu*, Stephen Vaughan* 译者: Zesheng Ye (叶泽生) 翻译审稿人: Zihong Xie (谢梓泓) * 发表时担任临床医生
注:此幻灯片基于截至 2020年3月30日的文献。 此处展示的药物是世卫 组织联合试验中涉及到 的药物,这些药物是根 据体外工作和MERS和 SARS的临床数据选择的。 机制研究尚处于初步阶 段,现并无足够的数据 支持或驳斥这些药物对 COVID-19的作用。研究 目前仍在进行中。
病毒复制 被终止
更少细胞被感染
炎症反应及免疫
反应的激活↓ 感染的临床症
状得以改善*
*See slide on pathophysiology and clinical findings of COVID19
究:假设作用机制
COVID-19 病毒复制途径
病毒与人体细胞表面的血管 紧张素转换酶2 (ACE-2)受体 结合
病毒被网格蛋白包被形成囊 泡,通过内吞作用进入细胞
囊泡通过内溶酶体途径成熟 病毒膜与成熟内溶酶体融合,
将病毒RNA释放到细胞质中 病毒RNA利用宿主细胞核糖
体合成新的病毒蛋白质,如 RNA聚合酶
病毒RNA聚合酶与宿主 细胞的核苷酸结合
新病毒RNA产生
氯喹或羟氯喹
(CQ, HCQ)
弱碱性导致内体及 溶酶体内的pH值↑
高尔基体中 ACE-2的N端糖 基化受抑制
细胞表面表达的 ACE-2受体结构 异常
病毒膜无法与未
成熟内体结合
病毒和ACE-2受 体结合受影响, 阻断病毒进入人 体宿主细胞
病毒内含物 无法释放
内体成 熟障碍
干扰素-β (IFNβ)
(联合洛匹那韦/ 利托那韦)
利托那韦 (RTV) (联合洛匹那韦)
抑制CYP450(一 种药物代谢酶)
激活
JAK/STA IFN-调控
与干扰素 受体结合
(机制尚未明确)
瑞德西韦 (RDV) 瑞德西韦磷酸化成
三磷酸-瑞德西韦
(RDV-TP)
洛匹那韦 (LPV)
LPV在血浆中半衰 抑制病毒3-糜蛋白酶 熟的病毒蛋白
T通路 基因转录 可能使LPV/RTV的抗病毒作用↑
抗病毒和免疫调节 蛋白的表达↑
抑制病毒复制(多种机制)
RDV-TP与ATP 竞争性结合 病毒的RNA聚 合酶
RDV-TP的掺 入终止了延 长中的RNA
洛匹那韦(LPV) 的降解速度↓
病毒蛋白前体
无法切割为成
病毒RNA及蛋白质组装成新 新形成的 的病毒颗粒
病毒颗粒
无法感染
新的细胞 病毒颗粒从细胞中释放出来
期及作用时间↑ 样蛋白酶
图注:
病理生理
机制
体征/症状/实验室检查
并发症
2020年3月30日发布于 www.thecalgaryguide.com")
肺高血压-病理生理特点和临床表现
 翻译审稿人: Yonglin Mai (麦泳琳) * 发表时担任临床医生
左心疾病(心力衰竭,心肌梗死) 心肌收缩能力 和/
或舒张能力↓ 左心室心输出量 ↓ 左心(心室、心房)血液充盈,充盈压升高 肺血管血液充盈,压力升高 肺毛细血管楔压↑– 估测左心房压力
慢性贫血 血浆血红蛋白↓
单位血液携氧能力 ↓ 心率代偿性 ↑,
以维持组织供氧
肺部疾病(慢性阻塞性肺疾病) 肺组织损失 ,肺弹性↓
肺通气表面 积↓à气体 交换↓
慢性血栓栓塞
肺血管疾病(肺动脉 高压, 硬皮病)
肺通气能 力↓
血管阻塞/纤维化
慢性低氧血症
局部肺泡氧分压↓
肺动脉血压力↑ 肺高血压
右心室后负荷↑ (心脏收缩射 血所承受的压力)
右室心输出量↓ 心脏收缩后右心剩余容积↑ 体循环淤血 静脉系统血容量↑ 毛细血管血容量↑及压力↑ 液体从血管进入组织间隙
血管顺应性↓
肺血管发生局部反应性 血管收缩,将血液分配 到通气较好的区域
血管壁增厚, 气体交换受损
血氧分压↓, 二 氧化碳分压↑
组织中氧气供应不足, 二氧化碳清除不足
心输出量↑ 慢性压力↑–血
管纤维化
左心循环血量↓, 左室充盈↓ 左心室输出量↓
肺血管阻力↑ (PVR)
随病程进展,发生心肌 肥厚(向心性&偏心性)
心脏体积↑
心肌需氧量↑
心肌缺血(供/需不匹配)
当需氧量↑时, 胸痛风险↑
外周水肿
心脏异常传导 通路和异位起 搏点形成
心律失常 心悸
组织灌注↓ 乏力
脑组织灌注↓ 晕厥
反射性触发代偿性深、 呼吸困难 快呼吸
t 图u : 注 :
病理生理学
发病机制
体征/症状/实验室检查
并发症
2020年9月8日发布于 www.thecalgaryguide.com")
gastroenteritis-patogenesis-y-hallazgos-clinicos

Rotavirus, norovirus, adenovirus, astrovirus, enterovirus
Bacterias (10-20%)
Campylobacter jejuni, Salmonella spp, Escherichia coli, Shigella, Yersinia, Vibrio cholerae
Infiltración patógena del tracto gastrointestinal
Otros (<10%)
Cryptosporidium, Giardia lamblia, Entamoeba histolytica, Strongyloides
Notas:
• La edad más frecuente para la gastroenteritis abarca de los 6 meses a los 2 años.
• La diseminación de la infección se produce por las vías respiratoria y fecal-oral.
Estímulos nocivos e inflamación del tracto gastrointestinal
Estimulación de nervios viscerales aferentes
Dolor abdominal Notas:
• Diarrea = aumento de la frecuencia y disminución de la consistencia de las heces, típicamente evacuación de ≥3 heces blandas/acuosas por día.
• Los mecanismos exactos para cada hallazgo clínico son probablemente multifactoriales y pueden variar según el patógeno involucrado.
**
Las toxinas patógenas estimulan la secreción entérica de cloruro
Interacción patogénica con el sistema nervioso entérico
Alteración de la actividad y/o estructura del borde en cepillo
Deterioro de la absorción de sustancias en el intestino delgado
Sustancias osmóticamente activas ingresan al intestino grueso
Capacidad de reabsorción de agua en el intestino grueso es sobrepasada
Liberación de 5HT por las células enterocromafines
Estimulación de la zona de activación
de los quimiorreceptores (área postrema)
Estimulación del centro del vómito (formación reticular lateral)
Náuseas/Vómitos
↑ Secreción del líquido GI
Diarrea
*mecanismo propuesto, mecanismo exacto desconocido
Leyenda: Patofisiología
Mecanismo
Signos/Síntomas/Hallazgos de Laboratorio
Complicaciones
Publicado el 28 Agosto, 2015 en www.thecalgaryguide.com")
faringitis-por-estreptococo-del-grupo-a-patogenesis-y-hallazgos-clinicos

Lesión celular inadvertida y hemólisis
Petequias palatinas + Eritema
Abreviaciones:
GAS – Estreptococo del Grupo A
IL – Interleucina
IFN – Interferón
SPE –Exotoxina Pirogénica Estreptocócica
SSA – Superantígeno estreptocócico
FNT –Factor de Necrosis Tumoral
GB – Glóbulos Blancos
Estimulan a las células mononucleares1
Liberación de IL-1
Estimula el centro de control de temperatura hipotalámico
Fiebre
↑ drenaje linfático a los ganglios regionales
Ganglios cervicales anteriores sensibles y agrandados>1cm
Liberación de FNTα
Facilta la infiltración de GB en el sitio de infección
Liberación de moduladores endoteliales2 ↑separación entre las células endoteliales
Líquido y proteínas de los vasos se filtran al espacio intersticial
Edema faríngeo
Notas
1. También conocidos como agranulocitos, incluidos los leucocitos que comprenden linfocitos, macrófagos y monocitos.
2. Histamina, bradicinina, sustancia P, leucotrienos.
3. Proceso sofisticado y multifacético que involucra la fagocitosis de bacterias por macrófagos y la posterior destrucción por exposición a
radicales libres, así como a compuestos bactericidas como lactoferrina, elastasa y lisozimas producidas dentro de los macrófagos.
4. Erupción papular fina descrita clásicamente como")
hipersensibilidad-tipo-i-patogenesis-y-hallazgos-clinicos
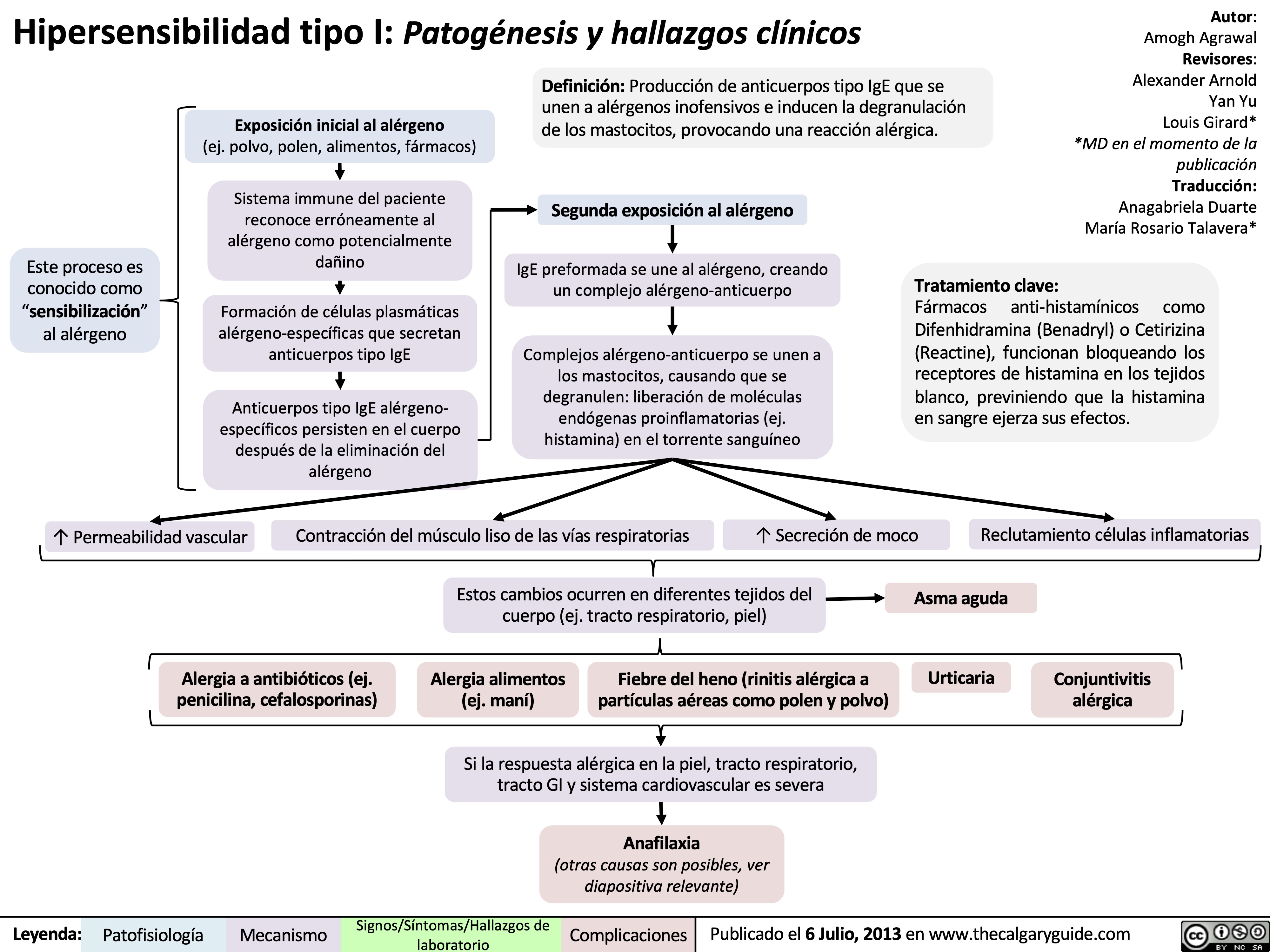
Celulitis

Organismos penetran los vasos sanguíneos
Bacteremia (presencia de bacterias en sangre)
Piel normal
Capa epidérmica
Unión dérmica- epidérmica Capa dérmica
Grasa subcutánea
Flora cutánea residente: Staphylococcus coagulasa negativos*
Flora cutánea transitoria:
Staphylococcus aureus*
Streptococcus pyogenes
Bacterias gram negativas Hongos
Patógeno en dermis profunda y grasa subcutánea
*patógenos más comunes
Rotura de la barrera cutánea (puede que no sea evidente) y entrada de patógenos
Virulencia del organismo supera los mecanismos de defensa del huésped (asociado a los factores de riesgo)
Celulitis: Una infección bacteriana en la que los patógenos penetran la dermis profunda y/o la grasa subcutánea
Citocinas activan la respuesta inmune
Acumulación de pus (bacterias, glóbulos blancos, piel muerta)
Formación de abscesos
Factores de riesgo:
Huésped inmunodeprimido: -Diabetes mellitus+
-Linfedema
-Desnutrición
-Paciente adulto mayor+ -Obesidad+
-Enfermedad vascular periférica Riesgo de infección general: -Historia de celulitis+
+factores de riesgo mayores
Factores de riesgo para celulitis por SARM:
Mayor exposición a SARM: -Deportes de contacto -Hacinamiento
-Trabajadores de la salud -Ascendencia indígena -Compartir toallas, equipos Mayor susceptibilidad: -Inmunodeficiencia
-Edad temprana
Infección se propaga a los ganglios linfáticos cercanos
Linfadenitis
infección se propaga a través de los vasos linfáticos
Linfangitis ascendente
Respuesta inflamatoria local en la piel
Diseminación a distancia en endocardio (revestimiento interno de las cámaras y válvulas del corazón)
Endocarditis SARM: Staphylococcus aureus resistente a meticilina
Dolor
Calor
Fiebre Malestar
Diseminación a distancia en el hueso
Osteomielitis
Escalofríos
Inflamación sistémica
Edema Eritema (enrojecimiento)
con márgenes indefinidos
Vesículas y ampollas
(poco frecuente)
Sepsis Abreviaturas:
Leyenda: Patofisiología
Mecanismo
Signos/Síntomas/Hallazgos de Laboratorio
Complicaciones
Publicado el 27 Septiembre, 2020 en www.thecalgaryguide.com")
Inmunidad Humoral
 o circulan en la sangre
Células B de memoria proliferan y se
diferencian en células plasmáticas en respuesta a la reexposición al antígeno.
↑ Tasa y amplitud de la respuesta inmune secundaria en exposiciones repetidas
Antígenos (Ag) son producidos por los patógenos (bacterias, virus, hongos, parásitos) o el paciente (a través de traumatismos, tumores, metabolismo), y circulan en el plasma, linfa u otros tejidos
Expansión clonal
(proliferación de células B activadas)
↑ GB (linfocitosis) Enfermedad autoinmune si
células B reconocen autoantígeno
Diferenciación (en células B de memoria o células plasmáticas)
Células plasmáticas producen anticuerpos, que contribuyen a la inmunidad de 3 formas:
Opsonización: Los Acs recubren a los patógenos, ayudando al reconocimiento por los fagocitos
Neutralización: Acs se unen a
las moléculas de superficie de los patógenos que se necesitan para invadir las células del huésped, neutralizándolas
Activación del complemento: Acs activan las proteínas del complemento a través de la vía clásica (consulte la diapositiva de Activación del complemento)
Eliminación de patógeno por la respuesta inmune adaptativa
Ag dependiente de células T:
Las células presentadoras de Ag (como las células dendríticas o los macrófagos) presentan el Ag a las células T auxiliares CD4+ y las activan. Las células T colaboradoras activadas luego estimulan a las células B
Ag independiente de células T:
Ags como péptidos, carbohidratos y lípidos pueden ser reconocidos directamente por las células B, lo que desencadena su activación
Complemento:
Proteínas del complemento circulantes en el plasma detectan y se unen a Ag. Los Ags marcados con el fragmento C3 del complemento se unen al complejo correceptor de las células B y mejoran la activación de las células B.
Abreviaturas:
Ac – Anticuerpo
Ag – Antígeno
Ig – Inmunoglobulina GB – Glóbulos blancos
Células B vírgenes
Células plasmáticas producen primero IgM
Citocinas y células T estimulan el cambio de clase de Ig de las células B (cambiando las regiones constantes de la cadena pesada de la molécula de Ig)
La producción de Ig cambia de IgM a IgG, IgA, IgE o IgD
IgG es la Ig más común en las reacciones inmunes. La IgA se concentra en la mucosa, la IgE desgranula a los mastocitos, la IgD ayuda a las células B maduras.
Células B activadas
(en órganos linfoides secundarios, como el bazo o los ganglios linfáticos)
↑ Ig plasmática
Leyenda: Patofisiología
Mecanismo
Signos/Síntomas/Hallazgos de laboratorio
Complicaciones
Publicado el 2 Mayo, 2020 en www.thecalgaryguide.com")
impetigo-patogenesis-y-hallazgos-clinicos

cascada-inflamatoria-patogenesis-y-hallazgos-clinicos

Activación de la respuesta inmune en el sitio de la lesión
Células endoteliales alteradas liberan citocinas proinflamatorias (e.j., IL’s, FNTα) y quimiocinas
↑ Permeabilidad vascular: extravasación de células inmunitarias/mediadores (e.j., GB, proteínas del complemento, plaquetas)
Vasodilatación:
↑ flujo sanguíneo al sitio lesionado
Quimiotaxis:
células inmunes migran al sitio de la lesión
Signos cardinales de la respuesta inflamatoria
Enrojecimiento (rubor) ↑Temperatura local (calor) Continuación Inmunidad innata
Dolor (dolor) Tumefacción (tumor)
Activación de la inmunidad adquirida
Fagocitosis:
(*mecanismo principal)
neutrófilos y macrófagos engullen patógenos extraños y tejido muerto
Pus
Sistema complemento:
opsonización del patógeno (marcado para ser destruido), promueve inflamación
Células Natural Killer:
ruptura de la pared mediada por perforinas (orificios), destrucción mediada por enzimas
Humoral:
(Respuesta de células B)
producción de anticuerpos, activación del sistema del complemento
Mediada por células:
(Respuesta de células T)
↑ actividad de células ayudadoras y células citotóxicas
Nota: células dendríticas en el sitio de la lesión ayudan en la fagocitosis y la presentación de antígenos a las células T.
Leyenda:
Patofisiología
Mecanismo
Signos/Síntomas/Hallazgos de laboratorio
Complicaciones
Publicado el 14 Diciembre, 2018 en www.thecalgaryguide.com")
respuesta-inmune-innata-patogenesis-y-hallazgos-clinicos

Patógenos superan las barreras físicas (e.j., epitelio, cilios)
Patrones moleculares asociados a patógenos
Trauma
Patrones moleculares asociados a daños
Ejemplos de macrófagos tisulares: • Macrófagos alveolares– Pulmón
• Histiocitos – Tejido conectivo • Células de Kupffer – Hígado
• Células mesangiales– Riñón
• Células microgliales– Cerebro • Osteoclastos – Hueso
Microbio es envuelto y expuesto al estallido respiratorio
Microbios destruidos
Pus
Reconocimiento por receptores de reconocimiento de patrones (e.j., receptores tipo toll)
Quimiocinas proinflamatorias
Reclutamiento de granulocitos y monocitos circulantes
Citoquinas proinflamatorias (e.j., IL- 1β, TNFα, IL-6)
Activación de macrófagos tisulares
Proteínas antimicrobianas
Infección/inflamación no resuelta
Antígeno es presentado a las células T
Reclutamiento de la respuesta inmune adaptativa
Respuestas immunes mejoradas
Producción hepática de proteína de fase aguda (es decir, proteína C reactiva)
Producción de prostaglandinas en el hipotálamo
Fiebre
Ruptura de las uniones
estrechas de las células endolteliales de la vasculatura
Fuga de líquido intravascular al espacio extravascular
Edema
Leyenda: Patofisiología
Mecanismo
Signos/Síntomas/Hallazgos de laboratorio
Complicaciones
Publicado el 19 Enero, 2020 en www.thecalgaryguide.com")
serologia-vhb-principios-basicos-e-interpretacion-de-patrones
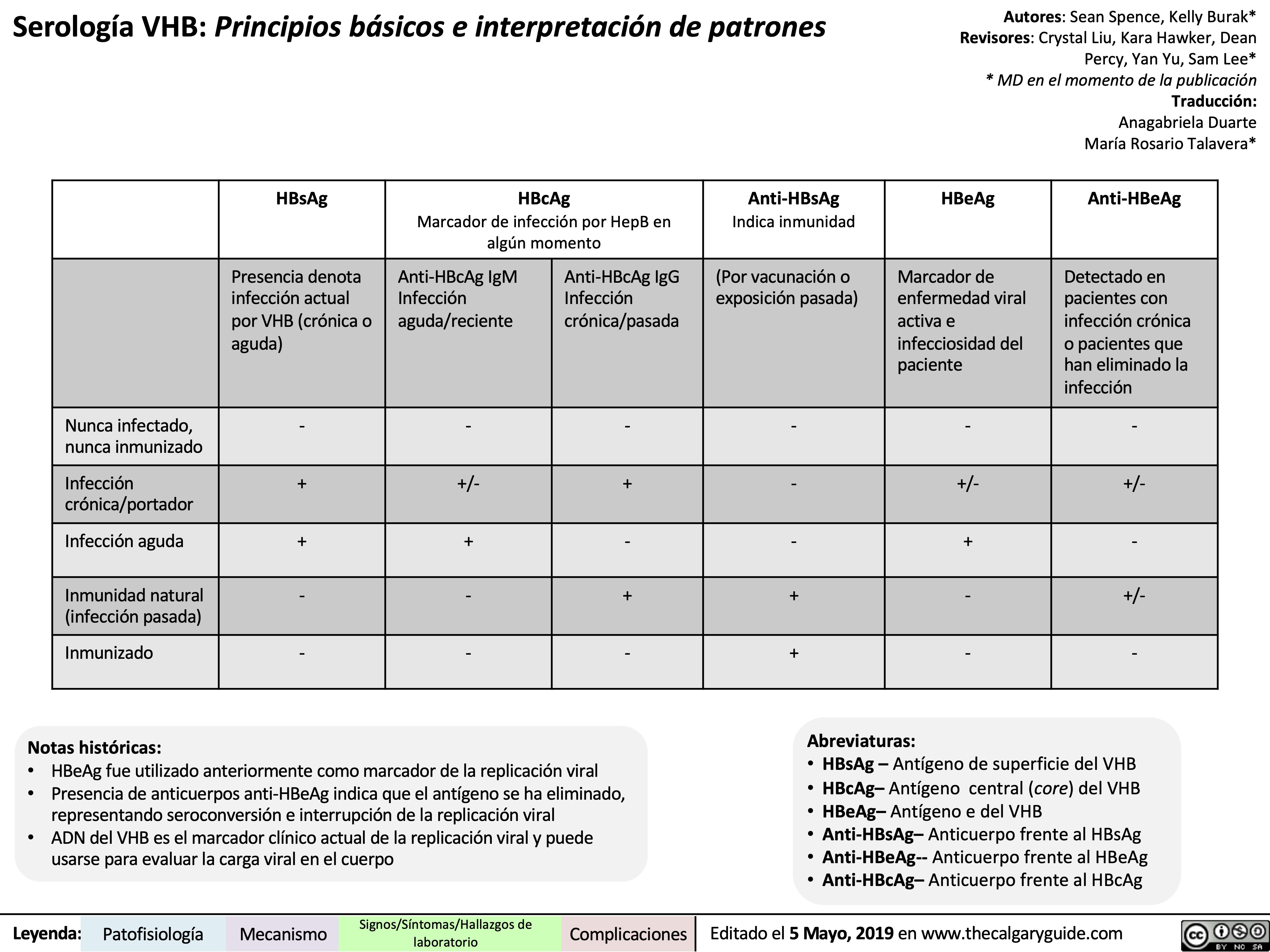
hepatitis-viral-patogenesis-y-hallazgos-clinicos
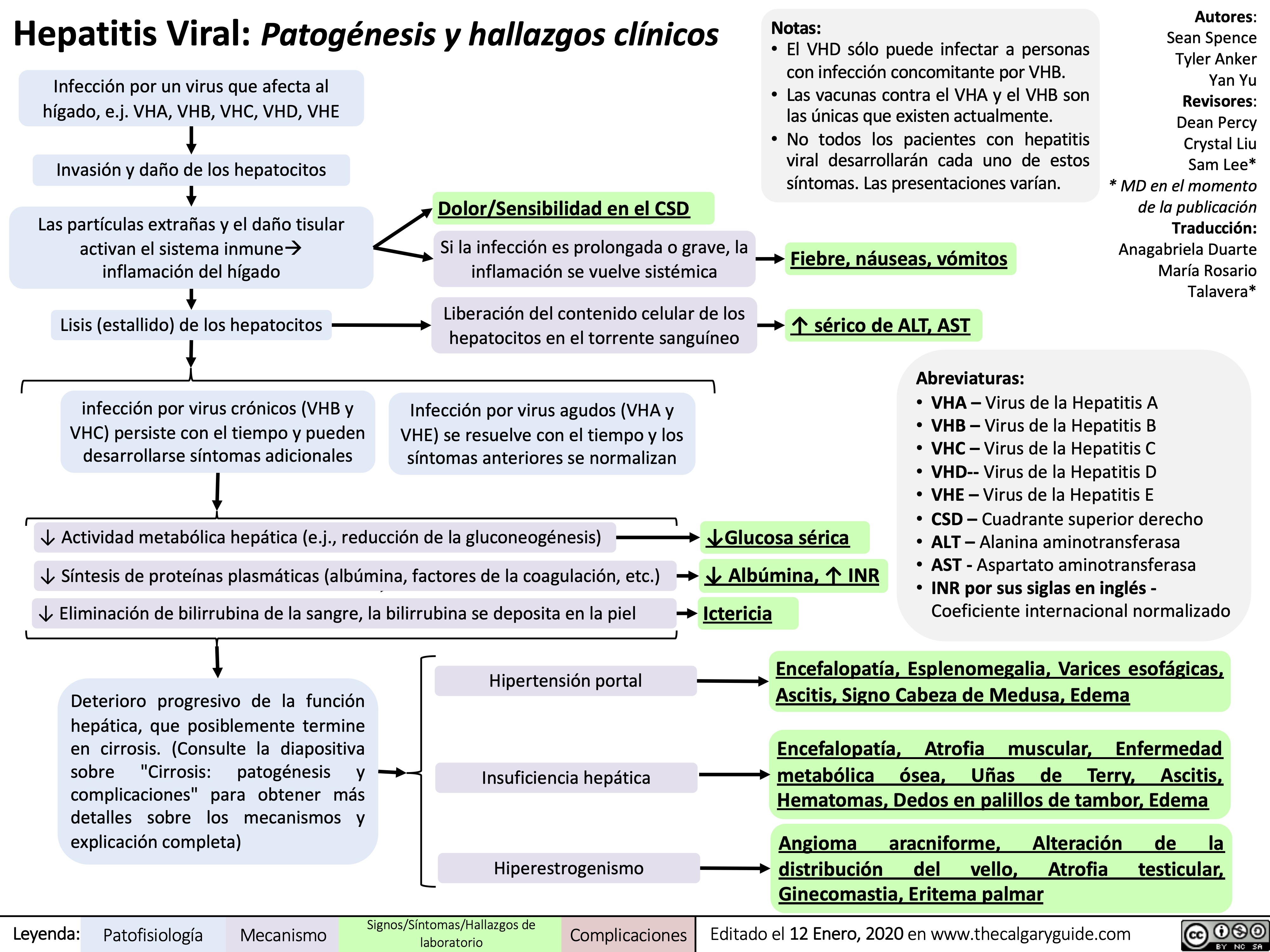
infeccion-por-hepatitis-a-vha-explicacion-de-los-patrones-serologicos
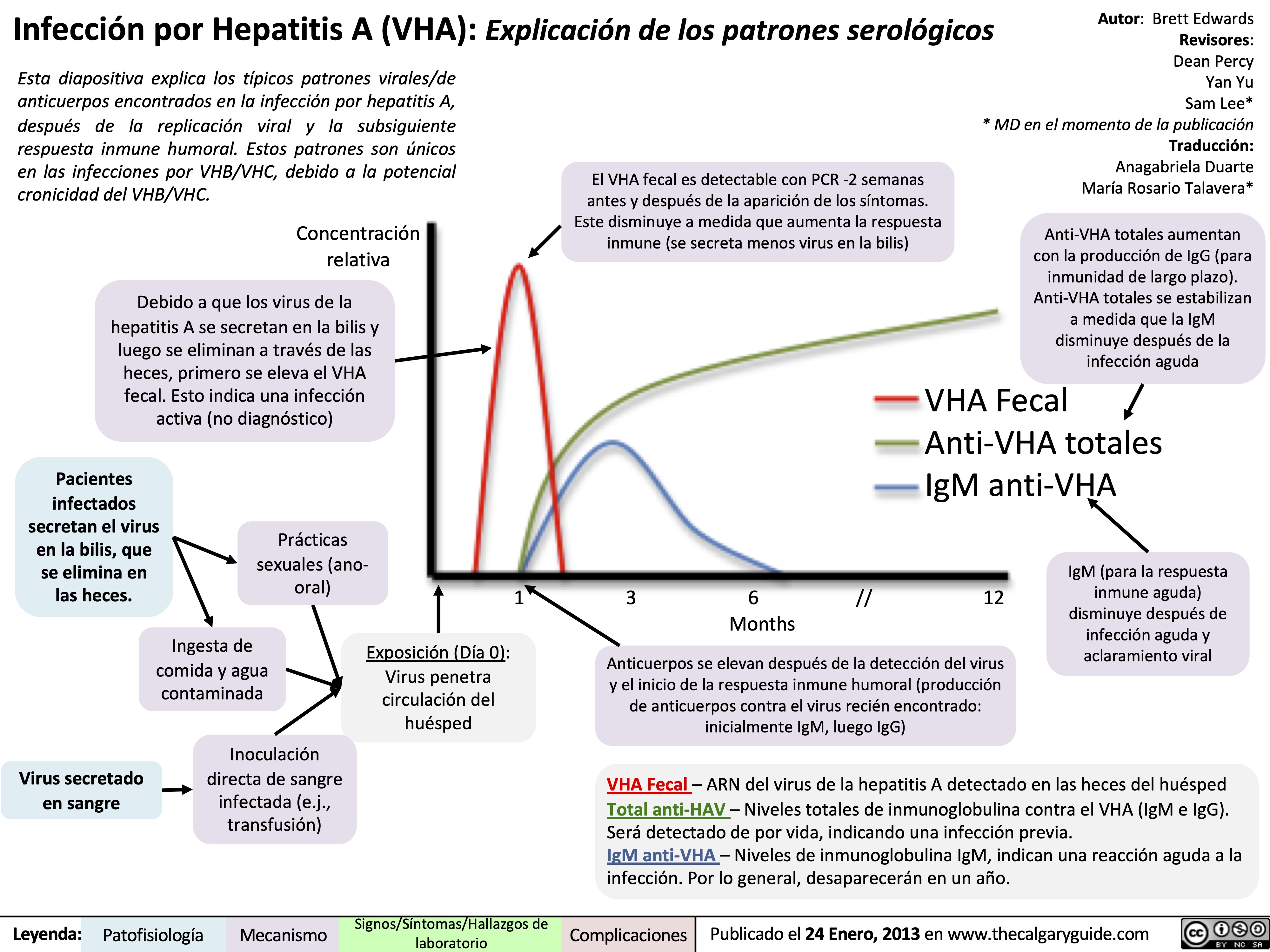
infeccion-por-hepatitis-c-vhc-explicacion-de-los-patrones-serologicos
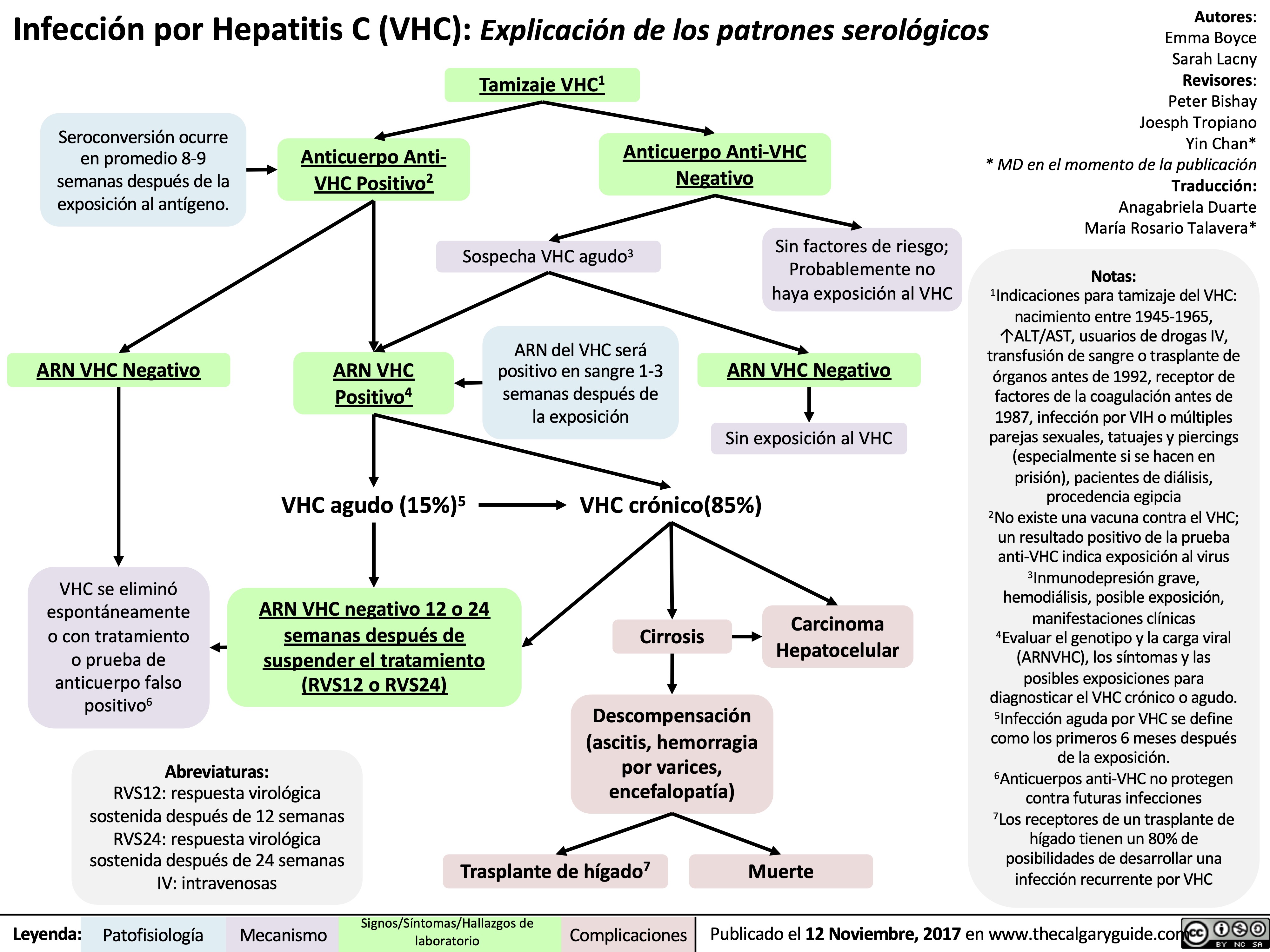
fiebre-reumatica-aguda-patogenesis-y-hallazgos-clinicos
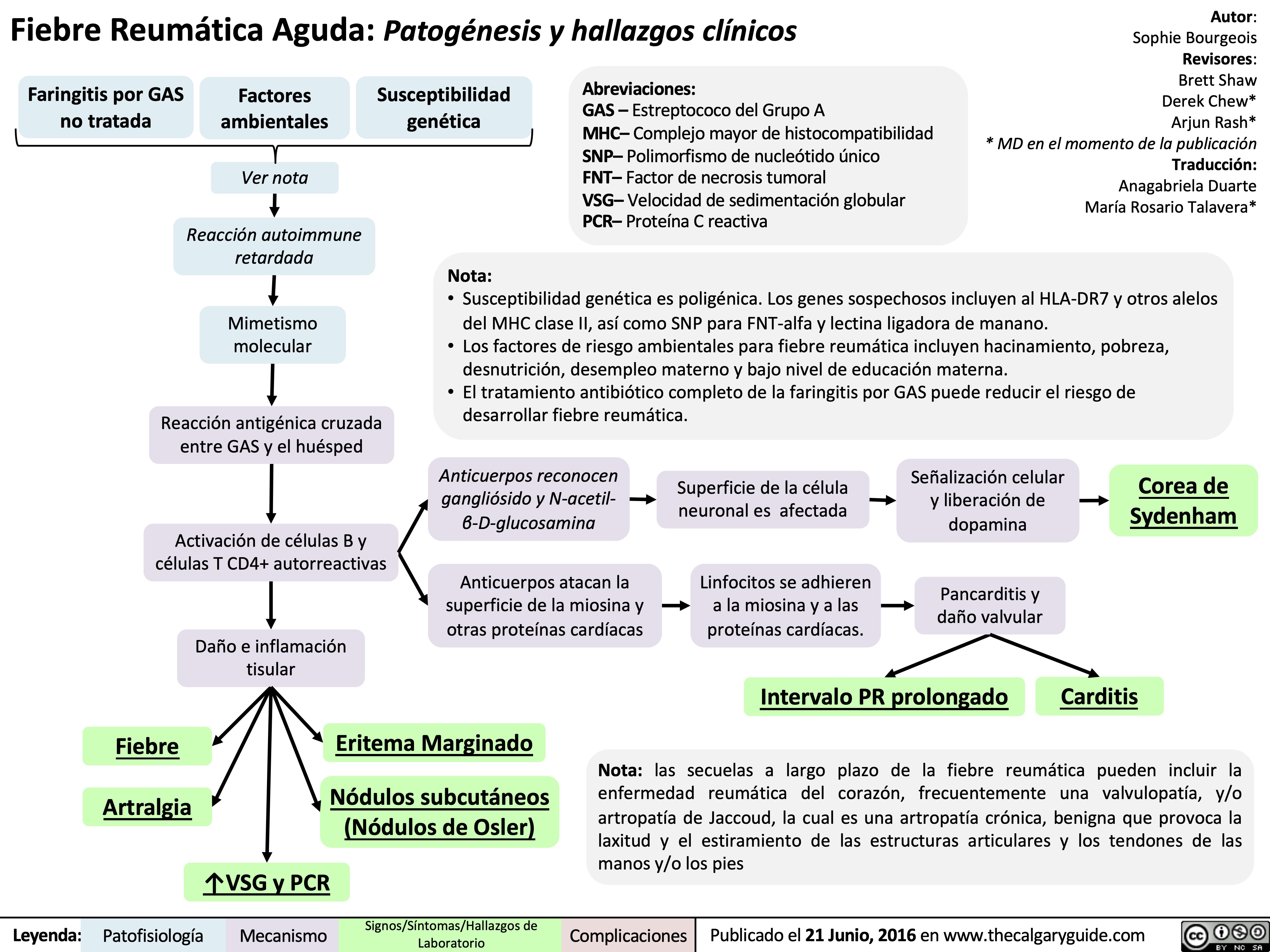
hemorroides-patogenesis-y-hallazgos-clinicos
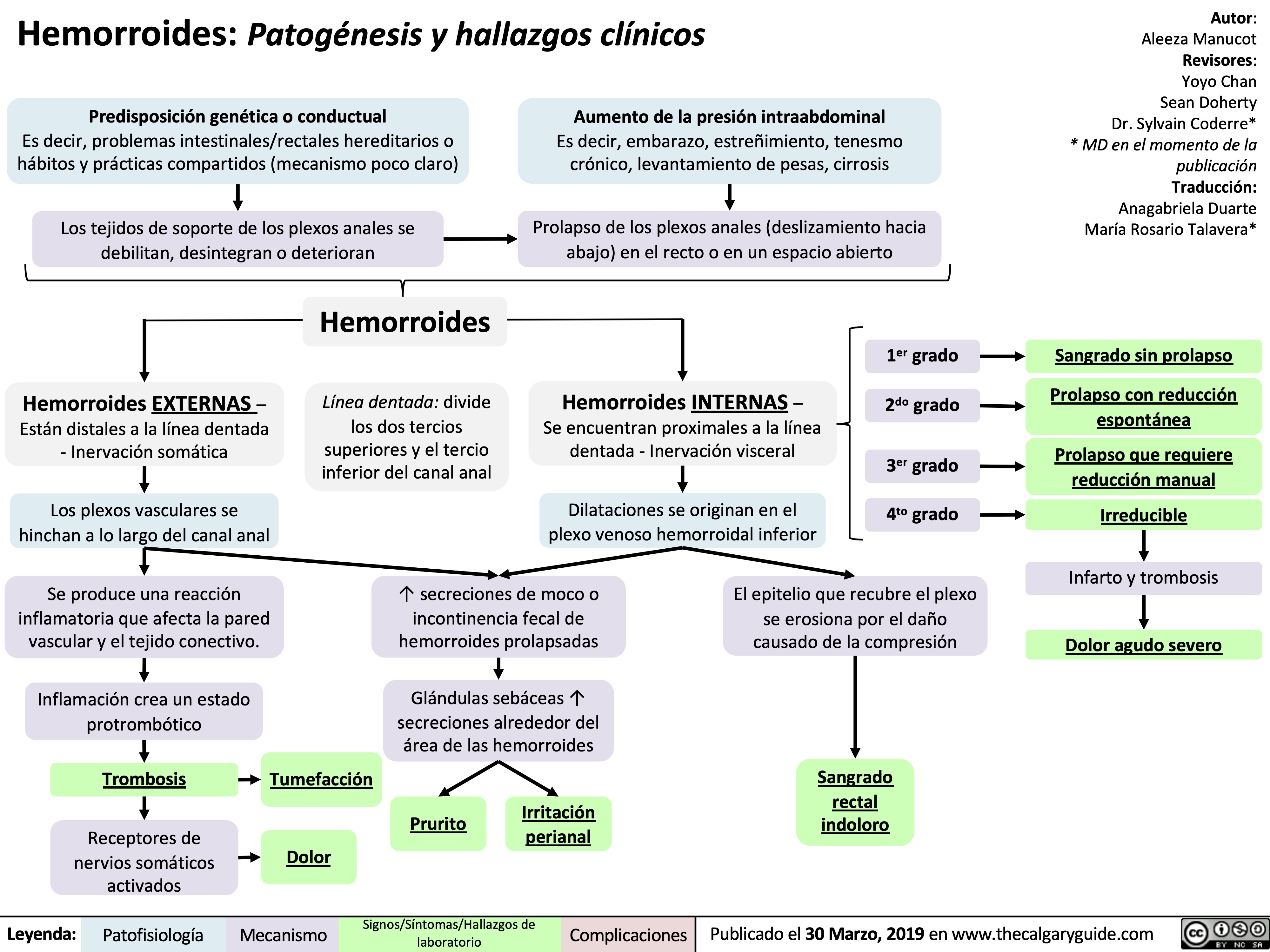
sangrado-gi-bajo-patogenesis-y-hallazgos-clinicos
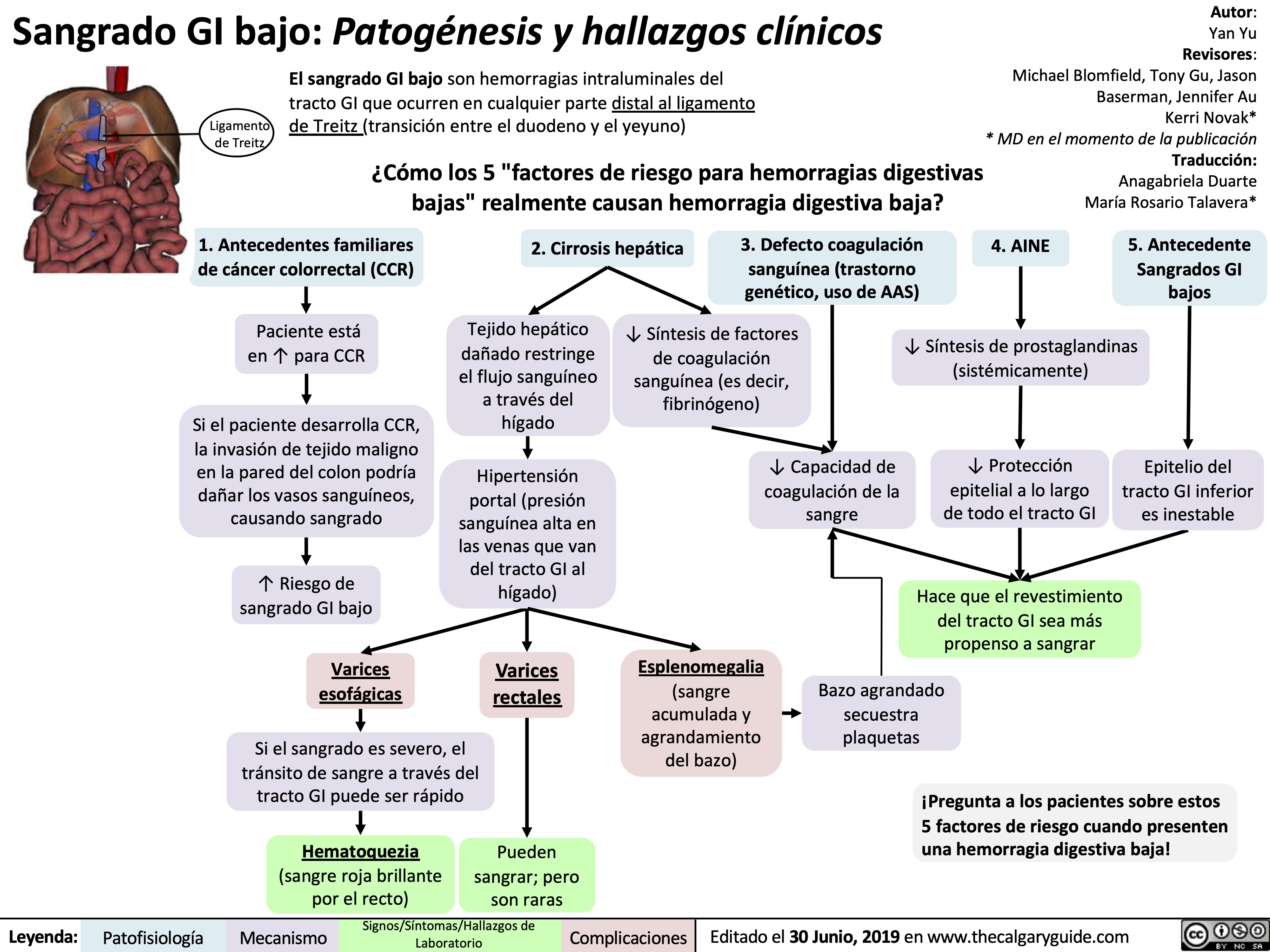
visceras-perforadas-alias-tracto-gi-intestinos
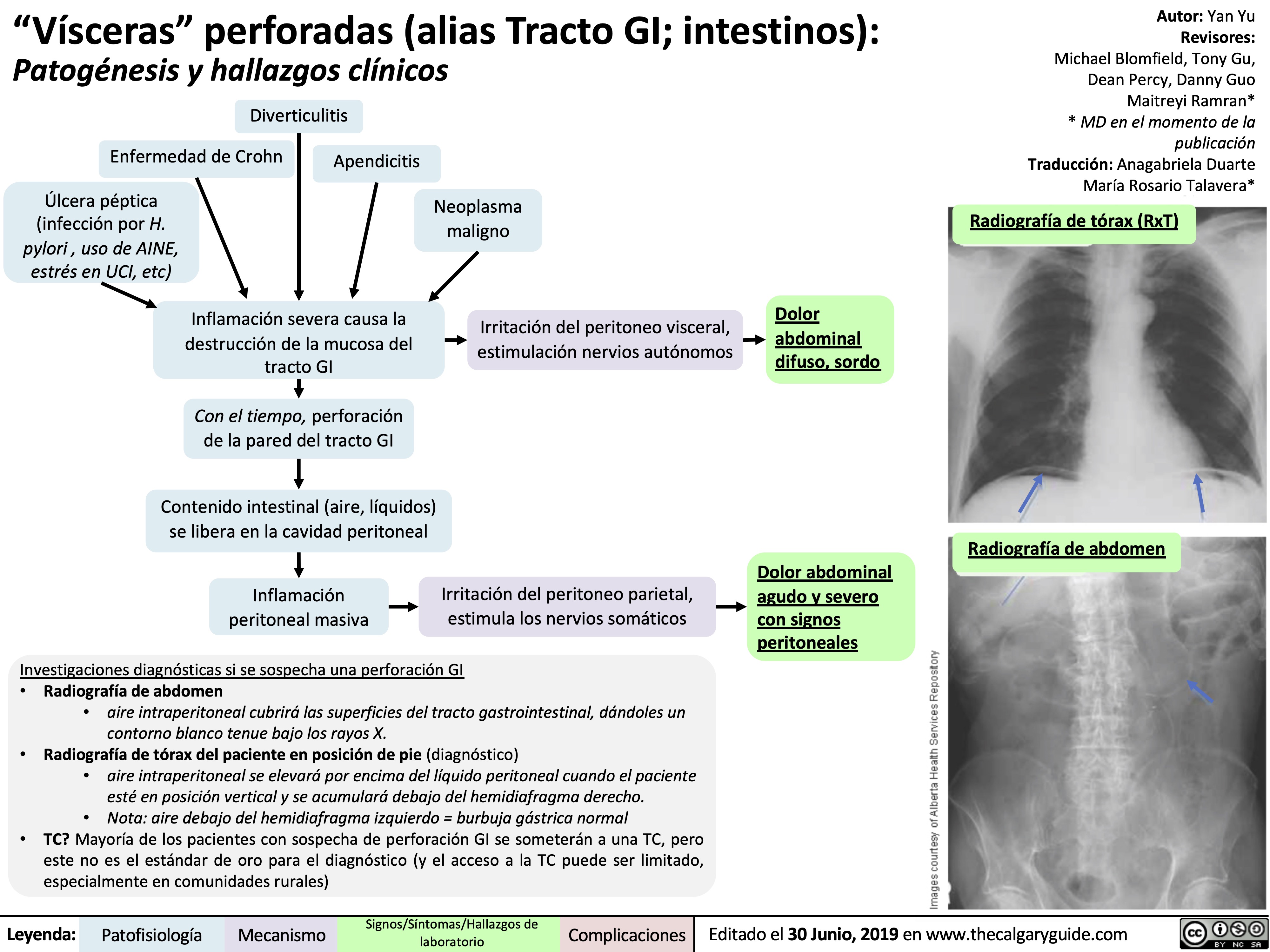
gastro-duodenale-ulkuskrankheit-pathogenese-und-klinische-befunde
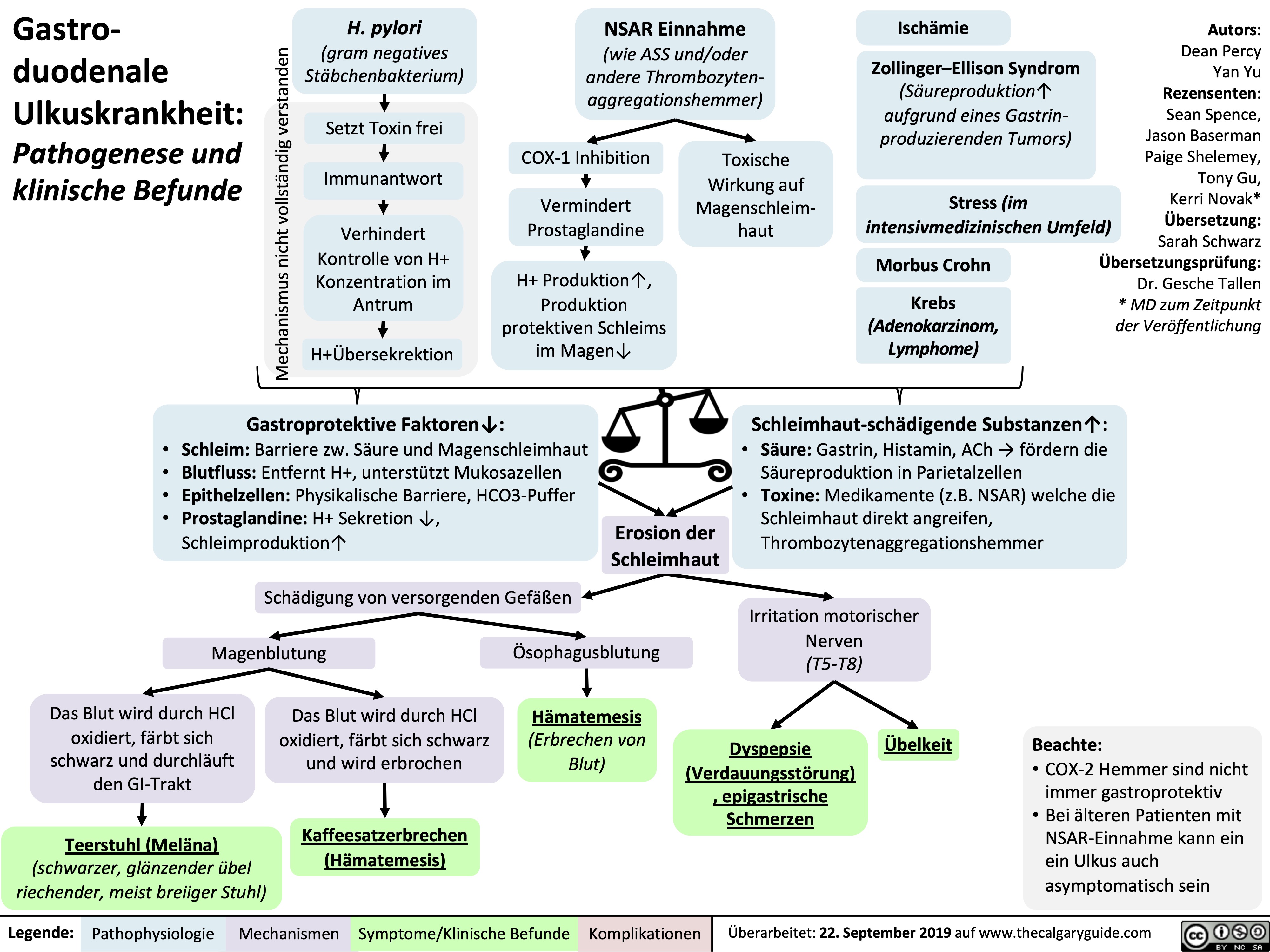
rechtsherzinsuffizienz-pathogenese-und-klinische-befunde
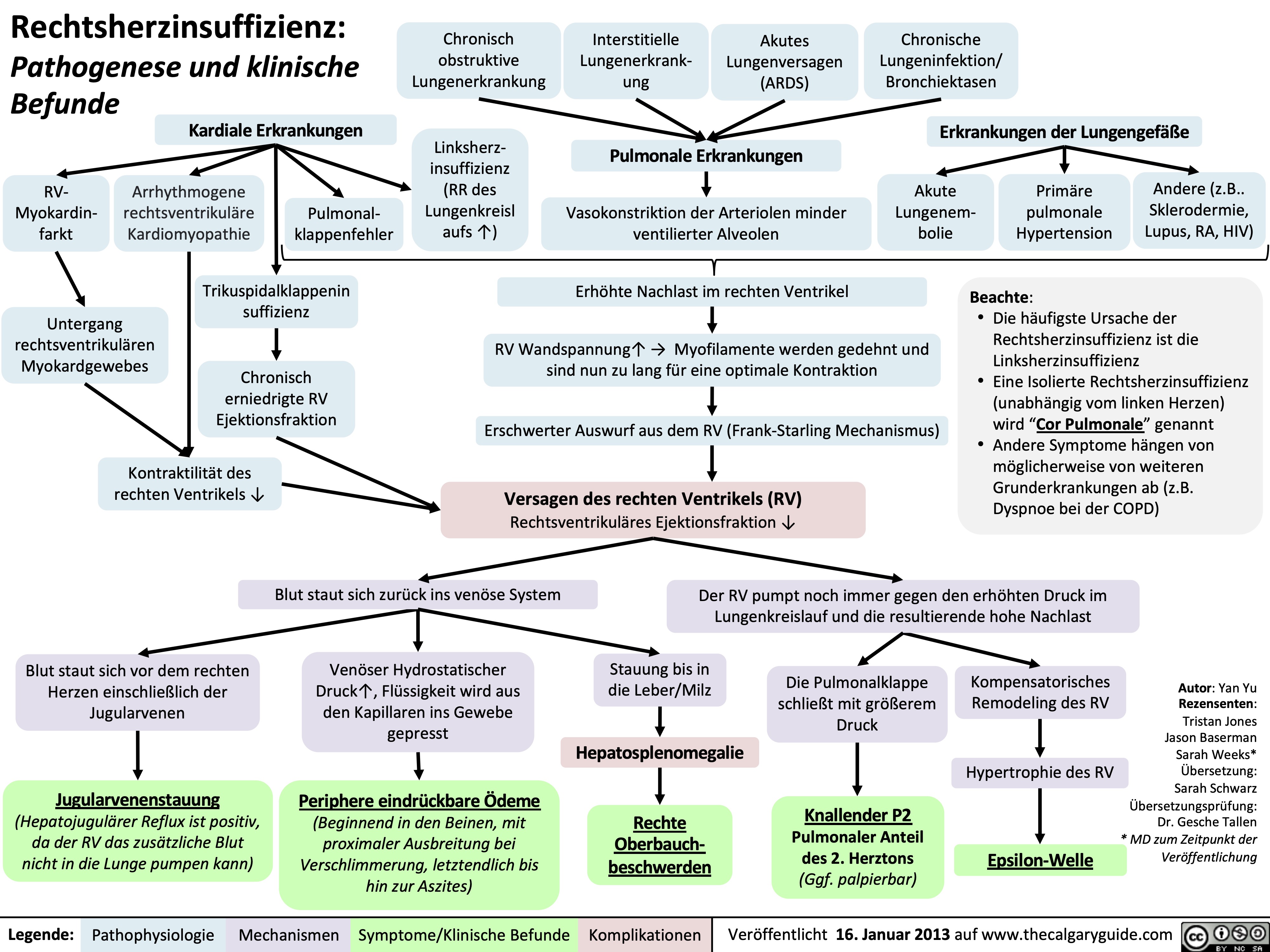
linksherzinsuffizienz-korperliche-untersuchungsbefunde
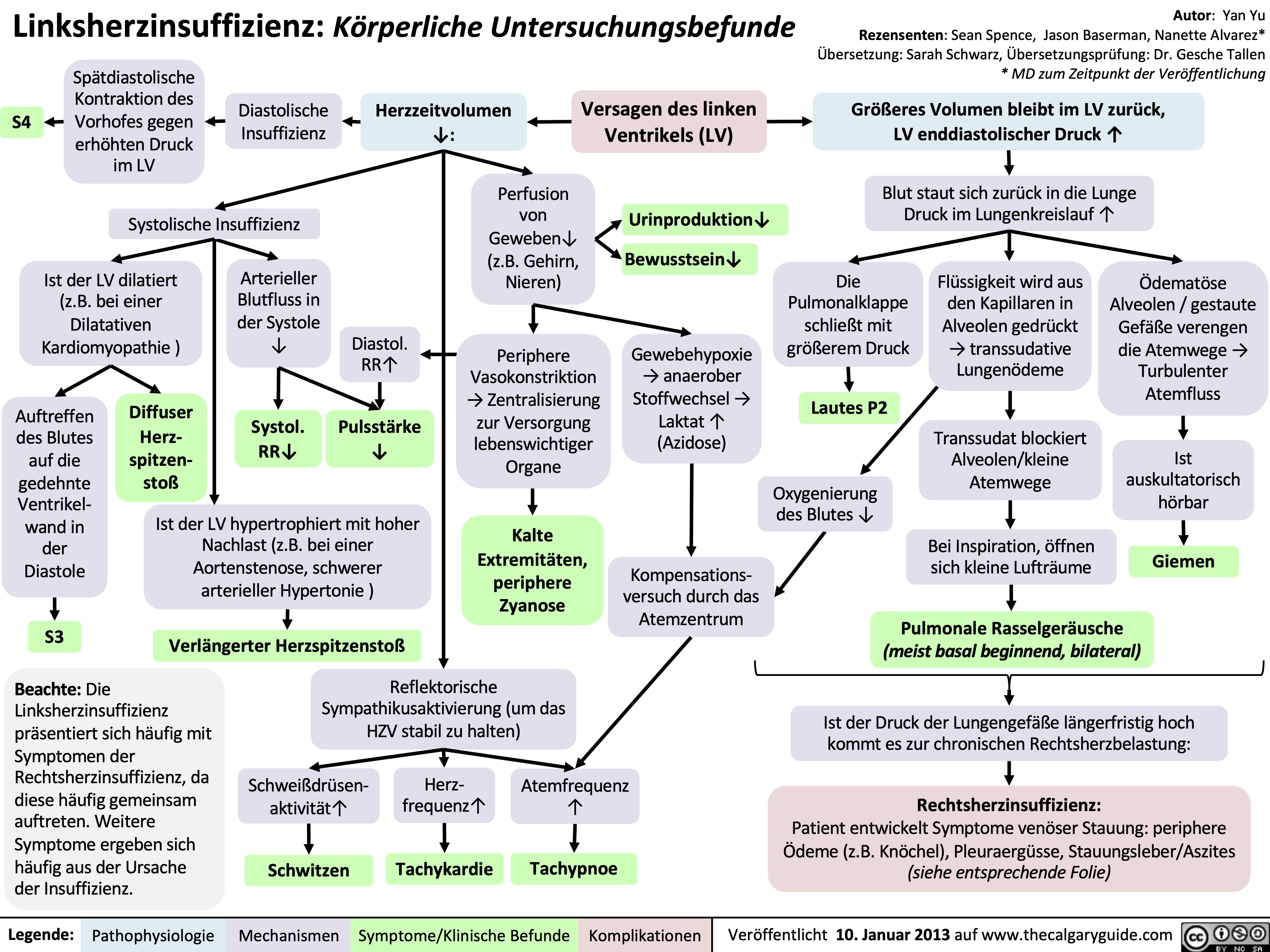
linksherzinsuffizienz-klinische-befunde
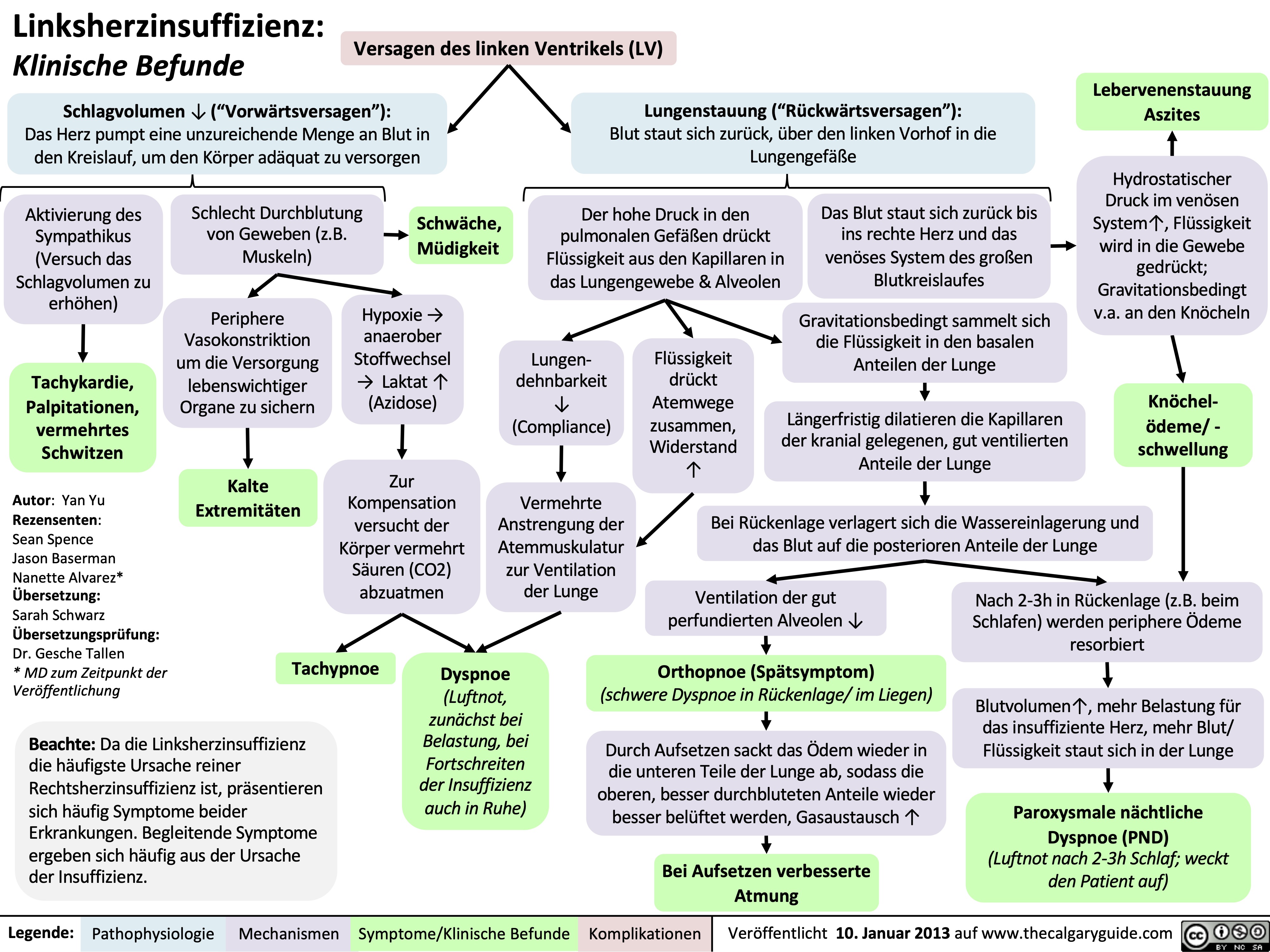
linksherzinsuffizienz-pathogenese
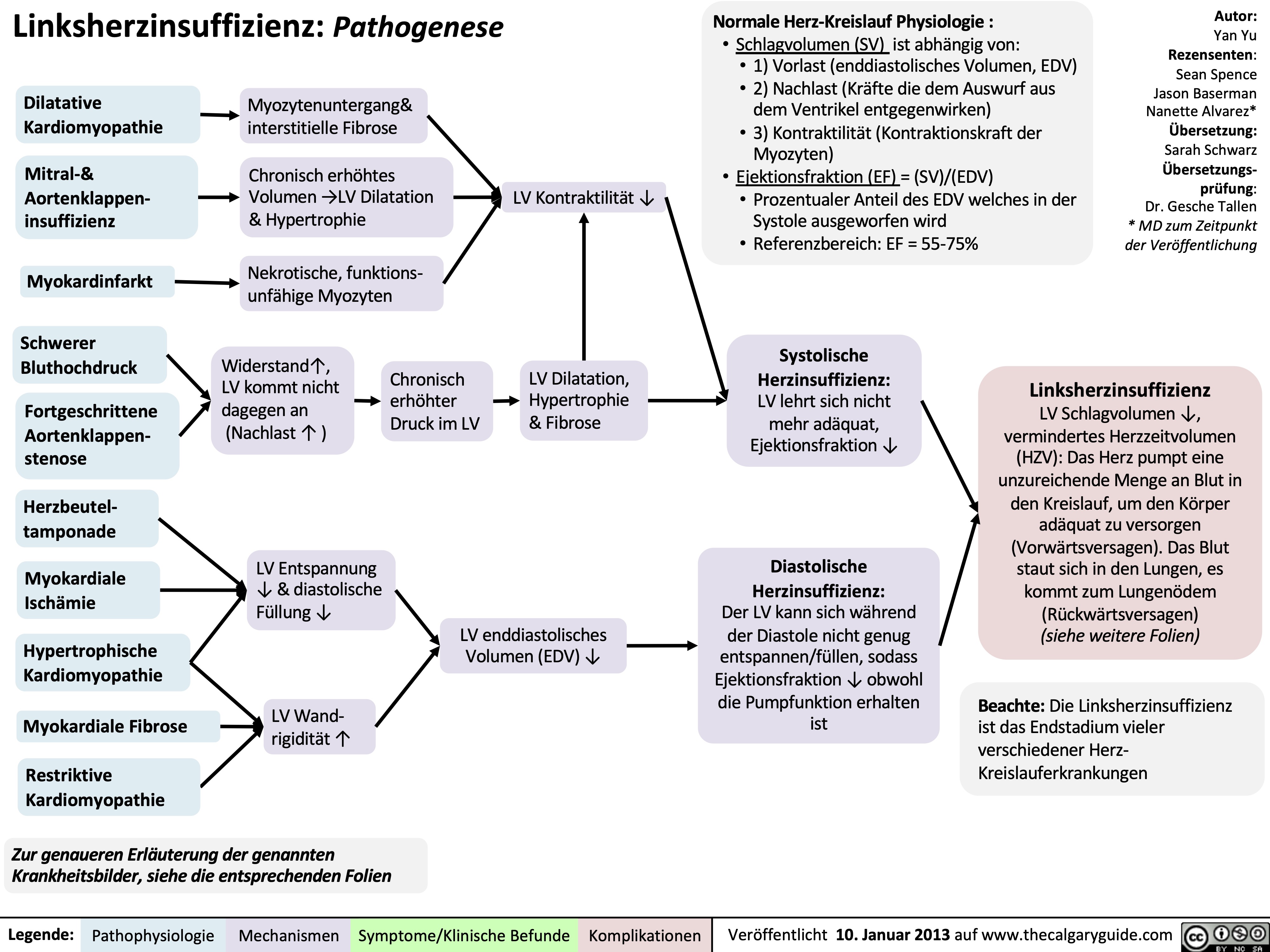
copd-komplikationen
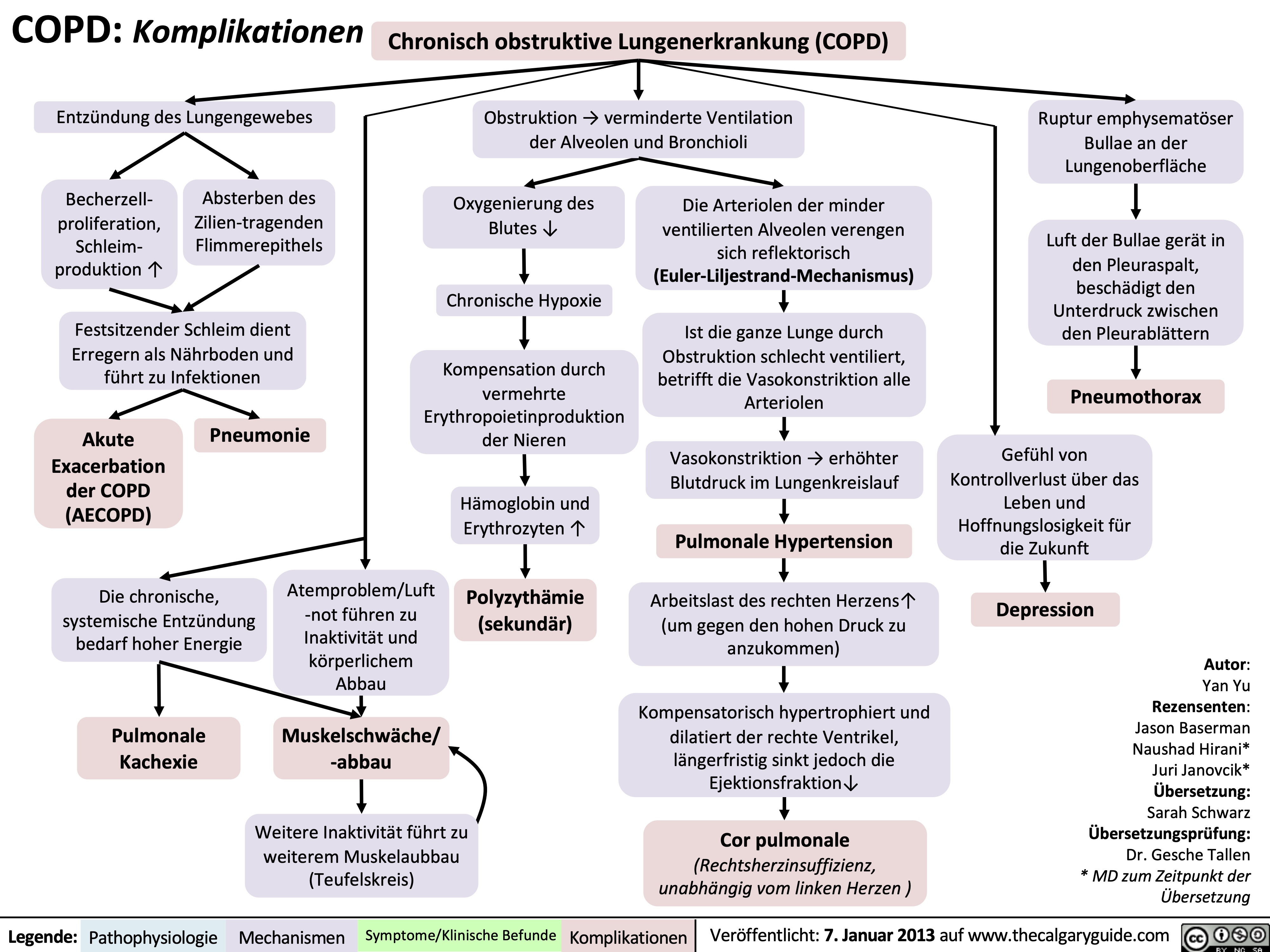
copd-diagnostik
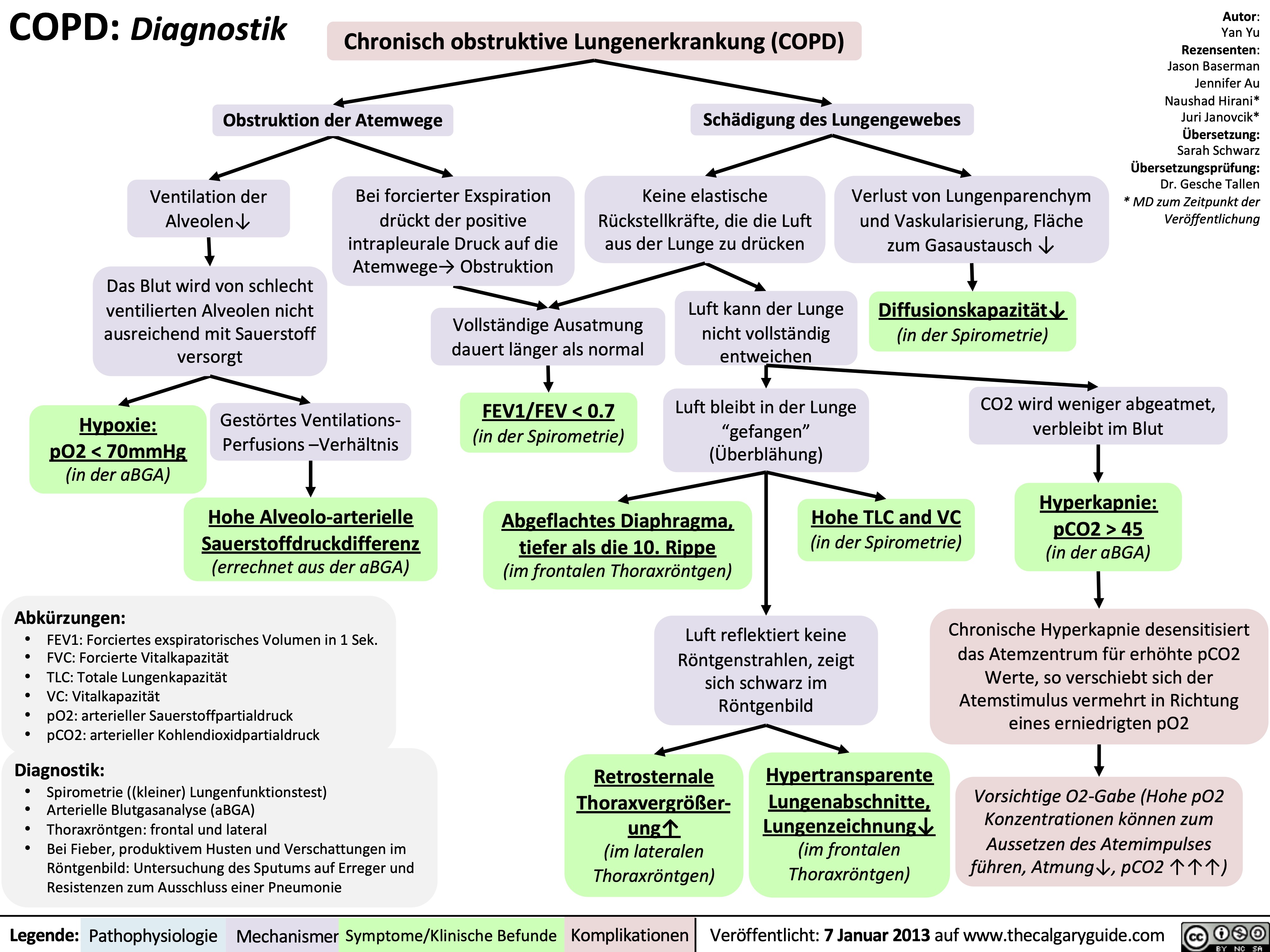
copd-klinische-befunde
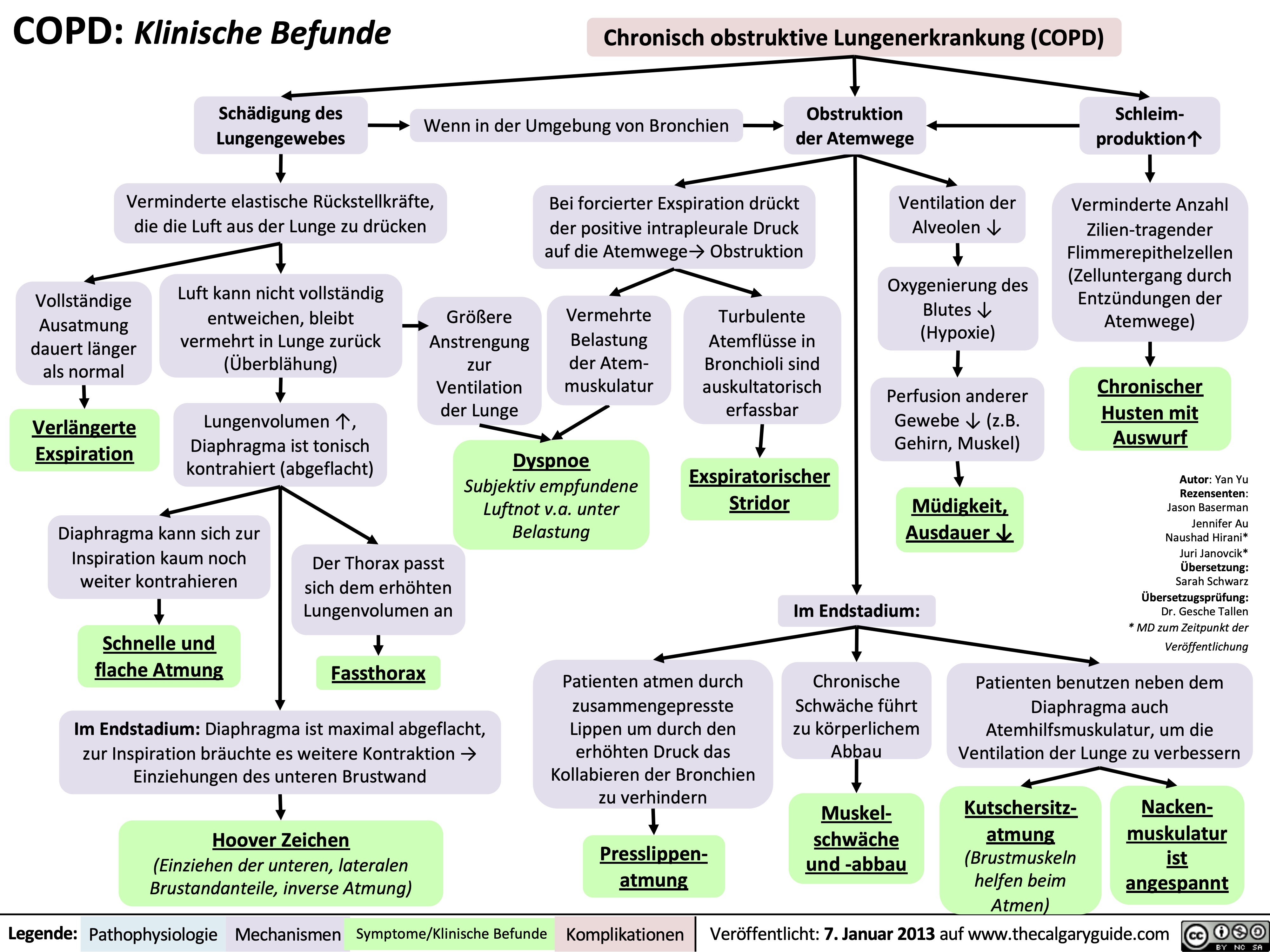
copd-pathogenese
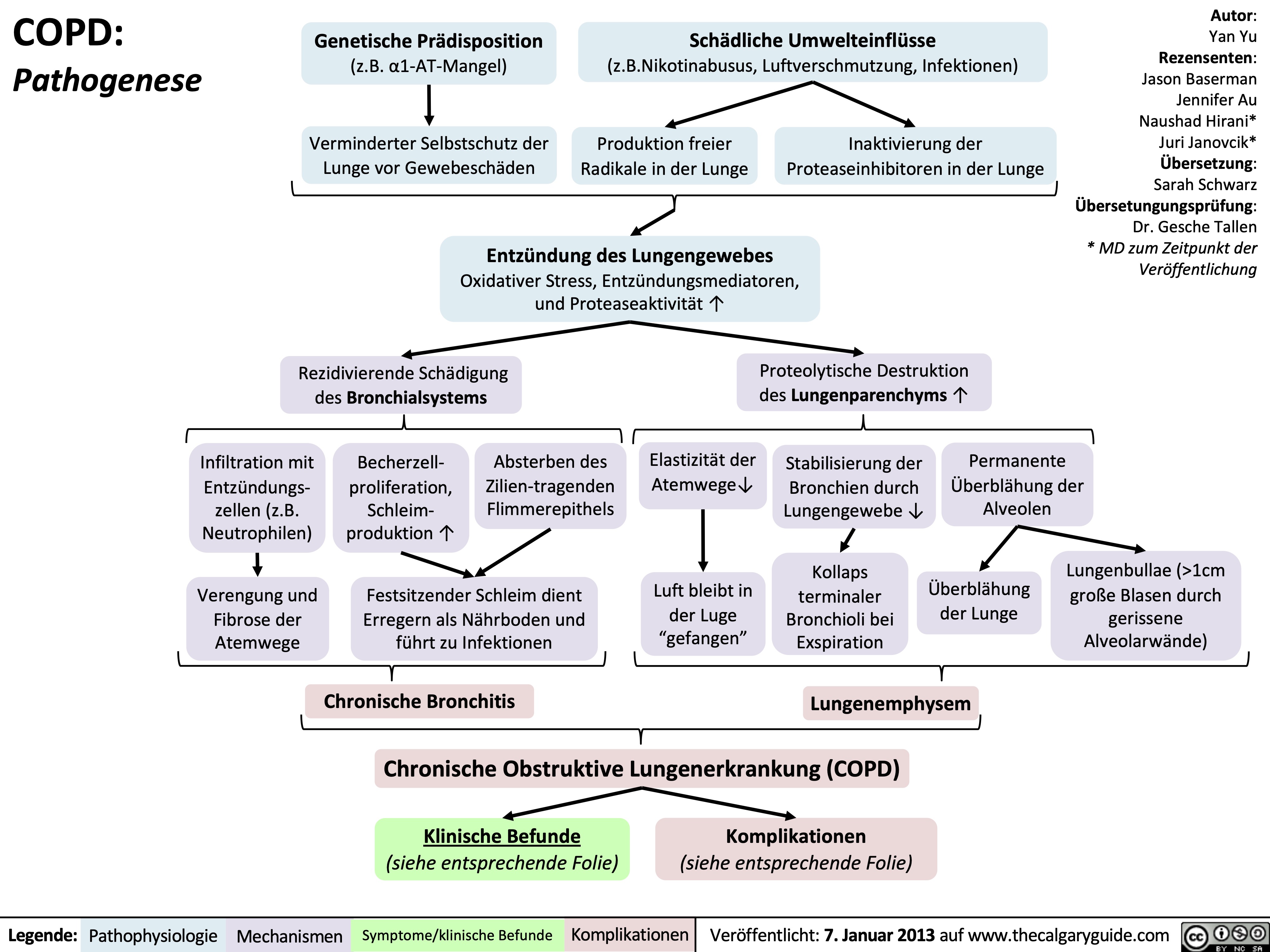
leberzirrhose-pathogenese-und-komplikationen
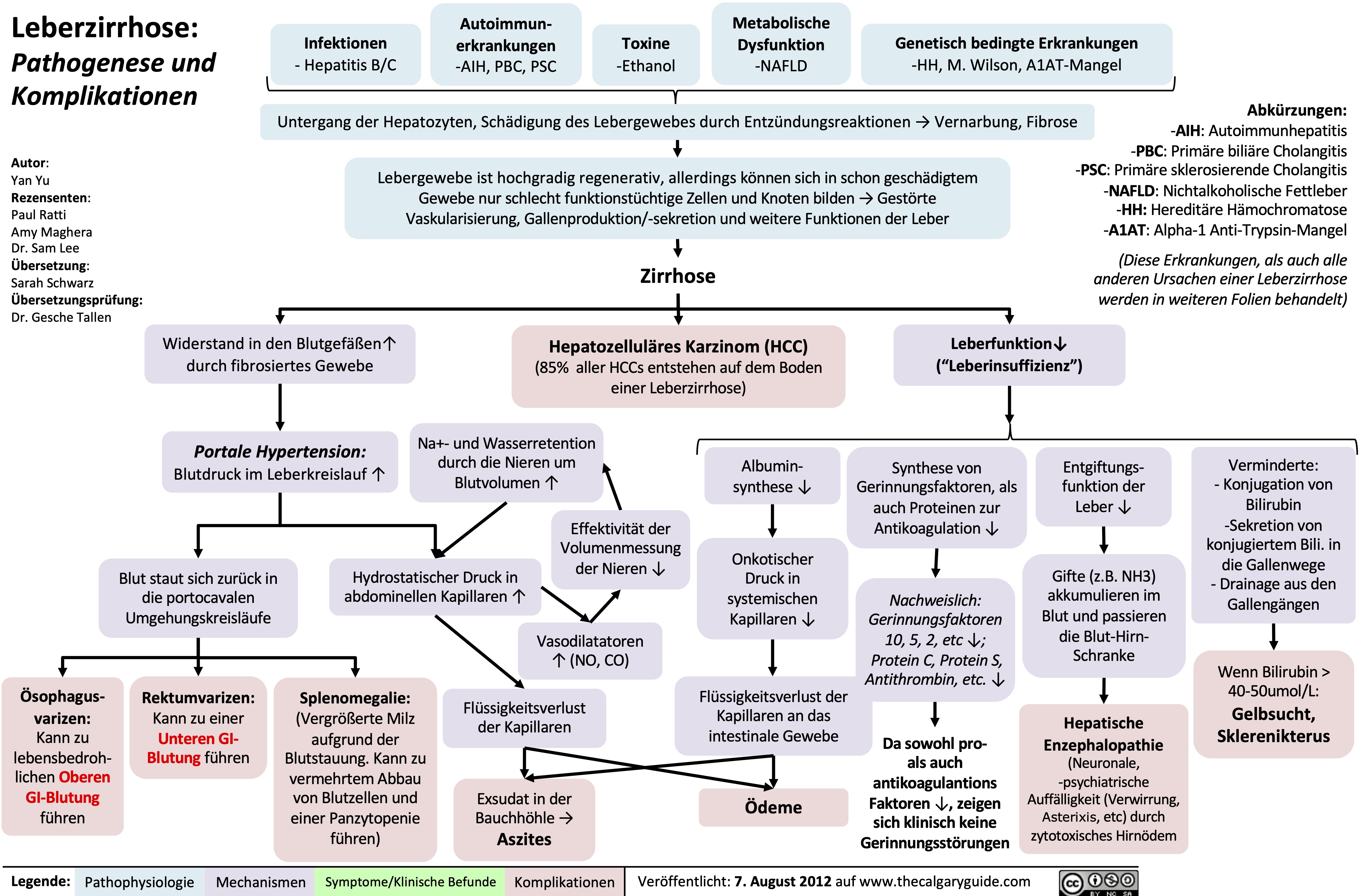
local-anesthetics-lidocaine-bupivacaine-ropivacaine-procaine-cocaine-mechanism-of-action-and-pharmacodynamics
: Mechanism of Action and Pharmacodynamics
Ester Class
Cocaine
Local Anesthetics
Lidocaine
Local infiltration of the drug in tissue
Amide Class
Ropivacaine
Authors: Evan Allarie Yan Yu* Reviewers: Stephen Chrusch Wendy Yao Brooke Fallis Melinda Davis* * MD at time of publication
These traits enable the anesthetic to be highly lipophilicà↑ anesthetic diffusion through nerve sheaths
↑ anesthetic potency
Anesthetics’ effect differ based on neuronal features. For instance: comparing small diameter
unmyelinated nociceptive neuron vs. larger diameter myelinated motor neurons:
Procaine
Bupivacaine
Addition of a vasoconstrictor (e.g. epinephrine) to the anesthetic solution
Local vasoconstriction at site of anesthetic infiltration
Aliphatic side chains and lipophilic linkages
High Molecular Weight
↓ systemic absorption of the anesthetic
↓ side effects Larger amounts
of local anesthetic can be safely administered
↑ anesthesia duration
↓ bleeding at site of infiltration
↑ pH of injected solution (e.g. adding sodium bicarbonate)
↓ pKa of local anestheticà↑ in unprotonated form of local anesthetic
↑ uptake of anesthetic into neurons
Short onset time
When the molecule’s tertiary amine is in its unprotonated form, the anesthetic is lipophilic and is able to diffuse across nerve cell lipid membranes
Anesthetic molecule becomes protonated within the neuron
Protonated form of anesthetic binds to transmembrane sodium channel
Sodium channels become impermeable to sodium ions, preventing sodium movement across the membrane
↓ nociceptive neuronal depolarization ↓ Pain sensation
Small diameter = fewer sodium channels for anesthetic to inhibit
Unmyelinated = anesthetic free
to diffuse anywhere along neuron
Differential nerve blockade
Pain sensation is blocked much more quickly than motor function
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published December 4, 2021 on www.thecalgaryguide.com")
local-anesthetics-lidocaine-bupivacaine-ropivacaine-procaine-cocaine-side-effects-and-complications
: Side Effects and Complications
Cocaine
Procaine
Ropivacaine
Bupivacaine
Amide Class
Lidocaine
Authors: Evan Allarie Yan Yu* Reviewers: Stephen Chrusch Wendy Yao Brooke Fallis Melinda Davis* * MD at time of publication
Ester Class
Both classes share a similar molecular structure, containing a lipophilic (hydrophobic) aromatic ring and a hydrophilic tertiary amine, making them amphipathic molecules (possessing both hydrophobic and hydrophilic components)
Local infiltration of the drug into tissue
Ester local anesthetics have a carboxylic ester bond between the aromatic ring and tertiary amine
Metabolized by plasma cholinesterases If the patient is afflicted with atypical
pseudocholinesterase or pseudocholinesterase deficiency, this condition will impair breakdown of ester local anesthetics
Amide local anesthetics have an amide bond between the aromatic ring and tertiary amine
Metabolized in liver by hepatic enzymes
If the patient is afflicted with liver disease, this condition will impair breakdown of amide local anesthetics
Local anesthetic can diffuse into sympathetic nervesàblocks sodium channels on neuronal membranes, inhibiting sympathetic nerve function
↓ vasoconstrictive signals to vascular smooth muscle
Vasodilation of systemic arteries
Hypotension
Some local anesthetic can enter systemic circulation and physically reach the heart
Anesthetic molecule will block ion channels on cardiac myocytes
If enough anesthetic reaches cardiac myocytes, their action will ↓ myocardial electrical signalling and thus ↓ cardiac rhythm and contractility
Prolonged clearance and ↑ duration of anesthetic blockade
Cardiac Arrest
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published December 4, 2021 on www.thecalgaryguide.com")
Psuedocholinesterase Deficiency

Break down or inhibit the pseudocholinesterase enzymeà↓ pseudo- cholinesterase activity
Note:
Pseudocholinesterase has many synonymous
names including butyrylcholinesterase, BChE, BuChE, plasma esterase, plasma cholinesterase, and serum cholinesterase
Malignancy Abnormal gene
expressionà↓ protein synthesis
Acquired
Liver disease
↓ liver’s ability to synthesize proteins
Malnutrition
↓ molecular precursors for protein production
Renal Disease
Mechanism unclear
Fluid Overload
Hemodilution of circulating proteins
Hereditary
BChE (Butyrylcholinesterase) gene mutation
↓ production or production of non-functional pseudo- cholinesterase by the liver
↓ synthesis of pseudocholinesterase by the liver
Pseudocholinesterase Deficiency: reduced levels of functional pseudocholinesterase in plasma and tissues
Impaired ability to hydrolyze ester linkages of substrates like neuromuscular blocking agents (e.g. succinylcholine, mivacurium, diamorphine, acetylsalicylic acid, methylprednisolone, cocaine, heroin )
Prolonged
binding of neuromuscular blocking agent to nicotinic cholinergic receptors in neuromuscular junctions
↑ patient’s susceptibility to side effects from drugs with ester linkages
Mivacurium administered to produce muscle paralysis
Succinylcholine administered
to produce muscle paralysis
Competitively binds to acetylcholine nicotinic receptor
Irreversibly binds to acetylcholine nicotinic receptor
Active site of post-synaptic acetylcholine receptor is blocked
Continuous depolarization
of skeletal muscle
Acetylcholine cannot act on receptor = skeletal muscle can’t depolarize
Skeletal muscle unable to repolarize
Skeletal muscle paralysis
Acetylcholine released cannot trigger an action potential
↑ duration of muscle fiber paralysis
Since respiratory muscles are affectedà Apnea requiring sedation and respiratory assistance for up to several hours
Extended paralysis and/or anesthesia
Authors: Evan Allarie Yan Yu* Reviewers: Stephen Chrusch Brooke Fallis Melinda Davis* * MD at time of publication
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published December 4, 2021 on www.thecalgaryguide.com")
chronic-hypertension-complications
 ≥ 135/85 (on ambulatory or home blood pressure measurement) in patients without diabetes, or BP ≥ 130/80 in patients with diabetes
Authors: Samin Dolatabadi, Yan Yu* Reviewers: Meena Assad, Jessica Krahn Juliya Hemmett* * MD at time of publication
↑ Afterloadà ↑ Resistance to left ventricle ejection
To overcome resistance and preserve cardiac
output, the myocardium undergoes structural and functional changes
Left ventricular hypertrophy and fibrosis
Stiff ventricle
↓ Contractility of the left ventricle
Impaired forward flow of blood from heart
Chronic stress on the endothelium of systemic blood vessels
↑ Blood pressure in retinal circulation
Hypertensive Retinopathy (See
slide on Chronic Hypertensive Retinopathy: Pathogenesis and Clinic Findings)
Smooth muscle of kidney’s
afferent arterioles constricts to prevent transmission of ↑ blood pressure to glomerulus
Overtime, smooth muscles of afferent arterioles hypertrophy from prolonged vasoconstriction
Chronic stress and trauma on endothelial and smooth muscle cells of kidney
Injury leads to excretion of cytokines and extracellular matrix such as fibrin and collagen into subendothelial layer
Endothelial dysfunction (See slide on Atherosclerosis: Pathogenesis)
Atherosclerosis
Loss of normal arterial architecture in the brain due to stress of ↑ blood pressure
Weakening of cerebral arteries
Formation and rupture of microaneurysms
Intracerebral Hemorrhage
Accumulation of plaques in
the walls of cerebral arteries
↓ Cerebral blood flow
Ischemic Stroke
Accumulation of plaques in
the walls of coronary arteries
↓ Myocardial blood flow
Oxygen supply- demand mismatch
Coronary Artery Disease
Thickening of arteriolar wall and narrowing of afferent arterioles
↓ Glomerular blood flow
Glomerular and tubular ischemia
Glomerular sclerosis and tubular atrophy
Blood backs up into lungs
↓ perfusion of blood throughout the bodyà inability of the heart to meet metabolic demands
Congestive Heart Failure
Hypertensive Nephrosclerosis
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published December 4, 2021 on www.thecalgaryguide.com")
Primary Aldosteronism Pathogenesis
à↑ aldosterone
Overproduction of aldosterone, independent from renin-angiotensin- aldosterone system (RAAS)
Ectopic aldosterone secreting tumor
adrenocortical tissue outside of the adrenal glands produce aldosterone
Familial hyperaldosteronism (FH) Type I
Rare mutation causing ACTH- sensitive aldosterone production in the zona fasciculata
Aldosterone binds its receptor on the principal cells located in the collecting duct of the kidney
↑ Na+ and K+ channel insertion on luminal surface of the principal cell and ↑ Na/K ATPase activity on basolateral surface
Na+ follows the concentration gradient and moves into the principal cell cytoplasmàK+ moves into the collecting duct to maintain electroneutrality
Aldosterone binds its receptor on the alpha intercalated cells located in the late distal tubule and collecting duct of the kidney
↑ H+ ATPase and Na/H+ ATPase activity on the luminal surface of alpha intercalated cellsà↑ H+ excretion
H+ loss permits HCO3- to move down the electrochemical gradient across the luminal surfaceà↑ HCO3- resorption into peritubular capillaries
Metabolic alkalosis
K+ efflux (or hypokalemia of any etiology) is counterbalanced by influx of H+ into tubular cells to maintain electroneutrality
↓ pH in tubular cells activates glutaminase (a pH dependent enzyme) generating glutamate from glutamine
Glutamate within renal tubules dissociates into NH4+, HCO3- Intracellular acidosis within tubular cells
Disrupted cellular signaling within the collecting duct results in reduced APQ2 translocation to luminal surface
Authors: Kyle Moxham Reviewers: Emily Wildman Austin Laing Yan Yu* Matthew Harding* * MD at time of publication
Hypertension
↑ Na+ reabsorption
H2O follows active reabsorption
of solute into circulationà ↑ effective arterial blood volume (EABV)
Chronic EABV expansion resets hypothalamic osmotic sensitive ADH releaseàdecreased ADH production
↑ K+ excretion
Hypokalemia (see our Hypokalemia: Clinical Findings Slide)
↓ Na+ delivery to macula densa in the distal convoluted tubule
↓Endogenous renin secretion by juxtaglomerular apparatus while aldosterone continues to be independently produced
Polydipsia & Polyuria
Mild hypernatremia
↓ Renin: Aldosterone ratio
Nephrogenic diabetes insipidus: ↓ urinary concentrating ability
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published December 4, 2021 on www.thecalgaryguide.com")
Asthma clinical findings

Author: Yan Yu Reviewers: Jason Baserman Jennifer Au Yonoglin Mai (麦泳琳) Naushad Hirani* * MD at time of publication
Variable, sporadic airway obstruction in response to triggers
Associated allergic eosinophil response
Eosinophils infiltrate: Skin
If severe:
↓ ventilation of alveoli
↓ oxygenation of blood (hypoxemia)
During expiration, positive pleural pressure squeezes on airwaysà↑↑ airway obstruction
Heart rate to improve
perfusion of tissue
Tachycardia
Respiratory centers rate of breathing to
compensate
Tachypnea
Gas is trapped within alveolià hyperinflates lungs
Ventilating larger lungs needs more effort
Patients need to voluntarily contract
their expiratory muscles faster and more forcefully to effectively expire
Narrower airways àturbulent
airflow, heard on auscultation
Expiratory Wheeze (high-pitched expiratory sound)
Nose
Rhinitis/ sinusitis
Runny nose, sneezing, etc
Atopic dermatitis
Skin rash, hives
Eyes
Conjunctivitis
Red itchy eyes, visual blurring
Episodic
dyspnea
(shortness of breath)
Chest tightness
During severe attacks:
Note: Asthma attacks often have two phases:
• An immediate attack (within 0-2 hours of the trigger, due to acute release of histamine from mast cells)
• A delayed attack (due to eosinophil infiltration of airways, presents within 3-4 hours after exposure to the trigger, peaks within 6-8 hours, and resolves within 24 hours).
Keep the possibility of a delayed attack in mind when treating patients in Emergency!
Note: Symptoms often worse at night or early in the morning.
Note: Asthma should be suspected in children experiencing dyspnea with multiple episodes of Upper Respiratory Tract Infections or Croup.
Patient compensates by activating accessory respiratory muscles to ↑ thoracic volume
Visible contraction of
neck muscles (Scalene, sternocleidomastoids)
↑↑↑ airway obstruction on
expiration, lungs take more time to empty
Prolonged expiratory phase of breathing
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published Dec 17, 2012 and updated Dec 4, 2021 on www.thecalgaryguide.com")
Chronic Cough Pathogenesis_2021

Infection
IP; likely chronic ↑ cough receptors commonly pertussis- related or post-viral
Asthma
↑ EDN
↑ MBP levels in RT
Neoplasm
Bronchiectasis
COPD
GERD
Allergic Rhinitis
↑ sputum production
and accumulation
Damage from chronic inflammation causes poor mucus clearance
Inhalants trigger ↑ cytokines ↑ mucus
Aspiration of refluxed microdroplets
Irritation by post- nasal drip
↑ inflammatory mediators in RT
Mechanical obstructions (i.e. mediastinal masses, neoplasms)
Foreign body Presence irritation of irritants
Stimulation of sensory nerve receptors expressed on nerve endings penetrating the epithelia of the upper RT
Abbreviations:
• RT: respiratory tract
• IP: indefinite
pathophysiology
• GERD: Gastro-esophageal
reflux disease
• EDN: Eosinophil-derived
neurotoxin
• MBP: Major basic protein
• COPD: Chronic
Obstructive Pulmonary Disease
Complications: Syncope, insomnia, hemoptysis, rib fractures
Slowly adapting receptors
Respond to mechanical forces during breathing
Stimulation of the vagus nerve
Activation of nucleus tractus solitarius in central respiratory generator (brainstem)
Generation of efferent cough signal
Chronic cough: Cough of over 8 weeks duration with no dyspnea or fever
C-fibre receptors
Respond to chemical stimuli (bradykinin, capsaicin, acid/alkaline solutions, mannitol, hypertonic saline, and other inhaled irritants)
Authors: Arsalan Ahmad Lance Bartel Reviewers: Midas (Kening) Kang Usama Malik Ciara Hanly Yonglin Mai (麦泳琳) Yan Yu*, Eric Leung* * MD at time of publication
Rapidly adapting receptors
Responds to mechanical stimuli, such as physical obstruction of the airways
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published Feb 06, 2018, updated Dec 4, 2021 on www.thecalgaryguide.com")
慢性咳嗽-发病机制

哮喘-发病机制

哮喘-临床表现

哮喘-检查结果

过敏反应-发病机制

肺栓塞并发症

慢性阻塞性肺疾病简称copd定义

copd-诱发因素和体征-急性加重的症状

咳嗽生理机制

应对covid-19公共卫生作用

肺栓塞-发病机制-及实验室检查

Mycobacterium-Tuberculosis

Pro-inflammatory cytokines further activate and recruit neutrophils, T- lymphocytes and monocytes to the site of respiratory infection
Intracellular lysosomal enzymes are released into the vesicle containing the bacilli
Lysosomal enzymes digest and destroy the bacilli M. tuberculosis infection is cleared from the body
Sputum culture and smear microscopy –ve for M. tuberculosis
Infected macrophages travel to thoracic lymph nodes via the lymphatic systemàimmune cell proliferation within thoracic nodes
Bacilli activate macrophage cell death programsàbacilli are
released and enter the lymphatic and circulatory system
Bacilli spread to other regions of the body
Extrapulmonary tuberculosis
Abbreviations TNF-a: tumor necrosis factor alpha
IL-1B: interleukin 1B
IL-6, IL-8: interleukin 6 & 8
MHC: Major histocompatibility complex IFN-y: Interferon gamma
Hilar + paratracheal lymphadenopathy
Recruited macrophages phagocytose extracellular bacilli and become infected
Macrophages form a granuloma to isolate and contain the bacilli to that area of the lung
Recruited dendritic cells phagocytose M. tuberculosis and present antigens to T-cells via MHC class II receptors
CD4+ T-cells are sensitized to M. tuberculosis antigens and release IFN-y
IFN-y activates infected macrophages within granuloma to more effectively destroy intracellular bacilli via lysosomal enzymes
Sputum culture and smear microscopy +ve for M. tuberculosis
Ghon focus visible on Chest X-Ray
+ve tuberculin skin test and +ve IFN-y Release Assay
Resilient bacilli within granuloma survive lysosomal enzymes and become dormant
Latent M. tuberculosis; +/- reactivation due to immune suppression (ie: TNF-a inhibitors)
T-cell response is delayed in an immunocompromised hostà prolonging infection
Center of the granuloma necroses and liquifies allowing the bacilli to escape back into the airways
Bacilli re-enter host’s airway, allowing droplet transmission to other hosts
Accumulation of inflammatory mediators and necrotic tissue in lung, irritating airways
Chronic cough
Active M. tuberculosis Multifactorial
response, most with unclear mechanisms
Fevers, night sweats, weight loss
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published December 6, 2021 on www.thecalgaryguide.com")
brulures-non-accidentelles-chez-les-enfants-resultats-de-lexamen-physique

psychodynamic-psychotherapy-principles-and-reasoning

Projection: Qualities felt about one person are directed towards another
Denial: Refusing to admit unpleasant emotion/experience
Repression: Blocking a threatening memory from entering consciousness
Maladaptive thoughts include worthlessness, hopelessness or grandiosity
Maladaptive emotions include explosive anger or extreme self-loathing
Maladaptive behaviours include passive aggressiveness or withdrawal from others
Recognition of maladaptation to external stressors motivates patient to engage in therapeutic process
Patient and therapist attempt co-construction of connections between maladaptive behaviours and past experiences
Initiation of therapy challenges patient’s unconscious psychic equilibrium
Transference
Patient’s transfers thoughts on therapist rooted in emotions from previously important individuals
Counter-transference
Therapist’s thoughts and feelings about the patient hinders co-constructive therapeutic process
Resistance
Interferes with therapy due to subjective sense of shame and judgement
Patient and Therapist work through therapeutic barriers
Supportive interventions
include advice and praise, psychoeducation, and empathic validation
Expressive interventions
include clarification, confrontation, and interpretation
Patient accepts therapist’s connection
Patient gains insight into how they feel and think and can then make better choices about their lives
Patient continues to engage in therapeutic processes, utilizing strategies to reduce maladaptive responses
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published December 9, 2021 on www.thecalgaryguide.com")
mechanical-bowel-obstruction-and-ileus-pathogenesis-and-clinical-findings

Post-abdominal surgery
Authors: Yan Yu, Wayne Rosen* Reviewers: Nicole Burma, Jason Baserman, Jennifer Au, Maitreyi Raman* * MD at time of publication
Adhesions Note:
• Complete obstruction typically
presents with acute abdominal
pain and related symptoms • Incomplete obstructions can present with either acute or
Congenital abnormality GI neoplasms
If partial obstruction: ↓ frequency of bowel movements
Since gas/air sounds hollow to percussion Abdomen tympanic to percussion
Mechanical obstruction: physical blockage of bowel lumen
Inflammatory bowel disease (IBD)
Gas (from swallowed air, bacterial fermentation, & CO2 made via HCO3- neutralization) & ingested gastro- intestinal (GI) contents accumulate before obstruction
Accumulated GI contents contain salts and other osmotically active solutes that osmotically draw water into the GI tract
Continued ↑ bowel distention & ↑ luminal pressure over time
↑ pressure squeezes shut intestinal blood vesselsà↓ bowel perfusion
chronic abdominal pain
If complete obstruction: Obstipation (no flatus) & absent bowel movements
Irritation of autonomic nerves in visceral peritoneum
Lower effective arterial blood volumeàdehydration
Continued peristalsis proximal to obstruction continues to push GI contents against the obstruction
If obstruction is proximal (closer to mouth), higher luminal pressure may force regurgitation of GI contents
Bloating, cramping, anorexia Diffuse visceral abdominal pain
Flat/low jugular venous pressure (JVP), resting tachycardia, orthostatic hypotension
Severe abdominal pain (may come in waves), peritonitis, guarding, rigidity
Hyperactive bowel sounds (proximal to obstruction) or absent bowel sounds (ileus)
Nausea/vomiting
Bowel ischemia and infarction, tissue necrosis, possible perforation +/- bacterial invasion (see relevant slides)
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Re-Published December 15, 2021 on thecalgaryguide.com")
hernias-inguinales-adquiridas-indirecta-directa

dolor-abdominal-agudo-relacionado-con-el-tracto-gi-patogenesis-y-caracteristicas

pancreatitis-aguda-patogenesis-y-hallazgos-clinicos

apendicitis-patogenesis-y-hallazgos-clinicos

culebrilla-herpes-zoster-patogenesis-y-hallazgos-clinicos

infeccion-por-herpes-simple-patogenesis-y-hallazgos-clinicos

enfermedad-de-huntington-patogenesis-y-hallazgos-clinicos

escabiosis-patogenesis-y-hallazgos-clinicos

sifilis-1ria-y-2ria-patogenesis-y-hallazgos-clinicos

necrotizing fasciitis

Laceration
Recent surgery
Injection
Burn
Blunt force trauma
Childbirth
Lower extremity wounds
Bacteria enters tissue through open wound
Infection of muscle fascia Local immune response
Production of exotoxins by bacteria
Disruptions of protective skin barrier
Bacteria introduced into tissue during injury
Necrotizing Fasciitis
Type I infection: mixed aerobic and anaerobic bacteria Type II infection: group A streptococcus
Type III infection: marine organisms, clostridial infections Type IV infection: fungal organisms
Poor blood supply of muscle fascia allows for progressive spread of infection
Systemic immune response
Pyrogens produced by immune system
Pyrogens travel through
the bloodstream to the hypothalamus and alters the body’s thermal setpoint
Transmission of bacteria from infected tissue to blood
Sepsis
Streptolysin (exotoxin) causes blood clot formation
Blood clots in vessels
Tissue ischemia in epidermis, dermis, subcutaneous fat, muscle fascia, and/or muscle
Stimulation of programmed cell death
Tissue destruction
Pain more severe than clinical findings
↓ blood flow fails to meet tissue’s needs
Tissue death
Build up of gas in subcutaneous
tissue from bacteria metabolism
Crepitus
↑ serum creatinine
kinase from protein breakdown
↑ blood flow to infected tissue
Warmth Erythema
Immune cells release vasoactive cytokines into the blood
Capillary vasodilation
Fluid and proteins shift from cells and capillaries to interstitial space
Blood
vessel dilation
↓ perfusion of vital organs
Organ failure
Hypotension
↑ heart rate to perfuse vital organs
Tachycardia
Bacteria releases toxins which are taken up into the bloodstream
Immune cells produce inflammatory cytokines
Circulating toxins activate T cells, over- activating the systemic immune response
Toxic Shock syndrome
Infection ↑ white blood cell production in bone marrow
↑ white blood cells
Destructionof peripheral nerve endings
Insensitivity to pain
Tissue hypoxia à anaerobic metabolism
Poor perfusion of lungs impairs gas exchange
Tachypnea
Cytokines affect dopamine production in the basal ganglia
Acute malaise
Production of non-specific acute phase reactants
↑ C reactive protein and erythrocyte sedimentation rate
Fluid-filled blisters
Edema
Fever Compartment syndrome (see relevant Calgary Guide slide)
Amputation ↑ serum
lactate
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
First published Nov 20, 2013, updated Dec 19, 2021 on www.thecalgaryguide.com")
generalized-seizures-pathogenesis-clinical-findings-and-complications

↑ CNS neuronal irritability or encephalopathy
Glycemic dysregulation
↓ ATP production
Impaired Na+/K+ ATPase function
Author: Erik Fraunberger Yan Yu* Reviewers: Davis Maclean Ephrem Zewdie Negar Tehrani Mehul Gupta Carlos Camara-Lemarroy* * MD at time of publication
Overall ↑ in excitatory tone producing aberrant cortical electrical activity
Cerebral hemispheres synchronously and spontaneously depolarize as electrical signals spread diffusely through brain tissue
Resting membrane potential maintained above threshold
Altered neuronal excitability
Altered communication between cortex and subcortical structures
See “Focal Seizures in the Adult” slide
Motor (tonic-clonic, tonic, clonic, other)
Generalized Seizures
Failure of seizure termination mechanisms and/or ↑ propensity for abnormally prolonged seizures
Physical injury, rhabdomyolysis, aspiration, fractures
(See “Status Epilepticus” slide)
Nonmotor (absence)
↑ corticothalamic signaling and inhibition of the default mode network
Impairment of consciousness
Abrupt cessation or impairment of activities
Learning difficulties, behavioural issues, progression to generalized tonic-clonic seizures
Dysregulation of autonomic control
Post-ictal cardiac dysrhythmias and respiratory dysfunction
↓ cerebral blood flow & oxygenation
Post-ictal Sudden Unexpected Death Confusion In Epilepsy (SUDEP)
Loss of consciousness &
synchronized muscular contractions
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published December 19, 2021 on www.thecalgaryguide.com")
obstruccion-del-intestino-delgado-hallazgos-en-los-rayos-x

bronquiolitis-patogenesis-y-hallazgos-clinicos

asma-patogenesis

otitis-media-aguda-complicaciones

otitis-media-aguda-oma-patogenesis-y-hallazgos-clinicos-en-ninos

diverticulitis-aguda-patogenesis-y-hallazgos-clinicos

meningitis-bacteriana-patogenesis

meningitis-bacteriana-hallazgos-clinicos

meningitis-bacteriana-complicaciones

carcinoma-colorectal-patogenesis-y-hallazgos-clinicos

crup-patogenesis-y-hallazgos-clinicos

trastornos-de-la-vesicula-biliar

enfermedad-por-reflujo-gastroesofagico-erge-complicaciones

hernia-incisional-patogenesis-y-hallazgos-clinicos

colitis-isquemica-patogenesis-y-hallazgos-clinicos

obstruccion-intestinal-mecanica-e-ileo-patogenesis-y-hallazgos-clinicos

sepsis-y-shock-septico-patogenesis-y-hallazgos-clinicos

prolapso-del-cordon-umbilical-patogenesis-y-hallazgos-clinicos

Normal anion gap metabolic acidosis
, the distal nephron secretes one corresponding urine Cl- anion
Urine NH4+ cannot be measured, so measured
urine Cl- exceeds measured urine (Na+ + K+)
Negative urine net charge (Urine (Na+ + K+) - Urine Cl-)
Type II RTA RTA: renal tubular acidosis ↓ activity of proximal
Type I RTA
↓ activity of H+ ATPase on luminal surface of ⍺- intercalated cell in the collecting duct
Impaired ⍺-intercalated cell function
↓ H+ secretion into urine by ⍺- intercalated cell
↑ H+ concentration in plasmaà↓ in blood pH
Activation of blood bicarbonate buffer system
Compensatory ↓ in plasma HCO3- (in response to ↑ in plasma H+)
Type IV RTA
↓ aldosterone production or aldosterone resistant state
↓ Na+ channels on luminal surface of principal cell in the collecting duct
Impaired principal cell function
↓ Na+ reabsorption by principal cell
↑ Na+ remains in collecting duct lumen
↓ negative charge in collecting duct lumen
↓ K+ secretion into urine by principal cell
↑ K+ in plasma
Hyperkalemia
Normal anion gap
tubule transporters, pumps or enzymes
Impaired proximal tubule cell function
reabsorption
Complex and Incompletely understood mechanisms
Hypokalemia
Distal nephron function is impairedàdistal nephron is unable to secrete excess acid into the urine in the form of NH4+
↓ in urinary NH + 4
secretion is accompanied by corresponding ↓ in urinary Cl- secretion
Measured urine (Na+ + K+) exceeds measured urine Cl-
Positive urine net charge (Urine (Na+ + K+) - Urine Cl-)
↓ HCO3
-
in proximal tubule
↑ HCO3- loss through urine
↓ HCO3- concentration in plasma
Relative ↑ in plasma H+ compared to plasma HCO3-
Compensatory ↑ in CO2 exhalation by lungs (since CO2 is acidic once metabolized in the blood)
Since Anion gap = Na+ - Cl- - HCO3-,
1 to 1 replacement of Cl- for HCO - in the 3
plasma results in anion gap unchanged
↓ ability to buffer excess acid
Activation of the membrane Cl-/HCO3- exchanger in the collecting duct
Exchanger works to ↑ Cl- levels in the plasma by
one Cl- for each ↓ of one HCO3- in the plasma
Metabolic Acidosis
↓ Plasma partial pressure of CO2
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published December 21, 2021 on www.thecalgaryguide.com")
Syndrome of Inappropriate Anti-Diuretic Hormone SIADH Pathogenesis and Clinical Findings
: Pathogenesis and clinical
Malignancy
(e.g. Small cell lung cancer, head and neck cancer)
Tumor originates from neuroendocrine cells
Tumor acts as an ectopic site of ADH production
ADH binds receptors on basolateral side (facing peritubular capillary) of principal cells in nephron
ADH ↑ principal cells’ production of Aquaporin type II channels on their apical surface (side facing tubule lumen)
↑ Reabsorption of water from the collecting ducts back into circulation
↑ Blood Volume (↑ extracellular fluid volume)
Blood Na+ levels diluted
Hyponatremia
(blood Na+ <135 mEq/L)
Brain injury
(e.g. Stroke, encephalitis hemorrhage, trauma)
↑ hypothalamic ADH production, storage in posterior pituitary, & pituitary secretion of ADH
↑ Uncontrolled ADH secretion
SIADH
(Syndrome of Inappropriate Anti-Diuretic Hormone)
Drugs
(e.g. Cyclophosphamide, SSRIs, vincristine)
Break down into active metabolites
Metabolites mimic ADH activity
findings
Authors: Krusang Patel Yan Yu* Reviewers: Davis Maclean Brooke Fallis Juliya Hemmett* * MD at time of publication
Atria and Ventricles of the heart stretch
Heart secretes ↑ atrial natriuretic and
B-type natriuretic peptides (ANP/BNP)
Peptides promotes natriuresis (excretion of Na+ into urine)
Loss of Na+ in serumàalters charge balance across neuron
membranesàImproper action potential firing:
↓ Renin secretion
↓ Release of Angiotensin II & Aldosterone
↓Reabsorption of Na+ into circulation
in area postrema of medulla
in hypothalamus in motor neurons
Extracellular fluid volume becomes hypotonic relative to intracellular fluid volume
Water moves from circulation into cells
Extracellular fluid volume normalizes
Euvolemia
Nausea
Headaches Muscle Cramps
↓ Urine volume
Cells, particularly neurons, swell up
Cerebral edema
Neurons burst and die
Severe Neurocognitive
Effects (confusion, mood swings, hallucinations, seizure, coma)
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published Dec 30, 2021 on www.thecalgaryguide.com")
Renal Artery Stenosis
 of the renal artery
Renal Artery Stenosis can be unilateral or bilateral
Authors: Samin Dolatabadi, Yan Yu* Reviewers: Meena Assad, Jessica Krahn Timothy Fu, Brooke Fallis, Juliya Hemmett* * MD at time of publication
↓ Pressure perfusing the kidney
↑ RAAS (renin-angiotensin- aldosterone system) activation
↓ pressure gradient in glomerulus
↓ Glomerular filtration rate (GFR)
↑ Secretion of aldosterone
Turbulent blood flow through area of stenosis
Abdominal bruit on side of affected kidney(s)
↑ Secretion of Angiotensin IIà ↑ Systemic vasoconstriction
Hypertension
↑ Expression of epithelial sodium channels in cortical collecting duct
↑ Blood volume within volume- constrained space of blood vessels
↓ Renal blood flowà ischemic renal injury
Atrophy and fibrosis of affected kidney(s)
Unilateral stenosis à Kidney size asymmetry (≥1.5cm difference)
↓ Positively charged Na+ in lumen à Electronegative lumen compared to the interstitial/tubular epithelial cells
K+ follows the electrical gradient and is secreted into the electronegative tubular lumen
↓ Serum K+ concentration
Hypokalemia
*Note: In unilateral renal artery stenosis, the contralateral (normal) kidney can compensate for the increase in renal perfusion pressure caused by hypertension by increasing sodium excretion (pressure natriuresis), preventing flash pulmonary edema.
↑ NHE3 (Sodium Hydrogen Exchanger 3) activity in proximal collecting tubule
↑ Na+ and water reabsorption from renal tubular lumen into blood vessels
Chronic left ventricle pressure overload àLeft ventricle hypertrophy (see Left Heart Failure: Pathogenesis Slide)
Systolic dysfunction → ↓ left ventricle stroke volume
Normally, ↑ in renal perfusion pressureà↑ Na+ excretion and water loss (pressure natriuresis)
In bilateral renal artery stenosis*, ↓ GFRàinability for kidney to ↑ renal Na+ & water excretion à volume overload
Acute increase in afterload or preload → sudden ↑ in left ventricular filling pressures → blood backup into lungs
Pulmonary vasculature hypertension → ↑ fluid filtration across the pulmonary endothelium into interstitium and alveolar spaces
Flash (rapid onset) pulmonary edema
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published December 30, 2021 on www.thecalgaryguide.com")
Membranous Nephropathy
 Membranous Nephropathy
Secondary Membranous Nephropathy
Drugs, infections, auto-immune diseases, and malignancies make antigens
Blood tests for Anti- PLA2R antibodies
are positive in ~70% of cases
Autoantibodies are made to antigens expressed near or on podocyte surfaces or planted in the glomerular capillary wall (ie. PLA2R; THSD7A; NELL-1)
IgG antibodies filter through the glomerulus
Autoimmune: (e.g. Systemic Lupus Erythematous, thyroiditis)
Drugs: (e.g. NSAIDs, gold, penicillamine, captopril)
Malignancies: (ie. Prostate, Colon)
Infections: (e.g. Hepatitis B, Syphilis)
Circulating antigens filter through the glomerulus and between the GBM and podocytes
Antibodies & antigens form immunocomplexes, which lodge Immunofluorescence shows diffuse “granular”
between the glomerular basement membrane & podocytes
deposits of immunocomplexes throughout GBM
Basement membrane is formed between and around immunocomplex deposits
Immunocomplexes activate complement and the assembly of the Membrane attack complex (MAC)
MAC creates holes in podocyte plasma membranes
Stimulates the release/activation of proteases and oxidases from glomerular podocytes and mesangial cells
GBM appears thick on light microscopy
“Spike and dome” pattern appears with silver stain
Podocyte effacement
Damage to negatively charged foot processes damages the charge barrier of the glomerulus that repels negatively charged molecules
Increased filtration of large negatively charged molecules, especially albumin
Induces ↑ hepatic lipoprotein synthesis and ↓ lipoprotein catabolism
Hyperlipidemia
(↑ serum LDL, VLDL, and triglycerides)
↑ lipid filtration through glomerulus
Lipiduria (fatty casts)
*Hypo-albuminemia
↓ oncotic pressure in capillaries
Fluid leaks into interstitial space
↑ Filtration of Proteins C and S and antithrombin
Hypercoagulable state
Thrombosis *Edema (especially
*Proteinuria
↑ Filtration of immunoglobulins
Immunosuppression
Infections
↑ Filtration of Plasminogen
Plasminogen converted to plasmin in the cortical collecting duct via urokinase- type plasminogen activator
Plasmin activates the epithelial sodium channel
↓ intravascular volume
Hypotension
Pre-Renal Acute Kidney Injury
Underfill edema
(see slide)
peri-orbital, scrotal, labial, and extremities)
Overfill
edema
(see slide)
↑ Na+ and water reabsorption
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published MONTH, DAY, YEAR on www.thecalgaryguide.com")
Overview of Calcium Phosphate Vitamin D Physiology
 and ↓ expression of osteoprotegerin (OPG), a decoy receptor for RANKL
RANKL binds to receptor activator of nuclear factor kappa-B (RANK) on osteoclasts
↑ Osteoclast differentiation and activity
↑ Ca2+ resorption from bone into blood
Sensed by Calcium-Sensing Receptor on Chief Cells of the Parathyroid Gland
− −
↑ Serum phosphate (PO4) ↓ Serum calcium (Ca2+)
Parathyroid Gland releases Parathyroid Hormone (PTH) into the blood, which acts on the kidneys and the bones
Kidney
↑ Activation of Transient Receptor Potential Vanilloid subfamily member 5 (Ca2+ channel)
in the distal convoluted tubule
↓ Expression of 24-hydroxylase enzyme (which functions to catabolize calcitriol)
↑ Expression of 1α- hydroxylase enzyme
↑ Conversion of 25-hydroxy vitamin D (Calcidiol) to the
active form, 1,25-dihydroxy vitamin D (Calcitriol)
↑ Calcitriol Small Intestine
↑ Endocytosis of sodium phosphate co-transporters NaPi-2a and NaPi-2c, which reabsorb PO4 from ultrafiltrate in the renal tubules
↓ Reabsorption of PO 4
↓ Serum PO
Calcitriol negatively feeds back on parathyroid gland to inhibit PTH production
↑ Expression of sodium phosphate co- transporters NaPi-2a and NaPi-2b that absorb PO4 from intestinal lumen
4
↑ Expression of apical epithelial Ca2+ channels (Transient Receptor Potential Vanilloid subfamily member 6)
↑ Entry of Ca2+ on apical side of enterocytes
↑ Reabsorption of Ca2+ in the distal convoluted tubule through Ca2+ channels
↑ Reabsorption of PO
↑ Expression of cytoplasmic Calbindin-D
↑ Transport of Ca2+ across enterocytes
↑ Absorption of Ca2+ in small intestine
↑ Serum Ca+2
↑ Expression of basolateral plasma membrane calcium ATPase
↑ Extrusion of Ca2+ from enterocytes into bloodstream
4
Authors: Samin Dolatabadi, Hannah Yaphe Reviewers: Amanda Henderson, Meena Assad, Brooke Fallis, Yan Yu*, Hanan Bassyouni* * MD at time of publication
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
First published Oct 15, 2017, updated Nov 11, 2021 on www.thecalgaryguide.com")
Minimal Change Disease
 present in serum
Immunofluorescence test negative
Idiopathic/Primary Minimal Change Disease
No identifiable extraglomerular disease process causes this condition
Secondary Minimal Change Disease
Infections, NSAIDS, neoplasms via unclear mechanisms
Minimal change to glomerulus seen on light microscopy
Podocyte effacement seen on electron microscopy
Abnormal T Cell activation and release of cytokines (sometimes called permeability factors) that are toxic to podocytes
Podocyte foot processes efface (become flattened) or fuse together
Damage to negatively charged foot processes damages the charge barrier of the glomerulus that repels negatively charged molecules
↑ filtration of larger negatively charged molecules, such as low-molecular weight proteins like albumin, from the blood into the renal tubular filtrate
Induces ↑ hepatic lipoprotein synthesis and ↓ lipoprotein catabolism
Hyperlipidemia
(↑ serum LDL, VLDL, and triglycerides)
↑ lipid filtration through glomerulus
Lipiduria (fatty casts)
Hypo-albuminemia
↓ oncotic pressure in capillaries
Fluid leaks into interstitial space
↑ Filtration of Proteins C and S and antithrombin
Hypercoagulable state
Proteinuria
↑ Filtration of immunoglobulins
Immunosuppression
Infections
↑ Filtration of Plasminogen
Plasminogen converted to plasmin in the cortical collecting duct via urokinase- type plasminogen activator
Plasmin activates the epithelial sodium channel
↓ intravascular volume
Hypotension
Pre-Renal Acute Kidney Injury
Underfill edema
(see slide)
Thrombosis Edema (especially peri-
orbital, scrotal, labial, and extremities)
Overfill
edema
(see slide)
↑ Na+ and water reabsorption
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published December 30, 2021 on www.thecalgaryguide.com")
Cerebral Edema

↑ Intra-Cranial Pressure (ICP) Cerebral vasculature compressed
↓ Cerebral Perfusion Pressure (CPP; CPP = Mean Arterial Pressure – Intracranial Pressure)
Cerebral hypoxia
↓ O2 detection in vasomotor center within medulla
↑ Sympathetic nervous system activity in attempt restore cerebral perfusion
Release of adrenergic and noradrenergic neurotransmitters from nerve terminals
Stimulation of α1, α2, β1, and β2 receptors located on the cell surface of target organ tissue
↑ Peripheral arteriole constriction
Optic Nerve swelling
Distortion of brainstem
Compression of the medulla
Nerve fibre dysfunction
Axoplasmic stasis within optic nerve fibers
Oculomotor nerves compressed
Vision loss
Papilledema
Dilation and fixation of ipsilateral pupil
Distortion of cortex
Brain Herniation (e.g. Subfalcine, Uncal, Tonsillar)
Massive cerebral malperfusion
Neuronal cell injury and death
Hypoxic Brain Injury and Brain Death
Compression of frontal cortex
Neuron dysfunction
Frontal cortex behaviour changes (lethargy, irritability, anxiety, seizures)
Meninges compressed between brain and skull
Meningeal pain receptors activated
Headache
Compression of the diencephalon and midbrain
Dysfunction of respiratory control center
Slow and irregular respirations
Area postrema (nausea control center) compressed
Nausea & Vomiting
Descending corticospinal tract compressed against tentorium
Upper Motor Neuron (UMN) dysfunction
Impaired function of the Reticular Activating System
Depressed level of consciousness
Cushing’s Triad
UMN disease (see relevant slide for full mechanisms)
Signs of increased ICP associated with impending herniation of the brain:
1. Irregular, slow respirations
2. Bradycardia
3. Systolic Hypertension
Author:
Stephen Chrusch Reviewers:
Emily Wildman
Austin Laing
*Scott Jarvis
*Yan Yu
* MD at time of publication
Muscle weakness
Hyperreflexia
Spasticity (Hypertonicity)
Extensor Plantar Response
Systolic Hypertension
↑ Heart rate and cardiomyocyte contractility
Carotid and aortic baroreceptors detect ↑ BP
Tachycardia (early)
Vagus
nerve stimulation
Parasympathetic response triggered
Reflex bradycardia (late)
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published Jan 5, 2022 on www.thecalgaryguide.com")
BPPV
: Pathogenesis and Clinical Findings
Authors: Ryan Chan, Jonathan Wong, Mehul Gupta, Yan Yu* Reviewers: Davis Maclean, Saud Sunba, Euna Hwang* * MD at time of publication
Up-beating, torsional geotropic (towards the
ground) fast-phase nystagmus (towards affected side)
Down-beating +/-torsional fast-phase nystagmus (towards opposite side)
Idiopathic Older Age
Head trauma
Recent Ear Surgery
Underlying Vestibular Disorders/Infections: Meniere’s Disease, Vestibular Neuritis, Labyrinthitis
Risk Factors of Labyrinth
Ischemia: Hypertension, Hyperlipidemia, Migraines
Dislodged otoliths/otoconia from the macula of the utricle
Posterior Canal BPPV (~95-99%)
Superior Canal BPPV (~1%)
Horizontal Canal BPPV (~5-20%)
Ocular muscles are stimulated to generate
a downward, torsional slow-phase movement
Ocular muscles are stimulated to generate
an upward, torsional slow-phase movement
Cupulolithiasis Theory: Otolith adheres to cupula of the semicircular canal (SCC)
Otolith displaces the cupula during head position changes resulting in prolonged sense of head rotation along the semicircular canal axis
OR
Canalithiasis Theory: Free-floating otolith in the semicircular canal (SCC)
Otolith induces inertial drag of the endolymph fluid during motion, displacing the cupula, resulting in prolonged sense of head rotation
Dix-Hallpike Maneuver: Sitting with head rotated laterally (45°), moving quickly to supine with head extended 30° off table. Observe for any nystagmus. Direction of nystagmus indicates which of the three semicircular canals is affected.
Ocular muscles are stimulated to generate a horizontal slow-phase movement away from affected side
Horizontal Nystagmus: Fast phase beats toward the affected side Supine Head Roll Test: lying supine, roll head laterally to each
side to move otoliths along horizontal SCC axis
BPPV: Episodic, positional bouts of vertigo and nystagmus not due to an underlying neurological or insidious reason
If the otolith is free-floating in the SCC, movement of the affected ear towards the table generates a net stimulatory endolymph flow in the affected horizontal SCC
The stimulatory signal is carried to brainstem nuclei, generating a reflexive slow movement
of the eyes away from the affected ear, and quick horizontal movements back towards the affected ear
Geotrophic Horizontal Nystagmus: fast-phase nystagmus beats horizontally towards the table
If the otolith is adherent to the SCC cupula,
movement of the affected ear towards the table generates a net inhibitory deflection of the horizontal SCC cupula
The inhibitory signal is carried to brainstem nuclei, generating a reflexive slow movement of the eyes towards the affected ear, and quick horizontal movements away from the affected ear
Apogeotropic Horizontal Nystagmus: fast-phase nystagmus beats horizontally towards the ceiling
Otoconia & otoliths only affect the
semicircular canal (SCC), not the cochlea
No tinnitus or hearing loss
Otoconia tend to settle quite quickly (<1 min) when body is still, resolving the mismatch of body movement & semicircular canal (SCC) excitation
Vertigo and nystagmus is transient (lasting ~1min or less)
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published January 9, 2022 on www.thecalgaryguide.com")
ACE inhibitors
 inhibitors: Mechanism of Action and Clinical Findings Angiotensin Converting Enzyme (ACE) Inhibitors
Inhibits the conversion of angiotensin I to angiotensin IIà↓ serum angiotensin II levels
Inhibits breakdown of bradykinin
↓ Activation of zona glomerulosa region of the adrenal cortex
↓ Aldosterone secretion from the adrenal cortex
↓ Expression of basolateral Na+/K+ pump
and ↓ insertion of luminal Na+ channels in principal cells of the cortical collecting duct in kidney
↓ K+ secretion into urine
↑ Serum K+ concentration
Hyperkalemia
↓ Activation of Na+/H+ exchanger in proximal convoluted tubule (PCT) of the kidney
↓ Na+ absorption into PCT and ↓ H+ excretion into lumen
↓ Na+ absorption into the blood
↓ Renal H2O reabsorption via osmosis
↓ Activation of paraventricular nuclei located in the hypothalamus
↓ Secretion of anti- diuretic hormone (ADH)
into the capillaries of the posterior pituitary gland
↓ Aquaporin channel insertion at cortical collecting duct in kidney
↓ Renal H2O reabsorption
à↑ serum bradykinin levels Vasodilation of efferent
arteriole in kidneys
↓ Hydrostatic pressure within the glomerular capillaries
↑ Activation of cyclooxygenase-2 pathway
↑ Prostaglandin production
↓ Activation of angiotensin II type 1 receptors on surface of arteriolar smooth muscle
↓ Activity of G protein-coupled receptor intracellular secondary messenger cascade
↓ Systemic arteriolar vasoconstriction
↓ Total peripheral resistance (resistance
to blood flow by systemic vasculature)
↓ Blood Pressure
↓ total blood flow perfusing kidneys
Renal hypoxia
↑ Systemic arteriolar vasodilation
↓ Total peripheral resistance
↓ Blood Pressure
↓ Concentration of protein
travelling through tubules for excretion
↓ Fraction of H2O filtered from glomerular capillaries into bowman’s capsule (start of the renal tubule)
↓ Filtration of blood contents through glomerular membranes
Sensitization of airway vagal afferent receptors
Stimulation of the cough
center located in the medulla oblongata
Cough
↓ Protein in urine
High K+ levels surrounding the heart muscle cellsàchronic depolarization
of the cardiomyocyte cell membrane àalters cardiomyocyte conduction (complex mechanisms)
Cardiac Arrythmias (see relevant slide on Hyperkalemia: Clinical Findings)
↓ Effective arterial blood volume
Authors:
Nicole Brockman Reviewers:
Emily Wildman, Austin Laing Adam Bass*, Yan Yu*
* MD at time of publication
Renal tubules have relatively less water compared with the peritubular capillaries
↓ H2O reabsorption from the renal tubule back into the blood
↑ Serum creatinine, ↓ estimated glomerular filtration rate
Renal nephron injury and dysfunction
Acute Kidney Injury
(rise in creatinine >26.5umol/L in <7 days)
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published January 10, 2022 on www.thecalgaryguide.com")
complications-of-chronic-kidney-disease-ckd

Authors: Samin Dolatabadi, Brooke Fallis Reviewers: Jessica Krahn, Meena Assad, Yan Yu* Juliya Hemmett* * MD at time of publication
Abnormalities of kidney structure or function that is present for 3 or more months
Kidney tubules atrophy & kidney interstitial tissue undergo fibrosis
Kidney damage ↓ erythropoietin production (normally a kidney function)
↓stimulation of bone marrow → ↓ red blood cell production
Build up of toxic substances (e.g. urea, guanidine, and indoxyl sulfate)
↓ Conversion of calcidiol to calcitriol in by kidney
↓ Calcitriol levels in blood
↓ Ca+2 absorption from small intestine
Hypocalcemia
↓ Glomerular Filtration Rate
↑ Vascular calcification and endothelial dysfunction (e.g. changes in permeability, clotting response to inflammation, amongst other mechanisms)
Atherosclerotic disease (see “Complications of Atherosclerosis” slide)
↓ Lipoprotein lipase, apoA-1, and Lecithin-cholesterol acyltransferase
activity acting on serum lipoproteins (complex mechanisms)
↓ Lipoprotein clearance from blood
Dyslipidemia (↑ LDL, ↑ triglycerides, ↓ HDL)
Toxic damage ↓ red blood cell survival
Anemia
Uremic Syndrome
↓ Renal excretion of phosphate
↑ phosphate remaining in bloodstream
Hyperphosphatemia
↑ serum phosphate binds with ionized calcium
↓ Renal excretion of K+
↑ K+ remaining in bloodstream
Hyperkalemia Edema
↓ Renal excretion of ammonium
Reduced filtration capability ↑ organic anions remaining in blood→ ↑ anion gap
↓ Renal excretion of salt and water
↑ in extracellular fluid → systemic volume overload
↓ calcium in blood ↓ inhibition of Parathyroid Hormone (PTH) release
↑ PTH in blood
Secondary hyperparathyroidism
Metabolic Acidosis
Hypertension
Pitting
Chronic kidney disease – mineral and bone disorder (CKD-MBD)
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published Jan 4, 2022 on www.thecalgaryguide.com")
NSAIDs and the Kidney Nephrotoxicity

Authors: Kyle Moxham Mehul Gupta Reviewers: Emily Wildman Yan Yu* Adam Bass* * MD at time of publication
Inhibition of Cyclooxygenase COX-1 (expressed in kidney) and COX-2 (expressed in kidney and sites of inflammation)
NSAID induced nephrotoxicity: associated with chronic NSAID usage independent of dosage
COX inhibition ↑ conversion of arachidonic acid (AA) to leukotrienes, causing systemic T-cell dysfunction (unknown mechanism)
Type IV systemic hypersensitivity (delayed
T helper cell mediated) reaction to drug exposure
T cells release inflammatory cytokines into the bloodstream
(see Calgary Guide slide on NSAIDs and the Kidney: Mechanism of Action and Side Effects)
T cells infiltrate the renal interstitium, sparing the glomeruli and blood vessels
Overproduction of cytokines by T cells causing inflammation,
tissue damage, and cell death, of the renal intersitium
Drug Induced Acute interstitial nephritis (AIN): a type of immune-mediated tubulointerstitial injury
Activated T-cells infiltrate the glomerulus and cause podocyte injury (epithelial cells attached to the glomerular basement membrane)
Membranous nephropathy:
nephrotic syndrome involving autoimmune glomerular basement membrane thickening & complete podocyte effacement (seen on kidney biopsy)
Minimal change disease: nephrotic syndrome caused by autoimmune podocyte effacement (seen on kidney biopsy)
Cytokine mediated activation and
proliferation of immune cells like macrophages and eosinophils
Cytokines travel to hypothalamus,
causing change in the body’s thermal set point
Fever
Podocyte effacement allows for serum proteins to across the glomerulus into the tubular lumen (see Calgary Guide slide on Nephrotic Syndrome for full mechanisms)
Repeated NSAID exposure causing
recurrent unrecognized AIN and damage of the kidney
Chronic interstitial nephritis /analgesic nephropathy
Infiltrating immune cells (predominantly neutrophils) are filtered into/enter the renal tubules, and form clumps (“casts”) within the tubulesà casts are then released into urine
WBC cast on urinalysis
↑ Blood eosinophils
Immune cells infiltrate the
dermis and epidermis of the skin
Rash
Abnormal quantities of protein present in urine
Protein present in the blood is improperly filtered into the filtrate at the glomerulus
Protein- Creatinine
Ratio >3.5mg/mg
Proteinuria >3.5g/d
Hypo- albuminemia
↓ plasma oncotic pressure resulting in fluid extravasation into the interstitium (see Calgary guide slide on Edema for full mechanisms)
Pitting Edema
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published January 13, 2022 on www.thecalgaryguide.com")
Langerhans Cell Histiocytosis
 CD1a+/CD207+ (Langerhans cell phenotype surface receptors) dendritic cells in tissue(s)
Somatic BRAFV600E mutation: BRAF is a kinase in the MAPK pathway
Other somatic (non- reproductive cell) mutations
Idiopathic (unknown cause)
Mutation(s) can occur in one of these precursor cell types*
Hematopoietic stem cell (earliest cell of blood cell differentiation) in bone marrow
Committed dendritic cell (type of myeloid antigen-presenting cell) precursor in bone marrow or blood
Committed dendritic cell precursor in tissue
Constitutive activation of the MAPK pathway (signalling pathway that regulates variety of cellular processes) in one of these precursor cell types
*Note: Based on the “misguided myeloid differentiation” modelàthe earlier the mutation(s) occur in the myeloid cell differentiation pathway, the more severe the disease.
Langerhans Cell Histiocytosis
Accumulation of abnormal CD1a+/CD207+ dendritic cells (Langerhans Cell Histiocytosis cells or LCH cells) with an inflammatory background in one or more organs
Authors:
Ran (Marissa) Zhang Reviewers:
Mehul Gupta
Kiera Pajunen
Yan Yu*
Lynn Savoie*
* MD at time of publication
↑ recruitment & activation of T cells, macrophages, eosinophils in tissue(s) around the body
↓ CCR7 & CXCR4 (chemokine receptors) expression on LCH cellsàinhibits migration of LCH cells to lymph nodes
↑ BCLXL (an apoptosis regulator protein) expression on LCH cellsà inhibits apoptosis of LCH cells
Immune cell infiltration & ↑ pro-inflammatory chemokine/cytokine release à dysregulated local & systemic inflammation
Accumulation of LCH cells in tissue(s) around the body
Inflammatory lesion (an area of abnormal tissue) formation in one or more organs:
In pituitary stalk
Mass effectà
obstruction of antidiuretic hormone (complex mechanisms)
In liver
Invasion & accumulation of cells foreign to liverà expands liver
Chronic local inflammation
Scarring of bile ducts
↓ bilirubin clearance from liveràbuildup into serum
↑ serum bilirubin
Jaundice
In cortical bone Cytokine production
à↑ osteoclast
(cells that break down bone) activity
↑ rate of bone loss
Osteolytic bone lesions
In bone marrow
Unclear mechanism but likely due to macrophage activation
↑ phagocytosis (ingestion & destruction) of blood cells
In spleen
Invasion & accumulation of cells foreign to spleen
Forms aggregates that expand the red pulp (functions as the blood filter in spleen)
Splenomegaly
In skin
Unclear mechanisms
Variable presentations: most commonly pinpoint erythematous (red) papules or erythematous plaques with crusting & scaling
Hepatomegaly • BRAF- B-Raf proto-oncogene, serine/threonine kinase
• CCR7- C-C motif chemokine receptor type 7
• CD1a- Cluster of differentiation 1A
• CD207- C-type lectin domain family 4 member k • CXCR4- C-X-C chemokine receptor type 4
• LCH- Langerhans Cell Histiocytosis
• MAPK- Mitogen-activated protein kinase
Diabetes Insipidus
Abbreviations:
• BCLXL- B-cell lymphoma-extra large
Anemia, thrombocytopenia &/or neutropenia
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published January 13, 2022 on www.thecalgaryguide.com")
Overfill Edema Pathogenesis

Aberrant filtration of proteins including plasminogen
(glycoprotein in the systemic circulation involved in the dissolution of fibrin blood clots)
↑ Plasminogen concentration in tubular fluid
Conversion of plasminogenà plasmin by urokinase-type plasminogen activator in the cortical collecting tubule
Plasmin activates epithelium
sodium channels (involved in Na+ reabsorption) in principal cells of the cortical collecting duct
Nonsteroidal Anti-inflammatory Drugs
Inhibition of cyclooxygenase throughout body (enzyme that converts arachidonic acidàprostaglandins)
↓ Prostaglandins throughout body
Thiazolidines
↓ Prostaglandin- mediated inhibition of Na+ and Cl- transport in ascending loop of Henle and collecting ducts
↑ Na+ reabsorption from kidney tubules back into blood
↓ Prostaglandin-mediated vasodilation in kidneys
↓ Pressure of blood perfusing kidneys → ↓ pressure gradient between glomerulus and bowman’s capsule
↓ Glomerular filtration rate
↓ Renal excretion of salt & water
↑ Salt and water retention
↑ Effective arterial blood volume
↑ blood pressure in veins
Unclear mechanism but possible theories include upregulation of epithelium sodium channels in the cortical collecting tubule and involvement of other transporters in the proximal tubule and cortical collecting tubule
Acute Renal Failure Chronic Renal Failure
↑ Hydrostatic pressure within capillariesàexceeds hydrostatic pressure within interstitial spaceàfluid moves from capillaries into interstitial space
Overfill Edema
Abnormal accumulation of fluid in the interstitial space where urine Na+ >40meq/L
Authors: Samin Dolatabadi Reviewers: Meena Assad, Jessica Krahn, Brooke Fallis, Ran (Marissa) Zhang, Yan Yu*, Juliya Hemmett* * MD at time of publication
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published January 23, 2022 on www.thecalgaryguide.com")
telangiectasie-hemorragique-hereditaire-maladie-rendu-osler-pathogenie-et-resultats-cliniques
:
Pathogénie et résultats cliniques")
rosacee-pathogenie-et-resultats-cliniques

molluscum-contagiosum-pathogenie-et-resultats-cliniques

NSAIDs and the Kidney mechanism of action and side effects
 or angiotensin II receptor blockers (ARB)
AECi and ARBs act to ↓ renin- angiotensin-aldosterone system (RAAS) activation
Reduced angiotensin (AT) 2 activity at its receptors on efferent arteriole of the nephron
Net vasodilation of efferent arterioles ↓ in glomerular pressure
↓ blood perfusing kidney tissueà↑ hypoxemia & renal ischemia
Pre-existing ↓effective arterial blood volume (EABV) from
dehydration, GI loss, diuretics, CKD, CHF, cirrhosis, etc.
Decreased EABV triggers endogenous renal autoregulation, resulting in norepinephrine (NE) mediated vasoconstriction of afferent arteriole of the nephron
Net ↓ in renal blood flow and ↓ in glomerular pressure
↓ volume of blood filtered by the glomeruli per unit time
Non-steroidal anti-inflammatory drugs (NSAIDs)
Inhibition of Cyclooxygenase COX-1 (expressed in kidney) and COX-2 (expressed in kidney and sites of inflammation)
↓ Renal prostaglandin (PG) synthesis: local hormones involved in renal homeostasis
NSAID induced nephrotoxicity:
associated with chronic usage independent of dosage
(see Calgary Guide slide on NSAIDs and the Kidney: NSAID induced nephrotoxicity)
↓ intrarenal PG reduces inhibitory effects of PG over ADH in cortical colleting duct (CCD) of the kidneyà↓ antagonism on ADH activity
↑ ADH activity causes insertion of more
aquaporins (water channels) in the collecting duct of renal tubules
Net ↑ in volume of water reabsorbed into the blood
↓ vasodilatory effect of PG at the afferent arteriole of the nephron
↓ Glomerular filtration rate (GFR)
↓ PG signalling results in ↓ renin secretion at juxtaglomerular apparatus
Low renin levelsà↓ conversion of angiotensinogen into its AT1 form and, by extension, ACE mediated conversion of AT1 to AT2
↓ ACE 2 signalling leads to ↓ levels of aldosterone in the serum
↓ Na+ and K+ channel insertion on apical surface and ↓ Na/K ATPase activity on basolateral surface of principal cells
↓ K+ excretion into urine, and ↓ Na+ reabsorption
back into the blood, at the late DCT and collecting duct of the kidney
Pre-renal Acute Kidney Injury (AKI): kidney injury due to renal hypoperfusion
Prolonged and/or severe ischemia causes cell death and aggregation of tubular epithelial cells of the kidney with subsequent inability to reabsorb luminal Na+
Renal dehydration predisposes
precipitation of uromodulin protein
Renal tubules mold uromodulin into cylindrical structures known as “casts”. Casts that contain only uromodulin protein are known as “hyaline casts”
Hyaline cast seen on urinalysis
Hypoperfusion of the kidney activates the renin- angiotensin- aldosterone system (RAAS)
↑ amount of sodium (Na) reabsorbed from the filtrate (less Na excreted)
Fractional excretion of Na <1%
Papillary necrosis: ureteral passage of sloughed ischemic tissue causing ureteral obstruction
Acute tubular necrosis: a type of kidney injury causing damage to the tubules
Hypertension
Post-renal AKI: a type of kidney injury due to
obstruction of the urinary tract
Distal distal obstruction of the urinary tract causes fluid to accumulate within the kidneysàenlarging the kidneys
Renal Ultrasound shows hydronephrosis (enlarged kidneys)
Damaged tubule epithelial cells slough into the tubular lumen
Epithelial cell breakdown in the tubular lumen
releases uromodulin & other proteins, which aggregate into “casts” (cylindrical imprints of the renal tubule). The varied protein content of these casts result in them having a coarse, granular appearance
Damaged tubular epithelial cells are unable to properly reabsorb sodium
↓ amount of sodium (Na) reabsorbed from filtrate into the blood (more Na excreted)
Fractional excretion of Na >2%
True excess of free water relative to Na+ in the blood
Excess free water ↑ venous hydrostatic pressure (see Calgary Guide slide on edema for full mechanisms)
Pitting Edema
Hyperkalemia
Hyponatremia
Coarse granular casts seen on urinalysis
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published January 29, 2022 on www.thecalgaryguide.com")
adult-pneumonia-pathogenesis-and-clinical-findings
 and can be further classified by location of exposure: community, health- care, hospital acquired
Inflammatory response varies depending on type of invading pathogen (i.e. S. Pneumonia causes a lobar pattern and Influenza A & B cause an interstitial pattern)
LOBAR: Accumulation of neutrophils and plasma exudate from capillaries into alveoli specific to a lung area/lobe
INTERSTITIAL: Accumulation of infiltrates (i.e. inflamed cellular debris) in the alveolar walls (i.e. space between the alveolar spaces and bloodstream)
Fever
Notes:
• Other signs and symptoms
of pneumonia exist such as chest pain, accessory muscle use, crackles on auscultation and fatigue
• These signs and symptoms are less specific to the ones outlined on this slide
Irritation and attempted clearance of airways
Fluid infiltrates are inside alveoli, airway clearance leads to phlegm production
Productive Cough
Fluid build up does not allow X-rays to pass through à white opacity on plain film at site of fluid buildup
Consolidation on CXR
Alveolar sacs blocked by fluid accumulation
Thickening of alveolar walls ↑ diffusion distance between alveoli & capillaries
Irritated alveolar walls trigger cough reflex
Since fluid infiltrates are NOT in the alveoli, attempts to empty the alveoli through coughing doesn’t lead to production of fluid
Dry Cough
Chills/Rigors
↓ Exchange of CO2 and O2
Hypoxemia
Triggers peripheral and central chemoreceptors to ↑ respiratory drive
Dyspnea
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published Sept 26 2016, updated Feb 9, 2022 on www.thecalgaryguide.com")
greffes-de-peau-physiologie-de-la-greffe-et-resultats-cliniques

Épiderme Derme
Follicule pileux Glande
sudoripare
Chronologie
1. Adhésion initiale (8 heures)
2. Imbibition
plasmatique (24-48 heures)
3. Inoculation (2-4 jours)
4. Croissance néovasculaire (5-7 jours)
5. Épithélialisation (4 jours – 4 semaines)
6. Contraction de la plaie (6-18 mois)
7. Régénération & Ré- innervation (mois- années)
Méchanisme
Un réseau de fibrine se forme pour permettre l'adhérence du greffon
Le greffon absorbe les nutriments et l'oxygène dissous de l'exsudat dans le lit tissulaire
Les bourgeons capillaires du site receveur poussent vers les vaisseaux ouverts sous la greffe
Les facteurs angiogéniques permettent la revascularisation ce qui permet un écoulement rapide et à volume élevé à travers les gros vaisseaux. Un drainage lymphatique est également mis en place
Dans les greffes de peau maillées, les kératinocytes basaux au bord de la plaie facilitent la prolifération et la migration des cellules épithéliales
L'actine dans les myofibroblastes se contracte et rapproche les bords de la plaie. La couche dermique profonde dans les greffes de pleine épaisseur peut mieux inhiber les myofibroblastes et réduire la contraction versus les greffes à épresseur fractionnée.
Les follicules pileux, les glandes sudoripares et sébacées se régénèrent dans les greffes plus épaise. L'innervation commence à la périphérie et se développe au centre")
Vitiligo Pathogenesis and Clinical Findings
 that, through complex mechanisms, stimulate the immune system’s CD8+ T-cells to destroy melanocytes
Melanocytes enter apoptotic or senescent state
↓ Functional melanocytes
↓ Melanin production
Overall loss of functional melanocytes
Vitiligo
Normal Skin
Pigmented Epidermis Dermal- Epidermal Junction
Dermis
Autoimmune destruction of melanocytes
Depigmented Epidermis
Melanocytes
Immune-mediated destruction of melanocytes (by both neutrophils and CD8+ T cells)
A depigmenting skin disorder characterized by selective loss of melanocytes
Depigmentation in areas of
↑ pressure, friction and/or trauma
Nonsegmental vitiligo
Smooth unpigmented macules or patches in bilateral, often symmetric pattern
Somatic mosaicism (mutation limited to a subset of cells) in zygote during development
Segmental vitiligo
Smooth unpigmented macules or patches in unilateral pattern not crossing midline
Vitiligo
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published February 15, 2022 on www.thecalgaryguide.com")
angor-instable-angine-de-poitrine-pathogenese-et-observations-cliniques

Hypokalemia Physiology
![Hypokalemia: Physiology
Authors: Samin Dolatabadi, Ran (Marissa) Zhang, Mannat Dhillon Reviewers: Meena Assad, Yan Yu*, Juliya Hemmett*
Beta-2 receptor stimulation
(e.g. Salbutamol)
↑ Red blood cell production
↑ Na+/K+ ATPase activity in skeletal muscle cells (moves K+ into the cell & Na+ out of cell)
↑ K+ entry into skeletal muscle cells
* MD at time of publication
Abbreviations:
• EABV – Effective Arterial
Blood Volume
• ENaC – Epithelial Sodium
Channel
• HCL – Hydrochloric acid • HCO3- –Bicarbonate ion
↑ K+ uptake by new red blood cells
↑ Intracellular shift of K+ into cells
Refeeding Syndrome Exogenous insulin
↓ K+ dietary intake
(rare cause in isolation)
↑ Insulin in response to carbohydrate load
↑ Na+/K+ ATPase activity in skeletal muscle & hepatic cells
↑ K+ entry into skeletal muscle & hepatic cells
↓ K+ availability for gastrointestinal absorption
Hypokalemia (Serum [K+] < 3.5 mmol/L)
↑ Renal K+ secretion
K+ follows the electrical gradient into tubular lumen
↑ Electronegativity of tubular lumen
↑ Na+ reabsorptionin principal cellsà Cl- left behind in tubular lumen of kidneys
Gastric acid depletionà ↓HCl
Loss of H+àShift in bicarbonate buffer system to ↑ plasma HCO3-
Plasma HCO3- above reabsorptive capacity of the proximal tubule
↑ HCO3- in the distal tubular lumen of kidneys
Vomiting
Diarrhea Laxatives
Renin secreting tumour Hyperaldosteronism Renal artery stenosis Loop and Thiazide
diuretics
Bartter’s and Gittelman’s syndrome
Liddle syndrome
Extracellular fluid volume depletion
↓ EABV ↑ Renin secretion
↓ Afferent arteriole pressure perfusing kidneys
Renin-Angiotensin- Aldosterone System (RAAS) activationà ↑ Aldosterone release from the adrenal cortex
↑ Expression of ENaC (Na+ reabsorption) in principal cells of the cortical collectingduct)
+ ↑Na &
water excretion in kidneys
↓ EABV
Genetic condition leading to inability to degrade ENaC channels in principal cells of the cortical collecting duct
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published March 6, 2019, updated Jan 23, 2022 on www.thecalgaryguide.com
Hypokalemia: Physiology
Authors: Samin Dolatabadi Reviewers: Meena Assad Dr. Juliya Hemmett* * MD at time of publication
TTKG > 4 with N/↑ EABV in hypokalemia is inappropriate and a principal cell problem.
β2 Stimulation (e.g., Salbutamol)
↑ Na+/K+ ATPase activity
↑ K+ entry into cell
↑ RBC Production
↑ Cell production
↑ K+ uptake by new cells
↓ Extracellular ↑ Insulin in response to H+
carbohydrate load
↑ Na+/H+ antiporter activity (movement of H+ out of cell and Na+ into cell)
↑ Intracellular Na+
↑ Na+/K+ ATPase activity ↑ K+ entry into cell
↑ Intracellular Shift of K+
Notes:
Refeeding Syndrome
Insulin
Alkalemia
•
Abbreviations:
• CCD – Cortical Collecting Duct
• EABV – Effective Arterial Blood Volume
• RAAS – Renin-Angiotensin-Aldosterone System • TTKG – Trans-tubular Potassium Gradient
• ENaC – Epithelial Sodium Channel
↓ K+ Intake (Rare cause in isolation)
↓ K+ availability
Diarrhea, Vomiting, Laxatives
↑ Gastrointestinal loss of K+
Polyuria
↑ Renal loss of K+ (TTKG < 4 as principal cell is working appropriately but small amount of K+ is lost per urination)
Hypokalemia (Serum [K+] < 3.5 mmol/L)
Liddle Syndrome Hyperaldosteronism Renin Secreting Tumour
Renal artery stenosis
Loop and Thiazide Diuretics
Bartter’s and Gittelman’s Syndrome
Genetic condition leading to inability to degrade ENaC channels ↑ Renin
↑ Renal K+ secretion
K+ follows the electrical gradient
Electronegative lumen
↓ Pressure perfusing the kidney
RAAS activation RAAS activation
↑ Aldosterone
↑ Na+ and water excretion
↓ EABV
↑ Expression of ENaC in principal cells of CCD
↑ Na+ reabsorption
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published March 6, 2019 on www.thecalgaryguide.com](https://calgaryguide.ucalgary.ca/wp-content/uploads/2019/03/Hypokalemia-Physiology.jpg "Hypokalemia: Physiology
Authors: Samin Dolatabadi, Ran (Marissa) Zhang, Mannat Dhillon Reviewers: Meena Assad, Yan Yu*, Juliya Hemmett*
Beta-2 receptor stimulation
(e.g. Salbutamol)
↑ Red blood cell production
↑ Na+/K+ ATPase activity in skeletal muscle cells (moves K+ into the cell & Na+ out of cell)
↑ K+ entry into skeletal muscle cells
* MD at time of publication
Abbreviations:
• EABV – Effective Arterial
Blood Volume
• ENaC – Epithelial Sodium
Channel
• HCL – Hydrochloric acid • HCO3- –Bicarbonate ion
↑ K+ uptake by new red blood cells
↑ Intracellular shift of K+ into cells
Refeeding Syndrome Exogenous insulin
↓ K+ dietary intake
(rare cause in isolation)
↑ Insulin in response to carbohydrate load
↑ Na+/K+ ATPase activity in skeletal muscle & hepatic cells
↑ K+ entry into skeletal muscle & hepatic cells
↓ K+ availability for gastrointestinal absorption
Hypokalemia (Serum [K+] < 3.5 mmol/L)
↑ Renal K+ secretion
K+ follows the electrical gradient into tubular lumen
↑ Electronegativity of tubular lumen
↑ Na+ reabsorptionin principal cellsà Cl- left behind in tubular lumen of kidneys
Gastric acid depletionà ↓HCl
Loss of H+àShift in bicarbonate buffer system to ↑ plasma HCO3-
Plasma HCO3- above reabsorptive capacity of the proximal tubule
↑ HCO3- in the distal tubular lumen of kidneys
Vomiting
Diarrhea Laxatives
Renin secreting tumour Hyperaldosteronism Renal artery stenosis Loop and Thiazide
diuretics
Bartter’s and Gittelman’s syndrome
Liddle syndrome
Extracellular fluid volume depletion
↓ EABV ↑ Renin secretion
↓ Afferent arteriole pressure perfusing kidneys
Renin-Angiotensin- Aldosterone System (RAAS) activationà ↑ Aldosterone release from the adrenal cortex
↑ Expression of ENaC (Na+ reabsorption) in principal cells of the cortical collectingduct)
+ ↑Na &
water excretion in kidneys
↓ EABV
Genetic condition leading to inability to degrade ENaC channels in principal cells of the cortical collecting duct
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published March 6, 2019, updated Jan 23, 2022 on www.thecalgaryguide.com
Hypokalemia: Physiology
Authors: Samin Dolatabadi Reviewers: Meena Assad Dr. Juliya Hemmett* * MD at time of publication
TTKG > 4 with N/↑ EABV in hypokalemia is inappropriate and a principal cell problem.
β2 Stimulation (e.g., Salbutamol)
↑ Na+/K+ ATPase activity
↑ K+ entry into cell
↑ RBC Production
↑ Cell production
↑ K+ uptake by new cells
↓ Extracellular ↑ Insulin in response to H+
carbohydrate load
↑ Na+/H+ antiporter activity (movement of H+ out of cell and Na+ into cell)
↑ Intracellular Na+
↑ Na+/K+ ATPase activity ↑ K+ entry into cell
↑ Intracellular Shift of K+
Notes:
Refeeding Syndrome
Insulin
Alkalemia
•
Abbreviations:
• CCD – Cortical Collecting Duct
• EABV – Effective Arterial Blood Volume
• RAAS – Renin-Angiotensin-Aldosterone System • TTKG – Trans-tubular Potassium Gradient
• ENaC – Epithelial Sodium Channel
↓ K+ Intake (Rare cause in isolation)
↓ K+ availability
Diarrhea, Vomiting, Laxatives
↑ Gastrointestinal loss of K+
Polyuria
↑ Renal loss of K+ (TTKG < 4 as principal cell is working appropriately but small amount of K+ is lost per urination)
Hypokalemia (Serum [K+] < 3.5 mmol/L)
Liddle Syndrome Hyperaldosteronism Renin Secreting Tumour
Renal artery stenosis
Loop and Thiazide Diuretics
Bartter’s and Gittelman’s Syndrome
Genetic condition leading to inability to degrade ENaC channels ↑ Renin
↑ Renal K+ secretion
K+ follows the electrical gradient
Electronegative lumen
↓ Pressure perfusing the kidney
RAAS activation RAAS activation
↑ Aldosterone
↑ Na+ and water excretion
↓ EABV
↑ Expression of ENaC in principal cells of CCD
↑ Na+ reabsorption
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published March 6, 2019 on www.thecalgaryguide.com")
Hypokalemia Physiology
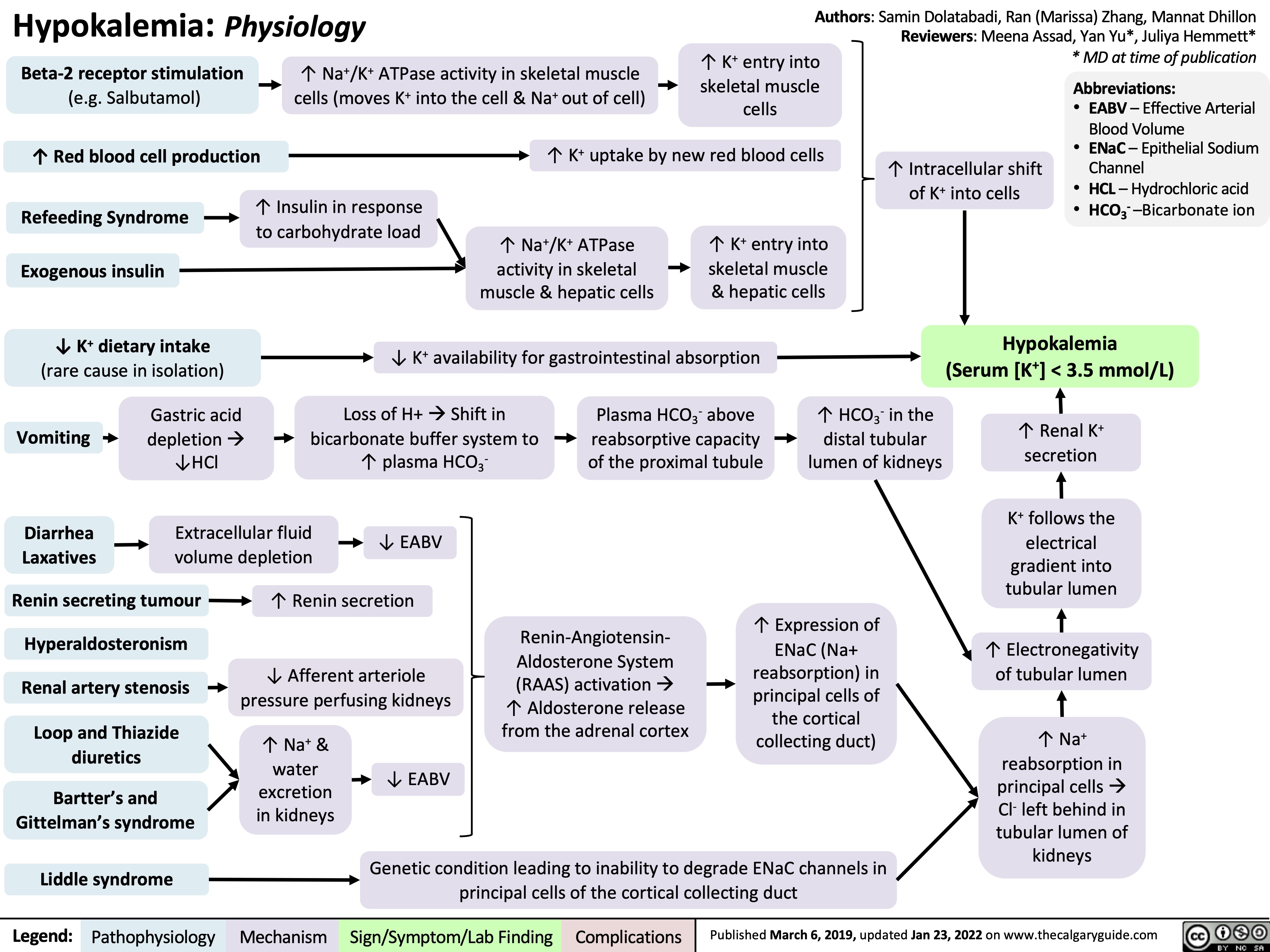
angine-de-poitrine-angor-stable-pathogenese-et-decouvertes-cliniques

hip-osteoarthritis-pathogenesis-and-clinical-findings

Trauma
Infection (e.g. septic arthritis)
Anatomical abnormality (e.g. developmental hip dysplasia)
↑ Cartilage stiffness and degeneration ↑ Friction in the hip joint with movement
Changes in normal hip architecture Abnormal load on the hip joint
Radiographic changes
See Osteoarthritis (OA): X-Ray Features slide
Repeated attempts to repair cartilage damage and bone
Bone spur (bony growth) formation in joint
Joint fluid accumulation from mild inflammation of the joint
Stiffness
Limited joint movement
Hip Osteoarthritis
Multifactorial condition that manifests as degeneration of cartilage, bone, and synovium in the hip joint
↓ Cartilage between femoral head and acetabulum ↑ Bone on bone contact
Mechanical symptoms (crepitus, locking, clicking, catching)
Bones rubbing together activates nociceptors within the hip joint
Nociceptors further aggravated by actions that ↑ bone on bone contact (e.g. prolonged loading, movements that ↓ joint space)
Worsening pain in the groin region
Favour the affected leg to avoid pain while walking
↓ Space for the femoral head to move
↓ Range of motion of affected hip joint
Limited range of motion when walking
Antalgic gait
↓ Use of muscles around the affected hip
Muscular atrophy around the hip
C Sign: patient identifies location of pain by cupping lateral hip with one hand, which creates a “C” shape with that hand
Pain in groin region
↑ Sensitivity of surrounding nerves
Radiating pain to buttocks and knee
Inactivity
Hip instability when walking
Muscle contractures
↑ Falls
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published March 6, 2022 on www.thecalgaryguide.com")
topical-retinoids-mechanisms-and-side-effects

↑ Corneocyte shedding
Excessive corneocyte sheddingà desquamation of the skin
Skin peeling
Moisture loss and irritation
Xerosis, pruritis, erythema (dryness, itchiness, redness)
Retinoid molecules bind nuclear receptors in epidermal keratinocytes
Bound nuclear receptors form heterodimers
Heterodimers bind retinoic acid response elements, which transcribe retinoic acid-responsive genes
Gene products inhibit leukocyte migration, toll-like receptors on monocytes, and transcription factor activator protein-1
Inhibits release of pro- inflammatory cytokines (IL-6, IL-12, TNF-α and IFN-γ)
Shed corneocytes expel comedonal contents (open and closed types)
Open comedone
Inhibits C. acnes induced pro- inflammatory pathways
Inhibits microcomedone formation (acne lesion precursor)
↑ Phagocytosis of vascular cell adhesion molecule 1 (mediates cytokine response)
Anti-inflammatory
Reduction of number and size of acne lesions
Comedolytic
Closed comedone
Epidermal layer
C. acnes
Normal Skin
Dermal-Epidermal Junction Dermal layer Pilosebaceous follicle
Healthy hair follicle
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published March 13, 2022 on www.thecalgaryguide.com")
cough-physiology
* Yan Yu* Stephen Field* * MD at time of publication
Systemic immune reaction
Inflammation of alveoli Cytokine release
Cytokines bind to sensory neurons
Neurons ↑ receptor sensitivity and number
↑ sensation of stimuli Ionic flux across neuronal
membrane
Vagus neurons depolarize
Thinly myelinated axons (Aδ fibers)
General chemicals, heat, cold, and hydrogen ions
Transient receptor potential vanilloid (TRPV)/Transient receptor potential ankyrin (TRPA) receptors activate
TRPV/TRPA activation opens ion channels on vagus neurons
Unmyelinated axons (C fibers)
carry depolarization
Vagus nerves conduct depolarization to
carry depolarization
SUMMARY
Afferent pathways activated
Cough center (medulla)
Efferent pathways Respiratory muscles
nucleus of the solitary tract
Medullary nuclei activated, stimulates:
Phrenic nerves
Diaphragm contraction
Spinal nerves
External intercostal contraction
Vagal nerves
Glottis closure
Spinal nerves
Thoracic, abdominal, and pelvic muscle contraction
Vagal nerves
Glottis opens, allowing forceful expiration
Intrathoracic pressure and volume decrease
Volume in lungs increases
Intrathoracic pressure increases
Mechanical Cough
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published Sept 1, 2019, updated Dec 5, 2021 on www.thecalgaryguide.com")
pancreatite-aigue-pathogenese-et-signes-cliniques

pression-veineuse-jugulaire-pvj-signe-de-kussmaul-explique
: signe de Kussmaul expliqué")
isotretinoin-systemic-retinoid-mechanisms-and-side-effects
:
Mechanisms and Side Effects
Isotretinoin
Authors: Ayaa Alkhaleefa Reviewers: Mehul Gupta, Ben Campbell Stephen Williams, Lauren Lee, Yan Yu*, Laurie Parsons* * MD at time of publication
↑ Apoptotic signaling in sebocytes
Inhibits androgen nuclear receptors responsible for sebum secretion
↓ Sebaceous lipogenesis
↑ Apoptotic signaling in neural crest cells during embryonic development
Alterations in hindbrain, neural crest, otic anlage, and reduced pharyngeal arch in embryo
Craniofacial, cardiac, thymic, and central nervous system malformations in fetus
Isotretinoin isomerizes to all-trans retinoic acid (ATRA)
ATRA enters cell nucleus and binds retinoic acid receptors and retinoic X receptors
ATRA induces tumour necrosis factor-related apoptosis-inducing ligand
↑ Apoptotic signaling in epidermal keratinocytes
↑ Expression of FoxO1
↑ Expression of p53 (tumour suppressor)
Release of caspases 3, 6, 7, and 9
↑ Cell cycle inhibitors p21 and p27
↓ Pro-survival proteins (Survivin)
Sebaceous gland involution
Sebum suppression
C. acnes unable to break down sebum into pro- inflammatory lipids
↓ Colonization with C. acnes
Teratogenicity
↑ Cornification (death) of epidermal keratinocytes
↓ Corneodesmosomes (main adhesive structures of the stratum corneum)
↓ Cohesion of corneocytes (dead keratinocytes)
↓ Corneocyte buildup in pilosebaceous follicles
C. acnes unable to populate and release cytokines in corneocytes
Epidermis
Dermis
Pilosebaceous follicle
↓ Stratum corneum thickness
↑ Trans-epidermal water loss
Dryness, peeling & inflammation
of lips (cheilitis), skin (dermatitis) & mucosa (mucositis)
↓ Comedogenesis
Sebum
Hair follicle Dermal-
Epidermal Junction
Sebaceous gland (sebocytes)
Death of sebocytes in pilosebaceous follicles
Reduction of number and size of acne lesions
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published March 20, 2022 on www.thecalgaryguide.com")
Diabetische Nephropathie: Pathogenese

Verdachtsdiagnose tiefe Beinvenenthrombose (TVT): Pathogenese und Komplikationen
: Pathogenese und Komplikationen")
Hypovolämischer Schock: Pathogenese, Komplikationen und klinische Befunde

Akute Pankreatitis: Pathogenese und klinische Befunde

Akute Pankreatitis: Komplikationen

complications-of-pulmonary-embolism
![Complications of Pulmonary Embolism
Authors:
Sravya Kakumanu, Dean Percy, Yan Yu
Reviewers:
Tristan Jones, Ciara Hanly, Jieling Ma (马杰羚), Ben Campbell, Dr. Man-Chiu Poon*, Dr. Lynn Savoie*, Dr. Tara Lohmann * * MD at time of publication
IF CHRONIC:
Unresolved clot after 2 years leading to fibrosis of pulmonary vasculature
Chronic Thromboembolic Pulmonary Hypertension (CTEPH)
(<5% of PE cases)
Venous Stasis Hypercoagulable state
Vessel Injury
Virchow’s Triad (*See Suspected Deep Vein Thrombosis slide)
Deep Vein Thrombosis
Clot migrates from deep limb veins à femoral àiliac veins
ACUTE/MASSIVE PE:
Clot obstructs pulmonary arterial or arteriolar flow
Lung infarction (tissue death) from ischemia
Inflammatory cells migrate to site and release cytokines
↑ Permeability of blood vessels
Permeability-driven (exudate) fluid leakage into pleural space
Pleural Effusion
Clot migratesàinferior vena cava àright atrium (RA) of heartà right ventricle (RV) à gets lodged in pulmonary arteries/arterioles
Pulmonary Embolism (PE)
↑ RV afterload
↑ RV pressure and expansion
Well-ventilated (V) areas of lung do not receive adequate blood supply (Q)
V/Q Mismatch
Leftward shift of ventricular septum
↓ Left ventricle filling in diastole
↓ Cardiac output
Obstructive Shock
Impaired heart filling
Pulseless Electrical Activity
(ECG activity in absence of palpable pulse)
Back up of pressure in systemic venous system
↑ Pressure in capillaries draining parietal pleura
Pressure-driven (transudate) fluid leakage into pleural space
For signs and symptoms, see the Obstructive Shock slide
For signs and symptoms refer to CTEPH slide
Chronic ↑ RV afterload
↑ Stretching of myocytes causing RV hypertrophy and dilation
↓ RV ejection fraction
Right Heart Failure
“Cor Pulmonale”
For signs and symptoms, see the Right Heart Failure slide
Failure to oxygenate blood
Type I Respiratory Failure
Hypoxemic: patient has ↓ blood [O2]
IF MASSIVE PE (less common):
↑ Alveolar dead space
Failure to ventilate
Type II Respiratory Failure Hypercapnic: patient has ↑ blood [CO2]
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published August 7, 2012, updated Mar 31, 2022 on www.thecalgaryguide.com](https://calgaryguide.ucalgary.ca/wp-content/uploads/2014/09/Complications-of-Pulmonary-Embolism-2022.jpg "Complications of Pulmonary Embolism
Authors:
Sravya Kakumanu, Dean Percy, Yan Yu
Reviewers:
Tristan Jones, Ciara Hanly, Jieling Ma (马杰羚), Ben Campbell, Dr. Man-Chiu Poon*, Dr. Lynn Savoie*, Dr. Tara Lohmann * * MD at time of publication
IF CHRONIC:
Unresolved clot after 2 years leading to fibrosis of pulmonary vasculature
Chronic Thromboembolic Pulmonary Hypertension (CTEPH)
(<5% of PE cases)
Venous Stasis Hypercoagulable state
Vessel Injury
Virchow’s Triad (*See Suspected Deep Vein Thrombosis slide)
Deep Vein Thrombosis
Clot migrates from deep limb veins à femoral àiliac veins
ACUTE/MASSIVE PE:
Clot obstructs pulmonary arterial or arteriolar flow
Lung infarction (tissue death) from ischemia
Inflammatory cells migrate to site and release cytokines
↑ Permeability of blood vessels
Permeability-driven (exudate) fluid leakage into pleural space
Pleural Effusion
Clot migratesàinferior vena cava àright atrium (RA) of heartà right ventricle (RV) à gets lodged in pulmonary arteries/arterioles
Pulmonary Embolism (PE)
↑ RV afterload
↑ RV pressure and expansion
Well-ventilated (V) areas of lung do not receive adequate blood supply (Q)
V/Q Mismatch
Leftward shift of ventricular septum
↓ Left ventricle filling in diastole
↓ Cardiac output
Obstructive Shock
Impaired heart filling
Pulseless Electrical Activity
(ECG activity in absence of palpable pulse)
Back up of pressure in systemic venous system
↑ Pressure in capillaries draining parietal pleura
Pressure-driven (transudate) fluid leakage into pleural space
For signs and symptoms, see the Obstructive Shock slide
For signs and symptoms refer to CTEPH slide
Chronic ↑ RV afterload
↑ Stretching of myocytes causing RV hypertrophy and dilation
↓ RV ejection fraction
Right Heart Failure
“Cor Pulmonale”
For signs and symptoms, see the Right Heart Failure slide
Failure to oxygenate blood
Type I Respiratory Failure
Hypoxemic: patient has ↓ blood [O2]
IF MASSIVE PE (less common):
↑ Alveolar dead space
Failure to ventilate
Type II Respiratory Failure Hypercapnic: patient has ↑ blood [CO2]
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published August 7, 2012, updated Mar 31, 2022 on www.thecalgaryguide.com")
proximal-biceps-tendon-rupture
 place more stress on the proximal biceps tendon
Repetitive microtrauma and inflammation of biceps tendon Compromised biceps tendon integrity from intra-tendinous collagen degeneration, disorientation, and thinning
Increased involvement in physical labour and hobbies
Impaired cell proliferation and healing after injury
Exact mechanisms remain unclear
Bicep tendon involved in shoulder stabilization, elbow flexion, and forearm supination
Biceps muscle cannot contract with as much force when one head is detached
Heavy lifting or fall on outstretched handà sudden tension in the proximal biceps tendon
Proximal biceps tendon rupture
Long head of proximal biceps tendon detaches from bony attachment on supraglenoid tubercle
Audible “pop” at time of injury
No tension to hold proximal bicep tendon head in place
Proximal bicep tendon head retracts distally
Biceps muscle fatigue and cramping
Weakness in shoulder flexion, elbow flexion & supination
Nerve damage (musculocutaneous nerve) during injury
Local inflammatory response at site of injury
Surrounding blood vessels damaged during injury
Blood collection under skin surface
Immediate bruising over rupture site
Sudden and sharp pain at shoulder with initial injury
Inflammatory response aggravates musculocutaneous nerve
Swelling over rupture site
+ Speed test: affected elbow extended and forearm supinated. Shoulder flexed against resistanceàpain at bicipital groove
+ Neer test: on affected side, scapula stabilized while arm passively flexed and internally rotatedàpain at bicipital groove
+ Yergeson test: affected elbow flexed at 90° and forearm pronated. Forearm supinated against resistance àpain at bicipital groove
“Popeye” deformity:
mass in distal upper arm due to excessive
retraction of biceps muscle distally, away from its origin
Palpable gap
between proximal biceps tendon and bicipital groove
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published April 20, 2022 on www.thecalgaryguide.com")
distal-biceps-tendon-rupture
 during forearm pronation
Repetitive compressionà tendon degeneration
↑ Involvement in physical labour and hobbies
Exact mechanisms remain unclear
Impaired cell proliferation
Biceps tendon inserts at the radial tuberosity to control forearm supination and flexion
Compromised biceps tendon integrity from intra-tendinous collagen degeneration, disorientation, and thinning
Sudden eccentric force to flexed elbow (forced lengthening of a contracted bicep muscle)àtension in the distal biceps tendon
Biceps muscle cannot contract when distal tendon is detached from insertion site
Distal biceps tendon rupture
Complete detachment of distal biceps tendon at the attachment site on the radial tuberosity
Audible “pop” at time of injury
Weak forearm supination and flexion
+ Ruland biceps squeeze test:
Affected elbow held in 60-80° of flexion, with forearm slightly pronated. Distal biceps muscle squeezed by practitioner à forearm does not supinate
Local inflammatory response at site of injury
Inflammatory response aggravates musculo- cutaneous nerve
Pain over rupture site after initial injury
Swelling over rupture site
Nerve damage (musculocut aneous nerve) during injury
Sudden and sharp pain at
elbow at initial injury
Surrounding blood vessels damaged during injury
Blood collects under skin surface
Immediate bruising over rupture site
Biceps tendon retracts proximally
High tension during tendon retraction
Fragment of bone torn off from tendon insertion site on radial tuberosity
Avulsion fracture of
radial tuberosity
Connective tissue attachment (aponeurosis) torn during injury
Palpable gap between distal end of biceps tendon & radial tuberosity
+ Hook test: Elbow actively flexed to 90° and forearm supinated. Index finger of examiner cannot be placed beneath distal aspect of biceps tendon (>1 cm deep)
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published April 20, 2022 on www.thecalgaryguide.com")
肺泡-动脉氧分压差-为何它会存在,为什么我们要

肺泡-动脉氧分压差-关于公式的说明-科学正确的解

肺泡-动脉氧分压差-关于公式的说明简易说明

cognitive-behavioural-therapy-cbt-principles-and-reasoning
: Principles and Reasoning
A stressor, e.g., failing a test, activates underlying maladaptive core beliefs
and personal rules (ie. schema) based in Beck’s cognitive triad:
1. Negative Views about the World 2. Negative Views about Oneself
e.g., “Everyone is against me because I am e.g., “I’m worthless and inadequate.” worthless.”
Authors: Erik Fraunberger, Alison Darnley Reviewers: Allen Vorobeichik, Brooke Fallis, Ran (Marissa) Zhang, Margaret Oakander* * MD at time of publication
3. Negative Views about the Future
e.g., “I’ll never be good at anything.”
Negative bias in thought patterns, attention, memory, and interpretation of events/stimuli
Negative automatic thoughts activated in certain situations e.g., ”I will never succeed in anything I do”
Emotional & behavioural reactions
e.g., Feeling hopeless (emotional reaction) and unmotivated in school (behavioural reaction)
Patient engages with the therapist to identify cognitive distortions about self, others, and the world (not an exhaustive list)
Therapist and patient problem solve together and challenge existing cognitive distortions using these techniques (not exhaustive list)
Therapist works with patient to improve awareness of cognitive distortions and employ techniques in daily life
Over time with continued support and practice, the patient’s negative schemas are addressed, emotional and behavioral symptoms improve
Personalization: assigning disproportionate personal blame
Disattribution of bad events to the individual and recognizing role of environmental factors
Catastrophizing:
dwelling on the worst possible outcome
Estimate actual probabilities, clearly state facts of situation & emphasize worst case scenario did not occur
Dichotomous thinking: absolute thinking in the form of two extremes
Demonstrate that a continuum of outcomes is possible
Overgeneralization: making conclusions based on one event/situation
Expose faulty logic and discuss similarities and differences in closely related events
Improved function and ability to adapt to life stressors
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published April 26, 2022 on www.thecalgaryguide.com")
irritable-bowel-syndrome-ibs-pathogenesis-and-clinical-findings
: Pathogenesis and clinical findings
Authors: Ben Campbell Reviewers: Mehul Gupta Kiana Hampton Yan Yu* Edwin Cheng* * MD at time of publication
Note:
*Several signaling pathways in the gut are believed to play a role in IBS. The serotonin pathway is represented here for illustrative purposes, and it is a common target of therapy.
Pathogenesis is multifactorial, not fully understood, and may arise from any one or a combination of these factors
Genetic factors
are believed to influence many of these pathways
Environmental factors
Gut infection, ischemia, injury, chronic low-level inflammation, altered microbiome
Abnormal gut chemical milieu (cytokines, lymphocytes, mast cells, hormones)
One common pathway: altered quantity or activity of serotonin-releasing enterochromaffin * cells and serotonin reuptake transporters in gut
Serotonin is a key stimulator of gut muscle contraction and sensory signaling
Altered bowel motility
Bowel motility can ↑, ↓ or alternate ↑/↓
Psychosocial factors
Anxiety, depression, adverse experiences, dysregulated stress response
Brain can alter normal reflexes of enteric nervous system (e.g. via hypothalamic stress hormone release)
↑ Activity of sensory receptors in gut + recruitment of formerly ”silent” receptors
Altered visceral bowel sensation
Visceral hypersensitivity
Complex mechanisms
↓ Brain’s ability to modulate pain signals from gut;
↑ vigilance to pain
Irritable Bowel Syndrome
Disorder of brain-gut interaction with gastrointestinal manifestations
Not a predominantly inflammatory condition
During times of ↑ bowel motility:
No lesions in gut mucosa
(Absence of ”red flags” for organic disease)
↑ Impacts of other factors that precipitate diarrhea
Dietary sensitivity
(e.g. to intake of fermentable oligo/di/ monosaccharides and polyols— FODMAPs—which draw water into gut lumen by osmosis)
↑ Impacts of other factors that precipitate constipation
Dietary sensitivity (e.g. to inadequate intake of motility- promoting fibre)
During times of ↓ bowel motility:
↑ Time for water absorption from feces and bacterial fermentation
↑ Hard stool and trapped gas in bowel
↑ Number and sensitivity of active pain receptors (nociceptors) in gut
Hyperalgesia
↑ Response to painful stimulus
Stretch receptors in gut stimulate pain in normally unconscious neural pathways
Allodynia
Pain in response to normal stimulus
↑ Peristalsis
↓ Time for water absorption from feces
↑ Water volume in gut lumen
↑ Stretch / stimulation of visceral afferent nerves in gut
Absence of bloody stool and anemia
Absence of nocturnal symptoms and inflammatory markers
Constipation
Bloating
Diffuse abdominal pain
All symptoms may exacerbate psychosocial factors
Bowel distension
Diarrhea
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published April 30, 2022 on www.thecalgaryguide.com")
成人肺炎发病机制和临床表现

过敏性肺炎-发病机制和临床发现

肺栓塞症状和体征

深静脉血栓形成——腘静脉、股静脉、髂静脉 栓子脱落移动à下腔静脉 IVC à右心房 à 右心室à
肺血管系统
大血块(鞍状栓塞)滞留在肺动脉中
小血块滞留在肺小动脉中
鞍状栓子(肺动脉阻塞)
血液回流至右心
右心组织受到牵拉
作者: Dean Percy Yan Yu 审稿人: Tristan Jones Julia Heighton Man-Chiu Poon* Lynn Savoie* 译者:Jieling Ma(马杰羚) 翻译审稿人: Ying Li(李颖), Yonglin Mai(麦泳琳) * 发表时担任临床医生
若肺外周循环阻断,可致胸膜
下肺组织缺血和梗塞
栓子导致肺动脉/小动脉血流↓
输送到肺部,呼 输送至肺泡进行氧
肺通气良好 (V) 区域没有获得足 够的血流供应 (Q); 反之亦然
肺V/Q扫描提示: V/Q 异常
信号传导至中枢 使心率↑
刺激壁层胸膜上躯体感觉
神经末梢
缺血组织发生炎症反
应,粘附胸膜
胸膜摩擦音
听诊时听到类似砂纸
的声音
出的CO2↓
合的低氧血液↓
主动脉/颈动脉化 学感受器检测到血 液低O2浓度(O2 饱和度↓)
信号传导至中枢 呼 吸频率↑
呼出二氧化碳减
少,血液中二氧
化碳潴留,刺激
延髓化学感受器,
呼吸频率↑
疼痛刺激肾上腺素系统反应
心动过速
胸膜炎性胸痛
呼吸困难/气短 (SOB)
PE最敏感指标,但特异性不高 伴随每次呼吸出现的局部胸痛
图注:
病理生理学
机制
体征/症状/实验室检查
并发症
2019年6月15日重新发布于 www.thecalgaryguide.com")
疑似深静脉血栓形成-dvt-发病机制和并发症
: 发病机制和并发症
作者: Dean Percy Yan Yu 审稿人: Tristan Jones Ryan Brenneis Man-Chiu Poon* Maitreyi Raman* 译者:Jieling Ma(马杰羚) 翻译审稿人: Ying Li(李颖), Yonglin Mai(麦泳琳) * 发表时担任临床医生
Notes:
恶性肿瘤 血小板活化
遗传性疾病
遗传性易栓症(如:PC、PS、抗 凝血酶III、因子V Leiden突变)血 液易凝性↑
雌激素促进血液高凝
,尤其是存在其他危
险因素的情况下
• •
静脉血栓引起肺栓塞, 动脉血栓可引起中风; 既往 DVT 病史是当前 DVT复发 的危险因素
创伤/手术
促凝细胞因子异常释放
血栓形成增加
全身性损伤 高凝状态 凝血级联反应激活 血液在外界刺激下,
凝固能力↑
血管损伤 Virchow 受损细胞和内皮组织 三要素
释放组织因子入血, 与vWF结合
脂肪含有大量芳香
酶,将更多的雄激
素转化为雌激素
久坐不动生活方式,
静脉回流不良
高血压
细菌
人工瓣膜
血管壁机械损伤
粘附/侵袭血管壁 瓣膜表面异常
静脉血淤滞
血管损伤部位血流慢, 凝血因子在该部位聚集
肥胖
1.血液回流至深静脉(血液淤滞,处于高 栓子导致静脉瓣膜功能受损 凝状态)
血栓形成通常发生在下肢静脉
肌肉运动↓ 静脉血流量↓
骨折、固定、卧床休息、 长时间乘坐汽车/飞机
肺部血栓栓塞
血栓栓塞
2.下肢静脉回流需对抗重力(血液淤滞) 3.下肢静脉瓣膜易发生返流(血液淤滞)
怀孕,口服避孕药
(OCP)
静脉功能不全
凝块阻碍血液回流至心脏。 腿部血液淤 积,导致不对称性腿部水肿和静脉炎症 (发红、发热、压痛)
-*肺栓塞(急性危及生命的并发症) -慢性血栓栓塞性肺动脉高压
图注:
病理生理
机制
体征/症状/实验室检查
并发症
2019年9月1日重新发表于 www.thecalgaryguide.com")
张力性气胸发病机制,临床表现和x线表现
, Yonglin Mai (麦泳琳)*, Naushad Hirani*, Yan Yu* 译者: Huiting Wang(王慧婷), Yang Xiang (向杨) 翻译审稿人: Ran Zhong(钟然), Yonglin Mai (麦泳琳), Zesheng Ye(叶泽生) * 发表时担任临床医生
继发性气胸 (有基础性肺疾病)
手术
(如: 胸腔穿刺术,中心导管 正压通气
置入,肺结节活检)
原发性气胸 (无基础性肺疾病)
气压性创伤
医源性气胸
刺激高敏感性 起病急骤,突发刀割样胸痛 壁层胸膜 (可放射至肩部)
对侧气管移位
对侧纵膈移位
肺纹理 缺失
肺萎陷(脏层胸膜线
肋间隙增大
膈肌变平
创伤性气胸
如穿通性损伤,肋骨骨折
壁层胸膜/脏层胸膜/支气管树的破坏 胸膜裂口形成气体进入胸膜
腔的单向阀/单向活瓣 (由于生理性胸膜负压的存在)
空气进入、填充并潴留于胸膜腔 患侧胸腔内气压↑
患侧肺因气压↑塌陷
较于萎陷的 肺实质,积
气部位叩诊 时发出叩诊 音音响↑
叩诊呈鼓音, 取决于病情
轻重程度 (用手在患侧 叩击)
对侧心影移位
可见脏层 胸膜线 (气胸线)
患侧横膈膜变平 空气无法进入 纵膈偏离患 健侧在影像学上
进入胸腔的气体将内脏和壁层胸膜
两层胸膜隔开,可见脏层胸膜线
患侧肋骨张开 (肋间隙↑)
患侧肺
侧
挤压健侧肺
可见心影和肺门
压迫上腔静脉和(或)下腔静脉 右心房血液回流受阻
声音震动无法像 胸壁扩张度 正常那样从喉部 ↓ 发出,沿着肺部 组织传至胸壁引
心输出量↓ 血液倒流回静脉系统
心动过速 低血压
颈静脉压↑ 血液氧合
↓(血氧分压)
呼吸急促
( 呼吸频率 ↑)
起共鸣
X光可见,脏 胸膜和边缘 肺纹理缺失
语音震颤↓ 呼吸音↓ (发声时胸壁可
触及的震颤) 呼吸困难
患侧肺塌陷
患侧肺泡内的
气体交换受损
通气/灌注失调、 及出现血液分流
低氧
图注:
病理生理
机制
体征/症状/实验室检查
X-光检查结果
2013年12月11日首次发表,2020年10月24日更新www.thecalgaryguide.com
)")
abnormal-uterine-bleeding-aub-pathogenesis-and-clinical-findings
 release from hypothalamus
↑Prolactin (see Feedback Loop: Prolactin)
Exogenous estrogen (e.g. estrogen-only birth control)
↑ Peripheral adipose tissue
↑ Aromatase (enzyme present in adipose tissue)
↑ Conversion of androgens to estrogens (aromatization)
Excessive stress, exercise, low body mass (mechanism unclear)
↓ Hypothalamic gonadotropin releasing hormone secretion
↓Luteinizing hormone and follicle stimulating hormone release from pituitary
↑ Estrogen
Heavier bleeding
Angiogenesis in tumour tissue
Polycystic ovarian syndrome (see slide)
Pelvic inflammatory disease
Immature hypothalamic- pituitary-ovarian axis
(an immature axis is transient in most females)
↓ Positive feedback of estrogen in late follicular stage
No LH surge
Adenomyosis (endometrial tissue grows into uterus muscular wall)
Endometrial polyps
Retained products of contraception
Infection
Inflammation of endometrium
Endometrium is more fragile
Anovulatory Bleeding
Leiomyoma (benign tumour in myometrium)
Tara Shannon, Hannah Yaphe, Dr. Sarah Glaze* * MD at time of publication
Ovarian Scarring
Foreign bodies
Intrauterine device perforation
Physical trauma
Impaired follicle maturation
Anovulation
Corpus luteum does not form
No ovarian progesterone production
↑ Estrogen to progesterone ratio
Proliferative effect of estrogen unopposed
Endometrial proliferation
No progesterone to organize growing endometrium
Disorganized endometrium overgrows and sloughs off
Irregular bleeding
Menopause
Premature ovarian insufficiency
Intermittent congestion of polyp blood supply
Transient ischemia and slight polyp necrosis
Altered growth factor production
Vascular dysregulation and leakier vessels
Coagulopathies (e.g. von Willebrand disease)
Impaired hemostasis
Invasion of normal tissue
↓ Estrogen (hypoestrogenism)
Atrophy of endometrium and vulvovaginal tissue
Dry endometrial surfaces (↓ fluid to prevent friction)
Endometrial carcinoma
Micro- erosions of epithelium
Inflammation
Evidence unclear
Heavier and more irregular bleeding
Spotting
Heavier and more irregular bleeding
Light bleeding and spotting
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published May 2, 2022 on www.thecalgaryguide.com")
rhumatisme-psoriasique-pathogenese-et-resultats-cliniques
![rhumatisme-psoriasique-pathogenese-et-resultats-cliniques
Psoriatic Arthritis: Pathogenesis and clinical findings
Auteur:
Payam Pournazari
Rédacteurs:
Yan Yu Scott Rapske Liam Martin* Traducteurs: Dianne Ganeswaran Stephen Williams Sylvain Coderre* * MD au moment de publication
Note:
L’arthrite rhumatoïde peut survenir avant ou après le psoriasis (la pathogenèse est la même).
Augmentation de l’activité ostéoclastique
Érosion de l’os sous- chondral mais formation de nouvelle osseuse ailleurs dans l’articulation
Radiographie: la périostite; les syndesmophytes, érosions, déformation du crayon en godets (les articulations IP), ankylose des articulations IP
HLA B27, Cw6 et d’autres sous- types
Antécédents familiaux positifs
Activation des cellules T (mécanisme inconnu)
Infiltration du tissu synovial de l’articulation par les cellules T et B, cellules tueuses naturelles et les macrophages
Production augmentée des molécules inflammatoires (les facteurs de nécrose tumorale [TNFs], les interleukines, etc.) qui agissent de façon systémique (dans tout le corps)
Dans les tendons et le tissu conjonctif
Inflammation des enthèses et du tissu conjonctif provoque le gonflement
Dans la peau
Réaction immunitaire détruisant les structures de la peau et des ongles (Voir diapositive Psoriasis)
Le psoriasis en plaques sur la peau, dépressions punctiformes, onychorrhexie, taches d’huile, onycholyse, décoloration et hyperkératose
Angiogenèse et vascularite dans les vaisseaux sanguins de la membrane synoviale
Dans les articulations
Les cytokines inflammatoires stimulent les nocicepteurs locaux
1. Oligoarthrites - asymétriques
2. Arthrite des articulations IPD
3. Polyarthrite rhumatoïde -
symétrique
4. Atteinte axiale (raison
inconnue, probablement en raison de « biomécanique », « innervation », et
« vascularisation ».
5. Arthrite mutilante (grave et destructrice)
L’enthésite
(Douleur/sensibilité à l’insertion du ligament dans l’os)
La dactylite (Inflammation de tout le doigt – les tissus mous et les articulations sont enflammés
Légende:
Physiopathologie
Mécanisme
Signe/Symptôme/Résultats de Laboratoire
Complications
Publié 10 November 2012 sur www.thecalgaryguide.com](https://calgaryguide.ucalgary.ca/wp-content/uploads/2022/05/Psoriatic-Arthritis-PsA-Pathclinical.jpg "rhumatisme-psoriasique-pathogenese-et-resultats-cliniques
Psoriatic Arthritis: Pathogenesis and clinical findings
Auteur:
Payam Pournazari
Rédacteurs:
Yan Yu Scott Rapske Liam Martin* Traducteurs: Dianne Ganeswaran Stephen Williams Sylvain Coderre* * MD au moment de publication
Note:
L’arthrite rhumatoïde peut survenir avant ou après le psoriasis (la pathogenèse est la même).
Augmentation de l’activité ostéoclastique
Érosion de l’os sous- chondral mais formation de nouvelle osseuse ailleurs dans l’articulation
Radiographie: la périostite; les syndesmophytes, érosions, déformation du crayon en godets (les articulations IP), ankylose des articulations IP
HLA B27, Cw6 et d’autres sous- types
Antécédents familiaux positifs
Activation des cellules T (mécanisme inconnu)
Infiltration du tissu synovial de l’articulation par les cellules T et B, cellules tueuses naturelles et les macrophages
Production augmentée des molécules inflammatoires (les facteurs de nécrose tumorale [TNFs], les interleukines, etc.) qui agissent de façon systémique (dans tout le corps)
Dans les tendons et le tissu conjonctif
Inflammation des enthèses et du tissu conjonctif provoque le gonflement
Dans la peau
Réaction immunitaire détruisant les structures de la peau et des ongles (Voir diapositive Psoriasis)
Le psoriasis en plaques sur la peau, dépressions punctiformes, onychorrhexie, taches d’huile, onycholyse, décoloration et hyperkératose
Angiogenèse et vascularite dans les vaisseaux sanguins de la membrane synoviale
Dans les articulations
Les cytokines inflammatoires stimulent les nocicepteurs locaux
1. Oligoarthrites - asymétriques
2. Arthrite des articulations IPD
3. Polyarthrite rhumatoïde -
symétrique
4. Atteinte axiale (raison
inconnue, probablement en raison de « biomécanique », « innervation », et
« vascularisation ».
5. Arthrite mutilante (grave et destructrice)
L’enthésite
(Douleur/sensibilité à l’insertion du ligament dans l’os)
La dactylite (Inflammation de tout le doigt – les tissus mous et les articulations sont enflammés
Légende:
Physiopathologie
Mécanisme
Signe/Symptôme/Résultats de Laboratoire
Complications
Publié 10 November 2012 sur www.thecalgaryguide.com")
tinea-capitis-tinea-corpora-and-tinea-pedis-pathogenese-et-resultats-cliniques
:
Contact direct avec des êtres humains, des animaux ou des objets inanimés qui sont infectés
Le dermatophyte envahit la couche superficielle de la peau (non vivante et kératinisée), la couche cornée
Le dermatophyte produit l’enzyme kératinase La kératinase catalyse la dégradation de
kératine (une protéine) dans la peau
Le dermatophyte s’infiltre dans les couches les plus profondes de la peau
Les kératinocytes libèrent des cytokines inflammatoires en réaction aux antigènes dermatophytes
Le dermatophyte peut se propager du site principal de l’infection aux sites avoisinants
Taches rondes roses à rouges, avec guérison au centre
Desquamation de la peau (écailles dans les espaces palmés entre les orteils: macération)
1. Trichophyton
2. Epidermophyton 3. Microsporum
Invasion du follicule pileux
La cassure de tige du cheveux
Alopécie (perte de cheveux)
Prurit (démangeaison) Érythème (rougeur)
Induration
(gonflement)
Chaleur
Réponse inflammatoire locale dans la peau
Tinea capitis Tinea corporis
Tinea pedis
Mycose du pied (pied d’athlète)
Infection du cuir chevelu Infection du tronc et des extrémités de corps
Légende:
Physiopathologie
Mécanisme
Signe/Symptôme/Résultats de Laboratoire
Complications
Publié 17 février 2020 sur www.thecalgaryguide.com")
acute-cholecystitis
![Acute Cholecystitis: Pathogenesis and clinical findings
Gallstone blocks the cystic duct, backing up bile into the gallbladder
Gallstones causing physical trauma to gallbladder wall
Irritation of adjacent diaphragm, stimulates phrenic nerve (C3-C5)
Activates stretch receptors of visceral peritoneum, stimulates foregut autonomic nerves (T5-T8)
Inflammatory mediator (i.e. prostaglandin) release by gallbladder and systemic inflammatory response
Thickened gallbladder wall on ultrasound (gold standard test)
On inspiration, the diaphragm pushes the gallbladder downward
Irritation of parietal peritoneum, stimulates somatic nerves
↑ Permeability of vessels with systemic inflammation, which leak
fluid from the blood into the interstitial space
Radiating pain to the back and right shoulder
Dull, diffuse abdominal pain referred to the epigastric region
Fever, nausea/vomiting, tachycardia
Positive Murphy’s sign (pain upon palpation of right upper quadrant [RUQ] on inspiration)
Persistent RUQ pain, abdominal guarding and peritoneal signs
Dehydration
Authors: Yan Yu, Vina Fan Reviewers: Dean Percy, Mirna Matta, Crystal Liu, Ben Campbell Maitreyi Raman* * MD at time of publication
Inflammation self-perpetuates
Irritation of inner gallbladder wall/mucosa
↑ Gallbladder lumen pressure
Intraluminal pressure exceeds arterial pressure
↓ Blood flow to gallbladder
Gallbladder ischemia
Local inflammation, loss of gallbladder mucosal integrity
Bacterial invasionàtransmural inflammation of gallbladder
Without treatment, prolonged ischemia and inflammation of the gallbladder
Gallbladder gangrene (20%)
Gallbladder perforation (20%)
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published August 4 2019, updated May 16 2022 on www.thecalgaryguide.com](https://calgaryguide.ucalgary.ca/wp-content/uploads/2015/05/Acute-Cholecystitis-2022.jpg "Acute Cholecystitis: Pathogenesis and clinical findings
Gallstone blocks the cystic duct, backing up bile into the gallbladder
Gallstones causing physical trauma to gallbladder wall
Irritation of adjacent diaphragm, stimulates phrenic nerve (C3-C5)
Activates stretch receptors of visceral peritoneum, stimulates foregut autonomic nerves (T5-T8)
Inflammatory mediator (i.e. prostaglandin) release by gallbladder and systemic inflammatory response
Thickened gallbladder wall on ultrasound (gold standard test)
On inspiration, the diaphragm pushes the gallbladder downward
Irritation of parietal peritoneum, stimulates somatic nerves
↑ Permeability of vessels with systemic inflammation, which leak
fluid from the blood into the interstitial space
Radiating pain to the back and right shoulder
Dull, diffuse abdominal pain referred to the epigastric region
Fever, nausea/vomiting, tachycardia
Positive Murphy’s sign (pain upon palpation of right upper quadrant [RUQ] on inspiration)
Persistent RUQ pain, abdominal guarding and peritoneal signs
Dehydration
Authors: Yan Yu, Vina Fan Reviewers: Dean Percy, Mirna Matta, Crystal Liu, Ben Campbell Maitreyi Raman* * MD at time of publication
Inflammation self-perpetuates
Irritation of inner gallbladder wall/mucosa
↑ Gallbladder lumen pressure
Intraluminal pressure exceeds arterial pressure
↓ Blood flow to gallbladder
Gallbladder ischemia
Local inflammation, loss of gallbladder mucosal integrity
Bacterial invasionàtransmural inflammation of gallbladder
Without treatment, prolonged ischemia and inflammation of the gallbladder
Gallbladder gangrene (20%)
Gallbladder perforation (20%)
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published August 4 2019, updated May 16 2022 on www.thecalgaryguide.com")
clostridium-difficile-infection-pathogenesis-and-clinical-findings
 Infection
Authors: Ryan Brenneis, Sravya Kakumanu Reviewers: Yoyo Chan, Sean Doherty, Vina Fan, Ben Campbell, Dr. Steve Vaughan*, Dr. Sylvain Coderre* * MD at time of publication
Community exposure
Infected close contacts
Nosocomial exposure (most common)
Poor hand hygiene and sanitization of surfaces and medical equipment
Nosocomial risk factors
Any antibiotic use
(Especially clindamycin, fluoroquinolones, penicillins, cephalosporins)
↑ Antibiotic resistant strains
Presence of pre-disposing risk factors
(Note: do not need to be present for infection)
Recent GI surgery
Chemotherapy that has antimicrobial and immunosuppressive effects
Usage of medications that reduce stomach acid (↑ pH)
↑ C. diff spores on surfaces and personnel
Contact exposure
Environmental exposure
to C. diff carriers
Inoculation of GI tract
Disruption of normal gut microbiome allowing C. diff overgrowth
Comorbidities
(>65 years old, cirrhosis, inflammatory bowel disease, enteral feeding, obesity)
via fecal-oral route
Clostridium difficile Infection of GI Tract
Spores unaffected by antibiotics germinate post-antibiotic treatment
Infection recurrence
Pseudomembranous colitis on endoscopy
(colonic ulcerations potentially seen with severe infection)
Hypotension Acute kidney injury
Release of C. diff toxin A and B inactivates Rho and Ras GTPases in colonic epithelial cells (colonocytes)
(Rho and Ras GTPases control cytoskeletal dynamics and gene expression)
Cytoskeletal disorganization and arrest of RNA synthesis causes necrosis of colonocytes and triggers host immune response
Neutrophil chemotaxis and activation
↑ Inflammation of colon
Disruption of tight junctions between colonocytes
Release of fluid into intestinal lumen and inability of colon to reabsorb it
Toxic megacolon
Bowel perforation
Bloody stool
(<10% of patients)
Abdominal cramps
Large bowel dilation from muscle paralysis
Inflammation and destruction of underlying smooth muscle
Breakdown of colonocyte cell membranes
Inflammation of visceral peritoneum
Volume depletion
Watery diarrhea: ↑ frequency, small volume
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published March 30, 2019, updated May 16, 2022 on www.thecalgaryguide.com")
Osteoporosis Pathogenesis and risk factors
.
Secondary Osteoporosis
Smoking, alcohol, ↓ exercise, chronic malnutrition, hyperthyroidism, hyperparathyroidism, corticosteroid use, ↓ vitamin D, bone malignancies, chronic kidney or liver disease, and malabsorption.
Reduced peak bone mass achieved during skeletal growth and/or an imbalance in the factors favoring bone resorption compared to bone formation in adult bone
↓ Serum vitamin D from malabsorption, poor nutrition, or chronic liver/ kidney disease
↓ Serum activated vitamin D (precursor for Ca2+ transporters in the gut)
↓ Ca2+ absorption from gut
↓ Ca2+ available for mineralization of bone matrix
Age related bone degeneration and ↓ exercise
↑ Reactive oxygen species (ROS) systemically
due to aging
↑ Oxidative stress on bone
↑ Osteocyte death in cortical bones
↓ Bone Formation
↓ Estrogen
in post-menopausal women
Hyperparathyroidism/ bone malignanciesà ↑ parathyroid hormone (PTH) or parathyroid hormone-related (PTHR) protein released
↑ PTH and PTHR peptide receptor signaling in osteocytes results in ↓ osteoprotegerin (OPG) expression
↓ OPG secretion
Hyperthyroidism à↑ T3 and T4
↑ Thyroid receptor- alpha-1 activation (located in bones)
↑ Osteoclasts (cells that break down bone)
↑ Bone Resorption
Corticosteroid use
↑ Corticosteroid binding to glucocorticoid receptors on bone suppresses OPG
expression in bone
Smoking
Multiple mechanisms (e.g. ↓ blood supply to bone, ↑ free radicals, direct effects on RANKL/RANK/ OPG bone remodeling axis leading to alterations in osteogenesis
↑ Bone Resorption and ↓ Bone Formation
Abbreviations:
• T3 - Triiodothronine • T4 - Thyroxine
↓ Stimulation of OPG transcription in osteoblasts
Authors: Ben Wajda, Arsalan Ahmad, Lance Bartel Reviewers: Alyssa Federico, Mehul Gupta,
Tara Shannon, Reza Ojaghi, Usama Malik,
Carol Hutchinson*, David Hogan*
* MD at time of publication
↓ Inhibition of receptor activator of nuclear factor kappa-B ligand (RANKL) (receptor that stimulates osteoclast growth)
Osteoporosis: Imbalance of bone formation and resorption leading to decreased bone mineral density and bone mass, or quality changes that lead to a decrease in bone strength and an increase risk of fractures
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published May 20, 2022 on www.thecalgaryguide.com")
constatations-classiques-de-sclerose-en-plaques-sep-sur-irm-cerebrale
 sur IRM cérébrale
Auteurs: Evan Allarie Davis Maclean Viesha Ciura* Rédacteurs: Yan Yu* Traducteurs: Liana Martel Eddy Lang* * MD au moment de la publication
Infiltrations dans la région infratentorielle
Perte de la densité neuronale/axonale dans la région affectée, remplacée par une gliose au fil du temps
Remarque : les constatations peuvent varier.
Les constatations présentées ici ne sont pas exhaustives, mais constituent certaines des zones les plus fréquemment mises en évidence par l'IRM du cerveau dans la SEP. Les sites les plus communément observés sont : les régions juxtacorticales, périventriculaires, infratentorielles, la moelle épinière et le nerf optique. Cependant, les lésions peuvent apparaître partout où il y a de la myéline dans le SNC.
L'inflammation et la destruction actives permettent au contraste de gadolinium de traverser la barrière hémato-encéphalique, ce qui peut être visualisé comme un marqueur de l'inflammation active dans la SEP
Lésion classique du nerf optique (CN II) en cas de névrite optique. Rehaussement par Gadolinium (↑ intensité du signal de la lésion après l’injection de gadolinium) sur cette image en T1 démontre une inflammation active
Image Credits: Dr. Viesha Ciura
Pour plus de détails sur la pathogenèse, voir la diapositive du Guide de Calgary: Multiple Sclerosis (MS): Pathogenesis and Clinical Findings
Inflammation dans le système nerveux central (SNC)
Les cellules T, les cellules B et les macrophages infiltrent le SNC
Infiltration se produit autour des veinsàinflammation locale Infiltrats périveineux autour des veines médullaires
(perpendiculaires aux ventricules)
Inflammation et destruction de la barrière hémato-encéphalique ↑ liquide inflammatoire extravasculaire autour des lésions, qui apparaît hyperintense/brillant
Lésions s’étendent autour des veines, créant les lésions ovoïdes charactéristiques
Plaques hyperintenses perpendiculaires périventriculaires classiques en T2/FLAIR suivant les veins médullaires – ‘Doigts de Dawson’
Lésion hyperintense du pédoncle cérebelleux moyen en T2/FLAIR dans une localisation classique de la SEP
Légende:
Physiopathologie
Mécanisme
Signe/Symptôme/Résultats de Laboratoire
Complications
Publié 25 octobre 2020 sur www.thecalgaryguide.com")
syndrome-de-guillain-barre

Mimétisme moléculaire : antigènes gangliosidiques partagés entre le nerf périphérique et les protéines d'enveloppe des agents pathogènes
Auteurs: Nissi Wei Rédacteurs: Owen Stechishin, Matthew Harding, Cory Toth* Traducteurs: Liana Martel, Eddy Lang* * MD au moment de la publication
↑ protéines dans le LCR
Déclencheurs mineurs
(chirurgie, trauma, greffe de moelle osseuse)
Anticorps IgG contre les gangliosides dans le sérum
ECN: ↓ vitesse de conduction, bloc de conduction
Polyneuropathie inflammatoire démylisante aiguë (PIDA) (80-90%)
Initiation de la réponse immunitaire (mécanisme inconnu)
La réponse immunitaire déclenchée réagit de manière croisée avec les nerfs périphériques, en commençant par les racines nerveuses
↑ perméabilité de la barrière hémato-nerveuse au niveau des racines nerveuses proximales
Démyélinisation: les anticorps attaquent les cellules de Schwann
dommage secondaire
Lésion axonale: les anticorps attaquent les noeuds de Ranvier
ECN: ↓ PAMC amplitude, vitesse de conduction normale
Anomalies des mouvements extraoculaires
(Syndrome de Miller Fisher – forme rare de PIDA à restriction régionale)
Neuropathie axonale motrice aiguë (NAMA)
Neuropathie axonale sensorielle motrice aiguë (NASMA)
Faiblesse oculomotrice (NC III, IV, VI)
Paralysie bulbaire (NC IX, X, XI,XII)
Atteinte du nerf phrénique
Polyneuropathie aiguë à médiation immunitaire
Atteinte des nerfs crâniens
Dysautonomie
Rétention urinaire
(transitoire, en fin de parcours)
↓ capacité à évacuer les sécrétions des voies respiratoires
Troubles de dégluttitionà pneumonie par aspiration
Paralysie du diaphragme
Insuffisance
respiratoire
(Menace la vie: besoin d’une surveillance respiatoire et possiblement une assistance respiratoire)
Abbreviations:
• ECN - examen de
Déficits moteurs
Faiblesse des membres (les jambes sont généralement touchées en premier)
Aréflexie universelle
Déficits sensoriels
Douleur & paresthésies au dos et dans les extrémités
Tachycardie, Dysrhythmies (besoin d’une surveillance cardiaque)
Fluctuation de la TA, hypotension • orthostatique
conduction nerveuse PAMC – potentiel d’action musculaire composé
Remarque : les fibres nerveuses périphériques Aα, Aβ (grands axones à conduction rapide et fortement myélinisés pour l'étirement des muscles, le toucher léger et la proprioception) sont plus touchées que les fibres Aδ et C (petites fibres moins myélinisées et à conduction lente pour la douleur et la température).
CMV - Cytomégalovirus • NC - Nerf cranien
• VEB - Virus Epstein-Barr
Mort soudaine •
Légende:
Physiopathologie
Mécanisme
Signe/Symptôme/Résultats de Laboratoire
Complications
Publié 1 novembre 2013 sur www.thecalgaryguide.com")
pleural-effusions-pathogenesis-and-anterior-posterior-chest-x-ray-findings

Excess fluid accumulation within pleural space that is gravity- dependent/free- flowing
Large accumulation of fluid pressing against lung tissue and mediastinum
Fluid layering (i.e. opacification) between lung and chest wall
Author: Sravya Kakumanu Reviewers: Reshma Sirajee, Tara Shannon *Stephanie Nguyen, *Shelley Spaner * MD at time of publication
Transudative
pleural effusion
*See Pathogenesis and Clinical Findings of
Transudative Pleural Effusions slide
Exudative pleural
effusion
*See Pathogenesis and Clinical Findings of
Exudative Pleural Effusions slide
Fluid accumulation at bottom of pleural space in anterior- posterior chest x- ray
Lung atelectasis (lung collapse)
Fluid is denser than air and appears white on x-ray
↓ Pressure and occupied space in thoracic cavity
Meniscus sign (concave line above opacification)
Opacification of affected pleural space
Blunting of costophrenic angles
Diaphragmatic silhouette sign (sharp diaphragm edge obscured by fluid)
Mediastinal shift towards atelectic lung
Contralateral mediastinal shift (when no lung atelectasis)
Contralateral tracheal deviation
Pleural infections
↑ Inflammation at infection site à↑ permeability of parietal pleura endothelium
Bacterial invasion into pleural space from pulmonary parenchyma
↑ Migration of neutrophils and release of inflammatory cytokines in pleural space
Inflammation ↑ activation of coagulation cascade and ↓ fibrinolytic activity
↑ Deposition of fibrin clots/membranes within pleural space creating septations
Loculated pleural effusion/empyema (fluid stuck within septations within the pleural space)
Opacification does not follow gravity
Effusion does not move in decubitus view
Pleural thickening (whitening of lung perimeter from fibrin deposition - more visible on CT scan)
More rounded margins with chest wall/fissures
Note: ~175-500mL of fluid may need to accumulate before being visible on an Anterior-Posterior Chest X-Ray
Image credit: https://radiopaedia.org/cases/pleural-effusion-7
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published May 22, 2022 on www.thecalgaryguide.com")
iliotibial-band-itb-syndrome-pathogenesis-and-clinical-findings
 Syndrome: Pathogenesis and clinical findings
Authors: Ciara Hanly, Alyssa Federico Reviewers: Tara Shannon, Hannah Koury, Mehul Gupta
Extrinsic Risk Factors
Intrinsic Risk Factors
Genu varum or valgum (deformity causing outward or inward bowing of knees, respectively)
↑ Lengthening of ITB at lateral femoral epicondyle
Ryan Shields*
* MD at time of publication
Leg length discrepancy
Compensatory
unilateral foot pronation and hip drop of shorter leg
↑ Stretch of ITB on longer leg
Sudden ↑ in training volume and overtraining
Repetitive flexion and extension of lower limb (i.e. running, cycling)
Running surface with horizontal or vertical gradient
↑ Knee flexion at foot strike
Excessively long strides
↑ Hip flexion
Hip abductor weakness (gluteus medius, gluteus minimus, tensor fascia latae)
↑ Hip adductionà internal rotation at the knee
Excessive foot pronation
↑ Stretch of ITB
ITB originates from the iliac crest, runs past the lateral femoral epicondyle, and inserts into the lateral aspect of the proximal tibia
Lower limb flexion and extension displaces the ITB over the lateral femoral condyle in an anterior-posterior direction
↑ Friction between ITB and lateral femoral condyle
Blood vessels, nerves, Pacinian corpuscles (pressure and vibration receptors), and vascularized fat pad are compressed in sub-ITB space
Activation of nociceptors (pain receptors) in femur
Micro-trauma to ITB
Inflammatory response and scar tissue formation
Fluid ITB
+ Noble’s compression test:
Patient supine with hip and knee flexed to 90o, pressure is applied to distal ITB while patient extends hip and knee àpain over distal ITB at 30o flexion
+ Ober test: Patient lays on unaffected side with knee flexed to 90o, hip abducted and extended, and patient asked to adduct hip as far as possibleà ↓ hip adduction on affected side
Tenderness to palpation over lateral femoral epicondyle (2- 3cm above lateral joint line)
Radiating pain to lateral thigh and calf
Pain over lateral femoral epicondyle during maximal point of ITB tension, just prior to or during foot strike (i.e. during downhill walking or running)
collection deep to ITB
thickens
Persistent pain throughout exercise
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published May 27, 2022 on www.thecalgaryguide.com")
metabolic-alkalosis-pathogenesis
![Metabolic Alkalosis: Pathogenesis
Authors: Wazaira Khan Reviewers: Jessica Krahn, Emily Wildman, Austin Laing, Huneza Nadeem, Ran (Marissa) Zhang, Adam Bass* * MD at time of publication
Primary hyperaldosteronism E.g., aldosterone- secreting mass, adrenal hyperplasia
Secondary hyperaldosteronism E.g., renin-secreting mass, renal artery stenosis
Pseudo- hypoaldosteronism E.g., Liddle’s Syndrome
Unregulated aldosterone production in adrenal cortex
Excess aldosterone suppresses renin production
Unregulated renin production by juxtaglomerular cells
Sustained ↑ in mineralocorticoid (aldosterone) activity
Insertion of epithelial sodium channels on principal cells in collecting duct
↑ Water retention by the kidney
↑ Na+ reabsorption by principal cells
↑ EABV
• ↑ Jugular venous pressure • Hypertension
• Urine Na+ > 40 mEq/L
Low renin, high aldosterone state
Activation of renin- angiotensin-aldosterone system (RAAS)
Release of aldosterone from adrenal cortex
High renin, high aldosterone state
↑ Negative charge in collecting duct lumen
K+ leaks into collecting duct lumen by principal cell to maintain electroneutrality
Hypokalemia
Intracellular K+ leaks out of any cell in the body to compensate for low serum K+ levels
Extracellular H+ enters cell to maintain electroneutrality
Intracellular acidosis
Activation of compensatory acid secreting mechanisms in kidney
Impaired tubular function
E.g., loop/thiazide diuretics, Bartter’s/ Gitelman’s Syndrome
Upper GI Loss
E.g., vomiting, nasogastric suction
Unregulated epithelial sodium channel activity in collecting duct mimicking aldosterone function
↑ in Na+ and water retention à RAAS inhibition
↓ Na+ and Cl- reabsorption in thick ascending limb or distal convoluted tubule
Low renin, low aldosterone state
↑ Na+, Cl- and water secretion through kidney
RAAS activation
See “Physiology of RAAS” slide
↓ EABV • • •
Temporary ↑ in mineralocorticoid (aldosterone) activity
↓ Jugular venous pressure Orthostatic hypotension Dry mucous membranes Urine Na+ < 20 mEq/L
•
Loss of fluid through GI tract
↓ HCl delivery to small intestine
↑ NH + secretion and 4
↑ HCO3- reabsorption in proximal tubule
↑ H+ secretion in cortical collecting duct
Loss of gastric contents, including HCl
↓ HCO3- secretion by pancreas, liver and intestines to neutralize HCl
Loss of intrinsic acid to neutralize HCO3- ↑ Plasma [HCO3-]
Metabolic Alkalosis
Arterial blood gas pH > 7.40 Plasma [HCO3-] > 24 mEq/L
Effective Arterial Blood Volume (EABV): component of arterial blood volume that is effectively perfusing organs
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published May 31, 2022 on www.thecalgaryguide.com](https://calgaryguide.ucalgary.ca/wp-content/uploads/2022/05/Metabolic-Alkalosis-1.jpg "Metabolic Alkalosis: Pathogenesis
Authors: Wazaira Khan Reviewers: Jessica Krahn, Emily Wildman, Austin Laing, Huneza Nadeem, Ran (Marissa) Zhang, Adam Bass* * MD at time of publication
Primary hyperaldosteronism E.g., aldosterone- secreting mass, adrenal hyperplasia
Secondary hyperaldosteronism E.g., renin-secreting mass, renal artery stenosis
Pseudo- hypoaldosteronism E.g., Liddle’s Syndrome
Unregulated aldosterone production in adrenal cortex
Excess aldosterone suppresses renin production
Unregulated renin production by juxtaglomerular cells
Sustained ↑ in mineralocorticoid (aldosterone) activity
Insertion of epithelial sodium channels on principal cells in collecting duct
↑ Water retention by the kidney
↑ Na+ reabsorption by principal cells
↑ EABV
• ↑ Jugular venous pressure • Hypertension
• Urine Na+ > 40 mEq/L
Low renin, high aldosterone state
Activation of renin- angiotensin-aldosterone system (RAAS)
Release of aldosterone from adrenal cortex
High renin, high aldosterone state
↑ Negative charge in collecting duct lumen
K+ leaks into collecting duct lumen by principal cell to maintain electroneutrality
Hypokalemia
Intracellular K+ leaks out of any cell in the body to compensate for low serum K+ levels
Extracellular H+ enters cell to maintain electroneutrality
Intracellular acidosis
Activation of compensatory acid secreting mechanisms in kidney
Impaired tubular function
E.g., loop/thiazide diuretics, Bartter’s/ Gitelman’s Syndrome
Upper GI Loss
E.g., vomiting, nasogastric suction
Unregulated epithelial sodium channel activity in collecting duct mimicking aldosterone function
↑ in Na+ and water retention à RAAS inhibition
↓ Na+ and Cl- reabsorption in thick ascending limb or distal convoluted tubule
Low renin, low aldosterone state
↑ Na+, Cl- and water secretion through kidney
RAAS activation
See “Physiology of RAAS” slide
↓ EABV • • •
Temporary ↑ in mineralocorticoid (aldosterone) activity
↓ Jugular venous pressure Orthostatic hypotension Dry mucous membranes Urine Na+ < 20 mEq/L
•
Loss of fluid through GI tract
↓ HCl delivery to small intestine
↑ NH + secretion and 4
↑ HCO3- reabsorption in proximal tubule
↑ H+ secretion in cortical collecting duct
Loss of gastric contents, including HCl
↓ HCO3- secretion by pancreas, liver and intestines to neutralize HCl
Loss of intrinsic acid to neutralize HCO3- ↑ Plasma [HCO3-]
Metabolic Alkalosis
Arterial blood gas pH > 7.40 Plasma [HCO3-] > 24 mEq/L
Effective Arterial Blood Volume (EABV): component of arterial blood volume that is effectively perfusing organs
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published May 31, 2022 on www.thecalgaryguide.com")
obstructive-sleep-apnea-pathogenesis-and-clinical-findings
![Obstructive Sleep Apnea: Pathogenesis and clinical findings
Vascular Factors: During recumbent sleep, more bodily fluids enter the head and neck area (compared to when the patient is standing/sitting)
↑ volume of head/neck tissue surrounding the upper airwayà possible airway obstruction
Authors: Ciara Hanly Austin Laing Alexander Arnold Reviewers: Steven Liu Amogh Agrawal Yonglin Mai (麦泳琳) Naushad Hirani* Yan Yu* *MD at time of publication
Neuromuscular Factors: Sleep onset and/or the sleeping state reduces the drive of respiratory muscles to breathe
↓ Upper airway neuromuscular activityà↓ upper airway caliber, ↑ upper airway resistance, ↑ upper airway collapsibility during sleep
Structural Factors: Obesity, tonsillar or adenoid hypertrophy, macroglossia, ↑ neck circumference, craniofacial abnormalities
Excess pressure on upper airway, or deformity to that area, ↑ risk of upper airway collapse
Polysomnography
Absence of airflow but persistent ventilatory effort
Hypopnea or Apnea
Paradoxical breathing Chest wall draws in and abdomen expands during inspiration
Ventilatory effort persists against closed airway
No air entry due to collapsed upper airway
↑ Negative intrathoracic pressure
↑ Venous return to right atrium
Stretching of right atrial myocardium à secretion of atrial natriuretic peptide (ANP)
ANP inhibits epithelial Na+ channels (ENaC) in the collecting ducts of the kidney from reabsorbing Na+ à Na+ excretion
↑ Na+ excretionà↑ water excretion
Nocturia
Complete or partial upper airway obstruction during sleep
↑ PCO2 & ̄ PO2
in the lungsà ̄ diffusion gradient of CO2 & O2 between lungs & arteries
↑ PaCO2,, ̄ PaO2
Respiratory acidosis (↑ [H+] in blood)àactivation of vascular endothelial voltage gated K+ channels
Cerebral blood vessel dilation to provide adequate O2 to brain
Morning Headaches
Activation of central (medulla oblongata) & peripheral (carotid body) chemoreceptors
↑ Respiratory drive à ↑ activation of respiratory muscles (ventilatory effort )
Transient arousal from sleep
↑ sympathetic nervous system activityà arterial vasoconstriction
↑ systemic vascular resistance
Systemic Hypertension
↑ intraluminal pressure within blood vesselsàadaptive vascular endothelial and smooth muscle changes
Artery walls thicken, harden and lose elasticityà ̄ perfusion to end organs (such as the brain)
Ischemic stroke
Hypoxia during the day and night
↑ pulmonary vascular resistance
Pulmonary Hypertension
Right heart pumps against higher pulmonary pressure àcardiomyocytes undergo concentric hypertrophy over time
Cor Pulmonale
(Right heart failure due to pulmonary hypertension, separate from left heart failure)
Respiratory muscles overcome upper airway obstructionà airway patency restored
Sleep fragmentation
̄ Daytime cognitive performance and attentiveness
↑ Risk of motor vehicle accidents
Daytime Sleepiness
Eg. Epworth Sleepiness Scale >10
Abbreviations:
PCO2: partial pressure of carbon dioxide PO2: partial pressure of oxygen PaCO2: partial pressure of carbon dioxide in arteries PaO2: partial pressure of oxygen in arteries
Ventilatory response overcompensatesà breathe out more CO2 than is required for homeostasisà ̄ PaCO2
̄ respiratory driveà ̄ ventilatory effort
Resuscitative Gasping
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published August 19, 2013, updated May 31, 2022 on www.thecalgaryguide.com
阻塞性睡眠呼吸暂停:发病机制及临床表现
作者:Ciara Hanly, Austin Laing, Alexander Arnold 审稿人: Steven Liu, Amogh Agrawal, Naushad Hirani*,Yan Yu* 译者: Zesheng Ye(叶泽生) 翻译审稿人: Yonglin Mai(麦泳琳) *发表时担任临床医生
神经肌肉因素: 睡眠状态下, 患者无法通过 适当增加上气道肌张力来维持气道通畅
上气道神经肌肉活动 ̄à上气道直径 ̄, 上气道 阻力↑, 睡眠时上气道塌陷
结构(解剖)因素: 肥胖、扁桃体或腺样体 肥大, 舌体肥大, 颈围增大, 颅面部畸形
上气道压力过大或上气道畸形, 上气道塌陷 的风险 ↑
血管因素: 仰卧位睡觉引起 夜间嘴侧液体移位
周围组织与压力 ↑à上气道阻塞
多导睡眠描记术
没有气流,但持
续通气
呼吸浅慢或 呼吸暂停
反常呼吸 吸气时胸壁凹陷, 腹部膨隆
持续通气以抵抗气道 闭合
上气道塌陷导致空气进
入气道受阻
腹膜腔负压↑ 静脉血回流右心室阻力↑
右心房心肌细胞拉伸 à心房利钠肽分泌 (ANP)
ANP抑制肾集合管的上 皮Na+通道(ENaC)对 Na+重吸收à Na+排出
Na+排出量↑ à 水排出量 ↑
睡眠时全部
或部分上呼
吸道阻塞
肺内PO2 ̄ 且 PCO2↑ à CO2 及 O2在肺和动脉 间的扩散梯度 ̄
↑ PaCO2, ̄ PaO2
呼吸性酸中毒 (血液中 [H+] ↑) à激活血管内皮电压
门控 K+
脑血管扩张为大 晨间头痛 脑提供足够的 O2
激活中央(延髓)和外周(颈动脉体)的化学感受器 呼吸驱动↑à呼吸肌活动 (呼吸做功 )↑
短暂的睡眠唤醒
通道 交感神经系统活动↑
全天缺氧 肺血管阻力↑
肺动脉高压
右心泵血以抵抗肺 动脉高压à 随着时 间推移,心肌向心 性肥大
肺心病(区别于左
心衰,右心衰是肺
动脉高压所致)
呼吸肌克服上气道阻力à 气道 明显恢复
睡眠过程不连续
白天的认知功能
及注意力 ̄
机动车辆事故风险↑
白天嗜睡
à 动脉收缩 全身血管阻力↑
高血压
血管内压力↑ à 血 管内皮和平滑肌发生 适应性改变
动脉壁增厚、硬化、失 去弹性à器官血液灌 注量 ̄ (如脑部)
缩写: PCO2:二氧化碳分压 PO2:氧分压 PaCO2:动脉二氧化 碳分压 PaO2:动脉血氧分压
通气过度 à呼出CO2 ↑ à PaCO2 ̄
呼吸驱动 ̄à 呼吸做功 ̄
复苏性鼾音
夜尿症
如:伊普沃斯嗜睡评分
>10
缺血性卒中
图注:
病理生理
机制
体征/临床表现/实验室检查
并发症
2013年8月19日发表 www.thecalgaryguide.com, 2022年5月31日更新](https://calgaryguide.ucalgary.ca/wp-content/uploads/2014/09/OSA-2021-1.jpg "Obstructive Sleep Apnea: Pathogenesis and clinical findings
Vascular Factors: During recumbent sleep, more bodily fluids enter the head and neck area (compared to when the patient is standing/sitting)
↑ volume of head/neck tissue surrounding the upper airwayà possible airway obstruction
Authors: Ciara Hanly Austin Laing Alexander Arnold Reviewers: Steven Liu Amogh Agrawal Yonglin Mai (麦泳琳) Naushad Hirani* Yan Yu* *MD at time of publication
Neuromuscular Factors: Sleep onset and/or the sleeping state reduces the drive of respiratory muscles to breathe
↓ Upper airway neuromuscular activityà↓ upper airway caliber, ↑ upper airway resistance, ↑ upper airway collapsibility during sleep
Structural Factors: Obesity, tonsillar or adenoid hypertrophy, macroglossia, ↑ neck circumference, craniofacial abnormalities
Excess pressure on upper airway, or deformity to that area, ↑ risk of upper airway collapse
Polysomnography
Absence of airflow but persistent ventilatory effort
Hypopnea or Apnea
Paradoxical breathing Chest wall draws in and abdomen expands during inspiration
Ventilatory effort persists against closed airway
No air entry due to collapsed upper airway
↑ Negative intrathoracic pressure
↑ Venous return to right atrium
Stretching of right atrial myocardium à secretion of atrial natriuretic peptide (ANP)
ANP inhibits epithelial Na+ channels (ENaC) in the collecting ducts of the kidney from reabsorbing Na+ à Na+ excretion
↑ Na+ excretionà↑ water excretion
Nocturia
Complete or partial upper airway obstruction during sleep
↑ PCO2 & ̄ PO2
in the lungsà ̄ diffusion gradient of CO2 & O2 between lungs & arteries
↑ PaCO2,, ̄ PaO2
Respiratory acidosis (↑ [H+] in blood)àactivation of vascular endothelial voltage gated K+ channels
Cerebral blood vessel dilation to provide adequate O2 to brain
Morning Headaches
Activation of central (medulla oblongata) & peripheral (carotid body) chemoreceptors
↑ Respiratory drive à ↑ activation of respiratory muscles (ventilatory effort )
Transient arousal from sleep
↑ sympathetic nervous system activityà arterial vasoconstriction
↑ systemic vascular resistance
Systemic Hypertension
↑ intraluminal pressure within blood vesselsàadaptive vascular endothelial and smooth muscle changes
Artery walls thicken, harden and lose elasticityà ̄ perfusion to end organs (such as the brain)
Ischemic stroke
Hypoxia during the day and night
↑ pulmonary vascular resistance
Pulmonary Hypertension
Right heart pumps against higher pulmonary pressure àcardiomyocytes undergo concentric hypertrophy over time
Cor Pulmonale
(Right heart failure due to pulmonary hypertension, separate from left heart failure)
Respiratory muscles overcome upper airway obstructionà airway patency restored
Sleep fragmentation
̄ Daytime cognitive performance and attentiveness
↑ Risk of motor vehicle accidents
Daytime Sleepiness
Eg. Epworth Sleepiness Scale >10
Abbreviations:
PCO2: partial pressure of carbon dioxide PO2: partial pressure of oxygen PaCO2: partial pressure of carbon dioxide in arteries PaO2: partial pressure of oxygen in arteries
Ventilatory response overcompensatesà breathe out more CO2 than is required for homeostasisà ̄ PaCO2
̄ respiratory driveà ̄ ventilatory effort
Resuscitative Gasping
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published August 19, 2013, updated May 31, 2022 on www.thecalgaryguide.com
阻塞性睡眠呼吸暂停:发病机制及临床表现
作者:Ciara Hanly, Austin Laing, Alexander Arnold 审稿人: Steven Liu, Amogh Agrawal, Naushad Hirani*,Yan Yu* 译者: Zesheng Ye(叶泽生) 翻译审稿人: Yonglin Mai(麦泳琳) *发表时担任临床医生
神经肌肉因素: 睡眠状态下, 患者无法通过 适当增加上气道肌张力来维持气道通畅
上气道神经肌肉活动 ̄à上气道直径 ̄, 上气道 阻力↑, 睡眠时上气道塌陷
结构(解剖)因素: 肥胖、扁桃体或腺样体 肥大, 舌体肥大, 颈围增大, 颅面部畸形
上气道压力过大或上气道畸形, 上气道塌陷 的风险 ↑
血管因素: 仰卧位睡觉引起 夜间嘴侧液体移位
周围组织与压力 ↑à上气道阻塞
多导睡眠描记术
没有气流,但持
续通气
呼吸浅慢或 呼吸暂停
反常呼吸 吸气时胸壁凹陷, 腹部膨隆
持续通气以抵抗气道 闭合
上气道塌陷导致空气进
入气道受阻
腹膜腔负压↑ 静脉血回流右心室阻力↑
右心房心肌细胞拉伸 à心房利钠肽分泌 (ANP)
ANP抑制肾集合管的上 皮Na+通道(ENaC)对 Na+重吸收à Na+排出
Na+排出量↑ à 水排出量 ↑
睡眠时全部
或部分上呼
吸道阻塞
肺内PO2 ̄ 且 PCO2↑ à CO2 及 O2在肺和动脉 间的扩散梯度 ̄
↑ PaCO2, ̄ PaO2
呼吸性酸中毒 (血液中 [H+] ↑) à激活血管内皮电压
门控 K+
脑血管扩张为大 晨间头痛 脑提供足够的 O2
激活中央(延髓)和外周(颈动脉体)的化学感受器 呼吸驱动↑à呼吸肌活动 (呼吸做功 )↑
短暂的睡眠唤醒
通道 交感神经系统活动↑
全天缺氧 肺血管阻力↑
肺动脉高压
右心泵血以抵抗肺 动脉高压à 随着时 间推移,心肌向心 性肥大
肺心病(区别于左
心衰,右心衰是肺
动脉高压所致)
呼吸肌克服上气道阻力à 气道 明显恢复
睡眠过程不连续
白天的认知功能
及注意力 ̄
机动车辆事故风险↑
白天嗜睡
à 动脉收缩 全身血管阻力↑
高血压
血管内压力↑ à 血 管内皮和平滑肌发生 适应性改变
动脉壁增厚、硬化、失 去弹性à器官血液灌 注量 ̄ (如脑部)
缩写: PCO2:二氧化碳分压 PO2:氧分压 PaCO2:动脉二氧化 碳分压 PaO2:动脉血氧分压
通气过度 à呼出CO2 ↑ à PaCO2 ̄
呼吸驱动 ̄à 呼吸做功 ̄
复苏性鼾音
夜尿症
如:伊普沃斯嗜睡评分
>10
缺血性卒中
图注:
病理生理
机制
体征/临床表现/实验室检查
并发症
2013年8月19日发表 www.thecalgaryguide.com, 2022年5月31日更新")
阻塞性睡眠呼吸暂停-发病机制及临床表现

囊性纤维化-胸部-x-线和肺窗-ct-扫描结果

gout-pathogenesis-of-x-ray-findings
 crystals
Uric acid crystallizes due to lower temperature, change in pH, mechanical stress and other synovial factors
Crystals found more commonly in 1st MTP joint > ankle > wrist
MSU deposits within the bone producing
intra-osseus tophi (stone-like deposits of crystals)
MSU deposits in soft tissue and bone
↑ Inflammatory marker recruitment causes granulomatous inflammation
Osteoclasts (↑ bone resorption) are activated, and osteoblasts (↑ bone formation) are inhibited at
site of inflammation Marked localized bone loss
‘Rat Bite’ erosions
Intra-osseus bone lesion
(rare and non-specific for gout)
Joint effusions may be seen on x-ray as an early finding in the acute phase of gout
Soft Tissue Tophi
(appear cloudy and can hide other x-ray findings)
Normal bone density and preserved joint space
(until late in the disease)
Overhanging
sclerotic margins
Bone remodelled in an outward fashion to create the edge
Punched out lytic bone lesions Appear as circular ‘hole punches’ in bone
Note: Repeated episodes of gout must occur for 5 to 10 years before most joint changes may be seen on x-ray
Image credit: Radiopaedia
Legend:
Pathophysiology
Mechanism
Sign/Radiographic Findings
Complications
Published June 7, 2022 on www.thecalgaryguide.com")
beta-thalassemia-minor
 Zhang Man-Chiu Poon* Lynn Savoie* * MD at time of publication
Composition of normal adult hemoglobin (Hb):
Ineffective erythropoiesis
Excessive free α-chains accumulate, and α-chains precipitate in RBC precursors and RBCs
These precipitants (inclusion bodies) give RBCs an abnormal shape
Spleen destroys abnormally shaped RBCs
Spleen accumulates destroyed red blood cells and enlarges to an abnormal size
Splenomegaly (*See Splenomegaly slide for clinical findings)
Mild microcytic anemia
Hb: ↓ MCV: ↓
• • •
96% HbA (ααββ=2 α chains + 2 β chains) 2% HbA2 (ααẟẟ)
2% HbF (αα!!; fetal hemoglobin)
Genetic point mutation on a single allele of the β globin gene on chromosome 11 (heterozygous with one normal alleleà HbA ααββ0 or ααββ+)
Mild ↓ of β-chain production relative to ⍺- chain
↓ β-chain available for HbA synthesis
Body compensates by ↑ production
of non-affected globins (HbA , HbF) 2
↑ HbF (nonspecific), ↑ HbA2 (hallmark sign specific to Beta Thalassemia Minor) on electrophoresis
• •
α-chain precipitates form structures visible under the microscope
Inclusion bodies
Heme from the destroyed RBCs is degraded into bilirubin and iron
Excess unconjugated bilirubin is released into the blood
Excess bilirubin is deposited in the skin
Jaundice
Excess iron is stored as ferritin
↑ or normal ferritin
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Re-Published June 8, 2022 on www.thecalgaryguide.com")
gastroenteritis-pathogenese-und-klinische-befunde

negativdruck-lungenodem-pathophysiologie

benzodiazepine-wirkmechanismus

succinylcholin-depolarisierende-muskelrelaxanzien
")
Status-Epilepticus
 Zhang Carlos Camara-Lemarroy* * MD at time of publication
Structural brain injury (stroke, trauma, hypoxia)
Drugs that lower seizure threshold
Antiseizure drug discontinuation
Alcohol, barbiturate, benzodiazepine withdrawal
Metabolic disturbance
Infection
See “Generalized Seizures”
Altered excitability and communication between neuronal structures
Isolated generalized seizures
Ongoing seizure activity and repetitive neuronal firing
Changes in receptor trafficking (seconds to minutes)
Changes in neuromodulator expression in hippocampus (minutes to hours)
Endocytosis of synaptic GABAA inhibitory receptors
↓ Number of inhibitory GABAA receptors
Progressive resistance to benzodiazepines (drugs that upregulate GABA receptors) as seizure continues
↑ Expression of excitatory peptides (substance P, neurokinin B)
Abbreviations:
• GABA- γ-aminobutyric acid
• NMDA- N-methyl-D-aspartic acid • AMPA- α-amino-3-hydroxy-5-
methyl-4-isoxazolepropionic acid
NMDA and AMPA excitatory receptors mobilize to synaptic membrane
↑ Number of excitatory NMDA and AMPA receptors
↓ Expression of inhibitory peptide (dynorphin, galanin, somatostatin, neuropeptide Y)
Seizure-induced failure of inhibitory mechanisms involved in seizure termination and increased neuronal excitability
Status Epilepticus (SE)
An abnormally prolonged seizure ≥ 5 minutes or 2+ sequential seizures without full recovery in between
↑↑ Glutamate release and activation of NMDA excitatory receptors
↑ Ca2+ entry into neurons
Mitochondrial dysfunction
↑ Reactive oxygen species (nitric oxide) production
Neuronal injury/death (↑ risk of developing chronic epilepsy)
↑ Autonomic activity
Intense, sustained muscle contractions
Persistent stimulus OR altered neuronal landscape
(i.e., Immune mediated)
Refractory SE:
SE that does not respond to 1st or 2nd line therapy
Prolonged seizures (≥30 mins) lead to failure of compensatory mechanisms
Circulatory collapse
↓ Cerebral blood flow
• Hypertension • Hyperglycemia • ↑ Cardiac
output
• ↑ Secretions
• Hypotension
• Hypoventilation
Energy demands > ATP produced through oxidative phosphorylation
Myocytes start utilizing anerobic glycolysis
↑ Lactic acid production
↑ Serum lactate
Sustained muscle activity produces body heat
Hyperpyrexia (axillary temperature ≥ 40° C)
Myocyte injury
Leakage of muscle contents into the circulation (Rhabdomyolysis)
↑ Serum creatine kinase
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published June 27, 2022 on www.thecalgaryguide.com")
pheochromocytoma-pathogenesis-and-clinical-findings
 Zhang Hanan Bassyouni* * MD at time of publication
↑ Blood pressure results in the activation of neural pain receptors
Sustained or paroxysmal 2o hypertension (↑ blood pressure)
Pallor
Tachycardia (↑ heart rate), palpitations
Diaphoresis (excessive sweating)
Hyperglycemia Weight loss, fatigue
Familial Disorders (10%)
E.g., Multiple Endocrine Neoplasm 2 Syndrome (MEN2) types A and B, Neurofibromatosis 1 (NF-1), Von Hippel Lindau Syndrome (VHL), familial pheochromocytoma
Dysfunction of various tumor suppressor and/or oncogene proteins
Uncontrolled proliferation of the chromaffin cells in the medulla of the adrenal gland(s)
Adenoma formation (10% bilateral)
Over-production of epinephrine and
norepinephrine from the adrenal adenoma(s) or extra- adrenal tumour (10%)
Detectable metanephrines (epinephrine/norepinephrine breakdown products) in both plasma and urine
Sporadic DNA mutations arising from
DNA damage from exposure to mutagens, malignancy (10%), or during DNA replication
Detectable mutations on the Von Hippel Linda (VHL), (Rearranged During Transcription) RET, (Succinate Dehydrogenase) SDH, and/or other tumour suppressor or oncogenes
Visible adrenal mass on CT scan Heart attack, stroke, or death (Note: These
tumors can be fatal therefore screening is essential in patients with adrenal masses or 2o hypertension)
Episodic hyper-activity of the sympathetic nervous system
Hyper-stimulation of G protein-coupled receptors involved in catabolic metabolic processes
Headaches
↑ Vasoconstriction of peripheral blood vessels
Hyper-stimulation of adrenergic receptors on cardiac myocytes
↑ Secretion from the eccrine sweat glands
Panic, tremor, anxiety
↑ Ability to mobilize glucose into the bloodstream through enhanced lipolysis, glycogenolysis and gluconeogenesis
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published February 20, 2014, updated June 27, 2022 on www.thecalgaryguide.com")
overdiagnosis-in-medicine-causes-and-complications

↑ The number of diagnosed individuals serves to ↑ profits by expanding consumer base for treatments
Authors: Davis Maclean Reviewers: Ben Campbell Eddy Lang* *MD at time of publication
Unintended consequences of disease screening programs
Direct to consumer testing (e.g home medical genetics testing kits)
Note: Misdiagnosis ≠ Overdiagnosis
Misdiagnosis refers to making an incorrect diagnosis in a symptomatic patient.
Defensive medical practices
Increased testing to ‘rule out’ pathology and reduce legal liability
↑
Availability of diagnostic tests
Technological advancements leading to ↑ sensitivity of diagnostic tests
Common public perceptions of health and healthcare
Public overestimation of benefit and underestimation of risk modern medical interventions
Assumption that more medical information (e.g testing) results in better health
↑ Number of findings on diagnostic tests (e.g Imaging and lab work)
Medicalization of normal life experiences
Overdetection: Identification of an abnormality that would not have caused any symptoms or harm in the patient’s lifetime if left undiscovered (e.g detection of subsegmental pulmonary embolism)
Overdefinition: Lowering thresholds for classification of diseases and what is considered abnormal without net benefit
for those diagnosed (this includes people with symptoms but who are more likely to be harmed than benefit from a diagnosis)
Overdiagnosis: making an accurate diagnosis in an patient where making the diagnosis does not produce a net benefit for the patient
Overtreatment: treatment that occurs in absence of evidence for net benefit to the patient
Treatment side effects
↓ Quality of life
(e.g nausea and fatigue secondary to chemotherapy)
Overtreatment and overutilization are common consequences of overdiagnosis, but can happen in the absence of overdiagnosis
Overutilization: ↑ Use of health services and systems without evidence of net benefit
↑ Health system spending
Unnecessary testing
Testing-related complications (e.g infection of surgical biopsy site)
↑ Risk for other Illness
(e.g ↑ risk of infection in the setting of immunotherapy)
Indirect costs: (e.g time off work for medical appointment, travel costs)
↑ Individual costs
Direct costs: paying for additional tests and treatment (e.g medications)
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published June 30, 2022 on www.thecalgaryguide.com")
kolorektales-karzinom-pathogenese-und-klinische-befunde

epidurales-hamatom-pathogenese-und-klinische-befunde

nicht-depolarisierende-muskelrelaxanzien

arteriosklerose-komplikationen

arteriosklerose-pathogenese

koronare-herzkrankheit-khk-pathogenese-verschiedener-formen-der-khk

orthostatische-hypotonie-pathogenese-und-klinische-befunde

perikarderguss-tamponade-pathogenese-und-klinische-befunde

induction-of-labour-ripening-of-the-cervix-mechanisms-and-methods

Please refer to “Induction of labour: Indications and contraindications” slide
Bishop Score > 6
Cervix favorable
↑ Natural release of prostaglandins from uterine wall
Bishop Score 4-5
Proceed based on clinical picture
Amniotic membranes intact
Amniotomy to artificially rupture amniotic membranes
Rush of amniotic fluid
Bishop Score < 3
Cervix unfavorable and requires ripening
Artificial prostaglandin into vagina (gel or vaginal insert)
Ripening options
Ripening balloon in cervix
Pressure applied to internal and external cervical os
Foley catheter in cervix
Pressure applied to internal cervical os
Act as calcium ionophores to áintracellular Ca2+
Activate EP1 and EP3 receptors on myometrial cells
Lack of fluid cushion causes more fetal head engagement
Fluid flow may carry umbilical cord with it
Cord prolapse (if fetal head not engaged/ low enough in pelvis to block exit of cord)
Degradation of
collagen in the connective tissue stroma of cervix
Cervix softens
Cervical dilation
Cervix stretches (until balloon falls out)
Author: Lindey Felske Reviewers: Ran (Marissa) Zhang Brianna Ghali Ingrid Kristensen* * MD at time of publication
Risk of uterine rupture
(if prostaglandins used as a ripening method after previous C- section)
Uterine contractions
↑ Myometrial contractility
↑ Pressure on cervix
Labour
If needed, proceed to amniotomy and/or oxytocin once cervix is favorable
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published July 4, 2022 on www.thecalgaryguide.com")
postpartum-puerperal-fever-pathogenesis-and-complications
 Fever: Pathogenesis and complications
Author: Lindey Felske Reviewers: Brianna Ghali Ran (Marissa) Zhang Ingrid Kristensen* * MD at time of publication
Breast Feeding
Delayed gastric emptying in pregnancy
↑ Risk of aspiration during delivery
Inhalation of gastric contents
Chemical burn of the airways from gastric acid
Tissue injury
Chemokines released by alveolar cells recruit neutrophils
Accumulation of neutrophils and plasma exudate in alveoli
Aspiration Pneumonia
Delivery
(Vaginal or Cesarean Section)
Tissue damage:
Urinary tract catheterization
Foreign body can: Introduce
bacteria into bladder Provide a biofilm surface for bacterial adhesion Cause mucosal irritation
Invasion of bacteria into urinary tract mucosa
• • • •
Perineal tear/episiotomy (perineal incision) Abdominal incision site
Uterine damage
Retained products of conception (RPOC)
Bacteria enter open tissue
Production of antimicrobial peptides and proinflammatory mediators in epidermis
Cellulitis
Necrosis of RPOC (good medium for bacterial growth)
Post-operative pain
Hypoventilation from shallow breathing
Low volume in alveoli
Alveolar collapse
Endogenous cervicovaginal flora migrate into the uterine cavity
Infiltration of bacteria into endometrium
Endometrial TLR4 receptors recognize the endotoxin of Gram-negative bacteria
Secretion of proinflammatory cytokines (IL-6, IL-8) and prostaglandin E(2)
Activation of coagulation cascade
Coagulation in areas of hemostasis (e.g., deep veins)
Deep vein thrombosis
Dislodged DVT travels to pulmonary arteries
Pulmonary embolism
• •
•
Skin openings in breasts (milk ducts +/- cracks)
Bacteria from skin and/or saliva enter body
Milk backup from blocked duct or poor breastfeeding technique
Milk stasis provides environment for bacterial growth
Upregulation of IFN- γ, and IL-12A cytokines in milk ducts
Mastitis
Collection of inflammatory exudate
Breast abscess
Cytokine expression and inflammatory cell infiltration
Sloughing of
urinary tract lining to reduce bacterial load
Atelectasis
Maternal fever (> 38.0°C) within 6 weeks of delivery
Urinary tract infection
Endometritis
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published July 4, 2022 on www.thecalgaryguide.com
The image part with relationship ID rId4 was not found in the file.")
Trendelenburg Gait: Pathogenesis and clinical findings

Degradation or damage to hip bones (e.g., acetabular or femur fracture)
Hip replacement surgery
Strain or trauma to hip abductors (gluteus medius and minimus)
Hip abductors detach from greater trochanter
↓ Structural integrity in bones that hip abductors use for leverage
Hip abductors fail to maintain a level pelvis during stance phase of gait cycle
Trendelenburg Gait
Failure to keep the pelvis level when the contralateral leg is raised during walking (contralateral hemi-pelvis drops)
Lesion at or above the superior gluteal nerve (innervates hip abductors)
Paralysis or paresis of hip abductors
Thorax doesn’t lean toward the affected hip to correct balance
Thorax leans toward the affected hip to correct balance
Trendelenburg sign
Unable to maintain a horizontal pelvis during 30 seconds of standing on affected leg
Can manifest as bilateral abductor dysfunction
(same mechanisms as unilateral dysfunction)
Knee valgus
Accentuated side-to- side trunk movement
Force of gait impact is directed to lateral knee joint
Stimulation of pain receptors in bone
Lateral Sacroiliac knee pain joint pain
Abnormal biomechanical forces are applied to the
affected hip and adjacent joints when walking
Hip joint is subjected to abnormal wear and tear
Degeneration of affected hip joint
Suprapelvic muscles and hip flexors (psoas and rectus
femoris) engage in an attempt to correct pelvis alignment
Prolonged abnormal force and function of suprapelvic and hip flexor muscles
Strain in suprapelvic, psoas and rectus femoris muscles
↓ Hip abductor activation in affected hip
Abductor muscle atrophy in affected hip
↓ Range of motion in hip abduction and internal rotation
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published Feb 15, 2016, updated July 5, 2022 on www.thecalgaryguide.com")
Epilepsy Pathogenesis

Neoplasm
Metabolic
Inborn errors of metabolism
Inherited enzyme deficiencies
Neurodegenerative
Alzheimer’s Disease
Protein-rich plaque build-up and brain atrophy
Inflammation and formation of scar tissue irritates neural tissue
Infiltration of mass, grey matter irritation
Damage to the brain and subsequent alteration of neuronal circuitry
Neurotransmitter imbalances (i.e. ↑ glutamate, ↓ GABA, ↓ serotonin, ↓ dopamine, ↓ noradrenaline)
Abbreviations:
• HSV1 – Herpes Simplex Virus 1 • VZV – Varicella Zoster Virus
↑ Inflammatory cytokines (e.g. interleukin-6, tumor necrosis factor-α)
Altered neurogenesis and gliosis
Ion channel and receptor dysfunction causes imbalance of ion channel charges
Authors: Keerthana Pasumarthi Christopher Li Reviewers: Negar Tehrani, Ephrem Zewdie, Ran (Marissa) Zhang, Carlos R. Camara-Lemarroy* * MD at time of publication
Neurons fire in burst activity (referred to as paroxysmal depolarization shift) and often in groups (hypersynchrony)
Abnormal neural activities eventually create self-reinforcing circuits and transform neural network over time
Injuries from
falls, bumps, operating heavy machinery
Involuntary contractions Loss of consciousness
Post-ictal confusion
Recurrent seizures
Sudden Unexplained Death of Epilepsy Patient (SUDEP)
Loss of airway reflexes
Aspiration of saliva or food contents
Aspiration Pneumonia
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published July 5, 2022 on www.thecalgaryguide.com")
Cervical Insufficiency
 Zhang Brianna Ghali Ingrid Kristensen* * MD at time of publication
Debris seen in amniotic fluid on U/S
Short cervix (<2.5cm
on trans-vaginal ultrasound)
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published July 5, 2022 on www.thecalgaryguide.com")
Mastitis
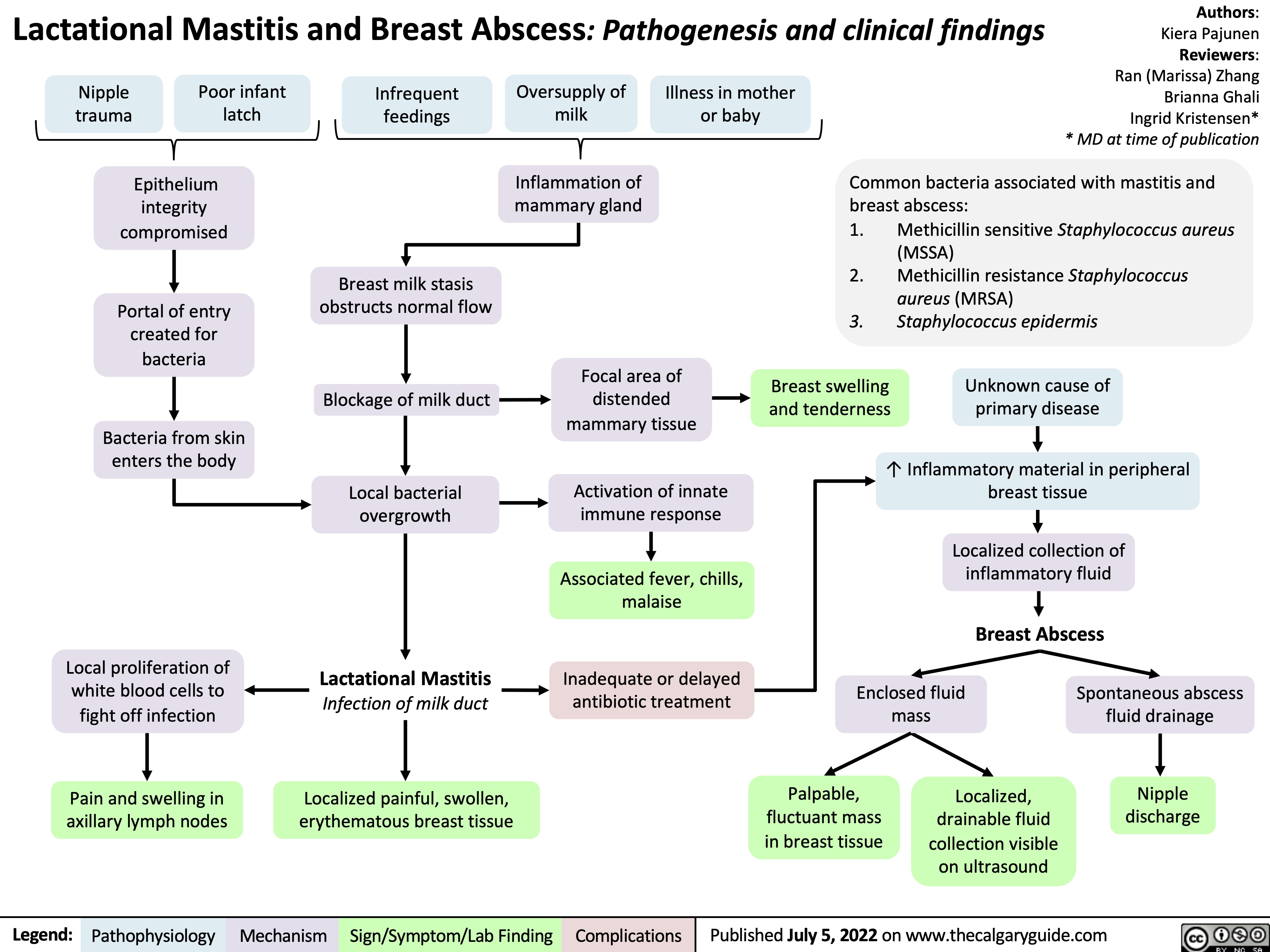
etomidate

↓ Adrenal cortisol production
↓ Serum cortisol available for stress response
Transient adrenal steroid insufficiency
↑ Morbidity & mortality in trauma/critically ill patients (Contraindicated in sepsis)
Inhibits nitric oxide signaling
Transient cerebral vasoconstriction
↓ Cerebral blood flow
↓ Oxygen delivery to brain
↓ Cerebral metabolic rate
Suppression of electrical activity in brain
Flat electroencephalogram
Binds to GABAA
(inhibitory CNS neurotransmitter) receptor
Authors:
Cindy Chang
Ran (Marissa) Zhang Reviewers:
Melissa King
Brooke Fallis
Julia Haber*
* MD at time of publication
Abbreviations:
• CNS - Central Nervous
System
• GABAA- Gamma-
aminobutyric acid type A • TRPA1- Transient
receptor potential type A1
Higher selectivity for specific GABAA receptor subtypes compared to other induction agents
Positively modulates GABAA receptor (GABAA receptor activated at lower concentrations of GABA )
↓ Cerebral blood volume
Activation of fewer GABAA receptor subtypes in neurons and cardiovascular structures compared to other induction agents
↓ Intracranial pressure ↑ Hemodynamic stability
(minimal changes in blood pressure or heart rate)
Myoclonus (brief, involuntary, irregular muscle twitch)
↑ Spontaneous CNS neuron firing to skeletal muscle
Prolonged opening of GABAA receptors (acts as ion channel for chloride)
Inhibits activity of nerve cells in the reticular activating system
Activation of peripheral nociceptor receptors
↑ Involuntary muscle contractions
Influx of chlorideàHyperpolarization of nerve membranes in reticular activating system
(brainstem neuronal network that regulates arousal and sleep- wake transitions)
Depression of arousal & loss of consciousness
Induction of general anesthesia
(no analgesic effect)
Direct activation of TRPA1 cation channels (key receptor in pain pathway)
Stinging pain with injection
(Treat with co-administration of local anesthetic)
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published July 9, 2022 on www.thecalgaryguide.com")
hyperkalemia-pathophysiology-intracellular-shift-and-intake
![Hyperkalemia (intracellular shift and ↑ intake): Pathophysiology
↑ K+ dietary intake (rarely causative)
β2 receptor inhibition (i.e. beta blockers)
Digoxin
α1 receptor stimulation (i.e. epinephrine, norepinephrine)
Insulin deficiency or resistance (i.e. diabetes mellitus)
↓ Stimulation of NHE1 (moves 1 Na+ into cell, 1 H+ out of cell) throughout body
Normal Anion- Gap Metabolic Acidosis (NAGMA)
Excess serum H+ results in ↓ NHE1 activity (See NAGMA: Pathogenesis and Laboratory Findings slide)
↑ Serum osmolarity (i.e. hyperglycemia)
Osmotic movement of water from cells to serum
Cell lysis (i.e. tumour lysis syndrome, rhabdomyolysis, hemolytic anemia)
↑ K+ release from lysed cells into the serum
Na+/K+ ATPase (moves 3K+ into cell, 2 Na+ out) activity inhibited on cells throughout body
↓ Activity of Na+/K+ ATPase on cells throughout the body
↓ Amount of K+ entering cells
↓ NHE1 activity prevents Na+ from entering the cell
Lack of high intracellular [Na+] needed to drive the Na+/K+ ATPase on cells
Loss of water from cells ↑ intracellular [K+]
K+ moves down concentration gradient from cell into serum
↑ K+ available for absorption
Since K+ is dissolved in water, some K+ is carried by water as water osmotically moves through water channels into the serum (phenomenon known as “solvent drag”)
See Hyperkalemia: Clinical Findings slide
Authors:
Mannat Dhillon, Joshua Low, Emily Wildman Reviewers:
Huneza Nadeem, Marissa (Ran) Zhang, Andrea Kuczynski, Yan Yu*, Kevin McLaughlin*, Adam Bass*
* MD at time of publication
↑ K+ in serum
Hyperkalemia
Serum [K+] > 5.1 mmol/L
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published July 17, 2022 on www.thecalgaryguide.com](https://calgaryguide.ucalgary.ca/wp-content/uploads/2022/07/Hyperkalemia-shift-1.jpg "Hyperkalemia (intracellular shift and ↑ intake): Pathophysiology
↑ K+ dietary intake (rarely causative)
β2 receptor inhibition (i.e. beta blockers)
Digoxin
α1 receptor stimulation (i.e. epinephrine, norepinephrine)
Insulin deficiency or resistance (i.e. diabetes mellitus)
↓ Stimulation of NHE1 (moves 1 Na+ into cell, 1 H+ out of cell) throughout body
Normal Anion- Gap Metabolic Acidosis (NAGMA)
Excess serum H+ results in ↓ NHE1 activity (See NAGMA: Pathogenesis and Laboratory Findings slide)
↑ Serum osmolarity (i.e. hyperglycemia)
Osmotic movement of water from cells to serum
Cell lysis (i.e. tumour lysis syndrome, rhabdomyolysis, hemolytic anemia)
↑ K+ release from lysed cells into the serum
Na+/K+ ATPase (moves 3K+ into cell, 2 Na+ out) activity inhibited on cells throughout body
↓ Activity of Na+/K+ ATPase on cells throughout the body
↓ Amount of K+ entering cells
↓ NHE1 activity prevents Na+ from entering the cell
Lack of high intracellular [Na+] needed to drive the Na+/K+ ATPase on cells
Loss of water from cells ↑ intracellular [K+]
K+ moves down concentration gradient from cell into serum
↑ K+ available for absorption
Since K+ is dissolved in water, some K+ is carried by water as water osmotically moves through water channels into the serum (phenomenon known as “solvent drag”)
See Hyperkalemia: Clinical Findings slide
Authors:
Mannat Dhillon, Joshua Low, Emily Wildman Reviewers:
Huneza Nadeem, Marissa (Ran) Zhang, Andrea Kuczynski, Yan Yu*, Kevin McLaughlin*, Adam Bass*
* MD at time of publication
↑ K+ in serum
Hyperkalemia
Serum [K+] > 5.1 mmol/L
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published July 17, 2022 on www.thecalgaryguide.com")
complicaciones-de-la-enfermedad-renal-cronica

fisiologia-del-sistema-renina-angiotensina-aldosterona-sraa

covid-19-新型冠状病毒肺炎-病理生理学和临床表现

hyperkalemia-↓-renal-excretion-pathophysiology
![Hyperkalemia (↓ Renal Excretion): Pathophysiology
Non-steroidal anti- inflammatory drugs (NSAIDs)
Inhibition of prostaglandins which promote renin secretion (see NSAIDs and the Kidney: Mechanism of Action and Side Effects slide)
Diabetic Nephropathy
Acute (AIN)and chronic (CIN) interstitial nephritis
Immune-mediated damage of the kidney tubule and interstitium
Damage to distal tubule leads to aldosterone resistance at principal cell
Aldosterone cannot ↑ EnaC insertion on principal cell of CCD
Epithelial sodium channel (ENaC) blockers
Acute kidney injury and chronic kidney disease
↓ Effective arterial blood volume (volume of blood effectively perfusing tissue)
↓ Oxygen perfusion to renal tissue causing renal ischemia
Autonomic neuropathy ↓ sympathetic drive to produce renin
Chronic juxtaglomerular cell damage ↓ synthesis of renin
Blockage of ENaC on the principal cells of the CCD
Angiotensin converting enzyme inhibitors (ACEi) and angiotensin receptor blockers (ARBs)
Angiotensin II is either not formed (ACEi), or blocked at its receptor (ARBs)
Angiotensin II cannot stimulate the release of aldosterone from the adrenal cortex
Adrenal Insufficiency
Adrenal gland cannot produce sufficient amounts of aldosterone
↓ Renin secretion by the afferent arteriole prevents RAAS activation
↓ Aldosterone release from adrenal cortex
↓ ENaC (Na+ reabsorption channel) expression on principal cells of the cortical collecting duct (CCD)
Damage to kidney causes renal impairment and ↓ glomerular filtration rate
↓ Glomerular filtrate production means ↓ tubular flow rate
↓ Na+ delivery to the distal tubule
↓ Na+ reabsorption by ENaCs at the principal cell in CCD
↓ Na+ and water reabsorption by ENaCs in CCD ↓ Effective arterial blood volume (EABV)
Activates renin-angiotensin-aldosterone system (RAAS) leads to ↑ renin secretion in the afferent arteriole (see Physiology of the renin-angiotensin-aldosterone system (RAAS) slide)
↑ Na+ and water loss in tubular lumen
↑ Positive charge in tubular lumen
↓ Electronegativity gradient in tubular lumen
of CCD
↓ K+ excretion by the principal cell in the CCD as there is less of a electronegative gradient
↑ Accumulation of K+ in blood Hyperkalemia
Serum [K+] > 5.1 mmol/L
See Hyperkalemia: Clinical Findings slide
In the case of diabetic nephropathy and NSAIDS ↓ EABV does not stimulate RAAS and therefore aldosterone production
↓ Renin
↓ Aldosterone
↑ Renin secretion activates an ↑ in aldosterone production but aldosterone action at the principal cell is blocked because of ENaCs or resistance in AIN and CIN
↑ Renin
↑ Aldosterone
In the case of ACEi, ARBs, or adrenal insufficiency ↑ renin secretion does not lead to ↑ aldosterone
↑ Renin
↓ Aldosterone
Note: as described in the above flow chart, measuring serum renin and aldosterone levels can be used to help diagnose the cause of hyperkalemia.
Authors: Mannat Dhillon, Joshua Low, Emily Wildman, Huneza Nadeem Reviewers: Andrea Kuczynski, Marissa (Ran) Zhang, Adam Bass*, Kevin McLaughlin* * MD at time of publication
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published Aug 2, 2022 on www.thecalgaryguide.com](https://calgaryguide.ucalgary.ca/wp-content/uploads/2022/08/Hyperkalemia-renal-excretion.jpg "Hyperkalemia (↓ Renal Excretion): Pathophysiology
Non-steroidal anti- inflammatory drugs (NSAIDs)
Inhibition of prostaglandins which promote renin secretion (see NSAIDs and the Kidney: Mechanism of Action and Side Effects slide)
Diabetic Nephropathy
Acute (AIN)and chronic (CIN) interstitial nephritis
Immune-mediated damage of the kidney tubule and interstitium
Damage to distal tubule leads to aldosterone resistance at principal cell
Aldosterone cannot ↑ EnaC insertion on principal cell of CCD
Epithelial sodium channel (ENaC) blockers
Acute kidney injury and chronic kidney disease
↓ Effective arterial blood volume (volume of blood effectively perfusing tissue)
↓ Oxygen perfusion to renal tissue causing renal ischemia
Autonomic neuropathy ↓ sympathetic drive to produce renin
Chronic juxtaglomerular cell damage ↓ synthesis of renin
Blockage of ENaC on the principal cells of the CCD
Angiotensin converting enzyme inhibitors (ACEi) and angiotensin receptor blockers (ARBs)
Angiotensin II is either not formed (ACEi), or blocked at its receptor (ARBs)
Angiotensin II cannot stimulate the release of aldosterone from the adrenal cortex
Adrenal Insufficiency
Adrenal gland cannot produce sufficient amounts of aldosterone
↓ Renin secretion by the afferent arteriole prevents RAAS activation
↓ Aldosterone release from adrenal cortex
↓ ENaC (Na+ reabsorption channel) expression on principal cells of the cortical collecting duct (CCD)
Damage to kidney causes renal impairment and ↓ glomerular filtration rate
↓ Glomerular filtrate production means ↓ tubular flow rate
↓ Na+ delivery to the distal tubule
↓ Na+ reabsorption by ENaCs at the principal cell in CCD
↓ Na+ and water reabsorption by ENaCs in CCD ↓ Effective arterial blood volume (EABV)
Activates renin-angiotensin-aldosterone system (RAAS) leads to ↑ renin secretion in the afferent arteriole (see Physiology of the renin-angiotensin-aldosterone system (RAAS) slide)
↑ Na+ and water loss in tubular lumen
↑ Positive charge in tubular lumen
↓ Electronegativity gradient in tubular lumen
of CCD
↓ K+ excretion by the principal cell in the CCD as there is less of a electronegative gradient
↑ Accumulation of K+ in blood Hyperkalemia
Serum [K+] > 5.1 mmol/L
See Hyperkalemia: Clinical Findings slide
In the case of diabetic nephropathy and NSAIDS ↓ EABV does not stimulate RAAS and therefore aldosterone production
↓ Renin
↓ Aldosterone
↑ Renin secretion activates an ↑ in aldosterone production but aldosterone action at the principal cell is blocked because of ENaCs or resistance in AIN and CIN
↑ Renin
↑ Aldosterone
In the case of ACEi, ARBs, or adrenal insufficiency ↑ renin secretion does not lead to ↑ aldosterone
↑ Renin
↓ Aldosterone
Note: as described in the above flow chart, measuring serum renin and aldosterone levels can be used to help diagnose the cause of hyperkalemia.
Authors: Mannat Dhillon, Joshua Low, Emily Wildman, Huneza Nadeem Reviewers: Andrea Kuczynski, Marissa (Ran) Zhang, Adam Bass*, Kevin McLaughlin* * MD at time of publication
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published Aug 2, 2022 on www.thecalgaryguide.com")
exudative-pleural-effusions-pathogenesis-and-lab-findings

Lung infection signals inflammatory response
Systemic Lupus Rheumatoid Erythematosus (SLE) arthritis (RA)
Autoimmune antibodies localize to pleura
Pleural tumors (primary or secondary from metastatic cancer)
Inflammatory cells migrate to affected site and release cytokines
↑ Permeability of pleural capillaries ↑ Fluid leakage across capillaries
Exudative Pleural Effusion
Cancer invades lymphatic drainage of pleural space (PS)
↓ Drainage of pleural fluid (PF) from pleural space
If infectious etiology
Tumor invasion = inflammatory response
See Pleural Effusions: X-ray Findings and Physical Exam Findings of Lung Diseases slides
↑ Permeable pleural capillaries allow ↑ protein and cell leakage into pleural space
If pleural tumour: Release of cancer cells into pleural space
Cancer cells on PF cytology 60-75% sensitive for malignancy
If rheumatoid arthritis:
Release of auto-antibodies into pleural space
Auto-antibodies initiate inflammatory response in pleural space
↑ Inflammatory cells have ↑ glucose metabolism in pleural space
Sterile PF with mildly elevated white blood cells, normal pH, normal glucose ↑ Inflammation at infection site damages endothelium of pleura
Pleural Infection Stage I: Simple Parapneumonic Effusion
↑ Inflammatory cells and bacterial cells in pleural space have ↑ glucose metabolism in pleural space
Bacterial invasion from infected parenchymaà pleural space
< 40mg/dL glucose in PF
↑ Activation of coagulation cascade and ↓ fibrinolytic activity
↑ Deposition of fibrin
clots/membranes within pleural
space creates loculated effusion
(compartmentalized effusion due to septations in pleural space)
↑ Production of
lactate
dehydrogenase
(LDH)
(LDH maintains NAD+ supply during ↑ glucose metabolism)
↑ CO2 production pH of PF < 7.20
↑ CO2 production pH of PF < 7.20
< 3.3mmol/L glucose in PF
Pleural Infection Stage II: Complicated Parapneumonic Effusion
Loculated effusion OR bacteria present OR ↓ pH + ↓ glucose
↑ Fibroblast proliferation creates thickened pleura ↑ Pus in pleural space Pleural Infection Stage III/Empyema: Loculated effusion and pus in pleural space
PF/serum protein ratio ≥ 0.5
PF/serum LDH ratio ≥ 0.6
Light’s Criteria: Any criteria can be met to be an exudative pleural effusion
PF LDH ≥ 2/3 upper limit of normal
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published August 9, 2022 on www.thecalgaryguide.com")
transudative-pleural-effusions-pathogenesis-and-lab-findings

Left ventricle unable to pump sufficient blood into systemic circulation
Backup of blood in pulmonary veins
↑ Hydrostatic pressure
in pulmonary veins
Pulmonary embolism
R ventricle unable to pump blood due to clot in pulmonary artery
Backup of blood in systemic veins
↑ Hydrostatic pressure
in veins draining parietal pleura
Nephrotic syndrome
Damaged glomerulus has ↑ permeability to plasma proteins in blood
↑ Loss of proteins through urine
↓ Oncotic pressure
in systemic capillaries (including within parietal pleura)
Normally, permeable pleural capillaries do not allow protein leakage into the pleural space
↑ Interstitial fluid leakage across intact pulmonary or pleural capillaries into pleural space
Transudative Pleural Effusion
Absence of bacteria and inflammatory cells in pleural space
No increase in cellular activity in pleural space
Normal levels of glucose metabolism in pleural space = low lactate dehydrogenase (LDH) (LDH increases when glucose metabolism, particularly glycolysis, increases to maintain supply of NAD+)
Large accumulation of pleural fluid (PF) pressing against lung tissue and mediastinum
Lung atelectasis (lung collapse)
See Pleural Effusions: X- ray Findings and Physical Exam Findings of Lung Diseases slides
PF/serum protein ratio < 0.5
PF LDH < 2/3 upper limit of normal
Light’s Criteria: All three criteria must be met to be a transudative pleural effusion
PF/serum LDH ratio < 0.6
See Hypoxemia: Pathogenesis and Clinical Findings slide for pathophysiology and signs of hypoxemia
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published August 9, 2022 on www.thecalgaryguide.com")
presentation-of-sah
![Subarachnoid Hemorrhage: Clinical Findings
Sudden bleeding into space surrounding the brain (for pathogenesis, see Subarachnoid Hemorrhage: Pathogenesis)
Authors: Jason An, M. Patrick Pankow Reviewers: Owen Stechishin, Dave Nicholl, Haotian Wang, Hannah Mathew, Ran (Marissa) Zhang, Yan Yu*, Cory Toth* * MD at time of publication
Bleed into subarachnoid space
Subarachnoid Hemorrhage (SAH)
Posterior hypothalamus ischemia (↓ Blood flow and oxygen)
Red blood cell lysis from energy depletion or complement activation
Release of spasmogens (spasm inducing agents)
Cerebral vasospasm (narrowing of arteries from persistent contraction) ↓ blood flow
Cerebral ischemia
Release catecholamines (hormones from the adrenal gland; e.g., epinephrine, norepinephrine)
↑ Intracellular calcium
Release of antidiuretic hormone
Antidiuretic hormone acts on the distal convoluted tubule and collecting duct in kidney to reabsorb water
Dilution of serum sodium
Hyponatremia (low blood sodium levels)
Release of epileptogenic (potential seizure causing agents) into cerebral circulation
Seizure
Products from blood breakdown in cerebral spinal fluid
Irritation of meninges (membranes surrounding the brain)
Aseptic meningitis (non-infectious inflammation)
Meningismus
(neck pain + rigidity)
Cerebral infarction (death of tissue)
Obstructs cerebral spinal fluid flow and absorption at subarachnoid granulations
Hydrocephalus (fluid build up in ventricles)
↓ Level of consciousness
Reduced cerebral blood flow
Dilation of cranial vessels to ↑ blood flow
Rapid ↑ internal carotid artery intracranial pressure
Refer to Increased Intracranial Pressure: Clinical Findings slide
Internal carotid artery
Pituitary ischemia
Hypopituitarism
[underactive pituitary gland, failing to produce 1+ pituitary hormone(s)]
Refer to hypopituitarism slides
Myocardial disruption
Left ventricle dysfunction
↑ Pressure in left heart
Blood forced backwards into pulmonary veins
↑ Pulmonary blood pressure
Fluid from blood vessels leaks into lungs
Dysrhythmias (disturbance in rate/rhythm of heart) causing ↓ cardiac output
Syncope
(loss of consciousness due to ↓ blood flow to the brain)
Pulmonary edema
(excess accumulation of fluid in lung)
Cerebral hypoperfusion
Sudden ↑in blood volume
Vessels and meninges suddenly stretch
Thunderclap Headache (worst headache of patient's life)
Shortness of breath
Reactive cerebral hyperemia (excess blood in vessels supplying the brain)
Artery specific findings:
Rapid ↑ internal carotid artery intracranial pressure
Middle cerebral artery
Posterior communicating artery
Compression of outer CN3 Compression of inner CN3
Anterior communicating artery
Nonreactive pupil
Gaze palsy
(eye deviates down and out)
Diplopia
(double vision)
Ptosis
(drooping of upper eyelid)
Frontal lobe ischemia
Avolition
(complete lack of motivation)
Ischemia of motor strip pertaining to the legs
Bilateral leg weakness
Motor strip ischemia
Hemiparesis
(weakness/ inability to move one side of the body)
Ischemia of parietal association areas (brain regions integral for motor control of the eyes, the extremities and spatial cognition)
Aphasia
(impaired ability to speak and/or understand language)/ neglect
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published July 1, 2014, updated August 10, 2022 on www.thecalgaryguide.com](https://calgaryguide.ucalgary.ca/wp-content/uploads/2015/05/SAH-Clinical-Findings-2022.jpg "Subarachnoid Hemorrhage: Clinical Findings
Sudden bleeding into space surrounding the brain (for pathogenesis, see Subarachnoid Hemorrhage: Pathogenesis)
Authors: Jason An, M. Patrick Pankow Reviewers: Owen Stechishin, Dave Nicholl, Haotian Wang, Hannah Mathew, Ran (Marissa) Zhang, Yan Yu*, Cory Toth* * MD at time of publication
Bleed into subarachnoid space
Subarachnoid Hemorrhage (SAH)
Posterior hypothalamus ischemia (↓ Blood flow and oxygen)
Red blood cell lysis from energy depletion or complement activation
Release of spasmogens (spasm inducing agents)
Cerebral vasospasm (narrowing of arteries from persistent contraction) ↓ blood flow
Cerebral ischemia
Release catecholamines (hormones from the adrenal gland; e.g., epinephrine, norepinephrine)
↑ Intracellular calcium
Release of antidiuretic hormone
Antidiuretic hormone acts on the distal convoluted tubule and collecting duct in kidney to reabsorb water
Dilution of serum sodium
Hyponatremia (low blood sodium levels)
Release of epileptogenic (potential seizure causing agents) into cerebral circulation
Seizure
Products from blood breakdown in cerebral spinal fluid
Irritation of meninges (membranes surrounding the brain)
Aseptic meningitis (non-infectious inflammation)
Meningismus
(neck pain + rigidity)
Cerebral infarction (death of tissue)
Obstructs cerebral spinal fluid flow and absorption at subarachnoid granulations
Hydrocephalus (fluid build up in ventricles)
↓ Level of consciousness
Reduced cerebral blood flow
Dilation of cranial vessels to ↑ blood flow
Rapid ↑ internal carotid artery intracranial pressure
Refer to Increased Intracranial Pressure: Clinical Findings slide
Internal carotid artery
Pituitary ischemia
Hypopituitarism
[underactive pituitary gland, failing to produce 1+ pituitary hormone(s)]
Refer to hypopituitarism slides
Myocardial disruption
Left ventricle dysfunction
↑ Pressure in left heart
Blood forced backwards into pulmonary veins
↑ Pulmonary blood pressure
Fluid from blood vessels leaks into lungs
Dysrhythmias (disturbance in rate/rhythm of heart) causing ↓ cardiac output
Syncope
(loss of consciousness due to ↓ blood flow to the brain)
Pulmonary edema
(excess accumulation of fluid in lung)
Cerebral hypoperfusion
Sudden ↑in blood volume
Vessels and meninges suddenly stretch
Thunderclap Headache (worst headache of patient's life)
Shortness of breath
Reactive cerebral hyperemia (excess blood in vessels supplying the brain)
Artery specific findings:
Rapid ↑ internal carotid artery intracranial pressure
Middle cerebral artery
Posterior communicating artery
Compression of outer CN3 Compression of inner CN3
Anterior communicating artery
Nonreactive pupil
Gaze palsy
(eye deviates down and out)
Diplopia
(double vision)
Ptosis
(drooping of upper eyelid)
Frontal lobe ischemia
Avolition
(complete lack of motivation)
Ischemia of motor strip pertaining to the legs
Bilateral leg weakness
Motor strip ischemia
Hemiparesis
(weakness/ inability to move one side of the body)
Ischemia of parietal association areas (brain regions integral for motor control of the eyes, the extremities and spatial cognition)
Aphasia
(impaired ability to speak and/or understand language)/ neglect
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published July 1, 2014, updated August 10, 2022 on www.thecalgaryguide.com")
hypercalcemia-pathogenesis

Ectopic (outside of kidney) production of 1-α-hydroxylase
1-α-hydroxylase converts calcidiol to calcitriol (aka 1,25-OH Vitamin D or active Vitamin D)
↑ 1-25 OH Vitamin D
↑ Absorption of Ca2+ in the small intestine
↑ Vitamin D Intake
Dietary Vitamin D is converted to calcidiol (aka 25- OH Vitamin D or inactive Vitamin D) in the liver
Primary Hyperparathyroidism
↑ or normal* serum parathyroid hormone (PTH) level
Tumour released Parathyroid Hormone related Peptide (PTHrP) mimics natural PTH (PTH level itself is low)
Tumour produces bone- reabsorbing substances (IL-6, IL-1, RANKL)
Paget’s Disease
Osteoblasts produce abnormally high levels of RANKL
Block Na+/Cl- cotransporter (NCC) on distal convoluted tubule (DCT) cells of the nephron
↓ Na+ and Cl- transport from lumen into DCT cells
↓ Intracellular Na+ in DCT cells
+ To compensate Na ,
↑ Ca-ATPase and 3:Na:Ca exchanger activity on DCT cells (moves 3 Na+ into cell, 1 Ca+ out into peritubular capillary)
↑ Ca+ moving into bloodstream
↓ Mechanical stimulation of osteocytes
↓ Signaling of osteoblasts (cells that form bone)
↓ New bone formation
↓ Ca2+ incorporation
into bone
↑ Ca2+ in plasma
↑ reabsorption of Ca2+ from the distal portion of nephron back into the blood
↑ 1-α-hydroxylase production in the proximal convoluted tubule
Milk Alkali Syndrome
Ca2+ intake exceeding 2000 mg per day leads to unregulated gut Ca2+ absorption into the blood
PTH binds to osteoblasts on the surface of bone
Osteoblasts produce RANKL to stimulate osteoclast function
↑ Osteoclast (cells that breakdown bone) activity
↑ Bone breakdown
Ca2+ within bone is released into the bloodstream
Osteolytic bone metastases
Hyperthyroidism
↑ Catabolic thyroid hormones
↑ RANKL: OP ratio, RANKL promotes bone breakdown by stimulating osteoclast growth while OP (osteogenic protein) promotes bone formation
Hypercalcemia Hypercalcemia
Authors: Alexander Arnold, Peter Vetere, Huneza Nadeem Reviewers: Mark Elliott, Ran (Marissa) Zhang, Yan Yu*, Hanan Bassyouni* * MD at time of publication
See Hypercalcemia: Clinical Findings slide
*Note: A normal PTH value is abnormal in the context of hypercalcemia, since hypercalcemia would normally negatively feed back to suppress PTH production.
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published February 11, 2014, updated Aug 11, 2022 on www.thecalgaryguide.com")
lower-gi-bleed-risk-factors

Rectal varices (large veins in the rectum that are at risk for bleeding)
Splenomegaly (enlargement of the spleen)
Large spleen sequesters (traps) platelets, reducing blood platelet counts
↓ Clotting ability of the blood
↓ Epithelial protection along entire GI tract
Pre-existing damage to the lower GI tract epithelium
Malignant tissue invades the colon wall and disrupts colonic blood vessels
New, extremely friable blood vessels develop within the tumor
Venous blood pressure exceeds vessel wall strength, and the vessel ruptures and bleeds
Blood moves rapidly through the GI tract (this is an upper GI bleed that can produce lower GI bleed symptoms)
Hematochezia
(bright red blood per rectum)
Damaged liver tissue restricts
blood flow through liver
hypertension (high blood pressure in the veins running from the GI tract to the liver)
Blood backs up in the splenic vein
Venous blood pressure exceeds vessel wall strength, and the vessel ruptures and bleeds
Liver cirrhosis
Authors: Yan Yu, Miranda Schmidt Illustrator: Mizuki Lopez Reviewers: Michael Blomfield, Tony Gu, Jason Baserman, Jennifer Au, Vina Fan, Ben Campbell, Kerri Novak* * MD at initial time of publication
↓ Synthesis of blood clotting factors that are normally produced in the liver (i.e. fibrinogen)
Bleeding of capillaries under the skin
Petechiae
(small red dots on skin)
Inferior Vena Cava
Blood clotting defect (genetic disorder, Acetylsalicylic Acid use)
Non-Steroidal Anti- Inflammatory Drug (NSAID) Use
Prior lower GI bleeds
Family history of colorectal cancer
Diaphragm
Esophagus
↓ Systemic prostaglandin synthesis
↑ Risk for lower GI bleed
↑ Risk for colorectal cancer
Development of colorectal cancer
Duodenum
Ligament of Treitz
Lower GI Bleeds are intra-luminal GI tract bleeds that occur anywhere distal to the ligament of Treitz (transition between duodenum and jejunum)
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Re-Published June 30, 2019, updated August 15, 2022 on www.thecalgaryguide.com")
burn-shock-pathogenesis-complications-and-clinical-findings

Endothelial cell lining in blood vessel walls is compromised
↑ Local vessel permeability
Shift of plasma + proteins from vessel into interstitial space
Direct vascular thermal injury (within burn wound)
↑ Production of circulating inflammatory mediators (ex. IL-1, IL-6, TNF-!)
↑ Systemic vessel permeability
↑ Circulating reactive oxygen species
Damage to DNA, proteins, and lipids throughout body, including myocardium (muscle cells of the heart)
↑ Myocardial stress
Myocardial dysfunction
↓ Cardiac contractility ↓ Stroke volume ↓ Cardiac output Cardiogenic
Shock
Refer to Cardiogenic Shock:
Pathogenesis, Complications and Clinical Findings
↓ Protein concentration in vessels causes ↓ intravascular oncotic pressure
Further shift of plasma from vessel into interstitial space (↑ interstitial proteins pull plasma into interstitium)
↓ Intravascular plasma
↑ RBCs per unit volume of plasma
↑ Systemic vasoconstriction to maintain blood pressure (↑ Afterload)
↓ Circulating blood volume leads to less venous return (↓ Preload)
↑ Hematocrit
Pitting edema (burned & unburned tissue)
↓ Circulating blood volume
Hypovolemic Shock
Refer to Hypovolemic Shock: Pathogenesis, Complications and Clinical Findings
Burn Shock: A complication of large burns causing end-organ hypoperfusion with resultant organ dysfunction
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published August 15, 2022 on www.thecalgaryguide.com")
thrombose-veineuse-profonde-soupconne-tvp-pathogenese-et-complications

Auteurs: Dean Percy Yan Yu Rédacteur: Tristan Jones Julia Heighton Man-Chiu Poon* Lynn Savoie* Traducteurs: Sophia Shah Sylvain Coderre* *MD à la publication
La grossesse, contraceptifs oraux
Pathogénèse et complications
Activation plaquettaire
↑ formation de caillots
Maladies héréditaires
Anomalie congénitale de la coagulation (p.e. facteur V Leiden, facteur II muté, déficit en protéine S/C) ↑ capacité de coagulation
Notes:
• TVP provoque une embolie pulmonaire, le thrombus artériel provoque un accident vasculaire cérébral
• TVP précédente est un facteur de risque de TVP courante
Malignité
Libération anormale de cytokines favorisant la coagulation
Traumatisme/opération
Blessure systémiqueà activation de la cascade de coagulation
État d'hypercoagulabilité↑ capacité du sang à coaguler lors de la stimulation
Oestrogène favorise l'hypercoagulabilité, surtout en présence d'autres risques
Hypertension Bactéries
Valve artificielle
Endommage les parois des vaisseaux
Adhèrent/ envahissent / vaisseaux
Surface anormale
Blessure de vaisseau
Expose le facteur tissulaire sur les cellules endommagées /sous-endothélium pour la liaison au FvW
Triade de Virchow
Stase veineuse
↓ débit sanguins au site de la lésion
vasculaireà concentration des facteurs de coagulation sanguine sur ce site
La graisse contient plus d'aromatase: ↑ d'androgènesà œstrogènes
mode de vie sédentaire, mauvais retour veineux
Obésité
Les caillots se produisent généralement dans les veines des jambes
1. Les veines larges et profondes permettent l'accumulation de sang
2. Retour veineux des jambes est contre la gravité
3. Valves dans les veines des jambes enclins au refoulement
↓ mouvement musculaire = ↓ débit sanguin
Fracture, immobilisation, alitement, long tour d’avion/ de véhicule
Destruction de la valve veineuse par un caillot
Insuffisance veineuse
Les caillots empêchent le sang à retourner au cœur. L'accumulation de sang dans une jambe entraîne l’œdème unilatéral et l’inflammation veineuse (rougeur, chaleur, sensibilité)
Le caillot s'embolie dans les poumons
Thromboembole
-*Embolie pulmonaire (complication aiguë mortelle)
- Hypertension pulmonaire thromboembolique chronique
Légende:
Physiopathologie
Mécanisme
Signe/Symptôme/Résultats de Laboratoire
Complications
Publié 15 juin 2019 sur www.thecalgaryguide.com")
anaphylaxie-traitements-aigu

alcoholic-fatty-liver-disease-pathogenesis-and-clinical-findings
 of
triglycerides in the liver
Abbreviations:
• CYP2E1 – Cytochrome P450, subtype
2E1
• NADH/NAD+ – Nicotinamide
Adenine Dinucleotide (in reduced and oxidized form respectively)
Ethanol upregulates hepatic cell fatty acid transporters
such as fatty acid transport protein 1 and 5 (FATP1,5)
↑ Fatty acid transporters in hepatic cells
↑ Uptake of free fatty acids from circulation into hepatic cells
Alcohol (ethanol) consumption
Ethanol is primarily metabolized in the liver by enzymes alcohol dehydrogenase (ADH) and CYP2E1 ADH converts ethanol into acetaldehyde & NADH CYP2E1 converts ethanol into acetaldehyde
Excessive alcohol consumedàthe liver cannot process ethanol and its metabolites quickly enoughà ethanol, acetaldehyde, and NADH accumulate in the liver and may enter the bloodstream
Acetaldehyde is a highly reactive and unstable molecule that can damage proteins, enzymes, and DNA
(Hepatic stellate cells (HSC) are sensitive to liver damage and release cytokines when activated)
Acetaldehyde activates HSCàHSC release tumor necrosis factor alpha (TNF-!), an inflammatory markerà↑ liver inflammation
Acetaldehyde upregulates the production of enzymes required for fatty acid synthesis
TNF-! stimulates fat production in liver cells
TNF-! enters circulation and stimulates adipose cells to break down, releasing triglyceride stores as free fatty acids
↑ Free fatty acids released into circulation
Liver biopsy shows liver inflammation
TNF-! causes the production of reactive oxygen species (unstable, highly reactive molecules)
Reactive oxygen species cause oxidative damage to hepatic cells’ mitochondria
Damaged mitochondria can no longer efficiently perform metabolic tasks
↑ Building blocks, catalysts, and direct stimulating factorsàliver cells ↑ fat production
↑ Lipogenesis in the liver (triglyceride production)
↑ Triglyceride accumulation in liver cells
Triglyceride-rich droplets in hepatic cells change the cells’ physical properties compared to normal liver cells, allowing imaging modalities to exploit these differences to detect lipid infiltration
Fatty livers appear darker on CT scans
Liver biopsy shows triglyceride- rich droplets inside liver cells
Fatty livers appear brighter and with poorer visualization of structures (like bile ducts and vessels) on ultrasounds
Imaging such as ultrasound, CT, and MRI can visualize and quantify liver fat
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published August 21, 2022 on www.thecalgaryguide.com")
tetanos-patogenesis-y-hallazgos-clinicos

neumonia-del-adulto-patogenesis-y-hallazgos-clinicos

infeccion-por-clostridium-difficile-c-diff

infeccion-por-clostridium-difficile-c-diff")
patellar-tendon-rupture-pathogenesis-and-clinical-findings

Repetitive activities involving rapid knee flexion and extension
Tendon inflammation and micro-tearing
Fluoroquinolone (antibiotic class) use
Smoking
Immobilization of knee
↓ Muscle and tendon flexibility and strength
Steroid use (including intraarticular injections)
Changes in collagen fibrils composing tendon (exact mechanism unknown)
Disrupted blood supply to tendon and poor healing
Compromised integrity and strength of the tendon
Sudden heavy load on flexed knee while foot is planted
Quadriceps muscles contract while lengthening (eccentric contraction)àforce exceeds tendon strength
Patellar tendon innervated by common peroneal (fibular) nerve
Nociceptors (pain receptors) within tendon activated by tendon rupture
Pain and tenderness below the patella
Collagen fibres in tendon tear
Tearing or popping sensation at time of injury
Patellar Tendon Rupture
Tendon/Ligament detaches from bony attachment on patella or tibial tubercle No connection of quadriceps to tibia via patellar tendon
↑ Force on tibial tubercle during tendon detachment
Avulsion fracture of tibial tubercle
Trauma to tendon ruptures surrounding blood vessels
Coagulation cascade and inflammatory mediators released at rupture site
Quadriceps muscles unable to stabilize patellar tracking
Instability of knee joint
Quadriceps muscle tension pulls patella upwards with no resistance from patellar tendon
Patellar tendon cannot use distal attachment site for leverage
Knee extensor muscles unable to effectively contract
Knee buckling
Abnormal gait
Patella alta
(high sitting patella) on X-ray
Palpable gap below the patella
Bruising below the patella
Swelling below the patella
↑ Risk of falls
Loss of patellar reflex
Inability to perform a straight leg raise (hip flexes and knee cannot remain straight)
Hematoma (blood collection under the skin)
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published August 28, 2022 on www.thecalgaryguide.com")
medial-epicondylitis-golfers-elbow-pathogenesis-and-clinical-findings
: Pathogenesis and clinical findings
Intrinsic Factors: age, body weight, nutrition, gender, anatomical variations, joint laxity, systemic disease, muscle weakness / imbalance, vascular perfusion
Micro-tears within flexor-pronator tendons initiating healing process: inflammation, proliferation, and remodeling (see acute wound healing slide)
Continued repetitive strains with inadequate recovery time between activitiesàhealing unable to meet tissue damage
Authors: Brett Lavender Reviewers: Alyssa Federico, Liam Thompson, Tara Shannon, *Gerhard Nikolaus Kiefer * MD at time of publication
Extrinsic Factors: activities involving repeated forceful use of the flexor-pronator muscle groups (sports including golf and baseball, activities such as shoveling, gardening or hammering nails)
Ineffective revascularization of damaged tissue
MRI or Ultrasound Findings: Tendon thickening, partial tears, disrupted vascular distribution +/- edema of surrounding tissues
Disorganized collagen formation and scarring à↑ type III collagen (most common collagen involved with wound healing)
↑ Tendon thickening
Decreased tensile strength of tendon
Weakness
of the flexor-pronator muscle groups
↑ Nerve growth within damaged tissue (consequence of healing response)
Local nerves are compressed by thickened tendonànociceptors within tendon are activated
(Ulnar nerve passes through cubital tunnel adjacent to medial epicondyle)
Ulnar paresthesias
may result to structures innervated distal to the cubital tunnel
Medial Epicondylitis
Tendinosis at the common flexor-pronator origin at the medial epicondyle of the humerus
Pain with passive wrist extension or resisted flexion
Tenderness
over the proximal wrist flexor-pronator muscles
Pain
localized to medial epicondyle
Severity ranges from mild to severe based on the effect on patient activities
Mild tendinopathy: patient continues most activities with minor pain
Moderate tendinopathy: patient continues some activities with modifications
Severe tendinopathy: patient’s daily activities are impacted by severe pain
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published August 30, 2022 on www.thecalgaryguide.com")
epithelial-ovarian-cancer-subtypes-molecular-alterations-risk-factors
 Mucinous carcinoma
Ovarian endometriosis (implantation of endometrial tissue on the ovaries)
↑ Chance of endometrioma (cyst comprised of endometrial tissue) formation in ovaries
See Endometriosis Slide
Familial BRCA1 or BRCA2 mutation
Impaired double strand DNA repair mechanism results in accumulation of DNA damage
5) High grade serous carcinoma
Lynch syndrome (autosomal dominant mutations in DNA repair genes)
Impaired DNA mismatch repair
mechanismà accumulation of DNA damage
1) Low grade serous carcinoma
3) Clear cell carcinoma
4) Endometrioid carcinoma
Either clear cell, endometrioid carcinomas, or a mix of both subtypes
General risk factors for developing all epithelial ovarian cancer (unknown pathogenesis)
1. Old age
2. Family history of breast or ovarian cancer
3. Post-menopausal hormone replacement therapy
4. Irregular age of menarche & menopause 5. High number of lifetime ovulation events /
nulliparity
Alteration in genes like KRAS, NRAS, BRAF, ERBB2 & CDKN2A
Alteration in genes like ARID1A, PIK3CA & ERBB2
Alteration in genes like POLE & TP53
Molecular alterations in genes like TP53 and/or CCNE1
Molecular alterations commonly associated with specific epithelial ovarian cancer subtypes
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Findings
Complications
Published August 30, 2022 on www.thecalgaryguide.com")
epoc-patogenesis

↓ Capacidad del pulmón para
prevenir daños en el tejido
Contaminantes ambientales
(consumo de tabaco a largo plazo, contaminación, infecciones)
Autores: Yan Yu Revisado por: Jason Baserman Jennifer Au Naushad Hirani* Juri Janovcik* Traductores Gloria Talavera Sandino María Rosario Talavera* * MD al momento de la publicación
Radicales libres producidos en los pulmones
Inflamación pulmonar
Inactivación de las anti proteasas del pulmón
↑ estrés oxidativo, citoquinas inflamatorias, y función de las proteasas
Daño continuo al árbol bronquial
↑ destrucción proteolítica del
parénquima pulmonar
↓ Soporte estructural para la permeabilidad de la vía aérea
Estrechamiento y colapso de la vía aérea
Infiltración de células inflamatorias, especialmente de neutrófilos
Estrechamiento y fibrosis de la vía aérea
Proliferación de cel. caliciformes,↑ producción de moco
Muerte de las células ciliadas de la vía aérea
↓ elasticidad de la vía aérea (Presión elástica de retroceso pulmonar)
Atrapamiento de aire en los pulmones
Agrandamiento permanente del alvéolo
El moco atrapado en la vía aérea provoca el desarrollo de infecciones
Pulmones hiper- insuflados
Ampollas en la superficie pulmonar
Bronquitis crónica
Enfermedad pulmonar obstructiva crónica (EPOC)
Enfisema
Hallazgos clínicos
(ver diapositiva correspondiente)
Complicaciones
(ver diapositiva correspondiente)
Leyenda:
Patofisiología
Mecanismo
Signos/Síntomas/Hallazgos de laboratorio
Complicaciones
Publicado el 7 de enero, 2013 en www.thecalgaryguide.com")
gonorrea-patogenesis

Autoinoculación
Transferencia inmediata de bacterias a la mucosa del paciente por medio del contacto indirecto con la mucosa infectada
Los componentes bacterianos incluyen: pili y otras proteínas permiten la adherencia a las células de la mucosa del huésped
Erradicación exitosa del patógeno. Patógeno eliminado del huésped
Supervivencia y replicación dentro de los fagolisosomas de los leucocitos
Diseminación del patógeno
La bacteria invade y se replica dentro de las células epiteliales columnares
Respuesta Imnune del Huésped
Fagocitosis bacteriana por los monocitos circulantes
Erradicación ineficaz del patógeno.
La proteínas bacterianas bloquean la fusion fagolisosomal y el estallido oxidativo de los PMN
Variación antigénica de proteínas y LPS en la bacteria
El enmascaramiento del antígeno gonocócico por los LPS evita la unión de anticuerpos bactericidas y provoca una activación incompleta del sistema del complemento
Inmunidad del huésped evitada
Proceso multifactorial que conduce a la capacidad de evitar la respuesta inmune del huésped
Mimetismo molecular (ejemplo, azúcares LPS terminales similares a los glicolípidos del huésped)
Secreción de IgA proteasas
Signos y síntomas de la infección por Gonorrea
(ver diapositivas de hallazgos clínicos)
Leyenda:
Patofisiología
Mecanismo
Signos/Síntomas/Hallazgos de laboratorio
Complicaciones
Publicado el 15 marzo, 2015 en www.thecalgaryguide.com")
Síndrome estafilocócico de piel escaldada
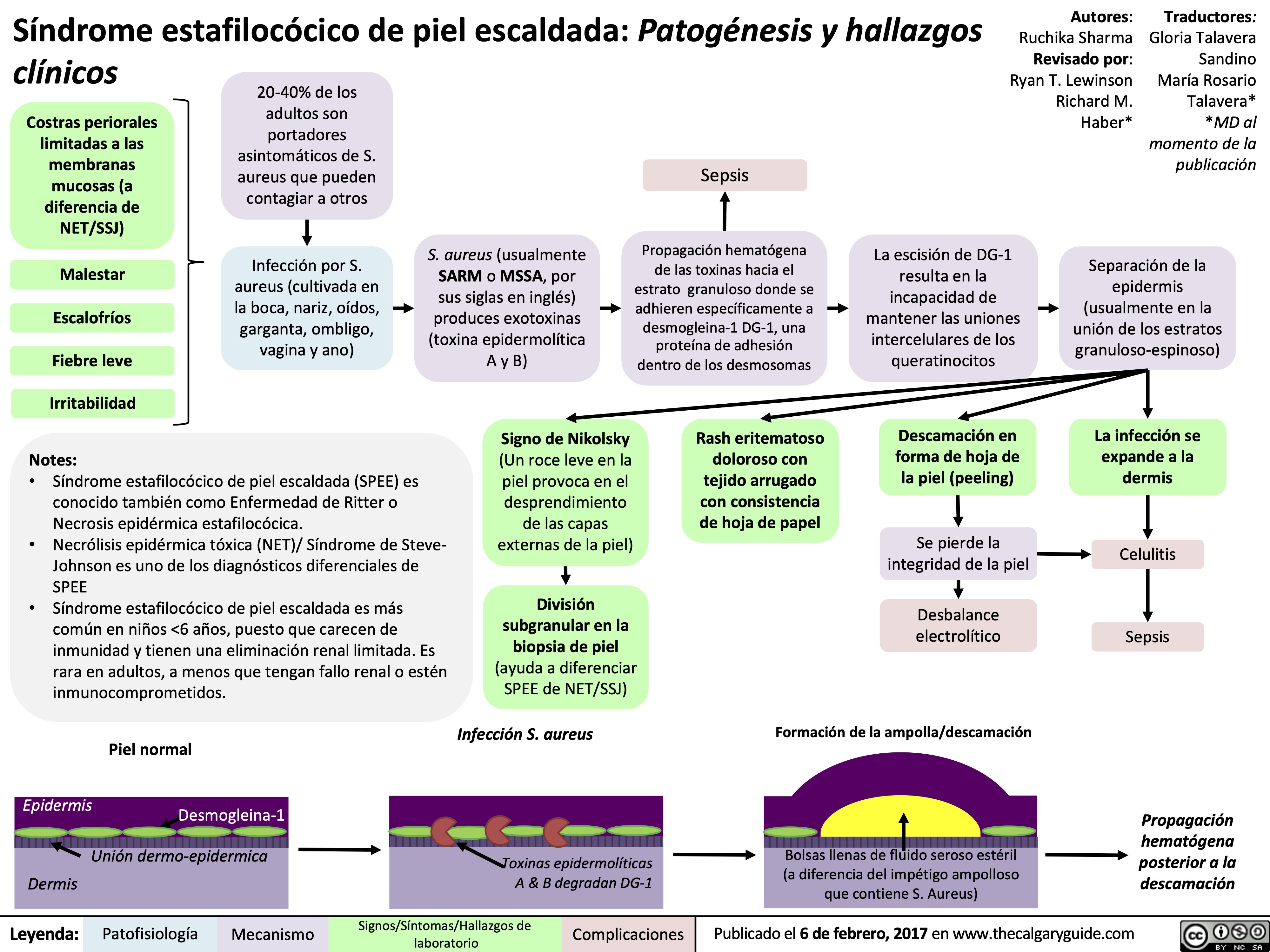
ascending-cholangitis-pathogenesis-clinical-findings

Bacteria ascends into biliary tract from duodenum via Sphincter of Oddi
Gallstone in the common bile duct
Stricture of biliary/hepatic ducts
Biliary / pancreatic duct malignancy
Biliary obstruction (partial bile duct obstruction)
↓ Excretion of bilirubin into
bileà ↑ bilirubin in blood
↑ Serum bilirubin
Deposition of bilirubin in the skin and mucous membranes
Jaundice*†
Parasites in bile duct
(E.g. Clonorchis)
Complication of endoscopic retrograde cholangiopancreatography (ERCP)
Bile accumulates in biliary tract
Bile duct dilation on ultrasound
Sludge (bile precipitants) impact bile ducts worsening obstruction
Obstruction & detergents in bile inflame ductal mucosa. Inflammation then spreads to adjacent structures
Stimulates phrenic (C3-C5) and foregut autonomic nerves (T5-T8)
Dull right upper quadrant pain*† radiating to back and right shoulder
↑ Intra-biliary pressure
Impaired forward flowà
↑ backflow of bile
Impaired bile secretion damages ductal epithelium of the biliary tract
Damaged ductal epithelium leaks ALP and GGT (enzymes) into blood
↑ Serum ALP and GGT
↑ Permeability of bile ductules
Reduced flushing of bile out to duodenum
Inflammatory response triggered
Fever*† ↑ WBCs
Tachycardia Hypotension† Confusion† Oliguria
Bacterial translocation from biliary tract into blood
Bacteremia
Massive systemic inflammatory response
Septic Shock
(see Distributive Shock slide)
* Charcot’s triad ~20% of cases | † Reynolds’ pentad ~7% of cases
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
First published November 18, 2015; Updated August 31, 2022 on www.thecalgaryguide.com")
acute-lower-gi-bleeds-pathogenesis-and-clinical-findings

Intestinal vessels are stretched over the domes of the diverticula
Angiodysplasia
Formation of
dilated, thin- walled vessels in GI tract mucosa/ submucosa
Colorectal Cancer
Infectious Colitis
Invasion of bacteria and/or bacterial toxins into intestinal wall
Cell damage and cell death
Sloughing off of intestinal epithelial cells
Ischemic Colitis
↓ Blood flow to a portion of the colon
Lack of oxygen delivery to colon wall
Necrosis (cell death) in the colon wall
Inflammatory Bowel Diseases (Note: these are chronic diseases and rarely present with acute lower GI bleed)
New, extremely friable blood vessels develop within the tumor
Malignant tissue invades the colon wall and disrupts colonic blood vessels
Crohn Disease
Immune- mediated full thickness inflammation of bowel wall
Ulcer formation and disruption of intestinal vessels
Ulcerative Colitis
Recurrent immune- mediated inflammation of colon mucosa
Authors:
Miranda Schmidt Illustrator:
Mizuki Lopez Reviewers:
Vina Fan,
Ben Campbell, Kerri Novak*
* MD at initial time of publication
Acute Lower GI Bleed
↑ Risk for vessel damage and rupture
Blood travels rapidly through GI tract
Hematochezia
(bright red blood per rectum)
May result in significant blood loss
Blood from the small intestine or right colon travels a longer distance through the GI tract
Bacteria in the GI tract has time to oxidize hemoglobin in the blood
Oxidization makes blood a darker color
Melena (rare in a lower GI bleed) (tarry black stool)
Inferior Vena Cava
Diaphragm
Esophagus
Loss of red
blood cells results in a loss of hemoglobin (a component of red blood cells)
Fluid from the extravascular space moves into the blood vessels to maintain vascular volume
Fluid is administered in a healthcare setting to compensate for blood loss
Hypovolemic Shock (rare in a lower GI bleed) (↓ Oxygen delivery to tissues due to low blood volume)
See “Hypovolemic Shock” slide for signs and symptoms
Lower GI Bleeds
are intra-luminal GI tract bleeds that occur anywhere distal to the ligament of Treitz (transition between duodenum and jejunum)
After 24 hours, the addition of fluid to the intravascular space dilutes hemoglobin in the blood
Normocytic Anemia
Duodenum
Ligament of Treitz
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published August 31, 2022 on www.thecalgaryguide.com")
melanoma-pathogenesis-and-clinical-findings
 and ultraviolet A radiation therapy used for psoriasis, vitiligo
Genetic risk factors like fair skin, multiple nevi on skin surface, extreme sun hypersensitivity
Intermittent intense sun exposure and sunburn in childhood
No/minimal sunscreen use (broad spectrum SPF 30+)
Minimal protective clothing e.g., sunglasses, hats
Geographic location e.g., equatorial regions
Tanning beds
Pyrimidine Dimer
Normal DNA
↑ Ultraviolet A and B exposure (UVA/B)
Formation of cyclobutene pyrimidine dimers in melanocytes (see illustration)
Radiant energy in UVA/B emits electromagnetic radiation
Melanin and reactive oxygen species activation in melanocytes
Oxidative DNA damage in melanocytes
Mutation in tumour protein 53 (a tumor suppressor gene) in melanocytes
↑ Cell division of mutated melanocytes
Melanoma
Asymmetry: One half of lesion does not look like the other Borders: Irregular, poorly defined, or scalloped appearance
Accelerated proto-oncogene serine/threonine kinase (BRAF)
Rapid proliferation of abnormal melanocytes at different stages of growth
Growth of mutated melanocytes at irregular and uncontrolled rates
Unregulated production of melanin by each melanocyte
Color: Varied throughout lesion e.g., black, blue, or red
Plaque with prolonged horizontal growth phase occurring anywhere on the body
Rapidly growing nodule occurring anywhere on the body
Evolving solar freckle in sun exposed areas (head, neck) found in older adults
Found in palms of hands and soles of feet
Superficial spreading melanoma
Nodular melanoma
Lentigo maligna melanoma
Acral lentiginous melanoma
Diameter: Usually greater than 5 mm
Evolving: Rapid change (few months) in shape, size, and color
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
First published August 14, 2017, updated September 3, 2022 on www.thecalgaryguide.com")
croup
 colonizes the nasopharyngeal mucosa
Virus migrates along epithelium and invades the upper airways in the larynx and trachea
Croup (laryngotracheobronchitis)
Cytokines disrupt hypothalamic thermoregulation
Fever Refer to
Fever: Pathogenesis slide
Laryngeal swelling compresses vocal cords
Hoarseness of voice
Infection of the larynx, trachea, and bronchi – typically in children 6 years or younger in the Fall and Winter
Host immune response to virus ↑ inflammatory cytokine release into circulation à Immune cells migrate to sites of infection and produce free radicals to fight off virus
Damage to healthy cells within the upper airways
↑ Permeability of blood vessels within the upper airways
Protein and fluid leak into nasopharyngeal, laryngopharyngeal, and/or tracheal interstitiums
↓ Ciliary cells (responsible for clearing mucous and dirt from the respiratory tract)
↓ Mucous movement across airway
Nasal mucous discharge
Nasal edema and swelling
Nasal congestion
↑ Tracheal mucous Trachea is narrowed and clogged
↓ Diameter of trachea
↓ Air pressure inside the tracheaàtracheal wall collapses (Bernoulli's principle)
↑ Nasal mucous
Immature/less rigid tracheal walls in young children susceptible to further structural narrowing
Breathing across a narrowed trachea à turbulent airstreams ↑ Work of breathing to adequately ventilate lungs with O2
Inspiratory stridor Expiratory Children rely on compensatory respiratory mechanisms as they fatigue overtime
(harsh, high- “barking” cough pitched sound)
Respiratory distress
Nasal flaring Chest-wall in-drawing Tachypnea Accessory respiratory muscle use
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
First published May 28, 2013, Updated September 3, 2022 on www.thecalgaryguide.com")
ventilator-associated-pneumonia-pathogenesis-and-clinical-findings
 for invasive mechanical ventilation to maintain respiration
Paralytics and
sedatives lead to inhibition of cough reflex
Damage to cilia on epithelial cells of trachea during insertion of ETT
↓ Mucociliary clearance
Insertion of nasogastric (NG) tube for feeding
Constant opening by NG tube ↓ esophageal sphincter function
Introduction of oropharyngeal microbes during intubation
Severe acute illness impairs phagocytosis and dysregulates T- cells (mechanism unclear)
Medications, chronic disease, or severe acute illness can weaken immune system
Desensitization of
pharyngoglottal
adduction reflex (PAR) (PAR normally induces closure of epiglottis to protect the airway when swallowing)
↑ Reflux of gastric contents
Accumulation of subglottic secretions containing microbes
Microbial colonization on inside of ETT
Development of biofilm on inside of ETT
Dislodgement of biofilm into lower airway
↑ Age or chronic disease can weaken respiratory function
Micro aspirations of subglottic and gastric contents
Impaired mechanisms to remove microbes from airway Positive pressure pushes microbes down
Fever/rigors ↑ White blood cell count
Introduction of pathogenic microbes to airway Susceptible patient (not required for infection)
Septic shock
See Distributive Shock slide
Sepsis
Microbes descend airway and infect lungs
Ventilator-Associated Pneumonia (VAP)
Occurs > 48-72 hours after intubation
↑ Inflammatory response at infection site promoting immune cell extravasation and cytokine release
Cytokine signalling ↑ permeability of capillaries leading to ↑ fluid leakage into interstitium and alveoli
Authors: Sravya Kakumanu Reviewers: Ben Campbell *Tara Lohmann *Bryan Yipp * MD at time of publication
Acute respiratory distress syndrome
See ARDS pathogenesis slide
Pleural effusion Lung consolidation on chest x-ray (CXR) ↑ Sputum production to clear fluid on CXR (Note: In patients with Acute Respiratory Distress Syndrome (ARDS), look for VAP consolidation in non- within alveoli/airways (may be
Positive microbial culture from sputum
dependent/upper regions of the lung where ARDS consolidation would be unexpected to extend to)
purulent in worse infections)
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published September 2, 2022 on www.thecalgaryguide.com")
syndrome-du-compartiment-deffort-chronique-scec

syndrome-du-compartiment-aigu-sca

pyoderma-gangrenosum-type-classique-pathogenese-et-resultats-cliniques

physiology-of-gene-transcription-translation
 Zhang, Yan Yu*
Translation: RNAàProtein * MD at time of publication Each 3-nucleotide sequence in the mRNA is a codon. Each
codon is recognized by a specific transfer RNA (tRNA) molecule through its complementary anticodon sequence (E.g. mRNA codon= AUG, tRNA anticodon= UAC)
Transcription: DNAàRNA
Ribosome attaches to mRNA (Ribosome= Cellular apparatus for translation made up of RNA and protein molecules)
Basal levels of cellular activity/gene expression to maintain basic cellular functions (e.g. structural proteins, enzymes of glycolysis and metabolism)
Changes to extracellular or intracellular environment (E.g. ↑ Blood glucose levels)
Cell needs to respond to stimulus
Stimulus produces a signal molecule (e.g. cytokines, hormones, other small molecules)
Each tRNA molecule has a specific nucleotide sequence to bind to a singular amino acid. Therefore, each mRNA codon
codes for a specific amino acid
(Amino acids= Building blocks of proteins)
The first transfer RNA (tRNA) enters the ribosome, carrying a specific amino acid, and binds to the first codon in the mRNA to initiate translation
Subsequent tRNAs carrying amino acids corresponding to the next codons enter the ribosome and attach their amino acid to previous amino acids to create a polypeptide chain
Ribosome reaches the last codon (translation ends). The polypeptide chain is released into cytosol or into other organelles (e.g. endoplasmic reticulum) for further processing
The polypeptide chain folds together based on biochemical interactions between the individual amino acids
Specific amino acid sequences within the nascent protein signal for further biochemical modifications (e.g. addition of phosphate, ions, sugar groups, other polypeptide chain) to make the protein functional
Functional protein can stay intracellularly or get transported to cell membrane or extracellularly based on its function
Amino Amino-acid acid
Cells surface receptor detects signal molecule and activates an intracellular cascade of signalling proteins
Signal molecule diffuses directly into cell nucleus
binding site
Anti-codon Ribosome
Growing polypeptide chain
tRNA
Signal proteins/molecules activate transcription-promoting proteins in nucleus
Transcription-promoting proteins help RNA polymerase (RNA building protein) bind to the appropriate gene sequence in DNA (complex mechanisms, simplified here)
RNA polymerase transcribes gene sequence and creates complementary messenger RNA (mRNA) strand (Complementary strand= If the DNA sequence is TTGC the complementary mRNA sequence
would be AACG)
RNA polymerase releases mRNA once entire gene sequence has been transcribed
mRNA gets transported from nucleus into the cell’s cytosol
RNA Polymerase
mRNA
Nucleus
Cytoplasm
Legend:
Pathophysiology
Mechanism
Published September 25, 2022 on www.thecalgaryguide.com")
lateral-epicondylitis-tennis-elbow-pathogenesis-and-clinical-findings
: Pathogenesis and clinical findings
Authors: Brett Lavender Reviewers: Alyssa Federico, Liam Thompson, Tara Shannon Gerhard Nikolaus Kiefe* * MD at time of publication
Extrinsic Factors: activities involving repeated forceful use of the extensor-supinator muscle groups (sports including tennis and squash, activities such as painting, carpentry or using certain hand tools)
Intrinsic Factors: age, body weight, nutrition, gender, anatomical variations, joint laxity, systemic disease, muscle weakness / imbalance, vascular perfusion
Micro-tears within extensor-supinator tendons initiating healing process: inflammation, proliferation, and remodeling (see acute wound healing slide)
Continued repetitive strains with inadequate recovery time between activitiesàhealing unable to meet tissue damage
Ineffective revascularization of damaged tissue
MRI or Ultrasound Findings:
Tendon thickening, partial tears, disrupted vascular distribution +/- edema of surrounding tissues
Disorganized collagen formation and scarringà↑ type III ↑ Nerve growth within damaged tissue collagen (most common collagen involved with wound healing) (consequence of healing response)
↑ Tendon thickening
Decreased tensile strength of tendon
Weakness
of the extensor- supinator muscle groups
Lateral Epicondylitis
Tendinosis at the common extensor-supinator origin at the lateral epicondyle of the humerus
Local nerves are compressed by thickened tendon ànociceptors within tendon are activated
Pain with passive wrist Pain localized to flexion or resisted extension lateral epicondyle
Tenderness over the proximal wrist extensor-supinator muscles
Severity ranges from mild to severe based on the effect on patient activities
Mild tendinopathy: patient continues Moderate tendinopathy: patient continues Severe tendinopathy: patient’s daily most activities with minor pain some activities with modifications activities are impacted by severe pain
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published September 25, 2022 on www.thecalgaryguide.com")
diabetes-insipidus-pathogenesis-and-clinical-findings
![Diabetes Insipidus: Pathogenesis and clinical findings
Hereditary
Autoimmune/ Idiopathic
Auto-antibodies destroy neurons that release antidiuretic hormone (ADH)
Mass Effect/ Tumor Invasion
Mass pressing on hypothalamus or pituitary
Electrolyte Imbalance
(mechanism unclear)
Hereditary
Lithium (Li)
(mechanism unclear)
Li enters principal cells of collecting ducts via ENaCs
Li inhibits GSK3β, reducing adenylyl cyclase activity
↓ cAMP- dependent phosphorylation of aquaporin-2
↑ Serum [Ca2+]
Activation of
CaSR in thick ascending limb of Loop of Henle
↓ NaCl reabsorption in thick ascending limb
↓ Generation of medullary osmotic gradient
↓ Serum [K+]
↑ Degradation of aquaporin-2 channels in collecting duct
↓ Aquaporin- 2 channels transporting water across apical membrane of collecting duct
Mutation of AVPR2 gene on X chromosome
Antidiuretic hormone (ADH) receptor cannot reach basolateral surface of principal cells of collecting duct
Mutation of aquaporin-2 gene on chromosome 12
↓ Fusion of aquaporins with apical membrane of collecting duct
Mutation of WFS1 gene on chromosome 4 (Wolfram syndrome)
↓ Processing of antidiuretic hormone (ADH) precursors and ↓ADH-releasing neurons
Surgery/ Trauma
Injury to hypothalamus or pituitary stalk
Mutation of PCSK1 gene on chromosome 5
Deficiency in PC1/3 (encoded by PCSK1)
↓ Processing of ADH by PC1/3
Aquaporin dysfunction
↓ Kidney response to ADH, which mediates reabsorption of water down its osmotic gradient through aquaporins
↓ Production of ADH by hypothalamus or ↓ secretion from ADH-releasing neurons in posterior pituitary (depending on location of lesion)
Central Diabetes Insipidus
Nephrogenic Diabetes Insipidus
Abbreviations:
AVPR2: arginine vasopressin receptor 2 CaSR: calcium-sensing receptor
ENaC: epithelial sodium channel
GSK3β: glycogen synthase kinase type 3 beta PC1/3: proprotein convertase
Diabetes Insipidus
Decreased ability of kidneys to concentrate urine
↓ Reabsorption of water from collecting duct into vasculature
Author:
Oswald Chen
Reviewers:
Huneza Nadeem,
Ran (Marissa) Zhang,
Yan Yu*
Sam Fineblit*
* MD at time of publication
Urine becomes more dilute
↓ Urine osmolality
↑ Urine output
↓ Blood volume
Blood becomes more concentrated
Occurs during late sleep period
Nocturia
Polyuria
(>3 L/day)
↑ Serum osmolality
Activation of hypothalamic osmoreceptors
Hypernatremia
(Serum [Na+] >145 mEq/L)
Polydipsia
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published September 25, 2022 on www.thecalgaryguide.com](https://calgaryguide.ucalgary.ca/wp-content/uploads/2022/09/Diabetes-Insipidus.jpg "Diabetes Insipidus: Pathogenesis and clinical findings
Hereditary
Autoimmune/ Idiopathic
Auto-antibodies destroy neurons that release antidiuretic hormone (ADH)
Mass Effect/ Tumor Invasion
Mass pressing on hypothalamus or pituitary
Electrolyte Imbalance
(mechanism unclear)
Hereditary
Lithium (Li)
(mechanism unclear)
Li enters principal cells of collecting ducts via ENaCs
Li inhibits GSK3β, reducing adenylyl cyclase activity
↓ cAMP- dependent phosphorylation of aquaporin-2
↑ Serum [Ca2+]
Activation of
CaSR in thick ascending limb of Loop of Henle
↓ NaCl reabsorption in thick ascending limb
↓ Generation of medullary osmotic gradient
↓ Serum [K+]
↑ Degradation of aquaporin-2 channels in collecting duct
↓ Aquaporin- 2 channels transporting water across apical membrane of collecting duct
Mutation of AVPR2 gene on X chromosome
Antidiuretic hormone (ADH) receptor cannot reach basolateral surface of principal cells of collecting duct
Mutation of aquaporin-2 gene on chromosome 12
↓ Fusion of aquaporins with apical membrane of collecting duct
Mutation of WFS1 gene on chromosome 4 (Wolfram syndrome)
↓ Processing of antidiuretic hormone (ADH) precursors and ↓ADH-releasing neurons
Surgery/ Trauma
Injury to hypothalamus or pituitary stalk
Mutation of PCSK1 gene on chromosome 5
Deficiency in PC1/3 (encoded by PCSK1)
↓ Processing of ADH by PC1/3
Aquaporin dysfunction
↓ Kidney response to ADH, which mediates reabsorption of water down its osmotic gradient through aquaporins
↓ Production of ADH by hypothalamus or ↓ secretion from ADH-releasing neurons in posterior pituitary (depending on location of lesion)
Central Diabetes Insipidus
Nephrogenic Diabetes Insipidus
Abbreviations:
AVPR2: arginine vasopressin receptor 2 CaSR: calcium-sensing receptor
ENaC: epithelial sodium channel
GSK3β: glycogen synthase kinase type 3 beta PC1/3: proprotein convertase
Diabetes Insipidus
Decreased ability of kidneys to concentrate urine
↓ Reabsorption of water from collecting duct into vasculature
Author:
Oswald Chen
Reviewers:
Huneza Nadeem,
Ran (Marissa) Zhang,
Yan Yu*
Sam Fineblit*
* MD at time of publication
Urine becomes more dilute
↓ Urine osmolality
↑ Urine output
↓ Blood volume
Blood becomes more concentrated
Occurs during late sleep period
Nocturia
Polyuria
(>3 L/day)
↑ Serum osmolality
Activation of hypothalamic osmoreceptors
Hypernatremia
(Serum [Na+] >145 mEq/L)
Polydipsia
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published September 25, 2022 on www.thecalgaryguide.com")
quadriceps-tendon-rupture-pathogenesis-and-clinical-findings

Quadricep muscles atrophy
↓ Quadricep muscle strength
Smoking
Chronic disease (e.g. renal failure, gout, rheumatoid arthritis, and diabetes)
Fluoroquinolone (antibiotic class) use
Changes in collagen fibrils composing tendonàimpaired tendon strength (exact mechanism unknown)
Quadriceps tendon shortens
↓ Quadricep tendon flexibility
Disrupted blood supply to quadriceps tendon and poor healing
Compromised integrity of the quadriceps tendon and ↓ strength
Sudden heavy load on flexed knee while foot is plantedàquadriceps muscles contract while lengthening (eccentric contraction)
Force on quadriceps tendon exceeds its strength
Quadriceps Tendon Rupture
Tendon detaches from bony attachment on patella
↑ Force on patella during quadriceps tendon detachment
Collagen fibres in quadriceps tendon tear
No connection of quadriceps to tibia via quadriceps tendon
Patellar tendon tension pulls patella downwards with no resistance from quadriceps muscles
Quadriceps tendon innervated by common peroneal (fibular) nerve
Nociceptors (pain receptors) within tendon activated by tendon rupture
Pain and tenderness above the patella
Trauma to tendon ruptures blood vessels
Coagulation cascade and inflammatory mediators released at rupture site
Quadriceps muscles unable to stabilize patellar tracking
Knee joint instability
Patella baja (low sitting patella) on X-ray
Palpable gap above the patella
Loss of patellar reflex
Patellar tendon cannot use proximal attachment site for leverage
Knee extensor muscles unable to effectively contract
Inability to perform a straight leg raise (hip flexes while knee cannot remain straight)
Bruising above the patella
Swelling above the patella
Avulsion fracture of patella
Tearing or popping
sensation at time of injury
Knee buckling
Abnormal gait
↑ Risk of falls
Hematoma (blood collection under the skin)
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published October 2, 2022 on www.thecalgaryguide.com")
unstable-angina-pathogenesis-and-clinical-findings
![Unstable Angina/Unstable Angina Pectoris: Pathogenesis and clinical findings Primary cause:
Secondary causes:
Coronary artery vasospasm - primary or drug induced (Ex: cocaine, triptans)
Coagulopathy
(Ex: antiphospholipid antibody syndrome)
Vasculitic syndromes (Ex: Takayasu arteritis)
Authors: Marisa Vigna Ryan Wilkie Yan Yu* Reviewers: Julena Foglia Davis Maclean Mehul Gupta Andrew Grant* * MD at time of publication
Atherosclerosis
Fatty plaque accumulates inside the intimal walls of arteries Coronary arterial atherosclerotic plaque rupture or erosion
Plaque disruption exposes subendothelial components of damaged vessel wall to platelets, initiating the coagulation cascade and platelet adhesion
Aggregation of platelets results in the formation of a thrombus Thrombus partially occludes blood flow through a coronary artery âmyocardial blood supply
Congenital anomalies (Ex: myocardial bridge, anomalous coronary)
Spontaneous coronary artery dissection
Increased blood viscosity (Ex: polycythemia, thrombocytopenia)
Factors thatámyocardial (cardiac muscle) oxygen demand (Ex: tachycardia, hypotension, hypertension, anemia, exertion, stress)
Coronary embolism (Ex: A. Fib, endocarditis, prosthetic valve thrombus)
áheart rate, contractility, and/or wall tension ámyocardial oxygen demand
Myocardial ischemia due to imbalance between blood supply and oxygen demand (insufficient blood/oxygen supply)
Unstable Angina/Unstable Angina Pectoris
Can be new onset angina; typically progressive in frequency, severity, or duration; can occur at rest
Subtotal occlusion of a coronary arteryà
reduced, but continued, myocardial blood supply
Maintained perfusion means cardiomyocytes are still alive and thus do not leak troponin into bloodstream
Normal serum troponin
Diaphoresis
(sweating)
Since bloodflow occurs from epicardium to endocardium, myocardial ischemia is more
pronounced in the subendocardium (region furthest away from heart’s external surface)
Sufficient blood flow is maintained in regions superficial to the subendocardium, resulting in non-transmural (partial thickness) heart wall ischemia
Non-inferior wall ischemia triggers a predominantáin sympathetic nervous system activity, given the proximity of cardiac sympathetic nerve innervation
Ischemiaâ cardiomyocyte resting membrane potential andâ action potential duration
Voltage gradient between normal and subendocardial ischemic zones creates injury currents, shifting the ST- vector on ECG
ECG: ST depression
and/or T wave inversion
Cardiac sensory nerve fibres mix with somatic sensory nerve
fibres and enter the spinal cord via the T1-T4 nerve roots
Brain perceives increased cardiac sensory nerve signaling as nerve pain coming from the skin of T1-T4 dermatomes (“Referred Pain”)
Myocardial ischemia causes hypoxic stress on cardiomyocytesàâaerobic (requiring oxygen) metabolism,áanaerobic (not requiring oxygen) metabolism
áanerobic respirationálactic acid production,á[H+], andâcellular pH which impairs cardiomyocyte function
Cardiomyocyte dysfunction impairs myocardial relaxation in diastole and/orâ left ventricular contractility in systole
âleft ventricular cardiac output àbackup of blood in the left ventricle, atrium, and pulmonary vasculature
ápulmonary capillary pressures pushes fluid out of the capillaries into the alveoli in the lungs
Fluid filled alveoliâgas exchange andâ oxygenation, triggering harder and faster breathing in order to compensate
Dyspnea
Activation of sweat glands via acetylcholine release
Hormones bind to cardiac β1 receptors
Tachycardia
(áheart rate)
Epinephrine/ Norepinephrine hormone release from the adrenal medulla
Hormones bind to arterial smooth muscle α1 receptors ávascular tone (vasoconstriction)
Hypertension
The Vagus nerve sits in close physical proximity to the inferior wall of the heart àinferior wall ischemia triggers involuntary Vagus nerve activation
Since the Vagus nerve coordinates parasympathetic activity,áVagus nerve activity leads to a variety of parasympathetic nervous system responses:
Retrosternal discomfort: May present as pain, heaviness, tightness, aching, pressure, burning or squeezing
Pain radiation to T1-T4 dermatomes:
Left shoulder and arm, lower jaw, neck, abdomen, upper back
Syncope
(fainting)
Bradycardia
Nausea Hypotension
(âheart rate)
(âblood pressure)
(áblood pressure)
(shortness of breath)
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Findings
Complications
Published Oct 18, 2015, updated Aug 29, 2021 on www.thecalgaryguide.com](https://calgaryguide.ucalgary.ca/wp-content/uploads/2015/10/Unstable-Angina-2021.jpg "Unstable Angina/Unstable Angina Pectoris: Pathogenesis and clinical findings Primary cause:
Secondary causes:
Coronary artery vasospasm - primary or drug induced (Ex: cocaine, triptans)
Coagulopathy
(Ex: antiphospholipid antibody syndrome)
Vasculitic syndromes (Ex: Takayasu arteritis)
Authors: Marisa Vigna Ryan Wilkie Yan Yu* Reviewers: Julena Foglia Davis Maclean Mehul Gupta Andrew Grant* * MD at time of publication
Atherosclerosis
Fatty plaque accumulates inside the intimal walls of arteries Coronary arterial atherosclerotic plaque rupture or erosion
Plaque disruption exposes subendothelial components of damaged vessel wall to platelets, initiating the coagulation cascade and platelet adhesion
Aggregation of platelets results in the formation of a thrombus Thrombus partially occludes blood flow through a coronary artery âmyocardial blood supply
Congenital anomalies (Ex: myocardial bridge, anomalous coronary)
Spontaneous coronary artery dissection
Increased blood viscosity (Ex: polycythemia, thrombocytopenia)
Factors thatámyocardial (cardiac muscle) oxygen demand (Ex: tachycardia, hypotension, hypertension, anemia, exertion, stress)
Coronary embolism (Ex: A. Fib, endocarditis, prosthetic valve thrombus)
áheart rate, contractility, and/or wall tension ámyocardial oxygen demand
Myocardial ischemia due to imbalance between blood supply and oxygen demand (insufficient blood/oxygen supply)
Unstable Angina/Unstable Angina Pectoris
Can be new onset angina; typically progressive in frequency, severity, or duration; can occur at rest
Subtotal occlusion of a coronary arteryà
reduced, but continued, myocardial blood supply
Maintained perfusion means cardiomyocytes are still alive and thus do not leak troponin into bloodstream
Normal serum troponin
Diaphoresis
(sweating)
Since bloodflow occurs from epicardium to endocardium, myocardial ischemia is more
pronounced in the subendocardium (region furthest away from heart’s external surface)
Sufficient blood flow is maintained in regions superficial to the subendocardium, resulting in non-transmural (partial thickness) heart wall ischemia
Non-inferior wall ischemia triggers a predominantáin sympathetic nervous system activity, given the proximity of cardiac sympathetic nerve innervation
Ischemiaâ cardiomyocyte resting membrane potential andâ action potential duration
Voltage gradient between normal and subendocardial ischemic zones creates injury currents, shifting the ST- vector on ECG
ECG: ST depression
and/or T wave inversion
Cardiac sensory nerve fibres mix with somatic sensory nerve
fibres and enter the spinal cord via the T1-T4 nerve roots
Brain perceives increased cardiac sensory nerve signaling as nerve pain coming from the skin of T1-T4 dermatomes (“Referred Pain”)
Myocardial ischemia causes hypoxic stress on cardiomyocytesàâaerobic (requiring oxygen) metabolism,áanaerobic (not requiring oxygen) metabolism
áanerobic respirationálactic acid production,á[H+], andâcellular pH which impairs cardiomyocyte function
Cardiomyocyte dysfunction impairs myocardial relaxation in diastole and/orâ left ventricular contractility in systole
âleft ventricular cardiac output àbackup of blood in the left ventricle, atrium, and pulmonary vasculature
ápulmonary capillary pressures pushes fluid out of the capillaries into the alveoli in the lungs
Fluid filled alveoliâgas exchange andâ oxygenation, triggering harder and faster breathing in order to compensate
Dyspnea
Activation of sweat glands via acetylcholine release
Hormones bind to cardiac β1 receptors
Tachycardia
(áheart rate)
Epinephrine/ Norepinephrine hormone release from the adrenal medulla
Hormones bind to arterial smooth muscle α1 receptors ávascular tone (vasoconstriction)
Hypertension
The Vagus nerve sits in close physical proximity to the inferior wall of the heart àinferior wall ischemia triggers involuntary Vagus nerve activation
Since the Vagus nerve coordinates parasympathetic activity,áVagus nerve activity leads to a variety of parasympathetic nervous system responses:
Retrosternal discomfort: May present as pain, heaviness, tightness, aching, pressure, burning or squeezing
Pain radiation to T1-T4 dermatomes:
Left shoulder and arm, lower jaw, neck, abdomen, upper back
Syncope
(fainting)
Bradycardia
Nausea Hypotension
(âheart rate)
(âblood pressure)
(áblood pressure)
(shortness of breath)
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Findings
Complications
Published Oct 18, 2015, updated Aug 29, 2021 on www.thecalgaryguide.com")
chest-exam-findings-of-lung-pleural-diseases
, Chronic Obstructive Pulmonary Disease (COPD), Heart Failure, and Kidney Disease
Authors: Sravya Kakumanu Reviewers: Ben Campbell, *Tara Lohmann, *Yan Yu * MD at time of publication
Interstitial Lung Disease
Pneumothorax
Rupture of visceral pleura
Build-up of air within pleural space
Pleural Effusion
Atelectasis
Consolidation
↑ Hydrostatic pressure pushes fluid into pleural space
↑ Capillary permeability = fluid leaks into pleural space
↓ Oncotic pressure = fluid moves into pleural space
Bronchus obstructed
Scarring/infiltration of lung tissue
Surfactant dysfunction (ex. ARDS)
Loss of contact between visceral and parietal pleura
(ex. effusion, pneumothorax)
Parenchymal compression (ex. loculated effusion, mass)
↑ Hydrostatic
pressure
pushing fluid
into alveoli (ex. cardiogenic pulmonary edema)
↑ Capillary
permeability
allowing fluid
to move into
alveoli (ex. pneumonia)
Idiopathic
Connective tissue diseases
Sarcoidosis, Amyloidosis
Chronic medical diseases
(ex. COPD, heart failure, kidney disease, etc)
Occupational or environmental exposures
(ex. silicosis, organic dusts, metals, gases, aerosols, etc)
Accumulation of fluid in pleural space
Collapse of alveoli
Accumulation of fluid within alveoli
Pulmonary fibrosis (scarring of alveoli bilaterally)
Breath Sounds
Lung unable to inflate fully Fluid in pleural space Collapsed lung unable to inflate Airspace filled with fluid and unable to fill with air dampens sound
↓ Breath sounds ipsilaterally
Scarred alveoli unable to expand to fill with air
↓ Breath sounds bilaterally (may not be noticeable)
Chest Rising on Inspiration
Chest cavity filled with air, but lung Lung unable to inflate fully Scarred alveoli unable to expand to fill with air unable to inflate fully
↑ Chest size, ↓ Rising ipsilaterally
↓ Chest rising ipsilaterally ↓ Chest rising bilaterally (may not be noticeable)
Percussion
Sound resonates through air in pleural space
Ipsilateral hyperresonance on percussion
Sound unable to resonate through pleural fluid
Sound unable to resonate through compacted lung tissue
Ipsilateral dullness on percussion
Sound unable to resonate through fluid-filled alveoli
Diffuse scarring of alveoli
No notable changes on percussion
Tracheal Deviation
Air pushes trachea Fluid pushes trachea ↓ Pressure within chest wall pulls trachea No pushing or pulling of trachea
Contralateral tracheal deviation Ipsilateral tracheal deviation No tracheal deviation
Adventitious Lung Sounds
Alveoli not impacted (i.e. not collapsed or fluid-filled)
Sudden opening of collapsed alveoli filling with air
Fine inspiratory crackles
Air movement through fluid-filled alveoli
Coarse inspiratory crackles
Fluid-filled alveoli can’t dampen breath sounds from larger central airways
Bronchial breath sounds all over consolidated area (harsh, ↑ pitch with expiratory phase > inspiratory; only normal when heard centrally over trachea + bronchi)
Scarred inelastic alveoli suddenly open on inspiration
Fine inspiratory crackles
No crackles
No abnormal breath sounds
Severe pneumothorax or effusion pushing against lung parenchyma
Other sounds
Amplification of patient voice through fluid-filled alveoli
Tactile fremitus (↑ vibrations felt by hand placed on chest wall when patient speaks) *Whispers also sound louder on auscultation
Fluid-filled alveoli only allow certain sound frequencies to be audible
Egophony (“E” sounds like “A” on auscultation)
Legend:
Pathophysiology
Mechanism
Physical Exam Findings
Complications
Published October 9, 2022 on www.thecalgaryguide.com")
isolated-anterior-cruciate-ligament-acl-injury-pathogenesis-and-clinical-findings
: Pathogenesis and clinical findings
Authors: Colleen Nesbitt, Amy Rudkoski Reviewers: M. Patrick Pankow, Tara Shannon, Reza Ojaghi, Usama Malik, Dr. Ryan Shields*, Dr. R. Buckley* * MD at time of publication
Female Gender
Extrinsic Risk Factors
High traction playing surface and/or footwear
↑ Torque on knee during change of direction/deceleration
Previous anterior cruciate ligament (ACL) injury
Intrinsic Risk Factors Physical Fatigue
Unknown mechanism – likely related to altered patterns in lower limb muscle activation
Incomplete healing due to poor ligament blood supply
Inadequate landing force attenuation after reconstruction/healing
Hamstring weakness and under recruitment
↑ Quadriceps (Q) angle (angle formed between quad muscle and patella tendon) à↑ lateral pull of quadriceps
+ Lachman Test:
Knee is placed in 30-degree flexion and an anterior force is applied to tibia. Excessive anterior translation of tibia relative to contralateral side is a positive result
+ Anterior Drawer Test:
Knee is placed in 90-degree flexion and an anterior force is applied to tibia. Excessive anterior translation of tibia relative to contralateral side is a positive result
+ Pivot Test:
Knee is internally rotated and a valgus stress applied. Reduction of tibia as it externally rotates from a subluxated position on the femoral condyles is a positive result
MRI can confirm the grade of tear. Knee arthroscopy is gold
standard to diagnose an ACL tear and grade the injury
↑ ACL vulnerability to injury
Contact event
(Ex: impact to lateral aspect of leg, motor vehicle accident)
Anterior translation of tibia on femur +/- valgus deformation
+/- hyperextension of the knee +/- internal rotation of tibia
↑ Forces on structures surrounding ACL
Non-contact event (Ex: sudden deceleration and/or direction change)
ACL stretches beyond capacity of collagen fibers in ligament
ACL Injury
Stretch of ligament, no tear
Incomplete tear Complete tear
Grade 1
Grade 2 Grade 3
Torn vessel inside the ACL bleeds into the joint capsule
Meniscus injury
Joint capsule Injury
Femoral subchondral bone injury
Antalgic gait
Acute hemarthrosis
Acute swollen knee
Audible “pop” at time of injury
Knee instability when full weight bearing
Development of abnormal knee biomechanics
↑ Risk of developing osteoarthritis
If all 3 findings presentàassociated with a likelihood ratio of 17.7 and positive predictive value of 81%*
* Wagemakers HP, et al. Diagnostic accuracy of history taking and physical examination for assessing anterior cruciate ligament lesions of the knee in primary care. Arch Phys Med Rehabil. 2010.
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
First published Sept 28, 2018, updated Oct 9, 2022 on www.thecalgaryguide.com")
asthma-how-treatments-work-and-common-side-effects

Long-acting Beta Agonists (Controller)
Short-acting Muscarinic Antagonists (Exacerbation)
Long-acting Muscarinic Antagonists (Controller)
Magnesium Sulfate (Exacerbation)
Monoclonal Antibodies (Controller)
Leukotriene Receptor Antagonists (Controller)
Inhaled Corticosteroids (ICS) (Controller)
Systemic Corticosteroids (Exacerbation)
Binding to beta-2 adrenergic receptors and subsequent intracellular signal cascade in bronchial smooth muscle
Off-target binding occurs in other systemic cells
Inhibition of muscarinic acetylcholine receptors in airway muscle cells
↓ Activation of the inositol triphosphate (IP3)
intracellular pathway
(IP3 pathway normally functions to mobilize intracellular Ca2+ stores)
Inhibition of Ca2+ channels on airway smooth muscle surface
↓ Influx of Ca2+
Binding and inactivation of inflammatory signal molecules (IgE, IL-5)
Inhibition of leukotriene receptors in lung and immune cells
Steroid binds to nuclear receptors within cells
↓ Gene expression/synthesis of immune mediators (ex. cytokines)
Stimulation of
Na/K ATPase (which transports K into cells)
Hypokalemia Palpitations Tachycardia Muscle cramps Tremor
Authors: Chunpeng Nie Reviewers: Sravya Kakumanu, Ben Campbell, *Tara Lohmann
* MD at time of publication
↓ Activation of mast cells (by ↓ IgE) and eosinophils (by ↓ IL-5/leukotrienes)
↓ Release of inflammatory cytokines by these cells (↓ Type 2 inflammatory response in airways)
↓ Permeability of airway vasculature
↓ Microvascular leakage into airway
Inadvertent sympathetic nervous system activation
Bronchial smooth muscle cells have ↓ Ca2+ release from intracellular stores (i.e. from sarcoplasmic reticulum)
↓↓ Cytoplasmic Ca2+
Myosin (muscle protein) unable to be activated for muscle contraction
↓ Smooth muscle contraction in bronchioles
Bronchodilation
ICS cause ↓
immune cell activity in the oropharynx
Susceptibility to infection and irritation of the oropharynx from inhaled particles and pathogens
Hoarseness Thrush
Systemic corticosteroids cause ↓ immune cell activity in whole body
↑ Susceptibility to any infection
Many other side effects
Similar effects on other muscles in the body outside bronchi
Dry mouth
Urinary retention
Constipation
↓ Mucosal edema
↓ Airway Mucus
Airflow improvement
↑ Peak flow, ↑ Oxygenation, ↓ Dyspnea
Legend:
Pathophysiology
Mechanism
Treatment Effect
Complications
Published October 9, 2022 on www.thecalgaryguide.com")
beta-bloker-mekanisme-kerja-dan-efek-samping

farmakokinetik-prinsip-dasar

syok-luka-bakar-patogenesis-komplikasi-dan-temuan-klinis

syok-hipovolemik-patogenesis-komplikasi-dan-temuan-klinis

chronic-pancreatitis-complications
 from acinar cells in exocrine pancreas
(early in disease course)
Proteins precipitate and form aggregates within pancreatic ducts
Accumulation of protein aggregates and localized fibrosis block pancreatic ducts
Rupture of acinar cells near blocked ducts → release of intracellular enzymes and fluid
Accumulation of enzyme- rich fluid within pancreas
Intra-pancreatic pseudocysts (differ from pseudocysts in acute pancreatitis, which are primarily extra-pancreatic)
Chronic Pancreatitis
Recurrent episodes of acute pancreatitis leading to irreversible fibroinflammatory pancreatic damage
Inflammatory cytokines are
continuously released from damaged pancreas over years
Cytokines damage endothelium of intra- and peri-pancreatic blood vessels (including splenic vein, which runs posteriorly behind pancreas and allows for its venous drainage)
Thin and weakened
vessel walls balloon outwards from pressure of blood flow
Pseudoaneurysms
Venous stasis (low blood flow) → ↑ concentration of clotting factors
Obstruction of peripancreatic ducts
Author: Ashar Memon Reviewers: Yan Yu*, Kiana Hampton, Sylvain Coderre* * MD at time of publication
Exocrine insufficiency
(↓ secretion of digestive enzymes, e.g., Lipase, into gastro-intestinal tract)
↓ digestion of foods and absorption of nutrients (including fats)
↓ absorption of fat-soluble vitamin D
Metabolic bone disease
(a group of disorders of decreased bone mineralization)
Blood vessels dilate and swell from increased blood flow
Gastric varices
Cytokines perpetually activate pancreatic stellate cells (stellate cells produce proteins that remodel extra- cellular matrix)
Pancreatic stellate cells increase amounts of collagen and other extra- cellular matrix molecules in pancreas → Fibrosis
Pancreatic proteolytic enzymes (e.g., trypsin) in fluid-filled pseudocysts digest walls of adjacent blood vessels
Fibrotic tissue and pseudocysts compress peripancreatic structures (including splenic vein)
Cytokines stimulate apoptosis of hormone- producing pancreatic Islet cells
(e.g., beta cells)
Endocrine insufficiency
(↓ production and secretion of pancreatic hormones)
Damaged endothelial cells of splenic vein trigger coagulation cascade
(See Calgary Guide slide on Coagulation Cascade)
Splenic vein thrombosis
↑ resistance to blood flow through splenic vein
Cytokines
stimulate apoptosis of acinar cells in exocrine pancreas
Malnutrition
↓ secretion of insulin
↓ cellular uptake and metabolism of glucose → hyperglycemia
Diabetes mellitus
Collateral blood vessels develop around stomach so blood can circumvent splenic vein and relieve splenic vein hypertension
Duodenal obstruction Biliary obstruction
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published October 18, 2022 on www.thecalgaryguide.com")
felty-syndrome
: a disease of systemic autoimmunity – refer to Rheumatoid Arthritis slides for detailed pathogenesis
Genetic susceptibility
Presence of a specific type of Human Leukocyte Antigen (HLA-DR4), a surface protein on immune cells that is known to be associated with or worsen autoimmune activity
Idiopathic autoimmunity
Neutrophil activation, apoptosis and adherence to endothelial cells in the spleen
Felty Syndrome
An uncommon extraarticular manifestation of seropositive Rheumatoid Arthritis characterized by the triad of splenomegaly, neutropenia and arthritis
Immunologic stress leads to proliferation of white pulp in spleen (responsible for initiating immune responses to foreign antigens)
Splenomegaly
↑ RBC and platelet sequestration within the spleen leading to a quantitative shortage of these cells in circulation
Enlarged spleen compresses stomach
Loss of appetite
Complex autoimmune phenomena lead to ↓ neutrophil production and ↑ removal from the circulating pool
Neutropenia
For a detailed mechanism, refer to Neutropenia slides
↓ Circulating neutrophils leads to ↑ susceptibility to infections
Severe, untreated Rheumatoid Arthritis will lead to systemic inflammation
See Rheumatoid Arthritis slides for detailed mechanism
Chronic erosion of synovial membranes in joints commonly affecting the hands, wrists and knees
Arthralgia (symmetric polyarthritis, usually with small joint distribution)
Thrombocytopenia
Refer to Thrombocytopenia slides for signs and symptoms
Normocytic Anemia Refer to Normocytic
Anemia slide for signs and symptoms
Host becomes infected
Fever
Recurrent infections
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published October 18, 2022 on www.thecalgaryguide.com")
microscopic-colitis-pathogenesis-and-clinical-findings
)
Downregulation of intestinal tight junction proteins
↓ Epithelial barrier function
↑ Transmucosal permeability
Infiltration of bacteria or antigens
↑ Expression of nuclear factor kappa B (NF-kB) in colonic epithelial cells
NF-kB increases transcriptional activity in specific genes
↑ Inflammatory molecules
(histamine, prostaglandins, and nitric oxide) in epithelium
predisposition Unknown trigger
↑ Transforming growth factor beta 1 (TGF- β1)
and vascular endothelial growth factor (VEGF) expression in colonic epithelial cells
Equilibrium shift to more production of collagen
(fibrinogenesis) than breakdown (fibrinolysis)
Drug exposure (e.g., non-
steroidal anti- (+)
inflammatory drugs (NSAIDs), proton pump inhibitors (PPIs))
Inflammation and accumulation of immature subepithelial collagen matrix
Subtype: Collagenous Colitis
Intestinal mucosal inflammation
Subtype: Lymphocytic Colitis
characterized by a colonic subepithelial characterized by intraepithelial
collagen band on histology
lymphocytic infiltrates on histology
Microscopic Colitis
Intestinal epithelial damage
Malabsorption of nutrients (including bile acids)
Weight loss
↑ Intestinal transmucosal permeability
Water drawn into lumen through osmosis
Ions and water back leak into intestinal lumen
Non-bloody watery diarrhea
Dehydration and electrolyte disturbances
Continuous release of inflammatory mediators
Peripheral afferent nerve sensitization
Visceral hypersensitivity
Abdominal pain
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published December 8, 2018, Updated October 18, 2022 on www.thecalgaryguide.com")
thiazide-diuretics-mechanism-of-action-and-adverse-side-effects
 Zhang, Julian Midgley* * MD at time of publication
Thiazides in circulation enter the proximal convoluted tubule (PCT) cells by basolateral Organic Acid Transporter 1 (OAT1)
↑ Intracellular thiazide availability
for apical OAT4 exchangers
OAT4 reabsorb urate in lumen in exchange
for thiazide that is excreted
↑ Urate in serum Hyperuricemia Uric acid build up in
joints Gout
Block the Na-Cl cotransporter (NCC) in the distal convoluted tubule (DCT) of the nephron
Impaired water excretion, vasopressin release, and ↑ water intake (mechanisms unclear)
Hyponatremia
Lack of intracellular Na+ needed to drive the Na+/K+ ATPase (moves 2 K+ into cell, 3 Na+ out into peritubular capillary)
↓ Intracellular Na+ drives Ca-ATPase and 3:Na:Ca exchanger activity (moves 3 Na+ into cell, 1 Ca+2 out into peritubular capillary)
↑ Ca+2 in serum
Hypercalcemia
See Hypercalcemia: Clinical Findings slide
↓ Na+ and Cl- reabsorption into proximal DCT cells
↑ Na+ in DCT filtrate by ∼3-5%
Water follows Na+ to maintain balanced osmotic pressure
↑ Water available for excretion
Mild Diuresis
↓ Blood volume Hypotension
↑ Na+ filtrate delivery to cortical collecting duct (CCD)
Epithelial sodium channels (ENaC)s on principal cells of the CCD transport Na+ from filtrate into the principal cells
↑ Intracellular Na+ drives Na/K+ ATPase activity on principal cells (moves 2 K+ into cell, 3 Na+ out into peritubular capillary)
↑ Intracellular K+ drives H/K+ ATPase activity on
intercalated cells (moves 1 H+ into cell, 1 K+ out into tubular filtrate)
↓ K+ in serum
Hypokalemia
See Hypokalemia: Clinical Findings slide
Acute response to
high dose thiazide diuretics (mechanism unclear)
Hyperlipidemia
Hypokalemia induces hyperpolarization of pancreatic beta cells
↓ Number of voltage gated Ca+2 channels
open given intracellular charge
↓ Ca+2 influx prevents exocytosis mediated insulin release
↓ Glucose uptake in the body
Hyperglycemia
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published October 18, 2022 on www.thecalgaryguide.com")
les-signes-et-symptomes-de-lembolie-pulmonaire-ep

les-signes-et-symptomes-de-lembolie-pulmonaire-ep")
infarctus-du-myocarde-signes-sur-examen-physique

bloc-de-branche-gauche-pathogenese-et-signes-cliniques

diverticulite-aigue-pathogenese-et-signes-cliniques

hernies-inguinales-acquises-indirect-directe

le-travail-comme-determinant-social-de-la-sante

type-ii-proximal-renal-tubular-acidosis-pathogenesis-and-laboratory-findings
 Zhang, Julian Midgley* * MD at time of publication
Overview of bicarbonate reabsorption in the proximal tubule (PT):
Serum bicarbonate (HCO3-) is filtered by the glomerulus and enters PT lumen
H+ ATPase and sodium- hydrogen exchanger 3
(NHE3) on luminal surface of PT cell secrete H+ into PT lumen
H+ combines with HCO3- in
PT lumen to form carbonic acid (H2CO3)
H2CO3 is broken down to H2O and CO2 by luminal membrane carbonic anhydrase 4
CO2 diffuses into PT cell
CO2 reacts with H2O to form H2CO3 in PT cell, catalyzed by cytosolic carbonic anhydrase 2
H2CO3 rapidly breaks down intracellularly to form HCO3- and H+
HCO3- is reabsorbed into plasma by sodium
bicarbonate transporter (NBCE1) on basolateral surface of PT cell
HCO3- is available for use to buffer plasma H+
H+ is available intracellularly for use by H+ ATPase and NHE3 exchanger
Mutation encoding NHE3 exchanger
↓ H+ secretion into PT lumen
Galactosemia, Wilson disease, cystinosis, tyrosinemia, glycogen storage disorders
Dent disease, Lowe syndrome
Infiltrative disorders
e.g., amyloidosis, multiple myeloma, monoclonal gammopathies
Drugs
e.g., tenofivir, ifosfamide, cisplatin
Carbonic anhydrase inhibitors
↓ H2CO3 breakdown to H2O and CO2
Mutation encoding CAII enzyme, carbonic anhydrase inhibitors
↓ H2CO3 production in PT lumen
↓ CO2 diffusion into PT cell
↓ H2CO3 production in PT cell
↓ HCO3- production in PT cell
↓ HCO3- reabsorption by PT cell into plasma
Type II/Proximal Renal Tubular Acidosis (RTA)
Defective HCO3- reabsorption in proximal tubule
Normal anion gap metabolic acidosis (NAGMA)
See NAGMA slide
Accumulation of toxic metabolites systemically, including in the PT
Disruption of endocytosis and intracellular transport systemically, including in the PT
Accumulation of light chain/amyloid deposits in PT
Interfere with PT’s ability to reabsorb ions/molecules
• • • •
Fanconi Syndrome:
generalized PT dysfunction
Defective tubular reabsorption of other ions/molecules to varying
degrees, including phosphate, glucose, sodium and amino acids
Tubular proteinuria Phosphaturia
Glucosuria
↑ Na+ secretionàhypovolemia
Mutation encoding NBCE1 transporter
↑ HCO3- delivery to distal nephron
K+ in distal nephron lumen binds to HCO3-
K+ is lost through osmotic diuresis
Hypokalemia
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published October 24, 2022 on www.thecalgaryguide.com")
copd-findings-on-posterior-anterior-and-lateral-chest-x-ray-findings
 See Pathogenesis of COPD Slides
Findings of COPD may not be apparent on chest X-ray early within the disease
Lung inflammation causes proteolytic destruction of lung parenchyma (part of lungs involved in gas exchange)
Alveolar walls are damagedàformation of fragile enlarged sacs of air called bullae
Rupture of bullae due to weak alveolar walls
Air leaves alveola and enters pleural space
Pneumothorax
See Primary Spontaneous Pneumothorax slide
Vasoconstriction of pulmonary arteries to shunt blood to better ventilated areas
Pulmonary hypertension
See Pathogenesis of Pulmonary Hypertension slide
↑ Pressure within pulmonary arteries
Enlargement of pulmonary arteries
Hilar enlargement Lung hyperinflation
(defined as 10 posterior ribs above the diaphragm level on the midclavicular line)
↑ Lucency of the lung
field, ↓ lung markings
(unless COPD is of chronic bronchitis phenotype)
↑ Size of retrosternal airspace
↑ Anterior – posterior (AP) diameter (“Barrel Chest”)
Flattening of hemidiaphragms
12 3 4
5 6
7
8
9 10
Poorly ventilated areas of lung
Hyperinflation as air becomes trapped in damaged lungs
↑ Total volume of air within the chest
Airway fibrosisà bronchial wall thickening (yellow arrow heads)
Air is less dense than soft tissue and vessels so it appears black
↑ Pressure anteriorly on sternum
↑ Pressure posteriorly on thoracic wall
↑ Pressure posteriorly on diaphragms
PA
Bullae sometimes visible on X-ray (focal areas of lucency with spread out vascular markings)
Image credit: Bronchial wall thickening image courtesy of Dr. Ashley Davidoff
Image credit: Radiopaedia
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published October 26, 2022 on www.thecalgaryguide.com")
Syok Obstruktif: Patogenesis, komplikasi, dan temuan klinis

Syok Kardiogenik: Patogenesis, komplikasi, dan temuan klinis

Syok Distributif: Patogenesis, komplikasi, dan temuan klinis

Hidrosefalus: Temuan Klinis pada Pediatri

Perubahan Normal pada Neonatus: Patogenesis dan temuan klinis

Granulomatosis with Polyangiitis Pathogenesis

Environmental exposures
Silica dust, cigarette smoke, infections (Staphylococcus aureus)
Genetic factors
Alpha-1 antitrypsin deficiency, proteinase 3 gene mutation
↑ Production of cytokines and antineutrophil cytoplasmic antibodies (ANCAs) (mechanism unknown) Cytokines bind to endothelial cells (that line blood vessels) and neutrophils, priming them
Proteinase 3 (PR3), an enzyme that degrades extracellular matrix proteins, migrates from neutrophil granules to neutrophil cell surface
Circulating ANCAs bind to PR3 on neutrophils
PR3 stimulates maturation of dendritic cells in lungs
Dendritic cells present antigen (PR3) to naïve CD4+ T cells in peripheral lymph nodes
T cells differentiate into type 1 and type 17 helper T cells (Th1 and Th17 cells)
Th1 and Th17 cells secrete cytokines (interferon γ (INF-γ) and tumor necrosis factor α (TNF-α)) in lungs
Secreted cytokines trigger macrophage maturation
Formation of granulomas (giant cells with central necrosis surrounded by plasma cells, lymphocytes, and dendritic cells) primarily in lungs and upper airways
ANCA-activated neutrophils release proinflammatory cytokines, attracting more neutrophils to endothelium (blood vessel wall)
ANCA-activated neutrophils undergo firm adhesion to endothelium
ANCAs stimulate ↑ secretion of proteolytic enzymes and reactive oxygen species from neutrophils
Endothelial damage and tissue injury
Granulomatosis with polyangiitis (GPA)
ANCA-associated vasculitis affecting medium and small-sized arteries, associated with necrotizing granulomas
Constitutional symptoms with involvement of multiple organ systems
(see slide on clinical findings)
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published December 4, 2022 on www.thecalgaryguide.com
Granulomatosis with polyangiitis: Clinical findings Inflammation-mediated endothelial damage and granuloma formation
(see slide on pathogenesis)
Granulomatosis with polyangiitis (GPA)
Author:
Oswald Chen
Reviewers:
Ben Campbell
*Liam Martin
* MD at time of publication
ANCA-associated vasculitis affecting medium and small-sized arteries, associated with necrotizing granulomas
Constitutional symptoms
Fever, unintentional weight loss, night sweats, arthralgias
Skin involvement
Inflammation of cutaneous vessels
Systemic inflammation obstructing blood flow, with granulomatous lesions primarily in upper airways and lungs
Ear, nose, and throat involvement
↑ C-reactive protein (CRP) and erythrocyte sedimentation rate (ESR) (markers of inflammation)
Necrotizing granulomas on biopsy of affected tissue
Positive PR3-ANCA/c-ANCA blood test (antibodies present in ~90% of patients, described in GPA Pathogenesis slide)
Renal involvement
Inflammation of renal vessels
Rupture of basement membrane (layer that filters blood from glomerular capillaries into Bowman’s capsule)
Pauci-immune glomerulonephritis (see Nephritic Syndrome slide)
Rapidly progressive glomerulonephritis
Eye involvement
Inflammation of ocular tissue
Conjunctivitis
Scleritis/ episcleritis (painful red eye)
Lower respiratory tract involvement
Inflammation of pulmonary vessels
Vessel occlusion and ischemia
Skin necrosis
Vessels burst and blood pools under skin
Round and retiform (net- like) palpable purpura of lower extremities
Granulomatous destruction of nasal cartilage
Collapse of nasal bridge
Saddle nose deformity
Inflammation of paranasal sinus and nasal cavity vessels
↓ Perfusion of lungs
Dyspnea
Damage to interstitial capillaries
Hemoptysis
Diffuse alveolar hemorrhage
Rhinitis/ sinusitis
Granulomatous obstruction of eustachian tube
Otitis media (see Otitis Media slide)
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published December 4, 2022 on www.thecalgaryguide.com")
Granulomatosis with polyangiitis: Clinical findings

Granulomatosis with polyangiitis (GPA)
Author:
Oswald Chen
Reviewers:
Ben Campbell
*Liam Martin
* MD at time of publication
ANCA-associated vasculitis affecting medium and small-sized arteries, associated with necrotizing granulomas
Constitutional symptoms
Fever, unintentional weight loss, night sweats, arthralgias
Skin involvement
Inflammation of cutaneous vessels
Systemic inflammation obstructing blood flow, with granulomatous lesions primarily in upper airways and lungs
Ear, nose, and throat involvement
↑ C-reactive protein (CRP) and erythrocyte sedimentation rate (ESR) (markers of inflammation)
Necrotizing granulomas on biopsy of affected tissue
Positive PR3-ANCA/c-ANCA blood test (antibodies present in ~90% of patients, described in GPA Pathogenesis slide)
Renal involvement
Inflammation of renal vessels
Rupture of basement membrane (layer that filters blood from glomerular capillaries into Bowman’s capsule)
Pauci-immune glomerulonephritis (see Nephritic Syndrome slide)
Rapidly progressive glomerulonephritis
Eye involvement
Inflammation of ocular tissue
Conjunctivitis
Scleritis/ episcleritis (painful red eye)
Lower respiratory tract involvement
Inflammation of pulmonary vessels
Vessel occlusion and ischemia
Skin necrosis
Vessels burst and blood pools under skin
Round and retiform (net- like) palpable purpura of lower extremities
Granulomatous destruction of nasal cartilage
Collapse of nasal bridge
Saddle nose deformity
Inflammation of paranasal sinus and nasal cavity vessels
↓ Perfusion of lungs
Dyspnea
Damage to interstitial capillaries
Hemoptysis
Diffuse alveolar hemorrhage
Rhinitis/ sinusitis
Granulomatous obstruction of eustachian tube
Otitis media (see Otitis Media slide)
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published December 4, 2022 on www.thecalgaryguide.com")
Erythema multiforme (EM): Pathophysiology and clinical findings
: Pathophysiology and clinical findings Infectious agents (cause >90% of EM)
Herpes simplex virus types 1 and 2
Mononuclear cells transport herpes simplex virus DNA fragments to keratinocytes in epidermis
Herpes simplex virus-specific CD4 T-cells of the immune system are recruited to the epidermis
CD4 T-cells release interferon-gamma (a pro-inflammatory cytokine) in response to viral antigens in keratinocytes in the epidermis
Lymphocytes infiltrate along the dermal-epidermal junction of the skin Lymphocytes create an inflammatory and edematous environment Keratinocytes irreversibly degenerate and undergo apoptosis and necrosis Formation of inflammatory epidermal lesions
Authors: Ayaa Alkhaleefa Reviewers: Ben Campbell Damilola Omotajo Jori Hardin* *MD at time of publication
Drugs (a rare cause of EM)
Non-steroidal anti- inflammatory drugs i.e. diclofenac sodium
Antibiotics i.e. TMP- SMX
Topical 5% Imiquimod, used to treat actinic keratoses
Activate tumour-necrosis-factor- alpha, an inflammatory cytokine
Skin
Epidermal layer
Dermal-Epidermal
Junction Dermal layer
Triggering agent (infection, drugs)
Immune-mediated inflammation and epidermal tissue damage
Erythema multiforme
Inflammation ↑ capillary blood flow in dermis
Red colouring of skin in the area
Centre of lesions
contain a necrotic core
Inflammatory edema causes margins around necrosis to form a raised and pale ring
Inflammation stimulates cutaneous itch receptors
Erythematous, papular, target-shaped, pruritic lesions on mucosal and acral (palms of hands, soles of feet) sites and face
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published November 29, 2022 on www.thecalgaryguide.com")
rheumatoid-pneumoconiosis-caplans-syndrome-pathogenesis-and-clinical-findings
: Pathogenesis and clinical findings
Authors: Christopher Li Keerthana Pasumarthi Reviewers: Daniela Urrego, Ben Campbell, Liam Martin* * MD at time of publication
Dust-exposed occupation (e.g. coal, asbestos, silica)
↑ Accumulation of dust particles in lungs
Dust (e.g. silica) mobilizes from the lungs to other organs such as the kidneys, lymph nodes, and spleen
Accumulation of silica in other organs which activate the immune system to secrete cytokines, chemokines, and lysosomal enzymes
Activation of antigen- presenting and antibody- producing cells
Increased risk to produce autoantibodies
Increased risk for autoimmune conditions such as rheumatoid arthritis
See slide: Rheumatoid Arthritis (RA): Extra- articular manifestations for mechanism of systemic inflammation
See slide Pneumoconioses: Pathogenesis and clinical findings for mechanism
Note: Rheumatoid arthritis may precede lung nodules or develop later in the course
Rheumatoid arthritis
Systemic autoimmune inflammatory disease that mainly involves synovial joints
(+)
Alveolar macrophages and neutrophils ingest dust particles
↑ Inflammation and cytokine production in pulmonary parenchyma (e.g. interleukin-1, tumor necrosis factor-α)
Rheumatoid pneumoconiosis
Interstitial lung disease, with characteristic nodules, resulting from the interaction between dust inhalation and rheumatoid arthritis inflammation
Cytokines recruit histiocytes, neutrophils, lymphocytes, and fibroblasts to the area to produce a zone of inflammation around the dust-containing cell
Necrosis and apoptosis of dust-containing cells, macrophages, and surrounding collagen
Necrotic cells are digested by new macrophages to restart the process
Production of nodules that contain a central necrotic area surrounded by alternate layers of dust and necrotic tissue (Caplan nodules)
Hyperactive immune response to foreign materials in lungs
Multiple concentric rings of dust seen histologically on light microscopy
0.5-5 cm rounded opacities on chest X-ray in periphery of lungs
See slide on
Pneumoconioses
for symptoms and pulmonary function test results
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published November 17, 2022 on www.thecalgaryguide.com")
Acute Liver Failure: Pathogenesis and clinical findings

NAPQI binds hepatocellular proteins
(see Acetaminophen Overdose: pathogenesis and clinical findings slide)
Drug-induced liver injury
Metabolism of drugs by the liver can produce reactive drug metabolites
Intracellular stress, mitochondrial injury, or immune response
Viral Hepatitis (i.e. HAV, HBV, HEV, HSV)
Acute infection or infection flare provokes an immune response against infected hepatocytes
Autoimmune Hepatitis
Autoimmune antibodies attack hepatocytes (see Auto-immune Hepatitis (AIH) slide)
Ischemia (i.e. from shock)
↓ O2 delivery to the liver
Hepatocellular hypoxia
Wilson’s Disease
Heritable mutation in the ATP7B gene
↓ Biliary excretion of copper
Hepatic copper accumulation injures hepatocytes (see Wilson’s Disease slide)
Accelerated rate of hepatocellular necrosis or apoptosis
Hepatocyte death exceeds regeneration such that liver function is compromised within a short amount of time
Acute Liver Failure
An illness of <26 weeks duration in the absence of pre-existing cirrhosis, characterized by INR ≥1.5 and evidence of altered mentation (hepatic encephalopathy)
Injured hepatocytes leak hepatic enzymes (AST, ALT, GGT) into circulation
↑ Liver enzymes
Hepatocellular inflammation
Stimulation of foregut
autonomic nerves
Right upper quadrant pain
↓ Toxin metabolism
Toxins build up and activate microglial cells (brain macrophage)
Oxidative stress and cerebral edema
Hepatic encephalopathy
Characteristic set of neuropsychiatric symptoms (see Hepatic Encephalopathy slide)
↓ Hepatocellular function and number
↓ Complement protein synthesis
↓ Ability to clear immune complexes and activate B cells
Accumulation of pigmented bilirubin
↓ Synthesis of coagulation factors
↑ INR
↓ Conjugation of bilirubin by the liver and ↓ transport into bile for excretion
↑ Serum bilirubin
Jaundice
Infection
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published November 15, 2022 on www.thecalgaryguide.com")
Acute and Chronic Gastritis: Pathogenesis and clinical findings

Noninfectious
NSAIDs, alcohol, gastric reflux
Stress
Critical illness, trauma
Gastric ulceration (see Peptic Ulcer Disease slide)
↓ Gastric mucosal defense
(↓ secretion of prostaglandins and mucus)
Acid erodes gastric mucosa
Inflammatory cells (primarily neutrophils) infiltrate site of injury
Acute Gastritis
Acute inflammation of gastric mucosa
Inflammatory cells (primarily lymphocytes and plasma cells) accumulate in gastric mucosa
Autoimmune Metaplastic Environmental Metaplastic Atrophic Gastritis (AMAG) Atrophic Gastritis (EMAG)
Atrophic Gastritis
Chronic inflammation of gastric mucosa
Inflammatory cells destroy gastric glandular epithelial cells
Gastric mucosal atrophy and metaplasia (replacement of gastric mucosal cells with intestinal epithelial cells, commonly goblet cells)
Hypochlorhydria
(parietal cell loss → ↓ hydrochloric acid secretion)
↓ Iron absorption
Iron deficiency anemia
(see Iron Deficiency Anemia slide)
Activation of chemoreceptors and mechanoreceptors
Activation
of visceral nociceptors
Visceral afferents stimulate chemoreceptor trigger zone of medulla
Nausea/ vomiting
Autoimmune
Associated with human leukocyte antigens HLA-B8 and HLA-DR3, which are proteins expressed on immune cell surface
Antibodies destroy parietal cells and intrinsic factor in gastric body and fundus
Epigastric pain (typically burning or gnawing) Dyspepsia (abdominal discomfort after
eating, often with early satiety and bloating)
Parietal cell loss →
↓ intrinsic factor →
↓ vitamin B12 absorption
Vitamin B12 deficiency
(see Vitamin B12 Deficiency slide)
Macrocytic Peripheral anemia neuropathy
↓ Gastric acidity leads to ↓ inhibition of G cells in antrum Hypergastrinemia (↑ gastrin release from G cells)
Gastrin binds to cholecystokinin-B (CCK-B) receptors on parietal and enterochromaffin-like (ECL) cells of gastric body
↑ CCK-B signaling leads to ↑ cell proliferation and ↓ cell death (↓ apoptotic activity)
Hyperplasia (↑ number of cells)
Low-grade dysplasia (disordered growth of epithelium) High-grade dysplasia
Carcinoid tumor (0.7% of cases) Malignancy arising from ECL cells
Gastric adenocarcinoma (0.3% of cases) Malignancy arising from gastric epithelial cells
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published November 15, 2022 on www.thecalgaryguide.com")
Pubic Rami Fracture: Pathogenesis and clinical findings

Bone loses calcium, becoming less dense weaker and more susceptible to fracture (See Osteoporosis: pathogenesis and clinical findings slide)
Athletic injury in skeletally immature athletes (e.g., soccer, gymnastics)
Open growth plates are weak and more susceptible to injury
Lateral compression or vertical shear fracture
Mild to moderate osteoporosis
Fragility pubic rami fracture from low-energy impact (e.g., falls from standing, falls in the bathtub)
Severe osteoporosis
Spontaneous pubic rami fracture
Sudden, forceful contraction of the hamstring muscles (e.g. sudden change of direction, sudden stop)
Hamstring pulls a piece of the ischial tuberosity from the pelvis boneàpubic rami avulsion fracture
(Posterior pelvic rim fracture
requires CT to diagnose and is often missed)
Missed or delayed diagnosis due to incomplete workup
Pubic Rami Fracture
Fracture of the anterior pelvic ring, can be the superior and/or inferior pubic rami
Co-existing posterior pelvic ring fracture (e.g. acetabulum, sacrum)
Fracture on both sides of pelvis
Unstable pelvis
Blood vessels in and surrounding the bone rupture during injury
Blood accumulates under the skin
Bruising around fracture site
Inflammatory response to injury
Recruitment of white blood cells and fluid to the area
Swelling around fracture site
Irritation at superior/inferior pubic rami muscle attachment sites (groin and hip abductor muscles)
Pain in groin near fracture site
↑ Displacement and incomplete healing of the posterior and anterior pelvic ring fracture
↑ Morbidity and mortality
↑ Disuse osteoporosis ex: lower limbs, pelvis, and back
↑ Muscle stiffness ex: lower limbs, pelvis, and back
↑ Joint stiffness ex: lower limbs, pelvis, and back
Fracture hematoma in pubic rami
Hematoma distends the periosteum, irritating nerves in the area
Moving/walking further irritates nerves in the area
Moving and walking ↑ pain àinadequate movement
↓ Mobility (to avoid pain)
Antalgic gait (stance phase of walking is shortened relative to swing phase)
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published December 4, 2022 on www.thecalgaryguide.com")
COPD: Treatments and Common Side Effects

Short-acting Beta Agonists (SABA) Reliever
Long-acting Beta Agonists (LABA) Controller
Long-acting Muscarinic Antagonists Controller
Inhaled Corticosteroids (ICS) Controller
Phosphodiesterase Inhibitors (PDE4i) Controller
Short-acting Muscarinic Antagonists Reliever
Systemic Corticosteroids Exacerbation
Antibiotic therapy Exacerbation + Controller
Binds to beta-2 adrenergic receptors in bronchial smooth muscle cells
Activation of intracellular signal cascade in bronchial smooth muscle
Off-target binding occurs in other systemic cells
Prevent phosphodiesterase enzymes from breaking down secondary messenger molecules such as cyclic adenosine monophosphate (cAMP)
cAMP binds to other cell signaling proteins
Bronchial smooth muscle cells have ↓ Ca2+ release from intracellular stores (i.e. from sarcoplasmic reticulum)
Inhibition of muscarinic acetylcholine receptors in airway muscle cells
↓ Activation of the inositol triphosphate (IP3)
intracellular pathway
(IP3 pathway normally functions to mobilize intracellular Ca2+ stores)
Steroid binds to nuclear receptors within cells
↓ Gene expression/ synthesis of immune mediators (ex. cytokines)
Frequent moderate-severe exacerbations despite maximal therapy
Prophylactic macrolide treatment in outpatients
Certain macrolides have anti- inflammatory properties
↓ Infection risk = ↓ exacerbation risk
COPD exacerbations often triggered by respiratory infections
Empiric antibiotic therapy in hospitalized patients with acute exacerbations
↓ Infection in lungs
↓ Sputum production
↓ Respiratory distress
Stimulation of
Na/K ATPase (which transports K into cells)
Hypokalemia Palpitations Tachycardia Muscle cramps Tremor
Inadvertent sympathetic nervous system activation
↓↓ Cytoplasmic Ca2+
Myosin (muscle protein) unable to be activated for muscle contraction
↓ Smooth muscle contraction in bronchioles
Bronchodilation
ICS ↓ immune cell activity in the oropharynx
Susceptibility to infection and irritation of the oropharynx due to inhaled particles and pathogens
Hoarseness Thrush
Systemic steroids cause ↓ immune cell activity in whole body
↑ Susceptibility to any infection (and many other side effects)
↓ Release of inflammatory cytokines (↓ Type 2 inflammatory response in airways)
↓ Permeability of airway vasculature
↓ Microvascular leakage into airway
Authors: Reshma Sirajee, Chunpeng Nie
Reviewers: Sravya Kakumanu, Ben Campbell, Tara Lohmann* * MD at time of publication
Similar effects on other muscles in the body outside bronchi
Dry mouth
Urinary retention
Constipation
↓ Airway mucus ↓ Mucosal edema
Legend:
Pathophysiology
Mechanism
Treatment Effect
Side Effects
Published November 15, 2022 on www.thecalgaryguide.com")
Achalasia: Findings on Fluoroscopy with Barium Swallow
 tract during fluoroscopy)
Primary Achalasia
(idiopathic)
Secondary (Pseudo) Achalasia
(due to another disease process such as tumor, Chaga’s disease, diabetes mellitus)
Achalasia
Secondary disease processes can cause denervation or nerve dysfunction of esophageal myenteric plexus (EMP). Tumors can obstruct the lumen and infiltrate the EMP.
Visual differences from primary achalasia
Look for fixed abnormalities and/or mucosal irregularities and shouldering in the setting of a tumor
Nerves controlling the lower esophagus are damaged, making it difficult for food and liquids to pass the esophagus
See “Achalasia: Pathogenesis and Clinical Findings” for full pathogenesis of primary and secondary achalasia
Inflammation and degeneration of nerves in the wall of esophagus
Dysfunction of the esophageal myenteric plexus (network of nerves in the GI tract responsible for peristalsis and sphincter relaxation)
Incomplete relaxation of the Loss of peristalsis in distal lower esophageal sphincter (LES) esophagus
Barium has difficulty passing through the LES to the stomach
Barium outlines the GI tract, following esophageal abnormalities
Normal findings
Esophageal Dilatation
Occurs upstream of narrow LES
Bird Beak Sign / Rat Tail Sign
Smooth narrowing of the distal esophagus
Incomplete LES relaxation
Barium Swallow may be normal in some patients with achalasia. Esophageal manometry (gold standard for diagnosis) or upper endoscopy can be considered instead.
Image Source: Radiopaedia
Legend:
Pathophysiology
Mechanism
Radiographic Findings
Complications
Published November 9, 2022 on www.thecalgaryguide.com")
popliteal-bakers-cyst-pathogenesis-and-clinical-findings
 Cyst: Pathogenesis and clinical findings
Authors: Liam Thompson, Megan Ure Reviewers: M. Patrick Pankow, Tara Shannon, Reza Ojaghi, Usama Malik, Dr. Gerhard Kiefer* * MD at time of publication
Repeated contraction of muscles surrounding the bursaàmicro trauma to bursa
Inflammatory response
Idiopathic
(common: kids 4-7 years old)
Bursa located between medial head of gastrocnemius and semimembranosus tendon
Accumulation of inflammatory fluid into the bursa forming a cyst
Meniscal Tear
Tear creates a channel between joint capsule and bursa posterior to the knee joint
Rheumatoid Arthritis
Auto-immune mediated inflammation of knee joint
Osteoarthritis
Destruction of articular cartilageà inflammation within knee joint See Osteoarthritis: Pathogenesis slide
Damage to joint capsule and accumulation of inflammatory fluid
Proteolytic enzyme dysregulation Local muscle action influences fluid flow and pressure changes
Extra- articular Popliteal Cyst
Fluid filled sac behind knee
Usually asymptomatic
(resolves spontaneously)
Synovial joint herniates into popliteal fossa (most commonly medially)àaccumulation of fluid (synovial + inflammatory) into synovial herniation, forming a cyst
Knee flexion
Intra-articular Popliteal Cyst
Known as “Baker’s cyst”
Knee extension
↑ Muscle tension around cyst
Channel between knee joint and cyst gradually closes with ↑ tensionàat full extension, no fluid flow between the joint and cyst (trapping the fluid in the cyst)
↓ Muscle tension around cyst
Channel between knee joint and cyst opens
Knee joint pressure becomes negative
↑ Fluid flow from cyst into knee joint
↓ Fluid in popliteal cyst
Knee joint pressure becomes positiveà ↑ Fluid flow from knee joint into cyst
(Posterior tibial nerve located lateral to the popliteal cyst and enervates posterior knee, calf, and bottom of foot)
Posterior tibial nerve becomes entrapped by mass (cyst)
Enlarging mass in posterior knee
Cyst reaches maximum volume
Pressure within cyst exceeds tensile strength of surrounding sac
Fluid drains distally into calf
Popliteal vein located lateral to cyst
↑ Venous pressure distal to cyst
Popliteal vein occluded by mass
Venous pooling distal to site of occlusion
↓ Space in posterior calf compartmentàcompression of calf muscles and posterior tibial nerve
Ankle dorsiflexion ↑ pressure in compartment
Positive Homan’s sign
(discomfort with passive ankle dorsiflexion)
Posterior plantar numbness
Pain receptors (nociceptors) activated
Posterior knee pain/pressure
Cyst Rupture Abrupt and intense pain
Stimulates inflammatory response
Fluid pushed from veins into interstitial space
Swelling and erythema (redness) in distal calf and foot
Pseudo-Thrombophlebitis
worse with extension or physical activity
(shares symptoms with deep vein thrombosis but no associated clot)
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published Feb 10, 2018, updated Nov 19, 2022 on www.thecalgaryguide.com")
hypovolemic-shock

Inflammation (pancreatitis, cirrhosis, post-operative, etc.)
Inflammatory mediators vessel permeability and fluid leaks out
Trauma
Ruptured vessels leak fluid into potential spaces
Hemorrhagic losses
(GI bleed, postpartum hemorrhage, etc.)
↓ Intravascular volume
↓ Venous return to the heart
↓ Cardiac output (blood pumped from the heart)
Hypovolemic Shock
↓ Oxygen delivery to tissues due to low blood volume
Insufficient organ perfusion
Non-Hemorrhagic losses
(dehydration, GI losses, skin losses / burns, renal losses, etc.)
‘Third Spacing’ of fluid
(fluid located outside the intravascular or intracellular space; large collections can occur in the pelvis, thorax, GI tract, long bones of children, intra-abdominally, retroperitoneally)
P = Q x R; less ‘flow’ in the vessels (Q), with vessels not constricting enough to maintain resistance (R)à pressure (P) will drop
↓ Blood Pressure
Caution: young, healthy individuals can maintain blood pressure during circulatory collapse with cardiac output and vasoconstriction; do not use blood pressure as an indicator of shock severity in children
Carotid sinus baroreceptors sense low blood pressure ↓ Carotid sinus inhibition of sympathetic nervous system Release of sympathetic catecholamines (epinephrine and
↓ Pressure in venous circulation
Brain
Heart
Kidneys
↓ Blood in the right internal jugular vein
↓ Oxygen delivery to the brain
↓ Myocardial contractility (from lactic acidosis)
↓ Blood flow to kidneys
↓ Jugular Venous Pressure
Catecholamines bind to beta-1 receptors in the sinoatrial node of the heart
Beta-1 receptor activation causes ↑ heart rate
Tachycardia
norepinephrine)
Catecholamines bind to and stimulate alpha-1 receptors in peripheral vessels
Vasoconstriction of peripheral vessels
↓ Blood flow to peripheral tissue
Catecholamines bind to and stimulate beta receptors in sweat glands
Diaphoresis
(sweating)
In all body tissues
Inadequate oxygen delivery
↓ ATP production
↑ Anaerobic metabolism
↓ Body temperature
Impaired neurological functioning
Renal ischemia
Activation of the renin-angiotensin aldosterone system
↓ Glomerular filtration rate
↓ Clearance of lactic acid by the kidney
↑ Lactic acid production
↓ Rate of activity of clotting enzymes
Lactic Acidosis
Unknown mechanism
Coagulopathy Hypothermia
Trauma Triad of Death
↑ Capillary Cold, mottled refill time extremities
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published January 24, 2013, updated December 4, 2022 on www.thecalgaryguide.com")
Ischemic Stroke: Pathogenesis
 of basal or brainstem penetrating arteries
Large artery atherosclerosis
Cholesterol plaque ↓ diameter of intra- or extracranial vessel
Cardiac embolism
Blood clot in heart breaks free, travels to brain
Other
E.g. volume loss, severe infection
Unknown
E.g. 2 or more mechanisms
Modest ↓ in O2 at penumbra (see figure)
Authors: Mizuki Lopez Andrea Kuczynski Illustrator: Mizuki Lopez Reviewers: Sina Marzoughi Usama Malik Hannah Mathew Ran (Marissa) Zhang Andrew M Demchuk* Gary M. Klein* * MD at time of publication
Significant ↓ in O2 at ischemic core (see figure)
↑ Anaerobic metabolism ↓ ATP
Production
Dysfunction of Na+/K+ ATPase pump (for 1 ATP molecule, 3 Na+ moved out of cell, 2 K+ moved into cell)
H2O influx following Na+ Cerebral edema
Compression of vessels and surrounding tissue damages blood-brain barrier
↑ Permeability of damaged blood-brain barrier
Infiltration by peripheral immune cells
Immune cells release inflammatory cytokines
↓ Cerebral Blood Flow
Penumbra Ischemic core
Metabolic demands are greater than supply of ATP
Cell death
Microglia (resident neural immune cells) activate to clean dead cell debris
Microglia release inflammatory cytokines (TNFα, IFγ, IL-1β)
Cytokines lead to astrocyte activation (support cells for neurons)
Astrocytes release more inflammatory cytokines
Inflammation of brain tissue
↑ Na+, Ca2+ influx, K+ outflux
↓ Glutamate (excitatory neurotransmitter) reuptake by astrocytes (support cells for neurons)
↑ Glutamate in extracellular fluid
Spreading depolarization from core (unclear mechanism)
Activate biochemical pathways including glutamate receptor activation
↑ Glutamate activity
Activate glutamate receptors that conduct Ca2+
↑ Ca2+ influx into neuron
Activation of catabolic proteases, lipases, nucleases in neuron
Dysfunction of neuronal protein synthesis and activity
Neuronal cell death
↑ Volume of dead (infarcted) brain tissue
Neurons depolarize and release glutamate
Reversal of Na+ Dependent Glutamate Reuptake Transporters on astrocytes (normally 3 Na++ 1 H+ + 1 glutamate into cell, for 2 K+ out)
↑ Glutamate in extracellular fluid
Stroke symptoms (e.g. weakness, slurred speech, visual field losses, autonomic dysfunction)
(see Ischemic Stroke: Impairment by Localization stroke slide)
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published November 14, 2017; updated November 6, 2022 on www.thecalgaryguide.com")
Cirrhosis
 Hepatitis D occurs with hepatitis B
Alcohol related liver disease
Non-alcoholic steatohepatitis
Autoimmune: autoimmune hepatitis, primary biliary & sclerosing cholangitis
Genetic: hemochromatosis, Wilson’s disease, α1- antitrypsin deficiency
Toxic: drugs may rarely cause chronic liver disease or cirrhosis
Severe or chronic hepatocyte injury overrides regenerative capacity → Fibrosis
Cirrhosis
Mild fibrosis may reverse with treatment of underlying cause
Nodular/shrunken liver on ultrasound
↑ Liver stiffness on transient elastography Diffuse fibrosis with nodular regeneration on biopsy
Fibrotic liver provides ↑ resistance to blood flow
Portal hypertension
Irreversible formation of fibrosis, within which hepatic cell regeneration is restricted to form nodules of poorly-functioning cells
Inflammation → epigenetic changes, oncogene mutations
Hepatocellular carcinoma
↑ Serum α-fetoprotein (sensitivity 50%, specificity 99%)
Fibrosis disrupts normal function of hepatic lobules
Hepatic insufficiency
↓ Liver synthetic function
↓ Thrombopoietin synthesis
Thrombocytopenia
↓ Conjugation of bilirubin,
↓ secretion of bilirubin into bile ducts, and
↓ drainage of bilirubin into hepatic duct
Accumulation of serum bilirubin >30 μmol/L
Jaundice
Scleral icterus, jaundiced frenulum
↑ Pressure in portal venous circulation
Portosystemic shunts
Increased flow to esophageal, rectal, and splenic veins
Vascular stretch → endothelial vasodilator (e.g. NO) release
Vasodilators enter systemic circulation
Pulmonary vasodilation
Blood cells in pulmonary vasculature have ↓ time for gas exchange
Hepatopulmonary syndrome (rare)
Dyspnea or hypoxemia, worsened when upright
↓ Synthesis of clotting factors (V, VII, IX, X, XI, XII, fibrinogen, prothrombin) and anticoagulant proteins (antithrombin, proteins C/S)
Unpredictable imbalance of hemostatic and anticoagulant factors
Elevated INR
± coagulopathy
Impaired metabolism of waste products and toxins
Toxins (mainly ammonia) accumulate and cross the blood- brain barrier
Hepatic encephalopathy
Day-night reversal, asterixis, delirium
Varices
(dilated veins in the esophagus, stomach, or rectum)
Variceal bleed
GI bleed and hypovolemia
Backflow of blood into spleen
Splenomegaly
Enlarged spleen sequesters (traps) blood cells
Cytopenias
(eg. thrombocytopenia or anemia), petechiae, easy bruising
↓ Blood flow to kidneys, which sense ↓ effective blood volume
Kidneys retain water and Na+
↑ Hydrostatic pressure in splanchnic vessels
↓ Albumin synthesis
↓ Capillary oncotic pressure
Net fluid flux out of vasculature into interstitial space
Ascites
(fluid in peritoneal cavity)
Peripheral edema
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published August 7, 2022, updated November 6, 2022 on www.thecalgaryguide.com")
Farmakodinamik: Prinsip Dasar

Aterosklerosis: Patogenesis

Aterosklerosis: Komplikasi

Approach to Arterial Blood Gases ABGs

Normal pO2 should be FiO2 x 4-5
(FiO2 in room air= 21%)
pH >7.40 = Alkalemia
1. Adequate oxygenation?
2. Define the acid-base disturbance
Diarrhea (Loss of electrolytes not reabsorbed through bowel)
Normal ABG on Room Air = 7.40 / 40 / 90 / 24
pO2 ↓ than expected
Possible hypoxemia
See slide on
Hypoxemia: Pathogenesis and Clinical Findings
(pH)
(Loss of acidic fluids from stomach)
(HCO3-)
(pCO2)
(pO2)
Renal Tubular Acidosis (RTA)
pH <7.40 = Acidemia
Metabolic derangement (Due to poisons, infection, or ketones)
Gain of acid (H+)
Brain unable to promote respiratory drive
(ex. drugs, trauma)
Inability for chest wall to expand (ex. obesity, neuromuscular weakness, pleural or chest wall abnormalities)
Vomiting or
nasogastric
suction
Impaired tubular transport of H+
(Due to loop/thiazide diuretics, hypomagnesemia, congenital abnormalities)
1o Hyperaldosteronism =
↑ H+ ion secretion at
distal tubule
(Due to tumours, congenital abnormalities , malignant hypertension, etc)
Type II RTA = HCO3- not reabsorbed
Type I or IV RTA = H+ not secreted
GI or renal loss of HCO3-
Hypoventilation
↑ CO2 from ↓ exhalation Respiratory Acidosis Acute= ↑10:↑1, Chronic= ↑10:↑3
GI or renal loss of acid (H+)
↑ HCO3- from ↓ H+ to bind with Metabolic Alkalosis ↑7:↑10
↑ Ventilation from stress, trauma, infection
↓ CO2 from ↑ exhalation Respiratory Alkalosis Acute= ↓10:↓2, Chronic= ↓10:↓4
3. Identify the primary (1o) process
4. Compensatory mechanisms in lungs and kidneys attempt to lose/retain CO2 or HCO3- to maintain normal pH. Determine if compensation is appropriate (approximate normal ratio of change in pCO2 : HCO3- is shown in green boxes). If inappropriate, there is a secondary (2o) process occurring
HCO3- binds with extra H+
↓ HCO3- Metabolic Acidosis ↓12 :↓10
Less CO2 than expected, due to ↑ exhalation = Less acid in serum
2o Respiratory alkalosis
More CO2 than expected, due to ↓ exhalation = More acid in serum
2o Respiratory acidosis
Less HCO3- than expected, due to 2o gain of H+ or loss of HCO3-
2o Metabolic acidosis
More HCO3- than expected, due to 2o loss of H+
2o Metabolic alkalosis
Less CO2 than expected, due to ↑ exhalation = Less acid in serum
2o Respiratory alkalosis
↑ CO2 than expected, due to ↓ exhalation = More acid in serum
2o Respiratory acidosis
Less HCO3- than expected, due to 2o gain of H+ or loss of HCO3-
2o Metabolic acidosis
More HCO3- than expected, due to 2o loss of H+
2o Metabolic alkalosis
5. Calculate anion gap (AG) to determine the presence and type of metabolic acidosis. This is done regardlessofthe1o or2oprocess, as there may also be a hidden metabolic acidosis
6. HAGMA only: In normal blood buffer system, acid gain should match bicarbonate lost. If not, identify if another process is causing an inappropriate loss or gain of HCO3-
Gain of acid (∆AG) = AG-12 LossofHCO3- (∆HCO3-)=24-HCO3-
7. Calculate Osmolar Gap (for HAGMA) or Urine net charge (for NAGMA) to narrow the etiology
Authors:
Sravya Kakumanu
Reviewers:
Huneza Nadeem, Ben Campbell *Adam Bass, *Yan Yu
* MD at time of publication
(normal is ~12)
Calculate AG = Na+- Cl- - HCO3-
If a metabolic acidosis is present, ↓ HCO3- is
compensatedby↑otherserumanionstomaintain neutral charge
If no metabolic acidosis was identified in steps 2-4:
No Metabolic acidosis present
Classically, the compensating anion is Cl-,maintaininganormalAG
AG ≤ 12
If a 1o or 2o metabolic acidosis was identified in steps 2-4:
Normal Anion Gap Metabolic Acidosis (NAGMA)
In this scenario, a normal distal nephron should be excreting excess acid
Excess acid is normally excreted as ammonium (NH +) with Cl- 4
The more Cl- in the urine relative to cations, the more acid is being excreted Calculate urine (U) net charge to determine cause of NAGMA = UNa+ + UK+ - UCl-
When the cause is addition of an abnormal acid, HCO3- is used up asabuffer,andthecompensatinganionistheabnormalacid's conjugate base – which is not measured in the AG calculation
↓ HCO3- is compensated by unmeasured anionsà the AG ↑ AG>12
High Anion Gap Metabolic Acidosis (HAGMA)
Calculate ∆AG = AG-12
Calculate ∆HCO3- = 24-HCO3-
∆HCO3- > ∆AG
Loss of HCO3- > Gain of acid
Additional process causing further loss of HCO3-
HAGMA + NAGMA
∆HCO3- = ∆AG
Loss of HCO3- = Gain of acid
Only acid gain from HAGMA causing HCO3- loss
HAGMA only
∆HCO3- < ∆AG
Loss of HCO3- < Gain of acid
Additional process causing gain of HCO3- despite losses due to HAGMA
HAGMA + Metabolic alkalosis
See slide on
Metabolic Alkalosis: Pathogenesis
-ve U net charge Appropriately large Cl- excretionà
Toxic alcohol ingestion ↑ serum osmolality (and H+) from metabolites produced, so osmolality helps identify etiology
Calculate osmolar gap to determine cause of HAGMA
= (measured serum osm) – (expected serum osm)
= (measured serum osm) – (2(Na+) + urea + serum glucose)
+ve U net charge
Inappropriately small Cl- excretionà impaired acid excretion is the issue
Type IV or Type I RTA See slide on Normal Anion Gap Metabolic Acidosis: Pathogenesis and Laboratory Findings
HCO3
GI losses or Type II RTA
-
loss is the issue
Osmolar gap > 10 (extra osmoles present)
Toxic alcohol poisoning
Osmolar gap ≤10 (normal)
Test for other causes
See slide on High Anion Gap Metabolic Acidosis: Pathogenesis and Laboratory Findings
Legend:
Disease State
Mechanism
Lab Finding/Calculation
Published January 29, 2023 on www.thecalgaryguide.com")
Hidradenitis Suppurativa

Defective notch signaling pathway (a regulator of many cell processes)
Hormones (excess androgen activity)
Smoking (nicotine exposure)
Skin to skin friction
Systemic inflammation
Hidradenitis Suppurativa:
Pathogenesis and Clinical Findings
Other unknown genetic factors
Follicular occlusion
HS nodule
Epidermal layer
Dermal- Epidermal Junction
Dermal layer
Inflammatory cytokine- mediated activation of nociceptors
Inflammatory cytokines
Sebaceous gland
Apocrine sweat gland
Hair follicle
Pilosebaceous- apocrine unit
Changes in gene expression of hair follicle and apocrine gland anti-microbial peptides
Accumulation of corneocytes (dead keratinocytes) and sebum àfollicular occlusion and rupture
↑ Interleukin-36 (IL-36, a cytokine) Altered keratinocyte differentiation
Hair follicle hyperkeratinization
Abnormal structure and function of the pilosebaceous- apocrine unit
Infiltration of normal skin flora microbes, macrophages, dendritic cells, and Th17 cells
Increased density of nicotinic acetylcholine receptors in pilosebaceous-apocrine unit (see figure)
Mechanism not fully understood
Hyperhidrosis
Pain
Innate immune cells produce inflammatory cytokines: tumour necrosis factor alpha (TNF-α) and various interleukins (IL-1, IL-6, IL-12, IL-17, IL-22 and IL-23)
IL-17 promotes neutrophil migration into the skin
Formation of neutrophil extracellular traps (NETs)
B-cell activation and IgG autoantibody formation against skin tissue antigens
Scarring
Ongoing inflammation impairs wound healing
Granulocyte infiltration and activation, release of granulocyte colony-stimulating factor
Accumulation of keratin, purulent and/or serosanguinous fluid in dermis
Inflammatory papules, nodules, and abscesses
Pro-inflammatory cytokine-mediated response, leading to epithelial hyperplasia with increased fibrosis and collagen remodelling in the dermis
Formation of epithelial tissue tunnels in the dermis with two cavities on either end that open to the skin surface and fill with fluid or keratin
Hidradenitis Suppurativa
Chronic Inflammatory skin disease that occurs in areas with a high density of apocrine sweat glands, including the axilla, underneath the breasts, groin, and buttocks
Sinus tracts (dermal connections between lesions)
Double-ended comedones
Authors: Leah Johnston Reviewers: Mehul Gupta Lauren Lee Stephen Williams Ben Campbell Laurie Parsons* * MD at time of publication
Wound drainage and odour
Psychological distress
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published January 30, 2023 on www.thecalgaryguide.com")
Pityriasis Rosea

Female sex
(2:1 female to male ratio)
Risk factors
Recent vaccinations: Bacillus Calmette-Guerin (BCG), influenza, H1N1, diphtheria, smallpox, hepatitis B, and Pneumococcus
Red blood cell lymphocytes extravasation
Perivascular dermal infiltrates: lymphocytes, monocytes, and eosinophils
Spongiosis (intercellular fluid accumulation in epidermis)
Erythematous, pink-salmon or brown coloured lesions
Altered production of melanin by epidermal melanocytes
Post-inflammatory hyperpigmentation or hypopigmentation
Prodromal symptoms: fatigue, nausea, headaches, joint pain, enlarged lymph nodes, fever, and/or sore throat
Age 10 to 35
Exact mechanism unknown
Activation of T-cell mediated host immune response
↑ Signal proteins in serum including interleukin 17 (IL-17), interferon gamma (IFN-γ), vascular endothelial growth factor (VEGF), and induced protein 10 (IP-10), leading to increased capillary permeability
Spring and fall seasons
Fluid extravasation from blood vessels
Red blood cell extravasation
Often asymptomatic, can be pruritic
Cell infiltration isn’t as intense or doesn’t produce as dramatic an inflammatory response as other skin conditions
↑ CD4 lymphocytes, monocytes, eosinophils and Langerhans cell infiltration into the dermis and epidermis
Pityriasis Rosea
Activation of B cells and production of anti- keratinocyte IgM antibodies
Parakeratosis (incomplete keratinocyte maturation) and proliferation of epidermal cells (known as psoriasiform hyperplasia)
Thickened epidermal layer
Scale
A self-limited, papulosquamous eruption that is characterized by round or oval-shaped, erythematous to brown patches or plaques with scaling borders that typically occur on the trunk and proximal extremities and tend to follow Langer’s lines. Often initially presents with a larger herald patch as the first sign.
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published January 30, 2023 on www.thecalgaryguide.com")
Kejang Demam Sederhana: Patogenesis dan temuan klinis

Inhalational Injury

Acute hypoxemia
Loss of consciousness
Death
Upper airway injury (above vocal cords)
Cell lysis & necrosis
Local release of inflammatory substances
↑ Vascular permeability à edema of tissues in upper airway
Damage to lower respiratory tract cilia
↓ Mucus clearance from alveoli
Parenchymal injury
(delayed reaction dependent on severity of burn)
Alveolar epithelial and endothelial barrier irritation/damage
Inflammatory response
Carbon monoxide poisoning
Cyanide poisoning
Cyanide binds to mitochondrial cytochrome oxidase a3
CO binds more strongly to hemoglobin than O2
↑ Carboxyhemoglobin in blood, ↓ free hemoglobin available to bind O2 in the lungs
↑ Affinity for O2 on remaining binding sites in hemoglobin (i.e. hemoglobin binds O2 more strongly and is slow to release it)
↓ O2 delivered to tissues
Inhibition of cytochrome c oxidase
Mucous obstructs airways
Air retained
distal to the obstruction is resorbed from nonventilated alveoli
Regions without gas collapse i.e atelectasis
↑ Risk of infection
↓ Mitochondrial respiratory chain function
Impaired oxidative phosphorylation & cellular energy production
Cellular dysfunction in high metabolic tissues
Nitric oxide acts as a vasodilator in alveolar arterioles
Loss of hypoxic vasoconstriction (constriction of arterioles in alveoli due to ↓ O2)
Reactive inflammation & bronchoconstriction
Stridor
Complete airway obstruction
↑ Vascular permeability leading to fluid leakage into interstitium and alveoli
Blood flow to poorly ventilated alveoli is maintained
Ventilation/perfusion (V/Q) mismatch (regions of lung not effectively ventilated despite being well perfused by blood)
Acute Respiratory Distress Syndrome See ARDS Pathogenesis slide
Hypoxemia
Lower airway edema
Wheezing Coughing
Impaired brain function
Muscle weakness
Impaired heart function
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
First published November 10, 2019; Updated on February 12, 2023 on www.thecalgaryguide.com")
Large Bowel Obstruction: Findings on Abdominal X-ray
 and cecal or sigmoid volvulus (11-15%)
Obstruction from mechanical causes such as physical blockade of bowel lumen or twisting of the large bowel
Bowel Obstruction
*See Mechanical Bowel Obstruction and Ileus: Pathogenesis and clinical findings
Large bowel contents cannot pass the obstructionàgas buildup in colon from swallowed air, bacterial fermentation, CO2 from acid + bicarbonate reaction
Colon intraluminal pressure overcomes venous and lymphatic pressure
Impaired venous outflowàbowel wall edema/thickening
↑ Vascular resistance eventually impedes arterial inflowàbowel wall ischemia
Ischemia can progress to bowel infarction and necrosis
Large bowel loops are anatomically peripheral to the small bowel
Evacuation of gas and water reabsorption distal to obstruction
If ileocecal (IC) valve incompetent or sufficient pressure buildup in large bowel can overcome IC valve
↑ intraluminal pressure
Gas dissects into
bowel wall from mucosal disruption
Pneumatosis coli (air within bowel wall)
Bowel wall perforation (especially when cecum diameter > 10-12cm)
Dilated large bowel loops located peripherally (highlighted yellow)
Collapsed distal colon (few or no air-fluid levels in the large bowel as water is reabsorbed)
Small bowel dilatation (> 3 cm)
Colonic dilatation (> 6 cm)
Cecum dilatation (> 9 cm)
Haustra (anatomical folds of the large bowel) become visible as it is distended
Pneumoperitoneum (sub-diaphragmatic air on erect chest x-ray)
Release of gas into peritoneum
PA
Radiopaedia, rID:17957
PA
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published February 16, 2023 on www.thecalgaryguide.com")
Ischemic Stroke Impairment by Localization
 in respective blood vessels
Ischemia: ↓ blood flow
(See Ischemic Stroke: Pathogenesis slide)
Left hemisphere damage
Right hemisphere damage
Motor and sensory cortices of upper limb and face damage
Urinary incontinence Aphasia (inability to comprehend or produce
Ischemia in
the middle cerebral artery (MCA)
MCA divides into segments
language) (See Aphasia slide)
Left sided agnosia (visual perceptual deficits)
Contralateral hemiparesis (weakness on side of body opposite to injury) & sensory deficits, visual field deficits, aphasia, agnosia (inability to process sensory information), apraxia (motor planning deficits) & agraphia (inability to communicate by writing)
M1-MCA (sphenoidal segment)
M2-MCA (insular segment)
Ischemia in the posterior cerebral artery
Spares the lower extremity, affects the upper extremity and face
Lesion to frontal lobe (Broca area) Infarction of occipital cortex
Lesion to superior temporal gyrus of temporal lobe (Wernicke area)
No homonymous hemianopsia (one-sided visual field loss) Expressive Broca’s/motor aphasia (inability to produce language)
Contralateral homonymous hemianopsia
(visual field loss on opposite side)
Receptive Wernicke’s/sensory aphasia
(inability to comprehend language)
Sensory loss, memory loss, contralateral homonymous hemianopsia & alexia (reading difficulty)
Ischemia of the occipital lobe, posteromedial temporal lobes, midbrain & thalamus
Ischemia in the vertebral basilar artery
Ischemia in the basilar artery
Ischemia of brainstem & medulla
Ischemia of midbrain, thalami, inferior temporal & occipital lobes
Cranial nerve disorders: dysarthria (slurred/slowed speech) (IX, X), diplopia (double vision), facial numbness or paresthesia (VII), Foville’s syndrome (ipsilateral cerebellar ataxia), Horner's syndrome, (paresis of conjugate gaze and contralateral hemiparesis, facial palsy, pain & thermal hypoesthesia)
Motor deficits: Millard-Gubler syndrome (pons lesion), Raymond’s syndrome (ipsilateral abducens impairment, contralateral central facial paresis & contralateral hemiparesis), Wallenburg syndrome (sensory deficits in the contralateral limb, ipsilateral face), ataxia (abnormal gait), unilateral or bilateral sensory loss of position & vibration
Cranial nerve disorders: dysconjugate gaze (unpaired eye movements) (III, IV, VI), ipsilateral facial hypoalgesia (↓ pain sensitivity) (V), unilateral lower motor neuron face paralysis (VII), vertigo (spinning sensation), dysarthria (weak speech muscles) (IX, X)
Motor deficits: contralateral hemiparesis, quadriplegia (paralysis of all 4 limbs), contralateral limb hypoalgesia
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
First published February 3, 2018, updated February 28, 2023 on www.thecalgaryguide.com")
Hemorrhagic Stroke
 Zhang Mao Ding Michael D Hill* Gary Klein* * MD at time of publication
Primary Intracerebral Hemorrhage (~75%)
Secondary Intracerebral Hemorrhage (~25%)
Amyloid Angiopathy
Amyloid deposits in blood vessels and weakens vessel walls
Hypertension
Lipohyalinosis (lipid and protein aggregation in arterial walls) weakens blood vessels
Unknown
Aneurysm
Dilation of a weakened blood vessel
Drugs (e.g., cocaine, crystal meth, decongestants, anticoagulants)
Vascular Malformations
Note: the pathophysiology and exact mechanism is not well known
Release of toxic blood plasma components (coagulation factors, immunoglobins)
Red blood cell lysis
Cytotoxic hemoglobin (heme, iron) release
Fenton-type free radical generation (Fe(II) + H2O2 → Fe(III) + OH− + OH•)
Oxidative damage to carbohydrates, lipids, nucleic acids, and proteins in brain
Necrosis of hypoxic brain tissue
Neurological signs: focal motor weakness, aphasia, vision loss, sensory loss, imbalance/incoordination, altered LOC
Rupture of blood vessel(s) Accumulation of blood → hematoma formation
↓ Cerebral tissue perfusion (↓ O2 availability)
↓ Mitochondrial oxidative phosphorylation (final step in aerobic glucose metabolism)
↓ Adenosine triphosphate (ATP) production
↑ Anaerobic glucose metabolism → ↑ Cerebral lactate production
Cerebral lactic acidosis Impaired cellular metabolism Death of neurons and glia
Microglia clear debris and release inflammatory markers (TNFα, IFγ, IL-1β)
↑ Endothelial cell apoptosis and ↑ blood-brain barrier permeability
Cerebral edema
Increased intracranial pressure: papilledema, sudden headache, non-reactive pupils, ↓ level of consciousness (LOC), nausea/vomiting
Astrocytes release glutamate (main excitatory neurotransmitter)
Activation of neuronal metabotropic glutamate receptors
↑ Ca2+ influx into neurons
Excitotoxicity (excess stimulation of glutamate receptors leading to neuronal death)
Dysfunction of Na+/K+ ATPase pump (moves 3 Na+ out of cell and 2 K+ into cell) on neurons
↓ Na+ efflux and ↓ K+ influx
Neuronal membrane potential becomes less negative (closer to threshold potential)
Neurons depolarize → ↑ Glutamate release
General findings: Seizures, lethargy
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
First published June 6, 2018, updated February 28, 2023 on www.thecalgaryguide.com")
Myelodysplastic Syndrome Pathogenesis and clinical findings
: Pathogenesis and clinical findings
Authors: Mao Ding Reviewers: Ashar Memon Man-Chiu Poon Yan Yu* * MD at time of publication
Stem cells differentiateà accumulation of bone marrow cells with aberrant morphology & maturation
Idiopathic (unknown cause)
Treatment related: exposure to ionizing radiation, chemotherapeutic agents (e.g., alkylating agents, anti- metabolite, topoisomerase II inhibitors)
Environmental toxins (e.g., tobacco, benzene & other organic solvents)
Familial predisposition to MDS
Acquired somatic (non- reproductive) gene mutations (DNA alterations)
Inherited
germline (reproductive cells) gene mutations
Recurrent mutations cause alteration(s) in one or more protein functions with/without chromosomal abnormalities in hematopoietic stem cell (earliest cell of blood cell differentiation):
Mutation in the transcription factor gene RUNX1 impairs regulation of normal hematopoietic (blood cell) development
Mutation(s) in epigenetic regulator genes (DNMT3A, TET2) impairs regulation of DNA methylation
Mutation in splicing regulator genes (SF3B1) causes mistakes in splicing mRNA moleculesàaberrant translation (production) of proteins
Chromosomal abnormalities: deletions (chromosome 5, 7, and/or 20), duplications (chromosome 8), structural abnormalities (inversion of a gene segment)
Myelodysplastic Syndrome
Mutation-associated clonal disorders of hematopoietic stem cells, causing dysplasia (abnormal development) of one or more myeloid cell lineages (granulocytes, monocytes, red blood cells & platelets) in the bone marrow
Genetic mutations or chromosomal abnormalities occur in hematopoietic stem cellsàclonal expansion of abnormal cells in bone marrow
Apoptosis (programed cell death) of clonal cells, inhibiting development of granulocytes, monocytes, red blood cells & platelets in the bone marrow
Neoplastic myeloid precursor cells (blasts) accumulate in the bone marrow
Acute Myeloid Leukemia (AML)
> 20% Blasts in the bone marrow
Excessive blasts displace other precursor cells & inhibit differentiation
Pancytopenia
(↓ number of cells of ALL 3 cell lines: platelets, white blood cells & red blood cells) (see Acute Myeloid Leukemia for signs/symptoms/complications)
↓ Number of mature functional blood cells leave the bone marrow to go to peripheral bloodà↓ number of cells of one or more cell lines
Bone marrow’s inability to make sufficient blood cells cause extramedullary hematopoiesis (formation/activation of blood cells outside the medulla of bone) at sites such as liver and/or spleen
Hematopoietic stem cells migrate to the spleen/liver & differentiate into blood cells
Expanding bone marrow physically pushes on bone’s cortex from within, activating nociceptors
Multifactorial causes mostly with unclear mechanisms
Intracellular granules precipitate inside blasts & the precipitate spills into the blood
↑Division & death of cancerous cells → ↑ cell lysis & release of intracellular contents into plasma
Bone pain
B symptoms: Weight
loss, fever, night sweats
Auer Rods (needle- like crystals) seen on blood smear
↑ Serum levels of uric acid, K+, LDH
↓ Red blood cells
Anemia
fatigue, pallor
↓ White blood cells
Leukopenia
Recurrent infections
↓ Platelets
Thrombo- cytopenia
Bruising, bleeding
Accumulation of cells within the spleen/liver increase the size of the organ
Splenomegaly Hepatomegaly
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published March 4, 2023 on www.thecalgaryguide.com")
Central Retinal Vein Occlusion

↓ Anticoagulation (e.g., Protein C or S def) and/or ↑ coagulation (e.g., malignancy, Polycythemia Vera)
Non-ischemic (perfused) CRVO
Glaucoma
(a disease characterized by ↑ intraocular pressure)
Optic disc drusen
(calcified nodules located within the optic disk – the most anterior part of the optic nerve)
Diabetes
Dyslipidemia
Hypertension
Vasculitis
(inflammation of blood vessels; e.g., sarcoid, Systemic Lupus Erythematosus)
Endothelial damage
Pupillary responses do not vary between both eyes when a light is shone in one eye at a time
Relative afferent pupillary defect (RAPD) mild or absent
Lost vascular wall integrity
Normal retinal electrical response to a light stimuli
Normal
electro- retinography (ERG) with b- wave >60%
Few scattered Intra-retinal hemorrhages
↓ In retinal electrical response to a light stimuli
Pupils respond differently to a light stimulus shone in one eye at a time
Severe ↓ in visual acuity (Snellen acuity of <20/400)
Visual field deficits
↓ b-wave amplitude (<60%) on ERG
RAPD present
↑ Pressure compromises retinal vein outflow
Central retinal atherosclerosis (build-up of fat & cholesterol plaque in arteries) & hardening
Atherosclerotic changes in the central retinal artery compress the central retinal vein (since they are both held together in region of the optic disc)
Venous stasis (stagnation of blood flow) ↑ Likelihood of thrombus formation
Central Retinal Vein Occlusion (CRVO)
A common retinal vascular disease characterized by blockage of the main vein that drains blood from the retina
CRVO classified based on the degree of perfusion (as seen through retinal angiography)
Ischemic (nonperfused) CRVO
↑ Intraluminal venous pressure
Dilated tortuous retinal veins
Normal visual fields
Vascular wall integrity lost
Four-quadrant hemorrhages described as “blood and thunder” on fundus exam
Obstruction due to thrombus
↓ Retinal capillary perfusion causes ischemia
Hypoxia in the retinal tissues
↑ Release of vascular endothelial growth factor to revascularize diseased tissue & overcome hypoxic conditions
Angiogenesis (formation of new blood vessels)
Intraretinal infarcts
“Cotton wool spots” on fundus exam
Venous capillary fluid/protein leakage
Mild retina, macula and disc edema
Moderate ↓in visual acuity often (Snellen acuity >20/400)
New vessel proliferation can occur in the anterior chamber of the eye
This change can block the outflow path of the aqueous humor in the eye
Neovascular glaucoma
Neovascular vessels are structurally different in comparison to regular retinal vasculature
Neovascular vessels are more fragile
↑ Likelihood of rupture Vitreous hemorrhage
Neovascular vessels lack tight junctions
↑ Fluid leakage
Retina, macula and disc edema
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
First Published June 28, 2017, updated March 4, 2023 on www.thecalgaryguide.com")
Syndrome de détresse respiratoire aigu: Pathogenèse et trouvailles cliniques
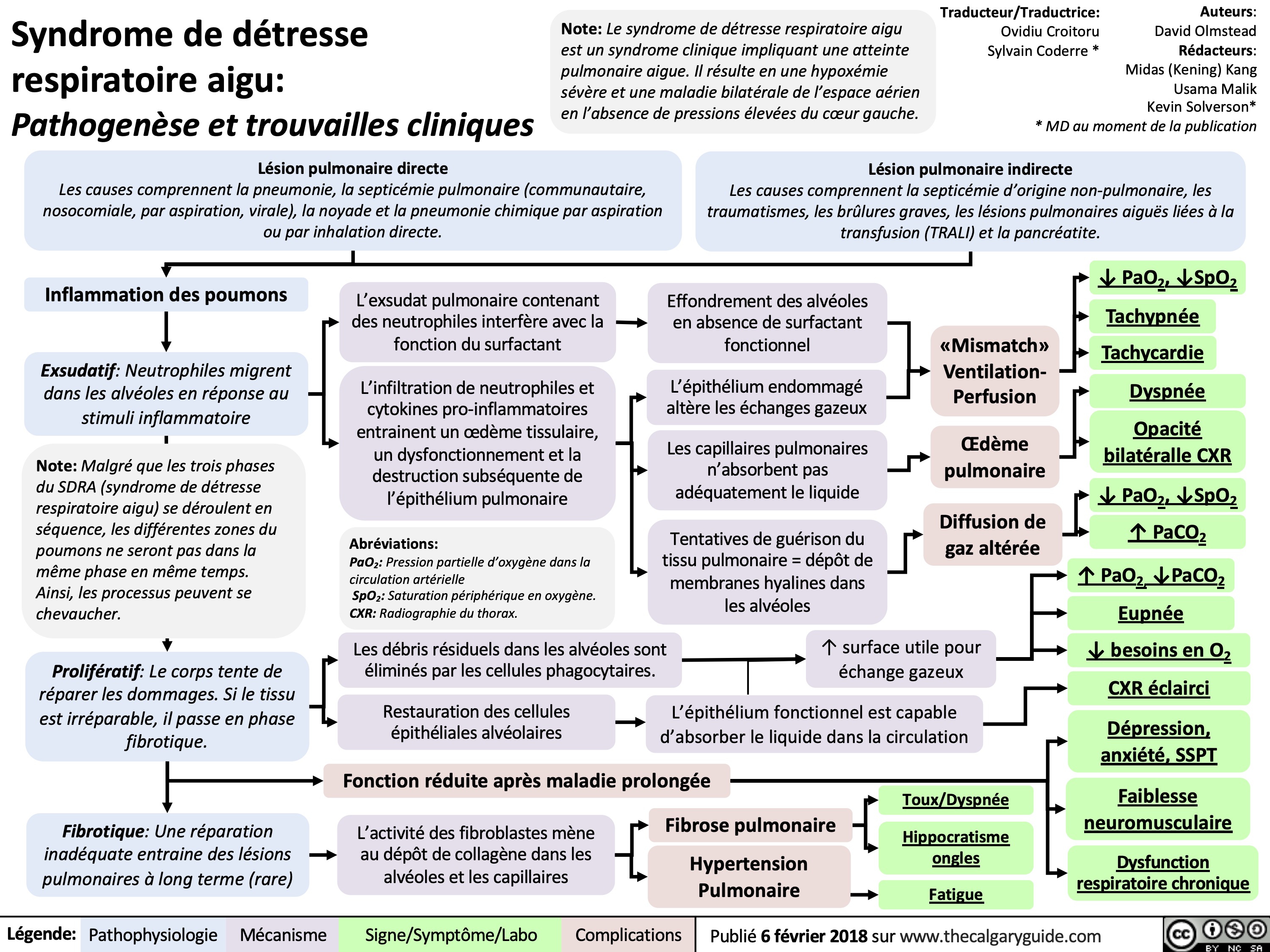
reaction-transfusionnelle-hemolytique-aigue-signes-et-symptomes

Douleur somatique aigue
 Thermique (ex: poêle chaude activé) Chimique (ex: inflammation)
Nocicepteurs activités au site de blessure (Neurones sensoriels de 1er ordre)
Fibres nociceptives (A∂ et C) transportent l’information sensorielle nocive vers la corne dorsale ipsilatérale de la moelle épinière
Les neurotransmetteurs excitateurs sont libérés et stimulent les neurones sensoriels de 2ème ordre.
Les neurones sensoriels de 2ème ordre décussent immédiatement dans la moelle épinière et remontent via les voies antérolatérales (spinothalamiques) du côté opposé, se terminant à divers endroits
Vers le thalamus
Les neurones de 2e ordre synapsent dans le thalamus
Dans le thalamus, les neurones sensoriels de 2ème ordre synapsent avec les neurones sensoriels de 3e ordre, qui transmettent le signal au cortex cérébral
Vers le tronc cérébral
Les neurones de 2e ordre
synapsent dans la formation réticulaire du tronc cérébral
Vers le mésencéphale
Les neurones de 2e ordre
synapsent dans la zone grise périaqueducale (PGA) du mésencéphale
Localisation et sensation douleur
Réponse émotionnelle et comportementale
Stimule les voies descendantes pour moduler le signal de douleur
Diminution ou augmentation de la perception de la douleur
↑ Rythme cardiaque
Nausée
Légende:
Pathophysiologie
Mécanisme
Signes/Symptôme/Labo
Complications
Publié 25 avril 2019 sur www.thecalgaryguide.com")
варикозное-расширение-вен-пищевода-ж

язвенная-болезнь-патогенез-и-клиниче

синдром-меллори-вейса-патогенез-и-кли

Skin Grafts Transplant physiology and clinical findings

Skin anatomy
Sebaceous gland alongside hair follicle
Hair Follicle Sweat gland
Blood vessels
Full thickness donor sites cannot be re-harvested
Epidermis
Dermis
Subcutaneous tissue
Full thickness skin graft (FTSG) including the epidermis and entire dermis
The full thickness donor site is closed by primary intention (sutured)
Skin grafts
Indicated when healing by secondary intention (natural closure) is not possible. The graft consists of avascular skin harvested from an uninjured donor site
Split thickness skin graft (STSG) including the epidermis and a portion of the dermis
The split thickness donor site heals by re- epithelialization
Donor grafts are applied to the injured area
1. Initial adherence (8 hrs)
A fibrin network fastens the graft to the wound bed
Fibroblasts, leukocytes & phagocytes from the wound bed enter the fibrin network, building a fibrous connection between the graft & wound bed
2. Plasmatic imbibition (24-48 hrs)
The graft absorbs nutrients and dissolved oxygen from plasma in the recipient wound bed
The graft appears white in color
3. Inosculation and capillary ingrowth (2-4 days)
Capillary buds from the recipient wound bed anastomose with vessels on underside of graft
Physiology of graft take
4. Revascularization (5-7 days)
5. Graft contraction (6-18 months)
Actin in myofibroblasts contract & approximate the wound edges
Angiogenic factors enable revascularization allowing rapid, high-volume flow through large vessels
Lymphatic drainage established
Dermal structures such as hair follicles, sweat & sebaceous glands may be partially restored
Grafts are pruritic (itchy), discoloured, and do not function or look like normal skin
Grafts are reinnervated from periphery to center
STSGs are reinnervated faster than FTSGs, though incompletely
Graft fails (does not adhere) with a poorly vascularized and unclean wound bed
Restoration of vasculature returns color to the graft
Successfully adhered grafts are pink (STSG) or marginally paler (FTSGs) and immobile
The deep dermal layer in FTSGs better inhibits myofibroblast action
STSGs heal with significant contracture, limiting function & mobility
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
First published February 6, 2017, updated March 12, 2023 on www.thecalgaryguide.com")
Primary Hemostasis

Disorders of primary hemostasis:
Collagen and microfibrils (subendothelial elements
that are normally protected) are exposed to circulating blood due to vessel injury
Storage granules in endothelial cells release large multimers of clotting factor von Willebrand (vWF) when damaged
Acquired or inherited reduction in vWF quantity or quality (see vWF deficiency slide)
von Willebrand Disease
Defect in expression of GpIb interferes with platelet ability to bind to vWF
Bernard-Soulier syndrome
Defect in platelet membrane glycoproteins IIb and IIIa
Glanzmann’s Thrombasthenia
Insufficient platelets to form a platelet plug
Thrombocytopenia
(low platelet count)
Disorders of primary hemostasis: problems with formation of a platelet plug
Platelet adhesion: vWF binds to collagen and interacts with the platelet surface receptor glycoprotein Ib (GpIb) allowing platelet adhesion
Platelet activation: mediated by agonists like ADP, thrombin and collagen
Mucocutaneous bleeding (bleeding of the skin and mucous membrane)
↑ Closure time, ↑ bleeding time
Platelets change shape and release substances from their granules
Conformational changes in platelet cell surface glycoprotein IIb/IIIa
↑Affinity for fibrinogen through GpIIb/IIIa
ADP
Fibrinogen, vWF, and other substances
Thromboxane A2 (see note)
Induces aggregation
Epistaxis
Menorrhagia Note:
Petechiae
Platelet aggregation: fibrinogen forms bridges between adjacent platelets by binding to activated glycoprotein IIb/IIIa
Interaction with coagulation factors: platelets provide a scaffold that allows the activation of phospholipid-dependent coagulation factors
Primary hemostasis
(formation of a platelet plug)
Secondary hemostasis (see slide for normal physiology)
Drugs like Aspirin and NSAIDs inhibit Thromboxane A2 synthesis (by inhibiting cyclooxygenase) and thereby inhibit platelet aggregation
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published March 30, 2023 on www.thecalgaryguide.com")
Concussion

Disruption of reticular activating system (brain area that regulates arousal)
Altered level of consciousness
Direct blow to the head or the body that causes an impulsive force to the head (i.e., falls, motor vehicle accidents, sports, assaults)
Skull acceleration / deceleration
Coup (brain strikes skull on side of impact)
Brain tissue swelling
Cerebral edema (fluid build up around brain)
↑ Intracranial pressure
Cerebral herniation
(shifting of brain tissue into adjacent space)
Contrecoup (brain strikes skull on opposite side of impact)
Anatomical damage
Skull fracture
Broken bone fragments ruptures blood vessels
Intracranial hemorrhage
(bleeding into brain tissue)
Papilledema (swelling around optic disk, where optic nerve enters eyeball)
Disruption of messages from eye to brain
Vision problems (i.e., blurred or double vision)
Cellular damage
Indiscriminate, rapid neurotransmitter release
↑Extracellular K+ and glutamate, accumulation of intracellular calcium
Ionic disequilibrium across neuronal membrane
Energy consumed by Na+/K+ ATPase pumps to re-establish ionic homeostasis
↑ Cerebral glucose metabolism
↑ Energy demand
Brain injury
Axonal stretch due to
biomechanical forces
Microtubule disruption
Structural (cytoskeletal) disturbance
Axonal degeneration
Impaired neural communication
Broken bone fragments
Ruptures blood vessels
Compresses blood vessels
↓ Cerebral blood flow
↓ Cerebral glucose supply
↓ Energy supply
↓ Oxygenated blood to brain
Brain cell death
Chronic brain atrophy
Persistent impaired cognition
Cellular energy crisis (mismatch between energy supply and demand due to effort in restoring homeostasis)
Nausea, vomiting
↓ Participation in daily Confusion, disorientation, unsteadiness, headache activities and work
Death Anxiety, depression
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published February 12, 2019, updated March 27, 2023 on www.thecalgaryguide.com")
Pyogenic Brain Abscess on MRI
 transcellularly, paracellularly or by a Trojan Horse mechanism
Authors: Omer Mansoor, Aly Valji, Nameerah Wajahat Reviewers: Mao Ding, Reshma Sirajee, James Scott* *MD at time of publication
R
Transcellular
Pathogen invades BBB cell directly to enter
Paracellular
Pathogen goes between BBB cells by disrupting tight junctions
Trojan Horse
Pathogen bypasses BBB by hiding inside a macrophage cell
Ring Enhancing Lesion Sensitive, but not specific for a Brain Abscess
Pathogen causes parenchymal inflammation and progresses through four stages of infection
Axial T1 + Gadolinium MRI Head. Pyogenic Brain Abscess showing infection at Stage 3 or 4. T1 highlights fat, whereas T2 highlights fat and water*.
R
Stage 1: Early Cerebritis Focal infection (2-3 days) with no pus formation
Stage 2: Late Cerebritis Progressive (1 week) infection of abscess
Stage 3: Early Encapsulation
In 1-2 weeks, pus is formed from the pathogen
Stage 4: Late Encapsulation After 2 weeks, abscess shrinks in size as it necroses
Pathogen causes acute inflammatory changes of swelling (edema) and vascular congestion
No fibrous capsule is formed yet and infection is not well localized on a T1 MRI
Increased edema could be seen on T2 MRI
Poor Demarcation and Vasogenic Edema
Abscess is not well-defined with minimal enhancement on T1 but can have increased signal on T2
Fibrous capsule is formed by surrounding healthy brain tissue that walls off abscess
Capsule is made of granulation tissue and fat, which lights up non-specifically on a T1 MRI
Use Diffusion Weighted Imaging (DWI) to distinguish abscess from other brain lesions
DWI measures water diffusion in different directions, specifically diseased areas where water movement is restricted
Restricted Diffusion Edema and inflammation allow water to move freely (hyperintense signal)
*Imaging Source: Feraco et al., 2020: https://jptcp.com/index.php/jptcp/article/download/688/685?inline=1
DWI Sequence Axial MRI Head. Same Pyogenic Brain Abscess as above. DWI is the mainstay imaging sequence for diagnosing a pyogenic brain abscess*.
Legend:
Pathophysiology
Mechanism
Radiographic Findings
Complications
Published Mar 25, 2023 on www.thecalgaryguide.com")
Carpal Tunnel Syndrome

↑ Blood sugar: deposition of advanced glycation end (AGE) products (proteins or lipids glycated when exposed to sugar)
AGE attaches to and prevents tendons from moving properly
Authors: Amanda Eslinger Yvette Ysabel Yao Reviewers: Matthew Harding Owen Stechishin Mao Ding, Cory Toth* * MD at time of publication
Wrist trauma (distal radius fractures, carpal/metacarpal fractures, tendon ruptures)
Repetitive Strain Injury (repetitive hand & wrist movements)
Irritation, swelling & thickening of tendons in carpal tunnel
Calcium deposits
Calcifica tion
Deposition Amyloidosis
Amyloid
(protein aggregates) deposition
Gout
(See Gout Slide)
Uric acid crystal deposition
Pregnancy
↑ Concentration of hormones & uterine pressure on inferior vena cava
Backup of blood into systemic circulation
Autoimmune
(i.e. Rheumatoid arthritis, scleroderma, lupus, Sjogren’s syndrome)
↑ Inflammatory cytokines causing inflammation
Hypo- thyroidism
Myxedema (swelling of skin and underlying tissues) in carpal tunnel
Idiopathic or Congenital
Edema
Narrowed carpal tunnel leads to ↑ internal pressure
Carpal Tunnel Syndrome
Vascular: median artery thrombosis in carpal tunnel
Median nerve compression inside the carpal tunnel Mechanical disruption of median nerve
Compression exacerbated with flexed wrists (i.e. sleep, driving, holding phone/cup)
Disruption of daily activities and sleep
Ischemia (↓ blood supply) to median nerve
Hypoxia (↓ oxygenated blood flow)
Metabolic conduction block (impaired axonal transport due to ischemia)
Nerve conduction study
(show sensory nerve impulses slowing across the wrist, followed by mild / moderate / severe loss of sensory nerve amplitude
Damage to the myelin sheath
↓ Saltatory conduction (action potential propagation along myelinated axons)
Neuropraxia (nerve compression blocks conduction)
Interruption in axonal continuity
Axonotmesis (endoneural tube stays intact but myelin & distal axon degenerates)
Recovery possible
Full disruption of myelin, axon & nerve sheath
Neurotmesis (axons no longer have an endoneural tube to guide regrowth)
Recovery impossible
↓ Ability to contract and use abductor pollicis brevis muscle
↓ Signals through median nerve
Interference with signals to the brain causes unusual sensations
Hypoalgesia (↓ pain Dysesthesia (tingling, burning, or sensitivity at 1st 3 1⁄2 digits) painful sensation at 1st 3 1⁄2 digits)
Thenar muscle wasting
Reduced hand dexterity
Weak thumb abduction
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published December 2nd, 2013, updated March 22, 2023 on www.thecalgaryguide.com")
Complications of Prematurity

Hyperoxia vasoconstricts retinal blood vessels leading to retinal ischemia
Ischemic retina produces vascular endothelial growth factor (VEGF) to generate new blood vessels + improve retinal perfusion
Excess VEGF causes abnormal blood vessel proliferation
Abnormal vessels are fragile + prone to hemorrhage which can lead to retinal distortion
Retinopathy of Prematurity (ROP)
(See relevant slide)
Poor structural support to blood vessels of the germinal matrix (the tissue surrounding the lateral ventricles of the brain)
Vessels are vulnerable to hemodynamic instability
Abrupt changes to cerebral blood flow damage blood vessels
Vessels bleed into germinal matrix and lateral ventricles
Intraventricular Hemorrhage (IVH) (See relevant slide)
Note: Pre-term complications not discussed here include hypothermia, hypoglycemia, apnea, patent ductus arteriosus
Low quantity: ↓ surfactant production
Low quality:
↓ surfactant activity (different lipid + protein composition compared to full-term infants)
↑
Infections
↓ Number of white blood cells and complement protein
↑ Bacterial colonization of GI tract + penetration of intestinal wall
Abnormally high surface tension of alveoli
Alveoli spontaneously collapse (diffuse alveolar atelectasis)
Ineffective ventilation of alveoli (V)ànot supplying pulmonary blood (Q) with oxygen (V-Q mismatch)
Respiratory Distress Syndrome (RDS) (See relevant slide)
Positive pressure ventilation (PPV) used therapeutically
PPV disrupts lung development & damages lung tissue
Bronchopulmonary Dysplasia (BPD) (See relevant slide)
Intestinal ischemia, or Formula Feeds (unclear mechanisms)
Activation of inflammatory cascade releases cytokines (peptides that help activate immune response)
Cytokines cause bowel necrosis (tissue death)
Necrotizing Enterocolitis (NEC)
(See relevant slide)
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published October 31, 2013, updated March 25, 2023 on www.thecalgaryguide.com")
Acute Pulmonary Embolism on CTPA

Virchow’s Triad:
Hypercoagulability, venous stasis, vascular endothelial injury
Image Source: European Society of Radiology
Image: Polo mint sign on axial CTPA.
Image Source: Journal of The Indian Academy of Echocardiography
Image: Railway sign on axial CTPA. Image Source: Moore et al. 2018
Image: Pleural effusion and pulmonary infarction on axial CTPA.
Authors: Aly Valji, Nameerah Wajahat, Omer Mansoor Reviewers: Reshma Sirajee, Sravya Kakumanu, Victória Silva, Mao Ding Vincent Dinculescu* *MD at time of publication
Deep Vein Thrombosis (DVT): Majority of pulmonary embolism (PE) arise from DVT: Clot travels via inferior vena cava → right atrium → right ventricle → pulmonary arteries/arterioles
See “Virchow’s Triad and Deep Vein Thrombosis” for full pathogenesis
Polo Mint Sign
Clot visualized in short axis, Filling defect entirely surrounded by IV contrast creating a circle (polo mint)
Railway Sign
Clot visualized in long axis, Filling defect surrounded by IV contrast on two sides creating the appearance of a railway track
Type I or II Ventilation/Perfusion respiratory failure (V/Q) mismatch
Reverse Halo Sign
Pulmonary infarction leads to wedge shaped opacity with a rim of consolidation (black arrows) surrounding “ground glass” (red arrows)
Pulmonary Embolism: Clot in pulmonary arteries
See “Signs and Symptoms of Pulmonary Embolism” for full presentation
Saddle Embolus: Large clot over the pulmonary trunk bifurcation Lobar/Segmental/Subsegmental Embolus: Clot within the pulmonary arteries of the lungs
Blockage of pulmonary arteries = ↓ Blood flow, ↑ Right heart pressure
↓ Gas exchange b/w lungs and blood
Ischemia of lung tissue → infarction → inflammation of dead tissue
↑ Right heart filling and expansion
Left heart filling impaired
↓ Cardiac output due to ↓ Left heart filling
“Massive PE” = sustained systemic hypotension or bradycardia (SBP < 90 mmhg, HR < 40 bpm)
Saddle Embolus
Filling defect due to blockage of bifurcation. IV contrast appears white, and embolus appears grey
Pleural Effusion
Exudative Pleural Effusion
Tissue inflammation → ↑ blood vessel permeability = Leakage of fluid into pleural space
Transudative Pleural Effusion
↑ Hydrostatic pressure from right heart congestion = Pushes fluid into pleural space
Image Source: Samra et al. 2017
Image: Saddle embolus on axial CTPA.
Legend:
Pathophysiology
Mechanism
Radiographic Findings
Complications
Published March 28, 2023 on www.thecalgaryguide.com")
Wstrząs obturacyjny: Patogeneza, powikłania oraz zmiany kliniczne

Wstrząs dystrybucyjny: Patogeneza, powikłania oraz zmiany kliniczne

Wstrząs kardiogenny: Patogeneza, powikłania oraz zmiany kliniczne

Wstrząs oparzeniowy: patogeneza, powikłania i zmiany kliniczne

Leczenie wstrząsu: wyjaśnienie podstawowych mechanizmów

Rozwarstwienie aorty: Patogeneza, powikłania i zmiany kliniczne

Tętniak aorty brzusznej: Patogeneza

Tętniak aorty brzusznej: Obraz kliniczny, powikłania

Sarcoidosis pathogenesis and CXR findings
 such as tumor necrosis factor to neighboring cells
Tumor necrosis factor is a pro- inflammatory cytokine promoting cell survival and proliferation, predominantly in the upper/middle lung for unknown causes
More macrophages and helper T cells are activated and accumulate in the mediastinum, peritracheal region, and the upper/middle lung
Macrophages and helper T cells promote fibroblasts (cells that produce collagen) in the upper and middle lungs
Macrophages in the lungs present the antigen to the helper T cells in lymph nodes
Helper T cells become activated and accumulate in the lungs
Image Credit: Radiopaedia
The right peritracheal and bilateral hilar lymph nodes becomes swollen
Inflammatory signals stimulate macrophages to form granulomas (clusters of white blood cells) in the lungs, peritracheal and bilateral hilar lymph nodes
X-ray images show visible white opacities in areas of increased densities, such as areas of macrophage and helper T cell concentration
Enlarged peritracheal lymph node Stage 1 and 2
Enlarged bilateral hilar lymph node Stage 1 and 2
Interstitial Opacities Stage 2 and 3
Bilateral and symmetric lacey lung markings in upper/middle lungs
Decreased lung volume on either sides (higher than normal diaphragm)
Collagen accumulate which causes fibrotic tissue (scarred and thickened), replacing healthy tissue
Pulmonary fibrosis of upper and middle lung Stage 4
The fibrotic tissue is stiffer, reduces amount of air held in lungs, and has a higher density (appearing white)
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published April 7, 2023 on www.thecalgaryguide.com")
Coronary Artery Bypass Graft CABG Indications
: Indications
Author: Breanne Gordulic Reviewers: Miranda Schmidt Ben Campbell Sunawer Aujla Angela Kealey* * MD at time of publication
Symptomatic multivessel (≥ 3 vessels) coronary artery disease (MVCAD) or complex MVCAD
Acute coronary syndrome (ACS)
Left main coronary artery disease
Multivessel (≥ 3 vessels) CAD and diabetes
Cardiac surgery required for other pathology
Multivessel CAD, LV dysfunction and congestive heart failure (CHF)
Complex CAD includes stenosed vein grafts, bifurcation lesions, calcified lesions, total occlusions, ostial lesions
STEMI initial treatment is PCI/thrombolysis
CABG outcomes compared to percutaneous coronary intervention (PCI) in MVCAD include ↑ survival in diabetes, ↑ survival with LV dysfunction, ↓ repeat revascularization, ↓ myocardial infarction, ↓ stroke
↑ Risk of failure in complex CAD with PCI
Rapid reperfusion to myocardium most important in STEMI to decrease myocardial damage
CABG can be considered for residual stenoses 6-8 weeks later
NSTEMI or unstable angina (UA) with MVCAD involving at least three vessels including the proximal left anterior descending (LAD)
Left main coronary artery divides into left anterior descending (LAD) and left circumflex (LCx) which supplies 2/3 of myocardium
↑ Survival Myocardial infarction from left main artery occlusion Death
Left main stenosis
Ventricular dysrhythmias
Ongoing ischemia
LV dysfunction Hemodynamic instability
↑ risk of PCI
CABG has mortality benefit
↓ Number of operations
↑ Risk of cardiovascular disease in diabetes
Revascularization indicated along with other cardiac surgery
Multivessel CAD with >90% stenosis and CHF
LV ejection fraction <35%
↑ Risk of atherosclerosis from hyperglycemia and dyslipidemia
CABG bypasses several atherosclerotic plaques in coronary arteries
↑ Durability
↑ Complete perfusion
Valve stenosis or regurgitation Septal defect
Aortic root or arch pathology
Combination procedure
Evidence of ischemia at rest
Evidence of
impaired LV function at rest
↓ All-cause mortality in CABG vs medical management
Chronic obstructive pulmonary disease
Abbreviations:
• ACS- acute coronary syndrome. Acute reduction in
blood flow to heart muscle resulting in cell death. • CAD- coronary artery disease. Narrowing or blockage of the coronary arteries by plaque
• NSTEMI- myocardial infarction (heart attack) with no ST elevation on electrocardiogram
• PCI- percutaneous coronary intervention. A balloon tipped catheter is used to open blocked coronary arteries; a stent may be placed.
• STEMI- myocardial infarction with ST segment elevation on electrocardiogram
Consider revascularization (restore blood flow to blocked or narrowed blood vessels) of coronary arteries to increase perfusion to myocardium (heart muscle)
Coronary artery bypass graft recommended
Assess surgical risk and comorbidities with evaluation by heart team that includes both a cardiac surgeon and interventional cardiologist (SYNTAX Trial)
Individual management plan for patients with comorbidities that increase mortality
Frailty
Chronic Renal Failure
↑ Inflammation and deregulated angiogenesis affects all organ systems
↓ Physiologic reserve and ↓ ability to recover from acute stress
↑ Pneumonia
↑ Respiratory and Renal Failure
↑ Stroke
↑ In hospital mortality
↓ Survival two years after surgery
Use Society of Thoracic Surgeons Score, EuroSCORE, or SYNTAX II Score to predict patient outcome with anatomy, disease severity, and preoperative characteristics
Coronary Artery Bypass Graft
Surgery to take healthy blood vessels from the body and connect them proximally and distally to blocked coronary arteries
Cardiopulmonary bypass, fluid overload, ↑ renal vasoconstriction and ↓ renal oxygenation from rewarming
Kidney injury
↑ End stage kidney disease
Blood flow restored to
ischemic myocardium
↓ Angina
↑ Quality of life ↑ LV function ↑ Survival
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published April 12, 2023 on www.thecalgaryguide.com")
Benzodiazepine Mechanism of Action
 receptor in vascular smooth muscle and the central nervous system (CNS)
APs inhibited in
vascular smooth muscle
Vascular smooth muscle relaxes
Vasodilation ↓ Blood pressure
Authors: Victoria Silva Travis Novak Reviewers: Billy Sun Mao Ding Melinda Davis* *MD at time of publication
↑ Frequency of chloride channel opening
Hyperpolarization of nerve membrane
Action potential (AP) inhibited
APs inhibited in the
medulla oblongata
(the respiratory center)
↓ Respiratory drive: the body fails to ↑ depth and rate of respirations when arterial CO2 ↑
General CNS inhibition
Anti-convulsion
(Seizures are caused by a burst of uncontrollable, electrical activity in the brain)
APs inhibited in the thalamus and hypothalamus (play a role in memory)
APs inhibited in the limbic system (the behavioral and emotional response centers in the brain)
Hypotension
↓ Cerebral blood flow
↓CO2 diffusion from arterial blood to alveoli
↓O2 diffusion from alveoli to arterial blood
Pharyngeal muscle relaxation
↑ Arterial CO2
↓ Arterial O2
Airway obstruction
Amnesia
↑ PaCO2
↓ PaO2
↓ Anxiety
Anxiolysis Hypercapnia
Hypoxemia
In high doses:
Depression of arousal and loss of consciousness
Induction of general anesthesia
(No analgesic effect)
↓ Intracranial pressure Benzodiazepine reversal:
Temporary cessation of breathing
Apnea
Flumazenil competitively binds to GABAA
Flumazenil reverses the binding of benzodiazepine to GABAA
↓ Frequency of chloride channel opening
Depolarization of nerve membrane
Benzodiazepine reversal
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published Aug 09, 2018, updated April 25, 2023 on www.thecalgaryguide.com")
急性胸部综合征镰状细胞贫血发病机制和临床表现
:发病机制和临床表现")
Subdural Hematoma on CT

Other (i.e. brain mass)
Stretching & tearing of bridging cortical veins that cross from the cortex to dural sinuses
Crescent shape
Blood accumulation in the potential space between the dura mater & the arachnoid mater
As the blood clots over time, its appearance varies on CT and is measured as different radiodensities (MRI may be needed to detect subtle bleeds)
Acute < 3 days
Subacute 3 days to 3 weeks
Chronic > 3 weeks Acute on Chronic
Acute blood (50-60 HU) is hyperdense to the surrounding cortex
As blood clot ages and protein degradation occurs, the density drops to 35-40 HU
The collection of blood becomes hypodense (~0 HU) to adjacent cortex
Both hypodense & hyperdense collections visible forming a hematocrit level
Hounsfield units (HU) are a measure of radiodensity (Air -1000 HU appears black, CSF is 0 HU appears dark, and cortical bone is >1000 HU appears white)
Hypodense collection
A pre-existing chronic hematoma
Hematocrit level
Fluid-fluid level in the case of an acute bleed into a pre-existing chronic subdural collection.
This can also be seen in patients with coagulopathy as blood clots improperly, allowing for dependent blood layering
Hyperdense collection
Acute blood sinks inferiorly (as CT is taken with patient supine)
Authors: Nameerah Wajahat, Aly Valji, Omer Mansoor Reviewers: Reshma Sirajee, Tara Shannon, Mao Ding, Petra Cimflova* *MD at time of publication
Subdural hemorrhages spread past suture lines to take on a crescent shape (seen bilaterally on this CT), but limited by dural reflections (falx cerebri, tentorium)
R
Intracranial pressure ↑
Mass effect on the brain tissue
Midline shift
Shift of brain tissue across the center line of the brain (dashed line shows ideal midline)
Ventricular effacement (partial)
Ventricles appear smaller as some cerebrospinal fluid (CSF) is pushed out
Sulcal effacement
CSF filled sulci become less apparent as CSF is squeezed out and gyri are lying on each other
Herniation
Protrusion of brain through rigid membranes or foramina of skull
Axial CT Head: Acute on Chronic Bilateral Subdural Hematoma showing features of both acute and chronic bleeding. Image Source: Radiopaedia.org
Legend:
Pathophysiology
Mechanism
Radiographic Findings
Complications
Published May 10, 2023 on www.thecalgaryguide.com")
Telogen Effluvium
: Causes, pathophysiology, and clinical findings Reviewers: Elise Hansen, Sunawer Aujla, Dr. Jori Hardin* *MD at time of publication
Telogen effluvium: non-scarring alopecia characterized by diffuse shedding of telogen-phase hair due to a reactive process
Hypothyroidism
↓ Thyroid hormones (T3,T4)
↓ Binding of thyroid hormone to receptors in the skin and hair
Cell division ceases in keratinocytes
The catagen phase of the hair cycle is triggered (involuting phase where hair enters apoptosis)
Delayed re-entry of telogen (resting)
hair into the anagen (growing) phase
Post-partum hair loss (telogen gravidarum)
↑ Circulating placental estrogen
Prolonged anagen phase
↑ Hair growth during pregnancy
Baby is delivered
↓ Estrogen and other trophic
hormones postpartum
The increased amount of anagen
hairs from pregnancy all enter catagen phase simultaneously
Nutritional deficiencies i.e. iron deficiency
Critical illness
Fever triggers various pro- inflammatory cytokines (tumor necrosis factor, interleukin 1b, interleukin 6, and interferon types 1 and 2)
Premature entry into catagen phase (the body induces cell-cycle arrest in all non-essential structures)
Hair follicle keratinocytes undergo apoptosis in response to inflammation
Ribonucleotide reductase (an enzyme involved in DNA synthesis) cannot utilize iron as a co-factor
↓Iron stores
↓ Expression of iron-
dependent genes (CDC2, NDRG1, ALAD, and RRM2)
↓ Expression of matrix genes of a healthy hair follicle (Decorin and DCT)
↓ Production of matrix keratinocytes (cells that form the hair shaft of growing hair)
Arrest of matrix proliferation
Hair shedding commonly occurs in the bitemporal areas 2-3 months after triggering event
Hair shaft Hair follicle
Telogen phase
Skin
Epidermis
Dermal- Anagen phase
Epidermal Junction Dermis
Catagen phase
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published May 10, 2023 on www.thecalgaryguide.com")
Diabetic Retinopathy

Ethnicity: White youth, African American, Hispanic, Asian-Pacific Islanders, and American Indigenous
Type 1 Diabetes Mellitus (T1DM) (see Diabetes Mellitus, Type I for pathogenesis)
Type 2 Diabetes Mellitus (T2DM) (see Diabetes Mellitus, Type II for pathogenesis)
Polycystic Ovarian Syndrome
Family history of T2DM
History of Gestational Diabetes
↑ Body Mass Index
↑ Age
Ethnicity: Indigenous Americans, African American, Hispanic
Poor glycemic control
Chronic high blood sugaràinflammatory response
↑ cytokines and growth factors including vascular endothelial growth factor (VEGF)
Vascular permeability Retinal neovascularization
Vascular endothelial dysfunction: basement membrane thickening, vascular cell death, vascular occlusion from platelet aggregation
Retinal hypoxia
Outpouchings of the weakened capillary walls or endothelial buds attempting to re-vascularize the ischemic retina
Weakened blood-retinal barrier (BRB) allows for rupture into the deeper retinal layers
Lipid deposits with sharp margins due to lipoproteins & other proteins leaking through the damaged blood-retinal barrier
Nerve fiber layer infarcts from occlusions of the precapillary arterioles
Retinal hemorrhages occur in the more superficial nerve layer
Focal areas of saccular venous bulges due to significant retinal ischemia & endothelial wall weakening and damage
Diabetic Retinopathy (DR)
A complication of diabetes due to chronic hyperglycemia resulting in abnormal permeability and ischemia of retinal vessels
Micro-aneurysms
Dot/blot hemorrhages Hard exudates (yellow
opaque solids on retina)
Cotton-wool spots (cloudy translucent patches on retina)
Flame hemorrhages
(multiple opaque red patches)
Venous beading (veins with areas of narrowing forming bead-like segments)
Traction retinal detachment
Bleeding into the vitreous humor
Pericyte death, breakdown of endothelial tight junctions & basement membrane thickening damages blood-retinal barrier
Damaged blood- retinal border leaks fluid into the retinal tissues
Macular edema
(swelling of macula)
Mild Non-proliferative DR
Moderate Non-proliferative DR
Severe Non-proliferative DR
Proliferative DR
Presence of neovascularization
Localized retinal ischemia causes upregulation of vascular endothelial growth factor causing fine, irregular & easily friable neovascularization in the disc, macula, and/or retina
Neovascularization
(fine loop networks of new blood vessels)
Neovascular membranes in vitreous gel form vitreoretinal adhesions and contract
Fragile vessels extending into the vitreous humor
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published June 28, 2017, updated May 18, 2023 on www.thecalgaryguide.com")
OA Clinical findings

Damage of normal cartilage under abnormal loading
Authors: Sean Spence Modhawi Alqanaie Reviewers: Yan Yu Jennifer Au Mao Ding Gary Morris* * MD at time of publication
Single large traumatic event or repeated microtrauma
Damage to normal articular cartilage under normal loading (force put on a joint)
Genetic anomalies in cartilage production and inborn errors of cartilage metabolism
Damage of abnormal cartilage under normal joint loading
Destruction/attrition of articular cartilage
Osteoarthritis
A degenerative joint disease that can affect both load bearing joints (knee, hip) as well as in smaller joints (proximal inter-phalangeal, carpometacarpal joints in hand)
Repeated physical joint trauma
Aberrant bone deposition secondary to wear on subchondral bone
Formation of osteophytes (bony projections) and subchondral sclerosis (bone thickening)
Lack of cartilage
Direct contact between bony processes with movement of the joint
Inflammation alters the chemical milieu of joint tissue
Synovial fluid forced into bone
Damage to subchondral (under cartilage) blood vessels
Subchondral fractures
cytokines regulate hyaluronic acid synthase
Synovium makes lower molecular weight hyaluronic acids (a potent proinflammatory molecule)
↓ synovial fluid viscosity
↑ risk of infection
Increased secretion of proteolytic enzymes
↑ joint fluid production
Joint effusion
Disruption of normal joint architecture
Palpable bony hypertrophy
(ex. Bouchard’s nodes (bony bumps on the middle joints of the finger))
Impaction of osteophyte with normal joint structures during movement
Physical disability
Crepitus
(grating sound in a joint)
Joint movement with reduced lubrication stimulates joint nociceptors
Stimulation of joint nociceptors (sensory receptors that detect damaging stimuli) in subchondral bone
Pain with use of joint
Pain with motion
↓ Range of motion
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published November 1, 2012, updated May 18, 2023 on www.thecalgaryguide.com")
Digestion and Absorption of Macromolecules
 Zhang, Shyla Bharadia, Mao Ding Sylvain Coderre* *MD at time of publication
Lipids (triglycerides)
Gastric lipase breaks down triglycerides into diglycerides and fatty acids (little digestion of triglycerides occur in stomach)
Bile, produced by the liver, and pancreatic lipase enter the small intestine through the common bile duct
The hydrophobic and hydrophilic properties of bile allow it to effectively bind hydrophobic fats with hydrophilic pancreatic lipases
Pancreatic lipases break down triglycerides into monoglycerides, fatty acids, and glycerol as a result of emulsification by bile
Monoglycerides and fatty acids are absorbed through bile-stabilized chylomicrons (lipid molecules arranged in spherical form)
Triglyceride-rich chylomicrons are transported through the lymphatic system and require the activity of tissue lipoprotein lipase when they arrive at the tissue
Triglycerides enter systemic circulation for metabolism, energy source, and fat storage
Ingestion of nutrition sources containing macromolecules
Carbohydrates (oligosaccharide)
Salivary amylases in the mouth break down oligosaccharides and starch into shorter polysaccharides
Pancreatic amylases break down polysaccharides into monosaccharides, disaccharides, and oligosaccharides in the stomach
Brush border enzymes in small intestinal villi break down disaccharides into
Proteins (peptides)
Pepsinogen is secreted by the stomach wall
Hydrochloric acid activates pepsinogen to pepsin in the stomach
Pepsin breaks down protein peptides into oligopeptides and amino acid chains
The pancreas generates trypsinogen and other enzymes to be released into the duodenum
Trypsinogen undergoes cleavage in the duodenum through the action of duodenal enteropeptidases to form trypsin
Intravenous proton pump inhibitors used in upper gastrointestinal bleeds stabilize pepsinogen, and prevent pepsin from breaking down clotting factors (proteins) enabling clotting
Maltose Maltase
monosaccharides: Sucrose
Sucrase
Glucose + fructose
Lactose Lactase
Glucose + galactose
Glucose + glucose
Conditions affecting the duodenum (e.g., Celiac disease) can lead to lower enteropeptidase levels, resulting in impaired protein digestion
Activation of trypsinogen to
trypsin within the pancreas, rather than in the duodenum, contributes to autodigestion observed in acute pancreatitis
Absorption of glucose and galactose through active transport via SGLT-1 carrier protein in the Jejunum and Ileum
Absorption of fructose through facilitative diffusion via transporter GLUT5 in the Jejunum and Ileum
Pancreatic enzymes (trypsin, chymotrypsin, carboxypeptidase) break down oligopeptides and amino acid chains into amino acids, dipeptides, and tripeptides in the duodenum
Monosaccharides enter systemic circulation for energy use and storage
Active Na+ cotransporters facilitate amino acid absorption in the jejunum and ileum
PEPT1 enzymes facilitate absorption of dipeptides and tripeptides, which are then immediately cleaved into amino acids in the jejunum and ileum
Amino acids enter systemic circulation for building & repairing muscles
Legend:
Pathophysiology
Mechanism
Treatment Effect
Complications
Published May 18, 2023 on www.thecalgaryguide.com")
Physiology of Anti-diuretic hormone
/Arginine Vasopressin (AVP)
Authors: Manaswi Yerrabattini Reviewers: Parker Lieb Mao Ding Shyla Bharadia Laura Hinz* * MD at time of publication
Limbic activation (e.g., pain, nausea)
Placental production of vasopressinase during pregnancy catalyzes the breakdown of ADH, leading to a temporary Diabetes Insipidus state (see Diabetes Insipidus: Pathogenesis and Clinical Findings slide)
Systemic arteriole vasoconstriction
Hypovolemia/Hypotension
Hyperosmolar state (i.e., extracellular fluid osmolarity above a certain threshold, most commonly due to hypernatremia)
Sensed by osmoreceptors in hypothalamus
Angiotensin II synthesized through activation of Renin- Angiotensin-Aldosterone System (RAAS) (see Physiology of RAAS slide)
Binds to receptors located in the hypothalamus
↓ pressure sensed by baroreceptors in the heart (left atrium and large veins)
Receptors transmit signals to brain via the vagus nerve
↓ Arterial baroreceptor firing
↑ Sympathetic activity of nerves innervating afferent arterioles
↑ Hypothalamic secretion of ADH (peptide hormone), transportation to posterior pituitary, and release from posterior pituitary into blood circulation
Blood Vessels (Minor role)
Kidneys (Main role)
ADH binds to to Vasopressin-2 receptors on basolateral side of principal cells in kidneys
↑ Insertion of aquaporin II channels onto apical membrane of late distal tubule and collecting ducts
↑ Water reabsorption
↓ Urine Output and Maintains narrow range of serum osmolarity ↑ Urine Osmolarity and preserves sodium homeostasis
ADH binds to Vasopressin-1 receptors on smooth muscle of blood vessels
ADH activates calcium signaling pathway
↑ Blood Pressure
Maintains overall fluid volume status
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published May 20, 2023 on www.thecalgaryguide.com")
Oxygen-Hemoglobin-Dissociation-Curve

Normal physiology of hemoglobin:
α α β β
Adult hemoglobin, Hemoglobin A (HbA), is made up of two α- subunits and two β- subunits
When partial pressure of O2 (pO2) is ↓, bonds between subunits are strainedàHbA subunits stabilized in this deoxygenated tense state (T- state)àHas ↓ O2 affinity
O2 O2
As pO2 ↑, O2 binds a T-state subunit àSubunit destabilizesàSubunit changes shapeàBecomes oxygenated relaxed state (R-state)à Has ↑ O2 affinity
O2 O2 O2 O2
O2 rapidly binds to R-state subunits, saturating HbA
Reviewers:
Parker Lieb, Sunawer Aujla, Marissa Zhang, Tara Lohmann* * MD at time of publication
Normal physiology of hemoglobin in the body: In lungs:
In tissues:
↑ O2 consumption due to cellular respiration
↑ pO2 during respiration (~100mmHg)
HbA is in its deoxygenated, T-state
HbA is in its oxygenated, R-state
O2 O2 O2 O2
T-state subunits favor O2 binding
When pO2 > 60, O2 binds to a circulating subunit in the T-state
Subunit destabilizesà Subunit changes shape to R- stateà↑ O2 affinity
Positive cooperativity:
All subunits transition from T- to R-state
O2 quickly binds remaining subunits
↓ pO2 (~40mmHg) R-statesubunitsfavorO2 unloading
O2
O2
O2
O2 O2 O2 O2
In some HbA molecules, O2 remains bound
Reserve oxyhemoglobin is available for higher O2 demand states
Certain disease processes (i.e., Hypoventilation, V/Q mismatch, impaired O2 diffusion) cause pathologically ↓ pO2 states
↑↑ Deoxygenated T-state HbA
Rapid desaturation of remaining oxyhemoglobin due to positive cooperativity
Rapid hypoxia onset at low pO2
When ↓ pO2 in tissues, O2 unloads from a R-state subunit
Subunit destabilizesà Subunit changes shape to T- stateà↓ O2 affinity
Positive cooperativity: All subunits transition from R- to T-state and release O2
O2 unloading in peripheral tissue
O2 O2
O
O2 O2
2
O
O2 O2
O
2
2
Sigmoid-shape oxyhemoglobin ↑↑ O2 loading in dissociation curve pulmonary capillaries
Legend:
Pathophysiology
Mechanism
Final effect
Complications
Published , 2022 on www.thecalgaryguide.com")
Acute Laryngitis

Malaise Fever
Fungal
Atopy (asthma, allergy)
Non-infectious
Gastroesophageal Reflux
Trauma or damage to larynx
Smoking
Yan Yu*
* MD at time of publication
Environmental Pollution/Inhalants
Bacterial (S. pneumoniae,
H. influenzae, M. catarrhalis)
Systemic immune response
Spread of infection to larynx through upper respiratory tract
Infection of the vocal folds and surrounding tissue
Mechanical
(vocal misuse/ trauma)
(Area in the neck that contains the structures for voice production, anatomically anterior to the esophagus, inferior to the pharynx and superior to the trachea)
Irritation of the vocal folds and surrounding tissue
Inflammatory cascade triggered
Acute Laryngitis
Symptoms for <3 weeks
Acute injury to vocal folds
Vocal fold
lesions (i.e., vocal polyps)
Laryngeal inflammation
Neutrophils and macrophages release inflammatory cytokines
Local laryngeal inflammationà↑ vascular permeability ↑ Secretion of mucous leading to airway congestion Cough reflex initiated to clear airway congestion Cough
Edema of vocal folds and surrounding tissue
Dysphagia (difficulty swallowing)
Dysphonia (difficulty speaking)
Odynophagia (painful swallowing)
Swelling impairs vocal cord vibration
Frank aphonia (loss of voice)
Progressive worsening of edema
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published May 24, 2023 on www.thecalgaryguide.com")
Neonatal Sepsis

Group B Strep vaginal colonization
Intra- amniotic infection
Prematurity (< 37 weeks) or low birth weight
(< 2500 gm)
Under- developed immune system
Predisposed to infection
Congenital anomaly that disrupts skin
Birth asphyxia (lack of oxygen and blood flow to brain)
Male gender (mechanism unclear)
Invasive Procedures
Direct introduction of bacteria to neonate’s blood
↑ likelihood of introducing bacteria to the fetus
Vertical transmission of maternal bacteria from lower genital tract to uterus
Contamination of amniotic fluid
Fetal bacteremia (presence of bacteria in bloodstream
Direct transmission of bacteria from maternal birth canal to fetal blood during delivery
Disruption in neonatal host defenses
Neonatal Sepsis
An invasive infection, usually bacterial, occurring during the neonatal period (<4 weeks of age for term infants, or <4 weeks after the due date for preterm infants)
Note:
*APGAR = appearance, pulse, grimace, activity and respirations at 1-, 5-, and 10-min post birth
Gastrointestinal
Poor feeding
Vomiting
Diarrhea, constipation, or bloody stool
Urological ↓ Urine output
Note: See relevant slide(s) for mechanisms of how each sign and symptom comes about.
CVS/RESP
Apnea/tachypnea Labored breathing Pallor or cyanosis Brady-/tachycardia Hypotension
Metabolic
Jaundice
Hypo- or hyper-glycemia
Metabolic acidosis
CNS
Lethargy Irritability
Focal neurological signs
Seizure
General
Low APGAR*
Temperature instability
Bulging or sunken fontanels
Rash
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published June 3, 2013, updated June 7, 2023 on www.thecalgaryguide.com")
Menopause
 See relevant slide: Menstrual Cycle Physiology: Ovarian Cycle – Brief Overview
Each cycle involves ovulation, during which an oocyte is released from the ovary’s dominant follicle into the Fallopian tube
Some non-dominant follicles degenerate in a process known as atresia
Menstrual cycle stops
Menopause marks 1 year since last menstrual cycle
↓ Fluid transudatio
n from blood vessels of vaginal wall
↓ Vaginal lubrication
Vaginal tissue becomes thinner and more easily irritated
Over time, fewer
follicles remain in the ovary
Some cycles become anovulatory (no oocyte is released from ovary)
↓ Ovulation causes prevents thickening of the endometrial lining
↓ regularity and frequency of periods
Ovaries eventually stop releasing oocytes
↑ Oxidative stress- induced apoptosis of dermal fibroblasts
Remaining non-dominant follicles become less sensitive to LH and FSH
Since follicular cells are responsible for estrogen production, less follicles result in reduced estrogen production
↓ Expression of serotonin receptors in the CNS
↓ LDL receptor expression and ↑HMG- CoA reductase activity
↓ Regulation of the production and clearance of LDL
↑ LDL Cholesterol levels
Author: Sunawer Aujla Reviewers: Ashar Memon Yan Yu* * MD at time of publication
↓ Serotonin activity
↓ Density of
↓ Healthy vaginal flora
↑ pH of vaginal fluid
↑ Spread of bacteria otherwise unable to survive in low pH environment
Recurrent urinary tract infections
↓ Calcitonin
↑ Sensitivity of bone mass to Parathyroid Hormone
↑ Activation of osteoclasts
Mechanism is likely
multifactorial and the subjective symptoms of menopause may contribute
Depression
5HT receptors in
thermoregulatory region of hypothalamus
↑ Inhibition of sexual responses initiated in prefrontal cortex
↓ Libido
2A
↓ Collagen, elastin, and hyaluronic acid
↓ Proliferation of smooth muscle fibers
↓ Inhibition of osteoclasts
Narrower thermoregulatory zone
Injury to epithelial tissue in multiple areas of the body
Atrophy of bladder and urethra epithelium
Urinary incontinence
More bone resorption than formation
Osteoporosis
See relevant slide: Osteoporosis: Pathogenesis and risk factors
Sometimes, for unknown reasons, core body temperature increases above upper threshold of narrowed thermoregulatory zone
Hot Flashes
Sudden, temporary onset of body warmth, flushing, and sweating
Sometimes, for unknown reasons, core body temperature decreases below lower threshold of narrowed thermoregulatory zone
Chills
Sudden, temporary onset of shivering, tingling, cold feeling
Atrophy of vaginal epithelium
Dyspareunia
Pain during sexual intercourse
↓ Integrity of of blood vessels
Atherosclerosis
↑ Risk for cardiovascular disease
Genitourinary Syndrome of Menopause
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published June 7, 2023 on www.thecalgaryguide.com")
otite-moyenne-aigue-oma-pathogenese-et-manifestations-cliniques-chez-les-enfants
 Pathogenèse et manifestations cliniques chez les enfants")
appendicite-pathogenese-and-trouvailles-cliniques

chutes-pathogenese-et-complications

granulomatose-avec-polyangeite-manifestations-cliniques

hernie-incisionnelle-pathogenese-et-trouvailles-cliniques

thyroidite-dhashimoto-pathogenese-et-signes-cliniques

commotions-cerebrales-pathophysiologie-aigue-et-resultats

otite-moyenne-aigue-complications

mpoc-declancheurs-et-signes-symptomes-des-exacerbations-aigues

mpoc-decouvertes-cliniques

mpoc-complications

mpoc-pathogenie

mpoc-conclusions-des-enquetes

Burns - Full Thickness - Pathogenesis and Clinical Findings 2023

Author and Illustrator: Amanda Eslinger Tracey Rice Reviewers:
Sunawer Aujla Alexander Arnold Duncan Nickerson* Jori Hardin*
* MD at time of publication
Epidermis
Dermis
Sub-cutaneous tissue
Fire
Contact Scald Chemical Electrical
Transfer of heat energy causes direct injury to keratinocytes
Hypoxic injury (lack of oxygen) causes ischemia-related cell death leading to necrosis
Full thickness burn
Non-uniform damage to the epidermal, dermal & subcutaneous layers at varying widths & depths due to unpredictable injury pattern
Degraded
epidermal
& dermal layers cover granulated tissue
Ulceration covered by eschar: a thick, dried, black (necrotic) layer
Long-term risk of ulcers & infections
↑ Vascular permeability in subcutaneous layers
↑ Intravascular fluid leaves capillaries, ↓ uptake by lymph vessels
Edema
Compression of surrounding muscles, nerves & vessels
Ischemia and/or necrosis
↓ Immunologic
response due to impaired epidermal barrier function
↑ Microbial growth creating biofilm & secretion of chemicals that inhibit natural protective process
Irritated,
inflamed wound bed +/- exudate
↑ Risk of infection & septic shock
Destruction
of somatosensory structures
Hypoesthesia (↓ sensation to stimuli)
↑ Risk of
repeated injury due to ↓ response to noxious stimuli
Destruction of cutaneous capillary beds
↓ Healing due to ↓ dermal structures throughout wound
White or leathery appearance
Chronic wounds require surgical interventions
↑ Risk of contractures & skin barrier weakness
Legend:
Mechanism of Injury
Pathophysiology
Sign/Symptom
Complication
Published December 2, 2013, updated June 26, 2023 on www.thecalgaryguide.com")
Burns - Full Thickness - Pathogenesis and Clinical Findings

Author and Illustrator: Amanda Eslinger Tracey Rice Reviewers:
Sunawer Aujla Alexander Arnold Duncan Nickerson* Jori Hardin*
* MD at time of publication
Epidermis
Dermis
Sub-cutaneous tissue
Fire
Contact Scald Chemical Electrical
Transfer of heat energy causes direct injury to keratinocytes
Hypoxic injury (lack of oxygen) causes ischemia-related cell death leading to necrosis
Full thickness burn
Non-uniform damage to the epidermal, dermal & subcutaneous layers at varying widths & depths due to unpredictable injury pattern
Degraded
epidermal
& dermal layers cover granulated tissue
Ulceration covered by eschar: a thick, dried, black (necrotic) layer
Long-term risk of ulcers & infections
↑ Vascular permeability in subcutaneous layers
↑ Intravascular fluid leaves capillaries, ↓ uptake by lymph vessels
Edema
Compression of surrounding muscles, nerves & vessels
Ischemia and/or necrosis
↓ Immunologic
response due to impaired epidermal barrier function
↑ Microbial growth creating biofilm & secretion of chemicals that inhibit natural protective process
Irritated,
inflamed wound bed +/- exudate
↑ Risk of infection & septic shock
Destruction
of somatosensory structures
Hypoesthesia (↓ sensation to stimuli)
↑ Risk of
repeated injury due to ↓ response to noxious stimuli
Destruction of cutaneous capillary beds
↓ Healing due to ↓ dermal structures throughout wound
White or leathery appearance
Chronic wounds require surgical interventions
↑ Risk of contractures & skin barrier weakness
Legend:
Mechanism of Injury
Pathophysiology
Sign/Symptom
Complication
Published December 2, 2013, updated June 26, 2023 on www.thecalgaryguide.com")
diverticulosis-vs-diverticulitis-distinguishing-features
 contributing to reduced gut motility
↑Intraluminal pressure in the colon
Herniation of colonic mucosa and submucosa through circular muscle at points of weakness to form outpouchings
Diverticulosis
Presence of outpouchings in the colon (diverticuli)
Note: In Western populations, most diverticulosis is left-sided, whereas in Asian populations, these outpouchings are more often right-sided.
Mucosal abrasion or micro-perforation by ↑ intraluminal pressure or dense food particles
Bacterial overgrowth,
dysbiosis and passage into the lamina propria
Feces collects in diverticuli
Gut bacteria metabolize undigested material and produce gas
Stretching of colon Bloating wall irritates and
visceral afferent flatulence nerves
Episodic abdominal discomfort and cramping
Blood vessels in
the mucosa and submucosa (vasa recta) are stretched over the diverticuli, and may rupture from ↑ pressure
Diverticular bleed
Painless hematochezia (passage of fresh blood from the rectum)
Inflammatory cytokines activate clotting factors
Clotting of blood in vessels supplying diverticula
Inflammatory cytokine release (IL-6, TNF-α)
Cytokines enter systemic circulation
Edema in the bowel wall
Irritation of adjacent parietal peritoneum and somatic nerves
Left lower quadrant (LLQ) pain, guarding
Small abrasions are walled off by pericolic fat and mesentery
↑WBC
Fever
Inflammation may spread to nearby organs, leading to ulceration and abnormal connections between organs
Pericolic abscess
No hematochezia
Local ischemia and focal necrosis resulting in loss of integrity of the bowel wall
Perforation
Generalized peritonitis
Colonic obstruction (rare)
Fistulae (rare): i.e. colovesical, coloenteric
Stricture/fibros is formation with healing
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published October 12, 2016; Updated July 30, 2023 on www.thecalgaryguide.com")
diverticulosis-vs-diverticulitis-caracteristicas-distintivas

Knee Osteoarthritis

Articular trauma
Inflammatory disease or infection (e.g. Rheumatoid or septic arthritis)
Obesity and ↑leptin ↑ Chondrocytes,
inflammatory mediators, and metalloproteinases
Extracellular matrix degradation
↑ Knee joint loading forces
Metabolic syndromes (e.g. diabetes mellitus)
↑ Oxidative stress and insulin resistance
Low-grade systemic inflammation
↓ Elasticity and ↑ degradation of cartilage
↑ Friction in knee joint with movement
↓ Cartilage along femoral groove and posterior surface of patella
Pain, catching, and crepitus (crackling/clicking sound) in the patellofemoral joint
Inability/difficulty with kneeling or climbing stairs
Abnormal distribution of forces accumulate and stress articular surface
↑ Damage/laxity to soft tissue structures stabilizing knee joint
Knee Osteoarthritis
(Multifactorial entity characterized by cartilage breakdown, deterioration of connective tissue, and bone deformities)
↓ Cartilage between distal femur and proximal tibia ↓ Joint spaceàto articular dysfunction
Radiographic changes
See Osteoarthritis (OA): X-Ray Features slide
Repeated attempts to repair cartilage and joint disruption
Subchondral bone thickening (sclerosis) under joint cartilage and bone spur (osteophyte) formation around joint line
Rotational/antero-posterior instability and ↑ external adduction moments during walking
Alterations in proteoglycans, fiber arrangement, and collagen composition in soft tissue structures within/around knee joint
↑ Shear forces and medial compartment narrowing erode and pinch soft tissue structures within the knee joint
Cruciate ligament degeneration
Weakened passive stabilizers of the knee joint
Knee giving way and instability (falls)
Meniscal tears, if large àprevents knee extension/flexion
Locking of the knee
Joint line tenderness:
Patient points to area of tenderness/pain reproducible upon palpation
Anatomical axis of hip, knee, and ankle joints ↑ loading medially
Medial > lateral joint line tenderness
↑ Joint friction activating nociceptors in the surrounding anatomical tissues
Injury and inflammation ↑ nociceptive responses in soft tissue structures and subchondral bone within knee joint
Nociceptive feedback to brain inhibits activity of motor cortex neurons controlling muscles around the knee
↓ Motor output and muscle activation over time
↓ Muscle strength/endurance, lower limb muscle use, functional ability (walking, stairs, etc.)
Joint inflammationà accumulation of fluid within joint
Stiffness, swelling, redness, and pain
Limited joint space reduces range of motion for femur to roll/slide on tibia
↓ Knee flexion and extension
Flexion contracture and antalgic gait
Reduced weight acceptance of the joint and surrounding muscles/tendons
↓ Mobility and physical dysfunction
Muscular atrophy
Reduced function of active stabilizers of the knee joint (quadriceps, adductors, hamstrings)
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published July 30, 2023 on www.thecalgaryguide.com")
Rotator Cuff Disease
 of glenohumeral joint
Tensile forces
Repeated eccentric tension from overhead activities
Trauma, sports, and occupation
↑ Torque, compression, and translational stresses
Metabolic syndromes
Reactive oxygen species
interact with ↑ glucose forming advanced glycation end-products (AGEs) which accumulate in soft tissues
Smoking
Impingement syndromes
Vessel damage, ischemia, tenocyte apoptosis
Macro-trauma causing an acute, complete tear in the rotator cuff muscle(s)
↓Dynamic stability (from rotator cuff and periscapular muscles) and range of motion of the shoulder at the glenohumeral joint
↑ Bone on bone contact of proximal humeral head and boney structures of the scapula
Subacromial bursa degeneration
↓ Protection of underlying supraspinatus muscle from attrition between humeral head and acromion
Rotator Cuff Syndrome
(Inflammation, impingement, or tearing of one or more of the four muscles/tendons of the rotator cuff: supraspinatus, subscapularis, infraspinatus, teres minor)
Repetitive loading and micro-tearing of tendon/muscle fibers
↑ Oxidative stressors and inflammatory cascades
↓ Vascularity of rotator cuff structures
Radiographic changes: See Rotator Cuff Disease: X-ray and ultrasound features slide, in addition to: calcific tendonitis, calcification of in the coracohumeral ligament, and hooked acromion (calcification from tendon pulling)
In some cases, soft tissues enclose/surround shoulder joint capsule thicken (fibrose) and tighten
Degenerative joint disease and rotator cuff arthropathy
Proximal humeral head migration and ↓ subacromial space
Inflammation and insufficient healing of rotator cuff structures, which may lead to:
Supraspinatus (shoulder abduction) degeneration
Pain, shoulder stiffness, ↓ active AND passive range of motion
Adhesive Capsulitis (frozen shoulder)
Infraspinatus and teres minor (external rotation) degeneration
Rotator cuff tendons become inflamed and irritated as they rub against acromion
Subacromial impingement
Subscapularis (internal rotation) degeneration
+Lift-off test: Inability to hold dorsum of the hand off lumbar spine while internally rotating shoulder
↓ Shoulder strength and muscular atrophy
Pain with passive shoulder flexion beyond 90°
Winging of the scapula during arm adduction
+Empty-can test: Weakness and/or arm depression with resisted abduction with arm internally rotated in 90°
+Drop-arm test: Inability to maintain shoulder in abducted position at 90° and/or adduct the arm in a controlled manner (resulting in ”dropping”)
Weakness to resisted external rotation with elbow in 90° flexion, inability to keep arm externally rotated (infraspinatus)
+Hornblower’s sign: decreased external rotation strength in arm abduction (suggests additional teres minor tear)
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published July 30, 2023 on www.thecalgaryguide.com")
Miocarditis

Shock por quemaduras

Angiotensin Converting Enzyme (ACE) inhibitors: Mekanisme Kerja dan Temuan Klinis
 inhibitors: Mekanisme Kerja dan Temuan Klinis")
Infark Miokard: Komplikasi

Angina Pektoris Angina Stabil: Patogenesis dan Temuan Klinis

Infark Miokard: Temuan Pemeriksaan Penunjang

Infark Miokard: Temuan pada Pemeriksaan Fisik

Epilepsi: Patogenesis

Penyakit Jantung Iskemik (PJI): Patogenesis berbagai jenis PJI
: Patogenesis berbagai jenis PJI")
Infark Miokard: Temuan pada Anamnesis

Anatomi Koroner pada EKG: Iskemia Lokal

Statin: Mekanisme & Efek Samping

Keratosis pilaris
: Risk factors, pathophysiology, and clinical findings
Authors:
Ayaa Alkhaleefa Reviewers: Tracey Rice Sunawer Aujla Jori Hardin* MD at time of publication*
Loss-of-function mutation in filaggrin (a multifunctional structural protein expressed in the epidermis)
Inhibits the release of specific amino acids that maintain
Keratin (a protein critical to the integrity of the skin barrier) cannot aggregate to align intermediate filaments within corneocytes (cells that comprise the stratum corneum)
the natural moisture of the skin ↑ Skin pH (normal pH is 5.5)
Distention of the follicular opening
↑ Surface area for hair to sprout
Growth of multiple hairs from the distended follicle
Development of thick, coiled hairs
Rupture of follicular epithelium by the circular hair shaft
Skin
Epidermal layer
Dermal-Epidermal Junction
Dermal layer
Mutation in filaggrin
Keratin accumulation in corneocytes with hair
Grouped keratotic follicular papules
Excessive keratin accumulates in the follicular spaces
↑ Keratinization of the follicular epithelium forms a keratinous plug in the infundibulum (funnel-shaped epithelial segment of the hair follicle)
Irritation of the hair follicle
Excess irritation triggers release of inflammatory cytokines
FLG FLG
Inflammatory cascade creates edematous environment
Symmetrical keratotic papules are commonly grouped along extensor surfaces of the upper arms, thighs, and buttocks
Perifollicular edema
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Published 27, JUNE, 2023 on www.thecalgaryguide.com")
Zystische Fibrose: Pathogenese, klinische Befunde und Komplikationen

Infektiose Endokarditis

Kindliche Asthma Exazerbation: Pathogenese und klinische Befunde bei Kindern

Mitralklappeninsuffizienz: Pathogenese und klinische Befunde

Extrauteringraviditat: Pathogenese und klinische Befunde

Ovarialtorsion: Pathogenese und klinische Befunde

Dermatitis herpetiformis
 in gut lumen is cross linked to gliadin
HLA-DQ2 on antigen presenting cells recognizes gliadin antigen
Gliadin antigen is presented to sensitized T helper cells and epitope spreading occurs
Authors: Elise Hansen Reviewers: Sunawer Aujla Jori Hardin* * MD at time of publication
Type 1 T helper cells and plasma cells produce IgA
antibody with gliadin antigen
Type 1 T helper cells and plasma cells produce IgA
antibody with gliadin crosslinked to TTG
Type 1 T helper cells and plasma cells produce IgA antibody with gliadin crosslinked to epidermal transglutaminase 3 (TG3)
IgA anti-TTG and IgA anti-TG3 circulate in bloodstream
IgA anti-TG3 antibodies reach dermis
TG3-IgA complex forms and deposits in papillary dermis TG3-IgA complex and Interleukin 8 stimulate neutrophil chemotaxis Neutrophils infiltrate papillary dermis
Neutrophils produce proteases
Proteases destroy the basement membrane of the dermis Epidermis no longer adheres to dermis
Epidermal transglutaminase 3 (TG3) is released from superficial keratinocytes
Vesicles filled with IgA deposits and neutrophils
Epidermal layer
Destruction of dermal papillae/ basement membrane
Dermal layer
Y
Grouped vesicles on extensor surfaces
Pruritus
TG3-IgA complex
Grouped excoriations on extensor surfaces
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published August 25, 2023 on www.thecalgaryguide.com")
Superficial thickness burns

Dermis
Sub-cutaneous tissue
Erythema
Radiation
Sunlight (most common), x-ray, nuclear emission/explosion
Specific to sunlight radiation, UV rays reach keratinocytes in the epidermis
p53 tumor suppressor protein is induced in keratinocytes
Transient cell cycle arrest Apoptotic pathway is
Fire
Contact
Scald
Chemical
Electrical
Direct damage to keratinocytes
Skin erosion and sloughing of skin cells
Direct stimulation of nociceptive nerve endings in the epidermis
DNA repair mechanisms are activated
Mistakes in repair process
Malignancy
(See ‘Basal Cell Carcinoma’ Slide & ‘Squamous Cell Carcinoma’ Slide)
Fluid leaves vasculature and enters interstitial tissues of the skin, causing it to swell
Edema
Triggers release of endothelin A proalgesic protein
Selective excitement of nociceptive nerve ending in epidermis
Pain
initiated in keratinocytes
Keratinocytes apoptose
Prostaglandins, arachidonic acid metabolites, substance P & proinflammatory cytokines are released into surrounding tissue
↑ Vascular permeability
↑ bloodflow carries warmth to body area
Irritation of endothelial cells in the dermal vascular plexus
Release of endothelium derived vasodilators such as nitric oxide
Vasodilation, ↑ blood flow through vessels
↑ blood under skin leads to skin appearing red
Warmth Pressing on skin occludes blood vessels temporarily, making skin Blanchable underneath appear white immediately after pressure is lifted
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published Dec 2, 2013, updated Aug 25, 2023 on www.thecalgaryguide.com")
Netzhautablosung: klinische Befunde

Netzhautablosung: Pathogenese

Uterusmyom (Leiomyom): Pathogenese und klinische Befunde
: Pathogenese und klinische Befunde")
Quadrizepssehnenruptur: Pathogenese und klinische Befunde

Klassifikation der Beckenringfrakturen: Mechanismen, klinische Merkmale und Komplikationen

Polyzystisches Ovarialsyndrom (PCOS): Pathogenese und klinische Befunde
: Pathogenese und klinische Befunde")
Ketamin

Epitheliale Ovarialkarzinome: Subtypen, Mutationen & Risikofaktoren

Beckenentzundung: Pathogenese und klinische Befunde
): Pathogenese und klinische Befunde")
Mastitis puerperalis und Mammaabszess: Pathogenese und klinische Befunde

Offene Frakturen: Mechanismen, klinische Befunde und Komplikationen

Subtrochantare Femurfrakturen: Pathogenese und klinische Befunde

Rotator Cuff Disease Xray and Ultrasound Features

Chronic (>3 months) tear with degenerative-type changes
Rotator cuff insufficiency, loss of supporting structures holding humeral head inferiorly
Displacement of the humeral head anterosuperiorly and instability of joint
Microtrauma affecting superior aspect of glenohumeral joint
“Acetabularization” or coracoacromial arch: concave acromial erosion and increased sclerosis (hardening)
Subscapularis tear (second most common)
Teres minor tear
Infraspinatus tear
Rotator Cuff Syndrome
(Inflammation, impingement, or tearing of one or more of the four muscles/tendons of the rotator cuff: supraspinatus, subscapularis, infraspinatus, teres minor)
Acute (partial or full thickness) tear of rotator cuff tendons
Humeral subluxation (partial displacement of humeral head relative to glenoid)
High riding humerus: decreased acromial humeral distance
Decreased acromial humeral interval/space
(impinging tendons of rotator cuff)
Full: Defect extends from the subacromial bursa (fluid filled sack beneath the acromion and above the rotator cuff tendons) to the articular surface of the glenohumeral joint
Tendon/muscle fibers completely separated from bone and/or muscle fiber connections severed
Partial: Focal defect affecting a portion of the tendon which may involve the bursa or glenohumeral articular surface
Non-visualization of the tendon
Acetabularization of glenoid
Fluid replaces empty space of tendon tear
Overlying fat around the sub acromial bursa falls into tendon gap
Sagging peribursal fat sign on ultrasound
“Femoralization” of the humerus: bone erosion (destruction) and rounding of greater tuberosity
Osteoarthritis of glenohumeral joint: See Osteoarthritis (OA): X-ray features slide
Hyperechoic (brightened) line between articular cartilage of humeral head and muscle tendon on ultrasound
Cartilage interface sign on ultrasound
Hypoechoic (darkened) tendon outline discontinuity on ultrasound imaging
Femoralization (rounding) of greater tuberosity
Subluxation of the humeral head relative to the glenoid
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published September 6, 2023 on www.thecalgaryguide.com")
5HT3 Antagonists
 receptors in the area postrema within the brainstem containing the chemoreceptor trigger zone
↓ Binding of serotonin which is released from the raphe nucleus in the brainstem
↓ Stimulation of vomiting center
(medulla) supressing the vomiting reflex
↓ Stimulation of vagus nerve resulting in ↓ signals to chemoreceptor trigger zone
↓ Abdominal pain signals to the brain
↓ Levels of serotonin uptake in gastrointestinal tract
↓ Stimulation of gastrointestinal
tract, diaphragm, and abdominal muscles
Peripheral Actions
Binds to 5-HT3 (serotonin) receptors on the vagus nerve terminals
Other Actions
Unknown mechanism for patients with significant cardiac history (e.g., congenital long QT syndrome, bradycardia, electrolyte abnormalities)
Unknown mechanism Unknown mechanism
↓ Binding of serotonin which is released from enterochromaffin cells in the gastrointestinal tract
Potential blockage of K+ channels in the heart
↓ Visceral sensation associated with irritable bowel syndrome
↓ Colonic motility slowing the rate of digestion
↓ Diarrhea
Constipation
Alters depolarization
and repolarization of the heart
Prolongation of QT interval
Headache Drowsiness
Torsades de Pointes arrhythmia
Cardiac arrest
Death
Rare complications
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published September 6, 2023 on www.thecalgaryguide.com")
Strabismus
![Strabismus: Pathogenesis and clinical findings
Microvascular dysfunction, trauma, or compression of oculomotor nerve
Oculomotor
nerve palsy: Dysfunction of the nerve innervating the superior rectus (elevation), inferior rectus (depression), medial rectus (adduction), and inferior oblique muscles (excyclotorsion)
Thyroid eye disease (see Thyroid Eye Disease slide for pathogenesis)
Inflammatory processes
Thickening & fibrosis of extraocular muscles, most commonly the inferior rectus muscle (functions to rotate the eye and depress the gaze)
Brown syndrome (congenital)
Anomalous
interaction between the trochlea and superior oblique muscle tendon
Restriction of normal movement of the superior oblique tendon through the trochlea
Trochlear palsy (Dysfunction of the trochlear nerve (CN IV))
Weakness of the superior oblique muscle innervated by CN IV (responsible for depression of the gaze and incyclotorsion & rotation of the eye)
Near reflex: convergence (eyes adduct), accommodati on (thickening of the lens) & miosis (constriction of the pupil)
Excessive accommodation in hyperopic (farsighted) eyes
Over-activation of near reflex
Accommodative esotropia
Aneurysm, infection, iatrogenic injury to cranial nerve (CN) VI
Abducens palsy: ocular motor paralysis
Failure of CN VI to develop normally in utero
Duane syndrome: congenital malformation of CN VI
Congenital fibrosis of the extraocular muscles (CFEOM)
Restrictive global paralysis of the extraocular muscles that control the movements of the eye
Phenotype CFEOM2
Phenotype CFEOM1 & 3
Dysfunction of the abducens
nerve (CN VI: innervates the ipsilateral lateral rectus muscle which abducts the eye [turns it laterally])
Orbital fracture (fracture of the orbital floor)
Intraorbital contents (inferior rectus muscle and/or surrounding tissue) herniate through the fractured site & are entrapped
Idiopathic infantile esotropia
Intermittent exotropia
Unclear process
Exotropia: affected eye is rotated laterally
Esotropia: affected eye is rotated medially
Hypotropia: affected eye is rotated downward compared to non-affected eye
Hypertropia: affected eye is rotated upward compared to non-affected eye
Horizontal Strabismus
Two different images are received by the eye that cannot be fused together Visual cortex suppresses the input from one eye in order to avoid having diplopia
Amblyopia/lazy eye: visual cortex diminishes neural inputs from the corresponding cortical areas of affected eye
↓ Spatial awareness
Vertical Strabismus
Binocular diplopia: double vision when both eyes are open, and absent when either eye is closed
Atypical alignment of the eye
Psychosocial consequences: negative impact to mental health due to social bias or abuse, social anxiety, and difficulties with self-image
Authors: Mina Mina Lucy Yang Reviewers: Mao Ding William Stell* * MD at time of publication
↓ Visual acuity
↓ Oculomotor control
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published September 6, 2023 on www.thecalgaryguide.com](https://calgaryguide.ucalgary.ca/wp-content/uploads/2023/09/Strabismus.jpg "Strabismus: Pathogenesis and clinical findings
Microvascular dysfunction, trauma, or compression of oculomotor nerve
Oculomotor
nerve palsy: Dysfunction of the nerve innervating the superior rectus (elevation), inferior rectus (depression), medial rectus (adduction), and inferior oblique muscles (excyclotorsion)
Thyroid eye disease (see Thyroid Eye Disease slide for pathogenesis)
Inflammatory processes
Thickening & fibrosis of extraocular muscles, most commonly the inferior rectus muscle (functions to rotate the eye and depress the gaze)
Brown syndrome (congenital)
Anomalous
interaction between the trochlea and superior oblique muscle tendon
Restriction of normal movement of the superior oblique tendon through the trochlea
Trochlear palsy (Dysfunction of the trochlear nerve (CN IV))
Weakness of the superior oblique muscle innervated by CN IV (responsible for depression of the gaze and incyclotorsion & rotation of the eye)
Near reflex: convergence (eyes adduct), accommodati on (thickening of the lens) & miosis (constriction of the pupil)
Excessive accommodation in hyperopic (farsighted) eyes
Over-activation of near reflex
Accommodative esotropia
Aneurysm, infection, iatrogenic injury to cranial nerve (CN) VI
Abducens palsy: ocular motor paralysis
Failure of CN VI to develop normally in utero
Duane syndrome: congenital malformation of CN VI
Congenital fibrosis of the extraocular muscles (CFEOM)
Restrictive global paralysis of the extraocular muscles that control the movements of the eye
Phenotype CFEOM2
Phenotype CFEOM1 & 3
Dysfunction of the abducens
nerve (CN VI: innervates the ipsilateral lateral rectus muscle which abducts the eye [turns it laterally])
Orbital fracture (fracture of the orbital floor)
Intraorbital contents (inferior rectus muscle and/or surrounding tissue) herniate through the fractured site & are entrapped
Idiopathic infantile esotropia
Intermittent exotropia
Unclear process
Exotropia: affected eye is rotated laterally
Esotropia: affected eye is rotated medially
Hypotropia: affected eye is rotated downward compared to non-affected eye
Hypertropia: affected eye is rotated upward compared to non-affected eye
Horizontal Strabismus
Two different images are received by the eye that cannot be fused together Visual cortex suppresses the input from one eye in order to avoid having diplopia
Amblyopia/lazy eye: visual cortex diminishes neural inputs from the corresponding cortical areas of affected eye
↓ Spatial awareness
Vertical Strabismus
Binocular diplopia: double vision when both eyes are open, and absent when either eye is closed
Atypical alignment of the eye
Psychosocial consequences: negative impact to mental health due to social bias or abuse, social anxiety, and difficulties with self-image
Authors: Mina Mina Lucy Yang Reviewers: Mao Ding William Stell* * MD at time of publication
↓ Visual acuity
↓ Oculomotor control
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published September 6, 2023 on www.thecalgaryguide.com")
سندرم-زجر-تنفسی-حاد-ards

کمبود-آلفا-یک-آنتیتریپسین-α1at

سرفه-مزمن-سیر-بیماری

سرفه-فیزیولوژی

ketamine-francais

hypersensibilite-resume

hypersensibilite-de-type-iv-pathogenese-et-resultats-cliniques

Rhumatisme psoriasique Complications

normal neonatal changes pathogenesis and clinical findings
.
Authors: Erin Auld, Dasha Mori Reviewers: Kayla Feragen Mao Ding Danielle Nelson* *MD at time of publication
Birth weight: Loss of birth weight up to 10%; should be re-gained within 10-14 days (30 g/day)
Stool: transitions from meconium (first stool of neonate that is black, tarry and sticky) to normal (green/brown or yellow mustard) within 2-3 days
Meconium passage: within 24 hours of birth
Growth
• Height: 2.5 cm/month
• Head Circumference: Average 1 cm/month in the first year with greatest growth in the first month • Weight: 20-30 g/day for the first 3 months
Urination: within the first 24 hours
Mother receives intravenous fluids during delivery
Colostrum (first milk secretion that contains antibodies) produced in first 2-3 days of lactation post-partum (Lactogenesis I)
GI tract maturation
Adequate dietary intake
Maturation of urinary tract
Adequate fluid intake
↑ maternal blood volume
Fluid moves between fetus and mother through placenta
Fetus’s fluid volume ↑
Infant’s urine output ↑ in first 24 hours post-partum
Low breast milk intake in first 2-3 days
↓ progesterone, ↑ prolactin in mother at birth
Infant ingests colostrum
Mostly water loss, some fat loss
↑ Breast Milk Production (Lactogenesis II)
Stomach stretches
↑ in gastrointestinal tract motility (gastrocolic reflex)
Ingestion of milk post-partum further stimulates GI maturation
110-120 kilocalories/kg/day
Gastrointestinal tract formation begins when the fetus is 4 weeks old. Maturation continues into infancy.
Normal gestational age (38-42 weeks)
Fetus ingests amniotic fluid in utero
Stimulates structural changes, enzymatic activity, and metabolic activity of GI tract
Mature detrusor sphincter complex, appropriate bladder capacity, proper renal perfusion, and regular arousal of the neonate
Breast milk: Feeding when infant demands it, approximately every 2-3 hours Formula: Feeding when infant demands it, approximately every 4 hours
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published Oct 24, 2015, updated Sept 28, 2023 on www.thecalgaryguide.com")
کارسینوم اولیھ ریھ
برونکوژنیک( - تومورھای پانکواست")
سرطان ریھ: یافتھ ھای بالینی و نشانگان ھای پارانئوپلاستیک

Hipoglikemia Diabetik: Patogenesis dan Temuan Klinis

Gambaran Umum tentang Keganasan Sel Darah

корь-осложнения

Ишемическии-колит

Obstructive Hydrocephalus on CT MRI Pathogenesis and findings

Acute:
Lateral walls and inferior surface of the third ventricle bulge out
Temporal horns Temporal horns
of the lateral of the lateral
ventricles dilate ventricles dilate
Cerebrospinal fluid accumulates upstream
↑ Ventricular pressure
Ventricles dilate (ventriculomegaly)
↑ Intracranial Pressure (See Increased Intracranial Pressure: Clinical Findings Slide)
Headache Papilledema
Impaired consciousness
Nausea and vomiting
Image Credit: Radiopaedia
Image Credit: radRounds
Ventricular ependymal lining is disrupted
Cerebrospinal fluid migrates into the surrounding brain parenchyma
Transependymal edema
Fourth ventricle may Fourth ventricle may
Chronic:
Septum pellucidum becomes fenestrated
Pronounced dilation of the ventricles, especially the lateral and third ventricles
Fornices become depressed
Corpus callosum thins and elevates
dilate, although the dilate, although the
posterior fossa often posterior fossa often
Distended ventricles compress overlying cortex
prevents it from prevents it from
getting too large getting too large
High T2 or FLAIR High T2 or FLAIR
signal around the signal around the
lateral ventricles on lateral ventricles on
MRI MRI
Image Credit: Cumming School of Medicine
Image Credit: Radiology Key
Third ventricle, pineal, infundibular and supra- optic recesses balloon out
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published Oct 10, 2023 on www.thecalgaryguide.com")
Acute Respiratory Distress Syndrome
 is a clinical syndrome involving acute lung injury. It results in severe hypoxemia and bilateral
Authors: David Olmstead Mao Ding Reviewers: Midas (Kening) Kang Usama Malik Kevin Solverson* * MD at time of publication
↓ PaO2 (Partial pressure of oxygen in arterial blood ↓SpO2 (Peripheral oxygen saturation)
Tachypnea (↑ RR) Tachycardia (↑ HR)
Dyspnea
Bilateral Opacity on chest radiograph
↓ PaO2, ↓SpO2
↑ PaCO 2
↑ PaO2, ↓PaCO2 Eupnea (normal
breathing)
↓ O2 Requirements Depression, Anxiety, PTSD Neuromuscular Weakness
Chronic Respiratory Dysfunction
airspace disease in the absence of elevated left-heart pressures.
Direct Lung Injury
Causes include pneumonia and pulmonary sepsis (community- acquired, hospital-acquired, aspiration, viral), drowning, and chemical pneumonitis from aspiration or direct inhalational injury
Indirect Lung Injury
Causes include sepsis with a non-pulmonary source, trauma, severe burns, transfusion- related acute lung injury (TRALI) and pancreatitis
Lung Tissue Inflammation
Exudative: Neutrophils migrate into the alveoli in response to inflammatory stimulus
Note: While the three phases of ARDS take place in sequence, all areas of the lung may not be in the same phase at the same time. For this reason, the processes can be thought of as overlapping.
Proliferative: Body attempts to heal damage. If it is not successful, the tissue transitions to the fibrotic phase
Neutrophil-containing pulmonary exudate interferes with surfactant function
Neutrophil infiltration and proinflammatory cytokines lead to tissue edema, dysfunction and subsequent destruction of pulmonary epithelium
Residual debris in alveoli are cleared by phagocytic cells
Restoration of alveolar epithelial cells.
Alveoli collapse in absence of working surfactant
Damaged epithelium impairs gas exchange
Pulmonary capillaries do not adequately absorb fluid
The body’s attempts to heal lung tissue result in deposition of hyaline membranes in the alveoli
Ventilation- Perfusion Mismatch
Pulmonary Edema
Impaired Gas Diffusion
Functional epithelium is able to absorb fluid back into circulation
↑ useful surface area for gas exchange
Clearing of CXR
Impaired Function After Prolonged Illness
Pulmonary Hypertension
Fibrotic: Inadequate healing results in long-term pulmonary damage (rare)
Fibroblast activity leads to deposition of collagen in alveoli and alveolar capillaries
Fatigue Pulmonary Fibrosis
Nail Clubbing (nails appear wider & swollen) Cough/Dyspnea
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published Feb 6, 2018, updated Oct 10, 2023 on www.thecalgaryguide.com
Acute Respiratory Distress Syndrome: Note: Acute respiratory distress syndrome is a clinical
Authors: David Olmstead Reviewers: Midas (Kening) Kang Usama Malik Kevin Solverson* * MD at time of publication
Pathogenesis and clinical findings
Direct Lung Injury
Causes include pneumonia and pulmonary sepsis (community-acquired, hospital-acquired, aspiration, viral), drowning, and chemical pneumonitis from aspiration or direct inhalational injury
Indirect Lung Injury
syndrome involving acute lung injury. It results in severe hypoxemia and bilateral airspace disease in the absence of elevated left-heart pressures.
Causes include sepsis with a non-pulmonary source, trauma, severe burns, transfusion-related acute lung injury (TRALI) and pancreatitis
Lung Tissue Inflammation
Exudative: Neutrophils migrate into the alveoli in response to inflammatory stimulus
Note: While the three phases of ARDS take place in sequence, all areas of the lung may not be in the same phase at the same time. For this reason, the processes can be thought of as overlapping.
Proliferative: Body attempts to heal damage. If it is not successful, the tissue transitions to the fibrotic phase
Neutrophil-containing pulmonary exudate interferes with surfactant function
Neutrophil infiltration and proinflammatory cytokines lead to tissue edema, dysfunction and subsequent destruction of pulmonary epithelium
Abbreviations:
PaO2: Partial pressure of oxygen in arterial blood
SpO2: Peripheral oxygen saturation.
CXR: Chest radiograph.
Residual debris in alveoli are cleared by phagocytic cells
Restoration of alveolar epithelial cells.
Alveoli collapse in absence of working surfactant
Damaged epithelium impairs gas exchange
Pulmonary capillaries do not adequately absorb fluid
The body’s attempts to heal lung tissue result in
deposition of hyaline membranes in the alveoli
Ventilation- Perfusion Mismatch
Pulmonary Edema
Impaired Gas Diffusion
↓ PaO2, ↓SpO2 Tachypnea
Tachycardia
Dyspnea
Bilateral Opacity on CXR
↓ PaO , ↓SpO 2 2
↑ PaCO2
↑ PaO2, ↓PaCO2 Eupnea
↓ O2 Requirements
Clearing of CXR
Depression, Anxiety, PTSD
Neuromuscular Weakness
Chronic Respiratory Dysfunction
↑ useful surface area for gas exchange
Functional epithelium is able to absorb fluid back into circulation
Impaired Function After Prolonged Illness
Fibrotic: Inadequate healing results in long-term pulmonary damage (rare)
Fibroblast activity leads to deposition of collagen in alveoli and alveolar capillaries
Pulmonary Fibrosis
Pulmonary Hypertension
Cough/Dyspnea Nail Clubbing Fatigue
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published February 06, 2018 on www.thecalgaryguide.com")
Bronkiolitis Patogenesis dan temuan klinis

Asma Eksaserbasi Patogenesis dan temuan klinis pada anak

Kelahiran Prematur Tinjauan Komplikasi

Pneumonia Pediatri Patogenesis dan Temuan klinis

Tatalaksana Syok Penjelasan dari mekanisme dasar

Spondilosis Patogenesis dan komplikasi

تشدید آسم حاد بیماریزایی و درمان

آسم بیماریزایی

آسم یافتھ ھای بالینی

آسم یافتھ ھای تحقیقاتی

Approach To Dementia

Authors: Iqra Rahamatullah Mahrukh Kaimkhani
Reviewers: Yvette Ysabel Yao Mao Ding Gary Michael Klein* *MD at time of publication
1) Changes noticed?
Modest ↓cognitive performance from previous, DOES NOT interfere with daily independence
MILD COGNITIVE IMPAIRMENT
More pronounced ↓cognitive performance from previous, DOES interfere with daily independence
MILD TO MODERATE DEMENTIA
↓Cognitive performance, difficulty with ≥1 basic activities of daily living (ADL) or ≥2 instrumental ADLs
MODERATE TO SEVERE DEMENTIA
DEMENTIA
Fluctuating course, acute onset, inattention WITH either disorganized thinking or altered level of consciousness
DELIRIUM
2) Is it dementia?
Normal, age-related: ↓focus, ↓cognitive speed, ↓reaction time, ↓memory
NORMAL COGNITIVE DECLINE
3) What is the cause of the dementia? (main causes discussed here)
Loss of cognitive functioning, including memory, language, problem solving, and other thinking abilities, that interferes with independence in everyday activities
Beta-secretase cleaves beta amyloid protein
Atherosclerosis or thrombosis
Misfolded alpha- synuclein
Toxic beta amyloid plaque and tau tangle (sticky) formation
Ischemia to areas of brain (strokes)
Build ups and deposition within neurons (Lewy bodies)
Disrupted signaling, inflammation, hippocampal and cerebral impairment
Necrosis of brain tissue in areas impacted by strokes
Neuronal impairment and atrophy (especially in substantia nigra)
Neuronal atrophyàfrontal + temporal lobe atrophy
Progressive atrophy of basal ganglia and dorsal striatum + lateral ventricles expanding
Death of dopaminergic neurons in substantia nigra
Alzheimer’s Dementia
Vascular Dementia
Lewy Body Dementia
Frontotemporal Dementia
Huntington’s Disease
Parkinson’s Disease
↓Memory, ↓learning, ↓language skills, disorientation, inattention
Total debilitation, fatal infections
Findings vary depending on area
Step-wise worsening impairment
Parkinsonism, hallucinations, REM- sleep behavior disorder
Total debilitation, dependence
Personality and behavioral changes
Mental status changes
Chorea, ↓cognition, mood changes
Aspiration, dementia, suicide
Resting tremor, rigidity, anosmia
Depression, dementia, falls
Abnormal protein inclusions and tangles (usually tau) form in neurons
Autosomal dominant disease (with anticipation) with ↑CAG repeats in Huntingtin gene
Genetic mutations, environmental exposures, or idiopathic cause
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published September 17, 2023 on www.thecalgaryguide.com
Approach to Dementia/Major Neurocognitive Disorder (NCD)
Authors: Iqra Rahamatullah Mahrukh Kaimkhani Reviewers: Yvette Ysabel Yao
Fluctuating course, acute onset, inattention WITH either disorganized thinking or altered level of consciousness (LOC)?
DELIRIUM
1) Changes noticed?
2) Is it dementia?
Normal, age-related: ↓focus, ↓cognitive speed, ↓reaction time, ↓memory
NORMAL COGNITIVE DECLINE
Modest ↓cognitive performance from previous, DOES NOT interfere with daily independence
MILD COGNITIVE IMPAIRMENT
More pronounced ↓cognitive performance from previous, DOES interfere with daily independence
MILD TO MODERATE DEMENTIA
↓Cognitive performance, difficulty with ≥1 basic activities of daily living (ADL) or ≥2 instrumental ADLs
MODERATE TO SEVERE DEMENTIA
3) What is the cause of the dementia? (main causes discussed here)
Beta-secretase cleaves beta amyloid protein
Atherosclerosis or thrombosis
Misfolded alpha-synuclein
Toxic beta amyloid plaque and tau tangle (sticky) formation
Ischemia to areas of brain (strokes)
Build ups and deposition within neurons (Lewy bodies)
Disrupted signaling, inflammation, hippocampal and cerebral impairment
Necrosis of brain tissue in areas impacted by strokes
Neuronal impairment and atrophy (especially in substantia nigra)
Neuronal atrophyàfrontal + temporal lobe atrophy
Progressive atrophy of basal ganglia and dorsal striatum + lateral ventricles expanding
Death of dopaminergic neurons in substantia nigra
Alzheimer’s Dementia
Vascular Dementia
Lewy Body Dementia
Frontotemporal Dementia
Huntington’s Disease
Parkinson’s Disease
↓Memory, ↓learning, ↓language skills, disorientation, inattention
Total debilitation, fatal infections
Findings vary depending on area
Step-wise worsening impairment
Parkinsonism, hallucinations, REM- sleep behavior disorder
Total debilitation, dependence
Personality and behavioral changes
Mental status changes
Chorea, ↓cognition, mood changes
Aspiration, dementia, suicide
Resting tremor, rigidity, anosmia
Depression, dementia, falls
DEMENTIA
Loss of cognitive functioning, including memory, language, problem solving, and other thinking abilities, that interferes with independence in everyday activities
Abnormal protein inclusions and tangles (usually tau) form in neurons
Autosomal dominant disease (with anticipation) with ↑CAG repeats in Huntingtin gene
Genetic mutations, environmental exposures, or idiopathic cause
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published September 17, 2023 on www.thecalgaryguide.com")
Guillain-Barre Syndrome

Minor triggers
surgery, trauma, bone marrow transplant
Initiate immune response (unknown mechanism)
GI/respiratory infection (1-3 weeks prior) Campylobacter jejuni, cytomegalovirus, HIV, Epstein-Barr Virus
Molecular mimicry: shared ganglioside antigens between peripheral nerve and pathogen coat proteins
IgG antibodies to ganglioside antibodies in serum
Nerve Conduction Study : ↓ conduction velocity, conduction block
Triggered immune response cross-reacts with peripheral nerves, beginning at nerve roots
↑ permeability of blood- nerve barrier at level of proximal nerve roots
Demyelination: antibodies attack Schwann cells
secondary damage
Axonal damage: antibodies attack nodes of Ranvier
Nerve Conduction Study:
↓ CMAP (compound muscle action potential) amplitude, normal conduction velocity
Acute inflammatory demyelinating polyneuropathy (AIDP) (80-90%)
Acute motor axonal neuropathy (AMAN)
Acute motor sensory axonal neuropathy (AMSAN)
Acute immune-mediated polyneuropathy
Tachycardia & Dysrhythmias (Needs cardiac monitoring)
Sudden Death
Dysautonomia: disruption of the autonomic nervous system responsible for involuntary functions
Sensory deficits
Motor deficits
Universal Areflexia (loss of deep tendon reflexes)
Phrenic nerve involvement
Diaphragm paralysis
Cranial Nerve (CN) involvement
Bulbar palsy (CN IX, X, XI,XII)
Oculomotor weakness (CN III, IV, VI)
Eye Movement Abnormalities (Miller Fisher Syndrome – rare form of regionally- restricted AIDP)
BP Fluctuation/ Orthostatic Hypotension (drop of blood pressure from seated/lying to standing)
Urinary Retention (transient, late-course)
Limb Weakness
(legs usually affected first)
Impaired swallowing àaspiration pneumonia
↓ ability to clear airway secretions
Pain & Paresthesia
(in back and extremities)
Respiratory Failure
(Life threatening: needs ventilatory observation and possibly support)
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published Nov 1, 2013, updated Oct 15, 2023 on www.thecalgaryguide.com
Guillain-Barré Syndrome
Minor triggers (surgery, trauma,
GI/respiratory infection
Campylobacter jejuni, CMV, HIV , EBV
(1-3 weeks prior)
Molecular mimicry: shared ganglioside antigens between peripheral nerve and pathogen coat proteins
Author: Nissi Wei Reviewers: Owen Stechishin Matthew Harding Cory Toth* * MD at time of publication
↑ Protein in CSF
bone marrow transplant)
Initiate immune response (unknown mechanism)
IgG antibodies to ganglioside antibodies in serum
NCS: ↓ conduction velocity, conduction block
Triggered immune response cross-reacts with peripheral nerves, beginning at nerve roots
↑ permeability of blood- nerve barrier at level of proximal nerve roots
NCS: ↓ CMAP amplitude, normal conduction velocity
Demyelination: antibodies attack Schwann cells
secondary damage
Axonal damage:
antibodies attack nodes of Ranvier
Acute inflammatory demyelinating polyneuropathy (AIDP) (80-90%)
Acute motor axonal neuropathy (AMAN)
Cranial nerve involvement
Dysautonomia
Acute motor sensory axonal neuropathy (AMSAN)
Eye Movement Abnormalities
(Miller Fisher Syndrome – rare form of regionally-restricted AIDP)
Acute immune-mediated polyneuropathy
Oculomotor weakness (CN III, IV, VI)
Bulbar palsy (CN IX, X, XI,XII)
Phrenic nerve involvement
↓ ability to clear airway secretions
Impaired swallowingà aspiration pneumonia
Diaphragm paralysis
Respiratory Failure
(Life threatening: needs ventilatory observation and possibly support)
Motor deficits
Sensory deficits
Pain & Paresthesias in back and extremities
Limb Weakness
(legs usually affected first)
Universal Areflexia
Urinary Retention
(transient, late-course)
Sudden Death
BP Fluctuation, Orthostatic Hypotension
Tachycardia, Dysrhythmias (Needs cardiac monitoring)
Abbreviations:
• NCS - nerve conduction
study
• CMAP - compound muscle
action potential
• EBV - Epstein-Barr Virus
• CMV - cytomegalovirus
• CN - cranial nerve
Note: Aα, Aβ peripheral nerve fibres (large, fast-conducting, heavily myelinated axons for muscle stretch, light touch & proprioception) are more affected than Aδ and C fibres (small, less myelinated, slowly-conducting fibres for pain and temperature)
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published November 1, 2013 on www.thecalgaryguide.com")
Perdarahan Intraventrikuler pada Bayi Prematur Patogenesis

Pulmonary Embolism Pathogenesis and Clinical Findings

Authors: Mackenzie Gault Mao Ding Reviewers: Midas (Kening) Kang Usama Malik Kevin Solverson* *MD at time of publication
Hypercoagulable State
Blood clot develops (commonly in deep veins of legs)
Venous stasis
= Deep Vein Thrombus (95% of PE)
Vessel injury
Ultrasound:
Presence of Clot in Deep Vein of Leg
Clot dislodges & migrates to inferior vena cava (IVC)àright atrium of heartàright ventricleàlodges in pulmonary arteries/arterioles
Pulmonary Embolism (PE)
Thromboembolic blockage of pulmonary vasculature
↓ perfusion to lung parenchyma
Clot occludes pulmonary arteries/ arterioles
↑ dead space ventilation and V/Q mismatching
Blood pumped from RV to pulmonary arteries cannot pass clot
↑ pulmonary and right ventricle (RV) pressure
RV Strain
↑ RV workload, ↓ right coronary artery perfusion
Computed Tomography- Pulmonary Angiogram (CTPA): Filling Defect (*see Radiology slide for CTPA findings)
Echo: ↑ RV size + ↓ RV function
ECG: S1Q3T3 Pattern (McGinn-White Sign: a large S wave in lead I, a Q wave in lead III and an inverted T wave in lead III together indicate acute right heart strain
Lab: ↑ Brain natriuretic peptide (BNP)
Chest Pain Tachycardia
Dyspnea
(shortness of breath)
Pleuritic chest pain (worsens during breathing)
Ischemia of lung tissue distal to clot
X-Ray: usually normal, except Hampton’s Hump (↑ opacity in pleural based area), a rare but specific sign of PE)
Air flow/ventilation to lungs unaffected
VQ Scan (performed when CT contrast is contraindicated): Ventilation- perfusion ratio (V/Q) mismatch
Chemoreceptors detect ↑ CO2 and ↓ O2
Signal brain to ↑ breathing rate
Tachypnea (rapid breathing)
↓ Arterial O2
Lab:
↑Troponin
BP:
Hypotension
Lab:
↑ Lactate
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published Feb 7, 2018, updated Oct 15, 2023 on www.thecalgaryguide.com
Pulmonary Embolism: Pathogenesis and Laboratory Findings Virchow’s Triad
Authors: Mackenzie Gault Reviewers: Midas (Kening) Kang Usama Malik Kevin Solverson * * MD at time of publication
Body attempts to break down clot
Fibrinogen breakdown products in blood
Positive D-Dimer
↓ perfusion to lung parenchyma
Vessel injury = Deep Vein Thrombus (95% of PE)
Ultrasound:
Presence of Clot in Deep Vein of Leg
Notes:
Hypercoagulable State Venous stasis
Blood clot develops (commonly in deep veins of legs)
Clot dislodges, migrates to IVCàright atrium of heartà right ventricleàlodges in pulmonary artery
Pulmonary Embolism (PE):
Thromboembolic blockage of pulmonary vasculature
Clot occludes pulmonary artery/ arterioles
• D-Dimer is only performed if clinical suspicion of PE low (Well’s Criteria)
• CT-PA is the current diagnostic test for PE
• V/Q Scan is performed when CT contrast is contraindicated
• X-Ray is usually normal in PE (Except Hampton’s Hump, a rare but specific sign of PE)
Ischemia of lung tissue distal to clot
X-Ray:
Hampton’s Hump pleural based area of ↑ opacity
Air flow/ ventilation to lungs unaffected
Pleuritic Chest Pain + Dyspnea
VQ Scan: V/Q Mismatch
↑ dead space ventilation and V/Q mismatching
Chemoreceptors detect ↑ CO2 and ↓ O2
Signal brain to ↑ breathing rate
Tachypnea
↓ Arterial O2
Blood pumped from RV to pulmonary arteries cannot pass clot
↑ pulmonary and RV pressure
RV Strain
CT-PA: Filling Defect
Echo: ↑ RV size + ↓ RV Function
ECG: S1Q3T3 Pattern
Lab:↑ BNP Chest Pain Tachycardia
↑ RV work load, ↓ right coronary artery perfusion
Abbreviations:
• BNP – Brain Natriuretic Peptide
• CT-PA – Computed Tomography-Pulmonary Angiogram
• ECG – Electrocardiogram
• IVC – Inferior Vena Cava
• RV – Right Ventricle
• V/Q – Ventilation-Perfusion ratio
↑ Troponin
Hypotension
↑ Lactate
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published February 07, 2018 on www.thecalgaryguide.com")
Mitral Regurgitation Pathogenesis and clinical findings

Myocarditis
Left ventricular dilation displaces papillary muscles
Dilation of the Tethering of mitral valve annulus chordae tendineae
↑ Volume and pressure in left atrium
↑ Volume pushed back into left ventricle
Dilated left ventricle
Apical impulse on palpation and auscultation
↓ Forward flow of blood out of heart
Blood backs up into pulmonary circulation
↑ Intravascular hydrostatic pressure in pulmonary vessels
Fluid extravasates out of vessels and into the lungs
Papillary muscle rupture
Mitral valve leaflets flail
Mitral valve prolapse
Structurally abnormal valve
Connective tissue disorders
Weak valve leaflets
Rheumatic heart disease
Dilatation of the mitral valve annulus, inflammation of leaflets
Infective endocarditis
Vegetations form on valve leaflets
Mitral Regurgitation
Blood consistently flows backward throughout systole
Holosystolic murmur, radiates to axilla, ↑ with afterload (e.g. making a fist)
Backflow of blood from left ventricle to left atrium due to impaired mitral valve closure
S3 heart sound
Myocardial remodeling
↓ Muscle efficiency
↓ Left ventricle systolic function
↓ O2 saturation, tachypnea, wheeze, ↑ work of breathing, crackles, frothy sputum (if severe)
Congestive heart failure
↓ Stroke volume ejected into aorta
↓ Cardiac output
↓ Organ perfusion ↓ O2 to kidney
Activation of renin-angiotensin- aldosterone system
↑ Reabsorption of water by kidneys
↑ Intravascular hydrostatic pressure systemically
Peripheral edema
Injury to kidney parenchyma
↓ ability for kidney to clear creatinine
↑ Serum creatinine
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published Feb 3, 2018, updated Oct 15, 2023 on www.thecalgaryguide.com")
Alcohol Withdrawal Syndrome
: Pathophysiology & clinical findings
Authors: Rupali Manek Gurreet Bhandal Erika Russell Reviewers: Harjot Atwal Yvette Ysabel Yao Mao Ding Nureen Pirbhai* * MD at time of publication
Chronic alcohol use
↓Autonomic adrenergic systems
↓ Dopamine in the nucleus accumbens
↑ EtOH depressant effects on brain
↓ Glutamate- induced excitation
↑ GABA-induced inhibition
↑ Glutamate receptors in attempt to maintain normal arousal state
GABA insensitivity (↑ GABA needed to maintain a constant inhibitory tone)
Long-term physical dependence
Abrupt alcohol cessation àabrupt ↓ in blood EtOH concentration
Alcohol withdrawal Syndrome
Symptoms that occur when patients stop drinking or significantly decrease their alcohol intake after long-term dependence
↓ GABA-induced inhibition &↑ glutamate-induced excitation relative to chronic alcohol use
Central nervous system overactivity
Withdrawal seizures: Generalized tonic-clonic convulsions 24-48 hours after alcohol cessation
Fluid and electrolyte abnormalities
↑ Autonomic adrenergic systems (rebound over-activity of the brain and noradrenergic systems)
↑ Sympathetic activity: ↑ Heart rate (HR), ↑respiratory rate (RR) ↑blood pressure (BP), tremor & diaphoresis (↑sweating)
Early symptoms: Insomnia, tremulousness, anxiety, digestive upset, anorexia, headache, sweating, palpitations 6-12 hours after alcohol cessation
↑ Dopamine in nucleus accumbens
Alcoholic hallucinosis: Usually visual (but can be auditory or tactile), normal vitals 12-24 hours after alcohol cessation, typically resolve within 24-48 hours
Alcohol withdrawal delirium (delirium tremens or DT):
Hallucinations (mostly visual), disorientation, tachycardia, hypertension, hyperthermia, agitation, and diaphoresis 48-96 hours after alcohol cessation and lasts 1-5 days
Hypovolemia
(from diaphoresis, hyperthermia, vomiting, ↑RR & ↓oral intake)
Metabolic acidosis
(from hypoperfusion, infection, alcoholic ketoacidosis, or ↓
thiamine & other B vitamins)
↓ Potassium (K) (renal & extrarenal K losses, alterations in aldosterone concentrations, and changes in K distribution across the cell membrane)
↓ Phosphate
(from malnutrition)
Cardiac failure, rhabdomyolysis or muscle breakdown
↓ Phosphate available to make ATP
↓ ATP
↓ Magnesium (common in patients with DT)
Impaired Na-K ATPase function
Dysrhythmias
↑ Glutamate-activated depolarization in the brain
↑ Neuronal excitability
Seizures
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published Apr 15, 2017, updated Oct 18, 2023 on www.thecalgaryguide.com
Alcohol Withdrawal Syndrome (AWS): Pathophysiology & clinical findings
Authors: Rupali Manek Gurreet Bhandal Erika Russell Reviewers: Harjot Atwal Yvette Ysabel Yao Mao Ding Nureen Pirbhai* * MD at time of publication
Chronic alcohol use
↓Autonomic adrenergic systems
↓ Dopamine in the nucleus accumbens
↑ EtOH depressant effects on brain
↓ Glutamate- induced excitation
↑ GABA-induced inhibition
↑ Glutamate receptors in attempt to maintain normal arousal state
GABA insensitivity (↑ GABA needed to maintain a constant inhibitory tone)
Long-term physical dependence
Abrupt alcohol cessation àabrupt ↓ in blood EtOH concentration
Alcohol withdrawal Syndrome
Symptoms that occur when patients stop drinking or significantly decrease their alcohol intake after long-term dependence
↓ GABA-induced inhibition &↑ glutamate-induced excitation relative to chronic alcohol use
Central nervous system overactivity
Withdrawal seizures: Generalized tonic-clonic convulsions 24-48 hours after alcohol cessation
Fluid and electrolyte abnormalities
↑ Autonomic adrenergic systems (rebound over-activity of the brain and noradrenergic systems)
↑ Sympathetic activity: ↑ Heart rate (HR), ↑respiratory rate (RR) ↑blood pressure (BP), tremor & diaphoresis (↑sweating)
Early symptoms: Insomnia, tremulousness, anxiety, digestive upset, anorexia, headache, sweating, palpitations 6-12 hours after alcohol cessation
↑ Dopamine in nucleus accumbens
Alcoholic hallucinosis: Usually visual (but can be auditory or tactile), normal vitals 12-24 hours after alcohol cessation, typically resolve within 24-48 hours
Alcohol withdrawal delirium (delirium tremens or DT):
Hallucinations (mostly visual), disorientation, tachycardia, hypertension, hyperthermia, agitation, and diaphoresis 48-96 hours after alcohol cessation and lasts 1-5 days
Hypovolemia
(from diaphoresis, hyperthermia, vomiting, ↑RR & ↓oral intake)
Metabolic acidosis
(from hypoperfusion, infection, alcoholic ketoacidosis, or ↓ thiamine & other B vitamins)
↓ Potassium (K) (renal & extrarenal K losses, alterations in aldosterone concentrations, and changes in K distribution across the cell membrane)
↓ Magnesium (common in patients with DT)
Dysrhythmias, seizures
↓ Phosphate
(from malnutrition)
↓ Phosphate available to make ATP
↓ ATP
Cardiac failure, rhabdomyolysis or muscle breakdown
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published April 28th, 2014 on www.thecalgaryguide.com
Alcohol Withdrawal: Pathophysiology & clinical findings
Authors: Erika Russell Rupali Manek Gurreet Bhandal Reviewers: Yvette Ysabel Yao Harjot Atwal Nureen Pirbhai* * MD at time of publication
↓Autonomic adrenergic systems
↓ Glutamate-induced excitation
Chronic alcohol use
↑ EtOH depressant effects on brain
↓ Dopamine in the nucleus accumbens
↑ GABA-induced inhibition
↑ Glutamate receptors in attempt to maintain normal arousal state GABA insensitivity (↑ GABA needed to maintain a constant inhibitory tone)
↑ Autonomic adrenergic systems (rebound over- activity of the brain and noradrenergic systems)
↑ Sympathetic activity: ↑ Heart rate (HR), ↑respiratory rate (RR) ↑blood pressure (BP), tremor & diaphoresis (↑sweating)
Physical dependence due to chronic alcohol use
Abrupt alcohol cessation
Abrupt ↓ in blood EtOH concentration
Alcohol withdrawal
↓ GABA-induced inhibition &↑ glutamate-induced excitation relative to chronic alcohol use
Central nervous system overactivity
Withdrawal seizures: Generalized tonic-clonic convulsions 24-48 hours after alcohol cessation
Fluid and electrolyte abnormalities
↑ Dopamine in nucleus accumbens
Alcoholic hallucinosis: Usually visual (but can be auditory or tactile), normal vitals
12-24 hours after alcohol cessation, typically resolve within 24-48 hours
Early symptoms: Insomnia, tremulousness, anxiety, digestive upset, anorexia, headache, sweating, palpitations 6-12 hours after alcohol cessation
Alcohol withdrawal delirium (delirium tremens or DT):
Hallucinations (mostly visual), disorientation, tachycardia, hypertension, hyperthermia, agitation, and diaphoresis 48-96 hours after alcohol cessation and lasts 1-5 days
Hypovolemia (from diaphoresis, hyperthermia, vomiting,
↑RR & ↓oral intake)
Metabolic acidosis (from
hypoperfusion, infection, alcoholic ketoacidosis, or ↓ thiamine & other B vitamins)
↓ Potassium (K) (renal & extrarenal K losses, alterations in aldosterone concentrations, and changes in K distribution across the cell membrane)
↓ Magnesium (common in patients with DT)
Dysrhythmias, seizures
↓ Phosphate (from malnutrition)
↓ Phosphate available to make ATP
↓ ATP
Cardiac failure, rhabdomyolysis or muscle breakdown
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published April 28th, 2014 on www.thecalgaryguide.com
Authors: Erika Russell Rupali Manek Gurreet Bhandal Reviewers: ↓Autonomic adrenergic systems ↓ Dopamine in the nucleus accumbens Yvette Ysabel Yao Harjot Atwal Nureen Pirbhai* * MD at time of publication
Alcohol Withdrawal: Pathophysiology & clinical findings Chronic alcohol use
↑ EtOH depressant effects on brain
↓ Glutamate-induced excitation ↑ GABA-induced inhibition
↑ Glutamate receptors in attempt to maintain normal arousal state GABA insensitivity (↑ GABA needed to maintain a constant inhibitory tone)
↑ Autonomic adrenergic systems (rebound over- activity of the brain and noradrenergic systems)
↑ Sympathetic activity: ↑ Heart rate (HR), ↑respiratory rate (RR) ↑blood pressure (BP), tremor & diaphoresis (↑sweating)
Physical dependence due to chronic alcohol use
Abrupt alcohol cessation
Abrupt ↓ in blood EtOH concentration Alcohol withdrawal
↓ GABA-induced inhibition &↑ Glutamate-induced excitation relative to chronic alcohol use
Central nervous system overactivity
Withdrawal seizures: Generalized tonic-clonic convulsions 24-48 hours after alcohol cessation
Fluid and electrolyte abnormalities
↓ Potassium (K) (renal & extrarenal K losses, alterations in aldosterone concentrations, and changes in K distribution across the cell membrane)
↑ Dopamine in nucleus accumbens
Alcoholic hallucinosis: Usually visual (but can be auditory or tactile), normal vitals
12-24 hours after alcohol cessation, typically resolve within 24-48 hours
Alcohol withdrawal delirium (delirium tremens or DT):
Hallucinations (mostly visual), disorientation, tachycardia, hypertension, hyperthermia, agitation, and diaphoresis 48-96 hours after alcohol cessation and lasts 1-5 days
Early symptoms: Insomnia, tremulousness, anxiety, digestive upset, anorexia, headache, sweating, palpitations 6-12 hours after alcohol cessation
Hypovolemia (from diaphoresis, hyperthermia, vomiting, ↑RR & ↓oral intake)
Metabolic acidosis (from
hypoperfusion, infection, alcoholic ketoacidosis, or ↓ thiamine & other B vitamins)
↓ Magnesium (common in patients with DT)
Dysrhythmias, seizures
↓ Phosphate (from malnutrition) Cardiac failure, rhabdomyolysis
or muscle breakdown
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published April 28th, 2014 on www.thecalgaryguide.com
Alcohol Withdrawal: Pathophysiology & clinical findings Chronic alcohol use
↓ Dopamine in the nucleus accumbens ↓Autonomic adrenergic systems
Authors: Erika Russell Rupali Manek Gurreet Bhandal Reviewers: Yvette Ysabel Yao Harjot Atwal ↑ GABA-induced inhibition Nureen Pirbhai* * MD at time of publication
↑ EtOH depressant effects on brain
↓ Glutamate-induced excitation
↑ Glutamate receptors in attempt to maintain normal arousal state
GABA insensitivity (↑ GABA needed to maintain a constant inhibitory tone)
↑ Autonomic adrenergic systems (rebound over- activity of the brain and noradrenergic systems)
↑ Sympathetic activity: ↑Heart rate (HR), ↑respiratory rate (RR) ↑blood pressure (BP), tremor & diaphoresis (↑sweating)
Physical dependence due to chronic alcohol use
Abrupt alcohol cessation
Abrupt ↓ in blood EtOH concentration Alcohol withdrawal
↓ GABA-induced inhibition &↑ Glutamate-induced excitation relative to chronic alcohol use
Central nervous system overactivity
Withdrawal seizures: Generalized tonic-clonic convulsions 24-48 hours after alcohol cessation
Fluid and electrolyte abnormalities
↑ Dopamine in nucleus accumbens
Alcoholic hallucinosis: Usually visual (but can be auditory or tactile), normal vitals
12-24 hours after alcohol cessation, typically resolve within 24-48 hours
Alcohol withdrawal delirium (delirium tremens or DT):
Hallucinations (mostly visual), disorientation, tachycardia, hypertension, hyperthermia, agitation, and diaphoresis 48-96 hours after alcohol cessation and lasts 1-5 days
Early symptoms: Insomnia, tremulousness, anxiety, digestive upset, anorexia, headache, sweating, palpitations 6-12 hours after alcohol cessation
Hypovolemia (from diaphoresis, hyperthermia, vomiting, ↑RR & ↓oral intake)
Metabolic acidosis (from
hypoperfusion, infection, alcoholic ketoacidosis, or ↓ thiamine & other B vitamins)
↓ Potassium (K) (renal & extrarenal K losses, alterations in aldosterone concentrations, and changes in K distribution across the cell membrane)
↓ Magnesium
(common in patients with DT)
Dysrhythmias, seizures
↓ Phosphate (from malnutrition)
Cardiac failure, rhabdomyolysis or muscle breakdown
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published April 28th, 2014 on www.thecalgaryguide.com
Alcohol Withdrawal: Pathophysiology & clinical findings
Authors: Erika Russell Rupali Manek Gurreet Bhandal Reviewers: Yvette Ysabel Yao Harjot Atwal Nureen Pirbhai* * MD at time of publication
↑ EtOH depressant effects on brain
↓ Glutamate-induced excitation
↑ Glutamate receptors in attempt to maintain normal arousal state
Chronic alcohol use
↑ GABA-induced inhibition
GABA insensitivity (↑ GABA needed to maintain a constant inhibitory tone)
↓ Dopamine in the nucleus accumbens ↓Autonomic adrenergic systems
Abrupt alcohol cessation
Physical dependence due to chronic alcohol use
Alcohol withdrawal
↓ GABA-induced inhibition &↑ Glutamate-induced excitation relative to chronic alcohol use
Central nervous system overactivity
Withdrawal seizures: Generalized tonic-clonic convulsions 24-48 hours after alcohol cessation
↑ Autonomic adrenergic systems (rebound over-activity of the brain and noradrenergic systems)
↑ Sympathetic activity: ↑HR, ↑RR ↑BP, tremor & diaphoresis (↑sweating)
Early symptoms: Insomnia, tremulousness, anxiety, digestive upset, anorexia, headache, sweating, palpitations
6-12 hours after alcohol cessation
↑ Dopamine in nucleus accumbens
Alcoholic hallucinosis: Usually visual (but can be auditory or tactile), normal vitals
12-24 hours after alcohol cessation, typically resolve within 24-48 hours
Alcohol withdrawal delirium (delirium tremens or DT):
Hallucinations (predominately visual), disorientation, tachycardia, hypertension, hyperthermia, agitation, and diaphoresis 48-96 hours after alcohol cessation and lasts 1-5 days
Fluid and electrolyte abnormalities
Hypovolemia (from diaphoresis, hyperthermia, vomiting, ↑ RR & ↓
oral intake)
Metabolic acidosis (from
hypoperfusion, infection, alcoholic ketoacidosis, or ↓ thiamine & other B vitamins)
↓ Potassium (K) (renal & extrarenal K losses, alterations in aldosterone concentrations, and changes in K distribution across the cell membrane)
↓ Magnesium
(common in patients with DT)
Dysrhythmias, seizures
↓ Phosphate (from malnutrition)
Cardiac failure, rhabdomyolysis or muscle breakdown
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published April 28th, 2014 on www.thecalgaryguide.com
Alcohol Withdrawal: Pathophysiology & clinical findings
Authors: Erika Russell Rupali Manek Gurreet Bhandal Reviewers: Yvette Ysabel Yao Harjot Atwal Nureen Pirbhai* * MD at time of publication
↓ GABA-induced inhibition &↑ Glutamate-induced excitation relative to chronic alcohol use
CNS overactivity
Alcohol withdrawal delirium (delirium tremens or DT): Hallucinations (predominately visual), disorientation, tachycardia, hypertension, hyperthermia, agitation, and diaphoresis
48-96 hours after alcohol cessation and lasts 1-5 days
Alcohol cessation ↓ Dopamine
Alcohol withdrawal
Chronic alcohol use
↑ EtOH depressant effects on brain
in the NAc
↓Autonomic adrenergic systems
↑ Dopamine in nucleus accumbens
(NAc) relative to chronic alcohol use
Alcoholic hallucinosis: Usually visual (but can be auditory or tactile), normal vitals 12-24 hours after alcohol cessation, typically resolve within 24-48 hours
↑ Autonomic adrenergic systems relative to chronic alcohol use
↑ Sympathetic activity: ↑HR, ↑RR ↑BP, tremor & diaphoresis (↑sweating)
↓ Glutamate- induced excitation
↑ Glutamate receptors in attempt to maintain normal arousal state
↑ GABA-induced inhibition
GABA insensitivity (↑ GABA needed to maintain a constant inhibitory tone)
Early symptoms: Insomnia, tremulousness, anxiety, digestive upset, anorexia, headache, sweating, palpitations
6-12 hours after alcohol cessation
Withdrawal seizures: Generalized tonic- clonic convulsions 24-48 hours after alcohol cessation
Fluid and electrolyte abnormalities
Abbreviations:
CNS – Central nervous system K – Potassium
DT – Delirium tremens
Mg – Magnesium
NAc – Nucleus accumbens NMDA – N-methyl-D-aspartate EtOH – Alcohol
NT – Neurotransmitter
Hypovolemia (from diaphoresis, hyperthermia, vomiting, ↑ RR & ↓
oral intake)
Metabolic acidosis (from hypoperfusion, infection, alcoholic ketoacidosis, or ↓
thiamine & other B vitamins)
↓ K (renal & extrarenal K losses, alterations in aldosterone concentrations, and changes in K distribution across the cell membrane)
↓ Mg (common in patients with DT)
Dysrhythmias, seizures
↓ Phosphate (from malnutrition)
Cardiac failure, rhabdomyolysis or muscle breakdown
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published April 28th, 2014 on www.thecalgaryguide.com
Alcohol Withdrawal: Clinical Findings and Complications
Authors: Erika Russell Reviewers: Harjot Atwal Nureen Pirbhai* * MD at time of publication
Note: *The onset of alcohol withdrawal generally begins 6-24 hours after the last drink with symptoms peaking between 24-36 hours after and gradually lessening.
Symptoms typically progress from early symptoms and increased sympathetic activityà hallucinationsàseizures àpotentially Delirium Tremens.
Alcohol withdrawal is mild-moderate in severity for 90% of patients. Those who progressively worsen however can enter Delirium Tremens (DT) which has a mortality rate of up to 20%. More likely to have DT if had DT before, age >30 years, concurrent illness, >2 days after EtOH cessation before seeking help, and history of sustained drinking.
Long term, heavy alcohol use that leads to physical dependence
Abrupt ↓ in blood EtOH concentration
Negative physiological reactions to ↓ alcohol intake
Adaptive suppression of GABA activity from chronic alcohol enhancement
Alcohol Withdrawal*
Withdrawal symptoms alleviated by ingesting alcohol
Alcohol taken to relieve withdrawal AND/OR
Social and internal relapse cues trigger urge to use alcohol
Blood EtOH levels >600 mg% can lead to lethal respiratory depression by suppressing the respiratory centers in the brainstem
Upregulated autonomic adrenergic systems from chronic alcohol inhibition
Discontinuation of alcohol leads to rebound over-
activity of the brain and noradrenergic systems
Increased Sympathetic Activity Tachycardia, hypertension, tremor and diaphoresis
Generalized Tonic-Clonic Seizures
Usually begin within 8-24 hours of alcohol cessation and peak after 24 hours. Risk of having seizures ↑ with repeated withdrawals. 1/3 of people can progress to DT if seizures left untreated.
Discontinuation of alcohol causes a sudden relative
deficiency in inhibitory GABA activity
Reduction in dopamine in the nucleus accumbens
(NAc) from chronic alcohol exposure
Discontinuation of alcohol causes a increase in dopamine levels in NAc
Hallucinations
Commonly visual (but can be auditory or tactile), develop 12-24 hours after alcohol cessation.
Early Symptoms
Anxiety, insomnia, vivid dreams, anorexia, nausea, headache and psychomotor agitation
Delirium Tremens (DT)
Life-threatening state of greatly exaggerated withdrawal symptoms (severe tachycardia, diaphoresis etc.) with confusion/disorientation and hallucinations that generally appears 72-96 hours after the last drink and lasts 2-3 days.
Legend:
Published MONTH, DAY, YEAR on www.thecalgaryguide.com
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications")
Sustained Monomorphic Ventricular Tachycardia Pathogenesis
 A ventricular scar forms (in the setting of coronary artery disease or cardiomyopathy) that cannot conduct electrical activity
The scar is surrounded by a circular conduction pathway consisting of an ⍺- limb (slow conduction with a fast refractory period) and a β-limb (fast conduction but a slow refractory period)
A correctly timed depolarization impulse arrives during the refractory period of the β-limb so it can only propagate through the ⍺-limb
The β-limb’s refractory period ends just before the impulse leaves the ⍺-limb of the circular pathway
Retrograde depolarization occurs into the ⍺ -limb, creating a self-sustaining closed- loop circuit within the ventricle
“Re-entry” cause of tachyarrhythmia
Idiopathic Causes: (10% of cases) Structurally normal heart on imaging
Trigger(s) such as catecholamines ↑ cyclic adenosine monophosphate
Intracellular calcium overload occurs in some ventricular myocytes
↑ Intracellular calcium activates sodium- calcium exchangers
Sodium influx into the myocytes
During normal myocyte repolarization, the net calcium-mediated depolarization reaches the myocyte threshold for an action potential
A triggered action potential (termed a “delayed afterdepolarization”) repeatedly occurs within the ventricle
“Triggered activity” cause of tachyarrhythmia
Authors:
Rahim Kanji
Reviewers:
Stephanie Happ, Raafi Ali, Derek Chew*
* MD at time of publication
Sustained Monomorphic Ventricular Tachycardia
A wide QRS complex tachycardia originating from the ventricles lasting > 30 seconds. Common
mechanisms include re-entry (e.g., scar-mediated) or a ventricular ectopic focus with increased automaticity. Refer to Sustained Monomorphic Ventricular Tachycardia: Clinical findings for details
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published October 22, 2023 on www.thecalgaryguide.com")
Sustained Monomorphic Ventricular Tachycardia Clinical findings
 or a ventricular ectopic focus with increased automaticity. Refer to Sustained Monomorphic Ventricular Tachycardia: Pathogenesis slide for more details.
Authors: Rahim Kanji Reviewers: Stephanie Happ, Raafi Ali, Derek Chew* * MD at time of publication
The sinoatrial node continues to depolarize the atria while the ventricles depolarize independently and more rapidly
Heart rate > 100 beats per minute
The re-entrant circuit/ectopic focus uniformly and consistently depolarizes ventricular myocytes
Occasionally, a sinoatrial impulse conducts to the ventricles
Loss of coordination between the contractions of the atria and ventricles
ECG Finding: AV Dissociation
The impulse conducts normally through the His-Purkinje pathway and coincides with abnormal ventricular depolarization
ECG Finding: Fusion beat
Patient feels a forceful and rapid heart rate
Palpitations
Right atrium periodically contracts against a closed tricuspid valve
Cannon A waves (intermittent irregular jugular venous pulsations with large amplitudes)
↓ Ventricular filling time
↓ Preload
↓ Stroke volume cardiac output
Inadequate perfusion to organs
ECG Finding: Uniform morphology of QRS complexes
Direct myocyte-to- myocyte spread of the electrical impulse proceeds slower than an impulse conducted via the His-Purkinje pathway
ECG Finding: Wide QRS complexes (≥ 120 milliseconds)
The impulse conducts normally through the His-Purkinje pathway in between abnormal ventricular depolarizations
ECG Finding: Capture beat
Muscles and other organs
General malaise
Heart
Chest pain
Brain
Presyncope/ syncope
Inability to adequately respond to increased cardiac demand
Shortness of breath
Hemodynamic collapse
Sudden cardiac arrest
Death
Hypotension
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published October 22, 2023 on www.thecalgaryguide.com")
Limfoma Hodgkin: Patogenesis dan Temuan Klinis

Statins Mechanisms and Side Effects

↓ Coenzyme Q10 (ubiquinone)
↑ Mitochondrial superoxide
↓ Hepatic membrane stability
↓ Conversion of HMG-CoA to mevalonic acid
↑ Hepatic VLDL uptake
↓ Hepatic apolipoprotein B-100 secretion
↓ Oxidative phosphorylation
Impaired mitochondrial function
↑ Liver enzyme leakage
↓ Mitochondrial ATP production
↑ Aminotransferases enzymes in liver
↑ Clearance of
LDL cholesterol from bloodstream
Myopathy/ myalgias (muscle aches)
Hepatotoxicity
↓ Circulating LDL cholesterol
↓ LDL, ↑ HDL, ↓ TG
↓ Hepatic cholesterol synthesis
↓ VLDL synthesis
↑ Cell surface LDL receptor expression
Statins
First line therapy for treating hypercholesterolemia (↑ LDL cholesterol in blood) Common examples: rosuvastatin, atorvastatin, simvastatin, pravastatin, etc.
↑ Apolipoprotein AI production & ↑ hepatic HDL neogenesis
↓ TG
↑ HDL
↑ Vasodilation
↓ C-reactive protein
↓ Impacts of coagulation cascade
↓ Atherosclerosis (plaque along walls of blood vessels)
↓ Cardiovascular disease & mortality
Pleotropic effects (i.e. non lipid related effects)
Abbreviations:
HMG-CoA – Hydroxymethylglutaryl-CoA LDL – Low-density lipoprotein
VLDL – Very low density lipoprotein HDL – High density lipoprotein
TG – Triglycerides
Inhibition of synthesis of isoprenoid intermediates in the mevalonate pathway
↑ Nitric oxide activity ↓ Inflammation
↓ Tissue factor expression
↓ Macrophage proliferation
↓ Tissue factor (promotes macrophage mediated thrombus formation)
↑ Blood flow & endothelial function
↓ Thrombin generation
↓ Metalloproteinases expression
↑ Inhibition of metalloproteinase-1
↓ Thrombogenicity (production of blood clot/thrombus)
Plaque stabilization (↓ risk of atherosclerotic plaque rupture, myocardial infarction, and stroke)
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Physiological Outcome
Published July 9, 2017, updated Nov 6, 2023 on www.thecalgaryguide.com")
Pharmacotherapy for Dyslipidemia Overview

Bile-acid sequestrants
Bind bile acids in intestinal lumen to prevent reabsorption by enterohepatic (gut-liver) circulation
↑ Excretion of bile acids and cholesterol in stool
↓ LDL in blood
Side effects: GI disturbances, commonly interact with other drugs by interfering with absorption
See Calgary guide slide on “Bile-acid sequestrants: Mechanisms of action & side effects” for complete description of mechanism and side effects
(clinical imbalance of lipids)
Hypertriglyceridemia (if VLDL mediated & in need of treatment for pancreatitis prevention)
Hypercholesteremia
(↑ LDL in blood)
Ezetimibe
Inhibits cholesterol absorption via NPC1L1 transporter
↑ Hepatic (liver) LDL receptor expression
↑ LDL clearance from blood
↓ LDL in blood
Avoid in pregnancy
See Calgary guide slide on “Ezetimibe: Mechanisms of action & side effects” for complete description of mechanism and side effects
Combined hyperlipidemia (↑ Triglycerides and ↑ cholesterol)
Statins (ex. rosuvastatin, atorvastatin, simvastatin, pravastatin)
Competitive inhibitors of HMG-CoA reductase (rate-limiting enzyme in cholesterol synthesis)
Fibrates (ex. fenofibrate, gemfibrozil)
Activate PPAR! (nuclear receptor)
↑ Lipolysis (breakdown of lipids) and free fatty acid oxidation
↓ Triglycerides in blood
Side effects: GI discomfort, rash, pruritis
Contraindicated in pregnancy, renal failure, liver & gallbladder disease
See Calgary guide slide on “Fibrates: Mechanisms of action & side effects” for complete description of mechanism and side effects
PCSK9 inhibitors (ex. evolocumab and alirocumab which are monoclonal antibodies)
Inhibit PCSK9 (holds the LDL:LDL receptor complex together as it is internalized into the cell for destruction of LDL)
LDL receptor returns to surface without being destroyed
↑ LDL receptor expression
↑ LDL clearance from blood
↓ LDL in blood
See Calgary guide slide on “PCSK9 Inhibitors: Mechanisms of action & side effects” for complete description of mechanism and side effects
↓ Cholesterol synthesis in liver
↑ LDL receptor expression in liver
LDL receptor recognizes apoB100 (structural protein on LDL) and apoE (structural protein found on chylomicron, VLDL, IDL)
↑ Clearance of LDL cholesterol from bloodstream
↓ LDL cholesterol in blood ↑ HDL in blood
↓ Triglycerides in blood
↓ Atherosclerosis (plaque along walls of blood vessels)
Abbreviations: HDL – High density lipoprotein; HMG-CoA – Hydroxymethylglutaryl- CoA; LDL – Low-density lipoprotein; PCSK9 – Proprotein convertase subtilisin/kexin type 9; PPAR! – Peroxisome proliferator-activated receptor alpha; NPC1L1 – Niemann-Pick C1-Like 1; VLDL – Very low-density lipoprotein
Side effects: Myalgias (muscular aches), rhabdomyolysis (muscle breakdown), transaminitis (liver inflammation), liver failure, ↑ risk of diabetes mellitus
Contraindicated in pregnancy
See Calgary guide slide on “Statins: Mechanisms of action & side effects” for complete description of mechanism and side effects
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published Nov 6, 2023 on www.thecalgaryguide.com")
Anesthetic Considerations for Obese Patients

Pre-oxygenate to ↑ oxygen reserve during intubation
Avoid supine positioning, in place of alternate positioning (i.e., reverse trendelenburg)
Lung-protective ventilation (↓ tidal volume, optimize oxygen levels, positive end
expiratory pressure & recruitment maneuvers)
Pre-operative fasting, gastric ultrasound to assess volume
Rapid sequence induction to reduce aspiration risk
Adjust drug dosages based on individual recommendations to account for altered distribution, metabolism & clearance
Excess body fat on chest wall
Excess intra- abdominal fat
↑ Gastric volume
Excess body fat
↑ Circulating blood volume
Obesity- related restrictive lung disease
↑ Load compressing chest wall
BMI
≥30
kg/m2
Note: effects vary with the severity of obesity
↑ Pressure on diaphragm and lungs
↓ Outward chest wall force
↑ Abdominal pressure
↑ Fat acts as a reservoir for lipophilic drugs
↑ Fat storage in hepatocytes
↑ Cardiac output
↑ Pressure on gastric contents
↑ Distribution half-life of lipophilic drugs
↑ Volume of distribution for lipophilic drugs
↑ Hepatic cytochrome transcription
↑ Glomerular filtration rate and hepatic blood flow
Authors: Brianna Rosgen
Reviewers: Kayleigh Yang
Ran Marissa Zhang
Karl Darcus*
* MD at time of publication
Legend:
Pathophysiology
Mechanism
Goal
Anesthetic Intervention
Published Nov 8, 2023 on www.thecalgaryguide.com")
Overview of burns
 Less common Burns from any source can result in 3 damage zones
Common
Fire Scald
Zone of coagulation:
innermost zone; maximum damage through necrosis and irreversible tissue loss
Zone of stasis: middle zone; decreased tissue perfusion, potentially salvageable
Zone of hyperemia:
outermost zone; tissue perfusion is increased, damage is reversible
Epidermis, dermis, and subcutaneous tissue
Full Thickness (3rd degree burn)
Normal Skin
Epidermis only
Superficial Thickness (1st degree burn)
Epidermis and superficial (papillary) dermis
Superficial Partial Thickness (2nd degree burn)
Epidermis and deep (reticular) dermis
Deep Partial Thickness (2nd degree burn)
Skin and deep tissues, muscle, fascia, nerves, blood vessels, bone
Composite tissue injury
(4th degree burn)
Compartment syndrome
Epidermal layer Dermal-Epidermal Junction
Superficial (Papillary) Dermis
Deep (Reticular) Dermis
Note: The total burn surface area (TBSA) can be estimated using the rule of nines (1st degree burns are not included): Head and Neck: 9% total, Chest and Upper Back: 9% each, Arm: 9% each, Leg: 18% each (front and back),
Abdomen and Lower Back: 9% each, Genital Area: 1%
Refer to Complications of Burns
Burn Shock
Refer to Burn Shock: Pathogenesis, Complications, and Clinical Findings
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published November 8, 2023 on www.thecalgaryguide.com")
Death Cardiovascular Respiratory and Neurologic Mechanisms

Hypoxemia (Type I Respiratory Failure): low dissolved oxygen in blood (PaO2)
Lungs can’t oxygenate blood fast enough
Lungs can’t rid blood of CO2 fast enough
Hypercapnia / hypercarbia (Type II Respiratory Failure): elevated dissolved CO2 in blood (PaCO2)
Cerebral vasodilation
Toxins: e.g. cyanide, pesticides, arsenic Severe anemia
Distributive problems:
Systemic inflammation (sepsis, anaphylaxis, pancreatitis), adrenal insufficiency, vasodilatory drugs
Obstructive problems: Cardiac tamponade*, tension pneumothorax* or massive pulmonary embolism*
Hypovolemic* problems (low blood volume): Hemorrhage, dehydration, widespread skin disruption or burns
Cardiac valve dysfunction
Myocardial infarction* or cardiomyopathy
Cardiac arrhythmia or heart block
Disturbed electrical activity in cardiomyocytes
Peripheral metabolic disturbances
Hypokalemia*, Hyperkalemia* Acidosis* (including renal failure) Hypothermia*
Toxins* (e.g. cocaine, beta blockers, tricyclics) Severe thyroid derangement
Inappropriate systemic vasodilation
Adjacent forces impair heart filling
Low cardiac preload
Low stroke volume (SV; depends on valves, contractility, preload)
Decreased systemic vascular resistance (SVR)
Low blood pressure (BP = CO x SVR)
Decreased cardiac output (CO = SV x HR)
Disseminated intravascular coagulationàwidespread thrombi that occlude blood flow (also causes hemorrhage, see relevant box at left)
Methemoglobinemia: some hemoglobin gets stuck in a state that can’t carry O2
Hemoglobin has reduced capacity to carry or release O2
Drugs: e.g. dapsone, nitrates
Carbon monoxide poisoning
Circulatory collapse / shock: inadequate perfusion of tissue with blood
Respiratory collapse: blood has insufficient useable O2 content
Ventricular fibrillation (VF) or pulseless ventricular tachycardia (VT)
Hypoxia*: inadequate O2 delivery or utilization in tissues
Hypoxia creates metabolic disturbances that impair cardiac cells. Alternatively, any of the preceding conditions marked with (*) can directly trigger cardiac arrest first
Pulseless Electrical Activity (PEA): organized activity on ECG with no cardiac output (can be preceded or mimicked by pseudo-PEA, in which there is still some output on ultrasound)
Low atmospheric pressure or oxygen content Severe lung disease
Asthma, COPD, interstitial lung disease, congestive heart failure, pulmonary hypertension, pulmonary embolism, lung collapse / atelectasis
Acute respiratory distress syndrome
Pneumonia, aspiration pneumonitis, inhalational injury, systemic inflammation, drowning
Severe hypoventilation
Respiratory fatigue, advanced COPD, chest wall disorders, neuromuscular disorders, upper airway obstruction, toxins (e.g. opioids, botulism)
Can degenerate at any time
Asystole: no cardiac electrical activity or output
Death
Respiratory arrest: cessation of breathing
Inability to protect airway
Decreased level of consciousness
Note
This is a broad overview of the many scenarios that can result in death. For detailed explanations of the various disease mechanisms, refer to the corresponding slides.
* = reversible causes of cardiac arrest (Hs and Ts)
Author:
Ben Campbell
Reviewers:
Yan Yu*
Huma Ali*
* MD at time of publication
Bradycardia
(low heart rate, HR)
Unopposed parasympathetic stimulation of heart (can also cause vasodilation, see Distributive problems)
Disruption of spinal cord sympathetic control
Injury to cervical or upper thoracic spinal cord
Irreversible cessation of cardiac, respiratory, and brain function
Prolonged seizure initially causes increased cardiovascular activity, until the system fatigues
Disruption of respiratory control center in medulla
Expanding skull contents squeeze brainstem (herniation)
Increased intracranial pressure
Edema from intracranial hemorrhage, trauma, brain mass
Edema, inflammation, hypoxia and/or metabolic derangements cause diffuse neuron dysfunction
Central nervous system infection
Dementia, particularly with delirium
Massive ischemic stroke
Seizure
activity prevents or alters breathing
Metabolic disturbances that affect the central nervous system Hypoglycemia Hypocalcemia, hypercalcemia Hyponatremia, hypernatremia Uremia
Acute liver failure (hyperNH4) Many drugs / toxins Withdrawal (e.g. EtOH)
Status epilepticus
Brainstem lesion (e.g. stroke, neoplasm, inflammatory)
Nervous System Insult
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published November 11, 2023 on www.thecalgaryguide.com
Respiratory System Insult
Cardiovascular System Insult Cardiogenic problems")
Turner Syndrome Pathogenesis and Clinical Findings

Partial or complete absence of second sex chromosome, leaving only one normal X-chromosome
Possible Chromosomal Profiles
Other meiotic error → deletion or misdivision of X-chromosomal material
Complete loss of one X- chromosome in all cells (45,X) (45%)
Skeletal Abnormalities
↓ Expression of SHOX gene (present on X- and Y-chromosomes)
↓ Cellular proliferation in growth plates of bones in extremities during embryonic development
Short stature
High-arched palate
Genu valgum (knock knees)
Micrognathia (smaller lower jaw)
Broad chest
Cubitus valgus (forearm angled outward)
Mosaicism (complete loss of one X-chromosome in some cells (e.g., 45,X/46,XXX, etc)) (50%)
Congenital Heart Defects (most serious)
Single copy of TIMP1 gene and presence of risk TIMP3 allele, and differential expression of KDM6A gene
Bicuspid aortic valve
Aortic coarctation
Aortic dilation (worsened by hypertension)
One X-chromosome and presence of Y-chromosomal material in some or all cells (e.g., 45,X/46,XY)
Presence of an
X- isochromosome (most commonly i(Xq))
Other structural abnormalities of X-chromsome (e.g., Ring Chromosome X, partial deletion of X, X or Y marker chromosome)
Endocrine Disorders
Turner Syndrome
The most common sex chromosomal abnormality in females (affects 1/2000-3000)
Results in deletion or non- functioning of one X chromosome
Clinical presentations vary depending on chromosome profile
Aortic dissection
Renal disease
Neurocognitive Deficits
Mechanism unknown
Deficit in social skills
Specific (non-verbal) learning disorder (otherwise normal intelligence)
↓ Executive function skills
↓ Visuospatial skills ↓ Attention
Accelerated follicular apoptosis (i.e., loss of oocytes from ovaries) → streak gonads
Ovaries are unable to respond to high gonadotropins (FSH, LH)
Hypergonadotropic hypogonadism
Premature ovarian insufficiency
↓ Expression of immune- associated genes on X- chromosome
↑ Autoimmunity
Autoimmune diseases (celiac, thyroiditis, IBD, metabolic abnormalities)
↓ Estrogen levels
Lack of breast development
↑ Liver enzymes (Additional mechanisms likely)
↓ Bone mineral density
Other Dysmorphic Features
Lymphedema Webbed (buildup of lymph neck
fluidàswelling)
Primary amenorrhea
Infertility
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published November 25, 2023 on www.thecalgaryguide.com")
Non-Alcoholic Fatty Liver Disease
)
Steatohepatitis: chronic inflammatory and apoptotic climate in the hepatocytes (in the absence of alcohol consumption, termed Non-Alcoholic Steatohepatitis (NASH))
Fibrosis of the Liver: excessive scarring of liver tissue resulting from chronic inflammation, although liver architecture is largely intact
Fat droplets form and grow in the hepatocytes
Hepatic mitochondria increase their workload in attempt to break down the excess free fatty acids through beta-oxidation
↑ in cellular workload creates more reactive oxygen speciesà Inflammation and apoptosis of hepatocytes
On-going inflammation damages hepatic stellate cells (the primary extracellular matrix–producing cells of the liver) causing the release of fibrinogenic cytokines
Cirrhosis of the liver: normal lobular structure distorts and is replaced by regenerating nodules and bridging septa, disrupting normal liver blood flow
Deposition of fibrotic
material and collagen within the perisinusoidal spaces of the liver
Decompensated Cirrhosis Hepatocellular carcinoma
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published November 25, 2023 on www.thecalgaryguide.com")
Complication of MI - Acute Mitral Regurgitation
 commonly in the posterior descending artery
Plaque ruptures
Exposed plaque contentsàPlatelet adhesion and aggregation
Artery becomes partially or completely occluded
Mitral Regurgitation**
(Back flow of blood from left ventricle to left atrium during systole)
↑ Left atrial pressure
Blood from left atrium backs up into pulmonary venous system
↑ Pulmonary venous pressure
↑ Hydrostatic pressure in alveolar capillaries
↑ Fluid leak from alveolar capillaries to interstitium (pulmonary edema**)
↓ Gas exchange
Attempt at
physiologic compensation à ↑ Respiratory rate
Tachypnea
Holosystolic murmur
Heard loudest over the mitral valve (5th intercostal space, mid-clavicular line), with radiation to the axilla
↑ Volume of blood to left ventricle during diastole
↓ Blood supply to the portion of the ventricle that supports the papillary muscle à↓ Muscle movement
Left ventricle dysfunction
↓ Blood supply to the posterior medial papillary muscle
Cell death (myocardial infarction)
Papillary muscle ischemia (muscle is intact but cannot contract)
↑ Blood in right atriumà↑ Blood in superior vena cava
↑ Blood in internal jugular vein
↑ Jugular venous pressure (JVP)
Redistribution of interstitial fluid when lying flat (reduced effect of gravity)
↓ Forward blood flow from left ventricle to aorta
↓ Stroke volume (SV)
↓ Cardiac output (CO) CO = SV x HR (heart rate)
Sympathetic nervous system attempts to physiologically compensate
↑ Heart rate
Tachycardia
↓ Blood pressure (BP) because BP = CO X SVR (systemic vascular resistance)
Cardiogenic shock**
Papillary muscle rupture
Papillary muscle unable to provide adequate tension on mitral valve
Mitral valve unable to stay closed during systole
Orthopnea
Difficulty breathing
Dyspnea
Paroxysmal nocturnal dyspnea
**See corresponding Calgary Guide slides for more details
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published November 25, 2023 on www.thecalgaryguide.com")
RICE mechanism of action
: Mechanism of action
Healing of an injury requires pain and inflammatory control to encourage activity. The goal of Rest, Ice, Compression, Elevation (RICE) is to
decrease inflammation. Though activity itself will contribute to pain and inflammation, it is integral to the rehabilitation process.
Authors: Matthew Roberts Emma Windfeld Reviewers: Alexander Arnold Amanda Eslinger Shyla Bharadia Mao Ding Bradley Jacobs* * MD at time of publication
Rest: (weight bearing or stressful motion discontinued)
Further damage to affected tissues from mechanical stress is prevented
Ice: (applied to injury)
Compression:
(wrap applied to injured area)
Mechanical force applied to tissue
Excess fluid is pushed back into capillaries and lymph network
Elevation:
(limb raised above heart)
Blood flow to tissue is constricted
Mechanism not well understood
↓ delivery of inflammatory mediators such as polymorphonuclear neutrophils and macrophages to injured site
↓ production of inflammatory cytokines (pro-inflammatory substances) such as Tumor Necrosis Factor-α, Platelet Derived Growth Factor, Epidermal Growth Factor, and Transforming Growth Factor-β
↓ Inflammation
Gravity ↑ venous blood return to systemic circulation
↓ edema (accumulation of fluid in interstitium)
Early initiation of injury-specific rehabilitative exercises improves range of motion, strength, and proprioception
Stress to targeted area induces inflammation that, when tightly regulated, is integral to repair
Injured muscle, tendon, bone, or ligament is strengthened
↓ Pain
↑Range of motion and therefore function
Early recovery from injury
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published Dec 9, 2013, updated Oct 15, 2023 on www.thecalgaryguide.com
Rest, Ice, Compression, Elevation (RICE): Mechanism of action
Healing of an injury requires pain and inflammatory control to encourage activity. The goal of Rest, Ice, Compression, Elevation (RICE) is to decrease
inflammation. Though activity itself will contribute to pain and inflammation, it is integral to the rehabilitation process.
Authors: Matthew Roberts Emma Windfeld Reviewers: Alexander Arnold Amanda Eslinger Shyla Bharadia Bradley Jacobs* * MD at time of publication
Rest: (weight bearing or stressful motion discontinued)
Further damage to affected tissues from mechanical stress is prevented
Ice: (applied to injury)
Compression: (wrap applied to injured area)
Mechanical force applied to tissue
Excess fluid is pushed back into capillaries and lymph network
Elevation: (limb raised above heart)
Gravity ↑ venous blood return to systemic circulation
Blood flow to tissue is constricted
↓ delivery of inflammatory mediators such as polymorphonuclear neutrophils and macrophages to injured site
Mechanism not well understood
↓ production of inflammatory cytokines (pro- inflammatory substances) such as Tumor Necorsis Factor-α, Platelet Derived Growth Factor, Epidermal Growth Factor, and Transforming Growth Factor-β
↓ Inflammation
↓ Pain
↓ edema (accumulation of fluid in interstitium)
↑Range of motion and therefore function
Early initiation of injury- specific rehabilitative exercises improves range of motion, strength, and proprioception
Stress to targeted area induces inflammation that, when tightly regulated, is integral to repair
Injured muscle, tendon, bone, or ligament is strengthened
Early recovery from injury
Legend: Published MONTH, DAY, YEAR on www.thecalgaryguide.com
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
RICE: Mechanism of action
Authors: Matthew Roberts Emma Windfeld Reviewers: Alexander Arnold Amanda Eslinger Shyla Bharadia Bradley Jacobs* * MD at time of publication
Rest
(weight bearing or stressful motion discontinued)
Ice
(applied to injury)
Compression
(wrap applied to injured area)
Elevation
(limb raised above heart)
Constricts blood flow to tissue
Mechanism not well understood
Mechanical force applied to tissue
↓ Delivery of polymorphonuclear neutrophils and macrophages to injured site
Excess fluid pushed back into capillaries and lymph network
Gravity ↑ venous blood return to systemic circulation
Prevents further damage to affected tissues from mechanical stress
↓ Production of inflammatory cytokines
↓ Edema (accumulation of fluid in interstitium)
↓ Inflammation
↓ Pain
↑Range of motion and therefore function
Early initiation of injury-specific rehabilitative exercises to improve range of motion, strength, and proprioception
Stress to targeted area induces inflammation that, when tightly regulated, is integral to repair
Injured muscle, tendon, bone, or ligament is strengthened
Early recovery from injury
Legend: Published MONTH, DAY, YEAR on www.thecalgaryguide.com
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications")
Mechanical Ventilation mechanisms of action and complications
 when they cannot breathe on their own.
Invasive: Delivery of positive pressure to the lungs via endotracheal or tracheostomy tube
Mechanical ventilation
Pressure support ventilation (PSV): Set inspiratory pressure & flow. Patient initiates all breaths unassisted
Non-invasive: Delivery of oxygen into the lungs via positive pressure through the mouth
No endotracheal or tracheostomy tube
Assist/control ventilation (AC): Set respiratory rate & tidal volume (amount of air delivered to the lungs with each breath). Patient can trigger additional assisted breaths
Synchronized intermittent mandatory ventilation (SIMV): Set tidal volume & respiratory rate. Patient can trigger additional unassisted breaths
02 mask delivery
Continuous positive airway pressure (CPAP)
Provides continuous positive ventilatory pressure
Bilevel positive airway pressure (BIPAP)
Provides positive pressure with two different pressure levels for inhalation and exhalation
↑ Swallow non- inspiratory flow
Aspiration
Acute rise in airway pressure
Barotrauma
Patient- triggered breath
Ventilator senses negative pressure from inflation of the lungs
Time-triggered breath
Respiratory rate set at x breaths per min
Patient-triggered breath
Ventilator senses negative pressure from inflation of the lungs
Tidal volume determined by patient’s strength & lung compliance
Time-triggered breath
Respiratory rate set at x breaths per min
Delivery of set tidal volume, inspiratory flow rate & pattern
Complete patient- triggered breaths
Ventilator senses negative pressure from inflation of the lungs
Breathes assisted by set inspiratory pressure
Inspiratory flow drops below set inhalational negative pressure threshold
Pressure support terminates as exhalation cycle begins
Combined with SIMV
Inspiratory pressure added to patient triggered breaths
Patient can overcome resistance of the endotracheal tube or ↑ volume of spontaneous breathes
Delivery of set tidal volume, inspiratory flow rate & pattern
Airways remain open & clear of obstruction
Forced air into nasal passages
Nose bleeds (epistaxis)
Maximum tidal volume reached
Exhale valve opens
Patient exhales actively or passively until set end expiratory pressure in the lungs is reached (PEEP) to prevent alveolar collapse
Patient exhales until PEEP reached
Patient achieves optimal ventilation throughout respiratory cycles
Mouth breathing
Dry mouth
(xerostomia)
Increased work of breathing & muscle fatigue
Prolonged weaning & extubation
Breath stacking
↑ Volume and pressure in lungs Lung tissue injury (barotrauma)
Microorganisms colonize artificial airway
Ventilator associated pneumonia if ventilation >48 hrs
Tachypnea
↓ CO2in circulation Respiratory alkalosis
Legend:
Pathophysiology
Mechanism
Sign/symptom/lab finding
Complications
Published Nov 25, 2023 on www.thecalgaryguide.com")
Acute Respiratory Distress Syndrome ARDS CXR findings
: Chest X-Ray Findings
Author: Iffat Naeem
Direct or indirect lung injury causing acute respiratory distress syndrome
(see Acute Respiratory Distress Syndrome slide for pathogenesis and clinical findings)
Activation of dysregulated inflammatory cascade
Absent pleural effusion
Normal heart size
Absent Kerly B lines
No perihilar infiltrate pattern
Bilateral infiltrate that can present in all regions of lung
Air bronchograms
Silhouette sign
Reviewers: Victória Silva, Mao Ding Tara Lohmann* *MD at the time of publication
Edema not due to a cardiogenic cause
Damage to alveolar epithelium
Necrosis of epithelial cells
Erosion of alveolar basement membrane
↑ Alveolar epithelium permeability
Damage to lung capillary endothelium
Release of inflammatory cytokines
Neutrophils migrate into alveoli
Fluid-filled alveoli show as white/grey opacities
Air-filled bronchi appear dark when surrounded by grey/white opacification of fluid-filled alveoli
Increased opacification from fluid-filled alveoli results in lack of differentiation of heart borders
Diffuse and
widespread damage to alveoli and interstitium that show as white/grey opacities
↑ Capillary endothelium permeability
Alveolar edema
Degradation of alveolar- capillary barrier
Proliferative phase
Alveolar epithelium attempts to recover
Chronic phase
Can either resolve or progress to fibrotic thickening and scaring of alveoli
↑ Leakage of fluid from capillaries into alveoli and lung interstitium
Pulmonary fibrosis (scarring)
‘White lung’ appearance
Image credit: Radiopaedia
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published November 25, 2023 on www.thecalgaryguide.com
Acute Respiratory Distress Syndrome (ARDS): Chest X-Ray Findings Direct or indirect lung injury causing acute
Author: Iffat Naeem Reviewers: Victória Silva
respiratory distress syndrome*
Activation of dysregulated inflammatory cascade
Bilateral infiltrate that show as white/grey can present in
Damage to alveolar epithelium
Necrosis of epithelial cells
Erosion of alveolar basement membrane
↑ Alveolar epithelium permeability
Damage to lung capillary endothelium
Fluid-filled alveoli opacities
Air-filled bronchi appear dark when surrounded by grey/white opacification of fluid-filled alveoli
Increased opacification from fluid-filled alveoli results in lack of differentiation of heart borders
Diffuse and
all regions of lung
Release of inflammatory cytokines
Neutrophils migrate into alveoli
Alveolar edema
Air bronchograms
↑ Capillary endothelium permeability
Degradation of alveolar-capillary barrier
Proliferative phase
Alveolar epithelium attempts to recover
Chronic phase
Can either resolve or progress to fibrotic thickening and scaring of alveoli
↑ Leakage of fluid from capillaries into alveoli and lung interstitium
Silhouette Sign
widespread Pulmonary damage to alveoli
‘White lung’ appearance
fibrosis (scarring)
and interstitium that show as white/grey opacities
*See corresponding Calgary Guide slides for more details
Image credit: Radiopaedia
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published X, 2023 on www.thecalgaryguide.com
Acute Respiratory Distress Syndrome (ARDS): Chest X-Ray Findings Direct or indirect lung injury causing acute respiratory
Author: Iffat Naeem Reviewers: Victória Silva
distress syndrome*
Activation of dysregulated inflammatory cascade
Bilateral infiltrate that show as white/grey can present in
Damage to alveolar epithelium
Necrosis of epithelial cells
Denudation of alveolar basement membrane
↑ epithelium permeability
Degradation of alveolar-capillary barrier
Alveolar epithelium
attempts to recover through (proliferative phase)
Damage to lung capillary endothelium
Fluid-filled alveoli opacities
Air-filled bronchi appear dark when surrounded by grey/white opacification of fluid-filled alveoli
Increased opacification from fluid-filled alveoli results in lack of differentiation of heart borders
Diffuse and
all regions of lung
Air bronchograms
Release of proinflamm atory cytokines
Neutrophil migration into airspace
Alveolar Edema
↑ capillary endothelium permeability
↑ leakage of fluid from vasculature into airspace and lung interstitium
Can either resolve or progress to fibrotic
Silhouette Sign
widespread Pulmonary damage to alveoli
‘White lung’ appearance
thickening and scaring of Fibrosis alveoli (chronic phase)
and interstitium that show as white/grey opacities
Image credit: Radiopaedia
*See corresponding Calgary Guide slides for more details
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published X, 2023 on www.thecalgaryguide.com
Acute Respiratory Distress Syndrome (ARDS): Chest X-Ray Findings
Absent pleural effusion
Normal heart size
Absent Kerly B lines
No perihilar infiltrate pattern
Author: Iffat Naeem Reviewers: Victória Silva
Acute Lung Injury (see ‘ARDS: Pathogenesis and Clinical findings’ slide) causing impaired oxygenation
Lung injury not due to cardiogenic cause
(see ‘ARDS: Pathogenesis and Clinical findings’ slide)
Alveolar endothelium damage promotes inflammatory marker release
Exudative phase (1-6 days): neutrophils adhere to damaged endothelium and release pro- inflammatory mediators
Accumulation of intra-alveolar fluid that is rich in neutrophils, macrophages, and red blood cells
Proliferative phase (7-14 days): proliferation of alveolar epithelial
Fibroblasts deposit collagen tissue in alveolar walls and spaces
Can either resolve or progress to fibrotic thickening and scaring
Alveolar Edema
Fluid-filled alveoli show as white/grey opacities
Air-filled bronchi appear dark when surrounded by grey/white opacification of fluid-filled alveoli
Increased opacification from fluid-filled alveoli
Bilateral infiltrate present in all regions
Air bronchograms
results in lack of Silhouette Sign differentiation of
heart borders
Diffuse alveolar damage
‘White lung’ appearance
Image credit: Radiopaedia
*See corresponding Calgary Guide slides for more details
Legend:
(chronic phase) Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published X, 2023 on www.thecalgaryguide.com
Lung injury not due to a cardiogenic cause
Absent pleural effusion
Normal heart size
Absent Kerly B lines
No perihilar infiltrate pattern
Acute Respiratory Distress Syndrome (ARDS): Chest X-Ray Findings")
Leukemia Mieloid Kronis Patogenesis dan Presentasi Klinis
: Patogenesis dan Presentasi Klinis")
Leukemia Limfoblastik Akut Patogenesis dan Temuan Klinis
: Patogenesis dan Temuan Klinis")
Leukemia Limfositik Kronis Patogenesis dan Temuan Klinis

COPD - بیماریزایی

COPD - یافتھ ھای بالینی

COPD - یافتھ ھای تشخیصی

COPD - عوارض و عواقب

Common Reversible Causes of Cardiac Arrest Hs
")
Anesthetic Considerations One Lung Ventilation
 with dependent lung ventilated
Shunt: Non- dependent lung unventilated
Perfusion but no ventilation to collapsed lung
Hypoxic vasoconstriction decreases but does not stop perfusion to non- dependent lung
Right to left intrapulmonary shunt with some perfusion to non- dependent lung
V/Q mismatch causes ↑ hypoxemia
Increase FiO2 to 1 to maintain SpO2 ≥ 90%
Increased FiO2 can allow for toleration of shunt
Optimize cardiac output and shunt fraction to maximize PaO2
Author:
Aly Valji
Reviewers:
Jasleen Brar
Ryden Armstrong*
* MD at time of publication
Relative indications
Surgical exposure for pulmonary resection, mediastinal, esophageal, vascular, thoracic spine, or cardiac valve surgery
Double lumen tube (gold standard)
Endotracheal tube (ETT) with two lumens (bronchial and tracheal)
Insert longer side to a mainstem bronchus, shorter side ends in distal trachea
Absolute Indications
Isolation of healthy from contaminated lung (unilateral infection, hemorrhage)
Control unilateral disruption of ventilation (bronchopleural fistula, unilateral bullae)
Video assisted thoracoscopic surgery
Unilateral lung lavage
Airway Technique
Anesthetic Technique
General anesthetic with neuromuscular blockade
↓ Inspiratory muscle tone
Intraabdominal contents push up on diaphragm
↓ Functional residual capacity (FRC)
↑ Atelectasis if closing capacity > FRC
↑ Hypoxemia Optimize
Altered gravitational forces on thorax
↓ Compliance of dependent lung
↑ Airway pressure required
↑ Risk of lung barotrauma due to ↑ pressure
↑ Perfusion to dependent lung. ↑ Ventilation to nondependent lung (prior to lung isolation)
Collapse of nondependent lung using lung isolation causes ↓ ventilation to this lung
Optimize tidal volume (6-8 mL/kg), respiratory rate (maintain PaCO2 35-40 mmHg), PEEP (5-10 cm H2O) based on clinical picture
Univent tube
Single lumen ETT with movable endobronchial blocker in wall
Blocker steered after intubation into a mainstem bronchus with fiberoptic bronchoscope
Endotracheal tube in mainstem bronchus
Single lumen ETT pushed into a mainstem bronchus
Bronchial blocker
Shaft with an inflatable balloon on distal end
Inserted through single lumen ETT into a mainstem bronchus, after intubation
positive end- expiratory pressure (PEEP) of 5-10 cm H2O
Recruitment of dependent, atelectatic lung with positive pressure
Optimize FiO2
↓ Absorptive atelectasis (from ↑ partial pressure O2 and ↓ N2)
Cuff inflated in a mainstem bronchus to isolate respective lung. Placement should be verified using fiberoptic bronchoscope if possible after positioning
↓ Atelectasis and ↑ FRC
↓ Hypoxemia
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Management
Published December 5, 2023 on www.thecalgaryguide.com
Anesthetic Considerations: One-lung ventilation Mechanical separation of the lungs to allow for individualized ventilation of only one lung
Positioning: Lateral decubitus position (patient on their side) with dependent lung ventilated
Shunt: Non- dependent lung unventilated
Perfusion but no ventilation to collapsed lung
Hypoxic vasoconstriction decreases but does not stop perfusion to non- dependent lung
Right to left intrapulmonary shunt with some perfusion to non- dependent lung
V/Q mismatch from shunt causes ↑ hypoxemia
Increase FiO2 to 1 to maintain SpO2 ≥ 90%
Vasodilation of dependent lung vasculature to compensate for shunt to non- dependent lung
↓ V/Q mismatch
Author:
Aly Valji
Reviewers:
Jasleen Brar
Dr. Armstrong*
* MD at time of publication
Relative indications
Surgical exposure for pulmonary resection, mediastinal, esophageal, vascular, thoracic spine, or cardiac valve surgery
Double lumen tube (DLT)
Two endotracheal tubes (ETT) bonded together
Insert longer side to a mainstem bronchus, shorter side ends in distal trachea
Absolute Indications
Isolation of healthy from contaminated lung (unilateral infection, hemorrhage)
Control unilateral disruption of ventilation (bronchopleural fistula, unilateral bullae)
Video assisted thoracoscopic surgery
Unilateral lung lavage
Airway Technique
Anesthetic Technique
General anesthetic with neuromuscular blockade
↓ Inspiratory muscle tone
Intraabdominal contents push up on diaphragm
↓ Functional residual capacity (FRC)
↑ Atelectasis if closing capacity > FRC
↑ Hypoxemia Optimize
Altered gravitational forces on thorax
↑ Elastance of dependent lung
↑ Airway pressure required
↑ Risk of lung barotrauma due to ↑ pressure
↑ Perfusion to dependent, ventilated lung
↓ Ventilation- perfusion (V/Q) mismatch
↓ Hypoxemia
Optimize tidal volume (6-8 mL/kg), respiratory rate (maintain PaCO2 35-40 mmHg), PEEP (5-10 cm H2O) based on clinical picture
Univent tube
Single lumen ETT with movable endobronchial blocker in wall
Blocker steered after intubation into a mainstem bronchus with fiberoptic bronchoscope
Endotracheal tube in mainstem bronchus
Single lumen ETT pushed into a mainstem bronchus
Bronchial blocker
Shaft with an inflatable balloon on distal end
Inserted through single lumen ETT into a mainstem bronchus, after intubation
positive end- expiratory pressure (PEEP) of 5-10 cm H2O
Recruitment of dependent, atelectatic lung with positive pressure
Optimize
FiO
2
↓ Absorptive atelectasis (from ↑ partial pressure O2 and ↓ N2)
Cuff inflated in a mainstem bronchus to isolate respective lung. Placement should be verified using fiberoptic bronchoscope if possible after positioning
↓ Atelectasis and ↑ FRC
↓ Hypoxemia
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Management
Published MONTH, DAY, YEAR on www.thecalgaryguide.com
Anesthetic Considerations: One-lung ventilation Mechanical separation of the lungs to allow for individualized ventilation of only one lung
Author:
Aly Valji Reviewers: Jasleen Brar Name* * MD at time of publication
Non-dependent lung unventilated
Hypoxic vasoconstriction decreases but does not stop perfusion to non- dependent lung
Right to left intrapulmonary shunt with some perfusion to non- dependent lung
V/Q mismatch from shunt causes ↑ hypoxemia
Increase FiO2 to maintain SpO2 ≥ 90%
Vasodilation of dependent lung vasculature to compensate for shunt to non- dependent lung
Positioning: Lateral decubitus position (patient on their side) with dependent lung ventilated
Indications
Anesthetic
General anesthetic with neuromuscular blockade
↓ Inspiratory muscle tone
Relative indications
Surgical exposure for pulmonary resection, mediastinal, esophageal, vascular, thoracic spine surgery
Absolute Indications
Isolation of healthy from contaminated lung (Unilateral infection or hemorrhage)
Control unilateral disruption of ventilation (Bronchopleural fistula, unilateral bullae)
Video assisted thoracoscopic surgery
Unilateral lung lavage
Intraabdominal contents push up on diaphragm
↓ FRC
↑ Atelectasis if closing capacity > FRC
↑ Hypoxemia
Altered gravitational forces on thorax
Shaft with an inflatable balloon on distal end. Inserted through a single lumen ETT after intubation into a mainstem bronchi
Single lumen ETT pushed into a mainstem bronchus
Optimize positive end-expiratory pressure (PEEP))
Recruitment of dependent, atelectatic lung with positive pressure
↑ Elastance of dependent lung
↑ Airway pressure required
↑ Risk of lung barotrauma due to ↑ pressure
↑ Perfusion to dependent, ventilated lung
↓ Ventilation- perfusion (V/Q) mismatch
↓ Hypoxemia
Optimize tidal volume, respiratory rate, PEEP based on clinical picture
Bronchial blocker
Endotracheal tube in mainstem bronchus
Technique
Univent tube
Double lumen tube (DLT)
Optimize FiO2
↓ Absorptive atelectasis (from ↑ partial pressure O2 and ↓ N2)
Single lumen ETT with movable endobronchial blocker housed in wall of ETT. Blocker maneuvered after intubation into a mainstem bronchus
Two endotracheal tubes (ETT) bonded together. Longer side goes into a mainstem bronchus, shorter side ends in distal trachea
↓ Atelectasis and ↑ FRC
↓ V/Q mismatch
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complication/Intervention
Published MONTH, DAY, YEAR on www.thecalgaryguide.com
Anesthetic Considerations: One-lung ventilation Mechanical separation of the lungs to allow for individualized ventilation of only one lung
Author:
Aly Valji Reviewers: Jasleen Brar Name* * MD at time of publication
Non-dependent lung unventilated
Hypoxic vasoconstriction decreases but does not stop perfusion to non- dependent lung
Right to left intrapulmonary shunt with some perfusion to non- dependent lung
V/Q mismatch from shunt causes ↑ hypoxemia
Increase FiO to 2
maintain SpO2 ≥ 90%
Vasodilation of dependent lung vasculature to compensate for shunt to non- dependent lung
↓ V/Q mismatch
Positioning: Lateral decubitus position (patient on their side) with dependent lung ventilated
Indications
Anesthetic
General anesthetic with neuromuscular blockade
↓ Inspiratory muscle tone
Comorbidity: Likely underlying pulmonary disease
Pre-operative evaluation
Pulmonary function testing
Overall clinical picture, forced expiratory volume (FEV1), and diffusion capacity (DLCO)
Multidisciplinary determination of fitness for surgery
Pulmonary hemorrhage Whole lung lavage Unilateral infection Bronchopleural fistula
Isolation of affected lung from unaffected lung
Pulmonary resection
Mediastinal, esophageal, vascular, thoracic spine, or cardiac valve surgery
Operative lung deflated to expose surgical site
Intraabdominal contents push up on diaphragm
↓ FRC
↑ Atelectasis if closing capacity > FRC
↑ Hypoxemia Optimize positive
Altered gravitational forces on thorax
Contraindications
↑ Elastance of dependent lung
↑ Airway pressure required
↑ Risk of lung barotrauma due to ↑ pressure
↑ Perfusion to dependent, ventilated lung
↓ Ventilation- perfusion (V/Q) mismatch
↓ Hypoxemia
Optimize tidal volume, respiratory rate, PEEP based on clinical picture
Bilateral lung ventilation dependency
Hemodynamic instability
Severe hypoxia Severe COPD
Severe pulmonary hypertension
Potentially unable to tolerate one lung ventilation
Intraluminal airway obstruction/mass
Known difficult airway
Risk of dislodging mass and inability to secure airway
Pursue more advanced airway techniques
end-expiratory pressure (PEEP)
Recruitment of dependent, atelectatic lung with positive pressure
Optimize FiO2
↓ Absorptive atelectasis (from ↑ partial pressure O and ↓ N )
2
2
↓ Atelectasis and ↑ FRC
Post-operative pain management
Thoracotomy or VATS procedure causing ↑ pain along thoracic dermatomes
Epidural
Paravertebral block
Anesthetic injected into epidural space
Anesthetic injected into paravertebral spaces
Bilateral spinal nerve blockade below desired spinal level
Ipsilateral spinal nerve and sympathetic chain blockade in thoracic dermatomes
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complication/Intervention
Published MONTH, DAY, YEAR on www.thecalgaryguide.com
Anesthetic Considerations: One-lung ventilation Mechanical separation of the lungs to allow for individualized ventilation of only one lung
Author:
Aly Valji Reviewers: Jasleen Brar Name* * MD at time of publication
Non-dependent lung unventilated
Hypoxic vasoconstriction decreases but does not stop perfusion to non- dependent lung
Right to left intrapulmonary shunt with some perfusion to non- dependent lung
V/Q mismatch from shunt causes ↑ hypoxemia
Increase FiO2 to maintain SpO2 ≥ 90%
Vasodilation of dependent lung vasculature to compensate for shunt to non- dependent lung
↓ V/Q mismatch
Indications
Anesthetic
General anesthetic with neuromuscular blockade
↓ Inspiratory muscle tone
Comorbidity: Likely underlying pulmonary disease
Pre-operative evaluation
Pulmonary function testing
Overall clinical picture, forced expiratory volume (FEV1), and diffusion capacity (DLCO)
Multidisciplinary determination of fitness for surgery
Anesthetic injected into epidural space
Anesthetic injected into paravertebral spaces
Positioning: Lateral decubitus position (patient on their side) with dependent lung ventilated
Pulmonary hemorrhage Whole lung lavage Unilateral infection Bronchopleural fistula
Isolation of affected lung and unaffected lung
Pulmonary resection
Mediastinal, esophageal, vascular, thoracic spine, or cardiac valve surgery
Operative lung deflated to expose surgical site
Intraabdominal contents push up on diaphragm
↓ FRC
↑ Atelectasis ↑ Hypoxemia
Optimize
positive end- expiratory pressure (PEEP)
Recruitment of dependent, atelectatic lung with positive pressure
↓ Atelectasis and ↑ FRC
Altered gravitational forces on thorax
Contraindications
↑ Elastance of dependent lung
↑ Airway pressure required
↑ Risk of lung barotrauma due to ↑ pressure
Optimize tidal volume, respiratory rate, PEEP based on clinical picture
↑ Perfusion to dependent, ventilated lung
↓ Ventilation- perfusion (V/Q) mismatch
↓ Hypoxemia
Bilateral lung ventilation dependency
Hemodynamically unstable
Severe hypoxia Severe COPD
Severe pulmonary hypertension
Unable to tolerate one lung ventilation
Intraluminal airway obstruction/mass
Known difficult airway
Risk of dislodging mass and inability to secure airway
Pursue more advanced airway techniques
Post-operative pain management
Thoracotomy or VATS procedure causing ↑ pain along thoracic dermatomes
Epidural
Paravertebral block
Bilateral spinal nerve blockade below desired spinal level
Ipsilateral spinal nerve and sympathetic chain blockade in thoracic dermatomes
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complication/Intervention
Published MONTH, DAY, YEAR on www.thecalgaryguide.com
Anesthetic Considerations: One-lung ventilation Mechanical separation of the lungs to allow for individualized ventilation of only one lung
Author:
Aly Valji Reviewers: Name* * MD at time of publication
Indication
Contraindications
Comorbidity: Likely underlying pulmonary disease
Positioning: Lateral decubitus position (patient on their side) with dependent lung ventilated
General anesthetic with neuromuscular blockade
Post-operative pain management
Pulmonary resection, mediastinal, esophageal, vascular, thoracic spine, or cardiac valve surgery
Pulmonary hemorrhage, whole lung lavage, bronchopleural fistula, or unilateral infection
Operative lung deflated to expose surgical site
Isolation of affected lung and unaffected lung
Dependency on bilateral lung ventilation, hemodynamically unstable, severe hypoxia, severe COPD, or severe pulmonary hypertension
Unable to tolerate one lung ventilation
Intraluminal airway obstruction/mass or known difficult Pursue more advanced
Risk of dislodging mass and inability to secure airway
Multidisciplinary determination of fitness for surgery
airway
Pulmonary function testing
Hypoxic vasoconstriction decreases but does not stop perfusion to non- dependent lung
airway techniques
Overall clinical picture, forced expiratory volume (FEV1), and diffusion capacity (DLCO)
Pre-operative evaluation
Non- dependent lung not ventilated
Altered gravitational forces on thorax
Intraabdominal contents push up on diaphragm
↓ Inspiratory muscle tone
Likely procedure is thoracotomy or VATS causing ↑ pain along thoracic dermatomes
Right to left intrapulmonary shunt with some perfusion to non- dependent lung still present
V/Q mismatch from shunt causes ↑ hypoxemia
Increase FiO2 to maintain SpO2 ≥ 90%
Vasodilation of dependent lung vasculature to compensate for shunt to non- dependent lung
↓ V/Q mismatch
↓ Hypoxemia
Intervention:
Optimize tidal volume, respiratory rate, PEEP based on clinical picture
↓ Atelectasis and ↑ FRC
↑ Perfusion to dependent, ventilated lung
↑ Elastance of dependent lung
↓ FRC
↓ Functional residual capacity (FRC)
↓ Ventilation-perfusion (V/Q) mismatch
↑ Airway pressure required
↑ Atelectasis ↑ Hypoxemia
↑ Risk of lung barotrauma due to ↑ pressure
Intervention:
Optimize positive end-expiratory pressure (PEEP)
Recruitment of dependent, atelectatic lung with positive pressure
Epidural
Paravertebral block
Anesthetic injected into epidural space
Bilateral spinal nerve blockade below desired spinal level
Anesthetic injected into Ipsilateral spinal nerve and sympathetic chain blockade in thoracic paravertebral spaces dermatomes
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complication/Intervention
Published MONTH, DAY, YEAR on www.thecalgaryguide.com
Anesthetic Considerations: One-lung ventilation
Author:
Aly Valji Reviewers: Name* * MD at time of publication
One lung ventilation: mechanical separation of the lungs to allow for individualized ventilation of only one lung
Indication
Pulmonary resection, mediastinal, esophageal, vascular, thoracic spine, or cardiac valve surgery
Pulmonary hemorrhage, whole lung lavage, bronchopleural fistula, or unilateral infection
Exposure of surgical site by deflation of operative lung
Isolation of affected lung and unaffected lung
Dependency on bilateral lung ventilation, Contraindications hemodynamically unstable, severe hypoxia, severe
COPD, or severe pulmonary hypertension
Intraluminal airway obstruction/mass or known difficult airway
Pursue more advanced airway techniques
Unable to tolerate one lung ventilation
Risk of dislodging mass and inability to secure airway
Likely underlying Pre-operative Pulmonary pulmonary disease evaluation function testing
Overall clinical picture, forced expiratory Determination of volume (FEV1), and diffusion capacity (DLCO) fitness for surgery
Right to left intrapulmonary shunt as some perfusion to non- dependent lung is still present
↑ Perfusion to dependent, ventilated lung
↑ Elastance of dependent lung
↓ FRC
Non- dependent lung not ventilated
Hypoxic vasoconstriction decreases but does not stop perfusion to non- dependent lung
V/Q mismatch from shunt increases hypoxemia
Intervention:
Increase FiO2 to maintain SpO2 ≥ 90%
Vasodilation of dependent lung vasculature to compensate for non-dependent lung
↓ V/Q mismatch
↓ Hypoxemia
Intervention:
Optimize tidal volume, respiratory rate, PEEP
↓ Atelectasis and ↑ FRC
Positioning: Lateral position with dependent lung ventilated
Altered gravitational forces on thorax
↓ Ventilation-perfusion (V/Q) mismatch
General anesthetic with neuromuscular blockade
Intraabdominal contents push up on diaphragm
↑ Airway pressure required
↑ Atelectasis ↑ Hypoxemia
↑ Risk of lung barotrauma
Intervention: Optimize positive end-expiratory pressure (PEEP)
Recruitment of dependent, atelectatic lung from PEEP
↓ Inspiratory muscle tone
↓ Functional residual capacity (FRC)
Post- operative pain management
Thoracotomy or VATS causes pain along thoracic dermatomes
Epidural
Paravertebral block
Bilateral spinal nerve blockade below desired Anesthetic injected into epidural space spinal level
Anesthetic injected into Ipsilateral spinal nerve and sympathetic chain blockade in paravertebral spaces thoracic dermatomes
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complication/Intervention
Published MONTH, DAY, YEAR on www.thecalgaryguide.com
Anesthetic considerations: one-lung ventilation
Author:
Aly Valji Reviewers: Name* * MD at time of publication
One lung ventilation: mechanical separation of the lungs to allow for individualized ventilation of only one lung
Indication
Pulmonary resection, mediastinal, esophageal, vascular, thoracic spine, or cardiac valve surgery
Pulmonary hemorrhage, whole lung lavage, bronchopleural fistula, or unilateral infection
Exposure of surgical site by deflation of one lung
Isolation of one lung from other
Dependency on bilateral lung ventilation, Contraindications hemodynamically unstable, severe hypoxia, severe
COPD, or severe pulmonary hypertension
Intraluminal airway obstruction/mass or known difficult airway
Pursue more advanced airway techniques
Unable to tolerate one lung ventilation
Risk of dislodging mass and inability to secure airway
Pre-operative evaluation given likely Pulmonary Forced expiratory volume (FEV1) Determination of underlying pulmonary disease function testing Diffusion capacity (DLCO) fitness for surgery
Non- dependent lung not ventilated
Hypoxic vasoconstriction decreases but does not stop perfusion of non- dependent lung
Vasodilation of dependent lung pulmonary vasculature
Right to left intrapulmonary shunt causes V/Q mismatch
↑ Perfusion to dependent, ventilated lung
↑ Elastance of dependent lung
↓ FRC
↑ Hypoxemia
Intervention:
Increase FiO2 to maintain SpO2 ≥ 90%
↓ V/Q mismatch
↓ Hypoxemia
Intervention:
Optimize tidal volume, respiratory rate, PEEP
↓ Atelectasis and ↑ FRC
Positioning: Lateral decubitus with dependent lung ventilated
Altered gravitational forces on thorax
↓ Ventilation-perfusion (V/Q) mismatch
General anesthetic with neuromuscular blockade
Intraabdominal contents push up on diaphragm
↑ Airway pressure required
↑ Atelectasis ↑ Hypoxemia
↑ Risk of lung barotrauma
Intervention: Optimize positive end-expiratory pressure (PEEP)
Recruitment of dependent lung
↓ Inspiratory muscle tone
↓ Functional residual capacity (FRC)
Post- operative pain management
Thoracotomy or VATS causes pain along thoracic dermatomes
Epidural
Paravertebral block
Bilateral spinal nerve blockade below desired Anesthetic injected into epidural space spinal level
Anesthetic injected into Ipsilateral spinal nerve and sympathetic chain blockade in paravertebral spaces thoracic dermatomes
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complication/Intervention
Published MONTH, DAY, YEAR on www.thecalgaryguide.com")
Zenkers Diverticulum Pathogenesis and Clinical Findings
 resting pressure
Inadequate relaxation of the cricopharyngeal muscle of the UES during swallowing
Lack of synchronization between UES and hypopharynx during swallowing
Outflow obstruction in the esophagus
Increased intrabolus pressure with swallowing
Increase in hypopharyngeal pressure
Note: the pathogenesis of Zenker’s Diverticulum is multifactorial, but this mechanism is thought to be a significant contributor
Herniation of the esophagus at a weak point
between the inferior pharyngeal constrictor muscle and the cricopharyngeal muscle (Killian’s triangle)
Zenker’s Diverticulum
Acquired mucosal herniation between the horizontal and oblique fibers of the cricopharyngeus muscle
Inability of the the upper esophageal sphincter to completely open
Dysphagia
Extrinsic compression of the cervical esophagus by the diverticulum
Esophageal Obstruction
Diverticulum compresses recurrent laryngeal nerve
Impaired
innervation to
the intrinsic
muscles of
the larynx
and other
contributing
factors
False diverticulum retains food and saliva
Feeling of needing to clear throat
Palpable lump in the neck
Halitosis (bad smelling breath)
Secretions and food spontaneously empty into the bronchial tree
Splashing of the fluid that has accumulated in large diverticula
Cricoid Cartilage
Cricopharyngeal muscle of the UES
Thyroid Gland
Boyce’s sign (gurgling sound heard as air passes through the diverticulum)
Zenker’s Diverticulum
Weight loss and malnutrition
Aspiration
Cough reflex
Chronic cough
Regurgitation Hoarseness
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published November 25, 2023 on www.thecalgaryguide.com")
Aspiration Pneumonia
 (e.g. stroke)
Altered level of consciousness and impaired cough/clearance
Tube Poor Alcohol and Substance Medications Neurologic diseases
Esophageal and gastric motility disorders
Impaired swallowing
Chronic obstructive pulmonary disorder
feeding oral health
Bacteria adhere to epithelial surfaces and ↑ risk of airway and lung bacterial colonization
Aspirated oropharyngeal and gastric contents can also contain bacteria
↓ Elimination and clearance of foreign bacteria from airway and lung
Macroaspiration (large volume aspiration) of oropharyngeal bacteria, during eating and drinking
Bacteria and fluid fill bronchi and alveolar space
Aspiration Pneumonia
Alterations to lung microbial flora
An infectious lung process caused by inhalation of foreign bacterial and oropharyngeal and gastric contents
Aspiration of acidic fluid and pneumonia causative pathogen (typically anaerobes or bacteria in normal oral flora) with resultant inflammation
Infiltrate develops in a gravity-dependent pattern in patches around bronchi segments.
Produces proinflammatory cytokines, (e.g. tumor necrosis factor-alpha, and interleukin-1)
Hypothalamic production of prostaglandin E2 results in thermogenesis
Fever
Authors: Luiza Radu
Reviewers: Mao Ding, *Yan Yu, *Jonathan Liu *MD at time of publication
Aspiration to the right lung more common due to large diameter and more vertical orientation of the right main bronchus
Crackles and ↑ lung vibrations (fremitus) on auscultation
Productive Cough
Impaired alveolar gas exchange
Chemoreceptor detection of ↓ pO2 triggers
↑ ventilation
Hypoxemia
Dyspnea
Consolidation in lower lobes (particularly superior segments) and posterior segments of upper lobes
If untreated, a
pus-filled lung cavity develops (e.g. abscess)
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published Jan 11, 2024 on www.thecalgaryguide.com")
Bacterial Tracheitis

Children at higher risk due physiologic narrowing of airway
Recent upper respiratory viral infection
(often in Fall/Winter; respiratory virus season) Damage to airway mucosa
Activation of systemic inflammatory response
Inflammatory cytokines release into systemic circulation
↑ Thermo-regulatory set- point at the hypothalamus
↑ Work of breathing to adequately ventilate lungs
Respiratory distress (nasal flaring, grunting)
Activation of local inflammatory response
Results in thick mucopurulent secretions, ulcerations, and shedding of tracheal mucosa
Mucopurulent discharge
secretion of fluid contains mucus and pus
↑ Production of mucous results in more accumulation
Predisposition to bacterial infection
Bacterial pathogen invades trachea Ex: S. aureus (common), S. pyogenes, M. catarrhalis, or H. influenzae
Often high fever
Trachea is narrowed with purulent debris
Upper airway obstruction causes turbulent airstreams
Hoarse voice Tachypnea
Stridor
(with inhalation & exhalation; may be biphasic)
Tracheal tenderness
↓ Mucous clearance from the airways
Excess airway mucous triggers cough reflex
Cough
(may be barky)
↑ Use of accessory
respiratory muscles
(sternocleidomastoid and scalene muscles)
Toxic appearance (lethargy, cyanosis)
↓ Level of consciousness (due to hypoxia & hypercarbia)
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published Jan 11, 2024 on www.thecalgaryguide.com")
Shoulder Impingement Syndrome
 on the anterolateral acromion and coracoacromial ligament
X-Ray: Normal Ultrasound: +/- Tendinopathy, muscle atrophy
Pain with
overhead movement
Pain at night
(e.g., sleep position, gravity)
Pain with lifting
(e.g., weight- training, groceries)
Pain (between 60°-120°) with passive shoulder abduction
+ Painful arc Test
Pain with passive shoulder flexion
+ Neer’s Test
Pain with passive shoulder flexion (to 90°) + internal rotation
+ Hawkins-Kennedy test
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
First published May 27, 2018; updated Jan 11, 2024 on www.thecalgaryguide.com")
Cranial Nerve IV Palsy

Dysgenesis (defective development) of CN IV
Microvascular Disease (e.g. Stroke)
Damage or occlusion (complete or partial blockage) to the blood vessels supplying CN IV
Trauma
Temporary or permanent damage to the nerve fibers
Neoplasm
Metastasis (e.g. Leptomeningeal)
Compression of the nerve fibers along the nerve tract
Primary (e.g. Schwannoma)
Tumor develops new blood vessels that redirect blood flow to the malignancy, away from the nerve
Ischemia of CN IV
Infection (a rare cause) (e.g. Ehrlichia chaffeensis, Tuberculosis meningitis)
Infectious process in the subarachnoid space
Damage to axons of CN IV
Cranial Nerve (CN) IV Palsy
Superior oblique musculature weakness due to CN IV dysfunction
Lesion to the fascicle of CN IV (extending from midbrain to cavernous sinus)
Impaired ability to conduct motor commands from nucleus to superior oblique muscle in the eye
Weak superior oblique innervation
Difficult abduction and intorsion of the eye
Contralateral superior
oblique weakness
Lesion to the nucleus of CN IV (located in the midbrain)
Disturbed signal production occurring prior to demarcation of fibers to contralateral side
The pathophysiology above can cause damage to structures surrounding CN IV in the midbrain
Impacting ipsilateral sympathetic chain descending from the
hypothalamus prior to reaching the superior cervical ganglion
Authors:
Shahab Marzoughi Reviewers:
Sunawer Aujla
Yvette Ysabel Yao
Yan Yu*
Gary Michael Klein*
* MD at time of publication
Vertical/oblique Diplopia (double vision)
Hypertropia (one eye is deviated upward compared to the other)
Perinaud’s Syndrome (upgaze palsy, convergence retraction nystagmus, and pupillary hyporeflexia)
See relevant Calgary Guide slide on Parinaud’s Syndrome
Loss of eye muscle movement coordination and function of other structures relating to gait
Ataxia
Ipsilateral Primary Horner’s Syndrome (miosis, anhidrosis, ptosis)
See relevant Calgary Guide slide on Horner Syndrome
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published January 16, 2024 on www.thecalgaryguide.com")
Sugammadex
) in plasma when administered IV
↓ Concentration of functional nNMBs in plasma
Creates a concentration gradient from muscle tissue (high) to plasma (low)
nNMBs move from muscle compartment to plasma
Sugammadex in plasma encapsulates nNMBs that moved to the plasma
↓ Concentration of functional nNMBs in the plasma
↓ Concentration of nNMBs at the nicotinic acetylcholine receptor within the skeletal neuromuscular junction
Reverses neuromuscular blockade created by nNMBs
Sugammadex
Progesterone is similar in structure to nNMBs
Sugammadex binds progesterone
↓ Progesterone activity in the body
Progesterone is critical for maintenance of early pregnancy
Unknown significance, avoid use in early pregnancy
Sugammadex-nNMB complex is cleared by the kidneys
Higher concentrations of sugammadex facilitate faster nNMB clearance
Unknown mechanisms
Post operative nausea and vomiting
Headache Bradycardia Cardiovascular Collapse
↓ Effectiveness of progesterone-based contraception for 7 days
↓ Clearance in patients with severe renal impairment
Reversal of profoundly deep neuromuscular blockade at higher doses
Binds to IgG or IgE receptors on sensitized basophils/mast cells in allergic reactions
Activation of basophils/mast cells
Degranulation of basophils/mast cells
Release of granulation products
Anaphylaxis Bronchospasm Hypotension
Authors: Arzina Jaffer, Kayleigh Yang Reviewers: Jasleen Brar, Mao Ding Joseph Ahn* * MD at time of publication
Movement of limbs or body during anesthesia
Coughing during anesthesia
Grimacing or suckling on the endotracheal tube
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published January 16, 2024 on www.thecalgaryguide.com")
تعریف-بیماری-انسدادی-مزمن-ریھ

آسم-چگونگی-اثر-درمان-ھا-و-عوارض-جانبی-ر

Arachnoid Cysts MRI Findings

Cerebrospinal Fluid (CSF) collection within arachnoid membrane adhesions following trauma, infection, inflammation or surgery (secondary)
Arachnoid cyst (fluid-filled sac) formation within the layers of the arachnoid membrane, outside of brain parenchyma (for full pathogenesis, see Calgary Guide slide Arachnoid Cysts: Pathogenesis and clinical findings)
Use Diffusion Weighted Imaging (DWI) and Fluid-Attenuated Inversion Recovery (FLAIR) to distinguish from other cysts
Isointense to CSF on T1 and T2
Arachnoid cyst contents should appear isointense to CSF on each MR sequence, including diffusion-weighted imaging
Axial T2 MRI Head. Clearly demarcated hyperintense (bright) arachnoid cyst is seen (red arrows)
Axial T1 MRI Head. Clearly demarcated hypointense (dark) arachnoid cyst is seen (red arrows)
FLAIR allows for suppression of free water signal to enhance fluid with ↑ protein concentration
Arachnoid cysts contain CSF-like fluid with very little to no protein
Complete suppression on FLAIR
Cysts are mostly fluid and contains no protein, so it is suppressed and appears darker in FLAIR images.
Axial FLAIR MRI Head. Hypointense cyst is seen on FLAIR (red arrows), showing low protein content in CSF-like fluid inside arachnoid cyst
DWI measures water diffusion in different directions and whether there is restriction on the direction of flow
Fluid within the cyst can flow freely, with no restriction on the direction of movement
Non-restricted diffusion
There is no directional restriction on flow within the cyst (unrestricted diffusion), so it appears dark on DWI.
Axial DWI MRI Head. Hypointense cyst is seen on DWI (red arrows), showing unrestricted diffusion within arachnoid cyst
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published Jan 20, 2024 on www.thecalgaryguide.com")
vWF Deficiency
 Deficiency:
Authors: Tristan Jones, Nimaya De Silva Reviewers: Sean Spence, Yan Yu, Paige Shelemey, Tony Gu, Raafi Ali, Man-Chiu Poon*, Lynn Savoie* * MD at time of publication
Pathogenesis and clinical findings
Inherited
Acquired
Autosomal dominant inheritance
Recessive inheritance
Type III: Complete quantitative vWF deficiency
Presence of antibodies to vWF (a blood clotting protein
involved with platelet adhesion), often in setting of autoimmune disease
Antibodies bind and inactivate existing vWF
Clonal lympho-proliferative disease (uncontrolled production of lymphocytes, a type of immune cell)
Adsorption of vWF multimers on to tumor cells
↑ Plasma clearance of vWF
↓ vWF to stabilize/carry plasma Factor VIII (blood clotting protein)
↓ Plasma Factor VIII
Impaired intrinsic pathway of coagulation cascade (see Calgary Guide slide on coagulation cascade)
Type I: Partial quantitative vWF deficiency
Type II: Impaired vWF function (qualitative deficiency)
↓ Binding to vWF-specific antibody on immunoassay
↓ vWF antigen assay (diagnostic test for quantitative vWF deficiency)
↑ Bleeding/closure time (bleeding time measures the length of time required to form a platelet plug)
Spontaneous hematomas (large collection of blood outside the blood vessels)
Menorrhagia (heavy menstrual bleeds)
vWF Deficiency
↓ vWF quantity and/or function
↓ vWF available to bind to damaged blood vessel collagen to anchor platelets
Impaired platelet plug formation
Bleeding from mucosal surfaces
Easy bruising
Gum bleeding
↑ Time for blood clot to form
Risk of severe hemorrhage
↑ Partial Thromboplastin Time (PTT) (measures the integrity of the intrinsic pathway)
Epistaxis (nose bleed)
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Re-published December 5, 2023 on www.thecalgaryguide.com")
Arachnoid Cysts Pathogenesis and clinical findings
-like fluid is trapped within the erroneous membrane
Formation of a primary, congenital arachnoid cyst (most common cause)
Head trauma, intracranial hemorrhage, or infection
Inflammation and deposition of cellular matrix Adhesion of the arachnoid membrane
CSF accumulates in the subarachnoid space (space between the arachnoid mater and pia mater)
Formation of a secondary arachnoid cyst (less common)
Cyst remains stable in size and does not expand (most common)
Patients are asymptomatic
Arachnoid cyst is diagnosed incidentally on unrelated neuroimaging (see Calgary Guide slide Arachnoid Cysts: Findings on MRI)
Arachnoid Cyst
Cyst grows in size and expands (rare but more common in children under four years of age)
Cyst exerts pressure on other structures (mass effect)
Suprasellar region
Cyst ruptures into the subdural space (rare)
CSF-like fluid accumulates in the subdural space
Subdural hygroma (collection of non-bloody CSF)
Intracranial hypertension (↑ intracranial pressure)
Generalized symptoms
Middle fossa
Compression and irritation of the temporal cortex
Seizures
Focal symptoms depending on cyst location
Cerebellopontine angle
Compression of vestibulocochlear nerve (Cranial Nerve VIII)
Compression and interruption of cochlear blood supply
Cyst presses on the third ventricle and aqueduct
buildup of CSF in the ventricles
Obstructive hydrocephalus
Cyst presses on the optic chiasm, hypothalamus, and pituitary
Visual impairments and endocrinopathies
Headache (most common)
Vomiting
Nausea
Progressive hearing loss
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published Jan 25, 2024 on www.thecalgaryguide.com")
Avascular Necrosis AVN of the Femoral Head Findings on MRI
 of the Femoral Head: Findings on MRI
Traumatic or atraumatic disruption of blood supply to the proximal femur (for full pathogenesis, see Calgary Guide slide Avascular Necrosis: Pathogenesis and clinical findings)
Ischemia of the femoral head (usually unilateral for traumatic and bilateral for non-traumatic)
Prolonged anoxia (total oxygen deprivation) within the femoral head
Cell death (necrosis) of osteocytes and marrow cells in the femoral head, forming a focal lesion (sequestrum)
Histiocytes and giant cells (immune cells) aggregate around the sequestrum, forming a “reactive zone” around the periphery of the sequestrum
Apoptotic osteocytes in the anoxic reactive zone cannot be phagocytosed leading to dysregulated bone remodeling and osteosclerosis (hardening of bone and ↑ bone mineralization and density due to ↓ resorption and ↑ bone formation)
Femoral head becomes progressively weaker while the mechanical load on it remains the same
Progressive femoral head/subchondral bone collapse Osteoarthritis
Areas with ↑ fluid content appear darker on T1w images
Areas with ↑ fluid content appear brighter on T2w images
Areas with ↑ bone density and ↓ fat content appear darker on T1w images
Areas with ↑ bone density and ↓ fat content appear darker on T2w images
Basic MRI Physiology
Edema and inflammation increases fluid content
Sclerotic areas have ↑ bone density and thus ↓ fat content
Location of Signs
Inflamed reactive zones are darker on T1w images
Inflamed reactive zones are brighter on T2w images
Sclerotic areas are darker on both T1w and T2w images
Authors: George S. Tadros Reviewers: Matthew Hobart, Mao Ding Shahab Marzoughi David Cornell* * MD at time of publication
T1w
Signs on both T1-weighted and T2-weighted images are most commonly seen on the superior anterolateral aspect of the femoral head
Single dark band on T1-weighted MRI
A single band-like crescentic lesion of low signal intensity is seen on T1-weighted MRI images (white arrows). This band represents the edematous reactive zone between the necrotic and normal tissue, and typically extends to the subchondral plate
Double Line Sign on T2-weighted fat-saturated MRI
An outer distal low signal intensity line is seen (white arrows) representing reactive bone sclerosis
Image source: radiologymasterclass
T2w
An inner proximal high signal intensity line is also seen (red arrows) representing vascular and repair tissue at the periphery of the sequestrum
Image source: radiologymasterclass
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published Jan 27, 2024 on www.thecalgaryguide.com")
Bacterial Infections from Transfusion
 recognize bacteria
Immune cells release pyrogens (either bacterial components or signalling molecules) upon recognition
Pyrogens bind to receptors on the hypothalamus
Hypothalamus ↑ body temperature set point
Body generates and conserves heat to reach set point
Immune cells release pro- inflammatory cytokines (messenger protein)
Cytokines activate sympathetic nervous system via hypothalamus
Adrenal glands release stress hormones (epinephrine and norepinephrine) into bloodstream
Stress hormones bind to receptors on cardiomyocytes (heart muscle cells)
↑ Heart contractility
↑ Heart rate
Cytokines circulate throughout the body
Cytokines interact with endothelial cells of the blood vessels
↑ Nitric oxide production
Relaxes smooth muscle cells of blood vessel walls
Vasodilation (↑ blood vessel diameter)
Hypotension
Systemic inflammation
Stimulate nerve endings in muscles
Stimulate nerve endings in gastrointestinal tract’s lining
Blood brain barrier’s integrity is compromised
Nitric oxide reacts with oxygen to form reactive nitrogen species
Tissue damage
Fatigue
Weakness Muscle aches
Nausea and vomiting
Immune and inflammatory cells enter the brain
Authors:
Arzina Jaffer
Kayleigh Yang
Reviewers:
Nimaya De Silva
Raafi Ali
Michelle J. Chen
Yan Yu*
Kareem Jamani*
* MD at time of publication
Inflammation in the brain
Activates pain processing centers in the brain
Headache
Damages neurons and disrupts cell communication
Confusion and altered mental state
Shivering
Fever
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published January 30, 2024 on www.thecalgaryguide.com")
Acute Wound Healing

Puncture (small piercing caused by sharp object)
Acute injury to the skin
Crush (damage by compression)
Acute Wound Healing:
Pathogenesis and clinical findings
Author: Amanda Eslinger Mina Youakim Reviewers: Heena Singh Shahab Marzoughi Yan Yu Laurie Parsons* * MD at time of publication
8 – 365+ days post-injury
Remodeling
(↓ blood vessels & organized collagen)
Extensively cross-linked type 1 collagen replaces the disorganized
collagen laid down in the proliferative phase
↑ Protein content in collagen
Scarring (fibrotic tissue replaces previously healthy tissue)
Disruption of structure and function of dermis, epidermis and subdermal tissues
Subendothelial and endothelial damage activates the coagulation pathway
Formation of a platelet plug
Bleeding is slowed or stopped by
hemostatic plug (hemostasis)
Clot unifies wound edges
0 – 7 days post-injury
Inflammation
(In disrupted skin layers)
4 - 14 days post-injury
Proliferation of collagen, extracellular matrix & blood vessels
TGF-β attracts fibroblasts to the site of the wound
Fibroblast & macrophage stimulate tissue growth &
angiogenesis which replaces hemostatic plug
Scabbing (protective crust overlying damaged tissue)
Re-epithelialization beneath the scab sloughs it off
Healing (newly replaced tissue replaces damaged one)
In response to irritant, mast cells release histamine
Complement activation causing nearby endothelial cells to release prostaglandins
Vasodilation occurs around the wound area
Localized ↑ vascular supply (reception of blood and fluid from vessels)
↑ Hydrostatic pressure forces fluid from vessels into surrounding tissue
Edema (swelling from fluid buildup)
Erythema (redness)
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
First Published Sept 19, 2013; updated Jan 30, 2024 on www.thecalgaryguide.com")
Carbonic Anhydrase Inhibitor Diuretics

Inhibition of carbonic anhydrase on the apical surface of the brush border cells in the proximal convoluted tubule (PCT)
Activation of the Renin- Angiotensin-Aldosterone Systemfromvolume depletion
Activation of principle cell
Epithelial sodium channels (ENaC) on principal cells of the CCD reabsorb ↑ Na+ and waste K+
↓ K+ in serum
Hypokalemia
See Hypokalemia: Clinical
Findings slide
↑ Na+ delivery to the cortical collecting duct (CCD)
H2O follows Na+ into the CCD to maintain a balanced osmotic pressure
↑ H2O available for excretion
Mild diuresis (increase in frequencyandvolumeof urine)
↓ Blood volume
Hypotension
↓ Na+ and HCO3- reabsorption in the PCT
↑ HCO3- delivery to cortical collecting duct
Urine alkalization (increased pH)
Chronic urine alkalization
↓Solubilityof citrate
↓ Urinary citrate
↓ Citrate binding with Ca2+à↑ Ca2+ complexing with oxalate
↑ Spontaneous nucleation, growth and agglomeration of calcium oxalate crystals
Formation of calcium oxalate renal calculi
↑ HCO3- is lost in the urine ↓ pH of the blood
Type II Renal Tubular Acidosis
See Type II/Proximal Renal Tubular Acidosis slide
CAI prevents the up- regulationofglutamine transporters in the PCT
Inability to correct the metabolic acidosis and impaired urinary NH3 excretion
Hyperammonemia (↑ serum NH3 )
↑ Risk of hepatic encephalopathy in individuals with cirrhosis
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published Feb 3, 2024 on www.thecalgaryguide.com
Carbonic Anhydrase Inhibitor Diuretics: Renal Mechanism and Side Effects Carbonic Anhydrase Inhibitors (CAI)
Inhibition of carbonic anhydrase on the apical surface of the brush border cells of the proximal convoluted tubule (PCT)
Authors: Stephanie Happ Reviewers: Matt Hobart Name Name* * MD at time of publication
↓ Na+ and HCO3- reabsorption in the PCT
↑ Na+ delivery to the cortical collecting duct (CCD)
H2O follows Na+ into the CCD to maintain a balanced osmotic pressure
↑ H O available for 2
excretion
Mild diuresis
↓ Blood volume Hypotension
↑ HCO3- delivery to cortical collecting duct
Epithelial sodium channels (ENaC) on principal cells of the CCD reabsorb ↑ Na+
↑ Intracellular Na+ drives Na+/K+ ATPase activity on the principal cells (moving 2 K+ into cell and 3 Na+ out into the peritubular capillary)
↑ Intracellular K+ drives H+/K+ ATPase activity on the intercalated cells (moving 1 H+ into cell and 1 K+ out into the tubular filtrate)
↓ K+ in serum
Hypokalemia
See Hypokalemia: Clinical Findings slide
Urine alkalization
↑ HCO3- is lost in the urine, leading to ↓ pH of the blood
Renal Tubular Acidosis Type II
See Type II/Proximal Renal Tubular Acidosis slide
CAI inhibit the up-regulation of glutamine transporters in the PCT
Inability to correct the metabolic acidosis and
impaired urinary NH3 excretion
Hyperammonemia
↑ Risk of hepatic encephalopathy in individuals with cirrhosis
Chronic urine alkalization leads to marked ↓ in urinary citrate
↓ Ability of citrate to bind to Ca2+ and calcium oxalate stones
↓ Inhibition of spontaneous nucleation
↓ Prevention of growth and agglomeration of crystals
Formation of calcium oxalate renal calculi
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published MONTH, DAY, YEAR on www.thecalgaryguide.com")
Epiglottitis
, viral or fungal
Authors: Alisha Ebrahim Reviewers: Simran Sandhu Mao Ding Michelle J. Chen Danielle Nelson* * MD at time of publication
Infectious agent invades the bloodstream and/or the epithelial layer of the epiglottis, aryepiglottic folds and adjacent structures, allowing for spread
Non-infectious cause: Ingestion of toxin or foreign body, thermal injury, or trauma
The potential space between the squamous epithelial layer and the epiglottal cartilage fills with inflammatory cells such as neutrophils and eosinophils
Exudate of inflammatory cells spreads through the lymphatic and blood vessels in the lingual surface of the epiglottis and periepiglottic tissues
Fluid and inflammatory cells accumulate between the squamous epithelial layer and epiglottal cartilage
Swelling of the entire supraglottic larynx
Tripod/sniffing position (Anxious- looking and sitting with trunk leaning
forward, neck hyper-extended and chin pushed forward to maximize airway diameter)
Stridor (High-pitched sound that is produced by obstruction in the larynx or just below)
Stertor (Low-pitched noise created in the nose or the back of the throat)
Retraction of the intercostal and suprasternal muscles
Tachypnea (Rapid breathing)
Increased weight and mass of the epiglottis Epiglottis curls posteriorly and inferiorly
Ball-valve effect (Airflow obstructed during inspiration as epiglottis is pulled over airway but not during expiration as epiglottis moves back into position)
↓ Diameter of upper airway
Epiglottis obstructs the esophagus
Dysphagia (Difficulty swallowing)
Cyanosis (Blue tint to skin)
Turbulent inspiratory airflow Aspiration of oropharyngeal secretions
Hypoxemia (Low oxygen levels in blood)
↓ Air entry to lungs
Airway obstruction
↑ Work of breathing
Drooling Pain when swallowing
Muffled/”hot potato” voice
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published Feb 5, 2024 on www.thecalgaryguide.com")
Stable Angina

↓ Blood vessel lumen diameter
↓ Volume of blood is supplied to the heart
Predictable period of physical activity or emotional stress
↑ Heart rate
↓ Time for coronary arteries to fill heart with blood (diastole)
↑ Heart contractility
↑ Oxygen demand of heart muscle tissue (myocardium)
↓ Myocardial blood supply
Imbalance between blood supply & oxygen demand causes myocardial ischemia
Angina Pectoris/Stable Angina
Myocardial ischemia causes cardiac muscle cells (cardiomyocytes) to switch from oxygen-dependent (aerobic) to oxygen-absent (anaerobic) metabolism
Anaerobic metabolism produces metabolites that stimulate cardiac spinal afferent nerves
Myocardial visceral afferent & somatic sensory nerve fibers mix & enter the spinal cord via T1-T4 nerve roots
Brain interprets ↑ nerve signaling as nerve pain coming from the skin of T1-T4 dermatomes (referred pain)
↑ lactic acid production & ↓ cellular pH impairs cardiomyocytes’ function
Damaged cardiomyocytes impair myocardial relaxation & cause ↓ left ventricular contractility & cardiac output
Blood backs up into left ventricle, atrium, & pulmonary vasculature
↑ Pulmonary capillary pressures pushes fluid out & into the lung’s alveoli
↓ Gas exchange & oxygenation
↑ Respiratory rate & Dyspnea (shortness of breath)
Blood flow begins at the epicardium (outer heart layer) & ends at endocardium (inner layer)
Subendocardium (innermost heart layer) receives the least blood flow causing non-transmural (partial thickness) heart wall ischemia
Anterior/septal & lateral wall ischemia triggers ↑ sympathetic nervous system (SNS) activity given the proximity of cardiac SNS innervation
Inferior wall ischemia triggers involuntary ↑ in Vagus nerve activity given the nerve’s proximity
Bradycardia (↓ heart rate)
Nausea
Adrenal medulla releases Norepinephrine hormone
Activation of sweat glands via SNS acetylcholine neurotransmitter release
Hypotension (↓ blood pressure)
Pain radiation to left arm, jaw, abdomen & upper back
Chest pain, pressure, or discomfort
Unstable Angina (unpredictable & worsening chest pain)
See relevant Calgary Guide slide on Unstable Angina
Binds arterial smooth muscle α1 receptors
↑ Coronary arteries’ vascular tone (vasoconstriction)
Hypertension (↑ blood pressure)
Activates β1 receptors in the heart
Tachycardia (↑ heart rate)
Diaphoresis (↑ sweating)
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published Aug 8, 2013; updated Feb 5, 2024 on www.thecalgaryguide.com")
Acute Otitis Externa Complications
: Complications
Acute Otitis Externa (AOE)
Authors: Charmaine Szalay-Anderson Vaneeza Moosa Reviewers: Shayan Hemmati Shahab Marzoughi Ben Campbell Justin Lui* * MD at time of publication
Spread to subcutaneous tissue
Chronic otitis externa (>6 weeks)
Chronic inflammation of the outer ear
Fibroblast activation for collagen and extracellular matrix components production for tissue repair
Excess accumulation of tissue
Ear canal fibrosis (thickening)
Ear canal stenosis (narrowing)
Damage/obstruction to ear canal structures with impaired fluid drainage & pressure buildup
Inflammation of the outer ear
Recurrent or non-resolving acute otitis externa Dissemination of infection
Spread to connective tissue and cartilage
Perichondritis (inflammation of ear cartilage)
Spread of Pseudomonas aeruginosa
in an immunocompromised host or due to antibiotic resistance
Rapid infectious spread through soft tissue to mastoid and/or temporal bone
Malignant (necrotizing) otitis externa *can be life threatening
Inflammation of connective tissue and bony structures
Spread to
tympanic membrane
Myringitis (inflammation of tympanic membrane)
Swelling and thinning of tissue
Tympanic membrane perforation (tear)
Immune reaction with inflammation
Dead white blood cell, bacteria & tissue debris accumulation in the ear canal
Pus formation with purulent otorrhea (discharge from ear)
Localized pus accumulation
Abscess
Ear canal blockage
Periauricular/ pinna (outer ear) cellulitis
Facial cellulitis
Erosion of temporal bone decreasing bony sound conduction
Permanent conductive hearing loss
Direct toxicity of pathogens to surrounding nerves
Cranial nerve (CN) VII (facial) palsy (+/- CN X, XI, XII)
Out-of- proportion primary otalgia (ear pain)
Sensation of fullness in the ear
Temporary hearing loss
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published Dec 4, 2022; updated Feb 7, 2024 on www.thecalgaryguide.com")
Chancroid

Micro-abrasions occur in the epidermal tissue of the genital area
Sexual Transmission of Haemophilus ducreyi (H. ducreyi) bacteria from an infected sexual partner
H. ducreyi enters the epidermis through micro abrasions H. ducreyi infects epithelial cells
H. ducreyi secretes Large Supernatant Proteins LspA1 and LspA2 (which inhibit phagocytosis by neutrophils and macrophages)
T cells activate and release cytokines IL-6 and IL-8 Neutrophils are recruited to the site of infection
Local macrophages form a collar around the base of the papule to try to reach and engulf H. ducreyi
↑ Localized buildup of immune cells
While infection persists, H. ducreyi release lipooligosaccharides (LOS)
LOS travels to lymph nodes in the inguinal region Lymph nodes synthesize and proliferate T
cells specific to H. ducreyi antigen Inguinal lymph nodes swell due
to ↑ number of T cells
Inguinal buboes (swollen inguinal lymph nodes)
Neutrophils surround the bacteria and attempt to engulf it
Localized epidermal buildup of neutrophils and H. ducreyi
Papules (small protrusions on the skin)
Continued build-up of H. ducreyi, neutrophils, dead white blood cells (pus), and macrophages
Pustules (pus-filled skin lesion)
↑ Local pressure from growing pustule compresses surrounding vessels
↓ Local blood flow erodes and sloughs off the local epidermal roof
Ulcers (open skin sore)
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published Feb 8, 2024 on www.thecalgaryguide.com")
Dantrolene
, and spasticity associated with various neurological disorders such as multiple sclerosis, cerebral palsy, and spinal cord injury.
Dantrolene
Authors: Madison Amyotte, Arzina Jaffer Reviewers: Jasleen Brar, Mao Ding Luiza Radu Joanna Moser* * MD at time of publication
Metabolized in the liver by the cytochrome P450 enzyme
Metabolic process forms a high concentration of hydroxylamine
Hydroxylamine is a highly toxic metabolite associated with dantrolene induced liver injury
Impaired Liver Function
Binds to ryanodine receptors (RYR1) in the sarcoplasmic reticulum of skeletal muscle cells
Prevents ryanodine channel from opening when triggered by the action potential in the muscle
Prevents calcium release from the sarcoplasmic reticulum
Prevents binding of calcium to troponin on the actin filaments in the cytosol of the skeletal muscle cells
Myosin-binding site on the actin remains covered by the tropomyosin
Prevents cross-bridge formation between myosin and actin within the sarcomere
Prevents muscle contraction
↓ Progression of muscle rigidity and spasms ↓ Heat produced by muscular contraction
Skeletal & respiratory muscle weakness ↓ Body temperature
↓ Inspiratory capacity
Dyspnea
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published Feb 18, 2024 on www.thecalgaryguide.com")
Infective endocarditis
 drug use (mostly causes right sided endocarditis)
Bacteria enter the bloodstream (bacteremia)
In subacute cases, valvular abnormality usually present beforehand
In all cases, vegetation forms on affected valve
Immune complex deposit in kidney
Immune complexes cause vasculitis in retinal vessels
Immune complexes deposit subcutaneously
↓ Blood flow to organs perfused by the obstructed arteries
Valve endothelium is damaged
Bacteria adhere to thrombi on the cardiac valve endothelium
Infective Endocarditis
Infection of the thrombus typically produces a vegetation on the flow surface of a valve
Immune complexes (complexes of antibody bound to antigen) form secondary to infection
Generalized immune response
Malaise Chills Fever (> 38°C)
Author:
Sean Spence
George S. Tadros Reviewers:
Yan Yu
Jason Baserman
Danny Guo
Steve Vaughan*
*MD at time of publication
Parts of vegetation embolize systemically, obstructing arteries
Infection destroys infected valve
Smaller emboli block smaller vessels on hands/feet
Microinfarctions
Mitral regurgitation, Aortic stenosis, Aortic insufficiency
(Valve involvement: Mitral > Aortic > Tricuspid)
Vegetation seen on ultrasound/echocardiogram Damage to Glomerulonephritis
glomeruli (Inflammation of glomeruli) Roth’s spots (retinal hemorrhages with pale centers
due to coagulated fibrin)
Osler nodes (tender, raised, red lesions found on the hands and feet)
Organ infarction (tissue death)
Splinter hemorrhages (small red streaks under nails)
Janeway lesions (non-tender, red macules/nodules on palms/soles – only a few millimeters wide)
Regurgitation (blood leaks back through the insufficient valve despite it being closed)
Valve unable to fulfill normal functions (valve insufficiency)
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published Aug 20, 2013, updated Feb 24, 2024 on www.thecalgaryguide.com")
Gestational Diabetes Risk factors and pathogenesis

Authors:
Amyna Fidai
Maharshi Gandhi Reviewers:
Laura Byford-Richardson Shahab Marzoughi
Yan Yu*
Hanan Bassyouni*
* MD at time of publication
High risk population (Aboriginal, Hispanic, South Asian, Asian, African)
Previous or current macrosomia (>4000g) or polyhydramnios
Other conditions associated with Diabetes Mellitus such as polycystic ovarian syndrome, hypertension, metabolic syndrome
Placental counter regulatory hormones (particularly Human Placental Growth Hormone) oppose the action of insulin
↑ Insulin resistance (liver, muscle, adipose tissues become less responsive to insulin)
↑ Fetal demands after 18 weeks gestation
(fetus requires 80% of its energy from maternal glucose)
↑ Carbohydrate intake to keep up with the demands
Previous history of gestational diabetes or glucose intolerance
Family history of diabetes
Advanced maternal age
Obesity
Previous unexplained stillbirth
Multiples (larger placental mass and activity)
Corticosteroid use
↑ Risk for pregnancy induced glucose intolerance (mechanism unclear and/or complex)
↑ Insulin requirements Initially pancreatic beta cells work
overtime to keep up with the ↑ insulin demands
Eventually insulin demands are not met due to the exhaustion of pancreatic beta cells
Plasma glucose rises (Fasting plasma glucose ≥5.3 mmol/L)
Gestational Diabetes (or exacerbation of pre-existing DM)
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published Jan 28, 2017, updated Feb 24, 2024 on www.thecalgaryguide.com")
Le zona

Propofol français

Therapie cognitivo-comportementale

MPOC Resultats des radiographies du thorax

Trouble de la personnalite antisociale

Trouble de la personnalite schizotypique

Granulomatose avec polyangeite

Oedeme pulmonaire a pression negative

Eisenmenger Syndrome

↑ Shear stress and circumferential stress on the pulmonary arteries and arterioles
Atrioventricular septal defect
Truncus arteriosus (Only one common artery arises from the heart rather aorta and pulmonary artery)
Long-standing “right-to-left” shunt with too much pulmonary blood flow
Structural changes occur in pulmonary arteries and arterioles to adapt to ↑ flow and pressure
Hypertrophy of the smooth muscles (media) of pulmonary arteries and arterioles Thickening of the intima (innermost layer) of pulmonary arteries and arterioles
↑ Pulmonary vascular resistance (pressure in the pulmonary arteries)
Pressure within the right ventricle gradually ↑
Right ventricular pressure is equal to, or exceeds left ventricular pressure
Shunt changes from left-to-right to right-to-left “Right-to-Left” Shunt
De-oxygenated blood originating from the right ventricle bypasses the lungs and goes into systemic circulation ↓ Oxygen delivery to tissue across the body
Pulmonary hypertension
(mean pulmonary artery pressure at rest ≥ 25mmHg)
Right ventricular hypertrophy (enlarging)
Hypertrophied right ventricle cannot contract effectively
Right ventricle loses ability to pump blood efficiently
Right heart failure
Megakaryocytes (platelet precursors) are shunted away from the capillary beds of the lungs, where they usually get fragmented into platelets
Chronic central cyanosis (generalized bluish discoloration)
Induction of vascular endothelial growth factor (VEGF) in fingers
Terminal digit clubbing (uniform swelling of the fingers and toes)
Hypoxemia (<90% O2 saturation)
Thrombocytopenia (↓ platelet count) Spontaneous bleeding events
Body tries to compensate for ↓ O2 by ↑ oxygen-carrying capacity of the blood
Polycythemia (↑ in red cell count) and ↑ hemoglobin concentration
↑ blood viscosity Hypercoagulable and prothrombotic state
Not enough O2 to meet the body’s demands
Fatigue
Epistaxis (nose bleeds)
Minor (non-life-threatening)
Major (life-threatening)
Pulmonary hemorrhage
Dental bleeds
Menorrhagia (heavy periods)
Pulmonary Embolism (clot in pulmonary vessels) Stroke Deep vein thrombosis (clot in deep veins)
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published Mar 5, 2024 on www.thecalgaryguide.com")
Pagets Disease pathogenesis and clinical findings

Epigenetic modifications of different genes
Bone marrow cells (precursor of osteoclasts) get infected with measles or respiratory syncytial virus
↑ Osteoclast formation from progenitor (precursor) cells
Malfunction of genes involved in bone remodeling and regulation
Genetic predisposition
↑ Osteoclast quantity (10- 100X), size, and activity
Release of bone matrix proteins and collagen breakdown products such as C-terminal telopeptide
↑ Serum C-terminal telopeptide (marker of bone resorption)
Osteoclasts secrete acid and enzymes that dissolve the mineralized bone and matrix (bone resorption)
The breakdown leaves physical holes in the bone that show up as radio-lucent spots on x-ray
Radiolucent lytic bone lesions on x-ray (mostly seen in pelvis, vertebral bodies, tibia, femur and skull)
↑ Osteoblast activity as a compensatory mechanism
Disorganized new bone formation
Abnormal new bone is weak, but bone continues to bear regular stresses
Skeletal deformities E.g., Skull involvement (↑ hat size), bowed tibias, kyphosis (excessive forward curve of the upper spine)
Osteoblasts release alkaline phosphatase from the bone into the blood
↑ Serum alkaline phosphatase (marker of bone formation)
Damage to the auditory labyrinth (bones in the ear)
Bilateral progressive hearing loss
Disorganized remodeling, lytic lesion expansion and bowing of bone all stimulate nociceptive (pain-sensing) nerve endings on the periosteum (membrane covering the surfaces of bones)
Bone pain (dull, often at night-time)
Damage to bone surrounding joints
Osteoarthritis
Bone fractures
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published Nov 26, 2012, updated Mar 5, 2024 on www.thecalgaryguide.com
Paget’s Disease: Pathogenesis and clinical findings
Author: Payam Pournazari George S. Tadros Reviewers: Yan Yu Spencer Montgomery David Hanley* * MD at time of publication
Mutations in different genes (SQSTM1 gene most common, but also TNFRSF11A, ZNF687 and PFN1)
Epigenetic modifications of different genes (including RANKL, OPG, HDAC2, DNMT1, and SQSTM1)
Malfunction of genes involved in bone remodeling and regulation
Possible viral exposure (measles or respiratory syncytial virus)
Genetic predisposition
↑ Osteoclast quantity (10- 100X), size, and activity
Release of bone matrix proteins and collagen breakdown products such as C-terminal telopeptide (CTx)
↑ Serum C-terminal telopeptide
marker of bone resorption
Osteoclasts secrete acid and enzymes that dissolve the mineralized bone and matrix (bone resorption)
The breakdown leaves physical holes in the bone
that show up as radio-lucent spots on x-ray
Radiolucent Lytic bone lesions on x-ray (mostly seen in pelvis, vertebral bodies, tibia, femur and skull)
↑ Osteoblast activity as a compensatory mechanism
Disorganized new bone formation
Abnormal new bone is weak, but bone continues to bear regular stresses
Skeletal deformities
E.g., Skull involvement (↑ hat size), Bowed tibias, kyphosis (excessive forward curve of the upper spine)
Osteoblasts release Alkaline Phosphatase (ALP) from the bone into the blood
↑ Serum Alkaline Phosphatase
marker of bone formation
Damage to the auditory labyrinth (bones in the ear)
Bilateral progressive hearing loss
Disorganized remodeling, lytic lesion expansion and bowing of bone all stimulate nociceptive (pain-sensing) nerve endings on the periosteum (membrane covering the surfaces of bones)
Bone pain (dull, often at night-time)
Damage to bone surrounding joints
Osteoarthritis
Bone fractures
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published November 26, 2012 on www.thecalgaryguide.com
Paget’s disease: Pathogenesis, Clinical Findings
Author: Payam Pournazari Reviewers: Yan Yu Spencer Montgomery David Hanley* * MD at time of publication
Genetic predisposition (possibly in RANK encoding gene)
Possible viral exposure (measles and respiratory syncytial virus)
↑ in number (10-100X), size, and activity of osteoclasts
Osteoclasts cause excessive bone resorption, which also stimulates osteoblasts
Release of bone matrix proteins and collagen breakdown products such as C-terminal telopeptide of pyridinoline crosslinks (CTx)
↑ serum CTx
marker of bone resorption
Osteoclasts secrete acid and enzymes that dissolve the mineralized bone and matrix
Leaves physical holes in the bone that show up as radio- lucent spots on x-ray
Lytic bone lesions
(mostly seen in pelvis, vertebral bodies, tibia, femur and skull)
Marked ↑ osteoblastic activity results in disorganized new bone formation
Abnormal new bone bone is weak, but bone continues to bear regular stresses
Skeletal deformities: e.g. Skull involvement (↑ hat size), Bowed tibias, kyphosis and fractures
Osteoblasts release Alkaline Phosphatase
(ALP) from the bone into the blood
↑ serum ALP
marker of bone formation
Disorganized bone remodelling, lytic lesion expansion, fracture and bowing of bone all stimulate nociceptive nerve endings on the periosteum
Bone pain
(dull, often night-time)
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published November 26, 2012 on www.thecalgaryguide.com")
Hematoma Epidural
: Patogenesis dan temuan klinis")
Hematoma Epidural Temuan pada CT scan
 Akut: Temuan pada CT scan")
Onychomycosis

Proximal Subungual
White Superficial
Tinea infection (e.g. Tinea Pedis, Corporis, Capitis)
Infection spreads to distal hyponychial space
Dermatophytes colonize local tissue in nail plate and nail bed
Dermatophytes feed on keratinized tissue
General Symptoms (All Subtypes)
Spongiosis (Intercellular edema)
Acanthosis (Thickening of stratum spinosum layer of epidermis)
Hyperkeratosis (Thickening of stratum corneum In effort to rid infection)
Papillomatosis (Projections of dermal papillae)
Secondary damage to nail matrix
Loss of nail
Keratinocytes produce an acute, low-grade inflammatory cytokine response
Onychomycosis
Dermatophytic infection of the nail bed
Inflammation promotes ↑ fluid to tissues for ↑ immune cell delivery
Widespread inflammation thickens parts of the epidermis in efforts to shed the infection
Inflammation and epidermal hyperplasia (↑ growth of cells) influence local dermal papillae (group of cells just beneath the hair follicle) to proliferate and project above the skin
Distal Subungual
Superinfecting bacteria or other fungi proliferate beneath the compromised nail imparting a yellowish appearance
Distal Subungual Subtype
(Thick yellow nails, keratin and debris accumulate distally underneath nail plate)
Dermatophytes invade the proximal end of the nail plate
Dermatophytes penetrate through the cuticle to the newly forming nail plate moving distally
Proximal Subungual Subtype (Whitish discolouration of nail plate that begins proximally and moves distally, indicative of immunosuppression)
Fungi predominantly invade various areas of the superficial nail plate layers eventually joining together
White Superficial Subtype (Chalky white scale that spreads slowly beneath nail plate, well-defined “white islands” that coalesce as disease progresses)
The entirety of the nail plate is infected by the dermatophytes
Widespread inflammation thickens the nail plate as well as beneath the nail (subungual hyperkeratosis) in efforts to shed the infection
Total Dystrophic Subtype (End-stage nail disease, entire nail becomes thick and dystrophic)
Local spread of infection Dermatophytes spread causing cracks in the skin deeper into toe
Abnormal keratinization in hyponychium
Keratin accumulates between nail plate and hyponychium
Fissure (splits in the skin)
Bacteria enters lymphatics and bloodstream
Cellulitis Sepsis
Onycholysis (nail plate separates from nail bed)
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published Mar 13, 2024 on www.thecalgaryguide.com
Dermatophyte Onychomycosis: Pathogenesis and clinical findings
Authors: Holly Zahary Loreman Reviewers: Mina Youakim Elise Hansen Shahab Marzoughi Jodi Hardin* * MD at time of publication
Host Risk Factors
Environmental Risk Factors
Immuno- compromised
↓ Immune response to infection
Older age
Peripheral vascular disease
Reduced blood circulation
Diabetes
Pre-existing nail dystrophy
Previous nail trauma
Integrity of nail unit is compromised
Micro- traumatic pressure on nail
Dark, warm shoe environment
Optimal conditions for fugal growth
Exposure to tinea pedis or onychomycosis
Direct spread of infection to nail unit
High blood sugar favoring infection
Dermatophytes invade corneocytes on stratum corneum, the uppermost non-living layer of keratinized skin
Compromise/breaking of hyponychial seal or cuticle (connection between hyponychium and nail plate)
Proximal Subungual
White Superficial
Distal Subungual
Superinfecting bacteria or other fungi proliferate beneath the compromised nail imparting a yellowish appearance
Distal Subungual Subtype
(Thick yellow nails, keratin and debris accumulate distally underneath nail plate)
Infection spreads to distal hyponychial space
Dermatophytes colonize local tissue in nail plate and nail bed
Dermatophytes feed on keratinized tissue
Keratinocytes produce an acute, low-grade inflammatory cytokine response
Onychomycosis
Dermatophytic infection of the nail bed
Inflammation promotes ↑ fluid to tissues for ↑ immune cell delivery
Widespread inflammation thickens parts of the epidermis in efforts to shed the infection
Inflammation and epidermal hyperplasia (↑ growth of cells) influence local dermal papillae (group of cells just beneath the hair follicle) to proliferate and project above the skin
General Symptoms (All Subtypes)
Spongiosis (Intercellular edema)
Acanthosis (Thickening of stratum spinosum layer of epidermis)
Hyperkeratosis (Thickening of stratum corneum In effort to rid infection)
Papillomatosis (Projections of dermal papillae)
Secondary damage to nail matrix
Loss of nail
Tinea infection (e.g. Tinea Pedis, Corporis, Capitis)
Dermatophytes invade the proximal end of the nail plate
Dermatophytes penetrate through the cuticle to the newly forming nail plate moving distally
Proximal Subungual Subtype (Whitish discolouration of nail plate that begins proximally and moves distally, indicative of immunosuppression)
Fungi predominantly invade various areas of the superficial nail plate layers eventually joining together
White Superficial Subtype (Chalky white scale that spreads slowly beneath nail plate, well-defined “white islands” that coalesce as disease progresses)
The entirety of the nail plate is infected by the dermatophytes
Widespread inflammation thickens the nail plate as well as beneath the nail (subungual hyperkeratosis) in efforts to shed the infection
Total Dystrophic Subtype (End-stage nail disease, entire nail becomes thick and dystrophic)
Dermatophytes spread deeper into toe
Bacteria enters lymphatics and bloodstream
Abnormal keratinization in hyponychium
Keratin accumulates between nail plate and hyponychium
Local spread of infection causing cracks in the skin
Fissure (splits in the skin)
Cellulitis Sepsis
Onycholysis (nail plate separates from nail bed)
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published MONTH, DAY, YEAR on www.thecalgaryguide.com
Dermatophyte Onychomycosis: Pathogenesis and clinical findings
Authors: Holly Zahary Loreman Reviewers: Mina Youakim Elise Hansen Shahab Marzoughi Jodi Hardin* * MD at time of publication
Host Risk Factors
Environmental Risk Factors
Immuno- Older compromised age
↓ Immune response to infection
Peripheral vascular disease
Reduced blood circulation
Diabetes
Pre-existing nail dystrophy
Previous nail trauma
Integrity of nail unit is compromised
Micro- traumatic pressure on nail
Dark, warm shoe environment
Optimal conditions for fugal growth
Exposure to tinea pedis or onychomycosis
Direct spread of infection to nail unit
High blood sugar favoring infection
Dermatophytes invade corneocytes on stratum corneum, the uppermost non-living layer of keratinized skin
Compromise/breaking of hyponychial seal or cuticle (connection between hyponychium and nail plate)
Tinea infection (e.g. Tinea Pedis, Corporis, Capitis)
Infection spreads to distal hyponychial space
Dermatophytes colonize local tissue in nail plate and nail bed
Dermatophytes feed on keratinized tissue
Keratinocytes produce an acute, low-grade inflammatory cytokine response
Onychomycosis
Dermatophytic infection of the nail bed
General Symptoms (All Subtypes)
Spongiosis (Intercellular edema)
Acanthosis (Thickening of stratum spinosum layer of epidermis)
Papillomatosis
(Projections of dermal papillae)
Hyperkeratosis (Thickening of stratum corneum In effort to rid infection)
Secondary damage to nail matrix
Loss of nail
Proximal Subungual
White Superficial
Distal Subungual
Distal Subungual Subtype
(Thick yellow nails, keratin and debris accumulate distally underneath nail plate)
Proximal Subungual Subtype (Whitish discolouration of nail plate that begins proximally and moves distally, indicative of immunosuppression)
White Superficial Subtype (Chalky white scale that spreads slowly beneath nail plate, well-defined “white islands” that coalesce as disease progresses)
Total Dystrophic Subtype (End-stage nail disease, entire nail becomes thick and dystrophic)
Local spread of infection causing cracks in the skin
Dermatophytes spread deeper into toe
Abnormal keratinization in hyponychium
Keratin accumulates between nail plate and hyponychium
Onycholysis (nail plate separates from nail bed)
Fissure (splits in the skin)
Bacteria enters lymphatics and bloodstream
Cellulitis Sepsis
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Legend:
Published MONTH, DAY, YEAR on www.thecalgaryguide.com
Dermatophyte Onychomycosis: Pathogenesis and clinical findings
Authors: Holly Zahary Loreman Reviewers: Mina Youakim Elise Hansen Shahab Marzoughi Jodi Hardin* * MD at time of publication
Host Risk Factors
Environmental Risk Factors
Immuno- Older compromised age
↓ Immune response to infection
Peripheral vascular disease
Reduced blood circulation
Diabetes
Pre-existing nail dystrophy
Previous nail trauma
Integrity of nail unit is compromised
Micro- traumatic pressure on nail
Dark, warm shoe environment
Optimal conditions for fugal growth
Exposure to tinea pedis or onychomycosis
Direct spread of infection to nail unit
High blood sugar favoring infection
Dermatophytes invade corneocytes on stratum corneum, the uppermost non-living layer of keratinized skin
Compromise/breaking of hyponychial seal or cuticle (connection between hyponychium and nail plate)
Tinea infection (e.g. Tinea Pedis, Corporis, Capitis)
Infection spreads to distal hyponychial space
Dermatophytes colonize local tissue in nail plate and nail bed
Dermatophytes feed on keratinized tissue
Keratinocytes produce an acute, low-grade inflammatory cytokine response
Onychomycosis
Dermatophytic infection of the nail bed
Proximal Subungual
White Superficial
General Symptoms (All Subtypes)
Spongiosis (Intercellular edema)
Acanthosis (Thickening of stratum spinosum layer of epidermis)
Papillomatosis
(Projections of dermal papillae)
Hyperkeratosis (Thickening of stratum corneum In effort to rid infection)
Secondary damage to nail matrix
Loss of nail
Distal Subungual
Distal Subungual Subtype
(Thick yellow nails, keratin and debris accumulate distally underneath nail plate)
Proximal Subungual Subtype (Whitish discolouration of nail plate that begins proximally and moves distally, indicative of immunosuppression)
White Superficial Subtype (Chalky white scale that spreads slowly beneath nail plate, well- defined “white islands” that coalesce as disease progresses)
Total Dystrophic Subtype (End-stage nail disease, entire nail becomes thick and dystrophic)
Local spread of infection causing cracks in the skin
Dermatophytes spread deeper into toe
Abnormal keratinization in hyponychium
Keratin accumulates between nail plate and hyponychium
Onycholysis (nail plate separates from nail bed)
Bacteria enters lymphatics and bloodstream
Fissure (splits in the skin)
Cellulitis
Sepsis
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Legend:
Published MONTH, DAY, YEAR on www.thecalgaryguide.com
Dermatophyte Onychomycosis (Tinea Unguium): Pathogenesis, clinical findings,
Authors: Holly Zahary Loreman Reviewers: Elise Hansen Name Name* * MD at time of publication
and complications
Host Risk Factors
Environmental Risk Factors
Immuno- compromised
↓ immune response to infection
Older age
Peripheral vascular disease
Diabetes
Pre-existing nail dystrophy
Previous Nail Trauma
Integrity of nail unit is compromised
Micro-traumatic pressure on nail
Dark, warm shoe environment
Optimal conditions for fugal growth
Exposure to tinea pedis or onychomycosis
Direct spread of infection to nail unit
Reduced blood circulation
High blood sugar, favoring infection
Tinea pedis infection (see ‘Tinea Capitis, Tinea Corporis, and Tinea Pedis’)
Infection spreads to distal hyponychial space Dermatophytes colonize local tissue in nail plate and nail bed Dermatophytes feed on keratinized tissue
Proximal Subungual
White Superficial
Dermatophytes invade corneocytes on stratum corneum, the uppermost non-living layer of keratinized skin
Compromise/breaking of hyponychial seal or cuticle (connection between hyponychium and nail plate)
Keratinocytes produce an acute, low-grade inflammatory cytokine response
Onychomycosis (Tinea Unguium)
(dermatophytic infection of the nail bed)
Distal Subungual
General Symptoms (All Subtypes)
Spongiosis
Intercellular edema
Acanthosis
Thickening of stratum spinosum layer of epidermis
Papillomatosis
Projections of dermal papillae
Hyperkeratosis
Thickening of stratum corneum In effort to rid infection
Secondary damage to nail matrix
Loss of nail
Distal Subungual Subtype
Thick yellow nails, keratin and debris accumulate distally underneath nail plate
Proximal Subungual Subtype
Whitish discolouration of nail plate that begins proximally and moves distally, indicative of immunosuppression
White Superficial Subtype
Chalky white scale that spreads slowly beneath nail plate, well-defined “white islands” that coalesce as disease progresses
Total Dystrophic Subtype End-stage nail disease, entire nail becomes thick and dystrophic
Local spread of infection causing cracks in the skin
Dermatophytes spread deeper into toe
Abnormal keratinization in hyponychium
Keratin accumulates between nail plate and hyponychium
Onycholysis (nail plate separates from nail bed)
Tissue Damage
Cellulitis
Sepsis
Bacteria enters lymphatics and bloodstream
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Legend:
Published MONTH, DAY, YEAR on www.thecalgaryguide.com")
MI Complication Ventricular Wall Rupture
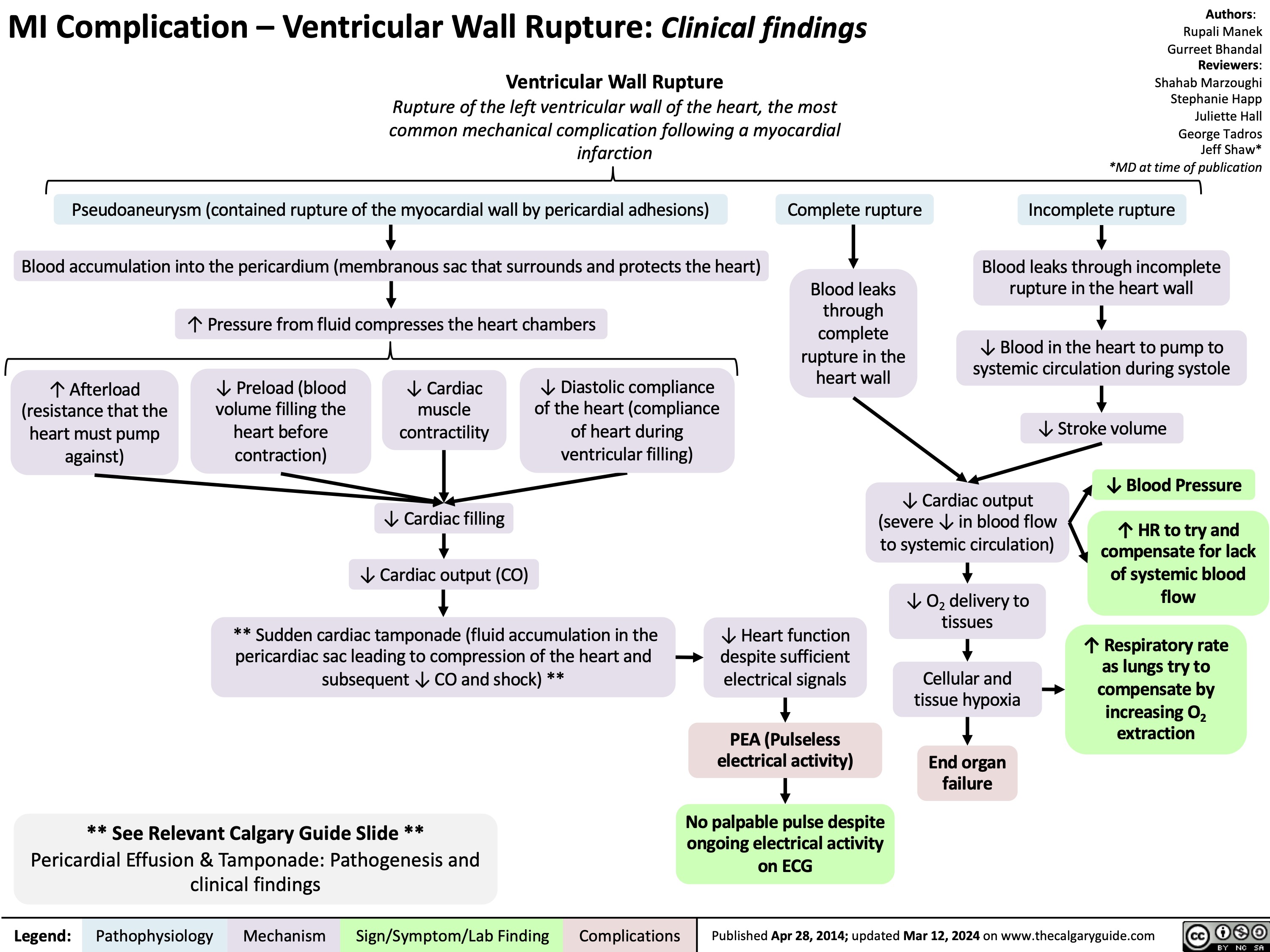
Atrial Septal Defect Pathogenesis and Clinical Findings
:
Pathogenesis and clinical findings
Genetic syndromes (e.g., Holt- Oram, Noonan, and Down)
Gene mutations (e.g., TBX5, GATA4, and NKX2-5)
Authors: Ryan Wilkie George Tadros Reviewers: Julena Foglia Haotian Wang Shahab Marzoughi Tim Prieur* * MD at time of publication
Spontaneous
Abnormal formation of the septum of the atria
Atrial Septal Defect (ASD)
An abnormal connection between the left and right atria of the heart
Following birth, lungs fill with air and resistance to blood flow in the lung vasculature ↓ Pressure within the right ventricle and right atrium ↓
Left atrial pressure exceeds right atrial pressure
Blood passes from left to right through the ASD (left-to-right shunt)
↑ Blood flow through the right heart
↑ Blood flow through
tricuspid valve
Mid-diastolic murmur
Right-sided heart dilation (enlargement of the right ventricle)
Enlarged ventricle cannot pump blood effectively
Congestive Heart Failure
↑ Blood flow through pulmonary valve and pulmonary vasculature
Right ventricular heave (visible or palpable chest wall impulse around sternum)
↑ Right atrial wall stress
Inspiration produces no net pressure change between communicating atria
Delayed closure of pulmonary valve (relative to aortic valve)
Morphologic changes in pulmonary vasculature from long standing exposure to high blood flow
Pulmonary vascular resistanceáover time, may surpass systemic vascular resistance
**Pulmonary Hypertension**
Mid-systolic ejection murmur
Heart is unable to pump enough blood to meet demand during activity (including feeding)
↑ Backup of blood in lungs
↑ Hydrostatic pressure in lung vasculature
Pulmonary edema
Damage to normal conduction of electrical signal from the atria to the ventricles
ECG Changes: Prolonged PR and QRS intervals, right bundle branch block, right axis deviation
Fixed split S2 (two distinct sounds are heard as part of S2)
Reduced exercise capacity
↓ Eating
Failure to thrive
Fatigue
Atrial arrhythmias (atrial fibrillation or flutter)
Irregular beats are felt in the chest wall
Palpitations
Dyspnea
↓ Ability to clear foreign particles from interstitium due to presence of extra fluid
Respiratory tract infections
** See Relevant Calgary Guide Slide **
Pulmonary Hypertension: Pathogenesis and clinical findings
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published Nov 1, 2014; updated Mar 21, 2024 on www.thecalgaryguide.com")
Angioedema Bradykinin Mediated
 & precursors (kininogen), triggered by ↑ systemic estrogen
Rheumatologic disorders & B- cell lymphoproliferative disease
Complement cascade activation results in ↑ C1 protease production
C1 esterase inhibitor is utilized to neutralize C1 protease, with its consumption exceeding its synthesis
Plasma cell proliferation (i.e., dyscrasia/ monoclonal gammopathy)
Immunoglobulin G antibodies act against C1 esterase inhibitor to render it non-functional
Genetic or spontaneous mutation in C1 esterase inhibitor gene
C1 esterase inhibitor deficiency
C1 esterase inhibitor dysfunction
Inhibition of angiotensin converting enzyme, dipeptidyl peptidase-4, or neprilysin induced metabolism of bradykinin
↓ Bradykinin (peptide hormone) degradation in plasma
Misfunctioning C1 esterase inhibiter is unable to inactivate bradykinin cascade members
↓ C1 esterase inhibiter results in inadequate inactivation of bradykinin cascade members
↑ Bradykinin protein production in plasma
Cutaneous Tissue
Mucosal Tissue
Epidermal layer
Dermal-Epidermal Junction
Dermal layer
Subcutaneous layer
Systemic bradykinin excess
Bradykinin-2 receptor binding on endothelial and vascular smooth muscle cells Hyperpermeability pathway activation, with the transcription of some signalling molecules taking hours Released pro-inflammatory mediators act on venules & arterioles in subcutaneous & submucosa tissues
Relaxation of vascular smooth muscle Dissociation of endothelial cell junctions
↑ Capillary blood flow ↑ Vascular permeability
↑ Plasma release into interstitial tissues (specific regions of the body hypothesized to be affected due to local differences in endothelial structure and its response to permeability inducing stimuli)
Dilation & ↑ permeability of vasculature results in fluid release into surrounding tissues
Mucosal Layer
Muscularis mucosae
Submucosal layer
Muscularis externa
Intestinal edema (Fluid buildup in Intestine tissues)
Intestinal swelling (↑ intra-abdominal pressure)
Laryngeal edema (Fluid buildup in larynx)
Swelling in larynx ↓ air flow into & out of lungs
Asphyxia (body is deprived of oxygen) Dyspnea (difficult in breathing)
Author: Aaron Varga Reviewers: Tracey Rice Sunawer Aujla Shahab Marzoughi Maharshi Gandhi Jori Hardin* Yan Yu* * MD at time of publication
Peripheral edema (Fluid buildup in extremities such as the hands, ankles, and feet)
Ascites, bowel obstruction, &/or hypovolemic shock
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published Mar 21, 2024 on www.thecalgaryguide.com")
Macrosomia Pathogenesis and Complications

Genes encoding cellular growth are mutated and induce ↑ cell proliferation
Fetus with XY chromosomes
Y chromosome predisposes fetus to excess growth factor
Pregnancy longer than 42 weeks
Birth weight ↑ as gestational age ↑
Pregnant parent with BMI > 30 kg/m2
↑ Central adipose tissue release insulin- desensitizing factors
↑ Insulin resistance promotes hepatic glucose production
Macrosomia
Parent has previously had ≥ 2 births
Average birth weight ↑ with each successive pregnancy
Pregnant parent with type 2 diabetes or gestational diabetes (1-hour 50 g glucose challenge test >140 mg/dL at 24-28 weeks gestation)
Parent’s glucose-rich blood is carried to the fetus through the placenta
↑ Levels of glucose present in fetal circulation promotes excessive growth
(Fetus grows beyond absolute birth weight (> 4000 g) regardless of gestational age)
Fetal dysregulation of glucose and fetal programming of later adiposity
Metabolic syndromes (e.g. hypoglycemia, hyperinsulinemia)
↑ Insulin levels delay pulmonary maturation
Respiratory distress
Large fetal size in the uterus
Cardiac mass ↑ in proportion to body size
Fetal cardiac remodeling (e.g. ↑ left ventricular mass)
Uterine muscle wall stretched beyond optimal range
Uterine rupture
Parent pushes fetus into birth canal
Maternal nutrition
supply is unable to meet fetus’ increased metabolic demands
↑ Uterine distension prevents uterine muscles from contracting (uterine atony)
Fetus takes longer to descend through the birth canal
Large fetal size overstretches pelvic structures
Perianal trauma (e.g. lacerations to pelvic floor, vagina, rectum)
Less space in birth canal prevents the parent from delivering the anterior fetal shoulder after the fetal head
Shoulder dystocia (baby’s shoulder stuck during birth)
Arrested labour (slow cervical dilation)
Insufficient space in birth canal to deliver fetus
Assisted vaginal birth/ cesarian section
Protracted labour (slow fetal descent)
Stillbirth
Fetal distress (↓ heart rate)
Lack of mechanical contraction of the spiral arteries, normally provided by uterine muscles
Blood loss (≥ 500-1000 mL 24 hours post birth)
Postpartum hemorrhage
Authors:
Akaya Blair
Reviewers:
Dasha Mori
Michelle J. Chen
Dr. Ian Mitchell*
* MD at time of publication
↑ Frequency/ prolonged admission (≥ 3 days) to neonatal intensive care unit
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published Mar 21, 2024 on www.thecalgaryguide.com")
Apnea of Prematurity
![Apnea of Prematurity: Pathogenesis, Signs & Symptoms, and Complications Physiologic immaturity from birth at < 37 weeks gestation
Authors: Akaya Blair Reviewers:
Dasha Mori Michelle J. Chen Danielle Nelson* * MD at time of publication
↓ Synaptic connection & poor myelination
Fetal brain areas responsible for breathing are poorly developed Immature neurologic respiratory function
Immature mechanical respiratory function
Poor hypopharyngeal muscle tone (soft upper airway helps with size and compliance of airway)
Nasal obstruction (e.g. anatomic and/or iatrogenic [suctioning, NG tubes])
Neonate is reliant on nose breathing
Airway is unable to remain open (patent)
Laryngeal/tracheal abnormalities (e.g. tracheomalacia, laryngeal edema, tracheal stenosis)
Anatomical narrowing leading to ↑ airway resistance
↑ Risk of mechanical airway obstruction
Disruption of central respiratory drive
↓ Sensitivity to increased CO2 in the ventral medulla oblongata
Disruption of peripheral respiratory reflex pathways
↓ Sensitivity to CO2 levels in peripheral carotid bodies and aortic bodies
Large head size forces neck into flexion when laying supine
Immature airway sensitive to collapse when in flexion
↑ Hypotonia (decreased muscle tone) in REM sleep
↓ Signaling to brainstem
Brainstem unable to mount appropriate ventilatory response to insufficient oxygen
Upper airway collapse
Apnea of prematurity
Respiratory pauses >20 sec or pauses <20 sec with bradycardia (<100 beats per minute), central cyanosis, and/or oxygen saturation <85% in neonates born at <37 weeks gestation and with no underlying disorders causing apnea. Most apneas in apnea of prematurity are central or mixed.
↓ Breathing rate
Bradycardia (<100 bpm)
↓ Oxygen to brain
Poor neurodevelopmental outcomes (e.g. cognitive function, brain adaptive potential and plasticity)
Hypoxemia (↓blood oxygen levels where SpO2 <85%)
↓ Oxygen and hemoglobin to mucous membranes (e.g. lips) & fingers and toes (periphery)
Central & peripheral cyanosis (bluish discoloration)
↓ Oxygen to retina
Abnormal growth of blood vessels in eyes
Retinopathy of prematurity (changes in visual acuity and possible blindness)
Death/impairment in cell function from lack of oxygen
↑ Risk of infant mortality
Imbalanced oxygen intake and CO2 output in lungs
Body transiently ↑ HR to unsuccessfully try to compensate for ↓ tissue oxygenation
Respiratory failure
Respiratory rate >60 ↓ Heart rate
↓ Blood pressure
Head bobbing Abdominal breathing
Skin mottling
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published Mar 21, 2024 on www.thecalgaryguide.com](https://calgaryguide.ucalgary.ca/wp-content/uploads/2024/03/Apnea-of-Prematurity.jpg "Apnea of Prematurity: Pathogenesis, Signs & Symptoms, and Complications Physiologic immaturity from birth at < 37 weeks gestation
Authors: Akaya Blair Reviewers:
Dasha Mori Michelle J. Chen Danielle Nelson* * MD at time of publication
↓ Synaptic connection & poor myelination
Fetal brain areas responsible for breathing are poorly developed Immature neurologic respiratory function
Immature mechanical respiratory function
Poor hypopharyngeal muscle tone (soft upper airway helps with size and compliance of airway)
Nasal obstruction (e.g. anatomic and/or iatrogenic [suctioning, NG tubes])
Neonate is reliant on nose breathing
Airway is unable to remain open (patent)
Laryngeal/tracheal abnormalities (e.g. tracheomalacia, laryngeal edema, tracheal stenosis)
Anatomical narrowing leading to ↑ airway resistance
↑ Risk of mechanical airway obstruction
Disruption of central respiratory drive
↓ Sensitivity to increased CO2 in the ventral medulla oblongata
Disruption of peripheral respiratory reflex pathways
↓ Sensitivity to CO2 levels in peripheral carotid bodies and aortic bodies
Large head size forces neck into flexion when laying supine
Immature airway sensitive to collapse when in flexion
↑ Hypotonia (decreased muscle tone) in REM sleep
↓ Signaling to brainstem
Brainstem unable to mount appropriate ventilatory response to insufficient oxygen
Upper airway collapse
Apnea of prematurity
Respiratory pauses >20 sec or pauses <20 sec with bradycardia (<100 beats per minute), central cyanosis, and/or oxygen saturation <85% in neonates born at <37 weeks gestation and with no underlying disorders causing apnea. Most apneas in apnea of prematurity are central or mixed.
↓ Breathing rate
Bradycardia (<100 bpm)
↓ Oxygen to brain
Poor neurodevelopmental outcomes (e.g. cognitive function, brain adaptive potential and plasticity)
Hypoxemia (↓blood oxygen levels where SpO2 <85%)
↓ Oxygen and hemoglobin to mucous membranes (e.g. lips) & fingers and toes (periphery)
Central & peripheral cyanosis (bluish discoloration)
↓ Oxygen to retina
Abnormal growth of blood vessels in eyes
Retinopathy of prematurity (changes in visual acuity and possible blindness)
Death/impairment in cell function from lack of oxygen
↑ Risk of infant mortality
Imbalanced oxygen intake and CO2 output in lungs
Body transiently ↑ HR to unsuccessfully try to compensate for ↓ tissue oxygenation
Respiratory failure
Respiratory rate >60 ↓ Heart rate
↓ Blood pressure
Head bobbing Abdominal breathing
Skin mottling
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published Mar 21, 2024 on www.thecalgaryguide.com")
Neonatal Necrotising Enterocolitis in Premature Neonates
 in Premature Neonates:
Pathogenesis and clinical findings
Prematurity risk factors ↓ Intestinal motility
↑ Intestinal stasis allows bacteria more time to proliferate
Bacterial overgrowth in gut
↓ Goblet cells in intestinal epithelium
↓ Intestinal mucus layer production leads to impaired mechanical defense against pathogenic bacteria
Immature tight junctions in intestinal epithelium
↑ Permeability of intestinal epithelial barrier
↑ Toll-like receptor 4 (TLR4) expression on intestinal epithelial cells
Aberrant bacterial colonization of gut
Impaired gut barrier allows for ↑ bacterial translocation across intestinal epithelium
TLR4 on intestinal mesentery endothelial cells bind lipopolysaccharides (LPS) on Gram-negative gut bacteria
Immune cells release proinflammatory mediators (TNF, IL-12, IL-18)
Cytokines mediate ↑ enterocyte apoptosis (including enteric stem cells) and ↓ enterocyte proliferation
Intestinal mucosa healing is impaired, leading to local inflammation & injury
TLR4 on intestinal epithelial cells binds LPS from Gram-negative gut bacteria
Authors: Rachel Bethune Naima Riaz Reviewers: Nicola Adderley Michelle J. Chen Kamran Yusuf* Jean Mah* * MD at time of publication
Endothelial nitric oxide synthase expression is reduced
Vasoconstriction from ↓ NO reduces blood flow to intestines
Prolonged ↓ in O2 perfusion results in irreversible intestinal mucosal cell death (necrosis)
Gas escapes into abdominal cavity
Leakage of intestinal contents irritates parietal peritoneum
Bacteria enter bloodstream
Pneumo- peritoneum
Abdominal distention
Peritonitis
Sepsis
Blood from tissue damage mixes with intestinal contents
Bloody stool
Intestinal sensory neurons detect damage and send signals to medullary vomiting centre
Bilious vomiting
Damaged intestinal cells are unable to absorb nutrients
Short gut syndrome
Persistent intestinal mucosal injury creates penetrating lesions through intestinal wall
Intestinal perforation
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published May 6, 2019; updated Mar 21, 2024 on www.thecalgaryguide.com")
Lichen Sclerosus
: Pathogenesis and clinical findings
Authors: Mina Youakim Reviewers: Elise Hansen Sunawer Aujla Shahab Marzoughi Jori Hardin* * MD at time of publication
Histamine receptor binding stimulates sensory nerve endings
Pruritus (itching)
Unknown triggers
Genetic predisposition (HLA-DQ7 and HLA-DR12)
Chronic inflammation (i.e. chronic infection, chronic toxin exposure) and trauma
Medications (e.g. carbamazepine, pembrolizumab, nivolumab, ipilimumab)
↑ Activation of CD4+ and CD8+ T cells released from macrophages (white blood cell) in the perineum and genital skin tissue infiltrate into the dermal-epidermal junction
T-cells proliferate in a horizontal linear formation Pro-inflammatory response activation
Fibroblasts (contributes to the formation of connective tissue) proliferate and persist producing altered collagen under the epidermis
Collagen deposits and hyalinizes (transforms into acellular translucent material) beneath the epidermal layer
Sclerotic plaques (localized areas of thickened skin)
T cells release of pro-inflammatory cytokines (interleukins and transforming growth factor β)
↑ Oxidative stress and cell damage
Progressive basal layer degeneration thins the overall skin thickness
Mast cells respond to increased need for immune cell flow to area
Nitric oxide is released when histamine binds to vascular receptors
Localized area appears red
Erythema (reddening of the skin)
Erosion/ulceration
Skin fissures (linear cleavage of skin)
Localized histamine release
Nitric oxide induces localized vasodilation (↑ blood flow)
Normal Skin
Lichen Sclerosus
Epidermal layer
Basal layer
Dermal-Epidermal Junction Dermal layer
Hypopigmented patches (localized, pale areas of skin)
Epidermal atrophy (crinkling paper-type skin appearance)
Thinned skin is weakened and prone to physical stress or trauma
↑ fluid in the extracellular space due to capillary leakage from ↑ blood flow
Superficial dermal edema (swelling)
Fibrotic tissue deposition following healing of damaged tissue
Atrophic Epidermal layer
Hyalinized collagen deposits
Basal layer degeneration Band of T-cell infiltrate
Dermal layer
Scarring
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published Mar 25, 2024 on www.thecalgaryguide.com
Lichen Sclerosus (genital manifestation): Pathogenesis and clinical findings
Authors: Mina Youakim Reviewers: Elise Hansen Sunawer Aujla Shahab Marzoughi Jori Hardin* * MD at time of publication
Histamine receptor binding stimulates sensory nerve endings
Pruritus (itching)
Unknown triggers
Genetic predisposition (HLA-DQ7 and HLA-DR12)
Chronic inflammation (i.e. chronic infection, chronic toxin exposure) and trauma
Medications (e.g. carbamazepine, pembrolizumab, nivolumab, ipilimumab)
↑ Activation of CD4+ and CD8+ T cells released from macrophages (white blood cell) in the perineum and genital skin tissue infiltrate into the dermal-epidermal junction
T-cells proliferate in a horizontal linear formation Pro-inflammatory response activation
Fibroblasts (contributes to the formation of connective tissue) proliferate and persist producing altered collagen under the epidermis
Collagen deposits and hyalinizes (transforms into acellular translucent material) beneath the epidermal layer
Sclerotic plaques (localized areas of thickened skin)
T cells release of pro-inflammatory cytokines (interleukins and transforming growth factor β)
↑ Oxidative stress and cell damage
Mast cells respond to increased need for immune cell flow to area
Nitric oxide is released when histamine binds to vascular receptors
↑ fluid in the extracellular space due to capillary leakage from ↑ blood flow
Superficial dermal edema (swelling)
Epidermal atrophy (crinkling paper- type skin appearance)
Thinned skin is weakened and prone to physical stress or trauma
Localized histamine release
Nitric oxide induces localized vasodilation (↑ blood flow)
Localized area appears red
Erythema (reddening of the skin)
Erosion/ulceration
Skin fissures (linear cleavage of skin)
Normal Skin
Lichen Sclerosus
Epidermal layer
Basal layer
Dermal-Epidermal Junction Dermal layer
Atrophic Epidermal layer
Hyalinized collagen deposits
Basal layer degeneration Band of T-cell infiltrate
Dermal layer
Progressive basal layer degeneration thins the overall skin thickness
Hypopigmented patches (localized, pale areas of skin)
Fibrotic tissue deposition following healing of damaged tissue
Scarring
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published MONTH, DAY, YEAR on www.thecalgaryguide.com
Chronic inflammation (i.e. chronic
Reviewers:
Dermal layer
Unknown triggers
Genetic predisposition (HLA-DQ7 and HLA-DR12)
infection, chronic toxin exposure) and trauma
Medications (e.g. carbamazepine, pembrolizumab, nivolumab, ipilimumab)
Elise Hansen Sunawer Aujla Shahab Marzoughi Jori Hardin* * MD at time of publication
↑ Activation of CD4+ and CD8+ T cells released from macrophages (white blood cell) in the perineum and genital skin tissue infiltrate into the dermal-epidermal junction
T-cells proliferate in a horizontal linear formation
Pro-inflammatory response activation
↑ Expression of MicroRNA-155 (enhances pro-inflammatory response and ↓ expression of tumor suppression genes)
Fibroblasts (contributes to the formation of connective tissue) proliferate and persist producing altered collagen
Collagen deposits and hyalinizes (transforms into acellular translucent material) beneath the epidermal layer
Sclerotic plaques (localized areas of thickened skin)
T cells release of pro-inflammatory cytokines (interleukins and transforming growth factor β)
↑ Oxidative stress and cell damage
Mast cells respond to increased need for immune cell flow to area
Nitric oxide is released when histamine binds to vascular receptors
↑ fluid in the extracellular space due to capillary leakage from ↑ blood flow
Superficial dermal edema (swelling)
Epidermal atrophy (crinkling paper- type skin appearance)
Thinned skin is weakened and prone to physical stress or trauma
Lichen Sclerosus
Localized histamine release
Nitric oxide induces localized vasodilation (↑ blood flow)
Localized area appears red
Erythema (reddening of the skin)
Erosion/ulceration
Skin fissures (linear cleavage of skin)
Atrophic Epidermal layer
Hyalinized collagen deposits
Basal layer degeneration Band of T-cell infiltrate
Histamine receptor binding stimulates sensory nerve endings
Pruritus (itching)
Normal Skin
Progressive basal layer degeneration thins the overall skin thickness
Hypopigmented patches (localized, pale areas of skin)
Epidermal layer
Basal layer
Dermal-Epidermal Junction
Fibrotic tissue deposition following healing of damaged tissue
Scarring
Chronic inflammation (i.e. chronic
Reviewers:
Dermal layer
Unknown triggers
Genetic predisposition (HLA-DQ7 and HLA-DR12)
infection, chronic toxin exposure) and trauma
Medications (e.g. carbamazepine, pembrolizumab, nivolumab, ipilimumab)
Elise Hansen Sunawer Aujla Shahab Marzoughi Jori Hardin* * MD at time of publication
↑ Activation of CD4+ and CD8+ T cells released from macrophages in the perineum and genital skin tissue infiltrate into the dermal-epidermal junction
T-cells proliferate in a horizontal linear formation Pro-inflammatory response activation
↑ Expression of MicroRNA-155 (short segment of RNA which enhances pro- inflammatory response and ↓ expression of tumor suppression genes)
Fibroblasts proliferate and persist producing altered collagen
Collagen deposits and hyalinizes (transforms into acellular translucent material) beneath the epidermal layer
Sclerotic plaques (localized areas of thickened skin)
T cells release of pro-inflammatory cytokines (interleukins and transforming growth factor β)
↑ Oxidative stress and cell damage
Progressive basal layer degeneration thins the overall skin thickness
Hypopigmented patches (localized, pale areas of skin)
Epidermal layer
Basal layer
Dermal-Epidermal Junction
Mast cells respond to increased need for immune cell flow to area
Nitric oxide is released when histamine binds to vascular receptors
Superficial dermal edema (swelling)
Epidermal atrophy (crinkling paper- type skin appearance)
Localized histamine release
Nitric oxide induces localized vasodilation (↑ blood flow)
Localized area appears red
Erythema (reddening of the skin)
Histamine receptor binding stimulates sensory nerve endings
Pruritus (itching)
Normal Skin
Lichen Sclerosus
Skin fissures (linear cleavage of skin)
Erosion/ulceration
Fibrotic tissue deposition following healing of damaged tissue
Scarring
Atrophic Epidermal layer
Hyalinized collagen deposits
Basal layer degeneration Band of T-cell infiltrate
Reviewers:
Dermal layer
Unknown triggers
Genetic predisposition (HLA-DQ7 and HLA-DR12)
Chronic inflammation (i.e. chronic infection, chronic toxin exposure) and trauma
Medications (e.g. carbamazepine, pembrolizumab, nivolumab, ipilimumab)
Elise Hansen Sunawer Aujla Shahab Marzoughi Jori Hardin* * MD at time of publication
↑ Activation and infiltration of CD4+ and CD8+ T cells into the dermal-epidermal junction
T-cells proliferate in a band (horizontal linear) formation Pro-inflammatory response activation
↑ Expression of MicroRNA-155 (short segment of RNA which enhances pro-inflammatory response and ↓ expression of tumor suppression genes)
Fibroblasts proliferate and persist producing altered collagen
Collagen deposits and hyalinizes (transforms into acellular translucent material) beneath the epidermal layer
Sclerotic plaques (localized areas of thickened skin)
T cells release of pro-inflammatory cytokines (interleukins and transforming growth factor β)
↑ Oxidative stress and cell damage
Progressive basal layer degeneration thins the epidermis
Hypopigmented patches (localized, pale areas of skin)
Epidermal layer
Basal layer
Dermal-Epidermal Junction
Mast cells respond to increased need for immune cell flow to area
Nitric oxide is released when histamine binds to vascular receptors
Superficial dermal edema (swelling)
Epidermal atrophy (crinkling paper- type skin appearance)
Localized histamine release
Histamine receptor binding stimulates sensory nerve endings
Pruritus (itching)
Skin fissures (linear cleavage of skin)
Nitric oxide induces localized vasodilation (↑ blood flow)
Localized area appears red
Erythema (reddening of the skin)
Scarring
Atrophic Epidermal layer
Hyalinized collagen deposits
Basal layer degeneration Band of T-cell infiltrate
Erosion/ulceration
Fibrotic tissue deposition following healing of damaged tissue
Normal Skin
Lichen Sclerosus
Chronic inflammation (i.e. chronic
Elise Hansen
Unknown triggers
Genetic predisposition infection, chronic toxin exposure) Medications (e.g. carbamazepine, (HLA-DQ7 and HLA-DR12) and trauma pembrolizumab, nivolumab, ipilimumab)
↑ Activation and infiltration of CD4+ and CD8+ T cells into the dermal-epidermal junction
Sunawer Aujla Shahab Marzoughi Name Name* * MD at time of publication
T-cells proliferate in a band (horizontal linear) formation
Pro-inflammatory response activation (increase in pro-inflammatory cytokines such as Interleukins 1-alpha and 1-beta)
↑ Expression of MicroRNA-155 (short segment of RNA which enhances pro-inflammatory response and ↓ expression of tumor suppression genes)
Fibroblasts proliferate and persist producing altered collagen
T cells release of pro-inflammatory cytokines (interleukins and transforming growth factor β)
↑ Oxidative stress and cell damage
Progressive basal layer degeneration thins the epidermis
Hypopigmented patches (localized, pale areas of skin)
Histamine receptor binding stimulates sensory nerve endings
Localized area appears red
Localized histamine release
Localized vasodilation (↑ blood flow)
Superficial dermal edema (swelling)
Pruritus
Erythema
Collagen deposits and hyalinizes (transforms into acellular translucent material) beneath the epidermal layer
Normal Skin
Sclerotic plaques (localized areas of thickened skin)
Epidermal layer
Basal layer
Dermal-Epidermal
Junction Dermal layer
Skin fissures (linear cleavage of skin)
Bleeding
Erosion/Ulceration
Epidermal atrophy (crinkling paper- type skin appearance)
Lichen Sclerosus
Atrophic Epidermal layer
Hyalinized collagen deposits
Basal layer degeneration
Band of T-cell infiltrate
Dermal layer
Fibrotic tissue deposition following healing of damaged tissue
Scarring
Lichen Sclerosus (genital manifestation): Pathogenesis and clinical findings
Authors: Mina Youakim Reviewers: Elise Hansen Sunawer Aujla Shahab Marzoughi Name Name* * MD at time of publication
Pruritus
Histamine receptor binding stimulates sensory nerve endings
Localized area appears red
Erythema
Unknown triggers
Genetic predisposition (HLA-DQ7 and HLA-DR12)
Chronic inflammation and trauma
Medications (e.g. carbamazepine, pembrolizumab, nivolumab, ipilimumab)
↑ Activation and infiltration of CD4+ and CD8+ T cells into the dermal-epidermal junction
T-cells proliferate in a band formation Pro-inflammatory response activation
↑ Expression of MicroRNA-155 (short segment of RNA which enhances pro-inflammatory response and ↓ expression of tumor suppression genes)
Fibroblasts proliferate and persist producing altered collagen
T cells release of pro-inflammatory cytokines (interleukins and transforming growth factor β)
Localized histamine release
Localized vasodilation (↑ blood flow)
Superficial dermal edema (swelling)
Collagen deposits and hyalinizes beneath the atrophic epidermal layer
Hypopigmented patches (localized, pale areas of skin)
↑ Oxidative stress and cell damage
Progressive basal layer degeneration thins the epidermis
Skin fissures
Lichen Sclerosus
Epidermal atrophy (crinkling paper- type skin appearance)
Sclerotic plaques (localized areas of thickened skin)
Bleeding Scarring
Erosion/Ulceration
Normal Skin
Epidermal layer
Basal layer
Dermal-Epidermal
Junction Dermal layer
Atrophic Epidermal layer
Hyalinized collagen deposits
Basal layer degeneration
Band of T-cell infiltrate
Dermal layer
Legend:
Published MONTH, DAY, YEAR on www.thecalgaryguide.com
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications")
Infectious Small Bowel Diarrhea

May produce Shiga toxins (a specific family of toxins that can lead to complications)
Authors: Noriyah Al Awadhi Yan Yu Sara Cho Reviewers: Paul Ratti Jason Baserman Shahab Marzoughi Kerri Novak* * Indicates MD at time of publication
Y. enterocolitica
(also milk, cheese)
Shellfish, undercooked seafood
V. parahaemolyticus V. cholerae
Enterohemorrhagic E. coli (EHEC)
Daycare centers/nurseries
Drinking/swimming in bad water — mountains/wells
S. aureus
B. cereus
Noroviruses
Rotavirus
G. lamblia
C. parvum
May produce emetic toxins (toxins that cause vomiting)
Adequate amount of organism and/or toxin is ingested
Organism adheres to the intestinal liningàorganism colonizes the small intestine Organism releases enterotoxin (a toxin that affects the intestines)
Binds to and disrupts intestinal transporters used in secretion and absorption of water and electrolytes
Toxin enters systemic vasculature
Shiga toxins inhibit ADAMTS13 (cleaving enzyme)
Failure to cleave von Willebrand Factor (vWF) multimers
Accumulation of vWF multimers
Platelets and thrombi accumulate in microvasculature
Hemolytic uremic syndrome (hemolytic anemia, thrombocytopenia, and acute renal damage)
↑ Water and electrolyte secretion
↓ Fluid absorption
Chemoreceptor trigger zone in medulla detects circulating emetic toxins
Nausea & vomiting
Triggers release of inflammatory mediators and cytokines that travel to the central nervous system
Prostaglandin is synthesized and released
Neurotransmitter cyclic AMP (cAMP) is released
cAMP ↑ hypothalamic thermoregulation set point
Fever
Large volume profuse diarrhea
Loss of water
Distention (swelling) of intestines stimulates visceral sensory pain fibers
Visceral pain fibers crosstalk with somatic nerves from the same spinal cord level
Occasional referred pain/cramping (typically diffuse pain around the abdomen)
Loss of bicarbonate, sodium, potassium, magnesium, and chloride
Dehydration
Electrolyte deficiency
Metabolic acidosis (pH < 7.4 & serum bicarbonate < 24)
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published Aug 7, 2012; updated Mar 25, 2024 on www.thecalgaryguide.com")
Post-Renal Acute Kidney Injury AKI
: Pathogenesis and clinical findings
Blood clot or cellular debris
Foreign body
Neurogenic bladder
Obstruction intrinsic to urine excretion system
Nephrolithiasis (kidney stones)
Anatomical defect(s)
Intra- abdominal adhesions
Retroperitoneal fibrosis (scar-like tissue)
Benign or malignant masses
Prostate cancer
Benign prostatic hyperplasia
↓ Urine flow across point of obstruction Obstruction extrinsic to urine excretion system
Urine buildup distends urine collecting system (hydronephrosis)
Compression of renal vasculature due to mass effect
↑ Volume/pressure proximal to obstruction
↑ Intratubular pressure
↓ Pressure gradient between glomerular afferent arteriole and Bowman’s space
Casts occlude tubules
↓ Filtration of plasma into nephrons
↓ Glomerular Filtration Rate (GFR)
Obstruction is relieved ↓ Intratubular pressure Rapid GFR ↑
Rapid diuresis of fluid and electrolytes
Dilated pelvicalyceal system on ultrasound
↓ Urine output
↓ Venous drainage
and arterial supply
Local ischemia and inflammation of kidney
Impaired resorption, excretion, and fine tuning by tubules
Acute Tubular Necrosis (ATN) with granular casts
↓ Urine output
↓ Clearance of free water and solutes
↑ Intravascular volume
↑ Serum creatinine
↓ Medullary solute concentration, ischemia, ↓ response of collecting ducts to antidiuretic hormone
Lasts > 24hrs
Post-obstructive diuresis causes hypovolemia and electrolyte derangements
Authors: David Campbell, Matthew Hobart Reviewers: Raafi Ali, Luiza Radu Huneza Nadeem, Marissa Zhang, Julian Midgley* * MD at time of publication
Resolves < 24hrs in euvolemia
Physiologic post- obstructive diuresis
↑ Na+ and Cl- delivery to distal convoluted tubule is sensed by macula densa
Secretion of adenosine by macula densa
Adenosine constricts afferent arterioles
↓ GFR
↓ Renal clearance of drugs and waste products
↑ Venous hydrostatic pressure
↑ Volume in arterial system overwhelms pressure regulation mechanisms
Hypertension
Fluid extravasation from veins and capillaries
Generalized Edema
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published Mar 25, 2024 on www.thecalgaryguide.com")
Irritant Contact Dermatitis Pathogenesis and Clinical Findings

Hyperkeratotic skin is less amenable to skin stretching and pressure
Skin fissures (cracks in the skin)
Irritant agents
(abrasives, cleaning solution, oxidative & reducing agents, dust, soils, water)
Acute exposure triggers inflammatory response
Keratinocytes undergo cytotoxic damage with ↑ neutrophil & cytokine release
Common occupational exposures (housekeeping, cleaning, catering, medical/dental, construction)
Risk factors
(atopy, fair skin, low temperature, low humidity)
Stimulation of local nociceptors (free nerve endings extend into the mid epidermis)
Perivascular (around the blood vessel) inflammation causes histamine release from mast cells
Damaged keratinocytes are destroyed via
apoptosis (programmed cell death)
Epidermal Necrosis (death of epidermal tissue)
Shedding of necrotic tissue
Ulceration (deep open wound on skin)
Burning
pain (uncomfortable stinging sensation)
Pruritus (itching)
Histamine causes local blood vessel dilation and ↑ blood flow to the area of skin affected
Erythema (area appears red from ↑ blood flow)
Burning & Itching Spongiosis
Neutrophils Neutrophils
Histamine causes local blood vessel walls to have
↑ permeability, thereby ↑ leakage of fluid
Spongiosis
(↑ fluid between keratinocytes in the epidermis)
Fluid continues to build up from ongoing inflammation
Vesiculation (fluid collections in the epidermis)
Long-term skin scratching causes chronic irritation which eventually hardens the skin
Lichenification
(thick, hardened patches of skin)
↑ Overall keratin production
Hyperkeratosis (thickening of the outermost skin layer)
Further fluid buildup bursts vesicles leaving behind erosions and dried crust on the epidermis
Crust (scaling over the skin)
Lichenification
Erosions (open sore on skin)
Ulcer
Epidermis
Perivascular Inflammation
Hyperkeratosis
Dermis
Dermal-epidermal junction
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published Oct 19, 2016; updated Mar 30, 2024 on www.thecalgaryguide.com")
Febrile Neutropenia Pathogenesis and clinical findings
: Pathogenesis and clinical findings
Administration of cytotoxic chemotherapy for cancer treatment
Acquired aplastic anemia
Autoreactive T cells destroy bone marrow stem cells
Congenital mutations in ELA2 gene (encodes neutrophil elastase)
↑ Neutrophil apoptosis
Chemotherapy eliminates beneficial bacterial species from gut microbiota
New microbiota composition allows for growth of colonizing bacteria
Indwelling catheters inserted to deliver chemotherapy
Skin-colonizing bacteria access tissues through catheters
Bacteria penetrate tissue barriers
Chemotherapy injures gastrointestinal mucosa
Broken mucosal barrier increases susceptibility to infections
Chemotherapy destroys circulating neutrophils
Chemotherapy impairs bone marrow stem cells
↓ Production of neutrophils
Neutropenia (Absolute Neutrophil Count (ANC) < 0.5x109 cells/L
↓ Immune cell ↓ Production of engulfment of microbes inflammatory mediators
Fewer circulating neutrophils blunts the innate immune response
Pathogens enter bloodstream from tissues Systemic infection
Authors: Max Lazar Braxton Phillips Reviewers: Naman Siddique Michelle J. Chen Lynn Savoie* * MD at time of publication
Dormant viral infections reactivate (e.g. cytomegalovirus, herpes simplex virus)
↑ Susceptibility to common bacterial infections
Positive blood bacterial cultures
Fever (T ≥ 38.3 oC or sustained T ≥ 38 oC for 1 hour)
Immune system mounts an excessive inflammatory response that damages tissues and organs
Sepsis
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published Dec 5, 2018; updated Mar 30, 2024 on www.thecalgaryguide.com")
Overview of Ischemic Heart Disease
: Pathogenesis of the various types of IHD
Authors:
Sean Spence Vaneeza Moosa Reviewers: Tristan Jones Jason Baserman Yan Yu
Michelle J. Chen Frank Spence* * MD at time of publication
↑ Serum low-density lipoprotein (LDL)
↑ Availability of lipids that deposit in arterial wall
↓ Serum high-density lipoprotein (HDL)
↓ LDL removal from coronary artery walls (transport of LDL to liver is impaired)
Atherosclerosis
Conditions predisposing to vessel wall endothelial cell dysfunction (e.g. metabolic syndrome, smoking, hypertension, physical inactivity)
Vessel wall vulnerable to infiltration by LDL and immune cells
Arterial wall degeneration, characterized by fat deposition (atheromatous plaque) in and fibrosis of the inner layer of arteries
Stable atheromatous plaque in coronary arteries
Fibromuscular cap (formed by smooth muscle cells) overlying fatty plaque contents remains intact & plaque contents are not released into vessel lumen
Plaque serves as a fixed lumenal obstruction to blood flow
If vessel stenosis (narrowing) is significant (≥70%) myocardial oxygen demand starts to exceed supply (especially with exertion)
Heart experiences a predictable & transient reduction in blood flow (myocardial ischemia)
Unstable atheromatous plaque in coronary arteries
The fibromuscular cap overlying fatty plaque ruptures
Thrombogenic plaque contents (especially tissue factor) are exposed to the coagulation factors in the vessel lumen
Activation of platelets & the clotting cascade at the site of rupture
Thrombus forms over already partially occlusive plaque and further partially or completely occludes lumen
↓ Perfusion (blood flow) of myocardium
Cardiomyocytes experience a transient decrease in blood flow (transient ischemia)
Unstable Angina
Cardiomyocytes experience death (infarction)
Myocardial Infarction (MI)
Stable Angina
Acute Coronary Syndromes (ACS)
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published Jan 8, 2013; updated Mar 30, 2024 on www.thecalgaryguide.com")
Bullous Pemphigoid

Author:
Danny Guo
Mina Youakim
Reviewers:
Yan Yu
Shahab Marzoughi
Jason Baserman
Laurie Parsons*
Régine Mydlarski*
* MD at time of publication
Preceding disease triggers Infections (e.g. Human Herpes Physical/Environmental triggers (e.g. (e.g. Psoriasis, Lichen Planus) virus, Epstein-Barr virus) Phototherapy, radiotherapy, burns)
Development of autoantibodies against the basement membrane antigens (i.e. BPAG1 and BPAG2) (can occur anywhere on the body)
Autoimmune destruction of the basement membrane
Antibodies activate the complement system and recruit inflammatory cells Further weakened adhesion between epidermis and dermis
Accumulation of extracellular serous fluids in a pseudo- pocket between the epidermis and the dermis
Unknown etiologies
Circulating Immunoglobulin G & E antibodies trigger either a prodrome or concurrent itch response (exact mechanism unknown)
Pruritis (itch)
Epidermal layer Serous fluid pocket
Urticaria (hives)
Widespread Bullae (serum-filled lesions)
Epidermal layer
Dermal-Epidermal
Junction Dermal layer
Antibody against keratinocytes and
Disrupted Dermal-Epidermal Junction
Y
hemidesmosomes
Normal Skin
Bullous Pemphigoid
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published Nov 1, 2012; updated Apr 20, 2024 on www.thecalgaryguide.com
Y Y
Y")
Deep Partial Thickness Burns Pathogenesis and Clinical Findings

Ionizing radiation gets into contact with DNA
Damage to keratinocytes
Fire (Flash fire or direct contact with flame)
Contact (Hot solid objects)
Scalding (Hot liquid via immersion, spill or splash)
Chemical (Strong acid, alkali or irritant gas)
Electrical (Contact with exposed electrical wiring/appliances)
Author and Illustrator: Amanda Eslinger Maharshi Gandhi Reviewers:
Alexander Arnold
Shahab Marzoughi
Duncan Nickerson*
* MD at time of publication
Direct transfer of heat energy
Coagulation necrosis (cell death due to ischemia) is induced
Deep Partial Thickness Burn
Injury to the epidermal layer and both the papillary and a portion of the reticular layer of the dermis
Transfer of heat energy & direct injury to cellular membranes
Epidermis
Papillary dermis Reticular dermis
Sub- cutaneous Tissue
↑ vascular permeability due to damage
Fluid leak results in edema between dermal & epidermal layer
Blisters (a bubble on the skin containing fluid)
Thin epidermal layer forming fluid-filled vesicle breaks open
Cutaneous capillary bed is destroyed
Blood flow to injured area is compromised
Pressing on the skin doesn’t easily force blood cells out of the area
Non-blanchable skin
Vasodilation in fascia (a thin casing of connective tissue) underlying subcutaneous tissue
Inflammatory mediators such as cytokines and prostaglandins activate fibroblasts
Collagen deposition and subsequently induration (thickening of skin)
Some healthy dermal appendages surrounded by islands of undamaged epithelial cells
Possible spontaneous re-epithelialization in 2-9 weeks
During re-epithelization of burns there can be excessive deposition of collagen and other extracellular matrix components
Thick raised scars in dermis due to excess collagen deposition
Somatosensory structures (nociceptors, thermoreceptors, and mechanoreceptors) are completely injured within the dermis
Analgesia of impacted area (the inability to feel pain)
Ongoing Inflammation and dilated blood vessels
Red Induration (Thickened skin appearing red)
Compromised blood flow and more extensive tissue damage
White Induration (Thickened skin appearing white)
Moist Wound
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published Dec 2, 2013, updated Apr 20, 2024 on www.thecalgaryguide.com")
Fetal Complications of Labour and Vaginal Delivery

Abnormal positioning increases risk of umbilical cord becoming entangled or dropping in front of the fetus in the birth canal
Passage of the fetus through birth canal compresses the umbilical cord
Edema
Perinatal hypoxia
Signs of fetal distress (e.g. late decelerations, changes in fetal heart rate, ↓ fetal movement)
Infant death
Uterine contractions are too weak to progress labour
Prolonged time in labour results in increased total number of contractions
Fractures (most commonly affecting the clavicle)
Uterine contractions compress blood vessels in the placenta, resulting in reduced blood flow
Severe obstetric hemorrhage causes circulating blood flow to decrease as blood volume is lost (see relevant slide for pathogenesis)
Small birth canal to baby size ratio
Lack of space traps the fetal shoulders following delivery of the head (shoulder dystocia)
Excessive lateral traction or stretching damages nerves in the brachial plexus
Erb’s palsy (paralysis of the arm caused by injury to C5-C6 of the brachial plexus)
Shearing force on the skull and scalp ruptures blood vessels
Cephalohematoma (blood accumulation below periosteum)
Boggy, soft lump on infant’s head
Soft tissue injury triggers release of inflammatory signaling molecules, which ↑ vascular permeability
Caput succedaneum (scalp swelling)
Boggy, soft swelling of infant’s head that extends across suture lines
Mild soft tissue injury
Hypoxic ischemic encephalopathy
Variable signs of brain injury: seizures, hypotonia, difficulty feeding, etc.
Cerebral palsy
Developmental delay, hypotonia, spasticity, etc.
Authors: Jasmine Nguyen Reviewers: Akanksha Bhargava Michelle J. Chen Sarah Glaze* * MD at time of publication
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published Apr 29, 2024 on www.thecalgaryguide.com")
Operative Vaginal Delivery Indications
: Indications
Authors: Taylor Pigott Akaya Blair Reviewers: Michelle J. Chen Sylvie Bowden* Stephanie Cooper* Sarah Glaze* * MD at time of publication
Abnormal fetal heart rate/fetal distress
↓ Risk of maternal and fetal morbidity/ morality
Rupture of membranes without fetal head adequately applied to maternal pelvis
Small pelvic outlet Narrow vaginal canal
Cardiovascular disease
Diabetes Hypertension Abdominal trauma
Short umbilical cord
Large for gestational age (LGA) fetus
Neurological/ muscular disease
↑ Pain during labor
Maternal contraindication to Valsalva maneuver (e.g., cardiac disease, cystic lung)
Rush of fluid past the fetal head out through cervix
Umbilical cord prolapse
Umbilical vasoconstriction
Umbilical cord compression
↓ Blood flow through umbilical cord
Fetal presenting part applies pressure on umbilical cord
Impaired vascular function and narrowed vessels ↓ blood flow around the body
↓ Oxygen transfer across the placenta
Fetal oxygen deprivation and hypoxia
Operative vaginal delivery with forceps or vacuum expedites delivery
Placenta partially or completely separates from uterine wall (abruption)
Increased tension of umbilical cord on placenta
Fetal shoulder is lodged behind maternal pubic symphysis
Delivery of the body is delayed
Inability to adequately bear down to push
Myometrium runs out of energy from repeated contractions and becomes less able to contract (↓ uterine tone)
Uterine spiral arteries dependent on contractions for vasoconstriction remain dilated, allowing blood flow
Postpartum hemorrhage
Uterine rupture
↑ Risk of future pelvic organ prolapse
↑ Risk of future incontinence
Prolonged labour
Repeated contraction/ relaxation of the uterus without progress tears and damages uterine muscles
↓ Maternal endurance
Inadequate fluid and food intake prior to/ during labor
Multiples
↑ Sympathetic nervous system activation during labor
Maternal exhaustion
Uterine muscles stretch to accommodate multiple growing fetuses
↑ Pushing against pelvic floor and perineum
Pushing force damages pelvic floor muscles and nerves
↓ Myometrial contractility
↑ Need for vaginal exams to check on labour progress
Bacterial overgrowth in vagina and/or uterus
Maternal infection
Fetal infection
↓ Strength of contractions
Epidural anesthetic
Inability to ambulate to urinate
↑ Urinary catheterizations
↑ Epinephrine secretion from adrenal cortex
Blood is shunted away from uterus/internal organs and towards heart and skeletal muscle
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published 03, ##, 2023 on www.thecalgaryguide.com")
Costochondritis

Infection spreads through blood to ribcage and chest wall
Age-related degeneration of cartilage within ribcage joints
Immune cells present in joints secrete proinflammatory cytokines
Cytokines sensitize nociceptors (pain receptors) in joints
Repetitive trauma or strain to chest wall muscles (e.g. chronic cough, chest wall injury, overexertion of chest wall muscles)
Excessive stretch and tearing of ribcage cartilage and ligaments
Autoimmune disease (e.g. rheumatoid arthritis)
Antigen presenting cells display self-antigens to CD4+ T cells
Mechanical stimuli (e.g. Damaged cells release cytokines Pathogens activate immune cells at site of stretch) activate nociceptors that recruit immune cells inflammation
Costochondritis
Benign inflammation of the ribcage (costal) cartilage, particularly at the costosternal junctions (connection between sternum and cartilage) and the costochondral junctions (connection between the cartilage and rib)
Authors:
Michelle J. Chen Reviewers:
Raafi Ali
Yan Yu*
Gerhard Kiefer*
* MD at time of publication
Cartilage, ligament, or muscle injury
Presence of foreign pathogen in costal cartilage
Injury or pathogen activates inflammatory cascade in the ribcage cartilage
Immune cells proliferate so that they exceed the available space in the cartilage matrix of the ribcage
Cartilage swelling compresses intercostal nerves
Compression acts as a mechanical stimulus to activate nociceptors
Sharp, localized pain reproducible upon palpation over ribcage
Immune cells secrete cytokines
Cytokines activate nociceptors so that they become more sensitive to mechanical stimuli
Muscle or ligament stretch from normal use activates sensitized nociceptors
Pain increases with inspiration or cough
Natural resolution of inflammation over time
Macrophages phagocytose apoptotic cells (immune and cartilage cells) and cellular debris
Immune cells release factors that degrade proinflammatory cytokines
Fewer cytokines reduce immune cell recruitment into costal cartilage
Pain usually self-resolves
Immune cells release lipid molecules that bind to the same cells (autocrine signaling) to inhibit further production of inflammatory cytokines and chemokines
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published Apr 29, 2024 on www.thecalgaryguide.com")
Acute Otitis Media Complications
 accumulates in mastoid air spaces
Untreated middle ear infection allows further bacterial proliferation
Persistent effusion
causes ion channel changes in inner ear
Composition of endolymph and perilymph in inner ear changes
Vestibular/Labyrinth dysfunction
Feelings of Vertigo imbalance
Purulent discharge from middle ear through perforation
Otorrhea (ear discharge)
↑ Pressure in middle ear
Pressure stretches tympanic membrane
Cytokines reach hypothalamus through the bloodstream
Hypothalamus responds to stimulation and ↑ thermoregulatory set-point
High-grade fever
Infection spreads into bloodstream
Sepsis
Cytokines alter metabolism pathways of neurotransmitters in the brain
Cerebral cortex dysfunction
Infection spreads to cranium
Intracranial complications (e.g., meningitis, brain abscess, thrombus)
Author: Jody Platt Stephanie de Waal Reviewers: Yan Yu Elizabeth De Klerk William Kim Annie Pham Michelle J. Chen Danielle Nelson* * MD at time of publication
Helper T cells and macrophages release inflammatory cytokines into the bloodstream
Perforation of tympanic membrane
↓ Conduction of sound waves
Conductive hearing loss
Pressure in middle ear diffuses out of perforation
↓ Tympanic membrane stretching
Otalgia (ear pain) fades
Accumulated debris compresses cranial nerve VII
Facial nerve palsy
Mastoid abscess
Febrile seizures
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published Feb 28, 2013, updated Apr 29, 2024 on www.thecalgaryguide.com")
Circle of Willis Anatomy and Physiology

Anterior Cerebral Artery (ACA)
Ischemic stroke (brain damage due to ischemia)
Inadequate blood flow & oxygenation of brain tissue
Occlusion or narrowing
Supplies midbrain, thalamus, & occipital lobe
Posterior Cerebral Artery (PCA) Vertebral Arteries (VA)
Inadequate blood flow & oxygenation of brain tissue
Occlusion or narrowing
Supplies lateral portions of frontal, temporal & parietal lobes (upper extremity & facial regions of motor/sensory cortices)
Middle Cerebral Artery (MCA) P2 segment
A1 segment
Supplies the MCA & ACA
A2 segment
Collateral circulation redistributes blood flow to maintain continuous blood supply
Occlusion or narrowing of vessels within the Circle of Willis
Anterior Communicating Artery (Acomm)
Weakness in the blood vessel walls
Aneurysm formation causes mass effect (displacement) on nearby structures
Supplies BA & PCA, lateral medulla & cerebellum via posterior inferior cerebellar artery (PICA), & upper spinal cord via anterior spinal artery (ASA)
Basilar Artery (BA)
Supplies occipital lobe, cerebellum & brainstem
Thromboembolism (blood clot obstruction)
Inadequate blood flow & oxygenation of brain tissue
Ischemic stroke
Damage to the ventral pons
Locked-in syndrome (quadriplegia, loss of voluntary breathing & speaking, intact cognition & blinking)
Internal Carotid Arteries (ICA)
Posterior Communicating Artery (Pcomm)
Weakness in the blood vessel walls
Aneurysm formation causes mass effect (displacement) on nearby structures
Oculomotor nerve (cranial nerve III): double vision & absent pupil response to light
Legend:
Pathophysiology
Mechanism Sign/Symptom/Lab Finding Complications Published Nov 5, 2018, updated Apr 29, 2024 on www.thecalgaryguide.com
P1 segment
Ruptured aneurysm: subarachnoid hemorrhage
Inadequate blood flow & oxygenation of brain tissue
Hemorrhagic stroke (brain damage due to bleeding)
Optic nerve (cranial nerve II): ↓ visual acuity
Frontal lobe: headache & psychological changes
Authors: Josh Kariath, Rafael Sanguinetti Reviewers: Andrea Kuczynski, Luiza Radu Gary Klein* * MD at time of publication")
Sickle Cell Disease Pathogenesis Clinical Findings and Complications
 Liu Priyanka Grewal Reviewers: Alexander Arnold Luiza Radu JoyAnne Krupa Yan Yu* Lynn Savoie* * MD at time of publication
Hemoglobin S (HbS) variant formed instead of normal Hemoglobin A (HbA)
Hb electrophoresis shows approximately 45% HbS, 52% HbA (ααββ), 2% HbA
Hb electrophoresis shows approximately 90% HbS, 8% HbF, & 2% HbA .
No HbA present
(ααδδ), & 1% HbF (ααγγ;2 fetal hemoglobin)
Heterozygous: point mutation in one of the two chromosomes (Hb AS)
Sickle cell trait
(asymptomatic unless severely hypoxic)
Homozygous: point mutation in both chromosomes (Hb SS)
Sickle cell disease
An inherited blood disorder characterized by defective hemoglobin that leads to red blood cells sickling
2
Dehydration Hypoxemia
Acidosis
↓ Volume of RBC cytoplasm
↓ O2 Saturation of Hb
O morereadily 2
released from Hb in low pH environment
↑ Concentration of deoxygenated HbSinred blood cells (RBCs)
Hydrophilic glutamate→ hydrophobic valine substitution makes HbS less soluble in the cytoplasm & more prone to polymerization & precipitation in its deoxygenated state
↑ Concentration of deoxygenated Hb in RBC leads to ↑ polymerization rate Polymerized & precipitated HbS forms long needle-like fibers
RBC shape becomes sickled
Sickle cells on peripheral blood smear
Vaso-occlusion
(sickled RBCs lodge in small vessels, blocking bloodflow to organs & tissues)
Blockage of venous outflow
Occlusion of vessels in lungs ↑ pulmonary blood pressure
Fluid extravasates into interstitial tissue leading to pulmonary edema
Acute chest syndrome (chest pain, hypoxemia (↓blood oxygen), etc.)**
to the penis:
to the spleen:
Priapism (persistent, painful erection)
Splenic Sequestration (blood pools in spleenà splenomegaly & hypotension)
Extravascular hemolysis (macrophages in the spleen phagocytose sickled RBCs)
Normocytic anemia
↑Marrow erythropoiesis (RBC production) to compensate for hemolysis
Infarction of bone
Pain crises
If occurring in hands
Dactylitis (inflammation of digits)
Blockage of arteries ↓ oxygenation of organs & tissues
Vaso-occlusion of the splenic artery
Splenic infarction (↓ blood supply leads to tissue death)
RBC inclusions (structures found in RBCs) not removed by spleen
Howell-Jolly bodies (RBC DNA remnants) on blood smear
Vaso-occlusion of other arteries (cranial, renal, etc)
Stroke, renal failure
↑ RBC breakdown
↑ Unconjugated bilirubin released from RBC breakdown
RBC precursors (reticulocytes) are released into the blood stream
Reticulocytosis (increased number of immature red blood cells) on peripheral blood smear
Patient’s RBC level becomes dependent on increased marrow activity
Bone marrow infarction or viral infections (i.e. Parvovirus B19) suppresses bone marrow activity
Aplastic crises (profound anemia)
** See corresponding Calgary Guide slide(s)
↑ Serum level of unconjugated bilirubin
Some of the circulating unconjugated bilirubin deposits in the skin
Jaundice (yellowing of skin)
↑ Conjugation of bilirubin in liver
↑Amounts of conjugated bilirubin released into bile
Gallstone formation
Cholelithiasis (presence of gallstones in the gallbladder)
Spleen releases invasive encapsulated bacteria (eg. Haemophilus influenzae, Streptococcus pneumoniae, Neisseria meningitidis) into circulation, which causes infections
Meningitis
Sepsis
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published Sept 16, 2013, updated Apr 29, 2024 on www.thecalgaryguide.com")
Wiskott-Aldrich Syndrome
: Pathogenesis and clinical findings X-linked recessive inheritance (mostly males impacted) Spontaneous DNA mutation
Genetic mutations in Wiskott-Aldrich Syndrome Protein (WASP) impair actin cytoskeleton remodeling in hematopoietic cells (immature blood cells)
Authors: Mina Mina Reviewers: Hana Osman Mao Ding Maharshi Gandhi Shahab Marzoughi Louis-Philippe Girard* * MD at time of publication
Megakaryocytes (platelet precursor cells) depend on the cytoskeleton changing shape to form pseudopodia (arm-like projections) which bud off forming cellular fragments (platelets)
Abnormal cytoskeleton reorganization leads to ineffective thrombocytopoiesis (production of platelets)
Microthrombocytopenia (↓ platelet size and quantity)
Issues with formation of a platelet plug due to abnormal platelets (dysfunction of primary hemostasis)
Reduced ability for platelet adhesion, activation, and/or aggregation
Natural killer cells depend on the cytoskeleton reorganization to form immunological synapses (communication) with body cells in order to have effective surveillance
T cells depend on the cytoskeleton remodeling to form pseudopods which allow them to synapse with other cells when a pathogen is encountered
Defective immune synapse formation
↓ Cancer immunosurveillance
↑ Risk of malignancy
Helper T cells cannot activate B cells which generate antibodies (immunoglobulins) to destroy pathogens
Regulatory T cells can not sufficiently downregulate effector cells which typically limit the immune response and prevent autoimmune conditions
↑ Risk of autoimmune diseases
Recurrent opportunistic infections
Immune dysregulation
Impaired immune response
↓ Number and function of T cells
Immune responses contribute to Type 2 immune responses
Atopic dermatitis (AD; chronic inflammation that causes itchy skin)
↑ Immunoglobulin (Ig) A
Epistaxis (nosebleed)
Red blood cells leak from capillaries
Menorrhagia (heavy menstrual bleeding)
Petechiae (small, flat, red spots that appear on the skin)
Inflammation in AD triggers release of interleukins (modulatory proteins during inflammatory and immune responses) leading to Immunoglobulin (Ig) class switching
↑ IgE levels
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published Apr 20, 2024 on www.thecalgaryguide.com")
Scabies pathogenesis and clinical findings

Authors: Heena Singh Amanda Eslinger Nirav Saini Reviewers: Shahab Marzoughi Danny Guo Yan Yu Richard Haber* * MD at time of publication
Mechanical irritation and immunological reaction
Mast cell activation and histamine release
Itch
(> 10 Mites)
Stratum corneum
Stratum lucidum Stratum granulosum
Stratum spinosum Stratum basale
Females lay 2-3 ova/day which hatch in the stratum corneum in pockets after 3 days
The larvae molt and mature for 2 weeks and mate within the pockets of the stratum corneum
Following mating, female mites begin burrowing
Cycle is propagated as new female mites create more burrows in stratum corneum
Superficial, Linear, Tortuous (i.e., Twisted) +/- Scaly Burrows
↑ Exposure to mite antigens such as Sar s 14.3 and Sar s 14.2
Type 2 helper T-cells produce proteins IL-4, IL-5, IL-13
↑ Serum Immunoglobulin G & E ↑ Eosinophil Count in Peripheral Blood Inappropriate T helper-type immune response in skin (mechanism unknown)
Antigens forming from mite feces, ova, or decomposing bodies
Type IV hypersensitivity reaction (delayed immune response 2-4 weeks following initial contact)
Skin barrier dysfunction from inflammation and histamine release
Pre-existing immunosuppression Chronic inflammatory response Allergic reaction to ↑ mite activity at night
Uncontrollable Itch (Worse At Night) + Excoriations (Scratch Marks)
Urticarial (Hive-Like) Crusted Papules Eczematous Plaques
Skin breakdown Opening for bacteria (commonly S. aureus) 2° Bacterial infection Pustules
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published Oct 17, 2013; updated Jun 9, 2024 on www.thecalgaryguide.com")
Peningkatan Tekanan Intrakranial TIK
 Temuan klinis")
Patofisiologi di Balik Leukemia secara Umum

Gynecomastia

Hyperthyroidism
Klinefelter Syndrome (males with > 1 X chromosome)
Liver cirrhosis
Certain tumors (e.g., germ cell, adrenal, Leydig cell, Sertoli cell)
Anabolic steroid usage (containing testosterone)
Finasteride (treatment for benign prostate hyperplasia and male pattern baldness)
Cimetidine - inhibits stomach acid production
Spironolactone (diuretic
used to treat high blood pressure and heart failure)
Ketoconazole (antifungal)
Cytotoxic agents (e.g. alkylating agents, vincristine, methotrexate)
Imbalance between estrogens and androgens
Estrogen stimulates breast tissue growth in newborn
Changes in metabolic rate ↑ fat production
Unclear mechanism
↑ Proinflammatory mediators and cytokines (e.g. prostaglandin E2, TNF⍺, IL-1, IL-6, cyclooxygenase-2)
Prostaglandin E2 and IL- 6 upregulate aromatase enzyme expression
Available estrogen is higher than available testosterone
↑ Aromatase enzyme activity, converting androgens to estrogen
↓ Testosterone release from the testes
↓ Testosterone
↑ Serum sex hormone binding globulin (SHBG)
SHBG binds estrogen with less affinity to testosterone
Thyroid hormone stimulates liver to express more sex hormone binding globulin
Thyroid hormone stimulates aromatase activity
Overexpression of aromatase enzyme
Seminiferous tubules in the testes hyalinize and fibrose
Suppression of the hypothalamic pituitary thyroid axis through an unclear mechanism
Tumor may produce estradiol
Tumor produces β- human chorionic gonadotropin (β-HCG)
↑ serum testosterone
Inhibits 5-α reductase
Blocks binding of 5-DHT to androgen receptors
↓ 2-hydroxylation of estradiol
Mimics structures of testosterone
Inhibits 17,20 desmolase and 17α-hydroxylase
Damage to Leydig cells in testes
↑ Estrogen to androgen ratio
Pathological causes
Impaired spermatogenesis and testosterone production
↓ GnRH secretion from hypothalamus
↓ Testosterone
↓ Luteinizing hormone (LH) release from anterior pituitary
↓ 5-DHT and/or testosterone binding to androgen receptors in chest tissue
↓ inhibition of breast development
Normal or increased estrogen acts on estrogen receptor on chest tissue
Estrogen receptors stimulate breast development
Estradiol negatively feedbacks on luteinizing hormone
β-HCG stimulates LH receptors on Leydig cells in the testes
Aromatase enzyme converts excess testosterone into estrogen and estradiol
↓ conversion of testosterone to 5- dihydrotestosterone (5-DHT), a more potent form of testosterone
Glandular proliferation in male breasts
Gynecomastia
(development of breast tissue in males)
Drug side- effects
↓ Metabolism of estradiol
Competitively binds to androgen receptors
↑ Serum estradiol levels
Exhibits physical attributes that do not align with gender identity
Psychological distress
In some cases, hormones stabilize
Involution and atrophy of ducts
Gynecomastia resolves
↓ Steroid synthesis
↓ Androstenedione produced (testosterone precursors)
↓ Serum testosterone levels
↓ Testosterone production
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published Jun 9, 2024 on www.thecalgaryguide.com")
Posterior Cruciate Ligament PCL Injury Pathogenesis and Clinical Findings
 Injury: Pathogenesis and clinical findings
Authors: Luc Wittig Stephanie de Waal Michelle J. Chen Reviewers: Reza Ojaghi Usama Malik Sunawer Aujla Nojan Mannani Mankirat Bhogal Dr. R. Buckley* Dr. Gerhard Kiefer* * MD at time of publication
Sport-related trauma
Dashboard knee injury from a motor vehicle collision
Tibia contacts dashboard at high velocity with knee in flexed position
Tibia is forced posteriorly, relative to knee joint The PCL undergoes sudden & forceful strain
Hyperextension injury (knee joint moves past its normal extension limit)
Extreme tibia movement anteriorly past knee joint overstretches PCL
Hard fall on flexed knee
Posterior Cruciate Ligament Injury
Strain & overstretching of the PCL, which connects the anterior distal femur to the posterior proximal tibia to prevent posterior translocation of the tibia relative to the femur, can result in a partial or complete tear. The tear can be isolated (rare) or be one of multiple ligaments that are torn (multi-ligamentous tear).
Mild injurious force causes a partial tear
Intact portion of PCL prevents extreme tibial translocation posterior to the femur. Torn portion still allows 1-5 mm of posterior tibial translocation (Grade 1)
Positive posterior drawer test
Knee injury tests
• Posterior drawer test – Push against leg below
the knee to test for posterior tibial translocation
relative to femur
• Lachman test – Pull leg below the knee to test
for anterior tibial translocation relative to femur • McMurray test – Tests for medial and lateral
meniscus tears
• Varus & valgus stress test – Tests for medial and
Strong injurious force causes a complete, isolated tear
PCL is unable to prevent posterior translocation of the tibia relative to the femur, allowing 6-10 mm of posterior tibial translocation (Grade 2)
Positive posterior Negative Lachman, McMurray, drawer test and varus & valgus stress test
Very strong injurious force causes a multi- ligamentous tear and damage to the knee joint capsule (capsuloligamentous injury)
Absence of stability from PCL combined with absence of stability from other knee ligaments allows for > 10 mm of posterior tibial translocation (Grade 3)
Positive posterior Positive Lachman OR McMurray drawer test OR varus & valgus stress test
PCL unable to stabilize knee by preventing posterior tibial translocation (chronic PCL insufficiency)
Body uses quadriceps tendon for knee stabilization to compensate for PCL insufficiency
Inflammation from injury contributes to progressive degenerative articular changes
lateral collateral ligament tears
Post-traumatic patellofemoral pain
Osteoarthritis
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published Sept 20, 2018; updated Jun 9, 2024 on www.thecalgaryguide.com")
Metastatic Bone Lesions
 into bones. Once tumours metastasize to bone, they are generally incurable and contribute to significant morbidity prior to a patient’s death
Authors: Curtis Ostertag Reviewers: Mankirat Bhogal Nojan Mannani Michelle J. Chen Dr. Gerhard Kiefer* * MD at time of publication
Cell-to-cell communication between tumour cells & bone cells (osteoclasts & osteoblasts)
Tumours release TNF-⍺, RANK-L, and PTHRP which ↑ osteoclast activity & ↓ osteoblast activity
Change in relative activity of bone cells results in osteolysis (breakdown of bone)
Calcium is released into the bloodstream
Hypercalcemia
Osteoblastic metastasis (common in prostate cancer)
Tumor growth
Secondary bone formation in response to bone destruction
TGF-β, PDGF, & IGF are released from the degraded bone matrix, which can stimulate tumors & osteoblasts
Weakened bone increases risk of fracture
Pathologic fracture
↑ Mortality
Bone tissue expands into surrounding space
Nerve compression
Disruption of cortical bone or surrounding soft tissues
Diffuse & achy rest/night pain
Long bone masses compress peripheral nerves
Neuropathy
Vertebral masses compress spinal nerves/cord
Radiculopathy /Myelopathy
↓ Quality of life
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published Jun 9, 2024 on www.thecalgaryguide.com")
Migraines and auras pathogenesis and clinical findings

Personal triggers (e.g., lack of food, emotional stress)
“Cortical Spreading Depolarization” followed by “Cortical Spreading Depression” across one cortical hemisphere
Cortical spreading depression
creates “auras” via unknown mechanism(s)
Expanding scotoma (area of visual blurriness)
Spreading paraesthesias (numbness that travels across body)
Dysphasic language (trouble finding words)
Brainstem symptoms (e.g., vertigo, tinnitus)
Motor symptoms (e.g., hemiplegia)
Triggering of a depolarization wave in a unilateral region of the cortex with associated ↑ blood flow to that region
Neurons and glia release K+ into extracellular space that spreads depolarization wave to nearby cortical areas
Ion gradient imbalances cause Prolonged vasoconstriction neurons in original cortical area to and ↓ blood flow
swell and become inhibited
Activation of the hypothalamus (responsible for maintaining homeostasis)
Prodromal symptoms (↑ thirst, hunger, yawning, ↓ cognitive function)
Author:
Yan Yu
Braxton Phillips
Reviewers:
Shahab Marzoughi
Owen Stechishin
Dustin Anderson
Scott Jarvis*
Sina Marzoughi*
* MD at time of publication
Release of hypothalamic neurotransmitters (e.g., orexins, neuropeptide Y) at the trigeminalcervical complex (TCC) of the brainstem and cervical spinal cord
Depolarizing wave passes over pseudounipolar neurons (neurons with two axons) of the trigeminal ganglion which synapse in brainstem & dura matter
These neurotransmitters reduce the activation threshold of spinal trigeminal nucleus neurons in the TCC
Neuropeptides (e.g., calcitonin gene-related peptide, pituitary adenylate cyclase-activating polypeptide)
are released at the TTC, triggering local inflammation (termed neurogenic inflammation)
Ongoing neurogenic inflammation activates secondary nociceptive neurons in the TTC
Central sensitization (↓ response threshold of secondary nociceptive neurons in the TTC)
Brain perceives referred pain from the face as the TTC also receives convergent nociceptive input from the face
Facial allodynia (pain with normally non-painful stimuli)
Serotonin & histamine are released on dural blood vessels triggering neurogenic inflammation in the dura matter
Ongoing neurogenic inflammation activates primary nociceptive neurons in the dura matter
Peripheral sensitization (↓ response threshold of primary nociceptive neurons around the dural blood vessels_
Unknown mechanisms no longer believed to be related to dural blood vessel dilation
Unilateral throbbing headache
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published Nov 22, 2012, updated Jul 1, 2024 on www.thecalgaryguide.com")
Anesthetic Considerations in Pregnancy

↑ Progesterone serum levels
↓ Esophageal tone & sphincter pressure
↑ Aspiration risk >20 weeks gestation & during labor
Aspiration prophylaxis
Rapid sequence induction
Nonparticulate antacid prophylaxis (e.g., sodium citrate, famotidine)
Pre-operative fasting & gastric ultrasound to assess volume
Breathing
↑ Minute ventilation
↓ PaCO2 at baseline
Uterine artery vasoconstriction with maternal PCO2 levels exceeding 32 mmHg
Impaired uteroplacental perfusion
Maintain physiologic alkalosis, avoid maternal hypercapnia
Maintain
maternal arterial PCO2 28–32 mmHg
Avoid permissive hypercapnia
↑ Progesterone serum levels
Disproportionate ↑ in plasma volume
↓ Serum colloid osmotic pressure
Drug-specific alterations in absorption, distribution, metabolism & excretion (e.g. ↑ distribution of acidic drugs due to ↓ albumin; ↑ clearance of renally- metabolized drugs)
Optimize anesthetic dosing for altered pharmacokinetics
Adjust dosages based on drug recommendations
Circulation
↑ Maternal blood volume
Peripheral vasodilation
↓In systemic vascular resistance by 25–30%
↓ Blood pressure
↑Abdominal pressure
Compression of the aorta & inferior vena cava
Supine hypotension
Impaired uteroplacental perfusion
Fetal ischemia
Optimize
placental blood flow
Position patient on left side to maintain uterine displacement
Diaphragmatic elevation & lung compression
↓ Functional residual capacity
↑ Risk of rapid desaturation
Fetal hypoxemia
Maintain maternal & fetal oxygenation
Optimize positioning (e.g., patient on left side to maintain uterine displacement)
& supplement oxygen as needed
Heart rate ↑ 15-25%
↑ Cardiac output
Manifestations of significant blood loss are delayed
Avoid hypotension
↑ Stroke volume
Avoid fluid overload
Treat abnormal variation in blood pressure at lower values
Fetus
Requirement to assess & treat two patients
Achieve optimal anesthetic dosing for mother while maintaining fetus safety
Maintain oxygenation of both mother & fetus & ensure adequate uteroplacental perfusion
Consider fetal-drug transfer & adjust agent & dose accordingly
Monitor maternal vital sign & fetal heart rate
Utilize a multidisciplinary team (e.g., pharmacy, nursing, physical & occupational therapy)
Legend:
Pathophysiology
Mechanism
Goal
Management
Published July 5, 2024 on www.thecalgaryguide.com")
Ketamine

Diabetes Mellitus Pathophysiology Behind Lab Findings

↓ Glucose uptake into cells
Autoimmune destruction of pancreatic beta cells (type 1 diabetes mellitus)
Insulin resistance (type 2 diabetes mellitus)
Other causes of diabetes mellitus (genetic, drug- induced, pregnancy, etc.)
↑ Lipolysis (fat breakdown) in adipose tissue
↑ Free fatty acids & glycerol in bloodstream
↑ Oxidation of fatty acids in liver to form acetyl CoA
↑ Conversion of acetyl CoA to ketone bodies (ketogenesis) to be used as fuel for the brain
Relative or absolute insulin deficiency
↓ Activation of adipose (fat tissue) & muscle cell transmembrane insulin receptors
↓ Activation of intracellular insulin signaling pathways (PI3K & MAP kinase)
Cells cannot use glucose as a source of energy; thus body reacts as if it is starving
↑ Protein breakdown in muscle tissue ↑ Amino acids & lactate in bloodstream
↑ Substrates (glycerol, amino acids & lactate) available to produce glucose in the liver
↑ Glycogenolysis (breakdown of stored glucose - glycogen) & gluconeogenesis (production of glucose) in liver
Glucose in bloodstream glycates (coats) hemoglobin in red blood cells; thus ↑ blood glucose leads to ↑ glycated hemoglobin
↑ Hemoglobin A1C
↑ Fasting blood glucose level ↑ Random blood glucose level
↑ Blood glucose following oral glucose tolerance test
Diabetic ketoacidosis (DKA) in absolute insulin deficiency *See DKA slide
Filtered ketone bodies exceed the reabsorption capacity of renal tubules
Ketonuria (↑ ketones in urine)
↑ Glucose in bloodstream
Build-up of glycation end products causes damage to glomerular tissue
Transient glomerular hyperfiltration followed by long-term ↓ in glomerular filtration rate
Albuminuria (↑ albumin in urine)
Diabetic nephropathy *See slide
Filtered glucose exceeds reabsorption capacity of renal tubules in the kidney
Glucosuria (↑ glucose in urine)
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published July 8, 2024 on www.thecalgaryguide.com")
Neonatal Hypoglycemia Pathogenesis
, glycogenolysis (breakdown of glycogen into glucose), and fatty acid oxidation (conversation of fatty acids to ATP)
Low glucose levels stimulate the neonate’s appetite, allowing them to adapt to intermittent feeds
Infant feeding regularly on a diet with sufficient carbohydrates creates a consistent plasma glucose concentration
Pregnant Parent Causes
Impaired Glucose Production
Inadequate Glucose Supply
Pregnant parent using β-blockers
β-blockers prevent epinephrine from binding to adrenergic receptors in skeletal muscle and liver
Blocked sympathetic signaling in these organs prevents glycogenolysis
↓ Breakdown of glycogen into glucose
Infant of a parent with diabetes
Parent has a high level of glucose in their blood
The fetus receives a glucose-rich blood supply
Fetal pancreatic β cells ↑ insulin production
Post-birth, glucose levels in the infant’s circulation ↓ but insulin levels
remain high
Excessive glucose uptake into cells
Fetus born with a metabolism disorder (e.g. organic amino acid disorder, disorder of glycogen metabolism, disorder of gluconeogenesis)
Genes that make proteins or hormones responsible for production, breakdown and regulation of glycogen are mutated
Fetus born with an endocrine disorder (e.g. congenital adrenal hyperplasia, hypopituitarism, Turner Syndrome)
Pituitary and/or adrenal gland dysfunction ↓ release of hormones that regulate glucose balance
Fetal growth is restricted, fetus is small for gestational age, or is premature (<37 weeks gestation)
Glycogen is deposited during the 3rd trimester of pregnancy. Infants born early have fewer stores; smaller infants born at term have ↓ stores, ↑ insulin sensitivity, & poorly coordinated counter- regulatory hormones
↑ Glucose demand for the transition to life out of the womb
Glycogen stores used up quickly
Perinatal stress (e.g. sepsis, asphyxia)
Stress stimulates fetal adrenal glands to ↑ epinephrine secretion
Fetal pancreatic ⍺- cells ↑ glucagon secretion
↑ Breakdown of muscles and fat for glucose- building blocks
Glycogen stores used up quickly
↑ Glucose usage as fetal metabolic demands ↑ to manage stress
Fetal β-cells secrete inappropriately high levels of insulin despite hypoglycemic state; this hyperinsulinism state can last for months & resolve spontaneously
↓ Serum glucose Neonatal hypoglycemia
Authors: Dasha Mori Reviewers: Michelle J. Chen *Dr. Danielle Nelson *MD at time of publication
Blood glucose < 2.6 mmol/L in term & preterm infants within 72 hours of birth or < 3.3 mmol/L after 72 hours of birth
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published July 19, 2024 on www.thecalgaryguide.com")
Legg Calve Perthes Disease

Early interruption of blood supply to femoral head
↓ Venous outflow from intraosseous vasculature ↑ pressure within bone
Fragmentation stage (8 months)
Body replaces resorbed necrotic bone with structurally weaker bone
Backup of blood within bone vasculature impedes arterial inflow
Temporary interruption of blood supply to proximal femoral epiphysis primarily during childhood
Residual stage
Differential growth of proximal femoral epiphysis
Overgrowth of greater trochanter
Healing stage (4 years)
Body gradually replaces necrotic bone with stronger, more dense bone
Lack of O2 & nutrients disrupts normal bone growth
Bone tissue begins dying from loss of O2 & nutrients (osteonecrosis)
Osteonecrosis is associated with inflammation of synovial tissue (synovitis) at the femoral head
Metaphyseal cyst (area of bone resorption) grows on proximal femoral neck metaphysis
Growth plate abnormalities
Femoral bone head deformity
Limited hip internal rotation & abduction
Antalgic gait
Femoral epiphysis collapses laterally and extrudes from acetabulum
Femoral head now composed of dense and less dense bone
Scattered radiolucency & radiodensity on plain radiographs
Femoral head deformity becomes set in place and limits hip range of motion
Femoral bone head deformity
Limb length discrepancy
Normal bone density on plain radiographs
Abnormal articulo- trochanteric distance (vertical distance between highest point of greater trochanter & highest point of femoral head)
Remodeling of femoral head & acetabulum seen on plain radiographs
Growth plate irregularity
↓ Range of motion
Widened medial joint space
Antalgic gait
Femoral head is displaced laterally
Fragmented epiphysis on plain radiographs
Groin pain & referred pain to anteromedial thigh or knee region
Authors: Janelle Wai Michelle J. Chen Reviewers: Patrick Pankow Nojan Mannani Kirat Bhogal Dr. Gerhard Kiefer* * MD at time of publication
Findings on magnetic resonance imaging (MRI) & plain radiographs
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published July 19, 2024 on www.thecalgaryguide.com")
Generalized Anxiety Disorder GAD
: Pathogenesis and clinical findings
Authors: Keira Britto Reviewers: Sara Cho, Luiza Radu, *Margaret Oakander *MD at time of publication
Altered cognition
Scan environment for perceived threats
Genetic predisposition & family history: polygenic, >30% heritability
Adverse childhood & life experiences: e.g., abuse, neglect, divorce, bereavement
Morbidity & stressors from chronic medical conditions
Hypersensitivity & reactivity to stimuli
áHPA axis activity
Neurobiological differences
Trait neuroticism (emotional instability & nervousness)
Reacts to unfamiliarity with withdrawal & fear
áLevel of uninhibited excitatory signaling
Psychomotor agitation
Frequent sympathetic nervous system (fight or flight) activation to perceived threats
Cycles of epinephrine surges
Inadequate recovery
State of hyperarousal
Disturbed sleep
Ongoing sleep deprivation
Easily fatigable
áACTH released from hypothalamus
áCortisol released from adrenal glands
áSerotonin uptake
âSerotonin Impaired
neuroplasticity &âcomplexity of synaptic connections in amygdala & hippocampus
âAbility to deal with emotional stressors
Impaired engagement of prefrontal cortex
âRegulation of emotional reactions
áAnticipatory emotional responses
âInhibition from GABA
áAmygdala volume & activity
áFear & anxiety circuits
Attentional focus directed towards worries & perceived threats
Divert blood to muscles
Skeletal muscle activation
Muscle tension
Psychomotor agitation (excessive motor activity associated with a feeling of inner tension)
áEmotional intensity
Chronic state of high physiological stress
âHippocampal BDNF (brain derived neurotrophic factor)
Hippocampal atrophy &â hippocampal neurogenesis
â Inhibitory control of the hippocampus over the HPA axis
áAlertness & wakefulness
Disturbed sleep
Strengthening of memories rooted in fear, to prevent encounters with future threats
Behavioural & cognitive changes to facilitate coping with threats
Irritability
**Excessive, persistent
Bottom up, stimulus driven attention
áDistractibility
Difficulty concentrating
anxiety & worry that is distressing &/or impairs functioning
Irritability
Generalized Anxiety Disorder:
**occurring more days than not for ≥ 6 months; **difficult to control; presence of ≥ 3 remaining symptoms
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published July 21, 2024 on www.thecalgaryguide.com")
Epitheliales Ovarialkarzinom Pathogenese und klinische Befunde

Endometriose Pathogenese und Komplikationen

Mitralklappenstenose Pathogenese und klinische Befunde

Virale Hepatitis Pathogenese und klinische Befunde

Pseudokrupp Pathogenese und klinische Befunde

Underfill Edema Pathogenesis

Vasodilatory medications
Various mechanisms
Right-sided heart failure
Compromised right heart function ↓ forward flow
↓ Hepatic albumin synthesis
Blood is unable to pass through hepatic vessels disrupted by cirrhosis and backs up in portal vein
↑ Blood pressure in portal vein (portal hypertension)
Less blood volume in hepatic veins and vena cava (underfilling)
Pregnancy
↑ Estrogen, progesterone and relaxin
Vasodilation
Gravity causes fluid accumulation in peripheral veins
↑ Capillary hydrostatic pressure
↑ Net fluid movement into interstitial space
↓ Serum albumin
↓ Capillary oncotic pressure
Fluid extravasation into interstitial space
More blood in portal vein ↑ capillary hydrostatic pressure in venous system
Pressure creates net fluid
movement from vascular space into interstitial space
Less blood volume in arteries (underfilling)
↓ Effective arterial blood volume (EABV)
↓ Renal blood flow activates the renin-angiotensin-aldosterone system (RAAS)
Angiotensin and aldosterone ↑ Anti-diuretic hormone released by tubular Na+ and H2O resorption posterior pituitary ↑ H2O resorption
↑ Fluid in circulation, worsening existing venous congestion
↑ Hydrostatic capillary pressure and fluid extravasation into interstitial space Underfill edema (edema worsened by activation of RAAS)
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published Aug 19, 2015; updated Aug 5, 2024 on www.thecalgaryguide.com")
Anterior Shoulder Dislocation Pathogenesis and clinical findings

Authors: Molly Joffe Miles Hunter Reviewers: Kristi Billard Stephanie de Waal Nojan Mannani Michelle J. Chen Jordan Raugust*, Ryan Shields* *MD at time of publication
Force travels proximally, forcing the head of the humerus anteriorly
Anterior force of humeral head exceeds tensile strength of soft tissue stabilizers Detachment of anterior band of inferior glenohumeral ligament
Anterior translation of humeral head relative to the glenoid
Humeral head causes the axial nerve to stretch
Anterior shoulder dislocation
Posterolateral aspect of humeral head impacts anterior portion of glenoid
Damage to surrounding soft tissues & sensory nerve branches
Numbness/altered sensation over lateral deltoid
Weakness with shoulder abduction
Visible deformity
Humeral head palpable anteriorly
Posterolateral humeral head fractures
Hill-Sachs lesion
Decreased range of motion
Detachment of antero-inferior glenoid labrum
Shoulder pain
Rupture of small surrounding blood vessels
Bruising Swelling Capsule stiffness
Permanent axillary nerve deficit
Avulsion of anterior glenoid rim
Bony Bankart lesion
Soft Bankart lesion (isolated soft tissue injury)
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published Apr 3, 2016; updated Aug 5, 2024 on www.thecalgaryguide.com")
Choque Hipovolemico

AVC Isquemico Patogenese

Aterosclerosi Patogenesi

Diabete Gestazionale fattori di rischio e patogenesi

Pelepasan Retina Patogenesis

Pelepasan Retina Temuan klinis

AVC Isquemico Comprometimento por localizacao
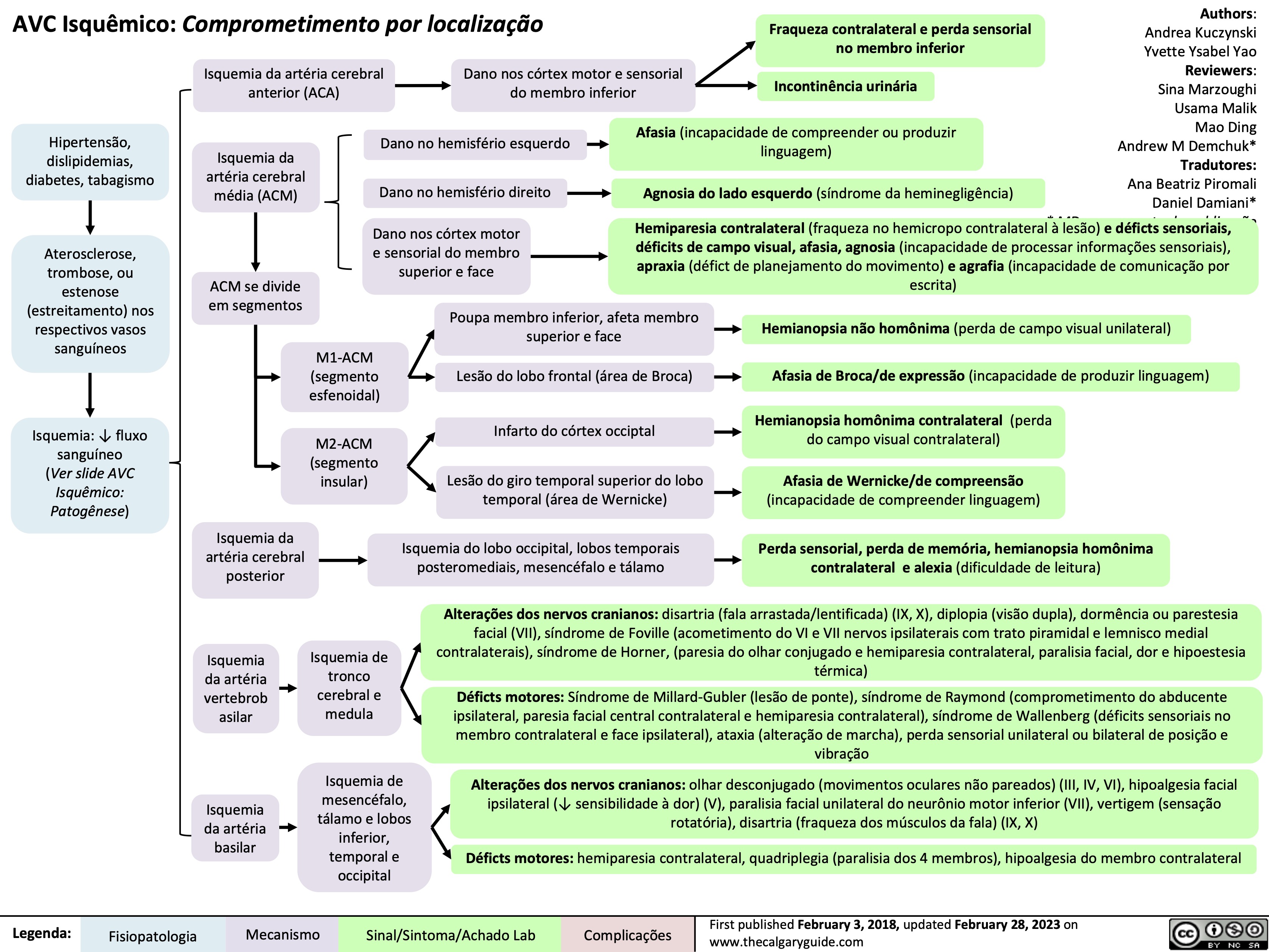
Tratamento do Choque: Explicacao dos mecanismos basicos
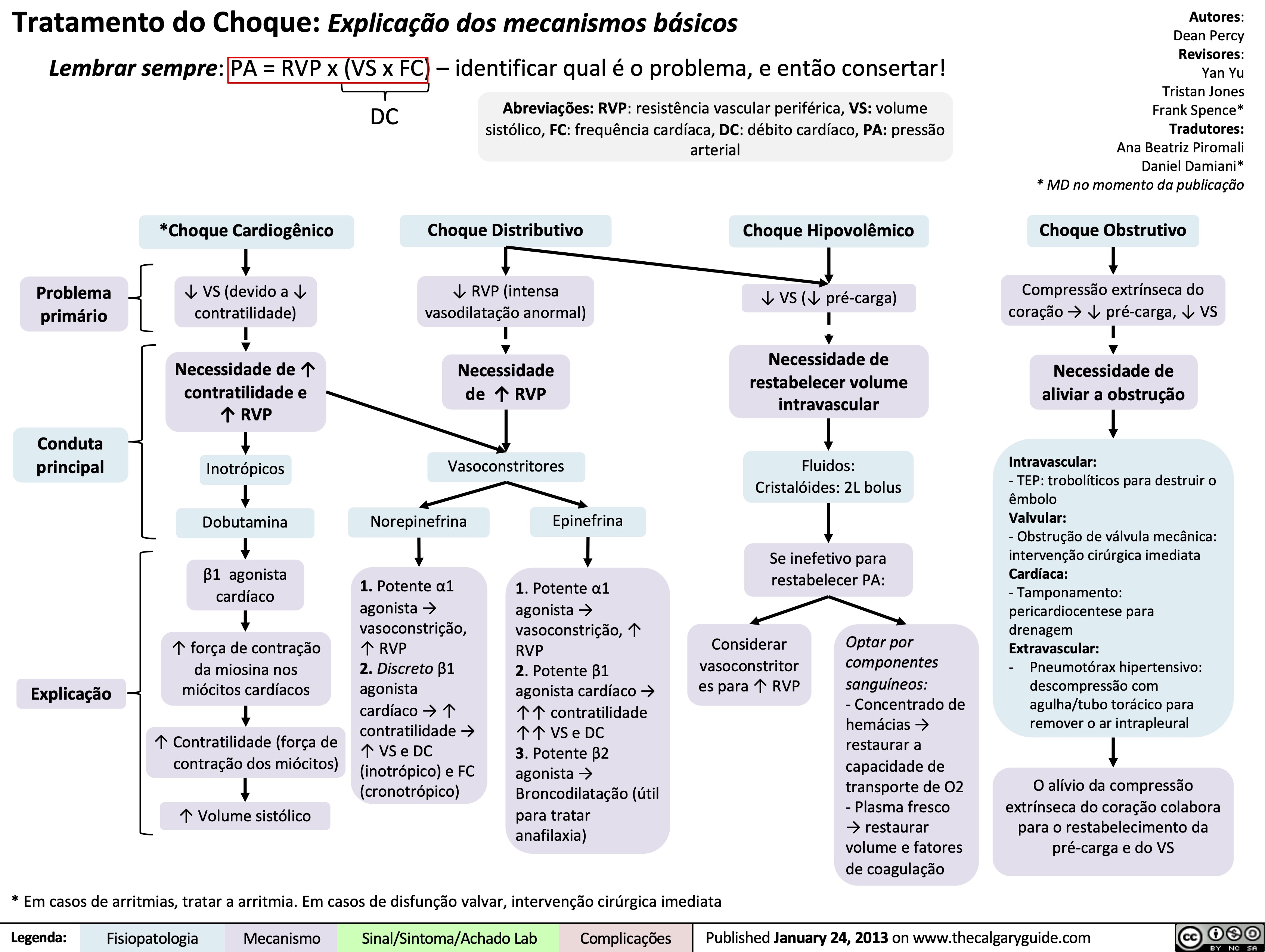
Acute Otitis Media Pathogenesis and Clinical Findings in Children

Exposure to tobacco smoke
Impaired macrophage function in nasopharynx
Lack of immunizations
Lack of breastfeeding
Infant does not receive antibodies from breast milk
Overcrowding
Age 6 – 16 months
Immune system deficiencies
Close proximity of kids & ↓ sanitation (e.g. daycare)
↑ Infection risk Upper Respiratory Tract Infection (i.e. bacterial (Streptococcus pneumoniae, Haemophilus influenzae, Moraxella catarrhalis), viral)
Immature Eustachian tube anatomy which facilitates pathogen transmission to middle ear
Author: Jody Platt Stephanie de Waal Reviewers: Yan Yu Elizabeth De Klerk William Kim Annie Pham Michelle J. Chen Danielle Nelson* * MD at time of publication
Abnormal anatomical structure (e.g. cleft palate)
↑ Nasopharyngeal streptococcus pneumoniae
Lack of immunity to pathogens
↑ Colonization of nasopharynx with bacterial pathogens
Inflammation & edema of respiratory mucosa including the nose, nasopharynx, and Eustachian tube
Obstruction of the Eustachian tube
Air from middle ear resorbs into circulation which creates a low-pressure environment
Negative pressure gradient pulls viral/bacterial pathogens into middle ear
Degenerating white blood cells, tissue debris, and microorganisms accumulate & develops into purulent effusion
Inflammation & infection of middle ear
Complications of acute otitis media**
↑ Pressure in middle ear
Stretching of tympanic membrane
Helper T cells & macrophages release cytokines into the bloodstream
Cytokines trigger hypothalamus to ↑ thermoregulatory set-point
Effusion behind tympanic membrane
Effusion obstructs visualization of ossicles
Neutrophils infiltrate middle ear & phagocytose pathogens
Pus accumulates behind the tympanic membrane
Blood vessels of the tympanic membrane vasodilate
Bulging tympanic membrane
Otalgia (ear pain)
Discomfort disrupts daily activities in young children
Fever
Irritable Poor feeding
Tympanic membrane erythema
Painful blisters on tympanic membrane (e.g. Bullous myringitis)
Can persist for up to 3 months after infection resolves
Loss of tympanic membrane landmarks (i.e. handle of malleus, light reflex)
** See corresponding Calgary Guide slide
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published Feb 28, 2013; updated Aug 25, 2024 on www.thecalgaryguide.com")
Pediatric Pneumonia Pathogenesis and Clinical Findings
 Kang Usama Malik Annie Pham Eric Leung* Jean Mah* * MD at time of publication
Immunological: unvaccinated, primary immunocompromise, pre-existing illness (e.g. HIV, measles), malnutrition
Environmental: smoke, air pollution, mold, crowded housing
Recent hospitalization or antibiotic-use
Physiological: neonates, low-birth weight, underlying lung disease
These factors make the host more susceptible to infection
Infection and proliferation of pathogen in lower respiratory tract/parenchyma
Pediatric pneumonia:
Inflammatory response to infection/proliferation of microbial pathogens at the alveolar level
Exposure to pathogen via inhalation, hematogenous, direct exposure, or aspiration
Epithelial cells in respiratory tract release cytokines that recruit neutrophils & plasma proteins to infection site, initiating a local inflammatory response
Cytokines released into the bloodstream (e.g. TNF, IL-1) initiate a systemic inflammatory response
↑ Vascular permeability
Accumulation of exudate, cellular debris, serous fluid, fibrin, or bacteria in the airway spaces
↑ Respiratory drive
Tachypnea
↑ Excitability of the peripheral somatosensory system
Circulating cytokines induce prostaglandin synthesis
Airway irritation as cilia are unable to efficiently clear fluid buildup
Crackles, ↓ breath sounds
Fluid, protein, or inflammatory cells leak into pleural space
Pleural effusion
Pulmonary edema
Fluid buildup in interstitial spaces ↑ gas diffusion distance
Bacteria enter the bloodstream (if bacterial pneumonia)
Sepsis
Fluid buildup in the alveoli ↓ available surface area for gas diffusion
↓ Efficiency of gas exchange
Intra- and extracranial arteries dilate
Headache
↑ Thermo-regulatory set-point of the hypothalamus
Fever
Myalgia
Hypoxemia
Malaise
Cough
Fluid accumulation in the pleural space prevents full lung expansion
↑ Work of breathing (tracheal tug, paradoxical abdominal breathing, subcostal/suprasternal indrawing)
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published May 28, 2018; updated Aug 25, 2024 on www.thecalgaryguide.com
Pediatric Pneumonia: Pathogenesis and clinical findings
Authors: Jasmine Nguyen Nicola Adderley Reviewers: Midas (Kening) Kang Usama Malik Annie Pham Eric Leung* * MD at time of publication
Immunological: unvaccinated, primary immunocompromise, pre-existing illness (e.g. HIV, measles), malnutrition
Environmental: smoke, air pollution, mold, crowded housing
Recent Hospitalization: length of stay, recent antibiotics, mechanical ventilation
Physiological: neonates, low-birth weight, underlying lung disease (ciliary dysfunction, asthma, cystic fibrosis, bronchiectasis)
Host is more susceptible to infection
Exposure to pathogen:
inhalation, hematogenous, direct, aspiration
Infection and proliferation of pathogen in lower respiratory tract/parenchyma
Pediatric pneumonia:
Inflammatory response to infection/proliferation of microbial pathogens at the alveolar level
Notes:
• Additional findings in pediatric pneumonia may include increased
irritability, nausea/vomiting, diarrhea,
otitis, and headache
• Viral pathogens most common in
children <2yrs; bacterial pathogens most common in children >2yrs
Local inflammatory response: epithelial cells release cytokines in response to infection, which recruit neutrophils and plasma proteins to site of infection
↑ Vascular permeability causes accumulation of plasma exudate, cellular debris, serous fluid, fibrin, or bacteria in the airway spaces
Systemic inflammatory response:
Cytokine release (eg. TNF, IL-1)
↑ respiratory drive
Airway irritation as cilia are unable to efficiently clear fluid buildup
Crackles, ↓ breath sounds
Fluid, protein, or inflammatory
cells leak into pleural space
Pleural effusion
Pulmonary edema
Fluid buildup in interstitial spaces increases gas diffusion distance
Fluid buildup in the alveoli decreases
available surface area for gas diffusion
↓ efficiency of gas exchange
Bacteria invade into the bloodstream (if bacterial pneumonia)
Sepsis
Hypoxemia
Circulating cytokines induce prostaglandin synthesis, which raise the thermoregulatory set-point of the hypothalamus
paradoxical abdominal breathing, subcostal/suprasternal indrawing)
Fever
Cough
Fluid accumulation in the pleural space prevents full
lung expansion, resulting in ↓ lung volumes
Tachypnea
↑ Work of breathing (tracheal tug,
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published Month Day, Year on www.thecalgaryguide.com
Pediatric Pneumonia: Pathogenesis and clinical findings
Immunological: immunization status, immune compromise
Environmental: second-hand smoke, air pollution
Hospitalization: length of stay, recent antibiotics, mechanical ventilation
Neonates, immunocompromise, underlying lung disease (ciliary dysfunction, Cystic Fibrosis, bronchiectasis)
Authors: Nicola Adderley Reviewers: Midas (Kening) Kang Usama Malik Eric Leung* * MD at time of publication
Additional findings in pediatric pneumonia may include nausea, otitis, headache
Viral pathogens most common in children <2yrs; bacterial pathogens most common in children >2yrs
Interstitial pattern: suspect Mycoplasma pneumoniae, Influenza A + B, Parainfluenza Lobar pattern: suspect S. pneumonia, H. influenzae, Moraxella, S. aureus
Systemic inflammatory response:
Cytokine release (eg. TNF, IL-1)
Exposure to pathogen: inhalation, hematogenous, direct, aspiration
Susceptible host and/or virulent pathogen
Infection and proliferation of pathogen in lower respiratory tract/parenchyma
Pediatric pneumonia:
Inflammatory response to proliferation of microbial pathogens at the alveolar level
Notes:
• •
• •
Local inflammatory response: neutrophils recruited to site of infection (LOBAR or INTERSTITIAL PATTERN, depending on pathogen) by epithelial cytokine release
Irritation of contiguous structures and/or referred pain (mechanism unclear)
Acute abdominal pain
Cough
Accumulation of plasma exudate (from capillary leakage at sites of inflammation), cell-debris, serous fluid, bacteria, fibrin
↑ respiratory drive
Disruption of hypothalamic thermoregulation
Fever/chills
Irritation of airways and failure of ciliary clearance to keep up with fluid buildup
Crackles, ↓ breath sounds
Fluid buildup in spaces between
alveoli (INTERSTITIAL PATTERN)
Interstitial opacity on CXR
Fluid buildup in alveoli (LOBAR PATTERN)
↓ efficiency of gas exchange (↑ diffusion distance in INTERSTITIAL, ↓ surface area in LOBAR)
Hypoxemia
Tachypnea
Lobar consolidation on CXR
Respiratory accessory muscle use (chest indrawing, paradoxical breathing, muscle retractions)
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published May 28, 2018 on www.thecalgaryguide.com
gin")
Pityriasis Tinea Versicolor
 Versicolor: Pathogenesis and clinical findings
Authors: Kerry Yang Reviewers: Mina Youakim Shahab Marzoughi Jori Hardin* * MD at time of publication
↑ Loosening of the stratum corneum
Scaling of skin
Hyperhidrosis (↑ sweating)
Poor ↑ Sebaceous nutrition gland secretions
Development of a lipid- rich environment
Use of oils on skin
↑ Environmental heat & humidity
Immunosuppression
Lipophilic Malassezia (normal constituent of skin flora) yeast cells transform into the pathogenic, mycelial (fungal thread) form, commonly occurring on the trunk, neck, and proximal extremities
Malassezia infect the stratum corneum (the outer layer) of the skin
↑ Malassezia – associated ligand – activated transcription factors
Rate of different gene transcriptions modified
↑ Melanin production by melanocytes
↑ In melanin more than the ↓ in melanin
Hyperpigmented macules, patches, and plaques
↑ Production of the enzyme keratinase
↑ Activation of Langerhans cells (macrophages in the skin)
Alteration in the expression of cytokines and chemokines in keratinocytes (epidermal skin cells)
↑ Inflammatory response
Activation of cutaneous nerve fibers
Itch signal transduction
Mild pruritis (itchiness)
Ligand-activated transcription factors produced by Malassezia
↑ Degradation of keratin in the skin
↑ Production of dicarboxylic acids by certain Malassezia species
Competitively inhibits the rate- limiting enzyme in melanin production tyrosinase
↓ Production of melanin
↓ In melanin more than the ↑ in melanin
Hypopigmented macules, patches, and plaques
↑ Production of inflammatory mediators
Dyspigmentation
Malassezia, particularly M. globosa, M. furfur, and M. sympodialis
Loosened Epidermal layer
Dermal-Epidermal Junction
Dermal layer
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published Aug 25, 2024 on www.thecalgaryguide.com")
Anti-Emetics Mechanism of Action

Migraineurs are hypersensitive to dopamine
D2 receptor antagonists bind to D2 (dopamine) receptors in brain
↓ Binding of endogenous dopamine to dopamine receptors
Emetogenic stimuli (e.g. chemotherapy) in blood and cerebrospinal fluid
Chemoreceptor trigger zone in area postrema of 4th ventricle that lies outside blood-brain barrier
Emetogenic stimuli in the bloodstream stimulate dopamine receptors in area postrema
D2 receptor antagonists bind to D2 (dopamine) receptors in chemoreceptor trigger zone
↓ Binding of dopamine in area postrema
5HT-3 receptor antagonists (e.g. ondansetron)
Emetogenic stimuli (e.g. chemotherapy) in blood and cerebrospinal fluid
Area postrema has no blood- brain barrier
Emetogenic stimuli in the bloodstream stimulate serotonin receptors in area postrema
5HT-3 receptor antagonists bind to 5HT-3 (serotonin) receptors in chemoreceptor trigger zone in area postrema of 4th ventricle
↓ Binding of serotonin in area postrema
Emetogenic stimuli (e.g. toxins, chemotherapy) and pathologies in gastrointestinal (GI) tract
↑ Local release of serotonin from enterochromaffin cells of GI tract
5HT-3 (serotonin) receptor antagonists bind to 5HT-3 receptors on vagal afferent (sensory) fibres in GI tract
↓ Binding of endogenous serotonin to vagal afferent fibres
↓ Innervation of medulla via vagal afferent fibres
H1 receptor antagonists (e.g. dimenhydrinate)
Brain receives conflicting information about body’s movement from visual, vestibular, proprioceptive senses
Motion sickness (nausea triggered by slow lateral or vertical movement)
↑ Activation of histaminergic neuronal system in hypothalamus
Antihistamine H1 receptor antagonists bind to H1 (histamine) receptors in chemoreceptors trigger zone in area postrema
↓ Binding of endogenous histamine in area postrema
Muscarinic antagonists (e.g. scopolamine)
Motion sickness activates vestibular afferent fibres
Vestibular afferents release acetylcholine
Muscarinic receptor antagonists bind to muscarinic receptors in posterior cerebellum
↓ Binding of endogenous acetylcholine to posterior cerebellum
↓ Diffusion of acetylcholine into 4th ventricle
↓ Binding of acetylcholine to area postrema
Cannabinoids* (e.g. synthetic tetrahydrocannabinol)
Emetogenic stimuli (e.g. chemotherapy) in blood and CSF
Area postrema has no blood- brain barrier
Emetogenic stimuli stimulate serotonin receptors in area postrema
Cannabinoids bind to 5HT-3 (serotonin) receptors in area postrema
↓ Binding of dopamine to area postrema
*Still under research
↓ Stimulation of vomiting center in medulla by emetogenic stimuli
Coordination of respiratory, gastrointestinal, abdominal muscle contraction
↓ Vomiting
Authors: Catherine Jung Reviewers: Claire Song Shahab Marzoughi Sylvain Coderre* * MD at time of publication
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published Aug 25, 2024 on www.thecalgaryguide.com")
Massive Transfusion Protocol
![Massive Transfusion Protocol: Considerations and rationale
Massive transfusion protocol (MTP) is a tool used by clinicians when there is a need to rapidly administer a large amount of blood products, including packed red blood cells (pRBCs), fresh frozen plasma (FFP), and platelets. Complications of MTP are commonly referred to as “The Lethal Triad” referring to hypothermia, acidosis and coagulopathy.
Authors: Kayleigh Yang Arzina Jaffer
Reviewers: Jasleen Brar,
Luiza Radu, Karl Darcus*
* MD at time of publication
Intervention
Indications Initial Response Pathophysiology Transfusion Targets
≥ 3 pRBCs unit transfusion requirement in 1 hour
Shock index (heart rate/systolic blood pressure) > 1
Blood volume loss >50% in ≤3 hours
ABC Score ≥ 3 of: 1. Penetrating mechanism of injury 2. Systolic blood pressure < 90 mmHg 3. Heart rate > 120 beats per minute 4. Evidence of hemoperitoneum or hemopericardium on ultrasound (positive FAST U/S exam)
RABT Score ≥ 2 of: 1. Penetrating mechanism of injury 2. Shock index > 1 3. Positive FAST U/S 4. Known or suspected pelvic fracture
Call for help
Activate institution's MTP protocol
Send for STAT type and screen
Establish large-bore intravenous access
Fluid resuscitation
Collect and send STAT bloodwork including hemoglobin, platelet, INR, fibrinogen, electrolytes, creatinine and arterial blood gas (ABG).
Citrate present in blood products to avoid clotting during storage
Stored pRBCs break down and release potassium due to time mediated degeneration
Temporary accumulation of citrate in patient's blood with rapid use of blood products
Citrate chelates calcium
Less negative cell membrane resting potential
Anaerobic metabolism
Promotes hypocalcaemia
Changes in membrane excitability
Lactic acid buildup
Coagulopathy
(see coagulation cascade slide)
Cardiac dysrhythmias (peaked T-waves, atrial block, “sine wave”, asystolic EKG changes)
Metabolic acidosis
End organ damage
Continued blood loss
Volume overload
Avoid hypocalcemia
Avoid hyperkalemia
pH 7.35-7.45
Bleeding source control
Hemoglobin >70-90
Platelets >50 INR <1.5 Fibrinogen >1.5
Avoid dilutional coagulopathy (clotting factor dilution)
Mean Arterial Pressure (MAP) >60mmHg
Temperature >35.0°C
Slow (over 5-10 minutes) IV calcium administration
Inhaled beta agonists
Insulin/Dextrose
EKG monitoring
Sodium bicarbonate
Increase minute ventilation
Fastest control method to prevent further blood loss (i.e., packing wounds)
Early tranexamic acid administration
Administer pRBCs, FFP, and platelets in a 1:1:1 ratio (fibrinogen replacement indicated if <1.5 despite FFP)
Minimize crystalloid use
Administer crystalloids in a 3:1 ratio to estimated blood loss until blood products available
Administer vasopressors to meet target, do not overshoot
Temperature monitoring Fluid warming
↑ [Potassium] in pRBCs solution
Administration of pRBCs ↑ potassium in patient's blood
Blood loss
↓ Hemoglobin
Tissue hypoperfusion
Tissue hypoxia
↑ Diluent volume
↓ Concentration of clotting factors
Tissue death
↓ Coagulation ability
↑ Transfusion requirements
Early fluid resuscitation
Rapid transfusion of cooled or room-temperature blood products/fluids
↑ Blood pressure
Development of hypothermia
↑ Bleeding and clot dislodgement potential
↓ Enzyme activity in the coagulation cascade
↓ Coagulation ability
Legend:
Pathophysiology
Mechanism
Targets
Intervention
Published Sept 5, 2024 on www.thecalgaryguide.com](https://calgaryguide.ucalgary.ca/wp-content/uploads/2024/09/Massive-Transfusion-Protocol.jpg "Massive Transfusion Protocol: Considerations and rationale
Massive transfusion protocol (MTP) is a tool used by clinicians when there is a need to rapidly administer a large amount of blood products, including packed red blood cells (pRBCs), fresh frozen plasma (FFP), and platelets. Complications of MTP are commonly referred to as “The Lethal Triad” referring to hypothermia, acidosis and coagulopathy.
Authors: Kayleigh Yang Arzina Jaffer
Reviewers: Jasleen Brar,
Luiza Radu, Karl Darcus*
* MD at time of publication
Intervention
Indications Initial Response Pathophysiology Transfusion Targets
≥ 3 pRBCs unit transfusion requirement in 1 hour
Shock index (heart rate/systolic blood pressure) > 1
Blood volume loss >50% in ≤3 hours
ABC Score ≥ 3 of: 1. Penetrating mechanism of injury 2. Systolic blood pressure < 90 mmHg 3. Heart rate > 120 beats per minute 4. Evidence of hemoperitoneum or hemopericardium on ultrasound (positive FAST U/S exam)
RABT Score ≥ 2 of: 1. Penetrating mechanism of injury 2. Shock index > 1 3. Positive FAST U/S 4. Known or suspected pelvic fracture
Call for help
Activate institution's MTP protocol
Send for STAT type and screen
Establish large-bore intravenous access
Fluid resuscitation
Collect and send STAT bloodwork including hemoglobin, platelet, INR, fibrinogen, electrolytes, creatinine and arterial blood gas (ABG).
Citrate present in blood products to avoid clotting during storage
Stored pRBCs break down and release potassium due to time mediated degeneration
Temporary accumulation of citrate in patient's blood with rapid use of blood products
Citrate chelates calcium
Less negative cell membrane resting potential
Anaerobic metabolism
Promotes hypocalcaemia
Changes in membrane excitability
Lactic acid buildup
Coagulopathy
(see coagulation cascade slide)
Cardiac dysrhythmias (peaked T-waves, atrial block, “sine wave”, asystolic EKG changes)
Metabolic acidosis
End organ damage
Continued blood loss
Volume overload
Avoid hypocalcemia
Avoid hyperkalemia
pH 7.35-7.45
Bleeding source control
Hemoglobin >70-90
Platelets >50 INR <1.5 Fibrinogen >1.5
Avoid dilutional coagulopathy (clotting factor dilution)
Mean Arterial Pressure (MAP) >60mmHg
Temperature >35.0°C
Slow (over 5-10 minutes) IV calcium administration
Inhaled beta agonists
Insulin/Dextrose
EKG monitoring
Sodium bicarbonate
Increase minute ventilation
Fastest control method to prevent further blood loss (i.e., packing wounds)
Early tranexamic acid administration
Administer pRBCs, FFP, and platelets in a 1:1:1 ratio (fibrinogen replacement indicated if <1.5 despite FFP)
Minimize crystalloid use
Administer crystalloids in a 3:1 ratio to estimated blood loss until blood products available
Administer vasopressors to meet target, do not overshoot
Temperature monitoring Fluid warming
↑ [Potassium] in pRBCs solution
Administration of pRBCs ↑ potassium in patient's blood
Blood loss
↓ Hemoglobin
Tissue hypoperfusion
Tissue hypoxia
↑ Diluent volume
↓ Concentration of clotting factors
Tissue death
↓ Coagulation ability
↑ Transfusion requirements
Early fluid resuscitation
Rapid transfusion of cooled or room-temperature blood products/fluids
↑ Blood pressure
Development of hypothermia
↑ Bleeding and clot dislodgement potential
↓ Enzyme activity in the coagulation cascade
↓ Coagulation ability
Legend:
Pathophysiology
Mechanism
Targets
Intervention
Published Sept 5, 2024 on www.thecalgaryguide.com")
IgA Nephropathy
 created by mucosa- bound IgA1 plasma cells is secreted into plasma instead of onto mucosal surface
IgA1 plasma cells hyper- responsive to triggers (eg. URTIs, gastroenteritis) ↑ synthesis of GD-IgA1 → spill-over into plasma
Immunoglobulin A1 (IgA1) plasma cells destined to reside in mucosa (eg. gut or respiratory tract) travel to and
reside in inappropriate site(s) (eg. bone marrow) releasing GD-IgA1 into plasma
GD-IgA1 is not cleared from plasma as quickly as IgA1 → ↑ plasma GD-IgA1 levels
Hit 3:
GD-IgA1-IgG complexes deposit in mesangium
C3 predominant complement activation amplifies inflammatory response
Renal biopsy:
IgA deposits in mesangium (100% sensitive)
Renal biopsy: Complement in mesangium (C3 predominant) (90-95% sensitive)
A cascade of multiple immunologic hits is initiated
Hit 1: ↑ Serum levels of GD-IgA1 multiple immunologic hits
GD-IgA1 hinge region is structurally distinct from IgA1 that would normally circulate in plasma (lack of galactosyl groups)
GD-IgA1 hinge region may mimic pathogens (ex. bacteria and viruses) or other antigens
Cross reactivity of IgG against GD-IgA1, or synthesis of anti-GD-IgA1 IgG antibodies
Immunoglobulin G (IgG) binds GD-IgA1 hinge region Hit 2: GD-IgA1-IgG immune complex formation
Circulating GD-IgA1-IgG complexes have high affinity for glomerular endothelial cells where they damage the glycocalyx → ↑ permeability of immunoglobulins into the mesangium
↑ Production of chemokines, cytokines and complement → ↑ mesangial cell proliferation and matrix expansion
Leukocyte recruitment and activation damages glomerulus and mesangium
Hit 4:
Inflammatory response to GD-IgA1 complexes in mesangium induce glomerular structure disruption (endothelium, basement membrane, podocytes, mesangium)
and impaired glomerular function
Loss of barrier functions of glomerulus allows for extravasation of blood & proteins into Bowman’s space and subsequently through tubules
Renal biopsy: Glomerulosclerosis, tubulointerstitial fibrosis, glomerular vasculitis, podocyte damage
Eventual end-Stage Renal Disease (ESRD)
Progressive ↓ of filtration surface area within glomeruli and ↓ number of functional glomeruli
Proteinuria
Synpharyngitic hematuria (hematuria with dysmorphic red cells co-occurring with pharyngitis)
↓ Glomerular Filtration Rate (GFR)
Nephrotic Syndrome
↑ Serum creatinine
Chronic kidney disease and eventually ESRD
IgAN is an autoimmune disease where IgA deposition in the glomerulus leads to an inflammatory cascade, endothelial dysfunction and mesangial expansion that damages glomeruli causing kidneys to leak blood and protein into urine and decreased kidney function. IgA nephropathy is a multifactorial disease requiring multiple immunologic hits
IgA Nephropathy (IgAN)
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published Sept 5, 2024 on www.thecalgaryguide.com")
Cystocele

Connective tissue disorders (e.g. Marfans, Ehlers- Danlos Syndrome)
Genetic susceptibility (e.g. Type III collagen gene abnormality
Menopause
Visceral fat places pressure on pelvic floor structures
Growing fetus places pressure on pelvic floor structures
Straining and bearing down on pelvic floor
Muscle tearing and damage
Disruption of nerves, loss of bladder structural support, and disruption of fascia and muscles
Collagen impairment
Depletion of ovarian follicles leading to ↓ in estrogen production
↑ Intra-abdominal pressure
Transfer of intra- abdominal pressure to pelvic floor
Pelvic floor muscles and pelvic floor fascia become weakened
Pelvic tissue and muscular atrophy
Loss of tissue function and structure support that collagen provided
↓ Stimulation of collagen production
↓ Estrogen levels
Pelvic Organ Prolapse Quantification (POP-Q) System: Grade 1: Bladder descends 1 cm above the hymen Grade 2: Bladder descends to ≤ 1 cm above or below hymen
Grade 3: Bladder descends past the hymen but 2 cm less than total vaginal length
Grade 4: Complete vaginal prolapse
Mechanical obstruction of bladder and urethra
Urinary retention
Descended bladder creates pressure in vagina
Pressure/bulging sensation
Symptoms of voiding dysfunction (e.g. incomplete emptying/frequency/ urgency/nocturia)
Vaginal intercourse puts pressure on descended bladder
Activates pain receptors
Dyspareunia (painful sex) for some patients
Cystocele
Descent of bladder through anterior vaginal wall
Hydronephrosis/ hydroureter
Recurrent UTI
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published Sept 5, 2024 on www.thecalgaryguide.com")
Cauda Equina Syndrome
 in lumbar spine
Mechanical compression of sacral and lumbar nerve roots between L2 and S1
Cauda Equina Syndrome
Damage to motor neurons within the compressed nerve roots
Acute ↓ stimulation/control of lower limb and pelvic floor muscles
Damage to fragile, smaller sacral nerves
Saddle anesthesia
Loss of sensation in perineum, perianal
area, and medial aspect of thighs
Patient unable to sense when bladder/ bowels are full
Damage to sensory neurons within the compressed nerve roots
Mechanically damaged nociceptive sensory neurons send ectopic impulses to the brain
Neuropathic pain in low back, radiating to one or both legs
Dermatomal pattern specific to the affected nerve root
Decreased rectal tone
Fecal incontinence
Flaccid paralysis in one or both legs
Permanent paralysis
Areflexia (absence of deep tendon reflexes)
Absent Achilles tendon reflex from compression of S1 nerve
Urinary detrusor muscle areflexia
Cannot overcome the residual tone of the internal urethral sphincters
Urinary retention
Compression of L2 nerve root
Sensory disturbance
along anterior and medial thigh
Compression of L3 nerve root
Sensory disturbance along anterior thigh, medial knee, and medial leg
Compression of L4 nerve root
Sensory disturbance along lateral knee, anterior leg, and dorsal aspect of foot
Compression of S1 nerve root
Sensory disturbance along posterior thigh, posterolateral aspect of leg, and lateral foot
Sexual dysfunction
Overflow incontinence
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published Nov 1, 2012; updated Sept 6, 2024 on www.thecalgaryguide.com")
Diffuse Axonal Injury

Increased intracellular sodium creates increased osmotic pressure between extracellular and intracellular space
Water moves passively along osmotic gradient into intracellular space
Neuron swelling
Hemorrhagic lesion on MRI
calcium exchangers
Activation of voltage gated calcium channels
Calcium enters the damaged neuron
Proteolytic activity increases with elevated intracellular calcium
Activated proteases cause delayed cytoskeleton damage Damage to neuron cytoskeleton impairs transport of axonal proteins Protein accumulates within axon creating bulb structure at the terminal Disconnection of axon terminal from adjacent dendrites and cell bodies
Diffuse Axonal Injury
Axonal bulbs on pathology
Damage to cortical, subcortical, and brainstem white matter tracts leads to a constellation of symptoms
Post-traumatic seizures Damage to temporal lobe Damage to primary motor cortex Lesions in ascending reticular activating system impact consciousness
Memory deficits Motor deficits
Coma & brain death
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published Sep 18, 2024 on www.thecalgaryguide.com")
S3 Pathogenesis

Author: Yvette Ysabel Yao Reviewers: Stephanie Happ George Tadros Shahab Marzoughi Jonathaan Howlett* * MD at time of publication
Athletes
Prolonged training causes ↑ left ventricle size and ↑ blood volume in ventricle
Tricuspid +/- mitral regurgitation (leaky tricuspid/ mitral valve)
Heart Failure (See Left Heart Failure slide)
Dilated cardiomyopathy (See Dilated cardiomyopathy slide)
Pulmonary +/- aortic regurgitation (leaky pulmonary / aortic valve)
Children and young adults
Compliant ventricle rapidly expands in early diastole
Diastole
S1
Systole
Diastole
↑ Blood volume flowing into ventricle from atrium after semilunar valves close (S2 heart sound)
Diastolic overload (↑ filling of ventricles)
Sudden intrinsic limitation of longitudinal expansion of the ventricular wall
Large volume of blood striking a very compliant ventricular wall
S3 (Low pitched, heard in early diastole, best heard apex in left lateral decubitus position with bell of stethoscope)
S2 S3
Left ventricle is located more posteriorly and is relatively unaffected by changes in intrathoracic pressure
Left ventricle S3 (unchanged or sometimes increased with expiration)
Anterior right ventricle more sensitive to negative intrathoracic pressure
↑ Venous return to right ventricle during inspiration
Right ventricle S3 (best heard along the lower left sternal border, intensity increases with inspiration)
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published Sep 18, 2024 on www.thecalgaryguide.com")
Low Ankle Sprain

Excessive stress to medial ankle deltoid ligament
Ankle plantar flexion and inversion beyond normal range (most common)
Excessive stress to lateral ankle ligament(s)
Associated fracture of malleolus or subtalar joint
Bone pain, inability to weight bear
Recruitment of inflammatory cells to damaged area
↑ Local pro- inflammatory cytokines
↑ Capillary permeability & vasodilation to damaged area
Collagen fibers in ligament(s) rupture
Low Ankle Sprain
(medial or lateral)
Local blood vessels tear
Blood leaks into surrounding tissue
Grade I
Mild injury
Grade II
Moderate injury
Grade III
Severe injury
Minimal ligament disruption
Incomplete ligament tear
Complete ligament tear
No joint laxity
Joint laxity
Gross joint laxity
Mechanical and functional instability
Decreased ankle joint space
Ankle Impingement
(compression of soft and/or bony structures in joint)
Chronic ankle instability
Talus subluxation with anterior force on heel
+ Anterior drawer test
Recurrent sprains
Synovial inflammation & hypertrophy
Remodeling of collagen and bone
Degenerative changes
(e.g., osteophytes, subchondral sclerosis)
Damage to mechanoreceptors in muscles surrounding ankle joint
Bruising
Swelling
Focal tenderness over torn ligament(s)
Pain on injury and with weight bearing
Impaired proprioception & neuromuscular control
Nociceptors activated by trauma and inflammatory factors
Falls Antalgic gait
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published Nov 9, 2014; updated Aug 15, 2023 on www.thecalgaryguide.com")
Malignant Hyperthermia
 & volatile anesthetic (e.g., sevoflurane, desflurane, isoflurane, halothane, NOT nitrous oxide)
Autosomal dominant mutation in ryanodine receptor (RyR1; RyR1 transports calcium out of the sarcoplasmic reticulum during muscle depolarization)
LowerthresholdforactivationofRyR1 state
Prolonged opening of RyR1
↑ Ca2+ in myocyte cytoplasm
Capacity of the reuptake protein to carry Ca2+ is overwhelmed
Sustained muscle contraction à hypermetabolic state
Malignant Hyperthermia
Vigorous exercise & heat (rare)
Rare life-threatening clinical syndrome that occurs in genetically susceptible patients upon exposure to a triggering agent
Skeletal muscle rigidity (i.e., masseter muscle spasm)
Sustained muscle contraction
Myocytes (muscle cells) deplete available ATP
Hypermetabolic state
↑ O2 consumption
↑ Heart rate to meet O2 demand
↑ CO2 production
Unexplained ↑ end tidal CO2 (early sign)
↑ Temperature
Hyperthermia (late sign)
↓ O2 supply
↑ Anaerobic metabolism
↑ Lactic acid production
Metabolic acidosis
Cell death
Leakage of muscle contents into circulation
↑ Serum creatinine kinase
Development of rhabdomyolysis (rapid breakdown of muscle tissue)
Myoglobinuria (presence of excess myoglobin in urine)
Acute kidney injury**
Hyperkalemia
Electrolyte imbalances (i.e., hyperkalemia)
Abnormal myocyte contraction
Cardiac dysfunction Dysrhythmias Cardiovascular collapse
Release of thromboplastin (converts prothrombinàthrombin) & other prothrombotic substances (promotes clot formation)
Imbalance between thrombotic & antithrombotic pathway
↑ Clot formation from activation of the extrinsic & common pathways
Disseminated intravascular coagulation**
Tachypnea (in absence of prominent mechanical ventilation)
Tachycardia (early sign)
Cardiovascular collapse
Vital organ failure Coma
**See corresponding Calgary Guide slide(s)
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published Oct 25, 2015; updated Oct 2, 2024 on www.thecalgaryguide.com")
Obesity Pathogenesis
 Zeng Reviewers: Gurreet Bhandal, Raafi Ali, Mizuki Lopez
Energy homeostasis dysregulation (Peptide hormones regulating hunger/satiety that influence the hypothalamic control of eating behaviours)
Luiza Radu, Dr. Sam Fineblit*
* MD at time of publication
Endocrine dysregulation
Neurological (↓ caloric reserve activates the neurological system)
Adiposity-related
Hypothalamus (hemostatic area)
↑ Production of ghrelin (peptide hormone)
↑ Activation of agouti-related protein (A Neuropeptide Y (NPY) neuron clusters in the hypothalamus via vagus nerve
↑ Appetite
Mesolimbic area (hedonic area)
Reward Driven Eating (meso-limbic region of brain)
↑ Food consumption
↑ Dopamine & endogenous opioid signals
↑ Pleasure & desire to consume more food
Prefrontal cortex (executive functioning)
↓ Glucagon-like peptide-1 (GLP-1) & pancreatic peptide YY (PYY) secretion post- meal
↓ Stimulation of Proopiomelanocortin (POMC) neuron clusters in hypothalamus
↓ Beta-melanocyte stimulating hormone (MSH) (endogenous peptide) production
Defect in leptin receptor gene
↓ Circulating
soluble leptin receptors (SLR)
↑ Serum leptin levels
Leptin resistance
Inactive leptin gene
↓ Adipocyte leptin production & secretion
↓ Leptin
transport across blood brain barrier
↓ Hypothalamus suppression of hunger
↓ Satiety sensation
↑ Caloric intake & weight gain
↑ Dietary carbohydrate intake
↑ Insulin production & secretion
↓ Cellular response to insulin
↑ Insulin resistance
↑ Insulin production & secretion to compensate for insulin resistance
Impaired glycemic control
Metabolic syndrome, type 2 diabetes mellitus and cardiovascular disease **
Metabolic adaptation to reduced-caloric intake efforts
↑ Body energy conservation
↓ Baseline energy expenditure
↓ Resting metabolic rate (the amount of energy body requires to function while at rest)
↓ Ability to lose weight & fat mass
↑ Hunger
↑ Caloric intake & weight gain
Obesity (A combination of metrics including ↑ BMI, ↑ weight circumference, ↑ waist-to-hip ratio, skinfold thickness, and other standardized measures varied by equipment available at various institutions resulting in a negative impact on the health of the individual)
**See slide on Pathogenesis of Type II Diabetes Mellitus
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published Aug 13, 2024 on www.thecalgaryguide.com")
Hyperthyroidism in Pregnancy
 hormone stimulates TSH receptors which ↑T3/T4 & ↓ physiologic TSH production in 1st trimester & normalizes in 2nd trimester
Transient hyperthyroxinemia in pregnancy (often benign)
Autoantibodies ↑ stimulation of thyroid stimulating hormone (TSH) receptors
Transplacental passage of TSH- receptor antibodies (can occur with normal thyroid function)
Graves’ disease
Transient ↓TSH & ↑T3/T4
Persistent ↓TSH & ↑T3/T4
Low birth weight Maternal congestive heart failure Pre-eclampsia (high blood pressure in
pregnancy)
Thyroid storm (excessive release of T3/T4 leading to a life-threatening hypermetabolic state)
↑ Triiodothyronine (T3) & thyroxine (T4) production independent of TSH
Abnormal differentiation of trophoblast embryonic cells ↑ hCG levels (cells that provide nutrition to the embryo)
Gestational trophoblastic disease
Toxic multinodular goiter
Toxic adenoma
Viral infection
Subacute thyroiditis (thyroid inflammation)
Hyperthyroidism in Pregnancy
Anterior pituitary gland releases stored TSH
↑Sympathetic nervous system stimulation
↑Thermogenesis
(heat production, regulated by thyroid & variousbraincentres)
↑Hyaluronic acid in dermis & subcutis tissue of the skin (Graves’ disease specific)
Transplacental passage of ↑T3/T4 to fetus
Gut hypermobility
Central nervous system overstimulation
↑Weight loss ↑Appetite
Heat intolerance
Diarrhea & ↑ bowel movements
Nervousness & anxiety
Hyperkinesia (excessive activity of a body part)
Hyperreflexia (overactive muscle reflex response)
Tremor
Poor attention span
↑ Heart rate
Palpitations (noticeable abnormal heartbeats)
Bruit (turbulent blood flow) heard over thyroid
↓ Exercise tolerance
↑ Cardiac output
De novo synthesis of TSH (synthesis of TSH independent of normal regulatory signals & processes as seen in toxic adenoma & toxic multinodular goiter)
Pregnancy Complications (abnormallyhighfreeT4&thyroid stimulating antibodies in the blood impacts fetal thyroid function)
↑ Renin angiotensin aldosterone system (RAAS) activation (important regulator of electrolytes, blood volume, & systemic vascular resistance)
↑ Erythropoietin (EPO, hormone made by kidneys that
stimulates red blood cell production)
Pretibial myxedema (condition causing skin lesions from deposition of hyaluronic acid)
Spontaneous abortion (pregnancy loss naturally ≤20 weeks gestation)
Premature labour (labour ≤37 weeks gestation)
Stillbirth (fetal death >20 weeks gestation)
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published September 29, 2024 on www.thecalgaryguide.com")
Secondary hypoglycemia Insulin Mediated

Non-insulinoma pancreatic islet cell disorder
(e.g. Non-insulinoma pancreatogenous hypoglycemia syndrome)
Nesidioblastosis (pancreatic islet tissues formed from pancreatic duct budding) formation
↑ In number & size of abnormal pancreatic beta islet cells throughout the entire pancreas
↑ Insulin production & secretion by abnormal pancreatic islet cells
↑ Glycogen synthesis
Insulin autoimmune syndrome
Autoantibodies bind to insulin molecules released post-meal
Insulin molecules unable to exert effects
Hyperglycemia Promotes insulin release
↓ Plasma glucose concentration
↓ Insulin release & ↓ total insulin levels
Insulin molecules dissociate from autoantibodies
↑ Free insulin levels (inappropriate for the ↓ plasma glucose levels)
↓ Glycogenolysis (glycogen breakdown to glucose)
Hypoglycemia
(serum glucose of <3mmol/L)
Intake of insulin secretagogues (e.g. Sulphonylureas, Meglitinide analogues)
Binds to adenosine triphosphate (ATP) dependant potassium channels on pancreatic islet cells
Inhibit potassium ion efflux through ATP-dependent potassium channels
Pancreatic islet cell depolarization
Opens voltage-gated calcium channel
Calcium influx into pancreatic islet cells
↑ Insulin release
↓ Gluconeogenesis (glucose synthesis from non-carbohydrate compounds)
Exogenous insulin intake
Suppress endogenous insulin production (by beta cells)
Low C-peptide levels
(byproduct of endogenous pro- insulin being converted to insulin)
90% caused by somatic mutations of pancreatic beta islet cells
10% due to genetic changes linked to MEN1(Multipl e endocrine neoplasia type-1) gene mutation
↑ Pancreatic islet cells proliferation
↑ Insulin secretion by hyperplastic (increasing in number) pancreatic islet cells
Neuroglycopenic symptoms (altered mental status, seizures)
Neurogenic symptoms (palpitations, tremulousness, diaphoresis
Authors: Iffat Naeem Run Xuan (Karen) Zeng Reviewers: Gurreet Bhandal Luiza Radu Yan Yu* Samuel Fineblit* * MD at time of publication
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published Sept 29, 2024 on www.thecalgaryguide.com")
Major Depressive Disorder 2024
: Pathogenesis and clinical findings
Self blaming tendency
Major stressor(s)
↑ Emotional reactivity Monoamine deficiency
Regional brain area alterations
↓ Behavioral activation
(engagement in meaningful activities to improve mood)
Hunger interoception (perception & awareness of hunger) changes
Attributes the occurrence of negative events to internal/personal factors
Expectation that future events are uncontrollable & internally caused
Feelings of worthlessness or excessive guilt
Recurrent thoughts of death or suicide
Insomnia (melancholic MDD)
Dysregulation of negative feedback control on the hypothalamus–pituitary– adrenal (HPA) axis
↓ Serotonin & norepinephrine signaling
↑ HPA axis sensitivity
Heightened responses to stressors
Major Depressive
Disorder
≥5 symptoms/complications during the same 2-week period; at least 1 of **symptoms; causing clinically significant distress or impairment in social, occupational, or other important areas of functioning
↑ Amygdala activity Changes to neuronal activity
Altered prefrontal cortex & limbic functioning
Depressed mood**
Mechanism unknown; can
precede MDD or be a symptom
Psychomotor agitation (restlessness)
↓ Dopaminergic transmission in ventral striatum
Overactivation of the insula (brain region involved in integrating activity in reward & interoception circuitry)
Under activation of the insula
Mechanism unknown
↓ Activation of mesolimbic system (reward system)
↓ Motivation & anticipation for rewards
↑ Activation of mesolimbic system
Psychomotor
retardation (slowing) Poor concentration
↑ Motivation & anticipation for rewards
Anhedonia (loss of interest/pleasure)**
Anergia (lack of energy)
↑ Eating (particularly high- sugar foods)
Hypersomnia (atypical MDD)
↓ or ↑ Appetite &/or weight
Authors: Keira Britto Sara Cho JoAnna Fay Reviewers: Luiza Radu Sara Meunier Jojo Jiang Alexander Arnold Brienne McLane* Phillip Stokes* *MD at time of publication
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
First published Mar 10, 2014, updated Sept 29, 2024 on www.thecalgaryguide.com")
Hypomagnesemia
![Hypomagnesemia: Physiology
Hyperglycemia
An increased amount of glucose enters renal tubules as glomerulus performs blood filtration
↑ [Solute] in renal tubules from ↑ glucose content exerts osmotic force that pulls water & electrolytes, including Mg2+, into renal tubules
↑ Urinary Mg2+ excretion
Lack of insulin
Lack of insulin receptor signaling in distal convoluted tubule (DCT) ↓ glucose uptake from renal tubules
Hypercalcemia
Ca2+ binds to Ca2+ sensing receptors on thick ascending limb (TAL) of loop of Henle, where resorption of Ca2+ & Mg2+ occurs
Receptor activation ↓ Na-K- 2Cl (NKCC) transporter activity which maintains electro- chemical gradient in TAL
Passive paracellular resorption of Ca2+ and Mg2+, dependent on electrochemical gradient, ↓
↑ Extra- cellular fluid
↓ Resorption of Na+ & H2O from renal tubules
Genetic disorders (e.g. Bartter syndrome, familial hypomagnesemia)
Medications (e.g. loop & thiazide diuretics, certain antibiotics, calcineurin inhibitors)
Some metabolic byproducts of these drugs are nephrotoxic
Inability to absorb free fatty acids (FFAs)
Mg2+, which associates with FFAs, is not absorbed through the gut
Steatorrhea (fat in the stool)
Mal- absorption (often due to inflammation or infection) & diarrhea
Acute pancreatitis
↓ Lipase secretion from pancreas ↑ levels of undigested fats in small intestine
↓ Passive Mg resorption from
tubules
Mg saponification in necrotic fat
2+
Varying mechanisms causing defective Mg2+ re-absorption (e.g. impacts to PCT, TAL, DCT disrupting transporters and ion shifting; ↓ gut resorption of Mg2+)
Nutrients & 2+
electrolytes are lost in stool
undergoes
Renal loss of magnesium
Gastrointestinal loss of magnesium
Hypomagnesemia
Serum [Mg2+] < 0.7 mmol/L
Impairs production and release of parathyroid hormone responsible for ↑ blood Ca2+ Hypocalcemia
Muscle cells are unable to activate Mg2+ dependent ATP hydrolysis
Impairs muscle relaxation and reduces the ability to stop muscular contraction
Lack of Ca2+ disrupts neurotransmitter release and neuronal signaling
Impairs rapid depolarization and repolarization during muscle contraction
Neuromuscular excitability (large, rapid change in membrane voltage due to small stimulus)
Delirium
Apathy
QRS widening and peaking of T waves on ECG
Torsade de Pointes
Constant muscle contraction compresses blood vessels
Reduced blood supply to hands, wrists, feet, and ankles
Trousseau sign (carpopedal spasm with inflation of BP cuff)
Authors: Caroline Kokorudz Reviewers: Shyla Bharadia Allesha Eman Michelle J. Chen Dr. Adam Bass* * MD at time of publication
Chvostek sign (facial muscle twitch with cheek touch)
Seizures
Tetany
Weakness
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published Oct 4, 2024 on www.thecalgaryguide.com](https://calgaryguide.ucalgary.ca/wp-content/uploads/2024/10/Hypomagnesia.jpg "Hypomagnesemia: Physiology
Hyperglycemia
An increased amount of glucose enters renal tubules as glomerulus performs blood filtration
↑ [Solute] in renal tubules from ↑ glucose content exerts osmotic force that pulls water & electrolytes, including Mg2+, into renal tubules
↑ Urinary Mg2+ excretion
Lack of insulin
Lack of insulin receptor signaling in distal convoluted tubule (DCT) ↓ glucose uptake from renal tubules
Hypercalcemia
Ca2+ binds to Ca2+ sensing receptors on thick ascending limb (TAL) of loop of Henle, where resorption of Ca2+ & Mg2+ occurs
Receptor activation ↓ Na-K- 2Cl (NKCC) transporter activity which maintains electro- chemical gradient in TAL
Passive paracellular resorption of Ca2+ and Mg2+, dependent on electrochemical gradient, ↓
↑ Extra- cellular fluid
↓ Resorption of Na+ & H2O from renal tubules
Genetic disorders (e.g. Bartter syndrome, familial hypomagnesemia)
Medications (e.g. loop & thiazide diuretics, certain antibiotics, calcineurin inhibitors)
Some metabolic byproducts of these drugs are nephrotoxic
Inability to absorb free fatty acids (FFAs)
Mg2+, which associates with FFAs, is not absorbed through the gut
Steatorrhea (fat in the stool)
Mal- absorption (often due to inflammation or infection) & diarrhea
Acute pancreatitis
↓ Lipase secretion from pancreas ↑ levels of undigested fats in small intestine
↓ Passive Mg resorption from
tubules
Mg saponification in necrotic fat
2+
Varying mechanisms causing defective Mg2+ re-absorption (e.g. impacts to PCT, TAL, DCT disrupting transporters and ion shifting; ↓ gut resorption of Mg2+)
Nutrients & 2+
electrolytes are lost in stool
undergoes
Renal loss of magnesium
Gastrointestinal loss of magnesium
Hypomagnesemia
Serum [Mg2+] < 0.7 mmol/L
Impairs production and release of parathyroid hormone responsible for ↑ blood Ca2+ Hypocalcemia
Muscle cells are unable to activate Mg2+ dependent ATP hydrolysis
Impairs muscle relaxation and reduces the ability to stop muscular contraction
Lack of Ca2+ disrupts neurotransmitter release and neuronal signaling
Impairs rapid depolarization and repolarization during muscle contraction
Neuromuscular excitability (large, rapid change in membrane voltage due to small stimulus)
Delirium
Apathy
QRS widening and peaking of T waves on ECG
Torsade de Pointes
Constant muscle contraction compresses blood vessels
Reduced blood supply to hands, wrists, feet, and ankles
Trousseau sign (carpopedal spasm with inflation of BP cuff)
Authors: Caroline Kokorudz Reviewers: Shyla Bharadia Allesha Eman Michelle J. Chen Dr. Adam Bass* * MD at time of publication
Chvostek sign (facial muscle twitch with cheek touch)
Seizures
Tetany
Weakness
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published Oct 4, 2024 on www.thecalgaryguide.com")
Transient Tachypnea of the Newborn

↑ Work of breathing
Maternal factors leading to impaired fetal lung development
Lack of labour-induced hormone changes (i.e. cortisol, catecholamines)
↓ Alveolar fluid reabsorption through epithelial aquaporin channels
↑ Alveolar fluid in lungs Disruption of laminar flow in airways
↑ Resistance to airflow
↑ Lung expansion to compensate
Limited surface area for gas exchange in the alveoli
↓ Ventilation
Fluid buildup in the major bronchi in the perihilar region
Prominent perihilar streaking on chest x-ray
↑ Intrathoracic pressure pushes the diaphragm down
Blunting of costophrenic angle on chest x-ray
Hyperinflation of lungs
Expanded lung fields on chest x-ray
↑ Deoxygenated hemoglobin in the bloodstream
Blue/purple discoloration of the skin particularly around the lips & fingers (cyanosis)
↓ Oxygenated hemoglobin in the bloodstream
↑ Nasal passage size allows for more air to enter the lungs with each breath
Nasal flaring
Scalene/intercostal/ sternocleidomastoid /abdominal muscle activation to assist with breathing
Accessory muscle use
↓Oxygen saturation (SpO2)
Neonate takes more breaths to compensate for limited oxygenation
Respiratory rate > 60 breaths/minute (tachypnea)
Transient Tachypnea of the Newborn
Temporary respiratory condition in newborns characterized by impaired lung function and rapid breathing
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published Oct 4, 2024 on www.thecalgaryguide.com")
Neonatal Hypoglycemia Clinical Presentation
 or was born to pregnant person with diabetes
Fetus was exposed to high levels of glucose through placental circulation
Fetus adapted to high blood glucose by producing high amounts of insulin
After birth, placental glucose supply is stopped but insulin remains high
↑ Insulin promotes inappropriately ↑ uptake of glucose into cells
put them at high risk for hypoglycemia
Cells cannot produce enough ATP from glucose to power physiological functions
Immature nervous system, use of fatty acids or proteins as an alternate ATP source, or adaptation to low glucose in utero can mask symptoms of hypoglycemia
Asymptomatic hypoglycemia
Neonates with low blood glucose often don’t show any symptoms and are detected by screening infants who are at a high risk for hypoglycemia
min are at risk for hypoglycemia
Infant is small for gestational age or experienced fetal growth restriction
Smaller neonates have smaller glycogen stores
Preterm infants born < 37 weeks
Glycogen is stored during the 3rd trimester; preterm infants did not have opportunity to build up stores
Loss of glucose source from placental circulation after birth puts preterm neonate at risk for low blood sugar
Body’s sympathetic system detects low glucose and triggers physical symptoms (neurogenic symptoms)
Symptomatic hypoglycemia
Neurons in brain are unable to produce enough ATP and thus function properly, triggering symptoms related to low sugar in the CNS (neuroglycopenic symptoms)
Non-specific symptoms are not exclusive to hypoglycemia and warrant further investigation to exclude other differential diagnosis (sepsis, inborn errors of metabolism, neonatal abstinence syndrome, neonatal encephalopathy, perinatal asphyxiation)
Jitteriness or tremors
Sweating Irritability Tachypnea Pallor
Pathologic poor feeding
Weak or a high- pitched cry
Change in level of consciousness (lethargy or coma)
Seizures
Pathologic hypotonia for gestational age
Apnea Bradycardia Cyanosis Hypothermia
Neonates with perinatal stress (e.g. birth asphyxia, meconium aspiration)
Body uses more glucose to produce ATP to manage condition causing physiological stress
Glucose stores are used up more quickly
Born to pregnant person with beta-blocker use
Body in stress releases epinephrine to promote sympathetic responses including glycogen breakdown into glucose
Beta blockers from placental circulation prevent epinephrine from binding to its receptor in infant’s body
Neonatal Hypoglycemia: Asymptomatic and symptomatic hypoglycemia satisfy the criteria of blood glucose < 2.6 mmol/L in both term and preterm infants within 72 hours of birth, and after 72 hours of age glucose < 3.3 mmol/L
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published Oct 4, 2024 on www.thecalgaryguide.com")
Spontaneous Rupture of Membranes

Authors: Wendy Xu Reviewers: Riya Prajapati Michelle J. Chen Dr. Jadine Paw* * MD at time of publication
Intrauterine inflammation
Fetal maturation
Fetal growth
Uterine contractions
↑ Pro-inflammatory cytokine & chemokine release in fetal membranes & amniotic fluid
↑ Stretch forces on fetal membranes
↑ Pro-apoptotic factors induces cellular apoptosis of fetal membranes
Changes in collagen and protein composition drive extracellular matrix remodeling in fetal membranes
↓ Tensile strength
Structural weakening of fetal membranes
Occurs primarily in the focal area of fetal membranes overlying the cervix
↑ Matrix metalloproteinases triggers extracellular matrix degradation in fetal membranes
Amnion and choriodecidua separation
Amniotic fluid flows from vagina
Amniotic fluid pools in posterior fornix on speculum exam
Pre-labour rupture of membranes
Membranes rupture before onset of uterine contractions
Chorioamnionitis (infection of the fetal membranes and amniotic fluid)
Neonatal infection
Endometritis (infection of the endometrium)
Amniotic fluid leaks through the cervix
Prolonged rupture of membranes (>18hrs) before delivery
Microbes ascend through vaginal canal
Low amniotic fluid volume on ultrasound
Amniotic fluid (pH 7.0-7.5) mixes with normal vaginal fluid (pH 4.5-6.0) which increases vaginal fluid pH to > 6.5
Positive nitrazine (pH indicator) test
Ion- and estrogen-containing amniotic fluid enters vaginal canal
Ferning (branching pattern) of vaginal fluid under microscope
Accompanies uterine contractions, cervical effacement & cervical dilation
Delivery/birth
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published Oct 4, 2024 on www.thecalgaryguide.com")
Precocious Puberty
 > chronological age
Gonadotropin releasing hormone (GnRH)-dependent causes involve activation of the hypothalamic-pituitary-gonadal (HPG) axis. Also known as central precocious puberty
GnRH-independent causes of precocious puberty are related to increased amounts of hormone in the body. Also known as peripheral precocious puberty
Neurogenic (previous CNS insult) and CNS tumors (e.g. hypothalamic hamartoma, germinoma, astrocytoma)
Idiopathic
Tumours causing ↑ hormone levels
Exogenous hormones causing ↑ hormone levels
Genetics causing ↑ hormone levels
21-hydroxylase enzyme deficiency
Growth of ovarian cysts/tumors (ex. germinoma, teratoma, choriocarcinoma)
Growth of β-HCG-secreting tumours (e.g. germinomas, dysgerminomas, hepatomas
Growth of benign or malignant adrenal tumours (e.g. adenoma, carcinoma)
↑ Estrogen and/or progesterone
↑ Beta-HCG stimulates ↑ testosterone production
↑ Cortisol, androgens, and/or aldosterone
Exposure to exogenous sex steroids (e.g. anabolic steroids, topical testosterone, oral contraceptives)
Steroid dependent features (e.g. exposure to estrogen creams in males may lead to breast development)
Missense mutation in LH receptor gene (male limited)
↑ Activation of LH receptors
↑ Testosterone production
Café au lait lesions Polycystic fibrous dysplasia
Puberty onset ages 1-4
↑ Activation and mutation of GNAS1 gene (McCune Albright Syndrome)
Various endocrinopathies
Growth acceleration
Congenital adrenal hyperplasia
↓ Cortisol, ↑ ACTH
Bone age > chronological age Absence of testicular enlargement
Female ambiguous genitalia
Authors: MacKenzie Horn Reviewers: Dasha Mori Michelle J. Chen Dr. Jean Mah* * MD at time of publication
Normal variations in hormone production
↑ Sensitivity to estrogen
Early adrenal androgen secretion
Idiopathic premature thelarche
Idiopathic premature adrenarche
Unilateral or bilateral breast development No other secondary sex characteristics Bone age = chronological age Development of pubic hair ± axillary hair No other secondary sex characteristics Bone age = chronological age
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published Oct 4, 2024 on www.thecalgaryguide.com")
Acute Diverticulitis

Obstipation (inability to pass any feces)
Inherent weakness in the muscle layers of the colonic wall associated with immature collagen fibers and diminished wall elasticity
Mucosal and submucosal layers of the colon wall push through a weak spot of the circular muscle layer
Formation of diverticulum (sac-like protrusion of the colonic wall)
Continued stress on diverticula causes micro- perforations of the diverticulum
Inflammation of diverticula
Mesentery and pericolic fat (fat surrounding the colon) attempt to wall off inflammation or perforations
Stool bacteria escapes the colon
Formation of abscess (accumulation of pus in response to the bacteria )
Pro-inflammatory cytokine release from nearby adipose tissue
Hypothalamic thermoregulatory center increases core temperature set point
Fever
Irritation of parietal peritoneum
Inflamed vessels are more permeable and fluid leaks from colonic vessels into the abdominal cavity
Chronic low-grade inflammation triggers activation of pro-fibrotic factors and fibroblasts
Excess production of extracellular matrix proteins
Stimulation of somatic nerves sends pain signals to the brain
Lower left quadrant abdominal pain
Peritoneal signs (abdominal guarding, rigidity, rebound tenderness)
↓ Total circulating blood volume
↑ heart rate
↓ Jugular venous pressure
Orthostatic hypotension (low blood pressure when standing after sitting/lying down)
Gastrointestinal strictures and/or colonic obstruction
Accumulation of micro- perforations further weakens intestinal wall
Complete bowel perforation (medical emergency)
Development of an abnormal connection (fistula) through the bladder, vagina, skin, or gut
Abscesses
Accumulation of fibrotic tissue narrows the intestinal lumen
Fibrosis of colon
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Re-Published Oct 4, 2024 on thecalgaryguide.com")
Tuberous Sclerosis Complex Dermatologic Manifestations
: Dermatologic Manifestations
De novo mutation of tumour suppressor genes (TSC1 (on chromosome 9q34) or TSC2 (on 16p13.3))
Encode hamartin & tuberin proteins that regulate cell division & growth
Hamartin & tuberin proteins are mediated by mammalian target of rapamycin (mTOR) Mutations ↑ mTOR signalling
Irregular, dysfunctional cell proliferation - often affecting embryonic cells
Author: Jasmine Gill Reviewers: Maharshi Gandhi Elise Hansen Sunawer Aujla Shahab Marzoughi Jodi Hardin* * MD at time of publication
Dysfunctional melanocytes that have an impaired ability to produce & transfer melanin
Dysregulated melanocyte migration during embryogenesis
Overgrowth of normal skin components (hamartomas)
Clonal patches of melanocytes produce increased pigment
Café-au-lait macules
Facial angiofibromas and fibrous cephalic plaque (red, pink papules)
Clonal patches of melanocytes are unable to produce sufficient pigment and/or there is impaired production and distribution of melanin
Hypopigmented macules
Affects dermal cells
Shagreen patch (cobblestone, yellow plaques often over lumbosacral area)
Affects epidermal cells
Molluscum pendulum (soft papules)
Affects epidermal & dermal cells
Ungual fibromas (flesh coloured or red papules around nails)
Insufficient melanin production by melanocytes leads to insufficient pigmentation
Ash leaf spot (rounded at 1 end and tapered at the other)
Overgrowth of dermis causes fibrous papules
Guttate/ ”confetti” macules
Epidermal layer
Melanocytes in basal layer
Fibroma expands and creates localized pressure and mass effect
Distortion of nail bed
Irregular, overgrowth of epidermal &/or dermal cells
Facial disfigurement
Dermal layer
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published Oct 4, 2024 on www.thecalgaryguide.com")
Chronic Subdural Hematoma
: Pathogenesis and Clinical Features Atraumatic SDH risk factors
Authors: Cora Laidlaw Reviewers: Braxton Phillips Shahab Marzoughi Gary Michael Klein* * MD at time of publication
For more information on acute subdural hematoma, see Calgary Guide slide - Acute Subdural Hematoma (SDH):
Traumatic SDH mechanisms
Intoxication leading to decreased balance and coordination
Neurodegenerative disease causes cerebral atrophy (such as ALS, MS, and dementia)
General cerebral atrophy with increased age
Chronic alcohol use dilates blood vessels, thinning the walls
Thinner, developing vessels in infants
Age related risks: decreased vision, decreased mobility, decreased balance
Low Impact trauma such as minor falls
Child abuse
Increased tension on bridging veins as ↓ brain volume ↑ distance they must span
Bridging veins are more delicate
Abusive head trauma
Cytokines increase the leaky nature of vessels
Blood degradation over time releases proinflammatory cytokines
Atraumatic risk factors combined with traumatic mechanisms results in the breaking of bridging veins
Low pressure venous blood slowly accumulates between the dura and arachnoid meningeal layers (increased with anticoagulation, hypertension, or other bleeding risk factors)
Damaged tissue release inflammatory factors that promote angiogenesis through secondary intention (the use of granular tissue to fill in the non-approximated edges of the blood vessels)
As the vessels are not approximated (connected to be rejoined), the granular tissue does not create a solid blood vessel wall
Vessels are partially repaired and leaky in nature
Recurrent bleeding due to small traumas and fluid accumulation due to leaky vessels results in expansion
Chronic Subdural Hematoma
(Bleeding within potential space between dura and arachnoid meningeal layers present >14 days)
Dural attachments limits fluid expansion
Local increased pressure and a mass effect on underlying brain tissue
Cerebral atrophy (specific areas and therefore symptoms will depend on lesion location)
Mesial temporal lobe
Impaired memory (including verbal)
Acute blood is present in small volumes with larger volume chronic hematoma
Damaged tissues release proinflammatory cytokines from immune cells (astrocytes, peripheral immune cells, neurons, and microglial cells)
Acute blood appears hyperintense on T2 MRI
Chronic blood appears hypointense on T2 MRI
Cytokines inflame nociceptive neurons (such as the trigeminal neuron)
Recurrent headaches
Altered mental status (confusion) Personality changes
Damage to neuronal tissue
Mass compression of underlying vasculature Hypoperfusion of brain tissue
T2 MRI Brain shows lesions with hyperintensive cores surrounded by hypointensive
Pressure placed indirectly onto cerebellum with inferior displacement of brain mass
Gait ataxia
Frontal lobe atrophy (specifically in orbitofrontal area) Orbitofrontal area Prefrontal cortex
Impaired working memory (short term memory used for rapid executive, phonological visuospatial thought processes)
Atrophy of focal brain structure
Contralateral homonymous hemianopsia (loss of the half visual fields in both eyes)
Somatic motor cortex
Contralateral hemiplegia (inability to move the body opposite to the side of lesion)
Primary visual cortex Pontine micturition centre
Urinary incontinence (uncontrolled leakage of urine)
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published Oct 4, 2024 on www.thecalgaryguide.com")
Epilepsy in Older Adults
, primarily ischemic stroke, intracerebral hemorrhage, subarachnoid hemorrhage
Ischemic and hemorrhagic injuries cause inflammation and nerve cell degeneration
Glial cells (astrocytes and oligodendrocytes) proliferate around the lesion area to repair the damaged tissue
Glial scar formation impedes neuronal reconnection and growth
Alzheimer’s or Vascular Dementia
Central nervous system disease (e.g. traumatic brain injury, prior meningitis, mass)
Medications associated with hyponatremia (e.g. diuretics, antidepressants, antipsychotics, etc.)
Cerebral edema
Increased intracranial pressure
Compression on structures and blood vessels
Sleep deprivation
Tau or amyloid deposition (abnormal protein aggregates in brain)
Small vessel disease
Increased delta wave activity
Heightened neural excitability
Decreased seizure threshold
Elevated stress hormones (e.g. cortisol)
Increased neuronal excitability and decreased inhibition
Areas of tissue death, white matter changes & cortical irritability
Structural and electrical brain changes
Epilepsy: Neurological disorder characterized by increased susceptibility to recurrent unprovoked seizures
Excessive, hypersynchronous & oscillatory network function Imbalance between excitatory and inhibitory activity
Resultant seizure activity
Atypical Seizure pattern; e.g. seem confused, stare into space, wander, make unusual movements, inability to answer questions
Often atypical location in brain (limbic or neocortical)
Focal seizures are more common than generalized
Postictal paresis can last for days & disorientation, hyperactivity, wandering and incontinence may persist for 1 week
Neurotransmitter dysregulation, neural network disruption, genetic factors, psychosocial factors
Psychiatric comorbidities
Authors: Anna Crone
Reviewers: Anika Zaman,
Rachel Carson, Raafi Ali, Luiza Radu, Gary Michael K Klein*
* MD at time of publication
Widespread structural changes and hippocampal atrophy
Dementia
Sub-optimal treatment results in ongoing and more frequent epileptic seizures
Status epilepticus (higher mortality among older adults)
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published Oct 4, 2024 on www.thecalgaryguide.com")
Cholesteatoma of middle ear
: Pathogenesis and clinical findings
Authors: Emma Holmes Angela Mak Reviewers: Stephanie Cote Vaneeza Moosa Shahab Marzoughi William Kim Sunawer Aujla Kristine Anne Smith* * MD at time of publication
Tympanic membrane perforation (e.g., acute otitis media, trauma)
Squamous epithelium invasion & migration into middle ear
Eustachian tube dysfunction (e.g., craniofacial abnormalities)
Negative pressure
Invagination of tympanic membrane
Retraction pocket with keratin trapping & ingrowth
Inflammation (e.g., upper respiratory tract infection, rhinitis)
Mucosal lining of middle ear become hyperproliferative
Squamous metaplasia
Basal cell hyperplasia
Invagination & epithelial ingrowth in basement membrane of the middle ear
Theory of implantation due to trauma (e.g., during surgery)
Implantation of skin into the
middle ear through a defect in the eardrum
Traps moisture
Bacterial infection
Erosion of external auditory canal
Conductive hearing loss
Labyrinthine fistula (abnormal communication between the inner ear and the surrounding structures)
Leakage of perilymph
Cholesteatoma
Destructive growth of keratinizing squamous epithelium in the middle ear
Bone erosion allows for secondary infection from outside the middle ear
Chronic otitis media
Migration of bacteria from middle to inner ear
Intracranial infection (e.g., meningitis, parenchymal abscess)
Acute otitis media
Accumulation of keratinous debris
Release of inflammatory molecules
Release of osteoclasts
Secretion of acid and proteinases
Erosion of temporal bone
Pressure change in the middle ear
Aural polyp (growth in external or middle ear canal)
Vertigo
Coalescent mastoiditis (bone is remodeled and resorbed from pressure necrosis, inflammation, and increased osteoclastic activity)
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published Oct 4, 2024 on www.thecalgaryguide.com")
Acute Subdural Hematoma
: Clinical Presentation Subdural Hematoma
Authors: Cora Laidlaw
Reviewers: Braxton Phillips Shahab Marzoughi Sina Marzoughi* * MD at time of publication
Ischemia of cranial nerve tissue in brainstem
Cranial nerve palsies
↓ Level of consciousness
Generalized or focal seizures
Uncal herniation
Compression of oculomotor nerve (CN III) – responsible for eye movement (adduction, elevation, and depression) and parasympathetic tone
Eyes fixed outwards and down with pupillary dilation
↑ Pressure compresses intracranial tissue
Compression of brain structures
Compression of arterial vasculature in brain
↓ Global perfusion to intracranial tissue
Uncus (mediobasal aspect of temporal lobe) herniates into the infratentorial via the tentorial notch
Cerebellar tonsils (part of posterior lobe of cerebellum) herniates through foramen magnum (opening in base of skull)
Cerebellar tonsil herniation
Tonsils compress brainstem
Brainstem loses ability to control vital functions of life (such as respiration, heart rate, blood pressure)
Coma or death
Blood accumulation results in ↑ intracranial pressure
(Bleeding within potential space between dura and arachnoid meningeal layers )
Compression of nociceptors in meninges and meningeal vasculature
Headache
Compression of medulla (contains emetic center)
Stimulation of emetic centers
Loss of function of primary sensory cortex (posterior to central sulcus)
Contralateral hemisensory loss (including proprioception, fine touch, and two point discrimination)
Loss of function of primary motor cortex (anterior to central sulcus)
Contralateral hemiparesis (upper motor neuron)
Ischemia and necrosis of brain parenchyma
Cerebral disturbances altering neuronal networks
Vomiting
Nausea
See Calgary Guide slide - Chronic Subdural Hematoma (SDH): Clinical Presentation
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published Oct 4, 2024 on www.thecalgaryguide.com")
Primary Combined Hyperlipidemia

Reviewers: Raafi Ali, Luiza Radu *Samuel Fineblit *MD at time of review
Familial dysbetalipoproteinemia or “Remnant Removal Disease” (autosomal recessive inheritance)
Apolipoprotein E (ApoE) mutation
(ApoE is found on triglyceride-rich lipoproteins, such as chylomicron & very low-density lipoprotein (VLDL))
Impaired triglyceride-rich lipoprotein particle binding to lipoprotein receptors
↓ Chylomicron & VLDL remnant clearance
Familial combined hyperlipidemia
Common, occurs in 1/50 to 1/100
(Polygenic, involving multiple genes, with variable expression & penetrance. Not all individuals will experience all clinical signs or complications)
Adipose tissue dysfunction
Impaired lipolysis (triglyceride breakdown in adipose tissue)
↓ Triglyceride storage in adipocytes
↑ Lipid levels
↓ Low-density lipoprotein (LDL) receptor function
Impaired LDL particle endocytosis into cells
↓ LDL clearance
↑ Plasma LDL levels
Hepatic fat accumulation
↑ Apolipoprotein B production
↑ Very low- density lipoprotein (VLDL) production
↑ Plasma VLDL levels
Lipoprotein lipase dysfunction
↓ Triglyceride breakdown
Delayed clearance of very low-density lipoprotein (VLDL), intermediate- density lipoproteins (IDL) & low-density lipoprotein (LDL) particles
↑ Plasma VLDL, IDL & LDL levels
↑ Triglycerides & cholesterol
↑ VLDL in serum
Xanthomas (build up of cholesterol deposits on skin, eyelids, palms or tendons of ankles, elbows & knees)
↓ Conversion of VLDL to low-density lipoproteins (LDL)
↓ LDL in serum
Atherosclerosis progression & ↑ risk of metabolic syndrome (↑Blood pressure, ↑ triglycerides, ↑ blood glucose, ↑ body mass index)
Coronary artery disease & peripheral vascular disease
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published Oct 4, 2024 on www.thecalgaryguide.com")
Bone Remodeling Physiology
 activity, ↑ osteoclast (bone cells that break down bone) activity
Hyperparathyroidism ↓ Blood Ca2+
↑ Parathyroid hormone (serum Ca2+ concentration increasing hormone)
Trauma
Osteoblasts detect breach in matrix integrity
Hyperthyroidism
↑ Triiodothyronine (T3)
↑ Bone turnover
Puberty
↑ Growth hormone
↑ Blood calcium (Ca2+)
↑ Calcitonin
(reduces serum Ca2+ by ↓ renal Ca2+ reabsorption & ↑ osteoclast activity)
Net bone resorption process (bone tissue released from bones)
Surveillance osteoblasts produce receptor activator of nuclear activator kappa beta (RANKL; osteoclast stimulating protein)
Monocytes fuse into osteoclasts (cells for bone breakdown)
Osteoclast actions
Secrete HCl to dissolve hydroxyapatite (bone matrix forming inorganic mineral)
Serum markers of bone resorption: C-Telopeptide (CTX) P-Telopeptide (PTX)
Net bone formation process
Hormones activate osteoblast
Osteoblast actions
Secrete osteoprotegerin that binds RANKL
Secrete osteoid seam (a new layer of unmineralized organic bone matrix)
Osteoblasts deposit hydroxyapatite on seam
Phagocytose osteocytes within bone matrix
Authors: Andrew Wu, Jason Kreutz Reviewers:Mizuki Lopez,
Gurreet Bhandal, Luiza Radu *Samuel Fineblit
* MD at time of publication
Secrete collagenase enzyme to help digest collagen
↓ Osteoclast activity
Osteoblasts become osteocytes (mature cells) within bone
Serum markers of bone formation:
↑ Alkaline phosphatase (ALP)
↑ Bone-specific alkaline phosphatase (BSAP; an enzyme produced by osteoblasts during mineralization of bone)
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published Oct 4, 2024 on www.thecalgaryguide.com")
Persistent Truncus Arteriosus

Spontaneous
Authors: Inioluwa Adeboye Reviewers: Juliette Hall George S. Tadros Shahab Marzoughi Kim Myers* * MD at time of publication
Persistent Truncus Arteriosus
Congenital heart defect
Cardiac neural crest (cells that form the outflow tract in fetal development) ablation defect
Dysplasia (abnormal cell growth) of the truncal valve
Common ventricular outflow tract (merged aorta and pulmonary trunk)
Valvular deformity: a single truncal valve
1-4 thick deformed leaflets
During normal fetal development and first week of life, pulmonary vascular resistance (PVR) is high to bypass undeveloped lungs
Peripheral vascular resistance (PVR) ↓ post-birth
Blood preferentially enters pulmonary circulation (left-to-right shunt)
Mixed oxygenated and deoxygenated blood is pumped to the pulmonary, systemic, and coronary circuits
Mild cyanosis and oxygen saturation <95%
Difficulty opening and
closing abnormally enlarged, stiff and/or rubbery valves
Ejection click (high-pitched early systolic sound)
Truncal valve regurgitation
(inadequate valve closure)
Turbulent backflow through valve into the ventricles
Diastolic murmur
Truncal valve stenosis
(inadequate valve opening)
↑ Ejection time
Systolic murmur
Runoff from aorta to pulmonary vasculature during diastole and systole
↓ Diastolic pressure in aorta
↑ Pulse pressure (difference between diastolic and systolic aortic pressure)
Bounding peripheral pulse
Pulmonary blood flow (PBF) ↑ while systemic and coronary blood flow ↓
↑ Blood return to left atrium
Volume overload of the left heart
Eccentric hypertrophy
↑ PBF causes vascular remodeling of the pulmonary vasculature
↑ PVR
↑ Pressure in the right ventricle and irreversible pulmonary hypertension
Right-to-left shunt development (Eisenmenger syndrome)
Congestive heart failure
↓ Cardiac output causing backup of blood into venous
system and ↑ heart rate and respiratory rate to attempt to compensate
↑ Heart rate, ↑ respiratory rate, and peripheral edema
Deoxygenated blood bypasses the lungs and is pumped systemically causing lower oxygen concentration in arterial circulation
Severe cyanosis
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published Oct 4, 2024 on www.thecalgaryguide.com")
Thyroid Eye Disease

Thyroid eye disease
Genetic factors (mechanism unknown)
An autoimmune condition wherein antibodies that target the thyroid & the periorbital tissues induce tissue swelling around the eye
Auto-antibody production
Type 1 insulin-like growth factor receptor (IGF-1, regulates cell growth) & thyroid stimulating hormone (TSH) receptor activation in CD34+ fibrocytes & orbital fibroblasts (cells part of the formation of connective tissue)
Chemokine production (cells part of the inflammatory response) ↑ Immune response in the tissue around the eye (orbit) Fibrosis (overgrowth/scarring) & swelling of orbital tissues
Eyelid retraction & lid lag on downward gaze
↑ Ocular surface exposure
Corneal trauma, dryness, & microbe exposure
Exposure keratopathy (corneal damage from ↑ exposure to air)
Mechanical force causes eye misalignment
Restrictive strabismus (both eyes do not line up in the same direction)
Double vision
Burning/foreign body sensation & epiphora (excess tears)
Exophthalmos (bulging of one or both eyes)
Mechanical strain on optic nerve
Compressive optic neuropathy (damage to the optic nerve)
Vision loss
Afferent pupillary defect (altered pupil dilation)
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published Oct 5, 2024 on www.thecalgaryguide.com")
Subtrochanteric Femur Fracture

Strong lateral or axial force directed through femur
Low energy mechanism of fracture
Low energy trauma typically seen in elderly or patients with osteoporotic (e.g. falls)
Atypical fractures
Chronic bisphosphonate use (drug that inhibits osteoclasts from resorbing bone)
Chronic suppression of bone resorption prevents natural bone building and breakdown (remodeling)
Microscopic damage accumulates, weakening bone Pathologic fracture
Primary or metastatic bone tumor
Cancer cells in bone disrupt physiological bone remodeling
Subtrochanteric Femur Fracture
Fracture in the region of the femur that is 5 cm distal to lesser trochanter
Muscle forces on proximal femur
Muscle forces on distal femur
Gracilis & adductor muscles exert shortening & adduction forces
Gluteus medius & gluteus minimus muscles exert abduction force
Iliopsoas muscles exert flexion forces
Short external rotator muscles exert external rotation forces
Bleeding from tissue & vascular damage
Swelling & ecchymosis at fracture site
Injury stimulates nerve fibres
Displaced proximal femur
Inability to weight bear
Authors:
Nojan Mannani
Jack Fu
Reviewers:
Annalise Abbott
Reza Ojaghi
Usama Malik
Michelle J. Chen
Dr. Richard Buckley*
Dr. Meredith Stadnyk*
* MD at time of publication
Shortened leg
Hip & thigh pain
Pain on motion
Surgery is required to fix fracture. Complications may arise after operation.
Femoral head & neck have limited blood supply which is required for fracture healing (union). Delay in surgery may ↑ time without supply
Non-union of fracture
Bone is unable to be aligned, leaving a space between broken bones
Malunion of fracture (limb length discrepancy, rotation of limb)
Surgical procedures ↑ risk of exposing incision to bacteria on contaminated instruments, skin, or in air
Infection (acute or chronic) often associated with malunion
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published Sept 24, 2017; updated Oct 14, 2024 on www.thecalgaryguide.com")
Complex Regional Pain Syndrome
: Pathogenesis and clinical findings
Type 1 origin: CRPS arises spontaneously or from trauma without confirmed damage to nerves (e.g. surgery, nerve compression, fracture, tissue trauma, ischemia, sprain)
Type 2 origin: CRPS arises from trauma with confirmed evidence of nerve damage
Intense psychological stress is associated with ↑ severity of CRPS
Peripheral nerve endings release ↑ levels of neuropeptides (e.g. substance P, bradykinin, calcitonin gene-related peptide)
Vasodilation & protein extravasation in tissues
↑ Perfusion to motor cortex
↓ Grey matter volume in cortical pain regions
↓ Cortical grey matter volume & ↓ perfusion of cortex in limbic & sensorimotor areas
Dysregulation of sympathetic nerve fibres Autonomic dysfunction
Central nerve fibres ↑ release of proinflammatory cytokines while ↓ release of anti- inflammatory cytokines
Central sensitization (threshold at which a central nerve transmits a signal is lowered so that it more readily transmits signals)
Mechanical hyperalgesia (hallmark of central sensitization)
Authors:
Jessica Hammal
Calvin Howard
Reviewers:
Sina Marzoughi
Michelle J. Chen
Scott Jarvis*
* MD at time of publication
Nociceptors (nerve endings detecting pain) on skin become activated
↑ Cytokine & nerve growth factor release
Primary afferent (sensory) neurons release ↑ levels of inflammatory neuropeptides
Peripheral sensitization (threshold at which a nerve transmits a signal is lowered so that it more readily transmits signals)
Widespread neurogenic inflammation (inflammation due to activation of peripheral nerve fibres)
Sustained and dysregulated pro-inflammatory state
Hyperalgesia (↑ sensitivity to feeling pain)
Sweat gland related changes: Edema, sweating changes, asymmetric sweating
Allodynia (pain from a stimulus that doesn’t normally cause pain)
Trophic changes (wasting of skin, muscle, tissues; thinning of bones; thickening/thinning of nails)
Motor dysfunction
↓ Range of motion
Immobile due to severity of pain
Impaired vasodilation & vasoconstriction
Temperature asymmetry
Altered skin color (red, blue, pale)
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published Oct 28, 2024 on www.thecalgaryguide.com")
Newborn Disorders of Sexual Development
 & Swyer syndrome (complete gonadal dysgenesis)
Lack or inactivation of sex determining region of the Y chromosome (SRY gene – responsible for testicular differentiation)
Testes produce ↓ testosterone & variable amounts (either ↓/ ↑) of anti- müllerian hormone (AMH – hormone produced by Sertoli cells in male fetuses that signals the regression of the müllerian ducts)
17-beta-hydroxysteroid dehydrogenase 3 deficiency (functional testes with variants in testosterone production)
Variant in any of 5 enzymes involved in the conversion of cholesterol to testosterone
Testes produce insufficient testosterone with no change to AMH production
Wolffian & Müllerian ducts regress
Development of female at birth external genitalia (clitoris, labia majora) or male at birth external genitalia (micropenis, hypospadias)
Puberty may develop secondary sexual characteristics typical of males at birth (deep voice, male pattern facial/body hair)
5-alpha reductase deficiency
Variation in the SRD5A2 gene that codes for 5-alpha-RD2 enzyme (responsible for differentiation of gonads into male reproductive system)
5-alpha-RD2 enzyme is impaired & unable to convert testosterone into dihydrotestosterone (DHT)
DHT unable to block formation of female at birth genitalia & promote development of penis & scrotum
Development of ambiguous external genitalia resembling female at birth (unfused labioscrotal folds, undescended testes)
Puberty may develop secondary sexual characteristics typical of males at birth
Complete androgen insensitivity
Impairment of androgen receptors causes unresponsiveness of testicles to testosterone & no change to AMH production
Testes unable to respond to exogenous testosterone treatment
Wolffian
& Müllerian ducts regress
Development of female at birth external genitalia
Puberty may develop secondary sexual characteristics typical of females at birth (breast enlargement, menarche)
Aromatase deficiency
Variations in the CYP19A1 gene that codes for aromatase enzyme
Aromatase enzyme is impaired & unable to convert androgens (testosterone) into estrogen in the placenta
↑ Testosterone levels & absence of anti-Mullerian hormone
Regression of Wolffian ducts & Mullerian ducts differentiate into fallopian tubes & uterus
Development of normal female at birth internal reproductive structures & ambiguous genitalia that is not clearly female or male at birth
Hypergonadotropic hypogonadism at puberty (↓ function of gonads & do not develop secondary sexual characteristics)
46XX Genotype
XX Ovotesticular disorder of sex development
Functional SOX9 gene that codes for testicular Sertoli cells differentiation despite lack of SRY gene
Fetus exposed to only X chromosome (responsible for female gonadal development) due to absence of testicular determinant (SRY)
Prescence of estradiol in developing ovarian follicles inhibits spermatogenesis (sperm production) & no exposure to AMH
Mullerian ducts differentiate into oviduct, uterus, cervix, upper vagina & regression of Wolffian ducts
Development of ambiguous external genitalia (labioscrotal fusion, hypospadias)
& bilateral ovotestes (both ovarian & testicular tissue) or combination of a unilateral ovary or testis with ovotestis on contralateral side
Puberty may develop secondary sexual characteristics typical of femalea at birth (breast enlargement, menarche)
*Samuel Fineblit *MD at time of publication
Ovaries with 21-hydroxylase deficiency
Congenital adrenal hyperplasia (↑ adrenal tissue cell production due to disruption in cortisol synthesis pathway)
↓ Cortisol/aldosterone production from adrenal glands & ↑ Adrenocorticotropic hormone (ACTH) to compensate
Incomplete development of Wolffian ducts (precursor to male internal genitalia) & Müllerian ducts (precursor to female reproductive tract)
Development of ambiguous external genitalia (one testicle undescended; hypospadias where the urethra position is atypical; clitoromegaly)
Puberty may develop secondary sexual characteristics typical of males or females at birth
Swyer syndrome specific: Wolffian & Müllerian ducts regress
Development of normal female internal reproductive structures (uterus & fallopian tubes) & gonadal streaks (underdeveloped ovaries replaced with fibrous scar tissue)
Puberty is stalled unless treated with hormone replacement therapy
ACTH stimulates adrenal gland to ↑ hormone production (both testosterone & estrogen)
Despite ↑ testosterone production, the levels are not sufficient for development of Wolffian ducts & ovaries cannot produce AMH
Wolffian ducts regress & Müllerian ducts develop
Male typical genitalia (male XY) with microorchidism, hyperpigmented scrotum, & enlarged penis
Signs of salt-wasting: low sodium, low potassium, low blood pressure, failure to thrive
Ambiguous external genitalia (female XX: fusion of labial fold, clitoromegaly, penile urethra, urogenital sinus abnormality, hyperpigmentation)
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published Nov 11, 2024 on www.thecalgaryguide.com")
Primary Hypercholesterolemia

Authors: Gurreet Bhandal Run Xuan (Karen) Zeng Rupali Manek Reviewers: Raafi Ali Luiza Radu Samuel Fineblit* *MD at time of publication
Autosomal co-dominant disease (disease will manifest with one mutated allele, but those with two mutated alleles have a more severe phenotype) impacting only one gene
Polygenic hypercholesterolemia (hypercholesterolemia involving multiple genetic abnormalities)
Multiple gene mutations
Accumulation of small effects on cholesterol synthesis, metabolism and clearance
↑ Low-density lipoprotein cholesterol (LDL-C) levels
Development of atherosclerosis** (plaque build up around coronary artery or peripheral blood vessels in the rest of the body)
Heterozygous familial hypercholesterolemia (one pathogenic variant allele; common, especially in French Canadians)
Homozygous familial hypercholesterolemia (both alleles are pathogenic variants; rare & presents in childhood)
Proprotein convertase subtilisin/ kexin type 9 (PCSK9, an enzyme holding low-density lipoprotein receptor complex together) gain- of-function mutation
↑ Breakdown of low-density lipoprotein (LDL) receptor in the liver
Low-density lipoprotein- receptor related protein- associated protein 1 (LDLRAP1;protein coding gene) mutation
↓ Internalization of LDL receptor from cell surface
↓ LDL clearance
Apolipoprotein B (ApoB; protein on LDL) mutation
↓ ApoB/LDL binding to LDL receptor
LDL receptor gene mutation (>90% of cases)
↓ LDL receptor production
↑ Hypercholesterolemia (↑ LDL-C in blood)
Development of aortic valve sclerosis (calcification & thickening of trileaflet aortic valve)
Cholesterol deposit accumulation in tendons & ligaments
Tendon xanthomas (nodules along tendons, typically at Achilles tendon & knuckles)
Cholesterol deposit accumulation on skin at pressured areas
Tuberous xanthomas (shiny red/orange nodules around knees, elbows & heels; less common)
Cholesterol deposit accumulation in the edges of cornea
Corneal arcus (white rings around iris)
Cholesterol deposit buildup under skin near corner of eyelids
Xanthelasmas (yellow plaques on or by the corners of eyelids)
↑ Risk of coronary artery disease
**See Calgary Guide slide on Atherosclerosis: Complications
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published Nov 11, 2024 on www.thecalgaryguide.com")
Suboxone & Methadone

Methadone (a long-acting, full agonist) acts at mu-opioid receptors
Buprenorphine (a long-acting, partial agonist with high affinity properties) displaces full-agonists at mu-opioid receptors (e.g., fentanyl).
Naloxone (a mu-opioid receptor antagonist) displaces buprenorphine at mu-opioid receptors
↓ Effects of buprenorphine in intramuscular and intranasal forms
↓ Potential for abuse of buprenorphine
Repeated intake of increasing doses of methadone
Accumulation of methadone
↑ Potency compared to partial agonist (e.g., buprenorphine)
↓ Inhibitory GABAergic neuronal activity
Disinhibition of dopamine at nucleus accumbens
↑ Dopamine levels at the nucleus accumbens and reward pathways
Euphoria
↑ Adherence
↑ Adherence especially in those with severe opioid dependence
Maintained stimulation of opioid receptors
↑ Buprenorphine required to occupy receptors to produce the same level of effect compared to full-agonists
Saturation of mu- opioid receptors (ceiling effect)
Further intake of buprenorphine does not ↑ stimulation of mu-opioid receptors at the locus coeruleus in the brainstem
Locus coeruleus maintains sensitivity to carbon dioxide concentrations
Ventilation is maintained
↓ Risk of overdose and respiratory depression
↓ Stimulation of mu-opioid receptors
↓ Analgesia
↑ Withdrawal
Potassium channels open
Post-synaptic hyperpolarization of cells
Presynaptic inhibition of neurotransmitter (glutamate and substance P) release
Inhibition of locus coeruleus which is in a hyperexcitable state from chronic opioid use
↓ Noradrenaline release from locus coeruleus
↓ Autonomic excitability throughout brain, spinal cord, peripheral nerves and GI tract.
↓ Withdrawal symptoms (restlessness, anxiety, lacrimation, nausea, vomiting, diarrhea)
↓ Euphoria
Methadone blocks channels involved in repolarization of myocytes
Prolonged repolarization of myocytes
Prolonged QTc interval & cardiac arrythmias
Inhibition of locus coeruleus’ ability to sense carbon dioxide and maintain ventilation
↑ Risk of overdose & respiratory depression
↓ Pain signal transmission
Analgesia
Opioid agonist therapy
Further intake of buprenorphine does not ↓ inhibitory GABAergic neuronal activity
GABAergic neurons maintain inhibitory effect on dopamine at the nucleus accumbens
No ↑ in euphoric effects
↓ Adherence to therapy especially in those with severe opioid dependence
(↓ cravings and withdrawal symptoms thereby reducing risk of relapse)
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published Nov 11, 2024 on www.thecalgaryguide.com")
Cannabis Use Disorder
 in cannabis binds human cannabinoid receptor 1 (CB1) receptors in ventral tegmental area
Biological factors: earlier age of cannabis use onset, male gender, genetic predisposition
Psychosocial factors:
stress, aggression, depression, anxiety, parent/peer cannabis or other substance use, ↓ SES, family dysfunction, bullying, occupational difficulties
Disinhibits dopaminergic signaling and reward response
↑ Motivation to continue cannabis use
Cannabis Use Disorder
Cannabis use that may lead to significant distress or impairment in psychological, physical, or social functioning
Cannabis (CB1 receptor agonist), especially with ↑ THC, acutely causes burst firing of ventral tegmental area
↑ Dopamine in striatal and prefrontal areas
↑ Dopaminergic transmission in the mesolimbic pathway (neuronal network involved in mediation of psychosis)
Delusions, hallucinations, & paranoia occurring with acute or chronic use
Cannabis induced psychotic disorder
↑ THC activates CB1 receptors on GABAergic terminals & ↓ THC activates CB1 receptors on glutaminergic terminals
GABA & glutamate dysregulation and imbalance (can occur with acute or chronic use)
Repeated exposures to ↑THC results in serotonin (5- HT) receptor upregulation
↑ Neuro-
endocrine responses of stress hormones, which may ↑ serotonin reuptake
Sedative action of low dose THC on CB1 receptors causes ↓ sleep latency and ↑ slow wave sleep with acute cannabis use
Tolerance with chronic usage causes reversal of acute effects
↑ Sleep latency & ↓ slow wave sleep
↓ Sleep time & quality; ↓ cerebral restoration & recovery
Cannabis induced sleep disorder
Chronic cannabinoid receptor overstimulation
Receptor dysregulation, desensitization, and downregulation
Hypothalamic pituitary adrenal (HPA) axis dysregulation
Inhibited gastrointestinal motility and digestion
Nausea & vomiting & abdominal pain after cannabis use
Cannabinoid hyperemesis syndrome (cyclic vomiting with cannabis use)
Down regulation of CB1 receptors due to long term cannabis usage, paired with sudden discontinuation of cannabis use
↓ Cannabinoid in ↑CB1 receptors disrupts emotional processing, sleep and appetite homeostasis within the endocannabinoid system
Anxiety, irritability, anger, depression, sleep disruption, appetite loss.
Cannabinoid withdrawal
Mood changes, anxiety and/or panic attacks
Cannabis induced mood and anxiety disorders
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published Nov 11, 2024 on www.thecalgaryguide.com
Sympathetic nervous system dysregulation")
Renal manifestations of SLE
: Renal Manifestations
Authors: Madison Turk Reviewers: Modhawi Alqanaie Mao Ding Luiza Radu Glen Hazlewood* * MD at time of publication
Genetic susceptibility and potential environmental triggers (smoking, silica, Epstein-Barr Virus, hormones, ect)
↓Clearance of dead cell debris in the body
(exact mechanisms unknown; See slide on pathogenesis of SLE)
Extracellular exposure of nuclear proteins (which bind DNA and regulate gene expression)
Immune cells respond to nuclear proteins as if they are non-self, as they usually don’t ‘see’ them
Systemic Lupus Erythematosus
An autoimmune disease characterized by anti-nuclear-antibody production resulting in widespread inflammation and tissue damage in varying affected organs, including one, or a combination of, joints, skin, brain, lungs, kidneys, and blood vessels
Production of auto-antibodies against self nuclear proteins
IC deposition in renal vessels
Activation of inflammatory cells against IC’s causing vascular injury/inflammation
↑Clot formation in vessels leading to ↓vessel lumen diameter and ↓blood flow
↑Reninàcleavage of angiotensinogen to angiotensin Ià cleavage by angiotensin converting enzyme into angiotensin II
Vessel constriction to ↑blood pressure (to ↑ renal blood flow)
IC deposition in tubule basement membrane of the kidney
Formation of immune complexes (IC) (collections of antigen(s), antibodies, and/or complement proteins bound together)
Tubulointerstitial nephritis: (Inflammation of the renal tubules and interstitium, sparing the glomeruli)
Renal ischemia
IC deposition in the glomerulus
Activation of complement, initiating an inflammatory response
Recruitment of myeloid cells (monocytes/ neutrophils)
Production of reactive oxygen species, cytokines and release of cytotoxic granules
Tubular inflammation and damage resulting in ↓sodium reabsorption
Renal release of reninà ↑ angiotensin I/II and resulting ↑ aldosterone
↑Sodium reabsorption, and potassium excretion in the collecting duct
↓Potassium secretion from the Distal Convoluted Tubule
Hyperkalemia Hypokalemia
⍺-intercalated cell damage
↓Acid excretion
Metabolic acidosis
End stage kidney disease
(all manifestations can result in this)
Hypertension
Tissue damage from the inflammatory response
Glomerular nephritis (inflammation/damage of the filtering part of the kidneys)
Fibrosis (thickening or scarring of tissue)
Mesangial and parietal cell proliferation
Podocyte injury
↑blood and protein excretion due to damage to the glomerular filtration membrane
↓protein in blood Edema
Proteinuria Hematuria
↓Functional renal tissue to filter creatinine from blood
↓ Glomerular filtration rate ↑ Plasma creatinine
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published Nov 11, 2024 on www.thecalgaryguide.com")
Learning Disability

E.g. Changes to neuronal migration genes (DCDC2 & ROBO1) in dyslexia
Co-morbid neurodevelopmental disorder, especially ADHD
Difficulty concentrating impedes learning
Home environment
↓ Reading at home, ↓ parental education, socio-economic status
Acquired (brain injury)
Learning Disability
Persistent impairment in reading (dyslexia), writing (dysgraphia) and/or arithmetic (dyscalculia) beyond what is expected for age
Differences in brain activation during learning
Dyslexia Dysgraphia Dyscalculia
↓ Activation of left temporal parietal cortex (sounding words out)
↓ White matter connections between brain regions important for reading
↓ Activation of visual-word form area (representing sounds as symbols)
↑ Activation of Broca’s area (spoken word)
↓ Letter & sound recall
Difficulty sounding out words
Poor spelling
Difficulty naming objects
Difficulty identifying words
↑ Sounding-out in later reading
Cerebellar activation differences
Cortical & subcortical damage to functional language regions
Anterior thalamic radiation differences (executive functioning)
Cingulum differences (cognitive control)
Illegible & very slow writing
Poor spelling & writing fatigue
↓ White matter in inferior longitudinal fasciculus (object recognition)
↓ Gray matter in inferior parietal lobe (understanding numbers & quantities)
↓ Gray matter in bilateral supramarginal gyri (retrieving arithmetic facts)
↓ White matter in fronto- occipital fasciculus & anterior thalamic radiation (goal-oriented behavior & executive functioning)
Difficulty recognizing patterns & numbers
Difficulty with basic arithmetic
Difficulty with mathematic operations
Difficulty with number processing
Work-avoidance ↓ Self-esteem Difficulty forming relationships Depression Impulsivity Opposition to authority
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published Nov 11, 2024 on www.thecalgaryguide.com")
Anticholingeric Syndrome in Older Adults

↓ Ability of the liver to metabolize drugs effectively
Breakdown of the blood- brain barrier
Relative cholinergic deficit compared to younger adults
Use of anticholinergic medication(s) (e.g. antihistamines, antipsychotics, antidepressants, urinary incontinence medications)
Higher anticholinergic burden in older adults
Anticholinergic Syndrome
Sequelae of symptoms following accidental or intentional overdose of compound(s) with anticholinergic activity
Anticholinergic drug competitively binds against acetylcholine at muscarinic cholinergic receptors
Inhibition of peripheral muscarinic cholinergic receptors
Inhibition of muscarinic cholinergic receptors in the central nervous system
Dysregulation of acetylcholine levels in the brain, which is responsible for memory, attention, learning, and arousal
Cardiovascular:
Sudoriferous glands: inability to produce or excrete sweat
Gastro- intestinal: impaired gut motility
Constipation
Blurry vision
Bladder: impaired detrusor muscle contraction & urethral
Eyes: impaired pupillary contraction & ability to accommodate
Inhibition of muscarinic- sensitive delayed rectifying potassium efflux channels on cardiomyocytes
Impaired
ability to slow heart rate & lower blood pressure
Impaired thermoregulation
Hyperthermia
Compensatory cutaneous vasodilation to release heat
Flushed skin
Dry sphincter skin opening
Urinary retention
Urinary stasis allows for bacterial overgrowth
Urinary tract infection
Abnormal pupillary dilation (mydriasis)
Hypertension
Delayed cardiomyocyte repolarization
Prolonged QT interval in cardiac cycle (on ECG)
Seizures Tachycardia
Inattention
Agitation/ Aggression
Delirium develops
Coma
Fluctuating cognition
Visual Hallucinations
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published Nov 11, 2024 on www.thecalgaryguide.com")
Benzodiazepine mechanism of action

Authors: Tracey Rice Usama Malik Amy Fowler Victoria Silva Reviewers: Sara Cho Keira Britto Luiza Radu Brienne McLane* Aaron Mackie* * MD at time of publication
Benzodiazepine binds to gamma-aminobutyric acid (GABAA) receptors in vascular smooth muscle & the central nervous system (CNS)
↑ Opening of chloride channels
Influx of chloride ions into the neuron Hyperpolarization of nerve membrane causing it to be more negative The cell membrane falls below the normal resting potential
Medulla oblongata inhibition
↓ Respiratory drive; ↓ depth & rate of respirations
Temporary cessation of breathing leads to reduced oxygen supply to the brain
↓ Level of consciousness
Pharyngeal muscle relaxation leading to obstruction
Hypoventilation and/or apnea
General CNS inhibition
↓ Neuronal activity
↓ Electrical brain activity
↓ Seizure activity & hypnotic effect
Thalamic & hypothalamic inhibition
Disruption of short & long- term memory consolidation
Limbic system inhibition
↓ Fear emotions (panic & phobia)
Anxiolytic effect (↓ Anxiety)
Smooth muscle inhibition
Smooth muscles become relaxed &/or less spastic
Vasodilation
↓ Preload Hypotension
↓ Cerebral blood flow
Pre-syncope or syncope
Anterograde amnesia
↓ Visuospatial ability, speed of processing & verbal learning
Confusion
↓ Deep stage of non-REM sleep & delayed REM sleep
Rapid sleep
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published July 21, 2024 on www.thecalgaryguide.com")
Pneumonie chez ladulte

Atherosclerose Pathogenese

Cardiopathie ischemique
 : Pathogenèse des différentes formes de CI")
Stenose aortique

Signes et symptomes dune Embolie Pulmonaire

Atherosclerose Complications

Malattia Infiammatoria Intestinale Reperti Clinici nella Colite Ulcerosa
: Reperti Clinici nella Colite Ulcerosa")
Sindrome da Inappropriata Secrezione di Ormone Antidiuretico
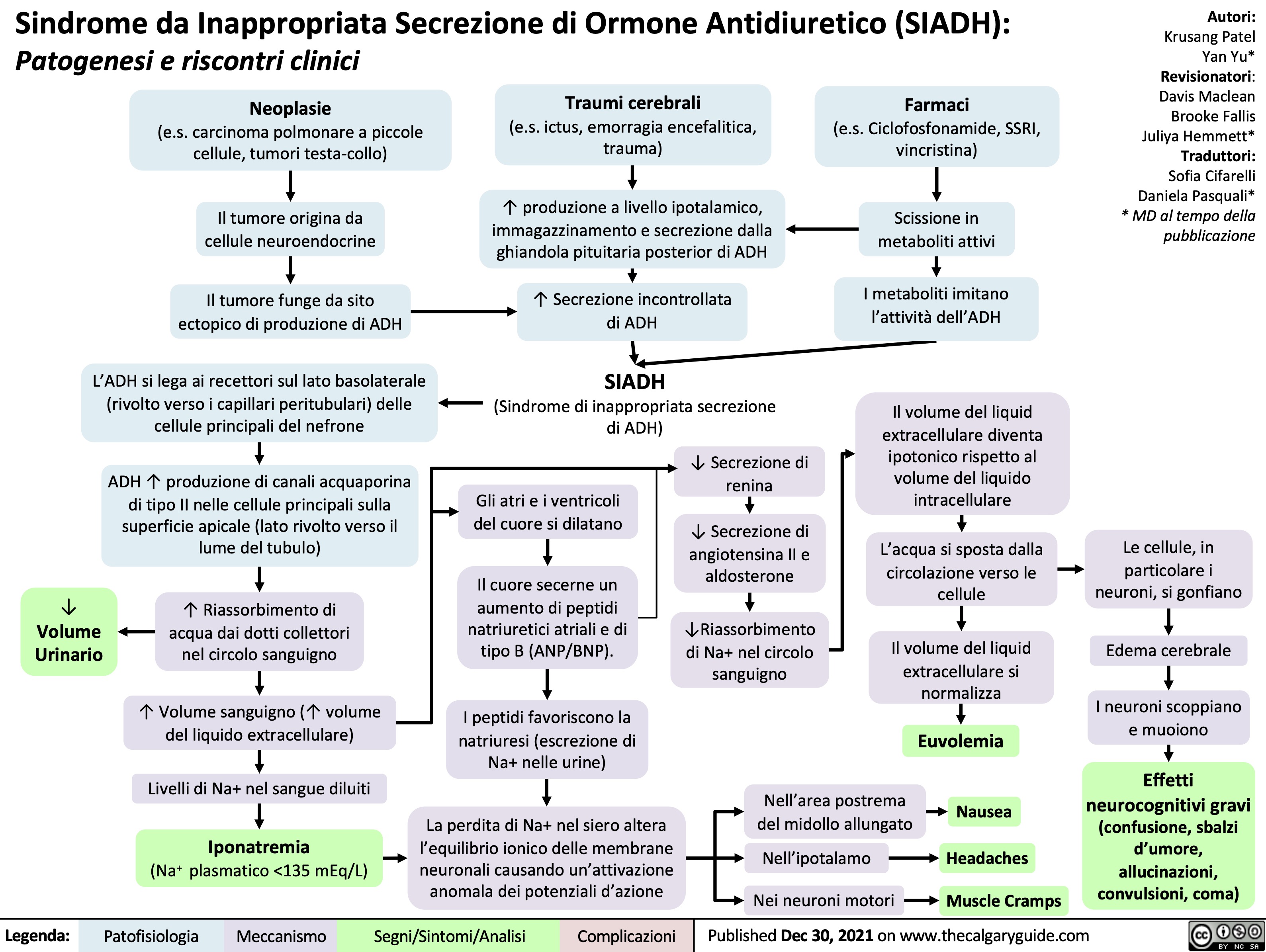
Ustioni Patofisiologia e Complicanze

Aterosclerosi Complicanze

Atresia biliar
: Patogenia y hallazgos clínicos")
Tumores oseos primarios benignos Patogenia del aspecto radiologico

Vang da o tre so sinh Benh sinh hoc

Diabetic Polyneuropathy

↓ Insulin induces expression of neurotrophic factors (i.e. nerve growth factor, brain-derived neurotrophic factor, neurotrophin-3, insulin-like growth factor (IGF), & vascular endothelial growth factor (VEGF))
↓ Stimulus for peripheral nerve maintenance & repair
Impaired peripheral nerve repair
Hyperglycemia
(↑ Intracellular glucose levels)
↑ Glycolysis (breakdown of glucose)
↑ Intermediates & products of glycolysis (i.e. Protein Kinase C, AGE, polyols (sorbitol & fructose), NADH)
Protein Kinase C Pathway activated & ↑ inflammation in endothelium, vascular smooth muscle, fibroblasts of blood vessels
Prothrombotic state with ↑ vascular fibrosis, ↑ smooth muscle proliferation, ↑ chance of blood clots, ↑ impaired endothelial- mediated vasodilation
Arterial stiffening & ischemia
Advanced Glycation End Products (AGEs): end products of glycolysis that have carbohydrates attached
AGEs bind AGE receptors on endothelium & white blood cells
↑ Inflammation, vascular permeability, procoagulant activity & monocyte influx
Immune cells generate toxic reactive oxygen species as part of their defense system
Polyol pathway activated in kidney, retina, nerves
Excess glucose converted to sorbitol & fructose
Metabolism of sorbitol & fructose produces intermediates that undergo auto-oxidation
↑ Reactive oxygen species
↑ NADH (reducing agent) overloads the electron transport chain
↑ Leakage of electrons in the electron transport chain
↑ Free radical formation (type of reactive oxygen species that is toxic to cells & tissues when in excess)
↑ Oxidative stress
Diabetic Polyneuropathy (damage to & loss of peripheral nerve function due to nerve hypoxia & impaired repair mechanisms)
Sensory axonal loss
Damage to small myelinated fibers
Impaired pain, light touch & temperature sensation
Pain, paresthesia (pins & needles feeling), dysthesia (burning, tingling, itching)
X-ray: destruction of weight-bearing foot joints
Damage to large myelinated fibers
Loss of vibratory sensation in glove & stocking pattern distribution; altered proprioception
Claw toe deformity
Charcot Foot: weakening of bones, joints, soft tissues causes pain insensitivity
Distal motor axonal loss
↓ Ability of axons to transmit signals to central nervous system to foot muscles
↓ Foot muscle strength,
↓ Coordination & imbalance between toe extensors & flexors
Weight shift creates pressure points
Fissures in skin of weight-bearing areas
Infection & chronic ulceration
Damage to nerves that control contractions of stomach muscles
Gastroparesis: stomach muscles weakened, ↓ motility & delayed transit of contents
Autonomic neuropathy
Damage to nerves that control heart blood flow, contractions & contractility
Damage to nerves that control blood flow and arousal responses for sexual function
Erectile dysfunction
Orthostatic or postural hypotension
Exercise intolerance
Resting tachycardia
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published Apr 28, 2014; updated Nov 16, 2024 on www.thecalgaryguide.com
Diabetic Polyneuropathy: Pathogenesis and clinical findings
Authors: Gurreet Bhandal Amanda Eslinger Reviewers: Raafi Ali Mark Elliott Jaye Platnich Alexander Arnold Haotian Wang Hanan Bassyouni* * MD at time of publication
Hypoinsulinemia
(↓ Intracellular insulin levels)
↓ Insulin induces expression of neurotrophic factors (i.e. nerve growth factor, brain-derived neurotrophic factor, neurotrophin-3, insulin-like growth factor (IGF), & vascular endothelial growth factor (VEGF))
↓ Stimulus for peripheral nerve maintenance & repair
Impaired peripheral nerve repair
Hyperglycemia
(↑ Intracellular glucose levels)
Protein Kinase C Pathway in endothelium, vascular smooth muscle, fibroblasts of blood vessels
↑ Inflammation
Prothrombotic state with ↑ chance of blood clots, ↑ vasoconstriction & ↑ arterial stiffening
Ischemia
↑ Glycolysis (breakdown of glucose)
Advanced Glycation End Products (AGEs): end products of glycolysis that have carbohydrates attached
AGEs bind cellular receptors
Inflammation, vascular permeability, procoagulant activity & monocyte influx
Immune cells generate toxic reactive oxygen species as part of their defense system
↑ Intermediates & products of glycolysis (i.e. Protein Kinase C, AGE, sorbitol & fructose, NADH)
Polyol Pathway in kidney, retina, nerves
Excess glucose converted to sorbitol & fructose
Metabolism of sorbitol & fructose produces intermediates that undergo auto-oxidation
↑ Reactive oxygen species
↑ Leakage of electrons when NADH (reducing agent) overloads the electron transport chain
↑ Free radical formation (type of reactive oxygen species that is toxic to cells & tissues when in excess)
↑ Oxidative stress
Diabetic Polyneuropathy (damage to & loss of peripheral nerve function due to nerve hypoxia & impaired repair mechanisms)
Sensory axonal loss
Damage to small myelinated fibers
Impaired pain, light touch & temperature sensation
Pain, paresthesia (pins & needles feeling), dysthesia (burning, tingling, itching)
X-ray: destruction of weight-bearing foot joints
Damage to large myelinated fibers
Loss of vibratory sensation in glove & stocking pattern distribution; altered proprioception
Claw toe deformity
Charcot Foot: weakening of bones, joints, soft tissues causes pain insensitivity
Distal motor axonal loss
↓ Ability of axons to transmit signals to central nervous system to foot muscles
↓ Foot muscle strength,
↓ Coordination & imbalance between toe extensors & flexors
Weight shift creates pressure points
Fissures in skin of weight-bearing areas
Infection & chronic ulceration
Damage to nerves that control contractions of stomach muscles
Gastroparesis: stomach muscles weakened, ↓ motility & delayed transit of contents
Autonomic neuropathy
Damage to nerves that control heart blood flow, contractions & contractility
Damage to nerves that control blood flow and arousal responses for sexual function
Erectile dysfunction
Orthostatic or postural hypotension
Exercise intolerance
Resting tachycardia
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published April 28th, 2014 on www.thecalgaryguide.com
Diabetic Polyneuropathy: Pathogenesis and clinical findings
Authors: Gurreet Bhandal Amanda Eslinger Reviewers: Mark Elliott Jaye Platnich Alexander Arnold Haotian Wang Hanan Bassyouni* * MD at time of publication
Hypoinsulinemia
↓ intracellular insulin levels
↓ Neurotrophic factors (i.e. nerve growth factor, brain-derived neurotrophic factor, neurotrophin-3, IGF, & VEGF)
↓ stimulus for peripheral nerve maintenance and repair
Impaired peripheral nerve repair
Sensory axonal loss Small myelinated fibers
Impaired pain, light touch & temperature sensation
Pain, paresthesias or tingling and numbness, dysthesias where sense of touch is distorted
Hyperglycemia
↑ Intracellular glucose levels
This produces an excess of glycolysis intermediates & products of glycolysis (i.e. sorbitol & fructose; AGE; Protein Kinase C)
PKC Pathway in endothelium, vascular
smooth muscle, fibroblasts of blood vessels
↑ inflammation
Prothrombotic state with ↑ chance of blood clots; vasoconstriction & arterial stiffening
Ischemia
Advanced Glycation End Products: The body “glycates” end products of glycolysis, which means that some end products have a carbohydrate added to them
Polyol Pathway
in kidney, retina, nerves
Excess glucose is converted to sorbitol and fructose, which accumulates in cells
↑ Oxidative stress
↑ Free Radical Formation
AGE’s bind cellular receptors inducing inflammation, vascular
permeability, procoagulant activity & monocyte influx
Abbreviations:
AGE – Advanced Glycation End Products
IGF - Insulin-like Growth Factor
VEGF - Vascular Endothelial Growth Factor PKC – Protein Kinase C
Autonomic neuropathy
Nerve hypoxia & impaired repair mechanisms leading to dysfunction & loss of peripheral nerves Distal motor axonal loss
Large myelinated fibers
Atrophy of intrinsic foot muscles
Fissures in skin of weight-bearing areas
Imbalance between toe extensors & flexors
Weight shift results in pressure points
Infection & chronic ulceration
Gastroparesis: stomach muscles weakened with ↓ motility
Claw toe deformity
Erectile dysfunction
Altered proprioception
X-ray: destruction of weight-bearing foot joints
Loss of vibratory sensation in glove & stocking pattern distribution
Charcot Foot: weakening of bones, joints, soft tissues causes pain insensitivity
Cardiac: Resting tachycardia, exercise intolerance, orthostatic or postural hypotension
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published April 28th, 2014 on www.thecalgaryguide.com
Diabetic Polyneuropathy: Pathogenesis and clinical findings
Authors: Amanda Eslinger Reviewers: Mark Elliott Jaye Platnich Alexander Arnold Haotian Wang Hanan Bassyouni* * MD at time of publication
Hypoinsulinemia
↓ Neurotrophic factors (i.e. nerve growth factor, brain-derived neurotrophic factor, neurotrophin-3, IGF, & VEGF)
↓ stimulus for peripheral nerve maintenance and repair
Impaired peripheral nerve repair
Sensory axonal loss
Small myelinated fibers
Impaired pain, light touch & temperature sensation
Pain, paresthesias, dysthesias
Hyperglycemia
↑ Intracellular glucose levels
This produces of glycolysis
an excess of glycolysis intermediates & products (i.e. sorbitol & fructose; AGE; Protein Kinase C)
PKC Pathway
PKC pathway activation results in ↑ inflammation
Prothrombotic state; vasoconstriction & arterial stiffening
Ischemia
Advanced Glycation End Products: The body “glycates” end products of glycolysis, which means that some end products have a carbohydrate added to them
AGE’s bind cellular receptors inducing inflammation, vascular
permeability, procoagulant activity & monocyte influx
Polyol Pathway
(i.e. sorbitol & fructose)
Excess glucose is converted to sorbitol, which accumulates in cells
↑ Oxidative stress
↑ Free Radical Formation
Nerve hypoxia & impaired repair mechanisms leading to dysfunction & loss of peripheral nerves Distal motor axonal loss
Abbreviations:
AGE – Advanced Glycation End Products
IGF - Insulin-like Growth Factor
VEGF - Vascular Endothelial Growth Factor PKC – Protein Kinase C
Autonomic neuropathy
Large myelinated fibers Altered
proprioception
Atrophy of intrinsic foot muscles
Fissures in skin of weight-bearing areas
Infection & chronic ulceration
Imbalance between toe extensors & flexors
Weight shift results in pressure points
Gastroparesis
Claw toe deformity
Erectile dysfunction
X-ray: destruction of weight-bearing foot joints
Loss of vibratory sensation in glove & stocking pattern distribution
Charcot Foot
Cardiac: Resting tachycardia, exercise intolerance, orthostatic hypotension
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published April 28th, 2014 on www.thecalgaryguide.com")
Rapid sequence induction and intubation
: Indications & considerations
“Full stomach”: ↑ risk of regurgitation, vomiting, aspiration Life-threatening injury or illness requiring immediate or rapid airway control
↓ Gastro- esophageal sphincter competence (elderly, pregnancy, hiatus hernia, obesity)
↑ Intragastric pressure (pregnancy, obesity, bowel obstruction, large abdominal tumors)
Delayed gastric emptying (narcotics, anticholinergics, pregnancy, renal failure, diabetes)
↓ Level of consciousness (drug/alcohol overdose, head injury, trauma or shock state)
Respiratory & ventilatory compromise (i.e., hypoxic or hypercapnic respiratory failure)
Achalasia (esophageal motility disorder resulting in impaired swallowing)
Dynamically deteriorating clinical situation (i.e., trauma)
GI bleed
Impaired airway reflexes
↓ Muscle tone of structures in the airway (i.e., tongue, pharyngeal walls, & soft palate)
Patients who did not stop GLP-1 agonist preoperatively as advised
Impaired clearance of secretions or vomitus
↓ Safe apnea time before hemodynamic decompensation
Unprotected airway
Need for rapidly securing airway while avoiding aspiration & hemodynamic compromise
Rapid sequence intubation (RSI): Simultaneous administration of induction agent (unconsciousness) & neuromuscular blocking agent (paralysis) to achieve intubation conditions (~45-60 seconds after IV push) for rapid control of an emergency airway
Preoxygenation
Deranged physiologic conditions (i.e., hypotension, acidosis, hypoxemia)
Reduced tolerance for
apnea (period with no ventilation or oxygenation)
Pre-oxygenate with high flow O2 (15L) to create a large pulmonary & tissue reservoir of oxygen
↓ Significant oxygen desaturation during apnea
↑ Oxygen saturation on pulse oximetry
Induction
Laryngoscopy & intubation are a potent sympathetic nervous system stimulus
Airway manipulation causes a surge in catecholamines
Paralysis
Visualization & passage of endotracheal tube requires relaxation of vocal cords & surrounding muscles
Neuromuscular blocking agents facilitate paralysis
Rescue
Some induction agents (i.e., propofol) are vasodilators
Hemodynamically unstable or patients in shock
Hypotension
Tachycardia
↑ Intracranial pressure (ICP)
Hypertension
Suppress cough & gag reflex
Prevent laryngospasm (involuntary closure of vocal cords to airway manipulation)
Minimize movement during procedure
Vasoactive agents (i.e., ephedrine, phenylephrine) ↑ systemic vascular resistance
Atropine & glycopyrrolate ↑ heart rate
Lidocaine (Na+ channel blocker) & opioids (μ receptor agonist) ↓ transmission of pain
↓ Sympathetic response, myocardial demand & physiologic stress
Anesthetics (i.e., propofol) achieve unconsciousness for paralysis & intubation
↓ Airway trauma & damage to vocal cords
Bag mask ventilation typically avoided in this step to ↓ gastric insufflation & risk of aspiration
Cricoid pressure (Sellick maneuver): posterior displacement of cricoid ring to compress esophagus against C-spine to prevent passive regurgitation of gastric contents to airway. Applied from start of induction, released when placement of endotracheal tube is confirmed by capnography.
Intubation
↑ Blood pressure and/or cardiac output
Authors: Jen Guo Reviewers: Priyanka Grewa Luiza Radu Leyla Baghirzada* * MD at time of publication
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published November 18, 2024 on www.thecalgaryguide.com")
Open Fractures
 or low-energy force on diseased bone
Force applied to bone exceeds strength of bone resulting in periosteal stripping and subsequent soft tissue and neurovascular destruction
Open Fractures
Also known as “compound fractures” and classified with the Gustilo-Anderson classification system (Types I, II, III), these are fractures in which the skin is penetrated and bone is exposed to the external environment. Comminuted fractures have ≥ 2 breaks in the bone.
Inside-out (bone) or outside-in (external) penetration of skin to create a wound
Skin tearing creates vacuum-like effect pulling debris into wound
Minimal comminution
Bone penetrates skin to create a wound < 1 cm in diameter (Type I)
Smaller wound creates minimal opportunities for pathogen entry and contamination
Moderate comminution
Bone penetrates skin to create a wound 1-10 cm in diameter (Type II)
Moderate wound creates some opportunity for pathogen entry and contamination
Extensive comminution
Bone penetrates skin to create a wound > 10 cm in diameter (Type III)
Large wound creates ample opportunity for pathogen entry and extensive contamination
Type IIIA (adequate soft tissue for bone coverage)
Type IIIB
(soft tissue damage with periosteal stripping)
Type IIIC (vascular injuries, potential amputation)
Displacement/shortening/ angulation/rotation of fracture fragment
Improper bone healing
Bone deformity
Authors: Meaghan MacKenzie Holly Zahary Loreman Nojan Mannani Reviewers: Annalise Abbott Usama Malik Michelle J. Chen Dr. Prism Schneider* Dr. Jared Topham* * MD at time of publication
Pain & lack of mechanical load bearing axis
Inability to weight bear
Decreased mobility promotes stasis of venous blood flow & intravascular vessel wall damage
Deep vein thrombosis
Potential progression to pulmonary embolism
Bleeding or inflammation within fascia
Muscle atrophy
Compartment syndrome (↑ pressure in muscle) **
Open wound exposes bone
Infiltration of debris & contaminants
Infection of soft tissues or bone (osteomyelitis)
Initial injury damages blood vessels
Initial injury damages nerves
↓ Sensation distal to injury
↓ Limb function & proprioception
↓ Pulses distal to injury
Amputation
↓ Blood flow to bone
Avascular necrosis (bone tissue death)
Limb ischemia
↓ Blood flow & oxygen delivery to tissues
Compartment syndrome**
Delayed union (bone healing) on serial radiographs
Non-union (bone fails to heal) on serial radiographs
**See corresponding Calgary Guide slide on Acute Compartment Syndrome
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published Nov 16, 2017; updated Nov 21, 2024 on www.thecalgaryguide.com")
Calcium Channel Blockers
![Calcium Channel Blockers: Mechanisms & side effects
Authors:
Caroline Kokorudz Reviewers:
Rafael Sanguinetti Andrew Wu
Luiza Radu
Timothy Pollak*
* MD at time of publication
Calcium channel blocker medications
inhibit Ca2+ channels in smooth muscle
Reduction of Ca2+ influx into smooth muscle cells
Inhibits calcium-dependent aldosterone synthesis reducing Na+ & H2O resorption in renal distal tubules
Negative feedback to pituitary gland causing ↑ ACTH (adrenocorticotropic hormone)
↑ Androgens (testosterone)
Testosterone acts on gingival cells (multiple cell types that support teeth) & connective tissue matrix
Gingival hyperplasia (gum overgrowth)
Non-dihydropyridines:
(Phenylalkylamines [verapamil], Benzothiazepines [diltiazem]) less potent vasodilators & selective for heart muscle
Prevents smooth muscle contraction
Dihydropyridines:
(amlodipine, felodipine, nifedipine) vasodilate vascular smooth muscle
↓ Arterial resistance and blood pressure in coronary & peripheral arteries
Coronary artery vasodilation
↓ Pressure in coronary arteries
↑ Blood flow through coronary arteries
Reduced
ischemia relieves angina
Inhibits L-type Ca2+ channels, preventing rapid nodal depolarization
Reduces excitation of sinoatrial (SA) & atrioventricular (AV) nodal tissues
↓ Conduction speed of electrical impulses
↓ Contractile strength of cardiomyocytes (heart muscle cell)
↓ Systemic vascular resistance & cardiac
afterload (heart pumping resistance)
↑ Blood volume flowing into significantly smaller vessels
↑ Capillary blood pressure
↑ Circulation to face
Flushes (red & warm)
↓ Cardiac output
↓ Tissue perfusion & attempt to ↑ cardiac output
Worsens heart failure
↓ Oxygen demand of heart muscle
More favorable oxygen supply to demand ratio
Relieves angina
↓ Blood pressure
↓ Cerebral perfusion
Syncope (fainting)
Relieves angina
Capillary fluid leak increased to interstitial space
Peripheral edema
↑ Intracranial pressure Compresses nerve endings Headache
↓ Heart rate Bradycardia
Suppresses dysrhythmias (abnormal heart rhythm)
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published Nov 21, 2024 on www.thecalgaryguide.com](https://calgaryguide.ucalgary.ca/wp-content/uploads/2024/11/Calcium-Channel-Blockers.jpg "Calcium Channel Blockers: Mechanisms & side effects
Authors:
Caroline Kokorudz Reviewers:
Rafael Sanguinetti Andrew Wu
Luiza Radu
Timothy Pollak*
* MD at time of publication
Calcium channel blocker medications
inhibit Ca2+ channels in smooth muscle
Reduction of Ca2+ influx into smooth muscle cells
Inhibits calcium-dependent aldosterone synthesis reducing Na+ & H2O resorption in renal distal tubules
Negative feedback to pituitary gland causing ↑ ACTH (adrenocorticotropic hormone)
↑ Androgens (testosterone)
Testosterone acts on gingival cells (multiple cell types that support teeth) & connective tissue matrix
Gingival hyperplasia (gum overgrowth)
Non-dihydropyridines:
(Phenylalkylamines [verapamil], Benzothiazepines [diltiazem]) less potent vasodilators & selective for heart muscle
Prevents smooth muscle contraction
Dihydropyridines:
(amlodipine, felodipine, nifedipine) vasodilate vascular smooth muscle
↓ Arterial resistance and blood pressure in coronary & peripheral arteries
Coronary artery vasodilation
↓ Pressure in coronary arteries
↑ Blood flow through coronary arteries
Reduced
ischemia relieves angina
Inhibits L-type Ca2+ channels, preventing rapid nodal depolarization
Reduces excitation of sinoatrial (SA) & atrioventricular (AV) nodal tissues
↓ Conduction speed of electrical impulses
↓ Contractile strength of cardiomyocytes (heart muscle cell)
↓ Systemic vascular resistance & cardiac
afterload (heart pumping resistance)
↑ Blood volume flowing into significantly smaller vessels
↑ Capillary blood pressure
↑ Circulation to face
Flushes (red & warm)
↓ Cardiac output
↓ Tissue perfusion & attempt to ↑ cardiac output
Worsens heart failure
↓ Oxygen demand of heart muscle
More favorable oxygen supply to demand ratio
Relieves angina
↓ Blood pressure
↓ Cerebral perfusion
Syncope (fainting)
Relieves angina
Capillary fluid leak increased to interstitial space
Peripheral edema
↑ Intracranial pressure Compresses nerve endings Headache
↓ Heart rate Bradycardia
Suppresses dysrhythmias (abnormal heart rhythm)
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published Nov 21, 2024 on www.thecalgaryguide.com")
Dexmedetomidine
 signal propagation
Analgesia
↑ Sedation Anxiety suppression
↓ Formation of cyclic AMP (cAMP)
↓ cAMP-dependent kinase activity
Neuronal firing suppression, mimicking endogenous sleep
↓Cerebral blood flow
↓ Sympathetic tone
Heart block and asystole
Maintenance of seizure foci on EEG in seizure surgeries
↑ Smooth muscle contraction
↑ Vasoconstriction
Transient hypertension and reflexive bradycardia on bolus administration
↓ Heart rate and systemic vascular resistance
Bradycardia & hypotension Authors: Kayleigh Yang, Arzina Jaffer
Reviewers: Priyanka Grewal, Luiza Radu Leyla Baghirzada * MD at time of publication
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published Nov 21, 2024 on www.thecalgaryguide.com")
Pre-Eclampsia Pathogenesis

Primigravida (1st pregnancy)
Advanced maternal age
Genetic factors (e.g. sFLT1)
Collagen vascular disease
Previous preeclamptic pregnancies
Multiple gestation
Other unclear causes
Dysregulated immune and non-immune decidual cells (specialized endometrial cells) promote abnormal placental trophoblast invasion and uterine spiral artery formation (exact mechanism unclear)
Multiple unclear, complicated mechanisms
Abnormal association of placental vasculature with uterine vasculature early in pregnancy (utero-placental mismatch) disrupts adequate blood perfusion from placenta to fetus (< 20 weeks gestation or early-onset)
Fetus experiences under- perfusion, hypoxia, ischemia, and oxidative stress
Placental cells release molecules toxic to vascular endothelium into the maternal circulation
Damaged maternal blood vessels are less able to perfuse the placenta
Systemic dysfunction of maternal blood vessel endothelium (endothelial cell activation) (>20 weeks gestation or late-onset)
Inflammatory cells retain memory postpartum which increases risk for developing preeclampsia in subsequent pregnancies
Hypoxia leads to placental cell death, which results in release of fetal antigens
Authors:
Yan Yu
Jasmine Nguyen Reviewers:
Kayla Nelson
Radhmila Parmar
Sean Spence
Monica Kidd*
Katrina Krakowski* Maryam Nasr-Esfahani* Riya Prajapati
Michelle J. Chen
Jadine Paw*
* MD at time of publication
Placental ischemia & injury
Pre-existing maternal diseases damage endothelial cells (e.g. vasculitis, diabetes, renal diseases, chronic hypertension)
Placenta is unable to supply sufficient O2 and nutrients to meet demands of growing fetus
Maternal Clinical Findings and Complications**
Fetal Clinical
Findings and Complications**
**See corresponding Calgary Guide slides
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published Feb 10, 2017; updated Nov 22, 2024 on www.thecalgaryguide.com")
Loop diuretics
 symporter at thick ascending limb of Loop of Henle (aLOH)
Authors: Andrew Wu, Rafael Sanguinetti Reviewers: Luiza Radu, Adam Bass* * MD at time of publication
Over-inhibition of ion symporter in ear at high concentrations
Damage to tight junctions in the blood vessel epithelium in the ear
Disruption of the blood- cochlear barrier
Hearing loss
Opening of ATP-sensitive K+ channels on pancreatic β- cells causes efflux of K+
↓ Intracellular K+ causes a negative intracellular charge
↓ Ability for pancreatic β- cells to depolarize & release insulin
Hyperglycemia (rare)
Stimulation of prostaglandin E2 (PGE2) synthesis in the cortical & medullary aLOH
Stimulation of endothelial nitric
oxide synthase releases nitric oxide
Systemic venodilation (dilation of veins)
↓ Systemic vascular resistance
↓ Blood pressure
↓ Na+ in renal interstitium
↓ Water reabsorption (water follows Na+)
Interstitial washout (↑ NaCl & water wasting in interstitium)
↓ Sodium (Na+) reabsorption
↑ Positive luminal charge at thick aLOH
↓ Electrochemical gradient reduces cellular transport of cations
↓ Ca2+ & Mg2+ reabsorption in the aLOH
Hypocalcaemia & hypomagnesemia
↑ Na+ delivery to the distal convoluted tubule (DCT)
↑ Intracellular Na+ at collecting duct
↑ K+ efflux via renal outer medullary potassium channel (ROMK)
↓ Fluid status
↓ Serum Na+
↓Serum Cl-
↑ Luminal K+ at alpha intercalated cell
Chemical gradient ↑ K+/H+ antiporter activity
↑ K+ excretion
Hypokalemia
Chemical gradient shifts K+ into cells creating a charge gradient to shift H+ out
Hyponatremia Hypovolemia Hypochloremia
↓ Volume status activates angiotensin II, which plays a role in sodium and volume retention
↑ Na+/H+ antiporter 3 (NHE3) activity in the proximal tubule
Kidneys excrete & conserve ions to preserve intracellular charge gradient
↑ H+ loss in the urine ↓ Serum H+
↑ Proximal Na+ reabsorption causes ↑ urate reabsorption via urate-hydroxyl exchangers
Hyperuricemia
Contraction alkalosis (loss of Na+-rich, HCO3- low fluid) & hypokalemia
↑ Serum HCO3-
Metabolic alkalosis (systemic pH > 7.45 due to metabolic process)
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published Nov 18, 2024 on www.thecalgaryguide.com")
Statins Mechanisms and Side Effects

↓ Mevalonate acid (cholesterol precursor) production in the mevalonate pathway (metabolic processes that produce cholesterol)
↓ Cholesterol ubiquinone (CoQ10) production
↓ Ubiquinone (protective of cellular oxidative effects)
Impaired mitochondrial function
↓ Cyclooxygenase-2 (COX2) expression (enzyme involved in the production of prostaglandins)
↓ Availability of
arachidonic acid (derivative of LDL cholesterol breakdown)
↓ Thromboxane A2 (lipid with prothrombotic properties)
↓ Platelet adhesion
↓ Thrombogenicity (tendency to generate blood clots)
Improved endothelial function
Improved myocardial blood flow
↓ Intrinsic cholesterol biosynthesis in the liver
Compensatory mechanisms
↓ Isoprenoid production (electron & proton carriers)
↓ Isoprenoid intermediates
↓Inflammatory signaling proteins
↓Proliferation of macrophages
↓ Inflammation
Upregulation of hepatic LDL receptors
↑ Hepatic LDL uptake
↓ Serum low-density lipoprotein (LDL)
↑ Apolipoprotein A1 (component of HDL) production
↑ High-density lipoprotein (HDL) Carries serum
cholesterol back to liver
↓ Serum apolipoprotein B levels (acts as a tag for LDL, facilitating uptake by LDL receptors)
↑ Oxidative Stress Hepatocyte damage & inflammation ↑ Serum liver enzymes Hepatotoxicity
↓ ATP
↓ Serum cholesterol
↓ Muscle membrane stability
Rhabdomyolysis (muscle breakdown)
Author: Rupali Manek, Andrew Wu, Rafael Sanguinetti, Luiza Radu Reviewers: Joshua Dian, Laura Byford-Richardson, Alexander Ah-Chi Leung* * MD at time of publication
Stabilization of plaques
↓ Atherosclerosis (plaque build-up in arteries)
↓ Coronary events & mortality
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Physiological outcome
Published Jan 9, 2017; updated Nov 22, 2024 on www.thecalgaryguide.com")
Fibromylagia
 in cerebrospinal fluid
↑ Glutamate (pro- nociceptive excitatory neurotransmitter)
↑ Nerve growth factor
↑ Propagation of pain signals
↓ Norepinephrine
↓ Serotonin
Hyperactive nociceptors (nerve cell endings for pain sensation)
↓ Polarization of afferent sensory neurons
↓ Suppression of pain transmission at the nociceptive spinal cord Underactive inhibitory pathway
Central sensitization
(amplifying neural signal to perceive pain from a non-painful stimuli)
Fibromyalgia Syndrome
Hypersensitivity to pressure (tender points)
Muscle spasm (involuntary contraction of a muscle)
Allodynia
(pain to a non-painful stimulus) Nocturnal awakening Sleep disturbance Chronic stress and tension Depression Prolonged cortisol elevation
Diffuse hyperalgesia (increased pain to pressure)
Persistent activation of peripheral pain receptors
Ongoing tissue injury
Chronic pain
Possible metabolic changes
Mitochondrial dysfunction
Oxidative stress (imbalance between free radicals and antioxidants)
Muscle fiber dysfunction
Reduced muscle oxidative capacity
Muscle fatigue Low energy
Systemic production of eotaxin chemokines and IL-10 cytokines
Disturbances in neural networks
↓ Attentional capacity
Mood fluctuations
↑ Irritability Social
isolation
Dysesthesia
(abnormal feelings of itching, stinging, tingling, or general tightening)
Muscle and joint stiffness
Changes in frontal cortex
Deficits in cognitive functions
Memory issues
Hippocampal atrophy Brain fog
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published Nov 22, 2024 on www.thecalgaryguide.com")
Hypocalcemia Pathogenesis

Autoimmune (e.g., autoimmune polyglandular syndrome)
Infiltrative (e.g., hemochro matosis, Wilson’s disease)
Congenital (e.g., DiGeorge)
Hypomagnesemia
Magnesium is needed to produce PTH
Malabsorption
↓ Small bowel surface area
↓ Absorption of 25-OH Vitamin D (calcidiol)
↓ Conversion of calcidiol to 1-25 (OH)2 Vitamin D (calcitriol) by 1-α- hydroxylase
↓ Calcitriol
(impacts GI absorption of calcium)
Chronic Renal Failure
↓ Renal parenchymal mass (site of 1-α- hydroxylase production)
Vitamin D Dependent Type 1 Rickets
Mutation in the gene that encodes 1-α- hydroxylase
↓ Sun exposure
↓ 1-α-hydroxylase Pancreatitis
Destruction or atrophy of parathyroid gland
↓ Parathyroid hormone (PTH)
Calcitriol resistance (e.g., Vitamin D Type 2 rickets)
Inflammation of pancreas
↑ Release of lipase enzyme into serum
↑ Breakdown of fat by lipase
↑ Free fatty acids in serum
↑ Ca2+ binding to negatively charged tail of free fatty acids
↑ Parathyroid hormone (PTH)
PTH resistance
Genetic mutation causes body to not respond to PTH
↓ Renal calcium reabsorption
↑ Calcium excretion
↓ Osteoclast activity
↓ Calcium release from bone
↓ Action of PTH on intestinal cells
↓ Absorption of calcium in the small intestine
↑ Serum phosphate (PO4) (e.g. tumor lysis, rhabdomyolysis)
↑ Ca2+
binding to
negatively charged PO4
↑ Serum pH
↓ H+ in serum binding to proteins
↑ Ca2+ binding to proteins
Calcium sequestration
↓ Absorption of calcium in the small intestine
Hypocalcemia
Author: Breanna Fang Reviewers: Gurreet Bhandal, Raafi Ali, Luiza Radu Samuel Fineblit* * MD at time of publication
Serum calcium levels below 2.10mmol/L
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published Nov 22, 2024 on www.thecalgaryguide.com")
Acute Compartment Syndrome
 Penetrating injuries
Crush injuries
High volume IV transfusion or punctured vein
Reperfusion syndrome
Venomous animal bites
Severe burns
Burn eschar (rigid dead tissue)
Prolonged limb compression
Injury resulting in swelling
Bleeding into muscle compartment
↑ Pressure compresses veins (thin walls make them easily compressible), preventing venous outflow from muscle compartment
Buildup of venous blood in capillaries & venules further ↑ pressure in compartment
Arterioles collapse as they are unable to withstand higher pressure
↑ Pressure in capillaries prevents/slows blood flow from arterioles to capillaries, reducing tissue perfusion
↓ Oxygen delivery to muscles
Muscle & nerve necrosis
Ability for somatic sensory fibres to transmit information to the brain is impaired
Fluids intended for IV space leak into surrounding tissues
Tissue damage triggers release of inflammatory mediators increasing vascular permeability
Accumulation of fluids ↑ pressure within a muscle compartment
Stimulation of nociceptors in muscle
Pain out of proportion to injury
Edema in muscle compartment
Muscle feels hard on palpation Swelling
Reduced arterial pulses (late finding)
Pallor
Muscle feels cold on palpation
Muscle weakness
Paresthesia Sensory deficits
Rigid eschar prevents compartment expansion
Ischemic muscle releases inflammatory mediators
Constrictive bandage/cast applied before swelling subsides
External compression prevents muscle compartment from expanding to accommodate intra-compartment swelling
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published Feb 4, 2014, Updated Nov 19, 2024 on www.thecalgaryguide.com")
Pelvic Ring Fractures

Diastasis (separation) of pubic symphysis
Anterior Posterior Compression (APC)
Axial shear force (e.g. fall)
Superior or inferior displacement of hemipelvis
Vertical Shear (VS)
Grade 3: APC 2 with additional disruption of posterior sacroiliac ligaments
Lateral compression force
(e.g. automobile collides with pedestrian)
Internal rotation of hemipelvis
Lateral Compression (LC)
Authors: Meaghan Mackenzie Stephanie de Waal Nojan Mannani Reviewers: Annalise Abbott Usama Malik Sunawer Aujla Mankirat Bhogal Michelle J. Chen Dr. Prism Schneider* Dr. Alyssa Federico* * MD at time of publication
Grade 1: pubic symphysis diastasis < 2.5 cm
Grade 2: pubic symphysis diastasis >2.5 cm, anterior sacroiliac diastasis, disruption of sacrospinous and sacrotuberous ligaments
Grade 1: pubic rami fracture with ipsilateral sacral compression fracture
Grade 2: pubic rami fracture with ipsilateral sacral and ilium fractures
Grade 3: ipsilateral LC 1 or 2 injury and contralateral APC injury (windswept pelvis)
Young-Burgess Classification of Pelvic Ring Fractures
(Combination of any of the three classes can also occur)
Blood vessels surrounding the fracture site rupture during injury
Pelvic has capacity of hold large volume of blood
Supporting ligaments are stretched or ruptured
Damage to adjacent urogenital structures
Gross hematuria
Posterior urethral tear or bladder rupture
Scrotal, labial or perineal hematoma
Displacement of hemipelvis
Limb-length discrepancy
Damage to adjacent neurological structures
Flank hematoma
Potential hemorrhagic shock
Loss of rectal tone or perirectal sensation
L5 or S1 dermatomal paresthesia
Chronic instability
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published Nov 16, 2017; updated Nov 22, 2024 on www.thecalgaryguide.com")
Vaccine-Mediated Immunity General Physiology
 phagocytose and process antigen
Adjuvants in vaccines activate local immune response
↑ Production of pro-inflammatory signals (chemokines & cytokines)
Chemokines & cytokines act as signaling molecules that attract APCs to draining lymph nodes
APCs present antigen on MHC I molecules to CD8+ T cells
APCs present antigen on major histocompatibility complex II (MHC-II) molecules to naïve CD4+ T helper cells
Recognition of antigenic material activates naïve CD4+ T helper cells
A subset of naïve B cells differentiate into short-lived plasma cells
Plasma cells produce antibodies
Antibodies cover the pathogen’s surface (neutralization)
Antibody-coated pathogens are phagocytosed by macrophages/neutrophils (opsonization)
↑ Pathogen-specific antibody titers
CD8+ T cells multiply to form an antigen- specific cytotoxic T cell population (clonal expansion)
↑ Lymphocyte counts
Authors: Tanis Orsetti Reviewers: Allesha Eman Michelle J. Chen Dr. Russell Sterrett* Prevention of specific pathogen from causing severe disease upon subsequent exposure * MD at time of publication
Some activated CD4+ T helper cells stimulate naïve B cells
A subset of activated CD4+ T helper cells differentiate into memory CD4+ T cells
A subset of CD8+ T cells differentiate into memory CD8+ T cells
A subset of naïve B cells differentiate into memory B cells
Memory B cells circulate the body and monitor for secondary exposure to the pathogen
Memory T cells circulate the body and monitor for secondary exposure
Memory T cells are activated upon secondary exposure to the pathogen
Activated memory T cells differentiate into effector T cells
Memory B cells differentiate into plasma cells when there is a secondary exposure to the pathogen
Effector CD4+ T helper cells stimulate B lymphocytes
CD8+ cytotoxic T cells induce apoptosis of infected cells
Vaccine-Mediated Immunity
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published Nov 23, 2024 on www.thecalgaryguide.com")
Anesthetic Considerations Laparoscopic Abdominal Surgery

Cardiovascular Effects
Positional
Pneumoperitoneum (introduction of gas eg. CO2 in the abdominal cavity for easier access to organs during laparoscopy)
Systemic vasodilation
↓ Systemic vascular resistance
↑ Systemic blood flow & oxygen delivery
Respiratory acidosis
Acidosis leads to ↓ cardiac contractility
Arrhythmia
Cardiac arrest
Reverse Trendelenburg positioning of patient (patient supine with head of OR table ↑)
↑ Venous pooling (accumulation of blood in veins)
↓ Venous return (blood returning to the heart)
↓ Preload
↓ Blood pressure
Trendelenburg positioning of patient (patient supine with head of OR table ↓)
Low fluid status
Compression of large abdominal vessels
↓ Preload
↓ Blood pressure
High fluid status
↑ Venous return (blood returning to the heart)
↑ Preload (ventricle filling pressure prior to contraction)
↑ Blood pressure
↑ Venous return
↑ Preload
↑ Blood pressure
Vagus nerve stimulation
Bradyarrhythmia (slow and irregular heart rate)
Asystole (absence of electrical activity and contractility of the heart)
↑ Cerebral blood flow
Cephalad (upward) displacement of diaphragm & compression of thoracic cavity
↓ Functional residual capacity (volume remaining after expiration) & pulmonary compliance
↓ Tidal volumes (air inhaled per breath)
Atelectasis (partial or full collapse of lung)
V/Q mismatch
(lung ventilation & perfusion imbalance)
Respiratory Effects
↑ Intraocular pressure (e.g. papilledema)
Ophthalmic compromise (visual system impairment)
↑ Intracranial pressure
Compression of renal vessels à↓ Renal blood flow
Release of catecholamines (norepinephrine & dopamine)
Renin angiotensin aldosterone system activation
Aldosterone & vasopressin release
↑ Blood pressure Renal System Effects
Neurologic Effects
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published Nov 25, 2024on www.thecalgaryguide.com")
Chronic Inflammatory Demyelinating Polyneuropathy

Systemic illness (e.g., hepatitis B, Hepatitis C, HIV, thyroid Disease, nephrotic syndrome, irritable bowel syndrome)
Authors: Andrea Soumbasis Reviewers: Braxton Phillips Shahab Marzoughi Sina Marzoughi* * MD at time of publication
Autoimmune response initiation
Lumbar Puncture: Cytoalbuminologic Dissociation CSF protein count elevated with normal cell count
Cell-mediated response: Putative antigen (unidentified immune target) presents to auto-reactive T cells
Activated T cells secrete inflammatory mediators (matrix metalloproteinases)
Inflammatory response ↑ peripheral nerve permeability & T cells cross blood-nerve barrier
Humoral Response: Immunoglobulin & complement deposits on compact myelin & nodal regions
Autoantibodies target unknown epitope (antigen molecule recognized by antibody) on outer surface of Schwann cells
IgG4 antibodies target axoglial junction (Contactin-1, Neurofascin 155) that maintain Node of Ranvier of myelinated axons
↑ Permeability of blood- nerve barrier allows large proteins to leak into CSF
CD8+ T cells, CD4+ T cells & macrophages infiltrate peripheral nerves
Macrophages form clusters around endoneurium, release proinflammatory cytokines & strip away myelin via phagocytosis
Abnormal expression & distribution of Contactin-1 & Neurofascin 155 antibodies along axoglial junction
Disrupted ion channel segregation & paranodal structure
↓ Saltatory conduction
Nodopathy & Paranodopothy: Neurofascin 155 – Distal sensorimotor ataxia
Contactin-1 – Sensorimotor ataxia Caspr-1 – Neuropathic pain & nephrotic syndrome
Onion bulb formation: Nerve biopsy showing concentric, circular layers of cells surrounding the axon
Segmental demyelination & remyelination of peripheral nerves
↑ Demyelination
Chronic Inflammatory Demyelinating Polyneuropathy
Progressive weakness & sensory loss over a period of greater than 8 weeks due to autoimmune dysfunction
Peripheral nerve degeneration & conduction velocity
Nerve Conduction Study: Partial nerve conduction block, conduction velocity slowing, prolonged distal motor latencies, delay or disappearance of CMAP (compound muscle action potential)
Lesions of larger myelinated fibers in the dorsal columns for vibration & position sense
Lesions of peripheral motor nerves in non-length dependent pattern (does not only affect longest nerves first)
Motor Deficits
↓ Sensory input and/or ↓ motor output
Abnormal Reflexes
Globally ↓ reflexes
Lesions affecting sympathetic & parasympathetic nerve fibers
Autonomic Deficits
Bladder & bowel dysfunction, heart rate irregularities, blood pressure fluctuation
Sensory Deficits
Gait ataxia
Proximal weakness (muscles closer to the trunk or center of the body): Difficulty climbing stairs, rising from seated position, lifting objects overhead, frequent falls
Distal weakness (muscles farther from the trunk or the extremities): Tripping due to foot drop, difficulty with fine motor tasks
↓ Vibration & proprioception
Paraesthesias (e.g., tingling, numbness)
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published Dec 29, 2024 on www.thecalgaryguide.com")
Acquired Inguinal Hernias

Collagen deficiency
↑ Protease activity Smoking Aging
Malnutrition Collagen breakdown
Long-term glucocorticoid use
Fibroblast (cells which produce collagen) suppression causing ↓ collagen production
Thinning of skin & soft tissues
Incomplete obliteration of the processus vaginalis (peritoneal tunnel through which the testes migrate toward the scrotum during embryological development)
Failed closure leaves an opening for organs to herniate through
Intestinal loops entrapped within herniation
Weakening of connective tissues
Indirect Inguinal Hernia
Protrusion of abdominal and/or pelvic contents through deep inguinal ring
Protrusion of abdominal and/or pelvic contents through the Hesselbach triangle (anatomic area defined by the rectus abdominis medially, inferior epigastric vessels laterally, and the inguinal ligament inferiorly)
Herniated contents become entrapped
Inguinal mass +/- pain that is worse with straining
Abdominal distension & tenderness
Rhythmic contractions of the intestine attempting to push contents past the blockage site
Colicky pain
Incarceration (Surgical Emergency)
Reduced vascular supply to entrapped contents
Strangulation (Surgical Emergency)
Constriction & ischemia of hernial contents
Gangrenous skin changes (swelling, blue/purple colour, ↑ temperature, ulceration, ↓ sensation)
Inflammation of the herniated contents to larger than the hernial opening
Irreducible hernia (a hernia which cannot be manually reduced)
Bowel Obstruction (Surgical Emergency)
Obstruction of intestinal lumen impairs passage of intestinal contents
Stimulation of stretch receptors in the bowel wall
Nausea +/- vomiting
Herniated tissue undergoes necrosis (irreversible cell death), perforating the gastrointestinal wall
Obstipation (complete inability to pass stool or gas)
Bowel perforation (surgical emergency)
Leakage of gastrointestinal contents allows bacteria to infect the abdominal cavity
Sudden ↑ pain
Abscess (localized collection of pus)
Peritonitis (inflammation of the peritoneum)
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published Jun 3, 2018; updated dec 29, 2024 on www.thecalgaryguide.com")
Polycystic Ovarian Syndrome
: Pathogenesis and clinical findings
Genetic Susceptibility:
↑ Expression of LH receptors in granulosa cells, and anterior pituitary ↑ production of luteinizing hormone (LH)
↑ Serum LH compared to FSHà higher levels of LH increasingly activate thecal cells
Excess nutrients (from overeating and sedentary behavior) is stored as visceral fat
↑ Central adiposity (accumulation of fat around abdominal area)
Adipose tissue ↑ secretion of estrogen, inflammatory mediators, adipokines, free fatty acids
̄ Hepatic synthesis of sex hormone-binding globulin
Acne
Hair loss on scalp, and less commonly eyebrows and eyelashes (alopecia)
Excessive hair growth around mouth, chin, chest, abdomen, upper arms, thighs, and upper & lower back (hirsutism)
Metabolic syndrome develops, including ↑ insulin resistance, obesity, dyslipidemia, liver disease, & cardiovascular disease
Theca cells in ovaries ↑ androgen secretion
↑ Serum androgens
State of hyperandrogenism
Androgen and estrogen levels are elevated too early in menstrual cycle (estrogen has negative feedback inhibition on anterior pituitary hormone production)
Early suppression of FSH Limited proliferation of follicles
Relative reduction in secretion of progesterone Imbalance between progesterone and estrogen
Unpredictable ovulation
↑ Circulating insulin- like growth factor
Growth factor promote keratinocyte and dermal fibroblast proliferation
Velvety, darkening of the skin fold areas such as back of neck, armpit area, groin (acanthosis nigricans)
↑ Risk of type 2 diabetes mellitus
Since granulosa cells normally convert testosterone to estrogen, ̄ granulosa cell function means more testosterone exists
Since follicle stimulating hormone (FSH) activates granulosa cells, ̄ FSH means ̄ granulosa cell function.
Follicles arrested in development accumulate fluid, becoming cysts
Polycystic ovaries visible on ultrasound
Infertility
Unpredictable uterine bleeding
↑ Risk of endometrial hyperplasia & endometrial cancer
Irregular menstruation (amenorrhea/oligomenorrhea)
Authors: Lauren Standerwick Claire Song Reviewers: Mackenzie Grisdale Amy Fowler Christina Schweitzer Michelle J. Chen Dr. Yan Yu* Dr. Bernard Corenblum* Dr. Sylvie Bowden* * MD at time of publication
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published Nov 20, 2017; updated Dec 15, 2024 on www.thecalgaryguide.com")
Barretts Esophagus

Authors: Sophia Khan Reviewers: Claire Song Shahab Marzoughi Sylvain Coderre* * MD at time of publication
Reflux of stomach acid, bile salts + digestive enzymes
through lower esophageal sphincter (located at junction between esophagus and stomach)
Chronic reflux exposure to squamous epithelium (cells lining distal esophagus)
Squamous epithelial cells release inflammatory cytokines: interleukin-8 and interleukin-1beta
Inflammation of squamous epithelium
Adaptive changes of squamous epithelium to prevent damage by acidic reflux
Migration of stem cells in gastric cardia (proximal region of the stomach) into distal esophagus
Replacement of esophageal squamous epithelium by gastric columnar epithelium
Conversion (metaplasia) of normal esophageal squamous epithelium into abnormal gastric columnar epithelium with interspersed goblet cells
Z line (the squamocolumnar junction between the esophagus and stomach) is irregular and displaced proximally on esophagus on endoscopy
Heartburn (retrosternal burning sensation from stomach reflux)
Regurgitation (involuntary expulsion
of stomach content back into esophagus)
Development of goblet cells (intestinal mucus-producing cells) within esophageal epithelium
Abnormal presence of goblet cells on histology of distal esophagus
Abnormal presence of columnar epithelial cells on histology of distal esophagus
↑ Atypical proliferation &
↓ apoptosis of Barrett’s epithelial cells
Dysplasia (disordered growth of cells with the
potential to develop into cancer) of Barrett’s epithelium
Esophageal adenocarcinoma
Improper division of nuclear chromatin (packaged DNA)
Cellular nuclei increase in size due to retained chromatin from improper division
Excess chromatin absorbs more stain during histology
Double- stranded DNA breaks
Nuclear enlargement on histology
Continued acidic reflux causes oxidative DNA damage
Hyperchromatic (darkly stained) nuclei on histology
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published Dec 15, 2024 on www.thecalgaryguide.com")
Ptosis

Gravitational effect from excess eyelid weight
Traumatic causes of low eyelid position
Direct injury limits function of LPS or conduction of generated force to the tarsal plate
Laceration of the LPS limiting its function or transection of the levator aponeurosis
Congenital causes of eyelid drooping
Fibrofatty replacement of the LPS muscle at birth
Reduced levator function
eyelid position
Insult to the oculomotor nerve (CNIII) (e.g., compressive, microvascular, demyelinating)
CNIII innervates levator palpebrae superioris, the main muscle responsible for upper eyelid elevation
Insult to the sympathetic innervation (e.g., Horner’s syndrome)
Ocular sympathetic innervate the Muller muscle, which provides approximately 2mm of the upper eyelid’s height
Neuromuscular or myogenic causes of low eyelid position (e.g., myasthenia gravis)
LPS muscle myopathy or defect at its neuromuscular junction that limits the elevation of the eyelid
Aponeurotic causes of low eyelid position
Involutional change (disinsertion) of the levator aponeurosis which connects the LPS to the tarsal plate
Ptosis (also called blepharoptosis)
Abnormally low position of the upper eyelid
↓ distance between the central corneal light reflex (as produced by an examiner’s penlight) and the level of the center of the upper-eyelid margin
↓ Margin-Reflex Distance (Normal: 4-6mm)
Obstruction of the pupil/visual axis
Decreased or occluded superior visual field
↓ distance between the upper eyelid margin and the lower eyelid margin
↓ Palpebral Fissure Height (Normal: 10-12mm)
Flattening of the peripheral cornea by eyelid pressure
Induced with-the-rule astigmatism
Authors: Mina Mina Reviewers: Aicha Djaoutkhanova Shahab Marzoughi Lucy Yang Mao Ding William Trask* * MD at time of publication
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published December 29, 2024 on www.thecalgaryguide.com")
Malignant Esophagitis

Chronic gastric reflux (acidic partially- digested stomach contents)
Conversion of normal esophageal squamous epithelium to metaplastic columnar epithelium
Gastroesophageal Reflux Disease
Recurrent regurgitation of gastric content
Chronic Irritation & damage to the esophageal mucosa
Lesions and plaques develop in distal esophagus
Malignancy develops in the mucus-producing glandular cells in the lower esophagus
Smoking
Carcinogenic tobacco condensate is swallowed
Irritation of the the esophageal lining
Heavy alcohol consumption
Mutation in alcohol- dehydrogenase, which breaks down ethanol
↓ Alcohol breakdown
Irritation of the esophageal mucosa & lining
↑ Inflammation of the squamous epithelium located in the upper & middle esophagus
Achalasia (Esophageal smooth muscle motility disorder)
Food stasis in the esophagus ↑ Bacteria production
Lactic acid production & fermentation damages esophageal mucosa
Chronic inflammation in the esophageal epithelium
Malignant cells invade the aorta & tracheobronchial region
↑ Spread & growth of cancer in other organ systems
Metastatic invasion into the aorta, lungs, liver, bones, & kidneys through the lymph nodes
↑ Spread & growth of cancer in other organ systems
↑ Metabolic demand
Unintentional weight loss
Generalized chest pain
Hoarseness Cough
Esophageal adenocarcinoma (malignancy of the glandular cells of the esophagus)
Esophageal squamous cell carcinoma (malignancy of the squamous epithelium of the esophagus)
↑ Metabolic demand
↑ Weight loss
Chronic GI bleeding
Iron-deficiency anemia
↑ Ulceration
Exposure to stomach acid
↑ Tumor- induced inflammation
↑ Fibrosis
Irregular stricture of the upper & middle esophagus
Dysphagia
Esophageal wall narrowing
Abnormal reflexes of GI tract
Dysphagia
Indigestion
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published Dec 15, 2024 on www.thecalgaryguide.com")
Pulsus Paradoxus

Thoracic cavity expands
Lungs expand and intrathoracic pressure ↓
Physiologic:
↑ Venous return to right (R) heart
↑ R heart preload (volume of blood inside the ventricle right before it contracts)
↑ Blood pools in the right side of the heart
Obstructive lung diseases (e.g., COPD**, asthma**)
Hyperinflated lungs
↑ Stretching of pulmonary vessels at rest
On inspiration, ↑↑ stretching of pulmonary vessels
↑↑ Blood pools within pulmonary vasculature
↓↓ Flow to L heart
Pathologic: Constrictive pathologies (e.g., cardiac
tamponade**, constrictive pericarditis**) Decreased pericardial compliance
Constriction of ventricles
On inspiration, ↑ venous return to R heart (normal)
R ventricle unable to fully expand due to ↓ compliance
Septum bows into L ventricle
L ventricle unable to fully expand ↓↓ Filling of L heart
↓↓ L ventricular end diastolic volume
↓↓ L heart stroke volume ↓↓ Cardiac output Pulsus Paradoxus
Exaggerated ↓in systolic BP on inspiration (>10mmHg)
Air flows into the lungs
Pulmonary vessels are physically stretched/pulled
↑ Blood pools in pulmonary vessels
↓ Return of blood to left (L) heart
↓ L heart preload
↓ L heart stroke volume
↓ Cardiac output
Obstruction of superior or inferior vena cava (e.g., clot)
↓↓ Venous return to R heart at rest
↓↓ Right heart filling
↓↓ Blood flow to pulmonary arteries
Pulmonary embolism**
Clot occludes pulmonary arteries/ arterioles
↓↓ Flow to pulmonary veins
At rest, ↓↓ flow to L heart
On inspiration, ↓↓↓ flow to L heart
↓ Systolic blood pressure (BP) of < 10mmHg on inspiration
BP = cardiac output x systemic vascular resistance
**See corresponding Calgary Guide slides
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published Jan 21, 2013; updated Dec 3, 2024 on www.thecalgaryguide.com")
Constipation in Children

Older Child (>1 years old)
Dietary (e.g., lack of fluids / fiber)
Mechanical (e.g.,. intestinal Atresia, anal atresia)
Congenital malformation (narrowing, absence, or malrotation) of structure of intestine / anus
Interrupted flow of bowel contents
Genetic (e.g., cystic Fibrosis)
Mucous blocks pancreatic duct
Inability of pancreatic enzymes to reach small intestine
↓ Digestion after a meal
Large, thickened stool
Neurologic (e.g., Hirschsprung's disease)
Congenital disruption of the migration of neural crest cells to the distal colon
Affected segment of the colon fails to relax
Progressive secondary dilation of the healthy proximal colon
Systemic (e.g., hypothyroidism)
Thyroid hormone deficiency
Reduction in the stimulation of gut tone & contractility by thyroid hormones
↓ Peristalsis (intestinal contractions) of the bowel
Dietary
(e.g., lack of fluids/ fiber)
Mucosal (e.g., celiac disease)
Inappropriate immune response against gluten
Intestinal mucosal injury
Malabsorpti on of water and other nutrients
Functional (e.g., pain)
Prolonged stool retention in bowel
↑ Time in bowel causing over- absorption of water from stool into the large intestine
Mechanical (e.g., bowel obstruction)
Mechanical obstruction in the intestines
Interrupted flow of bowel contents
Intestinal obstruction
Neurologic (e.g., neglect, physical abuse)
Disturbance in brain-intestine axis
Mechanisms not fully understood
Visceral hyper- sensitivity
(increased pain sensation)
Withholding behaviour
Lack of soluble fiber
↓ Attractive forces between water & stool
Prevention of secretion of water into stool
Lack of insoluble fiber
↓ Stool bulk and laxation
↓ Secretion of water &
mucous into stool
Lack of soluble fiber
↓ Attractive forces between water & stool
Prevention of secretion of water into stool
Lack of insoluble fiber
↓ Stool bulk and laxation
↓ Secretion of water & mucous into stool
Formation of dry & hard stool
Formation of dry & hard stool
Difficulty passing stool
Difficulty passing stool
Authors: Jennifer Wytsma Reviewers: Sophia Khan, Shahab Marzoughi, Sylvain Coderre* * MD at time of publication
Intestinal obstruction
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published Dec 15, 2024 on www.thecalgaryguide.com")
Avascular Necrosis of the Scaphoid
: Pathogenesis and clinical findings
The radial artery provides the primary blood supply to the scaphoid bone
Blood flows to the distal
end of the scaphoid and travels “retrograde” back along the bone towards the proximal end
Poor blood supply may lead to incomplete healing of the fracture
High risk of progressing to a non-union (non- healing) fracture
Further ↑ risk of developing osteonecrosis (previously known as avascular necrosis)
Trauma or injury to the wrist or hand
Scaphoid fracture, most commonly at the thin middle third of the bone, known as the waist
Retrograde blood supply to the proximal part of the scaphoid becomes disrupted
Poor blood supply to the proximal scaphoid
The body breaks down and absorbs necrotic bone tissue, commonly at the proximal part of the scaphoid with poor blood supply
Structural integrity of the scaphoid bone weakens
The scaphoid becomes more prone to collapse during normal wrist movement and the natural alignment of the carpal bones may become disrupted
Prolonged disruption in blood and oxygen supply
Ischemia of bone tissue Bone tissue necrosis
Osteonecrosis (AVN) of the Scaphoid
Disease resulting in death of bone tissue due to insufficient blood supply**
Authors: Sydney Guderyan Reviewers: Mankirat Bhogal Michelle J. Chen Dr. Gerhard Kiefer * MD at time of publication
Abnormal stress on the radiocarpal and intercarpal joints
Progressive degenerative changes
Proximal row collapse (proximal row of carpal bones destabilize)
Imaging shows bone collapse, sclerosis, or cystic changes
Tenderness at the anatomical snuffbox
Pain & swelling on the radial side of the wrist
↓ Range of motion at the wrist
↓ Grip strength & wrist function
Stiffness
** See corresponding Calgary Guide slide “Avascular Necrosis: Pathogenesis and Clinical Findings”
End-stage osteoarthritis
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published Dec 30, 2024 on www.thecalgaryguide.com")
Ankle Fracture

Crushing force (e.g. limb Loading force entrapment beneath heavy object) (e.g. fall)
Force exceeds mechanical strength of bone
Ankle eversion or inversion
Osteoporosis
Compromised bone scaffolding & repair impairs the structural integrity of bone. Force required for fracture is lowered
Ankle Fracture
(Fracture of the talus and/or the distal 6 cm of the tibia and/or fibula)
Fractured bone is displaced through the dermal layers
Open Fracture
Compromised dermal layers create an opportunity for pathogens to enter the wound site
Infection
Multiple malleoli are fractured within the ring of the ankle
Lack of ligamentous & bone support makes ankle joint unstable
Displacement of bone from fracture site
Misalignment of bone segments prevents regeneration & union
Malunion of unreduced fracture
Ligamentous injury occurs concurrently from excessive tensile force
Fractured bone disrupts surrounding vasculature
Hyaline cartilage of the articulating surface is damaged
Trauma induces synovitis, chondrocyte apoptosis, & necrosis
Fractured bone disrupts surrounding peripheral nerves
Numbness Localized Pain
Pain is induced when the patient attempts to weight bear
Inability to weight bear
Authors: Ethan Smith Reviewers: Nojan Mannani Michelle J. Chen Dr. Gerhard Kiefer* * MD at time of publication
Platelets are exposed to the extravascular environment, thereby releasing platelet derived factors & complement factors
Plasma coagulation cascade is activated
Chondrocyte dysfunction in proliferation
Reduced synovial functioning
↑Vascular permeability from inflammatory cytokines
Protective hematoma forms in the joint space
Hyaline cartilage loss
Lost cartilage over time degrades proper articulation & causes joint narrowing, osteophytes, subchondral sclerosis
Post traumatic osteoarthritis
Edema
Fluid in the joint space changes position of bony articulations
Bruising
Restricted range of movement
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published Dec 30, 2024 on www.thecalgaryguide.com")
Ewing Sarcoma
(q24;q12) detectable with a genetic molecular study called FISH
Authors: Curtis Ostertag Nojan Mannani Reviewers: Mankirat Bhogal Michelle J. Chen Dr. Michael Monument* * MD at time of publication
Well-defined histopathology
Biopsy: sheets of monotonous small round blue cells with increased nuclear to cytoplasmic ratio
Immunohistochemistry staining positive for CD99 (80% of cases)
Diagnostic confirmation by histopathology
Diagnostic confirmation by molecular genetics
X-Ray findings: moth-eaten
lesions, Codman triangle of elevated periosteum, or onion- skin periosteal reaction
MRI findings: solid mass in bone, low T1 intensity, high T2, gadolinium enhancement, surrounding edema & soft tissue mass
Malignant small round cell tumor; the second most common malignant bone tumor in adolescents and young adults. Most common in the axial skeleton (pelvis, shoulder girdle, and ribs) and diaphysis of long bones.
Rapid cell turnover
Tumor expansion in bone, disrupting normal tissue
Possible metastasis to surrounding soft tissue
Possible metastasis to lung/pleura
Nodule/malignant pleural effusion
Absent breath sounds, pleural signs, crackles/rales
Possible metastasis to bone marrow
Replacement of normal hematopoietic tissue, including megakaryocytes (source of platelets)
Thrombocytopenia
Petechiae/purpura
Peritumoral edema
Swelling Stiffness
Invasion of cancer cells to distant tissues via bloodstream
Tumor expands toward skin surface
Palpable lump
Tumor secretes ↑ vascular endothelial growth factor (VEGF)
↑ Generation of immature blood vessels which have ↑ permeability
↑ Expression of inflammatory cytokines (i.e. IL-6)
Massive metabolic demand from cancer cells
Energy starved state
Systemic constitutional symptoms including fevers, chills, weight loss, night sweats, fatigue
↓ Distractions at night brings ↑ attention to pain
Adrenal glands naturally release ↓ cortisol throughout the day with the lowest levels at night. Lack of anti- inflammatory properties from cortisol ↑ pain
Potential gravity- related ↑ in tumor- related pressure & ↑ in active bone remodeling
Tumour growth stretches periosteum (connective tissue around bone) causing osteolysis, inflammation, nerve infiltration, edema, structural weakening (microfractures)
Pain that worsens at night
Limp
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published Dec 30, 2024 on www.thecalgaryguide.com")
Salter Harris Fracture
: Crushing of physeal cells & premature closure of physis
No visible initial finding on imaging
Incomplete calcification of cartilage to bone in the zone of provisional calcification of the physis (growth plate)
The zone of provisional calcification is weak and connecting ligaments are stronger than the physis in pre-pubertal children
Trauma in a pediatric patient Damage occurs to the physis (growth plate)
Salter Harris Fracture
Rotational, shear, +/- longitudinal forces applied to the physis
Intra-articular damage
Longitudinal or shear force applied to the physis
Epiphysis splits from the metaphysis
Type I (5%): Complete or incomplete fracture through the physis only
Angular & longitudinal force exerted on the physis
Type II (75%): Partial fracture through physis continuing into the metaphysis
Creation of a Thurston- Holland fragment (metaphyseal fragment that is still connected to the epiphysis)
Thurston-Holland fragment is visible & palpable
Type III (10%): Fracture through the physis extending into the epiphysis
Palpable step- off over the epiphysis only
Type IV (10%): Fracture involving the epiphysis, physis, & metaphysis
Swelling around joint
Focal tenderness over the physis
Palpable step-off over the epiphysis & metaphysis
Risk of transphyseal vascularity development
Recruitment of osteoprogenitor cells
Bone bridge formation between the epiphysis & metaphysis
Widening & separation of epiphysis from metaphysis on imaging
Bone growth arrest
Limb length discrepancy
Asymmetric growth
Post-traumatic Altered joint arthritis mechanics
Growth arrest risk if the displacement is > 3mm
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published Jan 5, 2025 on www.thecalgaryguide.com")
Pituitary Tumour Classification and Clinical Outcomes
 (lack of hormone-producing cells or due to gene mutations)
Authors: Chris Oleynick Caroline Kokorudz Reviewers: Amyna Fidai Laura Byford- Richardson Joseph Tropiano Julia Gospodinov Luiza Radu Hanan Bassyouni* * MD at time of publication
hormones (70%)
Relative abundance & predisposition for lactotroph (prolactin-producing), somatotroph (growth hormone – producing), thyrotroph (TSH producing) & corticotroph (adrenocorticotropic hormone – producing) cells to form adenomas (epithelial cell tumours)
↑ Lactotroph cells
↑ Prolactin (PRL) (most common)
Hyper- prolactinemia (high prolactin blood levels)
↑ Somatotroph cells
↑ Growth hormone (GH)
Acromegaly* (excess tissue growth & metabolic dysfunction)
↑ Corticotroph cells
↑ Adrenocortico- tropic hormone (ACTH)
Cushing disease* (prolonged exposure to excess cortisol)
↑ Thyrotroph cells
↑ Thyroid stimulating hormone (TSH)
Central hyper- thyroidism (↑ T4 & T3)
Large size (macro) (>10mm on MRI) may cause mass effect (tissue compression from mass)
Small size (micro) (<10mm on MRI) unlikely to cause mass effect (compression to surrounding structures)
Asymptomatic
Headache
Nausea & vomiting
Compresses pituitary gland & impairs blood supply & function of pituitary cells
Impinges pituitary stalk & disrupts hormone transport from hypothalamus to the anterior and posterior pituitary
Compresses surrounding structures
↑ Pressure & stretching of dural mater
Stretching of the meninges activates mechanoreceptors (stretch receptors) in GI tract
Pituitary hormone deficiency (typically in left-right order with mass effect):
Dopamine release obstruction
↓ Inhibition of prolactin secretion
Antidiuretic hormone (ADH) obstruction
Inferior tumour growth
Growth into sphenoid sinus
Lateral growth of the tumor compresses surrounding cranial nerves
Superior tumour growth
↓ GH
↓ Protein production & muscle cell proliferation
↓ Luteinizing hormone (LH) (triggers ovulation & sex hormone synthesis) & follicle stimulating hormone (FSH) (stimulates ovarian follicles & sperm growth)
↓ (TSH)
↓ ACTH (stimulates cortisol & androgen release)
Cranial nerve (CN) II (optic)
↓ Visual acuity
Diplopia (double vision)
CN III, (oculomotor) IV (trochlear), or VI (abducens)
Ptosis* (drooping upper eyelid)
CN V (trigeminal) branches V1 & V2
Facial numbness
Ophthalmoplegia (eye muscle paralysis)
Compresses optic chiasma
Bitemporal hemianopsia (decreased lateral peripheral vision)
Tumour extends into hypothalamus
Hypothalamic dysfunction due to damage to hypothalamic cells
Disruption of multiple regulatory systems (i.e. sleep-wake cycles, appetite, temperature)
Tumour occludes ventricles
Tumour obstructs CSF flow
↑ Intracranial pressure
Hydrocephalus* (abnormal buildup of CSF in ventricles of the brain) and papilledema
Bacteria migrates from sinus flora into sphenoid sinus
Cerebrospinal fluid (CSF) leaks into throat
Post-nasal drip and nasopharyngeal mass
Stunted growth and short stature in children
↓ Muscle mass & fatigue
Diabetes insipidus* (excessive urination & extreme thirst)
Central hypothyroidism* (↓ T4 & T3)
Hyperprolactinemia
Meningitis* (inflammation of the meningeal layers of central nervous system)
*See relevant Calgary Guide slide
Hypogonadotropic hypogonadism (↓ Sex hormones)
Adrenal insufficiency* (↓ 8 AM cortisol)
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published Oct 1, 2027; updated Jan 5, 2025 on www.thecalgaryguide.com")
左向右先天性心脏分流

右束支传导阻滞

左束支传导阻滞

心包炎

不稳定型心绞痛

房颤并发症

房颤临床表现

心包积液及心包填塞

房扑

房间隔缺损
: 发病机制及临床表现")
前后束支阻滞

一度房室传导阻滞

二度房室传导阻滞莫氏I型
: 发病机制和临床表现")
二度房室传导阻滞莫氏二型

三度完全房室传导阻滞
房室传导阻滞: 发病机制和临床表现")
Insuficiencia Renal Aguda Post-Renal
: Patogenia y hallazgos clínicos")
Displasia Broncopulmonar
: Patogénesis y hallazgos clínicos")
Abces cerebral pyogene a lIRM

Blessures par inhalation

Mastoidite

Laryngite aigue

Hydrocele

Opioid Use Disorder

Neuronal injury or death
Myoclonus (involuntary muscle twitches/jerks)
In brain & brain stem
Parasympathetic stimulation of oculomotor nerve (cranial nerve 3)
Suppression of reticular activating system neurons
Cessation of respiration
Contraction of pupillary sphincter
↓ Excitatory input to the cortices
↓ Arousal
Lack of oxygen in the brain & other organs
Miosis (pupil constriction)
↓ Level of Consciousness
Death
↓ GABA release in ventral tegmental area
Activation of mesolimbic dopaminergic pathway
Release of dopamine in nucleus accumbens
↓ Traumatic memories
↓ Acetylcholine release in GI tract
↓ Peristalsis in GI tract
↓ GI transit & motility
↑ Time for colonic absorption of water
Constipation
Activation of mu & delta receptors in medulla’s chemoreceptor trigger zone
Nausea
Activation of mu receptors in the pons & medulla
↓ Neural drive for breathing
↓ Respiration
Euphoria
Analgesia
Craving, strong desire to use
More frequent opioid use
Authors:
Keira Britto
Kayla Marritt
Meera Grover*
Reviewers:
Erik Fraunberger
Sara Cho, Luiza Radu Spencer Krahn *
* MD at time of publication
Opioid Use Disorder:
Pattern of opioid use leading to clinically significant impairment or distress
Performing any given behaviour while intoxicated
cAMP Mechanism upregulation unknown
Mu opioid receptor desensitization
Physical dependence reinforces opioid use
Opioid withdrawal with cessation**
Unable to ↓ or stop opioid usage
With chronic use, ↓ response to opioids
Behaviour tolerance
↑ Opioid required to achieve euphoria & analgesia (i.e., tolerance)
↑ Quantity & prolonged opioid consumption
↑ Time spent obtaining, using, & recovering from opioids
Recurrent opioid use in physically hazardous situations despite physical, psychological, social & interpersonal consequences
↓ Participation in occupational social & recreational activities
↓ Fulfillment of work, school & personal obligations
**See relevant Calgary Guide slide
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published Jan 28, 2025 on www.thecalgaryguide.com")
Central Precocious Puberty
, trauma, infection, ischemia
Damage to hypothalamic & pituitary structures (i.e. infarct or traumatic sheering forces often causing inflammation and ultimately cell death)
Septo-optic dysplasia (early brain development disorder)
Abnormal pituitary & hypothalamus development (unknown pathogenesis)
Psychosocial stress
International adoption from developing countries
Rapid increase in nutrition
Rapid catch-up growth
Hormonal and metabolic changes
Endocrine disrupting chemicals (ex. Phthalates, bisphenol A)
Hypothalamic inflammation
Suppression of inhibitory and activation of stimulatory elements of the GnRH production pathway
Idiopathic familial central precocious puberty from different genetic mutations
primarily impacting tissues derived from ectoderm)
Makorin RING- finger protein 3( MKRN3) mutation (repressor of GnRH secretion)
Kisspeptin protein and/or receptor mutation (regulates puberty & reproductive function)
Extended Kisspeptin protein availability and/or ↑ production
Kisspeptin receptors signal directly to gonadotropin releasing hormone (GnRH) neurons to release GnRH into portal system
Early ↑ or inappropriate GnRH pulses
Neurofibromatos is T1 (NF1) (causes benign tumors to grow in the brain along nerves)*
Tumor development involving the optic chiasm (>15% of individuals with NF1) may impact hypothalamic function
Damage to and/or disruption of normal hypothalamic & pituitary function
Tuberous sclerosis autosomal dominant disorder (rare genetic disease that causes growth of benign tumors in brain & body)
Dysregulated cell division & growth
Hamartomas form as benign mass composed of cells similar to those surrounding it, occurs in many areas including hypothalamic hamartoma (HH)
HH acts as an accessory, unregulated hypothalamus
Autonomous (unregulated) pulsatile GnRH release
Sturge-Weber Syndrome (vascular disorder causing capillary-venus malformations impacting the face, brain, eyes)
Vascular malformation throughout the brain including the hypothalamus
Impaired blood flow to the hypothalamus
Hypothalamic ischemia
Hypothalamus dysregulation and premature activation of GnRH release
Disruption of excitation and inhibition balance in the CNS
Hypothalamic- pituitary dysregulation
Reduced inhibition of GnRH secretion
Increased corticotropin- releasing hormone
Adrenal gland production & release of androgens by unknown mechanism that play a role in maturation of the HPG axis
Adrenarche (start of androgen production). Including: acne, body odor, & pubarche (pubic hair growth)
Dysregulated GnRH pulses
Dysregulated luteinizing hormone (LH) & follicle simulating hormone (FSH) release
Early GnRH release
Early FSH secretion from pituitary gland
↑ Uterine volume & growth of ovarian follicles
Cyclical ovulation
Premature menarche (first menses < 7 years)
Early LH secretion from pituitary
↑ Gonadal androgen production
↑ Estrogen release from ovaries
gland
**See Calgary Guide slide - “Brain Neoplasms”
↑ Skeletal growth
↑ Bone mineral density
Thelarche (breast development)
Authors: Joanna Keough Reviewers: Run Xuan (Karen) Zeng Luiza Radu Samuel Fineblit* *MD at time of publication
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published Jan 28, 2025 on www.thecalgaryguide.com")
Vasoconstrictors in the OR
:
Mechanisms of action and clinical findings
Indirect effects of ephedrine
Ephedrine
Intraoperative use if patient becomes: -Hypotensive -Bradycardic
Inhibits reuptake of neuronal norepinephrine
Bind to alpha-1 receptors in peripheral vasculature
Bind to beta-1 receptors in cardiac tissue
Bind to beta-2 receptors in pulmonary tissue
Bind to alpha-1 receptors in peripheral vasculature
Bind to alpha-1 receptors in iris dilator muscle
(for opthalmic administration)
Norepinephrine remains in synapse longer
↑ Norepinephrine release from storage vesicles
Peripheral vein vasoconstriction
↑ Sinoatrial & atrioventricular nodal firing & inotropy (force of heart’s muscle contractions)
Relaxes airway smooth muscle
Vasoconstricts peripheral veins & arteries
Iris dilator (smooth muscle) contraction
Binds to alpha-1 receptors in peripheral vasculature
Binds to beta-1 receptors in cardiac tissue
Binds to beta-2 receptors in pulmonary tissue
↑ Blood pressure
↑ Systemic vascular resistance
↑ Heart rate
↑ Cardiac contractility
↑ Cardiac output Bronchodilation
↑ Blood pressure
↑ Preload
↑ Afterload
↑ Systemic vascular
↑ Blood pressure
↑ Heart rate
↑ Cardiac contractility Bronchodilation
Sustained tachycardia
(Heart rate >100 beats per minute)
Arrhythmias (abnormal heart rhythms)
Palpitations
Baroreceptor mediated reflex bradycardia
(Heart rate <60 beats per minute)
↓ Reduced cardiac output (when afterload > preload)
Direct effects ephedrine
of
Phenylephrine
Intraoperative use patient becomes:
-Hypotensive
if
resistance Pupil dilation
Authors: Rebecca Sugars Reviewers: Run Xuan (Karen) Zeng, Luiza Radu, Leyla Baghirzada* * MD at time of publication
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published Jan 28, 2025 on www.thecalgaryguide.com")
Dopamine Antagonists Metoclopramide & Domperidone
 Zeng Luiza Radu Esther Ho* * MD at time of publication
Binds to & blocks dopamine D2 receptors in the chemoreceptor trigger zone (CTZ) in medulla oblongata
Binds to & blocks
serotonin (5-HT3) receptors in the CTZ in medulla oblongata
Binds to & blocks dopamine D1 & D2 receptors in the nucleus accumbens of the basal forebrain
Binds to & blocks dopamine D2 receptors in the substantia nigra of the basal ganglia
Binds to & blocks dopamine D2 receptors in the anterior pituitary
Binds to & blocks dopamine D2 receptors in myenteric plexuses of the proximal gut
Binds to & blocks dopamine D2 receptors in the myocardium
Incoming vagal afferents (sensory nerve signals) reduce dopamine release at the CTZ
Incoming vagal afferents (sensory nerve signals) reduce serotonin release at the CTZ
↓ Binding of dopamine released by neurons in the ventral tegmental area (VTA) of the midbrain
↓ Dopamine’s inhibitory effect on neurons in the ventrolateral (VL) nucleus of the thalamus
↓ Binding of dopamine released by neurons in the hypothalamus
↑ Acetylcholine (Ach) release from parasympathetic neuronal nerve endings
↓ Dopamine binding at the CTZ
↓ Serotonin binding at CTZ
↓ Activation of wake-promoting VTA neurons
↑ Excitatory activity of neurons in the VL nucleus of the thalamus
↓ Dopamine’s inhibitory effect on prolactin secretion
Influx of calcium ions into smooth muscle
↓ Dorsal motor nucleus activity of the vagus nerve in the medulla oblongata
↓ Nucleus solitary tract activity (processes sensory signals from body)
↓ Vomiting center activity
Suppressed vomiting reflex
↓ Vomiting
Central Actions
↓ Excitatory signals to promote wakefulness
Drowsiness
Extrapyramidal symptoms (EPS; impaired motor control)
Dystonia (sustained muscle spasm & abnormal posture), akathisia (restlessness), chorea (rapid, irregular movement), parkinsonism (tremors, rigidity) EPS symptoms occur mostly with metoclopramide. Domperidone does not readily cross CNS and is less likely to produce EPS symptoms.
Continuous activation signal to corticospinal motor control system
Hyperprolactinemia
(↑ prolactin secretion)
↑ Involuntary movements
Peripheral Actions
Torsade de Pointes (polymorphic ventricular
tachycardia; rapid & irregular heart rhythm on ECG)
Membrane depolarization
↑ Velocity of excitatory waves on smooth muscle walls
↑ Gut tone & rhythm of contraction
↑ Gastric and small intestinal motility & ↑ lower esophageal sphincter tone
Blocks potassium channel
Inhibits potassium ion outflow
Delayed ventricular repolarization (>440 milliseconds in men
& >460 milliseconds in women)
Myocardium unable to compensate for arrhythmia
Ventricular fibrillation Cardiac Arrest Death
Bradycardia (Slow than normal heart rate)
Long QT Syndrome Extended QT interval seen on electrocardiogram (ECG)
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published Jan 28, 2025 on www.thecalgaryguide.com")
Osgood Schlatter Disease

Patellar tendon inserts more proximally or broadly on tibia
↓ Patellar tendon moment arm length causes ↑ biomechanical forces on tibial tubercle
Patella alta (superior displacement of the patella within the trochlear groove) *temporal relationship uncertain
Quadriceps require ↑ forces to achieve full extension
Patellar tendon overuse
↑ Patellar tendon tension
↑ Traction stress on the patellar tendon insertion at the cartilaginous tibial tubercle apophysis in one or both knees
Osgood-Schlatter Disease
Self-limiting osteochondrosis or traction apophysitis (inflammation of the growth plate) of the proximal tibial tubercle at the insertion of the patellar tendon
Tibial tubercle apophysis hypertrophies & develops chronic micro-avulsions (tibial apophyseal ossification centers & cartilage is pulled off the tibial metaphysis)
↑ Inflammation at injury site Proximal patellar tendon micro- Repetitive strain/forces on the proximal tibial apophysis
Tibial apophysis undergoes partial arrest
Asymmetric proximal tibial epiphyseal growth abnormality (posterior > anterior)
Genu recurvatum (knee hyperextension) (rare)
due to traction apophysitis avulses from the tibial apophysis
overcomes bone-tendon attachment strength
Activated immune cells release cytokines at injury site
Sensitization of nociceptors in periosteum and surrounding tissues
Tenderness and Antalgic swelling of gait
patellar tendon
Proximal tibial apophyseal fragmentation (visible on X-ray)
Avulsion fracture of tibial tubercle
Calcium is abnormally deposited in the tendon
Calcific tendinopathy in patellar tendon (visible on X-ray)
Anterior knee pain that worsens with exercise
Inflammatory mediators recruit osteoblast precursors to stabilize injury site
↑ Osteoblast activity drives bone remodeling and formation of ossicles (small pieces of bone)
Bone remodeling causes united ossicles (ossicle fusion with tibial tuberosity)
Ununited ossicle imbedded within patellar tendon (visible on X-ray)
Bony prominence of tibial tubercle (visible on X-ray)
Residual ununited ossicle remains after apophysis fuses with proximal tibial metaphysis
Persistent visible prominence after proximal tibial apophyseal closure
Authors: Emily J. Doucette Reviewers: Michelle J. Chan Yan Yu* Gerhard Kiefer* * MD at time of publication
Persistent pain & discomfort with ↓ strength & function
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published Feb 16, 2025 on www.thecalgaryguide.com")
Polycythemia Vera Pathogenesis
: Pathogenesis and Clinical Findings
Authors: Caitlin Bittman Noriyah Al Awadhi Yan Yu Peter Duggan* Reviewers: Maharshi Gandhi Kevin Zhan Michelle J. Chen Paul Ratti Merna Adly Crystal Liu Kareem Jamani* Man-Chiu Poon* Lynn Savoie* * MD at time of publication
Familial PV inherited as autosomal dominant incomplete penetrance (5% of PV cases)
Predisposition to acquire a second somatic mutation
Hematopoietic cells with no known mutations in their DNA
Acquired JAK2 (Janus kinase 2) mutation in a single hematopoietic pluripotent stem cell in the bone marrow
JAK2 mutated cells in the bone marrow ↑ production of hematopoietic cells (RBCs, WBCs, platelets) and cytokines
DNA changes leads to continuous replication of the mutated cell
Abnormal cell becomes the predominant hematopoietic stem cell in the bone marrow
↑ RBC production independent of erythropoietin (EPO)
↑ Mast cell production
↑ Leukocyte production (basophils, neutrophils, eosinophils, monocytes
↑ Platelet production (thrombocytosis)
Overproduced platelets often have abnormal function of activation and aggregation
Hypercellular bone marrow on biopsy with trilineage growth (erythroid, granulocytic, and megakaryocytic cell lines)
↑ Hemoglobin concentration
↑ Hematocrit (proportion of blood volume occupied by RBCs)
↑ RBC leads to down-regulation of EPO by kidneys
↓ Serum EPO
↑ Levels of histamine, which is stored in granules within basophils & mast cells
Contact with water causes degranulation and subsequent histamine release from mast cells & basophils
Aquagenic pruritis (itchy skin after contact with water)
Histamines released from mast cells in the stomach causes hypersecretion of gastric acid
Stomach ulcers
Acquired Von Willebrand syndrome (with platelets >1,000,000/microL)
Despite thrombocytosis, there is an ↑ risk of bleeding due to platelet dysfunction
↑ Risk of hemorrhage & abnormal bleeding (e.g. GI and mucosal bleeding)
Easy bruising, purpura, petechiae, dental bleeds, epistaxis
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published Aug 7, 2012; updated Feb 22, 2025 on www.thecalgaryguide.com")
Polycythemia Vera Complications
: Complications
High numbers of cells & platelets ↑ blood viscosity
Polycythemia Vera
Hematological disorder in which JAK2 mutations in hematopoietic cells result in increased RBC production. See corresponding Calgary Guide slide “Polycythemia Vera: Pathogenesis”
↑ Blood cell volume
Presence of increased numbers of platelets creates a hypercoagulable and prothrombotic state
↑ Risk of venous & arterial thrombosis
↑ Systemic vascular resistance Impaired/“sluggish” blood flow
↓ Perfusion to small vessels and ↓ oxygen delivery to throughout body
Arterial clots in arms
and legs or in vessels leading to brain prevent oxygen delivery to cells
Systemic hypertension
↑ Turnover of hematopoietic cells (RBCs, WBCs, platelets)
Fatigue
Transient visual disturbances
Neurological symptoms (e.g. headache, dizziness, tinnitus, concentration problems)
Breakdown of nucleic acids during cell turnover ↑ uric acid levels in blood
Lysing cells release lactate dehydrogenase (LDH) into bloodstream
↑ Spleen activity to filter and dispose of old blood cells
Splenomegaly
Authors: Caitlin Bittman Noriyah Al Awadhi Yan Yu Peter Duggan* Reviewers: Maharshi Gandhi Kevin Zhan Michelle J. Chen Paul Ratti Merna Adly Crystal Liu Kareem Jamani* Man-Chiu Poon* Lynn Savoie* * MD at time of publication
Extramedullary hematopoiesis
Pancytopenia
Clots in the
portal vein, splenic vein, & mesenteric vein are unusual & highly suggestive of PV
Clots in cardiac arteries prevent O2 delivery to cardiac tissue
Myocardial infarction
Venous blood moves more slowly and is less pressurized compared to arterial blood. Combined with hypercoagulable state, clots are likely to form in deep veins (usually of the legs)
Deep vein thrombosis
Clot breaks off and
travels through the inferior vena cava & right heart into the pulmonary arteries
Pulmonary embolism
Stimulation fibroblasts in the bone marrow
Uric acid accumulates & precipitates in blood
Hyperuricemia
↑ Serum LDH
Limb ischemia
Ischemic stroke & transient ischemic attacks
Erythromelalgias (burning pain in the extremities and erythema due to poor perfusion and transient microvascular occlusion)
Compensatory vasodilation in the capillaries of the skin
Plethora/rusted or “ruddy” complexion, particularly noticeable on the face, palms, nailbeds, & mucous membranes
Gout
Uric acid stones
Advanced Disease Progression
Abnormal blood cells produced in PV release various growth factors and cytokines
Production of excess collagen and other fibrous materials
Hematopoietic cells in bone marrow replaced with fibrous tissue over time
Bone marrow increasingly unable to produce healthy blood cells
Myelofibrosis (rare type of chronic blood cancer; considered late-stage progression of PV characterized by bone marrow fibrosis)
Bone marrow failure
Chronic bone marrow hyperactivity leads to exhaustion and/or damage of hematopoietic cells
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published Aug 7, 2012, updated Feb 22, 2025 on www.thecalgaryguide.com")
Aneurisma Aorta Abdomen Patogenesis
: Patogenesis")
Aneurisma Aorta Abdomen Temuan Klinis dan Komplikasi
: Temuan Klinis dan Komplikasi")
Hipertensi Kronik Komplikasi

Neuraxial anesthesia

Blood vessel punctured
Blood accumulates in epidural space
Epidural hematoma
Inhibits motor nerve conduction (least sensitive nerve fibers to anesthetics)
Anesthetic injected into cerebrospinal fluid (CSF) in the subarachnoid space
Punctured dura mater Cerebrospinal fluid leak Decrease cerebrospinal fluid pressure Vasodilation of cerebral blood vessels Post-dural puncture headache
Inhibits sympathetic nerve conduction (most sensitive nerve fibers to anesthetics)
Anesthetics diffuse across axon membranes & blocks voltage-gated sodium channel in nerve fibers
↓ Sodium ion flow into neuron
Nerve fibers unable to depolarize & reach threshold to trigger action potential
Inhibits sensory nerve conduction
Cardiac sympathetic nerve fibers inhibited
↓ Norepinephrine release
Unopposed parasympathetic (rest and digest) activity
Parasympathetic nerves release acetylcholine (Ach)
ACh binds to muscarinic receptors on smooth muscle
↑ Peristalsis
↑ Bowel motility
C-fibre nerve fibers (unmyelinated) inhibited
Loss of pain & temperature sensation
A-delta nerve fibers (thinly myelinated) inhibited
Loss of pinprick, pain & temperature sensation
A-beta nerve fibers (myelinated) inhibited
Loss of vibration, touch & pressure sensation
A-alpha nerve fibers (heavily myelinated) inhibited
Loss of proprioception
Phrenic nerve fibers inhibited
Diaphragm paralysis
Impaired diaphragm contraction
Apnea (temporary cessation of breathing)
Loss of motor function
↓ Cardiac pacemaker activity
Bradycardia
(slow heart rate)
Inhibits smooth muscle tone of blood vessels
↓ Systemic vascular resistance
Authors: Shiva Ivaturi Reviewers: Run Xuan (Karen) Zeng, Luiza Radu, Julia Haber* *MD at time of publication
Vasodilation
Hypotension
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published Feb 24, 2025 on www.thecalgaryguide.com")
Nitrous Oxide
 receptor
↑ Chloride ion influx into the neuron
Possible activation of Calmodulin–Nictric Oxide Synthase–Cyclic Guanosine Monophosphate dependent Protein Kinase pathway (exact mechanism unclear)
↑ Neurotransmission Inhibition
Minimal Sedation (Anxiolysis)
Authors: Parthiv Amin Andre Skipper Tracey Rice
Reviewers:
Billy Sun, Joseph Tropiano
Run Xuan (Karen) Zeng Luiza Radu
Michael Chong*
Leyla Baghirzada*
* MD at time of publication
N-methyl-D-aspartate (NMDA) receptor antagonist (inhibitor)
Closes NDMA receptor channels
Inhibit ionic currents
↓ Central nervous system excitability
Analgesia
Hyperhomocysteinemia
(Excessive build-up of homocysteine due to lack of conversion to methionine)
↑ Reactive oxygen species (ROS) & endothelial damage
↑ Coagulation & endothelial adhesion
↑ Atherosclerosis & thrombosis
↑ Risk of perioperative cardiovascular complications (Myocardial ischemia & myocardial infarction)
Pyramidal Cell Vacuole Reaction: Swelling of endoplasmic reticulum & mitochondria
Rapidly reversible pyramidal cell neurotoxic vacuole reaction in posterior cingulate/retrosplenial cortex (PC/RSC)
Neurotoxicity with prolonged use
Lack of methionine impairs ability to produce S- adenosylmethionine (SAM)
Lack of SAM (helps regulate folate production) causes diversion of folate to methionine production
Lack of folate impairs thymidine production
Lack of thymidine (crucial nucleotide in DNA synthesis) impairs DNA synthesis
Megaloblastic Anemia
Nociception modulation (Detection & transmission of harmful stimuli)
Neurons stimulation in periaqueductal grey area (PAG) of midbrain
Endogenous opioid peptides (EOPs) release
EOP activates peripheral opioid receptors GABA-ergic nuclei of pons
Inhibits activity of noradrenergic pathway (flight or flight response)
Descending noradrenergic pathway activation in dorsal horn of spine
Nitrous Oxide (N2O) irreversibly oxidizes (inactivates) vitamin B12 (cobalamin)
Vitamin B12 no longer available as cofactor for methionine synthase
Irreversible inhibition of methionine synthase (converts homocysteine to methionine)
↓ Pain signals between primary & second order afferent sensory neurons (direct pain inhibition)
Upregulation of GABA interneurons (Indirect pain inhibition)
GABA release further ↓ pain signals
Analgesia
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published Sept 20, 2017; updated Feb 24, 2025 on www.thecalgaryguide.com")
Considerations anesthesiques chez les patients souffrant dobesite

Agonistes Des Recepteurs Opioides

Neonatal Seizures

↑ Pro-inflammatory signaling
Neurocutaneous syndromes
(e.g. Tuberous sclerosis)
Brain lesions disrupt cortical formation
Metabolic disturbance (e.g. hypoglycemia, hyponatremia)
Electrolyte imbalances alter osmolality
Ion transport disruption
Malformations of cerebral development
Structural alteration in signal transmission
Medication induced (e.g. anesthetics, opioids, alcohol)
Direct toxicity or withdrawal symptom
Benign neonatal seizures
↓ Blood flow to the brain
Authors:
Pauline de Jesus Reviewers:
Annie Pham
Emily J. Doucette
Jean Mah*
*MD at time of publication
↑ Neuronal excitability
Non-Familial
No specific underlying genetic etiology (likely environmental)
Familial
Autosomal dominant epileptic syndrome
Derangement in the distribution of excitatory & inhibitory neurotransmitters and networks in the brain causes cortical dysfunction
↑ Influx of extracellular Ca2+
↑ Opening of voltage dependent Na+ channels ↑ Influx of Na+
Voltage-gated K+ channels lose their function
↓ of K+ current
Impaired repolarization of neuronal cell membranes
Subtle Seizures
Imitation of normal behaviours
Clonic Seizures
Alternating excitatory & inhibitory activity
Tonic Seizures
Excessive, widespread excitatory activity
Myoclonic Seizures
Rapid bursts of excitatory activity
High frequency bursts of action potentials, hyper-synchronized depolarization in neuronal populations, & ↓inhibitory signaling in surrounding neurons
Neonatal Seizures
Sudden, paroxysmal, abnormal alteration of electrographic activity lasting from seconds to minutes from birth to the end of the first 4 weeks of life
Paroxysmal alterations in behaviour
Paroxysmal alterations in autonomic function (apnea)
Focal: Altered activity in one neuronal population
Multifocal: Asynchronous migration of depolarization
Focal: Altered activity in one neuronal population
Generalized (rare): Altered activity propagating to the cortex of both hemispheres
Focal: Altered activity in one neuronal population
Rapid jerks of upper extremity flexor muscles
Multifocal: Asynchronous migration of depolarization
Asynchronous twitching of multiple muscle groups
Generalized (rare): Altered activity propagating to the cortex of both hemispheres
Bilateral jerks of upper & sometimes lower limb flexors
Paroxysmal alterations in motor function (ocular, oral-buccal-lingual)
Slow, rhythmic jerks in muscles of the face, extremities, or axial structures unilaterally
Time- synchronized EEG seizure activity
Sustained limb posturing or asymmetric posturing of axial structures
Sustained extension & flexion of upper extremities with extension of lower extremities
↑ Susceptibility to seizures later in life
Neurodevelopmental dysfunction in learning, memory & cognition
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published Mar 14 2025 on www.thecalgaryguide.com")
Tonsillitis Pathogenesis and clinical findings

Group A Streptococci (GAS) (most common bacteria)
Group B, C & G Strep,
Fusobacterium necrophorum
Age 5-15 (tonsils have ↑ role in immune function at this age)
Tonsillitis
Inflammation of the tonsils
Infectious agent exposure
Susceptible host Pathogen colonizes the oropharynx
Acute suppurative disease
Immune cells release proinflammatory cytokines & antibodies
Inflammatory mediators ↑ vascular permeability of tonsils
Leakage of protein & fluid into surrounding tissue
Regional nodes receive ↑ lymph
Enlarged anterior cervical nodes
Sinusitis**
Pharyngitis**
Local spread of pathogen
Acute otitis media**
Pneumonia**
Cervical lymphadenitis
Bacteria spread from
tonsils into lymphatic system & bloodstream
Bacteremia
F. necrophorum
invades lateral pharyngeal space & soft tissue in neck
Thrombosis forms in peritonsillar vein
Thrombosis extends into internal jugular vein
Lemierre’s syndrome
Systemic inflammatory cytokines disrupt hypothalamic regulation
Fever
Additional immune cells are recruited to facilitate immune response
Macrophages phagocytize pathogen
Bacteria invade distant tissue & elicit local inflammatory response
Hepatitis Osteomyelitis
Infective endocarditis
Bacteria illicit systemic response
Sepsis
Tonsillar tissue become swollen & irritated
Tonsillar hypertrophy
Localized collection of pus forms
Immune cells cause inadvertent cellular injury & hemolysis
Palatal petechiae
Meningitis
Products of immune response & cellular debris are deposited into tonsillar tissue
Peritonsillar or Tonsillar retropharyngeal abscess** exudate
** See corresponding Calgary Guide slide
Toxin-mediated disease
Bacteria release exotoxins into bloodstream
Inflammatory mediators & cytokines are overactivated (cytokine storm)
Skin has local inflammatory response
Toxic shock syndrome**
Scarlet fever**
Post-infectious disease
Antibodies to GAS cross react with host tissue
Acute rheumatic fever** Post-strep glomerulonephritis
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published Nov 5, 2018; updated Mar 14, 2025 on www.thecalgaryguide.com")
Tu chung Fallot

Chuyen vi Dai dong mach

Febrile Seizures
 activate host immune system
Immune cells release pro-inflammatory interleukin (IL)-1β, IL-6 & TNF-α cytokines that reach the central nervous system
Viral infections (primarily HHV-6 & H1N1) activate host immune system
Immature nervous system in children between 6 Family history of febrile seizures months & 5 years old (peak 12-18 months old) (genetic predisposition)
Hypothalamus releases prostaglandin E2 (PGE2) to ↑ body temperature
Fever reaches ≥380C
↑ Voltage gated Na+ & Ca2+ channel excitability
↑ Susceptibility to fevers
Fever ↑ metabolic rate & ↑ oxygen demand
↑ Respiratory rate
Hyperventilation ↑ volume of CO2 exhaled
↓ Serum CO2 leads to respiratory alkalosis
Inherited mutations in genes encoding the GABAA receptor subunit
↓ GABAA receptor expression
↓ GABAergic inhibition
Inherited mutations in genes encoding the Na+ channels
IL-1β ↑ glutamate release & ↓ GABAergic inhibition
Pro-inflammatory mediators ↑ sensitivity of N-methyl-D-aspartate (NMDA) (glutamate) receptors on medial temporal lobe neurons
Imbalances between excitatory glutamate & inhibitory gamma- aminobutyric acid (GABA) neurotransmission
↑ Neuronal Na+ channel activity
Neurons become more excitable & generate action potentials more readily
Large groups of neurons fire simultaneously (synchronize) in the medial temporal lobe (hippocampus) Activity spreads to other subcortical or cortical brain regions via synapses & gap junctions Excessive excitatory signaling disrupts normal brain activity
Authors: Merry Faye Graff Catherine Beyak, Dasha Mori Reviewers: Michelle J. Chen, Calvin Howard Emily J. Doucette, Gary Klein* Jean K. Mah* * MD at time of publication
Simple Febrile Seizure
Complex Febrile Seizure
Focal seizure, associated with a fever, that lasts > 15 minutes or has multiple recurrences in a 24-hour period
Generalized seizure, associated with a fever, that lasts < 15 minutes & does not recur in a 24-hour period
Generalized (bilateral cortical involvement) tonic-clonic seizure
Temporary post-ictal Temporary post-ictal weakness or
↓ Threshold for future febrile seizures
Prolonged (> 30 mins) or recurrent seizures without full return to consciousness in between (Febrile status epilepticus)
Focal (localized) seizures
Damages to neuronal circuits in affected regions
drowsiness or confusion
**See corresponding Calgary Guide slide
paralysis (Todd’s paralysis)
Epilepsy**
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complications
Published Jan 21, 2019; updated Mar 20, 2025 on www.thecalgaryguide.com")
Leucemia Linfatica Cronica

Angina Stabile

Polymyalgia Rheumatica
 is s9ll poorly understood. PMR is a rheuma9c condi9on characterized
Author: Anna Bobyn Reviewers: Anika Zaman, Kevin Zhan Luiza Radu, Emily J. Doucette Glen Hazlewood* *MD at time of publication
by pain and s9ffness around the neck, shoulder, and pelvic girdle.
Non-Modifiable factors
Assigned female at birth:
3♀ : 1♂
Environmental factors
Age-Related Changes: PMR almost exclusively affects people >50
Polymorphisms:
(eg. HLA-DR1, Interleukin (IL)-1, IL-6, TNF⍺)
Infectious Triggers: Epstein-Barr virus, Mycoplasma pneumoniae, parvovirus
Link to Vaccina=ons:
COVID-19, influenza, respiratory syncy=al virus
Innate immunity involvement: Macrophages & dendri=c cells recognize & respond to unknown an=gen
Immune cells ↑ production of cytokines (IL-6, IFN-γ, TNF-α)
Adap=ve immunity involvement: No strong evidence of autoan=body role
Early CD4+ T cell ac=va=on & infiltra=on of T cells into vascular =ssue
Subclinical vascular inflammation, particularly of medium & large arteries
↑ Vascular Endothelial Growth Factor & micro-vasculariza=on
Vascular dysfunction, ischemia, edema, & sensitization of pain receptors
Systemic inflamma=on
Local inflamma=on of synovial membrane, joint bursae, & tendon sheaths
B cell dysregula=on (↓ naïve B cells, ↑ memory B cells)
↑ Serum inflammatory
markers (CRP & ESR)
↑ Risk of cardiovascular
events (heart attack, stroke)
Low-grade fever (37.2- 38°C)
↓ Passive & ac>ve range of mo>on
Bilateral morning stiffness lasting >30 minutes
Fa>gue & malaise
Muscle pain in proximal joints
(shoulder & pelvic girdle)
Development of Giant Cell Arteri>s**
(15-20% of PMR pa>ents)
**See corresponding Calgary Guide slide
Legend:
Pathophysiology
Mechanism
Sign/Symptom/Lab Finding
Complica=ons
Published Mar 23, 2025 on www.thecalgaryguide.com")
Considerations anesthesiques pendant la grossesse

Benzodiazepine

Leukemia Mieloid Akut
: Patogenesis & presentasi klinis")
HIV Pathogenesis and Clinical Findings

Perinatal transmission during
pregnancy, childbirth or breast feeding
Direct contact of patient bloodstream or mucous membranes with HIV-1 infected blood, semen, rectal fluids, vaginal
discharge or breast milk (HIV-2 is transmitted in similar fashion but is less common & more likely to be seen in West Africa)
Authors:
Taylor Krawec
Ishjot Litt, Naima Riaz
Reviewers:
Candace Chan, Shahab Marzoughi
Emily J. Doucette, Sarah Smith*
* MD at time of publication
Glycoproteins in HIV viral envelope
bind to CD4 & chemokine receptors
on CD4+ T-cells & other immune cells
Viral & host cell
membranes fuse
Viral single-stranded RNA
(ssRNA) genome & enzymes
are released into the host cell
Host CD4+ cells are destroyed via direct cytotoxic
effects & immune cell-mediated cytotoxicity
CD4+ cell count ↓
Virions are disseminated throughout host
↑ Viral load
Viral proteins are transcribed
& assembled into virions
Virions bud off host
cell membrane
ssRNA is transcribed into proviral
DNA by viral reverse transcriptase
using host nucleotides
Infected cells revert to memory
state & establish latent HIV reservoir
Chronic HIV infection
Host is unable to keep up
with loss of CD4+ cells
Proviral DNA enters
nucleus & integrates
into host DNA
Acute HIV infection
Infected immune
cells release
proinflammatory
cytokines
Antibodies to HIV are generated
in attempt to control infection
(2-12 weeks post infection)
Positive antibody-antigen immunoassay
Cytokines disrupt
hypothalamic
temperature
regulation
Systemic immune
system activation
results in ↑
metabolic demand
Cytokines
extravasate
to the dermis
Fever Maculopapular
Chills
Fatigue
rash
Legend: Pathophysiology Mechanism
Sign/Symptom/Lab Finding Viral load ↓ &
stabilizes around
set-point viral load
Oral hairy leukoplakia
(oral infection with
Epstein-Barr Virus)
Complications
Viral load ↑ & CD4+ cell
count ↓ steadily over several
years without treatment
CD4+ cell count ↓
to 200-500 cells/μL
Impaired host immune response
↑ risk of opportunistic infections
Oropharyngeal
or vulvovaginal
candidiasis
Severe manifestations of
common infections (ex.
herpes simplex, HPV)
Published April 22, 2025 on www.thecalgaryguide.com
Virus evades immune
detection to allow
ongoing viral
replication, chronic
immune activation &
↓ thymic function
Vague viral
symptoms (ex.
fatigue, weight loss,
lymphadenopathy)
CD4+ count ↓
to < 200 cells/μL
Acquired
immunodeficiency
syndrome (AIDS)")
Varenicline

Activated α4β2
nicotinic receptors on
cerebral blood vessels
causes vasodilation
Activation of
gastrointestinal α4β2
nicotinic receptors
Excessive vasodilation
↑ intracranial pressure
Authors:
Trevor Low
Aliaksandr Savin
Reviewers:
Andrew Wu
Luiza Radu
Emily J. Doucette
Joel Tappay*
*MD at time
of publication
Headache
Varenicline binds central α4β2
nicotinic acetylcholine
receptors with higher affinity
than exogenous nicotine
↓ Binding of exogenous
nicotine to α4β2
nicotinic receptors
Ventral tegmental area of
midbrain releases ↓ dopamine
Nucleus accumbens of
ventral striatum receives ↓
dopaminergic signaling
↓ Sensations of pleasure or
euphoria with nicotine use
↓ Satisfaction from
nicotine consumption
↓ Positive reinforcement
with nicotine use
Legend: Selective activation of α4β2
nicotinic acetylcholine receptors
Partial activation of central
α4β2 nicotinic receptors
Partial but sustained release of
dopamine from ventral tegmental
area at a level lower than nicotine
Sustained partial release prevents
phasic dopamine signaling to the
nucleus accumbens
Tonic dopamine relieves
withdrawal symptoms
↓ Cravings & desire to
consume nicotine to relieve
withdrawal symptoms
↓ Nicotine use
(nicotine cessation)
Acetylcholine signaling to area
postrema (vomiting center)
↑ Basal levels of dopamine
disrupts signaling in nuclei
responsible for arousal &
sleep initiation
↑ Dopaminergic
signaling in the limbic
system during sleep
↑ Activation of amygdala
(emotions) & hippocampus
(memories) during sleep
↑ Intracranial pressure
activates meningeal
nociceptors
Nausea &
vomiting
↑ Wakefulness
Delay in
sleep onset
Fragmentation
of sleep cycles
↑ Activation causes
premature or
prolonged REM
episodes
Dopamine activates
cholinergic neurons in nuclei
responsible for rapid-eye-
movement (REM) sleep
Insomnia
Abnormal,
vivid &
emotional
dreams
Pathophysiology Mechanism
Pharmacologic effect Adverse effect
Published April 22, 2025 on www.thecalgaryguide.com")
Acute Infectious Mononucleosis
, Cytomegalovirus
(CMV), human immunodeficiency
virus (HIV), etc) through saliva
(most commonly in adolescents
& young adults aged 15-24)
Virus infects epithelial
cells of the oropharynx
Virus infects & immortalizes
circulating B lymphocytes
Virus replicates in infected B-cells
Immune cell proliferation
in lymphatic system
(primarily lymph nodes & spleen)
Infected B-cells enter
systemic circulation
Systemic inflammatory
response activated
Inflammation &
edema of sinuses
Bilateral periorbital &/or palpebral edema
Pharyngitis**
(often with grey/white
exudative secretions)
Palatal petechiae
Blood vessel damage
in the soft palate
Edema of soft palate & tonsils
Airway obstruction
↑ Immunoglobulin M (IgM) antibodies
to EBV viral capsid antigen (VCA) Positive IgM-VCA
Immortalized B-cells
produce ↑ antibodies
Production of heterophile antibodies
(weakly reactive & non-specific)
Positive monospot (heterophile antibody)
test (↓ sensitivity in children <4 years)
Virus may remain dormant in B-cells
Latent infection with periodic reactivation
Generalized lymphadenopathy (enlarged lymph nodes)
Massive cervical, mediastinal or hilar lymphadenopathy
Posterior cervical
lymphadenopathy
Platelets sequestered (trapped) within spleen &
overactive spleen (hypersplenism) discards ↑ platelets
Thrombocytopenia
(↓ platelets)
Weak reticular tissue in the spleen stretches
Splenomegaly Splenic rupture
& becomes more susceptible to injury
Inflammatory response ↑ energy demand
Fatigue
Longstanding impacts from
unknown mechanism Chronic Fatigue Syndrome
Inflammatory cytokines
released into circulation
↑ Thermo-regulatory
set-point at hypothalamus Fever
Lymphocytosis (↑ lymphocytes)
(markedly CD8+ T-cells & NK cells) Immune cells infiltrate the liver Hepatomegaly
↑ Aminotransferases
Leukocytosis (↑ leukocytes)
CD8+ T-cells & NK cells
indirectly damage hepatocytes
↑ Bilirubin & jaundice**
Systemic immune response &/or
antibiotic hypersensitivity reaction
CD8+ T-cells
respond to virus
Atypical lymphocytes
on blood smear
Generalized maculopapular rash
Authors:
Ayden Hansen, Griselle Leon
Reviewers:
Charissa Chen, Emily J. Doucette
Danielle Nelson*
* MD at time of publication
Published Sept 3, 2015; updated Apr 22, 2025 on www.thecalgaryguide.com
Pathophysiology Mechanism
Sign/Symptom/Lab Finding Complications")
Trachoma

Mechanical vectors: Eye-seeking
flies (e.g. Musca sorbens)
Chlamydia trachomatis infection (serotype A, B & C)
Trachoma
Chronic keratoconjunctivitis (inflammation of the conjunctiva & cornea) caused by recurrent infection with C. trachomatis
Active Phase
Bacteria infects the conjunctival epithelial cells
Redness, irritation & mucopurulent discharge
Immune response triggers inflammation of the upper tarsal conjunctiva
Herbert’s pits (small, sunken scars at the limbus (border
between cornea & sclera) from healed follicles
Corneal lesions
Pannus (invasion by superficial vessels) at the limbus
Lymphoid follicles consisting mainly of B & T cells form in the upper tarsal conjunctiva
Trachomatous inflammation - follicular: Presence of five or
more white or yellow follicles, each 0.5 mm or bigger in size
Inflammation leads to papillae formation with dilated blood vessels, infiltration
of lymphocytes, plasma cells & neutrophils & proliferation of epithelial cells
Small, raised bumps with a central blood vessel in the upper conjunctiva
Papillary hypertrophy causes conjunctival thickening & opaqueness Trachomatous inflammation - intense: Papillae obscure deep tarsal blood vessels
Repeated re-infection or untreated infections
Cicatricial (scarring) Phase
Proliferation of fibroblasts & deposition of collagen leads to scarring (fibrosis)
Trachomatous scarring: White, horizontal lines or bands on upper tarsal conjunctiva
Scarring blocks
meibomian gland ducts
Conjunctival epithelium atrophy
& goblet cell destruction Subconjunctival
fibrosis
Entropion (eyelid turns inward)
Cornea ulcers
& scarring
↑ Tear evaporation
↓ Tear production
Scar contraction distorts
upper tarsal plate
Dry eye
Trachomatous trichiasis
(eyelashes grow inward
toward the eye)
Corneal
abrasion
Corneal opacity
& blindness
Published Sept, 2 2015; updated May 7, 2025 on www.thecalgaryguide.com
Legend: Pathophysiology Mechanism
Sign/Symptom/Lab Finding Complications")
Wigger's Diagram

Atrial myocytes
repolarize and relax
Septum
depolarizes
Ventricular myocytes
depolarize and contract
(isovolumetric
contraction)
Ventricular myocytes
repolarize and relax
(isovolumetric relaxation)
↓ Left atrial
pressure
Left atrium squeezes blood
into left ventricle through
the open mitral valve
ECG: P-wave**
Left atria fills
with blood from
ECG: Q-wave ECG: R-wave ECG: T-wave
↑ Left
pulmonary
ventricular
circulation
pressure
Atrial pressure:
‘x’ descent
↓ Left ventricular
pressure
↑ Left atrial
pressure
↑ Left atrial
pressure
↑ Left ventricular
volume
Left ventricular pressure >
left atrial pressure
Mitral valve
bulges into
left atrium
Left ventricular
pressure >
aortic pressure
Atrial pressure:
‘v’ wave
Atrial
pressure:
‘a’ wave
↑ Left ventricular
pressure
Mitral valve closes
Atrial pressure:
‘c’ wave
Aortic valve opens
First heart sound (S1)
Left ventricle squeezes
blood into aorta (ejection)
↑ Aortic pressure Legend: Pathophysiology Mechanism
Left atrial pressure >
left ventricular pressure
Mitral valve opens
Rapid inflow of blood
into left ventricle from
left atria
↓ Left ventricular
volume
↑ Left
ventricular
volume
↓ Left atrial
pressure
↓ Left ventricular
pressure
Atrial pressure:
‘y’ descent
Aortic pressure >
left ventricular pressure
Aortic valve closes
Second heart sound (S2)
**See slide on PHYSIOLOGY OF THE NORMAL ECG WAVEFORM (LEAD II)
Author:
Nathan Archibald
Reviewers:
Stephanie Happ
Emily Kuervers
Sergio F. Sharif
Angela Kealey*
* MD at time of publication
Sign/Symptom/Lab Finding Complications
Published May 19, 2025 on www.thecalgaryguide.com")
PROP1-Related Combined Pituitary Hormone Deficiency
 from
lactotrophs
Impaired mammary
gland stimulation
prevents adequate
milk production
PROP1-Related Combined Pituitary Hormone Deficiency: Pathogenesis and clinical findings
Consanguinity
Inherited PROP1
mutation
Family history of
PROP1 mutation
Sporadic PROP1
mutation
Autosomal recessive or sporadic
mutation of PROP1 on chromosome
5q35 (various mutations identified;
ex. frameshift, insertion, deletion)
activation ability of pituitary-
specific transcription factor
encoded by PROP1 gene
PROP1-Related Combined Pituitary Hormone Deficiency
Genetic disorder resulting in combined pituitary hormone deficiency (CPHD) characterized by a
deficiency in growth hormone (GH) & ≥ 1 additional anterior pituitary hormone
↓ Follicle stimulating
hormone (FSH) from
somatotrophs
↓Luteinizing
hormone (LH) from
gonadotrophs
↓ GH from
gonadotrophs
↓ Thyroid stimulating
hormone (TSH) from
thyrotrophs
↓ Adrenocorticotropic
hormone (ACTH) from
corticotrophs
↓ Gonad
stimulation
↓ Sex hormone
production
Hypogonadotropic hypogonadism
(hypogonadism due to problem at
level of hypothalamus or pituitary)
↓ Secretion
of insulin-like
growth factor
1 (primarily
↓ Stimulation
of growth at
↓ Thyroid
stimulation
↓ Stimulation of
adrenal glands
epiphyseal
plate of long
bones
↓ Thyroid
by liver) ↓ Serum
hormone levels
cortisol
Absent secondary
sexual characteristics
Delayed or
absent puberty
Short stature
Secondary/central
hypothyroidism**
Secondary/central
adrenal insufficiency**
Postpartum
lactation failure
Authors:
Juliette Eshleman
Taylor Krawec
Reviewers:
Annie Pham
Emily J. Doucette
Danielle Nelson*
*MD at time of publication
Impaired glucose regulation
(↓ gluconeogenesis, ↓
glycogenolysis & ↓ lipolysis)
Altered stress
response to
illness or injury
↓ Vascular tone &
catecholamine response
(fight-or-flight response)
Impaired
glucose
metabolism
Neonatal hypoglycemia**
Adrenal crisis
Hypotension
Weakness
Fatigue
**See corresponding Calgary Guide slide
Legend: Pathophysiology Published May 19, 2025 on www.thecalgaryguide.com
Infertility
Mechanism
Sign/Symptom/Lab Finding Complications")
Brachial Plexus Injury

Fetal complications during labor &
delivery** (ex. shoulder dystocia)
Author:
Merry Faye Graff
Reviewers:
Taylor Krawec
Emily J. Doucette
Jean K. Mah*
*Indicates MD at time
of publication
Upward traction of arm with forced
widening of scapulohumeral angle
(lower brachial plexus injured first)
Downward traction of arm with forced
widening of shoulder-neck angle
(upper brachial plexus injured first)
Surgical
trauma
Direct trauma (i.e. gunshot
wound, penetrating injuries)
Pancoast
tumour**
Nerves are stretched or torn
Nerves are severed or crushed
Brachial Plexus Injury
Damage to brachial plexus nerves that may cause weakness, decreased
sensation or loss of movement in the shoulder, arm or hand
Injury or compression demyelinates nerves
without causing axonal injury (neuropraxia)
Injury demyelinates nerves &
severs axons (axonotmesis)
Injury completely transects
nerves (neurotmesis)
Demyelination slows & disrupts
nerve signal conduction
Basal lamina tubes (axon
scaffolds) are preserved
Basal lamina
tubes are severed
Nerves are torn proximal to attachment
at spine (post-ganglionic rupture)
Nerve roots are torn from spinal
cord (pre-ganglionic avulsion)
↓ Efferent
signals
(motor)
↓ Afferent
signals
(sensory)
Inflammatory mediators
sensitize primary
afferent neurons
Abnormal ectopic
firing of nerves at or
near nerve lesion
Neuroma (benign
growth or thickening of
nerve tissue at lesion)
during healing
Breakdown of axon(s) &
myelin distal to nerve lesion
(Wallerian degeneration)
Connection to
central nervous
system (CNS) lost
Muscle weakness
or loss of function
in affected
myotomes
Paresthesia
(numbness
or tingling)
in affected
dermatomes
Hyperalgesia (extreme
sensitivity to touch)
Neuropathic pain
(burning, stabbing,
electric-like pain)
Neuroma blocks distal
nerve conduction
No conduction in affected
nerves or nerve roots
Repeated pain
signals sensitize CNS
Afferent signals are
not transduced
Efferent signals are
not transduced
Paravertebral sympathetic chain
is disrupted (if C8 & T1 involved)
Central sensitization
Numbness
Paralysis
Horner’s syndrome**
Shortening & hardening
of muscles & tendons
↓ Muscle use
Widespread pain
Allodynia (normal touch
perceived as painful)
Contractures
Muscle atrophy
Note: Specific signs &
symptoms vary based
on nerves affected.
Complications
Published May 14, 2025 on www.thecalgaryguide.com
Pathophysiology Mechanism
Sign/Symptom/Lab Finding")
Acetylcholinesterase inhibitors

Reversible inhibition of
acetylcholinesterase activity
Cholinergic effects
↓ Breakdown of acetylcholine
in the synaptic cleft
Acetylcholine accumulates
in the synaptic cleft
Excessive
accumulation of
acetylcholine in
synapses
Acetylcholine competitively
displaces non-depolarizing
neuromuscular blockers**
from nicotinic receptors in
the neuromuscular junction
↑ Acetylcholine binding to
pre-synaptic nicotinic
receptors on neurons
↑ acetylcholine binding to
post-synaptic nicotinic
receptors on muscles
Pre-synaptic nicotinic
receptor depolarizes
terminal membrane
Post-synaptic nicotinic
receptor activation results
in action potential in muscle
↑ Vesicular release of
acetylcholine by voltage-
gated calcium signaling
Muscle contraction
occurs via excitation-
contraction coupling
Legend: Neurotransmission is
sustained during
Reversal of
neuromuscular block
repeated stimulation
**See corresponding Calgary Guide slide
Mechanism
↑ Activation
of muscarinic
receptors in
myocardium
↑ Activation of
muscarinic receptors
in endothelial tissue
Endothelial cells
release nitric oxide
Acetylcholine-
activated potassium
channels open Nitrous oxide causes
vascular smooth
muscle relaxation
Sinoatrial node of
the heart becomes
hyperpolarized
Vasodilation
↓ Vascular resistance
Bradycardia
Hypotension
Depolarization
block of
nicotinic
receptors in
muscles
Flaccid skeletal
muscle paralysis
Respiratory muscle
paralysis & failure
Bronchospasm
& excess
secretions
Prolonged period of
inadequate respiration
↓ Oxygen
saturation
↑ Carbon
dioxide
↑ Activation of gastrointestinal
muscarinic receptors
Stimulation of
gastrointestinal
muscles
Activation
of salivary
muscarinic
receptors
↑ Amplitude &
duration of gut
peristalsis
↑ Activity of
salivary glands
Overstimulation
of the enteric
nervous system
Nausea ± vomiting
↑ Salivation
Pathophysiology Sign/Symptom/Lab Finding Complications
Published May 31, 2025 on www.thecalgaryguide.com")
Meckels Diverticulum
 forms & is trapped in diverticulum
Foreign body enters diverticulum & becomes trapped inside
Incomplete vitelline duct obliteration (~5-7 weeks gestation)
Diverticulum sags into
Intussusception (section
bowel lumen & acts as lead
of intestine slides into
Meckel’s Diverticulum
point for intussusception
adjacent region)
Persistent remnant of vitelline duct containing all 3 layers of
ileal mucosa (asymptomatic in 96-98% of cases)
Diverticulum remains
attached to umbilicus by
persistent fibrous band
Volvulus (loop of ileum twists
around diverticulum)
Ectopic tissue present
within diverticulum
Diverticulum goes
through abdominal
wall defect
Littre’s Hernia
(diverticulum
in hernial sac)
Diverticulum
becomes
incarcerated
Ectopic gastric mucosa
produces acid secretions
Ectopic pancreatic tissue
secretes digestive enzymes
Ileal mucosa adjacent to
diverticulum become ulcerated
Inflammatory mediators
disrupt hypothalamic
regulation
Small bowel
becomes
obstructed
Perforation of
diverticulum
Brisk painless
bleeding from
small bowel
Chronic
inflammation
& ulceration
Fever
Nausea/
vomiting
Small bowel
necrosis
Hematochezia
(bright red blood
per rectum)
↓ Effective
arterial blood
volume
Mucosa undergoes
dysplastic & neoplastic
transformation
Complete
erosion through
ileal mucosa
Small bowel
perforation
Hypotension
Neuroendocrine
tumour or other
cancer
Diverticulum becomes
blocked & triggers
local inflammatory
response
Meckel’s Diverticulitis
Inflammation of Meckel’s
Diverticulum
Diverticulum is
distended due to
inflammation
Visceral afferent
nerves are stimulated
by intestinal stretch
Abdominal pain
Chronic painless
bleeding from
small bowel
Melena (partially
digested blood in
stool)
Iron-deficiency
anemia
Authors:
Taylor Krawec, Merry Faye Graff
Reviewers:
Emily J. Doucette, Sylvain Coderre*
* Indicates MD at time of publication
Legend: Pathophysiology Mechanism
Sign/Symptom/Lab Finding Complications
Intestinal contents
leak into peritoneum
Peritonitis
Systemic inflammatory
response to peritonitis
Published May 31, 2025 on www.thecalgaryguide.com
Sepsis")
SGLT2 Inhibitors
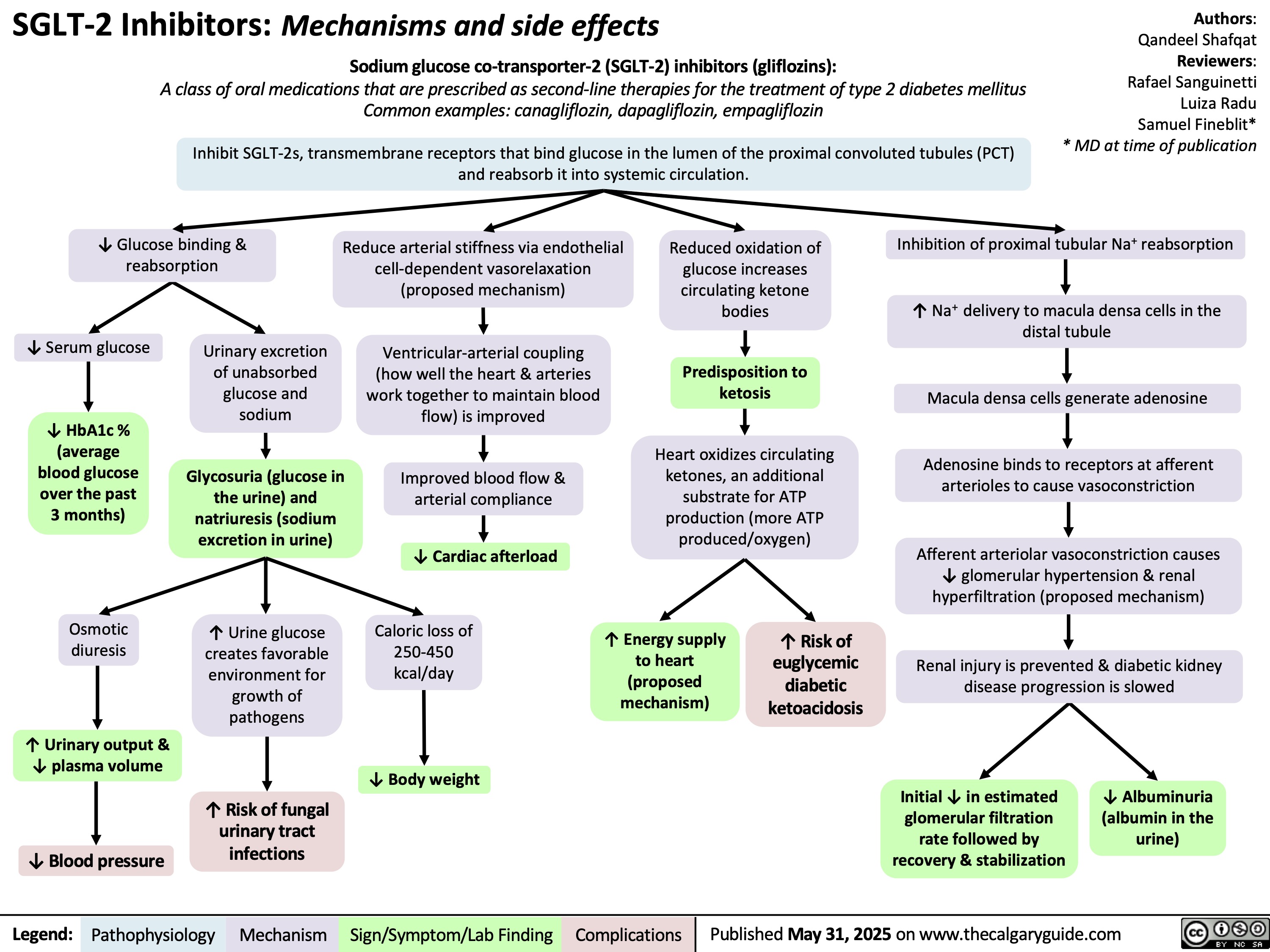
Vaccine Mediated Immunity Comparing Vaccine Subtypes
,
Varicella, Rotavirus, Influenza
COVID-19
Serial passage in sub-optimal
culture conditions weaken
pathogen
Pathogen maintains ability to
slowly replicate (attenuated)
Vaccine components (live
attenuated pathogen,
adjuvants, stabilizers,
preservatives) are
formulated & administered
Attenuated pathogen enters
host cells & replicates,
mimicking natural infection
Identify target protein of specific pathogen
RNA polymerase transcribes DNA for target
protein into mRNA (in vitro transcription)
mRNA is purified & concentrated
mRNA is encapsulated in lipid
nanoparticles to prevent degradation
Vaccine components (mRNA, lipid
nanoparticles, adjuvants, preservatives)
are formulated & administered
Host cells take up mRNA & synthesize the
antigenic protein
Diphtheria, Tetanus, Pertussis,
Pneumococcus, Meningococcus,
Haemophilus influenzae type b, Influenza,
Hepatitis B, Human Papilloma Virus
Polio (IPV), Hepatitis A
Antigen-presenting cells (APCs) take up
intracellular & extracellular antigens
Infected cells & APCs present intracellular
antigens to CD8+ T cells via major
histocompatibility complex-I (MHC-I) molecules
Robust cell-mediated immunity
↑ Lymphocyte counts
Legend: Pathophysiology Mechanism
Sign/Symptom/Lab Finding Complications
Authors:
Tanis Orsetti
Reviewers:
Jessica Hammal
Emily J. Doucette
James D. Kellner*
* MD at time of publication
Inactivated Whole Virus Vaccines
Identify target protein of specific
pathogen (antigenic protein)
Pathogen is inactivated using
chemicals/detergents/heat
Large quantity of antigenic protein
produced in a laboratory setting
Pathogen is unable
to replicate
Vaccine components (pathogen, adjuvants, stabilizers,
preservatives) are formulated & administered
APCs take up
extracellular antigens
APCs present extracellular antigens to CD4+
cells via major histocompatibility complex-II
(MHC-II) molecules
Activated CD4+ T helper cells stimulate B lymphocytes
B lymphocytes produce antibodies specific to pathogen
Robust humoral immunity
↑ Pathogen-specific antibody titers
Published May 31, 2025 on www.thecalgaryguide.com")
Chronic Mesenteric Ischemia

HLA-B52
allele &
other genetic
factors
Dyslipidemia
(abnormal blood
lipid levels)
Various etiologies (trauma,
coagulopathies, inflammatory
bowel disease, paraneoplastic
syndrome, surgery, portal
hypertension, cardiac)
High blood
pressure
Fibromuscular
dysplasia
(systemic vascular
disease)
Takayasu’s
arteritis (systemic
inflammatory
artery disease)
Median arcuate
ligament syndrome
Atherosclerosis
(plaque deposition in
the subendothelium;
cause of 90% of chronic
mesenteric ischemia
cases)
Arterial wall
thickening
Growth & hardening of
arterial wall
Compression of
celiac trunk by
the median
arcuate ligament
Thromboembolism
(clot formation)
Turbulent blood flow at
the level of the lesion
Mesenteric artery
stenosis (narrowing)
Abdominal bruit
(audible whoosh due to
artery narrowing)
Mucosal and
submucosal injury
& inflammation
Insufficient blood supply to the
gastrointestinal tract to meet
demand (i.e., “intestinal angina”)
Diarrhea ±
constipation
Gut motility
dysfunction
Activation of the
vomiting centre
in the brain
Chronic Mesenteric Ischemia
Chronic hypoperfusion of the bowel due to
(typically) multivessel mesenteric artery
stenosis or occlusion for >3 months
Aortic
dissection
(tear)
Radiation
exposure
Vascular injury
Exposure of
subendothelium
& disruption of
blood flow
Mesenteric artery
occlusion (blockage)
Compensation by the
mesenteric vasculature
through expansion of
collateral circulation
Majority of
patients are
asymptomatic
Nausea &
vomiting
Detectable
stenosis/occlusion of
mesenteric arteries
Intake of food increases metabolic
demand of bowel beyond levels
accommodated by strained blood supply
Mesenteric artery stenosis/
occlusion with medical
imaging (ie. CT Angiogram)
Postprandial abdominal discomfort
(typically epigastric pain ~30 minutes
following a meal, lasting for 1-4 hours)
Published May 19, 2025 on www.thecalgaryguide.com
Author: Liam Fitzgerald
Reviewers: Sophia Khan
Shahab Marzoughi
Sergio F. Sharif
Sylvain P. Coderre*
* MD at time of publication
Tobacco use
Buerger’s disease
(inflammatory
artery disease)
Formation of focal
inflammatory blood
clots
Weight loss
(>20 lbs)
Food aversion, smaller
meals, avoidance of fatty
foods (caloric restriction)
Legend: Pathophysiology Mechanism
Sign/Symptom/Lab Finding Complications")
Tetralogy of Fallot

Maternal exposure
to alcohol or
anticonvulsants
Maternal infections (e.g., hepatitis
B virus, coxsackievirus-B, human
cytomegalovirus virus, rubella)
Variable interaction between genetic associations and environmental risk factors
Antero-cephalad deviation of conal septum (muscular structure that forms the boundary
between right & left ventricular outflow tract during embryological development)
Tetralogy of Fallot
Cyanotic congenital heart disease with a combination of four cardiac defects
Ventricular septal
defect (VSD)**
Overriding aortic root
(aorta above VSD rather than left ventricle)
Right ventricular outflow
tract obstruction (RVOTO)
Right Ventricular
Hypertrophy
Hole in septum
Absent or reduced
Stressors (eg., crying,
between ventricles Right ventricle of
Oxygenated blood from the left
pulmonary valve
defecation, fever,
heart pumps
ventricle and deoxygenated blood
closure (P2)
dehydration) trigger acute
harder to push
from right ventricle flows into aorta
↑ Left ventricular
pressure
infundibular spasm (spasm of
blood through
conus arteriosus)
RVOTO
Chronic volume
overload
L-R ventricular
shunting
Single, loud S2
Spasm ↓ blood flow through
right ventricular outflow tract
Aortic
Dilated
aortic root
regurgitation** Hypercyanois or “tet” spells
Turbulent blood
flow from left
ventricle through
the hole in septum
to right ventricle
Turbulent diastolic
backflow through
incompetent aortic
valve
Deoxygenated
blood flows
through aorta to
systemic circulation
↓ O2 Saturation
Chemoreceptors in the carotid & aortic bodies
stimulate respiratory centre in the medulla oblongata
Harsh, holosystolic
murmur
Diastolic
decrescendo
murmur
↑ Deoxygenated blood
entering systemic circulation
↑ Catecholamine release from adrenal
medulla stimulating ↑ contractility of heart
↓ Blood flow
across VSD
↑ contraction against outflow obstruction
↑ pulmonary vascular resistance
↓ Intensity or
disappearance
of holosystolic
murmur
Peripheral & central
chemoreceptors detect ↓ arterial
O2 content, triggering brainstem
respiratory centre to stimulate ↑
work & rate of breathing
↓ Pulmonary blood flow
↑ Turbulence of blood
flow through RVOTO
↑ R-L shunting
↑ Deoxygenated hemoglobin (dark
red) relative to oxygenated hemoglobin
(bright red), reflecting more blue light
Cyanosis (blue discoloration of the
mucous membranes, nail beds, skin)
Paroxysmal
hyperpnea (rapid
and deep
respirations)
Irritability
and crying
Dyspnea
(shortness of
breath)
Poor feeding
Accentuation of systolic
crescendo/decrescendo
murmur with harsh ejection
**See corresponding
Calgary Guide slides
Legend: Mechanism
Published Jul 8, 2015; updated Jun 16, 2025 on www.thecalgaryguide.com
Pathophysiology Sign/Symptom/Lab Finding Complications")
Heparin-Induced Thrombocytopenia

Platelets aggregate
within the bloodstream
↓ Free unbound
platelets in bloodstream
Mild Thrombocytopenia:
platelet count between
100 x 109/L - 150 x 109/L
Fewer platelets
available for thrombosis
(formation of a
pathological blood clot)
Self limiting with
no thrombotic risk
Legend: Heparin-Induced Thrombocytopenia (HIT): Pathogenesis and clinical findings
Obesity Hypertension Macrovascular disease Hyperlipidemia
Heparin (often unfractionated)
complexes with platelet factor 4 (PF4)
on the platelet surface, due to
heparin’s negative charge & PF4’s
affinity for long-chain polysaccharides
Immune-Mediated:
Type 2 (within 5-15 days
of heparin administration)
Anti-heparin/PF4
Immunoglobulin G
antibodies binds to
the heparin-PF4
complex in the blood
Splenic macrophages
remove platelet-
immunoglobulin G
complexes in the spleen
Thrombocytopenia:
Platelet count < 100 x
109/L (though < 20 x
109/L should prompt
consideration of
other diagnoses)
↑ Vascular inflammation & endothelial
dysfunction activate platelets
Authors:
Kelle Edgar, Hadi Hassan
Sergio F. Sharif
Reviewers:
Haotian Wang, Jessica Asgarpour
Yan Yu*, Lynn Savoie*
Kareem Jamani*
* MD at time of publication
Activated platelets
release PF4
Anti-heparin/PF4
antibodies activate
adjacent platelets
Activated
platelets release
inflammatory
mediators
Circulating anti-
heparin/PF4 antibodies
bind adjacent cellular
targets
Anti-heparin/PF4
antibodies bind
endogenous heparan
sulfate on endothelium
Activated
endothelium
releases
procoagulants
thrombin &
tissue factor
Activated
platelets release
procoagulants
(factors
promoting blood
clotting)
Free platelets are
consumed from
formation of thrombi
Hypercoagulable state
(↑ risk of thrombosis)
↓ Platelets available
to clot at different
area of the body
Thrombosis in
coronary, peripheral
or cerebral arteries
Bleeding (rare)
Myocardial
infarction
Acute
ischemia
Stroke
Inflammatory
mediators disrupt
hypothalamic body
thermoregulation
Fever/
chills
Inflammatory mediators
promote vasodilation of
arterioles under the skin
↑ Blood to
surface of skin
Flushing of skin
Venous thrombosis
in lower leg or lung
Deep vein
thrombosis
Pulmonary
embolism
Microthrombi occlude
vessels surrounding
injection site, limiting
nutrient supply
Necrosis (tissue death)
at heparin injection site
Published Aug 29, 2014; updated Jun 16, 2025 on www.thecalgaryguide.com
Pathophysiology Mechanism
Sign/Symptom/Lab Finding Complications")
Obesity Hypoventilation Syndrome
![Obesity Hypoventilation Syndrome: Pathogenesis and clinical findings
Obesity (BMI ≥ 30 kg/m2) risk factors: Poor eating patterns, sedentary lifestyle, genetic predisposition,
hypothyroidism, Cushing’s syndrome, socio-economic factors, age
Sleep-disordered breathing risk factors: Family history, tonsillar or adenoidal
hypertrophy, ↑ neck circumference, type 2 diabetes, HTN
Authors: Mohammad Omer
Mujtaba Siddique
Reviewers:
Ali Babwani
Luiza Radu
Jonathan Liu*
MD at time of publication*
↑ Adipose deposition
in abdomen
Abdominal fat pushes
against diaphragm
↑ Diaphragmatic
displacement
↑ Resistance to chest
wall expansion
↑Leptin resistance
High pressure
Pharyngeal
on upper airway
dilations unable
Secondary depression
↓ Chest wall
↓ Leptins ability to stimulate
↑ lung
to compensate
Narrowing of
(compromised function) of
Poor ventilation to
expansion
ventilation (mechanism unknown)
collapsibility
for weight
upper airways
respiratory system
lower lobes of lungs
↓ Tidal volume (air
that moves in/out of
lungs in a respiratory
cycle)
↑ Respiratory rate
↑ Chest wall thickness ↑ Leptin (a hormone released by
adipose tissues that controls hunger by
signaling fullness)
↑ Adipose
deposition near
upper airways
↑ Buildup of
edema in lower
extremities
↑ Respiratory
workload
↓ Chest wall
compliance (ability to
stretch)
↓ Leptin receptor
expression
↓ Leptin through
blood-brain barrier
↓ Pharyngeal space
Respiratory system is
unable to compensate to ↑
Fluid shifts from
demands
legs to neck during
sleep
Hypoventilation in sleep
↓ Ventilation (air exchange in lungs)
↑ PaCO₂ (partial pressure of arterial carbon
dioxide)
↑ Serum [H+]
↑ Serum [HCO3
-] by renal
reabsorption buffers [H+] rise
↓ PaO₂
(partial pressure of arterial oxygen)
Hypoxia (low
O₂ in tissue)
Higher PaCO₂ required to
reduce pH
↓ O₂ levels in alveoli triggers pulmonary
vessel vasoconstriction
PaCO₂ > 45
mmHG
Respiratory
acidosis
↓ response to CO₂ in central
chemoreceptors in brain
Pulmonary hypertension (high pressure in
pulmonary arteries)
↓ Neural drive
↓ Ventilatory responsiveness
) Right heart pumps against higher
pulmonary pressure leading to
cardiomyocyte hypertrophy
Cor pulmonale
(right-sided heart
failure)
Fatigue
Chronic hypercapnia
(↑ CO2 retention)
Pathophysiology Legend: Mechanism
Sign/Symptom/Lab Finding Complications
Morning headaches
Daytime lethargy
Published Jun 16, 2025 on www.thecalgaryguide.com](https://calgaryguide.ucalgary.ca/wp-content/uploads/2025/06/Obesity-Hypoventilation-Syndrome.jpg "Obesity Hypoventilation Syndrome: Pathogenesis and clinical findings
Obesity (BMI ≥ 30 kg/m2) risk factors: Poor eating patterns, sedentary lifestyle, genetic predisposition,
hypothyroidism, Cushing’s syndrome, socio-economic factors, age
Sleep-disordered breathing risk factors: Family history, tonsillar or adenoidal
hypertrophy, ↑ neck circumference, type 2 diabetes, HTN
Authors: Mohammad Omer
Mujtaba Siddique
Reviewers:
Ali Babwani
Luiza Radu
Jonathan Liu*
MD at time of publication*
↑ Adipose deposition
in abdomen
Abdominal fat pushes
against diaphragm
↑ Diaphragmatic
displacement
↑ Resistance to chest
wall expansion
↑Leptin resistance
High pressure
Pharyngeal
on upper airway
dilations unable
Secondary depression
↓ Chest wall
↓ Leptins ability to stimulate
↑ lung
to compensate
Narrowing of
(compromised function) of
Poor ventilation to
expansion
ventilation (mechanism unknown)
collapsibility
for weight
upper airways
respiratory system
lower lobes of lungs
↓ Tidal volume (air
that moves in/out of
lungs in a respiratory
cycle)
↑ Respiratory rate
↑ Chest wall thickness ↑ Leptin (a hormone released by
adipose tissues that controls hunger by
signaling fullness)
↑ Adipose
deposition near
upper airways
↑ Buildup of
edema in lower
extremities
↑ Respiratory
workload
↓ Chest wall
compliance (ability to
stretch)
↓ Leptin receptor
expression
↓ Leptin through
blood-brain barrier
↓ Pharyngeal space
Respiratory system is
unable to compensate to ↑
Fluid shifts from
demands
legs to neck during
sleep
Hypoventilation in sleep
↓ Ventilation (air exchange in lungs)
↑ PaCO₂ (partial pressure of arterial carbon
dioxide)
↑ Serum [H+]
↑ Serum [HCO3
-] by renal
reabsorption buffers [H+] rise
↓ PaO₂
(partial pressure of arterial oxygen)
Hypoxia (low
O₂ in tissue)
Higher PaCO₂ required to
reduce pH
↓ O₂ levels in alveoli triggers pulmonary
vessel vasoconstriction
PaCO₂ > 45
mmHG
Respiratory
acidosis
↓ response to CO₂ in central
chemoreceptors in brain
Pulmonary hypertension (high pressure in
pulmonary arteries)
↓ Neural drive
↓ Ventilatory responsiveness
) Right heart pumps against higher
pulmonary pressure leading to
cardiomyocyte hypertrophy
Cor pulmonale
(right-sided heart
failure)
Fatigue
Chronic hypercapnia
(↑ CO2 retention)
Pathophysiology Legend: Mechanism
Sign/Symptom/Lab Finding Complications
Morning headaches
Daytime lethargy
Published Jun 16, 2025 on www.thecalgaryguide.com")
Varicella Zoster Virus
 - Chicken Pox: Pathogenesis and clinical findings Exposure to
individual with
VZV infection
VZV enters
respiratory tract
& conjunctiva
VZV enters
dendritic
cells (DC)
DCs travel to
regional
lymph nodes
Primary viremia
Authors:
Morgan Garland,
Sean Spence
VZV replicates
VZV proteins ↑ major
↓ MHCI
Reviewers:
in nasopharynx
histocompatibility
expression
Yan Yu, Danny Guo,
& CD4+ T cells
complex class I
on CD4+ T
Emily J. Doucette,
(MHCI) degradation
cell surface
Laurie Parsons*,
James D. Kellner*
*MD at time of publication
Acute phase
VZV evades the immune system
VZV travels through the blood in CD4+ T cells
Immune cells release
cytokines to fight the
virus (stronger in adults)
Fluid & cellular debris
accumulate between
epidermal layers
Generalized pruritic (itchy) vesicular
rash at different stages of development
(macule, papule, vesicle)
Immune system activates
& releases inflammatory
compounds
VZV replicates in liver,
spleen & lymph nodes
Erythema
(redness) & pain
Scratching lesions
introduces bacteria
Fibroblasts lay down
collagen to heal skin lesions
Resets thermostat
in hypothalamus
Systemic
response
Tissue damage
in oropharynx
Direct
inflammation in
neural tissues
Cerebellar ataxia
(more common
in children)
Bacterial superinfection from S.
aureus & Streptococcus pyogenes
(more common in children)
Scarring
Fever
(prodromal)
Malaise
(prodromal)
Sore throat
(prodromal)
Encephalitis (more
common in adults)
Abscess
Cellulitis
Toxic shock syndrome (TSS)
Secondary viremia
Immune cells release cytokines to
fight infection (stronger in adults)
Inflammation & fluid
build up in lungs
VZV travels through the blood in CD4+ T
cells expressing skin homing receptors
Fluid & cellular
debris accumulate
in damaged alveoli Lungs more susceptible to
secondary bacterial infection
Pneumonia** (more
common in adults)
VZV colonizes
keratinocytes
over several days
VZV colonizes
neurons & glial
cells in the brain
VZV colonizes
alveolar
epithelial cells
Resolution & latency
VZV tracks along
neurons from sites of
infection to dorsal root
ganglia & cranial nerves
↓ MHCI expression
in sensory ganglia
Adaptive
immune system
controls viremia
↓ Viral gene expression
Resolution of VZV
infection &
development of
lifelong immunity
Direct viral toxicity (DNA damage, mitochondrial
dysfunction, & protein misfolds in infected cells)
Apoptosis & dysfunction of infected cells
Latent
infection
Stress or immune suppression may
precipitate reactivation of VZV later in life Shingles**
**See corresponding Calgary Guide slide
Pathophysiology Mechanism
Published Nov 12, 2012; updated Jun 22, 2025 on www.thecalgaryguide.com
Legend: Sign/Symptom/Lab Finding Complications")
GLP-1 Receptor Agonists
 Receptor Agonists: Mechanism of action and side effects
Management of Type
2 Diabetes Mellitus**
Weight
management
Authors: Saania Tariq, Trevor Low
Reviewers: Rafael Sanguinetti
Luiza Radu, Emily J. Doucette
Activated GLP-1 receptors in
Activated GLP-1
Hanan Bassyouni*
* MD at time of publication
the myenteric plexus inhibit
receptors stimulate
gastric motor neurons
pancreatic islet cells
GLP-1 Receptor Agonists
Synthetic analogs (e.g., Semaglutide) of GLP-1 peptide hormone endogenously
produced by intestinal enteroendocrine L cells directly activate GLP-1 receptors
to modulate food intake & glucose metabolism.
↓ Gastric
motility
↑ Pyloric sphincter
tone via disinhibition
↓ Glucagon release
from α-cells
↑ Regeneration,
↑ growth &
↓ apoptosis of β-cells
Slowed gastric emptying
Intramuscular injection or oral ingestion
prolongs GLP-1 receptor activation
↑ β-cell function & glucose-
dependent insulin production
GLP-1 receptor activation on neurons in
the arcuate nucleus of the hypothalamus
modulates appetite & satiety
Activation of GLP-1 receptors
in the gastrointestinal system
regulates metabolism
Prolonged gastric
distension & stasis
activates
chemoreceptors &
mechanoreceptors
Delayed digestion of
gastric contents ↓
glucose spikes after
meals
↑ Glycogen synthesis &
glucose uptake in liver &
skeletal muscles
Neuropeptide Y &
agouti-related protein
expressing neurons are
hyperpolarized
Pro-opiomelanocortin
expressing neurons
become ↑ excitable
Activated GLP-1 receptors
signal to hypothalamus
via vagus nerve afferents
↑ Vagal afferent
signaling to area
postrema
(vomiting centre)
Nausea &
vomiting
↓ Release of
neuropeptide Y &
agouti-related protein
↑ Anorexigenic
signalling
↑ Parasympathetic
output via dorsal motor
nucleus of vagus nerve
GLP-1 agonists directly
activate receptors in
area postrema
↑ Satiety (feeling
of fullness)
↓ Adiposity
(weight loss)
↓ Orexigenic drive
↓ Inflammatory stress in
pancreatic β-cells
↓ Appetite
↓ Caloric intake
↓ Inflammatory
cytokines & lipotoxicity
↓ Systemic
inflammation
**See corresponding Calgary Guide slide
Legend: Sign/Symptom/Lab Finding Pathophysiology Mechanism
Complications
↓ Hepatic glucose
production
(glycogenolysis)
↓ Fasting & postprandial
blood glucose
↓ Red blood cell (RBC)
glycation (glucose
irreversibly binds to
hemoglobin in RBCs)
↓ Production
of glycated
proteins and
lipids
↓ % of glycated
RBCs over time
↑ β-cell function
improves insulin
secretion
↓ Vascular
endothelial
damage and
atherosclerosis
↓ Endothelial
damage to
glomeruli
↑ Insulin sensitivity in
skeletal muscle & liver
↓ Hemoglobin
A1c
↓ Risk of
cardiovascular
complications
↓ Risk of renal
complications
Published Jun 22, 2025 on www.thecalgaryguide.com")
Fluoroquinolones

Bactericidal antibiotics that inhibit bacterial DNA synthesis. Coverage depends on specific agent but includes gram-negatives
(e.g., E. coli, H. influenzae), atypicals (e.g., Legionella, Mycoplasma), anaerobes & selected gram-positives (e.g., Streptococcus)
High affinity for
connective tissue
FQ metabolism forms
toxic metabolites &
free radicals
↑ Concentration
of FQ in tendons &
connective tissue
Toxic metabolites &
free radicals
accumulate in nerve
tissue (mechanism
poorly understood)
↑ Metalloprotease
(MMP) activity &
magnesium chelation
(mechanism poorly
understood)
↑ Inflammation
& oxidative
stress causes
neuronal
dysfunction &
cell death
Tendinopathy &
tendon rupture
Peripheral neuropathy
(pain, numbness,
weakness, tingling)
Fluoroquinolones
can cross placental
membrane
Collagenolytic effect of ↑
MMP-9 & MMP-2 disrupts
aortic wall (mechanism
poorly understood)
Cartilage & bone
toxicity in fetus
Aortic dissection** &
aortic aneurysm**
**See corresponding Calgary Guide slide
Pathophysiology FQ targets bacterial
enzymes DNA gyrase
& topoisomerase IV
Structural similarity to
gamma-aminobutyric
acid (GABA)
Broad-spectrum
antibiotic
FQ-DNA-enzyme
complexes form &
become
irreversibly bound
DNA replication
fork movement
is blocked
causing DNA
strand breakage
DNA replication
inhibition &
bacterial cell
death
Bacterial clearance on
microbiological culture
Legend: Possible GABA
antagonist
(mechanism
unclear)
GABA binds less
effectively to
neurons in
central nervous
system (CNS) &
exerts a weaker
inhibitory effect
↑ Neuronal
excitability & CNS
stimulation
↓ Microbiome
diversity
↑ Antibiotic-
resistant strains
↑ Pathogenic
& ↓ protective
bacteria (gut
dysbiosis)
Gastritis
(inflammation
of the stomach
lining)
Clostridium
difficile
overgrowth
& infection
↓ Seizure threshold
Headache
Insomnia
Confusion
Mechanism
Sign/Symptom/Lab Finding Published Jun 22, 2025 on www.thecalgaryguide.com
QT interval
prolongation
Torsades de
Pointes**
Blockage of ATP-
sensitive potassium
channels in
pancreatic β cells
↑ β-cell
membrane
depolarization
↑ Insulin
secretion
Hypoglycemia**
Authors:
Ben Jackson
Reviewers:
Rafael Sanguinetti
Luiza Radu
Emily J. Doucette
Meagan Deviaene*
* MD at time of publication
Complications")
Polisitemia Sekunder Patogenesis

Polisitemia Vera Patogenesis dan Temuan Klinis
: Patogenesis dan Temuan Klinis")
Polisitemia Vera Komplikasi
: Komplikasi")
Traitement du choc

Choc cardiogenique

Choc distributif

Hyperthermie maligne

مرض القلب الإقفاري )م.ق.إ
م.ق.إ")
Unconjugated Hyperbilirubinemia
 factor
incompatibility
Maternal sensitization
against Rh-positive fetal cells
Maternal antibodies against Rh
factor attack fetal RBCs
ABO incompatibility
Native maternal antibodies against non-
native blood types attack fetal RBCs
Sepsis** Widespread systemic inflammation Cytokines & complement factors damage RBC membranes
Disseminated intravascular
coagulation** (clotting
proteins become overactive)
Clots form in
systemic circulation
Small clots shear & damage
RBCs in circulation
Red blood cell
(RBC) enzyme
defects
Glucose-6 phosphatase
dehydrogenase (G6PD)
deficiency**
G6PD protects RBCs against
oxidative damage
↑ RBC sensitivity to oxidative
stress (during acute stressors)
Pyruvate kinase
deficiency (PKD)
Abnormal glycolysis & cellular
energy production
↓ RBCs life spans
RBC membrane
defects
Hereditary spherocytosis
RBCs are abnormally round (spherocytes)
RBC
membranes
easily damaged
in circulation
Bilverdin reductase
Hereditary elliptocytosis
RBCs are abnormally elongated or oval
converts bilverdin to
Sickle cell
Abnormal hemoglobin (HbS)
RBCs become abnormally
unconjugated bilirubin
disease**
polymerizes under ↓ O2
sickle shaped
Hemoglobinopathies
Thalassemia's**
Defective hemoglobin
chains in RBCs
Irregularly
shaped RBCs
trapped in
spleen
RBC
sequestration
↑ Production of RBCs (Polycythemia**)
Accumulation of blood (i.e.
Cephalohematoma, hemorrhage)
↑ RBC load &
turnover
Causes of ↓
hepatocellular
bilirubin clearance
Genetic defects in
uridine diphosphate
glucuronosyltransfe
rase (UGT) enzyme
(conjugates
bilirubin in liver)
Gilbert syndrome
Unconjugated bilirubin
↓ UGT production
remains insoluble & cannot
Crigler–Najjar
be excreted in bile
syndrome Type II
Crigler–Najjar
syndrome Type I
No UGT production
Breast milk jaundice Breast milk contains β-glucuronidase
Causes of ↑
entero-hepatic
bilirubin circulation
Intestinal obstruction
Obstruction blocks bile flow from liver to intestines Breastfeeding
jaundice
Inadequate milk intake (volume
depletion/dehydration)
↑ Reabsorption of bilirubin
in the intestines
**See corresponding Calgary Guide slide
Legend: Macrophages engulf
old or damaged RBCs
in spleen & liver
↑ Hemolysis (RBC
breakdown)
RBCs release cellular
contents including heme
(from hemoglobin)
Heme oxygenase
converts heme to
bilverdin
↑ Unconjugated
(indirect) bilirubin
accumulation in blood
↑ Total serum
bilirubin levels
Excess bilirubin
deposition into elastin-
rich tissues (eg. skin,
sclera) due to ↑
affinity for elastin
Pathologic neonatal jaundice
(yellow discoloration of skin
within first 24 hours of life)
Pathophysiology Mechanism
Sign/Symptom/Lab Finding Complications
Published July 13, 2025 on www.thecalgaryguide.com
Authors:
Merry Faye Graff
Khushi Arora
Reviewers:
Annie Pham
Emily J. Doucette
Danielle Nelson*
* MD at time of publication
Hemolytic
anemia**
↓ Bilirubin
conjugation
β-glucuronidase deconjugates bilirubin
Bilirubin builds up
in hepatic system
Unconjugated
bilirubin crosses
blood-brain barrier
Bilirubin-induced
neurologic
dysfunction (BIND)
Kernicterus (bilirubin-
induced neurological
damage)
Scleral icterus
(yellowing of
sclera in eyes)")